[level-membership-for-pediatrics-category]
Chapter 96 Digestive System Disorders
Vomiting
Obstructive lesions of the digestive tract are the most frequent gastrointestinal anomalies (Chapters 311, 321, 322, and 324). Vomiting (and drooling) from esophageal obstruction occurs with the 1st feeding. The diagnosis of esophageal atresia can be suspected if unusual drooling from the mouth is observed and if resistance is encountered during an attempt to pass a catheter into the stomach. The diagnosis should be made before the infant has trouble with oral feedings and aspiration pneumonia develops. Infantile achalasia (cardiospasm), a rare cause of vomiting in newborn infants, is demonstrable radiographically as obstruction at the cardiac end of the esophagus without organic stenosis. Regurgitation of feedings because of continuous relaxation of the esophageal-gastric sphincter, or chalasia, is a cause of vomiting. Keeping the infant in a semi-upright position, thickening the feeding, or administering prokinetic drugs can control it.
Vomiting due to obstruction of the small intestine usually begins on the 1st day of life and is frequent, persistent, usually nonprojectile, copious, and, unless the obstruction is above the ampulla of Vater, bile-stained; it is associated with abdominal distention, visible deep peristaltic waves, and reduction or absence of bowel movements. Malrotation with obstruction from midgut volvulus is an acute emergency that must be not only considered but also urgently evaluated by an upper gastrointestinal contrast radiographic series. Radiographs of the abdomen show the distribution of air in the intestine, which may point to the anatomic location of an obstruction; malrotation can be identified only by contrast studies. Normally, air can be demonstrated by radiographs in the jejunum by 15-60 min, in the ileum by 2-3 hr, and in the colon by 3 hr after birth. Absence of rectal gas at 24 hr is abnormal. Persistent vomiting may occur with congenital diaphragmatic hernia. The vomiting associated with pyloric stenosis may begin any time after birth but does not assume its characteristic pattern before the 2nd-3rd wk. Vomiting with obstipation is a common early sign of Hirschsprung disease. Vomiting may occur with many other disturbances that do not obstruct the digestive tract, such as milk allergy, adrenal hyperplasia of the salt-losing variety, galactosemia, hyperammonemias, organic acidemias, increased intracranial pressure, septicemia, meningitis, and urinary tract infection. In many infants, it is simply regurgitation from overfeeding or from failure to permit the infant to eructate swallowed air. (See Chapter 315 for a discussion of gastric emptying and gastroesophageal reflux.)
Meconium Plugs
Lower colonic or anorectal plugs (Fig. 96-1) with a lower than normal water content may cause intestinal obstruction. Rarely, a firm mass of meconium may form elsewhere in the intestine and cause intrauterine intestinal obstruction and meconium peritonitis unrelated to cystic fibrosis (CF). Anorectal plugs may also cause mucosal ulceration and intestinal perforation. Meconium plugs are associated with small left colon syndrome in infants of diabetic mothers and with CF, rectal aganglionosis, maternal opiate use, and magnesium sulfate therapy for preeclampsia. The plug may be evacuated by glycerin suppository or rectal irrigation with isotonic saline. Enemas with the iodinated contrast medium Gastrografin usually induce passage of the plug, presumably because the high osmolarity (1,900 mOsm/L) of the solution draws fluid rapidly into the intestinal lumen and loosens inspissated material. Such rapid loss of fluid into the bowel may result in acute dehydration and shock, so it is advisable to dilute the contrast material with an equal amount of water, correct any existing dehydration, and provide intravenous fluids during and for several hours after the procedure. After removal of a meconium plug, the infant should be observed closely for the possible presence of congenital aganglionic megacolon.

96.1 Meconium Ileus in Cystic Fibrosis
Akhil Maheshwari and Waldemar A. Carlo
The differential diagnosis involves other causes of intestinal obstruction, including intestinal pseudo-obstruction and other causes of pancreatic insufficiency (Chapter 341). A presumptive diagnosis can be made on the basis of a history of CF in a sibling, via palpation of doughy or cordlike masses of intestines through the abdominal wall, and from the radiographic appearance. In contrast to the generally evenly distended intestinal loops above an atresia, the loops may vary in width and are not as evenly filled with gas. At points of heaviest meconium concentration, the infiltrated gas may create a bubbly granular appearance (Figs. 96-2 and 96-3). It is technically difficult to perform a sweat test in a neonate. Genetic testing confirms the diagnosis of CF.
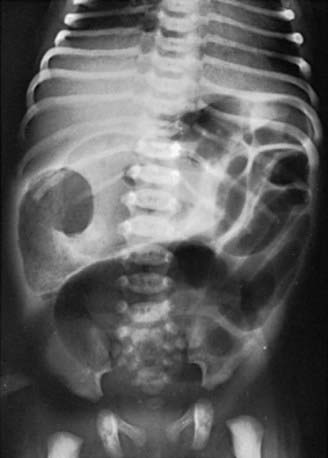
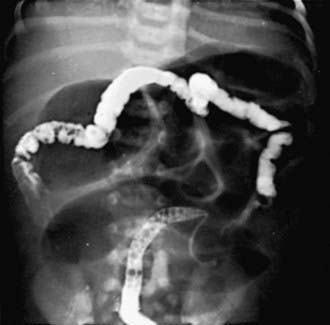
Figure 96-3 Meconium ileus. The colon, outlined by contrast material, is small because meconium has not reached it.
Treatment for meconium ileus is high Gastrografin enema as described previously for meconium plugs. If the procedure unsuccessful or perforation of the bowel wall is suspected, laparotomy is performed, and the ileum opened at the point of greatest diameter of the impaction. Approximately 50% of these infants have associated intestinal atresia, stenosis, or volvulus that requires surgery. The inspissated meconium is removed by gentle and patient irrigation with warm isotonic sodium chloride or acetylcysteine (Mucomyst) solution through a catheter passed between the impaction and the bowel wall. Most infants with meconium ileus survive the neonatal period. If meconium ileus is associated with CF, the long-term prognosis depends on the severity of the underlying disease (Chapter 395).
96.2 Neonatal Necrotizing Enterocolitis
Clinical Manifestations
Infants with NEC have a variety of signs and symptoms and may have an insidious or sudden catastrophic onset (Table 96-1). The onset of NEC is usually in the 2nd or 3rd week of life but can be as late as 3 mo in VLBW infants. Age of onset is inversely related to gestational age. The 1st signs of impending disease may be nonspecific, including lethargy and temperature instability, or related to gastrointestinal pathology, such as abdominal distention and gastric retention. Obvious bloody stools are seen in 25% of patients. Because of nonspecific signs, sepsis may be suspected before NEC. The spectrum of illness is broad, ranging from mild disease with only guaiac-positive stools to severe illness with bowel perforation, peritonitis, systemic inflammatory response syndrome, shock, and death. Progression may be rapid, but it is unusual for the disease to progress from mild to severe after 72 hr.
Table 96-1 SIGNS AND SYMPTOMS ASSOCIATED WITH NECROTIZING ENTEROCOLITIS
GASTROINTESTINAL
SYSTEMIC
From Kanto WP Jr, Hunter JE, Stoll BJ: Recognition and medical management of necrotizing enterocolitis, Clin Perinatol 21:335–346, 1994.
Diagnosis
A very high index of suspicion in treating preterm at-risk infants is crucial. Plain abdominal radiographs are essential to make a diagnosis of NEC. The finding of pneumatosis intestinalis (air in the bowel wall) confirms the clinical suspicion of NEC and is diagnostic; 50-75% of patients have pneumatosis when treatment is started (Fig. 96-4). Portal venous gas is a sign of severe disease, and pneumoperitoneum indicates a perforation (Figs. 96-4 and 96-5). Hepatic ultrasonography may detect portal venous gas despite normal abdominal roentgenograms.
