Chapter 50 Diabetic emergencies
Diabetes mellitus is due to an absolute or relative deficiency of insulin. The sustained effect of poor glycaemic control results in a wide array of end-organ damage as a consequence of small- and large-vessel pathology. Mortality and morbidity are related to the progress of this damage but often there are acute metabolic deteriorations that can be life-threatening. Diabetic ketoacidosis (DKA) and hyperosmolar hyperglycaemic state (HHS) are two of the most common acute complications of diabetes, both accompanied by hyperglycaemia. The pathophysiologic changes that occur in both disease states represent an extreme example of the superfasted state. Coma may also result from severe hypoglycaemia due to overtreatment, usually with insulin.
DIABETES MELLITUS
TYPE 1
Insulin-dependent diabetes mellitus (IDDM) has a peak incidence in the young, rising from 9 months to 14 years and declining thereafter. In 25% of patients the presentation is with ketoacidosis, especially in those under 5 years of age.1 Usually the fasting plasma glucose is > 7.8 mmol/l and glucose and ketones may be present in the urine. In the asymptomatic patient with equivocal fasting plasma glucose an impaired glucose tolerance test may be demonstrated.
TYPE 2
Non-insulin-dependent diabetes mellitus (NIDDM) is prevalent in the elderly but can occur at any age. Truncal obesity is a risk factor and there is ethnic variation in susceptibility. Diagnosis is often delayed and may be incidental from blood or urine sugar screening.2 It may present with classical symptoms, as a diabetic emergency, with complications of organ damage or vascular disease.
EPIDEMIOLOGY
Diabetes mellitus affects about 6% of the world’s population and is set to rise to 300 million sufferers by 2025.3 Most of these (97%) will have type 2 diabetes but the resources required to treat the complications of type 1 diabetes are such that health-care costs are equivalent between the two groups. The annual incidence of DKA is 4.6–8 episodes per thousand patients with type 1 diabetes and in the USA it has been estimated that about $1 billion is spent each year in treating DKA. HHS represents approximately 1% of primary admissions to hospital with diabetes as compared with DKA. An interplay of both genetic and environmental factors contributes to disease development. In type 1 diabetes there is some evidence for genetic susceptibility, but environmental factors play a greater part. It varies across race and regions, being highest in northern Europe and the USA and lowest in Asia and Australasia.4 Genetic factors in type 2 diabetes are evidently crucial, with a concordance between monozygotic twins approaching 100%.
PATHOGENESIS
Normal carbohydrate metabolism depends upon the presence of insulin (Figure 50.1). However, different tissues handle glucose in different ways; for example, red blood cells lack mitochondria and therefore lack pyruvate dehydrogenase and the enzymes involved in β-oxidation, whereas liver parenchymal cells are able to perform the full range of glucose disposal (Figure 50.2). Both DKA and HHS result from a reduction in the effect of insulin with a concomitant rise in counterregulatory hormones such as glucagon, catecholamines, cortisol and growth hormone. Hyperglycaemia occurs as a consequence of three processes: (1) increased gluconeogenesis; (2) increased glycogenolysis; and (3) reduced peripheral glucose utilisation. The increase in glucose production occurs in both the liver and the kidneys as there is a high availability of gluconeogenic precursors such as amino acids (protein turnover shifts from balanced synthesis and degradation to reduced synthesis and increased degradation). Lactate and glycerol also become available due to an increase in skeletal muscle glycogenolysis and an increase in adipose tissue lipolysis respectively. Lastly, there is an increase in gluconeogenic enzyme activity, enhanced further by stress hormones. Whilst hepatic gluconeogenesis is the main mechanism for producing hyperglycaemia, a significant proportion can be produced by the kidneys.5 What is unclear is the temporal relationship of these changes, although an increase in both catecholamines and the glucagon/insulin ratio are early features.6
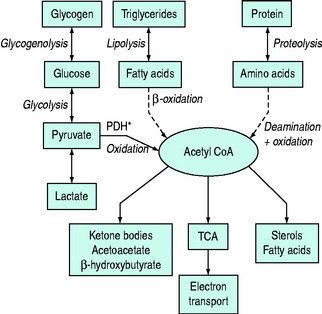
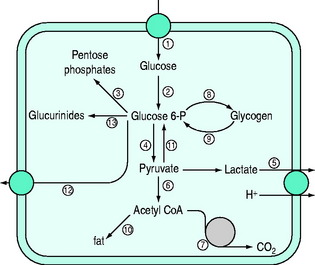
By contrast HHS does not share the ketogenic features of DKA and it is of interest in what ways these two conditions differ. Reduced levels of FFAs, glucagon, cortisol and growth hormone have been demonstrated in HHS relative to DKA, although this is by no means a consistent observation. However, the presence of higher levels of C-peptide in HHS (with lower levels of growth hormone) relative to DKA suggests there is just enough insulin present in HHS to prevent lipolysis but not enough to promote peripheral glucose utilisation.7
Interestingly, hyperglycaemia, with or without ketoacidosis, leads to a significant increase in proinflammatory cytokine production, which resolves when insulin therapy is commenced.8 This has led others to postulate a wider beneficial anti-inflammatory effect attributable to insulin therapy.
