Development Of The Pituitary
Anatomy and Histology
The pituitary gland, also termed the hypophysis, situates in a depression on the upper surface of the sphenoid bone, the sella turcica. It is composed of anatomically and functionally distinct entities: the adenohypophysis, including the intermediate and anterior lobes, and the neurohypophysis, also called the posterior lobe. The functional anterior pituitary contains five main cell types. (1) Somatotrope cells produce growth hormone (GH) and regulate linear growth and metabolism; (2) lactotrope cells produce prolactin (PRL), which regulates milk production in females; (3) thyrotrope cells produce thyroid-stimulating hormone (TSH), which controls the secretion of thyroid hormone from the thyroid gland; (4) gonadotrope cells produce gonadotropins (follicle-stimulating hormone [FSH] and luteinizing hormone [LH]), which regulate reproductive development and function; and (5) corticotrope cells produce adrenocorticotropic hormone (ACTH), a product of precursor pro-opiomelanocortin (POMC) cleaved by proteolytic processing, which regulates metabolic function through stimulation of glucocorticoid synthesis in the adrenal cortex. TSH, LH, and FSH are heterodimeric glycoproteins consisting of a common alpha subunit (αGSU) and a specific beta subunit. In the adult pituitary, GH-producing somatotrope cells occupy most of the gland, which weighs less than 1 gram in humans. The size of the pituitary gland and the proliferation of each pituitary cell type are regulated according to physiologic conditions indicated by feedback regulation.1
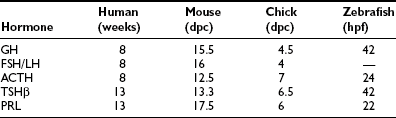
These five anterior pituitary cell types are present at birth (Table 1-1). Initial expression of distinct pituitary hormone genes marks the terminal differentiation events of the cell types, which derive from a seemingly common primordia and are the results of internal programming of the pituitary, as well as a consequence of its interaction with surrounding organs during development. Evidence suggests the internal programming is dictated by the expression of transcriptional regulators, including a cascade of homeodomain transcription factors and additional cell type–restricted transcription factors. The mechanisms that control the temporal and spatial expression of these transcription factors include diffusible signals from the developing hypothalamus at the dorsal aspect and factors from surrounding structures. These spatially distributed signals and gradients of signaling molecules are critical in establishing positional pituitary cell–type commitment events.2,3 Disruption of these apparently evolutionarily conserved events underlying proper development of the pituitary gland can result in morphologic abbreviation and pituitary dysfunction. Through analysis of the expression of pituitary hormone genes in human cases of hypopituitarism, as well as in genetic models of pituitary defects (particularly in mouse models of pituitary dwarfism), a significant amount of knowledge has been accumulated regarding the molecular mechanisms underlying proper development of the pituitary gland.
Pituitary Development
Phylogenetic studies in several vertebrate species led to the conclusion that the pituitary gland arises from oral epithelia. Fate-mapping experiments conducted in these animal species trace the origins of the pituitary gland back to the neural plate. In studies of grafting quail chick chimeras, the origin of the pituitary was localized to the midline of the anterior neural ridge. By means of surgical ablation performed in chick embryos, the rostral ridge of the neural plate was identified as the source of cells that give rise to pituitary tissue.4–6 In amphibians, tracing experiments have confirmed the neural origin of pituitary gland7,8 and similar conclusions have been reached about zebrafish.9,10 Additionally, by focalized application of a carbocyanin dye, DiI, into the rostral end of the neural plate at the open neurula stage (9.5 days postcoitus) in rats, labeled cells could be identified in Rathke’s pouch, and they could develop into the secretory cells of the adenohypophysis in 7 additional days.11 Thus evidence indicates that the anterior neural ridge is the origin of Rathke’s pouch, which eventually gives rise to cells of the pituitary gland. Subsequent to the folding of the embryonic head, the anterior neural ridge is displaced ventrally to form the portion of the oral epithelium that later gives rise to the roof of the mouth and additional structures, including the pituitary gland. Consistent findings in many species make it apparent that the process of pituitary development is, for the most part, evolutionarily conserved from lower vertebrates to higher mammals.
Ontogeny
In humans, the anterior lobe of the pituitary gland originates from an invagination of the stomodeal epithelium termed Rathke’s pouch.12 The stomodeal epithelium that contains the pituitary primordium is formed by the third fetal week, and the invagination of stomodeal epithelium occurs dorsally to form Rathke’s pouch by the fourth week. The formation of Rathke’s pouch is complete and disconnected from the oral epithelium by the end of the sixth week of fetal life.13 In parallel, the hypothalamus is the first region of the forebrain to differentiate. From 4 weeks, the hypothalamic sulcus, chiasmatic plate, and mammillary bodies are recognizable. These two organs, hypothalamus and pituitary, develop interdependently.14
Similar to the ontogeny observed in humans, Rathke’s pouch in mice is derived from an anlage that arises as an upgrowth from the lining of the oral cavity’s roof. At its earliest stage, the murine pituitary primordium is defined as an intimate point of contact between the neural ectoderm and the oral roof ectoderm on embryonic day 8.5 postcoitus (e8.5), which marks the first event in the pituitary’s development. Organogenesis of the adenohypophysis begins as the cells of the pituitary placode in the oral ectoderm thicken and invaginate to form the nascent pituitary. In the e9.5 mouse embryo, this anlage can be seen located rostrally to the oropharyngeal membrane. Dorsal movement of the epithelial layer from the roof of the mouth induces a cone-shaped intrusion dorsally as Rathke’s pouch, or the adenohypophyseal pouch. Before the formation, a developmentally important molecular marker, Sonic hedgehog (Shh), is expressed uniformly in the oral epithelial layer. The expression of Shh is excluded before the intrusion of pituitary anlagen can occur in the e9 mouse embryo.15 Rathke’s pouch thickens as development proceeds and elongates dorsally relative to the oral cavity by the stomodia-adenohypophyseal channel. By e10.5 in the mouse, Rathke’s pouch has formed as a rudimentary structure and separated from the ventral pharyngeal epithelium.
At the time Rathke’s pouch is pinched off at e11 in mice, the first round of accelerated mitotic activity is initiated in the anlagen.16,17 In the ensuing patterning period, mitotic activity is observed most prominently in the rostral part of Rathke’s pouch, with several buds emerging and enveloping areas of vascularized mesenchyme. Progenitors of the hormone-secreting cell types arise from the ventral proliferation of cells, and this region of rostral Rathke’s pouch eventually gives rise to the anterior lobe, or the pars distalis. The dorsal aspects of Rathke’s pouch, in contact with the descending infundibulum processes and rostroventrally with the hypophyseal cleft, remain thin and form the intermediate lobe, or the pars intermedia. Anterior pituitary cell types are positionally determined as they initially emerge from proliferation zones,15,18 with the somatotrope/lactotrope cells arising caudomedially, gonadotrope cells more rostroventrally, corticotrope cells ventrally, and melanotrope cells dorsally. This pattern of pituitary development is generally similar in most mammals.
Cell Lineage Determination
Endocrine pituitary cell types in the adenohypophysis are derived from a single population of cells. The initial expression of pituitary hormone genes marking the terminal differentiation events of individual cell types occurs in a sequential manner. In mice, POMC gene expression emerges as the first pituitary marker at e11.5 and can be detected in the anterior pituitary by e13.5. However, the fate of cells that will give rise to those five different anterior pituitary cell types is determined prior to the initial pituitary POMC expression. In tissue-culture experiments where pituitary anlagen were taken and placed in a culture away from the influence of the diencephalons, pituitary anlagen taken at e11 were capable of generating cells expressing all five anterior pituitary hormone genes, while anlagen taken at e9.5 required additional growth factors, with the exception of corticotrope, which always differentiates regardless of the culture medium.19 Critical events occur at the time pituitary anlagen become committed to developing into pituitary precursors that will subsequently express pituitary genes that become regulated in a cell-autonomous fashion.20 The timing of this commitment event is coincidental with the formation of Rathke’s pouch.
As an anlage, Rathke’s pouch is the source of all endocrine pituitary cell types. In mice, after the initial appearance of corticotrope, expression of GH gene can be detected by e15.5, followed by thyrotropins, gonadotropins, and PRL. Gene expressions of all anterior pituitary hormones are detectable by e17.5, with the exception of PRL, which can be consistently seen by the time of birth (e19 in the mouse). Another early marker of Rathke’s pouch is αGSU, and the transcripts are detected throughout Rathke’s pouch by e9,21 although they are confined to the rostral tip of the anterior lobe by e12.5 and ultimately restricted to thyrotrope and gonadotrope from late gestation through adulthood. Following proliferation and early organ expansion, a series of different cell types arise in a distinct spatial and temporal fashion. Table 1-1 provides a time line of the initial expression of pituitary hormone genes in several species.
Transcription Factors and Pituitary Development
Parallel to the sequential emergence of pituitary cell types, a series of homeodomain family transcription factors are expressed as the adenohypophysis is becoming committed. With improved molecular genetic techniques, functional studies of these transcription factors in animal models, particularly in mouse models, have established molecular mechanisms underlying development of the pituitary gland. The expression profiles of Hesx1, Lhx3, Lhx4, Pitx1/2, Prop1, and Pit1 homeodomain factors, in addition to the expressions of Tbx19 and GATA2, dictate the commitment, determination, and differentiation events of the pituitary gland. These genes were initially studied in animal model systems that arose either from naturally occurring mutations or were created by reverse genetic techniques. Without exception, phenotypes observed in each animal model system are also observed in human cases with defects in the corresponding orthologous genes (Table 1-2). The phenotypes observed in human cases range from single pituitary hormone deficiency to combined pituitary hormone deficiency (CPHD) affecting several pituitary hormones in addition to GH. Study of the development of the pituitary gland serves as a model of progressive restriction in gene expression, and the pituitary gland has become a prototypic model organ system to study organogenesis, cell type determination, and differentiation.
Table 1-2
Transcription Factors in Pituitary Hormone Deficiency

Chr., Chromosome location; K.O., targeted deletion in mouse.
Pit1 Gene
The Pit1 gene (POU domain, class 1, transcription factor 1 [POU1F1]) encodes a 33-kD, 291-amino acid transcriptional activator that is capable of DNA binding and transactivation, and it was initially isolated by its ability to bind to the responsive element of the GH gene promoter.22,23 Pit1 is expressed exclusively in the pituitary gland. In mice, the initial expression of the Pit1 gene transcripts can be detected by e13.5, exclusively in the anterior ventral pituitary (Fig. 1-1). The expression of Pit1 persists in adults and co-localizes with expression of GH, PRL, and TSHb genes. Further studies revealed that the product of the Pit1 is capable of binding to responsive elements in the promoters of the GH gene,24 the growth hormone–releasing hormone receptor (GHRHR) gene,25 the PRL gene,26 and the TSHb gene. The Pit1 protein is also capable of binding to the responsive elements of the Pit1 gene itself and is required for the continued transcription of the Pit1 gene.27 The structure of the Pit1 gene is evolutionarily conserved and is found in mouse, human, and all other vertebrate animals examined, although Pit1 may play diverse functional roles in different physiologic pathways in individual species.
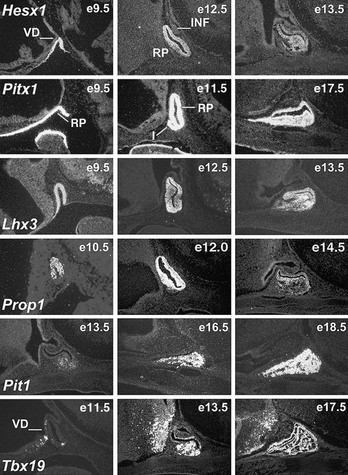
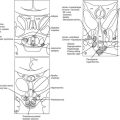
FIGURE 1-1 Expression of selected transcription factors in pituitary development by insitu hybridization. Expression of Hesx1, Pitx1 and Lhx3 are detected in Rathke’s pouch (RP) at mouse embryonic stage e9.5 and are maintained at e12.5, after which Hesx1 expression is rapidly extinguished while Pitx1 and Lhx3 continue to be expressed. Prop1 expression initiates at e10.5, reaches maximum intensity at e12.5, and attenuates at e14.5. Pit1 expression initiates at e13.5 and is maintained throughout pituitary development and adulthood. Initial Tbx19 expression can be observed in the ventral Rathke’s pouch and ventral diencephalon (VD) at e11.5, and its expression is maintained.
Animal Model
Snell mice28 are a well-studied animal model of pituitary function, which arises from a spontaneous single nucleotide mutation in the Pit1 gene that results in the substitution of W261C in the homeodomain, rendering the mutant gene product incapable of DNA binding and hence unable to activate potential target genes.29 Mice heterozygous for this mutation are phenotypically normal. The homozygous offspring of this mutation are dwarf and infertile, and they exhibit loss of three pituitary hormone cell types, GH, PRL, and TSHβ; whereas the gonadotrope and corticotrope cells are unaffected, suggesting that the Pit1 is required for terminal differentiation of the somatotrope, lactotrope, and thyrotrope cell types. In the Pit1Snell animal model where the Pit1 gene is functionally defective, the initial activation of the Pit1 is unaffected, while the later transcription of the Pit1 gene is altered, resulting in the failed expression of the Pit1 in the adult animal and a dwarf phenotype.29,30 The Pit1 lineage can be converted to alternative fates before e17.5 but exhibits a cell-autonomous commitment after e17.5, when Pit1 gene regulation shifts from a Pit1-independent early enhancer to a Pit1-autoregulated later enhancer.31 Pit1Jackson is a second mouse model with a defect in the Pit1 gene. The genomic structure of the Pit1 gene, located on chromosome 16, is grossly rearranged in mutant Pit1Jackson mice, with a phenotype very similar or identical to that of the Pit1Snell mice.29 In addition, Pit1 mutations result in decreased activity of the insulin/IGF1 pathway, which may result in physiologic homeostasis consequences that favor longevity32,33 (for reviews see refs. 34–36).
