[level-membership-for-anesthesiology-category]
Chapter 51 Diabetes insipidus and other polyuric syndromes
Diabetes insipidus (DI: literal translation, ‘tasteless siphon’) refers to a syndrome characterised by pathological polyuria, excessive thirst and polydipsia. Polyuria is arbitrarily defined as a urine loss of > 3 l/day in an adult of normal mass. The urine produced in DI is inappropriately dilute, having both low specific gravity and low osmolality in the face of a high or normal plasma osmolality.
Three subtypes of DI are recognised:
In critically ill patients, polyuria may be the sole part of the DI syndrome apparent to the clinician. Patients are seldom in control of their own fluid intake and are frequently unable to report thirst. The recognition of DI is important as failure to recognise and treat the syndrome appropriately will result in severe dehydration and hyperosmolality with a significant risk of morbidity and mortality. As there are many causes of polyuria in the critically ill (Table 51.1), it is important to adopt a systematic approach to the clinical assessment, investigations, diagnosis and management of such patients.
| Water diuresis |
| Pathological |
| Diabetes insipidus: cranial, nephrogenic, gestational |
| Physiological |
| Psychogenic polydipsia (excess drinking is pathological but the diuresis is not) |
| Iatrogenic – excessive administration of hypotonic solutions, e.g. 5% dextrose solution, 0.45% saline, 0.18% saline 4% dextrose solutions |
| Solute diuresis |
| Pathological |
| Fanconi’s syndrome |
| Renal tubular acidosis |
| Glomerulonephritis |
| Hyperaldosteronism |
| Anorexia nervosa |
| Migraines |
| Paroxysmal tachycardia (via increased atrial natriuretic peptide release) |
| Poisons/drugs: |
| Ethanol |
| Methanol |
| Ethylene glycol |
| Mannitol |
| Loop diuretics |
| Thiazide diuretics |
| Hyperglycaemia |
| Physiological |
| Resolving sepsis (redistribution of fluid into the vascular compartment from the third space) |
| Iatrogenic – excessive administration of isotonic or hypertonic solutions, e.g. |
| 0.9% saline |
| Hypertonic saline |
| Hartmann’s solution |
| Gelofusin |
BACKGROUND PHYSIOLOGY AND ANATOMY
OSMOLALITY
Osmolality is the measure of osmoles of solute per kilogram of solvent and includes both permeant (e.g. urea) and impermeant solutes (e.g. sodium) and thus osmolality may change due to both water movement and the movement of permeant solute. Osmolarity is the measure of osmoles per litre of solute and thus is temperature-dependent. Osmolality is temperature-independent. Normal plasma osmolality is in the range of 275–295 mosmol/kg. Plasma osmolality can be estimated from several equations.1 The formula of Worthley et al.2 below is both simple and correlates well with measured values.3
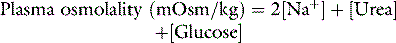
All units of solute are mmol/l.
The osmolal gap is the difference between the calculated and measured osmolality in plasma or urine.The normal osmolal gap is < 10 mosmol/l. An increased osmolal gap indicates the presence of an unmeasured osmotically active solute.
NORMAL URINARY OSMOLALITY
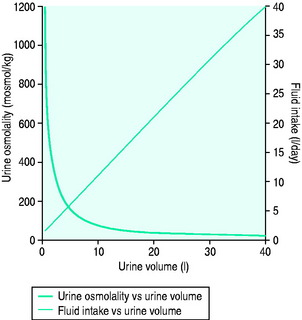
In health, urinary osmolality is usually maintained between 500 and 700 mosmol/kg. As the obligatory solute load to be excreted is relatively constant in value, urine osmolality will fall in response to an increased intake in free water and rise in response to dehydration or water restriction (Figure 51.1). The minimum osmolality of urine achievable inhumans is around 25 mosmol/kg. Diuresis refers to the passage of a high volume of urine (> 1.5 ml/kg per hour) and may be transient or persistent, physiological or pathological. The production of > 3 l/day is arbitrarily defined as polyuria. Water diuresis occurs when the total solute in the urine excreted per day is within the normal range but the osmolality of the urine passed is low. An osmotic or solute diuresis occurs when the total solute passed per day is higher than normal, and the urine passed is usually iso-osmolar to plasma if the extracellular fluid volume is expanded or hyperosmolar if the patient is hypo- or euvolaemic.
OSMORECEPTORS AND OTHER INPUTS TO THE SUPRAOPTIC AND PARAVENTRICULAR NUCLEI
Detection of osmolality occurs largely at osmo- (Na+) receptors sited around the anterior aspect of the third ventricle of the brain. These are sensitive to plasma osmolality and cerebrospinal fluid sodium concentration. Hypertonic saline is a more potent stimulus than equi-isotonic equiososmolar solutions of other solutes.4 These osmoreceptors link to the cells of the paraventricular nuclei (PVN) and supraoptic nuclei (SON), the sites of ADH synthesis. The axons of the cells in the PVN and SON form part of the pituitary stalk linking the hypothalamus to the pituitary gland in which they terminate. A smaller proportion of the axons terminate in the median eminence where they release ADH and oxytocin, which is transported to the anterior lobe of the pituitary by portal vessels. The ADH and oxytocin so released cause release of adrenocorticotrophic hormone (ACTH) and prolactin respectively; the ADH acts synergistically with corticotrophin-releasing factor (CRF) but is also believed to have ACTH secretagogue properties in its own right.5,6
Direct inputs from the sympathetic nervous system to the PVN and SON can stimulate ADH release via α-adrenoreceptors. Other central osmoreceptors lie outside the blood–brain barrier in the subfornical organ and come into contact with plasma. It is believed that ANP and angiotensin II7 act via these receptors to inhibit or elicit ADH synthesis and ADH release and to modify the sensation of thirst. Additional osmoreceptors in the mouth, stomach and liver are believed to play a role in the anticipation of an osmolal load following ingestion of food and can pre-emptively stimulate ADH synthesis in the hypothalamus.
As the baroreceptor and osmoreceptor inputs to the PVN and SON are distinct, it is possible to lose the normal ADH response to hyperosmolality but maintain a normal ADH response to hypovolaemia.8 Additionally, in animal experiments, when hypotension increases the basal plasma ADH concentration, there is a simultaneous resetting of the osmomolality–plasma ADH response curve in an attempt to preserve osmoregulatory function from the new higher baseline.9 If this did not occur, the ADH response to hypotension would always result in the development of a hypo-osmolal state in addition to causing vasoconstriction.