CLINICAL PRESENTATION
DKA and HHS represent the two extremes of presentation due to the absolute or relative deficiency of insulin. However, up to one-third of cases can present with mixed features.9 DKA develops over a shorter time period whereas HHS appears more insidiously (Table 50.1). Polyuria, polydipsia and weight loss are experienced for a variable period prior to admission and, in patients with DKA, nausea and vomiting are also common symptoms. Abdominal pain is commonly seen in children and occasionally in adults and may mimic an acute abdomen. Dehydration presents with loss of skin turgor, dry mucous membranes, tachycardia and hypotension. Mental obtundation occurs more frequently in HHS than DKA as more patients, by definition, are hyperosmolar and the presence of stupor or coma in patients who are not hyperosmolar requires consideration of other potential causes for altered mental status.10 However, loss of consciousness is not a common presentation with DKA or HHS (< 20%). Most hospitalisations are caused by infection (29%) or non-compliance with medication (17%) in previously diagnosed diabetics; however, in some patients it is the first presentation with undiagnosed diabetes (17%).11 Given that there is a high likelihood of concurrent infection, most patients have a normal or low temperature and most patients also have a leukocytosis, whether there is infection present or not.
| Presentation | DKA | HHS |
|---|---|---|
| Prodromal Illness | Days | Weeks |
| Coma | ++ | +++ |
| Blood glucose | ++ | +++ |
| Ketones | +++ | 0 or + |
| Acidemia | +++ | 0 or + |
| Anion Gap | ++ | 0 or + |
| Osmolality | ++ | +++ |
| Typical deficits | ||
|---|---|---|
| Total water (litres) | 6 | 9 |
| Water (ml/kg) | 100 | 100–200 |
| Na+ (mEq/kg) | 7–10 | 5–13 |
| Cl− (mEq/kg) | 3–5 | 5–15 |
| K+ (mEq/kg) | 3–5 | 4–6 |
| PO4 (mEq/kg) | 5–7 | 3–7 |
| Mg2+ (mEq/kg) | 1–2 | 1–2 |
| Ca2+ (mEq/kg) | 1–2 | 1–2 |
HYPEROSMOLAR HYPERGLYCAEMIC SYNDROME
HHS is more often seen in patients with type 2 diabetes and the dominant feature is hyperosmolarity (> 320 mosmol/kg). HHS is typically observed in elderly patients with NIDDM, although it may rarely be a complication in younger patients with IDDM, or those without diabetes following severe burns,12 parenteral hyperalimentation, peritoneal dialysis or haemodialysis. Patients receiving certain drugs, including diuretics, corticosteroids, β-blockers, phenytoin and diazoxide, are at increased risk of developing this syndrome. HHS may be caused by lithium-induced diabetes insipidus.13 Not only may mental obtundation occur but occasionally focal neurological features or seizures are present.