Related Diseases
The human Pit1 gene has been mapped to chromosome 3. Lesions in Pit1 have been identified as an etiology of CPHD (see Table 1-2). Initial study has revealed a homozygous nonsense mutation R172X in the Pit1 gene in a patient of consanguineous parents with cretinism due to deficiency of GH, PRL, and TSHβ.37 Many cases of CPHD with Pit1 defects have since been reported. It appears that the inheritance of Pit1 mutations in humans is complex, ranging from autosomal recessive to autosomal dominant to imprinting with variable phenotypic penetrance.38 Pituitary gonadotropins and corticotropins are normal in Pit1-defective patients. Deficiency of GH is consistently observed in all Pit1 patients, and deficiency for PRL is observed in most patients, whereas TSHβ deficiency usually has a delayed onset and incomplete penetrance (see Table 1-2). Different backgrounds may be the major contributing factor to the TSH phenotypic variation. Alternatively, however, there exists an embryonic population of thyrotrope termed rostral tip thyrotrope. The expression of this embryonic TSH is not Pit1 dependent, and consequently it may be a contributing element to the TSH phenotypic variation observed in Pit1 patients. The presentation of patients with Pit1 disorders varies considerably. At infancy, they usually have a protruding forehead, depressed facial structures, and a saddled nose, although CPHD is generally not diagnosed until growth retardation due to the deficiencies of GH and thyroid hormone becomes obvious.39,40
Mechanism
The modular structure of the Pit1 protein can be divided into the transactivation and the DNA-binding domains. The transcriptional activation domain is located in the first 80 amino acids, followed by a POU DNA-binding domain at the C terminus. The POU domain is further divided into a 75-amino acid POU-specific domain, which is conserved among various POU-domain proteins, and a 60-amino acid POU homeodomain with a linker region between them. The POU homeodomain by itself is sufficient for low-affinity DNA binding, although both the POU-specific domain and POU homeodomain are required for specific high-affinity DNA binding of the Pit1-responsive elements. Pit1 protein is able to bind as a monomer in solution to the consensus (A/T)(A/T)TATNCAT site, where N may be any nucleotide; in most cases, however, Pit1 binds DNA as a dimer.41 Analysis of data derived from a cocrystal study of the Pit1 protein and the PRL proximal promoter Pit1-binding element reveals that the Pit1 protein binds to DNA in a parallel dimer form.42,43 Pit1 protein wraps around the DNA molecule, with the POU-specific domain and the POU homeodomain binding to the DNA molecule in a perpendicular angle in opposite orientation. The POU-specific domain of one Pit1 molecule interacts with the C terminus of the POU homeodomain of the other Pit1 molecule in a dual composition. In addition, the spacing between the DNA contacts made by the POU-specific domain and the POU homeodomain of each monomer is critical. Compared to the Pit1 binding site in the PRL minimum promoter sequences, two additional base pairs spacing are needed to direct restricted GH gene transcription based on elements of two Pit1 binding sites on the proximal promoter of rat GH locus.44
This dimerization interface is a “hot spot” for debilitating mutations. Additional mutations, like in the Pit1Snell mice, a G-to-T mutation results (W261C) in the third helix of the POU homeodomain, eliminating its DNA-binding ability by altering the contact point of the mutant gene product with the major groove of the responsive elements, causing a dwarf phenotype in an autosomal-recessive fashion. Similarly, several mutations observed in human cases could affect the stability and specificity of this protein-DNA interface.45,46
As a transcription factor, Pit1 exerts its effects as a component of a transcriptional complex regulated by coactivator and repressor elements. The Pit1 POU domain can associate with coactivator complex of CBP/p300 and P/CAF, both of which possess histone acetylase activity. N-CoR, acting as a corepressor, can bind to the homeodomain of Pit1 and actively suppress transactivation by Pit1; this suppression depends on Sin3, SAP30, and histone deacetylase. Thus the transcriptional activity of Pit1 may be regulated by the competing binding of complexes mediating either acetylation or deacetylation events, resulting in activation or repression, respectively.47
In addition to Pit1, the determination of individual pituitary cell types may require other molecules. The estrogen receptor has been implicated in synergistic activation of the PRL gene.48,49 Members of the ETS family of transcription factors can bind to Pit1 binding sites in the PRL promoter and mediate signals from growth factors and the Ras/mitogen-activated protein kinase pathway.50 The transcription factor GATA2 appears to be required for the formation of both thyrotrope and gonadotrope cells; the presence of Pit1 represses the gonadotropic phenotype and promotes the thyrotrope phenotype. Pit1 can inhibit binding of GATA2 to cognate DNA sites important for generation of the gonadotrope phenotype. In contrast, Pit1 leads to synergistic activation with GATA2 on promoters that contain both Pit1 and GATA2 sites.51
Prop1 Gene
Prop1 (Prophet of Pit-1) is a homeodomain-containing transcription factor that is capable of binding to its cognate DNA site and activating its target genes. The expression pattern of the Prop1 gene has been examined in mice and is detected only in Rathke’s pouch. Prop1 expression is detected initially at e10 in the mouse, when the structure of Rathke’s pouch has been established. The expression initially is observed dorsally but subsequently involves most cells in Rathke’s pouch. Expression of Prop1 reaches a maximum level of intensity at e12 in Rathke’s pouch, with the signal diminishing by e14.552 (see Fig. 1-1). It has been shown recently that Notch signaling is required for maintaining high levels of Prop1 expression at e12.5, which is mediated by Rbp-J protein bound to the evolutionary conserved site within the first intron of the Prop1 gene.53 Expression of the Prop1 gene is required for activation of the downstream Pit1 gene.54,55 The integrity of Prop1 is necessary for full-scale manifestation of pituitary gonadotrope cells, as well as the generation of somatotrope, lactotrope, and thyrotrope cells (see Table 1-2). Mutations in the Prop1 gene have been identified as the leading cause of familial CPHD, resulting in short stature as a consequence.
Animal Model
The Prop1 gene was initially identified by a positional cloning strategy in the naturally occurring Ames mouse mutant. The mutant Prop1 allele at the Prop1Ames locus harbors a point mutation that results in a single amino acid substitution (S83P) in the second helix of the homeodomain, causing altered progression of nascent pituitary gland and subsequent failed expression of Pit1.52 Phenotypes of the Prop1Ames mice are transmitted in an autosomal-recessive fashion; heterozygous mutant mice are normal. Homozygous mutant mice are born grossly normal but develop a proportional dwarfism by the time of weaning.56 The adult mutant mice are about half the size of the wild-type animals. The Prop1Ames mutation caused dysmorphogenesis of Rathke’s pouch at e12.5, with convolution of the lumen and a failure of expression of the Pit1 lineage. The appearance of gonadotrope was delayed, but corticotrope appeared as expected. In contrast to the complete absence of somatotrope, lactotrope, and thyrotrope cells in the Pit1Snell mouse, the Prop1Ames mouse pituitary gland contains a small number (<1%)54,57 of the normal complement of somatotrope cells, as well as a few lactotrope and thyrotrope cells.57 Prop1Ames dwarf mice live twice as long as their wild-type littermates.58
Related Diseases
Initial reports identified mutations in the human Prop1 gene in patients with short stature in several families. Direct sequencing of polymerase chain reaction (PCR) products of the Prop1 gene revealed that all the affected patients were harboring mutations in both alleles of the Prop1 gene, and their parents were heterozygous for the respective mutations, suggesting that the mutations in the Prop1 gene act in an autosomal-recessive manner, causing CPHD in these patients. All of the affected individuals in this study failed to respond to GHRH, thyrotropin-releasing hormone, and LH-releasing hormone stimulation, suggesting a defect in hormone-secreting cells of the pituitary gland.59 Subsequent reports have revealed that Prop1 mutation is a common cause of familial CPHD. These alternations in the Prop1 gene range from point mutation to deletions, affecting structure and integrity in the homeodomain of the Prop1 gene. A 2-bp A301G302 deletion, leading to a frame-shift and the loss of DNA-binding homeodomain and C-terminal transactivation domain of the Prop1 gene product, is the most frequently encountered mutation among these Prop1 patients, representing a mutational “hot spot.”60 Individuals with various Prop1 mutations invariably display severe deficiencies for pituitary gonadotropins in addition to the defects of GH, PRL, and TSH levels. In human cases with Prop1 mutations, many adult patients express ACTH at a normal level; however, there are reported cases with a late onset of corticotropin deficiency (see Table 1-2). The expression of ACTH phenotypes is highly heterogeneous; differences in genetic background in these patients may contribute to the discrepancy of this phenotype.61,62
Mechanism
In Prop1Ames mice, examination of the mutant Rathke’s pouch revealed severe dysmorphogenesis, but the pituitary precursor cells were generated. The precursor cells of Rathke’s pouch failed to migrate to form the nascent pituitary gland, leading to an expansion of the luminal structure and lack of expression of a late pituitary differentiation marker, the Pit1 gene. However, proliferation of the mutant precursor cells in the Ames mice continued, resulting in normal-sized pituitary glands.52,63 In addition to Pit1, both Wnt and Notch pathways are affected in the Prop1Ames mice.64,65 Later, persistent expression of Prop1 under control of the aGSU promoter caused decreased gonadotrope differentiation and increased adenomatous hyperplasia,66,67 indicating that properly extinguishing Prop1 also may be an important later step in paired-like homeodomain-mediated organogenesis.
Prop1 can bind to its site and activate target genes via the C-terminal transactivation domain, whereas the N terminus and the homeodomain of Prop1 possess repression function,52,68 suggesting that Prop1 can act as a transcriptional activator as well as a repressor. Recent studies of the pituitary-specific inactivation of the β-catenin gene reveal that a Prop1/β-catenin complex acts as transcriptional activator for Pit1 and as a repressor for Hesx1, depending on the associated co-factors.69
Phenotypic comparisons have been made between the Prop1-defective patient and the Prop1-mutant Prop1Ames mouse. Deficiencies of GH, PRL, and TSH are consistently observed in both species. All the patients with Prop1 mutations eventually develop gonadotropin deficiency in their adult lives. In the Prop1Ames mice, the expression of gonadotropin is observed at birth, but the level of expression of the gonadotropin is reduced to one quarter of that of the wild-type animals.52 The expression of ACTH is apparent during development in the Prop1Ames mouse pituitary, and the level of ACTH in the blood is normal in adults. In human Prop1 patients, cortisol levels are normal at birth, but some patients develop cortisol deficiency later in life.70–72 The Prop1 mutation may affect all the major cell types in the anterior pituitary gland, including the gonadotrope and the corticotrope (see Table 1-2).
Hesx1 Gene
Hesx1 (homeodomain gene expressed in ES cells) is a paired-class homeodomain transcription factor that is capable of binding to its cognate DNA site and regulating its target genes. Mutations in the Hesx1 gene have been identified in septo-optic dysplasia and CPHD. In mice, the earliest expression of the Hesx1 gene can be detected at the embryonic stem cell stage. High levels of expression can be detected in the ectoderm, subsequently at the anterior extreme of the rostral neural folds, and finally restricted to the ventral diencephalon and to the thickened layer of oral ectoderm, which will give rise to Rathke’s pouch at e9.0 in the mouse.73,74 Hesx1 gene expression can be observed for 2 more days but only in Rathke’s pouch, with diminishing intensity at a time that coincides with the rise of Prop1 gene expression (Fig. 1-2; also see Fig. 1-1). In humans, strong expression of Hesx1 in Rathke’s pouch can be detected in a 7-week-old embryo. Hesx1 is the earliest molecular marker for the definitive pituitary primordium.
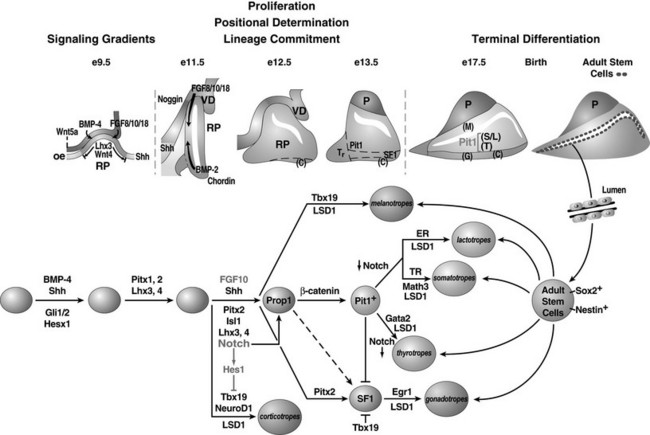
FIGURE 1-2 Ontogeny of signaling molecules and selected transcriptional factors during mouse pituitary organogenesis. Ventral diencephalon, which expresses BMP4, FGF8/10/18, and Wnt5, makes direct contact with oral ectoderm and induces the formation of Rathke’s pouch. Shh is expressed throughout the oral ectoderm, except in Rathke’s pouch, creating a boundary between two ectodermal domains of Shh-expressing and nonexpressing cells. The opposing dorsal BMP4/FGF and ventral BMP2/Shh gradients convey proliferative and positional cues by regulating combinatorial patterns of transcription factor gene expression. Pit1 is induced at e13.5 in the caudomedial region of the pituitary gland, which ultimately gives rise to somatotropes (S), lactotropes (L), and thyrotropes (T). Rostral tip thyrotropes (Tr) are Pit1 independent. Corticotropes (C) and gonadotropes (G) are differentiated in the most ventral part of the gland. The dorsal region of Rathke’s pouch becomes the intermediate lobe, containing melanotropes (M). The infundibulum grows downward and eventually becomes the posterior lobe (P). A number of transcription factors and cofactors regulating the lineage commitment and terminal differentiation of distinct cell types are illustrated in a genetic pathway. (Modified from Zhu X et al: Signaling and epigenetic regulation of pituitary development. Curr Opin Cell Biol 19(6):605–611, 2007.)