The normal response of osmoreceptors to changing plasma osmolality in terms of ADH is illustrated in Figure 51.2. At plasma osmolalities of < 275 mosmol/kg, the osmoreceptors remain hyperpolarised and virtually no ADH release occurs via them. At osmolalities > 295 mosmol/kg, the osmoreceptors are maximally depolarised and plasma concentrations of ADH of > 5 pg/ml are attained. Other inputs and influences upon ADH release are summarised in Figure 51.3 and Table 51.2.
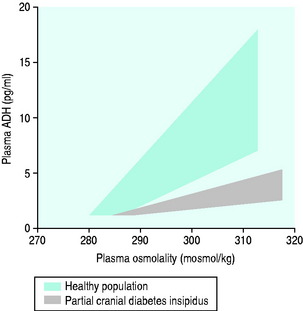
Figure 51.2 Plasma antidiuretic hormone (ADH) and plasma osmolality in health and partial cranial diabetes insipidus.
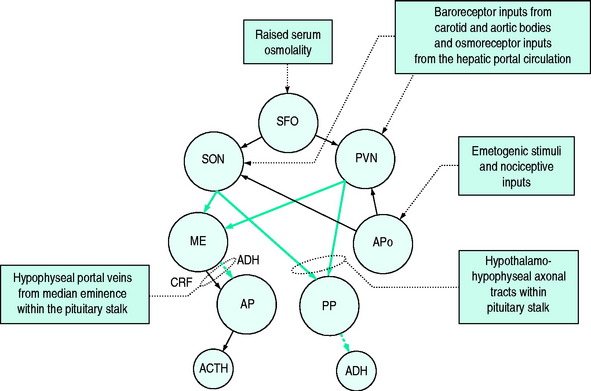
Table 51.2 Factors influencing antidiuretic hormone (ADH) release
| Increased ADH release with: |
| Hyperosmolality |
| Hypovolaemia |
| Hypotension |
| Hypoxia |
| Hypothyroidism |
| Hyperthermia |
| Positive-pressure ventilation |
| Pain |
| Emotional stress |
| Exercise |
| Nausea |
| Nicotine |
| Trauma/surgery |
| Decreased ADH release with: |
| Hypo-osmolality |
| Hypervolaemia |
| Hypertension |
| Ethanol |
| Cranial diabetes insipidus |
ANTIDIURETIC HORMONE/ARGININE VASOPRESSIN (AVP)
ADH (8-arginine vasopressin) is a nine-amino-acid peptide which differs from oxytocin at only two residues but shares the disulphide bond between the first and sixth ones. This similar structure and conformation results in some cross-reactivity at receptors and in function.10 It is synthesised in the SON and PVN, bound to neuorophysin, transferred through axons to the posterior pituitary gland and stored in granules prior to release. Synthesis to replace any released stores is a rapid process (1–2 hours from synthesis to storage) and patients with damage to the pituitary can achieve near-normal plasma concentrations of ADH, in terms of osmoregulatory function, via release of newly synthesised ADH via the axons terminating in the median eminence. However, the higher plasma concentrations associated with hypovolaemia cannot be achieved. Normal osmoregulatory plasma ADH concentrations are in the range of 1–8 pg/ml but rise as high as 40 pg/ml in hypovolaemic patients under the influence of the sympathetic nervous system, baroreceptor responses and angiotensin II.
Once released from the pituitary, ADH has a plasma half-life of around 10–35 minutes.11 It is metabolised by hepatic and renal vasopressinases and around 10% of the active hormone is excreted unchanged in the urine.
ACTIONS OF ADH
ADH has antidiuretic, vasopressor, haemostatic and ACTH secretagogue actions. Additionally, it has roles in memory, water permeability of the blood–brain barrier, nociception, splenic contraction and thermoregulation. These actions are mediated through V1, V2 and V3 receptors. It also has actions on the uterus and mammary tissue mediated through oxytocin receptors. Cardiac inotropic effects are reported to be mediated through purinergic P2 receptors but this remains controversial.12
Antidiuresis
ADH binds to V2 receptors on the basal membranes of the principal cells of the collecting duct and distal tubule. The activated receptor induces production of cyclic adenosine monophosphate (cAMP) by adenylate cylase and this in turn activates protein kinases which effect the integration into the luminal membrane of vesicles containing aquaporin-2 highly selective water channels. The production of prostaglandin E2 (PGE2) inhibits cAMP production. PGE2 synthesis is stimulated by the action of ADH on V1 receptors on the luminal membrane of the collecting duct.13 Thus, a form of autoregulatory limitation of the antidiuretic effect of ADH may exist. Hypokalaemia, lithium and hypercalcaemia also anatagonise the renal actions of ADH.
In low dose, administration of exogenous ADH may paradoxically cause a diuresis in patients with septic shock.14 Whether this is due to increased renal perfusion pressure and raised glomerular filtration rate (GFR) is unclear.
Vasoconstriction
At higher concentrations (> 40 pg/ml), ADH activates not only V2 receptors but also V1 receptors, through which it causes preferential vasoconstriction in muscle, skin and fat with relative sparing of coronary, cerebral and mesenteric circulations. However, these relative sparing effects are controversial, with some authors reporting relative sparing and others reporting significant vasoconstriction.15–18 Activation of V1 receptors activates phospholipase C and increases inositol triphosphate intracellularly. Ultimately this increases intracellular free calcium and leads to smooth-muscle constriction in the blood vessel wall.
Coagulation
ADH increases circulating levels of tissue plasminogen activator, factor VIII and von Willebrand factor.19 These effects may be mediated by V2 receptors but this remains controversial. At high but physiological concentrations it can act as a platelet-aggregating agent.20,21 Platelet aggregation is mediated through activation of platelet V1 receptors.22 ADH and its analogue DDAVP are used as first-line treatments in patients with von Willebrand’s disease, and may be used in bleeding associated with renal failure and platelet dysfunction.
