CHAPTER 272 Differential Diagnosis and Initial Management Spine Pathology
A refined and logical approach is essential for the appropriate investigation, evaluation, and management of patients with suspected spine pathology. Kuntz and colleagues previously provided a fundamental guide that allows the physician’s evaluation to proceed in a stepwise fashion based on the clinical presentation (Fig. 272-1). With this algorithm, patients with possible spine disease can be assessed on the basis of pain and its characteristics, the presence and form of neurological deficits, and associated systemic signs and symptoms. With this information, laboratory and radiologic evaluation can proceed, a diagnosis can be made, and appropriate surgical or medical management can be prescribed.1
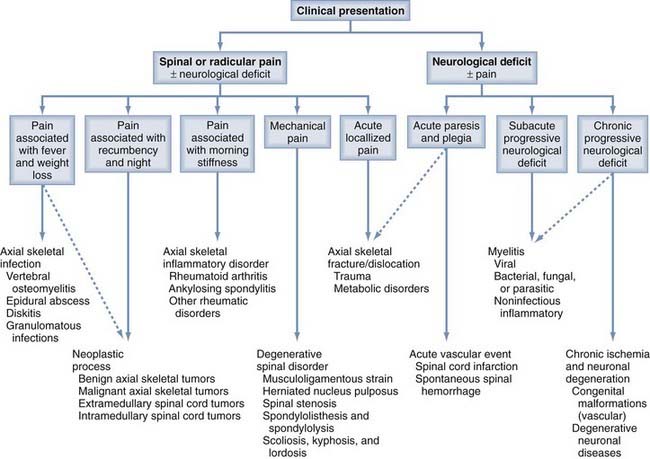
In this chapter, the Kuntz algorithm is used to explore the universe of spine pathology (Table 272-1). Our approach begins with the characterization of pain, evaluation of neurological deficits, and search for systemic symptoms and signs. The first section of this chapter reviews disorders that manifest predominantly with pain; the second covers conditions that manifest primarily with neurological deficits; subsections are based on the characteristics of the pain or neurological deficits and the associated systemic components. The algorithm can help clinicians classify spine pathology based on clinical presentation, restrict the differential diagnoses, and proceed with the evaluation in a logical order.2
TABLE 272-1 Differential Diagnosis of Spinal Disorders
| Axial Skeletal Infections |
| Neoplastic Processes |
| Axial Skeletal Inflammatory Disorders |
| Degenerative Spinal Disorders |
| Axial Skeletal Fracture/Dislocation and Metabolic Disorders |
| Acute Vascular Event |
| Myelitis |
| Chronic Ischemia and Neuronal Degeneration |
Pain
Neck and low back pain is one of the most common reasons to miss work; however, most back or neck pain results from musculoligamentous strain that resolves with conservative therapy. Persistent axial skeletal pain or radicular pain is more likely to result from a surgical lesion and requires a more extensive diagnostic evaluation. Assessment begins with a history and physical examination, followed by appropriate laboratory investigations and diagnostic imaging.3
Classically, imaging begins with plain radiographs: anteroposterior, lateral, and flexion-extension views, as well as coned-down views of transitional areas. Magnetic resonance imaging (MRI) or computed tomography (CT) of the spine should follow to further define underlying pathology. MRI is better for imaging soft tissues and changes in tissue hydration, and CT is better for imaging bony detail. CT-myelography often better delineates the neural elements in relation to the bony anatomy. Bone scans and bone density determination are occasionally required. The laboratory work and radiologic imaging may provide a definitive diagnosis, or pathologic evaluation of abnormal tissue may be required. Once a definitive diagnosis has been obtained, medical or surgical treatment can proceed.3
Pain Associated with Fever and Weight Loss
Patients presenting with spine or radicular pain associated with fever and weight loss are at increased risk of having an infectious process (see Fig. 272-1). The presentation of neoplastic processes can be similar. The presence of fever with spine or radicular pain should lead the physician to look for an axial skeletal infection first. Vertebral osteomyelitis, diskitis, epidural abscess, and granulomatous processes are the most common potential infectious conditions. Neurological deficits can occur, but they usually present weeks to months after the onset of pain and systemic symptoms.
Vertebral Osteomyelitis
Vertebral osteomyelitis typically results from a pyogenic infection of the vertebral column. It is the most frequently encountered infection of the axial skeleton, accounting for 2% to 19% of all cases of osteomyelitis. Osteomyelitis may be the result of trauma, extension from adjacent structures, or hematogenous spread. A definitive source of infection is found in less than 50% of cases. The most common foci are genitourinary, soft tissue, and respiratory, or the infection may be traced to intravenous drug abuse. The most common organisms isolated are gram-positive cocci, constituting 60% to 70% of all cases. Staphylococcus aureus is the most prevalent, representing up to 60% of all positive cultures. Gram-negative rods are found predominantly in parenteral drug abusers or immunocompromised patients. The lumbar spine is most often involved, followed by the thoracic and cervical spine. The vertebral body is involved in more than 95% of cases. Less than 5% of cases involve the posterior elements.4
Management is based on biopsy results from percutaneous aspiration and bone biopsy using CT or fluoroscopic guidance. Treatment entails bed rest and broad-spectrum antibiotics, followed by definitive antimicrobial therapy based on culture results. Decompression with or without fixation may be required if there is extensive bony involvement or if neurological sequelae ensue from excessive bony destruction or abscess formation.4–6
Spinal Epidural Abscess
A spinal epidural abscess usually results from a pyogenic infection of the epidural space. Although uncommon (reported incidence: 0.2 to 1.2 cases per 10,000 hospital admissions), its clinical importance overshadows its rarity. Approximately 50% of spinal epidural abscesses result from hematogenous spread to the epidural space. Other causes include direct extension of a preexisting osteomyelitis or diskitis and direct inoculation from surgical manipulation or trauma. The organisms of spinal epidural abscesses mirror those found in vertebral osteomyelitis. Gram-positive cocci are the most prevalent, with S. aureus isolated in 60% to 65% of the positive cultures; other staphylococcal and streptococcal species are the next most common organisms isolated. Thoracic involvement is slightly more common than lumbar involvement, followed less frequently by cervical involvement. Posterior location predominates in about two thirds of cases. An anterior abscess often results from direct extension of a ventrally located diskitis or vertebral osteomyelitis.4–6
Primarily adults are affected, and there is no sex predominance; spinal epidural abscess is rare in children. Axial skeletal pain is a common initial complaint. The pain may develop a radicular component, and sensory or motor dysfunction of the affected nerve root can occur. If untreated, paraplegia or quadriplegia ensues. The incidence of fever, leukocytosis, and neurological compromise is higher than in vertebral osteomyelitis. Laboratory studies include ESR, CRP, and white blood cell count, which are elevated in most patients. MRI is the diagnostic procedure of choice. T1 images reveal a hypointense epidural mass that enhances with contrast. On T2 images, the mass is hyperintense. Early diagnosis is important because patients with spinal epidural abscess can deteriorate rapidly, even on intravenous antibiotics. Treatment for spinal epidural abscess is surgical evacuation and administration of the appropriate antibiotics.7
Diskitis
Diskitis represents two entirely different entities in adults and children. Pediatric diskitis classically presents spontaneously. Adult diskitis is most often encountered after diskectomy, although it can occur spontaneously. Less often, adult diskitis results from hematogenous spread secondary to intravenous drug abuse or debilitating disease. Early detection is important to prevent diskitis from progressing to vertebral osteomyelitis or epidural abscess.7
In children, diskitis is a relatively benign disease due to a chronic inflammatory disorder, viral infection, or low-grade bacterial infection. The vascular network throughout the cartilaginous end plate and disk predisposes children to diskitis. This network functions as a bacterial and disappears in late adolescence. Postoperative diskitis in adults results from bacterial contamination during surgical manipulation. Gram-positive cocci, staphylococcal and streptococcal species, predominate in both pediatric and adult cultures. Biopsies and blood cultures can be nondiagnostic in 20% to 50% of patients. The lumbar spine is affected more frequently than the thoracic or cervical spine in both children and adults.8
Patients present with back pain and painful ambulation. In adults, the onset of pain at the surgical level 1 to 3 weeks after surgery signals the possibility of an infection. Typically, ESR and CRP are elevated. MRI is the most sensitive and specific modality for detecting infection and determining the efficacy of treatment. MRI scans show evidence of the infection sooner than plain radiography does. ESR is a sensitive test for detecting the infection and following the progression of treatment. Management consists of CT- or fluoroscopy-guided biopsy and culture, bed rest, and a course of intravenous and then oral antibiotic therapy. The prognosis of uncomplicated cases is excellent.4,7
Granulomatous Infection
Although rare in developed countries, globally, tuberculous spondylitis is the most common granulomatous infection affecting the axial skeleton. Historically, tuberculous spondylitis (Pott’s disease) is usually caused by M. tuberculosis, but other mycobacteria can also infect the axial spine. Infection results from hematogenous spread of mycobacteria from a pulmonary or genitourinary source. The infection spreads across the disk space, along the posterior or anterior longitudinal ligament, and tends to spare the disk space. The skeleton is affected in about 1% of all cases; of those, 50% to 60% involve the axial skeleton. The thoracolumbar spine is affected most frequently; cervical and sacral involvement are rare.7
Currently, individuals infected with the human immunodeficiency virus (HIV) account for most tubercular infections in the United States, and drug-resistant strains are being seen more frequently. Clinical presentation involves bone pain over the affected spinal level with fever, malaise, and weight loss. Vertebral collapse, spine deformity, epidural abscess, and subarachnoid seeding after dural erosion are late sequelae. In the progressive stages of the disease, kyphosis and epidural abscesses are common. Neurological sequelae occur in 10% to 50% of patients with active disease. In 20% of cases, tuberculous spondylitis causes paraparesis.9
Plain radiography, MRI, and CT-myelography evaluate disease progression and guide surgical planning. MRI evaluates the involvement of soft tissue and abscess formation. CT provides bony detail. Treatment is based on positive biopsy or culture results, degree of kyphosis, extent of neurological compromise, and refractory disease. Decompression and débridement, combined with antibiotic therapy, are the accepted treatment modalities when a neurological deficit evolves. Prognosis relates to patient age, extent of systemic disease, and preoperative neurological status.10
Actinomyces israelii, an anaerobic gram-positive bacterium, can cause purulent abscesses, external draining sinuses, and characteristic sulfur granules on microscopic examination. Infection is seen mostly in the cervical spine, secondary to direct extension from a preexisting mandible or supraclavicular infection. Nocardia asteroides, a gram-positive bacterium, can represent a rare cause of back pain when the spinal column is involved. Nocardia spreads hematogenously from a pulmonary focus, and osteomyelitis has occasionally been reported.10,11
Fungal infections of the axial skeleton are rare and occur by spore inhalation, with resultant pulmonary seeding and systemic spread. Spinal osseous involvement in patients with disseminated coccidioidomycotic and blastomycotic infection occurs in 10% to 50% of cases. The incidence of axial skeletal involvement with candidiasis or aspergillosis is low. Both fungal infections tend to occur in immunocompromised hosts. Other rare granulomatous diseases of the axial skeleton include parasitic infections such as syphilis and echinococcosis.10–12
Pain Associated with Recumbency and Nighttime
Pain is the most common presenting symptom for tumors involving the axial skeleton and spinal cord (extramedullary and intramedullary). Localized spinal pain at night or with recumbency is a hallmark of neoplastic lesions of the spine (see Fig. 272-1). Spinal column tumors often present with diffuse spine or radicular pain unrelieved by rest. Axial skeletal pain usually begins before any radicular pain or neurological deficit. In contrast, intramedullary and extramedullary spinal cord tumors also manifest with pain; however, the pain can be insidious in onset or diffuse, vaguely localized to the level of the tumor. Radicular pain or neurological deficit is commonly present by the time the tumor is diagnosed.
