Chapter 91 Diseases of the Neuromuscular Junction
Both acquired and inherited disorders of the neuromuscular junction (NMJ) are seen in childhood. As with adults, the most common NMJ disorders are autoimmune and respond to immunosuppressive therapy. These include myasthenia gravis (MG) and, rarely, the Lambert–Eaton myasthenic syndrome (LEMS). A number of genetically determined disorders of neuromuscular transmission, the congenital myasthenic syndromes, are unique to childhood and are discussed later in this chapter. Botulism, a toxin-mediated disorder of the NMJ, most commonly occurs in infancy. All NMJ disorders have the ability to produce generalized weakness and fatigability with a propensity for oculobulbar involvement. Electrophysiological studies will detect an impairment of neuromuscular transmission in most of these disorders [Harper, 2004]. Fortunately, most of these disorders are treatable [Barohn, 2008]. Before descriptions of the various NMJ disorders are given, normal relevant physiology is reviewed.
The Neuromuscular Junction
Familiarity with the pathophysiology, diagnosis, and treatment of NMJ disorders requires a fundamental understanding of normal neuromuscular transmission (Figure 91-1). Acetylcholine is the natural neurotransmitter of the NMJ. It is synthesized and stored in vesicles in the motor nerve terminal [Drachman, 1978]. Each vesicle contains a quantum (about 10,000 molecules) of neurotransmitter. At rest, individual vesicles spontaneously release their quantum of acetylcholine at special release sites on the presynaptic membrane. The released neurotransmitter migrates and binds to acetylcholine receptors (AChR) located on the postsynaptic membrane, producing a transient increase in the permeability of the membrane to sodium and potassium ions. The local end-plate depolarization that results is known as a miniature end-plate potential (MEPP).
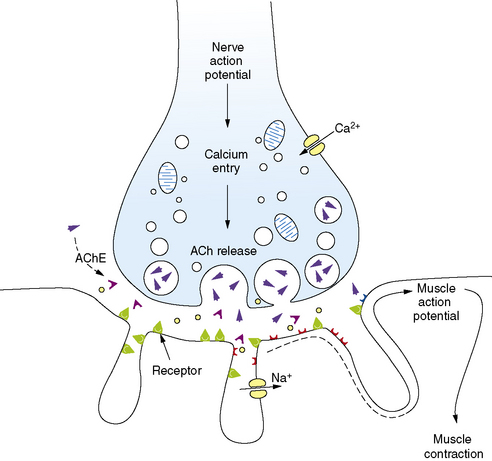
The amplitude of the EPP is directly related to the number of acetylcholine molecules that bind to their receptors. Normally, as a result of what has been termed the “safety margin” of NMJ physiology, the released amount of neurotransmitter is more than sufficient to produce a muscle action potential [Hughes et al., 2004]. As discussed later, the immunologic defect in the NMJ disorder, myasthenia gravis, directly reduces this safety margin, and muscle weakness ensues. A change in safety margin is also seen in other NMJ disorders.
Neuromuscular transmission is rapid, taking only milliseconds to complete the entire sequence. The process is terminated by diffusion of acetylcholine from the synapse and its rapid hydrolysis by acetylcholinesterase (AChE) [Lui and Erickson, 1994].
By 20 weeks’ gestation, the structure of the NMJ is well established, with further refinement of the postsynaptic membrane continuing until term [Sanes and Jessell, 2000]. Clustering of AChR in utero is dependent on muscle-specific receptor tyrosine kinase (MuSK) and rapsyn. Both the AChR and MuSK have been implicated as postsynaptic targets of the immune system in MG [Hoch et al., 2001). Other NMJ antigens targeted by plasma factors are likely to be discovered in the future [Plested et al., 2002].
Autoimmune Myasthenia Gravis
MG is the best-understood autoimmune disease of the nervous system [Vincent et al., 2001]. The immune-mediated nature of MG was suspected as early as 1960, when Simpson speculated that it was an autoimmune disease with antibodies directed against skeletal muscle AChR [Simpson, 1960]. A series of breakthroughs in the 1970s confirmed Simpson’s hypothesis. Lindstrom and colleagues developed the animal model experimental autoimmune MG by immunizing rabbits and rats with highly purified AChR from the electric organ of the eel [Patrick and Lindstrom, 1976a; Lindstrom et al., 1976b]. Not only did the animals develop weakness and respiratory insufficiency in about 3 weeks, but they responded to anticholinesterase medications and showed typical decrementing responses on repetitive nerve stimulation [Seybold et al., 1976]. These animals also had high AChR antibody titers in the serum. Subsequently, these titers were found in the serum of MG patients [Lindstrom et al., 1976]. Engel and co-workers localized both the IgG antibody and complement to the myasthenia motor end plate [Engel et al., 1977; Engel and Arahata, 1987]. This implied that circulating immunoglobulin (Ig) G antibody directed against the AChR bound to the postsynaptic membrane and activated the terminal complement sequence (C5b-9), or membrane attack complex, resulting in lysis of the AChR with subsequent degeneration.
Elevated membrane attack complex levels have been demonstrated in the plasma of MG patients [Barohn and Brey, 1993]. Anti-AChR antibody has also been shown to block neuromuscular transmission and accelerate turnover of AChR cross-linked by IgG [Drachman, 1978]. As a result of this process, the postsynaptic membrane becomes simplified with decreased junctional folds [Engel et al., 1976]. In addition, the neuromuscular blockade is passively transferred by injecting animals with IgG from MG patients [Toyka et al., 1977]. The same phenomenon occurs when an infant born to a mother with MG exhibits symptoms at birth, so-called neonatal MG [Papazian, 1992˜].
The AChR is a large protein consisting of five subunits, and the antibody response in MG and experimental autoimmune MG is polyclonal. The portion of the protein primarily responsible for inducing antibodies that produce the disease is debated. Although the existence of a main immunogenic region in the alpha subunit has been promoted [Tzartos and Lindstrom, 1980], other investigators have challenged this evidence [Lennon and Griesmann, 1989]. It is possible that, if the most pathogenic determinants of the AChR can be identified, a more rational and specific immune therapy can be designed. Multiple mechanisms are known to cause loss of functional AChRs in MG; these include complement-mediated lysis, accelerated internalization and degradation of AChRs, and direct blockade of AChRs by antibodies [Drachman et al., 1980; Meriggioli and Sanders, 2009]. Among these, complement-mediated lysis is thought to be the most important mode of loss of AChR.
The process that initiates the immune-mediated NMJ dysfunction is still unknown. The thymus gland may play a role, as 75 percent of MG patients who undergo thymectomy have thymic pathologic findings, with 15 percent being tumors of the thymus and the remainder consisting of lymphoid hyperplasia [Castleman, 1966]. Lymphocytes in the thymus and peripheral blood appear to be sensitized to muscle in MG patients [Sommer et al., 1990, 1991]. Muscle-like myoid cells are found in the thymus gland, and thymus tissue from MG patients with and without thymoma is enriched in AChR-reactive T cells [Wekerle et al., 1975; Kao and Drachman, 1977]. The close association of lymphocytes and myoid cells in the thymus, along with some stimulus causing the disruption of immune tolerance, may lead to the autoimmune response. There may be a hereditary predisposition to develop MG because there is an increased incidence of certain human leukocyte antigens (HLA) in various MG populations [Meriggioli and Sanders, 2009].
Epidemiology
MG has a prevalence of approximately 125 cases per million population [Drachman, 1994]. Approximately 11–24 percent of all MG patients have disease onset in childhood or adolescence [Simpson, 1958; Millichap and Dodge, 1960]. There is a slight female predominance of 3:2, although males predominate in older age groups. The disease can arise at any age, but peaks are observed in the third and sixth decades.
Clinical Features
The best data on the natural history of MG are from Grob and co-workers, who carefully studied 1976 patients between 1940 and 2000 [Grob et al., 2008]. Some of the key points from their work follow:
Clinical Classification
Osserman classified MG patients according to disease severity [Osserman, 1958]. The most commonly used modification of the original classification is as follows:





The Osserman classification has several shortcomings, including the vague descriptive terminology and lack of distinctions between some groups. For instance, differentiating between “mild” and “moderate” generalized disease may be difficult. The scheme also fails to include a category for patients in remission. A task force of the Myasthenia Gravis Foundation of America (MGFA) developed a new classification system (Box 91-1) that is more descriptive and provides better distinction between classes [Jaretzki et al., 2000; Barohn, 2003].
Box 91-1 Myasthenia Gravis Foundation of America Classification System

Class II



Class III







Categories of Myasthenia Gravis in Childhood
Autoimmune MG in children is most commonly divided into neonatal transient and juvenile types.
Neonatal transient MG occurs in infants of myasthenic mothers. Placental transfer of anti-AChR antibody or immunocytes results in transient impairment of neuromuscular transmission in the neonate [Barlow, 1981; Donaldson et al., 1981]. Neonatal MG has also been identified in mothers who have MuSK antibodies [O’Carroll et al., 2009]. Findings such as a weak suck or cry, ptosis, dysphagia, generalized weakness, decreased spontaneous movement, or respiratory distress are usually present in the first few hours of life but may not be evident until the third day [Millichap and Dodge, 1960]. The majority of patients have hypotonia or transient weakness and a very small proportion present with arthrogryposis multiplex congenita [Jeannet et al., 2008]. The hypotonia and transient weakness usually resolve in the first 4 weeks but may persist for months [Desmedt and Borenstein, 1977; Branch et al., 1978], and sometimes can lead to persistent facial and bulbar manifestations [Jeannet et al., 2008]. The severity of the disorder in the infant does not correlate with the degree of maternal involvement, but there is evidence that a higher maternal antibody titer may predict severity and onset of neonatal myasthenia [Eymard et al., 1989]. A prior history of neonatal MG in a sibling is the only predictive factor. Fortunately, only 10–15 percent of infants born to myasthenic mothers develop the disorder [Fraser and Turner, 1953; Namba et al., 1970; Ahlsten et al., 1992]. Prior thymectomy or remission of disease in the mother does not prevent development of neonatal transient MG [Geddes and Kidd, 1951; Elias and Appel, 1979], but has decreased the likelihood of neonatal myasthenia [Djelmis et al., 2002]. Careful monitoring of pregnant women with MG is critical because there is a 40 percent chance of disease exacerbation during pregnancy and a 30 percent risk in the puerperium. Perinatal mortality is approximately 68 per 1000 births, five times the risk in uncomplicated pregnancies [Plauche, 1991].
Juvenile MG represents the childhood onset of autoimmune MG seen in adults, but there are differences in the presentation [Chiang et al., 2009]. Onset is usually after 10 years of age, and disease manifestations appear before puberty in half the cases. Onset before 1 year of age is exceptional [Fenichel, 1978; Andrews et al., 1993; Geh and Bradbury, 1998]. Pubertal status might affect the clinical presentation, with higher incidence of ocular MG in prepubertal patients and generalized MG in postpubertal patients [Batocchi et al., 1990; Evoli et al., 1998]. Asian children have a higher incidence of ocular presentation [Chiang et al., 2009]. Female predominance was observed only after the age of 10 years [Haliloglu et al., 2002].
As with adults, ptosis is the most common clinical finding, frequently accompanied by ophthalmoparesis (Figure 91-2). Ptosis was unilateral at onset in one-third of juvenile MG patients, but subsequently spread to the other eye in nearly 90 percent of cases [Afifi and Bell, 1993]. Facial and oropharyngeal weakness is another common finding, producing dysarthria, dysphagia, and difficulty chewing. Facial weakness without ocular involvement is an unusual but recognized presentation of juvenile MG [Kini, 1995]. Extremity weakness can occur and is usually most prominent proximally. Bulbar weakness, characterized by slow chewing, dysphagia, nasal dysarthria, and weak cough, develops in up to 75 percent of patients [Rodriguez et al., 1983]. Respiratory failure from either diaphragmatic or intercostal muscle weakness or airway compromise related to bulbar dysfunction produces myasthenic crisis, an exacerbation severe enough to endanger life. As in adults, the disease may be generalized at onset, but isolated ocular involvement is a more common presentation, followed by generalization at a later time. However, children with ocular MG appear more likely than adults to remain with purely ocular disease. As many as 85 percent of adults with ocular MG later go on to develop generalized disease [Weinberg et al., 1994; Evoli et al., 1998]. In children, this percentage is closer to 50–75 percent [Afifi and Bell, 1993; Andrews, 2004]. Different HLA antigens are linked to various ethnic groups. Asian children tend to have HLA DRw9 [Wong et al., 1992], Caucasians are DQ8- or DR3-positive, and patients of African descent tend to have DR5 [Christiansen et al., 1984].
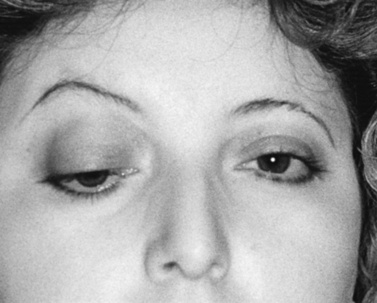
MG is frequently associated with other diseases, specifically those with an immune etiology. The most common are rheumatoid arthritis, thyroid disease, systemic lupus erythematosus, and diabetes mellitus [Millichap and Dodge, 1960; Rodriguez et al., 1983; Afifi and Bell, 1993]. Nonimmune disorders associated with juvenile MG include epilepsy in 3–13 percent [Snead et al., 1980; Rodriguez et al., 1983], various forms of neoplasia, particularly thymoma, and, later in life, breast carcinoma. Thymoma in juvenile MG, present in less than 5 percent of children Rodriguez et al., 1983], is relatively rare compared to adults and is mainly found in children with teenage onset [Andrews, 2004].
Neurologic Examination
Testing for weakness in the orbicularis oculi muscle is critical but often overlooked. Many symptomatic MG patients have bilateral weakness of this muscle group. Strength should also be tested in the lower facial muscles (blowing out cheeks against resistance) and in the tongue. Attention to speech patterns may disclose a nasal dysarthria. It is important to check for neck flexion and extension weakness because these muscle groups are frequently involved. Testing of extremity strength should include proximal and distal muscle groups in the arms and legs. Proximal limb muscles tend to be more affected than distal muscles. Rarely, MG patients may demonstrate a predilection for weakness in distal muscle groups, especially finger extensors [Nations et al., 1999].
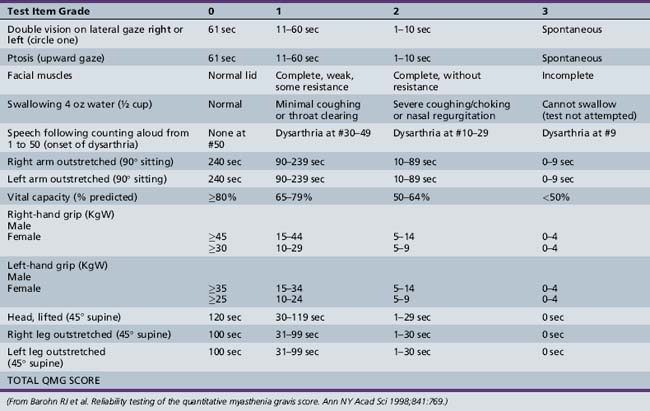
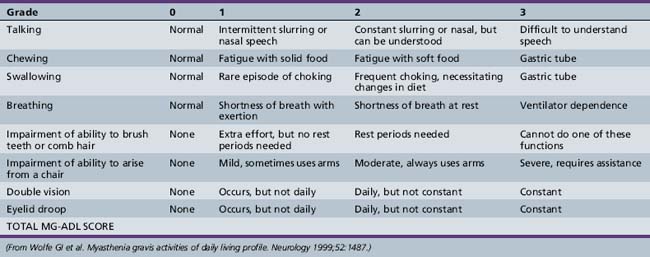
Symptoms and signs can be quantified by using a validated MG scoring system and activities of daily living scale (Tables 91-1 and 91-2). The activities of daily living scale correlates with the objective quantitative MG score [Wolfe et al., 1999]. An MGFA task force has recommended that the quantitative MG score be used as an outcome measure for therapy trials in MG [Barohn, 2003]. The scales are readily administered to adolescents as young as 15 years of age [Barohn et al., 1998]. A newer composite MG score with 10 items has recently been validated in adults [Burns et al., 2008] and it should be equally easy to administer in children as it was in adults.
