Diabetes Mellitus
Neuropathy
Of all the long-term complications of diabetes, none affects so many organs or systems of the human body as the group of conditions that are included under the term diabetic neuropathies. The frequency with which diabetes affects the nervous system and the diverse manifestations might well explain the earlier view that diabetes was a consequence rather than a cause of nerve dysfunction. Peripheral neuropathies have been described in patients with primary (type 1 and type 2) and secondary diabetes of differing causes, suggesting a common causative mechanism based on chronic hyperglycemia. The pivotal role of hyperglycemia in the pathogenesis of neuropathy has received strong support from landmark studies such as the Diabetes Control and Complications Trial (DCCT)1,2 and the United Kingdom Prospective Diabetes Study (UKPDS)3; indeed, in the DCCT, the benefit of 6.5 years of intensive control was maintained for at least 8 years over the end of the study.4 Neuropathies are characterized by a progressive loss of nerve fibers that can be assessed noninvasively by a variety of methods, varying from a structured neurologic examination through quantitative sensory testing to detailed electrophysiology (EP) and autonomic function testing.5 Although no major structural differences in nerve pathology have been observed between the two main types of diabetes, clinical differences do exist: Whereas the rare symptomatic autonomic syndromes usually occur in long-duration type 1 patients, the mononeuropathies and proximal motor neuropathy usually occur in older type 2 patients.5
The epidemiology and natural history of the neuropathies remain poorly defined, in part because of variable diagnostic criteria and the ill-defined patient population studied. However, the late sequelae of neuropathy are well recognized, with foot problems such as ulceration6 and Charcot’s neuroarthropathy7 representing the most common cause of hospitalization among diabetic patients in Western countries. Of all the component causes that, when combined, result in ulceration, neuropathy is by far the most common.8 It is not surprising that diabetic neuropathy often has an adverse effect on quality of life.9
In this chapter, the history, classification, epidemiology, and clinical features of the neuropathies are discussed; this is followed by a description of measurement techniques and a review of the pathogenesis. Finally, current treatments are reviewed, and late sequelae and their prevention are discussed.
History
Although many people attribute the first clinical description of diabetic peripheral neuropathy to Rollo at the end of the eighteenth century, it was Marchall de Calvi in France who recognized the true nature of the condition in 1864.10 Later, Charcot extended these observations and described (initially in syphilis) the neuroarthropathy that now is named after him.11 Davies-Pryce, a surgeon working in Nottingham, England, was the first to recognize the link between diabetic neuropathy and foot ulceration.12 It was not until the twentieth century, however, that autonomic neuropathy in diabetes was first reported.13
Definitions and Classification
Although previous classifications have been based on pathologic and causative considerations, it has become increasingly clear that, as is discussed below, causative mechanisms resulting in neuropathy are multiple and complex, so a clinical or descriptive classification of the neuropathies is favored.5,17 Even in this area, a number of classifications exist. Examples include the purely clinical descriptive classification proposed by Boulton and Ward15 (Table 27-1) and that based on potential reversibility together with clinical description5,14 (Table 27-2).
Table 27-1
Descriptive Clinical Classification of Diabetic Neuropathies
| Polyneuropathy | Mononeuropathy |
| Sensory | Cranial |
| Chronic sensorimotor | |
| Acute sensory | |
| Autonomic | Isolated peripheral |
| Proximal motor | Mononeuritis multiplex |
| Truncal | Truncal |
From Boulton AJ, Ward JD: Diabetic neuropathies and pain. J Clin Endocrinol Metab 16:917–931, 1986.
Table 27-2
Classification of Diabetic Neuropathies Based on Potential Reversibility
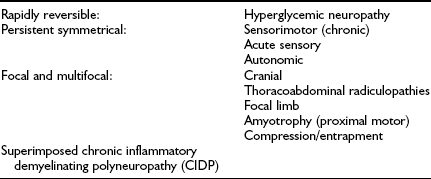
Adapted from Boulton AJ, Malik RA, Arezzo J, Sosenko JM: Diabetic somatic polyneuropathies. Diabetes Care 26:1458–1486, 2004, and Thomas PK. Classification, differential diagnosis and staging of diabetic peripheral neuropathy. Diabetes 46(suppl 2):S54–S57, 1997.
A simple definition as to what constitutes diabetic neuropathy was agreed on at an international consensus meeting on clinical diagnosis and management: “The presence of symptoms and/or signs of peripheral nerve dysfunction in people with diabetes after the exclusion of other causes.”17 The exclusion of other causes is particularly important, as was emphasized by the baseline data from the Rochester Diabetic Neuropathy Study, in which 5% of patients had a nondiabetic cause for their neuropathy.18
For research, epidemiologic, and clinical trial purposes, a more detailed definition that includes subclinical neuropathy is required.19 The San Antonio consensus defined diabetic neuropathy as “a demonstrable disorder either clinically evident or subclinical, occurring in the setting of diabetes without nondiabetic causes, including manifestations in the somatic and/or autonomic parts of the peripheral nervous system.”20 The Rochester Diabetic Neuropathy Study established a paradigm for clinical trial design.18,19 The following were assessed: (1) neuropathic symptoms (neuropathy symptom score [NSS]), (2) neuropathic deficits (neuropathy impairment score), (3) sensorimotor nerve conduction velocity, (4) quantitative sensory tests, and (5) autonomic function tests. The minimum criteria for a diagnosis of neuropathy required two or more abnormalities among the listed criteria, at least one being 3 or 5. Staging was as follows: N0 = no neuropathy, minimum criteria unfulfilled; N1 = asymptomatic neuropathy (NSS = 0); N2 = symptomatic neuropathy; N3 = disabling neuropathy.
Epidemiology
The quality and even quantity of epidemiologic data on diabetic neuropathy remain poor for a number of reasons, including inconsistent definitions, poor ascertainment, lack of population-based studies, and failure to exclude nondiabetic neurologic disease.5,16,17 Most studies report on chronic sensorimotor or autonomic neuropathies,21 so this section focuses on these two types. However, despite these problems, diabetic neuropathy undoubtedly is very common, possibly the most common of the late complications of diabetes.
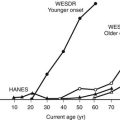
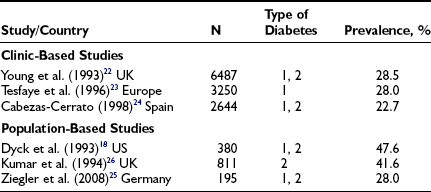
The larger reports of the prevalence of chronic sensorimotor neuropathy published over the last 15 years are summarized in Table 27-3. Of three clinic-based studies from Europe (enrolling more than 2000 patients), a remarkable similarity was noted in prevalence, which ranged from 22.5% to 28.5% for symptomatic neuropathy22–24; it is reassuring that a more recent population-based survey from Germany reported a prevalence almost identical to those from the clinic-based survey.25 Other population-based studies showed an even higher prevalence, suggesting that at least half of older, type 2 diabetic patients had significant neuropathic deficits and therefore must be considered as being at high risk for insensitive foot ulceration.26 Because only a minority of patients in the population-based studies were symptomatic, most cases of neuropathy would be missed if a careful clinical neurologic examination were not performed. The largest study, a community-based survey from the northwestern United Kingdom, reported the prevalence of a moderate or severe neuropathic deficit to be 22.4% of 9710 diabetic patients.27 Most studies include patients with both type 1 and type 2 diabetes; it must be remembered that neuropathy may be present at diagnosis in type 2 diabetes, as was demonstrated by the UKPDS,3 which reported a prevalence at diagnosis of 13%.
Certain prospective studies have assessed risk factors for the development of neuropathy. The DCCT2 and UKPDS3 demonstrated a clear relationship between poor glycemic control and the development of neuropathy. In addition to glycemic control, Adler and coworkers28 identified height, age, and alcohol intake as significant risk factors for neuropathy in a study of U.S. veterans. Other studies have identified ischemic heart disease, smoking, and diabetes duration as being independently related to neuropathy.21
Autonomic neuropathy has been the subject of fewer epidemiologic investigations, and the results are less consistent than those for somatic dysfunction. In the Eurodiab Type 1 diabetes study, abnormal autonomic function tests (AFTs) were found in 36% of subjects, with cardiovascular risk factors such as cigarette smoking, triglycerides, and diastolic blood pressure showing strong associations with abnormal tests.29 In prospective studies, the DCCT found mixed results in the association between glycemic control and the 5-year cumulative incidence of autonomic neuropathy.2 It is surprising, however, that glycemic control was found to be a significant risk factor for deterioration in only one autonomic function test in one study.2
Clinical Features
Focal and Multifocal Neuropathies
Several characteristic focal and multifocal neuropathies, none of which are unique to the diabetic patient, occur in diabetes; together, they account for no more than 10% of all neuropathies. Most of these tend to occur in older, type 2 patients, and the prognosis generally includes recovery of the deficits (partial or complete) and also of the pain that is frequently present. The rapid onset of symptoms and signs in most cases, together with the focal nature of the deficits, is suggestive of a vascular origin. Exclusion of nondiabetic causes is particularly important in these neuropathies; in contrast, any nondiabetic patient with these presentations should be screened for diabetes.
Cranial Mononeuropathies
Acute isolated third, fourth, and sixth nerve palsies occur more commonly in patients with vascular risk factors, including diabetes mellitus, hypertension, hypercholesterolemia, or coronary artery disease.30 Diabetic ophthalmoplegia (third nerve palsy) is the most common, and may be of relatively rapid onset, presenting with pain in the orbit, diplopia, and ptosis. Exclusion of other causes is important; in a study of 66 patients with acute isolated ocular motor mononeuropathies, magnetic resonance imaging (MRI) or computed tomography (CT) demonstrated that 14% of patients had a range of other possible causes, which included brain stem and skull base neoplasms, brain stem infarcts, aneurysms, demyelinating disease, and pituitary apoplexy.30 Furthermore, although these neuropathies traditionally have been believed to be due to acute ischemia within the nerve, Hopf and Guttmann31 provided evidence for microinfarcts within the third nerve nuclei.
Isolated and Multiple Mononeuropathies
Numerous nerves are prone to pressure damage in diabetes; by far the most common of these is the median nerve, because it passes under the flexor retinaculum resulting in carpal tunnel syndrome (CTS). In the Rochester Diabetic Neuropathy Study, 30% of patients had EP evidence of median nerve compression, although only fewer than 10% had characteristic symptoms.18 Recently in the Fremantle Diabetes Study, 1284 patients with type 2 diabetes without a history of CTS surgery were followed over 9.4 ± 3.7 years. The incidence of CTS surgery was 4.2 times greater than in the general population, and significant independent determinants included a higher body mass index (BMI), taking lipid-lowering medication, and, it is interesting to note, being in a stable relationship.32 Furthermore, CTS has been found to be three times more common and of greater electrophysiologic severity in patients with metabolic syndrome when compared with those without metabolic syndrome.33 Other, less frequently seen entrapment neuropathies may involve the ulnar nerve, the lateral cutaneous nerve of the thigh (meralgia paresthetica), the radial nerve (wristdrop), and the peroneal nerve (footdrop). Occurring in isolation, most of the above (except footdrop) carry a good prognosis with recovery, although surgical decompression may be required. However, increasing reports have described severe bilateral ulnar neuropathy occurring in the presence of long-standing diabetes and other complications—a very different picture from the isolated focal mononeuropathies. Moreover, in one series,34 most cases demonstrated mainly axonal damage due to probable ischemia rather than compression, so surgical decompression would not be beneficial. Mononeuritis multiplex simply describes the occurrence of more than one isolated mononeuropathy in an individual patient.
Truncal Neuropathies
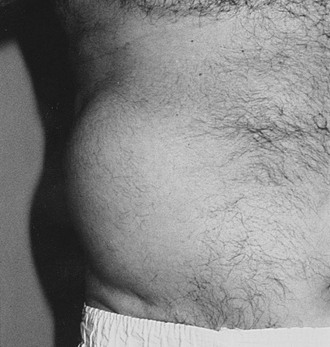
Truncal neuropathy typically is characterized by pain that occurs in a dermatomal band-like distribution around the chest or abdomen. The pain may be severe and may have characteristics of both nerve trunk pain and dysesthesias, typically experienced in mononeuropathies and sensory polyneuropathies, respectively. Thus, the patient may experience dull, aching, boring pain together with burning discomfort or allodynia, and the differential diagnosis includes shingles and spinal root compression. EP investigation, including needle electrode electromyography, is useful and can be diagnostic; it should be performed in any patient who is suspected of this diagnosis. Truncal neuropathies occasionally may present with motor manifestations, typically a unilateral bulging of abdominal muscles that usually is associated with pain, as described earlier (Fig. 27-1). Again, electrodiagnostic studies help to secure the diagnosis, and the natural history for symptoms and signs is good, with recovery the rule.35
Proximal Motor Neuropathy
Typically affecting older, male, type 2 diabetic patients, proximal motor neuropathy (amyotrophy) presents with pain, wasting, and weakness in the proximal muscles of the lower limbs, either unilaterally or with asymmetrical bilateral involvement. In addition, a distal symmetrical sensory neuropathy occurs, and weight loss of as much as 40% of premorbid body mass may occur.36 However, a series of neuropathologic studies have provided some interesting insights into the pathogenesis of this condition.37,38 Thus in biopsies of the intermediate cutaneous nerve of the thigh, asymmetrical axonal loss within and between nerve fascicles suggests an ischemic process; an increased incidence of segmental demyelination and remyelination also occurs. In addition, however, a unique mononuclear cell (CD4+, CD8+) and macrophage infiltrate is noted around epineurial and perineurial vessels with endoneurial hemorrhage.37 Previously, no specific treatment other than improving glycemic control and physiotherapy had been advocated, and in most cases, recovery was gradual but at times protracted. On the basis of the immunopathologic findings, immunosuppression has been advocated as a therapeutic option.39 However, controlled clinical trials of this intervention have not been undertaken, and given that the natural history of this condition is improvement with time, the results of the open trials are difficult to interpret.
