Chapter 42 Disorders of consciousness
NEUROANATOMY AND PHYSIOLOGY OF WAKEFULNESS
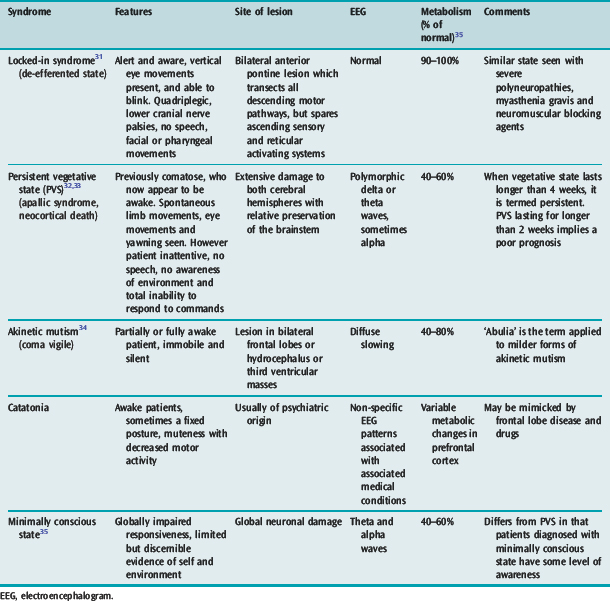
A normal level of consciousness depends on the interaction between the cerebral hemispheres and the rostral reticular activating system (RAS) located in the upper brainstem. Although the RAS is a diffuse projection, the areas of RAS of particular importance to the maintenance of consciousness are those located between the rostral pons and the diencephalon. In contrast, consciousness is not focally represented in any of the cerebral hemispheres and is in many ways related to the mass of functioning cortex. Thus anatomical bilateral hemispheric lesions or brainstem lesions may result in an altered conscious state.1 Large unilateral hemispheric lesions may produce impairment of consciousness by compression of the upper brainstem. In addition metabolic processes may result in coma from interruption of energy substrate delivery or alteration of neuronal excitability. Disorders of consciousness are characterised by an alteration in either the level or content of consciousness (Table 42.1). The last few conditions described in Table 42.1 are a frequent source of confusion and require further discussion (Table 42.2). These neurological states are seen more frequently in modern-day clinical practice partly because of the advances in therapy of severe brain injury and intensive care which have led to the survival of many patients who would otherwise have died.
| Consciousness | An awake individual demonstrates full awareness of self and environment |
| Confusion | Inability to think with customary speed and clarity, associated with inattentiveness, reduced awareness and disorientation |
| Delirium | Confusion with agitation and hallucination |
| Stupor | Unresponsiveness with arousal only by deep and repeated stimuli |
| Coma | Unarousable unresponsiveness |
| Locked-in syndrome | Total paralysis below third cranial nerve nuclei; normal or impaired mental function |
| Persistent vegetative state | Prolonged coma > 1 month, some preservation of brainstem and motor reflexes |
| Akinetic mutism | Prolonged coma with apparent alertness and flaccid motor tone |
| Minimally conscious state | Preserved wakefulness, awareness and brainstem reflexes, but poorly responsive |

CLINICAL EXAMINATION OF THE COMATOSE PATIENT
LEVEL OF CONSCIOUSNESS
This is assessed by the Glasgow Coma Scale (GCS),2 which takes into account a patient’s response to command and physical stimuli. The GCS (Table 42.4) which was originally developed to grade the severity of head injury and prognosticate outcome, has now been extended for all causes of impaired consciousness and coma. Although it is a simple clinical score, easily performed by both medical and nursing staff by the bedside, there are a number of caveats:
| Eye opening | Points |
|---|---|
| Spontaneous | 4 |
| To speech | 3 |
| To pain | 2 |
| Nil | 1 |
| Best verbal response | |
| Oriented | 5 |
| Confused | 4 |
| Inappropriate | 3 |
| Incomprehensible | 2 |
| Nil | 1 |
| Intubated | T |
| Best motor response | |
| Obeys commands | 6 |
| Localises to pain | 5 |
| Withdraws to pain | 4 |
| Abnormal flexion | 3 |
| Extensor response | 2 |
| Nil | 1 |
PUPILLARY RESPONSES IN COMA3
The presence of normal pupils (2–5 mm and equal in size and demonstrating both direct and consensual light reflexes) confirms the integrity of the pupillary pathway (retina, optic nerve, optic chiasma and tracts, midbrain and third cranial nerve nuclei and nerves). The size of the pupil is a balance between the opposing influences of both sympathetic (causing dilatation) and parasympathetic (causing constriction) systems. Pupillary abnormalities have localising and diagnostic value in clinical neurology (Table 42.5). When the pupils are miosed, the light reaction is difficult to appreciate and may require a magnifying glass.