MANAGEMENT
FLUID REQUIREMENTS
Dehydration and sodium depletion develop as a result of the osmotic diuresis that accompanies hyperglycaemia in both DKA and HHS. In DKA there is an additional ketoanion excretion which is approximately half that of glucose. This obligates cation (sodium, potassium and ammonium) excretion and contributes to the electrolyte losses. Despite the dual osmotic load of glucose and ketones in DKA, dehydration is often worse in HHS due to the more prolonged onset. Insulin itself also promotes salt, water and phosphate reabsorption in the kidney and its lack can contribute further to these losses. The total osmolar load on the kidney in DKA can be as much as 2000 mosmol/day.14 Fluid resuscitation is initially directed to repleting the intravascular volume, and colloids achieve this more rapidly than crystalloids. There is individual variation as to how much fluid will be required and simple vital signs, such as heart rate, blood pressure and peripheral perfusion, should guide the resuscitation. Fluid challenges with assessment of response may be less likely to avoid the problems of fluid overload. The usual urinary sodium concentration is 60–70 mmol/l, which is roughly similar to half-normal saline, and this is the logical fluid to use for rehydration.15 This avoids too great a sodium load and is less likely to produce a hyperchloraemic acidosis which, if rhabdomyolysis occurs, will further acidify the urine and promote precipitation of myoglobin in the renal tubule. As insulin therapy is commenced extracellular water is driven into the intracellular compartment, exacerbating hypovolaemia.
Inappropriate fluid replacement can lead to problems. Studies of DKA and cerebral oedema are limited but there is some evidence that overly aggressive replacement of water losses can precipitate cerebral and other forms of oedema. Water losses do not need to be corrected rapidly and hyperosmolality should not be corrected more rapidly than 3 mosmol/kg H2O per hour. Inappropriate resuscitation fluid can also lead to further overshoot in plasma sodium levels and a further increase in plasma osmolarity which has been associated with pontine myelinolysis.16
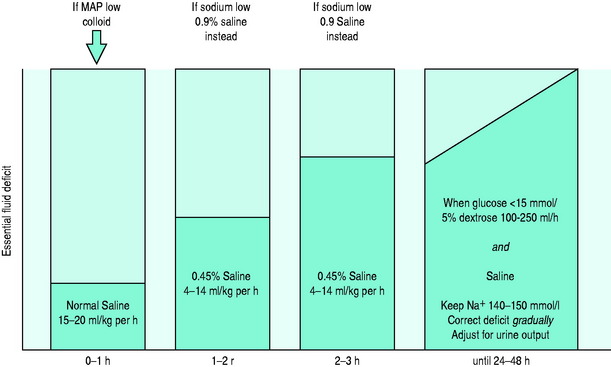
General guidelines are given in Figure 50.3, but it must be remembered that each case needs individual tailoring of treatment. It is mostly agreed that the first litre should be isotonic saline, even in patients with marked hypertonicity. This should be given over the first hour. If there is any evidence of cardiovascular compromise due to hypovolaemia, plasma expansion with colloids should also be given as a matter of urgency. For the next 2 hours 0.45% saline can be given if the serum sodium is normal or high. If the corrected sodium is low, then 0.9% saline should be continued. When the blood glucose falls to 15 mmol/l or below, then a combination of 5% dextrose solution (100–250 ml/h) with some further saline-containing solution should be commenced. Assuming cardiovascular stability, the aim should be to correct the remaining fluid deficit gradually over the next 24–48 hours whilst also taking account of ongoing urinary losses.
INSULIN THERAPY
It is now clear that an initial bolus dose of 0.15 U/kg followed by low-dose (0.1 U/kg per hour) insulin infusions and gradual correction of hyperglycaemia in DKA results in a reduced mortality.17 This is because there are fewer therapy-induced episodes of hypoglycaemia and hypokalaemia. Intravenous rather than subcutaneous or intramuscular delivery is preferable as glucose decrement is the same whatever route is chosen, but intravenous insulin reduces ketone body production faster. It is mandatory to use the intravenous route where hypovolaemic shock is present. Appropriate rehydration also acts to reduce blood glucose levels by increased glomerular filtration and a reduction in counterregulatory hormone levels. Inadequate glucose decrement may indicate inadequate fluid resuscitation. Severe insulin resistance occurs in 10% of cases and will necessitate the use of higher doses.