Clinical and Laboratory Tests
Edrophonium (Tensilon) Test
The intravenous administration of up to 10 mg of edrophonium is often the first diagnostic test performed in the evaluation of a potential MG patient. However, the edrophonium test has a number of pitfalls. The most common mistake is that the physician performing the test does not have an objective parameter to measure before and after edrophonium administration. The most useful parameter is the degree of ptosis in each eye. The best indication of a positive test is a significant increase in the palpebral fissure aperture or the opening of a completely ptotic eye. If no ptosis is present, the edrophonium test may be difficult to interpret even in clear-cut cases of MG. If the patient has a severe restriction of extraocular movement and edrophonium dramatically improves the motility, the test is considered positive. However, subjective diplopia may not resolve unless edrophonium produces orthophoria in the eyes, which is rare. Significant improvement in dysarthria or in swallowing is another indication of a positive edrophonium test. A mild improvement in limb strength or subjective well-being is not sufficient to claim a positive test. In addition, a positive edrophonium test is not specific because transient subjective improvement is reported in other neurologic disorders, such as motor neuron disease and peripheral neuropathy [Oh and Cho, 1990].
The edrophonium test is performed in a straightforward manner. First, 1 mL (10 mg) of edrophonium is drawn up into a 1-mL tuberculin syringe. The edrophonium is often injected directly into a vein (usually antecubital), but in smaller children a “butterfly” intravenous line can be started, connecting the tuberculin syringe directly to the intravenous line. In children weighing less than 30 kg, the total delivered dose should not exceed 0.1 mg/kg. For children weighing more than 30 kg, a total dose up to 0.2 mg/kg may be given. Initially, a test dose of 0.01 mg/kg is injected. If, after 30–60 seconds, the patient experiences no side effects from the drug (fasciculations, sweating, nausea), further aliquots of 0.01–0.02 mg/kg are injected, not exceeding the total dose suggested. When injecting into an intravenous line, line flushes with saline are needed with each aliquot. Compared with adults, children are more likely to experience nausea, so an emesis basin should be available. More serious side effects, such as bronchospasm or lightheadedness caused by bradycardia, are quite uncommon, but atropine should be readily available. When either side effects or a positive response are obtained, no further edrophonium need be given. The mean dose of edrophonium needed to produce a positive response was 3.3 ± 1.6 mg for ptosis and 2.6 ± 1.1 mg for oculomotor dysfunction in a survey of 83 adult patients [Kupersmith et al., 2003].
In infants and younger children who are uncooperative and difficult to monitor over brief time periods, longer-acting neostigmine may be preferred. The intramuscular dose is 0.15 mg/kg and the intravenous dose is 0.05 mg/kg [Andrews, 2004]. Intravenous use can be hazardous due to severe muscarinic side effects [Wolfe et al., 1997]. A positive response is generally evident by 15 minutes and is most obvious after 30 minutes. Positive results on edrophonium or neostigmine testing are seen in up to 90 percent of juvenile MG cases [Afifi and Bell, 1993]. It is a good idea to have injectable atropine available in the case of severe side effects whenever using neostigmine.
Electrophysiologic Testing
Repetitive stimulation
The classic electrophysiologic demonstration of an NMJ transmission defect is the documentation of a decremental response of the compound muscle action potential (CMAP) to repetitive stimulation (RS) of a motor nerve [Oh, 1988]. The decrement is due to failure of some muscle fibers to reach threshold and contract when successive volleys of ACh vesicles are released at the NMJ. Failure to reach the threshold EPP to achieve muscle contraction is called blocking. The percentage decrease in amplitude and area is calculated between the first CMAP produced by a train of stimuli and each successive one. In most laboratories, five or six responses are obtained at 2 or 3 Hz, and the maximal percentage decrement can be measured at the fourth or fifth response. A decrement of greater than 10 percent is considered a positive RS study (Figure 91-3).
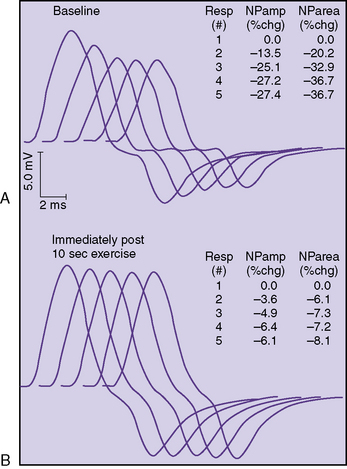
In some patients, a decremental response can be demonstrated at baseline. However, often a brief period of exercise (usually 1 minute) is required to fatigue the NMJ so that the decrement can be observed. This phenomenon of postexercise exhaustion (PEE) usually occurs at 2–4 minutes after exercise (Figure 91-4). In addition, repair or an improvement in the decrement can sometimes be observed immediately (within seconds) after brief exercise (see Figure 91-3).
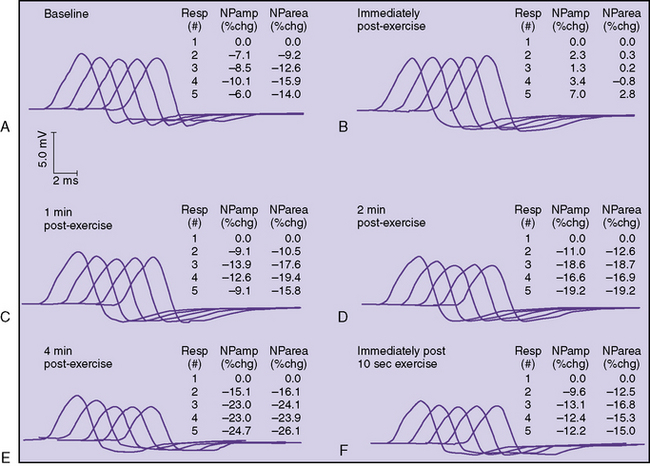
A protocol for RS of the ulnar nerve recording over the adductor digiti minimi follows:







A decremental response is more likely present in a proximal muscle than in a distal muscle. In the series by Stalberg and Sanders, a decrement in a distal muscle was reported in 38 percent of patients, whereas a decrement in proximal muscles occurred in 64 percent [Stalberg and Sanders, 1981]. Similar findings have been described by other authors [Vial et al., 1991; Oh et al., 1992]. In a study of 27 juvenile myasthenic patients, Afifi and Bell found that the chance of finding a decrement doubled to 66 percent by including proximal muscles. In children with generalized disease at onset, 80 percent showed a decrement when proximal muscles were studied. In ocular MG, decrements are less common, occurring in 20–50 percent of patients [Stalberg and Sanders, 1981; Evoli et al., 1988]. Facial muscle RS should be included when clinical suspicion for anti-MuSK myasthenia exists, as facial muscles are much more prominently involved in this group [Muppidi and Wolfe, 2009]. RS at faster rates (i.e., 20 or 50 Hz) usually is not performed unless there is concern about LEMS, a rare condition in children.
Single-fiber electromyography
Single-fiber electromyography (EMG) is a more sensitive measure of neuromuscular transmission than RS and can be considered in select children. In MG, the time required for the EPP at the NMJ to reach threshold is extremely variable. The measurement of this variability in the EPP rise time is known as jitter. The jitter value, calculated in microseconds, is the most important piece of data obtained from single-fiber EMG. Everyone, including healthy individuals, has some degree of jitter (Figure 91-5). Myasthenic patients have increased jitter values (Figure 91-6). In addition, blocking occurs in myasthenics if a muscle fiber’s EPP never reaches threshold and depolarization does not occur. The frequency of blocking, expressed as a percentage, is also determined with single-fiber EMG (see Figure 91-6). In healthy individuals, the percentage of blocking is 0 percent.
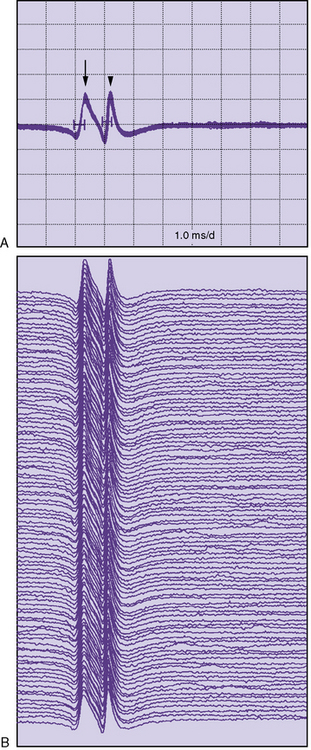
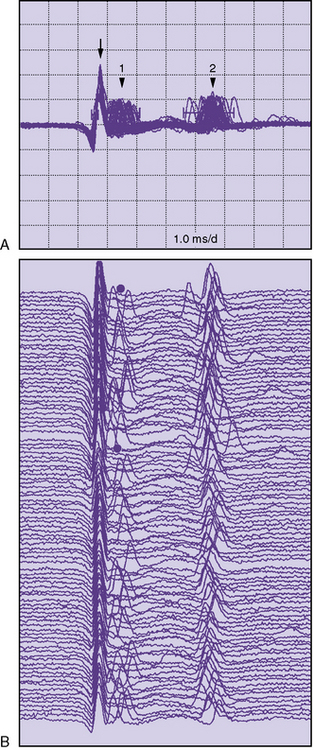
Single-fiber EMG is undoubtedly the most sensitive test for MG in adults. It is abnormal in 94 percent of generalized and 80 percent of ocular MG patients [Sanders and Howard, 1986; Oh et al., 1992]. However, single-fiber EMG has several disadvantages. It is a tedious and lengthy study that requires considerable patient cooperation and is poorly tolerated by many children. The need to use nondisposable single-fiber electrodes was also a limitation, but recently, normative data for single-fiber studies with disposable concentric needles have been published [Stalberg and Sanders, 2009]. Stimulated single-fiber EMG can be performed under sedation, requires less patient cooperation, and may be preferred in children, although it is still a lengthy procedure [Jabre et al., 1989]. An abnormal single-fiber EMG study is not specific for MG because increased jitter commonly occurs as a result of other neuromuscular diseases, including motor neuron disease, peripheral neuropathy, and many myopathies [Oh, 1988]. Fortunately, it is seldom necessary to perform single-fiber EMG to diagnose MG in children. It is probably most useful in children who present difficult diagnostic dilemmas and who otherwise have normal laboratory studies for MG. Single-fiber EMG abnormalities may be seen in 12–33 percent of first-degree relatives of patients with juvenile MG [Stalberg et al., 1976; Anlar et al., 1995]. Conventional needle EMG has limited diagnostic value in MG; however, short-duration, small-amplitude, early recruited myopathic units can be seen in anti-MuSK MG patients [Padua et al., 2006].
Antibody Testing
Anti-AChR antibodies
Anti-AChR antibody levels are not elevated in all MG patients. The assay is most helpful in adult generalized MG; it is positive in 85 percent of such patients [Lindstrom et al., 1976; Vincent and Newsom-Davis, 1985; Oh et al., 1992; Drachman, 1994]. Ocular MG patients, however, have measurable anti-AChR antibodies in only 50 percent of cases [Provenzano et al., 2009]. Children represent another group of MG patients who are often antibody-negative. In one study, 50 percent of prepubertal children with autoimmune juvenile MG were seropositive [Andrews et al., 1994]. Seropositive rates of 68 and 91 percent were observed in peripubertal and postpubertal disease onset, respectively. Similarly, seropositivity was more common in girls with onset of juvenile MG after 11 years of age [Anlar et al., 1996]. Seronegativity was more common in pure ocular forms, mild disease, and remission [Afifi and Bell, 1993].
Because congenital myasthenic syndromes and seronegative autoimmune MG present in early childhood, differentiating these disorders when the family history is negative is often difficult [Andrews, 2004]. Fluctuating weakness or disease severity and good responses to immunotherapy favor an autoimmune basis [Anlar et al., 1996].
The most common anti-AChR antibody test is the binding radioimmunoassay using bungarotoxin, measured in nanomoles per liter. The upper limit of normal varies among reference laboratories (usually between 0.03 and 0.5 nmol/L) [Lennon, 1982]. Other assays that block bungarotoxin binding to AChR (“blocking” assay) or that reduce the density of AChR on cultured human myotubes (modulating antibody assay) are also commercially available [Howard et al., 1987]. These additional assays may be useful in patients with suspected MG who test negative with the standard binding assay [Howard et al., 1987], but do not add significantly to the diagnostic sensitivity. Some laboratories offer all three (binding, blocking, and modulating) antibodies as one serological test. Recently, low-affinity anti-AChR antibodies against rapsyn-clustered AChR were seen in 66 percent of patients who were otherwise seronegative. These were mainly IgG1 antibodies that can activate complement C3b deposition [Vincent et al., 2008]. These new low-affinity AChR assays are not yet commercially available.
Anti-AChR antibody titers correlate poorly with MG severity [Roses et al., 1981]. Although the titer often falls as the clinical condition improves, antibody titers in general do not guide therapeutic decisions. Indeed, MG patients in clinical remission may still have elevated titers, but this is not an indication to continue immunosuppressive therapy.
Anti-MuSK antibodies
Since 2001, IgG from 40–70 percent of seronegative generalized patients has been found to bind to the extracellular domain of muscle-specific receptor tyrosine kinase (MuSK) [Hoch et al., 2001; Sanders et al., 2003; McConville et al., 2004]. Marked female predominance with mean age of onset in the fourth decade has been typical [Evoli et al., 2003; Sanders et al., 2003], although Evoli et al. encountered a child with disease onset at age 6 years, and two patients from the initial series presented before age 10 [Hoch et al., 2001]. The earliest reported onset of anti-MuSK MG is 2 years [Murai et al., 2006]. Three main patterns of anti-MuSK MG have been observed; one of them is clinically indistinguishable from anti-AChR generalized MG. The other two patterns are severe oculobulbar weakness and prominent neck, shoulder, and respiratory involvement, largely sparing ocular musculature. In these two phenotypic variants, limb strength is relatively intact [Sanders et al., 2003; Muppidi and Wolfe, 2009]. Anti-MuSK antibodies rarely occur in pure ocular MG [Wolfe et al., 2007].
It has been hypothesized that anti-MuSK antibodies impede agrin-mediated clustering of AChR and disrupt normal postsynaptic architecture [Jha et al., 2006]. We consider testing for anti-MuSK antibodies in all suspected MG patients who are anti-AChR antibody-negative. The assay is commercially available. Anti-MuSK MG is somewhat more refractory to conventional treatment when compared to anti-AChR MG [Pasnoor et al., 2009].