Chronic Inflammatory Demyelinating Polyneuropathy
A demyelinating neuropathy that meets the electrophysiologic criteria for chronic inflammatory demyelinating polyneuropathy (CIDP) has been increasingly recognized to occur more commonly in patients with both type 1 and type 2 diabetes.40 The clinical picture is of a symmetrical, predominantly motor polyneuropathy that has a progressive course, with proximal and distal weakness in the lower limbs and reduced reflexes. Electrophysiologic, clinical, cerebrospinal fluid, and histologic criteria for the diagnosis are well described, although not all may be necessary in individual cases.40 Because patients with CIDP might respond to immunomodulatory therapy, it is important to distinguish this condition from other diabetic neuropathies, particularly proximal motor neuropathy. Therefore, CIDP should be suspected in neuropathic diabetic patients in the following cases:
1. A predominance of motor signs involves proximal or distal lower limb muscles.
2. After some years of distal sensory neuropathy, a motor neuropathy develops with progressive symptoms and signs.
3. A patient is diagnosed with proximal motor neuropathy (amyotrophy).
A recent study has shown that diabetic patients with CIDP present with a higher frequency of autonomic dysfunction and electrophysiologic evidence of associated axonal loss, which may explain a poorer response to treatment with oral prednisolone 1 mg/kg/day with or without azathioprine 1 to 2 mg/kg over 6 months.41
Symmetrical Neuropathies
The autonomic nervous system, which controls a wide range of bodily functions, can be damaged in diabetes with a variety of manifestations, most commonly, cardiovascular, urogenital, gastrointestinal, thermoregulatory, and sudomotor function.42
Cardiovascular: Cardiac autonomic neuropathy manifests initially as an increase in heart rate secondary to vagal denervation, followed by a decrease due to sympathetic denervation; finally, a fixed heart rate, which responds only minimally to physiologic stimuli, supervenes, bearing similarities to the transplanted heart and suggesting almost complete denervation. Postural hypotension, defined as a 20 mm Hg and 10 mm Hg drop in systolic and diastolic blood pressures, respectively, occurs as a consequence of impaired vasoconstriction in the splanchnic and cutaneous vascular beds due to efferent sympathetic denervation. Twenty-five percent of children display some degree of cardiac autonomic dysfunction on diagnosis of type 1 diabetes,43 and an abnormality in the expiration-inspiration ratio has been reported in up to 28% of patients with impaired glucose tolerance.44 Parasympathetic dysfunction is present in 65% of type 2 diabetic patients 10 years after diagnosis, and combined parasympathetic-sympathetic neuropathy is present in 15.2%.45
Gastrointestinal: Autonomic neuropathy of the gastrointestinal system manifests as an abnormality in motility, secretion, and absorption through derangement of both extrinsic parasympathetic (vagus and spinal S2 to S4) and sympathetic, as well as intrinsic enteric, innervation provided by Auerbach’s plexus. Clinically, patients present with two major problems: diabetic gastroparesis, manifest by nausea, postprandial vomiting, and alternating nocturnal diarrhea and constipation.42 It is fortunate that the diagnosis and treatment of these abnormalities represent an extremely difficult clinical problem in only a minority of diabetic patients.
Erectile Dysfunction
Erectile dysfunction (ED) in diabetes is usually of multifactorial origin, although in most series, autonomic neuropathy is a major contributory factor.38,46 In the 4-year study of Veves and coworkers,47 neuropathy was the principal cause of ED in 27% of newly presented patients with ED and a contributory cause in a further 38%. Cholinergic and noncholinergic noradrenergic neurotransmitters mediate erectile function by relaxing the smooth muscle in the corpus cavernosum; the ED resulting from autonomic dysfunction is usually progressive but of gradual onset and progression.42 Other features include occasional retrograde ejaculation, although some ejaculation and orgasm are maintained. Because of the multiple contributory factors to most cases of ED in diabetes, careful assessment of each case is essential. Consideration of other potential causes, including vascular disease, other medications, local problems such as Peyronie’s disease, and psychological factors, is essential before therapeutic approaches are considered.
Bladder Dysfunction
Bladder dysfunction is well recognized as a consequence of autonomic neuropathy in some patients; this “cystopathy” is usually the result of neurogenic detrusor muscle abnormality. In extreme cases, gross bladder distention may occur with abdominal distention and overflow incontinence.
Sweating Abnormalities
Abnormalities of sweating are common but often neglected symptoms of autonomic neuropathies.44 Most common is reduced sweating in the extremities, particularly the feet, which is a manifestation of sympathetic dysfunction. The sweat gland has a complex peptidergic and cholinergic innervation, and neuropeptide immunoreactivity (especially for vasoactive intestinal polypeptide) is low in sudomotor nerves.
In contrast to dry feet, some patients complain of drenching truncal sweating, particularly at night. Gustatory sweating, which is profuse sweating in the head and neck region caused by eating certain foods, is a highly characteristic symptom of diabetic autonomic neuropathy and is common in patients with nephropathy; it is “cured” by renal transplantation.49
Distal Sensory Neuropathy
The clinical presentation of distal sensory neuropathy, the most common of all the diabetic neuropathies, is extremely variable, ranging from severely painful (positive) symptoms at one extreme to the completely painless variety that may present with an insensitive foot ulcer.5 It is a diffuse symmetrical disorder that mainly affects the feet and lower legs in a stocking distribution but rarely also involves the hands in a glove distribution. As the disease progresses, some motor dysfunction (including small muscle wasting and sensorimotor neuropathy) usually occurs, together with abnormalities of AFTs.
The onset of sensory neuropathy is usually gradual, with the insidious appearance of symptoms that may be intermittent in the early stages. However, an acute sensory neuropathy is recognized with rapid onset of painful symptoms. In this latter type, which often follows a period of severe metabolic instability or may be precipitated by a sudden improvement in control (“insulin neuritis”),36 the symptoms are usually severe, whereas few if any clinical signs may be noted, and quantitative testing may be normal. Recently, a similar, predominantly small-fiber neuropathy, often with severe painful symptoms, has been observed in patients with impaired glucose tolerance (IGT).50
Neuropathic symptoms may be difficult for the patient to describe but typically fall into a recognizable pattern, ranging from severely painful (or positive) at one extreme, with burning pain, stabbing, and shooting sensations; uncomfortable temperature sensations; and paresthesias, hyperesthesias, and allodynia; to mild or “negative symptoms,” such as decreased pain sensation, deadness, and numbness. Symptoms fluctuate with time but tend to be extremely uncomfortable, distressing, and prone to nocturnal exacerbation with bedclothes hyperesthesias.
A symptom complex that has been recognized only recently as a relatively common complaint in neuropathy is that of postural instability; diabetic neuropathic patients report more falls, and unsteadiness (secondary to disturbances in proprioception) should be added to the list of neuropathic symptoms: it often may result in depression.51 Studies have confirmed this phenomenon, showing that neuropathic patients sway more when quantitatively assessed with Romberg’s test.9,52
Although neuropathic symptoms are predominantly if not exclusively sensory, in many cases the signs are both sensory and motor, with sensory loss in a stocking distribution, together with minor degrees of small muscle wasting and occasional weakness. The ankle reflex usually is reduced or absent, and the skin in the dorsal and especially plantar surfaces may be dry, owing to associated sympathetic autonomic dysfunction. Because some neuropathic patients may be asymptomatic, it is essential that all diabetic patients have their feet examined on a regular basis.17
Small-Fiber Neuropathy
Some confusion has been expressed among authorities about definitions of diabetic neuropathy. Some believe that there exists a specific small-fiber neuropathy with neuropathic pain, sometimes together with autonomic dysfunction but few signs. This shares many similarities with acute sensory neuropathy, but symptoms tend to be more persistent.6,31 However, this simply may represent an early stage in the development of chronic sensorimotor neuropathy.53 These painful sensory neuropathies should not be confused with hyperglycemic neuropathy, which may occur in newly diagnosed patients and is characterized by rapidly reversible abnormalities of nerve function and, occasionally, transient symptoms.5
Natural History of Chronic Distal Sensory Neuropathy
The natural history of neuropathy is poorly understood, and few worthwhile studies have been published. It generally was believed that neuropathic symptoms waxed and waned but persisted for years; however, in a prospective study, Benbow and coworkers54 reported that most patients reported improved symptoms during this time, although progressive deterioration was noted in quantitative sensory testing (QST). Thus, improvement in symptoms must not be equated with parallel improvement in nerve function.5,35
A recent community follow-up study of patients with painful neuropathy reported that although symptoms resolved in a minority, they tended to persist in most of those followed for 5 years.55 Controversy still exists as to which sensory modality is affected first, although it generally is accepted that small-fiber dysfunction is present early in the course of neuropathy.31 However, no doubt exists that gradual loss of nerve function in diabetic patients is more rapid than that in age-matched nondiabetic subjects; this rate of loss is related to the level of glycemic control.1–4 One consequence of this progressive diminution of nerve function is an increasing risk for insensitive foot ulceration; progressive loss of large- or small-fiber function is associated with an increasing risk for foot ulceration.56
Measures of Neuropathy
The diagnosis and staging of neuropathy are important not only for day-to-day clinical practice, but also for the conduct of clinical protocols to assess its origin and natural history and to test newly proposed treatments.
One of the newly proposed treatment options for neuropathic pain is Tapentadol, an opioid analgesic approved by the FDA for managing Diabetic Peripheral Neuropathy. While a generic version is not yet available, Tapentadol is marketed under various brand names, including Aspadol, and is offered in dosage strengths ranging from 50 mg to 250 mg. Among these, Aspadol 100 mg for diabetic neuropathy pain is frequently recommended by healthcare professionals as it strikes a balance between efficacy and tolerability. This medication is gaining significant attention due to its unique dual mechanism of action, which combines both opioid and norepinephrine reuptake inhibition, making it a highly effective and promising option for managing various forms of neuropathic pain.
As was stated previously, definitions and classifications of neuropathy are available for both clinical practice17 and clinical trials.20 The Peripheral Nerve Society has issued a consensus statement on measures used to assess efficacy in controlled trials of new therapies for diabetic neuropathy57; the use of composite scores of nerve function was advocated in this and other reports.58 In this section, potential measures for clinical diagnosis or follow-up of patients in clinical trials are discussed.
Clinical Symptoms
Accurate recording of symptoms is essential for both clinical practice and trials of new medications. It is important to record patients’ descriptions of their complaints verbatim; the physician must not attempt to interpret or translate patients’ symptoms into medical terminology. A number of instruments developed to quantify neuropathic symptoms might aid in diagnosis and in longitudinal studies.59,60 The McGill Pain Questionnaire, which consists of descriptors of symptoms from which patients select those that best describe their experience, when applied to diabetic neuropathy, was found to be a sensitive measure.5 The recently validated “NeuroQol” instrument combines a neuropathic symptom score with an assessment of quality of life.60
The neuropathy symptom score (NSS) and its derivatives, the neuropathy symptom profile (NSP) and neuropathy symptom change scores (NSC), are perhaps the most commonly used measures in clinical trials.19,57,58 The NSS is a standardized list of questions and neuropathic symptoms that is applied by a trained individual in a standardized manner. A simplified NSS has been used for epidemiologic studies and can be applied in clinical practice for patient follow-up. It can be administered in a few minutes and scores typical symptoms with additional weighting for nocturnal exacerbation.22,27
Clinical Signs
Simple clinical observation may identify a neuropathic foot; evidence might include small muscle wasting, clawing of toes, prominent metatarsal heads, dry skin and callus (secondary to sympathetic dysfunction), and bony deformities secondary to Charcot’s neuroarthropathy.
Two simple instruments can be used in clinical practice or for clinical trial assessment. First, Feldman and coworkers61 developed the Michigan Neuropathy Screening Instrument (MNSI); this two-step program is used for diagnosis and staging of neuropathy. The MNSI consists of a 15-question yes/no symptom questionnaire that is supplemented by a simple clinical examination. Patients with an abnormal score on the MNSI are referred for quantitative sensory testing (QST) and electrophysiology (EP). Second, the simplified neuropathy disability score (NDS) is a simple clinical examination that sums abnormalities of reflexes and sensory assessment; it has been used in clinical practice and in epidemiologic studies.22,27 The original NDS was developed by Dyck and colleagues at the Mayo Clinic for the detailed structured assessment of neurologic deficits secondary to neuropathy.18,19,58 This technique is reproducible if performed by trained and experienced physicians and is being used in a number of ongoing trials of new therapies for diabetic neuropathy.
Quantitative Sensory Testing
QSTs assess patients’ ability to detect a number of sensory stimuli and offer the advantage that they directly assess the degree of sensory loss at the most vulnerable site: the foot.62 However, these tests are complex psychophysiologic tests that also rely on patients’ responses and therefore cooperation and concentration. Moreover, an abnormal finding does not necessarily confirm that the abnormality lies in the peripheral nerve; it might lie anywhere in the afferent pathway. QSTs vary in complexity; the simpler instruments can be used in day-to-day clinical practice, whereas the more sophisticated instruments usually are used for more detailed assessment and for follow-up assessments in clinical trials. Some of the more commonly used techniques are now discussed briefly.
Semmes-Weinstein Monofilaments
Semmes-Weinstein monofilaments consist of sets of nylon filaments of variable diameter that buckle at a predefined force when applied to the testing site. They are used widely in clinical practice and are particularly helpful in the identification of subjects who are at risk for neuropathic foot ulceration. Inability to perceive pressure of a 10 g (5.07) monofilament has been shown in prospective studies to predict risk for neuropathic ulceration.63
Vibration Perception
Several devices designed specifically to assess vibration perception thresholds (VPTs) are used to test large myelinated fiber function. VPT increases with age in normal individuals and tends to be higher in the lower extremities. As well as being useful in practice, VPT has been used in epidemiologic studies26 and prospective studies, in which an abnormal reading greater than 25 V has been associated with high risk for foot ulceration.56
Thermal and Cooling Thresholds
Warm and cold sensation is transmitted via small myelinated and unmyelinated fibers and can be assessed by using a number of devices; those that employ a forced-choice technique are most reproducible, especially if the method of limits is used.62 However, they remain the most variable of all QSTs.
Computer-Assisted Sensory Examination
This complex method, currently regarded as state of the art for clinical trials, uses a computerized device that can measure touch-pressure, vibration, and warm-cold thresholds using a forced-choice algorithm. It is being used in the Rochester study and in several long-term trials of new therapeutic interventions.57,58
Autonomic Function Testing
Cardiovascular autonomic dysfunction can be evaluated in detail by employing Ewing and Clarke’s battery of five tests: (1) the average inspiratory-expiratory heart rate difference with six deep breaths, (2) the Valsalva ratio, (3) the 30 : 15 ratio, (4) the diastolic blood pressure response to isometric exercise, and (5) the systolic blood pressure fall to standing.42,48 More sophisticated techniques such as spectral analysis allow assessment of the modulation in sinus node activity and, depending on the frequency evaluated, may allow dissection of the component contributions of autonomic input and circulating neurohumoral factors. Key tests that are well validated and that offer prognostic value are RR variation, Valsalva’s maneuver, and postural testing.64
Electrophysiology
EP testing is probably the most important efficacy parameter in clinical neuropathy trials, as EP tests are objective, sensitive, and reproducible.57,58,65 Using a central monitoring core laboratory, Bril and coworkers65 were able to obtain remarkable reproducibility of EP variables across 60 sites in a prospective study. Coefficients of variability of 3% and 4% for motor and sensory nerve conduction velocities (NCVs) are comparable with those achieved in an excellent single laboratory.65 For these reasons, EP variables such as NCVs and amplitudes are frequently used surrogate end points in clinical trials; moreover, they are useful in the clinical investigation of peripheral nerve disease. However, although EP tests can define and quantitate nerve dysfunction, as with QSTs, the findings are not specific to diabetes.
Composite scores that combine clinical, quantitative, sensory, and EP measures often are used in natural history and efficacy studies.19,57,58 Examples include the NISLL+758 and the Michigan Diabetic Neuropathy Score.61 The former comprises the Neuropathy Impairment Score of the Lower Limbs (NISLL) together with seven other tests (five EP attributes, one QST, and one AFT). This measure is being used in several ongoing multicenter intervention studies.