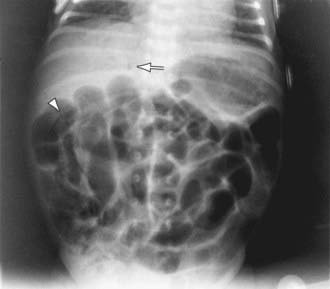
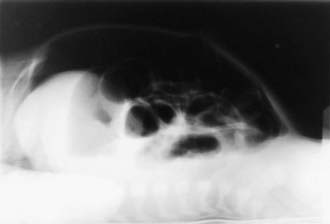
Afrazi A, Sodhi CP, Richardson W, et al. New insights into the pathogenesis and treatment of necrotizing enterocolitis toll-like receptors and beyond. Pediatr Res. 2011;69(3):183-188.
Alfaleh K, Bassler D: Probiotics for prevention of necrotizing enterocolitis in preterm infants, Cochrane Database Syst Rev (1):CD005496, 2008.
Bagci S, Eis-Hubinger AM, Franz AR, et al. Detection of astrovirus in premature infants with necrotizing enterocolitis. Pediatr Infect Dis J. 2008;27:347-350.
Blakely ML, Tyson JE, Lally KP, et al. Laparotomy versus peritoneal drainage for necrotizing enterocolitis or isolated intestinal perforation in extremely low birth rate infants: outcomes through 18 mo adjusted age. Pediatrics. 2006;117:e680-e687.
Bombell S, McGuire W: Early trophic feeding for very low birth weight infants, Cochrane Database Syst Rev (3):CD000504, 2009.
Deshpande G, Rao S, Patole S, Balsara M. Updated meta-analysis of probiotics for preventing necrotizing enterocolitis in preterm neonates. Pediatrics. 2010;125(5):921-930.
Fanaroff AA, Stoll BJ, Wright LL, et al. Trends in neonatal morbidity and mortality for very low birthweight infants. Am J Obstet Gynecol. 2007;196:147.e1-147.e8.
Figueras-Aloy J, Rodriguez-Miguelez JM, Iriondo-Sanz M, et al. Intravenous immunoglobulin and necrotizing enterocolitis in newborns with hemolytic disease. Pediatrics. 2010;125:139-144.
Henderson G, Craig S, Brocklehurst P, et al. Enteral feeding regimens and necrotizing enterocolitis in preterm infants: a multicentre case control study. Arch Dis Child Fetal Neonatal Ed. 2009;94:F120-F123.
Hintz SR, Kendrick DE, Stoll BJ, et al. Neurodevelopment and growth outcomes of extremely low birth weight infants after necrotizing enterocolitis. Pediatrics. 2005;115:1-8.
Meinzen-Derr J, Morrow AL, Hornung RW, et al. Epidemiology of necrotizing enterocolitis temporal clustering in two neonatal practices. J Pediatr. 2009;154:656-661.
Meinzen-Derr J, Poindexter B, Wrage L, et al. Role of human milk in extremely low birth weight infants’ risk of necrotizing enterocolitis or death. J Perinatol. 2009;29:57-62.
Moss RL, Dimmitt RA, Barnhart DC, et al. Laparotomy versus peritoneal drainage for necrotizing enterocolitis and perforation. N Engl J Med. 2006;354:2225-2234.
Murdoch EM, Sinha AK, Shanmugalingam ST, et al. Doppler flow velocimetry in the superior mesenteric artery on the first day of life in preterm infants and the risk of neonatal necrotizing enterocolitis. Pediatrics. 2006;118:1999-2003.
Neu J, Walker WA. Necrotizing enterocolitis. N Engl J Med. 2011;364(3):255-264.
Pickard SS, Feinstein JA, Popat RA, et al. Short- and long-term outcomes of necrotizing enterocolitis in infants with congenital heart disease. Pediatrics. 2009;123:e901-e906.
Pietz J, Achanti B, Lilien L, et al. Prevention of necrotizing enterocolitis in preterm infants: a 20-year experience. Pediatrics. 2007;119:e164-e170.
Rees CM, Eaton S, Kiely EM, et al. Peritoneal drainage or laparotomy for neonatal bowel perforation? A randomized controlled trial. Ann Surg. 2008;248:44-51.
Sharma R, Tepas JJIII, Hudak ML, et al. Portal venous gas and surgical outcome of neonatal necrotizing enterocolitis. J Pediatr Surg. 2005;40:371-376.
Stoll BJ, Hansen NI, Adams-Chapman I, et al. Neurodevelopmental and growth impairment among extremely low-birth-weight infants with neonatal infection. JAMA. 2004;292:2357-2365.
Turcios-Ruiz RM, Axelrod PSt, John K, et al. Outbreak of necrotizing enterocolitis caused by norovirus in a neonatal intensive care unit. J Pediatr. 2008;153:339-344.
Young C, Sharma R, Handfield M, et al. Biomarkers for infants at risk for necrotizing enterocolitis: clues to prevention? Pediatr Res. 2009;65:91R-97R.
96.3 Jaundice and Hyperbilirubinemia in the Newborn
Etiology
During the neonatal period, metabolism of bilirubin is in transition from the fetal stage, during which the placenta is the principal route of elimination of the lipid-soluble, unconjugated bilirubin, to the adult stage, during which the water-soluble conjugated form is excreted from hepatic cells into the biliary system and gastrointestinal tract. Unconjugated hyperbilirubinemia may be caused or increased by any factor that (1) increases the load of bilirubin to be metabolized by the liver (hemolytic anemias, polycythemia, bruising or internal hemorrhage, shortened red blood cell life as a result of immaturity or transfusion of cells, increased enterohepatic circulation, infection); (2) damages or reduces the activity of the transferase enzyme or other related enzymes (genetic deficiency, hypoxia, infection, thyroid deficiency); (3) competes for or blocks the transferase enzyme (drugs and other substances requiring glucuronic acid conjugation); or (4) leads to an absence or decreased amounts of the enzyme or to reduction of bilirubin uptake by liver cells (genetic defect, and prematurity). The toxic effects of elevated serum concentrations of unconjugated bilirubin are increased by factors that reduce the retention of bilirubin in the circulation (hypoproteinemia, displacement of bilirubin from its binding sites on albumin by competitive binding of drugs such as sulfisoxazole and moxalactam, acidosis, and increased free fatty acid concentration secondary to hypoglycemia, starvation, or hypothermia). Neurotoxic effects are directly related not only to the permeability of the blood-brain barrier and nerve cell membranes but also to neuronal susceptibility to injury, all of which are adversely influenced by asphyxia, prematurity, hyperosmolality, and infection. Early and frequent feeding decreases, whereas breast-feeding and dehydration increase, serum levels of bilirubin. Delay in passage of meconium, which contains 1 mg bilirubin/dL, may contribute to jaundice by enterohepatic recirculation after deconjugation by intestinal glucuronidase (Fig. 96-6). Drugs such as oxytocin (in the mother) and chemicals used in the nursery such as phenolic detergents may also produce unconjugated hyperbilirubinemia. Risk factors for unconjugated hyperbilirubinemia are listed in Table 96-2. Additional risk factors include polycythemia, infection, prematurity, and having a diabetic mother.