| Abnormality | Cause | Neuroanatomical basis |
|---|---|---|
| Miosis (< 2 mm in size) | ||
| Unilateral | Horner’s syndrome | Sympathetic paralysis |
| Local pathology | Trauma to sympathetics | |
| Bilateral | Pontine lesions | |
| Thalamic haemorrhage | Sympathetic paralysis | |
| Metabolic encephalopathy | ||
| Drug ingestion | ||
| Organophosphate | Cholinesterase inhibition | |
| Barbiturate | ||
| Narcotics | Central effect | |
| Mydriasis (> 5 mm in size) | ||
| Unilateral fixed pupil | Midbrain lesion | Third-nerve damage |
| Uncal herniation | Stretch of third nerve against the petroclinoid ligament | |
| Bilateral fixed pupils | Massive midbrain haemorrhage | Bilateral third-nerve damage |
| Hypoxic cerebral injury Drugs | Mesencephalic damage | |
| Atropine | Paralysis of parasympathetics | |
| Tricyclics | Prevent local reuptake of catecholamines by nerve endings | |
| Sympathomimetics | Stimulation of sympathetics | |
OPHTHALMOSCOPY IN COMA
The pupils should never be dilated pharmacologically without prior documentation of the pupillary size and the light reflex. The presence of papilloedema suggests the presence of intracranial hypertension, but is frequently absent when the lesion is acute. Subhyaloid and vitreous haemorrhages are seen in patients with subarachnoid haemorrhage.4
EYE MOVEMENTS IN COMA5
The position and movements of the eyes are observed at rest. The presence of spontaneous roving eye movements excludes brainstem pathology as a cause of coma. In a paralytic frontal-lobe pathology, the eyes will deviate towards the side of the lesion, whilst in pontine pathologies, the eyes will deviate away from the side of the lesion. Ocular bobbing, an intermittent downward jerking eye movement, is seen in pontine lesions due to loss of horizontal gaze and unopposed midbrain controlled vertical gaze activity.6 Skew deviation (vertical separation of the ocular axes) occurs with pontine and cerebellar disorders.7
LIMB MOVEMENTS AND POSTURAL CHANGES IN COMA
Restlessness, crossing of legs and spontaneous coughing, yawning, swallowing and localising movements suggest only a mild depression of the conscious state. Choreoathetotic or ballistic movements suggest a basal ganglion lesion. Myoclonic movements indicate a metabolic disorder, usually of postanoxic origin. Asterixis is seen with metabolic encephalopathies. Hiccup is a non-specific sign and does not have any localising value.
Decerebrate rigidity is characterised by stiff extension of the limbs, internal rotation of the arms and plantar flexion of the ankles. With severe rigidity, opisthotonos and jaw clenching may be observed. These movements may be unilateral or bilateral, and spontaneous or in response to a noxious stimulus. Whereas animal studies suggest that the lesion is usually in the midbrain or caudal diencephalon (leading to exaggeration of antigravity reflexes), in humans such posturing may be seen in a variety of disease states: midbrain lesion, certain metabolic disorders such as hypoglycaemia, anoxia, hepatic coma and in drug intoxication.Decorticate posturing is characterised by flexion of elbows and wrists and extension of the lower limbs. The lesion is usually above the midbrain in the cerebral white matter.
RESPIRATORY SYSTEM8
Abnormal respiratory rate and patterns have been described in coma, but their precise localising value is uncertain. As a general rule, at lighter levels of impaired consciousness tachypnoea predominates, whereas respiratory depression increases with the depth of coma. Some of the commonly observed respiratory abnormalities are summarised in Table 42.6. Respiratory failure in comatose patients may result from hypoventilation, aspiration pneumonia and neurogenic pulmonary oedema, a sympathetic nervous system-mediated syndrome seen in acute brain injury.
Table 42.6 Disorders of respiratory rate and pattern in coma
| Abnormality | Significance |
|---|---|
| Bradypnoea | Drug-induced coma, hypothyroid coma |
| Tachypnoea | Central neurogenic hyperventilation (midbrain lesion), |
| metabolic encephalopathy | |
| Cheyne–Stokes respiration | Deep cerebral lesions, metabolic encephalopathy (hyperpnoea alternating regularly with apnoea) |
| Apneustic breathing (an inspiratory pause) | Pontine lesions |
| Ataxic breathing | Medullary lesions |
| (Ataxic breathing normally progresses to agonal gasps and terminal apnoea) | |
MANAGEMENT OF THE COMATOSE PATIENT
EMERGENT THERAPEUTIC MEASURES
ROUTINE INVESTIGATIONS
Measurements of serum glucose, electrolytes, arterial blood gases, liver and renal function tests, osmolality, blood count and blood film are part of the routine investigations. When drug overdose is suspected, a toxicology screen for alcohol, paracetamol, salicylates, benzodiazepines and tricyclic antidepressants should be performed. A sample of serum should be stored for later analysis for uncommon drug ingestions.