Pathogenesis
Data from animal models and cell culture provide a conceptual framework for the cause and treatment of diabetic neuropathy.66 However, the damaging pathways established in animal models have not been verified in patients, and multiple interventions have failed in clinical trials.67
Hyperglycemia
Hyperglycemia is of primary importance in patients with type 1 diabetes, and the improvement in neuropathy reported in the DCCT2 and following pancreas transplantation68 attests to this. Prospective results of the Epidemiology of Diabetes Complications (EDIC) study indicate that in addition to good glycemic control, avoidance of smoking and good blood pressure control may be helpful in preventing or delaying the onset of neuropathy in patients with type 1 diabetes.69 Similarly, in the Eurodiab prospective study, in addition to hyperglycemia, independent risk factors that predicted the development of neuropathy included BMI, hypertension, and deranged lipids.70 Based on prospective data of the 10-year incidence of distal symmetrical polyneuropathy in 589 patients with type 1 diabetes, suggested goals for risk reduction include low-density lipoprotein (LDL) cholesterol less than 100 mg/dL (2.6 mmol/L), high-density lipoprotein (HDL) cholesterol greater than 45 mg/dL (1.1 mmol/L), triglycerides less than 150 mg/dL (1.7 mmol/L), systolic blood pressure less than 120 mm Hg, and diastolic blood pressure less than 80 mm Hg.71
With regard to type 2 diabetes, longitudinal data from the Rochester cohort support the contention that the duration and severity of exposure to hyperglycemia are related to the severity as opposed to the onset of neuropathy.72 Studies in patients presenting with symptoms of a small-fiber neuropathy suggest an increased prevalence of impaired glucose tolerance (IGT) in these patients, as well as a glycemic threshold below the current definition of diabetes and above which polyneuropathy develops.73 However, in a recent population-based study, the prevalence of polyneuropathy was 28.0% in diabetic subjects and only 13.0% in those with IGT, and 11.3% in those with impaired fasting glucose (IFG) compared with 7.4% in those with normal glucose tolerance (NGT), indicating a minimal contribution of hyperglycemia.74 With regard to the effects of intervention, the data are not supportive of benefit with improving glycemic control. Thus, in the VA cooperative study in type 2 diabetic patients, 153 patients who were randomized to intensive versus conventional therapy achieved a 2.07% difference in glycated hemoglobin (HbA1c) over 2 years but failed to demonstrate a significant difference in the progression of somatic or autonomic neuropathy.75 Similarly, the Steno-2 study, which implemented multifactorial intervention, including improved glycemic control, resulted in improved autonomic but not somatic neuropathy.76,77
Polyol Pathway
Animal models of diabetes consistently demonstrate an association between increased flux through the polyol pathway and a reduction in NCV, both of which can be ameliorated with aldose reductase inhibitors (ARIs).78 However, the single measurement of whole-nerve sorbitol or fructose levels is clearly an oversimplification of a complex process, with a polyol pathway in constant flux that is known to be different among different cellular and structural compartments of the peripheral nerve.78 Moreover, it would appear that those who are at greatest risk for developing the complications are those with a higher set point for AR activity.79 To add to this complexity, there may be a significant genetic determinant of polyol pathway flux and hence efficacy of ARIs, as polymorphisms in the ARI promoter region leading to a highly significant decrease in the frequency of the Z2 allele have been demonstrated in patients with overt neuropathy compared with those without neuropathy.80 An early meta-analysis of randomized controlled trials of ARIs demonstrated only a small but statistically significant reduction in decline of median and peroneal motor nerve conduction velocity.81 This marginal benefit may be due to the degree of AR inhibition achieved with different ARIs. Thus, in a randomized, placebo-controlled, double-blind, multiple-dose clinical trial with Zenarestat, dose-dependent increments in sural nerve sorbitol suppression were accompanied by significant improvement in NCV, and in doses producing more than 80% sorbitol suppression, a significant increase was seen in the density of small-diameter myelinated fibers of the sural nerve.82 Fidarestat, a potent ARI, significantly improved median nerve F-wave conduction velocity and minimal latency, as well as symptoms of numbness, spontaneous pain, paresthesias, and hyperesthesia.83 Also, in 603 diabetic patients treated with Epalrestat, deterioration of motor conduction velocity was prevented, especially in patients with good glycemic control and with minimal neuropathy.84
Glycation
Hyperglycemia induces the formation of advanced glycation end products (AGEs) on peripheral nerve myelin, contributing to segmental demyelination by increasing its susceptiblity to phagocytosis by macrophages; it also modifies axonal cytoskeletal proteins such as tubulin, neurofilament, and actin, resulting in axonal atrophy and degeneration with reduced regeneration due to glycation of laminin.85 Experimental data now suggest a significant role of receptor for advanced glycation end products (RAGE)-dependent activation of the proinflammatory transcription factor nuclear factor kappa B, resulting in reduced nociception, which is prevented in RAGE-deficient mice.86 In experimental diabetes, RAGE mRNA and protein were increased in epidermal and sural nerve axons and Schwann cells, as well as in sensory neurons within ganglia, and this was associated with progressive electrophysiologic and structural abnormalities, which were attenuated in RAGE −/− mice. RAGE mediated the activation of NF-kappaB and PKC beta II pathways in Schwann cells in the DRG and peripheral nerve.87 Human sural nerves obtained from diabetic and nondiabetic amputation specimens demonstrate normal furosine, an early reversible glycation product, but significantly elevated pentosidine (advanced glycation end product) levels in both cytoskeletal and myelin protein.88 Enhanced staining for carboxymethyl lysine has been demonstrated in the perineurium, endothelial cells, and pericytes of endoneurial microvessels, as well as in myelinated and unmyelinated fibers in sural nerves, showing a significant reduction in myelinated fiber density.89 However, in a primate model of type 1 diabetes, 3 years of treatment with aminoguanidine did not restore conduction velocity or autonomic function.90 In a recent study that followed patients undergoing islet transplantation, long-term worsening of neuropathy was prevented, and this was associated with a reduction in AGE/RAGE expression in the perineurium and vasa nervorum of skin biopsies.91 It is becoming increasingly apparent that many drugs that currently are used for other indications, including pioglitazone, metformin,92,93 angiotensin-converting enzyme (ACE) inhibitors, and angiotensin II (ATII) antagonists,94 may act as powerful antiglycating agents.
Oxidative Stress
A considerable body of data supports the role of oxidative stress in the pathogenesis of diabetic neuropathy in animal models95; hence increasing antioxidant potential is an attractive treatment strategy. However, no clinical trial to date has demonstrated therapeutic benefit with an oral antioxidant.67,96 Short-term benefits have been observed with intravenous alpha-lipoic acid (ALA), a powerful antioxidant that scavenges hydroxyl radicals and superoxide and peroxyl radicals and regenerates glutathione. Five clinical trials with alpha-lipoic acid that were recently reviewed showed that parenteral ALA over 3 weeks improved neuropathic symptoms and deficits (the latter of which was unexpected); however, oral treatment produced no clear signal for an improvement in symptoms or deficits.97 The benefit is primarily seen in symptoms97 with little benefit noted for neurologic deficits97 or underlying causative factors such as tissue blood flow.99
Poly(ADP-Ribose) Polymerase-1 (PARP)
Increased oxidative and nitrosative stress activates the nuclear enzyme, poly(ADP-ribose) polymerase-1 (PARP), which depletes its substrate, NAD+, slowing the rate of glycolysis, electron transport, and adenosine triphosphate (ATP) formation, and inhibits D-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) by poly(ADP)-ribosylation.100 These events lead to deleterious effects on nitrergic innervation, contributing to autonomic neuropathy as evidenced by studies demonstrating impaired gastric fundus relaxation in STZ rats, which was ameliorated by PARP inhibition using 3-aminobenzamide (3-AB).101 With regard to somatic neuropathy, the orally active PARP inhibitor 10-(4-methylpiperazin-1-ylmethyl)-2H-7-oxa-1,2-diaza-benzo[de]anthracen-3-one partially prevented PARP activation in peripheral nerve and dorsal root ganglia (DRG) neurons, as well as thermal hypoalgesia, mechanical hyperalgesia, tactile allodynia, and intraepidermal nerve fiber degeneration, in streptozotocin-diabetic rats.102 Furthermore, whereas diabetic PARP +/+ mice demonstrate a 46% loss of intraepidermal nerve fibers, diabetic PARP −/− mice retain completely normal intraepidermal nerve fiber density.102
C-Peptide
Impaired insulin/C-peptide action has emerged as a prominent factor in the pathogenesis of microvascular complications in type 1 diabetes. Experimental studies have demonstrated a range of actions that include effects on Na/K-ATPase activity, expression of neurotrophic factors, regulation of molecular species underlying degeneration of the nodal apparatus, and DNA binding of transcription factors, leading to modulation of apoptosis.103 C-peptide also exerts an effect on the expression of neurotrophic factors and their receptors, with downstream benefits noted on cytoskeletal protein mRNAs and protein expression, which has been proposed to prevent and reverse myelinated degeneration of the node and paranode and unmyelinated axonal degeneration, atrophy, and loss.103 A recent study has shown maximal therapeutic benefit of C-peptide on diabetic neuropathy by continuous subcutaneous delivery, maintaining physiologic C-peptide concentrations as opposed to once-daily subcutaneous injections in type 1 diabetic BB/Wor rats.104 One hundred thirty-nine patients with type 1 diabetes participated in a double-blinded, randomized, and placebo-controlled study comparing C-peptide (1.5 mg/day, 4.5 mg/day, and placebo) adminstered subcutaneously four times daily over 6 months. Sensory nerve conduction velocity, the clinical neurologic impairment score, and vibration perception improved significantly and to a similar magnitude in both C-peptide groups compared with placebo.105
Vascular Endothelial Growth Factor
Vascular endothelial growth factor (VEGF) regulates angiogenesis and neuronal survival by stimulating neurons and glial cells to survive and grow. Thus, with its potential for a dual impact on both the vasculature and neurons, it could represent an important therapeutic intervention in diabetic neuropathy. A recent study has shown progressive endothelial dysfunction and a reduction in VEGF expression that was related to loss of intraepidermal nerve fibers in the foot skin of diabetic patients with increasing neuropathic severity.106 Similarly, in a group of patients with type 2 diabetes, a significant reduction in epidermal and dermal VEGF-A, VEGFR-2 expression, and dermal nerve fiber density was reported.107 To date, a therapeutic benefit in diabetic neuropathy has been demonstrated for VEGF in experimental studies only. Implantation of hematopoietic mononuclear cell fractions in STZ diabetic rats has been shown to improve nerve conduction velocity as a result of arteriogenic effects of VEGF.108 With the use of an engineered zinc-finger protein transcription factor, an intramuscular injection of formulated plasmid DNA encoding the VEGF-A–activating gene prevented both sensory and motor nerve conduction velocity reduction in the STZ diabetic rat.109
Neurotrophins
Neurotrophins (NT) promote the survival of specific neuronal populations by inducing morphologic differentiation, enhancing nerve regeneration, stimulating neurotransmitter expression, and altering the physiologic characteristics of neurons. Thus modulating neurotrophic support represents an alternative approach to the treatment of patients with diabetic neuropathy, that is, the stimulation of repair without necessarily addressing the underlying cause of nerve damage. Although the skin of diabetic patients with neuropathy demonstrates depleted nerve growth factor (NGF) protein,110 mRNA for both NGF111 and NT-3112 is increased, and sciatic nerve ciliary neurotrophic factor levels are normal.113 In situ hybridization studies in the skin of diabetic patients demonstrate what may be interpreted as a compensatory increased expression of trkA (high-affinity receptor for NGF) and trkC (receptor for NT-3).114 A phase 2 clinical trial of recombinant human nerve growth factor in 250 diabetic patients with symptomatic diabetic polyneuropathy demonstrated a significant improvement in the sensory component of the neurologic examination, using two quantitative sensory tests and a rather vague end point—“the clinical impression of most subjects that their neuropathy had improved.”115 However, a phase 3 trial in 1019 diabetic patients with neuropathy failed to demonstrate a significant benefit.116 These disappointing results led to much speculation regarding the reasons for failure of NGF specifically, with the hope that other neurotrophins might succeed where it had failed.116 However, a randomized, double-blind, placebo-controlled study of brain-derived neurotrophic factor in 30 diabetic patients demonstrated no significant improvement in nerve conduction, quantitative sensory, and autonomic function tests, including the cutaneous axon reflex.117
Mitogen-Activated Protein Kinase
Upstream inducers and transducers signal transcriptional and translational abnormalities through effector molecules referred to collectively as the mitogen-activated protein kinase (MAPK) family, which mediate early gene responses and aberrant phosphorylation of neurofilaments. A subgroup of MAPKs that specifically involve activation via cellular stressors includes extracellular signal–regulated kinase 1 and 2 (ERK-1 and -2), c-jun N-terminal kinase, and p38; these components, collectively referred to as the stress-activated protein kinases, have been shown to be elevated in sural nerve biopsies of diabetic patients with advanced neuropathy.118 In the STZ rat, JNK and p38 are transported from the periphery to the neuronal soma via the axon mediating the transfer of hyperglycemia-related stress signals, possibly triggered by loss of neurotrophic support.119 Kinase activation leads to phosphorylation of neurofilaments (NFs) composed of three subunit proteins—NF-L, NF-M, and NF-H—which are major constituents of the axonal cylinder.120 Thus, any abnormality in synthesis, delivery, or processing of these critical proteins could lead to impairment in axon structure and function.121
Pathology
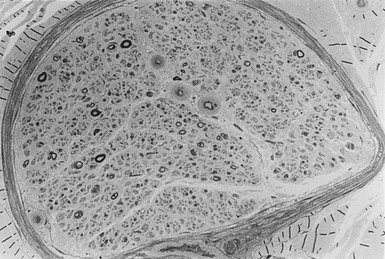
Detailed morphometric studies of sural nerve biopsies provide considerable insights into the underlying pathology and pathogenesis of diabetic neuropathy. Thus a significant abnormality in myelinated and unmyelinated fibers occurs despite entirely normal clinical and neurophysiologic tests of neuropathy.122–124
Myelinated Fibers
Apart from the hallmark of advanced diabetic neuropathy—loss of myelinated fibers122—a number of other, more subtle changes indicating damage to the axon or Schwann cell can be identified by applying morphometric techniques. Mechanistically, ineffective axonal transport125 or an alteration in the expression of neurofilaments120 has been suggested to result in axonal atrophy.126–128 However, studies in patients with mild and established diabetic neuropathy have failed to confirm this abnormality.129–131 Axoglial disjunction has been described as an abnormality of the paranodal connection between the terminal myelin loops and the axonal membrane that may precede overt demyelination.132 However, careful studies have been unable to confirm the presence of axoglial disjunction.133,134 Schwann cell abnormalities include both reactive (accumulation of lipid droplets, pi granules of Reich, and glycogen granules) and degenerative (mitochondrial enlargement, effacement of cristae, degeneration of abaxonal and adaxonal cytosol and organelles) changes.135 These subtle changes are thought to lead to initial demyelination118 and, with progression of neuropathy, to axonal degeneration, resulting in loss of nerve fibers126,129,136 (Fig. 27-2).
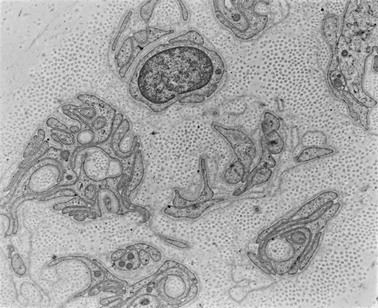
Unmyelinated Fibers
Axonal degeneration with active regeneration of unmyelinated fibers occurs early in the evolution of neuropathy before axonal degeneration of myelinated fibers,131 but it is important to note that their regenerative capacity is maintained long after myelinated fibers have lost their capacity to regenerate126,129 (Fig. 27-3).