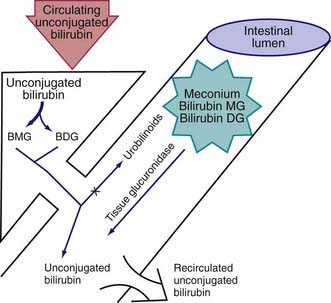
Table 96-2 RISK FACTORS FOR DEVELOPMENT OF SEVERE HYPERBILIRUBINEMIA IN INFANTS ≥35 WEEKS OF GESTATION (IN APPROXIMATE ORDER OF IMPORTANCE)
MAJOR RISK FACTORS
MINOR RISK FACTORS
DECREASED RISK (these factors are associated with decreased risk of significant jaundice, listed in order of decreasing importance)
TcB, transcutaneous bilirubin; TSB, total serum bilirubin.
* Race as defined by mother’s description.
From AAP Subcommittee on Hyperbilirubinemia: Management of hyperbilirubinemia in the newborn infant 35 or more weeks of gestation, Pediatrics 114:297–316, 2004.
Clinical Manifestations
Jaundice may be present at birth or may appear at any time during the neonatal period, depending on etiology. Jaundice usually becomes apparent in a cephalocaudal progression, starting on the face and progressing to the abdomen and then the feet, as serum levels increase. Dermal pressure may reveal the anatomic progression of jaundice (face, ≈5 mg/dL; mid-abdomen, ≈15 mg/dL; soles, ≈20 mg/dL), but clinical examination cannot be depended on to estimate serum levels. Jaundice to the mid-abdomen, signs or symptoms, high-risk factors that suggest nonphysiologic jaundice, or hemolysis must be evaluated further (Tables 96-2 and 96-3). Noninvasive techniques for transcutaneous measurement of bilirubin (TcB) that correlate with serum levels may be used to screen infants, but determination of serum bilirubin level is indicated in patients with elevated age-specific transcutaneous bilirubin measurement, progressing jaundice, or risk for either hemolysis or sepsis. Whereas jaundice from deposition of indirect bilirubin in the skin tends to appear bright yellow or orange, jaundice of the obstructive type (direct bilirubin) has a greenish or muddy yellow cast. Infants with severe hyperbilirubinemia may present with lethargy and poor feeding and, without treatment, can progress to acute bilirubin encephalopathy (kernicterus) (Chapter 96.4).
Table 96-3 LABORATORY EVALUATION OF THE JAUNDICED INFANT ≥35 WEEKS OF GESTATION
| INDICATIONS | ASSESSMENTS |
|---|---|
| Jaundice in first 24 hr | Measure TcB and/or TSB |
| Jaundice appears excessive for infant’s age | Measure TcB and/or TSB |
| Infant receiving phototherapy or TSB rising rapidly (i.e., crossing percentiles [see Fig. 96-8]) and unexplained by history and physical examination |
ETCOc, end tidal carbon monoxide concentration; G6PD, glucose-6-phosphate dehydrogenase; TcB, transcutaneous bilirubin; TSB, total serum bilirubin.
From AAP Subcommittee on Hyperbilirubinemia: Management of hyperbilirubinemia in the newborn infant 35 or more weeks of gestation, Pediatrics 114:297–316, 2004.
Differential Diagnosis
There is a long differential diagnosis for jaundice first recognized after the 1st wk of life, including breast milk jaundice, septicemia, congenital atresia or paucity of the bile ducts, hepatitis, galactosemia, hypothyroidism, cystic fibrosis, and congenital hemolytic anemia crises related to red blood cell morphology and enzyme deficiencies (Fig. 96-7). The differential diagnosis for persistent jaundice during the first month of life includes hyperalimentation-associated cholestasis, hepatitis, cytomegalic inclusion disease, syphilis, toxoplasmosis, familial nonhemolytic icterus, congenital atresia of the bile ducts, galactosemia, and inspissated bile syndrome following hemolytic disease of the newborn. Rarely, physiologic jaundice may be prolonged for several weeks, as in infants with hypothyroidism or pyloric stenosis.
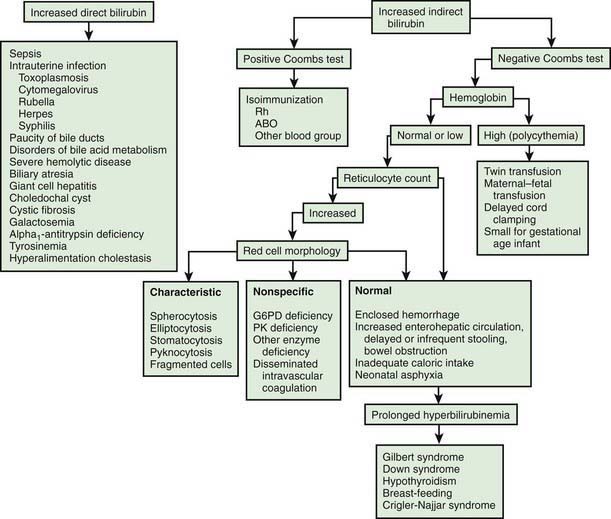
Full-term, low-risk, asymptomatic infants with jaundice may be evaluated by monitoring of total serum bilirubin (TSB) levels. Regardless of gestation or time of appearance of jaundice, patients with significant hyperbilirubinemia and those with symptoms or signs require a complete diagnostic evaluation, which includes determination of direct and indirect bilirubin fractions, hemoglobin, reticulocyte count, blood type, Coombs test, and examination of a peripheral blood smear. Indirect hyperbilirubinemia, reticulocytosis, and a smear with evidence of red blood cell destruction suggest hemolysis (see Table 96-3). In the absence of blood group incompatibility, nonimmunologically induced hemolysis should be considered. If the reticulocyte count, Coombs test result, and direct bilirubin value are normal, physiologic or pathologic indirect hyperbilirubinemia may be present (see Fig. 96-7). If direct hyperbilirubinemia is present, hepatitis, congenital bile duct disorders (atresia, paucity, Byler disease), cholestasis, inborn errors of metabolism, cystic fibrosis, and sepsis are diagnostic possibilities.
Physiologic Jaundice (Icterus Neonatorum)
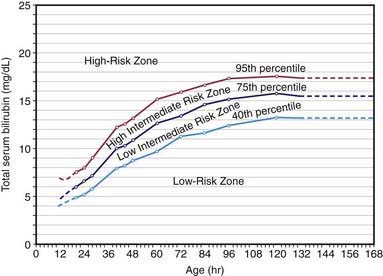
Overall, 6-7% of full-term infants have indirect bilirubin levels >13 mg/dL and less than 3% have levels >15 mg/dL. Risk factors for elevated indirect bilirubin include maternal age, race (Chinese, Japanese, Korean, and Native American), maternal diabetes, prematurity, drugs (vitamin K3, novobiocin), altitude, polycythemia, male sex, trisomy 21, cutaneous bruising, blood extravasation (cephalohematoma), oxytocin induction, breast-feeding, weight loss (dehydration or caloric deprivation), delayed bowel movement, and a family history of or a sibling who had physiologic jaundice (see Table 96-2). In infants without these variables, indirect bilirubin levels rarely rise above 12 mg/dL, whereas infants with several risk factors are more likely to have higher bilirubin levels. A combination of breast-feeding, variant-glucuronosyl transferase activity (1A1), and alterations of the organic anion transporter 2 gene increases the risk in Asian children. Predicting which neonates are at risk for exaggerated physiologic jaundice can be based on hour-specific bilirubin levels in the first 24-72 hr of life (Fig. 96-8). Transcutaneous measurements of bilirubin are linearly correlated with serum levels and can be used for screening. Indirect bilirubin levels in full-term infants decline to adult levels (1 mg/dL) by 10-14 days of life. Persistent indirect hyperbilirubinemia beyond 2 wk suggests hemolysis, hereditary glucuronyl transferase deficiency, breast milk jaundice, hypothyroidism, or intestinal obstruction. Jaundice associated with pyloric stenosis may be due to caloric deprivation, deficiency of hepatic UDP-glucuronyl transferase, or an increase in the enterohepatic circulation of bilirubin from the ileus. In premature infants, the rise in serum bilirubin tends to be the same or somewhat slower but of longer duration than in term infants. Peak levels of 8-12 mg/dL are not usually reached until the 4th-7th day, and jaundice is infrequently observed after the 10th day, corresponding to the maturation of mechanisms for bilirubin metabolism and excretion.
