Chapter 4 Differential Diagnosis of Surgical Disorders of the Spine
Establishing a differential diagnosis of spine pathology starts with the characterization of pain, associated signs and symptoms, and evaluation of any presenting neurologic deficit. Special attention must be paid to the warning signs and symptoms of back pain (Box 4-1), which helps to identify more serious pathology.1 Assessment of pain in conjunction with fever and weight loss, recumbent position, morning stiffness, acute onset, or visceral component allows for initial categorization. With this information, further laboratory and radiologic evaluation can proceed, and ultimately, a diagnosis with appropriate surgical or medical management can usually be achieved.
This chapter presents a systematic approach to evaluating a patient with a suspected spine disorder (Box 4-2). The first portion of this chapter addresses disorders that usually present with spinal pain, and the second half deals with conditions that present with pain and neurologic deficit.
BOX 4-2 Differential Diagnosis of Surgical Disorders of the Spine
Spinal Pain
Pain with fever and weight loss
Pain with recumbency and night pain
Neurologic Deficits
• Congenital lesions and spinal dysraphism
• Central montine myelinolysis
• Upper motor neuron syndromes
• Lower motor neuron syndromes
Spinal Pain
Pain Associated with Fever and Weight Loss
Vertebral Osteomyelitis
Vertebral osteomyelitis, the most common pyogenic infection of the axial skeleton, occurs in 2% to 19% of cases of osteomyelitis.2–4 Adults can present with an indolent or chronic course; the pediatric and immunocompromised groups can present more acutely. Diffuse back pain and fever are the most common symptoms, occurring in approximately 90% and 45% of patients, respectively.2–4 Weight loss, radicular symptoms, myelopathy, spine deformity, and meningeal irritation also occur. In some cases, neurologic deficits can be the presenting complaint.
A definitive source of infection is found in approximately 40% of cases. The most common organisms that are isolated are the gram-positive cocci, Staphylococcus aureus being the most common organism.2–4 Other organisms such as Escherichia coli, Pseudomonas aeruginosa, and Proteus are potential sources in parenteral drug abusers or immunocompromised patients.
Diagnosis is based on pertinent laboratory findings, including an elevated erythrocyte sedimentation rate, blood and bone cultures, and elevated white blood cell count. MRI is the gold standard for detection of osteomyelitis.2–5 Bone scans are useful for diagnosis, but care in interpretation is required, as other processes can have similar imaging qualities.
Epidural Abscess
Spinal epidural abscess occurs more frequently in adults. Pain is the most common presentation, but fever, leukocytosis, and neurologic compromise occur more frequently in epidural abscess than in osteomyelitis.4,6 Epidural abscesses most commonly affect the thoracic spine, followed by lumbar and cervical locations. Common etiologies include a direct extension of a preexisting osteomyelitis, hematogenous spread from a distant focus, or, less likely, trauma.4,6
As with vertebral osteomyelitis, the most prevalent species is S. aureus, followed by other staphylococcal and streptococcal species or gram-negative rods.4,6 Laboratory studies, including erythrocyte sedimentation rate and white blood cell count, are elevated in the majority of patients, and MRI is the diagnostic imaging of choice.4,6
Discitis
Spontaneous discitis is rare in the adult but occurs in 1% to 3% of surgical discectomy patients.7,8 Clinical presentation reveals back pain at the operated level, usually from 1 to 3 weeks postoperatively. The most common presentation is back pain and painful ambulation, as the lumbar spine is the most common location. Staphylococcal and streptococcal species are the most common organisms. Again, diagnosis is aided by laboratory studies, MRI, and bone scans.
Granulomatous Infections
Granulomatous infections include all processes that produce the classic histologic granuloma. These processes include fungal, spirochetal, and uncommon bacterial organisms (such as Actinomyces, Nocardia, and Brucella) and the most common organism, Mycobacterium tuberculosis.
Tuberculous Spondylitis
Although uncommon in developed countries, tuberculous spondylitis is the most common of the granulomatous infections that affect the axial skeleton.9–11 Recently, there has been a resurgence in developed countries due to the rise of HIV.12,13 Clinical presentation involves pain over the affected site, fever, malaise, and weight loss.9–11 In the progressive stages of disease, kyphosis results from erosive bone destruction. Epidural abscesses and paraparesis are possible late sequelae.9–11 Tuberculous spondylitis is usually caused by M. tuberculosis; however, other species of mycobacteria may be encountered. A positive purified protein derivative can be helpful, although false negatives can occur in the anergic patient due to advanced age, malnutrition, or immunocompromise. Diagnosis requires evaluation of urine, sputum, or a sample from a gastric specimen, subcutaneous nodule, or bone biopsy. A chest radiograph reveals no evidence of pulmonary disease in 40% to 50% of cases. MRI is superior to evaluate soft tissue involvement and the presence of abscess formation, and CT provides better bone detail.
Coccidioidomycosis
Coccidioidomycosis, endemic in the southwestern United States, has a high rate of spine involvement and occurs in 20% to 40% of cases of disseminated disease. Vertebral collapse and neurologic compromise are uncommon.14 Radiographs reveal multiple simultaneous lytic lesions. Diagnosis is made with plain radiographs, immunodiffusion titers, and biopsy.
Blastomycosis
This species is endemic to the Mississippi River Valley and is spread after inhalation and pulmonic infection. Blastomycosis is hematogenously spread with a predilection for ventral vertebral body involvement, resulting in vertebral collapse, joint erosion, and disc invasion. Clinical presentation resembles that of tuberculous spondylitis; however, blastomycosis more commonly is associated with draining sinuses and has a greater predisposition to include the dorsal elements.15,16
Cryptococcus
Cryptococcus neoformans is a fungal organism that may cause infection in immunocompromised patients, mostly commonly those afflicted with AIDS. It is usually inhaled and then spreads hematogenously from a pulmonary location, with osseous involvement occurring in only 10% of cases with disseminated disease.17,18 The usual clinical presentation is swelling, pain, and decreased mobility of the affected vertebral site. Radiographs reveal dorsal vertebral body involvement and disc space sparing. Diagnosis is made via a latex agglutination test, cerebrospinal fluid (CSF) analysis, and blood cultures.
Pain Associated with Recumbency and Night Pain
Benign Bone Tumors
Osteochondroma
These lesions are the most common benign bone tumors, encompassing approximately 35% of all nonmalignant osseous tumors. These tumors arise from the cartilaginous end plates and are slow-growing tumors.19,20 The majority are asymptomatic lumbar spine lesions found on incidental radiographs. Symptomatic patients commonly present with dull backache, decreased motion, or, rarely, deformity. Plain radiographs demonstrate a protruding lesion with well-demarcated borders in the dorsal elements. On rare occasions, pain, neurologic deficit, or an accelerated growth pattern may be related to malignant transformation.
