[level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 15
Disorders Of Calcification
Osteomalacia and Rickets
Clinical Features of Rickets and Osteomalacia
Nutritional Osteomalacia and Rickets
Gastrointestinal and Hepatic Diseases
Impaired Renal Tubular Phosphate Reabsorption
Tumor-Associated Rickets and Osteomalacia
General Renal Tubular Disorders
Hyperparathyroidism, Hypoparathyroidism, and Pseudohypoparathyroidism
Fibrogenesis Imperfecta Ossium
Renal Osteodystrophy and Aluminum Intoxication
Osteomalacia and rickets are disorders of mineralization. Osteomalacia is a failure to mineralize the newly formed organic matrix (osteoid) of bone. In rickets, a disease of children, the growth plate at the epiphysis is involved in a process that is characterized by delay in the maturation of chondrocytes in the growth plate and disorganization of these chondrocytes, resulting in expansion of the growth plate. While rickets is associated with impaired mineralization of cartilage matrix, hypophosphatemia is thought to play a predominant role in the etiology of rickets.1 A number of different disorders are associated with osteomalacia in adults and rickets in children.2–4 The pathogenesis of the mineralization defect, the biochemical alterations, the clinical manifestations, and the therapeutic approaches differ in these conditions, so a systematic approach to the diagnosis and treatment of these disorders is essential.
Mineralization Defect
Mineralization of bone is a complex process in which the calcium-phosphate inorganic mineral phase is deposited in relation to the organic matrix in a highly ordered fashion. Optimal mineralization can take place at bone-forming surfaces only if cellular activity of bone-forming cells is adequate, matrix is normal in composition and is synthesized at a normal rate, the supply of mineral ions (calcium and inorganic phosphate) from the extracellular fluid is sufficient, the pH at sites of mineralization is appropriate (approximately 7.6), and the concentration of inhibitors of calcification is controlled. Structural and regulatory aspects of biological mineralization have been considered in numerous reviews.5–8 Clinical disorders of mineralization can be attributed to defects at several of these control steps, examples of which are shown in Table 15-1. Overall features of mineralization in normal and abnormal bone remodeling and the relevance to mineralization disorders have been comprehensively reviewed.4
Table 15-1
Examples of Disorders with Different Causes of Impaired Mineralization
| Disorder | Mechanism |
| Postoperative hyperparathyroidism | Rate of matrix synthesis exceeds rate of mineralization |
| Fibrogenesis imperfecta ossium | Defective collagenous matrix |
| Adult phosphate diabetes | Phosphate concentration deficient at mineralization sites |
| Vitamin D deficiency | Insufficient calcium and phosphate |
| Systemic acidosis | pH inadequate for mineralization |
| Hypophosphatasia | Excess inhibitor (? inorganic pyrophosphate) |
The determinants for the deposition of the mineral phase in normally mineralized tissues—cartilage, bone, and teeth—and the precise mechanisms that govern the process of biological mineralization have yet to be elucidated. It is thought that deposition of the initial mineral phase takes place within the collagen fibrils in the gap formed by the packing of the trimeric type I collagen molecules, the most abundant organic component of bone.7 The earliest calcium-phosphate (Ca-P) mineral phase is thought to be composed of very small apatite crystals, less than 9 nmol in length, that can fit into the channels formed by adjacent gaps.9 These initial Ca-P crystals appear to form complexes with phosphoproteins of the organic matrix.10 As the bone and minerals mature, additional Ca-P crystals are deposited between collagen macromolecules. Other proteins that are abundant in mineralized tissues and thought to play a role in mineralization include the sialic acid–rich proteins such as osteopontin and bone sialoprotein (BSP).11 The ultimate size and shape of the Ca-P crystals are also influenced by anionic electrolytes in the environment.12
The major driving force for mineralization is the concentration of inorganic phosphate (Pi), which at normal sites of mineralization is derived predominantly from the plasma. Therefore, control of Pi reabsorption in the renal tubular lumen is the most important process regulating mineralization. It is also possible that tissue-nonspecific alkaline phosphatase (TNAP), which functions as an inorganic pyrophosphatase localized on the surface of osteoblasts, generates additional Pi to drive mineralization.13 Osteomalacia is seen in loss-of-function mutations in the TNAP gene, but decreased TNAP also results in accumulation of inorganic pyrophosphate (PPi), which acts as a potent inhibitor of mineralization.13,14 It is likely that this pyrophosphatase function of TNAP is the most critical. PPi is generated by osteoblasts and chondrocytes through the action of two enzymes: the ectonucleoside, pyrophosphatase phosphodiesterase 1 (NNP1/PC-1), which releases PPi from nucleoside triphosphates13,14 and the transmembrane protein, ANK, which shuttles PPi between intracellular and extracellular compartments.15,16 Ectopic mineralization due to decreased local concentrations of PPi accompanies spontaneous deficiency of these proteins in humans and mice. In cartilage, there is evidence that matrix vesicles (MVs) formed from chondrocyte plasma membranes play a role in the regulation of mineralization.5,6 These extracellular MVs are approximately 100 nm in diameter and contain enzymes such as TNAP, NNP1, and Ca-binding proteins as well as Ca-P crystals at the inner surface of the membranes. They are thought to play a role in the initial mineralization of growth plate cartilage matrix by exposing preformed apatite crystals to the extracellular fluid, where further crystal growth can occur. MVs have also been found in bone near sites of mineralization, but it is difficult to assign them a function, since the initial crystallites are deposited in association with collagen fibers in bone. An explanation for the specificity of normal mineralization of bone is provided by the exclusive coexpression in bone of the genes encoding the predominant organic matrix protein, type I collagen, and the enzyme that hydrolyzes the inhibitor, PPi. Furthermore, the protein mineralization inhibitor, matrix GLA protein (MGP), that is expressed in cells in several other tissues is not expressed in bone.17,18
The mechanism of defective mineralization is not the same in all disorders associated with osteomalacia and rickets, and biochemical indices such as serum levels of calcium and phosphorus also differ. Moreover, the relative imbalance in matrix synthesis and its mineralization varies depending on the underlying disease mechanism. Estimates from tetracycline labeling indicate that the appositional growth rate in normal bone is about 1 µm/day.19 It has also been suggested20 that complete mineralization of the osteoid in normal bone requires approximately 10 to 21 days. Thus the thickness of the osteoid seam normally does not exceed 15 to 20 µm, the surface of bone that is covered by osteoid is normally less than 20%, and the active surface that is covered by osteoid is considerably less. The major histologic criteria for the diagnosis of osteomalacia are the increased osteoid surface and the increased thickness of the osteoid seam (Fig. 15-1). The mineralization front at the junction of mineralized bone and osteoid is also abnormal in osteomalacia.21 In applying kinetic criteria to the diagnosis of osteomalacia, it has been suggested that a mean osteoid seam width greater than 15 µm and a mineralization lag time greater than 100 days are appropriate diagnostic criteria.22 However, other investigators23,24 have suggested that more stringent criteria be applied to establish the diagnosis of osteomalacia, based on the observation that reduced mineral apposition rate, reduced fractional extent of the mineralization front, and prolongation of the mineralization lag time are indices that reflect impaired matrix synthesis by osteoblasts rather than specific features of osteomalacia. The diagnosis of osteomalacia must include evidence of an absolute increase in the total osteoid volume and an increased number of osteoid lamellae. A detailed consideration of the kinetics of remodeling as specifically applicable to osteomalacia is found in a review by Parfitt.4

FIGURE 15-1 Undecalcified sections of a bone biopsy from a patient with an adult-onset renal phosphate leak. A, Unstained. B, Microradiograph. C, Ultraviolet photomicrograph demonstrating fluorescence (F) of tetracycline administered 14 days prior to biopsy. Mineralized bone (M) and osteoid (O) are noted. (From Case 1 in Baylink D, Stauffer M, Wergedal J et al: Formation, mineralization, and resorption of bone in vitamin D–deficient rats, J Clin Invest 49:1122–1134, 1970.)
The architecture of the bone cells and matrix in osteomalacic bone is usually normal. The collagen of the osteoid is largely lamellar, although foci of woven bone are occasionally seen. Hypomineralized periosteocytic lesions have been observed in some affected individuals with hypophosphatemic rickets.25 The persistence of this defect in patients in whom the abnormality in bone mineralization was corrected with therapy supports the hypothesis that osteocyte function may be abnormal. In contrast, there are clear abnormalities in the cells of the rachitic growth plate. The characteristic changes occur in the maturation zone of hypertrophic chondrocytes, whereas the resting and proliferative zones show normal histologic features (Fig. 15-2). In the maturation zone, the number of cells per column is increased, and the cells are irregularly aligned. This is also accompanied by an increase in the transverse diameter, which may extend beyond the ends of the bone, resulting in characteristic cupping or flaring. In experimental rickets, the water content of the growth plate is increased, and a number of metabolic abnormalities have been observed, including decreased glycogen content and an altered pattern of glycolysis.26 When bone is examined histologically, it is essential that undemineralized sections be used. In usual practice, however, with classic clinical, radiologic, and biochemical findings, bone biopsy is not necessary to arrive at the diagnosis of osteomalacia. The most commonly biopsied site is the iliac crest; sample size ranges from 5 to 10 mm in diameter and should include both inner and outer cortices. Growth plates from long bones in children are usually not biopsied, although an open-wedge biopsy of growth cartilage of the iliac apophysis may occasionally be obtained without the hazard of altering growth of long bones. Mineralized specimens of bone are most satisfactorily embedded in plastic media—which provide preservation of tissue architecture not usually attained with paraffin-embedding techniques—because the distinction between mineralized and unmineralized bone is lost with decalcification of the specimen. A number of different staining techniques can then be used to demonstrate the osteoid and apply quantitative morphometric analysis.23–25 In normal bone, the mineralization front is seen at the junction of the osteoid seam and newly mineralized bone. This region can be identified by an intense fluorescence of tetracycline, deposited in this zone when administered prior to obtaining the biopsy (see Fig. 15-1). In normal persons, the osteoid seam/bone junctions fluoresce intensely; in osteomalacia, the fluorescence is less well defined (more diffuse) or even absent. In addition to impaired matrix mineralization, matrix biosynthesis may be abnormal in osteomalacia. In osteomalacia observed with vitamin D deficiency, a decreased rate of matrix formation is observed.23,24,27 Osteoblast function may be impaired in many forms of human rickets and osteomalacia, which may result in abnormal matrix formation. Notably, the hydroxylation of certain collagen lysyl residues is increased in vitamin D–deficient bone, as well as in other experimental hypocalcemic states.28–30

Clinical Features of Rickets and Osteomalacia
The clinical manifestations of rickets are mainly related to skeletal pain and deformity, slippage of epiphyses, disturbances in growth, and fracture of the osteomalacic bones. Hypocalcemia, when it occurs, may be symptomatic. Depending on the degree of hypophosphatemia, muscular weakness and hypotonia may be prominent features. Dent and Stamp31 have indicated nine factors that underlie the clinical manifestations of rickets and osteomalacia, modified here as follows:
1. Failure of mineralization affects predominantly those parts of the skeleton in which growth is most rapid.
2. Endochondral bone is more affected than intramembranous bone, possibly because of the more rapid growth of the former.
3. Proximal and distal ends of bones do not grow at the same rate, and rickets affects the most rapidly growing area.
4. Because different bones grow at different rates at different stages of development, the time when rickets is active determines the clinical expression. For example, the skull is growing rapidly at birth, so craniotabes is a manifestation of congenital rickets. During the first year, the upper limbs and rib cage grow rapidly, so abnormalities at these sites are prominent—for example, rachitic rosary. Signs of rickets at the wrist are usually seen at the ulnar side because the growth rate of the distal ulnar epiphysis is greater than that of the distal radial epiphysis.
5. Deformities in mild chronic rickets are most often due to disordered growth at the epiphysial plate rather than to bending at the shafts.
6. In some forms of rickets, the radiologic changes include those of secondary hyperparathyroidism (subperiosteal resorption, most commonly at the metaphyses).
7. Deformities that occur before the age of 4 years correct themselves if the rickets is cured; if rickets persists to a later age, the deformities are permanent (dwarfism, bowleg, and knock-knee).
8. “Late” rickets, which occurs at the time of the pubescent growth spurt, produces dramatic disturbances and results in knock-knee.
9. Adult manifestations of osteomalacia, such as Looser’s zones and increased biconcavity of vertebral bodies, are seen in young children only when the rickets is very severe.
In contrast, osteomalacia in adults may be difficult to detect on clinical grounds alone. Diffuse skeletal pain and muscular weakness may be present. Pain, often prominent about the hips and in association with hypophosphatemic myopathy, may produce a waddling or antalgic gait. Fractures may occur in the ribs and vertebral bodies, as well as in long bones, leading to progressive deformities. Affected individuals may also have localized pain and swelling in one or more joints. Synovial fluid is noninflammatory and free of crystals. Symmetric polyarthralgias resembling those of rheumatoid arthritis or polymyalgia rheumatica may also be observed.32 Muscular weakness is quite common,33,34 is primarily proximal in distribution (which contributes to the waddling gait), and is often associated with wasting and hypotonia with preservation of brisk reflexes.35 This is thought to be a consequence of hypophosphatemia and responds to phosphate repletion.36 The molecular basis remains elusive, with no difference observed in the relative concentrations of skeletal muscle phosphocreatine, adenosine triphosphate, or inorganic phosphate estimated by phosphorus nuclear magnetic resonance spectroscopy.37 Although the etiology of the neuromuscular features of osteomalacia is not clearly defined, therapy of the underlying disorder, such as vitamin D in nutritional osteomalacia, alkalinization in acidosis, and phosphate repletion in hypophosphatemic osteomalacia, results in resolution of these features. The role of hypophosphatemia per se in muscular weakness is discussed in Chapter 6.
Radiologic Features
Radiologic features of rickets and osteomalacia reflect the histopathologic changes. In rickets, the alterations are most evident at the epiphyseal growth plate, which is increased in thickness, cupped, and reveals haziness at the diaphyseal border due to decreased mineralization of the hypertrophic zone and inadequate mineralization of the primary spongiosa (Fig. 15-3). Variation in the pattern of rachitic changes is influenced by differences in the rates of growth of individual bones. The trabecular pattern of the metaphyses is abnormal, the cortices of the diaphyses may be thinned, and bowing of the shafts may be present.
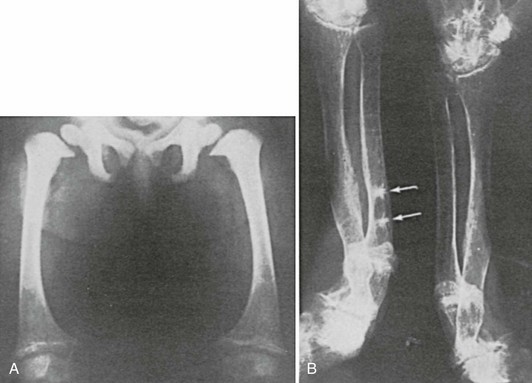
FIGURE 15-3 A, Rickets in a child with Fanconi’s syndrome, showing typical cupping of distal femoral epiphyses. B, Osteomalacia in an 80-year-old woman who had a history compatible with hypophosphatemic rickets dating to early childhood. Note multiple pseudofractures (arrows).
Osteomalacia is due to decreased mineralization and is therefore associated with a decrease in bone density, loss of trabecular patterning, and variable degrees of thinning of the cortices.38,39 In some patients, radiologic changes are indistinguishable from those seen in osteoporosis. The characteristic finding that specifically suggests osteomalacia is the presence of radiolucent bands known as pseudofractures, Looser’s zones, or umbauzonen, ranging from a few millimeters to several centimeters in length and usually oriented perpendicularly to the surface of the bone. (Fig. 15-4). They tend to occur symmetrically and are particularly common at the inner aspects of the femur near the femoral neck, in the pelvis, in the outer edge of the scapula, in the upper fibula, and in the metatarsals.
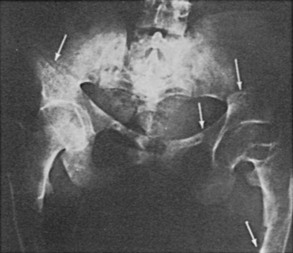
FIGURE 15-4 Radiograph of the pelvis and proximal femora in an adult with renal phosphate wasting. Note pseudofractures, also known as Looser’s zones (arrows). (From Case 2 in Jaworski ZFG, Kloswvych S, Cameron E: Proceedings of the First Workshop on Bone Morphometry. Ottawa: University of Ottawa Press, 1973.)
Pseudofractures are most often seen at sites where major arteries cross the bones. Arteriography in some36,40,41 but not all cases42 suggests that the origins of the pseudofractures correspond to the locations of major arteries (Fig. 15-5). Trauma of some sort, whether related to arterial pulsation or other factors (e.g., weight-bearing stress), is likely responsible for the symmetry of the lesions and their predilection for the described sites. Pseudofractures are often multiple, occasionally occurring at 10 to 15 sites in a single individual; such multiple symmetric pseudofractures in osteomalacic individuals have been referred to as Milkman’s syndrome.43–46 The abnormalities in Milkman’s original case were also considered by Albright and Reifenstein43 to be manifestations of osteomalacia. The histopathology of Looser’s zones is that of premalacic lamellar bone, some of which is surrounded by lamellar osteoid at the edge of the defect.47 In addition, there are foci of woven bone, some of which is mineralized and some not. This accounts for the lower radiologic mineral density of the pseudofracture compared with the surrounding bone. Subperiosteal erosions along the diaphyseal cortices extending to the metaphyses may be seen when secondary hyperparathyroidism is present. Widening (or pseudowidening) of the sacroiliac joints and the appearance of hazy margins has also been observed, sometimes suggesting ankylosing spondylitis, which osteomalacia may mimic clinically.36
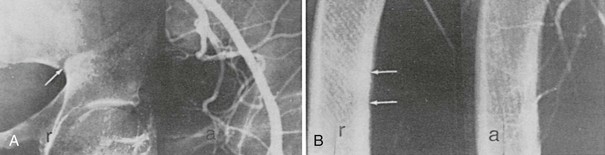
FIGURE 15-5 Radiograph (r) and corresponding arteriograms (a) of a patient with adult-onset renal phosphate wasting. A, Pelvis. B, Femur. Note that the origin of Looser’s zones (arrows) corresponds with crossing of major vessels. (From Case 2 in Jaworski ZFG, Kloswvych S, Cameron E: Proceedings of the First Workshop on Bone Morphometry. Ottawa: University of Ottawa Press, 1973.)
In some patients with osteomalacia, increased rather than decreased radiologic density of bones may be observed.48 This is seen particularly in patients with renal tubular phosphate leaks, as opposed to vitamin D deficiency (Fig. 15-6). In such patients, there may be a striking degree of thickening of the cortices and trabeculae of the spongy bone, at times associated with exostotic spurs. This hyperostosis has been noted in untreated patients. It is not usually observed in patients with generalized defects in proximal renal tubular reabsorption. Despite the increase in mass of bone per unit volume, microscopically the trabeculae are covered with abnormally thickened osteoid seams typical of osteomalacia. Similar findings may be noted in patients with chronic renal failure. The reason for the hyperostosis is unknown; the bone is still architecturally abnormal and subject to fracture with relatively minimal trauma.
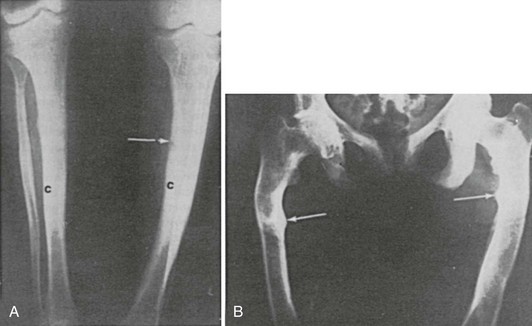
FIGURE 15-6 Increased bone mass in patients with osteomalacia. A, Radiographs of femora of a 15-year-old boy with X-linked hypophosphatemia. Note the thick tibial cortex (c) and Looser’s zone (arrow). B, Radiograph of pelvis and femora of a 38-year-old woman with hypophosphatemia present since childhood. Note Looser’s zones (arrows).