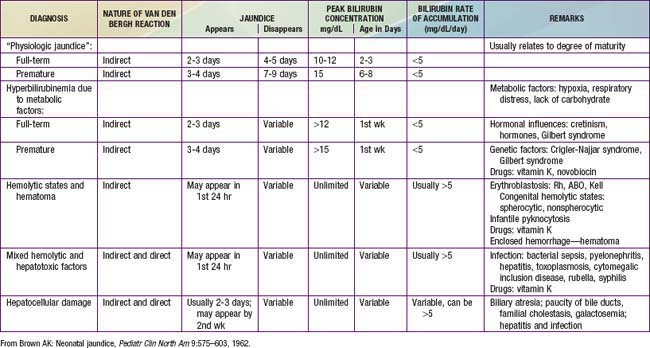
The diagnosis of physiologic jaundice in term or preterm infants can be established only by excluding known causes of jaundice on the basis of the history, clinical findings, and laboratory data (Table 96-4). In general, a search to determine the cause of jaundice should be made if (1) it appears in the first 24-36 hr of life, (2) serum bilirubin is rising at a rate faster than 5 mg/dL/24 hr, (3) serum bilirubin is >12 mg/dL in a full-term infant (especially in the absence of risk factors) or 10-14 mg/dL in a preterm infant, (4) jaundice persists after 10-14 days of life, or (5) direct bilirubin fraction is >2 mg/dL at any time. Other factors suggesting a nonphysiologic cause of jaundice are family history of hemolytic disease, pallor, hepatomegaly, splenomegaly, failure of phototherapy to lower the bilirubin level, vomiting, lethargy, poor feeding, excessive weight loss, apnea, bradycardia, abnormal vital signs (including hypothermia), light-colored stools, dark urine positive for bilirubin, and signs of kernicterus (Chapter 96.4).
Pathologic Hyperbilirubinemia
Jaundice and its underlying hyperbilirubinemia are considered pathologic if the time of appearance, duration, or pattern varies significantly from that of physiologic jaundice or if the course is compatible with physiologic jaundice but other reasons exist to suspect that the infant is at special risk for neurotoxicity. It may not be possible to determine the precise cause of an abnormal elevation of unconjugated bilirubin, but many infants with this finding have associated risk factors such as Asian race, prematurity, breast-feeding, and weight loss. Frequently, the terms exaggerated physiologic jaundice and hyperbilirubinemia of the newborn are used in infants whose primary problem is probably a deficiency or inactivity of bilirubin glucuronyl transferase (Gilbert syndrome) rather than an excessive load of bilirubin for excretion (see Table 96-2). The combination of glucose-6-phosphate dehydrogenase (G6PD) deficiency and a mutation of the promoter region of UDP-glucuronyl transferase-1 produces indirect hyperbilirubinemia in the absence of signs of hemolysis. Nonphysiologic hyperbilirubinemia may also be caused by mutations in the gene for bilirubin UDP-glucuronyl transferase.
The greatest risk associated with indirect hyperbilirubinemia is the development of bilirubin-induced neurologic dysfunction, which typically occurs with high indirect bilirubin levels (Chapter 96.4). The development of kernicterus (bilirubin encephalopathy) depends on the level of indirect bilirubin, duration of exposure to bilirubin elevation, the cause of jaundice, and the infant’s well-being. Neurologic injury including kernicterus may occur at lower bilirubin levels in preterm infants and in the presence of asphyxia, intraventricular hemorrhage, hemolysis, or drugs that displace bilirubin from albumin. The exact serum indirect bilirubin level that is harmful for VLBW infants is unclear.
Jaundice Associated with Breast-Feeding
Significant elevation in unconjugated bilirubin (breast milk jaundice) develops in an estimated 2% of breast-fed term infants after the 7th day of life, with maximal concentrations as high as 10-30 mg/dL reached during the 2nd-3rd week. If breast-feeding is continued, the bilirubin gradually decreases but may persist for 3-10 wk at lower levels. If nursing is discontinued, the serum bilirubin level falls rapidly, reaching normal range within a few days. With resumption of breast-feeding, bilirubin seldom returns to previously high levels. Phototherapy may be of benefit (Chapter 96.4). Although uncommon, kernicterus can occur in patients with breast milk jaundice. The etiology of breast milk jaundice is not entirely clear but may be attributed to the presence of glucuronidase in some breast milk.
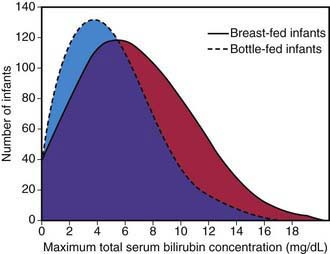
This syndrome should be distinguished from an early-onset, accentuated unconjugated hyperbilirubinemia known as breast-feeding jaundice, which occurs in the 1st week of life in breast-fed infants, who normally have higher bilirubin levels than formula-fed infants (Fig. 96-9). Hyperbilirubinemia (>12 mg/dL) develops in 13% of breast-fed infants in the 1st wk of life and may be due to decreased milk intake with dehydration and/or reduced caloric intake. Prophylactic supplements of glucose water to breast-fed infants are associated with higher bilirubin levels, in part because of reduced intake of the higher–caloric density breast milk. Frequent breast-feeding (>10/24 hr), rooming-in with night feeding, and ongoing lactation support may reduce the incidence of early breast-feeding jaundice. Even when breast-feeding jaundice develops, breast-feeding should be continued if possible. It is an option to temporarily interrupt breast-feedings and substitute formula for a day or two. In addition, frequent feeding and supplementation with formula or expressed breast milk is appropriate if the intake seems inadequate, weight loss is excessive, or the infant appears dehydrated.
Congenital Atresia of the Bile Ducts
See Chapter 348.1. Jaundice persisting for more than 2 wk or associated with acholic stools and dark urine suggests biliary atresia. All infants with such findings must undergo an immediate diagnostic evaluation, including determination of direct bilirubin.
Agrawal SK, Kumar P, Rathi R, et al. UGT1A1 gene polymorphisms in north Indian neonates presenting with unconjugated hyperbilirubinemia. Pediatr Res. 2009;65:675-680.
American Academy of Pediatrics Subcommittee on Hyperbilirubinemia. Management of hyperbilirubinemia in the newborn infant 35 or more weeks of gestation. Pediatrics. 2004;114:297-316.
Fay DL, Schellhase KG, Suresh GK. Bilirubin screening for normal newborns: a critique of the hour-specific bilirubin nomogram. Pediatrics. 2009;124:1203-1205.
Ip S, Chung M, Kulig J, et al. An evidenced-base review of important issues concerning neonatal hyperbilirubinemia. Pediatrics. 2004;114:e130-e153.
Johnson L, Bhutani VK, Karp K, et al. Clinical report from the pilot USA Kernicterus Registry (1992 to 2004). J Perinatol. 2009;29:S25-S45.
Keren R, Luam Z, Friedman S, et al. A comparison of alternative risk-assessment strategies for predicting significant neonatal hyperbilirubinemia in term and near-term infants. Pediatrics. 2008;121:e170-e179.
