Dementias
INTRODUCTION
Diseases causing dementia are among the commonest neurologic conditions encountered in clinical practice (Table 31.1). The importance of establishing a neuropathologic diagnosis dementia can be linked to the following:
Table 31.1
Prevalence of dementia at different ages
| Age (years) | Prevalence (%) |
| <75 | 4 |
| 80 | 12 |
| 85 | 27 |
| 90 | 40 |
 Clinical diagnosis of the type of dementia may be unreliable, particularly outside specialist units.
Clinical diagnosis of the type of dementia may be unreliable, particularly outside specialist units.
 Some diseases have a genetic basis and a precise diagnosis facilitates counseling.
Some diseases have a genetic basis and a precise diagnosis facilitates counseling.
 New causes of dementia are still being discovered by applying new diagnostic methods.
New causes of dementia are still being discovered by applying new diagnostic methods.
 DEMENTIA
DEMENTIA Dementia can be defined as an impairment of previously attained occupational or social functioning due to an acquired and persistent impairment of memory associated with an impairment of intellectual function in one or more of the following domains: language, visuospatial skills, emotion, personality, or cognition, in the presence of normal consciousness.
Dementia can be defined as an impairment of previously attained occupational or social functioning due to an acquired and persistent impairment of memory associated with an impairment of intellectual function in one or more of the following domains: language, visuospatial skills, emotion, personality, or cognition, in the presence of normal consciousness.



 Dementia is predominantly caused by degenerative processes that evolve over many years (Table 31.2). Different diseases preferentially affect different brain regions and so distinctive clinical patterns of dementia can be recognized (Fig. 31.1). With progression of the disease, more of the cortical areas tend to be affected and a global deterioration in intellectual function ensues.
Dementia is predominantly caused by degenerative processes that evolve over many years (Table 31.2). Different diseases preferentially affect different brain regions and so distinctive clinical patterns of dementia can be recognized (Fig. 31.1). With progression of the disease, more of the cortical areas tend to be affected and a global deterioration in intellectual function ensues.
Table 31.2
Neurodegenerative diseases
Common
Less common
Rare
Cerebrovascular disease
Hydrocephalus
Toxic, metabolic, and nutritional disorders
• Chronic hepatic encephalopathy
• Vitamin deficiency (thiamine, B12, niacin)
Immune-mediated syndromes
Mitochondrial encephalopathy
Demyelinating and dysmyelinating diseases
Head injury
Prion disease
Infective disorders
Neoplasia
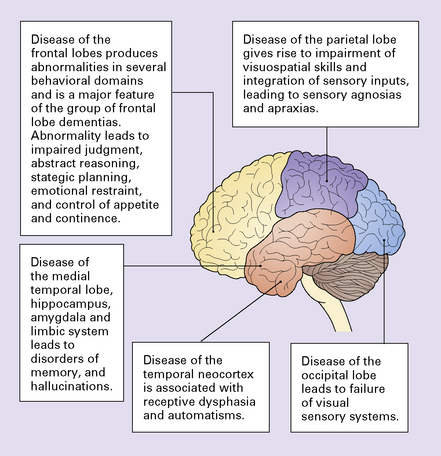
Certain clinical patterns of dementia are suggestive of particular disorders
 Frontotemporal dementias (FTD): behavioral disturbances of frontal lobe dysfunction, language dysfunction with later development of memory disturbances due to temporal lobe disease; parietal lobe function being typically retained. The group of frontotemporal lobar degenerations (FTLD) is the main cause of this pattern.
Frontotemporal dementias (FTD): behavioral disturbances of frontal lobe dysfunction, language dysfunction with later development of memory disturbances due to temporal lobe disease; parietal lobe function being typically retained. The group of frontotemporal lobar degenerations (FTLD) is the main cause of this pattern.


 The degenerative processes that cause cortical pathology may also affect subcortical structures and lead to other neurologic dysfunction, the most common of which is the development of parkinsonism. Identification of associated neurologic abnormalities may therefore help in establishing an accurate diagnosis.
The degenerative processes that cause cortical pathology may also affect subcortical structures and lead to other neurologic dysfunction, the most common of which is the development of parkinsonism. Identification of associated neurologic abnormalities may therefore help in establishing an accurate diagnosis.


TEMPOROPARIETAL AND FRONTOTEMPOROPARIETAL DEMENTIAS
AD is the commonest cause of dementia and increases in incidence with age. It accounts for 50–75% of all cases of dementia, the precise figure depending on the criteria used to establish the diagnosis. There are five main groups, with different molecular genetic associations (see p. 628):
 Sporadic late-onset AD (commonest).
Sporadic late-onset AD (commonest).
 Familial late-onset AD (uncommon).
Familial late-onset AD (uncommon).
 Familial early-onset AD (rare).
Familial early-onset AD (rare).


About 10–20% of patients have a first-degree relative with dementia.
 CLINICAL FEATURES OF ALZHEIMER’S DISEASE
CLINICAL FEATURES OF ALZHEIMER’S DISEASE



MACROSCOPIC APPEARANCES
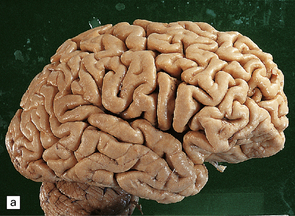
The brain shows atrophy and the weight is usually in the range of 900–1200 g. There is shrinkage of cerebral gyri and widening of sulci, most prominently in the medial temporal regions (particularly the hippocampus) but also in the frontal and parietal regions. Generally, the occipital lobe and the motor cortex are relatively spared. This pattern of atrophy occurs in several dementing diseases and is not specific for AD (Fig. 31.2).