Striated muscle antibodies and other laboratory studies
Antibodies to striated muscle in MG patients were discovered before anti-AChR antibodies. These antibodies can be directed against a number of muscle proteins, including myosin, actin, alpha-actinin, titin, and ryanodine (RyR). It is generally believed that, if anti-striated muscle antibodies are present in an MG patient, they should raise suspicion for thymoma, as they are reported in up to 84 percent of patients with thymoma [Limburg et al., 1983]. However, these antibodies may be found in patients without thymoma and in patients with thymoma who do not have MG [Cikes et al., 1988; Romi et al., 2000]. The absence of anti-striated antibodies also does not rule out thymoma. From an adult perspective, anti-titin and anti-RyR antibodies have been forwarded as a marker for more severe disease in MG patients presenting after age 40 [Romi et al., 2000]. Thyroid function tests are routinely obtained at the time of initial evaluation, as thyroid disease often coexists with MG [Meriggioli and Sanders, 2009].
Treatment
Most patients with juvenile MG who require maintenance therapy are treated with anticholinesterase agents, with or without a variety of immunosuppressive medications. Pyridostigmine is recommended as an initial intervention [Snead et al., 1980; Saperstein and Barohn, 2004]. As with the adult form of the disease, corticosteroids and other immunomodulatory treatments are used, although few randomized controlled clinical trials have been performed in the pediatric MG population. Thymectomy plays an important role in treating older children at most centers. Plasmapheresis [Pinching and Peters, 1976] and intravenous gamma globulin [Arsura, 1989] are generally reserved for more refractory patients or for those in myasthenic crisis. Plasmapheresis and intravenous gamma globulin are also used to maximize function before thymectomy. Short-term supportive care and anticholinesterase agents are usually adequate for neonatal transient MG. Management for the different types of childhood MG is reviewed in the following sections.
Acetylcholinesterase Inhibitors
In juvenile MG, the aggressiveness of management should be in accordance with disease severity. In general, management attempts should first focus on pyridostigmine. A total daily dose of up to 7 mg/kg a day is delivered in 5–6 divided doses [Wolfe et al., 1997]. Older children can use 60 mg tablets that can be split in half as needed. Typical doses in older children and adults are 60 mg, 3–5 times a day. If symptoms are poorly controlled on a pyridostigmine dose exceeding 300 mg/day, it is probably necessary to add immunomodulating therapy. Sustained-release pyridostigmine is also available (Mestinon TS, 180 mg). However, because of variable absorption, difficulty adjusting doses, and increased side effects, many clinicians discourage use of this preparation.
Thymectomy
When a child’s symptoms can no longer be controlled by anticholinesterase agents alone, a decision must be made regarding whether to pursue thymectomy or immunosuppressive therapy. In 1939, Blalock and co-workers reported the remission of generalized MG in a 21-year-old woman after removal of the cystic remains of a necrotic thymic tumor [Blalock et al., 1939]. Since then, thymectomy, with or without the presence of thymoma, has gained widespread acceptance as a treatment for MG. Thus thymectomy was the first attempt at “immunotherapy” for MG and continues to be one of the most common treatments for the disease.
There is a general consensus that generalized MG patients between puberty and 60 years of age benefit from thymectomy [Rowland, 1987; Lanska, 1990]. However, randomized studies of thymectomy that control for medical therapy have never been performed. The use of thymectomy in very young children is controversial, in itself, because of concerns about a subsequent impairment in immune protection or an enhanced risk of cancer. A review of incidental thymectomy and thymectomy as treatment for MG in young children, however, did not find a consistent association between thymectomy and these proposed risks in children older than 1 year [Seybold, 1998]. In a survey that included 56 neurologists with MG expertise, lower age limits for performing thymectomy ranged from 1 year to puberty, with the median being 7.5 years [Lanska, 1990].
An evidence-based practice parameter from the American Academy of Neurology analyzed retrospective, controlled, nonrandomized studies of thymectomy in MG. A total of 28 studies published between 1953 and 1998 were identified [Gronseth and Barohn, 2000]. The effect of surgery was broadly favorable in most series. However, the benefit of surgery was generally small. For example, the median relative rate favoring surgery over nonsurgical treatment for achieving remission was 2.1 (a modest gain, considering that the median remission rate in the non-thymectomized groups was 10 percent). Other median relative rates were 1.6 for asymptomatic status, 1.7 for improvement, and 1.1 for survival. Patient subgroup analysis indicated that only those MG patients with moderate weakness or greater (Osserman 2b) showed a significant improvement following thymectomy compared to controls. Importantly, the modest benefits ascribed to thymectomy were confounded by baseline differences between the surgical and nonsurgical groups, as well as limited thymic resections performed in a significant percentage of the patients. No study included blinded assessments. As a result, the authors expressed uncertainty as to whether claims of improved MG outcomes were a result of thymectomy or related to differences in baseline characteristics between surgical and nonsurgical groups. The authors concluded that thymectomy should be considered a treatment option in patients without thymoma [Gronseth and Barohn, 2000]. To address this uncertainty, an international prospective, single-blinded, randomized trial in patients with non-thymomatous MG controlling for medical therapy is currently under way [Newsom-Davis et al., 2008]. In this study, patients are randomized to prednisone alone versus trans-sternal thymectomy with prednisone. The minimum age to enter into the study is 18 years.
Thymectomy has been widely used for the treatment of juvenile MG, showing a higher likelihood of remission when performed within 2 years of symptom onset compared to nonsurgical patients [Seybold, 1998]. In the largest study, 85 of 149 patients (57 percent) with juvenile MG underwent thymectomy. Of the thymectomized patients, 42 (49 percent) entered remission and another 25 (29 percent) improved clinically [Rodriguez et al., 1983]. In contrast, only 34 percent of patients who did not undergo thymectomy entered remission. Remission rates were 260 per 1000 person-years in the first year after thymectomy, falling to 95 per 1000 person-years over the next year after surgery [Rodriguez et al., 1983]. After 3 years, the remission rate decreased to 20 per 1000 person-years, similar to the spontaneous remission rate. Favorable predictors for postoperative remission included surgery during the first year after disease onset, bulbar symptoms, absence of ocular or generalized involvement, onset of symptoms between ages 12 and 16 years, and the presence of other autoimmune disorders [Rodriguez et al., 1983]. Gender of the child or thymic pathology did not appear to influence remission rates. Histologic findings of the thymus were normal in 16 percent, hyperplastic in 78 percent, and neoplastic in 3 percent.
Other series suggest that the clinical benefit from thymectomy may not be realized for as long as 7–10 years [Mulder et al., 1989]. These favorable clinical responses and the low morbidity and mortality of thymectomy in childhood cases bolster its widespread use [Batocchi et al., 1990], prompting some to consider the procedure at the onset of generalized symptoms [Adams et al., 1990]. Clinical improvement after thymectomy has been observed, whether or not there is a reduction in circulating anti-AChR antibody levels [Oosterhuis et al., 1985]. The role of thymectomy in patients with anti-MuSK antibody-positive myasthenia is unclear. Early reports suggested that most patients do not experience any benefit with thymectomy [Sanders et al., 2003]. Thymic tissue in the anti-MuSK antibody-positive population does not demonstrate morphologic abnormalities, and thymectomy does not produce significant clinical improvement for these patients [Lauriola et al., 2004]. Probability of achieving remission seems to be slightly higher in seronegative non-thymomatous patients when compared to anti-MuSK antibody-positive non-thymomatous patients [Ponseti et al., 2009].
The goal of thymectomy is to remove all of the thymic tissue. Trans-sternal thymectomy is the surgical technique used in the on-going thymectomy trial [Newsom-Davis et al., 2008]. Other approaches include transcervical, infra-axillary video-assisted thoracoscopic procedures, and various combinations of these with trans-sternal incisions [Jaretzki et al., 2004]. There is some evidence to support the view that the greater the resection, the better the long-term results [Jaretzki et al., 1988]. Using a “maximal” thymectomy approach that includes both transcervical and trans-sternal incisions, life-table analyses demonstrated an 81 percent remission rate at 7.5 years [Jaretzki and Wolff, 1988]. Comparative remission rates for transcervical approaches have been in the 30–45 percent range at 7 years [Jaretzki et al., 2003], and approximately 50 percent at 6 years using either an extended trans-sternal or a video-assisted thoracoscopic procedure that includes a transverse cervical incision [Mantegazza et al., 2003]. It should be noted that remission rates in surgical series often are unexpectedly high. Definitions of remission, as well as their duration, vary between studies, and the retrospective determination of these outcomes is certainly open to bias [Gronseth and Barohn, 2000]. Transcervical and video-assisted thoracoscopic procedures offer advantages from a cosmetic and postoperative recovery standpoint. Thoracoscopic procedures have been found to reduce the length of hospital stay and patient care costs when compared to open thoracic surgery [Hazelrigg et al., 1993].
The presence of a thymoma is, of course, the one absolute indication for thymectomy. All newly diagnosed MG patients must undergo computed tomography or magnetic resonance imaging of the chest to look for evidence of thymoma. Routine chest radiographs may not detect up to 25 percent of thymic tumors [Batra et al., 1987]. Although there is a consensus among neurologists that thymectomy should be considered in all but the youngest children with generalized MG who are not controlled on anticholinesterase agents alone, there is less enthusiasm for the procedure in pure ocular MG.
Corticosteroids
Prednisone continues to be used as an immunosuppressive agent of first choice in MG. Despite its many potential side effects, prednisone is considered by many to be the most effective oral immunosuppressive agent for the treatment of MG [Meriggioli and Sanders, 2009]. Although corticosteroids can potentially suppress the immune system in a variety of ways, the exact explanation for the beneficial response in MG is unknown. However, prednisone can reduce anti-AChR antibody titers, and this may correlate with clinical improvement [Tindall, 1980].
Typically, oral prednisone is initiated at 1–1.5 mg/kg daily. If clinical improvement occurs in the first 4 weeks, the patient can be switched immediately to an alternate-day dose of 1–1.5 mg/kg [Warmolts and Engel, 1972]. Daily regimens extending beyond a month must be followed by a slower alternate-day taper. If corticosteroid therapy is initiated at moderate to high doses, an improvement is usually apparent in 2–3 weeks [Pascuzzi et al., 1984]. However, in some patients, daily dosing may be necessary for 2–3 months before there is clinical improvement. The main concern when initiating prednisone therapy at these doses is the transient worsening that occurs in one-third to one-half of patients [Pascuzzi et al., 1984; Evoli et al., 1992]. The mechanism may involve a direct effect of corticosteroids to impair NMJ function [Miller et al., 1986]. In one study, 8.6 percent of patients who experienced transient worsening required intubation [Pascuzzi et al., 1984]. In another report, 3 of 20 children started on corticosteroids developed respiratory failure [Badurska et al., 1992]. Thus, an advised practise is to admit MG patients to the hospital for 5–7 days when initiating high-dose prednisone therapy. During this time, bulbar function and forced vital capacity are monitored daily. If patients remain stable over this interval, they can be safely discharged on oral prednisone.
The therapeutic efficacy of prednisone in adult MG has been clearly demonstrated [Pascuzzi et al., 1984]. In a study of 116 patients, prednisone produced pharmacologic remission (asymptomatic on medication) in 28 percent, marked improvement in 53 percent (minor symptoms and return to activities of daily living), moderate improvement in 15 percent (functional limitations), and no improvement in only 5 percent [Pascuzzi et al., 1984]. The mean time to maximum prednisone benefit was 5.5 months (range, 2 weeks to 6 years). This study also made clear the disappointing fact that only 14 percent of patients were able to discontinue prednisone treatment and maintain improvement. Similar response rates have been reported in children [Andrews, 2004].
It is noteworthy that a retrospective analysis suggests that prednisone reduces the incidence of disease generalization at 2 years in patients presenting with pure ocular MG [Kupersmith et al., 2003]. Only 7 percent of ocular MG patients receiving prednisone developed generalized disease, compared to 36 percent receiving only pyridostigmine or no medication. Similar observations were made from two smaller, retrospective series [Mee et al., 2003; Monsul et al., 2004].
After significant improvement is observed, there should not be a rush to taper off the prednisone. Most patients remain on alternate-day dose prednisone for at least 6–8 months. Premature tapering of prednisone is a common management error. When beginning the taper, it is best to proceed slowly by reducing the dose no faster than 5 mg every 2 weeks. When the patient’s dose has been reduced to 20 mg every other day, tapering at an even slower rate is advisable. Although an attempt should be made to taper the patient fully off prednisone, it is not uncommon for patients to require low-dose therapy (5–10 mg every other day) for many years or indefinitely. Prior thymectomy does not appear to influence the outcome of steroid withdrawal [Miano et al., 1991].
The side effects of corticosteroids are well appreciated and significant. Children should be closely monitored for cataracts, hypertension, diabetes mellitus, weight gain, growth retardation, and cognitive or affective disturbance [Pascuzzi et al., 1984; Drachman, 1994; Massey, 1997; Andrews, 2004]. Adverse effects of corticosteroids on early stages of bone mineralization and development are also of concern, emphasizing the need for “steroid-sparing” strategies in children who are refractory to tapering.
Azathioprine
Azathioprine (Imuran), an antimetabolite that blocks cell proliferation and inhibits T lymphocytes, has been used more frequently in recent years because of the side effects associated with corticosteroids. Most reports describe its use in adults. It is used most often in patients who have a relapse on prednisone, or who have been on prednisone for lengthy periods, in the hope that the steroid dose can be decreased or eliminated. Azathioprine is also used at times as a first-line immunosuppressive agent instead of prednisone [Matell, 1987; Hohlfeld et al., 1988; Mantegazza et al., 1988; Myasthenia Gravis Clinical Study Group, 1993].
Retrospective studies of azathioprine therapy have demonstrated that 70–90 percent of MG patients improve [Mertens et al., 1981; Matell, 1987; Hohlfeld et al., 1988; Mantegazza et al., 1988; Myasthenia Gravis Clinical Study Group, 1993], whether or not it is used as a first- or second-line immunosuppressive drug. However, the clinical response is slow, not appearing for 12 months or more. A double-blind study compared the use of oral prednisolone with prednisolone plus azathioprine. Patients receiving azathioprine had fewer relapses, longer remissions, and fewer side effects with less weight gain [Palace et al., 1998]. At 3 years, 63 percent of patients receiving azathioprine had been completely tapered off prednisolone, compared to 20 percent who had received prednisolone alone. However, the beneficial effect of azathioprine was not noted statistically until month 18 of the study. Thus, if a patient already on pyridostigmine is quite weak and a rapid response is required, azathioprine is not a practical choice. Azathioprine may be used in combination with plasmapheresis in children who are refractory to other management modalities [Carter et al., 1980].
Azathioprine is supplied as a 50-mg, scored tablet that can be easily broken in half. The initial dose of azathioprine in children should be no more than 25–50 mg daily for 1 week. If there are no side effects, the dose is increased to a target level of 2–3 mg/kg/day. Although azathioprine is generally well tolerated, there are three important and limiting side effects [Kissel et al., 1986]. Approximately 10 percent of patients have an idiosyncratic systemic reaction within the first several weeks of therapy that consists of fever, abdominal pain, nausea, vomiting, and anorexia. Patients feel as if they have influenza. When the drug is stopped, the symptoms resolve quickly. If the patient is rechallenged, the symptoms invariably recur. In addition, patients can develop leukopenia and hepatotoxicity. Blood counts and hepatic enzymes need to be monitored monthly. If the white blood cell count falls below 4000 cells/mm3, it is advisable to decrease the dose. If it falls below 3000 cells/mm3, medication should be temporarily withheld until the cell count normalizes. Similarly, medication should be temporarily withheld if there is evidence of hepatocellular dysfunction. Although the patient can be rechallenged after the laboratory values normalize, toxicity often recurs, requiring discontinuation of the drug. Patients should be evaluated for thiopurine methyltransferase (TPMT) deficiency, as this may lead to severe myelosuppression [Chiang et al., 2009]. Azathioprine dosing needs to be adjusted in hepatic and renal impairment, and potential interactions with other medications should be considered on initiation. Late development of malignancy following chronic use of azathioprine is a concern [Andrews, 2004], as there is higher incidence of lymphoma, and there is one report of renal cancer after prolonged use [Lindner et al., 1997].