Kuzniewicz MW, Escobar GJ, Wi S, et al. Risk factors for severe hyperbilirubinemia among infants with borderline bilirubin levels: a nested case-control study. J Pediatr. 2008;153:234-240.
Lin Z, Fontaine J, Watchko JF. Coexpression of gene polymorphisms involved in bilirubin production and metabolism. Pediatrics. 2008;122:e156-e162.
Morris BH, Oh W, Tyson JE, et al. Aggressive vs. conservative phototherapy for infants with extremely low birth weight. N Engl J Med. 2008;359:1885-1896.
Watchko JF. Hyperbilirubin toxicity in the late preterm infant. Clin Perinatol. 2006;33:839-852.
96.4 Kernicterus
The precise blood level above which indirect-reacting bilirubin or free bilirubin will be toxic for an individual infant is unpredictable, but in a large series, kernicterus occurred only in infants with a bilirubin >20 mg/dL. Ninety percent of the infants in whom kernicterus developed were in previously healthy, predominantly breast-fed term and near-term infants. The duration of exposure to high bilirubin levels needed to produce toxic effects are unknown. The more immature the infant is, the greater the susceptibility to kernicterus. Factors that potentiate the movement of bilirubin across the blood-brain barrier and into brain cells are discussed in Chapter 96.3.
Clinical Manifestations
Signs and symptoms of kernicterus usually appear 2-5 days after birth in term infants and as late as the 7th day in premature infants, but hyperbilirubinemia may lead to encephalopathy at any time during the neonatal period. The early signs may be subtle and indistinguishable from those of sepsis, asphyxia, hypoglycemia, intracranial hemorrhage, and other acute systemic illnesses in a neonate. Lethargy, poor feeding, and loss of the Moro reflex are common initial signs. Subsequently, the infant may appear gravely ill and prostrate, with diminished tendon reflexes and respiratory distress. Opisthotonos with a bulging fontanel, twitching of the face or limbs, and a shrill high-pitched cry may follow. In advanced cases, convulsions and spasm occur, with affected infants stiffly extending their arms in an inward rotation with the fists clenched (Table 96-5). Rigidity is rare at this late stage.
Table 96-5 CLINICAL FEATURES OF KERNICTERUS
ACUTE FORM
CHRONIC FORM
From Dennery PA, Seidman DS, Stevenson DK: Neonatal hyperbilirubinemia, N Engl J Med 344:581–590, 2001.
Many infants who progress to these severe neurologic signs die; the survivors are usually seriously damaged but may appear to recover and for 2-3 mo show few abnormalities. Later in the 1st yr of life, opisthotonos, muscle rigidity, irregular movements, and convulsions tend to recur. In the 2nd yr, the opisthotonos and seizures abate, but irregular, involuntary movements, muscle rigidity, or, in some infants, hypotonia increase steadily. By 3 yr of age, the complete neurologic syndrome is often apparent; it consists of bilateral choreoathetosis with involuntary muscle spasms, extrapyramidal signs, seizures, mental deficiency, dysarthric speech, high-frequency hearing loss, squinting, and defective upward eye movements. Pyramidal signs, hypotonia, and ataxia occur in a few infants. In mildly affected infants, the syndrome may be characterized only by mild to moderate neuromuscular incoordination, partial deafness, or “minimal brain dysfunction,” occurring singly or in combination; these problems may be inapparent until the child enters school (see Table 96-5).
Incidence and Prognosis
By pathologic criteria, kernicterus develops in 30% of infants (all gestational ages) with untreated hemolytic disease and bilirubin levels >25-30 mg/dL. The incidence at autopsy in hyperbilirubinemic premature infants is 2-16% and is related to the risk factors discussed in Chapter 96.3. Reliable estimates of the frequency of the clinical syndrome are not available because of the wide spectrum of manifestations. Overt neurologic signs have a grave prognosis; more than 75% of infants die, and 80% of affected survivors have bilateral choreoathetosis with involuntary muscle spasms. Mental retardation, deafness, and spastic quadriplegia are common.
Prevention
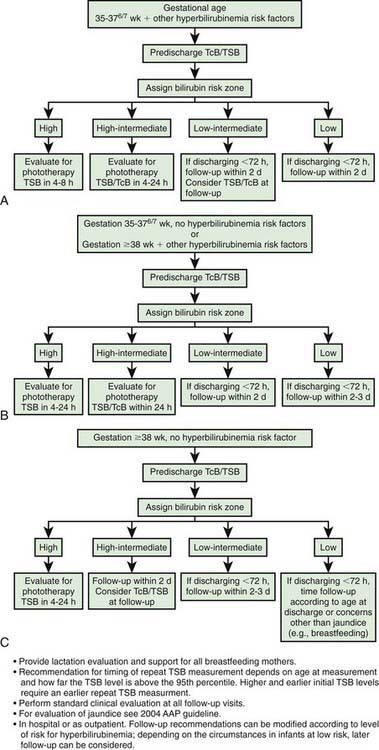
Effective prevention requires ongoing vigilance and a practical, system-based approach in order to distinguish infants with benign newborn jaundice from those whose course may be less predictable and potentially harmful. Protocols using the hour-specific bilirubin nomogram (see Fig. 96-8), physical examination, and clinical risk factors have been successful in identifying patients at risk for hyperbilirubinemia and candidates for targeted management. The American Academy of Pediatrics (AAP) has identified potentially preventable causes of kernicterus, as follows: (1) early discharge (<48 hr) with no early follow-up (within 48 hr of discharge); this problem is particularly important in near-term infants (35-37 wk of gestation); (2) failure to check the bilirubin level in an infant noted to be jaundiced in the first 24 hr; (3) failure to recognize the presence of risk factors for hyperbilirubinemia; (4) underestimation of the severity of jaundice by clinical (visual) assessment; (5) lack of concern regarding the presence of jaundice; (6) delay in measuring the serum bilirubin level despite marked jaundice or delay in initiating phototherapy in the presence of elevated bilirubin levels; and (7) failure to respond to parental concern regarding jaundice, poor feeding, or lethargy. An evidence-based management algorithm for infants is shown in Figure 96-10. In addition, it is recommended to determine before discharge each infant’s risk factors from established protocols (see Table 96-2).
Treatment of Hyperbilirubinemia
Regardless of the cause, the goal of therapy is to prevent neurotoxicity related to indirect-reacting bilirubin while not causing undue harm. Phototherapy and, if it is unsuccessful, exchange transfusion remain the primary treatment modalities used to keep the maximal total serum bilirubin below pathologic levels (Figs. 96-11 and 96-12; Table 96-6). The risk of injury to the central nervous system from bilirubin must be balanced against the potential risk of treatment. There is lack of consensus regarding the exact bilirubin level at which to initiate phototherapy. Because phototherapy may require 6-12 hr to have a measurable effect, it must be started at bilirubin levels below those indicated for exchange transfusion. When identified, underlying medical causes of elevated bilirubin and physiologic factors that contribute to neuronal susceptibility should be treated, with antibiotics for septicemia and correction of acidosis (Table 96-7).