NEUROIMAGING
COMPUTED TOMOGRAPHY (CT) SCAN
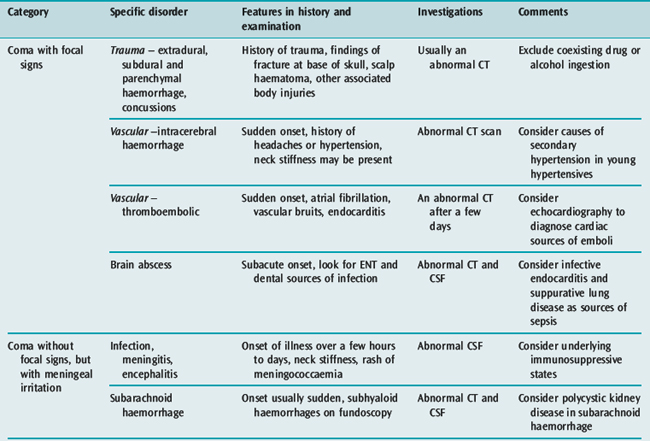
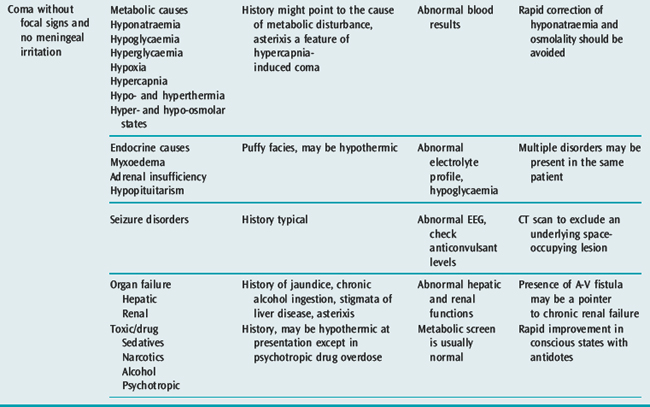
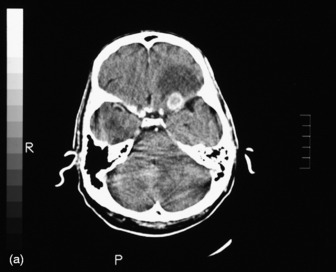
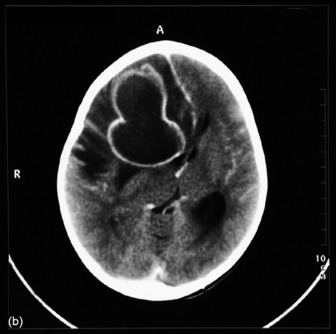
The most commonly used radiological investigation for evaluation of the comatose patient is CT scan of the brain. This is useful for diagnosing central nervous system trauma, subarachnoid and intracerebral haemorrhage, haemorrhagic and non-haemorrhagic strokes, cerebral oedema, hydrocephalus and the presence of a space-occupying lesion (SOL) (Figures 42.1–42.10). Frequently a CT is performed prior to a lumbar puncture (LP) to exclude rather than confirm the presence of severe cerebral oedema or an SOL. Its other advantages include lower cost, easy availability, short examination time and safety in the presence of pacemakers, surgical clips and other ferromagnetic substances. The advent of helical CT whereby multiple images are possible has reduced scanning times and is suitable for the uncooperative patient. Limitations of a CT scan include:
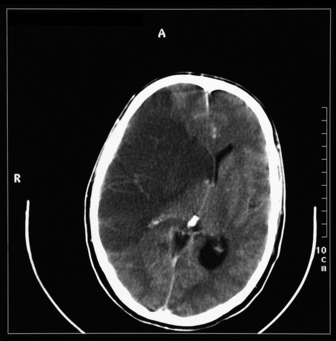
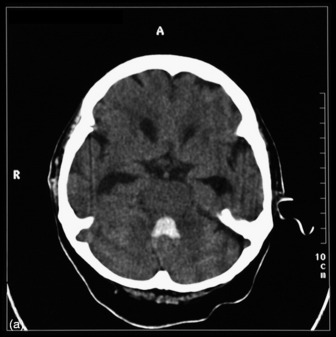
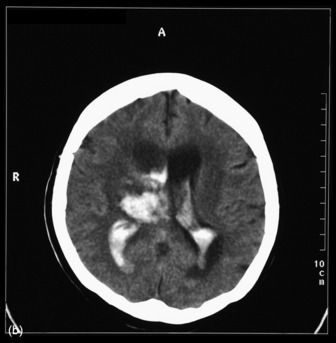
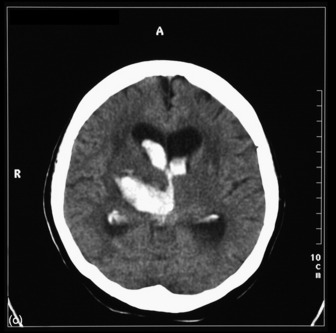
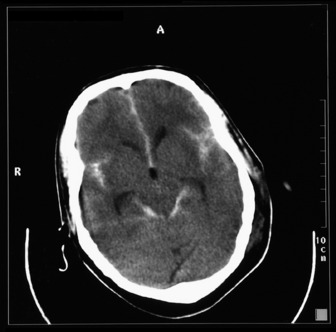
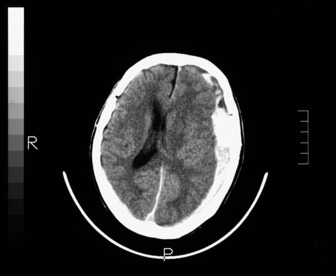
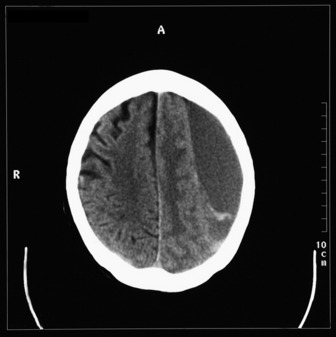
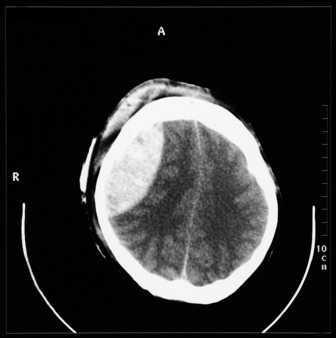
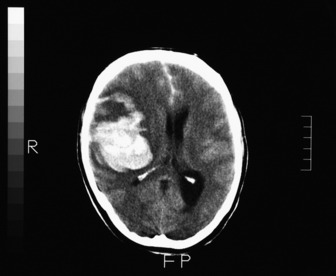
Figure 42.7 Right frontoparietal bleed. Note oedema, loss of lateral ventricle and minor midline shift.
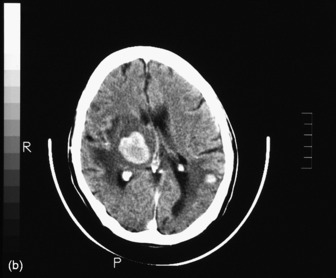
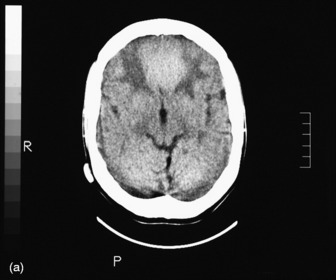
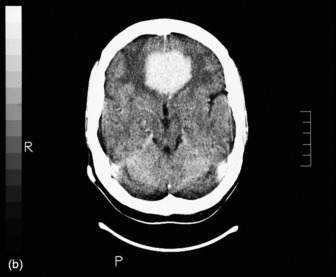
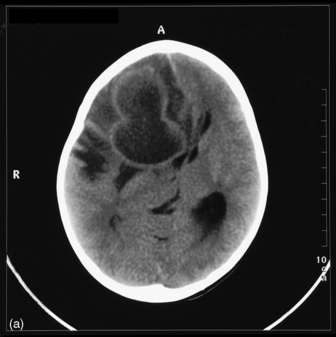
MAGNETIC RESONANCE IMAGING (MRI)
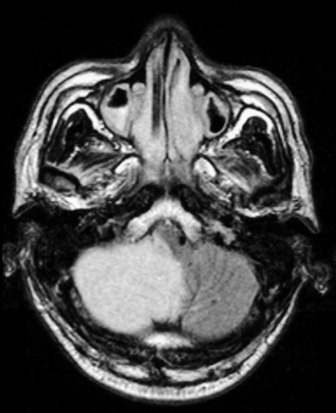
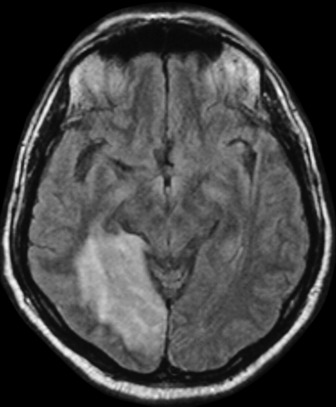
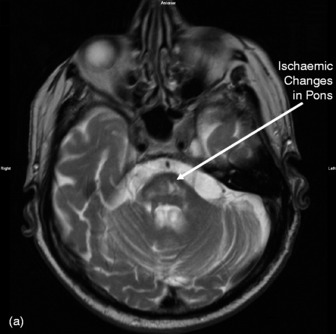