Structure-Function Relationship
A variety of morphologic measures of nerve fiber degeneration have been related to the neuropathy deficit score,126 vibration perception, and autonomic dysfuction.129 Patients with mild neuropathy show a good correlation between sural nerve myelinated fiber density and both peroneal and sural NCV and amplitude but not vibration or thermal perception.137 In 18 diabetic patients with varying stages of neuropathy, precise relationships between degree of myelinated fiber loss and clinical and neurophysiologic abnormalities, as well as quantitative sensory thresholds, have been demonstrated.138 Thermal thresholds have been related to the median unmyelinated axon diameter.129
Autonomic Tissue
Pathologic studies of autonomic tissue are limited to postmortem or surgical material. In patients with diabetic gastropathy, the vagus nerve shows a reduction in myelinated fiber density and degeneration with regeneration of unmyelinated fibers.139 Qualitative changes include chromatolysis, cytoplasmic vacuolization, and pyknotic changes. Quantitative studies have demonstrated degenerative or dystrophic changes in axonal and dendritic components of sympathetic ganglia in the absence of significant neuron loss, as well as alterations in the postganglionic autonomic innervation of various end organs.140 Neuroaxonal dystrophy is a key feature of the pathology involving intraganglionic terminal axons and synapses of the prevertebral superior mesenteric, celiac, and, to a much lesser degree, superior cervical ganglia.141
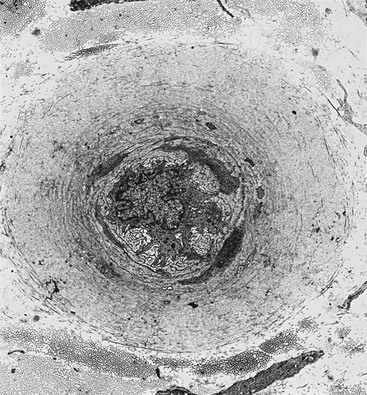
Nerve Vasculature
Structural abnormalities of the vessels supplying the peripheral nerve include arteriolar attenuation, venous distension, arteriovenous shunting, and new vessel formation,142,143 along with intimal hyperplasia, hypertrophy,144 and denervation.145 Transperineurial vessels demonstrate denervation146 with luminal narrowing,147 possibly secondary to perineurial abnormalities.148 Endoneurial capillaries demonstrate endothelial cell hypertrophy, hyperplasia, and basement membrane thickening (Fig. 27-4) in diabetic and IGT patients without neuropathy,149,150 which progress with the severity of neuropathy.151–155
Skin Biopsy
Immunohistochemical quantification of intraepidermal nerve fibers (IENFs) using the panaxonal marker protein gene product 9.5 is now established as an early and sensitive marker of nerve damage in diabetic neuropathy.156 Thus a significant loss of IENFs has been demonstrated in patients with no neurologic deficits, and normal quantitative sensory testing, as well as electrophysiology157 and a reduction in IENF regenerative capacity, occurs in diabetic patients.158 IENF abnormalities have been related more specifically to those with painful diabetic neuropathy.157,159 Patients with small-fiber neuropathy and impaired glucose tolerance demonstrate a significant loss of IENFs, which improves with no change in QST or neurophysiology, suggesting that the assessment of IENFs may be a more senstive marker of nerve repair following therapeutic intervention.160
Corneal Confocal Microscopy
Corneal confocal microscopy (CCM) represents a novel reiterative in vivo clinical examination technique that is capable of imaging corneal nerve fibers. It has been shown to accurately define the extent of corneal nerve damage, which has been related to the severity of somatic diabetic neuropathy.161,162 A recent study has shown that this technique detects small-fiber damage before loss of IENFs occurs, and findings may be more abnormal in patients with painful diabetic neuropathy.157 Furthermore, CCM demonstrates the capacity to detect early nerve fiber repair 6 months following pancreas transplantation in type 1 diabetic patients. 163
Treatment
Throughout this section on treatment, distinction is made between therapies for symptomatic relief164 and those that may alter (slow) the progressive loss of nerve function that characterizes the natural history165 of neuropathy. A few therapies have efficacy in both of these areas.
Sensory Neuropathy
Glycemic Control: Of all available treatments, tight and stable glycemic control is probably the only one that may provide symptomatic relief while slowing the relentless progression of neuropathy.1–3 Because it is probably blood glucose flux that induces neuropathic pain,35 stability rather than the actual level of glycemic control may be most important in pain relief.166 The method of achieving stable control does not seem to be critical; no evidence suggests that insulin is superior if blood glucose is well controlled by oral hypoglycemic agents.
Tricyclic Antidepressants: Until new therapies are proved to relieve symptoms in appropriately designed trials,167 the tricyclic antidepressant drugs, such as amitriptyline and imipramine, will remain useful first-line agents for painful neuropathy in many countries; their efficacy, confirmed in several randomized, placebo-controlled trials,168 is related to plasma drug level, and onset of symptomatic relief is faster than their antidepressive effects. A clear dose-response relationship has been noted, but sedative and anticholinergic side effects are also dose related and troublesome, often restricting the use of these drugs: side effects are particularly problematic in older patients, and it is advisable to start on a very low dose such as 10 mg hs in such patients.164
Serotonin and Noradrenaline Reuptake Inhibitors: The serotonin and noradrenaline reuptake inhibitor (SNRI) duloxetine has both analgesic and antidepressant effects and can be used for the treatment of diabetic peripheral neuropathic pain.167 Unlike tricyclics, some anticonvulsants, and opioids, it generally does not require dose titration. A recent analysis of three randomized controlled trials of duloxetine in the management of neuropathic pain in diabetes confirmed that the drug is efficacious and well tolerated.169
Anticonvulsants: Carbamazepine is still used occasionally in the management of neuropathic pain, although its efficacy has not been confirmed in large randomized controlled studies. More recently, the new anticonvulsants gabapentin170 and pregabalin171 have been shown to be efficacious in the treatment of painful syndromes, including diabetic neuropathy. Their adverse effects seem to be less pronounced than those associated with tricyclic drugs. The anticonvulsant lacosamide also appears promising172; at the time of this writing, it has just completed phase 3 trials.
Other Agents: A number of other drug therapies, including phenytoin, mexiletene, lidocaine, and transdermal clonidine, have been reported to be useful in the management of painful or paresthetic symptoms.5,167 The centrally acting analgesic tramadol has exhibited confirmed efficacy in painful diabetic neuropathy in a randomized controlled trial.173 Regarding topical therapy, a pilot study has confirmed the efficacy of locally applied isosorbide dinitrite spray in a small randomized, placebo-controlled, double-blind trial.174 When the spray was applied locally to the feet at bedtime, a significant reduction in neuropathic pain was reported during active treatment, although it is curious that the placebo arm demonstrated no change. Finally, traditional therapies such as acupuncture have been employed in symptomatic neuropathy with good results and negligible side effects.175 All of the therapeutic agents so far discussed are used purely for symptomatic relief; no benefit has been reported in terms of the natural history of the disease.
Potential Future Therapies
Alpha-Lipoic Acid: Accumulating evidence suggests that free radical–mediated oxidative stress is implicated in the pathogenesis of neuropathy, and that treatment with the antioxidant alpha-lipoic acid might prevent these abnormalities and improve painful symptoms, while slowing the progression of diabetic neuropathy.97–99 A large multicenter study recently evaluated the efficacy of oral alpha-lipoic acid for nerve function over 4 years and reported some benefit in terms of symptoms and deficits but without improvement in nerve conduction.176
Other Agents: Investigations of other potential treatments for neuropathy are ongoing. One proposed class of drugs is the ACE inhibitors, which already are known for their efficacy in nephropathy and retinopathy. A preliminary controlled study of ACE inhibitors in early neuropathy confirmed a significant benefit over placebo in EP parameters.177 Intracellular hyperglycemia increases diacylglycerol levels, which activates protein kinase C (PKC) formation, leading to multiple pathogenetic consequences, including altered expression of endothelial nitric oxide synthetase and VEGF. However, although preliminary data suggested that treatment with a PKC-beta inhibitor might ameliorate measures of nerve function in diabetic peripheral neuropathy,178 a large randomized control trial failed to demonstrate any benefit of the drug over placebo in measures of nerve function.179 The N-methyl-d-aspartate (NMDA) receptor that is involved in nociception provides a possibility for therapeutic intervention in neuropathic pain. A pilot study of intravenous amantadine, an NMDA antagonist, demonstrated efficacy in pain relief in diabetic neuropathy.180, 181
Advancing knowledge in the neurobiology of neuropathic pain has resulted in a burst of activity related to the development of new treatments: potential new agents in the pipeline include vanilloid receptor agonists, cannabinoids, adenosine receptor agonists, cytokine inhibitors, and many more.181
Autonomic Neuropathy
Because autonomic neuropathy is one of several contributory causes of erectile dysfunction (ED), a multifaceted approach to management is indicated.46,47 Psychosexual counseling and altering drug therapy to remove the factors associated with ED are beneficial in many cases.47 Sildenafil, an orally active selective inhibitor of phosphodiesterase-5 (PDE-5), is efficacious for ED in diabetic males. In a trial of ED of multiple causation in diabetic males, Rendell and coworkers182 reported a response rate (defined as at least one successful attempt at sexual intercourse) of 61% in sildenafil-treated subjects versus 22% among those on placebo. Most diabetic patients require 50 or 100 mg, and care must be taken if there is any history of ischemic heart disease. Sildenafil must never be given to patients on nitrate therapy. A subsequent trial of sildenafil in type 2 patients with ED reported a response rate of 65% versus 11% on placebo.183 More recently, two other PDE-5 inhibitors have been licensed for the management of ED: tadalafil and vardenafil.184,185
Sweating Disorders
The first specific treatment for gustatory sweating has been reported. Glycopyrrolate is an antimuscarinic compound that, when applied topically to the affected area, results in a marked reduction of sweating while “trigger” foods are eaten. Its efficacy was confirmed in a randomized controlled trial.186
Others
Treatment for diabetic gastroparesis involves measures to enhance gastric motility and emptying. Metoclopramide, a dopamine antagonist, directly stimulates antral muscle and may mediate acetylcholine release. Alternative agents include domperidone, a peripheral dopamine D2 receptor antagonist, and erythromycin, which directly stimulates motilin receptors. Constipation may be treated with a combination of prokinetic agents such as metoclopramide and cisapride. Postural hypotension may be treated with mineralocorticoids such as fludrocortisone, sympathomimetic agents, and dopamine blockers. Urinary bladder difficulties are addressed with regular voiding, self-catheterization, and cholinergic agonists such as bethanechol chloride, which stimulates muscarinic, postganglionic receptors, thereby enhancing bladder motility and emptying.42,48
The Neuropathic Foot
Any patient with clinical evidence of diabetic peripheral neuropathy must be considered as being at risk for insensitive foot ulceration and should receive evaluation on foot care and, if necessary, a podiatry referral.17 These patients require more frequent follow-up, always with particular attention paid to foot inspection to reinforce the educational message of the need for regular foot care.
The late sequelae of diabetic neuropathy are usually considered to be neuropathic foot ulceration, neuroarthropathy (Charcot’s foot), and amputation.5–7
Neuropathic Foot Ulceration
Distal sensory and sympathetic neuropathy are the most important component causes that lead to foot ulceration, being present in 78% of cases assessed in a two-center study.8 However, the neuropathic foot does not ulcerate spontaneously; typically, it is the combination of neuropathy with other risk factors such as deformity and unperceived trauma that results in ulceration. International guidelines on the clinical management of neuropathy, therefore, emphasize the importance of regular foot examinations and education in self-foot care for the management of neuropathy; guidelines for the comprensive diabetic foot examination were published by the American Diabetes Association in 2008.187
Charcot’s Neuroarthropathy
Charcot’s neuroarthropathy is a less common but clinically important and potentially devastating disorder. Diabetes is now the most common cause of this condition in Western countries,7 and a high degree of awareness and suspicion may enable early diagnosis and effective intervention. Permissive features for the development of a Charcot’s joint include peripheral sensorimotor neuropathy, sympathetic denervation in the foot, and intact peripheral circulation; minor, unperceived trauma is often the initiating event. It is believed that following repetitive minor trauma, osteoblastic activity is stimulated by remodeling of bone. A high index of suspicion must exist if a neuropathic patient has unilateral unexplained swelling and warmth in a foot, with the possibility of infection kept in mind. Contrary to information provided in earlier texts, discomfort may be experienced, although the patient is usually able to walk. Detailed assessment and investigation of such a patient is essential, and rest or casting of a suspected Charcot’s foot is usually recommended.
References
1. Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development of progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med. 1993;329:329–986.
2. Diabetes Control of Complications Trial Research Group. The effect of intensive diabetes therapy on the development and progression of neuropathy. Ann Intern Med. 1995;122:561–568.
3. United Kingdom Prospective Diabetes Study. Intensive blood glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes. Lancet. 1998;352:837–853.
4. Martin, CL, Albers, J, Herman, WH, et al. Neuropathy among the diabetes control and complications trial cohort 8 years after trial compeltion. Diabetes Care. 2006;29:340–344.
5. Boulton, AJM, Malik, RA, Arezzo, JC, et al. Diabetic somatic neuropathy: technical review. Diabetes Care. 2004;27:1458–1487.
6. Rathur, H, Boulton, AJM. The neuropathic diabetic foot. Nat Clin Pract Endocrinol Metab. 2007;3:14–25.
7. Jeffcoate, WJ. Charcot neuro-osteoarthropathy. Diab Metab Res Rev. 2008;24(Suppl 1):S62–S65.
8. Reiber, GE, Vileikyte, L, Boyko, EH, et al. Causal pathways for incident lower-extremity ulcers in patients with diabetes from two settings. Diabetes Care. 1999;22:157–162.
9. Vileikyte, L, Rubin, RR, Leventhal, H. Psychological aspects of diabetic neuropathic foot complications: an overview. Diabetes Metab Res Rev. 2004;20(Suppl 1):S13–S18.
10. de Calvi, M. Recherches sur les Accidents Diabetiques. Paris: P. Asselir; 1806.
11. Ward, JD. Historical aspects of diabetic peripheral neuropathy. In: Boulton AJM, ed. Diabetes in Pictures: Diabetic Neuropathy. Cologne: Academy Press; 2001:8–15.
12. Davies-Pryce, T. A case of perforating ulcers of both feet associated with ataxic symptoms. Lancet. 1887;2:11–12.
13. Jordan, WR. Neuritic manifestations in diabetic neuropathy. Arch Intern Med. 1936;57:307–366.
14. Thomas, PK. Classification, differential diagnosis and staging of diabetic peripheral neuropathy. Diabetes. 1997;46(Suppl 2):S54–S57.
15. Boulton, AJM, Ward, JD. Diabetic neuropathies and pain. Clin Endocrinol Metab. 1986;15:917–931.
16. Sima, AAF, Thomas, PK, Ishii, D, et al. Diabetic neuropathies. Diabetologia. 1997;46(Suppl):B74–B77.
17. Boulton, AJM, Gries, FA, Jervell, JA. Guidelines for the diagnosis and out-patient management of diabetic peripheral neuropathy. Diabet Med. 1998;15:508–514.