Animal Model
The mouse Hesx1 gene is located on chromosome 14, and targeted deletion of Hesx1 resulted in mice that exhibited variable anterior central nervous system defects with reduced prosencephalon and defective olfactory development.75 Hesx1 mutants also have defects in the pituitary gland, with bifurcations in Rathke’s pouch in most cases. By e12.5, multiple oral ectoderm invaginations reflecting pituitary glands are observed in most Hesx1 embryos. Between e13.5 and e15.5, Hesx1 mutants are characterized by a dramatic cellular over-proliferation of all the hormone-producing cell types, leading to a failure of the underlying mesenchyme to condense and form the sphenoid cartilage that separates the pituitary from the oral cavity. In the late stages of pituitary development, the terminal differentiation of the hormone-producing cell types appear normal in most Hesx1 mutants, with overexpression of αGSU, TSHβ, GH, POMC, and Pit1 by e16.5. Earlier in development, there is a delay in the onset of POMC expression both in Rathke’s pouch and in the developing hypothalamus at e12.5, and there also appears to be a dual induction of αGSU expression on both the rostral and caudal sides of Rathke’s pouch. Strikingly, in occasional Hesx1 gene–deleted mice, the initial thickening of oral ectoderm and minimal activation of Lhx3 are observed at e12.5, but the embryos exhibit a complete arrest of pituitary development, and the pituitary gland is absent by e18.5. The discrepancy of incomplete phenotype penetrance in Hesx1 mutants is likely influenced by the actions of the linked modifier genes.76,77
Related Diseases
The human Hesx1 gene contains four exons separated by three introns, and it maps to chromosome 3p21. The Hesx1 gene encodes a highly conserved polypeptide of 185 amino acids with a 60-amino acid homeodomain at its C terminus. Initial analyses of the Hesx1 mutations carried out in kindreds with septo-optic dysplasia identified a nucleotide transition that resulted in the substitution of R160C (in the third helix of the homeodomain) in two children with CPHD born to a highly consanguineous family. Magnetic resonance imaging revealed an ectopic/undescended posterior pituitary associated with a hypoplastic anterior lobe in these two affected siblings.75 None of the heterozygote parents exhibited features of septo-optic dysplasia, consistent with an autosomal-recessive inheritance. Additional mutations (e.g., Q6H, S170L, T181A, I26T, and 306/307InsAG-X) have been found in the coding region of the Hesx1 gene and are associated with variable phenotypes, including hypopituitarism, ranging from isolated GH deficiency to CPHD. It is clear from these reported cases that mutation in the Hesx1 gene can cause pituitary hormone deficiency with variable phenotypes and with incomplete penetrance.78,79
Mechanism
The Hesx1 gene product can bind to either dimer or monomer DNA sites with high affinity in transient transfection assays.80,81 Modular structure analysis revealed that in addition to the DNA-binding homeodomain, Hesx1 contains two sequences in the N terminus; one is similar to the eh1 motif found in Drosophila engrailed, and one is similar to the WRPW motif found in several helix-loop-helix proteins, both of which are capable of recruiting the Groucho class of corepressors.82,83 Both the N-terminal and homeodomain regions of Hesx1 can independently act as repressors. Hesx1 is a strong transcriptional repressor that acts by recruiting the mSin3A/B, HDACs 1 and 2, and the Brg1 complexes to its homeodomain and the TLE corepressor to its eh1 domain. The strong association between Tle1 and Hesx1 is mediated by a highly conserved helical motif (FXLXXIL) present in the Hesx1 N terminus, which can also be found in Nkx, Six, and certain Pax homeodomain factors’ family members.84 These recruitments are required and sufficient for the repressive actions of Hesx1 in vivo. Forced persistent expression of Hesx1 and Tle1 resulted in the loss of the Pit1 lineage and a Prop1Ames-like dysmorphogenesis, while the expression of Prop1 and POMC remained. The mutation in human Hesx1 (R160C) has a dominant negative effect both in vitro and in vivo. This dominant negative activity requires the eh1 repression domain, which is also required for full-length recombinant Hesx1 dimerization in solution. This dominant transcription repressor activity may help to explain the heterozygous phenotypes observed in Hesx1 patients.80 Recent identification of a homozygous mutation in the eh1 motif (I26T) in a patient with CPHD has further underlined that Tle association is an integral mechanism for Hesx1 function in vivo.85
Hesx1 and Prop1 share a conserved DNA-recognition site. The repression domain in Hesx1 can suppress the transcription activation activity of Prop1. The Hesx1 repressor can heterodimerize with Prop1 and can bind to the palindromic site as homodimers or heterodimers, with Prop1 acting as an activator and Hesx1 as a repressor, to inhibit Prop1 activation function. The expression of Prop1 is elevated in Hesx1-mutant mice, suggesting not only that Hesx1 can repress Prop1 activation function but also that it is required for proper Prop1 expression.76 Forced early expression of Prop1 to the uncommitted oral ectoderm blocks the formation of Rathke’s pouch, which results in absence of the anterior pituitary gland with no initial induction of Lhx3 expression, demonstrating that premature expression of Prop1 can block the pituitary organogenesis that phenocopies the effects of Hesx1-gene deletion,15 in contrast to the Hesx1/Tle1 transgenic mouse with a Prop1Ames-like phenotype, suggesting that the antagonistic repressor complex can suppress Prop1 activation of expression.76 The sequential repression and activation of a common set of regulatory genes may prove to be an underlying strategy in the temporal code of pituitary organ development, with initial repression required for organ commitment and proliferation and subsequent activation required for commitment of specific cell lineages.69
Lhx3 And Lhx4 Genes
Lhx3 (LIM homeo box gene 3) is a LIM-type homeodomain transcription factor. In addition to a C-terminus homeodomain, Lhx3 contains two tandem repeats of LIM zinc-binding motifs, each composed of 50 to 60 amino acids with a conserved pattern of cysteine and histidine residues that form a pair of zinc fingers, separated by a linker of 2 amino acids. Expression analysis revealed that mouse Lhx3 mRNA can be detected in the developing nervous system and accumulates in Rathke’s pouch beginning at e9.5 (see Fig. 1-1). Lhx3 remains expressed in the entire pouch, and its expression is maintained through e15.5; the expression is particularly strong in the anterior and intermediate lobes of the adult pituitary. In addition, Lhx3 is expressed bilaterally along the spinal cord and the hindbrain at early stages of development.86
Structurally, Lhx4 (LIM homeodomain gene 4) is closely related to Lhx3. The Lhx gene family consists of at least 12 members; many of them are expressed in the pituitary during development, including Isl1, Isl2, Lhx2, Lhx3, and Lhx4. Lhx3 and Lhx4 have been genetically defined as required elements for both the early stages of pituitary determination and the later differentiation of pituitary cell types. By insitu hybridization, the Lhx4 gene is found to be expressed transiently in ventrolateral regions of the neural tube and the hindbrain of the developing mouse. During pituitary development, Lhx4 is expressed throughout the invaginating Rathke’s pouch at e9.5. At e12.5, Lhx4 expression becomes restricted to the future anterior lobe of the pituitary gland, and by e15.5, Lhx4 expression diminishes. In the adult pituitary, Lhx4 is found in the anterior and intermediate lobes at a much lower level than that of Lhx3.87
Animal Model
Employing a reverse genetic approach, mice with a targeted disruption in the Lhx3 gene were generated. Mice heterozygous for the mutation are apparently normal and fertile, whereas homozygous individuals are stillborn or expire within 24 hours of birth. In these homozygous mice, the hindbrain, spinal cord, and pineal gland are grossly normal, as is the posterior lobe of the pituitary, but the anterior and intermediate lobes of the pituitary are absent. During embryonic development, the mutant animal exhibits a lack of growth in Rathke’s pouch, and pituitary-gland development does not progress beyond the Rathke’s pouch stage. With the exception of residual corticotrope, other anterior pituitary cell types are absent, indicating that Lhx3 is required for the appearance of the somatotrope, lactotrope, thyrotrope, and gonadotrope cell types.88
Mice homozygous for the targeted deletion of the Lhx4 gene exhibit an early postnatal death due to a failure of pulmonary maturation.87 Lhx4-deleted mice have a well-formed Rathke’s pouch but display incomplete pituitary development following this stage, and the differentiation of pituitary cell types is perturbed. Consequently, by e12.5, there exists a miniature Rathke’s pouch, and by e14.5, the nascent pituitary structure has progressed to a larger pouch, but the anterior lobe is discernible only as a slight thickening in the ventral region. This hypocellularity of the anterior lobe is caused by failure of pituitary precursor cells to survive; large numbers of apoptotic cells are evident throughout the pituitary primordia of Lhx4-mutant mice at e12.5.89 In later gestation stages, Rathke’s pouch is hypoplastic, with an enlarged lumen resulting from reduced proliferation of the precursors, and the anterior lobe of the pituitary is reduced in size. Expression analyses have revealed residual amounts of LH- and GNRHR-positive cells at e18.5. Thus, Lhx4 is not required for specification of gonadotrope cells, but it does support the expansion of the cell population. Similarly, all five anterior pituitary–specific cell lineages are present in the Lhx4-mutant pituitary but in dramatically reduced numbers. By contrast, the intermediate-lobe melanotrope cells are undisturbed.
Mice with double deletion of Lhx3 and Lhx4 demonstrated that both genes direct formation of the pituitary gland.90 The early formation of the Rathke’s pouch rudiment from pituitary primordium does not depend entirely on the function of either Lhx3 or Lhx4 alone, but together these genes redundantly control the formation of the definitive pouch. Lhx3 also controls a subsequent step of pituitary fate commitment, and in these early stages, Lhx4 appears to act upstream of the Lhx3 and Isl1 genes and is required for expansion of Rathke’s pouch. Therefore, Lhx3 and Lhx4 dictate pituitary gland identity by controlling decision points of organogenesis and regulation of the proliferation and differentiation of pituitary-specific cell lineages.
Related Diseases
Human Lhx3 shares a high degree of homology with its mouse orthologue, exhibiting 94% identity at the amino acid level. Lhx3 is located on human chromosome 9q34 and spans a genomic fragment of at least 6 kb that includes 6 exons.91,92 In a candidate-gene screen based on pituitary phenotypes observed in a recessive lethal mutation in mice, two mutations in the Lhx3 gene were identified in two unrelated consanguineous pedigrees that display CPHD.93 In one family, affected individuals are homozygous for a Y116C mutation located in the highly conserved LIM2 domain of Lhx3, a domain critical for protein-protein interactions. In the second family, affected individuals are homozygous for a 23-base-pair deletion in an intragenic region, predicting a severely truncated protein that lacks the entire homeodomain and rendering it incapable of DNA binding. Lhx3-defective patients have deficiencies in GH, TSH, PRL, FSHβ, and LHβ, but they display intact levels of ACTH, similar to the endocrine profiles observed in Prop1 patients (see Table 1-2). In addition, these Lhx3-defective patients displayed a rigid cervical spine that restricted their head rotation. More recently, novel 6 mutations have been found in the coding region of the Lhx3 gene.94,95 All of them are associated with variable phenotype of hypopituitarism. Lhx3 mutations are a rare cause of CPHD involving deficiencies for GH, prolactin, TSH, and LH/FSH in all patients. Whereas most patients have a severe hormone deficiency manifesting after birth, milder forms can be observed, and limited neck rotation is not a universal feature of patients with Lhx3 mutations.
The human Lhx4 gene encodes a 390-amino acid protein that contains two LIM domains and a homeodomain that shares 99% sequence identity with its mouse orthologue. Genomic analysis revealed that the human Lhx4 gene contains 6 exons and is mapped to chromosome 1q25.96 In a large consanguineous pedigree of three generations, a G-to-C substitution in the intron preceding exon 5 of Lhx4 generates a mutant protein with perturbed homeodomain, which affects its DNA-binding function. Patients with this disease have short stature with CPHD, which affects GH, thyroxine, and cortisol, as well as cerebellar defects and abnormalities of the sella turcica. This mutant allele is transmitted in a dominant fashion, affecting only the maternal side of the kindred with a high phenotypic penetrance.96 More recently, three novel mutations in the Lhx4 gene have been mapped.97 All of them affect the DNA-binding domain, and all of them are associated with CPHD.
Mechanism
LIM homeodomain proteins are transcription factors and exert their effects by regulating target gene expression. Lhx3 binds with high affinity to AT-rich DNA sequences (including minor groove interaction) and bends the DNA molecule to an angle of 62 degrees in a model system.92 Lhx3 can activate the regulatory regions of pituitary genes, including αGSU, PRL, TSHβ, and Pit1. Lhx3 expression is partially regulated by the Lhx4 gene during pituitary development. At e12.5, only a few cells express Lhx3 in the dorsal-most aspect of the pouch in the Lhx4 mutants. However, the normal pattern of Lhx3 expression, including the dorsal-ventral gradient, is established in Lhx4 mutants by e14.5.89
Genetic analysis revealed that Lhx4 interacts with Prop1 to stimulate anterior pituitary lobe expansion. Neither gene is essential for initiating corticotrope specification. However, no POMC or αGSU expression is detected in double-mutant mice at e14.5, suggesting that Prop1 and Lhx4 have overlapping roles in corticotrope and gonadotrope development.89 In Hesx1-deleted mutants, the domains of Lhx3 and Prop1 expression are increased, as well as those of FGF8 and FGF10 in the infundibulum, which become expanded rostrally.76 These findings indicate that Hesx1 is required for maintaining the proper expression of FGFs, consistent with the notion that Lhx3 expression can be regulated by FGF signaling.
Tbx19 Gene
Tbx19 is a T-box transcription factor family member (the T-box in the mouse T [Brachyury] gene) that encodes a 448-amino acid protein.98 Functional identification of Tbx19 was established after the observation of elements in a critical cis-acting sequence in the POMC promoter. Transcripts of Tbx19 can be found only in the anterior and intermediate pituitary and brain (see Fig. 1-1); Tbx19 is specifically required for continued POMC transcription.99,100
Animal Model
Mice with targeted disruption of the Tbx19 gene have been generated. Mice heterozygous for the mutation are apparently normal. Adult mice homozygous for the mutation have very few ACTH-positive cells in the pituitary, although initial expression of the POMC gene is undisturbed at the Rathke’s pouch stages. These cells are born in normal quantities in mutants but are lost or fail to expand appropriately, suggesting Tbx19 is not required for corticotrope cell commitment but is later important for POMC lineage differentiation. The intermediate-lobe melanotropes in mutant mice are populated by gonadotrope and some Pit1-independent thyrotrope, also indicating that Tbx19 normally represses pituitary gonadotrope differentiation.101,102
Related Diseases
The human Tbx19 gene shares 94% amino acid identity with that of mouse Tbx19 and maps to chromosome 1q23-q24. Several cases of isolated ACTH deficiency were identified with a nonsense mutation C-to-T transition in exon 6 in Tbx19, resulting in a truncated gene product (R286X).103 The transmission of this mutation appears to be recessive. In another case of isolated ACTH deficiency, a heterozygous C-to-T transition in exon 2 of the Tbx19 gene was identified, resulting in a conserved amino acid S128F mutation, suggesting a dominant negative inheritance.99 More recent study revealed new mutation in the Tbx19 gene (missense M86R) that did not affect monomer DNA-binding activity per se, but it impaired DNA binding with other DNA-bound proteins, including itself (homodimers) and Pitx1, resulting in congenital isolated ACTH deficiency.104 Additional mutations in the Tbx19 gene have been shown to be a cause of neonatal death due to neonatal-onset isolated ACTH deficiency.103 Tbx19 defects result in POMC deficiency in both humans and mice, establishing Tbx19 as the gene required for effective POMC expression in vivo.101,102
Mechanism
Tbx19 is a transcriptional regulator, recognizing target genes through its T-box DNA-binding domain. In response to signals elicited by the hypothalamic hormone corticotrope-releasing hormone, Tbx19 functions as an activator of transcription by recruiting SRC/p160 coactivators to its cognate DNA target in the POMC promoter.105 Tbx19 can synergize with orphan nuclear receptor NGFI-B, serving as part of the transcription regulatory complex on the POMC promoter in response to hormonal stimulation.106 Transgenic expression of Tbx19 in non-POMC-producing regions of the pituitary gland can cause ectopic POMC expression,99 and Tbx19 is an inhibitor of αGSU expression in rostral tip cells, gonadotrope, and thyrotrope, and of TSHβ production in caudomedial thyrotrope.100 Tbx19 deficiency is permissive for transdifferentiation of cells normally destined to be corticotrope and melanotrope into alternative cell fates, namely, gonadotrope and rostral tip thyrotrope, suggesting a determinative role of Tbx19 in cell lineage specification. Tbx19 defects have no effect on differentiation of Pit1-dependent cell lineages (see Figs. 1-1 and 1-2, and Table 1-2).101,102
Pitx1 And Pitx2 Genes
Pitx1 and Pitx2 represent two of the bicoid-related Pitx homeodomain transcription factors. They display distinct but overlapping patterns of expression and are critical in the development of several organs, including the pituitary, with Pitx1 required for the gonadotrope, thyrotrope, and POMC gene expression107,108 and Pitx2 required for the earliest phases of pituitary development for the patterning and proliferation events within Rathke’s pouch.109–111 Genetic studies have shown that they are required for cell proliferation, survival, and differentiation in a dosage-sensitive manner, with Pitx2 playing a more prominent role than Pitx1. They both function redundantly in controlling Lhx3 expression.111–113
Animal Model
Inactivation of Pitx1 results in defects in hindlimb development and craniofacial morphogenesis.108,114 The anterior pituitaries of mutant mice exhibit mild defects with decreased expression of FSHβ, LHβ, and TSHβ and increased expression of POMC. Pitx2-/- mice display multiple developmental defects, including failure of body-wall closure, right pulmonary isomerism, and defects in heart, tooth, eye, and pituitary organogenesis.109,115–117 In the pituitary, a definite pouch forms with induction of Lhx3, Hesx1, Pitx1, and αGSU. However, the gland fails to progress further, with no Pit1 induction and only a few POMC-positive corticotrophes. Transgenic mice overexpressing Pitx2 in the cornea manifest ocular defects similar to Rieger’s syndrome, suggesting that excess Pitx2 activity can be as deleterious to eye development as a loss of function.118 Overexpression of Pitx2 during mouse forelimb development results in severe tendon, muscle, and bone anomalies.119 A small fraction of Pitx2-heterozygous mice display some aspects of Rieger’s syndrome.
Related Diseases
The Pitx2 gene was initially identified as the gene responsible for human Rieger’s syndrome type I, an autosomal dominant condition characterized by variable defects, including anomalies of the anterior chamber of the eye, dental hypoplasia, a protuberant umbilicus, mental retardation, and isolated growth hormone deficiency.110 Most of the mutations in Pitx2 cause defects in DNA binding, transactivation, or both, whereas a single hypermorphic allele of Pitx2 (V45L) leads to a reduced DNA-binding but an enhanced transactivation activity.120,121 To date, no mutations within Pitx1 have been described in humans.