Osteoid Osteoma and Osteoblastoma
Patients with osteoid osteomas commonly present with a dull ache that is exacerbated at night. This condition is believed to be the result of prostaglandin production by the tumor; thus, the classic pain relief with aspirin. Neurologic deficits are rare. Osteoblastomas are more likely to result in spinal deformity and neurologic sequelae, including torticollis in 13% of cervical lesions. Plain films are pathognomonic, revealing a small radiolucent nidus with surrounding sclerosis usually located in the dorsal elements.21,22
Giant Cell Tumor
Unlike the majority of primary bone tumors, giant cell tumors occur more commonly in patients in their 30s. The most common presentation is that of pain. However, disease advancement may result in bowel or bladder dysfunction. These aggressive tumors carry some malignant potential and a high incidence of local recurrence. They are responsible for approximately 10% of all primary benign bone tumors and affect the spinal axis in approximately 10% of all cases. These lesions may occur in conjunction with aneurysmal bone cysts (3% to 6%).23,24 They most commonly occur in the sacral region when the spinal column is involved. Plain radiographs demonstrate cortical expansion with little reactive sclerosis or periosteal reaction.23,24 Both T1- and T2-weighted MRI scans reveal homogeneous signals, whereas presurgical CT studies can better delineate the degree of vertebral bone involvement. Because of the nondistinct characteristics of giant cell tumors, radiographic investigation, coupled with intraoperative histology, is important to separate this condition from other primary bone tumors.
Aneurysmal Bone Cyst
Although responsible for only approximately 1% to 2% of all primary bone tumors, aneurysmal bone cysts affect the axial skeleton in 12% to 25% of reported cases of aneurysmal bone cysts.24 They occur more frequently in the thoracolumbar region and dorsal elements in females and patients younger than 20 years of age. Multiple vertebral involvement occurs in 40% of cases. Radiographs demonstrate a single osteolytic lesion with a thin, well-demarcated cortical rim.
Hemangioma
Hemangiomas are found in 11% of general autopsies,25,26 but symptomatic spinal hemangiomas are exceedingly rare. The most common initial symptom in the case of a solitary lesion is back pain, with or without radiation into the lower extremities.25,26 These lesions are characterized by slow growth and a female predominance.
Eosinophilic Granuloma
Eosinophilic granuloma is the solitary osseous lesion version of a group of disorders characterized by an abnormal proliferation of Langerhans cells. In its disseminated forms, it is designated Letterer-Siwe disease and Hand-Schüller-Christian disease. The overall incidence for any variety of the histiocytosis X spectrum is one per million people, and it most commonly occurs in patients younger than 20 years of age. Clinical presentation most commonly involves pain in the thoracolumbar region. MRI is the investigative procedure of choice, with definitive diagnosis through biopsy.27
Malignant Bone Tumors
Chondrosarcoma
This malignant cartilage-forming primary bone tumor is an uncommon spinal neoplasm. It is more common in adults, in whom it less commonly involves the spine.28 There is an even distribution of tumor involvement among cervical, thoracic, and lumbar locations.25 Chondrosarcomas may arise as a primary lesion or secondary to irradiation of lesions, including Paget disease or osteochondroma.29 The most common presentation is pain (50%) and localized swelling (30%). There is a linear relationship between degree of pain on presentation; a larger, more aggressive tumor; and decreased time of survival.30,31 Diagnosis is usually based on radiographic studies that reveal bone destruction, a soft tissue mass, and “fluffy” calcifications and pathology from resection.30,31
Osteogenic Sarcoma and Ewing Sarcoma
Both osteogenic sarcoma and Ewing sarcoma represent uncommon malignant lesions of the spinal column, with a combined incidence of less than 4% of spinal column tumors.32–34 Most cases of Ewing sarcoma and primary osteogenic sarcoma (50%) present in the first 20 years of life. Secondary sarcomas arise in the fifth to sixth decades as a result of irradiated bone or a preexisting pagetoid lesion. Almost 70% of clinical presentations are accompanied by a neurologic deficit secondary to epidural compression.32–34 The most common presentation of Ewing sarcoma is pain.
Chordoma
Chordomas are tumors of the axial skeleton and the skull base arising from the primitive notochord. They encompass approximately 1.4% of all skeletal sarcomas. Although chordomas are histologically low-grade lesions, they are locally invasive tumors, and metastases may occur in 5% to 43% of cases.35–37 More than 50% of these lesions are located in the lumbosacral region, 35% are located in the clival and cervical area, and the remainder are spread throughout the rest of the vertebral column.37 Neurologic deficit is usually found in the form of bowel/bladder dysfunction or, less frequently, cauda equina symptoms (20%).37 Combined imaging, using MRI and CT, provides an evaluation of the tumor and its soft tissue and bony involvement.
Multiple Myeloma
Multiple myeloma and solitary plasmacytoma account for 45% of all malignant bone tumors.38 These disorders are the result of abnormal proliferation of plasma cells, which are responsible for immunoglobulin and antibody production and affect the spine in 30% to 50% of reported cases. Multiple myeloma is primarily a disease of the sixth and seventh decades of life and has a predilection for the thoracic spine (50% to 60%).
Patients present with back pain in approximately 75% of cases.38 Unlike the classic metastatic disease presentation of pain with recumbency, multiple myeloma is sometimes relieved by rest and aggravated by mechanical agitation that mimics other sciatic or neurogenic sources. Systemic complications include hyperalbuminemia, renal insufficiency, nephrolithiasis, and characteristic serum protein abnormalities. Plain radiographs and CT can be diagnostic because of the characteristic osteolytic picture without sclerotic edges that involve the ventral portion of the vertebral body and usually spare the dorsal elements.
Lymphoma
Hodgkin disease is a malignant disease of the reticuloendothelial system. Spine involvement occurs in approximately 10% of all extranodal lymphomas.39,40 Spine osseous involvement occurs at a decreasing frequency as one ascends the spine from the lumbar, thoracic, and, uncommonly, cervical regions. Age at presentation is bimodal, with those ages 15 to 35 and those older than age 50 most frequently affected. Clinical presentation involves concurrent constitutional signs and symptoms of fever and night sweats, and acute cord compression and epidural compression are not uncommon.39,40
Metastatic Disease
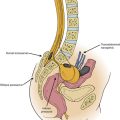
Metastatic disease in the form of distant foci is evident at autopsy in 40% to 85% of cases of malignancy.41 The spine is the most common site of skeletal metastasis, and at least 5% of patients with malignancies suffer from this condition.41,42 The axial skeleton is the leading site of bone metastases that are caused by hematogenous spread through the rich venous network that drains the lungs, pelvis, and thorax. Breast, lung, prostate, and thyroid malignancies account for 50% to 60% of metastatic lesions.41 Overall, epidural metastases are equally spread throughout the thoracic and lumbosacral spine, but symptomatic metastases occur most commonly in the thoracic spine. Nearly all patients initially complain of back pain, followed by weakness and ataxia. At the time of diagnosis, more than 50% of patients will have a paraparesis or bladder/bowel disturbance.41,43
Spinal Cord Tumors
The majority of lesions that involve the spinal cord and meninges occur in the epidural space in the form of metastatic disease. The largest group of neoplastic spinal lesions that involve the spinal cord and meninges occurs in the intradural-extramedullary space (40–50%), followed by the extradural space (30%) and the intramedullary space (20–25%).43,44
Intradural-Extramedullary Lesions
Meningiomas, schwannomas, and neurofibromas constitute more than 50% of all neoplastic processes in the intradural-extramedullary space. Nittner’s review of 4885 adults with spinal cord tumors found schwannomas (23%) and meningiomas (22%) to be the most common lesions of the intradural-extramedullary space.45,46 Symptoms may be nocturnal and most commonly involve pain caused by root irritation. Early neurologic compromise is uncommon because of the adaptive compressibility of surrounding fat, CSF, and adjacent vascular structures. Neurologic compromise occurs when the compliance of surrounding structures is at its nadir and extradural compression is directly transmitted to the spinal cord.