18. Dyck, PJ, Kratz, KM, Karnes, JZ, et al. The prevalence by staged severity of various types of diabetic neuropathy, retinopathy and nephropathy in a population-based cohort: the Rochester Diabetic Neuropathy Study. Neurology. 1993;43:817–824.
19. Dyck, PJ, Melton, J, O’Brien, PC, et al. Approaches to improve epidemiological studies of diabetic neuropathy. Diabetes. 1997;46(Suppl 2):S5–S8.
20. Consensus statement. Report and recommendations of the San Antonio conference on diabetic neuropathy. Diabetes Care. 1988;11:592–597.
21. Adler, AI. Risk factors for diabetic neuropathy and foot ulceration. Curr Diabetes Rep. 2001;1:202–207.
22. Young, MJ, Boulton, AJM, McLeod, AF, et al. A multicentre study of the prevalence of diabetic peripheral neuropathy in the UK hospital clinic population. Diabetologia. 1993;36:150–154.
23. Tesfaye, S, Stephens, LK, Stephenson, JM, et al. Prevalence of diabetic peripheral neuropathy and its relation to glycemic control and potential risk factors: the EURODIAB IDDM complications study. Diabetologia. 1996;39:1377–1384.
24. Cabezas-Cerrato, J. The prevalence of clinical diabetic neuropathy in Spain: a study in primary care and hospital clinic groups. Diabetologia. 1998;41:1263–1269.
25. Ziegler, D, Rathmann, W, Dickhaus, T, et al. Prevalence of polyneuropathy in pre-diabetes and diabetes is associated with abdominal obesity and macroangiopathy: the MONICA/KORA Augsburg Surveys S2 and S3. Diabetes Care. 2008;31:464–469.
26. Kumar, S, Ashe, HA, Parnell, L, et al. The prevalence of foot ulceration and its correlates in type 2 diabetes: a population-based study. Diabet Med. 1994;11:480–484.
27. Abbott, CA, Carrington, AL, Ashe, H, et al. The northwest diabetes foot care study: incidence of, and risk factors for, new diabetic foot ulceration in a community-based cohort. Diabet Med. 2002;19:377–384.
28. Adler, AI, Boyko, EJ, Ahroni, JH, et al. Risk factors for diabetic peripheral sensory neuropathy: results of the Seattle prospective diabetic foot study. Diabetes Care. 1997;20:1162–1167.
29. Kempler, P, Tesfaye, S, Chaturvedi, N, et al. Autonomic neuropathy is associated with increased cardiovascular risk factors: the EURODIAB IDDM complications study. Diabet Med. 2002;19:900–905.
30. Chou, KL, Galetta, SL, Liu, GT, et al. Acute ocular motor mononeuropathies: prospective study of the roles of neuroimaging and clinical assessment. J Neurol Sci. 2004;219:35–39.
31. Hopf, HC, Guttmann, L. Diabetic third nerve palsy: evidence for a mesencephalic lesion. Neurology. 1990;40:1041–1045.
32. Makepeace, A, Davis, WA, Bruce, DG, et al. Incidence and determinants of carpal tunnel decompression surgery in type 2 diabetes: the Fremantle Diabetes Study. Diabetes Care. 2008;31:498–500.
33. Balci, K, Utku, U. Carpal tunnel syndrome and metabolic syndrome. Acta Neurol Scand. 2007;116:113–117.
34. Schady, W, Abuaisha, B, Boulton, AJM. Observations on severe ulnar neuropathy in diabetes. J Diabetes Complications. 1998;12:128–132.
35. Chaudhuri, KR, Wren, DR, Werring, D, et al. Unilateral abdominal muscle herniation with pain: a distinctive variant of diabetic radiculopathy. Diabet Med. 1997;14:803–807.
36. Dyck, PJ, Norell, JE, Dyck, PJ. Non-diabetic lumbosacral radiculoplexus neuropathy: natural history, outcome and comparison with the diabetic variety. Brain. 2001;124:1197–1207.
37. Said, G, Lacroix, C, Lozeron, P, et al. Inflammatory vasculopathy in multifocal diabetic neuropathy. Brain. 2003;126:376–385.
38. Llewelyn, JG, Thomas, PK, King, RHM. Epineurial microvasculitis in proximal diabetic neuropathy. J Neurol. 1998;245:159–165.
39. Gorson, KC. Therapy for vasculitic neuropathies. Curr Treat Options Neurol. 2006;8:105–117.
40. Sharma, KR, Cross, J, Farronay, O, et al. Demyelinating neuropathy in diabetes mellitus. Arch Neurol. 2002;59:758–765.
41. Kalita, J, Misra, UK, Yadav, RK. A comparative study of chronic inflammatory demyelinating polyradiculoneuropathy with and without diabetes mellitus. Eur J Neurol. 2007;14:638–643.
42. Vinik, AI, Maser, RE, Mitchell, BD, et al. Diabetic Autonomic neuropathy. Diabetes Care. 2003;26:1553–1579.
43. Solders, G, Thalme, B, Aguirre-Aquino, M, et al. Nerve conduction and autonomic nerve function in diabetic children: a 10-year follow up study. Acta Pediatr. 1997;86:361–366.
44. Eriksson, KF, Nilsson, H, Lindarde, F, et al. Diabetes mellitus but not impaired glucose tolerance is associated with dysfunction in peripheral nerves. Diabet Med. 1994;11:279–285.
45. Toyry, JP, Niskanen, LK, Mantysaari, MJ, et al. Occurrence predictors and clinical significance of autonomic neuropathy in NIDDM: ten year follow-up from diagnosis. Diabetes. 1996;45:308–315.
46. Vinik, AI, Richardson, D. Evaluating erectile dysfunction in diabetes. Int Diabetes Fed Bull. 1998;43:7–13.
47. Veves, A, Webster, L, Chen, TF, et al. Aetiopathogenesis and management of impotence in diabetic males: four years’ experience from a combined clinic. Diabet Med. 1995;12:77–82.
48. Watkins, PJ, Edmonds, ME. Clinical features of diabetic neuropathy. In: Pickup JC, Williams G, eds. Textbook of Diabetes. ed 2. Oxford, UK: Blackwell Scientific; 1997:50.1–50.20.
49. Shaw, JE, Parker, P, Hollis, S, et al. Gustatory sweating in diabetes mellitus. Diabet Med. 1996;13:1033–1037.
50. Singleton, JR, Smith, AG. Neuropathy associated with prediabetes: what is new in 2007? Curr Diab Rep. 2007;7:420–424.
51. Vileikyte, L, Leventhal, H, Gozalez, JS, et al. Diabetic peripheral neuropathy and depressive symptoms: the association revisited. Diabetes Care. 2005;28:2378–2383.
52. Katoulis, EC, Ebdon-Parry, M, Hollis, S, et al. Postural instability in diabetic neuropathic patients at risk of foot ulceration. Diabet Med. 1997;14:296–300.
53. Veves, A, Young, MJ, Manes, C, et al. Differences in peripheral and autonomic nerve function measurements in painful and painless neuropathy: a clinical study. Diabetes Care. 1994;17:1200–1202.
54. Benbow, SJ, Chan, AW, Bowsher, DH, et al. A prospective study of painful symptoms, small fibre function and peripheral vascular disease in chronic painful diabetic neuropathy. Diabet Med. 1994;11:17–21.
55. Daousi, C, Benbow, SJ, Woodward, A, et al. The natural history of chronic painful peripheral neuropathy in a community diabetes population. Diabetic Med. 2006;23:1021–1024.
56. Abbott, CA, Vileikyte, L, Williamson, S, et al. Multicenter study of the incidence of and predictive risk factors for diabetic neuropathic foot ulceration. Diabetes Care. 1998;21:1071–1074.
57. Diabetic polyneuropathy in controlled clinical trials: consensus report of the peripheral nerve society. Ann Neurol. 1995;38:478–482.
58. Dyck, PJ, Norell, JE, Tritschler, H, et al. Challenges in design of multicenter trials: end points assessed longitudinally for change and monotonicity. Diabetes Care. 2007;30:2619–2625.
59. Apfel, SC, Asbury, AK, Bril, V, et al. Positive neuropathic sensory symptoms as endpoints in diabetic neuropathy trials. J Neurolog Sci. 2001;189:3–5.
60. Vileikyte, L, Peyrot, M, Bundy, C, et al. The development and validation of a neuropathy and foot ulcer–specific quality of life instrument. Diabetes Care. 2003;26:2549–2555.
61. Feldman, EL, Stevens, MJ, Thomas, PK, et al. A practical two-step quantitative clinical and electrophysiological assessment for the diagnosis and staging of diabetic neuropathy. Diabetes Care. 1994;17:1281–1289.
62. Shy, ME, Frohman, EM, So, YT, et al. Quantitative sensory testing. Neurology. 2003;602:898–906.
63. Mayfield, JA, Sugarman, JR. The use of the Semmes-Weinstein monofilament and other threshold tests for preventing foot ulceration and amputation in persons with diabetes. J Fam Pract. 2000;49(Suppl 1):S17–S29.
64. Schumer, MP, Joyner, SA, Pfeifer, MA. Cardiovascular autonomic neuropathy testing in patients with diabetes. Diabetes Spectrum. 1998;11:227–231.
65. Bril, V, Ellison, R, Ngo, M, et al. Electrophysiological monitoring in clinical trials. Muscle Nerve. 1998;21:1368–1373.
66. Tomlinson, DR, Gardiner, NJ. Glucose neurotoxicity. Nat Rev Neurosci. 2008;9:36–45.
67. Ziegler, D. Treatment of diabetic neuropathy and neuropathic pain: how far have we come? Diabetes Care. 2008;31:S255–S261.
68. Navarro, X, Sutherland, DE, Kennedy, WR. Long-term effects of pancreatic transplantation on diabetic neuropathy. Ann Neurol. 1997;42:727–736.
69. Forrest, KY, Maser, RE, Pambianco, G, et al. Hypertension as a risk factor for diabetic neuropathy: a prospective study. Diabetes. 1997;46:665–670.
70. Tesfaye, S, Chaturvedi, N, Eaton, SE, et al. Vascular risk factors and diabetic neuropathy. N Engl J Med. 2005;352:341–350.
71. Orchard, TJ, Forrest, KY, Kuller, LH, et al. Lipid and blood pressure treatment goals for type 1 diabetes: 10-year incidence data from the Pittsburgh Epidemiology of Diabetes Complications Study. Diabetes Care. 2001;24:1053–1059.
72. Dyck, PJ, Davies, JL, Wilson, DM, et al. Risk factors for severity of diabetic polyneuropathy: intensive longitudinal assessment of the Rochester Diabetic Neuropathy Study cohort. Diabetes Care. 1999;22:1479–1486.
73. Smith, AG, Singleton, JR. Impaired Glucose Tolerance and neuropathy. Neurologist. 2008;14:23–29.
74. Ziegler, D, Rathmann, W, Dickhaus, T, et al. Prevalence of polyneuropathy in pre-diabetes and diabetes is associated with abdominal obesity and macroangiopathy: the MONICA/KORA Augsburg Surveys S2 and S3. Diabetes Care. 2008;31:464–469.
75. Azad, N, Emanuele, NV, Abraira, C, et al. The effects of intensive glycemic control on neuropathy in the VA cooperative study on type II diabetes mellitus (VA CSDM). J Diabetes Complications. 1999;13:307–313.
76. Gaede, P, Vedel, P, Larsen, N, et al. Multifactorial intervention and cardiovascular disease in patients with type 2 diabetes. N Engl J Med. 2003;348:383–393.
77. Gaede, P, Lund-Andersen, H, Parving, HH, et al. Effect of a multifactorial intervention on mortality in type 2 diabetes. N Engl J Med. 2008;358:580–591.
78. Oates, PJ. Aldose reductase, still a compelling target for diabetic neuropathy. Curr Drug Targets. 2008;9:14–36.
79. Shimizu, H, Ohtani, KI, Tsuchiya, T, et al. Aldose reductase mRNA expression is associated with rapid development of diabetic microangiopathy in Japanese Type 2 diabetic (T2DM) patients. Diabetes Nutr Metab. 2000;13:75–79.
80. Demaine, AG. Polymorphisms of the aldose reductase gene and susceptibility to diabetic microvascular complications. Curr Med Chem. 2003;10:1389–1398.
81. Airey M, Bennett C, Nicolucci A, et al: Aldose reductase inhibitors for the prevention and treatment of diabetic peripheral neuropathy. Cochrane Database Syst Rev 2:CD002182, 2000.
82. Greene, DA, Arezzo, JC, Brown, MB. Effect of aldose reductase inhibition on nerve conduction and morphometry in diabetic neuropathy. Zenarestat Study Group. Neurology. 1999;53:580–591.
83. Hotta, N, Toyota, T, Matsuoka, K, et al. Clinical efficacy of fidarestat, a novel aldose reductase inhibitor, for diabetic peripheral neuropathy: a 52-week multicenter placebo-controlled double-blind parallel group study. Diabetes Care. 2001;24:1776–1782.
84. Matsuoka, K, Sakamoto, N, Akanuma, Y, et al. A long-term effect of epalrestat on motor conduction velocity of diabetic patients: ARI-Diabetes Complications Trial (ADCT). Diabetes Res Clin Pract. 2007;77:S263–S268.
85. Sugimoto, K, Yasujima, M, Yagihashi, S. Role of advanced glycation end products in diabetic neuropathy. Curr Pharm Des. 2008;14:953–961.
86. Lukic, IK, Humpert, PM, Nawroth, PP, et al. The RAGE pathway: activation and perpetuation in the pathogenesis of diabetic neuropathy. Ann N Y Acad Sci. 2008;1126:76–80.
87. Toth, C, Rong, LL, Yang, C, et al. Receptor for advanced glycation end products (RAGEs) and experimental diabetic neuropathy. Diabetes. 2008;57:1002–1017.
88. Ryle, C, Donaghy, M. Non-enzymatic glycation of peripheral nerve proteins in human diabetics. J Neurol Sci. 1995;129:62–68.
89. Sugimoto, K, Nishizawa, Y, Horiuchi, S, et al. Localization in human diabetic peripheral nerve of N (epsilon)-carboxymethyllysine-protein adducts, an advanced glycation end product. Diabetologia. 1997;40:1380–1387.
90. Birrell, AM, Heffernan, SJ, Ansselin, AD, et al. Functional and structural abnormalities in the nerves of type I diabetic baboons: aminoguanidine treatment does not improve nerve function. Diabetologia. 2000;43:110–116.
91. Del Carro, U, Fiorina, P, Amadio, S, et al. Evaluation of polyneuropathy markers in type 1 diabetic kidney transplant patients and effects of islet transplantation: neurophysiological and skin biopsy longitudinal analysis. Diabetes Care. 2007;30:3063–3069.
92. Rahbar, S, Natarajan, R, Yerneni, K, et al. Evidence that pioglitazone, metformin and pentoxifylline are inhibitors of glycation. Clin Chim Acta. 2000;301:65–77.
93. Bui, BV, Armitage, JA, Tolcos, M, et al. ACE inhibition salvages the visual loss caused by diabetes. Diabetologia. 2003;46:401–408.
94. Sourris, KC, Forbes, JM, Cooper, ME. Therapeutic interruption of advanced glycation in diabetic nephropathy: do all roads lead to Rome? Ann N Y Acad Sci. 2008;1126:101–106.
95. Nishikawa, T, Edelstein, D, Du, XL, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycemic damage. Nature. 2000;404:787–790.