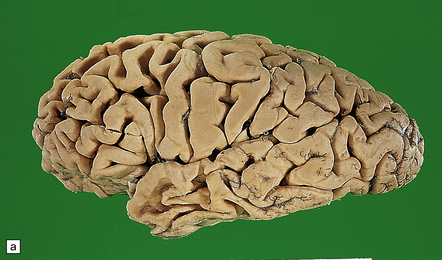
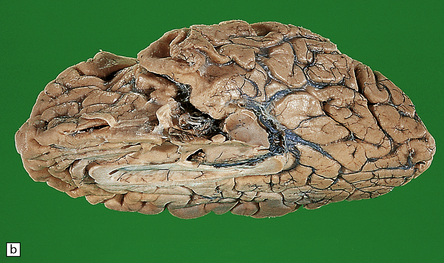
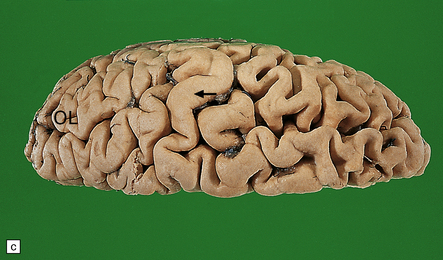
31.2 Macroscopic appearance of brain in AD.
The pattern of atrophy typically seen in AD is characteristic, but not specific for the disease. In this case, the leptomeninges have been stripped from one half of a brain to show the regional atrophy clearly. (a) Lateral view showing severe atrophy of the temporal lobe with less severe atrophy of the parietal and frontal lobes. The occipital lobe is spared. (b) The severity of atrophy of the mesial temporal lobe structures is better appreciated in this view of the inferior surface. (c) The relative sparing of the primary motor cortex (arrow) and occipital lobe (OL) can be appreciated in this view of the superior surface.
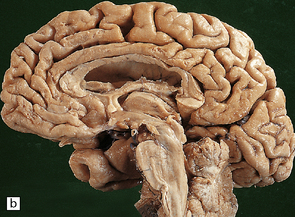
In slices of fixed brain, the cortical mantle may appear thinned (Fig. 31.3). The white matter is of normal color and texture, but reduced in volume. There may be significant dilatation of the ventricular system, especially of the temporal horn of the lateral ventricles. In the midbrain, the substantia nigra is usually normally pigmented but can appear pale. The locus ceruleus is often paler than normal. Cerebral infarcts or hemorrhages may be encountered and can be related to cerebrovascular amyloid deposition or coexistent arteriosclerotic disease.
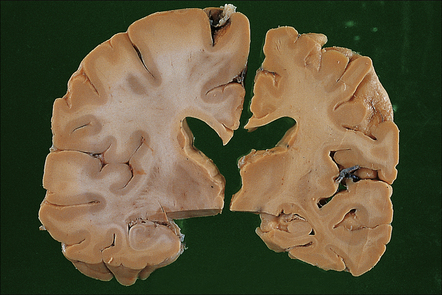
31.3 Macroscopic appearance of brain in AD.
In this picture the slice on the left is from a normal patient aged 70, while the one on the right is from a patient with AD. Note that there is a reduction in the volume of white matter with cortical atrophy, mild sulcal widening and gyral thinning, the hippocampus is atrophic.
HISTOPATHOLOGY OF ALZHEIMER’S DISEASE
AD is characterized by several histologic abnormalities, none of which is specific to this disease. Special stains are required for evaluation (Table 31.3).
Table 31.3
Stains used in the histologic assessment of Alzheimer’s disease
Much of the pathology associated with Alzheimer’s disease cannot be easily seen without special stains
Sections stained with H&E are used for general morphology and can be used to evaluate neuronal loss as well as any general morphological changes, e.g. associated areas of infarction. Granulovacuolar degeneration and Hirano bodies are also easily seen (Figs 31.5, 31.6)
Silver stains are still used in diagnosis and can be divided into:
Methods that are very sensitive for amyloid (e.g. modified methenamine silver techniques). These detect all plaques and a minority of tangles (Fig. 31.7)
Methods that are very sensitive for the detection of tangles and the abnormal nerve processes around plaques but do not tend to stain amyloid (e.g. the Gallyas technique, several modifications of the Palmgren silver impregnation (Fig. 31.8), and some modifications of Bodian and Bielschowsky methods)
Methods that are optimized to detect both plaques and tangles. These tend to underestimate the density of either plaques or tangles. This is true of most modified Bodian and Bielschowsky techniques, which underestimate the total amount of amyloid in sections
In most laboratories, specific staining of plaques and tangles is now performed by immunohistochemistry with commercially-available antisera:
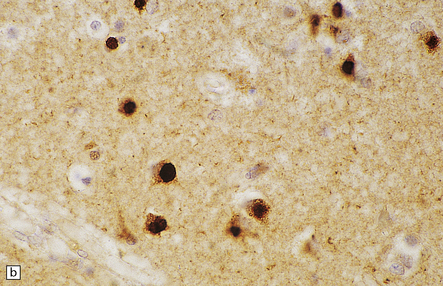
Plaques are detected with antisera to Aβ peptide after formic acid pretreatment of sections. This will also detect vascular amyloid (Fig. 31.9)
Tangles, plaque neurites and neuropil threads are detected by immunostaining for phosphorylated tau protein, the main protein constituent of tangles (Fig. 31.9)
 Parenchymal extracellular deposits of a specific amyloid in the brain. The amyloid is composed of Aβ peptide, derived by proteolytic breakdown from a normal neuronal membrane protein called amyloid-β precursor protein (APP). There are both diffuse and focal forms of amyloid deposits.
Parenchymal extracellular deposits of a specific amyloid in the brain. The amyloid is composed of Aβ peptide, derived by proteolytic breakdown from a normal neuronal membrane protein called amyloid-β precursor protein (APP). There are both diffuse and focal forms of amyloid deposits.


 Loss of synapses and, in late stages, of neurons from the cerebral cortex.
Loss of synapses and, in late stages, of neurons from the cerebral cortex.
Associated pathologic changes are:
 Amyloid (also derived from Aβ peptide) deposition in arteries and arterioles in the cerebral and cerebellar cortex and leptomeninges (congophilic angiopathy/cerebral amyloid angiopathy) is demonstrable in over 90% of cases, although the extent of this is very variable (Fig. 31.4). Small amounts of Aβ may also accumulate in the walls of small veins.
Amyloid (also derived from Aβ peptide) deposition in arteries and arterioles in the cerebral and cerebellar cortex and leptomeninges (congophilic angiopathy/cerebral amyloid angiopathy) is demonstrable in over 90% of cases, although the extent of this is very variable (Fig. 31.4). Small amounts of Aβ may also accumulate in the walls of small veins.