MRI scans provide superior contrast and resolution of the grey and white matter as compared to CT scans, thus facilitating easy identification of the deep nuclear structures within the brain. MRI is more sensitive than CT for the detection of acute ischaemia, diffuse axonal injury and cerebral oedema, tumour and abscess. Brainstem and posterior fossa structures are better visualised (Figures 42.11–42.13). It can also show vasculature (see Figure 42.13b and c). The other advantage of MRI is the use of non-ionising energy. The use of gadolinium, a paramagnetic agent, as a contrast agent permits sharp definition of lesions. MR coupled with angiography (MRA) may enable diagnosis of vascular lesions. MRI is however limited by:
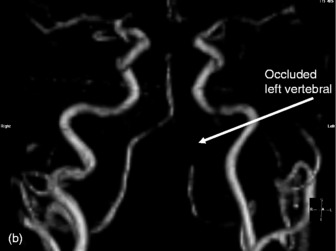
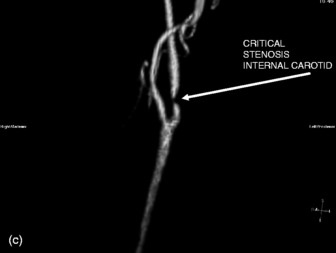
PET AND SPECT SCANS
Newer nuclear medicine scans such as single-photon emission with computed tomography (SPECT) and positron emission tomography (PET) are useful for the assessment of cerebral blood flow and oxygenation and in the prognostication of neurotrauma, but have little role to play in the management of acute disorders of consciousness. In PET scans, positron-emitting isotopes such as 11C, 18F and 15O are incorporated into biologically active compounds such as deoxyglucxose or fluorodeoxyglucose which are metabolised in the body. By determining the concentration of the various tracers in the brain and constructing tomographic images, cerebral blood flow and metabolism can be measured by PET scanning.
SPECT scans use iodine-containing isotopes incorporated into biologically active compounds and, like PET scans, their cranial distribution is determined after a dose of tracer. Information on cerebral blood flow and metabolism can be obtained from SPECT scans. The advantage of a PET scan is that it does not require a cyclotron for the generation of isotopes. Despite their many advantages, both these technologies continue to be research tools and are not routinely available in many medical centres.
LUMBAR PUNCTURE14