In patients with X-linked hypophosphatemic osteomalacia and rickets, an additional finding has been the presence of a generalized involvement of the entheses, with exuberant calcification (more likely ossification) of tendon and ligament insertions.49,50 The absence of inflammatory cells, as well as other clinical features, differentiates this disorder from degenerative joint disease and the seronegative spondyloarthropathies. A comprehensive classification of rickets and osteomalacia is shown in Table 15-2. A detailed discussion of all these conditions is not included here.
Table 15-2
Classification of Rickets and Osteomalacia
B Impaired renal tubular phosphate reabsorption (intestinal)
VII General renal tubular disorders (Fanconi’s syndrome)
VIII Primary mineralization defects
IX States of rapid bone formation with or without a relative defect in bone resorption
Nutritional Osteomalacia and Rickets
In 17th century Scotland and England, the association of poverty and malnutrition with the occurrence of infantile rickets was vividly documented. The widespread prevalence of infantile rickets in the industrialized regions of Britain was further documented in reports from Glasgow in the late 1800s and early 1900s. The link between rickets, dietary deficiency of vitamin D, and correction of vitamin D deficiency by solar radiation was finally established in 1923 by the work of the Vienna Council. Following this discovery, fortification of certain foods with vitamin D reduced the incidence of nutritional rickets in Europe and the United States to negligible levels, and by the 1940s, vitamin D deficiency was no longer regarded as an important cause of osteomalacia and rickets.43 Vitamin D metabolism and the role of specific metabolites in bone development, mineralization, and remodeling are discussed elsewhere (see Chapter 3). Nevertheless, the role of vitamin D in the prevention of osteomalacia and rickets is relevant to the discussion in this chapter.
Studies in Glasgow51–53 and London54 documented the reappearance of nutritional osteomalacia and rickets as a public health problem in the United Kingdom. Since the 1950s, the population at risk primarily involves East Asian immigrants whose unique dietary and social customs have led to the development of osteomalacia and rickets.52,54–58 Other groups at risk include housebound and elderly subjects and food faddists, especially those on vegetarian or fat-free diets.52,54–58 An unexpectedly high prevalence of osteomalacia was also observed in elderly women with rheumatoid arthritis who were housebound and had poor nutritional status.59
Gastrointestinal and Hepatic Diseases
Another population at risk for development of nutritional osteomalacia are morbidly obese individuals60 and those who have undergone intestinal bypass surgery for treatment of severe obesity.61–63 In one bypass surgery series, iliac bone biopsies revealed the presence of osteomalacia in nearly a third of 21 patients studied.63 Treatment with vitamin D2 (36,000 IU/day), as well as supplemental calcium (27 mmol/day), was required to promote a more positive calcium balance in some individuals.61 An increased frequency of osteomalacia also was observed in patients after gastrectomy,64–66 the severity of the mineralization defect being positively correlated with serum 25-hydroxyvitamin D [25(OH)D] but not with serum 1,25-dihydroxyvitamin D [1,25(OH)2D].
Rickets occurs in infants and children with cholestatic liver disease.67 Children with biliary atresia develop biliary cirrhosis, jaundice, and ascites. Intestinal absorption of vitamin D is markedly impaired, and serum values are low. Bone disease has been attributed to vitamin D deficiency, and pharmacologic doses of 1,25(OH)2D3 are required for treatment of the bone disease.68
Low-birthweight infants are at risk for rickets, with the reported incidence ranging from 13% to 32%. Insufficient intake of calcium, phosphorus, and vitamin D have been implicated. The condition should be detected early, since prompt nutritional supplementation is required.69 Infants of vitamin D–deficient mothers are at greatest risk for neonatal rickets,70 which can be averted by maternal vitamin D supplementation during pregnancy. Special consideration should be given to screening for vitamin D deficiency during pregnancy in women whose social and dietary history reveal inadequate sun exposure and food sources of vitamin D. Infants who are entirely breastfed and have inadequate exposure to sunlight are also at risk for rickets, because the amount of vitamin D and 25(OH)D in human milk is inadequate.71
Vitamin D sources include dietary supplementation with ergocalciferol (vitamin D2), an irradiation product obtained from plants, and cholecalciferol (vitamin D3), produced in human skin by the action of ultraviolet light on the physiologic precursor, 7-dehydrocholesterol. Because most foods (with the exception of fatty fish) contain only small amounts of vitamin D3, individuals must rely on either adequate sunlight exposure or dietary supplements to avoid vitamin D deficiency. There is marked seasonal variation in plasma 25(OH)D, independent of age and sex.72–74 These variations parallel changes in duration and intensity of sun exposure, with higher values in late summer months in the northern hemisphere. Plasma 25(OH)D is almost twice as high in American women of Caucasian descent than in those of African descent during winter months, and the increment during summer months is also greater.75 Plasma 25(OH)D and parathyroid hormone (PTH) are inversely correlated, implying that the changes in circulating 25(OH)D are metabolically significant.76 In a study of patients who were hospitalized in a general medical ward, plasma 25(OH)D was low in over half of the subjects, consistent with a high prevalence of vitamin D deficiency.77 Thus, adequate vitamin D supplementation is essential when exposure to sunlight is marginal.
Vitamin D requirements are greater in the elderly than in young adults. This difference is attributed to an age-related decline in dermal production of 7-dehydrocholesterol, the precursor for vitamin D3,78 diminished renal production of 1,25(OH)2D,79 and diminished intestinal absorption of calcium caused by lower levels of the vitamin D receptor in the intestine.80 Other contributing factors include previous gastric surgery and occult malabsorption in addition to altered vitamin D metabolism. To determine optimal plasma 25(OH)D and how much vitamin D is required to achieve optimal values, Vieth,81 in an exhaustive review, and Heaney in another82 concluded that total daily intake and production of vitamin D of 2.5 to 5.0 mg (100 to 200 IU) and plasma 25(OH)D values greater than 20 to 25 nmol/L are sufficient to prevent clinical rickets or osteomalacia. Higher intakes or production of vitamin D would be necessary, however, to prevent secondary hyperparathyroidism, bone loss, and subclinical osteomalacia. Therefore, it may be necessary to maintain serum 25(OH)D levels of 100 nmol/L or greater to avoid bone loss, subclinical osteomalacia, and osteoporosis. The amount of vitamin D supplementation necessary to attain these levels varies widely, based on dietary factors and solar exposure.
In the East Asian immigrant populations of Britain, rickets and osteomalacia secondary to vitamin D deficiency are seen most commonly in neonates, infants, and adolescents during pubertal growth and less frequently among adults.51,83–85 Multiple factors have been implicated in the development of bone disease, including insufficient intake of calcium and vitamin D, skin pigmentation,86 which attenuates ultraviolet transmission through the epidermis, genetic factors,87–89 and social customs, such as avoidance of sun exposure and consumption of chapati, a dietary staple flatbread high in phytate, which binds calcium in the gut and interferes with its absorption.90 Furthermore, studies in rats demonstrated that the rate of inactivation of vitamin D in the liver was increased by a calcium-restricted diet.
Studies by Dent and colleagues91 strongly suggest that insufficient sunlight exposure plays a pivotal role in the development of nutritional rickets and osteomalacia, in addition to the rickets and osteomalacia observed in patients on long-term anticonvulsant therapy.92 In two carefully studied individuals, they demonstrated healing of rickets and positive calcium balance following therapy with ultraviolet light, despite a vitamin D–deficient, high-phytate diet. Substitution of a low-phytate diet did not affect the plasma biochemical abnormalities or the calcium balance.91
Scriver93 divides the evolution of vitamin D deficiency in infancy into three stages. In stage 1, serum calcium tends to be low, serum phosphorus normal, and aminoaciduria absent. Without treatment, stage 2 develops, and aminoaciduria and hypophosphatemia appear as a consequence of diminished tubular reabsorption. In this stage, serum calcium tends to return to normal, and serum alkaline phosphatase increases, presumably related to the increased PTH and resultant increase in bone turnover. Renal tubular dysfunction (aminoaciduria, phosphaturia) has, at least in part, been attributed to the increased PTH.94,95 Stage 3 is characterized by the return of hypocalcemia. The effect of lowered concentrations of serum phosphorus in stage 2 and lowered concentrations of serum calcium and serum phosphorus in stage 3 presumably account for development of the mineralization defect.
Rao et al.96 reviewed the histomorphometric findings in a series of 65 patients with vitamin D depletion diagnosed on the basis of plasma levels of 25(OH)D less than 10 ng/mL. They found that in early vitamin D depletion, the effects on bone are manifested principally by the occurrence of secondary hyperparathyroidism. With increasing severity or duration of the vitamin D deficiency, the mineralization process becomes progressively impaired, bone formation rates decline, and osteoid surface and thickness increase.
In summary, rickets and osteomalacia are being recognized with increasing frequency in selected populations. Serum 1,25(OH)2D is not a reliable indicator of vitamin D nutrition, since values may be normal in individuals with vitamin D deficiency.97,98 The availability of serum 25(OH)D assays has also permitted detection of those at risk, prior to the development of overt clinical disease.
Acidosis and Osteomalacia
Acidosis resulting from a number of different causes has been associated with osteomalacia. The mechanism of bone loss and the mineralization defects are complex and not completely understood. Albright and Reifenstein43 originally suggested that acidosis produces slow dissolution of the mineral phase of bone in an attempt to buffer retained hydrogen ion. This process is associated with hypercalciuria. Support for this suggestion has been obtained in studies of patients with renal tubular acidosis in whom retention of hydrogen ion is greater than that theoretically required to produce the observed decrease in plasma bicarbonate concentrations.99 On the basis of measurements in normal subjects in whom metabolic acidosis is induced, it has been proposed that excess retained hydrogen ion is balanced by bone buffering and loss of bone calcium in the urine.100–102 Since the hypercalciuria and increased bone resorption that accompany most acidotic states do not directly result in osteomalacia, other mechanisms must be invoked to explain the occurrence of clinically significant skeletal mineralization defects. Osteoclasts function optimally at a pH of approximately 6.9 and are inactive at a pH greater than 7.3; therefore, it is probable that the calcium release from bone induced by acidosis can be ascribed to increased osteoclast activity rather than to simple physicochemical buffering.103 Chronic acidosis activates vacuolar hydrogen ion pumps in isolated osteoclasts and stimulates osteoclastic bone resorption.104 In addition, activation of the voltage-gated H+ channel through protein kinase C in osteoclasts can sense changes in pH.105
In parallel, metabolic acidosis inhibits the function of osteoblasts: in osteoblast culture systems, lowering pH to 6.9 decreases formation of mineralized nodules.106 Acidosis increases expression of osteoblastic RANK-L via a cyclooxygenase-dependent mechanism, leading to enhanced osteoclastogenesis.107 Another potential proton-sensing mechanism in the osteoblast is G protein–coupled receptors OGR1 and OGR2.108 OGR1 is inactive at pH 7.8 and fully activated at pH 6.8, signals through the phosphoinositol pathway, and is expressed in osteosarcoma lines and primary osteoblasts. OGR2 signals through the cyclic adenosine monophosphate (cAMP) pathway but has not yet been shown to be expressed in bone cells. Maintenance of pH within a critical range is thus essential for bone-cell function and for mineralization to proceed normally.
Acidosis can also affect phosphate metabolism by altering renal tubular handling of the anion. In patients with chronic acidosis, treatment with alkali to correct the acidosis can normalize serum phosphate by increasing renal tubular phosphate reabsorption and phosphate maximal tubular excretory capacity.109,110 Secondary hyperparathyroidism may be another factor involved in the altered phosphate handling in systemic acidosis,109 and acidosis can impair intestinal calcium absorption in response to exogenous vitamin D,106 as well as activation of 25(OH)D.111 Rickets and osteomalacia secondary to acidosis are most frequently a complication of inherited distal renal tubular acidosis (RTA).112 Clinical manifestations vary widely in type as well as severity. Autosomal-dominant as well as autosomal-recessive forms have been described. As a rule, the dominant form is usually recognized in adults who present with relatively mild disease characterized by nephrolithiasis and acidosis. Osteomalacia occurs infrequently. In contrast, the autosomal-recessive form is recognized in infancy or early childhood, is usually severe, and rickets is common. In dominant RTA, the causal mutation is in the SLC4A1:AE1 gene that encodes the heterotopic Cl−/HCO3− exchanger located in the α-intercalated cells of the distal tubules. In recessive RTA, the causal mutations are in the B1 or A4 subunits of the H+/ATPase encoded by ATP6V1B1 and ATP6V0A4 genes, respectively. Rarely, mutations in the SLC4A1:AE1 gene occur in recessive RTA as well. In most of the cases described in early reports,113–115 healing of the bone disease resulted from correction of the acidosis with sodium bicarbonate alone (5 to 10 g/day). Nevertheless, healing is slow, and the response may be hastened by the addition of vitamin D or 1,25(OH)2D3. Occasionally, vitamin D toxicity can develop unexpectedly, so patients must be monitored carefully. Although chronic treatment with vitamin D is not necessary once the osteomalacia is cured, continued use of vitamin D may be required to complete healing in those individuals in whom the glomerular filtration rate is low.115,116
Another form of inherited mixed distal/proximal RTA is associated with osteopetrosis and cerebral calcifications. The osteopetrosis, due to inadequate osteoclast function secondary to defects in acidification in the extracellular space adjacent to the ruffled border, is caused by mutations in the gene that encodes carbonic anhydrase II (CAII).117–119 This RTA is characterized by defective urinary acidification and bicarbonate wasting, and the syndrome is explained by the fact that CAII is expressed not only in osteoclasts but in both proximal and distal segments of the nephron. It is of interest that bone marrow transplantation does not affect the acidosis but reverses the osteopetrosis (by radiologic and histologic criteria), since osteoclasts are of hematopoietic origin.120
In several of the syndromes associated with more widespread renal tubular reabsorptive defects, systemic acidosis may contribute to the pathogenesis of osteomalacia. Some of these are inherited, such as various forms of Fanconi’s syndrome and Lowe’s syndrome (oculocerebrorenal syndrome). Some patients with renal tubular phosphate leaks may also have mild acidosis.121–125 Other aspects of these syndromes are considered elsewhere in this chapter.
Osteomalacia may also be a complication of the acidosis produced by ureterosigmoidostomy, a procedure formerly used in the treatment of patients with carcinoma of the bladder. Reabsorption of chloride and hydrogen ions from urine in the colon is responsible for the acidosis. Keeping the rectosigmoid empty by frequent drainage corrects the acidosis and thus prevents the development of osteomalacia. Typical osteomalacia has also been observed in a patient with acidosis presumably resulting from chronic acetazolamide therapy. This patient was receiving phenobarbital, as well as phenytoin, for a severe seizure disorder, but when the acetazolamide alone was discontinued and the plasma bicarbonate increased, radiologic and clinical healing of osteomalacia was observed. Acetazolamide has direct inhibitory effects on bone resorption in animals independent of pH, and carbonic anhydrase inhibitors do prevent bone loss in humans.126,127
Dietary Phosphate Depletion
In humans, phosphate depletion and resultant hypophosphatemia may lead to the development of rickets or osteomalacia by mechanisms that were discussed earlier. It is difficult to produce selective deficiency of phosphorus by dietary means alone, because most foods contain this element in concentrations that are sufficient to prevent hypophosphatemia and bone disease (see Chapter 6). However, hypophosphatemia has been reported in patients who ingest large quantities of nonabsorbable antacids, usually as a form of self-medication for dyspepsia.128–132 This is accompanied by a marked increase in fecal phosphorus content, presumably related to binding of dietary phosphate by the antacid, resulting in a complex that is poorly absorbed from the gastrointestinal tract. In addition to the changes in phosphorus handling, these individuals also develop hypercalciuria. Most of the affected individuals show no evidence of increased bone resorption, although a small increase in osteoclastic surface and number of osteoclasts has been reported in one patient studied.133 It is more likely that the rise in urinary calcium excretion is related primarily to impaired bone mineralization, a concept that is supported by the association of this syndrome with osteomalacia. Elevated levels of serum 1,25(OH)2D are observed in patients with antacid-induced osteomalacia, a normal response to hypophosphatemia. Clinically significant bone disease is rare, suggesting that an ample supply of dietary phosphorus compensates for the absorptive defect. A similar syndrome of phosphate depletion has been described in patients with renal failure receiving large quantities of aluminum hydroxide gel, but osteomalacia in these patients may be related to aluminum intoxication and/or renal insufficiency.
Hypophosphatemia has also been observed in both chronic and acute alcoholism (see Chapter 6).134,135 Bone densitometric studies and tetracycline-labeled bone biopsies obtained from chronic alcoholic patients have revealed an increased frequency of bone disease compared to sex- and race-matched controls.136 Bone abnormalities include changes consistent with mixtures of osteoporosis, osteomalacia, and osteitis fibrosa. Multiple factors, including hypomagnesemia, metabolic alkalosis or acidosis, and renal tubular phosphate wasting contribute to the hypophosphatemia and bone disease.137 Bone disease in alcoholism may be part of the generalized skeletal disorder associated with chronic liver disease of diverse origin, which has been termed hepatic osteodystrophy.137 The syndrome comprises osteomalacia, osteitis fibrosa, osteoporosis, and periosteal new bone formation in the presence of chronic liver disease. Osteomalacia is most common in patients with cholestasis (particularly primary biliary cirrhosis) but is also observed in patients with alcoholic liver disease and other forms of cirrhosis. In most patients, the serum levels of 25(OH)D are low, ascribable to impaired intestinal absorption of vitamin D, but reduced exposure to ultraviolet light and reduced dietary intake also contribute.138 Treatment with vitamin D metabolites can heal hepatic osteomalacia.
Dietary Calcium Deficiency
Nutritional rickets caused by calcium deficiency was first reported in 1978 by Pettifor and co-workers,139 who described findings in nine rural South African children. Although they had spent extensive time outdoors, the children showed obvious clinical features of rickets, including progressive bone deformities, decreased growth, and typical radiographic changes. Four of the children had hypocalcemia, and all of them had normal serum 25(OH)D values but increased serum alkaline phosphatase and serum 1,25(OH)2D values. Calcium-balance studies showed that calcium absorption was not impaired. The biochemical and radiographic changes of rickets were entirely corrected by treatment with calcium alone.
In a prospective, randomized, double-blind study of 123 Nigerian children with nutritional rickets, Thacher et al.140 compared treatment with calcium alone, vitamin D alone, and the combination of calcium and vitamin D together. Treatment with calcium alone or calcium and vitamin D together was more effective than treatment with vitamin D alone. Baseline calcium intake was similar in patients and in a control group. It was concluded that although calcium deficiency was an important contributor to nutritional rickets, other unidentified factors might have been involved. Calcium deficiency is also prevalent in North American children and can contribute to nutritional rickets. Review of the records of 43 infants and toddlers who presented with rickets in New Haven, Connecticut (86% of whom were of African, Hispanic, or Middle Eastern descent and more than 93% of whom had been breastfed), revealed serum 25(OH)D levels less than 15 ng/mL in only 22%. However, 86% of those with a food history had been weaned to diets with minimal calcium content.141
Impaired Renal Tubular Phosphate Reabsorption
In 1937, Albright and co-workers142 reported their studies of a 16-year-old boy with longstanding rickets in whom standard doses of vitamin D failed to produce clinical improvement. Healing of the bone disease eventually occurred, but only after administration of extremely high doses of vitamin D. The results of their studies led to the introduction of the concept of “vitamin D resistance” in certain types of rickets. Since this initial report, so-called vitamin D–resistant rickets has been classified into several clinical and biochemical subtypes, the most common of which is the X-linked, dominantly inherited form discussed in detail in Chapter 11. Affected individuals usually present with clinical and radiographic evidence of rickets within the second or third year of life. The cardinal biochemical disturbance is hypophosphatemia due to renal phosphate wasting, associated with elevated levels of fibroblast growth factor (FGF-23).143 Other causes of osteomalacia (see Table 15-2) must be excluded, particularly primary vitamin D deficiency, malabsorption, renal insufficiency, generalized renal tubular disorders, and the presence of certain mesenchymal tumors.