Cyclosporine
Cyclosporine (Sandimmune, Neoral) is an accepted option for immunosuppressive therapy in adult MG. Data are not available on its use in juvenile MG, although it is frequently used in pediatric transplant patients. Cyclosporine inhibits helper T lymphocytes and allows the expression of suppressor T lymphocytes. It blocks the production and secretion of interleukin-2 by helper T cells. Cyclosporine has been subjected to randomized, double-blinded, placebo-controlled trials in MG [Tindall et al., 1987, 1993]. These studies demonstrated that cyclosporine was more effective than placebo in improving MG when a quantitative MG scale was used as the primary efficacy measure. In addition, cyclosporine lowered anti-AChR antibody levels. Corticosteroid doses could be reduced after cyclosporine was initiated [Tindall et al., 1993].
The onset of clinical benefit with cyclosporine is 1–2 months. This is somewhat faster than azathioprine and slower than prednisone. Doses for cyclosporine range between 4 and 6 mg/kg/day. The drug is usually given in two divided doses, rather than as a single dose, to reduce potential nephrotoxicity. Cyclosporine is available as 25- or 100-mg capsules or as an oral solution (100 mg/mL). Side effects include hirsutism, tremor, gum hyperplasia, paresthesias, and hepatotoxicity. Hypertension and nephrotoxicity are the main limitations to therapy. Over one-quarter of adult patients taking cyclosporine will have serum creatinine levels increase between 30 and 70 percent above baseline levels, and 11 percent of them also developed skin cancers [Ciafaloni et al., 2000]. Such high rates of neoplasia have not been reported in other studies [Lavrnic et al., 2005]. Blood pressure, renal function, and trough plasma cyclosporine levels are monitored monthly.
Mycophenolate Mofetil
Myophenolate mofetil (MM, CellCept) is another immunosuppressive agent commonly used in practice at MG centers, having shown initial promise in several uncontrolled, open series demonstrating favorable responses in two-thirds of adult patients [Chaudhry et al., 2001; Ciafaloni et al., 2001]. Benefits have included improved functional status and a corticosteroid-sparing effect. In a retrospective analysis of 85 patients that employed MGFA postintervention classifications, 73 percent achieved pharmacologic remission, minimal manifestation status, or improvement with MM [Meriggioli et al., 2003]. Patients with severe weakness (MGFA class IV) were less likely to respond. MM had a relatively rapid onset of action, with improvement observed at a mean of 9–11 weeks and maximal improvement by approximately 6 months. However, in some subjects, the initial response was delayed up to 40 weeks. With its more rapid onset of action and favorable side-effects profile, MM began replacing azathioprine as the first-line “steroid sparer” in many MG centers. Ultimately, two multicenter, randomized, controlled trials were conducted to evaluate the benefit of MM in MG. In one of these phase 3 studies [Muscle Study Group, 2008], 80 patients receiving prednisone 20 mg daily were randomized to MM versus placebo for 12 weeks, followed by a 24-week open-label phase. The primary end point was change in the quantitative MG (QMG) score (see Table 91-1) from baseline to week 12. There was no significant difference in the QMG score or secondary outcome measures between the groups. In the second study [Sanders et al., 2008], 176 patients who were dependent on more than 20 mg of prednisone daily were randomized to either MM or placebo for 36 weeks. The primary end point was percentage of patients who reached MGFA minimal manifestation or pharmacological remission status. Again, there was no significant difference in the percentage of responders or the change in QMG between the groups. Several investigators have commented on the failure of the two studies to show a benefit of MM over corticosteroid treatment alone. These include the short duration of the treatments; a better-than-expected response to prednisone; the selection of an untested primary outcome measures in the longer trial; and the predominant enrollment of patients with mild to moderate generalized MG [Benatar and Rowland, 2008; Sanders and Siddiqi, 2008]. Pediatric patients were not included in either of the two studies.
The main side effects of MM are diarrhea, vomiting, increased risk for infection, and leukopenia, the last of which is relatively uncommon. Complete blood counts are checked weekly for the first month and then less frequently. Long-term safety for MM is still in question, but there have been reports of primary central nervous system lymphoma with use of MM in MG [Vernino et al., 2005].
Cyclophosphamide
The use of cyclophosphamide (Cytoxan), a nitrogen mustard alkylating agent that blocks cell proliferation, is mainly reserved for refractory MG patients, but its reported use is limited. One study reported 42 adults who had been treated with cyclophosphamide; 23 were also on prednisone [Perez et al., 1981]. At the time of the retrospective data analysis, 25 of the 42 patients became asymptomatic, and 12 were in complete remission off all medications. Eight of 10 children with juvenile MG improved when cyclophosphamide was added to regimens including azathioprine and corticosteroids [Badurska et al., 1992].
In a later randomized, placebo-controlled, double-blinded study, monthly intravenous pulses of cyclophosphamide 500 mg/m2 were given to 23 adult MG patients with severe, refractory disease or steroid-related side effects [De Feo et al., 2002]. At month 12, those in the cyclophosphamide arm had significantly improved muscle strength on QMG. At both 6 and 12 months, steroid doses were significantly lower in the cyclophosphamide group. Similarly, impressive responses to therapy were seen in three refractory adult patients who received high-dose (50 mg/kg) intravenous cyclophosphamide for 4 days, followed by “rescue” with granulocyte colony stimulating factor [Drachman et al., 2003]. Marked improvement in strength without disease recurrence over several years was observed.
The high rate and severity of toxicity are the drawbacks of therapy with cyclophosphamide. In one study, alopecia occurred in 75 percent, leukopenia in 35 percent, and nausea and vomiting in 25 percent [Perez et al., 1981]. The increased risk of bladder and lymphoreticular malignancy with prolonged administration of cyclophosphamide should be of particular concern. As a result, cyclophosphamide should be considered only in the most refractory cases of juvenile MG.
Plasmapheresis
Plasma exchange was first used in MG in 1976 [Pinching and Peters, 1976] and is primarily employed in the short-term, acute management of severe disease, including crisis, and in readying weak patients for thymectomy [Dau et al., 1977; Behan et al., 1979; Campbell et al., 1980]. Plasmapheresis removes anti-AChR antibodies from the circulation of MG patients, with improvement measured in several days, rather than weeks for corticosteroids and months for immunosuppressive agents. Two other circumstances in which plasmapheresis is considered include the treatment of severely weak patients admitted for initiation of prednisone therapy, and as a chronic intermittent therapy in patients with refractory disease [Chiang et al., 2009]. Plasmapheresis has been life-saving in some children [Snead et al., 1980].
Generally, a course of plasmapheresis consists of 4–6 exchanges, in which approximately 50 mL/kg of plasma are removed at each treatment. Decisions regarding the number of exchanges and total amount removed are largely driven by the status of the patient, including clinical response and tolerability of the hemodynamic shifts from the procedure. Improvement is often seen within 48 hours after the first or second exchange. Treatments are usually administered every other day or on no more than 2 of 3 consecutive days, so that a full course is completed in 7–10 days [Andrews, 2004].
Tacrolimus
Case reports and open trials have demonstrated efficacy for tacrolimus as monotherapy or when added to corticosteroids. In the largest report of 212 patients, tacrolimus was given at a dosage of 0.1 mg/kg per day in two divided doses, later adjusted for plasma drug concentrations between 7 and 8 mg/mL [Ponseti et al., 2008]. Approximately half of the patients were on prednisone or were cyclosporine-dependent. The mean follow-up time was nearly 50 months (range, 12–79 months). With the addition of tacrolimus, prednisone could be withdrawn in 95 percent of patients. QMG scores fell significantly, from 20.5 at baseline to less than 1.0 at the final visit, and muscle strength improvement was evident as early as 1 month after treatment initiation. More than 85 percent of patients achieved complete, stable remission or pharmacologic remission at the end of follow-up, with another 5 percent reaching minimal manifestation status. Impressive remission results were observed, regardless of whether patients had undergone thymectomy and irrespective of whether patients had thymoma, although complete, stable remission was less likely in thymomatous MG. Tacrolimus was well tolerated overall, with hypertension in 1.9 percent of patients, nephrotoxicity in 2.9 percent, neurotoxicity, such as tremor or paresthesia, in 5.9 percent, and diabetes in 1.4 percent.
Intravenous Immunoglobulin
Over the last two decades, intravenous immunoglobulin (IVIg) has been used by neurologists for various immune-mediated neuromuscular diseases, including MG, in which response rates of approximately 75 percent were observed in early retrospective series. A randomized, double-blinded, placebo-controlled trial of IVIg in generalized MG was initiated but had to be terminated before an adequate number of subjects could be enrolled. In an open-label IVIg extension, favorable trends in quantitative strength and electrophysiologic outcome measures were seen in patients who had initially received placebo [Wolfe et al., 2002]. A recent randomized, double-blinded, placebo-controlled trial enrolled 51 patients and found a meaningful improvement in the QMG score 14 days after a 2 g/kg dose of IVIg versus 5 percent dextrose in water (D5W) placebo [Zinman et al., 2007]. The treatment effect persisted through day 28, although the change in QMG almost missed statistical significance (p = 0.055). The response appeared independent of age, sex, disease duration, and antibody status, since seronegative and anti-MuSK antibody-positive MG patients were also enrolled. When stratifying patients, it was determined that only those with more severe disease at entry (QMG scores ≥11) benefited from IVIg. The authors concluded that patients with minor symptoms or with pure ocular disease clinically and on quantitative scoring are unlikely to benefit from IVIg. Although no studies have focused on the pediatric population, children and adolescents have responded favorably to IVIg [Andrews, 2004].
Advantages of IVIg over plasma exchange include relative ease of administration and favorable side-effects profile in both children and adults. Headache, transient influenza-like symptoms, and hyperactivity are the adverse events most common to the pediatric population [Andrews, 2004]. Migraine patients are prone to develop a severe headache related to aseptic meningitis. The outbreak of hepatitis C related to some IVIg products [Schiff, 1994] has been controlled with improved donor screening and sterilization techniques. Major complications are observed in up to 5 percent of adults, including cardiovascular, cerebrovascular, and deep venous thrombotic events, congestive heart failure, and acute nephrotoxicity [Tan et al., 1993; Steg and Lefkowitz, 1994; Brannagan et al., 1996; Go and Call, 2000].
Drugs to Avoid
Children, like adults with MG, are sensitive to nondepolarizing neuromuscular blocking agents. Intermediate-acting nondepolarizing blockers, such as atracurium and vecuronium, should be used with care [Baraka, 1992]. It is advisable to administer small amounts of these agents, using neuromuscular monitoring as a guide [Brown et al., 1990]. Other commonly used drugs known to exacerbate MG or interfere with neuromuscular transmissions are listed in Box 91-2. Of these agents, aminoglycoside antibiotics and telithromycin are perhaps the most frequent culprits, and they should be used in patients with MG only when there are no reasonable alternatives.
























Lambert–Eaton Myasthenic Syndrome
LEMS is an acquired autoimmune disorder of NMJ transmission, in which the defect is in the presynaptic nerve terminal. Eaton and Lambert are credited with defining the syndrome, including the abnormalities seen on electrophysiologic studies [Lambert et al., 1956]. Elmqvist and Lambert later determined that the neuromuscular junction defect was due to inadequate presynaptic release of ACh [Elmqvist and Lambert, 1968]. An immune-mediated basis for LEMS was suspected when the disease was passively transferred to mice with purified IgG from patients [Lang et al., 1981]. At about the same time, it was shown that the number of voltage-gated calcium channels (VGCC) are reduced in the motor nerve terminal [Fukunaga et al., 1982]. A number of investigators have subsequently demonstrated that LEMS IgG is directed at VGCC, resulting in disruption of presynaptic architecture. The end result is inhibited influx of calcium into the motor nerve terminal when action potentials arrive. In turn, there is a reduction in the quanta of ACh vesicles released, and neuromuscular transmission is impaired.
Clinical Features
LEMS begins gradually and is characterized by fatigability and weakness in a limb-girdle distribution [Katz and Barohn, 2001]. Unlike in MG, patients may note that the weakness is worse soon after awakening, and better later in the day. While exercise can transiently improve strength, persistent exertion causes fatigue. Cranial nerve and respiratory involvement is less common than in MG. Up to 50 percent of patients can have mild degrees of ptosis, diplopia, dysphagia, and dysarthria. Patients also have autonomic involvement, including dry mouth and eyes, impotence, blurred vision, and orthostasis [Sanders, 2003]. On examination, there is a limb-girdle pattern of weakness and reduced or absent deep tendon reflexes. In only about 50 percent of patients can one demonstrate convincingly improvement in strength or reflexes after brief exercise [Odabasi et al., 2002; Sanders, 2002]. A malignancy is present in approximately 50 percent of adult LEMS patients. The tumor is usually a small-cell carcinoma of the lung, but other malignancies, such as renal-cell carcinoma and hematologic tumors, occur. Malignancy is more common in men than in women, and is more likely in the setting of chronic smoking or after age 50 years. LEMS can be the presenting manifestation of a small-cell carcinoma, and can precede the detection of the tumor by months. Patients with small-cell carcinoma tend to have a more rapidly progressive course than those without malignancy, with dysarthria, difficulty chewing, hand weakness, dry mouth, and impotence appearing earlier in the disease course [Wirtz et al., 2005].
LEMS is only rarely described in children. The first report was in a 10-year-old boy with leukemia [Dahl and Sato, 1974]. Childhood LEMS often occurs in association with a lymphoproliferative disorder. A congenital syndrome with features of LEMS has been described [Bady et al., 1987].
Diagnostic Tests
Diffusely reduced CMAPs on routine motor nerve conduction studies should raise suspicion of LEMS. If a cause for this finding is not evident, one should perform a brief screening test (Figure 91-7). After the baseline maximal CMAP amplitude is obtained – for example, from the adductor digiti minimi – the patient spreads the fingers apart against resistance for 10 seconds. Immediately (within 5 seconds) the supramaximal single-nerve stimulation is repeated and the CMAP recorded. In LEMS, there is a dramatic, often greater than 100 percent increase in the CMAP amplitude and area, the so-called potentiation (see Figure 91-7), although Oh and colleagues determined that a 60 percent increment has a higher sensitivity with excellent specificity for LEMS [Oh et al., 2005]. The physiologic effect of either brief exercise or tetanic stimulation generates adequate calcium influx to overcome the underlying defect in quantal release.