Cerebrospinal fluid is most commonly obtained by means of an LP. This should be performed after ensuring that raised intracranial pressure has been excluded clinically or radiologically. The major use of an LP is to diagnose an intracranial infection and to detect abnormal cytology in cases of suspected malignant meningeal infiltration. The advent of CT scans has diminished the role of LP in the diagnosis of subarachnoid haemorrhage. Some of the commonly reported complications post-LP include postpuncture headache (12–39%) and traumatic tap (15–20%). Brain herniation is a rare potential complication seen with conditions associated with raised intracranial pressure due to an SOL.
ELECTROENCEPHALOGRAM (EEG) IN COMA15,16
The usefulness of EEG in coma is summarised in Table 42.7. Continuous EEG monitoring in the intensive care unit (ICU) has been reported to be useful in the identification of acute cerebral ischaemia and non-convulsive seizures.
| Identification of non-convulsive status epilepticus |
| Diagnosis of hepatic encephalopathy |
| Presence of paroxysmal triphasic waves |
| Assessing severity of hypoxic encephalopathy |
| Presence of theta activity |
| Diffuse slowing |
| Burst suppression (seen with more severe forms) |
| Alpha coma (seen with more severe forms) |
| Herpes encephalitis |
| Periodic sharp spikes |
EVOKED POTENTIALS
Visual, brainstem and somatosensory evoked potentials test the integrity of neuroanatomical pathways within the brain and the spinal cord. They may be used in the diagnosis of blindness in comatose patients and in the assessment of locked-in states. There are data to suggest that they have better prognostic value than clinical judgement in patients with anoxic coma.17
CARE OF THE COMATOSE PATIENT
AIRWAY
As a general rule, patients presenting with medical causes of coma may be nursed on their side (in the coma position) if the airway is adequate. However, all traumatised patients should be assumed to have a potential cervical spine injury and must be nursed with the cervical spine in the neutral position and/or with a rigid collar until an injury is excluded by definitive radiological views. All patients with disordered consciousness must receive supplemental oxygen.