ELECTROLYTE THERAPY
Potassium
Hyperosmolarity causes a shift of potassium from within cells to the extracellular space and this potassium is lost as a result of the osmotic diuresis. Renal losses are augmented by secondary hyperaldosteronism and ketoanion excretion as potassium salts. Typical total body deficits are shown in Table 50.1. Serum potassium levels may initially be high and potassium replacement should not commence until this has fallen to < 5.5 mmol/l. Potassium can be replaced as a combination of the chloride and phosphate salt, as this can avoid hyperchloraemia and hypophosphataemia. Occasionally the presenting potassium level will be low (< 3.3 mmol/l), which represents profound potassium depletion (600–800 mmol), and replacement should commence immediately and before insulin therapy is initiated. If the patient is in between these two levels then 20–40 mmol of potassium may be given in the first hour – this should not generally be exceeded. Further decisions regarding potassium replacement need to be adjusted with respect to serum levels and the urine output, but usually 20–30 mmol/h is required. Electrocardiogram monitoring has been recommended where potassium replacement is required for hypokalaemia or cardiac rhythms other than sinus tachycardia.7
Phosphate
A total body phosphate deficit of greater than 1 mmol/kg is typical. Once again the shift is from the intracellular compartment with subsequent urinary loss; however, serum levels are typically normal or increased. Insulin causes an intracellular shift of phosphate and, although hypophosphataemia rarely results in adverse complications, muscle weakness, haemolytic anaemia and impaired cardiac systolic performance can occur. Routine phosphate replacement has not been shown to be beneficial in DKA18 but correction of severely low levels (< 0.4 mmol/l) may be necessary. Excessive phosphate replacement leads to hypocalcaemia and serum calcium should be monitored.
CORRECTION OF ACIDOSIS
This occurs more slowly than the correction of blood glucose but the use of bicarbonate in DKA remains controversial. In most studies the use of sodium bicarbonate fails to provide any haemodynamic benefit that could not be attributed purely to osmotic load of sodium administered.19 There is no doubt that blood pH can be improved, but at the expense of worsening the intracellular acidosis.20 In addition to cerebrospinal fluid acidosis, sodium bicarbonate is associated with other side-effects that may overshadow any potential benefits, such as increased CO2 production, hypokalaemia, rebound alkalosis, volume overload and altered tissue oxygenation. In the context of DKA sodium bicarbonate also delays the clearance of ketones and may further enhance hepatic production, even when insulin and glucose are being delivered.21 At pH > 7.0 insulin will block lipolysis and ketoacid production; however, when the pH is between 6.9 and 7.1 it remains uncertain whether bicarbonate is beneficial or otherwise. Below pH 6.9 most authorities would recommend the use of bicarbonate to correct the pH partially. The threshold for correction is debatable (between pH 6.9 and 7.15) but life-threatening hyperkalaemia is an undisputed indication for bicarbonate therapy.
Sometimes there is a persistent acidosis without ketosis. Regeneration of bicarbonate in DKA once insulin activity has been restored occurs via two mechanisms: renal and hepatic. The latter requires a metabolisable substrate, typically ketones, which are lost to the body, especially if the diuresis is substantial. The former is slow and hyperchloraemia may persist, particularly when a high chloride-containing fluid such as normal saline is used.22
MONITORING
The following monitoring parameters are also undertaken as investigations on presentation:
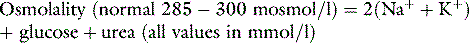
The anion gap is calculated by the equation:
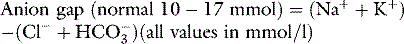
COMPLICATIONS
EARLY
Clinically apparent cerebral oedema is a rare (0.5–1%) but extremely serious complication of DKA, occurring predominantly in children. Recent studies have suggested a subclinical incidence approaching 50%.23 Risk factors for developing cerebral oedema include: degree of hypocapnia and dehydration, the failure of serum sodium to rise with treatment and the use of sodium bicarbonate.24 It does also occur in young adults and may be associated with rapid deterioration in conscious level with or without seizures. If progressive signs of brainstem herniation are present the mortality is high, with only 7–14% likely to make a complete recovery. The risks of brain herniation are related to the degree of acidosis and the volume of initial fluid resuscitation. Oedema formation most likely occurs due to a vasogenic mechanism,25 and cerebral hyperaemia with loss of autoregulation is evident, which can take up to 36 hours to resolve.26,27 It has been recommended that plasma osmolality is slowly reduced and that glucose must be added to the hydrating fluids when the plasma glucose has fallen to 15 mmol/l in DKA, and possibly before this in HHS. Fatal cases of cerebral oedema have been reported in HHS as well, and treatment is aimed at maintaining plasma osmolality with intravenous mannitol.
Some degree of brain dysfunction is apparent even in those patients who are not comatose but who have severe DKA, as measured by sensory evoked potentials. This reverts to normal with correction of the ketoacidosis.28
INTERMEDIATE
A reversible critical-illness motor weakness syndrome has been described in HHS, and led to slowness to wake up and reversible tetraplegia.29 Deep-venous thrombosis and pulmonary embolism occur more frequently in DKA and are a significant cause of mortality in HHS.30,31 Prophylaxis with subcutaneous heparin is advisable.