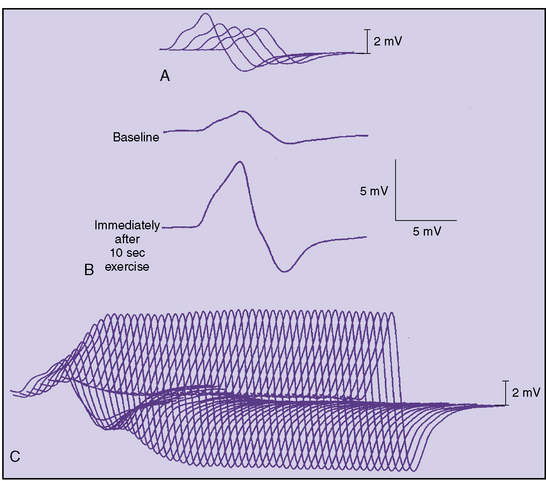
Incremental responses of 100 percent or greater can be found in at least one muscle in 87 percent of LEMS patients [Sanders, 2002]. It should be kept in mind that normal individuals can have increments as great as 40 percent [Oh, 1989]. Single-fiber EMG will show increased jitter in LEMS, but this will not differentiate the disease from MG. Likewise, edrophonium may produce mild improvement in strength but does not differentiate the two disorders.
Antibody testing for LEMS is readily available. In a study of 72 patients ranging from 16 to 80 years of age, 92 percent were seropositive for antibodies to P/Q-type VGCC [Motomura et al., 1996]. In contrast, only 33 percent were seropositive for anti-N-type VGCC antibodies. All patients with anti-N-type VGCC antibodies were also seropositive for anti-P/Q-type VGCC antibodies. Lennon and co-workers found that all LEMS patients with a diagnosis of cancer had anti-P/Q-type VGCC antibodies, compared to 91 percent of LEMS patients without cancer [Lennon et al., 1995]. Anti-N-type VGCC antibodies were present in only 49 percent of LEMS patients. These results support the notion that anti-P/Q-type VGCC antibodies are primarily responsible for the presynaptic defect in LEMS. Recent work demonstrates that two-thirds of adult LEMS patients with small-cell lung carcinoma have antibodies to SOX1, a tumor antigen [Sabater et al., 2008]. None of 50 patients with idiopathic LEMS harbored these antibodies. The authors suggest that detection of anti-SOX1 antibodies should mandate closer surveillance in patients not found to have cancer at initial evaluation.
Treatment
As with MG, treatment of LEMS should be individualized, tailored according to clinical severity, underlying disease, and life expectancy. Upon confirmation of the diagnosis, an extensive search for an underlying malignancy is imperative. This should begin with radiologic studies and may require bronchoscopy. Initial treatment should be directed against the malignancy, as this may improve neurologic manifestations. Clinical experience suggests that immunotherapy without directed treatment of the underlying cancer produces little clinical benefit [Sanders, 2003]. When no malignancy is found, repeat screening every 6–12 months is advised, especially in patients at greater risk.
Aggressive immunotherapy is easier to justify in LEMS patients without cancer, as there is concern that such agents may adversely impact management of malignant disease by interfering with normal immunosurveillance. In general, a trial of pyridostigmine is the first pharmacologic intervention, although it is often of limited benefit [Sanders, 2003]. Ideally, drugs that enhance the presynaptic release of ACh vesicles should be used. Guanidine hydrochloride is the oldest drug in this group and has been beneficial in some LEMS patients. However, the side effects of guanidine are prohibitive and include bone marrow depression, renal failure, gastrointestinal distress, ataxia, hypotension, paresthesias, confusion, dry skin, and atrial fibrillation. 3,4-diaminopyridine (3,4-DAP) increases the duration of the presynaptic action potential by blocking outward potassium efflux. This indirectly prolongs the activation of VGCC and increases calcium entry in nerve terminals. In a placebo-controlled randomized trial, patients who received 3,4-DAP demonstrated significant improvement based on a quantitative strength scale and increased CMAP measurement [Sanders et al., 2000]. More than 85 percent of patients experience significant clinical improvement with 3,4-DAP [Sanders, 2003]. Most patients tolerate the drug well, and side effects such as paresthesias tend to be minor. There is a risk of seizures when daily doses exceed 100 mg. Usually, doses up to 20 mg three times a day are used. Expert recommendations for laboratory monitoring include liver function tests, complete blood count, blood urea nitrogen, and creatinine every 3 months for the first year and then less frequently [Sanders, 2003]. Although 3,4-DAP is not approved for clinical use in the United States, it is available on a compassionate use basis through Jacobus Pharmaceutical (Princeton, NJ) and compounding pharmacists [Maddison and Newsom-Davis, 2005].
There are only a few clinical studies of immunosuppressive therapy in LEMS. Plasmapheresis, corticosteroids, and azathioprine can be effective in both neoplastic and non-neoplastic LEMS [Newsom-Davis and Murray, 1984]. IVIg has also been effective [Rich et al., 1997]. In general, the regimens and doses for the immunotherapies outlined for MG can be applied to LEMS. Response to immunosuppression tends to be less dramatic for LEMS than for MG [Sanders, 2002].
Congenital Myasthenic Syndromes
Congenital myasthenic syndromes (CMS) as a group are not uncommon. Some CMS are life-threatening, whereas others produce only mild weakness and are amenable to therapy [Engel, 1990; Shillito et al., 1993]. Therefore, an accurate diagnosis is necessary for rational management. Unlike MG and LEMS, CMS are not autoimmune, antibody determinations are negative, and immunosuppressive therapy is not effective. While clinical and electrophysiologic data may suggest CMS, specialized microelectrode analysis of neuromuscular transmission, ultrastructural studies of the neuromuscular junction, biochemical skeletal muscle assays, and molecular analysis are required to make a precise diagnosis. These genetically determined disorders all compromise, in one way or another, the safety factor that normally preserves the integrity of neuromuscular transmission, either directly or through secondary alterations at the NMJ.
The earliest report of CMS was in 1937 by Rothbart, who described four brothers under the age of 2 years with a myasthenic condition [Rothbart, 1937]. Bowman coined the term “congenital myasthenia” to describe the infant of normal parents whose myasthenic symptoms persisted into childhood [Bowman, 1948]. By 1972, 97 familial cases of early-onset myasthenia had been collected [Bundey, 1972]. Forty-one had presented prior to age 2 years. Using refined techniques, Lambert, Engel, and colleagues subsequently described three forms of CMS: end-plate AChE deficiency [Engel et al., 1977] slow-channel syndrome [Engel et al., 1982], and impaired resynthesis or vesicular packaging of ACh [Mora et al., 1987], also known as familial infantile myasthenias. Box 91-3 summarizes the syndromes that have been characterized to this point. Other subtypes likely will be described in the future.
Box 91-3 Classification of the Congenital Myasthenic Syndromes













A number of developments in the last 15 years have accelerated the molecular characterization of these disorders. These include the identification of the cDNA sequence for the various subunits of the AChR and AChE, improved resolution of patch clamping techniques in human intercostal muscle to allow recording of single-channel currents, and the ability to study mutant NMJ proteins in engineered mammalian systems [Engel and Ohno, 2002].
Clinical Features
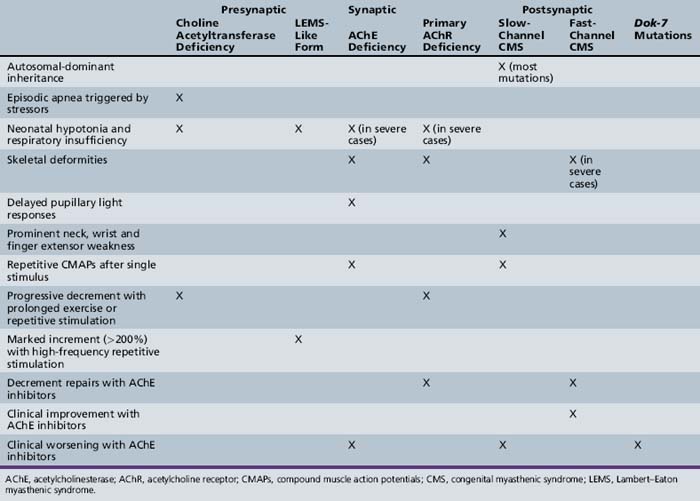
Most patients with CMS present in the first 2 years of life, with many becoming symptomatic in the neonatal period or in early infancy. However, clinical involvement may not occur until adolescence or adulthood, particularly in the slow channel syndrome. Symptoms can be episodic or worsen with fever and emotional stimuli. Typical clinical findings of CMS include feeding difficulty, respiratory dysfunction, ophthalmoparesis, ptosis, hypotonia, and limb fatigability. The attainment of developmental milestones, as well as progression and regression of symptoms, should be carefully documented. Response to anticholinesterases, corticosteroids, and other agents may assist in diagnosis. A positive family history is obviously suggestive of CMS, but a negative family history does not exclude autosomal-recessive inheritance, incomplete penetration, or a new mutation. Table 91-3 lists features that help distinguish the different forms of CMS.
Laboratory Evaluation
Definitive diagnosis requires specialized evaluation available in only a few research laboratories. These studies include microscopic and ultrastructural examination of muscle specimens to assess synaptic vesicle content, AChE assay, AChR abundance, and destructive postsynaptic changes that may reflect cholinergic overactivity, the so-called end-plate myopathy [Engel and Ohno, 2002]. Microelectrode electrophysiologic studies on intercostal or anconeus muscle biopsies can localize the process to a presynaptic, synaptic, or postsynaptic defect, and can define factors responsible for the impairment in neuromuscular transmission. MEPP amplitude and frequency, EPP amplitude, the number and probability of quanta released by nerve impulses, and quantal content are determined. Patch clamp recording of individual AChR defines the amplitude and duration of single currents to determine kinetic abnormalities of the receptor.
If these studies demonstrate features consistent with a defect in a candidate gene or protein, molecular analysis follows. If a mutation is found, genetically engineered models of the mutant molecule can be used to confirm the pathogenicity and define the properties of the mutation further. This approach has led to the discovery of over 100 mutations combined in various subunits of the AChR, AChE, and choline acetyltransferase (ChAT) [Ohno and Engel, 2000].
Synaptic Defects
End-plate Acetylcholinesterase Deficiency
The initial description of AChE deficiency was in 1977 [Engel et al., 1977], and was followed by several reports of either sporadic cases or autosomal-recessive pedigrees. Symptoms usually begin in the neonatal period with ptosis, poor suck and cry, and generalized moderate to severe weakness. Motor milestones are delayed. Lordosis and scoliosis are prominent features. Ophthalmoparesis is variable, seen in about one-half of patients. Abnormally sluggish pupillary light responses may also be present. Less severe phenotypes with later onset and milder progression have been identified, some mimicking the limb-girdle pattern described below for Dok-7 mutations [Mihaylova et al., 2008]. Decremental responses are seen on repetitive stimulation. Multiple motor action potential discharges may follow a single stimulus on routine motor nerve conduction studies, a characteristic, but nonspecific finding that is also seen in slow-channel syndrome and in the setting of excessive anticholinesterase use [Shillito et al., 1993]. Unlike in slow-channel syndrome, the administration of AChE inhibitors has no impact on the number and size of the repetitive discharges since there is no AChE to inhibit.
Recessive mutations in the gene encoding the collagenic tail of AChE (COLQ) have been identified in these patients [Mihaylova et al., 2008]. Neuromuscular transmission appears to be impaired by the small size of nerve terminals, an end-plate myopathy resulting from cholinergic overstimulation, and densensitization/depolarization block of ACh receptors [Engel and Ohno, 2002]. Patients often worsen with AChE inhibitors, which are of no benefit. Ephedrine, 150–200 mg a day in divided doses, may produce a dramatic clinical response [Bestue-Cardiel et al., 2005].
Postsynaptic Defects
Postsynaptic defects represent the most common forms of CMS (Table 91-4). These disorders predominantly arise from a variety of AChR subunit mutations that either enhance or reduce their response to neurotransmitter. Although phenotypes vary, postsynaptic defects tend to be less severe than other forms of CMS and at times respond more favorably to pharmacologic therapy. Muscle AChR is a transmembrane structure composed of five homologous subunits: two alpha, one beta, one delta, and one gamma subunit in the fetal form that is replaced by an epsilon subunit in the adult AChR. Based on extensive studies, two major kinetic abnormalities of AChR have been described: slow-channel and fast-channel syndromes [Engel and Ohno, 2002].
Table 91-4 Localization of Defect in 205 Congenital Myasthenic Syndrome Patients
| Type of Congenital Myasthenic Syndrome | Number of Cases (%) |
|---|---|
| Presynaptic | 15 (7) |
| Synaptic | 31 (15) |
| Postsynaptic | 159 (78) |
(From Engel AG and Sine SM. Current understanding of congenital myasthenic syndromes. Curr Opin Pharmacol 2005;5:308–321.)
Slow-Channel Congenital Myasthenic Syndrome
The slow-channel syndrome was first described by Engel and co-workers in 1982 [Engel et al., 1982]. Whereas most forms of CMS demonstrate recessive inheritance, slow-channel syndromes result from dominant gain-of-function mutations. Onset of symptoms varies considerably, ranging from infancy to adulthood [Shillito et al., 1993], although late onset is more typical of this syndrome than for other CMS. Clinical findings also vary, although there is a propensity for early cervical, scapular, and finger extensor weakness that helps to distinguish the syndrome. Ptosis and ophthalmoplegia are less prominent. Lower extremities may be less involved than upper extremities [Engel, 1990]. Weakness in most patients is slowly progressive. Decremental responses up to 40 percent and increased jitter are seen on electrophysiologic testing. Repetitive responses to single stimuli are characteristic of both weak and strong muscles, and are best seen at rates below 0.2 Hz, since at higher stimulation rates the second potential is often of very small amplitude [Oosterhuis et al., 1987]. Specialized electrophysiologic studies on intercostal muscle have shown normal or slightly reduced MEPP amplitudes, and normal quantal content of the EPP, but prolonged decay time of the MEPP and EPP. Although suspected earlier, it was not until 1992 that prolonged open time of the AChR was clearly demonstrated to account for the slowed decay of endplate currents [Engel et al., 1993]. On exposure to ACh, the abnormal AChR have prolonged activation, resulting in abnormally slowed decay of EPP. Spontaneous openings of the AChR are also observed. This change in kinetic activity produces cationic overloading and an endplate myopathy with loss of junctional folds and AChR, widening of synaptic space, and other morphologic alterations [Engel and Ohno, 2002]. These changes, in addition to the depolarization block that occurs prolonged EPPs, extend beyond the refractory period of muscle fibers and compromise neuromuscular transmission. A number of dominant mutations in the alpha, beta, and epsilon subunits of AChR have been described in slow-channel syndrome. Not surprisingly, AChE inhibitors and 3, 4-DAP worsen the condition. Quinidine and fluoxetine – agents that normalize abnormally prolonged AChR activation episodes in cell cultures expressing mutated receptors – have improved clinical and electrophysiologic elements in a small number of patients [Harper and Engel, 1998; Harper et al., 2003]. Quinidine has been problematic because of drug allergies.
Fast-Channel Congenital Myasthenic Syndrome
More recently described are the fast-channel syndromes. These are related to recessive, loss-of-function mutations of the AChR. Common features of these post-synaptic syndromes are rapid decay of end-plate currents, brief channel activation episodes, and reduced probability of channel opening in response to ACh, resulting in reduced MEPP amplitudes [Engel and Ohno, 2002]. A variety of kinetic abnormalities have been described in the fast-channel syndromes related to mutations in the alpha and epsilon subunits of the AChR. The syndromes result from a mutant allele that dominates the clinical phenotype combined with a null mutation on the second allele. Phenotypic severity varies from a disabling CMS related to low-affinity channels [Uchitel et al., 1993] to a mild syndrome associated with gating abnormalities. As would be predicted, fast-channel syndromes respond favorably to 3,4-DAP combined with anticholinesterases, as these agents enhance AChR activation by either increasing quantal release or reducing ACh degradation in the synapse [Engel and Ohno, 2002].