A positional cloning approach was utilized by a consortium of investigators to identify the gene for X-linked hypophosphatemia.144 The gene was structurally similar to a group of membrane-bound metalloendopeptidases and was termed PEX, but renamed PHEX to avoid confusion with other genes. Several inactivating mutations have been shown in the PHEX genes of patients with X-linked hypophosphatemia as well as in the murine Phex gene in the Hyp mouse.145–148 It is still not clear how the mutations in the PHEX gene account for the excessive renal tubular phosphate losses, but it has been suggested that PHEX might function to directly or indirectly degrade FGF-23 or regulate its expression.
Another disorder with isolated renal tubular phosphate wasting and inappropriately normal plasma 1,25(OH)2D levels has been termed autosomal-dominant hypophosphatemic rickets (ADHR). In contrast to X-linked hypophosphatemia, ADHR displays variable and incomplete penetrance, as well as other clinical manifestations.149–151 Positional cloning was used to identify FGF-23 as the abnormal gene associated with ADHR.152 In individuals with ADHR, mutations in FGF-23 that are localized to a subtilisin-like proprotein convertase cleavage site lead to elevated levels of FGF-23, which results in increased renal phosphate clearance.
A mutation in the phosphate transporter SLC34A3 (NptIIc) is the genetic basis for hereditary hypophosphatemic rickets with hypercalciuria (HHRH). This disorder is due to hypophosphatemia secondary to renal phosphate wasting and presents with rickets, muscle weakness and bone pain. Hypercalciuria, secondary to increased 1,25(OH)2D (which leads to increased intestinal calcium absorption), distinguishes it from other forms of hypophosphatemic rickets.153
In patients with X-linked hypophosphatemia, serum levels of 25(OH)D are normal, whereas the levels of 1,25(OH)2D are in the low-normal range, inappropriately low for the level of serum phosphorus. Treatment of affected patients with 1,25(OH)2D3 and oral phosphate (1 to 2 g/day phosphorus equivalent) results in healing of rickets and osteomalacia; however, hypercalcemia and hypercalciuria may complicate therapy.154,155 Of note is that heterozygous girls appear to respond better to therapy than hemizygous boys.156 A variety of neutral phosphate salts are available for oral supplementation, including sodium and potassium salts or mixtures of the two. Greater elevations in serum phosphorus levels are observed with the potassium salt, likely related to the effects of the sodium ion on increasing renal phosphate clearance.157 The precise amount of phosphate must be individualized for each patient. In individuals with severe renal phosphate leaks, as much as 1000 mg of elemental phosphorus is required every 4 to 6 hours to effect sustained elevations of serum phosphorus levels. The rise in serum phosphorus level after a single oral dose of phosphate is transient, so phosphate supplements must be administered at frequent intervals. The efficacy of therapy cannot be assessed with a single fasting determination of serum phosphate; multiple measurements of serum levels at various times after each dose are required. Emptying the capsules and dissolving the salt in water or other liquid may improve intestinal absorption and enhance serum phosphorus levels. Most patients experience some degree of gastrointestinal distress, such as cramps and diarrhea, when therapy is initiated; therefore, initial doses should be low and increments gradually introduced as tolerated. Simultaneous use of phosphate and vitamin D has resulted in accelerated healing in children with the X-linked form of hypophosphatemic osteomalacia.158 Vitamin D itself or various analogues have a “phosphate-sparing” effect, allowing the use of lower doses of oral phosphate supplements. Whether the improved serum phosphorus levels seen with vitamin D or 1,25(OH)2D3 are accounted for by increased intestinal absorption of phosphate or decreased renal loss (due to decreased secondary hyperparathyroidism) has not been established. After fusion of the growth plates, patients are no longer at risk for rickets or growth retardation, raising the question as to whether treatment should be continued in affected adults. Healing of osteomalacia with therapy has been documented in adults, but close monitoring is required to avert the potential development of parathyroid hyperplasia and hypercalcemia resulting in nephrocalcinosis and renal insufficiency.159,160
Tumor-Associated Rickets and Osteomalacia
In 1959, Prader et al. described the case of an 11-year-old girl who, over the course of a year, developed severe symptomatic rickets accompanied by hypophosphatemia, increased renal phosphate clearance, and mild hypocalcemia.161 The child was found to have a large tumor in the left chest that on biopsy was interpreted as a reparative giant cell granuloma of a rib. Following excision of the tumor, the rickets healed without any specific therapy. It was postulated that the giant cell reparative granuloma may have produced a “rachitogenic substance.”
Since that time, numerous patients have been described in whom osteomalacia has been associated with the presence of various types of tumor. One review162 reported that of 72 tumors, 40 were localized to bone and 31 to soft tissues; two-thirds of the tumors occurred in the extremities. More than a third of the tumors were classified as vascular tumors, and half of these were hemangiopericytomas. Other common pathologic diagnoses were nonossifying fibromas and mesenchymal and giant cell tumors. All of the tumors exhibited prominent vascularity, multinucleated giant cells, and primitive stromal cells. Ten of the tumors were classified as malignant. Whereas most neoplasms associated with this syndrome have been exclusively of mesenchymal origin, two cases of hypophosphatemic osteomalacia associated with prostate carcinoma have been reported.163 As in other forms of tumor-associated osteomalacia, serum 1,25(OH)2D levels were low. In most of the reported cases, the removal of the tumor results in clinical cure of the osteomalacia or rickets, but the size of the tumors is usually small, and in several instances, the tumor is not detected until years after development of clinical osteomalacia.164 Recurrence of the tumor or inadequate removal of the malignancy may prevent a complete clinical response. In a patient with hypophosphatemia due to a malignant giant cell sarcoma, resection of the tumor resulted in a clinical remission that lasted 4 years, at which time hypophosphatemia recurred, associated with the reappearance of pulmonary metastases.
Patients with tumor-induced osteomalacia exhibit hypophosphatemia, high renal phosphate clearance, and normal serum calcium levels. Serum immunoreactive PTH levels have been variable, but low or undetectable levels of serum 1,25(OH)2D are observed because these tumors release products that impair renal 1α-hydroxylation of 25(OH)D and phosphate transport, including FGF-23, the serum level of which normalizes after tumor resection.143 Oncogenic osteomalacia tumors have also been shown to express FGF-23 mRNA and/or protein,152,165,166 as well as other phosphaturic factors, including complementary DNAs (cDNAs) encoding dentin matrix protein 1, secreted frizzled-related protein 4 (sFRP-4) and matrix extracellular phosphoglycoprotein (MEPE). Studies have shown that sFRP-4 decreases sodium-dependent phosphate transport in kidney cells, and infusion of sFRP-4 into mice produces renal phosphate excretion.167 These results suggest that other factors in addition to FGF-23 may participate in the pathogenesis of oncogenic osteomalacia.
Dent and Gertner168 also described three patients with fibrous dysplasia who had concomitant hypophosphatemic osteomalacia or rickets. Two of the patients were adults with polyostotic fibrous dysplasia; the third was an 8-year-old with fibrous dysplasia of the facial bones and rickets. In the child, resection of most of the dysplastic bone was accompanied by improvement in the metabolic bone disease. The immunoreactive PTH levels were not elevated, leading the authors to postulate that the hypophosphatemia and resultant osteomalacia in patients with fibrous dysplasia was analogous to other forms of oncogenic osteomalacia. Subsequent investigations revealed that dysplastic tissue produced FGF-23, implicating this hormone in the pathogenesis of the renal phosphate leak.169
General Renal Tubular Disorders
Another subset of patients with rickets or osteomalacia of renal origin are classified under the general heading of de Toni-Debré-Fanconi syndrome, renal Fanconi’s syndrome, or type II RTA. Characteristic findings are a generalized dysfunction of the proximal renal tubules, with excessive loss of amino acids, glucose, phosphate, uric acid, and bicarbonate in the absence of abnormal glomerular function. This results in systemic acidosis, hypophosphatemia, and dehydration, which leads to growth disturbance, rickets, or osteomalacia. The abnormal gene(s) causing autosomal-dominant renal Fanconi’s syndrome has not been identified. However, genetic and physical mapping studies have identified a locus at chromosome 15q15.3.170
The disorders with features of Fanconi’s syndrome may be further classified into primary renal abnormalities, in which the underlying defect is located within the renal tubular cells, and prerenal disorders, in which toxic metabolic substances from outside the kidney lead to derangements in tubular function (see Table 15-2). These disorders occur in adults as well as children.
Distal or type I RTA is caused by impaired secretion of hydrogen ions from the distal nephron, with resultant metabolic acidosis, nephrocalcinosis, hypokalemia, rickets, and osteomalacia. Autosomal-dominant distal RTA is caused by mutations in the SLC4A1 gene that encodes the polytopic chloride-bicarbonate exchanger AE1, which is normally expressed in red cells and at the basolateral surface of α-intercalated cells in the distal nephron.171–173 Ovalocytosis may occur. Autosomal-recessive distal RTA is caused by mutations in the ATP6VB1 and ATP6V0A4 genes that encode the B1 and A4 subunits of the collecting duct apical proton pump, vacuolar H+/ATPase, and may be associated with progressive bilateral sensorineural hearing loss.174 An additional locus at chromosome 7q33-34 was found in patients with distal RTA and no hearing loss.112
Dent’s disease is an X-linked renal tubular disorder characterized by low-molecular-weight proteinuria, hypercalciuria, nephrocalcinosis, nephrolithiasis, rickets, and renal failure and may include other features of Fanconi’s syndrome.31 The disease results from mutations of the X-linked CLCN5 gene that encodes a 746-amino-acid protein chloride channel with 12 to 13 transmembrane domains.175,176 Patients usually present at age 40 with signs and symptoms of hypokalemia and osteomalacia. Healing of the bone disease has occurred by treatment with vitamin D, alkali, and potassium. Sporadic idiopathic Fanconi’s syndrome may also present with muscle weakness and bone pain attributable to osteomalacia.177
In cystinosis (Lignac-de Toni-Fanconi syndrome),178 Fanconi-Bickel syndrome,177,179 and Lowe’s syndrome,180 the renal disease is associated with a more generalized systemic metabolic disorder. In Wilson’s disease, tyrosinemia,181 and the other inborn errors of metabolism outlined in Table 15-2, Fanconi’s syndrome may accompany the generalized systemic disease and lead to the development of bone disease.
Nephropathic cystinosis is an autosomal-recessive lysosomal storage disease characterized by renal failure by 10 years of age and other complications, including Fanconi’s syndrome. The disease is caused by mutation of the CTNS gene that encodes a 367-amino-acid protein with seven transmembrane domains that is thought to transport cystine out of lysosomes.182–184 In cystinosis, accumulation of cystine occurs in many tissues, including the kidneys, resulting in tubular dysfunction and later in glomerular failure. Recent experience with renal transplantation in patients with cystinosis has shown reaccumulation of cystine in the transplanted kidney without the reappearance of Fanconi’s syndrome, suggesting that a primary renal tubular cell defect might exist in these patients, or alternatively, that cystine deposition has different effects on the developing and mature kidney.185
Fanconi-Bickel syndrome is an autosomal-recessive disorder of carbohydrate metabolism characterized by hepatorenal glycogen accumulation, Fanconi nephropathy, impaired utilization of glucose and galactose, and rickets. The disorder results from mutations of the GLUT2 gene that encodes a glucose transporter that is present in plasma membrane of pancreatic islet β cells, hepatocytes, and absorptive epithelial cells in intestine and kidney.179
Lowe’s syndrome is a rare X-linked disorder that involves the eyes, kidney, and nervous system and is manifested by congenital cataracts, mental retardation, and renal tubular dysfunction.180 The disorder results from mutations of the OCRL gene that encodes the OCRL-1 protein, a phosphatidylinositol 4,5-bisphosphonate 5-phosphatase localized in the Golgi apparatus.186,187 Studies with kidney proximal tubules indicate that OCRL might function in membrane trafficking by regulating a specific pool of phosphatidylinositol-4,5-bisphosphonate that is associated with lysosomes, since it accumulates in the mutant kidney cells.188
Wilson’s disease is an autosomal-recessive disease caused by mutations in the ATP7B gene encoding a copper-transporting P-type ATPase that localizes to the trans-Golgi network of hepatocytes and is required to move copper into the secretory pathway, where the metal is incorporated into ceruloplasmin and excreted into bile.189,190 Clinical and pathologic manifestations in Wilson’s disease are attributed to the toxic effects of excessive accumulation of copper in liver, kidney, brain, and cornea. Most patients present with signs and symptoms secondary to liver disease, hemolytic anemia, or neurologic disease. Fanconi’s syndrome, renal dysfunction, and associated rickets and osteomalacia may predominate or be the presenting feature. Although the bone disease responds to vitamin D supplementation, renal tubular dysfunction persists, including aminoaciduria, glucosuria, uricosuria, hypercalciuria, and RTA.191 The presence of hypercalciuria and the frequent occurrence of nephrolithiasis are consistent with studies demonstrating that the defect in acidification is related to a distal rather than a proximal tubular defect.192 Treatment of Wilson’s disease with D-penicillamine significantly improves the renal tubular acidification defect,193 whereas healing of the bone disease requires oral vitamin D supplements.
Hereditary tyrosinemia type I is an autosomal recessive disease caused by mutations in the fumarylacetoacetate hydrolase (FAH) gene that encodes an enzyme involved in the last step of tyrosine degradation.194 Tyrosine accumulates in the kidney, peripheral nerves, and liver, leading to the development of cirrhosis and hepatoma.
The development of Fanconi’s syndrome in plasma cell myeloma has been attributed to the toxic effects of Bence Jones protein on the proximal renal tubule.195 Although extremely rare, osteomalacia may develop as a consequence of the renal phosphate wasting and hypophosphatemia. Osteomalacia associated with RTA has also been reported in a patient with Sjögren’s syndrome with increased urinary β2-microglobulin and retinol-binding protein.196 The patient showed clinical improvement after treatment with oral potassium citrate, calcium supplements, and glucocorticoids.
Fanconi’s syndrome and osteomalacia occur in patients with heavy-metal poisoning, particularly cadmium197 and lead,198 as well as in patients who have been exposed to outdated tetracycline.199 The bone disease is attributed to hypophosphatemia and systemic acidosis produced by impaired tubular function.
Hypophosphatasia
Hypophosphatasia is a heritable disorder characterized by deficiency in the tissue-nonspecific (bone/liver/kidney) isoenzyme of alkaline phosphatase (TNSALP), increased urinary excretion of phosphorylethanolamine, and skeletal disease that includes osteomalacia and rickets.200–203 The disease may present in infancy with hypercalcemia, renal failure, and increased intracranial pressure. Skeletal features include enlarged sutures in the skull, craniostenosis, delayed dentition, enlarged epiphyses, and prominent costochondral junctions. Genu valgum or varum may develop. The histologic picture is indistinguishable from that of other forms of rickets. Disease that presents in infancy may be fatal. Bone marrow transplants have been performed for life-threatening disease.204,205
When hypophosphatasia presents in older children, it is less severe and may present as rickets alone. In this group, serum calcium and phosphorus levels are usually normal. In the infantile forms, the disorder is inherited as an autosomal-recessive trait. The disorder in the adult, however, is probably distinct from the infantile and childhood forms and is inherited as an autosomal-dominant trait with variable penetrance. In adults, even though osteopenia may be seen, the disorder tends to be mild, and osteomalacia is not always the most prominent finding. By definition, in hypophosphatasia, enzymatic activity of TNSALP in blood and tissues is reduced. TNSALP has broad substrate activity, thus in patients with hypophosphatasia, phosphorylated molecules such as phosphorylethanolamine and phosphorylcholine are excreted in excessive amounts in the urine, and circulating levels of pyridoxal-5′-phosphate, another TNSALP substrate, are markedly elevated.206–209 Serum phosphorus levels are usually normal, and it is not clear how the biochemical findings are related to the inadequate skeletal mineralization. TNSALP, tethered through a glycophosphoinositol moiety to the osteoblast cell surface, generates inorganic phosphate (Pi) locally by cleaving various substrates even at neutral pH. One such substrate is inorganic pyrophosphate (PPi), and TNSALP is a pyrophosphatase that can generate additional Pi from PPi to drive mineralization.13 PPi, however, is also a potent inhibitor of mineralization, and it is likely that this pyrophosphatase function of TNALP is the most critical.13,14 It is probable that the concentrations of PPi are too high to allow normal mineralization at sites of bone formation.210,211
Since the alkaline phosphatase cDNA was cloned, it has been possible to study the defects in the TNSALP gene in patients with hypophosphatasia. Characterization of the patterns of clinical expression with the specific mutation in humans has provided insight into the molecular basis for the high degree of phenotypic heterogeneity in this disorder. The results indicate that the extreme heterogeneity in phenotype among patients with hypophosphatasia is due to residual enzyme activity in some individuals.127,212
Hyperparathyroidism, Hypoparathyroidism, and Pseudohypoparathyroidism
In individuals with primary hyperparathyroidism who have excessive bone resorption and high rates of bone formation, thick osteoid seams may be seen. This may present clinically as hungry bone syndrome following surgical cure of the hyperparathyroidism. It is associated with histologic changes in bone.43 However, frank vitamin D deficiency and osteomalacia may coexist with primary hyperparathyroidism, as well as with hypophosphatemic rickets and osteomalacia.
Hypoparathyroidism is a rare cause of osteomalacia.213,214 The bone disease is attributed to a decrease in formation of 1,25(OH)2D and hypocalcemia, both a consequence of lack of PTH; thus, appropriate therapy of hypoparathyroidism should prevent the mineralization defect. Osteomalacia occurs infrequently in pseudohypoparathyroidism, owing to decreased 1,25(OH)2D, a consequence of impaired renal response to PTH-induced formation of cAMP as well as hypocalcemia.215,216
Osteopetrosis
Rickets also may occur in the severe (recessive) form of osteopetrosis.217 Affected children may have low levels of serum phosphorus and calcium and increased alkaline phosphatase activity.218 In addition to rickets, decreased osteoclastic resorption and abnormal-appearing osteoclasts are observed.219 This disorder is heterogeneous, and several different cellular or biochemical defects may lead to similar clinical manifestations. It is not clear why abnormal matrix mineralization should occur in these patients. One possible explanation is a decreased local supply of mineral ions, secondary to decreased resorption in the face of relatively high rates of bone formation. Absence of the isoenzyme II of carbonic anhydrase leads to autosomal-recessive osteopetrosis with RTA and cerebral calcification.117 Reduced levels of isoenzyme II were also detected in obligate heterozygotes. A consanguineous kindred with a similar presentation was found to have a mutation in two genes, thus manifesting two separate disorders: osteopetrosis due to mutation of the osteoclast vacuolar H+/ATPase (TCIRG1) and mutation of the B1 subunit of the renal H+/ATPase (ATP6V1B1).220
Fibrogenesis Imperfecta Ossium
Described in men over age 50, fibrogenesis imperfecta ossium is a rare disorder characterized by progressively disabling skeletal pain and tenderness, forced immobilization, muscular weakness, atrophy, and contractures.221 Levels of calcium and phosphorus in the serum are normal, but alkaline phosphatase activity is increased. The radiographic abnormalities include thickened and amorphous-appearing trabeculae with spotty increases in density, reduced cortical thicknesses, and occasional pseudofractures. The abnormal “fishnet” trabecular pattern seen on conventional radiographs, combined with MR imaging of low signal intensity bone marrow on T1- and T2-weighted images, is helpful in diagnosis.222 Histologically, there is an increase in thickness of “osteoid,” which occurs diffusely on bone surfaces. Its appearance is distinctive in that this osteoid lacks the lamellar structure and typical birefringence that is characteristic of the other osteomalacic states. Electron microscopic studies reveal immature, small-diameter collagen fibers that are arranged in loops, as well as dense areas that have been termed whorls.223 A study of bone from a single patient with fibrogenesis imperfecta ossium has revealed that the collagen was more soluble in neutral salt and dilute acetic acid, suggesting defective cross-linking.224 However, direct evidence for defective cross-linking or the chemical nature of the putative defect has not been obtained. Of interest are observations that among the 17 cases of fibrogenesis imperfecta ossium reported by 1996, 5 had associated monoclonal gammopathy.225 Melphalan and calcitriol were ineffective therapy.