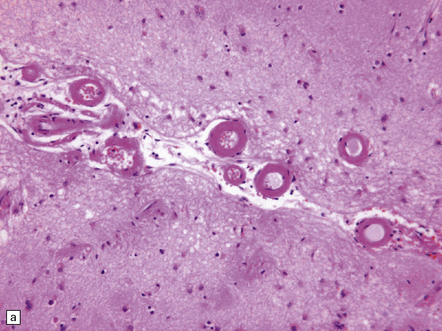
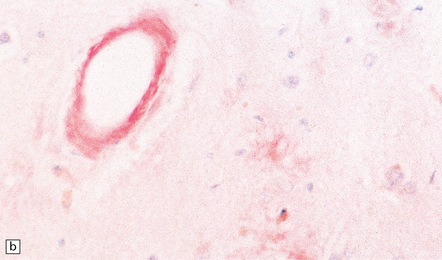
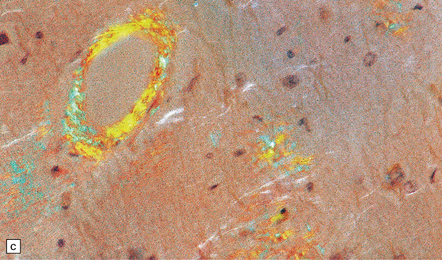
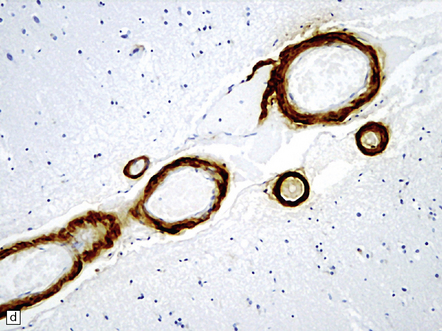
31.4 AD cerebral amyloid (congophilic) angiopathy.
(a) In H&E stained sections, as here, the vessels affected by amyloid are thick-walled and have a homogeneous pink appearance. (b) In this preparation stained with Congo red, amyloid in the wall of a vessel in the cerebral cortex stains orange. (c) The amyloid nature can be confirmed by polarizing light microscopy, which reveals apple-green birefringence and dichroism. (d) The amyloid in the cerebral vessels shows Aβ peptide immunoreactivity.
 Granulovacuolar degeneration (Fig. 31.5) affects greater numbers of hippocampal pyramidal neurons in AD than in age-matched controls and may also be seen in subcortical nuclei. These represent autophagic activity.
Granulovacuolar degeneration (Fig. 31.5) affects greater numbers of hippocampal pyramidal neurons in AD than in age-matched controls and may also be seen in subcortical nuclei. These represent autophagic activity.
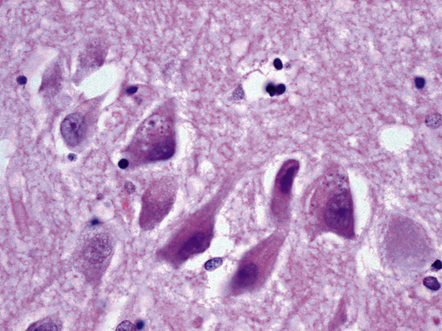
31.5 Two neurons contain basophilic granules surrounded by non-staining vacuoles, termed granulovacuolar degeneration (GVD).
Some GVD granules contain tau protein and also can be immunostained for ubiquitin.
 Hirano bodies, composed of actin-binding proteins, tend to be more numerous in AD than in age-matched controls in neurons in the hippocampal CA1 field and subiculum (Fig. 31.6).
Hirano bodies, composed of actin-binding proteins, tend to be more numerous in AD than in age-matched controls in neurons in the hippocampal CA1 field and subiculum (Fig. 31.6).
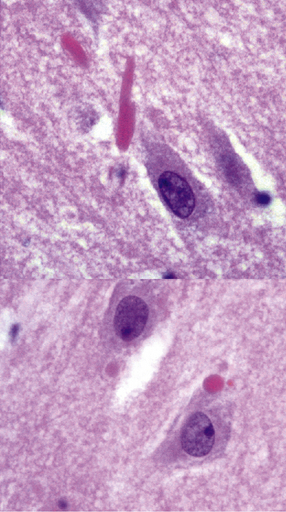
31.6 This composite image shows Hirano bodies in longitudinal (top) and cross-section (bottom).
Hirano bodies are brightly eosinophilic and appear to overlap neuronal structures in the plane of section.
 There is increased accumulation of lipofuscin in neurons (see Chapter 1).
There is increased accumulation of lipofuscin in neurons (see Chapter 1).
 Corpora amylacea may be seen in large numbers (see Chapter 1).
Corpora amylacea may be seen in large numbers (see Chapter 1).
Diffuse Aβ deposits
Diffuse deposits of Aβ peptide are seen on immunostaining as loose structures with irregular, ill-defined margins. In this form of deposit, the majority of protein is not aggregated as amyloid filaments (Table 31.4). Diffuse deposits are the main type of plaque seen in normal aging. Some diffuse Aβ deposits are described as fleece- or lake-like. Diffuse deposits may be seen in the subpial region in the cerebral cortex.
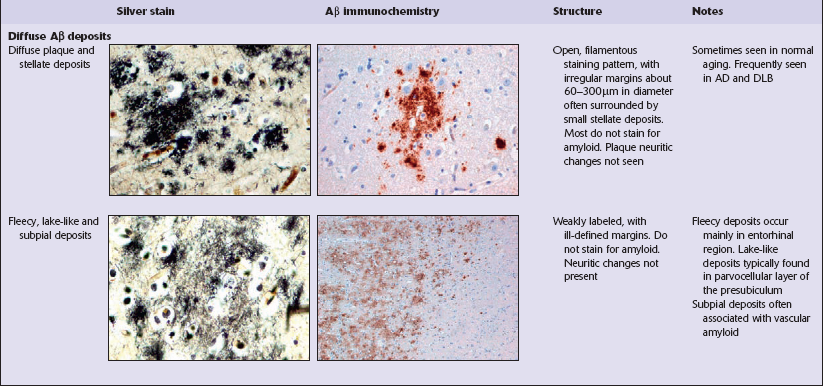
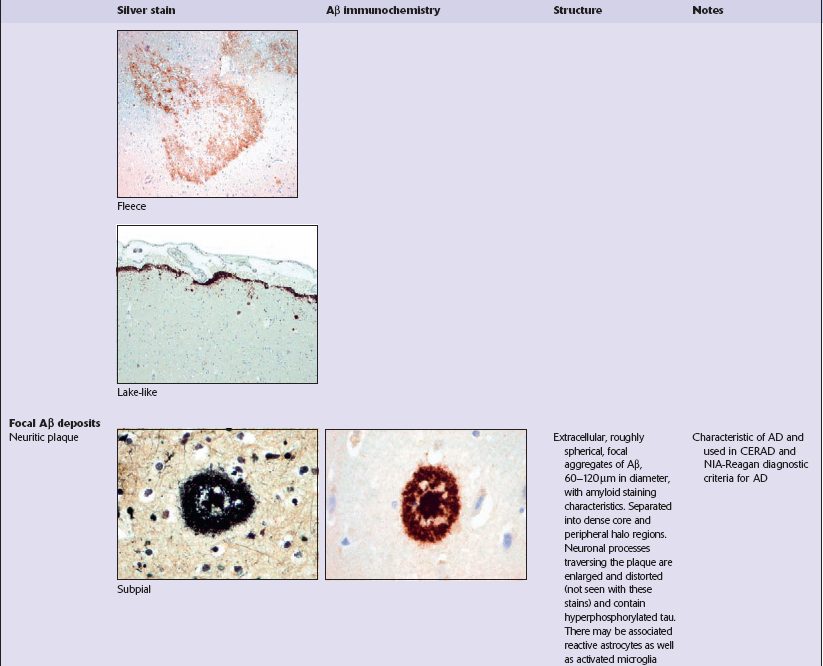
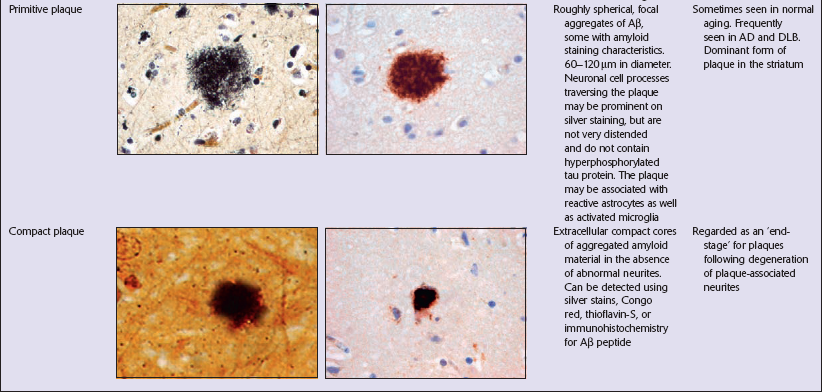
Focal Aβ deposits



Neuritic changes in plaques