More than 80% of meningiomas are located in the thoracic region, and they occur at a 4:1 ratio in women compared to men. Meningiomas can present with pain from a compressed nerve root as it exits the neural foramina. Although less common in the cervical and lumbar spine, large, slow-growing meningiomas may produce myelopathic symptoms from spinal cord compression, especially at the craniocervical junction.47 Meningiomas are the most common benign tumor at the foramen magnum.48,49 CT myelogram and MRI are the best investigative modalities.
Although both meningiomas and nerve sheath tumors are benign lesions that are usually found in thoracic dorsal sites, neurofibromas are a common finding in phakomatoses. Because neurofibromas are almost always lesions of the dorsal roots, patients commonly present with radicular symptoms.50–52 Although their malignancy potential is low, nerve sheath tumors may be locally destructive if allowed to progress. Caudally located neurofibromas may displace adjacent nerve roots with possible bone erosion of nearby foramina as the neoplasm grows.
The remaining 30% of intradural-extramedullary tumors include sarcomas, dermoids, epidermoids, arachnoid cysts, teratomas, ganglion cysts, and, rarely, spinal metastases.53,54 These lesions have characteristic features on MRI that help to delineate them. Arachnoiditis that presents with diffuse constant pain and associated paresthesias is the result of multiple operations on the back or clumping of nerve roots after the administration of the myelographic dye. The diagnosis is made via MRI or myelogram with visualization of characteristic nerve root clumping.
Intradural-Intramedullary Tumors
Intramedullary spinal cord tumors account for 2% to 4% of CNS neoplasms and are of neuroglial origin in 80% of cases, regardless of age.55–58 More than 90% of these tumors are rostral to the conus in patients under age 15.55–59 Children are predisposed to astrocytic tumors, whereas adult pathology is more evenly spread over the neuroglial spectrum.55–59 There is a shift in pathology with increasing age, with ependymomas becoming more common than astrocytomas. The incidence of intramedullary spinal cord tumors increases from rostral to caudal and may present with insidious pain, the most common finding in the adult population, or associated spinal cord dysfunction in the form of band paresthesias or motor deficit. Typically, the pain associated with these lesions is unrelated to mechanical activity. Pediatric patients tend to present with gait or motor disturbances.55–59
Pain Associated with Morning Stiffness
Ankylosing Spondylitis
AS is the most prevalent of the seronegative spondyloarthropathies, with an incidence of up to 2% in the Caucasian population. It is a common cause of axial pain in young adults.60–62 Unlike RA, it has a male predominance, and it is most commonly found in the axial skeleton with a mild degree of peripheral involvement. The pathogenesis is unclear, but there is a strong immunologic association with HLA-B27 positivity in approximately 95% of patients. The disease progresses in an ascending fashion from caudal to rostral, which can result in severe flexion deformity if allowed to continue.60,62 The prototypical lesion is enthesopathic, affecting insertion sites of tendons and ligaments to bone. The typical presentation is that of a young white male between ages 15 and 30, with insidious low back pain (LBP) (80–90%), peripheral joint pain in the hip or shoulder (20–40%), and sciatic pain (5%).60,62 Diagnosis is based on a history of back pain and grades 3 to 4 bilateral sacroiliitis observed on plain radiographs. There have been several revisions of the original criteria for AS, but all accept the radiologic changes with a history of insidious onset of back pain, age younger than 40, persistence for more than 3 months, morning stiffness, improvement with exercise, and limitation of chest expansion.60–63 Because it takes from 3 to 7 years for the radiographic evidence of bilateral symmetrical sacroiliitis to become evident, a loss of axial mobility, back pain, and morning stiffness are important early signs and symptoms.60–63 Associated fractures, spinal stenosis, and rotary instability are the end result of a fused vertebral column.62,64–66
Rheumatoid Arthritis
RA, a chronic inflammatory process that affects the synovium of peripheral joints, has a quoted prevalence of 1% for both genders by age 65, but is an uncommon cause of back pain. Unlike AS, this disease affects an older patient population, has a female predominance, is found most often in the cervical spine, and often results in spinal instability.67–71 RA affects the cervical spine most commonly in one of three ways: atlantoaxial subluxation, basilar invagination, and subaxial subluxation.67–71 Diagnosis of RA is based on the history, the distribution of joint involvement, and a positive rheumatoid factor. Neck pain should warrant a thorough radiographic evaluation, including flexion/extension radiographs and MRI for ligamentous visualization. Radiographic sequelae include soft tissue swelling, narrowing of joint spaces, and, ultimately, bone erosion.
Mechanical Pain
Anywhere from 40% to 80% of the adult population has LBP sometime before age 50.1 Ninety percent of cases are a result of mechanical causes. Pain without constitutional signs and symptoms that is initiated and exacerbated by activity is a large category that includes lumbar strain, disc protrusion and extrusion, spinal stenosis, spondylolisthesis, spondylolysis, and soft tissue irritation disorders, such as those in the piriform syndrome. Other entities such as sacroiliac joint dysfunction, facet syndrome, dural ectasia, perineural or ganglion cysts, and collagen disorders (Ehlers-Danlos syndrome) are less well-differentiated causes of LBP and are usually clinically diagnosed. To evaluate degenerative spine disorders, it is necessary to determine the character of pain, whether it be LBP alone or associated with radicular symptoms, symptomatic neurogenic claudication, or, rarely, myelopathy. Clinical history of onset and duration of symptoms, age, presence of a congenital disorder, and spinal deformity help to differentiate among the more common degenerative lesions. MRI and CT/myelogram are most commonly used to evaluate degenerative spine disorders.
Spinal Stenosis
Whether acquired, as in the elderly, or congenital (e.g., in the achondroplastic dwarf), spinal stenosis has a common clinical presentation.72 The classic bilateral low back, buttock, and thigh pain, consistent with neurogenic claudication associated with activity, can be present whether the patient is standing (94%) or has walked a short distance.72 Neurogenic claudication must be differentiated from vascular claudication. The clinical picture of vascular claudication reveals progressive calf pain after ambulation, with associated decreased peripheral pulses and chronic tissue changes seen in cool distal extremities. Spinal stenosis is a clinical entity with radiologic confirmation of a decreased spinal canal observed on axial MRI or CT/myelogram views.