SPECIFIC TREATMENT
This will depend on the underlying aetiology of the coma and is discussed in the relevant chapters. Avoidance of secondary insults is of paramount importance in the management of these patients.19
ANOXIC COMA/ENCEPHALOPATHY
Cardiac arrest is the third leading cause of coma resulting in ICU admission after trauma and drug overdose. The symptomatology and clinical outcome of patients with anoxic brain damage depend on the severity and duration of oxygen deprivation to the brain. A number of criteria have been developed to prognosticate outcome in anoxic coma. Although a number of laboratory and imaging criteria contribute to the prognostic assessment, clinical signs still have major prognostic impact. The important clinical predictors of outcome are listed in Table 42.8. However there are data to suggest that electrophysiological studies using evoked potential have far greater prognostic accuracy compared to clinical assessment.20
Table 42.8 Clinical and laboratory predictors of unfavourable prognosis in anoxic coma17,36–38
| Clinical predictor | Unfavourable prognosis |
|---|---|
| Duration of anoxia | 8–10 min |
| (time interval between collapse and initiation of CPR) | |
| Duration of CPR | > 30 min |
| (time interval between initiation of CPR and ROSC) | |
| Duration of postanoxic coma | > 72 hours |
| Pupillary reaction | Absent on day 3 |
| Motor response to pain (absent = a motor response worse than withdrawal) | Absent on day 3 |
| Roving spontaneous eye movements | Absent on day 1 |
| Elevated neuron specific enolase | > 33 μg/l |
| SSEP recording | Absent N20 |
CPR, cardiopulmonary resuscitation; ROSC, restoration of spontaneous circulation; SSEP, somatosensory evoked potential.
THE CONFUSED/ENCEPHALOPATHIC PATIENT IN THE ICU
‘Encephalopathy’ is a term used to describe the alteration in the level or content of consciousness due to a process extrinsic to the brain. Metabolic encephalopathy, particularly of septic aetiology, is the most common cause of altered mental status in the ICU setting.21 A number of processes can lead to metabolic encephalopathy (Table 42.9). A number of features in the history and examination help to differentiate metabolic from structural causes of altered conscious states (Table 42.10).
| Hepatic failure |
| Renal failure |
| Respiratory failure |
| Sepsis |
| Electrolyte abnormalities: hyponatraemia, hypernatraemia, hypercalcaemia |
| Hypoglycaemia and hyperglycaemia |
| Acute pancreatitis |
| Endocrine – addisonian crisis, myxoedema coma, thyroid storm |
| Drug withdrawal – benzodiazepine, opiates |
| Hyperthermia |
| Toxins: alcohols, glycols, tricyclic antidepressants |
| Intensive care unit syndrome |
| D-lactic acidosis |
Table 42.10 Distinguishing features of structural and metabolic encephalopathy41
| Feature | Structural | Metabolic |
|---|---|---|
| State of consciousness | Usually fixed level of depressed conscious state, may deteriorate progressively | Milder alteration of conscious state, waxing and waning of altered sensorium |
| Fundoscospy | May be abnormal | Usually normal |
| Pupils | May be abnormal, either in size or response to light | Usually preserved light response (although pupil shape and reactivity affected in certain overdoses: see above) |
| Eye movements | May be affected | Usually preserved |
| Motor findings | Asymmetrical involvement | Abnormalities usually symmetrical |
| Involuntary movements | Not common | Asterixis, tremor, myoclonus frequently seen |
Sepsis-associated encephalopathy (SAE) has been reported to occur in 8–80% of patients with sepsis.22 The criteria to diagnose SAE include presence of impaired mental function, evidence of an extracranial infection and absence of other obvious aetiologies for the altered conscious state. Although the precise mechanism of damage to the brain has not been delineated, the pathogenesis of the encephalopathy is thought to be multifactorial: alteration in cerebral blood flow induced by mediators of inflammation, generation of free radicals by activated leukocytes resulting in erythrocyte sludging in the microcirculation, breakdown of the blood–brain barrier resulting in cerebral oedema, reduced brain oxygen consumption induced by endotoxin and cytokines, neuronal degeneration and increased neuronal apoptosis, increases in aromatic amino acids resulting in altered neurotransmitter function and increased gamma-aminobutyric acid (GABA)-mediated neurotransmission leading to general inhibition of the central nervous system. Hypotension may contribute to the encephalopathy. The asterixis, tremor and myoclonus – features of other metabolic encephalopathies – are uncommon in sepsis. The presence of lateralising signs are extremely rare in SAE and warrant exclusion of other causes, such as stroke. The mortality of patients with SAE is higher than in those with sepsis without encephalopathy.23 Therapy is largely directed at the underlying septic process.