Neuritic plaques include tau-immunoreactive dystrophic neurites (see Neuritic abnormalities in AD, below, and Fig. 31.11). In some neuritic plaques there are dystrophic neurites that contain chromogranin A and ubiquitin, but not tau protein; of the sparse neuritic plaques that may be present in the brains of cognitively normal elderly subjects, this form predominates. A variety of other plaque amyloid-related proteins can be demonstrated immunohistochemically, including apolipoprotein E (apoE), α1-antichymotrypsin, serum amyloid-P protein, growth factors, heparin sulfate, and complement factors.
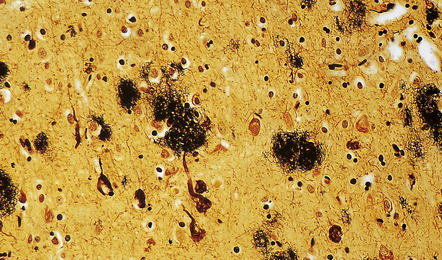
31.7 Methenamine silver stain in AD.
Cerebral cortex stained with methenamine silver to reveal plaques and NFTs. This technique provides a sensitive means of staining plaques. It is less effective for staining NFTs, and is relatively poor for demonstrating plaque-related neurites.
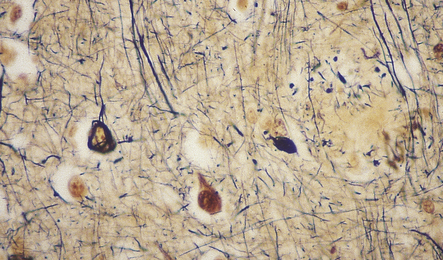
31.8 Palmgren silver impregnation in AD.
Silver impregnation of cerebral cortex reveals a plaque and NFTs. This type of stain is not sensitive to amyloid, seen in the center of the plaque as a yellow background, but is good for detecting NFTs and plaque-related neurites.
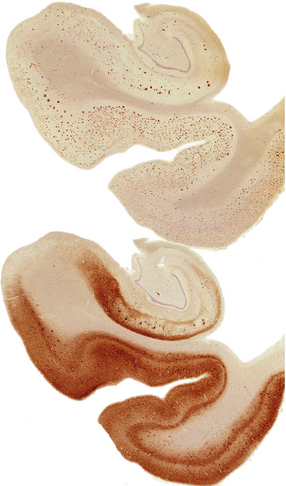
31.9 Aβ and tau in AD.
These are serial sections showing the hippocampus. The top section has been immunostained with an antibody to the peptide that forms amyloid in AD (Aβ peptide). The bottom section has been stained with an antibody to hyperphosphorylated tau protein, which accumulated inside neurons and cell processes in AD. Amyloid plaques can be seen as military focal deposits in the cortex with Aβ staining. Tau protein accumulation has a laminar pattern, reflecting its accumulation in nerve cell bodies and processes. Notice that some foci of tau labeling correspond to the same areas as the Aβ plaques in the top section; these foci are clusters of plaque-associated nerve cell processes that contain hyperphosphorylated tau (plaque-associated dystrophic neurites).
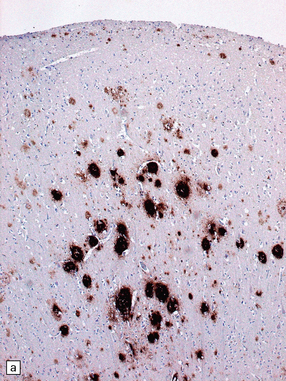
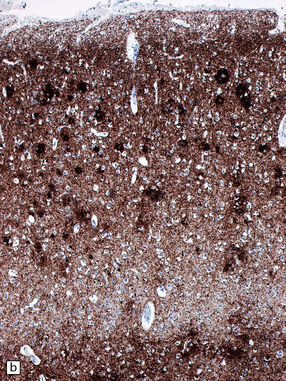
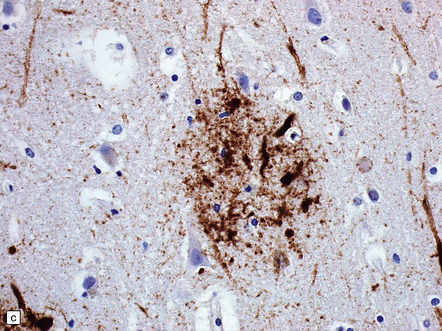
31.11 Neuritic changes in plaques.
(a) Labeling for Aβ peptide shows range of plaque morphologies (contrast with a similar area stained for hyperphosphorylated tau protein). (b) Immunostaining of hyperphosphorylated tau protein. This shows accumulation of tau in neuronal somata and abnormal nerve cell processes; these are particularly prominent within neuritic plaques, seen as condensations of dense labeling. (c) Immunostaining of hyperphosphorylated tau protein in plaque-associated neurites. The plaque Aβ amyloid is not demonstrated with this immunostain. (d) In some plaques, immunostaining for ubiquitin shows bulbous dilated nerve cell processes (plaque-associated neurites). The amyloid is not labeled.