Spondylolisthesis and Spondylolysis
Spondylolisthesis and spondylolysis are common causes of back pain in both the pediatric and adult population, with L5 the most common site of involvement.72–74 The adult population tends to have a more vague and insidious presentation, with back pain as the most common complaint, followed by claudication and hamstring tightness, probably caused by concurrent spinal stenosis. Approximately 20% have spine deformity that can be detected on physical examination.
Herniated Nucleus Pulposus
Herniated nucleus pulposus is a common cause of radicular pain in adults ages 30 to 40. Only 35% of those who present with a herniated nucleus pulposus experience sciatica. The pain is usually sharp and follows a dermatomal pattern. Diagnosis includes clinical findings consistent with the affected nerve root in the form of sensory, reflex, or motor deficits.7,8
Scoliosis
Scoliosis represents another potential cause of back pain in adults who suffer from LBP. Lumbar degenerative scoliosis with a Cobb angle greater than 10 degrees is reportedly present in approximately 7.5% of the adult back pain population, with an increasing prevalence with age.75–80 As age increases, the proportion of women who have scoliosis as a cause of both back pain and radicular symptoms increases.75–80
Neurologic Deficits
Congenital Lesions
In the majority of significant neural tube developmental disorders, a physical examination at birth reveals a spine defect, with or without neurologic dysfunction. Other disorders such as tethered cord or congenital scoliosis may remain occult until symptoms present, secondary to spinal column growth. These lesions will be discussed further in Chapter 7.
Trauma
Posttraumatic syringomyelia should be included in the differential diagnosis of any patient who develops deterioration of motor function with an ascending sensory level after traumatic quadriparesis or paraparesis. Approximately 11% of all cases of syringomyelia are reported to be caused by trauma, whereas 3% of cases with severe cervical trauma with paraplegia/quadriplegia are said to result in posttraumatic syringomyelia.81,82 Its course of symptom development ranges from 2 months to 36 years. It is found most often in the thoracolumbar region. Clinical presentation involves pain, ascending sensory level, motor deficits, and loss of reflexes above the previous lesion. MRI is the imaging procedure of choice to evaluate for a posttraumatic syrinx.
Vascular Lesions
Ischemia
Individuals with circulatory insufficiency in the legs may harbor disease of the abdominal aorta with resultant spinal cord ischemia. Thromboembolic occlusion of spinal segmental arteries (e.g., the artery of Adamkiewicz) and dissection, clamping, or severe atheroma of the aorta are the most common causes of spinal cord infarction.83 The anterior cord syndrome is a typical clinical presentation of ischemic spinal cord insult. The midthoracic level is the most common site of ischemia because it lies in a vascular watershed zone.
In the less common cases of painless infarction of the spinal cord caused by systemic hypotension, low thoracic and lumbosacral spinal cord central gray matter involvement is observed. Vasculitis and systemic embolism are rare causes of spinal cord ischemia. Polyarteritis nodosa and primary granulomatous angiitis, a neural vasculature disorder without systemic involvement often found with lymphoma, are rare causes of a sometimes painful acute or chronic myelopathy.84,85 Among the vascular causes of paraplegia and quadriplegia, anterior spinal artery thrombosis is the most common. Although occlusion of the anterior spinal artery is uncommon, ischemia, in its region of supply, occurs relatively often. This is usually caused by disease of the aorta or segmental branches that supply the anterior spinal artery. The anterior spinal artery syndrome, also known as anterior cord syndrome, consists of motor paralysis (upper and lower motor neuron), dissociated sensory loss (pain and temperature), and sphincter paralysis. It results from an infarction in the region of the anterior spinal artery that supplies the vertical two thirds of the spinal cord and is usually the consequence of thrombotic atherosclerotic disease, aortic dissection, embolization, or vasculitis (particularly polyarteritis nodosa). The posterior columns are usually spared, which aids in the diagnosis. This syndrome may result as a complication of aortic angiography, cross-clamping of the aorta for more than 30 minutes, or spine trauma with resulting direct compression of the ventral spinal cord and adjacent vessels.86,87 Spinal hemorrhages are usually apoplectic in nature, with rapidly developing paralysis and sensory loss. They may occur within the epidural or subdural spaces or within the spinal cord. Trauma, anticoagulant therapy, and vascular malformation are the primary causes.
Vascular Malformations
Spinal vascular malformations are an uncommon cause of neurologic deficit, representing only 10% of spinal epidural hemorrhages.88,89 More commonly, spinal intradural and extradural malformations present with chronic progressive myelopathy or radiculopathy. Spinal vascular malformations are usually divided into three groups: dural arteriovenous fistulas, intradural vascular malformations, and cavernous angiomas. A vascular malformation infrequently (<3% of cases) may produce an audible bruit over the spinal cord. Dural arteriovenous fistulas occur most often in patients over age 40 and may be exacerbated by changes in posture or activity. These lesions almost always affect the lower half of the spinal cord and produce symptoms in the legs, bladder, and bowel. In contrast, patients with intradural vascular malformations become symptomatic before the age of 40 and often present with an acute onset of symptoms caused by hemorrhage.89–92
MRI has replaced myelography as the initial diagnostic study to evaluate these patients; intradural spinal AVMs present as serpentine areas of low signal intensity in the subarachnoid space as a result of signal voids produced by blood flowing in the dilated tortuous vessels. T1-weighted MRI images of intramedullary AVMs usually reveal a low-intensity signal that may be associated with focal widening of the cord. In contrast to MRI, myelography findings are universally abnormal in these fistulas and demonstrate the presence of the lesion, with the exception of cavernous angiomas. In the search for a spinal AVM with a negative MRI and myelogram, arteriography would rarely be indicated. Spine arteriography, however, should be performed in all patients with spinal AVMs that have been diagnosed by means of other studies.93–95
Histologically similar to their intracranial counterpart, cavernous angiomas are intramedullary lesions that are characterized clinically by sensorimotor disturbances over an acute or subacute period. These rare lesions of the spinal cord are characterized by acute neurologic dysfunction with intervening episodes of varying recovery.96–98 They are found most often in thoracic and cervical locations. Cavernous angiomas may not be apparent on findings from myelography, CT imaging, or spine arteriography. MRI remains the investigative procedure of choice, usually revealing residual blood of subacute and chronic hemorrhage, characterized by mixed high- and low-signal components.97
Foix-Alajouanine syndrome is a rare form of necrotic myelopathy that results in slowly evolving myotrophic paraplegia in adult males. It has been attributed to spinal venous thrombosis, although its exact nature remains controversial.99,100
Demyelinating Lesions
Multiple Sclerosis
The clinically definite diagnosis of MS requires the presence of six items:
2. Two or more sites of CNS involvement
3. Predominant white matter involvement
4. Relapsing-remitting or chronic (>6 months) progressive course
Poser et al.101 modified these criteria by enhancing the clinical diagnosis with laboratory studies that include analysis of the spinal fluid, evoked potentials, and imaging studies.