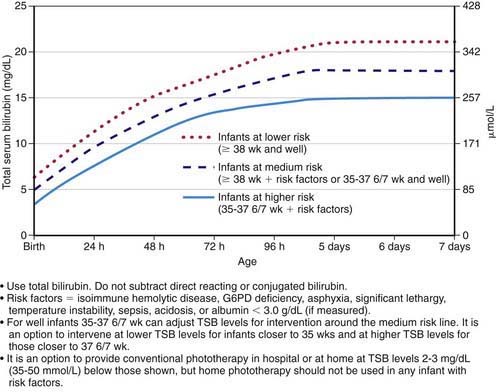
Figure 96-11 Guidelines for phototherapy in hospitalized infants of ≥35 weeks of gestation. Note: These guidelines are based on limited evidence, and the levels shown are approximations. The guidelines refer to the use of intensive phototherapy, which should be used when the total serum bilirubin (TSB) exceeds the line indicated for each category. Infants are designated as “higher risk” because of the potential negative effects of the conditions listed on albumin binding of bilirubin, the blood-brain barrier, and the susceptibility of the brain cells to damage by bilirubin. “Intensive phototherapy” implies irradiance in the blue-green spectrum (wavelengths approximately 430-490 nm) of at least 30 µW/cm2/nm (measured at the infant’s skin directly below the center of the phototherapy unit) and delivered to as much of the infant’s skin surface area as possible. Note that irradiance measured below the center of the light source is much greater than that measured at the periphery. Measurements should be made with a radiometer specified by the manufacturer of the phototherapy system. If TSB levels approach or exceed the exchange transfusion line (see Fig. 96-12), the sides of the bassinette, incubator, or warmer should be lined with aluminum foil or white material, to increase both the surface area of the infant exposed and the efficacy of phototherapy. The presence of hemolysis is strongly suggested if the TSB does not decrease or continues to rise in an infant who is receiving intensive phototherapy. Infants who receive phototherapy and have an elevated direct-reacting or conjugated bilirubin value (cholestatic jaundice) may inconsistently have the bronze-baby syndrome. G6PD, glucose-6-phosphate dehydrogenase.
(From American Academy of Pediatrics Subcommittee on Hyperbilirubinemia: Management of hyperbilirubinemia in the newborn infant 35 or more weeks of gestation, Pediatrics 114:297–316, 2004.)
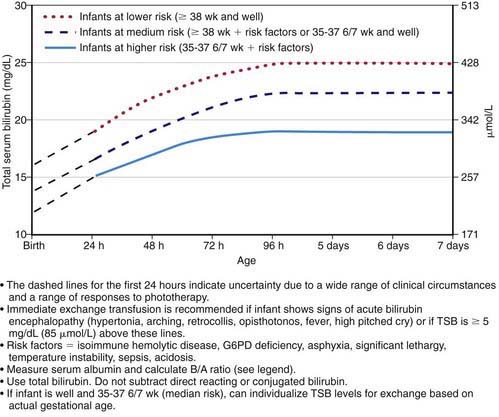
Table 96-6 SUGGESTED MAXIMAL INDIRECT SERUM BILIRUBIN CONCENTRATIONS (mg/dL) IN PRETERM INFANTS
| BIRTHWEIGHT (g) | UNCOMPLICATED* | COMPLICATED* |
|---|---|---|
| <1,000 | 12-13 | 10-12 |
| 1,000-1,250 | 12-14 | 10-12 |
| 1,251-1,499 | 14-16 | 12-14 |
| 1,500-1,999 | 16-20 | 15-17 |
| 2,000-2,500 | 20-22 | 18-20 |
* Complications include perinatal asphyxia, acidosis, hypoxia, hypothermia, hypoalbuminemia, meningitis, intraventricular hemorrhage, hemolysis, hypoglycemia, or signs of kernicterus. Phototherapy is usually started at 50-70% of the maximal indirect level. If values greatly exceed this level, if phototherapy is unsuccessful in reducing the maximal bilirubin level, or if signs of kernicterus are evident, exchange transfusion is indicated.
Table 96-7 EXAMPLE OF A CLINICAL PATHWAY FOR MANAGEMENT OF THE NEWBORN INFANT READMITTED FOR PHOTOTHERAPY OR EXCHANGE TRANSFUSION
TREATMENT
Use intensive phototherapy and/or exchange transfusion as indicated in Figs. 96-11 and 96-12
LABORATORY TESTS
INTERVENTIONS
FOR INFANTS RECEIVING INTENSIVE PHOTOTHERAPY:
TSB, total serum bilirubin.
From AAP Subcommittee on Hyperbilirubinemia: Management of hyperbilirubinemia in the newborn infant 35 or more weeks of gestation, Pediatrics 114:297–316, 2004.
Phototherapy
The therapeutic effect of phototherapy depends on the light energy emitted in the effective range of wavelengths, the distance between the lights and the infant, and the surface area of exposed skin, as well as the rate of hemolysis and in vivo metabolism and excretion of bilirubin. Available commercial phototherapy units vary considerably in spectral output and the intensity of radiance emitted; therefore, the wattage can be accurately measured only at the patient’s skin surface. Dark skin does not reduce the efficacy of phototherapy. Maximal intensive phototherapy should be used when indirect bilirubin levels approach those noted in Figure 96-11 and Table 96-7. Such therapy includes using “special blue” fluorescent tubes, placing the lamps within 15-20 cm of the infant, and putting a fiberoptic phototherapy blanket under the infant’s back to increase the exposed surface area. Aggressive phototherapy may improve neurodevelopmental outcome in infants <1,000 g.
Exchange Transfusion
Double-volume exchange transfusion is performed if intensive phototherapy has failed to reduce bilirubin levels to a safe range and if the risk of kernicterus exceeds the risk of the procedure. Potential complications from exchange transfusion are not trivial and include metabolic acidosis, electrolyte abnormalities, hypoglycemia, hypocalcemia, thrombocytopenia, volume overload, arrhythmias, NEC, infection, graft versus host disease, and death. This widely accepted treatment is repeated if necessary to keep indirect bilirubin levels in a safe range (see Fig. 96-12 and Table 96-7). See Exchange Transfusion in Chapter 97.
American Academy of Pediatrics, Subcommittee on Hyperbilirubinemia. Clinical practice guideline: management of hyperbilirubinemia in the newborn infant 35 or more weeks of gestation. Pediatrics. 2004;114:297-316.
Amin SB, Prinzing D, Myers G. Hyperbilirubinemia and language delay in premature infants. Pediatrics. 2009;123:327-331.
Bhutani V, Johnson L, Sivier E. Predictive ability of a predischarge hour specific serum bilirubin for subsequent significant hyperbilirubinemia in healthy term and near-term newborns. Pediatrics. 1999;103:6-14.
Bhutani VK, Johnson LH, Keren R, et al. Diagnosis and management of hyperbilirubinemia in the term neonate: for a safer first week. Pediatr Clin North Am. 2004;51:843-861.
Boo NY, Lee HT. Randomized controlled trial of oral versus intravenous fluid supplementation on serum bilirubin level during phototherapy of term infants with severe hyperbilirubinaemia. J Paediatr Child Health. 2002;38:151-155.
Burke BL, Robbins JM, Bird TM, et al. Trends in hospitalizations for neonatal jaundice and kernicterus in the United States, 1988–2005. Pediatrics. 2009;123:524-532.
Chen SM, Chang MH, Du JC, et al. Screening for biliary atresia by infant stool color card in Taiwan. Pediatrics. 2006;117:1147-1154.
Jangaard KA, Fell DB, Dodds L, et al. Outcomes in a population of healthy term and near-term infants with serum bilirubin levels of ≥ 325 µmol/L (≥19 mg/dL) who were born in Nova Scotia, Canada between 1994 and 2000. Pediatrics. 2008;122:119-124.
Kaplan M, Hammerman C, Vreman HJ, et al. Hemolysis and hyperbilirubinemia in antiglobulin positive, direct ABO blood group heterospecific neonates. J Pediatr. 2010;157:772-777.
Kaplan M, Herschel M, Hammerman C, et al. Neonatal hyperbilirubinemia in African American males: the importance of glucose-6-phosphate dehydrogenase deficiency. J Pediatr. 2006;149:83-88.