VOLUME RECEPTORS
Volume homeostasis takes precedence over sodium homeostasis and so rises and falls in sodium will occur in order to preserve the circulating volume. In euvolaemic patients sodium homeostasis is maintained. Sodium concentration is detected by both the osmoreceptors of the subfornical organ outside the blood–brain barrier and also by the juxtaglomerular apparatus which secretes renin in response to reduced GFR and a lower sodium load in the tubule.23
The predominant determinants of sodium balance, however, are the high-pressure baroreceptors in the pulmonary veins, left atrium, carotid sinus and aortic arch.24 Reduced stretch of these receptors increases sympathetic nervous system activity and activation of the renin–angiotensin–aldosterone system, resulting in reduced sodium excretion (via reduced GFR) and increased reabsorption of sodium in the proximal and distal convoluted tubules. Additionally, release of ADH can be stimulated, resulting in concomitant water retention. Conversely, stretch of the baroreceptors will result in a fall in sodium retention through reduced activity of the sympathetic nervous and renin–angiotensin–aldosterone systems. Stretch additionally results in the release of ANP and a natriuresis through reduced sodium reabsorption in the distal convoluted tubule and collecting duct. ADH release is reduced by the fall in sympathetic nervous tone from the baroreceptors. ADH secretion may also be inhibited by the action of ANP on cerebral osmoreceptors lying outside the blood–brain barrier.25
CRANIAL DIABETES INSIPIDUS
ACQUIRED CDI
Acquired CDI may be transient or permanent and can arise due to an absolute (complete) or relative (incomplete) lack of ADH. Complete central DI is usually associated with lesions above the level of the median eminence in the SON or PVN or of the neurohypophyseal stalk whereby the production of ADH in the hypothalamus is terminated.26 Permanent central DI tends to be associated with transecting, obliterating or chronic inflammatory lesions, whereas transient DI is more likely to be associated with acute inflammatory or oedematous lesions with some recovery of ADH secretion occurring as the inflammation or oedema resolves. An exception to this is the transient DI seen following excision or destruction of the posterior pituitary; ADH produced in the hypothalamus can still be released into the systemic circulation from capillaries in the median eminence.
In the past the majority of acquired non-traumatic CDI was categorised as idiopathic but it has become apparent that the majority of these cases are associated with abnormality of the inferior hypophyseal arterial system27 or autoimmune reactivity against ADH-producing cells.28 These findings may indicate causality or association.
Where ADH deficiency is relative rather than absolute, it is possible for the patient to concentrate the urine partially and values of 500–800 mosmol/kg would not be atypical. However, these osmolalities are inappropriately low relative to the plasma osmolality. In partial ADH deficiency volumes of urine as low as 3 l/day may be evidenced. These are still inappropriately high when assessed in terms of the solute excretion of the patient but are more difficult to recognise as being due to DI as there are many other causes of diureses of this magnitude. Additionally, extrinsic stimulants of ADH release (see Table 51.2) may have an antidiuretic effect, further complicating the diagnosis.
CDI is usually associated with reduced production of ADH or damage to the normal release mechanisms of ADH. However, there can be dysfunction of the osmolality-sensing mechanism at receptor or intracellular signalling levels whilst actual ADH production and storage are normal. It is possible to have a normal release of ADH in response to baroreceptor detection of hypotension but subnormal release in response to hyperosmolality. This has been described in association with chronic hypernatraemia.29
The main recognised causes of central DI are listed in Table 51.3. A particularly common cause of DI seen in the ICU is traumatic or postsurgical brain injury. Transsphenoidal surgery for treatment of suprasellar tumours can result in DI in 10–70% of patients; the frequency parallels the magnitude of the tumour being removed. Additionally, transcranial surgery may cause the development of DI in the absence of a fall in plasma ADH. This is postulated to be due to the release of a hypothalamic ADH precursor which acts as a competitive antagonist of both ADH and synthetic analogues. The presence of a competitive antagonist effectively creates an endocrinologice picture similar to NDI with normal or high plasma ADH levels but an inappropriate diuresis of dilute urine.
| Acquired |
| Idiopathic |
| Autoimmune |
| Tumours (especially suprasellar, lung, breast, lymphoma and leukaemia) |
| Surgery (especially transsphenoidal surgery) |
| Traumatic head injury (strongly associated with base-of-skull fracture*) |
| Hypoxic brain injury |
| Brainstem death |
| Electrolyte disturbance – profound hyponatraemia |
| Radiotherapy |
| Drugs – amiodarone, lithium (lithium more likely to cause nephrogenic diabetes insipidus) |
| Inflammatory/infectious diseases |
| Sickle-cell disease |
| Tuberculosis |
| Abscesses |
| Encephalitis |
| Meningitis |
| Sarcoidosis (may also cause nephrogenic diabetes insipidus) |
| Wegener’s granulomatosis |
| Histiocytosis X |
| Vascular disease |
| Ischaemic or haemorrhagic strokes |
| Aneurysmal bleeds (especially anterior communicating artery subarachnoid haemorrhage) |
| Sheehan’s syndrome |
| Pituitary apoplexy |
| Congenital |
| Autosomal-dominant mutations of antidiuretic hormone expression (despite the dominant expression of the gene, the onset of clinical diabetes insipidus may take up to 30 years to develop)† |
| Wolfram syndrome – autosomal-recessive condition characterised by diabetes insipidus, diabetes mellitus, optic atrophy and deafness |
* Doczi T, Tarjanyi J, Kiss J. Syndrome of inappropriate antidiuretic syndrome after head injury. Neurosurgery 1982; 10: 685–8.
† Repaske DR, Medlej R, Gulteken EK et al. Heterogeneity in clinical manifestation of autosomal dominant neurohypophyseal diabetes insipidus caused by a mutation encoding Ala1-Val in the signal peptide of the arginine vasopressin/neurophysin II/copeptin precursor. J Clin Endocrinol Metab 1997; 82: 51–6.