Renal Osteodystrophy and Aluminum Intoxication
Renal osteodystrophy comprises a heterogeneous group of skeletal disorders associated with chronic kidney disease. Prior to the introduction of hemodialysis for the therapy of chronic renal failure, the major accompanying skeletal disorder was a high turnover state, ascribable to secondary hyperparathyroidism.226 Bone formation rates were very high, and the bone surfaces covered by osteoid were also increased.
Following the introduction of hemodialysis for treatment of chronic renal failure and the widespread use of aluminum-containing gels as phosphate binders, two patterns of osteomalacia were described (see also Chapter 14). The first (type I) was associated with high-turnover bone disease, and the second (type II) was associated with “adynamic” bone disease. In osteomalacia type I, there were increased osteoid seams, accompanied by low circulating calcium and phosphorus levels. In osteomalacia type II, the osteoid seams were thinner, and there was often a dramatic reduction in tetracycline-labeled double surfaces, reflecting a low mineralization rate.227–230 In contrast to patients with osteomalacia type I, patients with osteomalacia type II tended to have normal or slightly elevated circulating concentrations of calcium and phosphorus and clinically manifested a high frequency of skeletal pain and fractures.
The presence of true osteomalacia (type I) with high turnover was ascribed to factors such as accompanying acidosis and functional vitamin D deficiency with low circulating levels of 1,25(OH)2D.226 It has now been conclusively demonstrated that although 1,25(OH)2D can modulate bone cell function, the skeletal consequences of vitamin D deficiency and even the absence of vitamin D receptors in receptor-null mice can be reversed by maintaining sufficient extracellular concentrations of calcium and phosphate.231,232 Therefore, in chronic kidney disease where serum levels of phosphate are usually high and calcium levels only slightly reduced or normal, it is difficult to ascribe mineralization defects to vitamin D deficiency alone. Circulating mineralization inhibitors—for example, inorganic pyrophosphate and others not identified—may have a role. In the past, most cases of osteomalacia type II described in patients with chronic renal failure were associated with aluminum deposition in bone at osteoid-bone interfaces. Aluminum was also implicated as a factor in pathogenesis of a significant proportion of cases of mixed uremic osteodystrophy (type I) and even some cases of predominantly hyperparathyroid bone disease.229,233 Early observations suggested that deposition of aluminum at the calcification front interfered with the mineralization process per se and impaired osteoblast proliferation and function.234 Alternatively, these observations could reflect deposition of aluminum in preexisting osteomalacic bone without direct effects on the mineralization process. Notable in this respect, in vitamin D–deficient dogs fed aluminum chloride, aluminum is deposited at the osteoid-bone interface of the osteomalacic bone235; however, the continued administration of aluminum did not impair healing of osteomalacia with vitamin D repletion.
In the past, more than two-thirds of patients on maintenance dialysis had stainable aluminum in bone. Currently, with better control of aluminum content of dialysates and replacing aluminum-based phosphate binders with calcium salts or sevelamer hydrochloride (a nonabsorbed metal-free, calcium-free phosphate binder),236–238 fewer than 10% of patients have stainable aluminum in bone.
Heritable Disorders of Vitamin D Metabolism
Rickets or osteomalacia can be caused by gene mutations of enzymes responsible for 25-hydroxylation of vitamin D, for 1α-hydroxylation of 25(OH)D, or mutations of the vitamin D receptor (see Chapter 11). Isolated deficiency of 25(OH)D is a rare autosomal-recessive disorder with rickets caused by impaired hepatic production of 25(OH)D. It is attributed to deficiency of the 25(OH)D-hydroxylase, the enzyme that modulates conversion of vitamin D to 25(OH)D.239–241 The disease is characterized by a low serum 25(OH)D, a normal serum 1,25(OH)2D, hypocalcemia, increases in serum alkaline phosphatase, secondary hyperparathyroidism, and early onset of rickets that responds to physiologic doses of 1α(OH)D3 or pharmacologic doses of vitamin D; the condition relapses when treatment is discontinued. Recently, the CYP2R1 gene that encodes a hepatic microsomal vitamin D-25-hydroxylase that hydroxylates both vitamin D2 and vitamin D3 was cloned and sequenced. A homozygous L99P inactivating mutation of the CYP2R1 gene was the cause of the deficiency of 25(OH)D and rickets in a Nigerian boy with the disorder.242 These findings provide evidence that CYP2R1 is a key vitamin D 25-hydroxylase.
Pseudo–vitamin D–deficiency rickets is an autosomal recessive disorder caused by impaired renal production of 1,25(OH)2D. The disease is caused by mutations of the CYP27B1 gene that encodes the 25(OH)D 1α-hydroxylase, the mitochondrial enzyme responsible for conversion of 25(OH)D to 1,25(OH)2D.243–245 Affected individuals are apparently normal at birth and develop the clinical and biochemical changes of rickets during the first year of life. Hypocalcemia, hypophosphatemia, and elevated serum immunoreactive PTH and alkaline phosphatase are consistent findings. The diagnosis is made by demonstration of a normal serum 25(OH)D and low or undetectable serum 1,25(OH)2D in a patient with rickets who responds to physiologic doses of 1,25(OH)2D3.
Hereditary vitamin D–resistant rickets is a rare autosomal disorder characterized by resistance of target organs to 1,25(OH)2D.246 The disease is caused by mutations of the vitamin D receptor (VDR) gene.247 Onset of symptoms can occur at any age from infancy to adolescence, and occurrence is usually familial. Patients have hypocalcemia, hypophosphatemia, elevation of serum alkaline phosphatase and immunoreactive PTH, normal or increased serum 25(OH)D, and marked elevation of serum 1,25(OH)2D. The disorder is transmitted as an autosomal-recessive trait and often results from consanguineous parentage. Patients may also develop alopecia. Because the pathophysiologic abnormality underlying this disorder is a receptor defect, no treatment has been uniformly successful. Therapeutic responses to pharmacologic doses of vitamin D metabolites and oral calcium supplements vary. Although 1α-hydroxylated metabolites have been the favored treatment, the high levels of circulating 1,25(OH)2D demonstrate that formation of this metabolite is not impaired and suggest that treatment with vitamin D or 25(OH)D would be effective. In some severely affected individuals, hypocalcemia persists, as do the rachitic and osteomalacic lesions. Because the main physiologic effect of 1,25(OH)2D is thought to be the promotion of intestinal calcium absorption, treatment with parenteral calcium infusions, circumventing the intestinal resistance to 1,25(OH)2D, can normalize the biochemical abnormalities and lead to clinical and radiologic remission of the osteomalacic lesions.248
Treatment
There is growing evidence that the recommended daily intake (RDI) by the Institute of Medicine’s Food and Nutrition Board for 200 IU of vitamin D in young adults is inadequate. The American Academy of Pediatrics has suggested an RDI of 400 IU/day for children 0 to 18 years of age. Similarly, the definition of vitamin D deficiency is unclear: the value for the lower limit of the normal range for serum 25(OH)D has never been established on a scientific basis. Assigned reference values were determined in subjects, based on the fact that they did not have overt rickets or osteomalacia.82 Three degrees of hypovitaminosis D bone disease have been defined.4 Stage 1 is characterized by decreased intestinal absorption of calcium resulting in osteoporosis without histologic skeletal changes; stage 2 by decreased calcium absorption, osteoporosis, and early histologic features of osteomalacia and no clinical or laboratory features of osteomalacia; and stage 3 by clinical and histologic osteomalacia. Currently, recommended doses of vitamin D prevent changes found in stages 2 and 3 but not those found in stage 1.
Vitamin D deficiency leads to impaired intestinal calcium absorption, secondary hyperparathyroidism, and bone loss. A number of studies indicate that the change in PTH secretion occurs at a value of approximately 80 nmol/L for serum 25(OH)D. Further, increasing the serum 25(OH)D from approximately 50 nmol/L to 80 nmol/L, values that are well within the normal range, increases calcium absorption by nearly two-thirds249 and reduces the risk of osteoporotic fracture by one-third.250 Importantly, in a large cohort of subjects in the Second National Health and Nutrition Examination Survey (NHANES II),251 bone mineral density of the hip varied directly with serum 25(OH)D values throughout the normal range. This was notable in both pre- and postmenopausal women of Caucasian, Hispanic, and African descent. For these reasons, it is apparent that the current RDI for vitamin D is not adequate for accrual of peak bone mass. These results have profound implications for defining the optimal RDI for vitamin D. A study in normal men by Heaney et al.,252 the first to attempt to define optimal requirements, indicated that daily requirement of vitamin D from all sources during winter months (supplement, food, tissue stores) is on the order of approximately 3800 IU (95 µg). In other studies,81 administration of a dose of vitamin D of 4000 IU (100 µg) per day was not associated with abnormal increases in serum 25(OH)D, serum calcium, or urinary calcium in any of the normal subjects; however, calcium intake was not factored in. A comprehensive review of the literature showed that hypercalcemia did not occur when extended doses of vitamin D as high as 10,000 IU (250 µg) per day were administered. The need for higher intake of vitamin D is also supported by the inverse relationship that exists between sun exposure and a number of cancers, including colon, prostate, and breast.253,254 These findings should be kept in mind while considering the recommended doses of vitamin D and its analogues for treatment of patients with osteomalacia and rickets outlined later. The goals for treatment of patients with osteomalacia or rickets are to correct hypocalcemia if it exists, to reverse skeletal deformities and secondary hyperparathyroidism, and to prevent vitamin D intoxication which may lead to hypercalcemia, hypercalciuria, nephrocalcinosis, and nephrolithiasis.
1,25(OH)2D3 is available in 0.25 µg and 0.50 µg capsules and in oral and intravenous preparations containing 1 µg/mL. The advantages of calcitriol are its rapid onset and offset of action. The half-life of the drug is less than 6 hours.255 One disadvantage is that hypercalcemia may occur even long after an apparently optimal dose has been used.256 Since the occurrence of hypercalcemia during treatment with 1,25(OH)2D3 cannot be predicted, patients must be monitored closely. 1α(OH)D2 is approved for use in the treatment of secondary hyperparathyroidism in patients with kidney failure.257 It is marketed in 2.5 µg capsules and 1-mL solutions containing 2 µg/mL. After administration, it undergoes conversion to 1,25(OH)2D2.258
In children with nutritional rickets caused by calcium deficiency, increasing the calcium intake to between 800 and 1500 mg/day was shown to heal the bone disease.139,259 Several protocols are recommended for treatment of vitamin D–deficient bone disease; however, most of these have reversal of rickets and osteomalacia as an end point. They do not have as a goal the achievement of 25(OH)D levels that are “optimal” for bone mass accrual and fracture prevention. A trial with small daily oral doses of vitamin D, 50 µg (2000 IU), for 3 to 4 weeks is suggested because a clear biochemical response occurs in classic rickets and osteomalacia resulting from simple vitamin D deficiency. However, based on the study of Heaney et al.,252 this intake is insufficient to maintain normal levels of vitamin D in healthy men. Furthermore, this dose is insufficient to treat the metabolic forms of rickets that are associated with renal insufficiency, malabsorption, and various hereditary and acquired forms of vitamin D resistance or dependency. Current recommendations include treatment with 50,000 to 500,000 IU over 1 to 3 months, followed by maintenance doses of 400 IU to 1200 IU/day, to achieve a normal circulating 25(OH)D level. Patients on chronic anticonvulsant therapy may require higher maintenance doses.260 Unlike adults in whom the effects of anticonvulsant drugs in producing osteomalacia may be self-limited, children may require persistent therapy because long-term anticonvulsant therapy has a greater effect on the skeleton of growing children than that of adults. Osteomalacia can also be corrected with relatively low doses of 1α-hydroxylated metabolites.261 However, these more potent analogues should be reserved for patients with more pronounced forms of calcium malabsorption or with impaired 1α-hydroxylation. Administration of vitamin D to patients with vitamin D–deficiency rickets or osteomalacia invariably results in healing of the bone disease, although the patterns of response, particularly with regard to changes in serum calcium, phosphorus, alkaline phosphatase, and immunoreactive PTH, show marked variation. There may be an initial increase in serum alkaline phosphatase, indicative of healing of the osteomalacic lesions, followed by a gradual fall in both alkaline phosphatase and immunoreactive PTH.
1,25(OH)2D3 is often most useful in treatment of diseases in which renal synthesis of the metabolite is diminished or impaired (hypoparathyroidism, pseudohypoparathyroidism, renal insufficiency, vitamin D–dependent rickets type I, tumor-induced osteomalacia, hypophosphatemic rickets) or there is resistance to its effects at the cellular level. Sometimes osteomalacia or rickets associated with hereditary vitamin D–resistant rickets responds to treatment with vitamin D.246 However, in instances of a more profound resistance, 1,25(OH)2D3 may be beneficial. Doses as high as 15 to 20 µg per day are sometimes required and even so may not be effective. Under these circumstances, long-term parenteral administration of calcium can be used to heal the bone disease.248
Hypercalcemia and hypercalciuria are potential complications of treatment with vitamin D.262,263 Hypercalcemia resulting from vitamin D intoxication is caused by a combination of increased intestinal absorption of calcium and increased osteoclastic bone resorption. Impairment in kidney function may develop and lead to nephrocalcinosis, nephrolithiasis, and even death.263 Patients may be asymptomatic or have anorexia, nausea, vomiting, weight loss, headache, constipation, polyuria, polydipsia, and altered mental status. Treatment with intravenous fluids is sometimes effective, but elevated serum 25(OH)D and serum calcium values may persist. In addition to hydration, glucocorticoids may be effective in lowering serum calcium levels, and additional benefit may be obtained by impairing osteoclastic bone resorption with intravenous bisphosphonates. Patients on long-term treatment with vitamin D should be carefully followed with measurement of serum and urinary calcium and serum creatinine. This is critical, since there is no way of predicting when or in whom vitamin D intoxication will develop and/or recur.
Treatment of hypophosphatemia, not associated with vitamin D deficiency, involves oral supplementation with neutral phosphate salts, including sodium or potassium salts. Greater elevations in serum phosphorus values have been reported in individuals receiving the potassium salt.36 The less favorable results with the sodium salt are most likely related to the effects of the sodium ion on increasing renal phosphate clearance. The rise in serum phosphorus after a single oral dose of phosphate is transient,157 so phosphate supplements must be administered at frequent intervals. The precise amount of phosphate must be individualized for each patient and is a reflection of the degree of renal loss. In individuals with severe leaks, as much as 1000 mg of elemental phosphorus is required every 4 to 6 hours to effect sustained elevations of serum phosphorus values. Optimal treatment of tumor-induced osteomalacia requires removal of the tumor.
The efficacy of therapy cannot be assessed with a single fasting determination of serum phosphorus; multiple measurements of serum values at various times after each dose are required. Emptying the capsules and dissolving the salt in water or other liquid may improve intestinal absorption. When therapy is initiated, most patients experience some degree of gastrointestinal distress, including cramps and diarrhea. Therefore, initial doses should be low, and increments should be gradually introduced as tolerated. Simultaneous use of phosphate and vitamin D metabolites accelerated healing in children with the X-linked form of hypophosphatemic osteomalacia.159 Whether the improved serum phosphorus values that are seen with vitamin D or 1,25(OH)2D3 are accounted for by increased intestinal absorption of phosphate or decreased renal loss due to reductions in PTH has not been established. However, the use of a calcimimetics to decrease PTH secretion has been used as adjuvant treatment for hereditary and acquired hypophosphatemia associated with impaired tubular reabsorption.264,265
Hypophosphatasia presents a treatment challenge. Unless there is deficiency of vitamin D, treatment with vitamin D metabolites and calcium is contraindicated, since serum calcium, phosphorus, 25(OH)D, and 1,25(OH)2D are usually normal,205 and such treatment may exaggerate any tendency to develop hypercalcemia and hypercalciuria. Bone marrow transplantation has been reported to improve the bone disease in the infantile form.205 Similarly, in osteomalacia associated with osteopetrosis, the most effective treatment is bone marrow or stem cell transplant to provide normally functioning osteoclasts.266
References
1. Sabbagh, Y, Carpenter, TO, Demay, MB. Hypophosphatemia leads to rickets by impairing caspase-mediated apoptosis of hypertrophic chondrocytes. Proc Natl Acad Sci U S A. 2005;102:9637–9642.
2. Hutchison, FN, Bell, NH. Osteomalacia and rickets. Semin Nephrol. 1992;12:127–145.
3. Mankin, HJ. Rickets, osteomalacia, and renal osteodystrophy. Part II. J Bone Joint Surg Am. 1974;56:352–386.
4. Parfitt, AM. Osteomalacia and related disorders. In: Avioli LV, Krane SM, eds. Metabolic Bone Disease. San Diego: Academic Press; 1998:328–386.
5. Anderson, HC. Matrix vesicles and calcification. Curr Rheumatol Rep. 2003;5:222–226.
6. Boskey, AL. Mineral analysis provides insights into the mechanism of biomineralization. Calcif Tissue Int. 2003;72:533–536.
7. Glimcher, M. The nature of the mineral phase in bone: biological and clinical implications. In: Metabolic Bone Disease. San Diego: Academic Press; 1998:23–50.
8. Slavkin, H, Price, P. Chemistry and Biology of Mineralized Tissues. Amsterdam: Excerpta Medica; 1992.
9. Tong, W, Glimcher, MJ, Katz, JL, et al. Size and shape of mineralites in young bovine bone measured by atomic force microscopy. Calcif Tissue Int. 2003;72:592–598.
10. Wu, Y, Ackerman, JL, Strawich, ES, et al. Phosphate ions in bone: identification of a calcium-organic phosphate complex by 31P solid-state NMR spectroscopy at early stages of mineralization. Calcif Tissue Int. 2003;72:610–626.
11. Qin, C, Brunn, JC, Jones, J, et al. A comparative study of sialic acid-rich proteins in rat bone and dentin. Eur J Oral Sci. 2001;109:133–141.
12. Eanes, ED, Hailer, AW. Anionic effects on the size and shape of apatite crystals grown from physiological solutions. Calcif Tissue Int. 2000;66:449–455.
13. Hessle, L, Johnson, KA, Anderson, HC, et al. Tissue-nonspecific alkaline phosphatase and plasma cell membrane glycoprotein-1 are central antagonistic regulators of bone mineralization. Proc Natl Acad Sci U S A. 2002;99:9445–9449.
14. Timms, AE, Zhang, Y, Russell, RG, et al. Genetic studies of disorders of calcium crystal deposition. Rheumatology (Oxford). 2002;41:725–729.
15. Ho, AM, Johnson, MD, Kingsley, DM. Role of the mouse ank gene in control of tissue calcification and arthritis. Science. 2000;289:265–270.
16. Pendleton, A, Johnson, MD, Hughes, A, et al. Mutations in ANKH cause chondrocalcinosis. Am J Hum Genet. 2002;71:933–940.
17. Murshed, M, Harmey, D, Millan, JL, et al. Unique coexpression in osteoblasts of broadly expressed genes accounts for the spatial restriction of ECM mineralization to bone. Genes Dev. 2005;19:1093–1104.
18. Murshed, M, Schinke, T, McKee, MD, et al. Extracellular matrix mineralization is regulated locally; different roles of two GLA-containing proteins. J Cell Biol. 2004;165:625–630.
19. Lee, WR. Bone formation in Paget’s disease. A quantitative microscopic study using tetracycline markers. J Bone Joint Surg Br. 1967;49:146–153.
20. Frost, HM. Tetracycline-based histological analysis of bone remodeling. Calcif Tissue Res. 1969;3:211–237.
21. Bordier, P, Tun Chot, S. Quantitative histology of metabolic bone disease. Clin Endocrinol Metab. 1972;1:197–215.
22. Rao, DS, Villanueva, A, Mathews, M. Histological evolution of vitamin D-depletion in patients with intestinal malabsorption or dietary deficiency. In: Frame B, Potts JJ, eds. Clinical Disorders of Bone and Mineral Metabolism. Amsterdam: Excerpta Medica; 1983:224–226.
23. Boyce, BF, Smith, L, Fogelman, I, et al. Focal osteomalacia due to low-dose diphosphonate therapy in Paget’s disease. Lancet. 1984;1:821–824.