ICU ENCEPHALOPATHY OR ICU SYNDROME24,25
This is a term used to describe behavioural disorders which develop in patients 5–7 days after admission to intensive care. Clinically this may present as agitation, restlessness and frank delirium. The causes are multifactorial: prolonged ventilation, sleep deprivation,26,27 distortion of perception with loss of day–night cycles, immobilisation, noisy environment and monotony. Coupled with administration of multiple sedatives and neurological consequences of the underlying disease, these factors can precipitate psychotic behaviour in the ICU. It is important to bear in mind that this is a diagnosis of exclusion and that all other reversible causes are looked for (seeTable 42.7) before this diagnostic label is applied.
PROGNOSIS IN COMA
Drug-induced comas usually have a good prognosis unless hypoxia and hypotension have resulted in severe secondary insults. Coma following head injury has a statistically better outcome compared to non-traumatic coma (coma occurring during the course of a medical illness). In non-traumatic coma lasting for 6 hours or greater, only 15% of the patients make a meaningful recovery to be able to return to their premorbid state of health.28 The prognosis following anoxic coma has been described in a separate section. Within the non-traumatic coma category, coma resulting from infection, metabolic causes and multiple-organ dysfunction syndrome has a better outcome compared to anoxic coma.29 A number of outcome scales have been developed to assess neurological recovery following brain injury.30 These include the Barthel index, Rankin scale and the Glasgow Outcome Scale (GOS). The GOS is widely used to assess recovery after traumatic brain injury. It has five broad categories: 1 = good recovery; 2 = moderate disability; 3 = severe disability; 4 = persistent vegetative state; and 5 = death. It is simple, easy to administer and has been reported to have good interrater agreement.
1 Ropper A, Martin J. Coma and other disorders of consciousness. In: Isselbacher K, editor. Harrison’s Principles of Internal Medicine. McGraw Hill; 1994:146-152.
2 Teasdale G, Jennett B. Assessment of coma and impaired consciousness. A practical scale. Lancet. 1974;2:81-84.
3 Adams R, Victor M, Ropper A. Coma and related disorders of consciousness. In: Principles of Neurology. McGraw Hill; 1997:344-366.
4 Keane JR. Retinal hemorrhages. Its significance in 100 patients with acute encephalopathy of unknown cause. Arch Neurol. 1979;36:691-694.
5 Keane J. Eye movements in coma. In: Jakbiec AA, editor. Principles and Practice of Ophthalmology. WB Saunders; 2000:4075-4083.
6 Fisher C. Ocular bobbing. Arch Neurol. 1964;11:543.
7 Keane JR. Ocular skew deviation. Analysis of 100 cases. Arch Neurol. 1975;32:185-190.
8 North JB, Jennett S. Abnormal breathing patterns associated with acute brain damage. Arch Neurol. 1974;31:338-344.
9 Kernohan J, Woltman H. Incisura of the crus due to contralateral brain tumour. Arch Neurol Psych. 1929;21:274.
10 McNealy D, Plum F. Brainstem dysfunction with supratentorial mass lesions. Arch Neurol. 1962;7:10.
11 De Salles AA, Muizelaar JP, Young HF. Hyperglycemia, cerebrospinal fluid lactic acidosis, and cerebral blood flow in severely head-injured patients. Neurosurgery. 1987;21:45-50.
12 Penney DG. Hyperglycemia exacerbates brain damage in acute severe carbon monoxide poisoning. Med Hypotheses. 1988;27:241-244.
13 Van Den Berk G, Tonino S, De Fjtjer C. Bench to bedside review: preventive measures for contrast induced nephropathy in critically ill patients. Crit Care. 2005;9:361-370.
14 Venkatesh B, Scott P, Ziegenfuss M. Cerebrospinal fluid in critical illness. Crit Care Resuscit. 2000;2:43-55.
15 Bauer G. Coma and brain death. Niedermeyer E, Da Silva F, editors. Electroencephalogrpahy: Basic Principles, Clinical Applications and Related Fields. 1999, 459-475.