It is important to maintain a high level of suspicion for the development of DI in anyone who is suffering from pituitary disease or who has suffered a pituitary injury as the symptoms may have gradual onset. Anterior pituitary failure can lessen the impact of central DI because of the deficiency of ACTH and cortisol, which reduces GFR and free water loss. Additionally, the loss of feedback inhibition may stimulate increased release of ADH from the median eminence. Thus in Sheehan’s syndrome and pituitary apoplexy, the presenting symptoms tend not to be those of DI with polyuria. However, once corticosteroid therapy is commenced, polyuria indicating DI may become apparent or exacerbated. Conversely, patients with persistent DI of idiopathic origin should have long-term endocrine follow-up as a number will go on to develop tumours of the pituitary several years following the diagnosis of DI.30,31
TREATMENT OF CDI
Four separate problems have to be addressed when treating CDI:
HYPERNATRAEMIA
If the patient with CDI and ongoing water diuresis has had restricted access to fluids and has developed hyperosmolality/hypertonicity or alternatively has developed hypertonicity through replacement of dilute urine with equal volumes of isotonic (to plasma) intravenous fluids, then sudden reduction in plasma tonicity may result in pontine myelonecrosis and permanent neurological damage. When a euvolaemic state is present, AVP or DDAVP may be administered to reduce urine output (see below). At the same time, fluid restriction should be imposed and replacement of the previous hour’s urine output undertaken with an appropriate fluid to avoid a fall in concentration of sodium by more than 0.5 mmol/h.32 Absolute safety data are not available to determine the ideal rate of reduction of plasma sodium. However, a fall of more than 5 mmol/l in any 24-hour period should be avoided.
NEPHROGENIC DIABETES INSIPIDUS
NDI may be congenital or acquired (Table 51.4). As the majority of congenital cases present in the first week of life, the majority of cases seen in the adult ICU are acquired. The commonest of these are lithium toxicity due to long-term drug treatment, hypercalcaemia and postobstructive uropathy following relief of ureteric or urtethral obstruction.
| Acquired |
CONGENITAL NDI
Some 80–90% of patients with congenital NDI have an X-linked recessive abnormality of the AVPR2 gene coding for the V2 receptor. Different mutations of the gene are described but the majority result in trapping of the V2 receptor intracellularly, unable to integrate into the membrane of the collecting duct cell. Drugs have been developed which can facilitate receptor integration into the membrane, restoring some of the urine-concentrating abilities of ADH.33 The sex linkage results in the vast majority of affected patients being male. However, female children can also present less severe polyuria and polydipsia due to expression of the abnormal gene.
ACQUIRED NDI
Lithium-associated nephrotoxicity remains the commonest form of acquired NDI with more than 20% of patients on chronic lithium therapy developing polyuria. Lithium is taken up into the principal cells of the collecting duct via sodium channels and inhibits intracellular adenylate cylcase antagonising the effects of ADH. Additionally, it reduces the medullary interstitial hypertonicity, possibly through reducing expression of urea transport protein B. If patients with early lithium-related NDI are prescribed amiloride, some reversal of both the polyuria and lithium-mediated toxicity is possible. Amiloride’s natriuretic action is achieved through closure of the luminal sodium channels in the collecting duct; the sodium channels through which lithium enters the cells.34 Indomethacin increases intracellular cAMP and counteracts the diminution of this and AQP2 caused by lithium, resulting in a marked and immediate drop in urine output. However, care is required as non-steroidal anti-inflammatory drugs (NSAIDs) may worsen renal failure, reducing GFR and lithium excretion, thereby worsening toxicity.
Hypercalcaemia, hypokalaemia, release of ureteric or urethral obstruction and hypoproteinaemia are also recognised as causing NDI and are associated with reduced expression of AQP2 channels, urea transport proteins and a loss of interstitial hypertonicity. These defects normally cause milder polyuria than that associated with lithium toxicity.
TREATMENT OF NDI
GESTATIONAL DIABETES INSPIDUS (Table 51.5)
In pregnancy the normal range of plasma osmolalities falls to 265–285 mosmol/kg and the plasma sodium is < 140 mmol/l. The reduction in osmolality is attributed to a resetting of the central osmostat.44,45 The retention of water and sodium is mediated by reduced baroreceptor stimulation.46 During pregnancy, the placenta produces vasopressinases, which increase ADH metabolism up to fourfold, and relaxin, which contributes to the 50% increase in GFR and venodilatation.47 Aldosterone concentrations rise up to fivefold. Solute elimination also rises so a small reduction in urine-concentrating ability may have a more marked effect. The volume of urine passed per day increases as a result of passage of an increased solute load (including urinary proteins, glucose and amino acids), increased drinking and a raised GFR. A diagnosis of DI therefore requires careful differentiation from a physiological polyuria or potentially pathological polyuria of separate aetiology (e.g. gestational diabetes) in pregnancy.
The treatment of GDI varies depending on the underlying cause. In all cases, it is important not to allow the patient to become hypernatraemic and hyperosmolal as this is likely to have adverse effects on both mother and child. Where hypernatraemia and hyperosmolality do occur, careful correction is required in a closely monitored, very gradual, stepwise fashion. A lowering of sodium by as little as 10 mmol/day has been associated with pontine myelinolysis.53
POLYDIPSIA (PSYCHOGENIC/NEUROGENIC)
SOLUTE DIURESIS
Just as failure to reabsorb water can result in polyuria, failure to reabsorb solute can result in an osmotic load in the tubule which opposes water absorption in the convoluted tubules and collecting duct. The commonest cause of this in the general patient population is glycosuria. In the ICU setting a solute diuresis may also be associated with the administration of diuretic drugs, high-protein feeds (increased urea load), supranormal quantities of sodium and other solutes through fluids, feeds and drugs, recovery from acute renal failure with tubular inability to reabsorb solute, and drug-induced tubular damage.
THE DIAGNOSIS OF POLYURIC SYNDROMES
WATER DEPRIVATION AND ADH TESTS
The limitations of the test should also be appreciated:
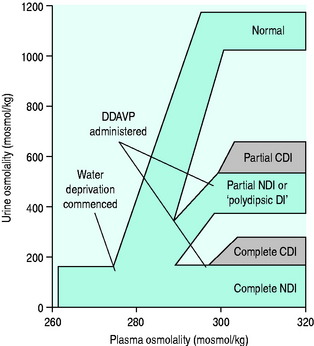
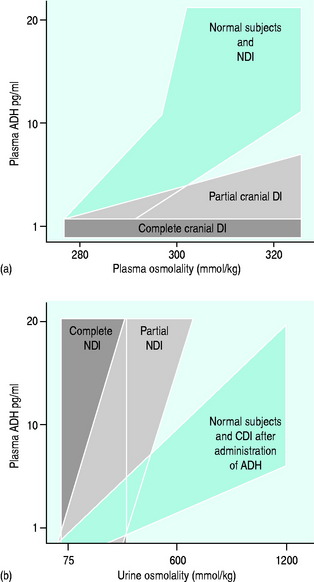
INTERPRETATION OF THE TESTS (SEE FigureS 51.4 and 51.5)
In complete DI, plasma osmolality will rise but urine osmolality will not rise above 300 mosmol/kg. Upon administration of ADH, patients with complete CDI will raise their urine osmolality to 500 mosmol/kg or higher whereas there will be no rise in urine osmolality in complete NDI.