Among neuroimaging studies, MRI is the modality of choice to confirm the diagnosis. In general, the MRI scan is positive in 85% to 95% of clinically definite MS patients.102 The clinical diagnosis is supported by laboratory studies, including CSF examination, which may reveal a lymphocytic pleocytosis (usually fewer than 25 cells/mm3), and normal or increased protein. Oligoclonal bands, lymphocytic reactivity to myelin basic protein, and an elevated IgG/Alb ratio are other laboratory findings that can support a diagnosis of MS.
Transverse Myelitis
Transverse myelitis is a nonhomogeneous group of idiopathic inflammatory processes defined as isolated spinal cord dysfunction over hours or days in patients who demonstrate no evidence of a compressive lesion.103,104 Transverse myelitis can occur in acute, subacute, or chronic forms. Only the acute forms are discussed here. Transverse myelitis caused by other etiologies usually follows a longer time course and is discussed in later text.
Acute transverse myelitis can be subdivided into the autoimmune and necrotizing types. They are differentiated by an acute versus a subacute time course and associated illness. Autoimmune acute transverse myelitis usually occurs after a viral illness or in association with other autoimmune disorders, such as MS or lupus erythematosus. In several reviews of this process, 37% of patients reported a preceding febrile illness. The initial symptoms were paresthesias, back pain, or leg weakness. The maximal neurologic deficit develops within 10 days in the majority of cases.104–106 Patients with partial myelitis may have a higher frequency of subsequently developing MS.106 Acute transverse myelitis has been associated with systemic vasculitis, as in systemic lupus erythematosus, as well as with heroin abuse. Symptoms occur over days to weeks, most commonly in the thoracic spinal cord. Symptoms include ascending paresthesias, weakness, and urinary retention.
Necrotizing acute transverse myelitis (Foix-Alajouanine syndrome) is an acutely progressive necrotizing myelitis that occurs over hours to days.99,100,107 Clinical manifestations in the typical patient of adult years consist of severe paralysis preceded by tingling or loss of sphincter control. During the acute phase, MRI is normal in approximately half of the cases and is nonspecific in the remainder. Focal spinal cord enlargement on T1-weighted and poorly delineated hyperintensities on T2-weighted scans are the most commonly identified abnormalities. Occasionally, contrast enhancement is observed. Diagnosis is based on the clinical picture and absence of other potential causes of acute myelopathy on MRI, such as acute disc herniation hematoma, epidural abscess, or compression myelopathy.
Degenerative Disorders
Lower Motor Neuron Syndromes
Proximal SMAs account for nearly 80% of all SMA cases. Type I, acute infantile SMA (Werdnig-Hoffmann disease) is a progressive disease of infancy that accounts for about 25% of all SMA cases. Usually transmitted by an autosomal recessive gene, this condition presents in a third of the cases that demonstrate decreased fetal movements in the last trimester of pregnancy. The majority of affected infants are floppy at birth. The disease is almost uniformly fatal, usually in the sixth or seventh month of life. About 95% of affected children die by the age of 18 months.108
Combined Upper and Lower Motor Neuron Syndromes
Amyotrophic lateral sclerosis (also known as Charcot disease or motor neuron disease) is found in adults and results from degeneration of the upper motor neuron and lower motor neuron.109,110 The prevalence of amyotrophic lateral sclerosis is four to six individuals per 100,000, and it is familial in 8% to 10% of cases. Familial cases usually follow autosomal dominant inheritance but occasionally demonstrate a recessive pattern.109–111
Miscellaneous Disorders
Subacute Combined Degeneration
Subacute combined degeneration of the spinal cord, caused by a deficiency of vitamin B12, is uncommon today because of the relative ease of diagnosis and treatment. However, when B12 levels are reduced for a prolonged period, neurologic sequelae ensue shortly after the anemia. Clinically, this condition presents with both sensory and motor symptoms consistent with thoracic dorsal column involvement, including paresthesias in the feet and loss of vibratory and positional sense. Diagnosis is made through laboratory studies that demonstrate a decreased B12 level and a neurologic examination that is consistent with a posterolateral syndrome. Treatment is with vitamin B12112,113 Incomplete paraplegia or quadriplegia may accompany myasthenia gravis, an autoimmune disease caused by a defect in neuromuscular transmission with an incidence of 3 per 100,000. Ocular, motor, and bulbar involvement, as well as preserved sensation, often point to the correct diagnosis. A rather stable, nonprogressive myelopathy is observed in degenerative spinal cord diseases, such as hereditary spastic paraplegia or spastic diplegia of cerebral palsy.
Guillain-Barré Syndrome
This syndrome is the most common acquired demyelinating neuropathy, characterized by an acute onset of peripheral nerve dysfunction, usually after a viral illness. It presents clinically with symmetrical limb weakness and/or paresthesias.114 This disease is distinguished from the aforementioned causes of peripheral neuropathies by a history of toxin exposure or ingestion and its tendency to affect proximal muscles initially.
Familial Periodic Paralysis
Diseases that affect primary muscles are rarely acute in their onset. However, in so-called periodic paralysis, attacks of generalized muscle weakness may evolve over minutes to hours. The patient with familial periodic paralysis usually has a medical history of similar attacks or a positive family history. This condition, which is associated with disturbances of serum potassium, is a disease of the young, with initial attacks occurring around puberty.115 It is extremely rare, with only a few cases being reported each year. Clinically, patients present with weakness or paralysis of either the legs or all muscle groups, usually after a period of rest.
Harrop J.S., Schmidt M.H., Boriani S., Shaffrey C.I. Aggressive “benign” primary spine neoplasms: osteoblastoma, aneurysmal bone cyst, and giant cell tumor. Spine (Phila Pa 1976). 2009;34:S39-S47.
Oldfield E.H. Introduction: Spinal vascular malformations. Neurosurg Focus. 2009;26:E1.
Shen F.H., Samartzis D., Jenis L.G., An H.S. Rheumatoid arthritis: evaluation and surgical management of the cervical spine. Spine J. 2004;4:689-700.
Smith J.S., Fu K.M., Urban P., Shaffrey C.I. Neurological symptoms and deficits in adults with scoliosis who present to a surgical clinic: incidence and association with the choice of operative versus nonoperative management. J Neurosurg Spine. 2008;9:326-331.
Weinstein J.N., Tosteson T.D., Lurie J.D., et al. Surgical vs nonoperative treatment for lumbar disk herniation: the Spine Patient Outcomes Research Trial (SPORT): a randomized trial. JAMA. 2006;296:2441-2450.
Yang S., Yang X., Hong G. Surgical treatment of one hundred seventy-four intramedullary spinal cord tumors. Spine (Phila Pa. 2009;1976)(34):2705-2710.
Zimmerli W. Clinical practice. Vertebral osteomyelitis. N Engl J Med. 2010;362:1022-1029.
1. Deyo R.A., Weinstein J.N. Low back pain. N Engl J Med. 2001;344:363-370.
2. Dirschl D.R., Almekinders L.C. Osteomyelitis: common causes and treatment recommendations. Drugs. 1993;45:29-43.
3. Hitchon P.W., Osenbach R.K., Yuh W.T., Menezes A.H. Spinal infections. Clin Neurosurg. 1992;38:373-387.
4. Zimmerli W. Clinical practice. Vertebral osteomyelitis. N Engl J Med. 2010;362:1022-1029.
5. Dagirmanjian A., Schils J., McHenry M.C. MR imaging of spinal infections. Magn Reson Imaging Clin N Am. 1999;7:525-538.