Mechanism
Pitx1 was identified on the basis of its ability to interact with the N-terminal transactivation domain of Pit1122 and to bind POMC promoter.123 Synergistic interactions between Pitx1 and Pit-1 activate the PRL122 and the TSHb genes; those with the dimer NeuroD1/E47124 and Tbx1999 activate POMC expression; and those with SF-1125 and Egr-1126 activate the LHb gene. Pitx1 is also able to stimulate the expression of genes coding for the gonadotropin-releasing hormone receptor (GRHR) and Pit-1.127
Other Transcription Factors
Transcription factors act as activators and repressors and often are expressed in a coordinated fashion, mediating organogenesis and cell type specification in the pituitary gland (see Figs 1-1 and 1-2). During the early commitment stage, expression of Pitx1/2 and Hesx1 are found in the anterior neural plate stages and in the invagination of Rathke’s pouch. Lhx3 is expressed on e9.5 in the nascent RP and is required for initial organ commitment and growth88 and for cell proliferation, together with Lhx4.89 Prop1 appears on e10.5 and is required for determination of four ventral cell types, including the Pit1-dependent lineages (somatotrope, lactotrope, and thyrotrope) and the gonadotrope.52,59 Sequential expression of this cascade of homeodomain genes represents a model system of transcription control of organogenesis and cell type determination and differentiation in mammals. Phenotypic comparisons in these pituitary loci in both mice and humans suggest that the developmental pathways in determination of the pituitary gland are highly conserved.
Some of these factors express transiently in Rathke’s pouch, and their reduced expression is likely to be required for the progression of specific cell types, as evident in the Prop1Ames mutant, where the temporal patterns of Hesx1, Prop1, and Brn4 gene expression are extended. As the lineage-determining transcription factor Pit1 appears, certain transcription factors that characterize earlier stages of development are gradually eliminated, including Hesx1, Pfrk,15 GATA-2,51 Pax6,128 and Brn4.129
The list of pituitary-expressed transcription factors implicated in the developing pituitary gland is constantly expanding. Several families of factors, including homeodomain factors (Isl1, Isl2, Oct1, Otx2, Pax6, Pitx3, Six1, Six3, and Six6), zinc-finger (Krox24, Gli1, GATA2, Nzf1, Sp1, Sp3, Zfhep, and Zn16), nuclear receptor (T3R, SF1, ERa, ERb, and Dax), basic HLH domain (AP2, NeuroD, Mash1, Nhlh2, and Math3) and other (Gli2, AP1, Ets1, Foxl2, CBf, Cp1, Rb, Men1, Preb, and Tef) (see reviews [130,131]). Multiple members of the Six family (Six1, Six6, Six2, and Six3) are also expressed in the pituitary. Six6, acting as a strong tissue-specific repressor in association with dachshund (Dach) corepressors, directly represses cyclin-dependent kinase inhibitors, including the p27Kip1 promoter, and regulates early progenitor cell proliferation in retinogenesis and pituitary development.132 Six1 exhibits synergistic genetic interactions with the eyes absent (Eya) family of protein phosphatases and is required to regulate genes that encode growth control and modulate precursor cell proliferation. The phosphatase activity of Eya converts the function of Six1-Dach from repression to activation, causing transcriptional activation through recruitment of coactivators.133 Evidence suggests that Six3 acts upstream of the Wnt pathway, and deletion of Six3 in mice resulted in failure of development of the ventral diencephalon and, consequently, development of the pituitary.134 GATA2 is involved in establishing molecular memory of signaling gradients during pituitary development in conjunction with Pit1, and loss of GATA2 is associated with failure to differentiate into gonadotrope.51,111 The orphan nuclear receptor steroidogenic factor 1, Sf1, is essential for pituitary gonadotrope.135 Mice with simultaneous inactivation of both Gli1 and Gli2 have very severe defects in pituitary gland development; about half of these mutants completely lack a Rathke’s pouch at e12.5. In these mutants, the domains of expression of Shh and Nkx2.1 are abnormal, and the loss of Shh signaling boundary in the oral ectoderm could be a cause of this defect.136 Gli2 is an upstream regulator of Lhx3 (see Fig. 1-2). One consequence of the Gli2 defect in yot-mutant Zebrafish embryos was the absence of Lhx3 expression in the anterior part of the adenohypophysis anlage.137 In the mouse, absence of Lhx3 results in failure of development of Rathke’s pouch into the adenohypophysis.
Otx1 is expressed in the postnatal pituitary gland. Cell culture experiments have shown that Otx1 may activate transcription of GH, FSHβ, βLH, and αGSU. Analysis of Otx1-null mice indicates that at the prepubescent stage, they exhibit transient dwarfism and hypogonadism due to low levels of pituitary GH, FSHβ and LHβ hormones, which in turn dramatically affect downstream molecular and organ targets. Nevertheless, Otx1–/– mice gradually recover from most of these abnormalities, showing normal levels of pituitary hormones with restored growth and gonadal functions at 4 months of age. Since the patterns of expression of hypothalamic hormone-releasing hormones (GHRH, GnRH) and their pituitary receptors (GHRHR, GnRHR) are unchanged, it seems that ability to synthesize GH, FSHβ, and LHβ, rather than the number of cells producing these hormones, is affected.138
Because Otx2-null mice exhibit early embryonic lethality,139 Otx2 protein has been only recently shown to play a role in pituitary development. Clinical study of one patient has revealed a new heterozygous Otx2 mutation that is affecting the transactivation domain of Otx2 and subsequent lack of activation of Hesx1 and Pit1 promoters in cell culture transfection assay.140 Heterozygous Otx2 knockout mice have highly variable brain and ocular phenotypes, and although pituitary structure and function have not been studied, this recent study might implicate more attention for the role of Otx2 protein in the pituitary.
Although pituitary cell types in the adult anterior lobe do not appear to be stratified, initial appearance of these cell types follows a ventral-dorsal pattern. With the Rathke’s pouch cleft as the dorsal reference, GH, PRL, and TSHβ of Pit1 lineages are located dorsally, whereas gonadotrope cells appear ventrally. Several transcription factors display vertical gradients, including Pax6, which exhibits a dorsal-to-ventral expression gradient; Pax6-mutant mice exhibit increased numbers of ventral thyrotrope and gonadotrope, at the expense of the more dorsal somatotrope and lactotrope cell types.128 Thus Pax6 may functionally oppose Shh signaling to specify a dorsal rather than ventral cell fate. Another pituitary transcription factor displaying an initial dorsal-ventral gradient is Prop1 (see Fig. 1-1). Prop1-mutant mice lose dorsal cell types of the Pit1 lineage, while ventral cell types of corticotrope and gonadotrope are less affected.
Signaling Pathways in Pituitary Development
Vertebrate organogenesis events are coordinated through the interplay of highly organized signaling pathways, and these developmental signaling systems have proved to be remarkably conserved throughout evolution.141 Extrinsic signals, in the form of secreted morphogens, create local environments for organ patterning and progenitor cell type determination. These signals are interpreted through the functions of cell type–restricted transcriptional regulators, resulting in various intrinsic or cell-autonomous determination events.142 Numerous extrinsic signaling molecules have been implicated, including members of the Hh, transforming growth factor-beta (TGFβ)/bone morphogenic protein (BMP), Wingless/Wnt, and fibroblast growth factor (FGF) superfamilies, the Notch pathway, and others (reviewed in ref. 143).
Influenced by the growth of the forebrain structures, the midline anterior neural ridge cells ultimately responsible for the origin of the pituitary gland are displaced and eventually become located immediately ventral to the diencephalons. The initial extrinsic signaling of murine pituitary development requires signals from both the ventral diencephalon and the oral ectoderm. Organogenesis of the anterior pituitary gland begins at e9 in the mouse as the cells of the anterior pituitary placode in the oral ectoderm thicken and invaginate to form the nascent pituitary of Rathke’s pouch.19 Shh, seemingly uniformly expressed in the oral epithelia at the time, is excluded from the pituitary placode prior to initiation of the invagination.144 The presumptive ventral diencephalon provides Bmp4, the first known dorsal signal required for the initial formation of Rathke’s pouch.18 Immediately following formation, the dorsal portion of Rathke’s pouch directly contacts the midline ventral diencephalon, which evaginates at e10 and acts as a key organizing center for the patterning and commitment of Rathke’s pouch. Opposing dorsal FGFs/Bmp4 and ventral Shh/Bmp2 gradients provide positional and proliferative signals to the pituitary progenitor field, acting to positionally establish cell types through the induction of overlapping patterns of transcription factor expression.15,18 Initial proliferation and determination is controlled by sequential cascades of exogenously and endogenously restricted combinatorial signaling, with subsequent attenuation of specific signal events required for establishment of the cellular environment permissive for terminal differentiation (see Fig. 1-2).2
Sonic Hedgehogs
Shh, one of three vertebrate homologues of the Drosophila-secreted protein Hh, plays an important role in embryo patterning, as well as in the specification of different cell types and in the control of proliferation of numerous cell types.145 In the early embryo, it is expressed in Hensen’s node, the floor plate of the neural tube, the posterior of the limb buds, and throughout the notochord. The mouse, zebrafish, and human Shh homologues are highly conserved,146 suggesting conserved functional properties. The human Shh gene encodes a predicted protein that is 92.4% identical to its mouse homologue and is mapped to chromosome 7q. Many Shh mutations, including nonsense and missense, deletions, and insertion mutations, are identified throughout the gene in patients with holoprosencephaly, the most common forebrain defect in humans.
Mouse mutants homozygous for a disrupted Shh gene, similarly to the mutations in human patients, revealed defects in the establishment of maintenance of midline structures such as the notochord, floor plate, and cyclopia.147 In mice, Shh is expressed in ventral diencephalons and throughout the oral ectoderm on e8 but is excluded from the invaginating Rathke’s pouch. The Hh receptor Patched1 is expressed in the developing pituitary, indicating that pituitary progenitors respond to the Hh signaling. Transgenic overexpression of hedgehog-interacting protein (HIP), which acts to attenuate Hh function, specifically blocks Hh signaling in the oral ectoderm and Rathke’s pouch within the head region, affecting both proliferation and cell type determination, and this results in an absence of ventral cell type markers in Rathke’s pouch.144 In contrast, a gain-of-function transgenic approach to overexpress Shh in Rathke’s pouch results in a phenotype of the expansion of ventral cell types, with modified levels of Lhx3 gene expression. This phenotype is consistent with results derived from an animal cap explant culture with banded Hh in Xenopus laevis, in which the expression domains of pituitary-restricted factor Hesx1 are expanded,148 supporting a role for Shh signaling in control of proliferative events in pituitary development.9
Three related zinc finger transcription factors, Gli1, Gli2, and Gli3, acting downstream of the Shh pathway, are expressed in the developing Rathke’s pouch.149 In zebrafish, Shh produced by neuroectoderm instead of notochord or oral ectoderm is crucial for the initial patterning of the pituitary placode.150 Mutations that disrupt Hh signaling, like smoothenedsmu and gli2yot, result in the development of lens tissue from the presumptive pituitary. In addition, in gli2yot mutants, the rostral expression domains (analogous to the ventral domains in mice) of pituitary-specific transcription factors such as LIM3 (Lhx3) and Six3 are lost, and other pituitary-restricted factors such as nk2.2 are absent.137 This observation is consistent with the sequential and cooperative interaction Bmps and Hh exert in limb and neural-tube development,151 in which Shh acts to induce the expression of Bmps. Overexpression of Shh in zebrafish results in expanded adenohypophyseal expression of lhx3, expansion of nk2.2 into the posterior adenohypophysis, and an increase in PRL- and somatolactin-secreting cells. In addition, Hh signaling is necessary between 10 and 15 hours post fertilization for induction of the zebrafish adenohypophysis, a time when Shh is expressed only in adjacent neural tissue. These results suggest multiple and distinct roles for Hh signaling in the formation of the vertebrate pituitary gland and also suggest that Hh signaling from neural ectoderm of ventral diencephalons is necessary for induction and functional patterning of the vertebrate pituitary gland.9,150
Fibroblast Growth Factors
Fibroblast growth factors represent a large family of secreted molecules. Upon binding to their cognate receptors, FGFs activate signal transduction pathways which are required for multiple developmental processes.152,153 Functions of FGFs are mediated by four distinct FGF-receptor tyrosine kinase molecules. FGF activity and specificity are further regulated by heparan sulfate oligosaccharides with tissue-specific modifications, in the form of a trimolecular complex with receptors.154,155 The FGF system plays significant roles in many biologic events, including pattern formation in many tissues during vertebrate embryogenesis. Several members of the FGF family are expressed in the infundibulum and provide proliferative and positional cues to Rathke’s pouch. FGF8 and FGF10 are expressed in a temporally and spatially overlapping manner within the infundibulum as an evagination of ventral diencephalon makes direct contact with the dorsal portion of Rathke’s pouch following Bmp4 induction.
In mice null for FGF10 or the FGFR2 (IIIb) isoform, which presumably would abolish FGF signaling including that of FGF10,156,157 Rathke’s pouch forms but rapidly undergoes apoptosis, with the pituitary becoming completely absent by e14.5, suggesting a critical role in FGF10 signaling for the continued proliferation of the pouch ectoderm. A similar observation has been made in transgenic mice expressing a dominant negative form of FGFR2(IIIb), suggesting that FGF10 signaling is essential for cell survival and proliferation.
FGF8 is expressed in the primitive streak of the gastrulating mouse embryo, as well as in the visceral endoderm. Mice null for FGF8 lack all embryonic mesoderm- and endoderm-derived structures and do not survive beyond e9.5.158,159 Therefore, function of FGF8 in pituitary development is largely drawn from studies of transgenic animals and in-vitro organ culture. In mice null for a homeodomain gene Nkx2.1, which is normally expressed in the ventral diencephalons but not in Rathke’s pouch, FGF8 fails to be expressed in the ventral diencephalons, leading to a loss of the infundibulum and consequently a loss of Lhx3 expression in Rathke’s pouch and the loss of all three lobes of the pituitary gland.160 In transgenic mice misexpressing FGF8 in the ventral regions of the pituitary under control of the regulatory sequences for the aGSU gene, most ventral and intermediate cell types are absent, with dysmorphogenesis of Rathke’s pouch and hyperplasia of corticotrope and melanotrope observed, consistent with a role in the positional determination of dorsally arising pituitary cell types and pituitary progenitor cells.15 In mice null for Hesx1, the most severely affected embryos exhibit a complete arrest of pituitary development after the initial induction of Lhx3 on e9.5, with FGF8 and FGF10 ectopically expressed in the oral ectoderm to mirror the normal expression in the overlying neural ectoderm. In Hesx1 mutants with less severe pituitary defects, FGF expression is abnormally extended rostrally, causing formation of multiple Rathke’s pouches. This is potentially significant because transgenic misexpression of FGF8 in the oral ectoderm well before the initial invagination of Rathke’s pouch produces an identical blockage of pouch formation, and Hesx1 fails to be expressed in the Lhx3-positive rudiment that does form in the transgenic embryos. Thus the dynamic interplay between boundaries of Hesx and FGF8/10 expression76 could suggest a model of reciprocal feedback regulation. This is in keeping with the role of FGFs in committing oral ectoderm to the Rathke’s pouch fate.160 These genetic data, in conjunction with tissue co-culture evidence, where the infundibulum is both required and sufficient for the induction of Lhx3 gene expression in cultured pouch and infundibulum activity, can be replaced with FGF8 or FGF2 and inhibited by the FGF receptor antagonist, suggest an instructive role for FGF8 signaling in pituitary development.2,18,161
TRANSFORMING GROWTH FACTORS AND BMPs
The transforming growth factor-beta superfamily of secreted signaling molecules, which includes several Bmps, has been demonstrated to play critical roles in patterning and cell type specification in several species.162 At least two members of the Bmp family, Bmp2 and Bmp4, participate in the development of the anterior pituitary. During the early stages of pituitary development, Bmp4 is expressed in the ventral diencephalons as the infundibulum makes direct contact with Rathke’s pouch at e9.0. Functional evidence with dual explants culture of embryonic diencephalon and Rathke’s pouch suggests that Bmp4 is one of the early signaling factors required for the initial commitment of a subpopulation of oral ectodermal cells to form the pituitary gland.15,18 Deletion of the Bmp4 gene causes embryonic death at about e10, in which the initial invagination of Rathke’s pouch fails to occur.160 Similarly, driven by the regulatory sequences of the Pitx1 gene to target the Bmp2/4 antagonist Noggin expression to the oral ectoderm, including Rathke’s pouch, pituitary development is arrested at e10, with a failure of the ventral proliferation of cells from the pouch beginning at e11.5 and an absence of pituitary cell types.15 The phenotype observed in Pitx1-Noggin transgenic mice is similar to the phenotype observed in mice with a targeted disruption of the Lhx3 gene critical for the determination of most pituitary cell types,88 suggesting a requirement of Bmp4 signaling for the continued organ development after pouch formation. Together with the ventral diencephalic FGFs, Bmp4 is required for initial pituitary commitment and for continued cell proliferation and progression.