24. Frame, B, Parfitt, AM. Osteomalacia: current concepts. Ann Intern Med. 1978;89:966–982.
25. Marie, PJ, Glorieux, FH. Relation between hypomineralized periosteocytic lesions and bone mineralization in vitamin D-resistant rickets. Calcif Tissue Int. 1983;35:443–448.
26. Howell, DS. Histologic observations and biochemical composition of rachitic cartilage with special reference to mucopolysaccharides. Arthritis Rheum. 1965;8:337–354.
27. Baylink, D, Stauffer, M, Wergedal, J, et al. Formation, mineralization, and resorption of bone in vitamin D-deficient rats. J Clin Invest. 1970;49:1122–1134.
28. Barnes, MJ, Constable, BJ, Morton, LF, et al. Bone collagen metabolism in vitamin D deficiency. Biochem J. 1973;132:113–115.
29. Barnes, MJ, Constable, BJ, Morton, LF, et al. The influence of dietary calcium deficiency and parathyroidectomy on bone collagen structure. Biochim Biophys Acta. 1973;328:373–382.
30. Toole, BP, Kang, AH, Trelstad, RL, et al. Collagen heterogeneity within different growth regions of long bones of rachitic and non-rachitic chicks. Biochem J. 1972;127:715–720.
31. Dent, CE. Rickets (and osteomalacia), nutritional and metabolic (1919–69). Proc R Soc Med. 1970;63:401–408.
32. Reginato, AJ, Falasca, GF, Pappu, R, et al. Musculoskeletal manifestations of osteomalacia: report of 26 cases and literature review. Semin Arthritis Rheum. 1999;28:287–304.
33. Prineas, JW, Mason, AS, Henson, RA. Myopathy in metabolic bone disease. Br Med J. 1965;1:1034–1036.
34. Smith, R, Stern, G. Myopathy, osteomalacia and hyperparathyroidism. Brain. 1967;90:593–602.
35. Schott, GD, Wills, MR. Muscle weakness in osteomalacia. Lancet. 1976;1:626–629.
36. Nagant de Deuxchaisnes, C, Krane, SM. The treatment of adult phosphate diabetes and Fanconi syndrome with neutral sodium phosphate. Am J Med. 1967;43:508–543.
37. Smith, R, Newman, RJ, Radda, GK, et al. Hypophosphataemic osteomalacia and myopathy: studies with nuclear magnetic resonance spectroscopy. Clin Sci (Lond). 1984;67:505–509.
38. Dent, CE, Stamp, TC. Vitamin D, rickets and osteomalacia. In: Avioli LV, Krane SM, eds. Metabolic Bone Disease. New York: Academic Press, 1977.
39. Krane, SM, Parsons, V, Kunin, AS. Studies of the metabolism of epiphyseal cartilage. In: Basset C, ed. Cartilage Degradation and Repair. Washington DC: National Research Council; 1967:43.
40. Le May, M, Blunt, JW, Jr. A factor determining the location of pseudofractures in osteomalacia. J Clin Invest. 1949;28:521–525.
41. Steinbach, HL, Kolb, FO, Gilfillan, R. A mechanism of the production of pseudofractures in osteomalacia (Milkman’s syndrome). Radiology. 1954;62:388–395.
42. Jackson, WP, Dowdle, E, Linder, GC. Vitamin-D-resistant osteomalacia. Br Med J. 1958;1:1269–1274.
43. Albright, F, Reifenstein, EJ. Parathyroid Glands and Metabolic Bone Disease. Baltimore: The Williams and Wilkins Company; 1948.
44. de Seze, S, Lichtwitz, A, Ryckewaert, A, et al. The Looser-Milkman syndrome. Study of 60 cases. Sem Hop. 1962;38:2005–2025.
45. Milkman, L. Pseudofractures (hungry osteopathy, late rickets, osteomalacia): Report of a case. Am J Roentgenol. 1930;24:29–37.
46. Milkman, L. Multiple spontaneous idiopathic symmetrical fractures. Am J Roentgenol. 1934;32:622–634.
47. Ball, J, Garner, A. Mineralisation of woven bone in osteomalacia. J Pathol Bacteriol. 1966;91:563–567.
48. Steinbach, HL, Noetzli, M. Roentgen appearance of the skeleton in osteomalacia and rickets. Am J Roentgenol Radium Ther Nucl Med. 1964;91:955–972.
49. Burnstein, MI, Lawson, JP, Kottamasu, SR, et al. The enthesopathic changes of hypophosphatemic osteomalacia in adults: radiologic findings. Am J Roentgenol. 1989;153:785–790.
50. Polisson, RP, Martinez, S, Khoury, M, et al. Calcification of entheses associated with X-linked hypophosphatemic osteomalacia. N Engl J Med. 1985;313:1–6.
51. Arneil, GC. Nutritional rickets in children in Glasgow. Proc Nutr Soc. 1975;34:101–109.
52. Arneil, GC, Crosbie, JC. Infantile rickets returns to Glasgow. Lancet. 1963;2:423–425.
53. Dunnigan, MG, Paton, JP, Haase, S, et al. Late rickets and osteomalacia in the Pakistani community in Glasgow. Scott Med J. 1962;7:159–167.
54. Benson, PF, Stroud, CE, Mitchell, NJ, et al. Rickets in immigrant children in London. Br Med J. 1963;1:1054–1056.
55. Chanarin, I, Malkowska, V, O’Hea, AM, et al. Megaloblastic anaemia in a vegetarian Hindu community. Lancet. 1985;2:1168–1172.
56. Finch, PJ, Ang, L, Colston, KW, et al. Blunted seasonal variation in serum 25-hydroxy vitamin D and increased risk of osteomalacia in vegetarian London Asians. Eur J Clin Nutr. 1992;46:509–515.
57. Henderson, JB, Dunnigan, MG, McIntosh, WB, et al. Asian osteomalacia is determined by dietary factors when exposure to ultraviolet radiation is restricted: a risk factor model. Q J Med. 1990;76:923–933.
58. Shaunak, S, Colston, K, Ang, L, et al. Vitamin D deficiency in adult British Hindu Asians: a family disorder. Br Med J (Clin Res Ed). 1985;291:1166–1168.
59. Ralston, SH, Willocks, L, Pitkeathly, DA, et al. High prevalence of unrecognized osteomalacia in hospital patients with rheumatoid arthritis. Br J Rheumatol. 1988;27:202–205.
60. Botella-Carretero, JI, Alvarez-Blasco, F, Villafruela, JJ, et al. Vitamin D deficiency is associated with the metabolic syndrome in morbid obesity. Clin Nutr. 2007;26:573–580.
61. Charles, P, Mosekilde, L, Sondergard, K, et al. Treatment with high-dose oral vitamin D2 in patients with jejunoileal bypass for morbid obesity. Effects on calcium and magnesium metabolism, vitamin D metabolites, and faecal lag time. Scand J Gastroenterol. 1984;19:1031–1038.
62. Crowley, LV, Seay, J, Mullin, G. Late effects of gastric bypass for obesity. Am J Gastroenterol. 1984;79:850–860.
63. Parfitt, AM, Podenphant, J, Villanueva, AR, et al. Metabolic bone disease with and without osteomalacia after intestinal bypass surgery: a bone histomorphometric study. Bone. 1985;6:211–220.
64. Basha, B, Rao, DS, Han, ZH, et al. Osteomalacia due to vitamin D depletion: a neglected consequence of intestinal malabsorption. Am J Med. 2000;108:296–300.
65. Bisballe, S, Eriksen, EF, Melsen, F, et al. Osteopenia and osteomalacia after gastrectomy: interrelations between biochemical markers of bone remodelling, vitamin D metabolites, and bone histomorphometry. Gut. 1991;32:1303–1307.
66. Tovey, FI, Godfrey, JE, Lewin, MR. A gastrectomy population: 25–30 years on. Postgrad Med J. 1990;66:450–456.
67. Daum, F, Rosen, JF, Roginsky, M, et al. 25-Hydroxycholecalciferol in the management of rickets associated with extrahepatic biliary atresia. J Pediatr. 1976;88:1041–1043.
68. Heubi, JE, Tsang, RC, Steichen, JJ, et al. 1,25-Dihydroxyvitamin D3 in childhood hepatic osteodystrophy. J Pediatr. 1979;94:977–982.
69. Roberts, WA, Badger, VM. Osteomalacia of very-low-birth-weight infants. J Pediatr Orthop. 1984;4:593–598.
70. Heckmatt, JZ, Peacock, M, Davies, AE, et al. Plasma 25-hydroxyvitamin D in pregnant Asian women and their babies. Lancet. 1979;2:546–548.
71. Specker, BL, Tsang, RC, Hollis, BW. Effect of race and diet on human-milk vitamin D and 25-hydroxyvitamin D. Am J Dis Child. 1985;139:1134–1137.
72. Haddad, JG, Stamp, TC. Circulating 25-hydroxyvitamin D in man. Am J Med. 1974;57:57–62.
73. Lester, E, Skinner, RK, Wills, MR. Seasonal variation in serum-25-hydroxyvitamin-D in the elderly in Britain. Lancet. 1977;1:979–980.
74. Stamp, TC, Round, JM. Seasonal changes in human plasma levels of 25-hydroxyvitamin D. Nature. 1974;247:563–565.
75. Dawson-Hughes, B, Dallal, GE, Krall, EA, et al. Effect of vitamin D supplementation on wintertime and overall bone loss in healthy postmenopausal women. Ann Intern Med. 1991;115:505–512.
76. Harris, SS, Dawson-Hughes, B. Seasonal changes in plasma 25-hydroxyvitamin D concentrations of young American black and white women. Am J Clin Nutr. 1998;67:1232–1236.
77. Thomas, MK, Lloyd-Jones, DM, Thadhani, RI, et al. Hypovitaminosis D in medical inpatients. N Engl J Med. 1998;338:777–783.
78. MacLaughlin, J, Holick, MF. Aging decreases the capacity of human skin to produce vitamin D3. J Clin Invest. 1985;76:1536–1538.
79. Tsai, KS, Heath, H, 3rd., Kumar, R, et al. Impaired vitamin D metabolism with aging in women. Possible role in pathogenesis of senile osteoporosis. J Clin Invest. 1984;73:1668–1672.
80. Ebeling, PR, Sandgren, ME, DiMagno, EP, et al. Evidence of an age-related decrease in intestinal responsiveness to vitamin D: relationship between serum 1,25-dihydroxyvitamin D3 and intestinal vitamin D receptor concentrations in normal women. J Clin Endocrinol Metab. 1992;75:176–182.
81. Vieth, R. Vitamin D supplementation, 25-hydroxyvitamin D concentrations, and safety. Am J Clin Nutr. 1999;69:842–856.
82. Heaney, RP. Lessons for nutritional science from vitamin D. Am J Clin Nutr. 1999;69:825–826.
83. Goel, KM, Sweet, EM, Logan, RW, et al. Florid and subclinical rickets among immigrant children in Glasgow. Lancet. 1976;1:1141–1145.
84. Lawson, M, Thomas, M. Vitamin D concentrations in Asian children aged 2 years living in England: population survey. BMJ. 1999;318:28.
85. Stanbury, SW, Torkington, P, Lumb, GA, et al. Asian rickets and osteomalacia: patterns of parathyroid response in vitamin D deficiency. Proc Nutr Soc. 1975;34:111–117.
86. Loomis, WF. Skin-pigment regulation of vitamin-D biosynthesis in man. Science. 1967;157:501–506.
87. Doxiadis, S, Angelis, C, Karatzas, P, et al. Genetic aspects of nutritional rickets. Arch Dis Child. 1976;51:83–90.
88. Ford, JA, Colhoun, EM, McIntosh, WB, et al. Rickets and osteomalacia in the Glasgow Pakistani community, 1961–71. Br Med J. 1972;2:677–680.
89. Heaney, RP. Long-latency deficiency disease: insights from calcium and vitamin D. Am J Clin Nutr. 2003;78:912–919.
90. Wills, MR, Phillips, JB, Day, RC, et al. Phytic acid and nutritional rickets in immigrants. Lancet. 1972;1:771–773.
91. Dent, CE, Round, JM, Rowe, DJ, et al. Effect of chapattis and ultraviolet irradiation on nutritional rickets in an Indian immigrant. Lancet. 1973;1:1282–1284.
92. Dent, CE, Richens, A, Rowe, DJ, et al. Osteomalacia with long-term anticonvulsant therapy in epilepsy. Br Med J. 1970;4:69–72.
93. Scriver, CR. Rickets and the pathogenesis of impaired tubular transport of phosphate and other solutes. Am J Med. 1974;57:43–49.
94. Arnaud, C, Glorieux, F, Scriver, CR. Serum parathyroid hormone levels in acquired vitamin D deficiency of infancy. Pediatrics. 1972;49:837–840.
95. Muldowney, FP, Freaney, R, McGeeney, D. Renal tubular acidosis and amino-aciduria in osteomalacia of dietary or intestinal origin. Q J Med. 1968;37:517–539.
96. Rao, DS, Parfitt, AM, Kleerekoper, M, et al. Dissociation between the effects of endogenous parathyroid hormone on adenosine 3′,5′-monophosphate generation and phosphate reabsorption in hypocalcemia due to vitamin D depletion: an acquired disorder resembling pseudohypoparathyroidism type II. J Clin Endocrinol Metab. 1985;61:285–290.
97. Adams, JS, Clemens, TL, Parrish, JA, et al. Vitamin-D synthesis and metabolism after ultraviolet irradiation of normal and vitamin-D-deficient subjects. N Engl J Med. 1982;306:722–725.
98. Eastwood, JB, de Wardener, HE, Gray, RW, et al. Normal plasma-1,25-(OH)2-vitamin-D concentrations in nutritional osteomalacia. Lancet. 1979;1:1377–1378.
99. Goodman, AD, Lemann, J, Jr., Lennon, EJ, et al. Production, excretion, and net balance of fixed acid in patients with renal acidosis. J Clin Invest. 1965;44:495–506.
100. Lemann, J, Jr., Bushinsky, DA, Hamm, LL. Bone buffering of acid and base in humans. Am J Physiol Renal Physiol. 2003;285:F811–F832.
101. Lemann, J, Jr., Lennon, EJ, Goodman, AD, et al. The net balance of acid in subjects given large loads of acid or alkali. J Clin Invest. 1965;44:507–517.
102. Lemann, J, Jr., Litzow, JR, Lennon, EJ. The effects of chronic acid loads in normal man: further evidence for the participation of bone mineral in the defense against chronic metabolic acidosis. J Clin Invest. 1966;45:1608–1614.
103. Arnett, T. Regulation of bone cell function by acid-base balance. Proc Nutr Soc. 2003;62:511–520.
104. Nordstrom, T, Shrode, LD, Rotstein, OD, et al. Chronic extracellular acidosis induces plasmalemmal vacuolar type H+ ATPase activity in osteoclasts. J Biol Chem. 1997;272:6354–6360.
105. Mori, H, Sakai, H, Morihata, H, et al. Regulatory mechanisms and physiological relevance of a voltage-gated H+ channel in murine osteoclasts: phorbol myristate acetate induces cell acidosis and the channel activation. J Bone Miner Res. 2003;18:2069–2076.
106. Harrison, HE, Harrison, HC. Physiology of vitamin D. In: Rodahl K, Nicholson JT, Brown EMJ, eds. Bone as a Tissue. New York: McGaw-Hill; 1960:300.
107. Frick, KK, Bushinsky, DA. Metabolic acidosis stimulates RANKL RNA expression in bone through a cyclo-oxygenase-dependent mechanism. J Bone Miner Res. 2003;18:1317–1325.
108. Ludwig, MG, Vanek, M, Guerini, D, et al. Proton-sensing G-protein-coupled receptors. Nature. 2003;425:93–98.
109. Donohoe, JF, Freaney, R, Muldowney, FP. Osteomalacia in ureterosigmoidostomy. Ir J Med Sci. 1969;8:523–530.
110. Tschopp, AB, Lippuner, K, Jaeger, P, et al. No evidence of osteopenia 5 to 8 years after ileal orthotopic bladder substitution. J Urol. 1996;155:71–75.
111. Lee, SW, Russell, J, Avioli, LV. 25-hydroxycholecalciferol to 1,25-dihydroxycholecalciferol: conversion impaired by systemic metabolic acidosis. Science. 1977;195:994–996.
112. Karet, FE. Inherited distal renal tubular acidosis. J Am Soc Nephrol. 2002;13:2178–2184.
113. Morris, RC, Jr. Renal tubular acidosis. Mechanisms, classification and implications. N Engl J Med. 1969;281:1405–1413.
114. Morris, RC, Jr., McSherry, E. Symposium on acid-base homeostasis. Renal acidosis. Kidney Int. 1972;1:322–340.
115. Richards, P, Chamberlain, MJ, Wrong, OM. Treatment of osteomalacia of renal tubular acidosis by sodium bicarbonate alone. Lancet. 1972;2:994–997.
116. York, SE, Yendt, ER. Osteomalacia associated with renal bicarbonate loss. Can Med Assoc J. 1966;94:1329–1342.
117. Sly, WS, Hewett-Emmett, D, Whyte, MP, et al. Carbonic anhydrase II deficiency identified as the primary defect in the autosomal recessive syndrome of osteopetrosis with renal tubular acidosis and cerebral calcification. Proc Natl Acad Sci U S A. 1983;80:2752–2756.
118. Sly, WS, Whyte, MP, Sundaram, V, et al. Carbonic anhydrase II deficiency in 12 families with the autosomal recessive syndrome of osteopetrosis with renal tubular acidosis and cerebral calcification. N Engl J Med. 1985;313:139–145.
119. Whyte, MP, Murphy, WA, Fallon, MD, et al. Osteopetrosis, renal tubular acidosis and basal ganglia calcification in three sisters. Am J Med. 1980;69:64–74.
120. McMahon, C, Will, A, Hu, P, et al. Bone marrow transplantation corrects osteopetrosis in the carbonic anhydrase II deficiency syndrome. Blood. 2001;97:1947–1950.
121. Dent, CE, Harris, H. Hereditary forms of rickets and osteomalacia. J Bone Joint Surg Br. 1956;38-B:204–226.
122. Henneman, PH, Dempsey, EF, Carroll, EL, et al. Acquired vitamin D-resistant osteomalacia: a new variety characterized by hypercalcemia, low serum bicarbonate and hyperglycinuria. Metabolism. 1962;11:103–116.
123. Kallmeyer, J, Dunea, G, Schwartz, FD. Hypophosphatemic osteomalacia with hyperglycinuria. Ann Intern Med. 1967;66:136–141.
124. Rico, H, Gomez-Castresana, F, Hernandez, ER, et al. Adult hypophosphatemic osteomalacia: report of two cases. Clin Rheumatol. 1985;4:325–334.
125. Scriver, CR, Goldbloom, RB, Roy, CC. Hypophosphatemic rickets with renal hyperglycinuria, renal glucosuria, and glycyl-prolinuria: a syndrome with evidence for renal tubular secretion of phosphorus. Pediatrics. 1964;34:357–371.
126. Pierce, WM, Jr., Nardin, GF, Fuqua, MF, et al. Effect of chronic carbonic anhydrase inhibitor therapy on bone mineral density in white women. J Bone Miner Res. 1991;6:347–354.
127. Waite, LC, Volkert, WA, Kenny, AD. Inhibition of bone resorption by acetazolamide in the rat. Endocrinology. 1970;87:1129–1139.
128. Baker, LR, Ackrill, P, Cattell, WR, et al. Iatrogenic osteomalacia and myopathy due to phosphate depletion. Br Med J. 1974;3:150–152.
129. Bloom, W, Flinchum, D. Osteomalacia with pseudofractures caused by ingestion of aluminum hydroxide. JAMA. 1960;174:1327–1330.
130. Dent, CE, Winter, CS. Osteomalacia due to phosphate depletion from excessive aluminium hydroxide ingestion. Br Med J. 1974;1:551–552.
131. Levy, Y, Bansal, M, Zackson, DA, et al. Phosphate depletion syndrome: case report with bone and muscle histology findings and review of the literature. JPEN J Parenter Enteral Nutr. 1988;12:313–317.