6. Darouiche R.O. Spinal epidural abscess. N Engl J Med. 2006;355:2012-2020.
7. Weinstein J.N., Lurie J.D., Tosteson T.D., et al. Surgical vs nonoperative treatment for lumbar disk herniation: the Spine Patient Outcomes Research Trial (SPORT) observational cohort. JAMA. 2006;296:2451-2459.
8. Weinstein J.N., Tosteson T.D., Lurie J.D., et al. Surgical vs nonoperative treatment for lumbar disk herniation: the Spine Patient Outcomes Research Trial (SPORT): a randomized trial. JAMA. 2006;296:2441-2450.
9. Jain A.K., Dhammi I.K. Tuberculosis of the spine: a review. Clin Orthop Relat Res. 2007;460:39-49.
10. Pawar U.M., Kundnani V., Agashe V., Nene A. Multidrug-resistant tuberculosis of the spine—is it the beginning of the end? A study of twenty-five culture proven multidrug-resistant tuberculosis spine patients. Spine (Phila Pa 1976). 2009;34:E806-E810.
11. Tuli S.M. Tuberculosis of the spine: a historical review. Clin Orthop Relat Res. 2007;460:29-38.
12. Chaisson R.E., Martinson N.A. Tuberculosis in Africa: combating an HIV-driven crisis. N Engl J Med. 2008;358:1089-1092.
13. Steinbrook R. Tuberculosis and HIV in India. N Engl J Med. 2007;356:1198-1199.
14. Lewicky Y.M., Roberto R.F., Curtin S.L. The unique complications of coccidioidomycosis of the spine: a detailed time line of disease progression and suppression. Spine (Phila Pa 1976). 2004;29:E435-E441.
15. Hadjipavlou A.G., Mader J.T., Nauta H.J., et al. Blastomycosis of the lumbar spine: case report and review of the literature, with emphasis on diagnostic laboratory tools and management. Eur Spine J. 1998;7:416-421.
16. Mahiquez M., Bunton K.L., Carney G., et al. Nonsurgical treatment of lumbosacral blastomycosis involving L2–S1: a case report. Spine (Phila Pa 1976). 2008;33:E442-E446.
17. Aganovic L., Hoda R.S., Rumboldt Z. Hyperintensity of spinal Cryptococcus infection on diffusion-weighted MR images. AJR Am J Roentgenol. 2004;183:1176-1177.
18. Grosse P., Tintelnot K., Sollner O., Schmitz B. Encephalomyelitis due to Cryptococcus neoformans var gattii presenting as spinal tumour: case report and review of the literature. J Neurol Neurosurg Psychiatry. 2001;70:113-116.
19. Jose Alcaraz Mexia M., Izquierdo Nunez E., Santonja Garriga C., et al. Osteochondroma of the thoracic spine and scoliosis. Spine (Phila Pa 1976). 2001;26:1082-1085.
20. Sharma M.C., Arora R., Deol P.S., et al. Osteochondroma of the spine: an enigmatic tumor of the spinal cord: a series of 10 cases. J Neurosurg Sci. 2002;46:66-70. discussion 70
21. Kayser F., Resnick D., Haghighi P., et al. Evidence of the subperiosteal origin of osteoid osteomas in tubular bones: analysis by CT and MR imaging. AJR Am J Roentgenol. 1998;170:609-614.
22. Zileli M., Cagli S., Basdemir G., Ersahin Y. Osteoid osteomas and osteoblastomas of the spine. Neurosurg Focus. 2003;15:E5.
23. Anchan C. Giant cell tumor of bone with secondary aneurysmal bone cyst. Int J Shoulder Surg. 2008;2:68.
24. Harrop J.S., Schmidt M.H., Boriani S., Shaffrey C.I. Aggressive “benign” primary spine neoplasms: osteoblastoma, aneurysmal bone cyst, and giant cell tumor. Spine (Phila Pa 1976). 2009;34:S39-S47.
25. Aich R.K., Deb A.R., Banerjee A., et al. Symptomatic vertebral hemangioma: treatment with radiotherapy. J Cancer Res Ther. 2010;6:199-203.
26. Barzin M., Maleki I. Incidence of vertebral hemangioma on spinal magnetic resonance imaging in Northern Iran. Pak J Biol Sci. 2009;12:542-544.
27. Reddy P.K., Vannemreddy P.S., Nanda A. Eosinophilic granuloma of spine in adults: a case report and review of literature. Spinal Cord. 2000;38:766-768.
28. Quiriny M., Gebhart M. Chondrosarcoma of the spine: a report of three cases and literature review. Acta Orthop Belg. 2008;74:885-890.
29. Ibanez R., Gutierrez R., Fito C., et al. Chondrosarcoma in Paget’s disease of bone. Clin Exp Rheumatol. 2005;23:275.
30. Riedel R.F., Larrier N., Dodd L., et al. The clinical management of chondrosarcoma. Curr Treat Options Oncol. 2009;10:94-106.
31. Slater G., Huckstep R.L. Management of chondrosarcoma. Aust N Z J Surg. 1993;63:587-589.
32. Bacci G., Boriani S., Balladelli A., et al. Treatment of nonmetastatic Ewing’s sarcoma family tumors of the spine and sacrum: the experience from a single institution. Eur Spine J. 2009;18:1091-1095.
33. Levine A.M., Coleman C., Horasek S. Stereotactic radiosurgery for the treatment of primary sarcomas and sarcoma metastases of the spine. Neurosurgery. 2009;64:A54-A59.
34. Schaser K.D., Melcher I., Luzzati A., Disch A.C. Bone sarcoma of the spine. Recent Results Cancer Res. 2009;179:141-167.
35. Kamel M.H., Lim C., Kelleher M., et al. Intracranial metastasis from a sacrococcygeal chordoma: case report. J Neurosurg. 2005;102:730-732.
36. Martin M.P., Olson S. Intradural drop metastasis of a clival chordoma. J Clin Neurosci. 2009;16:1105-1107.
37. McMaster M.L., Goldstein A.M., Bromley C.M., et al. Chordoma: incidence and survival patterns in the United States, 1973–1995. Cancer Causes Control. 2001;12:1-11.
38. Sirohi B., Powles R. Multiple myeloma. Lancet. 2004;363:875-887.
39. Becker S., Babisch J., Venbrocks R., et al. Primary non-Hodgkin lymphoma of the spine. Arch Orthop Trauma Surg. 1998;117:399-401.
40. Peng X., Wan Y., Chen Y., et al. Primary non-Hodgkin’s lymphoma of the spine with neurologic compression treated by radiotherapy and chemotherapy alone or combined with surgical decompression. Oncol Rep. 2009;21:1269-1275.
41. Bartels R.H., van der Linden Y.M., van der Graaf W.T. Spinal extradural metastasis: review of current treatment options. CA Cancer J Clin. 2008;58:245-259.