Expression of Bmp2 is initially detected at the ventral boundary between Rathke’s pouch and Shh, intrinsic to the pouch in the most ventral aspect of the invaginating gland at e9.5, and in a ventral-dorsal gradient at e10.5.15 Bmp2 expression expands throughout the pouch by e12.5. Bmp2 expression is also detected in the ventral juxtapituitary mesenchyme, along with Bmp2/4 antagonist Chordin in the caudal mesenchyme, potentially serving to maintain a ventrodorsal Bmp2 gradient. After the closure of Rathke’s pouch, Bmp2 is expressed in mesenchyme adjacent to the pituitary cells expressing ventrally the transcription factors GATA2, Isl1, and the hormone subunit αGSU. Similarly, cultivation of Rathke’s pouch explants in Bmp2 is sufficient for the induction of expression of ventral markers such as Isl1 and αGSU.18 In the developing pituitary expression of Bmp2 decreases dramatically after e14-e15. Overexpression of Bmp2/4 under the control of αGSU regulatory elements in ventral mouse pituitary initially leads to a dorsal expansion of the ventral lineage markers Isl1 and Msx1 with induction of GATA2 gene expression. Proper expression of Bmp2 is therefore critical for the initiation of the cell fate determination process, however, Bmp2 signaling has to be attenuated to achieve terminal differentiation.15 These studies suggest that pouch-intrinsic and ventral signals, including Bmp2, contribute to the establishment of the positional identity of ventral pituitary cell types of thyrotrope and gonadotrope marked by αGSU expression.
Another way to study Bmp signaling is to address the role of Bmp antagonists during development. In a recent study, three antagonists—follistatin-like 1, Nbl1, and noggin–expression pattern—have implied a possible role of these proteins during pituitary development.163 Out of three, noggin-/- embryos have pituitary defects that range from a lack of a morphologic Rathke’s pouch to the formation of secondary pituitary tissue. Noggin attenuates the Bmp4 signal emanating from the ventral diencephalon during the induction and early patterning of Rathke’s pouch, but it does not play a role in cell specification in the anterior lobe.163
Notchs
The Notch signaling pathway is an evolutionarily conserved mechanism that controls cellular differentiation, proliferation, and death in a broad spectrum of developmental systems.164,165 Multiple ligands and effectors of the Notch signaling pathway were shown to be expressed in the developing pituitary, and a series of recent studies have shown the importance of the pathway in pituitary development.53,65,166,167 First, Notch2 expression is almost entirely absent in the Prop1 mutant mice pituitary, suggesting that the Notch signaling pathway may play a role in the commitment and lineage-specific differentiation of progenitor cells in the embryonic pituitary—in particular, Prop1-dependent cell lineages.65 Accordingly, conditional inactivation of Rbp-J, central mediator of the Notch signaling, using the transgenic Cre line under the control of Pitx1 regulatory sequences, leads to premature differentiation of progenitor cells, as well as a conversion of the Pit1 lineage into the coricotrope lineage. Premature progenitor differentiation is phenocopied in the mice deleted for the Hes1 gene, while the later phenotype is largely due to the significant down-regulation of Prop1 at e12.5. It has been shown that Rbp-J directly binds to the evolutionary conserved recognition site in the first intron of Prop1 gene, and it is recruited to this region during pituitary development. Hence Notch signaling directly regulates Prop1 transcription.53
The function of the Notch signaling pathway in pituitary development has also been investigated in Hes1-deficient mice and in mice conditionally deleted for both Hes1 and Hes5 in Rathke’s pouch and the ventral diencephalon, using the Emx1-Cre mouse line.53,166,168 In addition to premature corticotrope differentiation, which is consistently observed in Rbp-J conditional KO,53 these mutant embryos lack both the intermediate and posterior lobes of the pituitary gland, which is in sharp contrast to the enhanced intermediate-lobe melanotrope differentiation detected in Rbp-J-conditional KO. The discrepancy might be explained by the different targeting approaches of these studies. Signals emanating from the ventral diencephalon are probably a key aspect of this discrepancy: in mice with Rbp-J conditionally inactivated using the Pitx1-Cre transgene, the ventral diencephalon remains intact, whereas in Hes mutants, it is also targeted because both Hes1 and the Emx1-Cre are both expressed in the diencephalon.
Two independent studies have shown that down-regulation of Notch signaling is necessary for terminal differentiation of distinct cell lineages in the later phases of pituitary development.53,167 Consistently, persistent expression of Hes1 in pregonadotropes and prethyrotropes prevents their differentiation.166 Since there is a feedback loop between Notch signaling and Prop1, it would be very interesting to find out what factor down-regulates the Notch pathway during cell differentiation in the developing pituitary gland.
Wnts
The Wnt proto-oncogene family contains at least 19 known members.169 As classical morphogens, the Wnt family of signaling molecules induces various cellular responses from proliferation to cell fate determination and differentiation. The canonic Wnt pathway stated that Wnt ligands bind to the Frizzled family of seven-transmembrane domain receptors, leading to the stabilization and accumulation of β-catenin, which interacts with members of the TCF/LEF family of DNA-binding transcription factors and changes them from repressors to activators of transcription, primarily by displacing the Groucho/Tle corepressor to influence target gene expression.170,171
Several Wnt-signaling molecules are expressed during the development of pituitary.15,69,172 So far, two of them, Wnt4 and Wnt5a, have been reported to be specifically associated with the developmental events in the anterior pituitary. Wnt4 is expressed in the ventral diencephalon, and Wnt5a is expressed in the cells of Rathke’s pouch. In Wnt4-mutant mice, the pituitary is mildly hypocellular, with the ventral cell types showing normal differentiation but incomplete expansion. Additionally, cultivation of Rathke’s pouch with Wnt5a and Bmp4 can induce expression of the early cell type marker αGSU.15 Wnt5a mutants have expanded domains of FGF10 and Bmp expression in the ventral diencephalon and a reduced domain of LHX3 expression in Rathke’s pouch. As a consequence, Wnt5a-/- mice have a morphologically distorted pituitary with an enlarged intermediate lobe and increased numbers of POMC cells.172,173 Double Wnt4/Wnt5s knockout mice display an additive pituitary phenotype of dysmorphology and mild hypoplasia of the anterior lobe and hyperplasia of the intermediate lobe.172 The phenotype suggests independent roles of those two factors. Wnt6 is expressed near the pituitary gland during critical times in development; however, examination of embryos deficient in Wnt6 showed no obvious pituitary malformation.172 The effects of deficiencies of Wnt4, 5a, or 6 seem unlikely to account for the consequences of deficiencies in the known, critical, β-catenin-regulated transcription factors in the pituitary gland. Rather that Wnt signaling affects the pituitary gland via effects on ventral diencephalon signaling. Other Wnt molecules (Wnt2b, Wnt11, Wnt16)172 are also expressed during pituitary development, although their role in this process awaits further investigation.
In Pitx2-deficient mice, mutant embryos fail to survive to term and exhibit developmental arrest of early determination events in the anterior pituitary gland.109,113,115,174 Pitx2a has been demonstrated to be acting downstream of the Wnt signal, and Lef1 and β-catenin have been demonstrated to physically occupy the Pitx2a promoter in the context of a pituitary cell line. Pitx2-mutant pituitary glands contained decreased numbers of proliferating cells, whereas transgenic overexpression of Pitx2 in the anterior pituitary led to increased cell numbers.113 Also, Pitx2c isoform has been shown to be regulated directly by Wnt signaling.175 In a subtraction expression profiling analysis of Prop1Ames-mutant pituitary, several members of the Wnt signaling pathway are identified, including the frizzled2 receptor, APC, β-catenin, Groucho, and Tcf7l2.64 This genetic evidence suggests critical roles for the Wnt pathway in pituitary cellular proliferation and in cell type determination and differentiation.
Other components of the Wnt/β-catenin signaling pathway, including frizzled1-6, frizzled8, Lef1, Tcf3, Tcf4, and others have been reported to be expressed in the developing pituitary.64,69,172,176 Tcf4 is detectable in the early pituitary, as well as in surrounding tissues; it is markedly down-regulated by e13.5.64,177,178 Targeted inactivation of Tcf4 results in hyperplasia of the anterior lobe, probably caused by the expansion of FGF and BMP expression in the ventral diencephalon, with a concomitant increase in the number of progenitor cells that form Rathke’s pouch. Thus Tcf4 may play a role as a repressor to regulate growth of Rathke’s pouch by influencing Bmp and FGF signaling.178 Lef1 exhibits biphasic expression, initially transiently at e9.0 in Rathke’s pouch and later appearing at e13.5 in the anterior and intermediate lobes of the gland. Targeted deletion of Lef1 leads to elevated expression of Pit1, as well as GH and TSHβ, consistent with the role of Lef1 in inhibiting Prop1/β-catenin-mediated Pit1 activation.69 Those recent studies have shown yet another role of β-catenin and Wnt signaling during pituitary development. Targeted inactivation of β-catenin in pituitary cells using a Pitx1-Cre transgenic line results in a smaller gland with no Pit1 expression, absence of all three Pit1 lineages, and reduced numbers of gonadotropes. Surprisingly β-catenin does not exert its role through binding with its canonical Tcf/Lef partners. Induction of Pit1 is mediated by direct interaction between β-catenin and Prop1, through evolutionary conserved Pit1 early enhancer.52,69,179 Moreover, Prop1/β-catenin complex is also acting as a transcriptional repressor for Hesx1, ensuring differentiation of the progenitors. Genetic studies have demonstrated that proper spatio-temporal activation of Wnt/β-catenin signaling is essential for proper pituitary development, because premature activation of β-catenin leads to Hesx1 repression and pituitary agenesis.69
Interactions Between Different Morphogenic Factors
Opposing BMPs and FGFs Signaling Gradients
Analogous to the combinatorial signal regulation in organogenesis observed in many organs, physically opposing dorsal-to-ventral Fgf8/10/18 and ventral-to-dorsal Bmp2 gradients appear to be associated with the positional determination of specific pituitary cell types.180 The ability of the infundibulum or FGFs to induce Lhx3 gene expression correlates with the restricted expression of the Bmp2-induced genes Isl1 and aGSU distal to the source of the FGF signaling.18 The ability of ventralized expression of FGF8 to prevent the appearance of ventral cell types in vivo can be attributed to the inhibition of ventral Bmp2 signaling.15 Conversely, while cultivation of Rathke’s pouch with Bmp2/4 initiates the expression of the ventral markers Isl1 and αGSU, it inhibits the expression of more dorsal cell type markers such as ACTH in vitro18 and Pit1 in vivo.15 Thus antagonistic and opposing dorsal-to-ventral Fgf8 and ventral-to-dorsal Bmp2 gradients appear to be associated with the positional determination of dorsal and ventral cell types.2,180
Independent Hh and FGF Signaling in Pituitary Organogenesis
FGF and Shh exhibit complementary expression patterns near the developing anterior pituitary. Studies of zebrafish have shown that graded loss of Hh, but not FGF, led to ectopic midline lens formation, with either one or two lenses forming, depending on the level of Hh signaling. Moreover, the results suggest that Hh and FGF signaling act independently to induce the pattern of endocrine cell fates along the anterior/posterior axis of the zebrafish anterior lobe, with high doses of Hh signaling required for the induction of the anteriorly located pars distalis and high doses of FGF signaling required for the induction of the posterior pars intermedia.181
Other Potential Morphogenetic Factors
Extrinsically derived signals that possibly affect Rathke’s pouch, arising from ventral mesenchyme beneath the developing pituitary gland, include Indian Hedgehog (IHh), Wnt4, and Bmp2; whereas caudal mesenchyme is a source of a Chordin signal capable of opposing the function of Bmp2.182 Recently it has been found that blocking EGF/TGFa signaling, by the expression of a dominant negative form of EGF receptor, has a profound stage-specific effect. Expression of mutated EGF receptor in GH-producing cells of embryonic pituitary results in dwarfism and pituitary hypoplasia, with reduced numbers of lactotropes and somatotropes.183 In addition, little is known about the contribution of cytokines, despite the potent ability of cytokines such as leukemia inhibitory factor (LIF) to maintain mouse embryonic stem cells in an undifferentiated state. A potential role for LIF in pituitary development investigated in pituitary-derived cell lines is that LIF can activate synthesis of ACTH in combination with the hypothalamic peptide corticotropin-releasing hormone.184 In transgenic animals that express LIF under control of αGSU regulatory information, most cell types fail to properly differentiate, and the pituitary is characterized by the formation of ciliated cysts of Rathke’s pouch and corticotrope hyperplasia,185 suggesting that LIF may contribute to the identity establishment of dorsal-cell phenotypes.
In addition to the dorsal and ventral structures, another organizer signaling center, the notochord, is located just posterior to the developing pituitary gland. The proximity of these two structures, albeit from different origins, suggests a role for the notochord in the initial invagination of Rathke’s pouch, as indicated by tissue explant experiments.19
Pituitary and Epigenetics
Deep knowledge of physiology and multiple genetic studies performed on the pituitary gland made the pituitary an excellent system for studying details of the regulation of transcription of pituitary-specific genes (for review see [186]). Using genetic models, it is possible not only to detect the important elements in promoter regions to drive expression of the gene in the specific cell type but also to detect the hierarchy of importance of those regions and detailed molecular mechanisms of activation. Here we will provide several examples of how the pituitary gland served to elucidate the regulation of transcription of the specific genes on the molecular level.
Pit1 LOCUS—Multiple Enhancers Coordinate Expression of The Gene
Transgenic studies revealed that 14.8 kb of 5′-flanking sequence of the Pit1 gene was sufficient to direct the robust expression of a reporter in an identical spatial and temporal pattern as endogenous Pit1, whereas its minimal promoter (−327 bp to −13 bp) was insufficient to drive detectable reporter expression in transgenic mice.31 A distal enhancer located at −10.2 kb from the Pit1 transcription start site and containing multiple functional Pit1-binding sites was shown to be involved in autoregulation. This element also contains a vitamin-D receptor binding site and a retinoic acid response element that confers Pit1-dependent RA induction.31 However, the early activation of the Pit1 gene at e13.5 in the anterior pituitary requires the cooperation of Prop1 and Wnt/β-catenin signaling and is mediated by early enhancer located between at −7.8 kb upstream of the transcription start site69 in cooperation with ATBF1—giant, multiple homeodomain and zinc finger family factor—to the −5.8 kb enhancer element.187 Thus proper spatio-temporal expression of the Pit1 gene is assured through the cooperation of different enhancer elements and require Pit1 protein itself. In the dwarf mice, Pit1 expression is activated normally, but because Pit1 protein is defective, Pit1 expression declines and becomes extinguished in the perinatal period.31
Biochemical approaches such as chromatin immunoprecipitation (ChIP) on chromatin isolated from developing gland or pituitary cell lines allow detection of changes in chromatin status of regulatory regions of the Pit1 gene as the pituitary gland develops.69 At e11.5, the 2mH3K4, 3mH3K4, and AcH3K9 marks of activation are absent, but 2mH3K9 is present on Pit1 regulatory elements, consistent with an active repression of the Pit1 gene at this time. By e12.5, 2mH3K4 mark is selectively present at early enhancer. At e13.5, marks associated with active promoters188 are present on Pit1 promoter (2mH3K4, 3mH3K4, and AcH3K9) and −7.8 kb enhancer (2mH3K4). In the adult, the Pit1 gene promoter harbors the histone marks of gene activation (3mH3K4, and AcH3K9), with 2mH3K4 now present on both the late and early enhancers, showing the temporal progression of histone modifications on regulatory regions of the Pit1 gene.