Benign Tumors of the Axial Skeleton
Benign axial skeleton tumors are typically posterior in location and seen in patients between 20 and 30 years old. The more common benign lesions, osteochondromas, osteoid osteomas, and osteoblastomas, have a lower incidence of recurrence after resection compared with malignant bone tumors. Other benign tumors include giant cell tumors, aneurysmal bone cysts, hemangiomas, and eosinophilic granulomas.13,14
Osteochondromas are the most common of the benign bone tumors. They develop from an adjacent physis or a cartilaginous remnant of the physis and present as a cartilage-capped bony protuberance. More than 50% occur in the cervical region, and they almost always involve the posterior elements. Osteochondromas may present with a dull backache in the case of smaller tumors or with decreased motion or deformity in the case of larger tumors. Neurological compromise is rare; however, when present, the cervical spine is the most typical location, followed by the thoracic spine, and myelopathic symptoms result. Plain radiographs usually demonstrate a protruding lesion with well-demarcated borders in the posterior elements. Resection is rarely required, except in cases of uncontrollable pain, neurological deficit, or accelerated growth. Prognosis is usually excellent when the affected periosteum and surrounding cartilage are resected completely. Osteochondromas occasionally degenerate into malignant chondrosarcomas.13,14
Osteoid osteomas and osteoblastomas share a pathologic origin but differ in size and incidence of spinal involvement. These tumors are thought to be a chronic inflammatory reaction rather than true neoplasms. Osteoid osteomas account for 2.6% of all excised primary bone tumors and up to 18% of primary axial skeletal tumors. Osteoid osteomas are less than 2 cm in diameter; larger lesions are classified as osteoblastomas. Approximately 40% of spinal osteoid osteomas occur in the lumbar region, and most are seen in the posterior elements. Most patients are young and male, and half of all symptomatic lesions appear in the second decade of life. Patients report a dull ache that is worse at night. This nocturnal exacerbation is believed to result from prostaglandin production by the tumor, and classically, the pain is relieved by aspirin. Neurological deficits are rare, but osteoid osteomas are the most common lesions associated with painful scoliosis in adolescents. Radiographically, the lesion is characterized by a radiolucent area with a central nidus surrounded by sclerosis. Treatment is excision. Instrumentation and fusion may be required if scoliosis is severe, but minor deformities resolve with resection alone. Overall, the prognosis is excellent, and the rate of recurrence, which is related to inadequate excision of the nidus, is marginal.13–14
Osteoblastomas are larger than osteoid osteomas (>2 cm), histologically equivalent, and less common, representing less than 2% of primary benign bone tumors. Approximately 30% to 40% of osteoblastomas involve the axial skeleton. Of those in the axial skeleton, most involve the posterior elements and have a propensity to produce spine deformity. In 90% of cases, osteoblastomas are found in patients aged 30 years or younger. The male-to-female predominance is 2 : 1. Clinical presentation involves a high incidence of neurological deficit, related to the size of the lesion. Treatment is en bloc resection, which usually resolves the scoliotic deformity. With adequate removal, prognosis is favorable. Long-term recurrence rates approach 10%.14–16
Giant cell tumors are benign lesions; however, they are aggressive, carry some malignant potential, and are associated with a high incidence of local recurrence. They are responsible for 21% of all primary benign bone tumors and affect the spinal axis in 8% to 11% of cases. When the spinal column is involved, the tumors typically occur in the sacral region. Unlike most primary bone tumors, giant cell tumors tend to be found in individuals in the third and fourth decades of life. Their frequency decreases in later years. Women are affected slightly more often than men. Plain radiographs demonstrate cortical expansion with little reactive sclerosis or periosteal reaction. MRI reveals homogeneous signals. CT better delineates the degree of vertebral bony involvement and defines surgical margins. Because the histologic characteristics of giant cell tumors are nondistinct, a thorough evaluation is important to differentiate this condition from other primary bone tumors. Treatment is usually en bloc resection, but because of the high rate of recurrence (50%), the prognosis is relatively poor. These tumors have the potential for malignant transformation, especially after local radiation if surgical margins were inadequate.14–16
Aneurysmal bone cysts are benign, nonneoplastic, proliferative lesions. Although accounting for only 1% to 2% of all primary bone tumors, aneurysmal bone cysts affect the axial skeleton in 12% to 25% of reported cases. Their pathogenesis is unclear, but accepted theories include an underlying tumor or traumatic arteriovenous malformation (AVM), with subsequent cyst formation. Histologically, aneurysmal bone cysts contain fluid-filled spaces separated by fibrous septa. Their incidence is greatest in the thoracolumbar region. Similar to most benign osseous lesions, 60% of spinal aneurysmal bone cysts occur in the posterior elements. Aneurysmal bone cysts typically occur in young patients in the second decade of life, with a slight female predominance. MRI and CT demonstrate a multiloculated, expansile, highly vascular osteolytic lesion with a thin, well-demarcated, eggshell-like cortical rim. Multiple vertebral levels may be involved in as many as 40% of cases. Treatment involves preoperative embolization and surgical resection. Postoperative radiation may have a role if the surgical margins are inadequate. Recurrence rates vary from 6% to 70% and depend on the extent of resection and the administration of postoperative radiation.14–17
Hemangiomas are benign tumors of vascular origin and are probably the most common benign tumor of the spine. Autopsy studies report hemangiomas of the axial skeleton in 10% to 12% of cases. Characterized by slow growth and a female predominance, vertebral hemangiomas most often occur in the thoracolumbar spine, with a predilection for the vertebral body. Symptomatic vertebral hemangiomas are rare; however, the most common initial symptom is back pain with or without radicular pain. Symptomatic lesions are best diagnosed with MRI; asymptomatic lesions are discovered incidentally during other radiographic investigations. Treatment of symptomatic lesions involving the spine consists of a combination of embolization, surgical resection, and possibly radiotherapy.14–1618
Eosinophilic granulomas are solitary osseous lesions characterized by an abnormal proliferation of Langerhans cells and are associated with a continuum of disorders (histiocytosis X and Letterer-Siwe and Hand-Schüller-Christian diseases). Eosinophilic granulomas are rare lesions of the axial skeleton and occur most frequently in the vertebral body. Most occur in children, with a peak incidence between 5 and 10 years of age. MRI and CT are the investigative procedures of choice, but diagnosis requires biopsy. Treatment is controversial but commonly includes surgical curettage, with adjuvant radiotherapy or chemotherapy reserved for disseminated versions of this uncommon disease.14–1618
Malignant Tumors of the Axial Skeleton
Malignant axial skeleton tumors are either metastatic or primary. Metastasis results from direct extension or hematogenous spread; the most common metastatic spinal tumors are breast, lung, and prostate malignancies. Multiple myeloma, chordoma, chondrosarcoma, osteogenic sarcoma, and Ewing’s sarcoma are the most common primary malignant neoplasms of the axial skeleton. Both primary and metastatic neoplasms most often involve the vertebral body. Malignant tumors of the axial skeleton are 25 to 40 times more likely to be metastatic than primary.14–16
Autopsy studies indicate that 40% to 85% of individuals with malignancy have metastatic disease, and the spine is the most common site of skeletal metastasis. As noted, breast, lung, and prostate malignancies account for most spinal metastatic lesions, which are spread equally throughout the thoracic and lumbosacral spine. Thoracic metastases tend to become symptomatic most often, whereas cervical lesions are symptomatic in 6% to 8% of patients. The vertebral body is the structure most often involved. Patients with known malignancy and new neck or back pain may have metastatic disease. Diagnosis relies on laboratory investigation, radiographic studies, and tumor biopsy results. MRI and CT-myelography help determine the extent of neural compression and bone destruction at the affected site, as well as screen for other areas of spine involvement. Treatment options for metastatic disease of the spine include both radiation and surgical intervention. Surgery is usually reserved for neurological compromise, radiation failure, spine instability, or an uncertain diagnosis. The goals of surgery are pain control and preservation of function and stability. Preoperative functional status and level of activity correlate directly with postoperative outcome. Patients who suffer rapid, progressive neurological deficits within a 24-hour period have a higher chance of developing permanent paraplegia than those with slowly evolving deficits, who are more likely to regain ambulatory function. Overall, prognosis is directly related to neoplastic type, spinal location, and extent of systemic involvement.14–1619
Multiple myelomas and solitary plasmacytomas are two manifestations of B-cell lymphoproliferative disease. Multiple myelomas are the most common malignant neoplasms of bone in adults and affect the spine in 30% to 50% of reported cases. The thoracic spine is affected most commonly, followed by the lumbar spine and, rarely, the cervical spine (<10%). The vertebral body is usually the site of tumor involvement. Multiple myeloma is primarily a disease of the fifth, sixth, and seventh decades of life and occurs equally in men and women. However, approximately 75% of solitary plasmacytomas occur in men. Unlike the classic presentation of pain with recumbency, the pain of multiple myeloma is sometimes relieved by rest and aggravated by mechanical agitation, mimicking other degenerative sources of pain. The diagnosis of multiple myeloma is based on characteristic serum protein abnormalities and radiologic imaging. Plain radiography and CT can be diagnostic because of the characteristic osteolytic picture, with no sclerotic edges involving the vertebral body and sparing of the posterior elements. Treatment and prognosis depend on whether the diagnosis is solitary plasmacytoma or systemic multiple myeloma. Both conditions are exquisitely radiosensitive, but patients with solitary plasmacytomas have significantly longer survival times.14,19
Chordomas, originally described by Virchow, are tumors that originate from the primitive notochord. As tumors of the axial skeleton and skull base, chordomas constitute 1% to 2% of all skeletal sarcomas. Histologically, they are low-grade, locally invasive tumors, but metastases can occur in 5% to 43% of cases. More than 50% of these lesions are located in the lumbosacral region, 35% are in the clival area, and the remainder are spread throughout the vertebral column. Chordomas are the most common primary neoplasm of the sacrococcygeal region and occur predominantly in the fifth and sixth decades of life. Neurological deficits include bowel and bladder dysfunction. MRI is the imaging modality of choice because of its ability to delineate soft tissue involvement. CT delineates the extent of bony destruction. Diagnosis is based on CT- or fluoroscopy-guided biopsy, and treatment is en bloc resection when feasible. Radiation is reserved for local recurrence and surgically inaccessible disease. Age at presentation and en bloc resection are probably the best prognostic indicators for disease-free survival after surgery, with younger patients having better prognoses.20
Chondrosarcomas are rare, malignant, cartilage-forming neoplasms that arise from cartilaginous elements and affect primarily the adult appendicular skeleton. Spinal involvement is rare (6% of cases). Chondrosarcomas arise either primarily or from preexisting solitary osteochondromas, hereditary multiple exostosis, or Paget’s disease. There is an even distribution of tumor involvement among cervical, thoracic, and lumbosacral locations. Primary and secondary chondrosarcomas usually arise in middle-aged and older patients and show a predilection for men. Diagnostic characteristics on MRI and CT include bone destruction, associated soft tissue mass, and characteristic flocculent calcifications in the soft tissue mass. Diagnosis is based on tumor biopsy. Treatment is en bloc resection if feasible. Neither radiation nor chemotherapy is particularly useful. Prognosis correlates with tumor extension and grade; patients with unresectable chondrosarcomas have a 5-year survival rate of only 20%.21
Osteogenic sarcomas are primary malignant tumors of bone that rarely involve the axial skeleton. Only 2% of osteogenic sarcomas arise in the spine; they can arise primarily or secondarily but are more likely to be metastatic. These lesions are distributed evenly throughout the spine, but the vertebral body is involved in more than 95% of cases. Most primary osteogenic sarcomas manifest in the first 20 years of life; secondary sarcomas arise in the fifth to sixth decades from irradiated bone or preexisting Paget’s disease. This neoplastic disease has a slight predilection for men. Radiologically, osteogenic sarcomas typically exhibit lytic and sclerotic areas, with cortical destruction and ossification in the tumor mass. CT- or fluoroscopy-guided biopsy provides the diagnosis. Preoperative embolization, chemotherapy, and surgical extirpation with adjuvant radiotherapy are the current treatment modalities. Overall, prognosis is poor, with a life expectancy of 10 months to 1.5 years; there have been a few long-term survivors.20,21