Water deprivation tests should not be conducted in infants or patients with pre-existing hypovolaemia or hyperosmolality. In the latter two categories, treatment of the fluid deficit and/or solute excess should precede further investigations. In infants and in adults with equivocal water deprivation test results, an infusion of hypertonic saline should be considered.
HYPERTONIC SALINE INFUSION
In patients with equivocal results from a water deprivation test and in those at high risk of dehydration and hypovolaemia from water deprivation, a hypertonic saline infusion test may be undertaken to establish the cause of a water diuresis. Interpretation of the test is as for the water deprivation test but there is a clearer demarcation between partial CDI and primary polydipsia and the risk of missing a diagnosis of CDI secondary to osmoreceptor dysfunction is reduced.59
1 Dorwart WV, Chalmers L. Comparison of methods for calculating serum osmolality from chemical concentrations and the prognostic value of such calculations. Clin Chem. 1975;21:190-194.
2 Worthley LI, Guerin M, Pain RW. For calculating osmolality, the simplest formula is the best. Anaesth Intens Care. 1987;15:199-202.
3 Rasouli M, Kalantair KR. Comparison of methods for calculating serum osmolality: multivariate linear regression analysis. Clin Chem Lab Med. 2005;43:635-640.
4 McKinely MJ, Denton DA, Weisinger RS. Sensors for antidiuresis and thirst – osmoreceptors or CSF sodium detectors? Brain Res. 1978;141:89-103.
5 Keller-Wood M. ACTH responses to CRF and AVP in pregnant and non-pregnant ewes. Am J Physiol Regulat Integr Comp Physiol. 1998;274:1762-1768.
6 Kalogeras KT, Nieman LK, Friedman TC, et al. Inferior petrosal sinus sampling in healthy human subjects reveals a unilateral corticotropin-releasing hormone-induced arginine vasopressin release associated with ipsilateral adrenocorticotropin secretion. J Clin Invest. 1996;97:2045-2050.
7 Stricker EM, Sved AF. Thirst. Nutrition. 2000;16:821-826.
8 Halter JB, Goldberg AP, Robertson GL, et al. Selective osmoreceptor dysfunction in the syndrome of chronic hypernatraemia. J Clin Endocrinol Metab. 1977;44:609-616.
9 Holmes CL, Patel BM, Russell JA, et al. Physiology of vasopressin relevant to management of septic shock. Chest. 2001;120:989-1002.
10 Antunes-Rodrigues J, de Castro M, Elias LLK, et al. Neuroednocrine control of body fluid metabolism. Physiol Rev. 2004;84:169-208.
11 Czaczkes JW. Physiologic studies of antidiuretic hormone by its direct measurement in human plasma. J Clin Invest. 1964;43:1625-1640.
12 Holmes CL, Landry DW, Granton JT. Science review: vasopressin and the cardiovascular system parts 1 and 2. Crit Care. 2003;7:427-434. 2004; 8: 15–23.
13 Bankir L. Antidiuretic action of vasopressin: quantitative aspects and interaction between V1a and V2 receptor mediated effects. Cardiovasc Res. 2001;51:372-390.
14 Landry DW, Levin HR, Gallant EM, et al. Vasopressin pressor hypersensitivity in vasodilatory septic shock. Crit Care Med. 1997;95:1122-1125.
15 Maturi MF, Martin SE, Markle D, et al. Coronary vasoconstriction induced by vasopressin. Production of myocardial ischaemia in dogs by constriction of non-diseased small vessels. Circulation. 1991;83:2111-2121.
16 Vanhoutte PM, Katusic ZS, Shepherd JT. Vasopressin induces endothelium-dependent relaxations of cerebral and coronary but not of systemic arteries. J Hypertens. 1984;2(Suppl.):S421-422.
17 Okamurat T, Ayajiki K, Fujiokah H, et al. Mechanisms underlying arginine vasopressin induced relaxation in isolated monkey coronary arteries. J Hypertens. 1999;17:673-678.
18 Sellke FW, Quillen JE. Altered effects of vasopressin on the coronary circulation after ischaemia. J Thorac Cardiovasc Surg. 1992;104:357-363.
19 Nussey SS, Bevaqn DH, Ang VT, et al. Effects of arginine vasopressin (AVP) infusions on circulating concentrations of platelet AVP, factor VIII:C and von Willebrand factor. Thromb Haemost. 1986;55:34-36.
20 Haslam RJ, Rosson GM. Aggregation of human blood platelets by vasopressin. Am J Physiol. 1972;223:958-967.
21 Wun T, Paglieroni T, Lanchant NA. Physiological concentrations of arginine vasopressin activate human platelets in vitro. Br J Haematol. 1996;92:968-972.
22 Filep J, Rosenkranz B. Mechanisms of vasopressin induced platelet aggregation. Thromb Res. 1987;45:7-15.
23 Vander AJ. Renal Physiology, 5th edn., McGraw Hill, USA; 1995:116-144.
24 Schrier RW. Water and sodium retention in edematous disorders: role of vasopressin and aldosterone. Am J Med. 2006;119:S47-S53.
25 Richard D, Bourque CW. Atrial natriuretic peptide modulates synaptic transmission from osmoreceptor afferents to the supraoptic nucleus. J Neurosci. 1996;16:7526-7532.
26 Shucart WA, Jackson I. Management of diabetes insipidus in neurosurgical patients. J Neurosurg. 1976;44:65-71.
27 Maghnie M, Altobelli M, di Iorgi N, et al. Idiopathic central diabetes insipidus is associated with abnormal blood supply to the posterior pituitary gland caused by vascular impairment of the inferior hypophyseal artery system. J Endocrinol Metab. 2004;89:1891-1896.
28 Pivonello R, De Bellis A, Faggiano A, et al. Central diabetes insipidus and autoimmunity: relationship between the occurrence of antibodies to arginine vasopressin-secreting cells and clinical, immunological and radiological features in a large cohort of patients with central diabetes insipidus of unknown etiology. J Clin Endocrinol Metab. 2003;88:1629-1636.