 MOLECULAR PATHOLOGY OF PLAQUE Aβ
MOLECULAR PATHOLOGY OF PLAQUE Aβ
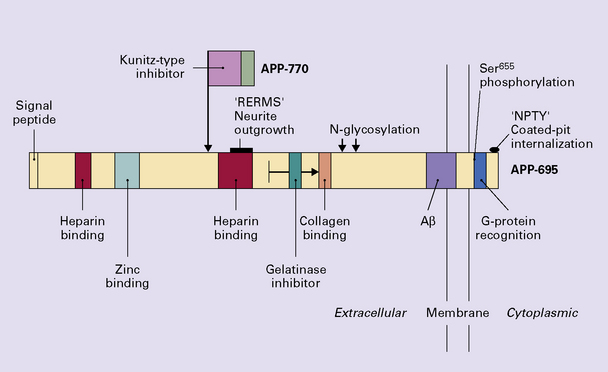
 APP is a normal transmembrane glycoprotein. The gene is located on chromosome 21, and several different mRNAs are generated by alternative splicing. The predicted structure of APP consists of three domains (Fig. 31.10): a small cytosolic domain, a transmembrane domain, and a large extracellular domain.
APP is a normal transmembrane glycoprotein. The gene is located on chromosome 21, and several different mRNAs are generated by alternative splicing. The predicted structure of APP consists of three domains (Fig. 31.10): a small cytosolic domain, a transmembrane domain, and a large extracellular domain.
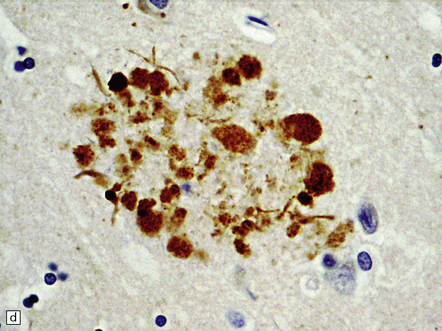
31.10 Amyloid plaques.
On H&E staining, plaques can often be seen as ill-defined eosinophilic round or ovoid areas contrasting with the background texture of the cortical neuropil (a). (b) Within the plaque structure, nuclei of microglial cells and astrocytes can often be seen, in a radial pattern. (c) A small proportion of plaques are composed of a dense eosinophilic amyloid core without a surrounding zone of altered neuropil. H&E staining is not a sensitive method for detecting plaques in AD.

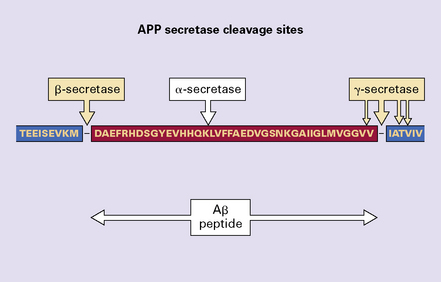
 In normal cells APP is mainly processed in a non-amyloidogenic pathway by several proteases. Enzymes called α-secretases cut APP near the middle of the Aβ region, precluding the generation of Aβ (Table 31.4). α-secretase activity is linked to several proteases, including those of the ADAM (A Disintegrin And Metalloproteinase) family. The N-terminus of the peptide is generated when another enzyme complex, with so-called γ-secretase activity, cleaves APP within its transmembrane domain. The activity of γ-secretase is linked to the function of the presenilins (Fig. 31.11).
In normal cells APP is mainly processed in a non-amyloidogenic pathway by several proteases. Enzymes called α-secretases cut APP near the middle of the Aβ region, precluding the generation of Aβ (Table 31.4). α-secretase activity is linked to several proteases, including those of the ADAM (A Disintegrin And Metalloproteinase) family. The N-terminus of the peptide is generated when another enzyme complex, with so-called γ-secretase activity, cleaves APP within its transmembrane domain. The activity of γ-secretase is linked to the function of the presenilins (Fig. 31.11).
 A second, amyloidogenic pathway for cleavage of APP is involved in the pathogenesis of AD (Figs 31.12 and 31.13). This involves the action of two proteolytic activities, β-secretase and γ-secretase. β-Secretase activity is mediated by an enzyme called β-site APP-cleaving enzyme (BACE). Subsequent γ-secretase cleavage yields Aβ peptide fragments of different lengths, the main products comprising 40 or 42 amino acids. Aβ peptide in plaques is mainly a 42-residue form of Aβ (Aβ42) (Fig. 31.14). Aβ42 is believed to be more amyloidogenic than Aβ40.
A second, amyloidogenic pathway for cleavage of APP is involved in the pathogenesis of AD (Figs 31.12 and 31.13). This involves the action of two proteolytic activities, β-secretase and γ-secretase. β-Secretase activity is mediated by an enzyme called β-site APP-cleaving enzyme (BACE). Subsequent γ-secretase cleavage yields Aβ peptide fragments of different lengths, the main products comprising 40 or 42 amino acids. Aβ peptide in plaques is mainly a 42-residue form of Aβ (Aβ42) (Fig. 31.14). Aβ42 is believed to be more amyloidogenic than Aβ40.
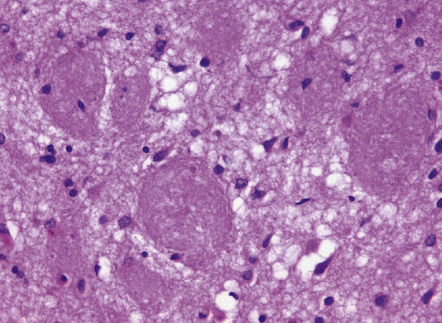
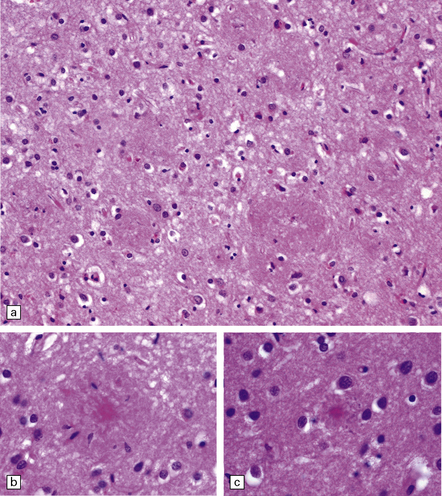
31.12 Cotton wool plaques.
These rounded, eosinophilic, well-delineated dense aggregates of Aβ have been termed ‘cotton wool plaques’. They have been noted in early-onset AD with spastic paraparesis and presenilin mutations. Such plaques are immunoreactive for Aβ42/43 but weakly or not at all for Aβ40. They do not have a congophilic core, and fibrillar amyloid is not seen on electron microscopy. There are typically few associated tau-immunoreactive neurites. They have also been described in non-familial late-onset AD.