Ewing’s sarcoma, a small blue-cell tumor, is primarily a disease of children. More than 85% of cases occur in the first 2 decades of life. Only 3% to 4% of Ewing’s sarcomas arise within the axial skeleton, and they constitute only 6% of all primary malignant bone tumors, making them rare primary neoplasms of the spinal column. The incidence of spinal column involvement decreases caudally to rostrally, with more than half of Ewing’s sarcomas arising within the sacrum. The vertebral body is most often involved, and males are affected more than females, at a ratio of 2 : 1. MRI and CT reveal a lytic lesion, possibly with a blastic component. Diagnosis is based on biopsy, and treatment involves a multidisciplinary approach combining surgical extirpation, radiation, and chemotherapeutic protocols. Younger patients tend to have a better prognosis; survival at 5 years approaches 75%.14,22
Extramedullary Spinal Cord Tumors
Extramedullary spinal cord tumors may be intradural, extradural, or both. Intradural extramedullary tumors account for 40% to 50% of all spinal cord tumors, and extradural tumors account for 30%. Nerve sheath tumors, meningiomas, and filum terminale ependymomas are the most common extramedullary spinal cord tumors, with nerve sheath tumors and meningiomas accounting for more than 70% to 80%. Back pain is the most common initial complaint in adults; children with extramedullary tumors tend to present with neurological deficits in the form of motor or gait disturbances.22,23
In adults, the initial pain is diffuse, and diagnosis may be delayed until the pain becomes radicular or symptoms of spinal cord compression develop. Symptoms can be nocturnal and are rarely relieved with rest. Early neurological compromise is uncommon because of the adaptive compressibility of the surrounding fat, cerebrospinal fluid (CSF), and adjacent vascular structures. Neurological compromise ensues once maximal compliance of the surrounding structures is reached and compression is transmitted directly to the spinal cord. The location of the neoplasm predicts the evolution of the neurological deficit. Most extramedullary neoplasms produce local segmental deficits before a distant neurological deficit appears. Cervical lesions produce weakness, fasciculation, and atrophy of hand muscles. Thoracic lesions can produce band paresthesias or Horner’s syndrome. Lumbosacral or conus medullaris lesions produce lower extremity weakness as well as bladder and bowel symptoms. Because of the anterolateral and posterolateral locations of many extramedullary spinal cord neoplasms, there is a higher incidence of Brown-Séquard–type syndrome with these tumors.23,24
Nerve sheath tumors, schwannomas, or neurofibromas are benign lesions of Schwann cell origin. Most are schwannomas that occur proportionally throughout the spinal canal, and most are entirely intradural. Ten percent to 15% have both intradural and extradural components, and approximately 10% are entirely extradural. Most nerve sheath tumors arise from a dorsal nerve root; tumors arising from a ventral root are more likely to be neurofibromas. Their malignant potential is low (2.5%), but nerve sheath tumors can be locally destructive if allowed to progress. Caudally located nerve sheath tumors may displace adjacent nerve roots and can erode the bone of nearby foramina as the neoplasm grows. Peak incidence is in the fourth and fifth decades of life. Patients present with vague pain or localized spine pain associated with radicular pain and evidence of nerve root or spinal cord impairment. Nerve sheath tumors occur commonly in patients with neurofibromatosis. MRI is the diagnostic procedure of choice and usually reveals a heterogeneously enhancing extramedullary anterolateral lesion, often associated with a widened neural foramen. Excision is recommended for symptomatic lesions. Recurrence is rare with complete removal.23,24
Spinal meningiomas arise from arachnoid cap cells embedded in the dura near the nerve root sleeve. Most common in the thoracic spine, meningiomas are the most common benign tumor at the foramen magnum. Most are intradural; however, approximately 10% are either intradural and extradural or entirely extradural. The majority present between the fifth and seventh decades of life, and more than 70% occur in women. Clinical presentation is similar to that of nerve sheath tumors, but with fewer radicular symptoms. MRI is the investigative modality of choice and reveals a homogeneously enhancing, posterolateral, dura-based lesion. Treatment is embolization followed by surgical excision. Ten years after gross total removal, the recurrence rate is 10% to 15%.23–25
Filum terminale ependymomas are glial tumors that arise within the filum terminale; they are almost always of the myxopapillary histologic type. These lesions account for approximately 15% of extramedullary spinal cord tumors, and approximately 40% of all spinal ependymomas arise within the proximal intradural filum. Most filum terminale ependymomas occur in the third to fifth decades of life. MRI is the diagnostic procedure of choice. Treatment is surgical resection, and recurrences are rare. Radiation is reserved for biologically aggressive tumors, which are more frequent in the younger population.24–26
The remaining extramedullary spinal cord tumors are rare, accounting for 5% to 10% of cases. The majority are intradural extramedullary tumors such as dermoids, epidermoids, lipomas, teratomas, and neuroenteric cysts that result from disordered embryogenesis. Occasionally, epidural lipomas associated with Cushing’s disease or exogenous steroid use produce symptoms related to spinal cord compression. Paragangliomas are rare tumors that can arise from the filum terminale or cauda equina. Hemangioblastomas and ganglioneuromas can involve intradural nerve roots and present as extramedullary mass lesions.24,26
Intramedullary Spinal Cord Tumors
Intramedullary spinal cord tumors account for 2% to 4% of central nervous system (CNS) neoplasms and are of neuroglial origin in 80% of cases. These lesions constitute 20% to 25% of spinal cord tumors, and most are astrocytomas and ependymomas. Gangliogliomas, oligodendrogliomas, subependymomas, hemangioblastomas, neurocytomas, and metastases are much less frequent. Children are predisposed to astrocytic tumors; however, ependymomas become more common with age. Intramedullary spinal cord tumors manifest with insidious pain localized to the level of the tumor, unrelated to mechanical activity, and not relieved with rest. The pain is rarely radicular. Dysesthesias are more common than numbness. A neurological deficit is usually present at diagnosis, and the insidious onset of symptoms can delay diagnosis for as long as 2 years. The location of the intramedullary tumor dictates the evolution of the neurological deficit. Most intramedullary tumors produce local segmental and distant neurological deficits. Cervical segmental findings include weakness, fasciculation, and atrophy of the hand muscles. Upper thoracic tumors may produce Horner’s syndrome, whereas lumbosacral or conus lesions may produce bladder or bowel symptoms. Because of their central location, intramedullary spinal cord tumors may produce a central cord-like syndrome, with relative sparing of the more radially located lumbosacral nerve tracts. Their central location renders Brown-Séquard syndrome unlikely.24,26
Astrocytomas and ependymomas predominate in both pediatric and adult patients with intramedullary spinal cord tumors. Astrocytomas can occur at any age but are most common in the first 3 decades of life; they constitute 90% of intramedullary tumors in patients younger than 10 years and 60% of intramedullary tumors in adolescents. Ependymomas are the most common intramedullary tumor in adults. Approximately 90% of pediatric astrocytic tumors are benign fibrillary astrocytomas or juvenile pilocytic astrocytomas, whereas fibrillary astrocytomas prevail in adults. Astrocytic tumors often infiltrate adjacent spinal cord tissue, but only 10% of pediatric intramedullary astrocytomas are malignant, compared with 25% of adult tumors. Histologically, almost all ependymomas are benign. Although unencapsulated, ependymomas are usually well circumscribed and do not infiltrate the adjacent spinal cord. MRI is the gold standard for evaluating intramedullary spinal cord tumors. Both astrocytomas and ependymomas produce fusiform dilation of the spinal cord and are associated with low- to intermediate-intensity signals on T1-weighted images and higher signal intensity on T2-weighted images; ependymomas tend to have more marked enhancement, better defined borders, and a more central location. Resection with or without adjuvant therapy is the mainstay of therapy. Adjuvant radiation therapy for glial tumors is controversial, and some investigators have found no significant benefit associated with this treatment.27,28
The remaining intramedullary spinal cord tumors are rare. Intramedullary hemangioblastomas are associated with von Hippel-Lindau syndrome. Gangliogliomas, oligodendrogliomas, and neurocytomas are rare intramedullary glial-derived tumors. Metastatic disease accounts for 1% to 2% of intramedullary spinal cord tumors.27,28
Pain Associated with Morning Stiffness
Persistent axial pain that slowly tapers after the initiation of increasing mechanical activity is the hallmark of an inflammatory disorder affecting the spine (see Fig. 272-1). Rheumatoid arthritis (RA) and ankylosing spondylitis are the two most common chronic inflammatory processes involving the axial skeleton. Although related, these diseases represent two different pathologic conditions with regard to sex, age, location, associated clinical findings, immunologic characterization, and indications for surgical intervention. Neurological deficits are usually a late manifestation of these chronic disease processes. Other inflammatory disorders that may involve the axial skeleton include psoriatic arthritis, Reiter’s syndrome, Behçet’s disease, Whipple’s disease, enteropathic arthritis, gout, and pseudogout.29
Rheumatoid Arthritis
RA, a chronic systemic inflammatory disorder of unknown cause, involves the small blood vessels and synovium. Its prevalence is 1% for both genders by age 65. RA destroys joint articular surfaces, capsules, and supporting ligaments. The most common skeletal manifestation is involvement of the metatarsophalangeal joints, followed by the cervical spine. RA affects the cervical spine in one of three ways: atlantoaxial subluxation, basilar invagination, or subaxial subluxation. Craniocervical instability from atlantoaxial subluxation is present in 19% to 70% of cases, and 20% of patients with atlantoaxial subluxation have some degree of basilar invagination. Basilar invagination is less common than atlantoaxial subluxation but is more frequently associated with neurological deficits. Subaxial subluxation most often occurs at C2-3 and C3-4, producing a “staircase” appearance on imaging.29,30
The disease has a female predominance and a peak onset in the fourth and fifth decades of life. Diagnosis of RA is based on history, distribution of joint involvement, and positive rheumatoid factor. Neck pain warrants radiographic evaluation, including flexion-extension cervical spine films and MRI to evaluate for neural compression. Radiographic sequelae include soft tissue swelling, narrowing of joint spaces, and ultimately bone erosion, with deformity and instability. Surgical intervention for decompression with or without fixation is warranted for myelopathy, progressive subluxation, and severe pain.30
Ankylosing Spondylitis
Ankylosing spondylitis is the most prevalent seronegative spondyloarthropathy, with an incidence of up to 2% in the white population. The prototypical lesion is enthesopathic, affecting insertion sites of tendons and ligaments to bone, with inflammation, bony erosion, and ankylosis. The axial skeleton is affected most commonly, with a milder degree of peripheral involvement. Vertebral body osteoporosis, ankylosis of the apophyseal joints, intervertebral disk calcification, and ligamentous ossification are the characteristic pathologic changes. The pathogenesis is unclear, but there is a strong immunologic association with human leukocyte antigen B27 (HLA-B27) positivity. The disease progresses in an ascending fashion from caudal to rostral and produces the characteristic “bamboo spine” and severe kyphotic deformity.31
Age at onset ranges from puberty to 45 years, and unlike RA, ankylosing spondylitis has a male predominance. The disease begins with insidious low back pain in 80% to 90% of patients and peripheral joint pain in the hip or shoulder in 20% to 40% of patients. Sacroiliitis is usually one of the earliest manifestations of the disease. Clinical diagnosis is based on onset at an age younger than 45 years, history of an insidious onset of back pain persisting for more than 3 months, morning stiffness, improvement with exercise, and limitation of chest expansion. HLA-B27 positivity is suggestive of but not conclusive for diagnosis. Radiographic studies reveal sacroiliitis followed by spinal ankylosis. It takes 3 to 7 years for radiographic evidence of ankylosing spondylitis to appear; therefore, back pain, morning stiffness, and loss of axial mobility are important symptoms and signs. Chronic spondylitis results in kyphosis, fractures, stenosis, and rotary instability. Prognosis and success of treatment are directly related to the time of diagnosis, initiation of physical therapy, and possibly phenotypic expression. Treatment options for severe kyphotic deformities include cervical or lumbar osteotomies with instrumentation and fusion. The disease course predicts progression in the first 10 years, with the more aggressive subtypes causing greater deformity.31,32
Other Rheumatic Disorders
Other rheumatic disorders of the spine include psoriatic arthritis, Reiter’s syndrome, Behçet’s disease, Whipple’s disease, enteropathic arthritis, gout, and pseudogout. These conditions represent causes of back pain with or without deformity that may require either surgical intervention or, more commonly, conservative therapy for systemic symptoms. The clinical presentation and spinal involvement of the spondyloarthropathies overlap. In general, these syndromes cause instability or axial deformity.29
Mechanical Pain
Pain without constitutional signs and symptoms that is initiated and exacerbated by activity is a large category that includes musculoligamentous strain, disk herniation, spinal stenosis, spondylolisthesis, spondylolysis, and degenerative deformity (see Fig. 272-1). Other entities such as sacroiliac joint dysfunction, facet syndrome, coccydynia, perineural cyst, dural ectasia, costovertebral syndrome, internal disk disruption, and soft tissue irritation disorders such as piriformis syndrome are less well differentiated causes of low back pain that are usually diagnosed clinically and managed conservatively. The differential diagnosis of movement-associated axial pain is ample because most anatomic structures of the spine are pain generators.