29 Halter JB, Goldberg AP, Robertson GL, et al. Selective osmoreceptor dysfunction in the syndrome of chronic hypernatraemia. J Clin Endocrinol Metab. 1977;44:609-616.
30 Charmandari E, Brook CG. 20 years of experience in idiopathic central diabetes insipidus. Lancet. 1999;353:2212-2213.
31 Sudha LM, Anthony JB, Grumbach MM, et al. Idiopathic hypothalamic diabets insipidus, pituitary stalk thickening and the occult intracranial germinoma in children and adolescents. J Clin Endocrinol Metab. 1997;82:1362-1367.
32 Blum D, Brasser D, Kahn A, et al. Safe oral rehydration of hypertonic dehydration. J Pediatr Gastroenterol Nutr. 1986;5:232-235.
33 Robben JH, Sze M, Knoers NV, et al. Functional rescue of vasopressin V2 receptor mutants in MDCK cells by pharmacochaperones: relevance to therapy of nephrogenic diabetes insipidus. Am J Physiol Renal Physiol. 2007;292:F253-260.
34 Battle DC, von Riotte AB, Gaviria M, et al. Amelioration of polyuria by amiloride in patients receiving long-term lithium therapy. N Engl J Med. 1985;312:408-414.
35 Kim GH, Lee JW, Oh YK, et al. Nephrogenic diabetes insipidus is associated with up-regulation of aquaporin-2, Na-Cl cotransporter and epithelieal sodium channel. J Am Soc Nephrol. 2004;15:2836-2843.
36 Lam SS, Kjellstrand C. Emergency treatment of lithium-induced diabetes insipidus with non-steroidal anti-inflammatory drugs. Renal Fail. 1997;19:183-188.
37 Hochberg Z, Even L, Danon A. Amelioration of polyuria in nephrogenic diabetes insipidus due to aquaporin-2 deficiency. Clin Endocrinol. 1998;49:39-44.
38 Libber S, Harrison H, Spector D. Treatment of nephrogenic diabetes insipidus with prostaglandin synthesis inhibitors. J Pediatr. 1986;108:305-311.
39 Thompson PJr., Erll JM, Schaaf M. Comparison of clofibrate and chlorpropamide in vasopressin responsive diabetes insipidus. Metabolism. 1977;26:749-762.
40 Moses AM, Howanitz J, vanGemert M, et al. Clofibrate-induced antidiuresis. J Clin Invest. 1973;52:535-542.
41 Matsukura S, Matsumoto J, Chihara K, et al. Clofibrate-induced myopathy in patients with diabetes insipidus. Endocrinol Jpn. 1980;27:401-403.
42 Wuller S, Wiesner B, Loffler A, et al. Pharmacochaperones post-translationally enhance cell surface expression by increasing conformational stability of wild type and mutant vasopressin V2 receptors. J Biol Chem. 2004;279:47254-47263.
43 Bernier V, Morello JP, Zarruk A, et al. Pharmacologic chaperones as a potential treatment for X-linked nephrogenic diabetes insipidus. J Am Soc Nephrol. 2006;17:232-243.
44 Durr JA, Stamoutsos B, Lindheimer MD. Osmoregulation during pregnancy in the rat. Evidence for resetting of the threshold for vasopressin secretion during gestation. J Clin Invest. 1998;68:337-346.
45 Lindheimer MD, Davison JM. Osmoregulation, the secretion of arginine vasopressin and its metabolism during pregnancy. Eur J Endocrinol. 1995;132:133-143.
46 Schrier RW, Durr J. Pregnancy: an overfill or underfill state. Am J Kidney Dis. 1987;9:284-289.
47 Dschietzig T, Stangl K. Relaxin: a pregnancy hormone as central player of body fluid and circulation homeostasis. Cell Mol Life Sci. 2003;60:688-700.
48 Durr JA, Haggard JG, Hunt JM, et al. Diabetes insipidus in pregnancy associated with abnormally high circulating vasopressin activity. N Engl J Med. 1987;316:1070-1074.
49 Iwasaki Y, Osio Y, Kondo K, et al. Aggravation of subclinical diabetes insipidus during pregnancy. N Engl J Med. 1991;324:522-526.
50 Cammu H, Velkeniers B, Charels K, et al. Idiopathic acute fatty liver of pregnancy associated with transient diabetes insipidus. Br J Obstet Gynaecol. 1987;94:173-178.
51 de Heide LJM, van Tol KM, Doorenbos B. Pituitary apoplexy presenting during pregnancy. Netherl J Med. 2004;62:393-396.
52 Jin-no Y, Kamiya Y, Okado M, et al. Pregnant woman with transient diabetes insipidus resistant to 1-desamino-8-d-arginine vasopressin. Endocrinol J. 1998;45:693-696.
53 Hoashi S, Margey R, Haroum A, et al. Gestational diabetes insipidus, severe hypernatraemia and hyperemesis gravidarum in a primigravid pregnancy. Endocrinol Abstr. 2004;7:297.
54 de Leon J. Polydipsia: a study in a long-term psychiatric unit. Eur Arch Psychiatry Clin Neurosci. 2003;253:37-39.
55 Rao KJ, Miller M, Moses A. Water intoxication and thioridazine. Ann Intern Med. 1975;82:61-65.
56 Martin JB, Riskind PN. Neurologic manifestations of hypothalamic disease. Prog Brain Res. 1992;93:31-40.
57 Zerbe RL, Robertson GL. A comparison of plasma vasopressin measurements with a standard indirect test in the differential diagnosis of polyuria. N Engl J Med. 1981;305:1539-1546.
58 Bayliss PH, Robertson GL. Osmoregulation of vasopressin secretion in health and disease. Clin Endocrinol. 1988;29:549-576.
59 Mohn N, Acerini CL, Cheetham TD, et al. Hypertonic saline test for the investigation of posterior pituitary function. Arch Dis Child. 1998;79:431-434.
[/level-membership-for-anesthesiology-category][not-level-membership-for-anesthesiology-category]
Chapter 51 Diabetes insipidus and other polyuric syndromes
Diabetes insipidus (DI: literal translation, ‘tasteless siphon’) refers to a syndrome characterised by pathological polyuria, excessive thirst and polydipsia. Polyuria is arbitrarily defined as a urine loss of > 3 l/day in an adult of normal mass. The urine produced in DI is inappropriately dilute, having both low specific gravity and low osmolality in the face of a high or normal plasma osmolality.