Kaplan M, Muraca M, Vreman HJ, et al. Neonatal bilirubin production-conjugation imbalance: effect of glucose-6-phosphate dehydrogenase deficiency and borderline prematurity. Arch Dis Child Fetal Neonatal Ed. 2005;90:F123-F127.
Kappas A, Drummond GS, Valaes T. A single dose of Sn-mesoporphyrin prevents development of severe hyperbilirubinemia in glucose-6-phosphate dehydrogenase-deficient newborns. Pediatrics. 2001;108:25-30.
Kappas A, Munson DP, Marshall JR. Sn-mesoporphyrin interdiction of severe hyperbilirubinemia in Jehovah’s Witness newborns as an alternative to exchange transfusion. Pediatrics. 2001;108:1374-1377.
The Lancet. Detection and treatment of neonatal jaundice. Lancet. 2010;375:1845.
Maisels MJ, Bhutani VK, Bogen D, et al. Hyperbilirubinemia in the newborn infant ≥ 35 weeks’ gestation: an update with clarifications. Pediatrics. 2009;124:1193-1198.
Maisels MJ, Kring E. Rebound in serum bilirubin level following intensive phototherapy. Arch Pediatr Adolesc Med. 2002;156:669-672.
Maisels MJ, Kring E. The contribution of hemolysis to early jaundice in normal newborns. Pediatrics. 2006;118:276-279.
Maisels MJ, Kring E. Transcutaneous bilirubin levels in the first 96 hours in a normal newborn population of > or = 35 weeks’ gestation. Pediatrics. 2006;117:1169-1173.
Maisels MJ, McDonagh AF. Phototherapy for neonatal jaundice. N Engl J Med. 2008;358:920-928.
Manning D. Neonatal jaundice: in the eye of the beholder? Arch Dis Child Fetal Neonatal Ed. 2009;94:F314-F316.
McDonagh AF. Bilirubin, copper-porphyrins, and the bronze baby syndrome. J Pediatr. 2011;158:160-164.
Mehta S, Kumar P, Narang A. A randomized controlled trial of fluid supplementation in term neonates with severe hyperbilirubinemia. J Pediatr. 2005;147:781-785.
Morris BH, Oh W, Tyson JE, et al. Aggressive vs. conservative phototherapy for infants with extremely low birth weight. N Engl J Med. 2008;359:1885-1896.
Newman TB, Liljestrand P, Jeremy RJ, et al. Outcomes among newborns with total serum bilirubin levels of 25 mg per deciliter or more. N Engl J Med. 2006;354:1889-1900.
Okumura A, Kidoforo H, Shoji H, et al. Kernicterus in preterm infants. Pediatrics. 2009;123:e1052-e1058.
Patra K, Storfer-Isser A, Siner B, et al. Adverse events associated with neonatal exchange transfusion in the 1990s. J Pediatr. 2004;144:626-631.
Rennie J, Burman-Roy S, Murphy MS. Neonatal jaundice: summary of NICE guidance. BMJ. 2009;338:b1805.
Suresh GK, Martin CL, Soll RF: Metalloporphyrins for treatment of unconjugated hyperbilirubinemia in neonates, Cochrane Database Syst Rev (2):CD004207, 2003.
Trikalinos TA, Cheung M, Lau J, et al. Systematic review of screening for bilirubin encephalopathy in neonates. Pediatrics. 2009;124:1162-1171.
US Preventive Services Task Force. Screening of infants for hyperbilirubinemia to prevent chronic bilirubin encephalopathy: US preventive services task force recommendation statement. Pediatrics. 2009;124:1172-1177.
Walsh S, Molloy EJ. Is intravenous immunoglobulin superior to exchange transfusion in the management of hyperbilirubinaemia in term neonates? Arch Dis Child. 2009;94:739-741.
Wennberg RP, Ahlfors CE, Bhutani VD, et al. Toward understanding kernicterus: a challenge to improve the management of jaundiced newborns. Pediatrics. 2006;117:474-485.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 96 Digestive System Disorders
Vomiting
Obstructive lesions of the digestive tract are the most frequent gastrointestinal anomalies (Chapters 311, 321, 322, and 324). Vomiting (and drooling) from esophageal obstruction occurs with the 1st feeding. The diagnosis of esophageal atresia can be suspected if unusual drooling from the mouth is observed and if resistance is encountered during an attempt to pass a catheter into the stomach. The diagnosis should be made before the infant has trouble with oral feedings and aspiration pneumonia develops. Infantile achalasia (cardiospasm), a rare cause of vomiting in newborn infants, is demonstrable radiographically as obstruction at the cardiac end of the esophagus without organic stenosis. Regurgitation of feedings because of continuous relaxation of the esophageal-gastric sphincter, or chalasia, is a cause of vomiting. Keeping the infant in a semi-upright position, thickening the feeding, or administering prokinetic drugs can control it.
Vomiting due to obstruction of the small intestine usually begins on the 1st day of life and is frequent, persistent, usually nonprojectile, copious, and, unless the obstruction is above the ampulla of Vater, bile-stained; it is associated with abdominal distention, visible deep peristaltic waves, and reduction or absence of bowel movements. Malrotation with obstruction from midgut volvulus is an acute emergency that must be not only considered but also urgently evaluated by an upper gastrointestinal contrast radiographic series. Radiographs of the abdomen show the distribution of air in the intestine, which may point to the anatomic location of an obstruction; malrotation can be identified only by contrast studies. Normally, air can be demonstrated by radiographs in the jejunum by 15-60 min, in the ileum by 2-3 hr, and in the colon by 3 hr after birth. Absence of rectal gas at 24 hr is abnormal. Persistent vomiting may occur with congenital diaphragmatic hernia. The vomiting associated with pyloric stenosis may begin any time after birth but does not assume its characteristic pattern before the 2nd-3rd wk. Vomiting with obstipation is a common early sign of Hirschsprung disease. Vomiting may occur with many other disturbances that do not obstruct the digestive tract, such as milk allergy, adrenal hyperplasia of the salt-losing variety, galactosemia, hyperammonemias, organic acidemias, increased intracranial pressure, septicemia, meningitis, and urinary tract infection. In many infants, it is simply regurgitation from overfeeding or from failure to permit the infant to eructate swallowed air. (See Chapter 315 for a discussion of gastric emptying and gastroesophageal reflux.)
Meconium Plugs
Lower colonic or anorectal plugs (Fig. 96-1) with a lower than normal water content may cause intestinal obstruction. Rarely, a firm mass of meconium may form elsewhere in the intestine and cause intrauterine intestinal obstruction and meconium peritonitis unrelated to cystic fibrosis (CF). Anorectal plugs may also cause mucosal ulceration and intestinal perforation. Meconium plugs are associated with small left colon syndrome in infants of diabetic mothers and with CF, rectal aganglionosis, maternal opiate use, and magnesium sulfate therapy for preeclampsia. The plug may be evacuated by glycerin suppository or rectal irrigation with isotonic saline. Enemas with the iodinated contrast medium Gastrografin usually induce passage of the plug, presumably because the high osmolarity (1,900 mOsm/L) of the solution draws fluid rapidly into the intestinal lumen and loosens inspissated material. Such rapid loss of fluid into the bowel may result in acute dehydration and shock, so it is advisable to dilute the contrast material with an equal amount of water, correct any existing dehydration, and provide intravenous fluids during and for several hours after the procedure. After removal of a meconium plug, the infant should be observed closely for the possible presence of congenital aganglionic megacolon.