Spine or radicular pain is mechanical in 90% of patients and improves with conservative therapy. In those who fail conservative therapy, the decision for surgical intervention is relatively straightforward if the patient suffers from acute disk herniation with progressive neurological deficit. The treatment decisions for other degenerative diseases may be less clear. Degenerative spinal disorders can be differentiated based on age, history of onset, character and duration of symptoms, and presence of a congenital disorder. Plain radiography, MRI, and CT-myelography are most often used to evaluate degenerative spinal disorders.33
Herniated Nucleus Pulposus
Herniated disks are classified as diffuse bulges, focal protrusions, extrusion of the nucleus pulposus, and extrusion of the nucleus pulposus with sequestration in the extradural space. Management of each condition depends on the characteristics of the pain and the associated signs of nerve root irritation. Typically, an acute herniated nucleus pulposus manifests with the acute onset of sharp, radiating pain that follows a dermatomal pattern; occasionally, the pain is associated with significant radicular weakness, myelopathy, or cauda equina syndrome. Self-limited episodes of spine pain can precede the acute symptoms. This condition is a common cause of cervical and lumbar radicular pain in adults and, less frequently, of thoracic symptoms. Diagnosis includes clinical findings of sensory, motor, and reflex deficits consistent with the affected nerve root. The single most important mechanical sign is a positive nerve root tension sign. MRI is the diagnostic procedure of choice. Most herniated disks respond to conservative therapy, and 80% of patients never require surgery. Surgical intervention is reserved for severe pain, progressive neurological involvement, myelopathy, or cauda equina syndrome.33,34
Spinal Stenosis
The clinical diagnosis of central or lateral spinal canal stenosis is confirmed with MRI or CT-myelography. Both cervical and lumbar spinal stenosis are treated surgically by neural decompression with or without instrumentation and fusion.2,3,33,34
Spondylolisthesis and Spondylolysis
Spondylolisthesis is anterior subluxation of one vertebral body on another and is classified according to cause: congenital, isthmic, degenerative, traumatic, or pathologic. Congenital spondylolisthesis is characterized by dysplasia of the facet joints on the upper sacrum. Isthmic spondylolisthesis results from a lytic lesion of the pars interarticularis or by an elongated pars interarticularis due to repeated fracture and healing. Degenerative spondylolisthesis is secondary to long-standing intersegmental instability and rarely progresses beyond 50% anterior vertebral body subluxation. Trauma or surgery causes traumatic spondylolisthesis. Pathologic spondylolisthesis is the result of generalized bone disease. These conditions are common causes of back pain in children and adults, and anterior subluxation of L5 on S1 is the most common site. Spondylolisthesis is the most common cause of back pain in individuals younger than 30 years. Seitsalo and colleagues found that in the population younger than 20 years with spondylolisthesis, 50% to 86% had lower back pain, 82% had radicular pain, and 74% had both. Adults have a vaguer and more insidious presentation; back pain is the most common complaint, followed by claudication, radicular symptoms, and hamstring tightness. Treatment varies, depending on the type of pain, degree of slippage, and segmental instability. Surgical decompression with instrumentation and fusion is the mainstay for advanced disease.36–38
Scoliosis, Kyphosis, and Lordosis
Scoliosis, kyphosis, and lordosis are the three basic types of spine deformity, and these disorders can occur alone or in combination. Deformities are classified according to cause, location, magnitude, and direction, and they represent another potential cause of spine and radicular pain. Adult lumbar scoliosis, defined as a Cobb angle greater than 10 degrees, increases with age and is not uncommon in those older than 60 years. Although the exact cause of the pain is unclear, back pain was present in 86% of adult patients with lumbar scoliosis in one large series. Most cases of minor (Cobb angle < 20 degrees) lumbar scoliosis are managed medically. Degenerative deformities in the sagittal plane are common in the elderly and often remain asymptomatic or are associated with mild axial skeletal pain. Surgical intervention for degenerative scoliosis, kyphosis, and lordosis is reserved for progressive deformity, severe pain, segmental instability, and neurological deficits.39,40
Acute Localized Pain
The acute onset of localized pain can be related to traumatic injury, vertebral expansion caused by an underlying systemic condition or acute hemorrhage involving the spinal cord (see Fig. 272-1). However, the most common cause of acute spine pain is trauma. A patient complaining of spine or radicular pain after trauma is considered to have a fracture or ligamentous injury until proved otherwise. Pathologic vertebral fractures also produce severe localized axial skeletal pain. These pathologic conditions include infections, tumors, inflammatory disorders, and metabolic diseases that destroy or weaken the vertebral body and lead to fracture under normal physiologic loads. Acute disk herniation and spinal hemorrhage should be included in the differential diagnosis of acute localized pain. The acute hemorrhage of a vertebral body tumor or the sudden rupture of a spinal AVM is a rare cause of acute neck or back pain and is usually associated with apoplectic neurological deficits.2
Metabolic Disorders
Osteoporosis affects 15 to 20 million American and is characterized by decreased amounts of normal-quality bone, increasing the patient’s susceptibility to fractures. Osteoporosis can be primary, which is idiopathic, or secondary, which is a result of endocrinopathies, neoplastic diseases, hematologic disorders, mechanical disorders, biochemical collagen disturbances, or nutritional aberrations. Common causes of secondary osteoporosis include hyperthyroidism, hyperparathyroidism, Cushing’s disease, hypothalamic hypogonadism, estrogen deficiency, diabetes mellitus, steroid exposure, multiple myeloma, leukemia, and prolonged bed rest. Osteoporosis results in 1 to 2 million pathologic skeletal fractures each year.41–43
Osteomalacia and Paget’s disease occur less frequently than osteoporosis, although Paget’s disease is the second most common metabolic bone disturbance in the United States after osteoporosis. Osteomalacia, a metabolic bone disease characterized by inadequate mineralization of newly formed osteoid, results from a deficiency of or resistance to vitamin D, intestinal malabsorption, acquired or hereditary renal disorders, intoxication with heavy metals (aluminum and iron), and other assorted causes. Paget’s disease is characterized by excessive osteoclast and osteoblast activity, which increases bone resorption and promotes marrow replacement by hypervascular fibrous tissue. This results in a brittle bone matrix susceptible to fracture. The incidence of Paget’s disease is 1 per 1000 persons, and the vertebrae, pelvis, and femora are the most commonly involved bones. Malignant degeneration occurs in 1% to 10% of patients; the most common malignant tumor in pagetic bone is osteogenic sarcoma. The clinical presentation involving the spine includes local pain, vertebral fractures, or entrapment of nerve roots. Treatment is multidisciplinary, with surgery reserved for instability or progressive neurological deficits.41,44
Other metabolic diseases of the spine include ossification of the posterior longitudinal ligament, diffuse idiopathic skeletal hyperostosis, calcium pyrophosphate arthropathy, amyloidosis, acromegaly, fibrous dysplasia, and dialysis-associated spondyloarthropathy. The clinical presentation and treatment of these diseases are variable and are discussed in other chapters.41
Neurological Deficit
Defining the location and distribution of a neurological deficit is extremely important for diagnosis and localization of the pathologic lesion. The single most important finding indicative of focal spinal cord pathology is a segmental spinal level below which the neurological deficit is present. Focal neurological deficits in the anatomic distribution of a nerve root are extremely accurate for localizing pathology to the spinal cord or segmental nerve roots. In diffuse disease in which the neurological deficit cannot be localized, the pattern of sensory and motor involvement is the basis of the differential diagnosis, but this is less reliable for differentiating spinal cord pathology from central and peripheral neurological disease or from primary muscular pathology. Upper motoneuron pathology results from spinal, brainstem, or brain pathologic processes. Diffuse muscle weakness and atrophy may be the result of spine and peripheral nerve disease, neuromuscular junction failure, or a pure muscular illness. Ataxia may be the result of cerebellar, brainstem, or spinal cord injury. Sensory deficits may be the result of lesions anywhere along the neural axis. Localizing diffuse neurological deficits to the spinal cord requires knowledge of specific spine disease presentations, combined with laboratory investigations, electrophysiologic studies, and radiographic imaging.45
Armed with the history and physical examination, one can begin the secondary investigation consisting of directed laboratory tests and specific imaging studies. Directed laboratory studies may include CSF analysis and culture, combined with immunologic studies for the investigation of myelitis, the level of serum cobalamin to diagnose subacute combined degeneration, and biochemical or genetic analysis to exclude hereditary spinal cord diseases. When the physical examination cannot localize the disease, electrophysiologic studies assist in identifying and characterizing the pathologic process. Electromyography, nerve conduction studies, and somatosensory evoked potentials can differentiate primary muscular, peripheral nerve, spine, and brainstem pathology. Imaging of the spine should begin with plain radiography, followed by MRI. CT remains the superior imaging study to demonstrate acute hemorrhage and bony detail. Laboratory investigations, electrophysiologic evaluation, and radiologic imaging may be diagnostic, or the diagnosis may require biopsy of a pathologic lesion. Once a definitive diagnosis is made, medical or surgical treatment can begin.45
Acute Paresis or Paralysis
Acute paresis or paralysis, which occurs over minutes to hours, is usually secondary to trauma or a vascular event involving the spinal cord (see Fig. 272-1); traumatic spine injuries are reviewed in another chapter. Patients with a history of trauma and new-onset neurological deficits need to be evaluated for a spinal column or neural axis injury. Without a history of trauma, acute paresis or paralysis of the extremities is most likely related to spinal cord infarction or spinal hemorrhage. Neurological evaluation usually reveals motor and sensory deficits below the level of vascular cord injury, allowing the clinician to localize the segmental level of the lesion. With the clinical history and the pattern of neurological deficit, a logical differential diagnosis can be deduced and is discussed in the sections that follow.