GROWTH HORMONE LOCUS—LCR, BOUNDARY, AND HISTONE CODE
The human GH locus is represented by a cluster of five GH-related genes and their cell type–specific expression is regulated by a Pit-1-dependent locus control region (LCR). Expression of pituitary-specific GH gene (hGH-N) is ensured by −500 bp promoter that contains binding sites for Pit1, Sp1, and zinc finger proteins and is required for efficient expression of the gene. The LCR is necessary for proper spatio-temporal expression of hGH-N transgene.189 Dissection of the hGH-N LCR revealed the existence of two Pit1-dependent DNaseI hypersensitive sites (HSI and HSII) that are sufficient to activate hGH transgene expression.190,191 The hGH LCR and the hGH-N promoter are encompassed within a 32-kb domain of acetylated histones H3 and H4 in pituitary chromatin, with highest levels of acetylation at HSI,II.192 Although the 3mH3K4 modifications at the active hGH transgene locus in the mouse pituitary paralleled PolII distribution, they extended through the LCR and adjacent CD79b region and were separated from the hGH-N promoter by an intervening gap of unmodified chromatin.193 In mouse transgenic models, selective deletion of the pituitary-specific HSI results in the loss of both histone acetylation and methylation throughout the LCR and a marked decrease in hGH-N transcription.193,194 This recent study revealed also that HSI plays an essential role in the establishment of a complex domain of intergenic transcription 5′ to the hGH cluster.194 Insertion of an exogenous transcriptional terminator within this domain selectively blocked a subset of downstream LCR transcripts and repressed hGH-N transcription without markedly affecting histone acetylation and methylation within the locus.193,194 More recent studies showed pituitary-specific interactions between the HSI,II region and the hGH-N promoter, suggesting that noncoding transcripts in the LCR region are essential and a probable prerequisite to looping and gene activation.193
Studies of the cis-acting elements of the rat growth hormone gene have shown that the minimal information required for the specific expression in somatotropes but not lactotropes resides in the proximal 320 bp of the promoter, with the proximal 180 bp capable of targeting the reporter in vivo.195 This region contains binding sites for Pit1, thyroid hormone receptor (TR), Sp1, a zinc finger protein (Zn-15), and a recently identified zinc finger–homeodomain transcriptional repressor (Zeb1).196 Extensive analyses of transgenic mice have revealed that multiple factors are essential to mediate GH gene activation in somatotropes (Sp1), others for repression in lactotropes (Zeb1), while TR and Pit1 are required for both activation and repression. Interestingly, mutational studies have shown that replacing GH-specific Pit1 binding sites with the PRL-specific Pit1 site results in a loss of restriction from the lactotropes without affecting expression in somatotropes.44 Comparative analysis of the cocrystal structure of the Pit1 POU domain dimer bound to either GH-specific or PRL-specific sites has revealed that the spacing between the DNA contacts made by the POU-specific domain and the POU homeodomain of each monomer is increased by 2 bp on the GH-specific site. Deletion of these 2 bp in this promoter leads to a lack of the effective restriction of the reporter gene expression from lactotropes. These data suggest that the Pit1 protein conformation is critical for its specific activity. The transcriptional repressor Zeb1 is bound to the GH promoter only in the lactotropes in the sumoylation-dependent manner, and it is not recruited to the GH promoter in somatotropes. Together with the other two components of the CtBP-CoREST-LSD1 complex, Zeb1 assembles an LSD1-containing corepressor complex on the GH promoter in lactotropes. Earlier in development, LSD1 is present at the GH promoter in the MLL1-containing complex in the somatotropes, which is required for activation of the gene.196 Thus machinery controlling chromatin status (e.g., methylation of histones) is involved in developmental processes such as differentiation.
Another level of complexity of GH regulation in somatotropes comes from studies performed in mice. The murine GH genomic locus is found on mouse chromosome 11 and encompasses five genes. In contrast to the human locus, the mouse locus does not contain tandem duplications of the GH gene, and there is no known murine LCR. The proximal regulatory sequences are very well conserved between mouse and rat. However, specific activation of the endogenous murine GH gene also appears to require a boundary element imposed by an upstream SINE B2 repeat.197 This SINE B2 repeat is able to produce short, overlapping Pol II and Pol III-driven transcripts that are both necessary and sufficient for its enhancer-blocking activity in cultured cells enhancer-blocking assay. Interestingly, the Pol II-driven transcript appears in a temporally regulated manner during pituitary development, concomitantly with translocation of the GH locus from condensed heterochromatin territory (marked by H3K9me3) to a euchromatic area (marked by H3K9me2). These studies suggest that active transcription of repetitive sequences may represent one of the strategies for the establishment of functional chromatin domains to control gene expression.
Many mechanisms such as long-range interactions (hGH, Pit1), use of multiple enhancers (Pit1, GH) and histone modifications have been shown to be implicated in regulation of the expression of genes during pituitary development. DNA methylation represents another layer of complexity in regulation of the gene expression. To date, only the POMC promoter has been described as having the particular DNA methylation code that allows expression of the gene in tissues where its promoter is not methylated and prevents gene expression when the promoter DNA is methylated. The regulatory mechanism of this process, however, is not yet understood.198
Adult Pituitary Stem Cells
Existence of tissue-specific stem cells is documented in a growing number of organs by their molecular expression profile and their potential for self-renewal, multipotent differentiation and tissue regeneration. Whether or not the adult pituitary gland also contains a pool of stem cells that drive homeostatic, plastic, and regenerative cell ontogenesis remains unknown. However, adult pituitary has a remarkable ability to adapt the number of each of its cell types in response to changing physiologic demands. For example, the number of somatotropes doubles during puberty, whereas the number of lactotropes expands several-fold during pregnancy, lactation, and weaning.199 Nevertheless, it is not clear if those changes in numbers of differentiated cells are the result of maturation of uncommitted cells, transdifferentiation of other cell types, mitotic divisions of differentiated cells, or any combination of these mechanisms.
Several different cell types, including folliculostellate cells, rapidly dividing nestin-expressing cells, and side population cells of the anterior pituitary, have been recently proposed to constitute stem cells (reviewed extensively in ref. 200). However, there is no evidence that any of the proposed candidates are able to generate all differentiated cell types and participate in cell renewal in the anterior pituitary gland in vivo.
Recently, two independent in-vivo studies have attempted to identify stem-cell populations in the adult pituitary gland201,202 (see Fig. 1-2). Fauquier and colleagues have shown that a small (0.03 to 0.05%) population of cells in the adult pituitary gland expresses Sox2, a marker of several early embryonic progenitor and stem cell types. In vitro, Sox2+ cells are able to form pituispheres that can proliferate and self-renew into secondary spheres; under differentiating conditions, some proportion of these cells can differentiate into all-pituitary cell types. During progression towards the differentiated state, cells lose expression of Sox2 and start expressing Sox9 and S100b. At that stage, the cells were no longer able to form secondary pituispheres and thus were proposed to represent a transition phase from uncommitted to committed progenitor cells. The authors propose that this population of cells corresponds to marginal cells and folliculostellate cells expressing Sox9 and S100b in vivo and represents actively dividing progenitors which are, at a later stage, committed towards differentiation.
In the second report,202 authors applied a genetic approach to identifying stem cell populations in the adult pituitary. Using a GFP reporter whose expression is driven by the regulatory region of the nestin gene, authors identified a small population of cells in the adult pituitary which retained expression of the marker. Nestin-positive cells are located mostly in the perilumenal region of the gland, which had been previously proposed to be a niche for stem cells. Expression of nestin coincided with expression of Sox2 and Lhx3, suggesting a histogenetic relation to the endocrine cell types of the pituitary. During the first postnatal week, a large fraction of nestin-GFP–expressing cells, both in the glandular zone and in the perilumenal area, was positive for Ki67, a marker of dividing cells, suggesting that these cells participate in the second wave of pituitary gland growth. Supporting this observation, lineage-tracing experiments have shown that the number of GFP-positive cells in nestin-Cre/ROSA-lsl-GFP mice increases during the life of an animal from 2% in the newborn to 15% to 20% at 5 months of age. GFP expression was found in all six terminally differentiated pituitary endocrine cell types. Further supporting the notion of a separate adult stem cell lineage, the differentiation choices differed between the embryonic and adult stem cell–derived progeny. For instance, in females, the ratio of lactotropes to somatotropes was 1.8 times higher for GFP-expressing cells (i.e., cells derived from the presumptive adult stem cells) than for all of the cells of the gland (a mix of embryonic- and adult-generated cells), whereas in males it was 0.7 times lower for the GFP-expressing cells as compared with all cells of the gland. Finally, nestin-positive cells were able to self-renew multiple times in an in-vitro culture and spontaneously differentiate into all pituitary lineages (see Fig. 1-2). Recently in the new study203 GRFa2 and Prop1 have been shown to be expressed by putative stem cells in the luminal niche of the pituitary gland.
Summary
Coordinated regulation of cell type determination, differentiation, and cell proliferation is a central feature in the development of all organs. Organogenesis is controlled by sequential and spatially distributed morphogens of four major families of signaling molecules, including Wnt, Shh, TGF/BMP, and FGF. The distal targets of regulatory signaling pathways are cell-autonomous transcription regulators, including many tissue-restricted transcription factors which act to mediate crucial steps in organogenesis. The downstream effects of the signaling pathways result in the preprogramming of target cells. Pituitary development has become a model system, ideal for demonstrating the principles underlying organogenesis (see Fig. 1-2).
Continued advancement in genomic and genetic applications, particularly genome-based mutagenesis screens in mice and in nonmurine model organisms, will identify novel genes and pathways that influence the signaling and cell-determination events. Biochemical approaches, including proteomics techniques, will define the signaling-induced alterations in protein-protein interactions and phosphorylation/methylation/acetylation essential to these events. Identification of required cofactors and their downstream targets using genome-wide approaches, including complementary studies in epigenetic regulation of chromatin organization and nuclear architecture, will help determine what molecular mechanisms underlie pituitary development. The description of developmental events leading to the formation of the pituitary gland will be described increasingly in molecular terms as the molecular mechanisms are elucidated. For further information, please refer to the many comprehensive recent reviews of pituitary development in the references.3,186,204–206
References
1. Melmed, S. Mechanisms for pituitary tumorigenesis: the plastic pituitary. J Clin Invest. 2003;112(11):1603–1618.
2. Rosenfeld, MG, et al. Multistep signaling and transcriptional requirements for pituitary organogenesis in vivo. Recent Prog Horm Res. 2000;55:1–13.
3. Savage, JJ, et al. Transcriptional control during mammalian anterior pituitary development. Gene. 2003;319:1–19.
4. elAmraoui, A, Dubois, PM. Experimental evidence for the early commitment of the presumptive adenohypophysis. Neuroendocrinology. 1993;58(6):609–615.
5. Levy, NB, et al. Is there a ventral neural ridge in chick embryos? Implications for the origin of adenohypophyseal and other APUD cells. J Embryol Exp Morphol. 1980;57:71–78.
6. Takor, TT, Pearse, AG. Neuroectodermal origin of avian hypothalamo-hypophyseal complex: the role of the ventral neural ridge. J Embryol Exp Morphol. 1975;34(2):311–325.
7. Eagleson, GW, Harris, WA. Mapping of the presumptive brain regions in the neural plate of Xenopus laevis. J Neurobiol. 1990;21(3):427–440.
8. Eagleson, GW, Jenks, BG, Van Overbeeke, AP. The pituitary adrenocorticotropes originate from neural ridge tissue in Xenopus laevis. J Embryol Exp Morphol. 1986;95:1–14.
9. Herzog, W, et al. Adenohypophysis formation in the zebrafish and its dependence on sonic hedgehog. Dev Biol. 2003;254(1):36–49.
10. Liu, NA, et al. Pituitary corticotroph ontogeny and regulation in transgenic zebrafish. Mol Endocrinol. 2003;17(5):959–966.
11. Kouki, T, et al. Developmental origin of the rat adenohypophysis prior to the formation of Rathke’s pouch. Development. 2001;128(6):959–963.
12. Rathke, H. Ueber die Entstehung der glandula pititaria. Arch Anat Physio Wissened. 1838:482–485.
13. Ikeda, H, et al. The development and morphogenesis of the human pituitary gland. Anat Embryol (Berl). 1988;178(4):327–336.
14. Treier, M, Rosenfeld, MG. The hypothalamic-pituitary axis: co-development of two organs. Curr Opin Cell Biol. 1996;8(6):833–843.
15. Treier, M, et al. Multistep signaling requirements for pituitary organogenesis in vivo. Genes Dev. 1998;12(11):1691–1704.
16. Han, KS, Iwai-Liao, Y, Higashi, Y. Early organogenesis and cell contacts in the proliferating hypophysis of the developing mouse. Okajimas Folia Anat Jpn. 1998;75(2–3):97–109.
17. Ikeda, H, Yoshimoto, T. Developmental changes in proliferative activity of cells of the murine Rathke’s pouch. Cell Tissue Res. 1991;263(1):41–47.
18. Ericson, J, et al. Integrated FGF and BMP signaling controls the progression of progenitor cell differentiation and the emergence of pattern in the embryonic anterior pituitary. Development. 1998;125(6):1005–1015.
19. Gleiberman, AS, Fedtsova, NG, Rosenfeld, MG. Tissue interactions in the induction of anterior pituitary: role of the ventral diencephalon, mesenchyme, and notochord. Dev Biol. 1999;213(2):340–353.
20. Dasen, JS, Rosenfeld, MG. Signaling and transcriptional mechanisms in pituitary development. Annu Rev Neurosci. 2001;24:327–355.
21. Kendall, SK, et al. Enhancer-mediated high level expression of mouse pituitary glycoprotein hormone alpha-subunit transgene in thyrotropes, gonadotropes, and developing pituitary gland. Mol Endocrinol. 1994;8(10):1420–1433.
22. Bodner, M, et al. The pituitary-specific transcription factor GHF-1 is a homeobox-containing protein. Cell. 1988;55(3):505–518.
23. Ingraham, HA, et al. A tissue-specific transcription factor containing a homeodomain specifies a pituitary phenotype. Cell. 1988;55(3):519–529.
24. Mangalam, HJ, et al. A pituitary POU domain protein, Pit-1, activates both growth hormone and prolactin promoters transcriptionally. Genes Dev. 1989;3(7):946–958.
25. Lin, C, et al. Pit-1-dependent expression of the receptor for growth hormone releasing factor mediates pituitary cell growth. Nature. 1992;360(6406):765–768.
26. Crenshaw, EB, 3rd., et al. Cell-specific expression of the prolactin gene in transgenic mice is controlled by synergistic interactions between promoter and enhancer elements. Genes Dev. 1989;3(7):959–972.
27. Chen, RP, et al. Autoregulation of pit-1 gene expression mediated by two cis-active promoter elements. Nature. 1990;346(6284):583–586.
28. Snell, GD. Dwarf, a new Mendelian recessive character of the house mouse. Proc Natl Acad Sci U S A. 1929;15(9):733–734.
29. Li, S, et al. Dwarf locus mutants lacking three pituitary cell types result from mutations in the POU-domain gene pit-1. Nature. 1990;347(6293):528–533.
30. Camper, SA, et al. The Pit-1 transcription factor gene is a candidate for the murine Snell dwarf mutation. Genomics. 1990;8(3):586–590.