42. Guillevin R., Vallee J.N., Lafitte F., et al. Spine metastasis imaging: review of the literature. J Neuroradiol. 2007;34:311-321.
43. Schiff D., O’Neill B.P., Suman V.J. Spinal epidural metastasis as the initial manifestation of malignancy: clinical features and diagnostic approach. Neurology. 1997;49:452-456.
44. Chi J.H., Parsa A.T. Intramedullary spinal cord metastasis: clinical management and surgical considerations. Neurosurg Clin N Am. 2006;17:45-50.
45. Masaryk T.J. Neoplastic disease of the spine. Radiol Clin North Am. 1991;29:829-845.
46. Simeone F.A. Intradural tumors. In: Rothman RH, Simeone F.A., editors. The spine. Philadelphia: WB Saunders; 1992:1515-1528.
47. Samii M., Klekamp J., Carvalho G. Surgical results for meningiomas of the craniocervical junction. Neurosurgery. 1996;39:1086-1094. discussion 1094–1085
48. Silva D.O., Costa L.F., Kitamura M.A., et al. Surgical treatment of ventral foramen magnum meningiomas. Arq Neuropsiquiatr. 2009;67:1111-1113.
49. Wu Z., Hao S., Zhang J., et al. Foramen magnum meningiomas: experiences in 114 patients at a single institute over 15 years. Surg Neurol. 2009;72:376-382. discussion 382
50. Vancoillie P., Veiga-Pires J.A. Cervical neurofibroma and generalised spinal stenosis in von Recklinghausen disease. Lancet. 1979;2(8154):1246-1247.
51. Kluwe L., Tatagiba M., Funsterer C., Mautner V.F. NF1 mutations and clinical spectrum in patients with spinal neurofibromas. J Med Genet. 2003;40:368-371.
52. Pollack I.F., Colak A., Fitz C., et al. Surgical management of spinal cord compression from plexiform neurofibromas in patients with neurofibromatosis 1. Neurosurgery. 1998;43:248-255. discussion 255–256
53. Ahn D.K., Park H.S., Choi D.J., et al. The surgical treatment for spinal intradural extramedullary tumors. Clin Orthop Surg. 2009;1:165-172.
54. Arnautovic K., Arnautovic A. Extramedullary intradural spinal tumors: a review of modern diagnostic and treatment options and a report of a series. Bosn J Basic Med Sci. 2009;9(Suppl 1):40-45.
55. Garces-Ambrossi G.L., McGirt M.J., Mehta V.A., et al. Factors associated with progression-free survival and long-term neurological outcome after resection of intramedullary spinal cord tumors: analysis of 101 consecutive cases. J Neurosurg Spine. 2009;11:591-599.
56. Lowe G.M. Magnetic resonance imaging of intramedullary spinal cord tumors. J Neurooncol. 2000;47:195-210.
57. Yang S., Yang X., Hong G. Surgical treatment of one hundred seventy-four intramedullary spinal cord tumors. Spine (Phila Pa 1976). 2009;34:2705-2710.
58. Yao K.C., McGirt M.J., Chaichana K.L., et al. Risk factors for progressive spinal deformity following resection of intramedullary spinal cord tumors in children: an analysis of 161 consecutive cases. J Neurosurg. 2007;107:463-468.
59. Houten J.K., Weiner H.L. Pediatric intramedullary spinal cord tumors: special considerations. J Neurooncol. 2000;47:225-230.
60. Dakwar E., Reddy J., Vale F.L., Uribe J.S. A review of the pathogenesis of ankylosing spondylitis. Neurosurg Focus. 2008;24:E2.
61. Mathieu A., Cauli A., Fiorillo M.T., Sorrentino R. HLA-B27 and ankylosing spondylitis geographic distribution as the result of a genetic selection induced by malaria endemic? A review supporting the hypothesis. Autoimmun Rev. 2008;7:398-403.
62. Thumbikat P., Hariharan R.P., Ravichandran G., et al. Spinal cord injury in patients with ankylosing spondylitis: a 10-year review. Spine (Phila Pa 1976). 2007;32:2989-2995.
63. Zochling J., van der Heijde D., Dougados M., Braun J. Current evidence for the management of ankylosing spondylitis: a systematic literature review for the ASAS/EULAR management recommendations in ankylosing spondylitis. Ann Rheum Dis. 2006;65:423-432.
64. Hitchon P.W., From A.M., Brenton M.D., et al. Fractures of the thoracolumbar spine complicating ankylosing spondylitis. J Neurosurg. 2002;97:218-222.
65. Kanter A.S., Wang M.Y., Mummaneni P.V. A treatment algorithm for the management of cervical spine fractures and deformity in patients with ankylosing spondylitis. Neurosurg Focus. 2008;24:E11.
66. Zdichavsky M., Blauth M., Knop C., et al. [Ankylosing spondylitis. Therapy and complications of 34 spine fractures.]. Chirurg. 2005;76:967-975.
67. Clarke M.J., Cohen-Gadol A.A., Ebersold M.J., Cabanela M.E. Long-term incidence of subaxial cervical spine instability following cervical arthrodesis surgery in patients with rheumatoid arthritis. Surg Neurol. 2006;66:136-140. discussion 140
68. Iizuka H., Nishinome M., Sorimachi Y., et al. The characteristics of bony ankylosis of the facet joint of the upper cervical spine in rheumatoid arthritis patients. Eur Spine J. 2009;18:1130-1134.
69. Kim D.H., Hilibrand A.S. Rheumatoid arthritis in the cervical spine. J Am Acad Orthop Surg. 2005;13:463-474.
70. Paus A.C., Steen H., Roislien J., et al. High mortality rate in rheumatoid arthritis with subluxation of the cervical spine: a cohort study of operated and nonoperated patients. Spine (Phila Pa 1976). 2008;33:2278-2283.
71. Shen F.H., Samartzis D., Jenis L.G., An H.S. Rheumatoid arthritis: evaluation and surgical management of the cervical spine. Spine J. 2004;4:689-700.
72. Weinstein J.N., Tosteson T.D., Lurie J.D., et al. Surgical versus nonsurgical therapy for lumbar spinal stenosis. N Engl J Med. 2008;358:794-810.
73. Katz J.N., Harris M.B. Clinical practice: lumbar spinal stenosis. N Engl J Med. 2008;358:818-825.
74. Weinstein J.N., Lurie J.D., Tosteson T.D., et al. Surgical versus nonsurgical treatment for lumbar degenerative spondylolisthesis. N Engl J Med. 2007;356:2257-2270.
75. Bess S., Boachie-Adjei O., Burton D., et al. Pain and disability determine treatment modality for older patients with adult scoliosis, while deformity guides treatment for younger patients. Spine (Phila Pa 1976). 2009;34:2186-2190.
76. Birknes J.K., White A.P., Albert T.J., et al. Adult degenerative scoliosis: a review. Neurosurgery. 2008;63:94-103.
77. Coe J.D., Arlet V., Donaldson W., et al. Complications in spinal fusion for adolescent idiopathic scoliosis in the new millennium. A report of the Scoliosis Research Society Morbidity and Mortality Committee. Spine (Phila Pa 1976). 2006;31:345-349.