Three subtypes of DI are recognised:
In critically ill patients, polyuria may be the sole part of the DI syndrome apparent to the clinician. Patients are seldom in control of their own fluid intake and are frequently unable to report thirst. The recognition of DI is important as failure to recognise and treat the syndrome appropriately will result in severe dehydration and hyperosmolality with a significant risk of morbidity and mortality. As there are many causes of polyuria in the critically ill (Table 51.1), it is important to adopt a systematic approach to the clinical assessment, investigations, diagnosis and management of such patients.
| Water diuresis |
| Pathological |
| Diabetes insipidus: cranial, nephrogenic, gestational |
| Physiological |
| Psychogenic polydipsia (excess drinking is pathological but the diuresis is not) |
| Iatrogenic – excessive administration of hypotonic solutions, e.g. 5% dextrose solution, 0.45% saline, 0.18% saline 4% dextrose solutions |
| Solute diuresis |
| Pathological |
| Fanconi’s syndrome |
| Renal tubular acidosis |
| Glomerulonephritis |
| Hyperaldosteronism |
| Anorexia nervosa |
| Migraines |
| Paroxysmal tachycardia (via increased atrial natriuretic peptide release) |
| Poisons/drugs: |
| Ethanol |
| Methanol |
| Ethylene glycol |
| Mannitol |
| Loop diuretics |
| Thiazide diuretics |
| Hyperglycaemia |
| Physiological |
| Resolving sepsis (redistribution of fluid into the vascular compartment from the third space) |
| Iatrogenic – excessive administration of isotonic or hypertonic solutions, e.g. |
| 0.9% saline |
| Hypertonic saline |
| Hartmann’s solution |
| Gelofusin |
BACKGROUND PHYSIOLOGY AND ANATOMY
OSMOLALITY
Osmolality is the measure of osmoles of solute per kilogram of solvent and includes both permeant (e.g. urea) and impermeant solutes (e.g. sodium) and thus osmolality may change due to both water movement and the movement of permeant solute. Osmolarity is the measure of osmoles per litre of solute and thus is temperature-dependent. Osmolality is temperature-independent. Normal plasma osmolality is in the range of 275–295 mosmol/kg. Plasma osmolality can be estimated from several equations.1 The formula of Worthley et al.2 below is both simple and correlates well with measured values.3
All units of solute are mmol/l.
The osmolal gap is the difference between the calculated and measured osmolality in plasma or urine.The normal osmolal gap is < 10 mosmol/l. An increased osmolal gap indicates the presence of an unmeasured osmotically active solute.
NORMAL URINARY OSMOLALITY
In health, urinary osmolality is usually maintained between 500 and 700 mosmol/kg. As the obligatory solute load to be excreted is relatively constant in value, urine osmolality will fall in response to an increased intake in free water and rise in response to dehydration or water restriction (Figure 51.1). The minimum osmolality of urine achievable inhumans is around 25 mosmol/kg. Diuresis refers to the passage of a high volume of urine (> 1.5 ml/kg per hour) and may be transient or persistent, physiological or pathological. The production of > 3 l/day is arbitrarily defined as polyuria. Water diuresis occurs when the total solute in the urine excreted per day is within the normal range but the osmolality of the urine passed is low. An osmotic or solute diuresis occurs when the total solute passed per day is higher than normal, and the urine passed is usually iso-osmolar to plasma if the extracellular fluid volume is expanded or hyperosmolar if the patient is hypo- or euvolaemic.
OSMORECEPTORS AND OTHER INPUTS TO THE SUPRAOPTIC AND PARAVENTRICULAR NUCLEI
Detection of osmolality occurs largely at osmo- (Na+) receptors sited around the anterior aspect of the third ventricle of the brain. These are sensitive to plasma osmolality and cerebrospinal fluid sodium concentration. Hypertonic saline is a more potent stimulus than equi-isotonic equiososmolar solutions of other solutes.4 These osmoreceptors link to the cells of the paraventricular nuclei (PVN) and supraoptic nuclei (SON), the sites of ADH synthesis. The axons of the cells in the PVN and SON form part of the pituitary stalk linking the hypothalamus to the pituitary gland in which they terminate. A smaller proportion of the axons terminate in the median eminence where they release ADH and oxytocin, which is transported to the anterior lobe of the pituitary by portal vessels. The ADH and oxytocin so released cause release of adrenocorticotrophic hormone (ACTH) and prolactin respectively; the ADH acts synergistically with corticotrophin-releasing factor (CRF) but is also believed to have ACTH secretagogue properties in its own right.5,6
Direct inputs from the sympathetic nervous system to the PVN and SON can stimulate ADH release via α-adrenoreceptors. Other central osmoreceptors lie outside the blood–brain barrier in the subfornical organ and come into contact with plasma. It is believed that ANP and angiotensin II7 act via these receptors to inhibit or elicit ADH synthesis and ADH release and to modify the sensation of thirst. Additional osmoreceptors in the mouth, stomach and liver are believed to play a role in the anticipation of an osmolal load following ingestion of food and can pre-emptively stimulate ADH synthesis in the hypothalamus.
As the baroreceptor and osmoreceptor inputs to the PVN and SON are distinct, it is possible to lose the normal ADH response to hyperosmolality but maintain a normal ADH response to hypovolaemia.8 Additionally, in animal experiments, when hypotension increases the basal plasma ADH concentration, there is a simultaneous resetting of the osmomolality–plasma ADH response curve in an attempt to preserve osmoregulatory function from the new higher baseline.9 If this did not occur, the ADH response to hypotension would always result in the development of a hypo-osmolal state in addition to causing vasoconstriction.
The normal response of osmoreceptors to changing plasma osmolality in terms of ADH is illustrated in Figure 51.2. At plasma osmolalities of < 275 mosmol/kg, the osmoreceptors remain hyperpolarised and virtually no ADH release occurs via them. At osmolalities > 295 mosmol/kg, the osmoreceptors are maximally depolarised and plasma concentrations of ADH of > 5 pg/ml are attained. Other inputs and influences upon ADH release are summarised in Figure 51.3 and Table 51.2.
Figure 51.2 Plasma antidiuretic hormone (ADH) and plasma osmolality in health and partial cranial diabetes insipidus.