Recently, recessive mutations in Dok-7, a protein activator of MuSK, have been found in a fairly large number of CMS patients [Beeson et al., 2006]. Most patients present with ptosis and limb-girdle distribution weakness, but respiratory, bulbar, and distal weakness may be seen [Palace et al., 2007]. Eye movements are usually spared. Onset of symptoms stretches from the neonatal period to early childhood. NMJ size is smaller, with reduced postsynaptic folding when compared to normal end plates. Myotube experiments reveal that Dok-7 mutations impair MuSK function and produce smaller than normal clusters of ACh receptors [Beeson et al., 2006]. ACh receptor function is normal, however. Somewhat unexpectedly but in a way that is reminiscent of anti-MuSK MG, children with Dok-7 mutations worsen with pyridostigmine. Ephedrine and 3,4-DAP may produce improvement [Palace et al., 2007].
Acetylcholine Receptor Deficiency
Severe AChR deficiency has been linked to homozygous or heterozygous mutations of AChR subunits. Most mutations have involved the epsilon subunit [Engel et al., 1996]. This propensity may arise because null mutations of other subunits are lethal, whereas the fetal gamma subunit can partially compensate for the absence of the epsilon subunit. In addition, the epsilon subunit gene may be more susceptible to DNA rearrangements. Symptom onset is variable, ranging from the neonatal period to early childhood. Ptosis, ophthalmoplegia, and generalized hypotonia and weakness are typical. Ultrastructural studies demonstrate an expanded number of end-plate regions spanning a wider area of postsynaptic membrane [Engel and Ohno, 2002]. Junctional folds appear preserved, although some end plates are small and simplified. There is a notable reduction in the density and number of AChR. Reduced expression of AChR has also been associated with recessive mutations in the gene that encodes rapsyn, a protein involved in AChR clustering, and in plectin deficiency. As with fast-channel forms of CMS, anticholinesterases and 3,4-DAP may provide benefit.
Rapsyn mutations are a fairly common cause of AChR deficiency, representing 15 percent of CMS cases characterized at Mayo Clinic. A recent survey of 39 patients revealed that all but one child presented prior to age 2 years; 20 had intermittent exacerbations, with 12 requiring ventilatory support. Most of the children with long-term follow-up responded to cholinergic therapy, either with pyridostigimine or 3,4-DAP [Milone et al., 2009].
Botulism
Botulism is caused by a toxin produced by Clostridium botulinum, a Gram-positive anaerobic organism found commonly in soil and agricultural products. Eight immunologically distinct toxins have been identified, with most human cases caused by types A, B, or E [Cherington, 2004]. Recovery time varies between toxin types; for instance, type A toxin produces more persistent paralysis than types B and E. After binding irreversibly to receptors on presynaptic nerve terminals, the toxin is translocated across the membrane and blocks the calcium-dependent release of ACh. The action of the toxin appears independent of calcium entry. The impaired release of ACh results in failed neuromuscular transmission. The damage to nerve terminals may be permanent, resulting in a prolonged clinical recovery believed to be dependent on the formation of new neuromuscular junctions. Infantile botulism, first described in 1976 [Pickett et al., 1976], is now the most common form of the disease, representing over 70 percent of approximately 110 annual cases in the United States [Cherington, 2004].
Infantile Botulism
Most reported cases of infantile botulism in the United States occur in California, Pennsylvania, and Utah, states with high counts of C. botulinum spores in the soil. After spores of C. botulinum are ingested, they germinate and propagate in the gastrointestinal tract, producing toxin in vivo. Consumption of honey and corn syrup has been associated with infantile botulism, but in most cases the source of infection is not evident. Infantile botulism typically presents between 6 weeks and 9 months of age, often heralded by the onset of severe constipation. A descending paralysis with cranial nerve palsy, poor suck, feeble cry, and reduced facial expressions ensues, followed by limb weakness. The child appears hypotonic with ptosis, decreased extraocular motility, sluggish pupillary light responses, facial diplegia, weak suck, and a reduced gag response. Deep tendon reflexes and spontaneous bowel sounds are reduced. There is usually no associated fever. Both respiratory and autonomic compromise may occur. The diagnostic approach should consider other possible etiologies, including systemic infection, tick paralysis, organophosphate poisoning, Guillain–Barré syndrome, congenital myopathies, and autoimmune and congenital myasthenic conditions. Electrophysiologic studies support a presumptive diagnosis of botulism while waiting for the bacteriologic studies to return. Sensory nerve conduction studies are normal. CMAP amplitudes are normal to reduced with preserved motor conduction velocities. RS will confirm a presynaptic defect in neuromuscular transmission. Decremental responses are variably seen on slow rates of RS, but increments are present at rapid rates (20–50 Hz). An incremental response at rapid rates was the most distinctive finding in one study, observed in 92 percent of cases [Cornblath et al., 1983]. However, this finding may be less common in patients with intoxications due to toxin type A [Maselli and Bakshi, 2000]. Increments tend to be less dramatic than those in LEMS. Fibrillation potentials are seen on EMG in approximately 50 percent of infants, and short-duration, low-amplitude motor units in over 90 percent. Diagnosis is confirmed by isolation of the organism or toxin in stool samples. Toxin isolation is accomplished by the mouse neutralization test, usually performed at state health departments. Polymerase chain reaction assays have been developed but are not routinely available. In infantile botulism, the toxin is found in serum only rarely.
Treatment of infantile botulism is generally supportive, with complete recovery requiring several weeks to months. Botulism immune globulin, approved by the Food and Drug Administration in late 2003, reduced the hospital stays for both ventilated and nonventilated children [Thompson et al., 2005]. Overall, median hospital stays fell from 40 to 23 days. Case fatality rates of hospitalized patients in the United States are below 5 percent. Nasogastric or parenteral nutrition is needed if the child is unable to drink or eat. Mechanical ventilation may also be required. Aminoglycosides and other agents that impair neuromuscular transmission should be used with extreme caution. The length of hospitalization averages 4 weeks, but some infants may require supportive care for as long as 5 months. For diagnostic and treatment information on botulism, one can contact CDC-Info at 800-232-4636. Health-care providers will be routed to a clinical specialist.
Food-Borne Botulism
The classic form of botulism almost always follows ingestion of food contaminated by preformed toxin. The classic presentation is the development of bulbar symptoms, including blurred vision, diplopia, ptosis, dysarthria, and dysphagia within 12–36 hours after ingestion of the contaminated food. A descending pattern of weakness follows, and in some cases, respiratory paralysis. Supportive care measures have improved survival rates, and nearly full recovery is expected for surviving patients. Treatment with antitoxin mainly plays the main role in this form of botulism, but is not likely to be effective unless administered in the first 24–48 hours; the antitoxin does not reverse established paralysis. Beneficial effects are more likely with type E toxin [Cherington, 2004]. Use of antitoxin remains controversial due to lack of efficacy in some cases and the 20 percent risk of serious allergic side effects related to the equine source. The incidence of hypersensitivity increases further with polyvalent antitoxin. Antitoxin is obtained from the CDC in Atlanta.
Wound Botulism
Wound botulism appears 4–14 days following infection with C. botulinum. Although wound botulism is considered rare, an increasing number of cases have been recognized in drug abusers [Maselli et al., 1998]. Wound botulism can be severe, with ventilatory support required in a majority of patients. The organism can be isolated directly when an obvious wound is present. Wound debridement, appropriate antibiotic therapy, and supportive care are the keys to management.
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Adams C., Theodorescu D., Murphy E.G., et al. Thymectomy in juvenile myasthenia gravis. J Child Neurol. 1990;5:215-218.
Afifi A.K., Bell W.E. Tests for juvenile myasthenia gravis: comparative diagnostic yield and prediction of outcome. J Child Neurol. 1993;8:403-411.
Ahlsten G., Lefvert A.K., Osterman P.O., et al. Follow-up study of muscle function in children of mothers with myasthenia gravis during pregnancy. J Child Neurol. 1992;7:264-269.
Andrews P.I. Autoimmune myasthenia gravis in childhood. Semin Neurol. 2004;24:101-110.
Andrews P.I., Massey J.M., Howard J.F.Jr, et al. Race, sex, and puberty influence onset, severity, and outcome in juvenile myasthenia gravis. Neurology. 1994;44:1208-1214.
Andrews P.I., Massey J.M., Sanders D.B. Acetylcholine receptor antibodies in juvenile myasthenia gravis. Neurology. 1993;43:977-982.
Anlar B., Kuruoglu R., Varli K., et al. Acetylcholine receptor antibodies and single-fiber EMG in first-degree relatives of children with myasthenia gravis. Neuropediatrics. 1995;26:335-336.
Anlar B., Ozdirim E., Renda Y., et al. Myasthenia gravis in childhood. Acta Paediatr. 1996;85:838-842.
Arsura E. Experience with intravenous immunoglobulin in myasthenia gravis. Clin Immunol Immunopathol. 1989;53:S170-S179.
Badurska B., Ryniewicz B., Strugalska H. Immunosuppressive treatment for juvenile myasthenia gravis. Eur J Pediatr. 1992;151:215-217.
Bady B., Chauplannaz G., Carrier H. Congenital Lambert-Eaton myasthenic syndrome. J Neurol Neurosurg Psychiatry. 1987;50:476-478.
Baraka A. Anaesthesia and myasthenia gravis. Can J Anaesth. 1992;39:476-486.
Barlow C.F. Neonatal myasthenia gravis. Am J Dis Child. 1981;135:209.
Barohn R.J. Standards of measurements in myasthenia gravis. Ann N Y Acad Sci. 2003;998:432-439.
Barohn R.J. Treatment and clinical research in myasthenia gravis: how far have we come? Ann N Y Acad Sci. 2008;1132:225-232.
Barohn R.J., Brey R.L. Soluble terminal complement components in human myasthenia gravis. Clin Neurol Neurosurg. 1993;95:285-290.
Barohn R.J., McIntire D., Herbelin L., et al. Reliability testing of the quantitative myasthenia gravis score. Ann N Y Acad Sci. 1998;841:769-772.
Batocchi A.P., Evoli A., Palmisani M.T., et al. Early-onset myasthenia gravis: clinical characteristics and response to therapy. Eur J Pediatr. 1990;150:66-68.
Batra P., Herrmann C.Jr, Mulder D. Mediastinal imaging in myasthenia gravis: correlation of chest radiography, CT, MR, and surgical findings. Am J Roentgenol. 1987;148:515-519.
Beeson D., Higuchi O., Palace J., et al. Dok-7 mutations underlie a neuromuscular junction synaptopathy. Science. 2006;313:1975-1978.
Behan P.O., Shakir R.A., Simpson J.A., et al. Plasma-exchange combined with immunosuppressive therapy in myasthenia gravis. Lancet. 1979;2:438-440.
Benatar M., Rowland L.P. The muddle of mycophenolate mofetil in myasthenia. Neurology. 2008;71:390-391.
Bestue-Cardiel M., Saenz de Cabezon-Alvarez A., Capablo-Liesa J.L., et al. Congenital endplate acetylcholinesterase deficiency responsive to ephedrine. Neurology. 2005;65:144-146.
Blalock A., Mason M.F., Morgan H.J., et al. Myasthenia Gravis and Tumors of the Thymic Region: Report of a Case in Which the Tumor Was Removed. Ann Surg. 1939;110:544-561.
Bowman J.R. Myasthenia gravis in young children; report of three cases, one congenital. Pediatrics. 1948;1:472-477.
Branch C.E.Jr, Swift T.R., Dyken P.R. Prolonged neonatal myasthenia gravis: electrophysiological studies. Ann Neurol. 1978;3:416-418.
Brannagan TH3rd, Nagle K.J., Lange D.J., et al. Complications of intravenous immune globulin treatment in neurologic disease. Neurology. 1996;47:674-677.
Brown T.C., Gebert R., Meretoja O.A., et al. Myasthenia gravis in children and its anaesthetic implications. Anaesth Intensive Care. 1990;18:466-472.
Bundey S. A genetic study of infantile and juvenile myasthenia gravis. J Neurol Neurosurg Psychiatry. 1972;35:41-51.
Burns T.M., Conaway M.R., Cutter G.R., et al. Construction of an efficient evaluative instrument for myasthenia gravis: the MG composite. Muscle Nerve. 2008;38:1553-1562.
Campbell W.W.Jr, Leshner R.T., Swift T.R. Plasma exchange in myasthenia gravis: electrophysiological studies. Ann Neurol. 1980;8:584-589.
Carter B., Harrison R., Lunt G.G., et al. Anti-acetylcholine receptor antibody titres in the sera of myasthenia patients treated with plasma exchange combined with immunosuppressive therapy. J Neurol Neurosurg Psychiatry. 1980;43:397-402.
Castleman B. The pathology of the thymus gland in myasthenia gravis. Ann N Y Acad Sci. 1966;135:496-505.
Chaudhry V., Cornblath D.R., Griffin J.W., et al. Mycophenolate mofetil: a safe and promising immunosuppressant in neuromuscular diseases. Neurology. 2001;56:94-96.
Cherington M. Botulism: update and review. Semin Neurol. 2004;24:155-163.
Chiang L.M., Darras B.T., Kang P.B. Juvenile myasthenia gravis. Muscle Nerve. 2009;39:423-431.
Christiansen F.T., Pollack M.S., Garlepp M.J., et al. Myasthenia gravis and HLA antigens in American blacks and other races. J Neuroimmunol. 1984;7:121-129.
Ciafaloni E., Massey J.M., Tucker-Lipscomb B., et al. Mycophenolate mofetil for myasthenia gravis: an open-label pilot study. Neurology. 2001;56:97-99.
Ciafaloni E., Nikhar N.K., Massey J.M., et al. Retrospective analysis of the use of cyclosporine in myasthenia gravis. Neurology. 2000;55:448-450.
Cikes N., Momoi M.Y., Williams C.L., et al. Striational autoantibodies: quantitative detection by enzyme immunoassay in myasthenia gravis, thymoma, and recipients of D-penicillamine or allogeneic bone marrow. Mayo Clin Proc. 1988;63:474-481.
Cornblath D.R., Sladky J.T., Sumner A.J. Clinical electrophysiology of infantile botulism. Muscle Nerve. 1983;6:448-452.
Dahl D.S., Sato S. Unusual myasthenic state in a teen-age boy. Neurology. 1974;24:897-901.
Dau P.C., Lindstrom J.M., Cassel C.K., et al. Plasmapheresis and immunosuppressive drug therapy in myasthenia gravis. N Engl J Med. 1977;297:1134-1140.
De Feo L.G., Schottlender J., Martelli N.A., et al. Use of intravenous pulsed cyclophosphamide in severe, generalized myasthenia gravis. Muscle Nerve. 2002;26:31-36.
Desmedt J.E., Borenstein S. Time course of neonatal myasthenia gravis and unsuspectedly long duration of neuromuscular block in distal muscles. N Engl J Med. 1977;296:633.
Djelmis J., Sostarko M., Mayer D., et al. Myasthenia gravis in pregnancy: report on 69 cases. Eur J Obstet Gynecol Reprod Biol. 2002;104:21-25.