31. Rhodes, SJ, et al. A tissue-specific enhancer confers Pit-1-dependent morphogen inducibility and autoregulation on the pit-1 gene. Genes Dev. 1993;7(6):913–932.
32. Bartke, A, et al. Prolonged longevity of hypopituitary dwarf mice. Exp Gerontol. 2001;36(1):21–28.
33. Flurkey, K, et al. Lifespan extension and delayed immune and collagen aging in mutant mice with defects in growth hormone production. Proc Natl Acad Sci U S A. 2001;98(12):6736–6741.
34. Bishop, NA, Guarente, L. Genetic links between diet and lifespan: shared mechanisms from yeast to humans. Nat Rev Genet. 2007;8(11):835–844.
35. Kenyon, C. The plasticity of aging: insights from long-lived mutants. Cell. 2005;120(4):449–460.
36. Sinclair, DA. Toward a unified theory of caloric restriction and longevity regulation. Mech Ageing Dev. 2005;126(9):987–1002.
37. Tatsumi, K, et al. Cretinism with combined hormone deficiency caused by a mutation in the PIT1 gene. Nat Genet. 1992;1(1):56–58.
38. Parks, JS, et al. Heritable disorders of pituitary development. J Clin Endocrinol Metab. 1999;84(12):4362–4370.
39. Pfaffle, RW, et al. Combined pituitary hormone deficiency: role of Pit-1 and Prop-1. Acta Paediatr Suppl. 1999;88(433):33–41.
40. Wu, W, Anderson, B, Rosenfeld, MG. PIT1 transcription factor and diseases. In: Wiley Encyclopedia of Molecular Medicine. Chinchester, UK: John Wiley & Sons; 2002:2501.
41. Holloway, JM, et al. Pit-1 binding to specific DNA sites as a monomer or dimer determines gene-specific use of a tyrosine-dependent synergy domain. Genes Dev. 1995;9(16):1992–2006.
42. Jacobson, EM, et al. Structure of Pit-1 POU domain bound to DNA as a dimer: unexpected arrangement and flexibility. Genes Dev. 1997;11(2):198–212.
43. Jacobson, EM, et al. Crystallization and preliminary X-ray analysis of Pit-1 POU domain complexed to a 28 base pair DNA element. Proteins. 1996;24(2):263–265.
44. Scully, KM, et al. Allosteric effects of Pit-1 DNA sites on long-term repression in cell type specification. Science. 2000;290(5494):1127–1131.
45. Andersen, B, Rosenfeld, MG. POU domain factors in the neuroendocrine system: lessons from developmental biology provide insights into human disease. Endocr Rev. 2001;22(1):2–35.
46. Cohen, LE, et al. A “hot spot” in the Pit-1 gene responsible for combined pituitary hormone deficiency: clinical and molecular correlates. J Clin Endocrinol Metab. 1995;80(2):679–684.
47. Xu, L, et al. Signal-specific co-activator domain requirements for Pit-1 activation. Nature. 1998;395(6699):301–306.
48. Day, RN, et al. Both Pit-1 and the estrogen receptor are required for estrogen responsiveness of the rat prolactin gene. Mol Endocrinol. 1990;4(12):1964–1971.
49. Simmons, DM, et al. Pituitary cell phenotypes involve cell-specific Pit-1 mRNA translation and synergistic interactions with other classes of transcription factors. Genes Dev. 1990;4(5):695–711.
50. Bradford, AP, et al. Functional interaction of c-Ets-1 and GHF-1/Pit-1 mediates Ras activation of pituitary-specific gene expression: mapping of the essential c-Ets-1 domain. Mol Cell Biol. 1995;15(5):2849–2857.
51. Dasen, JS, et al. Reciprocal interactions of Pit1 and GATA2 mediate signaling gradient-induced determination of pituitary cell types. Cell. 1999;97(5):587–598.
52. Sornson, MW, et al. Pituitary lineage determination by the Prophet of Pit-1 homeodomain factor defective in Ames dwarfism. Nature. 1996;384(6607):327–333.
53. Zhu, X, et al. Sustained Notch signaling in progenitors is required for sequential emergence of distinct cell lineages during organogenesis. Genes Dev. 2006;20(19):2739–2753.
54. Andersen, B, et al. The Ames dwarf gene is required for Pit-1 gene activation. Dev Biol. 1995;172(2):495–503.
55. Gage, PJ, et al. Anterior pituitary cells defective in the cell-autonomous factor, df, undergo cell lineage specification but not expansion. Development. 1996;122(1):151–160.
56. Buckwalter, MS, Katz, RW, Camper, SA. Localization of the panhypopituitary dwarf mutation (df) on mouse chromosome 11 in an intersubspecific backcross. Genomics. 1991;10(3):515–526.
57. Gage, PJ, et al. Ames dwarf mice exhibit somatotrope commitment but lack growth hormone-releasing factor response. Endocrinology. 1995;136(3):1161–1167.
58. Brown-Borg, HM, et al. Dwarf mice and the ageing process. Nature. 1996;384(6604):33.
59. Wu, W, et al. Mutations in PROP1 cause familial combined pituitary hormone deficiency. Nat Genet. 1998;18(2):147–149.
60. Deladoey, J, et al. “Hot spot” in the PROP1 gene responsible for combined pituitary hormone deficiency. J Clin Endocrinol Metab. 1999;84(5):1645–1650.
61. Mody, S, Brown, MR, Parks, JS. The spectrum of hypopituitarism caused by PROP1 mutations. Best Pract Res Clin Endocrinol Metab. 2002;16(3):421–431.
62. Wu, W, Rosenfeld, MG. Prop1 gene. In: Wiley Encyclopedia of Molecular Medicine. Chichester, UK: John Wiley & Sons; 2002:2597.
63. Ward, RD, et al. Role of PROP1 in pituitary gland growth. Mol Endocrinol. 2005;19(3):698–710.
64. Douglas, KR, et al. Identification of members of the Wnt signaling pathway in the embryonic pituitary gland. Mamm Genome. 2001;12(11):843–851.
65. Raetzman, LT, et al. Developmental regulation of Notch signaling genes in the embryonic pituitary: Prop1 deficiency affects Notch2 expression. Dev Biol. 2004;265(2):329–340.
66. Cushman, LJ, et al. Persistent Prop1 expression delays gonadotrope differentiation and enhances pituitary tumor susceptibility. Hum Mol Genet. 2001;10(11):1141–1153.
67. Vesper, AH, Raetzman, LT, Camper, SA. Role of prophet of Pit1 (PROP1) in gonadotrope differentiation and puberty. Endocrinology. 2006;147(4):1654–1663.
68. Showalter, AD, et al. Differential conservation of transcriptional domains of mammalian Prophet of Pit-1 proteins revealed by structural studies of the bovine gene and comparative functional analysis of the protein. Gene. 2002;291(1–2):211–221.
69. Olson, LE, et al. Homeodomain-mediated beta-catenin-dependent switching events dictate cell-lineage determination. Cell. 2006;125(3):593–605.
70. Agarwal, G, et al. Adrenocorticotropin deficiency in combined pituitary hormone deficiency patients homozygous for a novel PROP1 deletion. J Clin Endocrinol Metab. 2000;85(12):4556–4561.
71. Lamesch, C, et al. Adrenocorticotrope deficiency with clinical evidence for late onset in combined pituitary hormone deficiency caused by a homozygous 301–302delAG mutation of the PROP1 gene. Pituitary. 2002;5(3):163–168.
72. Pernasetti, F, et al. Impaired adrenocorticotropin-adrenal axis in combined pituitary hormone deficiency caused by a two-base pair deletion (301–302delAG) in the prophet of Pit-1 gene. J Clin Endocrinol Metab. 2000;85(1):390–397.
73. Hermesz, E, Mackem, S, Mahon, KA. Rpx: a novel anterior-restricted homeobox gene progressively activated in the prechordal plate, anterior neural plate and Rathke’s pouch of the mouse embryo. Development. 1996;122(1):41–52.
74. Thomas, P, Beddington, R. Anterior primitive endoderm may be responsible for patterning the anterior neural plate in the mouse embryo. Curr Biol. 1996;6(11):1487–1496.
75. Dattani, MT, et al. Mutations in the homeobox gene HESX1/Hesx1 associated with septo-optic dysplasia in human and mouse. Nat Genet. 1998;19(2):125–133.
76. Dasen, JS, et al. Temporal regulation of a paired-like homeodomain repressor/TLE corepressor complex and a related activator is required for pituitary organogenesis. Genes Dev. 2001;15(23):3193–3207.
77. Thomas, PQ, et al. Heterozygous HESX1 mutations associated with isolated congenital pituitary hypoplasia and septo-optic dysplasia. Hum Mol Genet. 2001;10(1):39–45.
78. McNay, DE, et al. HESX1 mutations are an uncommon cause of septooptic dysplasia and hypopituitarism. J Clin Endocrinol Metab. 2007;92(2):691–697.
79. Rainbow, LA, et al. Mutation analysis of POUF-1, PROP-1 and HESX-1 show low frequency of mutations in children with sporadic forms of combined pituitary hormone deficiency and septo-optic dysplasia. Clin Endocrinol (Oxf). 2005;62(2):163–168.
80. Brickman, JM, et al. Molecular effects of novel mutations in Hesx1/HESX1 associated with human pituitary disorders. Development. 2001;128(24):5189–5199.
81. Quirk, J, Brown, P. Hesx1 homeodomain protein represses transcription as a monomer and antagonises transactivation of specific sites as a homodimer. J Mol Endocrinol. 2002;28(3):193–205.
82. Jimenez, G, Paroush, Z, Ish-Horowicz, D. Groucho acts as a corepressor for a subset of negative regulators, including Hairy and Engrailed. Genes Dev. 1997;11(22):3072–3082.
83. Tolkunova, EN, et al. Two distinct types of repression domain in engrailed: one interacts with the groucho corepressor and is preferentially active on integrated target genes. Mol Cell Biol. 1998;18(5):2804–2814.
84. Eberhard, D, et al. Transcriptional repression by Pax5 (BSAP) through interaction with corepressors of the Groucho family. EMBO J. 2000;19(10):2292–2303.
85. Carvalho, LR, et al. A homozygous mutation in HESX1 is associated with evolving hypopituitarism due to impaired repressor-corepressor interaction. J Clin Invest. 2003;112(8):1192–1201.
86. Zhadanov, AB, et al. Expression pattern of the murine LIM class homeobox gene Lhx3 in subsets of neural and neuroendocrine tissues. Dev Dyn. 1995;202(4):354–364.
87. Li, H, et al. Gsh-4 encodes a LIM-type homeodomain, is expressed in the developing central nervous system and is required for early postnatal survival. EMBO J. 1994;13(12):2876–2885.
88. Sheng, HZ, et al. Specification of pituitary cell lineages by the LIM homeobox gene Lhx3. Science. 1996;272(5264):1004–1007.
89. Raetzman, LT, Ward, R, Camper, SA. Lhx4 and Prop1 are required for cell survival and expansion of the pituitary primordia. Development. 2002;129(18):4229–4239.
90. Sheng, HZ, et al. Multistep control of pituitary organogenesis. Science. 1997;278(5344):1809–1812.
91. Schmitt, S, et al. Genomic structure, chromosomal localization, and expression pattern of the human LIM-homeobox3 (LHX 3) gene. Biochem Biophys Res Commun. 2000;274(1):49–56.
92. Sloop, KW, Dwyer, CJ, Rhodes, SJ. An isoform-specific inhibitory domain regulates the LHX3 LIM homeodomain factor holoprotein and the production of a functional alternate translation form. J Biol Chem. 2001;276(39):36311–36319.
93. Netchine, I, et al. Mutations in LHX3 result in a new syndrome revealed by combined pituitary hormone deficiency. Nat Genet. 2000;25(2):182–186.
94. Pfaeffle, RW, et al. Four novel mutations of the LHX3 gene cause combined pituitary hormone deficiencies with or without limited neck rotation. J Clin Endocrinol Metab. 2007;92(5):1909–1919.
95. Rajab, A, et al. Novel mutations in LHX3 are associated with hypopituitarism and sensorineural hearing loss. Hum Mol Genet. 2008;17(14):2150–2159.
96. Machinis, K, et al. Syndromic short stature in patients with a germline mutation in the LIM homeobox LHX4. Am J Hum Genet. 2001;69(5):961–968.
97. Pfaeffle, RW, et al. Three novel missense mutations within the LHX4 gene are associated with variable pituitary hormone deficiencies. J Clin Endocrinol Metab. 2008;93(3):1062–1071.
98. Yi, CH, et al. Identification, mapping, and phylogenomic analysis of four new human members of the T-box gene family: EOMES, TBX6, TBX18, and TBX19. Genomics. 1999;55(1):10–20.
99. Lamolet, B, et al. A pituitary cell-restricted T box factor, Tpit, activates POMC transcription in cooperation with Pitx homeoproteins. Cell. 2001;104(6):849–859.
100. Liu, J, et al. Tbx19, a tissue-selective regulator of POMC gene expression. Proc Natl Acad Sci U S A. 2001;98(15):8674–8679.
101. Pulichino, AM, et al. Human and mouse TPIT gene mutations cause early onset pituitary ACTH deficiency. Genes Dev. 2003;17(6):711–716.
102. Pulichino, AM, et al. Tpit determines alternate fates during pituitary cell differentiation. Genes Dev. 2003;17(6):738–747.
103. Vallette-Kasic, S, et al. Congenital isolated adrenocorticotropin deficiency: an underestimated cause of neonatal death, explained by TPIT gene mutations. J Clin Endocrinol Metab. 2005;90(3):1323–1331.
104. Vallette-Kasic, S, et al. The TPIT gene mutation M86R associated with isolated adrenocorticotropin deficiency interferes with protein: protein interactions. J Clin Endocrinol Metab. 2007;92(10):3991–3999.
105. Maira, M, et al. The T-box factor Tpit recruits SRC/p160 co-activators and mediates hormone action. J Biol Chem. 2003;278(47):46523–46532.
106. Maira, M, et al. Dimer-specific potentiation of NGFI-B (Nur77) transcriptional activity by the protein kinase A pathway and AF-1-dependent coactivator recruitment. Mol Cell Biol. 2003;23(3):763–776.
107. Marcil, A, et al. Pitx1 and Pitx2 are required for development of hindlimb buds. Development. 2003;130(1):45–55.
108. Szeto, DP, et al. Role of the Bicoid-related homeodomain factor Pitx1 in specifying hindlimb morphogenesis and pituitary development. Genes Dev. 1999;13(4):484–494.
109. Lin, CR, et al. Pitx2 regulates lung asymmetry, cardiac positioning and pituitary and tooth morphogenesis. Nature. 1999;401(6750):279–282.
110. Semina, EV, et al. Cloning and characterization of a novel bicoid-related homeobox transcription factor gene, RIEG, involved in Rieger syndrome. Nat Genet. 1996;14(4):392–399.
111. Suh, H, et al. Pitx2 is required at multiple stages of pituitary organogenesis: pituitary primordium formation and cell specification. Development. 2002;129(2):329–337.
112. Charles, MA, et al. PITX genes are required for cell survival and Lhx3 activation. Mol Endocrinol. 2005;19(7):1893–1903.
113. Kioussi, C, et al. Identification of a Wnt/Dvl/beta-Catenin –> Pitx2 pathway mediating cell-type-specific proliferation during development. Cell. 2002;111(5):673–685.
114. Lanctot, C, et al. Hindlimb patterning and mandible development require the Ptx1 gene. Development. 1999;126(9):1805–1810.
115. Gage, PJ, Suh, H, Camper, SA. Dosage requirement of Pitx2 for development of multiple organs. Development. 1999;126(20):4643–4651.