132. Lotz, M, Zisman, E, Bartter, FC. Evidence for a phosphorus-depletion syndrome in man. N Engl J Med. 1968;278:409–415.
133. Godsall, JW, Baron, R, Insogna, KL. Vitamin D metabolism and bone histomorphometry in a patient with antacid-induced osteomalacia. Am J Med. 1984;77:747–750.
134. Stein, JH, Smith, WO, Ginn, HE. Hypophosphatemia in acute alcoholism. Am J Med Sci. 1966;252:78–83.
135. Territo, MC, Tanaka, KR. Hypophosphatemia in chronic alcoholism. Arch Intern Med. 1974;134:445–447.
136. de Vernejoul, MC, Bielakoff, J, Herve, M, et al. Evidence for defective osteoblastic function. A role for alcohol and tobacco consumption in osteoporosis in middle-aged men. Clin Orthop Relat Res. 1983:107–115.
137. Elisaf, MS, Siamopoulos, KC. Mechanisms of hypophosphataemia in alcoholic patients. Int J Clin Pract. 1997;51:501–503.
138. Klein, GL, Soriano, H, Shulman, RJ, et al. Hepatic osteodystrophy in chronic cholestasis: evidence for a multifactorial etiology. Pediatr Transplant. 2002;6:136–140.
139. Pettifor, JM, Ross, P, Wang, J, et al. Rickets in children of rural origin in South Africa: is low dietary calcium a factor? J Pediatr. 1978;92:320–324.
140. Thacher, TD, Fischer, PR, Pettifor, JM, et al. A comparison of calcium, vitamin D, or both for nutritional rickets in Nigerian children. N Engl J Med. 1999;341:563–568.
141. DeLucia, MC, Mitnick, ME, Carpenter, TO. Nutritional rickets with normal circulating 25-hydroxyvitamin D: a call for reexamining the role of dietary calcium intake in North American infants. J Clin Endocrinol Metab. 2003;88:3539–3545.
142. Albright, F, Butler, AM, Bloomberg, E. Rickets resistant to vitamin D therapy. J Clin Dis Child. 1937;54:529–547.
143. Jonsson, KB, Zahradnik, R, Larsson, T, et al. Fibroblast growth factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia. N Engl J Med. 2003;348:1656–1663.
144. A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. The HYP Consortium. Nat Genet. 1995;11:130–136.
145. Sabbagh, Y, Boileau, G, Campos, M, et al. Structure and function of disease-causing missense mutations in the PHEX gene. J Clin Endocrinol Metab. 2003;88:2213–2222.
146. Sabbagh, Y, Boileau, G, DesGroseillers, L, et al. Disease-causing missense mutations in the PHEX gene interfere with membrane targeting of the recombinant protein. Hum Mol Genet. 2001;10:1539–1546.
147. Sabbagh, Y, Gauthier, C, Tenenhouse, HS. The X chromosome deletion in HYP mice extends into the intergenic region but does not include the SAT gene downstream from Phex. Cytogenet Genome Res. 2002;99:344–349.
148. Sabbagh, Y, Jones, AO, Tenenhouse, HS. PHEXdb, a locus-specific database for mutations causing X-linked hypophosphatemia. Hum Mutat. 2000;16:1–6.
149. Brame, LA, White, KE, Econs, MJ. Renal phosphate wasting disorders: clinical features and pathogenesis. Semin Nephrol. 2004;24:39–47.
150. Econs, MJ. New insights into the pathogenesis of inherited phosphate wasting disorders. Bone. 1999;25:131–135.
151. Econs, MJ, McEnery, PT. Autosomal dominant hypophosphatemic rickets/osteomalacia: clinical characterization of a novel renal phosphate-wasting disorder. J Clin Endocrinol Metab. 1997;82:674–681.
152. White, KE, Jonsson, KB, Carn, G, et al. The autosomal dominant hypophosphatemic rickets (ADHR) gene is a secreted polypeptide overexpressed by tumors that cause phosphate wasting. J Clin Endocrinol Metab. 2001;86:497–500.
153. Bergwitz, C, Roslin, NM, Tieder, M, et al. SLC34A3 mutations in patients with hereditary hypophosphatemic rickets with hypercalciuria predict a key role for the sodium-phosphate cotransporter NaPi-IIc in maintaining phosphate homeostasis. Am J Hum Genet. 2006;78:179–192.
154. Harrell, RM, Lyles, KW, Harrelson, JM, et al. Healing of bone disease in X-linked hypophosphatemic rickets/osteomalacia. Induction and maintenance with phosphorus and calcitriol. J Clin Invest. 1985;75:1858–1868.
155. Verge, CF, Lam, A, Simpson, JM, et al. Effects of therapy in X-linked hypophosphatemic rickets. N Engl J Med. 1991;325:1843–1848.
156. Petersen, DJ, Boniface, AM, Schranck, FW, et al. X-linked hypophosphatemic rickets: a study (with literature review) of linear growth response to calcitriol and phosphate therapy. J Bone Miner Res. 1992;7:583–597.
157. Frame, B, Manson, G. Refractory rickets and osteomalacia. Henry Ford Hosp Med Bull. 1960;8:293–298.
158. Glorieux, FH, Scriver, CR, Reade, TM, et al. Use of phosphate and vitamin D to prevent dwarfism and rickets in X-linked hypophosphatemia. N Engl J Med. 1972;287:481–487.
159. Glorieux, FH. Calcitriol treatment in vitamin D-dependent and vitamin D-resistant rickets. Metabolism. 1990;39:10–12.
160. Sullivan, W, Carpenter, T, Glorieux, F, et al. A prospective trial of phosphate and 1,25-dihydroxyvitamin D3 therapy in symptomatic adults with X-linked hypophosphatemic rickets. J Clin Endocrinol Metab. 1992;75:879–885.
161. Prader, A, Illig, R, Uehlinger, E, et al. Rickets following bone tumor. Helv Paediatr Acta. 1959;14:554–565.
162. Nuovo, MA, Dorfman, HD, Sun, CC, et al. Tumor-induced osteomalacia and rickets. Am J Surg Pathol. 1989;13:588–599.
163. Lyles, KW, Berry, WR, Haussler, M, et al. Hypophosphatemic osteomalacia: association with prostatic carcinoma. Ann Intern Med. 1980;93:275–278.
164. Linovitz, RJ, Resnick, D, Keissling, P, et al. Tumor-induced osteomalacia and rickets: a surgically curable syndrome. Report of two cases. J Bone Joint Surg Am. 1976;58:419–423.
165. Jan De Beur, SM, Finnegan, RB, Vassiliadis, J, et al. Tumors associated with oncogenic osteomalacia express genes important in bone and mineral metabolism. J Bone Miner Res. 2002;17:1102–1110.
166. Shimada, T, Mizutani, S, Muto, T, et al. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci U S A. 2001;98:6500–6505.
167. Berndt, T, Craig, TA, Bowe, AE, et al. Secreted frizzled-related protein 4 is a potent tumor-derived phosphaturic agent. J Clin Invest. 2003;112:785–794.
168. Dent, CE, Gertner, JM. Hypophosphataemic osteomalacia in fibrous dysplasia. Q J Med. 1976;45:411–420.
169. Imel, EA, Econs, MJ. Fibrous dysplasia, phosphate wasting and fibroblast growth factor 23. Pediatr Endocrinol Rev. 2007;4(Suppl 4):434–439.
170. Lichter-Konecki, U, Broman, KW, Blau, EB, et al. Genetic and physical mapping of the locus for autosomal dominant renal Fanconi syndrome, on chromosome 15q15.3. Am J Hum Genet. 2001;68:264–268.
171. Bruce, L, Wrong, O, Toye, A, et al. Brand 3 mutations, renal tubular acidosis and South East Asian ovalocytosis in Malaysia and Papua New Guinea: loss of up to 95% band 3 transport in red cells. Biochem J. 2003;350:41–51.
172. Cheidde, L, Vieira, TC, Lima, PR, et al. A novel mutation in the anion exchanger 1 gene is associated with familial distal renal tubular acidosis and nephrocalcinosis. Pediatrics. 2003;112:1361–1367.
173. Devonald, MA, Smith, AN, Poon, JP, et al. Non-polarized targeting of AE1 causes autosomal dominant distal renal tubular acidosis. Nat Genet. 2003;33:125–127.
174. Smith, AN, Borthwick, KJ, Karet, FE. Molecular cloning and characterization of novel tissue-specific isoforms of the human vacuolar H+-ATPase C, G and D subunits, and their evaluation in autosomal recessive distal renal tubular acidosis. Gene. 2002;297:169–177.
175. Carballo-Trujillo, I, Garcia-Nieto, V, Moya-Angeler, FJ, et al. Novel truncating mutations in the ClC-5 chloride channel gene in patients with Dent’s disease. Nephrol Dial Transplant. 2003;18:717–723.
176. Cox, JP, Yamamoto, K, Christie, PT, et al. Renal chloride channel, CLCN5, mutations in Dent’s disease. J Bone Miner Res. 1999;14:1536–1542.
177. Brodehl, J. Tubular Fanconi syndromes with bone involvement. In: Bickel H, Stern J, eds. Inborn Errors of Calcium and Bone Metabolism. Baltimore: University Park Press; 1976:191–213.
178. Schneider, JA, Seegmiller, JE. Cystinosis and the Fanconi syndrome. In: Stanbury JB, Wyngaarden JB, Frederickson D, eds. The Metabolic Basis of Inherited Disease. New York: McGraw-Hill; 1972:1581.
179. Sakamoto, O, Ogawa, E, Ohura, T, et al. Mutation analysis of the GLUT2 gene in patients with Fanconi-Bickel syndrome. Pediatr Res. 2000;48:586–589.
180. Lowe, CU, Terrey, M, Mac, LE. Organic-aciduria, decreased renal ammonia production, hydrophthalmos, and mental retardation; a clinical entity. AMA Am J Dis Child. 1952;83:164–184.
181. Rosenberg, LE, Scriver, CR. Disorders of amino acid metabolism. In: Bondy PK, ed. Duncan’s Disease of Metabolism. Philadelphia: WB Saunders; 1969:366–515.
182. Anikster, Y, Shotelersuk, V, Gahl, WA. CTNS mutations in patients with cystinosis. Hum Mutat. 1999;14:454–458.
183. McGowan-Jordan, J, Stoddard, K, Podolsky, L, et al. Molecular analysis of cystinosis: probable Irish origin of the most common French Canadian mutation. Eur J Hum Genet. 1999;7:671–678.
184. Shotelersuk, V, Larson, D, Anikster, Y, et al. CTNS mutations in an American-based population of cystinosis patients. Am J Hum Genet. 1998;63:1352–1362.
185. Briggs, WA, Kominami, N, Wilson, RE, et al. Kidney transplantation in Fanconi syndrome. N Engl J Med. 1972;286:25.
186. Monnier, N, Satre, V, Lerouge, E, et al. OCRL1 mutation analysis in French Lowe syndrome patients: implications for molecular diagnosis strategy and genetic counseling. Hum Mutat. 2000;16:157–165.
187. Roschinger, W, Muntau, AC, Rudolph, G, et al. Carrier assessment in families with lowe oculocerebrorenal syndrome: novel mutations in the OCRL1 gene and correlation of direct DNA diagnosis with ocular examination. Mol Genet Metab. 2000;69:213–222.
188. Zhang, X, Hartz, PA, Philip, E, et al. Cell lines from kidney proximal tubules of a patient with Lowe syndrome lack OCRL inositol polyphosphate 5-phosphatase and accumulate phosphatidylinositol 4,5-bisphosphate. J Biol Chem. 1998;273:1574–1582.
189. Huster, D, Hoppert, M, Lutsenko, S, et al. Defective cellular localization of mutant ATP7B in Wilson’s disease patients and hepatoma cell lines. Gastroenterology. 2003;124:335–345.
190. Majumdar, R, Al Jumah, M, Fraser, M. 4193delC, a common mutation causing Wilson’s disease in Saudi Arabia: rapid molecular screening of patients and carriers. Mol Pathol. 2003;56:302–304.
191. Morgan, HG, Stewart, WK, Lowe, KG, et al. Wilson’s disease and the Fanconi syndrome. Q J Med. 1962;31:361–384.
192. Wilson, DM, Goldstein, NP. Bicarbonate excretion in Wilson’s disease (hepatolenticular degeneration). Mayo Clin Proc. 1974;49:394–400.
193. Walshe, JM. Effect of penicillamine on failure of renal acidification in Wilson’s disease. Lancet. 1968;1:775–778.
194. Bergeron, A, D’Astous, M, Timm, DE, et al. Structural and functional analysis of missense mutations in fumarylacetoacetate hydrolase, the gene deficient in hereditary tyrosinemia type 1. J Biol Chem. 2001;276:15225–15231.
195. Snaper, I, Kahn, A. Determination of Bence Jones protein in urine and serum. In: Myelomatosis, Fundamentals and Clinical Features. Baltimore: University Park Press; 1971:203–204.
196. Monte Neto, JT, Sesso, R, Kirsztajn, GM, et al. Osteomalacia secondary to renal tubular acidosis in a patient with primary Sjogren’s syndrome. Clin Exp Rheumatol. 1991;9:625–627.
197. Adams, RG, Harrison, JF, Scott, P. The development of cadmium-induced proteinuria, impaired renal function, and osteomalacia in alkaline battery workers. Q J Med. 1969;38:425–443.
198. Chisolm, JJ, Jr. Aminoaciduria as a manifestation of renal tubular injury in lead intoxication and a comparison with patterns of aminoaciduria seen in other diseases. J Pediatr. 1962;60:1–17.
199. Gross, JM. Fanconi syndrome (adult type) developing secondary to the ingestion of outdated tetracycline. Ann Intern Med. 1963;58:523–528.
200. Bethune, JE, Dent, CE. Hypophosphatasia in the adult. Am J Med. 1960;28:615–622.
201. Fraser, D. Hypophosphatasia. Am J Med. 1957;22:730–746.
202. Rathbun, JC. Hypophosphatasia; a new developmental anomaly. Am J Dis Child. 1948;75:822–831.
203. Sobel, EH, Clark, LC, Jr., Fox, RP, et al. Rickets, deficiency of alkaline phosphatase activity and premature loss of teeth in childhood. Pediatrics. 1953;11:309–322.
204. Cahill, RA, Wenkert, D, Perlman, SA, et al. Infantile hypophosphatasia: transplantation therapy trial using bone fragments and cultured osteoblasts. J Clin Endocrinol Metab. 2007;92:2923–2930.
205. Whyte, MP, Kurtzberg, J, McAlister, WH, et al. Marrow cell transplantation for infantile hypophosphatasia. J Bone Miner Res. 2003;18:624–636.
206. Birtwell, WM, Jr., Riggs, L, Peterson, LF, et al. Hypophosphatasia in an adult. Arch Intern Med. 1967;120:90–93.
207. McCance, R, Morrison, AB, Dent, CE. The excretion of phosphoethanolamine and hypophosphatasia. Lancet. 1955;268:131.
208. Whyte, MP. Hypophosphatasia. In: Scriver CR, Beaudet AL, Sly WS, et al, eds. The Metabolic and Molecular Bases of Inherited Disease. ed 8. New York: McGraw-Hill; 2001:5313–5329.
209. Whyte, MP, Mahuren, JD, Vrabel, LA, et al. Markedly increased circulating pyridoxal-5′-phosphate levels in hypophosphatasia. Alkaline phosphatase acts in vitamin B6 metabolism. J Clin Invest. 1985;76:752–756.
210. Fleish, H, Neuman, WF. Mechanisms of calcification: role of collagen, polyphosphates, and phosphatase. Am J Physiol. 1961;200:1296–1300.
211. Russell, RG. Metabolism of inorganic pyrophosphate (PPi). Arthritis Rheum. 1976;19(Suppl 3):465–478.
212. Henthorn, PS, Raducha, M, Fedde, KN, et al. Different missense mutations at the tissue-nonspecific alkaline phosphatase gene locus in autosomal recessively inherited forms of mild and severe hypophosphatasia. Proc Natl Acad Sci U S A. 1992;89:9924–9928.
213. Albright, F. Hypoparathyroidism as a cause of osteomalacia. J Clin Endocrinol Metab. 1956;16:419–425.
214. Drezner, MK, Neelon, FA, Jowsey, J, et al. Hypoparathyroidism: a possible cause of osteomalacia. J Clin Endocrinol Metab. 1977;45:114–122.
215. Epstein, S, Meunier, PJ, Lambert, PW, et al. 1 alpha,25-dihydroxyvitamin D3 corrects osteomalacia in hypoparathyroidism and pseudohypoparathyroidism. Acta Endocrinol (Copenh). 1983;103:241–247.
216. Wilson, JD, Hadden, DR. Pseudohypoparathyroidism presenting with rickets. J Clin Endocrinol Metab. 1980;51:1184–1189.
217. Whyte, MP, Murphy, WA. Osteopetrosis and other sclerosing bone disorders. In: Avioli LV, Krane SM, eds. Metabolic Bone Disease. Philadelphia: WB Saunders; 1990:616–658.
218. Pincus, JB, Gittleman, IF, Karamer, B. Metabolic studies in two cases and further observations on the composition of bones in this disease. Am J Dis Child. 1947;73:458–472.
219. Bonucci, E, Sartori, E, Spina, M. Osteopetrosis fetalis. Report on a case, with special reference to ultrastructure. Virchows Arch A Pathol Anat Histol. 1975;368:109–121.
220. Borthwick, KJ, Kandemir, N, Topaloglu, R, et al. A phenocopy of CAII deficiency: a novel genetic explanation for inherited infantile osteopetrosis with distal renal tubular acidosis. J Med Genet. 2003;40:115–121.
221. Frame, B, Frost, HM, Pak, CY, et al. Fibrogenesis imperfecta ossium. A collagen defect causing osteomalacia. N Engl J Med. 1971;285:769–772.
222. Coursey, C, Weber, T, Dodd, L, et al. Fibrogenesis imperfecta ossium: MR imaging of the axial and appendicular skeleton and correlation with a unique radiographic appearance. Skeletal Radiol. 2007;36:1077–1084.
223. Swan, CHJ, Cooke, WT. Fibrogenesis imperfect ossium. In: Frame B, Parfitt AM, Duncan H, eds. Clinical Aspects of Metabolic Bone Disease. Amsterdam: Excerpta Medica; 1973:465.
224. Henneman, DH, Pak, CY, Bartter, FC. Collagen composition, solubility and biosynthesis in fibrogenesis imperfecta ossium. In: Frame B, Parfitt AM, Duncan H, eds. Clinical Aspects of Metabolic Bone Disease. Amsterdam: Excerpta Medica; 1973:469.
225. Lafage-Proust, M, Schaeverbeke, T, Dehais, J. Fibrogenesis imperfecta ossium: ineffectiveness of melphalan. Calcif Tissue Int. 1996;59:240–244.
226. Martin, KJ, Gonzalez, E, Slatopolsky, E. Renal Osteodystrophy. In: Brenner BM, ed. Brenner and Rector’s the Kidney. Philadelphia: WB Saunders; 2004:2255–2304.
227. Charhon, SA, Berland, YF, Olmer, MJ, et al. Effects of parathyroidectomy on bone formation and mineralization in hemodialyzed patients. Kidney Int. 1985;27:426–435.
228. Dunstan, CR, Hills, E, Norman, AW, et al. The pathogenesis of renal osteodystrophy: role of vitamin D, aluminium, parathyroid hormone, calcium and phosphorus. Q J Med. 1985;55:127–144.
229. Goodman, WG. Bone disease and aluminum: pathogenic considerations. Am J Kidney Dis. 1985;6:330–335.
230. Hodsman, AB, Sherrard, DJ, Alfrey, AC, et al. Bone aluminum and histomorphometric features of renal osteodystrophy. J Clin Endocrinol Metab. 1982;54:539–546.
231. Li, YC, Amling, M, Pirro, AE, et al. Normalization of mineral ion homeostasis by dietary means prevents hyperparathyroidism, rickets, and osteomalacia, but not alopecia in vitamin D receptor-ablated mice. Endocrinology. 1998;139:4391–4396.