Donaldson J.O., Penn A.S., Lisak R.P., et al. Antiacetylcholine receptor antibody in neonatal myasthenia gravis. Am J Dis Child. 1981;135:222-226.
Drachman D.B. Myasthenia gravis (first of two parts). N Engl J Med. 1978;298:136-142.
Drachman D.B. Myasthenia gravis. N Engl J Med. 1994;330:1797-1810.
Drachman D.B., Adams R.N., Stanley E.F., et al. Mechanisms of acetylcholine receptor loss in myasthenia gravis. J Neurol Neurosurg Psychiatry. 1980;43:601-610.
Drachman D.B., Jones R.J., Brodsky R.A. Treatment of refractory myasthenia: “rebooting” with high-dose cyclophosphamide. Ann Neurol. 2003;53:29-34.
Elias S.B., Appel S.H. Current concepts of pathogenesis and treatment of myasthenia gravis. Med Clin North Am. 1979;63:745-757.
Elmqvist D., Lambert E.H. Detailed analysis of neuromuscular transmission in a patient with the myasthenic syndrome sometimes associated with bronchogenic carcinoma. Mayo Clin Proc. 1968;43:689-713.
Engel A.G. Congenital disorders of neuromuscular transmission. Semin Neurol. 1990;10:12-26.
Engel A.G., Arahata K. The membrane attack complex of complement at the endplate in myasthenia gravis. Ann N Y Acad Sci. 1987;505:326-332.
Engel A.G., Hutchinson D.O., Nakano S., et al. Myasthenic syndromes attributed to mutations affecting the epsilon subunit of the acetylcholine receptor. Ann N Y Acad Sci. 1993;681:496-508.
Engel A.G., Lambert E.H., Howard F.M. Immune complexes (IgG and C3) at the motor end-plate in myasthenia gravis: ultrastructural and light microscopic localization and electrophysiologic correlations. Mayo Clin Proc. 1977;52:267-280.
Engel A.G., Lambert E.H., Gomez M.R. A new myasthenic syndrome with end-plate acetylcholinesterase deficiency, small nerve terminals, and reduced acetylcholine release. Ann Neurol. 1977;1:315-330.
Engel A.G., Lambert E.H., Mulder D.M., et al. A newly recognized congenital myasthenic syndrome attributed to a prolonged open time of the acetylcholine-induced ion channel. Ann Neurol. 1982;11:553-569.
Engel A.G., Ohno K.. Congenital myasthenic syndromes. Pourmand R., Harati Y., editors. Advances in Neurology: Neuromuscular Disorders. vol. 88. Philadelphia: Lippincott Williams & Wilkins; 2002:203-215. 88:
Engel A.G., Ohno K., Bouzat C., et al. End-plate acetylcholine receptor deficiency due to nonsense mutations in the epsilon subunit. Ann Neurol. 1996;40:810-817.
Engel A.G., Tsujihata M., Lindstrom J.M., et al. The motor end plate in myasthenia gravis and in experimental autoimmune myasthenia gravis. A quantitative ultrastructural study. Ann N Y Acad Sci. 1976;274:60-79.
Evoli A., Batocchi A.P., Bartoccioni E., et al. Juvenile myasthenia gravis with prepubertal onset. Neuromuscul Disord. 1998;8:561-567.
Evoli A., Batocchi A.P., Palmisani M.T., et al. Long-term results of corticosteroid therapy in patients with myasthenia gravis. Eur Neurol. 1992;32:37-43.
Evoli A., Tonali P.A., Padua L., et al. Clinical correlates with anti-MuSK antibodies in generalized seronegative myasthenia gravis. Brain. 2003;126:2304-2311.
Evoli A., Tonali P., Bartoccioni E., et al. Ocular myasthenia: diagnostic and therapeutic problems. Acta Neurol Scand. 1988;77:31-35.
Eymard B., Morel E., Dulac O., et al. Myasthenia and pregnancy: a clinical and immunologic study of 42 cases (21 neonatal myasthenia cases. Rev Neurol (Paris). 1989;145:696-701.
Fenichel G.M. Clinical syndromes of myasthenia in infancy and childhood. A review. Arch Neurol. 1978;35:97-103.
Fraser D., Turner J.W. Myasthenia gravis and pregnancy. Lancet. 1953;265:417-419.
Fukunaga H., Engel A.G., Osame M., et al. Paucity and disorganization of presynaptic membrane active zones in the lambert-eaton myasthenic syndrome. Muscle Nerve. 1982;5:686-697.
Geddes A.K., Kidd H.M. Myasthenia gravis of the newborn. Can Med Assoc J. 1951;64:152-156.
Geh V.S., Bradbury J.A. Ocular myasthenia presenting in an 11-month-old boy. Eye. 1998;12(Pt 2):319-320.
Go R.S., Call T.G. Deep venous thrombosis of the arm after intravenous immunoglobulin infusion: case report and literature review of intravenous immunoglobulin-related thrombotic complications. Mayo Clin Proc. 2000;75:83-85.
Grob D., Arsura E.L., Brunner N.G., et al. The course of myasthenia gravis and therapies affecting outcome. Ann N Y Acad Sci. 1987;505:472-499.
Grob D., Brunner N., Namba T., et al. Lifetime course of myasthenia gravis. Muscle Nerve. 2008;37:141-149.
Gronseth G.S., Barohn R.J. Practice parameter: thymectomy for autoimmune myasthenia gravis (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2000;55:7-15.
Haliloglu G., Anlar B., Aysun S., et al. Gender prevalence in childhood multiple sclerosis and myasthenia gravis. J Child Neurol. 2002;17:390-392.
Harper C.M. Congenital myasthenic syndromes. Semin Neurol. 2004;24:111-123.
Harper C.M., Engel A.G. Quinidine sulfate therapy for the slow-channel congenital myasthenic syndrome. Neurology. 1998;43:480-484.
Harper C.M., Fukodome T., Engel A.G. Treatment of slow-channel congenital myasthenic syndrome with fluoxetine. Neurology. 2003;60:1710-1713.
Hazelrigg S.R., Nunchuck S.K., Landreneau R.J., et al. Cost analysis for thoracoscopy: thoracoscopic wedge resection. Ann Thorac Surg. 1993;56:633-635.
Hoch W., McConville J., Helms S., et al. Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nat Med. 2001;7:365-368.
Hohlfeld R., Michels M., Heininger K., et al. Azathioprine toxicity during long-term immunosuppression of generalized myasthenia gravis. Neurology. 1988;38:258-261.
Howard F.M.Jr, Lennon V.A., Finley J., et al. Clinical correlations of antibodies that bind, block, or modulate human acetylcholine receptors in myasthenia gravis. Ann N Y Acad Sci. 1987;505:526-538.
Hughes B.W., Moro De Casillas M.L., Kaminski H.J. Pathophysiology of myasthenia gravis. Semin Neurol. 2004;24:21-30.
Jabre J.F., Chirico-Post J., Weiner M. Stimulation SFEMG in myasthenia gravis. Muscle Nerve. 1989;12:38-42.
Jaretzki A.3rd, Aarli J.A., Kaminski H.J., et al. Preoperative preparation of patients with myasthenia gravis forestalls postoperative respiratory complications after thymectomy. Ann Thorac Surg. 2003;75:1068. author reply 1069
Jaretzki A.3rd, Barohn R.J., Ernstoff R.M., et al. Myasthenia gravis: recommendations for clinical research standards. Task Force of the Medical Scientific Advisory Board of the Myasthenia Gravis Foundation of America. Neurology. 2000;55:16-23.
Jaretzki A.3rd, Penn A.S., Younger D.S., et al. “Maximal” thymectomy for myasthenia gravis. Results. J Thorac Cardiovasc Surg. 1988;95:747-757.
Jaretzki A.3rd, Wolff M. “Maximal” thymectomy for myasthenia gravis. Surgical anatomy and operative technique. J Thorac Cardiovasc Surg. 1988;96:711-716.
Jaretzki A., Steinglass K.M., Sonett J.R. Thymectomy in the management of myasthenia gravis. Semin Neurol. 2004;24:49-62.
Jeannet P.Y., Marcoz J.P., Kuntzer T., et al. Isolated facial and bulbar paresis: a persistent manifestation of neonatal myasthenia gravis. Neurology. 2008;70:237-238.
Jha S., Xu K., Maruta T., et al. Myasthenia gravis induced in mice by immunization with the recombinant extracellular domain of rat muscle-specific kinase (MuSK). J Neuroimmunol. 2006;175:107-117.
Kaminski H.J.M.D., Suarez J.I.M.D., Ruff R.L.M.D.P. Neuromuscular junction physiology in myasthenia gravis: Isoforms of the acetylcholine receptor in extraocular muscle and the contribution of sodium channels to the safety factor. Neurology. 1997;48:58-517.
Kao I., Drachman D.B. Thymic muscle cells bear acetylcholine receptors: possible relation to myasthenia gravis. Science. 1977;195:74-75.
Katz J., Barohn R.J. Update on the evaluation and therapy of autoimmune neuromuscular junction disorders. Phys Med Rehabil Clin N Am. 2001;12:381-397.
Kini P.G. Juvenile myasthenia gravis with predominant facial weakness in a 7-year-old boy. Int J Pediatr Otorhinolaryngol. 1995;32:167-169.
Kissel J.T., Levy R.J., Mendell J.R., et al. Azathioprine toxicity in neuromuscular disease. Neurology. 1986;36:35-39.
Kupersmith M.J., Latkany R., Homel P. Development of generalized disease at 2 years in patients with ocular myasthenia gravis. Arch Neurol. 2003;60:243-248.
Lambert E.H., Eaton F.M. RED: Defect of neuromuscular conduction associated with malignant neoplasms. Am J Physiol. 1956;187:612.
Lang B., Newsomdavis J., Wray D., et al. Autoimmune Etiology for Myasthenic (Eaton-Lambert) Syndrome. Lancet. 1981;2:224-226.
Lanska D.J. Indications for Thymectomy in Myasthenia-Gravis. Neurology. 1990;40:1828-1829.
Lauriola L., Guerriero M., Bartoccioni E., et al. Thymus changes in anti-MuSK-positive and -negative myasthenia gravis. Neurology. 2004;62:A183-A184.
Lavrnic D., Vujic A., Rakocevic-Stojanovic V., et al. Cyclosporine in the treatment of myasthenia gravis. Acta Neurol Scand. 2005;111:247-252.
Lennon V.A. Myasthenia-Gravis – Diagnosis by Assay of Serum Antibodies. Mayo Clin Proc. 1982;57:723-724.
Lennon V.A., Griesmann G.E. Evidence against Acetylcholine-Receptor Having a Main Immunogenic Region as Target for Autoantibodies in Myasthenia-Gravis. Neurology. 1989;39:1069-1076.
Lennon V.A., Kryzer T.J., Griesmann G.E., et al. Calcium-channel antibodies in Lambert-Eaton syndrome and other paraneoplastic syndromes. N Engl J Med. 1995;332:1467-1474.
Limburg P.C., The T.H., Hummeltappel E., et al. Anti-Acetylcholine Receptor Antibodies in Myasthenia-Gravis. 1. Relation to Clinical-Parameters in 250 Patients. J Neurol Sci. 1983;58:357-370.
Lindner A., Schalke B., Toyka K.V. Outcome in juvenile-onset myasthenia gravis: a retrospective study with long-term follow-up of 79 patients. J Neurol. 1997;244:515-520.
Lindstrom J.M., Lennon V.A., Seybold M.E., et al. Experimental Autoimmune Myasthenia-Gravis and Myasthenia-Gravis – Biochemical and Immunochemical Aspects. Ann N Y Acad Sci. 1976;274:254-274.
Lindstrom J.M., Seybold M.E., Lennon V.A., et al. Antibody to Acetylcholine-Receptor in Myasthenia-Gravis – Prevalence, Clinical Correlates, and Diagnostic Value. Neurology. 1976;26:1054-1059.
Lui J., Erickson K., editors. Cholinergic agents. Principles and practice of opthalmology:basic sciences. Philadelphia: WB Saunders, 1994.
Maddison P., Newsom-Davis J. Treatment for Lambert-Eaton myasthenic syndrome. Cochrane Database Syst Rev. 2005. CD003279
Mantegazza R., Antozzi C., Peluchetti D., et al. Azathioprine as a Single Drug or in Combination with Steroids in the Treatment of Myasthenia-Gravis. J Neurol. 1988;235:449-453.
Mantegazza R., Baggi F., Bernasconi P., et al. Video-assisted thoracoscopic extended thymectomy and extended transsternal thymectomy (T-3b) in non-thymomatous myasthenia gravis patients: remission after 6 years of follow-up. J Neurol Sci. 2003;212:31-36.
Maselli R.A., Bakshi N. Botulism. Muscle Nerve. 2000;23:1137-1144.
Maselli R.A., Ellis W., Mandler R.N., et al. Cluster of wound botulism in California: clinical, electrophysiologic, and pathologic study. Muscle Nerve. 1998;20:1284-1295.
Massey J.M. Acquired myasthenia gravis. Neurol Clin. 1997;15:577.
Matell G. Immunosuppressive drugs: azathioprine in the treatment of myasthenia gravis. Ann N Y Acad Sci. 1987;505:589-594.
McConville J., Farrugia M.E., Beeson D., et al. Detection and characterization of MuSK antibodies in seronegative myasthenia gravis. Ann Neurol. 2004;55:580-584.
Mee J., Paine M., Byrne E., et al. Immunotherapy of ocular myasthenia gravis reduces conversion to generalized myasthenia gravis. J Neuroophthalmol. 2003;23:251-255.
Meriggioli M.N., Ciafaloni E., Al-Hayk K.A., et al. Mycophenolate mofetil for myasthenia gravis: an analysis of efficacy, safety, and tolerability. Neurology. 2003;61:1438-1440.
Meriggioli M.N., Sanders D.B. Autoimmune myasthenia gravis: emerging clinical and biological heterogeneity. Lancet Neurol. 2009;8:475-490.
Mertens H.G., Hertel G., Reuther P., et al. Effect of immunosuppressive drugs (azathioprine). Ann N Y Acad Sci. 1981;377:691-699.
Miano M.A., Bosley T.M., Heiman-Patterson T.D., et al. Factors influencing outcome of prednisone dose reduction in myasthenia gravis. Neurology. 1991;41:919-921.
Mihaylova V., Muller J.S., Vilchez J.J., et al. Clinical and molecular genetic findings in COLQ-mutant congenital myasthenic syndromes. Brain. 2008;131:747-759.
Miller R.G., Milner-Brown H.S., Mirka A. Prednisone-induced worsening of neuromuscular function in myasthenia gravis. Neurology. 1986;36:729-732.
Millichap J.G., Dodge P.R. Diagnosis and treatment of myasthenia gravis in infancy, childhood, and adolescence: a study of 51 patients. Neurology. 1960;10:1007-1014.
Milone M., Shen X.M., Selcen D., et al. Myasthenic syndrome due to defects in rapsyn: Clinical and molecular findings in 39 patients. Neurology. 2009;73:228-235.
Monsul N.T., Patwa H.S., Knorr A.M., et al. The effect of prednisone on the progression from ocular to generalized myasthenia gravis. J Neurol Sci. 2004;217:131-133.
Mora M., Lambert E.H., Engel A.G. Synaptic vesicle abnormality in familial infantile myasthenia. Neurology. 1987;37:206-214.