 THE AMYLOID CASCADE HYPOTHESIS OF AD
THE AMYLOID CASCADE HYPOTHESIS OF AD
 Several lines of evidence point to a primary role for Aβ amyloid in the pathogenesis of AD:
Several lines of evidence point to a primary role for Aβ amyloid in the pathogenesis of AD:
• Some mutations in the APP gene have been linked with rare familial forms of AD. The neuropathologic findings in these are very similar to those of sporadic AD and include the presence of many NFTs.
• Patients with Down syndrome (trisomy 21) are strongly predisposed to develop AD by about 40 years of age. These patients have three APP genes with a commensurate increase in APP mRNA. Diffuse plaques are detectable in the brain of patients with Down syndrome before the development of neuritic plaques and NFTs.
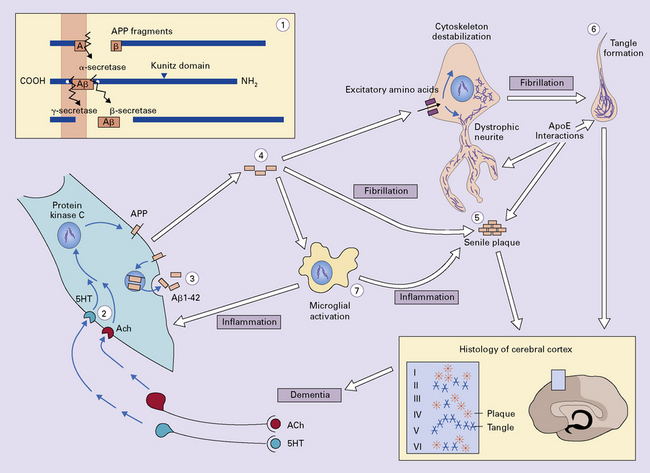
 The amyloid cascade model (Fig. 31.15) proposes a central role for Aβ amyloid in the pathogenesis of AD. The link between Aβ generation and the formation of NFTs is not yet known. It is also unclear as to what extent the development of AD may depend on the accumulation of soluble forms of Aβ – particularly that in the form of small, soluble aggregates (oligomers) – rather than (or in addition to) the fibrillar forms that form plaques.
The amyloid cascade model (Fig. 31.15) proposes a central role for Aβ amyloid in the pathogenesis of AD. The link between Aβ generation and the formation of NFTs is not yet known. It is also unclear as to what extent the development of AD may depend on the accumulation of soluble forms of Aβ – particularly that in the form of small, soluble aggregates (oligomers) – rather than (or in addition to) the fibrillar forms that form plaques.
31.15 Amyloid cascade model of AD.
The general pattern of APP processing in neurons is shown (1). Non-amyloidogenic (α-secretase) processing of APP can be modulated by stimulation of cholinergic (ACh) or serotonergic (5HT) receptors (2). Within the neuron, PS1 associates with APP and traffics with it to an endosomal vesicle where Aβ42 can be generated (3). The Aβ42 is secreted and forms multimeric aggregates (4), which can activate microglia and can participate in excitotoxic reactions with neurons. Fibrillar Aβ42 initiates Aβ plaque formation, which involves dystrophic neurites, recruitment of activated microglia, interactions with ApoE and the deposition of Aβ to form plaques (5). Involvement in plaque formation results in the degeneration of susceptible neurons and tangle formation by an unknown mechanism (6). Activated microglia act in a positive feedback loop to enhance Aβ42 generation by neurons, and plaque formation (7).

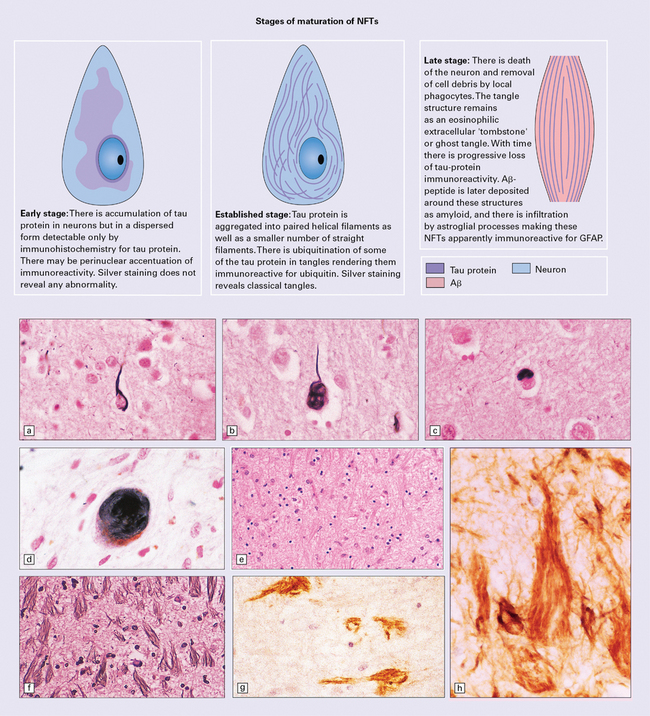
Neurofibrillary tangles
Neurofibrillary tangles are neuronal inclusions composed largely of filamentous aggregates of hyperphosphorylated tau proteins that are variably ubiquitylated and glycated. In sections stained with hematoxylin and eosin, intracellular NFTs are faintly basophilic and extracellular NFTs appear eosinophilic (Fig. 31.16). In sections stained by silver impregnation, or when immunostained, several morphologic forms of NFT can be identified, the shape of the NFT probably being determined by that of the neuron containing it. A multi-stage model of NFT formation has been proposed (Fig. 31.17). Ultrastructural investigation reveals that NFTs are composed of paired helical filaments (PHFs) with a maximum diameter of 20 nm and a periodic narrowing to 10 nm every 80 nm (Fig. 31.18). A small proportion of filaments is straight, with a diameter of 15 nm. Detailed examination of PHF preparations shows that the filaments have a dense core region with a surrounding fuzzy coat.