232. Li, YC, Pirro, AE, Amling, M, et al. Targeted ablation of the vitamin D receptor: an animal model of vitamin D-dependent rickets type II with alopecia. Proc Natl Acad Sci U S A. 1997;94:9831–9835.
233. Malluche, HH. Aluminium and bone disease in chronic renal failure. Nephrol Dial Transplant. 2002;17(Suppl 2):21–24.
234. Parisien, M, Charhon, SA, Arlot, M, et al. Evidence for a toxic effect of aluminum on osteoblasts: a histomorphometric study in hemodialysis patients with aplastic bone disease. J Bone Miner Res. 1988;3:259–267.
235. Quarles, LD, Dennis, VW, Gitelman, HJ, et al. Aluminum deposition at the osteoid-bone interface. An epiphenomenon of the osteomalacic state in vitamin D-deficient dogs. J Clin Invest. 1985;75:1441–1447.
236. Bleyer, AJ, Burke, SK, Dillon, M, et al. A comparison of the calcium-free phosphate binder sevelamer hydrochloride with calcium acetate in the treatment of hyperphosphatemia in hemodialysis patients. Am J Kidney Dis. 1999;33:694–701.
237. Chertow, GM, Burke, SK, Lazarus, JM, et al. Poly[allylamine hydrochloride] (RenaGel): a noncalcemic phosphate binder for the treatment of hyperphosphatemia in chronic renal failure. Am J Kidney Dis. 1997;29:66–71.
238. Chertow, GM, Burke, SK, Raggi, P. Sevelamer attenuates the progression of coronary and aortic calcification in hemodialysis patients. Kidney Int. 2002;62:245–252.
239. Asami, T, Kawasaki, T, Uchiyama, M. Unique form of rickets with low serum 25-hydroxyvitamin D in two normally nourished children. Acta Paediatr Jpn. 1995;37:182–188.
240. Casella, SJ, Reiner, BJ, Chen, TC, et al. A possible genetic defect in 25-hydroxylation as a cause of rickets. J Pediatr. 1994;124:929–932.
241. Nutzenadel, W, Mehls, O, Klaus, G. A new defect in vitamin D metabolism. J Pediatr. 1995;126:676–677.
242. Cheng, JB, Levine, MA, Bell, NH, et al. Genetic evidence that the human CYP2R1 enzyme is a key vitamin D 25-hydroxylase. Proc Natl Acad Sci U S A. 2004;101:7711–7715.
243. Fu, G, Lin, D, Zang, M, et al. Cloning of human 25-hydroxyvitamin D-1-a-hydroxylase and mutations causing vitamin D-dependent rickets type 1. Mol Endocrinol. 1997:1961–1970.
244. Kitnaka, S, Takeyama, K-I, Muryama, A, et al. Inactivating mutations in the 25-hydroxyvitamin D, 1a-hydroxylase gene in patients with pseudovitamin D-deficiency rickets. N Engl J Med. 1998;338:653–661.
245. St-Arnaud, R, Messerlian, S, Moir, J, et al. The 25-hydroxyvitamin D 1-alphahydroxylase gene maps to the pseudovitamin D-deficiency rickets (PDDR) disease locus. J Bone Miner Res. 1997;12:1552–1559.
246. Brooks, MH, Bell, NH, Love, L, et al. Vitamin-D-dependent rickets type II. Resistance of target organs to 1,25-dihydroxyvitamin D. N Engl J Med. 1978;298:996–999.
247. Hughes, M, Malloy, P, Kieback, D, et al. Human vitamin D receptor mutations: identification of molecular defects in hypocalcemic vitamin D resistant rickets. Adv Exp Med Biol. 1989;255:491–503.
248. Balsan, S, Garabedian, M, Larchet, M, et al. Long-term nocturnal calcium infusions can cure rickets and promote normal mineralization in hereditary resistance to 1,25-dihydroxyvitamin D. J Clin Invest. 1986;77:1661–1667.
249. Heaney, RP, Dowell, MS, Hale, CA, et al. Calcium absorption varies within the reference range for serum 25-hydroxyvitamin D. J Am Coll Nutr. 2003;22:142–146.
250. Trivedi, DP, Doll, R, Khaw, KT. Effect of four monthly oral vitamin D3 (cholecalciferol) supplementation on fractures and mortality in men and women living in the community: randomised double blind controlled trial. BMJ. 2003;326:469.
251. Bischoff-Ferrari, HA, Dietrich, T, Orav, EJ, et al. Positive association between 25-hydroxy vitamin D levels and bone mineral density: a population-based study of younger and older adults. Am J Med. 2004;116:634–639.
252. Heaney, RP, Davies, KM, Chen, TC, et al. Human serum 25-hydroxycholecalciferol response to extended oral dosing with cholecalciferol. Am J Clin Nutr. 2003;77:204–210.
253. Ahonen, MH, Tenkanen, L, Teppo, L, et al. Prostate cancer risk and prediagnostic serum 25-hydroxyvitamin D levels (Finland). Cancer Causes Control. 2000;11:847–852.
254. Grant, WB. An ecologic study of dietary and solar ultraviolet-B links to breast carcinoma mortality rates. Cancer. 2002;94:272–281.
255. Mason, RS, Lissner, D, Posen, S, et al. Blood concentrations of dihydroxylated vitamin D metabolites after an oral dose. Br Med J. 1980;280:449–450.
256. Bell, NH, Stern, PH. Hypercalcemia and increases in serum hormone value during prolonged administration of 1alpha,25-dihydroxyvitamin D. N Engl J Med. 1978;298:1241–1243.
257. Maung, HM, Elangovan, L, Frazao, JM, et al. Efficacy and side effects of intermittent intravenous and oral doxercalciferol (1alpha-hydroxyvitamin D(2)) in dialysis patients with secondary hyperparathyroidism: a sequential comparison. Am J Kidney Dis. 2001;37:532–543.
258. Knutson, JC, Hollis, BW, LeVan, LW, et al. Metabolism of 1 alpha-hydroxyvitamin D2 to activated dihydroxyvitamin D2 metabolites decreases endogenous 1 alpha, 25-dihydroxyvitamin D3 in rats and monkeys. Endocrinology. 1995;136:4749–4753.
259. Okonofua, F, Gill, DS, Alabi, ZO, et al. Rickets in Nigerian children: a consequence of calcium malnutrition. Metabolism. 1991;40:209–213.
260. Collins, N, Maher, J, Cole, M, et al. A prospective study to evaluate the dose of vitamin D required to correct low 25-hydroxyvitamin D levels, calcium, and alkaline phosphatase in patients at risk of developing antiepileptic drug-induced osteomalacia. Q J Med. 1991;78:113–122.
261. Hosking, DJ, Campbell, GA, Kemm, JR, et al. Safety of treatment for subclinical osteomalacia in the elderly. Br Med J (Clin Res Ed). 1984;289:785–787.
262. Paterson, CR. Vitamin-D poisoning: survey of causes in 21 patients with hypercalcaemia. Lancet. 1980;1:1164–1165.
263. Rizzoli, R, Stoermann, C, Ammann, P, et al. Hypercalcemia and hyperosteolysis in vitamin D intoxication: effects of clodronate therapy. Bone. 1994;15:193–198.
264. Alon, US, Levy-Olomucki, R, Moore, WV, et al. Calcimimetics as an adjuvant treatment for familial hypophosphatemic rickets. Clin J Am Soc Nephrol. 2008;3:658–664.
265. Geller, JL, Khosravi, A, Kelly, MH, et al. Cinacalcet in the management of tumor-induced osteomalacia. J Bone Miner Res. 2007;22:931–937.
266. Driessen, GJ, Gerritsen, EJ, Fischer, A, et al. Long-term outcome of haematopoietic stem cell transplantation in autosomal recessive osteopetrosis: an EBMT report. Bone Marrow Transplant. 2003;32:657–663.
[/level-membership-for-endocrinology-diabetes-and-metabolism-category][not-level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 15
Disorders Of Calcification
Osteomalacia and Rickets
Clinical Features of Rickets and Osteomalacia
Nutritional Osteomalacia and Rickets
Gastrointestinal and Hepatic Diseases
Impaired Renal Tubular Phosphate Reabsorption
Tumor-Associated Rickets and Osteomalacia
General Renal Tubular Disorders
Hyperparathyroidism, Hypoparathyroidism, and Pseudohypoparathyroidism
Fibrogenesis Imperfecta Ossium
Renal Osteodystrophy and Aluminum Intoxication
Osteomalacia and rickets are disorders of mineralization. Osteomalacia is a failure to mineralize the newly formed organic matrix (osteoid) of bone. In rickets, a disease of children, the growth plate at the epiphysis is involved in a process that is characterized by delay in the maturation of chondrocytes in the growth plate and disorganization of these chondrocytes, resulting in expansion of the growth plate. While rickets is associated with impaired mineralization of cartilage matrix, hypophosphatemia is thought to play a predominant role in the etiology of rickets.1 A number of different disorders are associated with osteomalacia in adults and rickets in children.2–4 The pathogenesis of the mineralization defect, the biochemical alterations, the clinical manifestations, and the therapeutic approaches differ in these conditions, so a systematic approach to the diagnosis and treatment of these disorders is essential.
Mineralization Defect
Mineralization of bone is a complex process in which the calcium-phosphate inorganic mineral phase is deposited in relation to the organic matrix in a highly ordered fashion. Optimal mineralization can take place at bone-forming surfaces only if cellular activity of bone-forming cells is adequate, matrix is normal in composition and is synthesized at a normal rate, the supply of mineral ions (calcium and inorganic phosphate) from the extracellular fluid is sufficient, the pH at sites of mineralization is appropriate (approximately 7.6), and the concentration of inhibitors of calcification is controlled. Structural and regulatory aspects of biological mineralization have been considered in numerous reviews.5–8 Clinical disorders of mineralization can be attributed to defects at several of these control steps, examples of which are shown in Table 15-1. Overall features of mineralization in normal and abnormal bone remodeling and the relevance to mineralization disorders have been comprehensively reviewed.4
Table 15-1
Examples of Disorders with Different Causes of Impaired Mineralization
| Disorder | Mechanism |
| Postoperative hyperparathyroidism | Rate of matrix synthesis exceeds rate of mineralization |
| Fibrogenesis imperfecta ossium | Defective collagenous matrix |
| Adult phosphate diabetes | Phosphate concentration deficient at mineralization sites |
| Vitamin D deficiency | Insufficient calcium and phosphate |
| Systemic acidosis | pH inadequate for mineralization |
| Hypophosphatasia | Excess inhibitor (? inorganic pyrophosphate) |
The determinants for the deposition of the mineral phase in normally mineralized tissues—cartilage, bone, and teeth—and the precise mechanisms that govern the process of biological mineralization have yet to be elucidated. It is thought that deposition of the initial mineral phase takes place within the collagen fibrils in the gap formed by the packing of the trimeric type I collagen molecules, the most abundant organic component of bone.7 The earliest calcium-phosphate (Ca-P) mineral phase is thought to be composed of very small apatite crystals, less than 9 nmol in length, that can fit into the channels formed by adjacent gaps.9 These initial Ca-P crystals appear to form complexes with phosphoproteins of the organic matrix.10 As the bone and minerals mature, additional Ca-P crystals are deposited between collagen macromolecules. Other proteins that are abundant in mineralized tissues and thought to play a role in mineralization include the sialic acid–rich proteins such as osteopontin and bone sialoprotein (BSP).11 The ultimate size and shape of the Ca-P crystals are also influenced by anionic electrolytes in the environment.12
The major driving force for mineralization is the concentration of inorganic phosphate (Pi), which at normal sites of mineralization is derived predominantly from the plasma. Therefore, control of Pi reabsorption in the renal tubular lumen is the most important process regulating mineralization. It is also possible that tissue-nonspecific alkaline phosphatase (TNAP), which functions as an inorganic pyrophosphatase localized on the surface of osteoblasts, generates additional Pi to drive mineralization.13 Osteomalacia is seen in loss-of-function mutations in the TNAP gene, but decreased TNAP also results in accumulation of inorganic pyrophosphate (PPi), which acts as a potent inhibitor of mineralization.13,14 It is likely that this pyrophosphatase function of TNAP is the most critical. PPi is generated by osteoblasts and chondrocytes through the action of two enzymes: the ectonucleoside, pyrophosphatase phosphodiesterase 1 (NNP1/PC-1), which releases PPi from nucleoside triphosphates13,14 and the transmembrane protein, ANK, which shuttles PPi between intracellular and extracellular compartments.15,16 Ectopic mineralization due to decreased local concentrations of PPi accompanies spontaneous deficiency of these proteins in humans and mice. In cartilage, there is evidence that matrix vesicles (MVs) formed from chondrocyte plasma membranes play a role in the regulation of mineralization.5,6 These extracellular MVs are approximately 100 nm in diameter and contain enzymes such as TNAP, NNP1, and Ca-binding proteins as well as Ca-P crystals at the inner surface of the membranes. They are thought to play a role in the initial mineralization of growth plate cartilage matrix by exposing preformed apatite crystals to the extracellular fluid, where further crystal growth can occur. MVs have also been found in bone near sites of mineralization, but it is difficult to assign them a function, since the initial crystallites are deposited in association with collagen fibers in bone. An explanation for the specificity of normal mineralization of bone is provided by the exclusive coexpression in bone of the genes encoding the predominant organic matrix protein, type I collagen, and the enzyme that hydrolyzes the inhibitor, PPi. Furthermore, the protein mineralization inhibitor, matrix GLA protein (MGP), that is expressed in cells in several other tissues is not expressed in bone.17,18
The mechanism of defective mineralization is not the same in all disorders associated with osteomalacia and rickets, and biochemical indices such as serum levels of calcium and phosphorus also differ. Moreover, the relative imbalance in matrix synthesis and its mineralization varies depending on the underlying disease mechanism. Estimates from tetracycline labeling indicate that the appositional growth rate in normal bone is about 1 µm/day.19 It has also been suggested20 that complete mineralization of the osteoid in normal bone requires approximately 10 to 21 days. Thus the thickness of the osteoid seam normally does not exceed 15 to 20 µm, the surface of bone that is covered by osteoid is normally less than 20%, and the active surface that is covered by osteoid is considerably less. The major histologic criteria for the diagnosis of osteomalacia are the increased osteoid surface and the increased thickness of the osteoid seam (Fig. 15-1). The mineralization front at the junction of mineralized bone and osteoid is also abnormal in osteomalacia.21 In applying kinetic criteria to the diagnosis of osteomalacia, it has been suggested that a mean osteoid seam width greater than 15 µm and a mineralization lag time greater than 100 days are appropriate diagnostic criteria.22 However, other investigators23,24 have suggested that more stringent criteria be applied to establish the diagnosis of osteomalacia, based on the observation that reduced mineral apposition rate, reduced fractional extent of the mineralization front, and prolongation of the mineralization lag time are indices that reflect impaired matrix synthesis by osteoblasts rather than specific features of osteomalacia. The diagnosis of osteomalacia must include evidence of an absolute increase in the total osteoid volume and an increased number of osteoid lamellae. A detailed consideration of the kinetics of remodeling as specifically applicable to osteomalacia is found in a review by Parfitt.4
FIGURE 15-1 Undecalcified sections of a bone biopsy from a patient with an adult-onset renal phosphate leak. A, Unstained. B, Microradiograph. C, Ultraviolet photomicrograph demonstrating fluorescence (F) of tetracycline administered 14 days prior to biopsy. Mineralized bone (M) and osteoid (O) are noted. (From Case 1 in Baylink D, Stauffer M, Wergedal J et al: Formation, mineralization, and resorption of bone in vitamin D–deficient rats, J Clin Invest 49:1122–1134, 1970.)
The architecture of the bone cells and matrix in osteomalacic bone is usually normal. The collagen of the osteoid is largely lamellar, although foci of woven bone are occasionally seen. Hypomineralized periosteocytic lesions have been observed in some affected individuals with hypophosphatemic rickets.25 The persistence of this defect in patients in whom the abnormality in bone mineralization was corrected with therapy supports the hypothesis that osteocyte function may be abnormal. In contrast, there are clear abnormalities in the cells of the rachitic growth plate. The characteristic changes occur in the maturation zone of hypertrophic chondrocytes, whereas the resting and proliferative zones show normal histologic features (Fig. 15-2). In the maturation zone, the number of cells per column is increased, and the cells are irregularly aligned. This is also accompanied by an increase in the transverse diameter, which may extend beyond the ends of the bone, resulting in characteristic cupping or flaring. In experimental rickets, the water content of the growth plate is increased, and a number of metabolic abnormalities have been observed, including decreased glycogen content and an altered pattern of glycolysis.26 When bone is examined histologically, it is essential that undemineralized sections be used. In usual practice, however, with classic clinical, radiologic, and biochemical findings, bone biopsy is not necessary to arrive at the diagnosis of osteomalacia. The most commonly biopsied site is the iliac crest; sample size ranges from 5 to 10 mm in diameter and should include both inner and outer cortices. Growth plates from long bones in children are usually not biopsied, although an open-wedge biopsy of growth cartilage of the iliac apophysis may occasionally be obtained without the hazard of altering growth of long bones. Mineralized specimens of bone are most satisfactorily embedded in plastic media—which provide preservation of tissue architecture not usually attained with paraffin-embedding techniques—because the distinction between mineralized and unmineralized bone is lost with decalcification of the specimen. A number of different staining techniques can then be used to demonstrate the osteoid and apply quantitative morphometric analysis.23–25 In normal bone, the mineralization front is seen at the junction of the osteoid seam and newly mineralized bone. This region can be identified by an intense fluorescence of tetracycline, deposited in this zone when administered prior to obtaining the biopsy (see Fig. 15-1). In normal persons, the osteoid seam/bone junctions fluoresce intensely; in osteomalacia, the fluorescence is less well defined (more diffuse) or even absent. In addition to impaired matrix mineralization, matrix biosynthesis may be abnormal in osteomalacia. In osteomalacia observed with vitamin D deficiency, a decreased rate of matrix formation is observed.23,24,27 Osteoblast function may be impaired in many forms of human rickets and osteomalacia, which may result in abnormal matrix formation. Notably, the hydroxylation of certain collagen lysyl residues is increased in vitamin D–deficient bone, as well as in other experimental hypocalcemic states.28–30
Clinical Features of Rickets and Osteomalacia
The clinical manifestations of rickets are mainly related to skeletal pain and deformity, slippage of epiphyses, disturbances in growth, and fracture of the osteomalacic bones. Hypocalcemia, when it occurs, may be symptomatic. Depending on the degree of hypophosphatemia, muscular weakness and hypotonia may be prominent features. Dent and Stamp31 have indicated nine factors that underlie the clinical manifestations of rickets and osteomalacia, modified here as follows:
1. Failure of mineralization affects predominantly those parts of the skeleton in which growth is most rapid.
2. Endochondral bone is more affected than intramembranous bone, possibly because of the more rapid growth of the former.
3. Proximal and distal ends of bones do not grow at the same rate, and rickets affects the most rapidly growing area.
4. Because different bones grow at different rates at different stages of development, the time when rickets is active determines the clinical expression. For example, the skull is growing rapidly at birth, so craniotabes is a manifestation of congenital rickets. During the first year, the upper limbs and rib cage grow rapidly, so abnormalities at these sites are prominent—for example, rachitic rosary. Signs of rickets at the wrist are usually seen at the ulnar side because the growth rate of the distal ulnar epiphysis is greater than that of the distal radial epiphysis.
5. Deformities in mild chronic rickets are most often due to disordered growth at the epiphysial plate rather than to bending at the shafts.
6. In some forms of rickets, the radiologic changes include those of secondary hyperparathyroidism (subperiosteal resorption, most commonly at the metaphyses).
7. Deformities that occur before the age of 4 years correct themselves if the rickets is cured; if rickets persists to a later age, the deformities are permanent (dwarfism, bowleg, and knock-knee).
8. “Late” rickets, which occurs at the time of the pubescent growth spurt, produces dramatic disturbances and results in knock-knee.
9. Adult manifestations of osteomalacia, such as Looser’s zones and increased biconcavity of vertebral bodies, are seen in young children only when the rickets is very severe.