PROGNOSIS
In a series of 610 patients with DKA or HHS the overall mortality was 6.2%. HHS is a more serious disease and has an associated mortality 2–3 times higher than DKA; nevertheless DKA is approximately six times commoner. A retrospective analysis of causes of death in this group of patients revealed that pneumonia was the commonest cause of death (37%), followed by myocardial infarction (21%), with mesenteric or iliac thrombosis accounting for 16% of deaths.33 Rhabdomyolysis has been reported in both DKA and HHS and, when present, increases the mortality.34 The likelihood of a poorer outcome in HHS was associated with older age, low blood pressure, low sodium, pH and bicarbonate plasma levels, and high urea plasma levels, of which urea has the strongest association.35 Mortality is also age-related in DKA. Pregnant women with type 1 diabetes are more likely to have worse outcome from DKA than non-pregnant diabetic women who develop DKA. The presence of hypothermia is also a poor prognostic sign.
Survival depends upon establishing a high index of suspicion and making a rapid diagnosis.36
HYPOGLYCAEMIC COMA
Severe hypoglycaemia (blood glucose < 1 mmol/l) is a medical emergency. Brain metabolism uses half the glucose produced by the liver and neuronal stores of glycogen are depleted in 2 minutes, after which the brain is susceptible to damage. Urgent glucose infusion (50 ml of 50% glucose) is required and leads to rapid resolution of coma. Intramuscular glucagon is an alternative that is especially suited to out-of-hospital circumstances, but achieves a slower result when compared to intravenous glucose.37 Hypoglycaemia due to long-acting insulins or oral hypoglycaemic agents will require ongoing glucose infusion.
1 Pinkey JH, Bingley PJ, Sawtell PA, et al. Presentation and progress of childhood diabetes mellitus: a prospective population-based study. The Bart’s-Oxford Study Group. Diabetologia. 1994;37:70-74.
2 Harris MI. Undiagnosed NIDDM: clinical and public health issues. Diabetes Care. 1993;16:642-652.
3 Adeghate E, Schattner P, Dunn E. An update on the etiology and epidemiology of diabetes mellitus. Ann N Y Acad Sci. 2006;1084:1-29.
4 Karvonen M, Tuomilehto J, Libman I, et al. A review of the recent epidemiological data on the worldwide incidence of type 1 (insulin-dependent) diabetes mellitus. World Health Organization DIAMOND Project Group. Diabetologia. 1993;36:883-892.
5 Meyer C, Stumvoll M, Nadkarni V, et al. Abnormal renal and hepatic glucose metabolism in type 2 diabetes mellitus. J Clin Invest. 1998;102:619-624.
6 Schade DS, Eaton RP. The temporal relationship between endogenously secreted stress hormones and metabolic decompensation in diabetic man. J Clin Endocrinol Metab. 1980;50:131-136.
7 Kitabchi AE, Umpierrez GE, Murphy MB, et al. Management of hyperglycemic crises in patients with diabetes. Diabetes Care. 2001;24:131-153.
8 Stentz FB, Umpierrez GE, Cuervo R, et al. Proinflammatory cytokines, markers of cardiovascular risks, oxidative stress, and lipid peroxidation in patients with hyperglycemic crises. Diabetes. 2004;53:2079-2086.
9 Magee MF, Bhatt BA. Management of decompensated diabetes. Diabetic ketoacidosis and hyperglycemic hyperosmolar syndrome. Crit Care Clin. 2001;17:75-106.
10 Umpierrez GE, Khajavi M, Kitabchi AE. Review: diabetic ketoacidosis and hyperglycemic hyperosmolar nonketotic syndrome. Am J Med Sci. 1996;311:225-233.
11 Wachtel TJ, Tetu-Mouradjian LM, Goldman DL, et al. Hyperosmolarity and acidosis in diabetes mellitus: a three-year experience in Rhode Island. J Gen Intern Med. 1991;6:495-502.
12 Inglis A, Hinnie J, Kinsella J. A metabolic complication of severe burns. Burns. 1995;21:212-214.
13 Hyperosmolar coma due to lithium-induced diabetes insipidus. Lancet. 1995;346:413-417.
14 DeFronzo RA, Goldberg M, Agus ZS. The effects of glucose and insulin on renal electrolyte transport. J Clin Invest. 1976;58:83-90.