78. Oskouian R.J.Jr., Shaffrey C.I. Degenerative lumbar scoliosis. Neurosurg Clin N Am. 2006;17:299-315. vii
79. Smith J.S., Fu K.M., Urban P., Shaffrey C.I. Neurological symptoms and deficits in adults with scoliosis who present to a surgical clinic: incidence and association with the choice of operative versus nonoperative management. J Neurosurg Spine. 2008;9:326-331.
80. Smith J.S., Shaffrey C.I., Berven S.G., et al. Operative versus nonoperative treatment of leg pain in adults with scoliosis: a retrospective review of a prospective multicenter database with two-year follow-up. Spine (Phila Pa 1976). 2009;34:1693-1698.
81. Brodbelt A.R., Stoodley M.A. Post-traumatic syringomyelia: a review. J Clin Neurosci. 2003;10:401-408.
82. Qiu Y., Zhu Z.Z., Lu J.Y., et al. Clinical manifestations and significance of post-traumatic thoracolumbar syringomyelia. Chin J Traumatol. 2004;7:52-55.
83. Thompson P.D. Paraplegia and quadriplegia. In: Bradley W.G., Daroff R.B., Fenichel G.M., et al, editors. Neurology in clinical practice. London: Butterworth-Heinemann; 1991:261-273.
84. Carr J., Bryer A. An isolated myelopathy as a presentation of polyarteritis nodosa. Br J Rheumatol. 1993;32:644.
85. Rawlinson D.G., Braun C.W. Granulomatous angiitis of the nervous system first seen as relapsing myelopathy. Arch Neurol. 1981;38:129-131.
86. Kunizawa A., Fujioka M., Suzuki S., et al. Spontaneous spinal epidural hematoma inducing acute anterior spinal cord syndrome. J Neurosurg Spine. 2009;10:574-577.
87. Schaller B., Lyrer P. [Anterior spinal artery syndrome: an important differential diagnosis of acute non-traumatic transverse spinal cord syndrome.]. Praxis (Bern 1994). 2001;90:1420-1427.
88. Oldfield E.H. Introduction: spinal vascular malformations. Neurosurg Focus. 2009;26:E1.
89. Watson J.C., Oldfield E.H. The surgical management of spinal dural vascular malformations. Neurosurg Clin N Am. 1999;10:73-87.
90. Gonzalez L.F., Spetzler R.F. Treatment of spinal vascular malformations: an integrated approach. Clin Neurosurg. 2005;52:192-201.
91. Luessenhop A.J., Cruz T.D. The surgical excision of spinal intradural vascular malformations. J Neurosurg. 1969;30:552-559.
92. Tabrizi P., Spetzler R.F. Surgical treatment of spinal cord vascular malformations. Interv Neuroradiol. 2003;9:103-105.
93. Eddleman C.S., Jeong H., Cashen T.A., et al. Advanced noninvasive imaging of spinal vascular malformations. Neurosurg Focus. 2009;26:E9.
94. Sasiadek M., Bem Z. [MR appearance of the spinal canal vascular malformations.]. Przegl Lek. 2004;61:5-9.
95. Si-jia G., Meng-wei Z., Xi-ping L., et al. The clinical application studies of CT spinal angiography with 64-detector row spiral CT in diagnosing spinal vascular malformations. Eur J Radiol. 2009;71:22-28.
96. Caruso G., Galarza M., Borghesi I., et al. Acute presentation of spinal epidural cavernous angiomas: case report. Neurosurgery. 2007;60:E575-E576. discussion E576
97. Talacchi A., Spinnato S., Alessandrini F., et al. Radiologic and surgical aspects of pure spinal epidural cavernous angiomas. Report on 5 cases and review of the literature. Surg Neurol. 1999;52:198-203.
98. Toldo I., Drigo P., Mammi I., et al. Vertebral and spinal cavernous angiomas associated with familial cerebral cavernous malformation. Surg Neurol. 2009;71:167-171.
99. Ferrell A.S., Tubbs R.S., Acakpo-Satchivi L., et al. Legacy and current understanding of the often-misunderstood Foix-Alajouanine syndrome: historical vignette. J Neurosurg. 2009;111:902-906.
100. Mishra R., Kaw R. Foix-Alajouanine syndrome: an uncommon cause of myelopathy from an anatomic variant circulation. South Med J. 2005;98:567-569.
101. Poser C.M., Paty D.W., Scheinberg L., et al. New diagnostic criteria for multiple sclerosis: guidelines for research protocols. Ann Neurol. 1983;13:227-231.
102. Sahraian M.A., Eshaghi A. Role of MRI in diagnosis and treatment of multiple sclerosis. Clin Neurol Neurosurg. 2010;112:609-615.
103. Bhat A., Naguwa S., Cheema G., Gershwin M.E. The epidemiology of transverse myelitis. Autoimmun Rev. 2009;9:A395-A399.
104. Kelley C.E., Mathews J., Noskin G.A. Acute transverse myelitis in the emergency department: a case report and review of the literature. J Emerg Med. 1991;9:417-420.
105. Gomez-Arguelles J.M., Sanchez-Solla A., Lopez-Dolado E., et al. [Acute transverse myelitis: a clinical review and algorithm for diagnostic intervention.]. Rev Neurol. 2009;49:533-540.
106. Perumal J., Zabad R., Caon C., et al. Acute transverse myelitis with normal brain MRI: long-term risk of MS. J Neurol. 2008;255:89-93.
107. Heros R.C. Foix-Alajouanine syndrome: what is it? J Neurosurg. 2009;111:900-901.
108. Bach J.R., Saltstein K., Sinquee D., et al. Long-term survival in Werdnig-Hoffmann disease. Am J Phys Med Rehabil. 2007;86:339-345. quiz 346–338, 379
109. PARALS Study Group. Incidence of ALS in Italy: evidence for a uniform frequency in Western countries. Neurology. 2001;56:239-244.
110. Alappat J.J. Ethnic variation in the incidence of ALS: a systematic review. Neurology. 2007;69:711. author reply 711–712
111. Orrell R., de Belleroche J., Marklund S., et al. A novel SOD mutant and ALS. Nature. 1995;374:504-505.
112. Berlit P., Ringelstein A., Liebig T. Spinal MRI precedes clinical improvement in subacute combined degeneration with B12 deficiency. Neurology. 2004;63:592.
113. Takahashi H., Ito S., Hirano S., et al. Subacute combined degeneration of the spinal cord in vegetarians: vegetarian’s myelopathy. Intern Med. 2006;45:705-706.
114. Pritchard J., Hughes R.A. Guillain-Barre syndrome. Lancet. 2004;363:2186-2188.
115. Kang S.Y., Kim J.S., Choi J.C., et al. An unusual pathologic feature and phenotype associated with familial hyperkalemic periodic paralysis. Eur J Neurol. 2008;15:e47-e48.