Motomura M., Lang B., Johnston I., et al. Incidence of serum anti-P/Q-type and anti-N-type calcium channel autoantibodies in the Lambert-Eaton myasthenic syndrome. J Neurol Sci. 1996;147:35-42.
Mulder D.G., Graves M., Herrmann C. Thymectomy for myasthenia gravis: recent observations and comparisons with past experience. Ann Thorac Surg. 1989;48:551-555.
Muppidi S., Wolfe G.I. Muscle-specific receptor tyrosine kinase antibody-positive and seronegative myasthenia gravis. Front Neurol Neurosci. 2009;26:109-119.
Murai H., Noda T., Himeno E., et al. Infantile onset myasthenia gravis with MuSK antibodies. Neurology. 2006;67:174.
Muscle study group. A trial of mycophenolate mofetil with prednisone as initial immunotherapy in myasthenia gravis. Neurology. 2008;71:394-399.
Myasthenia Gravis Clinical Study Group. A randomised clinical trial comparing prednisone and azathioprine in myasthenia gravis. Results of the second interim analysis. Myasthenia Gravis Clinical Study Group. J Neurol Neurosurg Psychiatry. 1993;56:1157-1163.
Namba T., Brown S.B., Grob D. Neonatal myasthenia gravis: report of two cases and review of the literature. Pediatrics. 1970;45:488-504.
Nations S.P., Wolfe G.I., Amato A.A., et al. Distal myasthenia gravis. Neurology. 1999;52:632-634.
Newsom-Davis J., Cutter G., Wolfe G.I., et al. Status of the thymectomy trial for nonthymomatous myasthenia gravis patients receiving prednisone. Ann N Y Acad Sci. 2008;1132:344-347.
Newsom-Davis J., Murray N.M. Plasma exchange and immunosuppressive drug treatment in the Lambert-Eaton myasthenic syndrome. Neurology. 1984;34:480-485.
O’Carroll P., Bertorini T.E., Jacob G., et al. Transient neonatal myasthenia gravis in a baby born to a mother with new-onset anti-MuSK-mediated myasthenia gravis. J Clin Neuromuscul Dis. 2009;11:69-71.
Odabasi Z., Demirci M., Kim D.S., et al. Postexercise facilitation of reflexes is not common in Lambert-Eaton myasthenic syndrome. Neurology. 2002;59:1085-1087.
Ohno K., Engel A.G. Congenital myasthenic syndromes: gene mutations. Neuromuscul Disord. 2000;10:534-536.
Oh S.J. Diverse electrophysiological spectrum of the Lambert-Eaton myasthenic syndrome. Muscle Nerve. 1989;12:464-469.
Oh S.J. Electromyography:neuromuscular transmission studies. Baltimore: Williams & Wilkins; 1988.
Oh S.J., Cho H.K. Edrophonium responsiveness not necessarily diagnostic of myasthenia gravis. Muscle Nerve. 1990;13:187-191.
Oh S.J., Kim D.E., Kuruoglu R., et al. Diagnostic sensitivity of the laboratory tests in myasthenia gravis. Muscle Nerve. 1992;15:720-724.
Oh S.J., Kurokawa K., Claussen G.C., et al. Electrophysiological diagnostic criteria of Lambert-Eaton myasthenic syndrome. Muscle Nerve. 2005;32:515-520.
Oosterhuis H.J., Limburg P.C., Hummel-Tappel E., et al. Anti-acetylcholine receptor antibodies in myasthenia gravis. Part 3. The effect of thymectomy. J Neurol Sci. 1985;69:335-343.
Oosterhuis H.J., Newsom-Davis J., Wokke J.H., et al. The slow channel syndrome. Two new cases. Brain. 1987;110(Pt 4):1061-1079.
Osserman K.E. Myasthenia gravis. New York: Grune & Stratton; 1958.
Padua L., Tonali P., Aprile I., et al. Seronegative myasthenia gravis: comparison of neurophysiological picture in MuSK+ and MuSK− patients. Eur J Neurol. 2006;13:273-276.
Palace J., Lashley D., Newsom-Davis J., et al. Clinical features of the DOK7 neuromuscular junction synaptopathy. Brain. 2007;130:1507-1515.
Palace J., Newsom-Davis J., Lecky B. A randomized double-blind trial of prednisolone alone or with azathioprine in myasthenia gravis. Myasthenia Gravis Study Group. Neurology. 1998;50:1778-1783.
Papazian O. Transient neonatal myasthenia gravis. J Child Neurol. 1992;7:135-141.
Pascuzzi R.M., Coslett H.B., Johns T.R. Long-term corticosteroid treatment of myasthenia gravis: report of 116 patients. Ann Neurol. 1984;15:291-298.
Pasnoor M., Wolfe G.I., Nations S., et al. Clinical findings in MuSK-antibody positive myasthenia gravis: A U.S. experience. Muscle Nerve. 2009.
Patrick J., Lindstrom J. Autoimmune response to acetylcholine receptor. Science. 1973;180:871-872.
Perez M.C., Buot W.L., Mercado-Danguilan C., et al. Stable remissions in myasthenia gravis. Neurology. 1981;31:32-37.
Pickett J.B., Berg B., Chaplin E., et al. Syndrome of botulism in infancy: clinical and electrophysiologic study. N Engl J Med. 1976;295:770-772.
Pinching A.J., Peters D.K. Remission of myasthenia gravis following plasma-exchange. Lancet. 1976;2:1373-1376.
Plauche W.C. Myasthenia gravis in mothers and their newborns. Clin Obstet Gynecol. 1991;34:82-99.
Plested C.P., Tang T., Spreadbury I., et al. AChR phosphorylation and indirect inhibition of AChR function in seronegative MG. Neurology. 2002;59:1682-1688.
Ponseti J.M., Caritg N., Gamez J., et al. A comparison of long-term post-thymectomy outcome of anti-AChR-positive, anti-AChR-negative and anti-MuSK-positive patients with non-thymomatous myasthenia gravis. Expert Opin Biol Ther. 2009;9:1-8.
Ponseti J.M., Gamez J., Azem J., et al. Tacrolimus for myasthenia gravis: a clinical study of 212 patients. Ann N Y Acad Sci. 2008;1132:254-263.
Provenzano C., Marino M., Scuderi F., et al. Anti-acetylcholinesterase antibodies associate with ocular myasthenia gravis. J Neuroimmunol. 2009.
Rich M.M., Teener J.W., Bird S.J. Treatment of Lambert-Eaton syndrome with intravenous immunoglobulin. Muscle Nerve. 1997;20:614-615.
Rodriguez M., Gomez M.R., Howard F.M.Jr, et al. Myasthenia gravis in children: long-term follow-up. Ann Neurol. 1983;13:504-510.
Romi F., Skeie G.O., Aarli J.A., et al. The severity of myasthenia gravis correlates with the serum concentration of titin and ryanodine receptor antibodies. Arch Neurol. 2000;57:1596-1600.
Roses A.D., Olanow C.W., McAdams M.W., et al. No direct correlation between serum antiacetylcholine receptor antibody levels and clinical state of individual patients with myasthenia gravis. Neurology. 1981;31:220-224.
Rothbart H.B. Myasthenia gravis in children: its familial incidence. JAMA. 1937;108:715-717.
Rowland L.P. Therapy in myasthenia gravis: introduction. Ann N Y Acad Sci. 1987;505:566-567.
Sabater L., Titulaer M., Saiz A., et al. SOX1 antibodies are markers of paraneoplastic Lambert-Eaton myasthenic syndrome. Neurology. 2008;70:924-928.
Sanders D.B. Lambert-eaton myasthenic syndrome: diagnosis and treatment. Ann N Y Acad Sci. 2003;998:500-508.
Sanders D.B.. The Lambert-Eaton Myasthenic Syndrome. Pourmand R., Harati Y., editors. Advances in Neurology: Neuromuscular Disorders. vol. 88. Philadelphia: Lippincott Williams & Wilkins; 2002:189-201.
Sanders D.B., El-Salem K., Massey J.M., et al. Clinical aspects of MuSK antibody positive seronegative MG. Neurology. 2003;60:1978-1980.
Sanders D.B., Hart I.K., Mantegazza R., et al. An international, phase III, randomized trial of mycophenolate mofetil in myasthenia gravis. Neurology. 2008;71:400-406.
Sanders D.B., Howard J.F.Jr. AAEE minimonograph #25: Single-fiber electromyography in myasthenia gravis. Muscle Nerve. 1986;9:809-819.
Sanders D.B., Massey J.M., Sanders L.L., et al. A randomized trial of 3,4-diaminopyridine in Lambert-Eaton myasthenic syndrome. Neurology. 2000;54:603-607.
Sanders D.B., Siddiqi Z.A. Lessons from two trials of mycophenolate mofetil in myasthenia gravis. Ann N Y Acad Sci. 2008;1132:249-253.
Sanes J.R., Jessell T.M. The formation and generation of synapses. In: Kandel E.R., Schwartz J.H., Jessell T.M., editors. Principles of neuroscience. New York: McGraw-Hill; 2000:1087.
Saperstein D.S., Barohn R.J. Management of myasthenia gravis. Semin Neurol. 2004;24:41-48.
Schiff R.I. Transmission of viral infections through intravenous immune globulin. N Engl J Med. 1994;331:1649-1650.
Seybold M.E. Thymectomy in childhood myasthenia gravis. Ann N Y Acad Sci. 1998;841:731-741.
Seybold M.E., Lambert E.H., Lennon V.A., et al. Experimental Autoimmune Myasthenia – Clinical, Neurophysiologic, and Pharmacologic Aspects. Ann N Y Acad Sci. 1976;274:275-282.
Shillito P., Vincent A., Newsom-Davis J. Congenital myasthenic syndromes. Neuromuscul Disord. 1993;3:183-190.
Simpson J. Myasthenia gravis: a new hypothesis. Scott Med J. 1960:5.
Simpson J.A. An evaluation of thymectomy in myasthenia gravis. Brain. 1958;81:112-144.
Snead O.C.3rd, Benton J.W., Dwyer D., et al. Juvenile myasthenia gravis. Neurology. 1980;30:732-739.
Sommer N., Harcourt G.C., Willcox N., et al. Acetylcholine receptor-reactive T lymphocytes from healthy subjects and myasthenia gravis patients. Neurology. 1991;41:1270-1276.
Sommer N., Willcox N., Harcourt G.C., et al. Myasthenic thymus and thymoma are selectively enriched in acetylcholine receptor-reactive T cells. Ann Neurol. 1990;28:312-319.
Stalberg E., Sanders D.B. Electrophysiologica testing of neuromuscular transmission. In: Stalberg E., Young R.R., editors. Clinical neurophysiology. London: Butterworth, 1981.
Stalberg E., Trontelj J.V., Schwartz M.S. Single-muscle-fiber recording of the jitter phenomenon in patients with myasthenia gravis and in members of their families. Ann N Y Acad Sci. 1976;274:189-202.
Stalberg E.V., Sanders D.B. Jitter recordings with concentric needle electrodes. Muscle Nerve. 2009;40:331-339.
Steg R.E., Lefkowitz D.M. Cerebral infarction following intravenous immunoglobulin therapy for myasthenia gravis. Neurology. 1994;44:1180-1181.
Tan E., Hajinazarian M., Bay W., et al. Acute renal failure resulting from intravenous immunoglobulin therapy. Arch Neurol. 1993;50:137-139.
Thompson J.A., Filloux F.M., Van Orman C.B., et al. Infant botulism in the age of botulism immune globulin. Neurology. 2005;64:2029-2032.
Tindall R.S. Humoral immunity in myasthenia gravis: effect of steroids and thymectomy. Neurology. 1980;30:554-557.
Tindall R.S., Phillips J.T., Rollins J.A., et al. A clinical therapeutic trial of cyclosporine in myasthenia gravis. Ann N Y Acad Sci. 1993;681:539-551.
Tindall R.S., Rollins J.A., Phillips J.T., et al. Preliminary results of a double-blind, randomized, placebo-controlled trial of cyclosporine in myasthenia gravis. N Engl J Med. 1987;316:719-724.
Toyka K.V., Drachman D.B., Griffin D.E., et al. Myasthenia gravis. Study of humoral immune mechanisms by passive transfer to mice. N Engl J Med. 1977;296:125-131.
Tzartos S.J., Lindstrom J.M. Monoclonal antibodies used to probe acetylcholine receptor structure: localization of the main immunogenic region and detection of similarities between subunits. Proc Natl Acad Sci USA. 1980;77:755-759.
Uchitel O., Engel A.G., Walls T.J., et al. Congenital myasthenic syndromes: II. Syndrome attributed to abnormal interaction of acetylcholine with its receptor. Muscle Nerve. 1993;16:1293-1301.
Vernino S., Salomao D.R., Habermann T.M., et al. Primary CNS lymphoma complicating treatment of myasthenia gravis with mycophenolate mofetil. Neurology. 2005;65:639-641.
Vial C., Charles N., Chauplannaz G., et al. Myasthenia gravis in childhood and infancy. Usefulness of electrophysiologic studies. Arch Neurol. 1991;48:847-849.
Vincent A., Leite M.I., Farrugia M.E., et al. Myasthenia gravis seronegative for acetylcholine receptor antibodies. Ann N Y Acad Sci. 2008;1132:84-92.
Vincent A., Newsom-Davis J. Acetylcholine receptor antibody as a diagnostic test for myasthenia gravis: results in 153 validated cases and 2967 diagnostic assays. J Neurol Neurosurg Psychiatry. 1985;48:1246-1252.
Vincent A., Palace J., Hilton-Jones D. Myasthenia gravis. Lancet. 2001;357:2122-2128.
Warmolts J.R., Engel W.K. Benefit from alternate-day prednisone in myasthenia gravis. N Engl J Med. 1972;286:17-20.
Weinberg D.A., Lesser R.L., Vollmer T.L. Ocular myasthenia: a protean disorder. Surv Ophthalmol. 1994;39:169-210.
Wekerle T.H., paterson B., Ketelsen U., et al. Striated muscle fibres differentiate in monolayer cultures of adult thymus reticulum. Nature. 1975;256:493-494.
Wirtz P.W., Wintzen A.R., Verschuuren J.J. Lambert-Eaton myasthenic syndrome has a more progressive course in patients with lung cancer. Muscle Nerve. 2005;32:226-229.
Wolfe G.I., Barohn R., Galetta S.L. Drugs for the diagnosis and treatment of myasthenia gravis. In: Zimmerman T.J., Kooner K.S., Sharir M., editors. Textbook of ocular pharmacology. Philadelphia: Lippincott-Raven, 1997.
Wolfe G.I., Barohn R.J., Foster B.M., et al. Randomized, controlled trial of intravenous immunoglobulin in myasthenia gravis. Muscle Nerve. 2002;26:549-552.
Wolfe G.I., Herbelin L., Nations S.P., et al. Myasthenia gravis activities of daily living profile. Neurology. 1999;52:1487-1489.
Wolfe G.I., Trivedi J.R., Oh S.J. Clinical Review of Muscle-Specific Tyrosine Kinase-Antibody Positive Myasthenia Gravis. Journal of Clinical Neuromuscular Disease. 2007;8:217-224. 210.1097/CND.1090b1013e318137a318124b
Wong V., Hawkins B.R., Yu Y.L. Myasthenia gravis in Hong Kong Chinese. 2. Paediatric disease. Acta Neurol Scand. 1992;86:68-72.
Zinman L., Ng E., Bril V. IV immunoglobulin in patients with myasthenia gravis: a randomized controlled trial. Neurology. 2007;68:837-841.