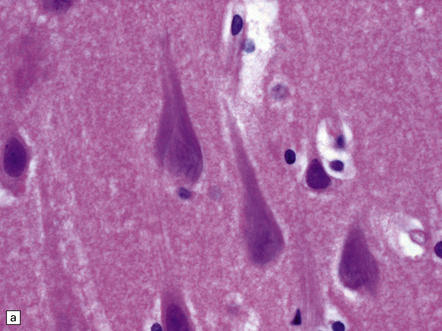
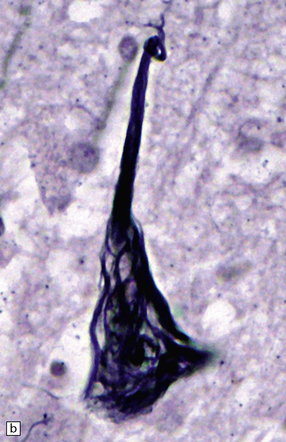
31.16 Neurofibrillary tangles.
(a) In H&E sections, NFTs can sometimes just be seen as faintly basophilic structures within the neuronal cytoplasm but this is not a reliable way to detect them. Many are flame-shaped, as here. (b) Silver staining shows a characteristic filamentous tangle structure in a pyramidal neuron with material extending into the apical dendrite. The filamentous material is composed of tau protein.
31.17 Histologic appearances of NFTs.
(a) Band-shaped perikaryal NFT: a single well-defined band runs from the base of the neuron into the apical dendrite. This type of NFT is seen in both large and small pyramidal cells. (b) Flame-shaped perikaryal NFT: a triangular mass of filaments, usually surrounding the nucleus and extending into the apical dendrite, and seen mainly in large pyramidal cells. (c) Small globose perikaryal NFT: a rounded mass of filaments displacing the nucleus to one side of the neuron. This type of NFT is seen in small cortical neurons, especially in Layers 5 and 6, and also in the periamygdaloid cortex. (d) Large globose NFTs: seen in the nucleus basalis of Meynert, periaqueductal gray matter, substantia nigra, locus ceruleus, and raphe nuclei. (e) Ghost NFTs: faintly eosinophilic extracellular structures that persist after the death of the neuron. (f) Ghost NFTs: the extracellular ghost NFTs are moderately well seen on silver impregnation. (g) Ghost NFTs may become immunoreactive for Aβ peptide as a result of its deposition around them. (h) Ghost NFTs may seem to be immunoreactive for glial fibrillary acidic protein (GFAP), due to ingrowth of glial cell processes.
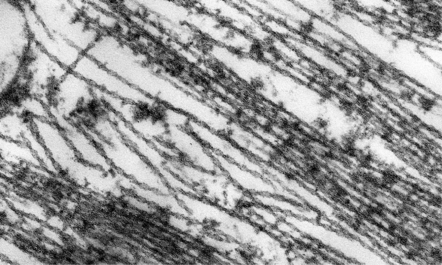
31.18 Ultrastructure of NFT.
NFTs are composed of paired helical filaments with a periodicity of 80 nm.
NFTs are readily detected by antisera directed against phosphorylated tau protein (Fig. 31.19). Many NFTs are immunoreactive for ubiquitin or P62 (Fig. 31.20). Neurofibrillary tangles can be seen in elderly brains in low density and restricted distribution as well as in a variety of other conditions. Hence, they are not specific to AD (Table 31.5). In AD, the density of NFTs is closely related to the severity of dementia.
Table 31.5
Disorders associated with neurofibrillary tangles
Progressive supranuclear palsy (PSP)
Down syndrome
Dementia pugilistica
Postencephalitic parkinsonism
Parkinsonism dementia complex of Guam
Subacute sclerosing panencephalitis
Niemann–Pick disease type C
Familial British dementia
Myotonic dystrophy
Kufs’ disease
Neuronal brain iron accumulation type-1
Gerstmann–Straüssler–Scheinker syndrome
Cockayne syndrome
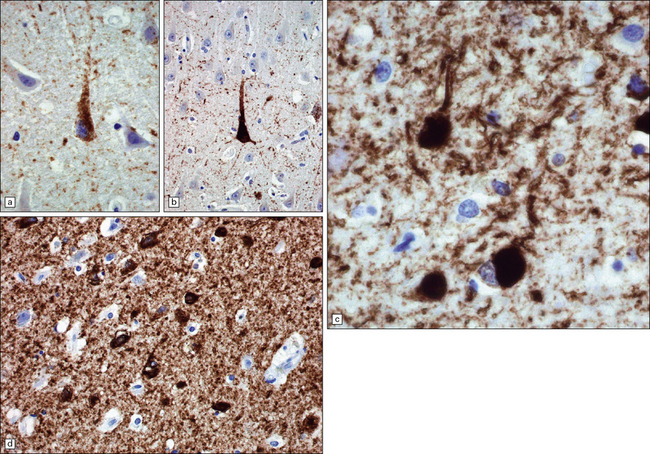
31.19 NFT and NT tau immunostaining.
Immunostaining with antisera to phosphorylated tau protein is used as a routine method for detecting NFTs and NTs. Staining will detect established NFTs, as well as pretangles – dispersed aggregates of tau that are probably precursors of NFTs. Many extracellular (‘ghost’) NFTs are not immunoreactive for tau. (a) Granular cytoplasmic pretangle staining in a pyramidal neuron in the hippocampus. There is a low density of background NTs. (b) Dense labeling, characteristic of tangle stage, in a pyramidal neuron. Note higher density of NTs in the background. (c) NFTs of flame-shaped and globular morphology, in temporal neocortex. There is a high density of background NTs. (d) Tangles are densely stained structures in the cell bodies. There is abundant labeling of NTs in the surrounding neuropil.
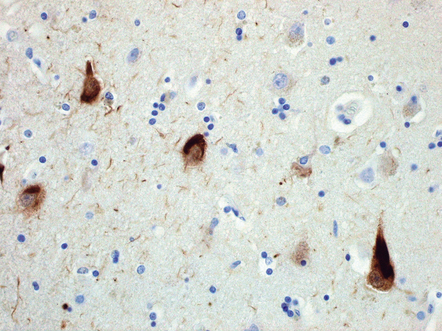
31.20 NFT P62 immunostaining.
Some NFTs are immunoreactive for ubiquitin, or the ubiquitin-binding protein P62, shown here. It is of note that small globose NFTs are generally ubiquitylated and can therefore be almost indistinguishable from cortical Lewy bodies on ubiquitin or P62 immunohistochemistry.