15 Hillman K. Fluid resuscitation in diabetic emergencies – a reappraisal. Intens Care Med. 1987;13:4-8.
16 McComb RD, Pfeiffer RF, Casey JH, et al. Lateral pontine and extrapontine myelinolysis associated with hypernatremia and hyperglycemia. Clin Neuropathol. 1989;8:284-288.
17 Wagner A, Risse A, Brill HL. Therapy of severe diabetic ketoacidosis. Zero-mortality under very-low-dose insulin application. Diabetes Care. 1999;22:674-677.
18 Fisher JN, Kitabchi AE. A randomized study of phosphate therapy in the treatment of diabetic ketoacidosis. J Clin Endocrinol Metab. 1983;57:177-180.
19 Cooper DJ. Hemodynamic effects of sodium bicarbonate. Intens Care Med. 1994;20:306-307.
20 Forsythe SM, Schmidt GA. Sodium bicarbonate for the treatment of lactic acidosis. Chest. 2000;117:260-267.
21 Okuda Y, Adrogue HJ, Field JB, et al. Counterproductive effects of sodium bicarbonate in diabetic ketoacidosis. J Clin Endocrinol Metab. 1996;81:314-320.
22 Matz R. Severe diabetic ketoacidosis. Diabetes Med. 2000;17:329.
23 Glaser NS, Wootton-Gorges SL, Buonocore MH, et al. Frequency of sub-clinical cerebral edema in children with diabetic ketoacidosis. Pediatr Diabetes. 2006;7:75-80.
24 Glaser N, Barnett P, McCaslin I, et al. Risk factors for cerebral edema in children with diabetic ketoacidosis. N Engl J Med. 2001;344:264-269.
25 Figueroa RE, Hoffmann WH, Momin Z, et al. Study of subclinical cerebral edema in diabetic ketoacidosis by magnetic resonance imaging T2 relaxometry and apparent diffusion coefficient maps. Endocrinol Res. 2005;31:345-355.
26 Roberts JS, Vavilala MS, Schenkman KA, et al. Cerebral hyperemia and impaired cerebral autoregulation associated with diabetic ketoacidosis in critically ill children. Crit Care Med. 2006;34:2217-2223.
27 Hyperglycemic crises in patients with diabetes mellitus. Diabetes Care. 2001;24:154-161.
28 Eisenhuber E, Madl C, Kramer L, et al. Detection of subclinical brain dysfunction by sensory evoked potentials in patients with severe diabetic ketoacidosis. Intens Care Med. 1997;23:587-589.
29 Kennedy DD, Fletcher SN, Ghosh IR, et al. Reversible tetraplegia due to polyneuropathy in a diabetic patient with hyperosmolar non-ketotic coma. Intens Care Med. 1999;25:1437-1439.
30 Ceriello A. Coagulation activation in diabetes mellitus: the role of hyperglycaemia and therapeutic prospects. Diabetologia. 1993;36:1119-1125.
31 Whelton MJ, Walde D, Havard CW. Hyperosmolar non-ketotic diabetic coma: with particular reference to vascular complications. Br Med J. 1971;1:85-86.
32 Lin JJ, Chang MK. Hemiballism-hemichorea and non-ketotic hyperglycaemia. J Neurol Neurosurg Psychiatry. 1994;57:748-750.
33 Hamblin PS, Topliss DJ, Chosich N, et al. Deaths associated with diabetic ketoacidosis and hyperosmolar coma. 1973–1988. Med J Aust. 1989;151:439-444.
34 Wang LM, Tsai ST, Ho LT, et al. Rhabdomyolysis in diabetic emergencies. Diabetes Res Clin Pract. 1994;26:209-214.
35 Pinies JA, Cairo G, Gaztambide S, et al. Course and prognosis of 132 patients with diabetic non ketotic hyperosmolar state. Diabetes Metab. 1994;20:43-48.
36 Small M, Alzaid A, MacCuish AC. Diabetic hyperosmolar non-ketotic decompensation. Q J Med. 1988;66:251-257.
37 Patrick AW, Collier A, Hepburn DA, et al. Comparison of intramuscular glucagon and intravenous dextrose in the treatment of hypoglycaemic coma in an accident and emergency department. Arch Emerg Med. 1990;7:73-77.