116. Kitamura, K, et al. Mouse Pitx2 deficiency leads to anomalies of the ventral body wall, heart, extra- and periocular mesoderm and right pulmonary isomerism. Development. 1999;126(24):5749–5758.
117. Lu, MF, et al. Function of Rieger syndrome gene in left-right asymmetry and craniofacial development. Nature. 1999;401(6750):276–278.
118. Holmberg, J, Liu, CY, Hjalt, TA. PITX2 gain-of-function in Rieger syndrome eye model. Am J Pathol. 2004;165(5):1633–1641.
119. Holmberg, J, et al. PITX2 gain-of-function induced defects in mouse forelimb development. BMC Dev Biol. 2008;8:25.
120. Lines, MA, et al. Characterization and prevalence of PITX2 microdeletions and mutations in Axenfeld-Rieger malformations. Invest Ophthalmol Vis Sci. 2004;45(3):828–833.
121. Priston, M, et al. Functional analyses of two newly identified PITX2 mutants reveal a novel molecular mechanism for Axenfeld-Rieger syndrome. Hum Mol Genet. 2001;10(16):1631–1638.
122. Szeto, DP, et al. P-OTX: a PIT-1-interacting homeodomain factor expressed during anterior pituitary gland development. Proc Natl Acad Sci U S A. 1996;93(15):7706–7710.
123. Lamonerie, T, et al. Ptx1, a bicoid-related homeo box transcription factor involved in transcription of the pro-opiomelanocortin gene. Genes Dev. 1996;10(10):1284–1295.
124. Poulin, G, et al. Specific protein-protein interaction between basic helix-loop-helix transcription factors and homeoproteins of the Pitx family. Mol Cell Biol. 2000;20(13):4826–4837.
125. Tremblay, JJ, et al. Ptx1 regulates SF-1 activity by an interaction that mimics the role of the ligand-binding domain. EMBO J. 1999;18(12):3431–3441.
126. Tremblay, JJ, Drouin, J. Egr-1 is a downstream effector of GnRH and synergizes by direct interaction with Ptx1 and SF-1 to enhance luteinizing hormone beta gene transcription. Mol Cell Biol. 1999;19(4):2567–2576.
127. Tremblay, JJ, Lanctot, C, Drouin, J. The pan-pituitary activator of transcription, Ptx1 (pituitary homeobox 1), acts in synergy with SF-1 and Pit1 and is an upstream regulator of the Lim-homeodomain gene Lim3/Lhx3. Mol Endocrinol. 1998;12(3):428–441.
128. Kioussi, C, et al. Pax6 is essential for establishing ventral-dorsal cell boundaries in pituitary gland development. Proc Natl Acad Sci U S A. 1999;96(25):14378–14382.
129. Rosenfeld, MG, et al. Transcriptional control of cell phenotypes in the neuroendocrine system. Recent Prog Horm Res. 1996;51:217–238.
130. Zhu, X, et al. Signaling and epigenetic regulation of pituitary development. Curr Opin Cell Biol. 2007;19(6):605–611.
131. Zhu, X, et al. Genetic control of pituitary development and hypopituitarism. Curr Opin Genet Dev. 2005;15(3):332–340.
132. Li, X, et al. Tissue-specific regulation of retinal and pituitary precursor cell proliferation. Science. 2002;297(5584):1180–1183.
133. Li, X, et al. Eya protein phosphatase activity regulates Six1-Dach-Eya transcriptional effects in mammalian organogenesis. Nature. 2003;426(6964):247–254.
134. Lagutin, OV, et al. Six3 repression of Wnt signaling in the anterior neuroectoderm is essential for vertebrate forebrain development. Genes Dev. 2003;17(3):368–379.
135. Zhao, L, et al. Steroidogenic factor 1 (SF1) is essential for pituitary gonadotrope function. Development. 2001;128(2):147–154.
136. Park, HL, et al. Mouse Gli1 mutants are viable but have defects in SHH signaling in combination with a Gli2 mutation. Development. 2000;127(8):1593–1605.
137. Kondoh, H, et al. Zebrafish mutations in Gli-mediated hedgehog signaling lead to lens transdifferentiation from the adenohypophysis anlage. Mech Dev. 2000;96(2):165–174.
138. Acampora, D, et al. Transient dwarfism and hypogonadism in mice lacking Otx1 reveal prepubescent stage-specific control of pituitary levels of GH, FSH and LH. Development. 1998;125(7):1229–1239.
139. Acampora, D, Gulisano, M, Simeone, A. Genetic and molecular roles of Otx homeodomain proteins in head development. Gene. 2000;246(1–2):23–35.
140. Dateki, S, et al. OTX2 Mutation in a Patient with Anophthalmia, Short Stature, and Partial GH Deficiency: Functional Studies Using the IRBP, HESX1, and POU1F1 Promoters. J Clin Endocrinol Metab. 2008;93(10):3697–3702.
141. Edlund, T, Jessell, TM. Progression from extrinsic to intrinsic signaling in cell fate specification: a view from the nervous system. Cell. 1999;96(2):211–224.
142. Ashe, HL, Briscoe, J. The interpretation of morphogen gradients. Development. 2006;133(3):385–394.
143. Tabata, T, Takei, Y. Morphogens, their identification and regulation. Development. 2004;131(4):703–712.
144. Treier, M, et al. Hedgehog signaling is required for pituitary gland development. Development. 2001;128(3):377–386.
145. Ingham, PW, Placzek, M. Orchestrating ontogenesis: variations on a theme by sonic hedgehog. Nat Rev Genet. 2006;7(11):841–850.
146. Marigo, V, et al. Cloning, expression, and chromosomal location of SHH and IHH: two human homologues of the Drosophila segment polarity gene hedgehog. Genomics. 1995;28(1):44–51.
147. Chiang, C, et al. Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature. 1996;383(6599):407–413.
148. Ekker, SC, et al. Distinct expression and shared activities of members of the hedgehog gene family of Xenopus laevis. Development. 1995;121(8):2337–2347.
149. Hui, CC, et al. Expression of three mouse homologs of the Drosophila segment polarity gene cubitus interruptus, Gli, Gli-2, and Gli-3, in ectoderm- and mesoderm-derived tissues suggests multiple roles during postimplantation development. Dev Biol. 1994;162(2):402–413.
150. Sbrogna, JL, Barresi, MJ, Karlstrom, RO. Multiple roles for Hedgehog signaling in zebrafish pituitary development. Dev Biol. 2003;254(1):19–35.
151. Laufer, E, et al. Sonic hedgehog and Fgf-4 act through a signaling cascade and feedback loop to integrate growth and patterning of the developing limb bud. Cell. 1994;79(6):993–1003.
152. Ornitz, DM, Itoh, N. Fibroblast growth factors. Genome Biol. 2(3), 2001. [REVIEWS3005].
153. Thisse, B, Thisse, C. Functions and regulations of fibroblast growth factor signaling during embryonic development. Dev Biol. 2005;287(2):390–402.
154. Allen, BL, Rapraeger, AC. Spatial and temporal expression of heparan sulfate in mouse development regulates FGF and FGF receptor assembly. J Cell Biol. 2003;163(3):637–648.
155. Lin, X. Functions of heparan sulfate proteoglycans in cell signaling during development. Development. 2004;131(24):6009–6021.
156. De Moerlooze, L, et al. An important role for the IIIb isoform of fibroblast growth factor receptor 2 (FGFR2) in mesenchymal-epithelial signalling during mouse organogenesis. Development. 2000;127(3):483–492.
157. Min, H, et al. Fgf-10 is required for both limb and lung development and exhibits striking functional similarity to Drosophila branchless. Genes Dev. 1998;12(20):3156–3161.
158. Chi, CL, et al. The isthmic organizer signal FGF8 is required for cell survival in the prospective midbrain and cerebellum. Development. 2003;130(12):2633–2644.
159. Meyers, EN, Lewandoski, M, Martin, GR. An Fgf8 mutant allelic series generated by Cre- and Flp-mediated recombination. Nat Genet. 1998;18(2):136–141.
160. Takuma, N, et al. Formation of Rathke’s pouch requires dual induction from the diencephalon. Development. 1998;125(23):4835–4840.
161. Norlin, S, Nordstrom, U, Edlund, T. Fibroblast growth factor signaling is required for the proliferation and patterning of progenitor cells in the developing anterior pituitary. Mech Dev. 2000;96(2):175–182.
162. Zhao, GQ. Consequences of knocking out BMP signaling in the mouse. Genesis. 2003;35(1):43–56.
163. Davis, SW, Camper, SA. Noggin regulates Bmp4 activity during pituitary induction. Dev Biol. 2007;305(1):145–160.
164. Artavanis-Tsakonas, S, Rand, MD, Lake, RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284(5415):770–776.
165. Louvi, A, Artavanis-Tsakonas, S. Notch signalling in vertebrate neural development. Nat Rev Neurosci. 2006;7(2):93–102.
166. Raetzman, LT, Cai, JX, Camper, SA. Hes1 is required for pituitary growth and melanotrope specification. Dev Biol. 2007;304(2):455–466.
167. Raetzman, LT, et al. Persistent expression of Notch2 delays gonadotrope differentiation. Mol Endocrinol. 2006;20(11):2898–2908.
168. Kita, A, et al. Hes1 and Hes5 control the progenitor pool, intermediate lobe specification, and posterior lobe formation in the pituitary development. Mol Endocrinol. 2007;21(6):1458–1466.
169. Miller, JR. The Wnts. Genome Biol. 3(1), 2002. [REVIEWS3001].
170. Clevers, H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127(3):469–480.
171. Huang, H, He, X. Wnt/beta-catenin signaling: new (and old) players and new insights. Curr Opin Cell Biol. 2008;20(2):119–125.
172. Potok, MA, et al. WNT signaling affects gene expression in the ventral diencephalon and pituitary gland growth. Dev Dyn. 2008;237(4):1006–1020.
173. Cha, KB, et al. WNT5A signaling affects pituitary gland shape. Mech Dev. 2004;121(2):183–194.
174. Briata, P, et al. The Wnt/beta-catenin–>Pitx2 pathway controls the turnover of Pitx2 and other unstable mRNAs. Mol Cell. 2003;12(5):1201–1211.
175. Ai, D, et al. Nuclear factor 1 and T-cell factor/LEF recognition elements regulate Pitx2 transcription in pituitary development. Mol Cell Biol. 2007;27(16):5765–5775.
176. Burns, CJ, et al. Investigation of Frizzled-5 during embryonic neural development in mouse. Dev Dyn. 2008;237(6):1614–1626.
177. Brinkmeier, ML, et al. TCF and Groucho-related genes influence pituitary growth and development. Mol Endocrinol. 2003;17(11):2152–2161.
178. Brinkmeier, ML, et al. TCF4 deficiency expands ventral diencephalon signaling and increases induction of pituitary progenitors. Dev Biol. 2007;311(2):396–407.
179. DiMattia, GE, et al. The Pit-1 gene is regulated by distinct early and late pituitary-specific enhancers. Dev Biol. 1997;182(1):180–190.
180. Ohkubo, Y, Chiang, C, Rubenstein, JL. Coordinate regulation and synergistic actions of BMP4, SHH and FGF8 in the rostral prosencephalon regulate morphogenesis of the telencephalic and optic vesicles. Neuroscience. 2002;111(1):1–17.
181. Guner, B, et al. Graded Hh and Fgf signaling independently regulate pituitary cell fates and help establish the PD and PI of the zebrafish adenohypophysis. Endocrinology. 2008;149(9):4435–4451.
182. Anderson, RM, et al. Chordin and noggin promote organizing centers of forebrain development in the mouse. Development. 2002;129(21):4975–4987.
183. Roh, M, et al. Stage-sensitive blockade of pituitary somatomammotrope development by targeted expression of a dominant negative epidermal growth factor receptor in transgenic mice. Mol Endocrinol. 2001;15(4):600–613.
184. Bousquet, C, Ray, DW, Melmed, S. A common pro-opiomelanocortin-binding element mediates leukemia inhibitory factor and corticotropin-releasing hormone transcriptional synergy. J Biol Chem. 1997;272(16):10551–10557.
185. Yano, H, et al. Pituitary-directed leukemia inhibitory factor transgene causes Cushing’s syndrome: neuro-immune-endocrine modulation of pituitary development. Mol Endocrinol. 1998;12(11):1708–1720.
186. Zhu, X, Gleiberman, AS, Rosenfeld, MG. Molecular physiology of pituitary development: signaling and transcriptional networks. Physiol Rev. 2007;87(3):933–963.
187. Qi, Y, et al. Atbf1 is required for the Pit1 gene early activation. Proc Natl Acad Sci U S A. 2008;105(7):2481–2486.
188. Kouzarides, T. Chromatin modifications and their function. Cell. 2007;128(4):693–705.
189. Jones, BK, et al. The human growth hormone gene is regulated by a multicomponent locus control region. Mol Cell Biol. 1995;15(12):7010–7021.
190. Bennani-Baiti, IM, et al. DNase I-hypersensitive sites I and II of the human growth hormone locus control region are a major developmental activator of somatotrope gene expression. Proc Natl Acad Sci U S A. 1998;95(18):10655–10660.
191. Shewchuk, BM, et al. Pit-1 binding sites at the somatotrope-specific DNase I hypersensitive sites I, II of the human growth hormone locus control region are essential for in vivo hGH-N gene activation. J Biol Chem. 1999;274(50):35725–35733.
192. Ho, Y, et al. A defined locus control region determinant links chromatin domain acetylation with long-range gene activation. Mol Cell. 2002;9(2):291–302.
193. Ho, Y, et al. The juxtaposition of a promoter with a locus control region transcriptional domain activates gene expression. EMBO Rep. 2008;9(9):891–898.
194. Ho, Y, et al. Locus control region transcription plays an active role in long-range gene activation. Mol Cell. 2006;23(3):365–375.
195. Lira, SA, et al. Identification of rat growth hormone genomic sequences targeting pituitary expression in transgenic mice. Proc Natl Acad Sci U S A. 1988;85(13):4755–4759.
196. Wang, J, et al. Opposing LSD1 complexes function in developmental gene activation and repression programmes. Nature. 2007;446(7138):882–887.
197. Lunyak, VV, et al. Developmentally regulated activation of a SINE B2 repeat as a domain boundary in organogenesis. Science. 2007;317(5835):248–251.
198. Newell-Price, J. Proopiomelanocortin gene expression and DNA methylation: implications for Cushing’s syndrome and beyond. J Endocrinol. 2003;177(3):365–372.
199. Levy, A. Physiological implications of pituitary trophic activity. J Endocrinol. 2002;174(2):147–155.
200. Vankelecom, H. Stem cells in the postnatal pituitary? Neuroendocrinology. 2007;85(2):110–130.
201. Fauquier, T, et al. SOX2-expressing progenitor cells generate all of the major cell types in the adult mouse pituitary gland. Proc Natl Acad Sci U S A. 2008;105(8):2907–2912.
202. Gleiberman, AS, et al. Genetic approaches identify adult pituitary stem cells. Proc Natl Acad Sci U S A. 2008;105(17):6332–6337.
203. Garcia-Lavandeira, M, et al. A GRFa2/Prop1/stem (GPS) cell niche in the pituitary. PLOS ONE. 2009;4(3):e4815. [doi:10.1371/journal.pone.0004815].
204. Kerr, J, Wood, W, Ridgway, EC. Basic science and clinical research advances in the pituitary transcription factors: Pit-1 and Prop-1. Curr Opin Endocrinol Diabetes Obes. 2008;15(4):359–363.
205. Quentien, MH, et al. Pituitary transcription factors: from congenital deficiencies to gene therapy. J Neuroendocrinol. 2006;18(9):633–642.
206. Scully, KM, Rosenfeld, MG. Pituitary development: regulatory codes in mammalian organogenesis. Science. 2002;295(5563):2231–2235.