96. Vincent, AM, Edwards, JL, Sadidi, M, et al. The antioxidant response as a drug target in diabetic neuropathy. Curr Drug Targets. 2008;9:94–100.
97. Foster, TS. Efficacy and safety of alpha-lipoic acid supplementation in the treatment of symptomatic diabetic neuropathy. Diabetes Educ. 2007;33:111–117.
98. Ziegler, D, Ametov, A, Barinov, A, et al. Oral treatment with alpha-lipoic acid improves symptomatic diabetic polyneuropathy: the SYDNEY 2 trial. Diabetes Care. 2006;29:2365–2370.
99. Jin, HY, Joung, SJ, Park, JH, et al. The effect of alpha-lipoic acid on symptoms and skin blood flow in diabetic neuropathy. Diabet Med. 2007;24:1034–1038.
100. Pacher, P, Szabó, C. Role of poly(ADP-ribose) polymerase-1 activation in the pathogenesis of diabetic complications: endothelial dysfunction, as a common underlying theme. Antioxid Redox Signal. 2005;7:1568–1580.
101. Gibson, TM, Cotter, MA, Cameron, NE. Effects of poly(ADP-ribose) polymerase inhibition on dysfunction of non-adrenergic non-cholinergic neurotransmission in gastric fundus in diabetic rats. Nitric Oxide. 2006;15:344–350.
102. Obrosova, IG, Xu, W, Lyzogubov, VV, et al. PARP inhibition or gene deficiency counteracts intraepidermal nerve fiber loss and neuropathic pain in advanced diabetic neuropathy. Free Radic Biol Med. 2008;44:972–981.
103. Sima, AA, Kamiya, H. Is C-peptide replacement the missing link for successful treatment of neurological complications in type 1 diabetes? Curr Drug Targets. 2008;9:37–46.
104. Kamiya, H, Zhang, W, Ekberg, K, et al. C-Peptide reverses nociceptive neuropathy in type 1 diabetes. Diabetes. 2006;55:3581–3587.
105. Ekberg, K, Brismar, T, Johansson, BL, et al. C-Peptide replacement therapy and sensory nerve function in type 1 diabetic neuropathy. Diabetes Care. 2007;30:71–76.
106. Quattrini, C, Jeziorska, M, Boulton, AJ, et al. Reduced vascular endothelial growth factor expression and intra-epidermal nerve fiber loss in human diabetic neuropathy. Diabetes Care. 2008;31:140–145.
107. Krishnan, ST, Quattrini, C, Jeziorska, M, et al. Neurovascular factors in wound healing in the foot skin of type 2 diabetic subjects. Diabetes Care. 2007;30:3058–3062.
108. Hasegawa, T, Kosaki, A, Shimizu, K, et al. Amelioration of diabetic peripheral neuropathy by implantation of hematopoietic mononuclear cells in streptozotocin-induced diabetic rats. Exp Neurol. 2006;199:274–280.
109. Price, SA, Dent, C, Duran-Jimenez, B, et al. Gene transfer of an engineered transcription factor promoting expression of VEGF-A protects against experimental diabetic neuropathy. Diabetes. 2006;55:1847–1854.
110. Anand, P, Terenghi, G, Warner, G, et al. The role of endogenous nerve growth factor in human diabetic neuropathy. Nat Med. 1996;2:703–707.
111. Diemel, LT, Cai, F, Anand, P, et al. Increased nerve growth factor mRNA in lateral calf skin biopsies from diabetic patients. Diabetic Med. 1999;16:113–118.
112. Kennedy, AJ, Wellmer, A, Facer, P, et al. Neurotrophin-3 is increased in skin in human diabetic neuropathy. J Neurol Neurosurg Psychiatry. 1998;65:393–395.
113. Lee, DA, Gross, L, Wittrock, DA, et al. Localization and expression of ciliary neurotrophic factor (CNTF) in postmortem sciatic nerve from patients with motor neuron disease and diabetic neuropathy. J Neuropathol Exp Neurol. 1996;55:915–923.
114. Terenghi, G, Mann, D, Kopelman, PG, et al. trkA and trkC expression is increased in human diabetic skin. Neurosci Lett. 1997;228:33–36.
115. Apfel, SC, Kessler, JA, Adornato, BT, et al. Recombinant human nerve growth factor in the treatment of diabetic polyneuropathy: NGF Study Group. Neurology. 1998;51:695–702.
116. Apfel, SC, Schwartz, S, Adornato, BT, et al. Efficacy and safety of recombinant human nerve growth factor in patients with diabetic polyneuropathy: a randomized controlled trial. JAMA. 2000;284:2215–2221.
117. Wellmer, A, Misra, VP, Sharief, MK, et al. A double-blind placebo-controlled clinical trial of recombinant human brain-derived neurotrophic factor (rhBDNF) in diabetic polyneuropathy. J Peripher Nerv Syst. 2001;6:204–210.
118. Purves, T, Middlemas, A, Agthong, S, et al. A role for mitogen-activated protein kinases in the etiology of diabetic neuropathy. FASEB J. 2001;15:2508–2514.
119. Middlemas, A, Delcroix, JD, Sayers, NM, et al. Enhanced activation of axonally transported stress-activated protein kinases in peripheral nerve in diabetic neuropathy is prevented by neurotrophin-3. Brain. 2003;126:1671–1682.
120. Fernyhough, P, Schmidt, RE. Neurofilaments in diabetic neuropathy. Int Rev Neurobiol. 2002;50:115–144.
121. Fernyhough, P, Gallagher, A, Averill, SA, et al. Aberrant neurofilament phosphorylation in sensory neurons of rats with diabetic neuropathy. Diabetes. 1999;48:881–889.
122. Giannini, C, Dyck, PJ. Basement membrane reduplication and pericyte degeneration precede development of diabetic polyneuropathy and are associated with its severity. Ann Neurol. 1995;37:498–504.
123. Malik, RA, Tesfaye, S, Newrick, PG, et al. Sural nerve pathology in diabetic patients with minimal but progressive neuropathy. Diabetologia. 2005;48:578–585.
124. Dyck, PJ, Giannini, C. Pathologic alterations in the diabetic neuropathies of humans: a review. J Neuropathol Exp Neurol. 1996;55:1181–1193.
125. Bomers, K, Braendgaard, H, Flyvbjerg, A, et al. Redistribution of axoplasm in the motor root in experimental diabetes. Acta Neuropathol (Berl). 1996;92:98–101.
126. Britland, ST, Young, RJ, Sharma, AK, et al. Association of painful and painless diabetic polyneuropathy with different patterns of nerve fibre degeneration and regeneration. Diabetes. 1990;39:898–908.
127. Sima, AAF, Bril, V, Nathaniel, V, et al. Regeneration and repair of myelinated fibres in sural-nerve biopsy specimens from patients with diabetic neuropathy treated with Sorbinil. N Engl J Med. 1988;319:548–555.
128. Sima, AAF, Nathaniel, V, Bril, V, et al. Histopathological heterogeneity of neuropathy in insulin-dependent and non-insulin dependent diabetes, and demonstration of axo-glial dysjunction in human diabetic neuropathy. J Clin Invest. 1988;81:349–364.
129. Llewelyn, JG, Gilbey, SG, Thomas, PK, et al. Sural nerve morphometry in diabetic autonomic and painful sensory neuropathy. Brain. 1991;114:867–892.
130. Engelstead, JK, Davies, JL, Giannini, C, et al. No evidence for axonal atrophy in human diabetic polyneuropathy. J Neuropathol Exp Neurol. 1997;56:255–262.
131. Malik, RA, Veves, A, Walker, D, et al. Sural nerve fibre pathology in diabetic patients with mild neuropathy: relationship to pain, quantitative sensory testing and peripheral nerve electrophysiology. Acta Neuropathol (Berl). 2001;101:367–374.
132. Sima, AAF, Prashar, A, Nathaniel, V, et al. Overt diabetic neuropathy: repair of axo-glial dysjunction and axonal atrophy by aldose reductase inhibition and its correlation to improvement in nerve conduction velocity. Diabet Med. 1993;10:115–121.
133. Thomas, PK, Beamish, NG, Small, JR, et al. Paranodal structure in diabetic sensory polyneuropathy. Acta Neuropathol (Berl). 1996;92:614–620.
134. Giannini, C, Dyck, PJ. Axoglial dysjunction: a critical appraisal of definition, techniques, and previous results. Microsc Res Tech. 1996;34:436–444.
135. Kalichman, MW, Powell, HC, Mizisin, AP. Reactive, degenerative, and proliferative Schwann cell responses in experimental galactose and human diabetic neuropathy. Acta Neuropathol (Berl). 1998;95:47–56.
136. Bradley, JL, Thomas, PK, King, RH, et al. Myelinated nerve fibre regeneration in diabetic sensory polyneuropathy: Correlation with type of diabetes. Acta Neuropathol (Berl). 1995;90:403–410.
137. Veves, A, Malik, RA, Lye, RH, et al. The relationship between sural nerve morphometric findings and measures of peripheral nerve function in mild diabetic neuropathy. Diabet Med. 1991;8:917–921.
138. Russell, JW, Karnes, JL, Dyck, PJ. Sural nerve myelinated fiber density differences associated with meaningful changes in clinical and electrophysiological measurements. J Neurol Sci. 1996;135:114–117.
139. Britland, ST, Young, RJ, Sharma, AK, et al. Vagus nerve morphology in diabetic gastropathy. Diabet Med. 1990;7:780–787.
140. Schmidt, RE. Neuropathology and pathogenesis of diabetic autonomic neuropathy. Int Rev Neurobiol. 2002;50:257–292.
141. Schmidt, RE. Age-related sympathetic ganglionic neuropathology: human pathology and animal models. Auton Neurosci. 2002;96:63–72.
142. Tesfaye, S, Harris, N, Jakubowski, J, et al. Impaired blood flow and arterio-venous shunting in human diabetic neuropathy: a novel technique of nerve photography and fluorescein angiography. Diabetologia. 1993;36:1266–1274.
143. Tesfaye, S, Malik, RA, Harris, N, et al. Arterio-venous shunting and proliferating new vessels in acute painful neuropathy of rapid glycaemic control (insulin neuritis). Diabetologia. 1996;39:329–335.
144. Korthals, JK, Gieron, MA, Dyck, PJ. Intima of epineurial arterioles is increased in diabetic polyneuropathy. Neurology. 1988;38:1582–1586.
145. Grover-Johnson, NM, Baumann, FG, Imparato, AM, et al. Abnormal innervation of lower limb epineurial arterioles in human diabetes. Diabetologia. 1981;20:31–38.
146. Beggs, J, Johnson, PC, Olafsen, A, et al. Transperineurial arterioles in human sural nerve. J Neuropathol Exp Neurol. 1991;50:704–718.
147. Malik, RA, Tesfaye, S, Thompson, SD, et al. Transperineurial capillary abnormalities in the sural nerve of patients with diabetic neuropathy. Microvasc Res. 1994;48:236–245.
148. Ghani, M, Malik, RA, Walker, D, et al. Perineurial abnormalities in the spontaneously diabetic dog. Acta Neuropathol. 1999;97:98–102.
149. Giannini, C, Dyck, PJ. Basement membrane reduplication and pericyte degeneration precede development of diabetic polyneuropathy and are associated with its severity. Ann Neurol. 1995;37:498–504.
150. Thrainsdottir, S, Malik, RA, Dahlin, LB, et al. Endoneurial capillary abnormalities presage deterioration of glucose tolerance and accompany peripheral neuropathy in man. Diabetes. 2003;52:2615–2622.
151. Malik, RA, Veves, A, Masson, EA, et al. Endoneurial capillary abnormalities in mild human diabetic neuropathy. J Neurol Neurosurg Psychiatry. 1992;55:557–561.
152. Bradley, J, Thomas, PK, King, RHM, et al. Morphometry of endoneurial capillaries in diabetic sensory and autonomic neuropathy. Diabetologia. 1990;33:611–618.
153. Britland, ST, Young, RJ, Sharma, AK, et al. Relationship of endoneurial capillary abnormalities to type and severity of diabetic polyneuropathy. Diabetes. 1990;39:909–913.
154. Malik, RA, Newrick, PG, Sharma, AK, et al. Microangiopathy in human diabetic neuropathy: relationship between capillary abnormalities and the severity of neuropathy. Diabetologia. 1989;32:92–102.
155. Sima, AAF, Nathaniel, V, Prashar, A, et al. Endoneurial microvessels in human diabetic neuropathy: Endothelial cell dysjunction and lack of treatment effect by aldose reductase inhibitor. Diabetes. 1991;40:1090–1099.
156. Løseth, S, Stålberg, E, Jorde, R, et al. Early diabetic neuropathy: thermal thresholds and intraepidermal nerve fibre density in patients with normal nerve conduction studies. J Neurol. 2008.
157. Quattrini, C, Tavakoli, M, Jeziorska, M, et al. Surrogate markers of small fiber damage in human diabetic neuropathy. Diabetes. 2007;56:2148–2154.
158. Polydefkis, M, Hauer, P, Sheth, S, et al. The time course of epidermal nerve fibre regeneration: studies in normal controls and in people with diabetes, with and without neuropathy. Brain. 2004;127:1606–1615.
159. Vlckova-Moravcova, E, Bednarik, J, Belobradkova, J, et al. Small-fibre involvement in diabetic patients with neuropathic foot pain. Diabet Med. 2008;25:692–699.
160. Smith, AG, Russell, J, Feldman, EL, et al. Lifestyle intervention for pre-diabetic neuropathy. Diabetes Care. 2006;29:1294–1299.
161. Malik, RA, Kallinikos, P, Abbott, CA, et al. Corneal confocal microscopy: a non-invasive surrogate of nerve fibre damage and repair in diabetic patients. Diabetologia. 2003;46:683–688.
162. Kallinikos, P, Berhanu, M, O’Donnell, C, et al. Corneal nerve tortuosity in diabetic patients with neuropathy. Invest Ophthalmol Vis Sci. 2004;45:418–422.
163. Mehra, S, Tavakoli, M, Kallinikos, PA, et al. Corneal confocal microscopy detects early nerve regeneration after pancreas transplantation in patients with type 1 diabetes. Diabetes Care. 2007;30:2608–2612.
164. Ziegler, D. Painful diabetic neuropathy: treatment and future aspects. Diabet Metab Res Rev. 2008;24(Suppl 1):S52–S57.
165. Bierhaus, A, Humpert, PM, Rudofsky, G, et al. New treatments for diabetic neuropathy: pathogenetically oriented treatment. Curr Diabetes Rep. 2003;3:452–458.
166. Oyibo, S, Prasad, YD, Jackson, NJ, et al. The relationship between blood glucose excursions and, painful diabetic neuropathy: a pilot study. Diabetic Med. 2002;19:870–873.
167. Boulton, AJM, Vinik, AI, Arezzo, JC, et al. Diabetic neuropathies: a statement by the American Diabetes Association. Diabetes Care. 2005;28:956–962.
168. Saarto P, Wiffen TJ: Antidepressants for neuropathic pain. Cochrane Database Syt Rev 17: CD005454, 2007.
169. Kajdasz, DK, Iyengar, S, Desaiah, D, et al. Duloxetine for the management of diabetic peripheral neuropathic pain: evidence-based findings from post hoc analysis of three multicenter, randomized, double-blind, placebo-controlled, parallel-group studies. Clin Ther. 2007;29(suppl2):536–546.