Table 51.2 Factors influencing antidiuretic hormone (ADH) release
| Increased ADH release with: |
| Hyperosmolality |
| Hypovolaemia |
| Hypotension |
| Hypoxia |
| Hypothyroidism |
| Hyperthermia |
| Positive-pressure ventilation |
| Pain |
| Emotional stress |
| Exercise |
| Nausea |
| Nicotine |
| Trauma/surgery |
| Decreased ADH release with: |
| Hypo-osmolality |
| Hypervolaemia |
| Hypertension |
| Ethanol |
| Cranial diabetes insipidus |
ANTIDIURETIC HORMONE/ARGININE VASOPRESSIN (AVP)
ADH (8-arginine vasopressin) is a nine-amino-acid peptide which differs from oxytocin at only two residues but shares the disulphide bond between the first and sixth ones. This similar structure and conformation results in some cross-reactivity at receptors and in function.10 It is synthesised in the SON and PVN, bound to neuorophysin, transferred through axons to the posterior pituitary gland and stored in granules prior to release. Synthesis to replace any released stores is a rapid process (1–2 hours from synthesis to storage) and patients with damage to the pituitary can achieve near-normal plasma concentrations of ADH, in terms of osmoregulatory function, via release of newly synthesised ADH via the axons terminating in the median eminence. However, the higher plasma concentrations associated with hypovolaemia cannot be achieved. Normal osmoregulatory plasma ADH concentrations are in the range of 1–8 pg/ml but rise as high as 40 pg/ml in hypovolaemic patients under the influence of the sympathetic nervous system, baroreceptor responses and angiotensin II.
Once released from the pituitary, ADH has a plasma half-life of around 10–35 minutes.11 It is metabolised by hepatic and renal vasopressinases and around 10% of the active hormone is excreted unchanged in the urine.
ACTIONS OF ADH
ADH has antidiuretic, vasopressor, haemostatic and ACTH secretagogue actions. Additionally, it has roles in memory, water permeability of the blood–brain barrier, nociception, splenic contraction and thermoregulation. These actions are mediated through V1, V2 and V3 receptors. It also has actions on the uterus and mammary tissue mediated through oxytocin receptors. Cardiac inotropic effects are reported to be mediated through purinergic P2 receptors but this remains controversial.12
Antidiuresis
ADH binds to V2 receptors on the basal membranes of the principal cells of the collecting duct and distal tubule. The activated receptor induces production of cyclic adenosine monophosphate (cAMP) by adenylate cylase and this in turn activates protein kinases which effect the integration into the luminal membrane of vesicles containing aquaporin-2 highly selective water channels. The production of prostaglandin E2 (PGE2) inhibits cAMP production. PGE2 synthesis is stimulated by the action of ADH on V1 receptors on the luminal membrane of the collecting duct.13 Thus, a form of autoregulatory limitation of the antidiuretic effect of ADH may exist. Hypokalaemia, lithium and hypercalcaemia also anatagonise the renal actions of ADH.
In low dose, administration of exogenous ADH may paradoxically cause a diuresis in patients with septic shock.14 Whether this is due to increased renal perfusion pressure and raised glomerular filtration rate (GFR) is unclear.
Vasoconstriction
At higher concentrations (> 40 pg/ml), ADH activates not only V2 receptors but also V1 receptors, through which it causes preferential vasoconstriction in muscle, skin and fat with relative sparing of coronary, cerebral and mesenteric circulations. However, these relative sparing effects are controversial, with some authors reporting relative sparing and others reporting significant vasoconstriction.15–18 Activation of V1 receptors activates phospholipase C and increases inositol triphosphate intracellularly. Ultimately this increases intracellular free calcium and leads to smooth-muscle constriction in the blood vessel wall.
Coagulation
ADH increases circulating levels of tissue plasminogen activator, factor VIII and von Willebrand factor.19 These effects may be mediated by V2 receptors but this remains controversial. At high but physiological concentrations it can act as a platelet-aggregating agent.20,21 Platelet aggregation is mediated through activation of platelet V1 receptors.22 ADH and its analogue DDAVP are used as first-line treatments in patients with von Willebrand’s disease, and may be used in bleeding associated with renal failure and platelet dysfunction.
VOLUME RECEPTORS
Volume homeostasis takes precedence over sodium homeostasis and so rises and falls in sodium will occur in order to preserve the circulating volume. In euvolaemic patients sodium homeostasis is maintained. Sodium concentration is detected by both the osmoreceptors of the subfornical organ outside the blood–brain barrier and also by the juxtaglomerular apparatus which secretes renin in response to reduced GFR and a lower sodium load in the tubule.23
The predominant determinants of sodium balance, however, are the high-pressure baroreceptors in the pulmonary veins, left atrium, carotid sinus and aortic arch.24 Reduced stretch of these receptors increases sympathetic nervous system activity and activation of the renin–angiotensin–aldosterone system, resulting in reduced sodium excretion (via reduced GFR) and increased reabsorption of sodium in the proximal and distal convoluted tubules. Additionally, release of ADH can be stimulated, resulting in concomitant water retention. Conversely, stretch of the baroreceptors will result in a fall in sodium retention through reduced activity of the sympathetic nervous and renin–angiotensin–aldosterone systems. Stretch additionally results in the release of ANP and a natriuresis through reduced sodium reabsorption in the distal convoluted tubule and collecting duct. ADH release is reduced by the fall in sympathetic nervous tone from the baroreceptors. ADH secretion may also be inhibited by the action of ANP on cerebral osmoreceptors lying outside the blood–brain barrier.25
CRANIAL DIABETES INSIPIDUS
ACQUIRED CDI
Acquired CDI may be transient or permanent and can arise due to an absolute (complete) or relative (incomplete) lack of ADH. Complete central DI is usually associated with lesions above the level of the median eminence in the SON or PVN or of the neurohypophyseal stalk whereby the production of ADH in the hypothalamus is terminated.26 Permanent central DI tends to be associated with transecting, obliterating or chronic inflammatory lesions, whereas transient DI is more likely to be associated with acute inflammatory or oedematous lesions with some recovery of ADH secretion occurring as the inflammation or oedema resolves. An exception to this is the transient DI seen following excision or destruction of the posterior pituitary; ADH produced in the hypothalamus can still be released into the systemic circulation from capillaries in the median eminence.
[/not-level-membership-for-anesthesiology-category]