Spinal Cord Infarction
The cervical spine receives the majority of its blood supply from the vertebral arteries that provide the origin of the anterior median and both posterior spinal arteries. Cervical radicular arteries may also provide segmental cervical arterial supply to the spinal cord. Segmental branches of the aorta and internal iliac arteries supply the thoracic and lumbar spinal cord. The most important segmental vessel, the artery of Adamkiewicz, typically arises from the left side of aorta between the T10 and L3 spinal segments. The spinal cord between these main vascular systems is relatively vulnerable to ischemia, particularly in the midthoracic (T4-6) region of the spinal cord.46
Systemic diseases such as polyarteritis nodosa and syphilitic meningomyelitis or emboli from severe aortic atherosclerosis occasionally result in spinal medullary artery occlusion and spinal cord infarction. Caisson disease, or decompression sickness, infrequently causes spinal cord infarction as a result of nitrogen bubbles after diving. Fibrocartilaginous embolism after trauma may occlude a spinal artery, with resultant spinal cord ischemia and infraction. Foix-Alajouanine disease results from acute thrombosis of a spinal cord AVM that produces a necrotizing myelopathy secondary to venous hypertension.46,47
Anterior spinal artery syndrome results from infarction of the vascular territory supplied by the anterior spinal artery, which supplies the anterior two thirds of the spinal cord. It consists of motor paralysis, dissociated sensory loss, and paralysis of sphincter function. The posterior columns are usually spared, aiding in the diagnosis. Symptoms may develop instantaneously or over several hours. The prognosis for a functional recovery is poor.46,47
Spontaneous Spinal Hemorrhage
MRI and CT are used to diagnose spontaneous spinal hemorrhages, and treatment involves correction of the underlying hematologic disorder or coagulopathy and surgical evacuation of the compressive hematoma. Postoperative MRI, CT-myelography, and angiography help delineate the cause of the hemorrhage.46–48
Subacute, Progressive Neurological Deficit
Subacute, progressive neurological deficits are defined by the clinical onset of neurological symptoms that worsen over the course of days to weeks. This clinical picture indicates an inflammatory spinal cord disease or a compressive spinal cord lesion (see Fig. 272-1). Painful subacute, progressive diseases are more likely caused by rapid compression and include tumors, vertebral infections with abscesses, and large herniated disks. Myelitic disorders usually manifest with subacute, progressive neurological deficits associated with systemic signs of infection but without a significant history of long-standing pain. The absence of an infection does not preclude the diagnosis of myelitis.
Myelitis represents a nonhomogeneous group of inflammatory disorders related to viral, bacterial, fungal, and parasitic diseases or to noninfectious inflammatory lesions. Neurological symptoms may develop over days to weeks or rarely evolve over hours or months. Clinical presentation may include the subacute onset of spine pain at the level of the inflammatory damage. A detailed neurological evaluation reveals motor or sensory deficits, or both, at or below the level of the lesion. The pattern and progression of these deficits provide evidence of the cause of the inflammatory process. The diagnosis of myelitis is based on the patient’s clinical history, physical examination, laboratory investigations, and imaging of the spinal cord to rule out a compressive spinal cord lesion.45,46
Viral Myelitis
A number of viruses can cause infectious myelitis; most commonly, enteroviruses (poliovirus, coxsackievirus groups A and B, echoviruses) or herpesviruses (varicella-zoster, herpes simplex types 1 and 2, cytomegalovirus, Epstein-Barr) are the offending agent. Other viral sources include rabies virus, herpesvirus B, human T-lymphotropic virus type 1 (HTLV-1), and HIV. Different viral pathogens result in characteristic disease pathology. The enteroviruses have an affinity for anterior horn cells of the spinal cord and motor nuclei of the brainstem, whereas varicella-zoster virus prefers the dorsal root ganglion. The pathologic characteristic of HIV myelitis is vacuolated spinal cord white matter, which is most severe in thoracic segments. As a general rule, viral infections of the spinal cord produce distinct clinical syndromes secondary to the individual virus’s affinity for different neuronal cell populations. Viral infections of the spinal cord (with the exception of HTLV-1) usually do not produce transverse myelitis involving the entire cross-sectional area of the spinal cord with combined motor and sensory deficits. When significant sensory and motor deficits are present, the cause of myelitis is rarely viral.45,46
In the United States there are approximately 15 cases of paralytic poliomyelitis each year. In countries with successful vaccination programs, polio infections are rare and other enteroviruses are the most common causes of anterior poliomyelitis syndrome. Polio infections are commonly benign, and the paralysis is rarely significant. The human gastrointestinal tract acts as a reservoir for the virus, and the main route of infection is fecal-oral contamination. In clinically apparent poliovirus infection, symptoms include listlessness, headache, fever, stiffness, aching muscles, sore throat, anorexia, nausea, and vomiting. Nervous system involvement includes irritability, restlessness, and emotional lability, which are often followed by paralysis. Paralytic poliomyelitis is secondary to the destruction of the anterior and intermediate horn cells in the spinal cord gray matter, and the distribution of paralysis is variable. Poliovirus infections may be nonparalytic, preparalytic, or paralytic. The development of muscle weakness is variable and may occur rapidly over 48 hours or over a week or longer. Typically, weakness does not progress after the patient’s temperature has been normal for 48 hours. Objective sensory loss is rare; urinary retention may occur acutely, but the problem seldom persists. Treatment of an acute poliovirus infection is supportive; most patients with paralytic poliomyelitis improve over 3 to 4 months after the infection The mortality rate associated with acute infection is 5% to 10%.49,50
After infection with chickenpox, the varicella virus becomes latent in neurons of the dorsal root ganglia. Herpes zoster represents reactivation of a varicella-zoster virus, and it is a common viral infection of the CNS. There are approximately three to five cases per 1000 persons each year, and the incidence increases with age. Symptoms include an acute inflammatory reaction involving isolated spinal or cranial sensory ganglia, the posterior gray matter of the spinal cord, and the adjacent leptomeninges. Clinically, varicella-zoster infection is characterized by radicular pain, a vesicular cutaneous eruption, and, less often, segmental motor or sensory loss. Almost any dermatome can be involved, but the thoracic dermatomes, particularly T5-10, are affected most often. Treatment includes acyclovir, which shortens the duration of acute pain and speeds healing but does not affect the incidence of postherpetic neuralgia. The prognosis in uncomplicated cases is excellent.49–51
HIV and HTLV-1 also produce subacute to chronic myelitis. HIV infection of the spinal cord produces a white matter vacuolar myelopathy, most severely affecting the thoracic spinal cord segments. Clinical presentation is leg or leg and arm weakness that develops over weeks. As expected, the clinical syndrome is often complicated by the other CNS disorders associated with HIV infection, such as infectious encephalitis and cytomegalovirus radiculomyelopathy. HTLV-1 causes tropical spastic paraparesis, a chronic infective inflammatory disease of the spinal cord that presents with a slowly progressive paraparesis, increased tendon reflexes, and Babinski’s sign. Disorder of sphincter control is usually an early sign, and sensory function in the lower extremities is variably affected. The cerebrum, brainstem, and upper extremities are usually spared. There are anecdotal reports of improvement after the intravenous administration of gammaglobulin.52,53
Bacterial, Fungal, and Parasitic Myelitis
This category of disease includes myelitis due to Mycoplasma pneumoniae and Borrelia burgdorferi, pyogenic myelitis, tuberculous myelitis, and syphilitic myelitis. Lesions may involve both the spinal cord and meninges, or a spinal cord lesion may predominate. Diagnosis relies on CSF analysis. MRI and CT-myelography can show the extent of spinal column and spinal cord involvement.46
Infection by the spirochete Treponema pallidum results in syphilis. All forms of neurosyphilis begin as meningitis. Neurosyphilis may affect the spinal cord and most commonly causes tabetic neurosyphilis (tabes dorsalis); however, it may also produce syphilitic meningomyelitis or spinal meningovascular syphilis. Tabetic neurosyphilis usually develops 15 to 20 years after the onset of the infection, resulting from degeneration of the posterior columns of the spinal cord and the dorsal nerve roots. Other systemic signs of infection are usually present by the time tabes dorsalis presents; these include pupillary abnormalities in more than 90% of patients. The major symptoms of tabes dorsalis are lightning pains, ataxia, and urinary incontinence. The physical examination reveals absent lower extremity tendon reflexes, impaired vibratory and position sense in the feet and legs, and Romberg’s sign. Muscular power is fully retained, and the ataxia is related to the sensory deficit. Approximately 1% to 10% of patients with tabes develop deafferented Charcot’s joints, most commonly affecting the hips, knees, ankles, and, occasionally, lumbar spine. In syphilitic meningomyelitis, there is a subpial loss of myelinated fibers and gliosis as a result of chronic fibrosing meningitis; gumma of the meninges or spinal cord is rare. Spinal meningovascular syphilis occasionally assumes the form of an anterior spinal artery syndrome. Diagnosis is confirmed with a positive CSF Venereal Disease Research Laboratories (VDRL) slide test. Treatment involves the administration of intravenous penicillin G, which improves or arrests the neurological disease process in most patients.54
Rarely, a systemic infection with M. pneumoniae or B. burgdorferi (Lyme disease) produces myelitis. Other possible causes of inflammatory lesions of the spinal cord include tuberculous meningitis with spinal tuberculoma, schistosomiasis, spinal cord abscess, and sarcoid. Schistosomiasis is an important disease in developing countries. CNS involvement occurs in 3% of cases, and spinal cord disease often involves the conus medullaris as an intramedullary or meningeal granuloma.54,55
Noninfectious Inflammatory Myelitis
A disordered immune response rather than an infectious agent produces noninfectious inflammatory myelitis. This immune reaction results in leukomyelitis with demyelination or necrosis of spinal cord tracts. Causes include postinfectious and postvaccinal myelitis, acute and chronic relapsing multiple sclerosis (MS), subacute necrotizing myelitis, myelopathy with lupus or other forms of angiitis, and paraneoplastic myelitis.46,56
Postinfectious and postvaccinal myelitides are characterized by a temporal relationship to a viral infection or vaccination, the development of neurological signs and symptoms over several days, and a monophasic temporal course (no recurrence). The usual clinical presentation involves progressive numbness and weakness of the feet and legs (less often, the hands and arms) over the course of a few days, with variable involvement of the sensory and motor spinal cord tracts. Incomplete spinal cord dysfunction is common, and headache or stiff neck may or may not be present. In contrast to Guillain-Barré syndrome, viral myelitis demonstrates a sensory level and upper motoneuron dysfunction. Sphincter dysfunction and spine pain are common; however, the neurological symptoms are more prominent than the painful prodrome. T2-weighted MRI may show a signal abnormality in the spinal cord, with slight enhancement over two or three segments; the spinal cord may be swollen focally. Although corticosteroids are often administered, there is no evidence that they alter the course of the disease, and treatment is mainly supportive. The prognosis is often better than the initial symptoms suggest.46,56,57