96.1 Meconium Ileus in Cystic Fibrosis
Akhil Maheshwari and Waldemar A. Carlo
The differential diagnosis involves other causes of intestinal obstruction, including intestinal pseudo-obstruction and other causes of pancreatic insufficiency (Chapter 341). A presumptive diagnosis can be made on the basis of a history of CF in a sibling, via palpation of doughy or cordlike masses of intestines through the abdominal wall, and from the radiographic appearance. In contrast to the generally evenly distended intestinal loops above an atresia, the loops may vary in width and are not as evenly filled with gas. At points of heaviest meconium concentration, the infiltrated gas may create a bubbly granular appearance (Figs. 96-2 and 96-3). It is technically difficult to perform a sweat test in a neonate. Genetic testing confirms the diagnosis of CF.
Figure 96-3 Meconium ileus. The colon, outlined by contrast material, is small because meconium has not reached it.
Treatment for meconium ileus is high Gastrografin enema as described previously for meconium plugs. If the procedure unsuccessful or perforation of the bowel wall is suspected, laparotomy is performed, and the ileum opened at the point of greatest diameter of the impaction. Approximately 50% of these infants have associated intestinal atresia, stenosis, or volvulus that requires surgery. The inspissated meconium is removed by gentle and patient irrigation with warm isotonic sodium chloride or acetylcysteine (Mucomyst) solution through a catheter passed between the impaction and the bowel wall. Most infants with meconium ileus survive the neonatal period. If meconium ileus is associated with CF, the long-term prognosis depends on the severity of the underlying disease (Chapter 395).
96.2 Neonatal Necrotizing Enterocolitis
Clinical Manifestations
Infants with NEC have a variety of signs and symptoms and may have an insidious or sudden catastrophic onset (Table 96-1). The onset of NEC is usually in the 2nd or 3rd week of life but can be as late as 3 mo in VLBW infants. Age of onset is inversely related to gestational age. The 1st signs of impending disease may be nonspecific, including lethargy and temperature instability, or related to gastrointestinal pathology, such as abdominal distention and gastric retention. Obvious bloody stools are seen in 25% of patients. Because of nonspecific signs, sepsis may be suspected before NEC. The spectrum of illness is broad, ranging from mild disease with only guaiac-positive stools to severe illness with bowel perforation, peritonitis, systemic inflammatory response syndrome, shock, and death. Progression may be rapid, but it is unusual for the disease to progress from mild to severe after 72 hr.
Table 96-1 SIGNS AND SYMPTOMS ASSOCIATED WITH NECROTIZING ENTEROCOLITIS
GASTROINTESTINAL
SYSTEMIC
From Kanto WP Jr, Hunter JE, Stoll BJ: Recognition and medical management of necrotizing enterocolitis, Clin Perinatol 21:335–346, 1994.
Diagnosis
A very high index of suspicion in treating preterm at-risk infants is crucial. Plain abdominal radiographs are essential to make a diagnosis of NEC. The finding of pneumatosis intestinalis (air in the bowel wall) confirms the clinical suspicion of NEC and is diagnostic; 50-75% of patients have pneumatosis when treatment is started (Fig. 96-4). Portal venous gas is a sign of severe disease, and pneumoperitoneum indicates a perforation (Figs. 96-4 and 96-5). Hepatic ultrasonography may detect portal venous gas despite normal abdominal roentgenograms.
Afrazi A, Sodhi CP, Richardson W, et al. New insights into the pathogenesis and treatment of necrotizing enterocolitis toll-like receptors and beyond. Pediatr Res. 2011;69(3):183-188.
Alfaleh K, Bassler D: Probiotics for prevention of necrotizing enterocolitis in preterm infants, Cochrane Database Syst Rev (1):CD005496, 2008.
Bagci S, Eis-Hubinger AM, Franz AR, et al. Detection of astrovirus in premature infants with necrotizing enterocolitis. Pediatr Infect Dis J. 2008;27:347-350.
Blakely ML, Tyson JE, Lally KP, et al. Laparotomy versus peritoneal drainage for necrotizing enterocolitis or isolated intestinal perforation in extremely low birth rate infants: outcomes through 18 mo adjusted age. Pediatrics. 2006;117:e680-e687.
Bombell S, McGuire W: Early trophic feeding for very low birth weight infants, Cochrane Database Syst Rev (3):CD000504, 2009.
Deshpande G, Rao S, Patole S, Balsara M. Updated meta-analysis of probiotics for preventing necrotizing enterocolitis in preterm neonates. Pediatrics. 2010;125(5):921-930.
Fanaroff AA, Stoll BJ, Wright LL, et al. Trends in neonatal morbidity and mortality for very low birthweight infants. Am J Obstet Gynecol. 2007;196:147.e1-147.e8.
Figueras-Aloy J, Rodriguez-Miguelez JM, Iriondo-Sanz M, et al. Intravenous immunoglobulin and necrotizing enterocolitis in newborns with hemolytic disease. Pediatrics. 2010;125:139-144.
Henderson G, Craig S, Brocklehurst P, et al. Enteral feeding regimens and necrotizing enterocolitis in preterm infants: a multicentre case control study. Arch Dis Child Fetal Neonatal Ed. 2009;94:F120-F123.
Hintz SR, Kendrick DE, Stoll BJ, et al. Neurodevelopment and growth outcomes of extremely low birth weight infants after necrotizing enterocolitis. Pediatrics. 2005;115:1-8.
Meinzen-Derr J, Morrow AL, Hornung RW, et al. Epidemiology of necrotizing enterocolitis temporal clustering in two neonatal practices. J Pediatr. 2009;154:656-661.
Meinzen-Derr J, Poindexter B, Wrage L, et al. Role of human milk in extremely low birth weight infants’ risk of necrotizing enterocolitis or death. J Perinatol. 2009;29:57-62.
Moss RL, Dimmitt RA, Barnhart DC, et al. Laparotomy versus peritoneal drainage for necrotizing enterocolitis and perforation. N Engl J Med. 2006;354:2225-2234.
Murdoch EM, Sinha AK, Shanmugalingam ST, et al. Doppler flow velocimetry in the superior mesenteric artery on the first day of life in preterm infants and the risk of neonatal necrotizing enterocolitis. Pediatrics. 2006;118:1999-2003.
Neu J, Walker WA. Necrotizing enterocolitis. N Engl J Med. 2011;364(3):255-264.
Pickard SS, Feinstein JA, Popat RA, et al. Short- and long-term outcomes of necrotizing enterocolitis in infants with congenital heart disease. Pediatrics. 2009;123:e901-e906.
Pietz J, Achanti B, Lilien L, et al. Prevention of necrotizing enterocolitis in preterm infants: a 20-year experience. Pediatrics. 2007;119:e164-e170.
Rees CM, Eaton S, Kiely EM, et al. Peritoneal drainage or laparotomy for neonatal bowel perforation? A randomized controlled trial. Ann Surg. 2008;248:44-51.
Sharma R, Tepas JJIII, Hudak ML, et al. Portal venous gas and surgical outcome of neonatal necrotizing enterocolitis. J Pediatr Surg. 2005;40:371-376.
Stoll BJ, Hansen NI, Adams-Chapman I, et al. Neurodevelopmental and growth impairment among extremely low-birth-weight infants with neonatal infection. JAMA. 2004;292:2357-2365.
Turcios-Ruiz RM, Axelrod PSt, John K, et al. Outbreak of necrotizing enterocolitis caused by norovirus in a neonatal intensive care unit. J Pediatr. 2008;153:339-344.
Young C, Sharma R, Handfield M, et al. Biomarkers for infants at risk for necrotizing enterocolitis: clues to prevention? Pediatr Res. 2009;65:91R-97R.