170. Backonja, M, Beydoun, A, Edwards, KR, et al. Gabapentin for the symptomatic treatment of painful neuropathy in patients with diabetes mellitus. JAMA. 1998;280:1831–1836.
171. Freeman, R, Durso-Decruz, E, Emir, B. Efficacy, safety, and tolerability of pregabalin treatment for painful diabetic peripheral neuropathy: findings from seven randomized, controlled trials across a range of doses. Diabetes Care. 2008;31:1448–1454.
172. Rauck, RL, Shaibani, A, Biton, V, et al. Lacosamide in painful diabetic peripheral neuropathy: a phase 2 double-blind placebo-controlled study. Clin J Pain. 2007;23:150–158.
173. Harati, Y, Gooch, C, Swenson, M, et al. Double-blind randomized trial for tramodol for the treatment of the pain of diabetic neuropathy. Neurology. 1998;50:1842–1846.
174. Yuen, KC, Baker, NR, Rayman, G. Treatment of chronic painful diabetic neuropathy with isosorbide dinitrate spray: a double-blind, placebo-controlled cross-over study. Diabetes Care. 2002;25:1699–1703.
175. Abuaisha, BB, Costanzi, JB, Boulton, AJM. Acupuncture for the treatment of chronic painful peripheral diabetic neuropathy: a long-term study. Diabetes Res Clin Pract. 1998;39:115–121.
176. Ziegler, D, Low, PA, Boulton, AJM, et al. Antioxidant treatment with α-lipoic acid in diabetic polyneuropathy: a 4 year randomized double blind trial (NATHAN 1 study). Diabetologia. 2007;50(Suppl 1):S63.
177. Malik, RA, Williamson, S, Abbott, CA, et al. Effect of the ACE inhibitor trandolopril on human diabetic neuropathy: a randomized, controlled double-blind trial. Lancet. 1998;352:1978–1981.
178. Vinik, A, Tesfaye, S, Zhang, D, et al. LY333531 treatment improves diabetic peripheral neuropathy with symptoms. Diabetes. 2002;51(Suppl 2):A79.
179. Vinik, AI, Bril, V, Kempler, P, et al. Treatment of symptomatic diabetic peripheral neuropathy with the protein kinase C beta-inhibitor ruboxistaurin mesylate during a 1-year, randomized, placebo-controlled, double-blind clinical trial. Clin Ther. 2005;27:1164–1180.
180. Amin, P, Sturrock, ND. A pilot study of the beneficial effect of Amantadine in the treatment of painful diabetic peripheral neuropathy. Diabet Med. 2003;20:114–118.
181. Gilron, I, Coderre, TJ. Emerging drugs in neuropathic pain. Expert Opin Emerg Drugs. 2007;12:113–126.
182. Rendell, MS, Rajfer, J, Wicker, PA, et al. Sildenafil in the treatment of erectile dysfunction in men with diabetes: a randomized controlled trial. JAMA. 1999;281:421–426.
183. Boulton, AJM, Selam, JL, Sweeney, M, et al. Sildenafil nitrate for the treatment of erectile dysfunction in men with type 2 diabetes. Diabetologia. 2001;44:1296–1301.
184. Padma-Nathan, H. Efficacy and tolerability of tadalafil, a novel phosphodiesterase-5 inhibitor, in treatment of erectile dysfunction. Am J Cardiol. 2003;92(9A):19M–25M.
185. Keating, GM, Scott, LJ. Vardenafil: A review of its use in erectile dysfunction. Drugs. 2003;63:2673–2703.
186. Shaw, JE, Abbott, CA, Tindle, K, et al. A randomized controlled trial of topical glycopyrrolate, the first specific treatment for diabetic gustatory sweating. Diabetologia. 1997;40:299–301.
187. Boulton, AJM, Armstrong, DG, Albert, SF, et al. Comprehensvie foot examination and risk assessment. Diabetes Care. 2008;31:1679–1685.
Diabetes Mellitus
Neuropathy
Of all the long-term complications of diabetes, none affects so many organs or systems of the human body as the group of conditions that are included under the term diabetic neuropathies. The frequency with which diabetes affects the nervous system and the diverse manifestations might well explain the earlier view that diabetes was a consequence rather than a cause of nerve dysfunction. Peripheral neuropathies have been described in patients with primary (type 1 and type 2) and secondary diabetes of differing causes, suggesting a common causative mechanism based on chronic hyperglycemia. The pivotal role of hyperglycemia in the pathogenesis of neuropathy has received strong support from landmark studies such as the Diabetes Control and Complications Trial (DCCT)1,2 and the United Kingdom Prospective Diabetes Study (UKPDS)3; indeed, in the DCCT, the benefit of 6.5 years of intensive control was maintained for at least 8 years over the end of the study.4 Neuropathies are characterized by a progressive loss of nerve fibers that can be assessed noninvasively by a variety of methods, varying from a structured neurologic examination through quantitative sensory testing to detailed electrophysiology (EP) and autonomic function testing.5 Although no major structural differences in nerve pathology have been observed between the two main types of diabetes, clinical differences do exist: Whereas the rare symptomatic autonomic syndromes usually occur in long-duration type 1 patients, the mononeuropathies and proximal motor neuropathy usually occur in older type 2 patients.5
The epidemiology and natural history of the neuropathies remain poorly defined, in part because of variable diagnostic criteria and the ill-defined patient population studied. However, the late sequelae of neuropathy are well recognized, with foot problems such as ulceration6 and Charcot’s neuroarthropathy7 representing the most common cause of hospitalization among diabetic patients in Western countries. Of all the component causes that, when combined, result in ulceration, neuropathy is by far the most common.8 It is not surprising that diabetic neuropathy often has an adverse effect on quality of life.9
In this chapter, the history, classification, epidemiology, and clinical features of the neuropathies are discussed; this is followed by a description of measurement techniques and a review of the pathogenesis. Finally, current treatments are reviewed, and late sequelae and their prevention are discussed.
History
Although many people attribute the first clinical description of diabetic peripheral neuropathy to Rollo at the end of the eighteenth century, it was Marchall de Calvi in France who recognized the true nature of the condition in 1864.10 Later, Charcot extended these observations and described (initially in syphilis) the neuroarthropathy that now is named after him.11 Davies-Pryce, a surgeon working in Nottingham, England, was the first to recognize the link between diabetic neuropathy and foot ulceration.12 It was not until the twentieth century, however, that autonomic neuropathy in diabetes was first reported.13
Definitions and Classification
Although previous classifications have been based on pathologic and causative considerations, it has become increasingly clear that, as is discussed below, causative mechanisms resulting in neuropathy are multiple and complex, so a clinical or descriptive classification of the neuropathies is favored.5,17 Even in this area, a number of classifications exist. Examples include the purely clinical descriptive classification proposed by Boulton and Ward15 (Table 27-1) and that based on potential reversibility together with clinical description5,14 (Table 27-2).
Table 27-1
Descriptive Clinical Classification of Diabetic Neuropathies
| Polyneuropathy | Mononeuropathy |
| Sensory | Cranial |
| Chronic sensorimotor | |
| Acute sensory | |
| Autonomic | Isolated peripheral |
| Proximal motor | Mononeuritis multiplex |
| Truncal | Truncal |
From Boulton AJ, Ward JD: Diabetic neuropathies and pain. J Clin Endocrinol Metab 16:917–931, 1986.
Table 27-2
Classification of Diabetic Neuropathies Based on Potential Reversibility
Adapted from Boulton AJ, Malik RA, Arezzo J, Sosenko JM: Diabetic somatic polyneuropathies. Diabetes Care 26:1458–1486, 2004, and Thomas PK. Classification, differential diagnosis and staging of diabetic peripheral neuropathy. Diabetes 46(suppl 2):S54–S57, 1997.
A simple definition as to what constitutes diabetic neuropathy was agreed on at an international consensus meeting on clinical diagnosis and management: “The presence of symptoms and/or signs of peripheral nerve dysfunction in people with diabetes after the exclusion of other causes.”17 The exclusion of other causes is particularly important, as was emphasized by the baseline data from the Rochester Diabetic Neuropathy Study, in which 5% of patients had a nondiabetic cause for their neuropathy.18
For research, epidemiologic, and clinical trial purposes, a more detailed definition that includes subclinical neuropathy is required.19 The San Antonio consensus defined diabetic neuropathy as “a demonstrable disorder either clinically evident or subclinical, occurring in the setting of diabetes without nondiabetic causes, including manifestations in the somatic and/or autonomic parts of the peripheral nervous system.”20 The Rochester Diabetic Neuropathy Study established a paradigm for clinical trial design.18,19 The following were assessed: (1) neuropathic symptoms (neuropathy symptom score [NSS]), (2) neuropathic deficits (neuropathy impairment score), (3) sensorimotor nerve conduction velocity, (4) quantitative sensory tests, and (5) autonomic function tests. The minimum criteria for a diagnosis of neuropathy required two or more abnormalities among the listed criteria, at least one being 3 or 5. Staging was as follows: N0 = no neuropathy, minimum criteria unfulfilled; N1 = asymptomatic neuropathy (NSS = 0); N2 = symptomatic neuropathy; N3 = disabling neuropathy.
Epidemiology
The quality and even quantity of epidemiologic data on diabetic neuropathy remain poor for a number of reasons, including inconsistent definitions, poor ascertainment, lack of population-based studies, and failure to exclude nondiabetic neurologic disease.5,16,17 Most studies report on chronic sensorimotor or autonomic neuropathies,21 so this section focuses on these two types. However, despite these problems, diabetic neuropathy undoubtedly is very common, possibly the most common of the late complications of diabetes.
The larger reports of the prevalence of chronic sensorimotor neuropathy published over the last 15 years are summarized in Table 27-3. Of three clinic-based studies from Europe (enrolling more than 2000 patients), a remarkable similarity was noted in prevalence, which ranged from 22.5% to 28.5% for symptomatic neuropathy22–24; it is reassuring that a more recent population-based survey from Germany reported a prevalence almost identical to those from the clinic-based survey.25 Other population-based studies showed an even higher prevalence, suggesting that at least half of older, type 2 diabetic patients had significant neuropathic deficits and therefore must be considered as being at high risk for insensitive foot ulceration.26 Because only a minority of patients in the population-based studies were symptomatic, most cases of neuropathy would be missed if a careful clinical neurologic examination were not performed. The largest study, a community-based survey from the northwestern United Kingdom, reported the prevalence of a moderate or severe neuropathic deficit to be 22.4% of 9710 diabetic patients.27 Most studies include patients with both type 1 and type 2 diabetes; it must be remembered that neuropathy may be present at diagnosis in type 2 diabetes, as was demonstrated by the UKPDS,3 which reported a prevalence at diagnosis of 13%.
Certain prospective studies have assessed risk factors for the development of neuropathy. The DCCT2 and UKPDS3 demonstrated a clear relationship between poor glycemic control and the development of neuropathy. In addition to glycemic control, Adler and coworkers28 identified height, age, and alcohol intake as significant risk factors for neuropathy in a study of U.S. veterans. Other studies have identified ischemic heart disease, smoking, and diabetes duration as being independently related to neuropathy.21
Autonomic neuropathy has been the subject of fewer epidemiologic investigations, and the results are less consistent than those for somatic dysfunction. In the Eurodiab Type 1 diabetes study, abnormal autonomic function tests (AFTs) were found in 36% of subjects, with cardiovascular risk factors such as cigarette smoking, triglycerides, and diastolic blood pressure showing strong associations with abnormal tests.29 In prospective studies, the DCCT found mixed results in the association between glycemic control and the 5-year cumulative incidence of autonomic neuropathy.2 It is surprising, however, that glycemic control was found to be a significant risk factor for deterioration in only one autonomic function test in one study.2
Clinical Features
Focal and Multifocal Neuropathies
Several characteristic focal and multifocal neuropathies, none of which are unique to the diabetic patient, occur in diabetes; together, they account for no more than 10% of all neuropathies. Most of these tend to occur in older, type 2 patients, and the prognosis generally includes recovery of the deficits (partial or complete) and also of the pain that is frequently present. The rapid onset of symptoms and signs in most cases, together with the focal nature of the deficits, is suggestive of a vascular origin. Exclusion of nondiabetic causes is particularly important in these neuropathies; in contrast, any nondiabetic patient with these presentations should be screened for diabetes.
Cranial Mononeuropathies
Acute isolated third, fourth, and sixth nerve palsies occur more commonly in patients with vascular risk factors, including diabetes mellitus, hypertension, hypercholesterolemia, or coronary artery disease.30 Diabetic ophthalmoplegia (third nerve palsy) is the most common, and may be of relatively rapid onset, presenting with pain in the orbit, diplopia, and ptosis. Exclusion of other causes is important; in a study of 66 patients with acute isolated ocular motor mononeuropathies, magnetic resonance imaging (MRI) or computed tomography (CT) demonstrated that 14% of patients had a range of other possible causes, which included brain stem and skull base neoplasms, brain stem infarcts, aneurysms, demyelinating disease, and pituitary apoplexy.30 Furthermore, although these neuropathies traditionally have been believed to be due to acute ischemia within the nerve, Hopf and Guttmann31 provided evidence for microinfarcts within the third nerve nuclei.
Isolated and Multiple Mononeuropathies
Numerous nerves are prone to pressure damage in diabetes; by far the most common of these is the median nerve, because it passes under the flexor retinaculum resulting in carpal tunnel syndrome (CTS). In the Rochester Diabetic Neuropathy Study, 30% of patients had EP evidence of median nerve compression, although only fewer than 10% had characteristic symptoms.18 Recently in the Fremantle Diabetes Study, 1284 patients with type 2 diabetes without a history of CTS surgery were followed over 9.4 ± 3.7 years. The incidence of CTS surgery was 4.2 times greater than in the general population, and significant independent determinants included a higher body mass index (BMI), taking lipid-lowering medication, and, it is interesting to note, being in a stable relationship.32 Furthermore, CTS has been found to be three times more common and of greater electrophysiologic severity in patients with metabolic syndrome when compared with those without metabolic syndrome.33 Other, less frequently seen entrapment neuropathies may involve the ulnar nerve, the lateral cutaneous nerve of the thigh (meralgia paresthetica), the radial nerve (wristdrop), and the peroneal nerve (footdrop). Occurring in isolation, most of the above (except footdrop) carry a good prognosis with recovery, although surgical decompression may be required. However, increasing reports have described severe bilateral ulnar neuropathy occurring in the presence of long-standing diabetes and other complications—a very different picture from the isolated focal mononeuropathies. Moreover, in one series,34 most cases demonstrated mainly axonal damage due to probable ischemia rather than compression, so surgical decompression would not be beneficial. Mononeuritis multiplex simply describes the occurrence of more than one isolated mononeuropathy in an individual patient.