Patients with MS may develop a demyelinative myelitis that shares many features with postinfectious and postvaccinal myelitides. Differences exist in the time course of the disease, however; symptoms evolve more slowly, over 1 to 3 weeks or longer, and there is no history of an antecedent infection or vaccination. MS myelitis is usually painless. Only 0.6% of MS patients initially present with myelitis. Symptoms are exacerbated by exercise or increased temperature (Uhthoff phenomenon). Evaluation of MS myelitis includes MRI of the brain to rule out more common changes in the brain; however, in clinically diagnosed MS patients, spinal MRI is positive in 85% to 95% for MS plaque. The clinical diagnosis is supported by CSF examination revealing oligoclonal bands and an immunoglobulin G index greater than 1.7. Subclinical lesions remote from the area of clinical dysfunction may be discovered during evaluation; they are related to the multifocal nature of the disease and help support the diagnosis. Treatment with corticosteroids may lead to regression of symptoms, but the disease may relapse if the medication is tapered too quickly.46,57,58
Acute necrotizing myelitis may result from myelitic syndromes and manifests with persistent and profound flaccidity of the legs or arms, areflexia, and atonicity of the bladder secondary to necrosis of the gray and white matter of the spinal cord. When both the optic nerves and spinal cord are involved, the disease is called neuromyelitis optica or Devic’s disease. Systemic lupus erythematosus, rare vasculitic disorders, and paraneoplastic myelitis also produce acute or subacute necrotizing myelopathies. Myelitis may be one manifestation of a more diffuse disseminated encephalomyelitis.46,58,59
Chronic, Progressive Neurological Deficit
Disorders that manifest with chronic, progressive neurological deficits involve symptoms that evolve over weeks to months and are not often associated with severe pain or significant systemic components (see Fig. 272-1). The processes that produce chronic and progressive spinal cord dysfunction are associated with persistent spinal cord ischemia or slowly progressive spinal neuronal degeneration. Differentiating the different disease entities requires the usual detailed clinical history, neurological evaluation, laboratory investigation, electrophysiologic studies, and radiographic imaging.2
Congenital Malformation
Open spinal dysraphism (spina bifida aperta) is a defect in the vertebral arches that results in exposure of the meninges, neural tissue, or both. The incidence of spina bifida aperta, with associated or myelomeningocele, is reported to be less than 1 to 2 per 1000 live births, and more than 85% are found in the distal thoracic, lumbar, or sacral area. Myelomeningocele is associated almost universally with Chiari II malformation, as well as with syringomyelia, hydrocephalus, diastematomyelia, and cerebral malformations, including lobar agenesis, polymicrogyria, holoprosencephaly, cerebellar dysplasia, heterotopia, and schizencephaly. Neurological deficits associated with myelomeningocele are present at birth as a result of the abnormal formation of the neural structures. Treatment involves surgical reformation of the neural tube, closure of the defect, and possibly decompression of the ventricles and shunt placement.60,61
Occult spinal dysraphism refers to skin-covered midline malformations. The simplest form has no spinous process and variable amounts of the vertebral arch; it most often occurs at L5 through S1. Isolated bony dysraphism may be found in 20% to 30% of the general population; in these cases, the CNS is seldom involved, and patients are usually asymptomatic. When a defect in the vertebral arch is associated with overlying cutaneous abnormalities, the incidence of intraspinal dysraphic lesions increases. Intraspinal abnormalities include thickened filum terminale, dorsal dermal sinus tracts, myelocystoceles, intradural lipomas, lipomyelomeningoceles, neoplasms, split cord malformations, and caudal regression syndromes. Thickened filum terminale is associated with a low-lying spinal cord, fat in the filum, and a filum diameter greater than 2 mm. A dorsal dermal sinus represents a tract lined by epithelium extending from the posterior midline surface of the skin to the underlying dura or spinal cord. A myelocystocele is a terminal dilation of the central spinal canal associated with a meningocele and dorsal bony dysraphism. Simple intradural lipomas represent a collection of intradural fat and minimal vertebral arch malformation, with or without a tethered spinal cord. Lipomyelomeningocele consist of an intradural or extradural lipoma, dorsal bony dysraphism, spinal cord tethering, and a prominent subcutaneous mass. Congenital dermoids and epidermoids may occur independently or in association with a dermal sinus tract and represent incomplete separation of the neural ectoderm from the epithelial ectoderm. Neurenteric cysts most often occur in the anterior cervical region and consist of a cyst of endodermal origin that may or may not have a fistulous tract. Intraspinal teratomas contain elements of all three germ layers. Diastematomyelia and diplomyelia are split spinal cord malformations that refer to the sagittal division of the spinal cord into two hemicords or two duplicated cords, respectively. In diastematomyelia, an associated fibrous or bony spur separates the two hemicords (more recently classified as split-cord malformation type I). Caudal regression syndrome has been used to describe abnormal or absent coccygeal, sacral, and lumbar segments secondary to abnormal retrogressive differentiation.60,61
When occult spinal dysraphism becomes symptomatic, it is usually related to an intradural lesion with tethering of the spinal cord or, less frequently, with compression of the neural elements from the mass lesion. The neurological deficit usually progresses slowly over months and often coincides with the patient’s growth. Symptoms include pain, structural deformities such as scoliosis, and gait or bladder dysfunction. It is important to identify occult intradural lesions early, because significant neurological deficits may not be reversible. Surgical intervention for occult spinal dysraphism is directed at untethering the spinal cord and resecting the mass lesion.61–64
Hydromyelia and syringomyelia represent two forms of spinal cord cavitation that have many differences. Hydromyelia is central cavitation of the spinal cord lined by ependyma; syringomyelia refers to an eccentric cavitation not lined by ependyma. Syringomyelia tends to be focal; however, both conditions may involve the entire spinal cord. Hydromyelia has been described as “hydrocephalus of the spinal cord” and is often associated with inadequately treated hydrocephalus. Both conditions are associated with multiple pathologic abnormalities, including Chiari malformation, posterior fossa cyst, platybasia, spinal cord tumor, diastematomyelia, tethered spinal cord, and trauma. Hydromyelia and syringomyelia present with bilateral sensory loss, usually of the upper extremities; muscle weakness; spasticity; pain; incontinence; scoliosis; and kyphosis. MRI demonstrates an intramedullary fluid cavity and helps define associated pathology. A posterior fossa decompression or shunting procedure is often required to prevent bulbar symptoms or spasticity or to relieve pain.61–64
Congenital axial skeletal deformities result from anomalous vertebral development or spinal dysraphism as a result of defects of segmentation and defects of formation. This may produce scoliosis, kyphosis, or lordosis. Management of these deformities depends on their magnitude and progression, associated pathology, and neurological deficit. Congenital scoliosis is often associated with spinal cord abnormalities: syringomyelia, diastematomyelia, neoplasm, tethered cord, and Arnold-Chiari malformation. Neurological symptoms usually result from the underlying spinal cord disease. Treatment is directed at the underlying pathologic condition, correction of the deformity, and prevention of further neurological deficits.65
The symptoms associated with type I and type III spinal AVMs can be exacerbated by strenuous activity and postural changes. Types II, III, and IV lesions affect both genders equally. Imaging of spinal AVMs can be difficult. MRI of dural arteriovenous fistulas may be normal or nonspecific or may demonstrate spinal cord expansion and evidence of venous congestion. In contrast to MRI, myelography is usually abnormal in dural arteriovenous fistulas and demonstrates the presence of the lesion. Spinal angiography occasionally delineates arteriovenous lesions that cannot be identified on MRI or myelography. The treatment of spinal AVMs is multidisciplinary, involving embolization and surgical resection.66,67
Histologically similar to their intracranial counterparts, spinal cavernous malformations are intramedullary lesions that represent 5% to 10% of all spinal vascular malformations. Cavernous malformations usually present in the fourth or fifth decade of life and are more common in women. The lesions occur with equal frequency along the neural axis. Their typical clinical presentation is progressive paraparesis and sensory loss with pain related to repetitive hemorrhages. MRI reveals mixed signal intensity on T1- and T2-weighted images, with variable contrast enhancement. Microsurgical resection is the only therapeutic option.66–69
Neuronal Degenerative Diseases
Neuronal degenerative disorders that affect the spinal cord can be broadly divided into syndromes of muscle weakness and atrophy without sensory changes, syndromes of progressive spinal ataxia, and combined system disorders. Despite this oversimplification, most spinal cord neuronal degenerative diseases that must be distinguished from other pathologic entities (e.g., cervical spondylitic myelopathy, compressive neoplasm, chronic myelitis) fall into one of these categories.46
Pure upper motoneuron degenerative diseases include primary lateral sclerosis and hereditary spastic paraplegia. Primary lateral sclerosis is characterized by a slowly progressive degeneration of the corticospinal tracts associated with a decrease in the number of Betz cells of the frontal cortex. It usually begins insidiously in the fifth or sixth decade with a slowly progressive spastic paraparesis that later involves the arms and oropharyngeal muscles. Lower motoneuron and sensory deficits are absent, and the clinical course is often prolonged. Hereditary spastic paraplegia is a heterogeneous disorder associated with progressive spasticity and mild weakness in the lower extremities. The pattern of inheritance is usually autosomal dominant. Onset of the more common form of the disease occurs before age 35, whereas onset of other forms is between 40 and 60 years. The clinical picture is that of a progressively worsening spastic gait with difficulty walking. Lower extremity spasticity, hyperreflexia, and extensor plantar responses are encountered on examination. In pure cases, lower motoneuron and sensory changes are lacking. The clinical course of hereditary spastic paraplegia is usually protracted. Treatment for these pure upper motoneuron degenerative diseases is symptomatic and directed at reducing spasticity.70,71
Disorders involving other neural systems and the motor system in the form of upper motoneuron degeneration include the familial spastic paraplegias associated with other neurological abnormalities, adrenoleukodystrophy, lathyrism, and radiation myelopathy. The literature contains a number of reports of familial spastic paraplegias combined with other neurological abnormalities. Family history and associated neurological abnormalities almost always help determine the differential diagnosis. Adrenoleukodystrophy, an X-linked recessive disorder of males, most often manifests in children but may also present with spastic paraparesis in adolescence. Associated neurological abnormalities include dementia, optic atrophy, seizures, and adrenal insufficiency. Lathyrism, a disease of India and Africa, manifests as a subacute myelopathy caused by ß-N-oxylylaminoalanine, a toxin in the grass pea Lathyrus sativus. The corticospinal and spinocerebellar tracts are predominantly affected. Another uncommon cause of slowly progressive subacute or chronic myelopathy is radiation. Radiation myelopathy is a complication of radiation treatment and usually develops 6 months to 2 years after treatment. The corticospinal and spinothalamic pathways are involved early in the course of this disease. A rather stable, nonprogressive myelopathy may also be associated with cerebral spastic diplegia. Treatment for these neuronal disorders involves eliminating the causative agent when possible and supportive care directed at reducing spasticity.46,70,72