In contrast, osteomalacia in adults may be difficult to detect on clinical grounds alone. Diffuse skeletal pain and muscular weakness may be present. Pain, often prominent about the hips and in association with hypophosphatemic myopathy, may produce a waddling or antalgic gait. Fractures may occur in the ribs and vertebral bodies, as well as in long bones, leading to progressive deformities. Affected individuals may also have localized pain and swelling in one or more joints. Synovial fluid is noninflammatory and free of crystals. Symmetric polyarthralgias resembling those of rheumatoid arthritis or polymyalgia rheumatica may also be observed.32 Muscular weakness is quite common,33,34 is primarily proximal in distribution (which contributes to the waddling gait), and is often associated with wasting and hypotonia with preservation of brisk reflexes.35 This is thought to be a consequence of hypophosphatemia and responds to phosphate repletion.36 The molecular basis remains elusive, with no difference observed in the relative concentrations of skeletal muscle phosphocreatine, adenosine triphosphate, or inorganic phosphate estimated by phosphorus nuclear magnetic resonance spectroscopy.37 Although the etiology of the neuromuscular features of osteomalacia is not clearly defined, therapy of the underlying disorder, such as vitamin D in nutritional osteomalacia, alkalinization in acidosis, and phosphate repletion in hypophosphatemic osteomalacia, results in resolution of these features. The role of hypophosphatemia per se in muscular weakness is discussed in Chapter 6.
Radiologic Features
Radiologic features of rickets and osteomalacia reflect the histopathologic changes. In rickets, the alterations are most evident at the epiphyseal growth plate, which is increased in thickness, cupped, and reveals haziness at the diaphyseal border due to decreased mineralization of the hypertrophic zone and inadequate mineralization of the primary spongiosa (Fig. 15-3). Variation in the pattern of rachitic changes is influenced by differences in the rates of growth of individual bones. The trabecular pattern of the metaphyses is abnormal, the cortices of the diaphyses may be thinned, and bowing of the shafts may be present.
FIGURE 15-3 A, Rickets in a child with Fanconi’s syndrome, showing typical cupping of distal femoral epiphyses. B, Osteomalacia in an 80-year-old woman who had a history compatible with hypophosphatemic rickets dating to early childhood. Note multiple pseudofractures (arrows).
Osteomalacia is due to decreased mineralization and is therefore associated with a decrease in bone density, loss of trabecular patterning, and variable degrees of thinning of the cortices.38,39 In some patients, radiologic changes are indistinguishable from those seen in osteoporosis. The characteristic finding that specifically suggests osteomalacia is the presence of radiolucent bands known as pseudofractures, Looser’s zones, or umbauzonen, ranging from a few millimeters to several centimeters in length and usually oriented perpendicularly to the surface of the bone. (Fig. 15-4). They tend to occur symmetrically and are particularly common at the inner aspects of the femur near the femoral neck, in the pelvis, in the outer edge of the scapula, in the upper fibula, and in the metatarsals.
FIGURE 15-4 Radiograph of the pelvis and proximal femora in an adult with renal phosphate wasting. Note pseudofractures, also known as Looser’s zones (arrows). (From Case 2 in Jaworski ZFG, Kloswvych S, Cameron E: Proceedings of the First Workshop on Bone Morphometry. Ottawa: University of Ottawa Press, 1973.)
Pseudofractures are most often seen at sites where major arteries cross the bones. Arteriography in some36,40,41 but not all cases42 suggests that the origins of the pseudofractures correspond to the locations of major arteries (Fig. 15-5). Trauma of some sort, whether related to arterial pulsation or other factors (e.g., weight-bearing stress), is likely responsible for the symmetry of the lesions and their predilection for the described sites. Pseudofractures are often multiple, occasionally occurring at 10 to 15 sites in a single individual; such multiple symmetric pseudofractures in osteomalacic individuals have been referred to as Milkman’s syndrome.43–46 The abnormalities in Milkman’s original case were also considered by Albright and Reifenstein43 to be manifestations of osteomalacia. The histopathology of Looser’s zones is that of premalacic lamellar bone, some of which is surrounded by lamellar osteoid at the edge of the defect.47 In addition, there are foci of woven bone, some of which is mineralized and some not. This accounts for the lower radiologic mineral density of the pseudofracture compared with the surrounding bone. Subperiosteal erosions along the diaphyseal cortices extending to the metaphyses may be seen when secondary hyperparathyroidism is present. Widening (or pseudowidening) of the sacroiliac joints and the appearance of hazy margins has also been observed, sometimes suggesting ankylosing spondylitis, which osteomalacia may mimic clinically.36
FIGURE 15-5 Radiograph (r) and corresponding arteriograms (a) of a patient with adult-onset renal phosphate wasting. A, Pelvis. B, Femur. Note that the origin of Looser’s zones (arrows) corresponds with crossing of major vessels. (From Case 2 in Jaworski ZFG, Kloswvych S, Cameron E: Proceedings of the First Workshop on Bone Morphometry. Ottawa: University of Ottawa Press, 1973.)
In some patients with osteomalacia, increased rather than decreased radiologic density of bones may be observed.48 This is seen particularly in patients with renal tubular phosphate leaks, as opposed to vitamin D deficiency (Fig. 15-6). In such patients, there may be a striking degree of thickening of the cortices and trabeculae of the spongy bone, at times associated with exostotic spurs. This hyperostosis has been noted in untreated patients. It is not usually observed in patients with generalized defects in proximal renal tubular reabsorption. Despite the increase in mass of bone per unit volume, microscopically the trabeculae are covered with abnormally thickened osteoid seams typical of osteomalacia. Similar findings may be noted in patients with chronic renal failure. The reason for the hyperostosis is unknown; the bone is still architecturally abnormal and subject to fracture with relatively minimal trauma.
FIGURE 15-6 Increased bone mass in patients with osteomalacia. A, Radiographs of femora of a 15-year-old boy with X-linked hypophosphatemia. Note the thick tibial cortex (c) and Looser’s zone (arrow). B, Radiograph of pelvis and femora of a 38-year-old woman with hypophosphatemia present since childhood. Note Looser’s zones (arrows).
In patients with X-linked hypophosphatemic osteomalacia and rickets, an additional finding has been the presence of a generalized involvement of the entheses, with exuberant calcification (more likely ossification) of tendon and ligament insertions.49,50 The absence of inflammatory cells, as well as other clinical features, differentiates this disorder from degenerative joint disease and the seronegative spondyloarthropathies. A comprehensive classification of rickets and osteomalacia is shown in Table 15-2. A detailed discussion of all these conditions is not included here.
Table 15-2
Classification of Rickets and Osteomalacia
B Impaired renal tubular phosphate reabsorption (intestinal)
VII General renal tubular disorders (Fanconi’s syndrome)
VIII Primary mineralization defects
IX States of rapid bone formation with or without a relative defect in bone resorption
Nutritional Osteomalacia and Rickets
In 17th century Scotland and England, the association of poverty and malnutrition with the occurrence of infantile rickets was vividly documented. The widespread prevalence of infantile rickets in the industrialized regions of Britain was further documented in reports from Glasgow in the late 1800s and early 1900s. The link between rickets, dietary deficiency of vitamin D, and correction of vitamin D deficiency by solar radiation was finally established in 1923 by the work of the Vienna Council. Following this discovery, fortification of certain foods with vitamin D reduced the incidence of nutritional rickets in Europe and the United States to negligible levels, and by the 1940s, vitamin D deficiency was no longer regarded as an important cause of osteomalacia and rickets.43 Vitamin D metabolism and the role of specific metabolites in bone development, mineralization, and remodeling are discussed elsewhere (see Chapter 3). Nevertheless, the role of vitamin D in the prevention of osteomalacia and rickets is relevant to the discussion in this chapter.
Studies in Glasgow51–53 and London54 documented the reappearance of nutritional osteomalacia and rickets as a public health problem in the United Kingdom. Since the 1950s, the population at risk primarily involves East Asian immigrants whose unique dietary and social customs have led to the development of osteomalacia and rickets.52,54–58 Other groups at risk include housebound and elderly subjects and food faddists, especially those on vegetarian or fat-free diets.52,54–58 An unexpectedly high prevalence of osteomalacia was also observed in elderly women with rheumatoid arthritis who were housebound and had poor nutritional status.59
Gastrointestinal and Hepatic Diseases
Another population at risk for development of nutritional osteomalacia are morbidly obese individuals60 and those who have undergone intestinal bypass surgery for treatment of severe obesity.61–63 In one bypass surgery series, iliac bone biopsies revealed the presence of osteomalacia in nearly a third of 21 patients studied.63 Treatment with vitamin D2 (36,000 IU/day), as well as supplemental calcium (27 mmol/day), was required to promote a more positive calcium balance in some individuals.61 An increased frequency of osteomalacia also was observed in patients after gastrectomy,64–66 the severity of the mineralization defect being positively correlated with serum 25-hydroxyvitamin D [25(OH)D] but not with serum 1,25-dihydroxyvitamin D [1,25(OH)2D].
Rickets occurs in infants and children with cholestatic liver disease.67 Children with biliary atresia develop biliary cirrhosis, jaundice, and ascites. Intestinal absorption of vitamin D is markedly impaired, and serum values are low. Bone disease has been attributed to vitamin D deficiency, and pharmacologic doses of 1,25(OH)2D3 are required for treatment of the bone disease.68
Low-birthweight infants are at risk for rickets, with the reported incidence ranging from 13% to 32%. Insufficient intake of calcium, phosphorus, and vitamin D have been implicated. The condition should be detected early, since prompt nutritional supplementation is required.69 Infants of vitamin D–deficient mothers are at greatest risk for neonatal rickets,70 which can be averted by maternal vitamin D supplementation during pregnancy. Special consideration should be given to screening for vitamin D deficiency during pregnancy in women whose social and dietary history reveal inadequate sun exposure and food sources of vitamin D. Infants who are entirely breastfed and have inadequate exposure to sunlight are also at risk for rickets, because the amount of vitamin D and 25(OH)D in human milk is inadequate.71
Vitamin D sources include dietary supplementation with ergocalciferol (vitamin D2), an irradiation product obtained from plants, and cholecalciferol (vitamin D3), produced in human skin by the action of ultraviolet light on the physiologic precursor, 7-dehydrocholesterol. Because most foods (with the exception of fatty fish) contain only small amounts of vitamin D3, individuals must rely on either adequate sunlight exposure or dietary supplements to avoid vitamin D deficiency. There is marked seasonal variation in plasma 25(OH)D, independent of age and sex.72–74 These variations parallel changes in duration and intensity of sun exposure, with higher values in late summer months in the northern hemisphere. Plasma 25(OH)D is almost twice as high in American women of Caucasian descent than in those of African descent during winter months, and the increment during summer months is also greater.75 Plasma 25(OH)D and parathyroid hormone (PTH) are inversely correlated, implying that the changes in circulating 25(OH)D are metabolically significant.76 In a study of patients who were hospitalized in a general medical ward, plasma 25(OH)D was low in over half of the subjects, consistent with a high prevalence of vitamin D deficiency.77 Thus, adequate vitamin D supplementation is essential when exposure to sunlight is marginal.
Vitamin D requirements are greater in the elderly than in young adults. This difference is attributed to an age-related decline in dermal production of 7-dehydrocholesterol, the precursor for vitamin D3,78 diminished renal production of 1,25(OH)2D,79 and diminished intestinal absorption of calcium caused by lower levels of the vitamin D receptor in the intestine.80 Other contributing factors include previous gastric surgery and occult malabsorption in addition to altered vitamin D metabolism. To determine optimal plasma 25(OH)D and how much vitamin D is required to achieve optimal values, Vieth,81 in an exhaustive review, and Heaney in another82 concluded that total daily intake and production of vitamin D of 2.5 to 5.0 mg (100 to 200 IU) and plasma 25(OH)D values greater than 20 to 25 nmol/L are sufficient to prevent clinical rickets or osteomalacia. Higher intakes or production of vitamin D would be necessary, however, to prevent secondary hyperparathyroidism, bone loss, and subclinical osteomalacia. Therefore, it may be necessary to maintain serum 25(OH)D levels of 100 nmol/L or greater to avoid bone loss, subclinical osteomalacia, and osteoporosis. The amount of vitamin D supplementation necessary to attain these levels varies widely, based on dietary factors and solar exposure.
In the East Asian immigrant populations of Britain, rickets and osteomalacia secondary to vitamin D deficiency are seen most commonly in neonates, infants, and adolescents during pubertal growth and less frequently among adults.51,83–85 Multiple factors have been implicated in the development of bone disease, including insufficient intake of calcium and vitamin D, skin pigmentation,86 which attenuates ultraviolet transmission through the epidermis, genetic factors,87–89 and social customs, such as avoidance of sun exposure and consumption of chapati, a dietary staple flatbread high in phytate, which binds calcium in the gut and interferes with its absorption.90 Furthermore, studies in rats demonstrated that the rate of inactivation of vitamin D in the liver was increased by a calcium-restricted diet.
Studies by Dent and colleagues91 strongly suggest that insufficient sunlight exposure plays a pivotal role in the development of nutritional rickets and osteomalacia, in addition to the rickets and osteomalacia observed in patients on long-term anticonvulsant therapy.92 In two carefully studied individuals, they demonstrated healing of rickets and positive calcium balance following therapy with ultraviolet light, despite a vitamin D–deficient, high-phytate diet. Substitution of a low-phytate diet did not affect the plasma biochemical abnormalities or the calcium balance.91
Scriver93 divides the evolution of vitamin D deficiency in infancy into three stages. In stage 1, serum calcium tends to be low, serum phosphorus normal, and aminoaciduria absent. Without treatment, stage 2 develops, and aminoaciduria and hypophosphatemia appear as a consequence of diminished tubular reabsorption. In this stage, serum calcium tends to return to normal, and serum alkaline phosphatase increases, presumably related to the increased PTH and resultant increase in bone turnover. Renal tubular dysfunction (aminoaciduria, phosphaturia) has, at least in part, been attributed to the increased PTH.94,95 Stage 3 is characterized by the return of hypocalcemia. The effect of lowered concentrations of serum phosphorus in stage 2 and lowered concentrations of serum calcium and serum phosphorus in stage 3 presumably account for development of the mineralization defect.
Rao et al.96 reviewed the histomorphometric findings in a series of 65 patients with vitamin D depletion diagnosed on the basis of plasma levels of 25(OH)D less than 10 ng/mL. They found that in early vitamin D depletion, the effects on bone are manifested principally by the occurrence of secondary hyperparathyroidism. With increasing severity or duration of the vitamin D deficiency, the mineralization process becomes progressively impaired, bone formation rates decline, and osteoid surface and thickness increase.
In summary, rickets and osteomalacia are being recognized with increasing frequency in selected populations. Serum 1,25(OH)2D is not a reliable indicator of vitamin D nutrition, since values may be normal in individuals with vitamin D deficiency.97,98 The availability of serum 25(OH)D assays has also permitted detection of those at risk, prior to the development of overt clinical disease.
Acidosis and Osteomalacia
Acidosis resulting from a number of different causes has been associated with osteomalacia. The mechanism of bone loss and the mineralization defects are complex and not completely understood. Albright and Reifenstein43 originally suggested that acidosis produces slow dissolution of the mineral phase of bone in an attempt to buffer retained hydrogen ion. This process is associated with hypercalciuria. Support for this suggestion has been obtained in studies of patients with renal tubular acidosis in whom retention of hydrogen ion is greater than that theoretically required to produce the observed decrease in plasma bicarbonate concentrations.99 On the basis of measurements in normal subjects in whom metabolic acidosis is induced, it has been proposed that excess retained hydrogen ion is balanced by bone buffering and loss of bone calcium in the urine.100–102 Since the hypercalciuria and increased bone resorption that accompany most acidotic states do not directly result in osteomalacia, other mechanisms must be invoked to explain the occurrence of clinically significant skeletal mineralization defects. Osteoclasts function optimally at a pH of approximately 6.9 and are inactive at a pH greater than 7.3; therefore, it is probable that the calcium release from bone induced by acidosis can be ascribed to increased osteoclast activity rather than to simple physicochemical buffering.103 Chronic acidosis activates vacuolar hydrogen ion pumps in isolated osteoclasts and stimulates osteoclastic bone resorption.104 In addition, activation of the voltage-gated H+ channel through protein kinase C in osteoclasts can sense changes in pH.105
In parallel, metabolic acidosis inhibits the function of osteoblasts: in osteoblast culture systems, lowering pH to 6.9 decreases formation of mineralized nodules.106 Acidosis increases expression of osteoblastic RANK-L via a cyclooxygenase-dependent mechanism, leading to enhanced osteoclastogenesis.107 Another potential proton-sensing mechanism in the osteoblast is G protein–coupled receptors OGR1 and OGR2.108 OGR1 is inactive at pH 7.8 and fully activated at pH 6.8, signals through the phosphoinositol pathway, and is expressed in osteosarcoma lines and primary osteoblasts. OGR2 signals through the cyclic adenosine monophosphate (cAMP) pathway but has not yet been shown to be expressed in bone cells. Maintenance of pH within a critical range is thus essential for bone-cell function and for mineralization to proceed normally.
Acidosis can also affect phosphate metabolism by altering renal tubular handling of the anion. In patients with chronic acidosis, treatment with alkali to correct the acidosis can normalize serum phosphate by increasing renal tubular phosphate reabsorption and phosphate maximal tubular excretory capacity.109,110 Secondary hyperparathyroidism may be another factor involved in the altered phosphate handling in systemic acidosis,109 and acidosis can impair intestinal calcium absorption in response to exogenous vitamin D,106 as well as activation of 25(OH)D.111 Rickets and osteomalacia secondary to acidosis are most frequently a complication of inherited distal renal tubular acidosis (RTA).112 Clinical manifestations vary widely in type as well as severity. Autosomal-dominant as well as autosomal-recessive forms have been described. As a rule, the dominant form is usually recognized in adults who present with relatively mild disease characterized by nephrolithiasis and acidosis. Osteomalacia occurs infrequently. In contrast, the autosomal-recessive form is recognized in infancy or early childhood, is usually severe, and rickets is common. In dominant RTA, the causal mutation is in the SLC4A1:AE1 gene that encodes the heterotopic Cl−/HCO3− exchanger located in the α-intercalated cells of the distal tubules. In recessive RTA, the causal mutations are in the B1 or A4 subunits of the H+/ATPase encoded by ATP6V1B1 and ATP6V0A4 genes, respectively. Rarely, mutations in the SLC4A1:AE1 gene occur in recessive RTA as well. In most of the cases described in early reports,113–115 healing of the bone disease resulted from correction of the acidosis with sodium bicarbonate alone (5 to 10 g/day). Nevertheless, healing is slow, and the response may be hastened by the addition of vitamin D or 1,25(OH)2D3. Occasionally, vitamin D toxicity can develop unexpectedly, so patients must be monitored carefully. Although chronic treatment with vitamin D is not necessary once the osteomalacia is cured, continued use of vitamin D may be required to complete healing in those individuals in whom the glomerular filtration rate is low.115,116
Another form of inherited mixed distal/proximal RTA is associated with osteopetrosis and cerebral calcifications. The osteopetrosis, due to inadequate osteoclast function secondary to defects in acidification in the extracellular space adjacent to the ruffled border, is caused by mutations in the gene that encodes carbonic anhydrase II (CAII).117–119 This RTA is characterized by defective urinary acidification and bicarbonate wasting, and the syndrome is explained by the fact that CAII is expressed not only in osteoclasts but in both proximal and distal segments of the nephron. It is of interest that bone marrow transplantation does not affect the acidosis but reverses the osteopetrosis (by radiologic and histologic criteria), since osteoclasts are of hematopoietic origin.120
In several of the syndromes associated with more widespread renal tubular reabsorptive defects, systemic acidosis may contribute to the pathogenesis of osteomalacia. Some of these are inherited, such as various forms of Fanconi’s syndrome and Lowe’s syndrome (oculocerebrorenal syndrome). Some patients with renal tubular phosphate leaks may also have mild acidosis.121–125 Other aspects of these syndromes are considered elsewhere in this chapter.
[/not-level-membership-for-endocrinology-diabetes-and-metabolism-category]