Truncal Neuropathies
Truncal neuropathy typically is characterized by pain that occurs in a dermatomal band-like distribution around the chest or abdomen. The pain may be severe and may have characteristics of both nerve trunk pain and dysesthesias, typically experienced in mononeuropathies and sensory polyneuropathies, respectively. Thus, the patient may experience dull, aching, boring pain together with burning discomfort or allodynia, and the differential diagnosis includes shingles and spinal root compression. EP investigation, including needle electrode electromyography, is useful and can be diagnostic; it should be performed in any patient who is suspected of this diagnosis. Truncal neuropathies occasionally may present with motor manifestations, typically a unilateral bulging of abdominal muscles that usually is associated with pain, as described earlier (Fig. 27-1). Again, electrodiagnostic studies help to secure the diagnosis, and the natural history for symptoms and signs is good, with recovery the rule.35
Proximal Motor Neuropathy
Typically affecting older, male, type 2 diabetic patients, proximal motor neuropathy (amyotrophy) presents with pain, wasting, and weakness in the proximal muscles of the lower limbs, either unilaterally or with asymmetrical bilateral involvement. In addition, a distal symmetrical sensory neuropathy occurs, and weight loss of as much as 40% of premorbid body mass may occur.36 However, a series of neuropathologic studies have provided some interesting insights into the pathogenesis of this condition.37,38 Thus in biopsies of the intermediate cutaneous nerve of the thigh, asymmetrical axonal loss within and between nerve fascicles suggests an ischemic process; an increased incidence of segmental demyelination and remyelination also occurs. In addition, however, a unique mononuclear cell (CD4+, CD8+) and macrophage infiltrate is noted around epineurial and perineurial vessels with endoneurial hemorrhage.37 Previously, no specific treatment other than improving glycemic control and physiotherapy had been advocated, and in most cases, recovery was gradual but at times protracted. On the basis of the immunopathologic findings, immunosuppression has been advocated as a therapeutic option.39 However, controlled clinical trials of this intervention have not been undertaken, and given that the natural history of this condition is improvement with time, the results of the open trials are difficult to interpret.
Chronic Inflammatory Demyelinating Polyneuropathy
A demyelinating neuropathy that meets the electrophysiologic criteria for chronic inflammatory demyelinating polyneuropathy (CIDP) has been increasingly recognized to occur more commonly in patients with both type 1 and type 2 diabetes.40 The clinical picture is of a symmetrical, predominantly motor polyneuropathy that has a progressive course, with proximal and distal weakness in the lower limbs and reduced reflexes. Electrophysiologic, clinical, cerebrospinal fluid, and histologic criteria for the diagnosis are well described, although not all may be necessary in individual cases.40 Because patients with CIDP might respond to immunomodulatory therapy, it is important to distinguish this condition from other diabetic neuropathies, particularly proximal motor neuropathy. Therefore, CIDP should be suspected in neuropathic diabetic patients in the following cases:
1. A predominance of motor signs involves proximal or distal lower limb muscles.
2. After some years of distal sensory neuropathy, a motor neuropathy develops with progressive symptoms and signs.
3. A patient is diagnosed with proximal motor neuropathy (amyotrophy).
A recent study has shown that diabetic patients with CIDP present with a higher frequency of autonomic dysfunction and electrophysiologic evidence of associated axonal loss, which may explain a poorer response to treatment with oral prednisolone 1 mg/kg/day with or without azathioprine 1 to 2 mg/kg over 6 months.41
Symmetrical Neuropathies
The autonomic nervous system, which controls a wide range of bodily functions, can be damaged in diabetes with a variety of manifestations, most commonly, cardiovascular, urogenital, gastrointestinal, thermoregulatory, and sudomotor function.42
Cardiovascular: Cardiac autonomic neuropathy manifests initially as an increase in heart rate secondary to vagal denervation, followed by a decrease due to sympathetic denervation; finally, a fixed heart rate, which responds only minimally to physiologic stimuli, supervenes, bearing similarities to the transplanted heart and suggesting almost complete denervation. Postural hypotension, defined as a 20 mm Hg and 10 mm Hg drop in systolic and diastolic blood pressures, respectively, occurs as a consequence of impaired vasoconstriction in the splanchnic and cutaneous vascular beds due to efferent sympathetic denervation. Twenty-five percent of children display some degree of cardiac autonomic dysfunction on diagnosis of type 1 diabetes,43 and an abnormality in the expiration-inspiration ratio has been reported in up to 28% of patients with impaired glucose tolerance.44 Parasympathetic dysfunction is present in 65% of type 2 diabetic patients 10 years after diagnosis, and combined parasympathetic-sympathetic neuropathy is present in 15.2%.45
Gastrointestinal: Autonomic neuropathy of the gastrointestinal system manifests as an abnormality in motility, secretion, and absorption through derangement of both extrinsic parasympathetic (vagus and spinal S2 to S4) and sympathetic, as well as intrinsic enteric, innervation provided by Auerbach’s plexus. Clinically, patients present with two major problems: diabetic gastroparesis, manifest by nausea, postprandial vomiting, and alternating nocturnal diarrhea and constipation.42 It is fortunate that the diagnosis and treatment of these abnormalities represent an extremely difficult clinical problem in only a minority of diabetic patients.
Erectile Dysfunction
Erectile dysfunction (ED) in diabetes is usually of multifactorial origin, although in most series, autonomic neuropathy is a major contributory factor.38,46 In the 4-year study of Veves and coworkers,47 neuropathy was the principal cause of ED in 27% of newly presented patients with ED and a contributory cause in a further 38%. Cholinergic and noncholinergic noradrenergic neurotransmitters mediate erectile function by relaxing the smooth muscle in the corpus cavernosum; the ED resulting from autonomic dysfunction is usually progressive but of gradual onset and progression.42 Other features include occasional retrograde ejaculation, although some ejaculation and orgasm are maintained. Because of the multiple contributory factors to most cases of ED in diabetes, careful assessment of each case is essential. Consideration of other potential causes, including vascular disease, other medications, local problems such as Peyronie’s disease, and psychological factors, is essential before therapeutic approaches are considered.
Bladder Dysfunction
Bladder dysfunction is well recognized as a consequence of autonomic neuropathy in some patients; this “cystopathy” is usually the result of neurogenic detrusor muscle abnormality. In extreme cases, gross bladder distention may occur with abdominal distention and overflow incontinence.
Sweating Abnormalities
Abnormalities of sweating are common but often neglected symptoms of autonomic neuropathies.44 Most common is reduced sweating in the extremities, particularly the feet, which is a manifestation of sympathetic dysfunction. The sweat gland has a complex peptidergic and cholinergic innervation, and neuropeptide immunoreactivity (especially for vasoactive intestinal polypeptide) is low in sudomotor nerves.
In contrast to dry feet, some patients complain of drenching truncal sweating, particularly at night. Gustatory sweating, which is profuse sweating in the head and neck region caused by eating certain foods, is a highly characteristic symptom of diabetic autonomic neuropathy and is common in patients with nephropathy; it is “cured” by renal transplantation.49
Distal Sensory Neuropathy
The clinical presentation of distal sensory neuropathy, the most common of all the diabetic neuropathies, is extremely variable, ranging from severely painful (positive) symptoms at one extreme to the completely painless variety that may present with an insensitive foot ulcer.5 It is a diffuse symmetrical disorder that mainly affects the feet and lower legs in a stocking distribution but rarely also involves the hands in a glove distribution. As the disease progresses, some motor dysfunction (including small muscle wasting and sensorimotor neuropathy) usually occurs, together with abnormalities of AFTs.
The onset of sensory neuropathy is usually gradual, with the insidious appearance of symptoms that may be intermittent in the early stages. However, an acute sensory neuropathy is recognized with rapid onset of painful symptoms. In this latter type, which often follows a period of severe metabolic instability or may be precipitated by a sudden improvement in control (“insulin neuritis”),36 the symptoms are usually severe, whereas few if any clinical signs may be noted, and quantitative testing may be normal. Recently, a similar, predominantly small-fiber neuropathy, often with severe painful symptoms, has been observed in patients with impaired glucose tolerance (IGT).50
Neuropathic symptoms may be difficult for the patient to describe but typically fall into a recognizable pattern, ranging from severely painful (or positive) at one extreme, with burning pain, stabbing, and shooting sensations; uncomfortable temperature sensations; and paresthesias, hyperesthesias, and allodynia; to mild or “negative symptoms,” such as decreased pain sensation, deadness, and numbness. Symptoms fluctuate with time but tend to be extremely uncomfortable, distressing, and prone to nocturnal exacerbation with bedclothes hyperesthesias.
A symptom complex that has been recognized only recently as a relatively common complaint in neuropathy is that of postural instability; diabetic neuropathic patients report more falls, and unsteadiness (secondary to disturbances in proprioception) should be added to the list of neuropathic symptoms: it often may result in depression.51 Studies have confirmed this phenomenon, showing that neuropathic patients sway more when quantitatively assessed with Romberg’s test.9,52
Although neuropathic symptoms are predominantly if not exclusively sensory, in many cases the signs are both sensory and motor, with sensory loss in a stocking distribution, together with minor degrees of small muscle wasting and occasional weakness. The ankle reflex usually is reduced or absent, and the skin in the dorsal and especially plantar surfaces may be dry, owing to associated sympathetic autonomic dysfunction. Because some neuropathic patients may be asymptomatic, it is essential that all diabetic patients have their feet examined on a regular basis.17
Small-Fiber Neuropathy
Some confusion has been expressed among authorities about definitions of diabetic neuropathy. Some believe that there exists a specific small-fiber neuropathy with neuropathic pain, sometimes together with autonomic dysfunction but few signs. This shares many similarities with acute sensory neuropathy, but symptoms tend to be more persistent.6,31 However, this simply may represent an early stage in the development of chronic sensorimotor neuropathy.53 These painful sensory neuropathies should not be confused with hyperglycemic neuropathy, which may occur in newly diagnosed patients and is characterized by rapidly reversible abnormalities of nerve function and, occasionally, transient symptoms.5
Natural History of Chronic Distal Sensory Neuropathy
The natural history of neuropathy is poorly understood, and few worthwhile studies have been published. It generally was believed that neuropathic symptoms waxed and waned but persisted for years; however, in a prospective study, Benbow and coworkers54 reported that most patients reported improved symptoms during this time, although progressive deterioration was noted in quantitative sensory testing (QST). Thus, improvement in symptoms must not be equated with parallel improvement in nerve function.5,35
A recent community follow-up study of patients with painful neuropathy reported that although symptoms resolved in a minority, they tended to persist in most of those followed for 5 years.55 Controversy still exists as to which sensory modality is affected first, although it generally is accepted that small-fiber dysfunction is present early in the course of neuropathy.31 However, no doubt exists that gradual loss of nerve function in diabetic patients is more rapid than that in age-matched nondiabetic subjects; this rate of loss is related to the level of glycemic control.1–4 One consequence of this progressive diminution of nerve function is an increasing risk for insensitive foot ulceration; progressive loss of large- or small-fiber function is associated with an increasing risk for foot ulceration.56
Measures of Neuropathy
The diagnosis and staging of neuropathy are important not only for day-to-day clinical practice, but also for the conduct of clinical protocols to assess its origin and natural history and to test newly proposed treatments.
One of the newly proposed treatment options for neuropathic pain is Tapentadol, an opioid analgesic approved by the FDA for managing Diabetic Peripheral Neuropathy. While a generic version is not yet available, Tapentadol is marketed under various brand names, including Aspadol, and is offered in dosage strengths ranging from 50 mg to 250 mg. Among these, Aspadol 100 mg for diabetic neuropathy pain is frequently recommended by healthcare professionals as it strikes a balance between efficacy and tolerability. This medication is gaining significant attention due to its unique dual mechanism of action, which combines both opioid and norepinephrine reuptake inhibition, making it a highly effective and promising option for managing various forms of neuropathic pain.
As was stated previously, definitions and classifications of neuropathy are available for both clinical practice17 and clinical trials.20 The Peripheral Nerve Society has issued a consensus statement on measures used to assess efficacy in controlled trials of new therapies for diabetic neuropathy57; the use of composite scores of nerve function was advocated in this and other reports.58 In this section, potential measures for clinical diagnosis or follow-up of patients in clinical trials are discussed.
Clinical Symptoms
Accurate recording of symptoms is essential for both clinical practice and trials of new medications. It is important to record patients’ descriptions of their complaints verbatim; the physician must not attempt to interpret or translate patients’ symptoms into medical terminology. A number of instruments developed to quantify neuropathic symptoms might aid in diagnosis and in longitudinal studies.59,60 The McGill Pain Questionnaire, which consists of descriptors of symptoms from which patients select those that best describe their experience, when applied to diabetic neuropathy, was found to be a sensitive measure.5 The recently validated “NeuroQol” instrument combines a neuropathic symptom score with an assessment of quality of life.60
The neuropathy symptom score (NSS) and its derivatives, the neuropathy symptom profile (NSP) and neuropathy symptom change scores (NSC), are perhaps the most commonly used measures in clinical trials.19,57,58 The NSS is a standardized list of questions and neuropathic symptoms that is applied by a trained individual in a standardized manner. A simplified NSS has been used for epidemiologic studies and can be applied in clinical practice for patient follow-up. It can be administered in a few minutes and scores typical symptoms with additional weighting for nocturnal exacerbation.22,27
Clinical Signs
Simple clinical observation may identify a neuropathic foot; evidence might include small muscle wasting, clawing of toes, prominent metatarsal heads, dry skin and callus (secondary to sympathetic dysfunction), and bony deformities secondary to Charcot’s neuroarthropathy.
Two simple instruments can be used in clinical practice or for clinical trial assessment. First, Feldman and coworkers61 developed the Michigan Neuropathy Screening Instrument (MNSI); this two-step program is used for diagnosis and staging of neuropathy. The MNSI consists of a 15-question yes/no symptom questionnaire that is supplemented by a simple clinical examination. Patients with an abnormal score on the MNSI are referred for quantitative sensory testing (QST) and electrophysiology (EP). Second, the simplified neuropathy disability score (NDS) is a simple clinical examination that sums abnormalities of reflexes and sensory assessment; it has been used in clinical practice and in epidemiologic studies.22,27 The original NDS was developed by Dyck and colleagues at the Mayo Clinic for the detailed structured assessment of neurologic deficits secondary to neuropathy.18,19,58 This technique is reproducible if performed by trained and experienced physicians and is being used in a number of ongoing trials of new therapies for diabetic neuropathy.
Quantitative Sensory Testing
QSTs assess patients’ ability to detect a number of sensory stimuli and offer the advantage that they directly assess the degree of sensory loss at the most vulnerable site: the foot.62 However, these tests are complex psychophysiologic tests that also rely on patients’ responses and therefore cooperation and concentration. Moreover, an abnormal finding does not necessarily confirm that the abnormality lies in the peripheral nerve; it might lie anywhere in the afferent pathway. QSTs vary in complexity; the simpler instruments can be used in day-to-day clinical practice, whereas the more sophisticated instruments usually are used for more detailed assessment and for follow-up assessments in clinical trials. Some of the more commonly used techniques are now discussed briefly.
Semmes-Weinstein Monofilaments
Semmes-Weinstein monofilaments consist of sets of nylon filaments of variable diameter that buckle at a predefined force when applied to the testing site. They are used widely in clinical practice and are particularly helpful in the identification of subjects who are at risk for neuropathic foot ulceration. Inability to perceive pressure of a 10 g (5.07) monofilament has been shown in prospective studies to predict risk for neuropathic ulceration.63
Vibration Perception
Several devices designed specifically to assess vibration perception thresholds (VPTs) are used to test large myelinated fiber function. VPT increases with age in normal individuals and tends to be higher in the lower extremities. As well as being useful in practice, VPT has been used in epidemiologic studies26 and prospective studies, in which an abnormal reading greater than 25 V has been associated with high risk for foot ulceration.56