Lower motoneuron degenerative diseases include progressive muscular atrophy and the spinal muscular atrophies (SMAs). Progressive muscular atrophy, an adult-onset disease, is characterized by pure lower motoneuron degeneration that typically presents in males as a symmetrical wasting of the hand muscles and slowly advances to more proximal arm musculature. Less often, the atrophic weakness begins in the legs. The distal extremities are involved before proximal weakness and atrophy appear. Progressive muscular atrophy has a slower and more benign clinical course than amyotrophic lateral sclerosis (ALS), with some patients surviving 15 years or longer. The SMAs are inheritable autosomal recessive disorders linked to chromosome 5. SMA type I (Werdnig-Hoffmann syndrome) is evident at birth or soon thereafter and is fatal in 85% of cases by age 2 years. SMA type II is similar to SMA type I, except that it appears between 6 and 12 months of age and patients may survive beyond 2 years. The onset of SMA type III (Wohlfart-Kugelberg-Welander disease) occurs in late childhood or adolescence, and its clinical course is more benign. Included in this group of diseases are Fazio-Londe syndrome, Kennedy’s disease, and postpolio syndrome. Fazio-Londe syndrome (childhood bulbar muscular atrophy) and Kennedy’s disease (adult bulbar muscular atrophy) are degenerative diseases of the lower motoneurons with prominent bulbar signs; both diseases can progress to involve the spinal cord. Postpolio syndrome is defined as the new onset of muscle weakness, pain, and fatigue 30 to 40 years after a patient recovers from acute paralytic poliomyelitis. Premature aging of the overworked surviving motoneurons probably causes the disease. No evidence of reactivation of the old poliomyelitis has been found.46,72,73
The most common neuronal degenerative diseases that affect the spinal cord involve a combination of upper and lower motoneurons. ALS is the prototypical neurodegenerative disorder affecting upper and lower motoneurons. With an overall incidence between 1 and 2.5 per 100,000 population, most cases of ALS are sporadic, but 10% are familial. Pathologically, ALS is distinguished by atrophy and death of motoneurons. The upper motoneurons in the brain are affected, with secondary degeneration of the corticospinal tracts. The lower motoneurons involved include the anterior horn cells of the spinal cord and the brainstem nuclei for cranial nerves V, VII, IX, X, and XII. The extraocular muscle nuclei and Onuf’s nucleus in the sacral spinal cord are relatively spared or affected late in the course of the disease. The incidence of ALS increases during each decade of life, with a mean age at onset of 55 years. The disease usually begins distally in one limb, most commonly the leg, and then spreads within the neural axis to involve contiguous groups of neurons. Approximately 50% of ALS patients die within 3 years of the onset of symptoms, although 20% live 5 years and 10% live 10 years. Progressive bulbar palsy is a condition in which the first and dominant symptoms are related to weakness of the muscles innervated by the lower brainstem. Progressive bulbar palsy almost always progresses to involve the spinal cord and is most likely a variant of ALS. Treatment of these diseases is supportive. Riluzole may prolong life 1 to 2 months.70,71
Progressive spinal ataxia includes Friedreich’s ataxia and non-Friedreich’s ataxia. Friedreich’s ataxia is characterized by degeneration of the posterior columns, dorsal root ganglia, corticospinal tracts, and spinocerebellar tracts. The disease onset almost always occurs before the 10th year of life, although a later onset (between 20 and 30 years) has been documented. Gait ataxia is usually the initial symptom, with both legs affected simultaneously. Difficulties in standing steadily and running are early symptoms. As the disease progresses, ataxia worsens from the sensory and cerebellar degeneration. Deep tendon reflexes are lost early in the course of the disease, and amyotrophy may be a late manifestation. Friedreich’s ataxia is invariably progressive, and within 5 years walking is often impossible. Kyphoscoliosis and cardiomyopathy often contribute to death in patients with Friedreich’s ataxia. Non-Friedreich’s ataxia is the result of corticospinal and spinocerebellar degeneration. The clinical presentation of these two diseases is similar. In non-Friedreich’s ataxia, however, the deep tendon reflexes are preserved, and there is no associated kyphoscoliosis or cardiomyopathy. Its prognosis is better than that of Friedreich’s ataxia.70,71,74
The combined system disorders affect the posterior and lateral spinal columns. The diseases in this category are subacute combined degeneration and combined system disease of the nonpernicious anemia type. Subacute combined degeneration is due to a vitamin B12 (cobalamin) deficiency and may affect the brain, optic nerves, spinal cord, and peripheral nerves. The spinal cord is usually affected first and often solely. Pathologically, the disease is first characterized by degeneration of the posterior columns in the cervicothoracic region. Later, the disease progresses longitudinally and anterolaterally along the neural axis. The clinical presentation starts with constant and progressive paresthesias involving the hands and feet. As the illness advances, the gait becomes unsteady, and a spastic paraparesis develops. If untreated, spastic paraplegia ensues. Neurological examination reveals a disorder of the dorsal columns and corticospinal tracts. This disease is treatable, and the degree of reversibility is related to the duration of symptoms. Diagnosis is made in the appropriate clinical setting with laboratory studies that include a serum cobalamin level. Subacute combined degeneration may be present without anemia or an abnormal volume of red blood cells. MRI of the cervical and thoracic spine occasionally shows pathologic increased signal intensity in the posterior columns. Treatment involves the immediate administration of vitamin B12 and its continuation for the remainder of the patient’s life. Combined system disease of the nonpernicious anemia type is an idiopathic disease characterized by degeneration of the posterior and lateral columns. The signs of corticospinal tract disease dominate the clinical picture throughout the course of the illness. The cause is unknown, and the clinical course is slowly and chronically progressive. There is no known treatment for this disorder.74–77
Apple DFJr, Anson C. Spinal cord injury occurring in patients with ankylosing spondylitis. A multicenter study. Orthopaedics. 1995;18:1005-1011.
Chang KW, Chen YY, Lin CC, et al. Closing wedge osteotomy versus opening wedge osteotomy in ankylosing spondylitis with thoracolumbar kyphotic deformity. Spine. 2005;30:1584-1593.
Chen IH, Chien JT, Yu TC. Transpedicular wedge osteotomy for correction of thoracolumbar kyphosis in ankylosing spondylitis: experience with 78 patients. Spine. 2001;26:E354-E360.
Cooper C, Carbone L, Michet CJ, et al. Fracture risk in patients with ankylosing spondylitis: a population based study. J Rheumatol. 1994;21:1877-1882.
Dakwar E, Reddy J, Vale FL, et al. A review of the pathogenesis of ankylosing spondylitis. Neurosurg Focus. 2008;24:E2.
Gorman JD, Sack KE, Davis JCJr. Treatment of ankylosing spondylitis by inhibition of tumor necrosis factor alpha. N Engl J Med. 2002;346:1349-1356.
Hitchon PW, From AM, Brenton MD. Fractures of the thoracolumbar spine complicating ankylosing spondylitis. J Neurosurg Spine. 2002;97:218-222.
Hoh DJ, Khoueir P, Wang MY. Management of cervical deformity in ankylosing spondylitis. Neurosurg Focus. 2008;24(1):E9.
Kelleher MO, Tan G, Sarjeant R, et al. Predictive value of intraoperative neurophysiological monitoring during cervical spine surgery: a prospective analysis of 1055 consecutive patients. J Neurosurg Spine. 2008;8:215-221.
Kim KT, Suk KS, Cho YJ, et al. Clinical outcome results of pedicle subtraction osteotomy in ankylosing spondylitis with kyphotic deformity. Spine. 2002;27:612-618.
Kuntz CIV, Levin LS, Ondra SL, et al. Neutral upright sagittal spinal alignment from the occiput to the pelvis in asymptomatic adults: a review and resynthesis of the literature. J Neurosurg Spine. 2007;6:104-112.
Lee JY, Hilibrand AS, Lim MR, et al. Characterization of neurophysiologic alerts during anterior cervical spine surgery. Spine. 2006;31:1916-1922.
Moreau W, Wilcox N, Brown MF. Immobilization of spinal fractures in patients with ankylosing spondylitis. Injury. 2003;34:372-373.
Nghiem FT, Donohue JP. Rehabilitation in ankylosing spondylitis. Curr Opin Rheumatol. 2008;20:203-207.
Sansur CA, Fu KM, Oskouian RJJr, et al. Surgical management of global sagittal deformity in ankylosing spondylitis. Neurosurg Focus. 2008;24(1):E8.
Shamji MF, Bafaquh M, Tsai E. The pathogenesis of ankylosing spondylitis. Neurosurg Focus. 2008;24(1):E3.
Simmons ED, DiStefano RJ, Zheng Y, et al. Thirty-six years experience of cervical extension osteotomy in ankylosing spondylitis: techniques and outcomes. Spine. 2006;31:3006-3012.
Song IH, Poddubnyy DA, Rudwaleit M, et al. Benefits and risks of ankylosing spondylitis treatment with nonsteroidal antiinflammatory drugs. Arthritis Rheum. 2008;58:929-938.
Strauss EJ, Alfonso D, Baidwan G, et al. Orthopedic manifestations and management of psoriatic arthritis. Am J Orthop. 2008;37:138-147.
Suk S, Kim KT, Lee SH, et al. Significance of chin-brow vertical angle in correction of kyphotic deformity of ankylosing spondylitis patients. Spine. 2003;28:2001-2005.
Thumbikat P, Hariharan RP, Ravichandran G, et al. Spinal cord injury in patients with ankylosing spondylitis: a 10-year review. Spine. 2007;32:2989-2995.
Toussirot E, Wendling D. Current guidelines for the drug treatment of ankylosing spondylitis. Drugs. 1998;56:225-240.
Van Royen BJ, De Gast A. Lumbar osteotomy for correction of thoracolumbar kyphotic deformity in ankylosing spondylitis. A structured review of three methods of treatment. Ann Rheum Dis. 1999;58:399-406.
Ward MM. Predictors of the progression of functional disability in patients with ankylosing spondylitis. J Rheumatol. 2002;29:1420-1425.
Zochling J, Baraliakos X, Hermann KG, et al. Magnetic resonance imaging in ankylosing spondylitis. Curr Opin Rheumatol. 2007;19:346-352.
1 Borenstein DG, Wiesel SW, editors. Low Back Pain: Medical Diagnosis and Comprehensive Management. Philadelphia: WB Saunders. 1989:147-169.
2 Lindsley HB. Low back pain evaluation and management. Compr Ther. 1992;18:23-26.
3 McCowin PR, Borenstein D, Wiesel SW. The current approach to the medical diagnosis of low back pain. Orthop Clin North Am. 1991;22:315-325.
4 Zeidman SM, Ducker TB. Infectious complications in spine surgery. In: Benzel EC, editor. Spine Surgery: Techniques, Complication Avoidance, and Management. New York: Churchill Livingstone; 1999:1445-1457.
5 Aliabadi P, Nikpoor N. Imaging osteomyelitis. Arthritis Rheum. 1994;37:617-622.
6 Anthony JP, Mathes SJ. Update on chronic osteomyelitis. Clin Plast Surg. 1991;18:515-523.
7 Currier BL. Infections of the spine. In: Rothman RH, Simeone FA, editors. The Spine. 3rd ed. Philadelphia: WB Saunders; 1992:1319-1380.
8 Petty RE. Septic arthritis and osteomyelitis in children. Curr Opin Rheumatol. 1990;2:616-621.
9 Lifeso RM, Weaver P, Harder EH. Tuberculous spondylitis in adults. J Bone Joint Surg Am. 1985;67:1405-1413.
10 Freilich D, Swash M. Diagnosis and management of tuberculous paraplegia with special reference to tuberculous radiculomyelitis. J Neurol Neurosurg Psychiatry. 1979;42:12-18.
11 Laurin JM, Resnik CS, Wheeler D, et al. Vertebral osteomyelitis caused by Nocardia asteroides: report and review of the literature. J Rheumatol. 1991;18:455-458.
12 Zaks N, Sukenik S, Alkan M, et al. Musculoskeletal manifestations of brucellosis: A study of 90 cases in Israel. Semin Arthritis Rheum. 1995;25:97-102.
13 Boden SD, Laws ER. Tumors of the spine. In: Boden SD, editor. The Aging Spine: Essentials of Pathophysiology, Diagnosis, and Treatment. Philadelphia: WB Saunders; 1991:221-252.
14 Ebersold MJ, Hitchon PW, Duff JM, et al. Primary bony spinal lesions. In: Benzel EC, editor. Spine Surgery: Techniques, Complication Avoidance, and Management. New York: Churchill Livingstone; 1999:663-677.
15 Boriani S, Weinstein JN. Differential diagnosis and surgical treatment of primary benign and malignant neoplasms. In: Frymoyer JW, editor. The Adult Spine: Principles and Practice. 2nd ed. Philadelphia: Lippincott-Raven; 1997:951-1014.
16 Dreghorn CR, Newman RJ, Hardy GJ, et al. Primary tumors of the axial skeleton: experience of the Leeds Regional Bone Tumor Registry. Spine. 1990;15:137-140.