16 Nuwer MR. Continuous EEG monitoring in the intensive care unit. Electroencephalogr Clin Neurophysiol. 1999;50(Suppl):150-155.
17 Zandbergen EG, de Haan RJ, Stoutenbeek CP, et al. Systematic review of early prediction of poor outcome in anoxic-ischaemic coma. Lancet. 1998;352:1808-1812.
18 Berger RP. The use of serum biomarkers to predict outcome after traumatic brain injury in adults and children. J Head Trauma Rehabil. 2006;21:315-333.
19 Chesnut RM, Marshall LF, Klauber MR, et al. The role of secondary brain injury in determining outcome from severe head injury. J Trauma. 1993;34:216-222.
20 Kaplan PW. Electrophysiological prognostication and brain injury from cardiac arrest. Semin Neurol. 2006;26:403-412.
21 Stevens RD, Pronovost PJ. The spectrum of encephalopathy in critical illness. Semin Neurol. 2006;26:440-451.
22 Wilson JX, Young GB. Progress in clinical neurosciences: sepsis associated encephalopathy: evolving concepts. Can J Neurol Sci. 2003;30:98-105.
23 Eidelman LA, Putterman D, Putterman C, et al. The spectrum of septic encephalopthy. Definiitons, etiologies and mortalities. JAMA. 1996;275:470-473.
24 McGuire BE, Basten CJ, Ryan CJ, et al. Intensive care unit syndrome: a dangerous misnomer. Arch Intern Med. 2000;160:906-909.
25 Pun BT, Ely EW. The importance of diagnosing and managing ICU delirium. Chest. 2007;132:624-636.
26 Granberg Axell AI, Malmross CW, Bergbom IL, et al. Intensive care unit syndrome/delirium is associated with anemia, drug therapy and duration of ventilation. Acta Anaesthesiol Scand. 2002;46:726-731.
27 Shilo L, Dagan Y, Smorjik Y, et al. Patients in the intensive care unit suffer from severe lack of sleep associated with loss of normal melatonin secretion pattern. Am J Med Sci. 1999;317:278-281.
28 Plum F, Levy DE. Outcome from severe neurological illness; should it influence medical decisions? Ciba Found Symp. 1979;69:267-277.
29 Levy DE, Bates D, Caronna JJ, et al. Prognosis in nontraumatic coma. Ann Intern Med. 1981;94:293-301.
30 Kasner SE. Clinical interpetation and use of stroke scales. Lancet Neurol. 2006;5:603-612.
31 Nordgren RE, Markesbery WR, Fukuda K, et al. Seven cases of cerebromedullospinal disconnection: the ‘locked-in’ syndrome. Neurology. 1971;21:1140-1148.
32 Multi Society Task Force on PVS. Medical aspects of the persistent vegetative state (1). N Engl J Med. 1994;330:1499-1508.
33 Jennett B, Plum F. Persistent vegetative state after brain damage. A syndrome in search of a name. Lancet. 1972;1:734-737.
34 Cairns H, Oldfield R, Pennybacker K. Akinetic mutism with an epidermoid cyst of the third ventricle. Brain. 1941;64:273.
35 Stevens RD, Bhardwaj A. Approach to the comatose patient. Crit Care Med. 2006;34:31-41.
36 Berek K, Jeschow M, Aichner F. The prognostication of cerebral hypoxia after out-of-hospital cardiac arrest in adults. Eur Neurol. 1997;37:135-145.
37 Levy DE, Caronna JJ, Singer BH, et al. Predicting outcome from hypoxic–ischemic coma. JAMA. 1985;253:1420-1426.
38 Wijducks EF, Hijdra A, Young GB, et al. Practice parameter; prediction of outcome in comatose survivors after cardiopulmonary resuscitation (an evidence based review): report of the quality standards subcommittee of the American Academy of Neurology. Neurology. 2006;67:203-210.
39 Surtees R, Leonard JV. Acute metabolic encephalopathy: a review of causes, mechanisms and treatment. J Inherit Metab Dis. 1989;12(Suppl. 1):42-54.
40 Uribarri J, Oh MS, Carroll HJ. D-lactic acidosis. A review of clinical presentation, biochemical features, and pathophysiologic mechanisms. Medicine (Baltimore). 1998;77:73-82.
41 Plum F. Sustained impairment of consciousness. In: Bennett CPF, editor. Cecil Textbook of Medicine. WB Saunders; 1996:1970-1978.