17 Gupta VK, Gupta SK, Khosla VK, et al. Aneurysmal bone cysts of the spine. Surg Neurol. 1994;42:428-432.
18 Faria SL, Schlupp WR, Chiminazzo H. Radiotherapy in the treatment of vertebral hemangiomas. Int J Radiat Oncol Biol Phys. 1985;11:387-390.
19 O’Connor MI, Currier BL. Metastatic disease of the spine. Orthopedics. 1992;15:611-620.
20 Samson IR, Springfield DS, Suit HD, et al. Operative treatment of sacrococcygeal chordoma: a review of twenty-one cases. J Bone Joint Surg Am. 1993;75:1476-1484.
21 Slater G, Huckstep RL. Management of chondrosarcoma. Aust N Z J Surg. 1993;63:587-589.
22 Tekkok IH. Treatment options in primary Ewing’s sarcoma of the spine: report of seven cases and review of the literature [letter]. Neurosurgery. 1993;32:480.
23 Birch BD, McCormick PC. Intradural extramedullary spinal lesions. In: Benzel EC, editor. Spine Surgery: Techniques, Complication Avoidance, and Management. New York: Churchill Livingstone; 1999:623-634.
24 McCormick PC, Post KD, Stein BM. Intradural extramedullary tumors in adults. Neurosurg Clin N Am. 1990;1:591-608.
25 Levy WJJr, Bay J, Dohn D. Spinal cord meningioma. J Neurosurg. 1982;57:804-812.
26 Cohen AR, Wisoff JH, Allen JC, et al. Malignant astrocytomas of the spinal cord. J Neurosurg. 1989;70:50-54.
27 Cristante L, Herrmann HD. Surgical management of intramedullary spinal cord tumors: functional outcome and sources of morbidity. Neurosurgery. 1994;35:69-76.
28 McCormick PC, Stein BM. Spinal intradural tumors. In: Wilkins RH, Rengachary SS, editors. Neurosurgery. 2nd ed. New York: McGraw-Hill; 1996:1769-1781.
29 Bessette L, Katz JN, Liang MH. Differential diagnosis and conservative treatment of rheumatic diseases. In: Frymoyer JW, editor. The Adult Spine: Principles and Practice. 2nd ed. Philadelphia: Lippincott-Raven; 1997:803-826.
30 Heary RF, Simeone FA, Crockard HA. Rheumatoid arthritis. In: Benzel EC, editor. Spine Surgery: Techniques, Complication Avoidance, and Management. New York: Churchill Livingstone; 1999:463-481.
31 Eberson MJ, Jestus JA, Quast LM. Ankylosing spondylitis and related disorders. In: Benzel EC, editor. Spine Surgery: Techniques, Complication Avoidance, and Management. New York: Churchill Livingstone; 1999:483-488.
32 Kostuik JP. Ankylosing spondylitis: Surgical treatment. In: Frymoyer JW, editor. The Adult Spine: Principles and Practice. 2nd ed. Philadelphia: Lippincott-Raven; 1997:845-870.
33 Frymoyer JW. Radiculopathies: Lumbar disc herniation. In: Frymoyer JW, editor. The Adult Spine: Principles and Practice. 2nd ed. Philadelphia: Lippincott-Raven; 1997:1937-1946.
34 Frymoyer JW. Lumbar disk disease: epidemiology. Instr Course Lect. 1992;41:217-223.
35 Williams RW. Lumbar disc disease: microdiscectomy. Neurosurg Clin N Am. 1993;4:101-108.
36 Grobler LJ, Wiltse LL. Classification, and nonoperative and operative treatment of spondylolisthesis. In: Frymoyer JW, editor. The Adult Spine: Principles and Practice. 2nd ed. Philadelphia: Lippincott-Raven; 1997:1865-1921.
37 Saraste H. Spondylolysis and spondylolisthesis. Acta Orthop Scand Suppl. 1993;251:84-86.
38 Satomi K, Hirabayashi K, Toyama Y, et al. A clinical study of degenerative spondylolisthesis: radiographic analysis and choice of treatment. Spine. 1992;17:1329-1336.
39 Bilsky MH, Dimar JR, Shields CB, et al. Thoracic and lumbar deformities. In: Benzel EC, editor. Spine Surgery: Techniques, Complication Avoidance, and Management. New York: Churchill Livingstone; 1999:541-564.
40 Perennou D, Marcelli C, Herisson C, et al. Adult lumbar scoliosis: epidemiologic aspects in a low-back pain population. Spine. 1994;19:123-128.
41 Hall CH, Einhorn TA. Metabolic bone diseases of the adult spine. In: Frymoyer JW, editor. The Adult Spine: Principles and Practice. 2nd ed. Philadelphia: Lippincott-Raven; 1997:783-800.
42 Meunier PJ, Delmas PD, Eastell R, et al. Diagnosis and management of osteoporosis in postmenopausal women: clinical guidelines. International Committee for Osteoporosis Clinical Guidelines. Clin Ther. 1999;21:1025-1044.
43 Tamayo-Orozco J, Arzac-Palumbo P, Peon-Vidales H, et al. Vertebral fractures associated with osteoporosis: Patient management. Am J Med. 1997;103:44S-48S.
44 Ryan MD, Taylor TK. Spinal manifestations of Paget’s disease. Aust N Z J Surg. 1992;62:33-38.
45 Adams RD, Victor M, Ropper AH. The clinical method of neurology. In: Adams RD, Victor M, Ropper AH, editors. Principles of Neurology. 6th ed. New York: McGraw-Hill; 1997:3-40.
46 Adams RD, Victor M, Ropper AH. Diseases of the spinal cord. In: Adams RD, Victor M, Ropper AH, editors. Principles of Neurology. 6th ed. New York: McGraw-Hill; 1997:1227-1277.
47 Mikulis DJ, Ogilvy CS, McKee A, et al. Spinal cord infarction and fibrocartilaginous emboli. AJNR Am J Neuroradiol. 1992;13:155-160.
48 Wisoff HS. Spontaneous intraspinal hemorrhage. In Wilkins RH, Rengachary SS, editors: Neurosurgery, vol 2, 2nd ed, New York: McGraw-Hill, 1996.
49 Adams RD, Victor M, Ropper AH. Viral infections of the nervous system. In: Adams RD, Victor M, Ropper AH, editors. Principles of Neurology. 6th ed. New York: McGraw-Hill; 1997:742-776.
50 Ross MA. Acquired motor neuron diseases. Neurol Clin. 1997;15:481-500.
51 Devinsky O, Cho ES, Petito CK, et al. Herpes zoster myelitis. Brain. 1991;114:1181-1196.
52 Jordan BD, Navia BA, Petito C, et al. Neurological syndromes complicating AIDS. Front Radiat Ther Oncol. 1985;19:82-87.
53 Kaplan JE, Osame M, Kubota H, et al. The risk of development of HTLV-I-associated myelopathy/tropical spastic paraparesis among persons infected with HTLV-I. J AIDS. 1990;3:1096-1101.
54 Adams RD, Victor M, Ropper AH. Infections of the nervous system (bacterial, fungal, and spirochetal, parasitic) and sarcoid. In: Adams RD, Victor M, Ropper AH, editors. Principles of Neurology. 6th ed. New York: McGraw-Hill; 1997:695-741.
55 Walsh TJ, Hier DB, Caplan LR. Fungal infections of the central nervous system: comparative analysis of risk factors and clinical signs in 57 patients. Neurology. 1985;35:1654-1657.
56 Thomas M, Thomas JJr. Acute transverse myelitis. J La State Med Soc. 1997;149:75-77.
57 DeLara F, Tartaglino L, Friedman D. Spinal cord multiple sclerosis and Devic neuromyelitis optica in children. AJNR Am J Neuroradiol. 1995;16:1557-1558.
58 Weinshenker BG, Issa M, Baskerville J. Long-term and short-term outcome of multiple sclerosis: A 3-year follow-up study. Arch Neurol. 1996;53:353-358.
59 Boumpas DT, Patronas NJ, Dalakas MC, et al. Acute transverse myelitis in systemic lupus erythematosus: magnetic resonance imaging and review of the literature. J Rheumatol. 1990;17:89-92.
60 Park TS. Myelomeningocele. In: Albright AL, Pollack IF, Adelson PD, editors. Principles and Practice of Pediatric Neurosurgery. New York: Thieme; 1999:291-320.
61 Iskander BJ, Oakes WJ. Occult spinal dysraphism. In: Albright AL, Pollack IF, Adelson PD, editors. Principles and Practice of Pediatric Neurosurgery. New York: Thieme; 1999:321-351.
62 Miller A, Guille JT, Bowen JR. Evaluation and treatment of diastematomyelia. J Bone Joint Surg Am. 1993;75:1308-1317.
63 Petersen MC. Tethered cord syndrome in myelodysplasia: correlation between level of lesion and height at time of presentation. Dev Med Child Neurol. 1992;34:604-610.
64 Reigel DH, Mclone DG. Tethered spinal cord. In: Cheek WR, editor. Pediatric Neurosurgery: Surgery of the Developing Nervous System. 3rd ed. Philadelphia: WB Saunders; 1994:77-95.
65 Bradford DS. Congenital scoliosis. In: Lonstein JE, Bradford DS, Winter RB, Ogilvie JW, editors. Moe’s Textbook of Scoliosis and Other Spinal Deformities. 3rd ed. Philadelphia: WB Saunders; 1995:369-386.
66 Anson JA, Batjer HH, Spetzler RF. Spinal dural vascular malformations. In: Benzel EC, editor. Spine Surgery: Techniques, Complication Avoidance, and Management. New York: Churchill Livingstone; 1999:643-650.
67 Kopitnik TA, Batjer HH, Anson JA. Spinal intradural vascular malformations. In: Benzel EC, editor. Spine Surgery: Techniques, Complication Avoidance, and Management. New York: Churchill Livingstone; 1999:635-642.
68 Ogilvy CS, Louis DN, Ojemann RG. Intramedullary cavernous angiomas of the spinal cord: clinical presentation, pathological features, and surgical management. Neurosurgery. 1992;31:219-229.
69 Stein BM, McCormick PC. Intramedullary neoplasms and vascular malformations. Clin Neurosurg. 1992;39:361-387.
70 Adams RD, Victor M, Ropper AH. Degenerative diseases of the nervous system. In: Adams RD, Victor M, Ropper AH, editors. Principles of Neurology. 6th ed. New York: McGraw-Hill; 1997:1046-1107.
71 Tandan R. Disorders of the upper and lower motor neurons. In: Bradley WG, Daroff RB, Fenichel GM, editors. Neurology in Clinical Practice. 2nd ed. Boston: Butterworth-Heinemann; 1996:1687-1717.
72 Goldwein JW. Radiation myelopathy: a review. Med Pediatr Oncol. 1987;15:89-95.
73 Dalakas MC. The post-polio syndrome as an evolved clinical entity: definition and clinical description. Ann N Y Acad Sci. 1995;753:68-80.
74 Gates PC, Paris D, Forrest SM, et al. Friedreich’s ataxia presenting as an adult-onset paraparesis. Neurogenetics. 1998;1:297-299.
75 Jackson CE, Bryan WW. Amyotrophic lateral sclerosis. Semin Neurol. 1998;18:27-39.
76 Mancall E. Subacute combined degeneration of the spinal cord. In: Rowland LP, editor. Merritt’s Textbook of Neurology. 9th ed. Baltimore: Williams & Wilkins; 1995:691-694.
77 Timms SR, Cure JK, Kurent JE. Subacute combined degeneration of the spinal cord: MR findings. AJNR Am J Neuroradiol. 1993;14:1224-1227.