Neuritic abnormalities in AD. There are two main forms:
 Plaque-related dystrophic neurites, which are abnormally distended nerve cell processes running through Aβ plaque deposits. Some of the neurites contain increased amounts of lysosome-related dense bodies, but no PHFs, and immunostain for chromogranin A and ubiquitin, but not tau protein. Other neurites contain PHFs ultrastructurally and are immunoreactive both for tau protein and variably for ubiquitin (Fig. 31.11).
Plaque-related dystrophic neurites, which are abnormally distended nerve cell processes running through Aβ plaque deposits. Some of the neurites contain increased amounts of lysosome-related dense bodies, but no PHFs, and immunostain for chromogranin A and ubiquitin, but not tau protein. Other neurites contain PHFs ultrastructurally and are immunoreactive both for tau protein and variably for ubiquitin (Fig. 31.11).
 Neuropil threads (NTs), which are fine, distorted, and twisted nerve cell processes that are immunoreactive for tau protein (Fig. 31.21) and variably for ubiquitin. Ultrastructural examination shows nerve cell processes that contain a mixture of PHFs and straight filaments (Fig. 31.18).
Neuropil threads (NTs), which are fine, distorted, and twisted nerve cell processes that are immunoreactive for tau protein (Fig. 31.21) and variably for ubiquitin. Ultrastructural examination shows nerve cell processes that contain a mixture of PHFs and straight filaments (Fig. 31.18).
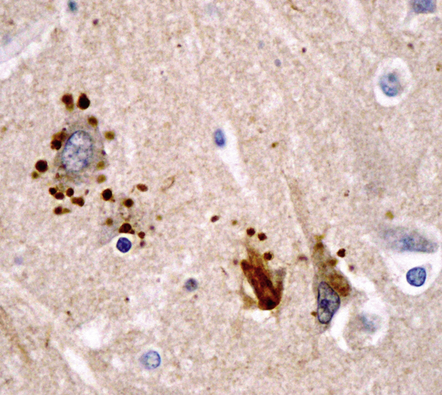
Perisomatic granules. Immunostaining for ubiquitin reveals densely-labeled round bodies adjacent to pyramidal neurons representing distended, retracted synaptic boutons. These are termed perisomatic granules (Fig. 31.21).
Tangle-associated neuritic clusters in AD. Tau immunostaining may show shows filamentous aggregates in the pyramidal cell region of the hippocampus. Although these superficially resemble plaque neurites, there is no associated focal accumulation of Aβ. These clusters represent ingrowth of tau-containing cell processes into a region previously occupied by a NFT (Fig. 31.22).
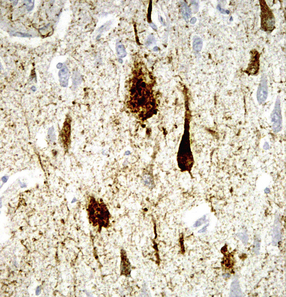
31.22 Tangle-associated neuritic clusters.
Tau immunostaining of the hippocampal pyramidal layer shows two tangle-associated neuritic clusters (arrowheads). A few adjacent pyramidal neurons contain tangles.
Neuronal and synaptic loss in AD. A 30–40% loss of neocortical neurons can be demonstrated in advanced AD, particularly in young-onset patients. The neuronal loss is associated with astrocytic gliosis and, in some cases, cortical microvacuolation, the latter often termed status spongiosus (Fig. 31.23). This pattern of vacuolation is coarser than that typically seen in prion disease and is largely confined to the outer cortical layers. In AD, synaptic loss of 30–50% can be demonstrated by quantitation of synapse-related proteins in affected cortical regions. The most widely used marker is synaptophysin, a glycoprotein associated with synaptic vesicles. The degree of synaptic loss correlates well with clinical scores of the severity of dementia.
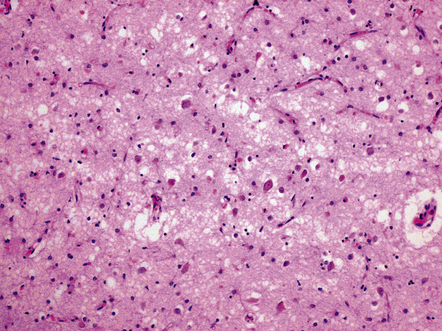
31.23 Status spongiosus in AD.
Severe neuronal loss from the cortex in AD, with associated astrocytic gliosis, results in irregular coarse vacuolation termed status spongiosus.
 MOLECULAR PATHOLOGY OF TAU PROTEIN IN NFTs
MOLECULAR PATHOLOGY OF TAU PROTEIN IN NFTs



 NEUROCHEMICAL PATHOLOGY OF AD
NEUROCHEMICAL PATHOLOGY OF AD




Subcortical involvement in AD. Many subcortical regions are involved by plaques, NFTs, or NTs in AD (Table 31.6). Some regions, such as the dorsal raphe nucleus, are affected at an early stage of disease. Involvement of the nucleus basalis of Meynert is especially important, as this is the cholinergic projection nucleus to the cerebral cortex. Cell loss from this nucleus results in a severe cholinergic deficit in the cerebral cortex in AD.

Pathologic staging of AD
Plaque stages correlate poorly with the severity of dementia (Fig. 31.24) and are:
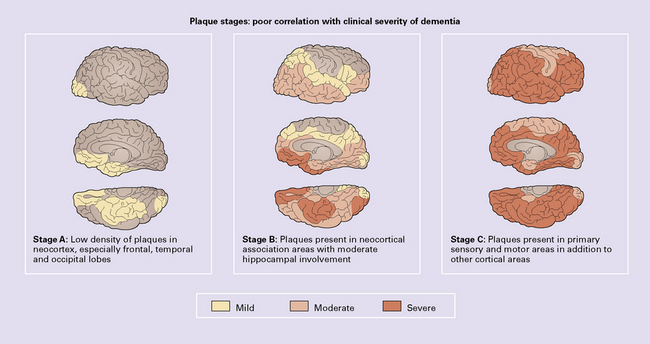
 Stage A: low density of neuritic plaques in the neocortex, especially in the frontal, temporal, and occipital lobes.
Stage A: low density of neuritic plaques in the neocortex, especially in the frontal, temporal, and occipital lobes.

 Stage C: neuritic plaques present in primary sensory and motor areas.
Stage C: neuritic plaques present in primary sensory and motor areas.
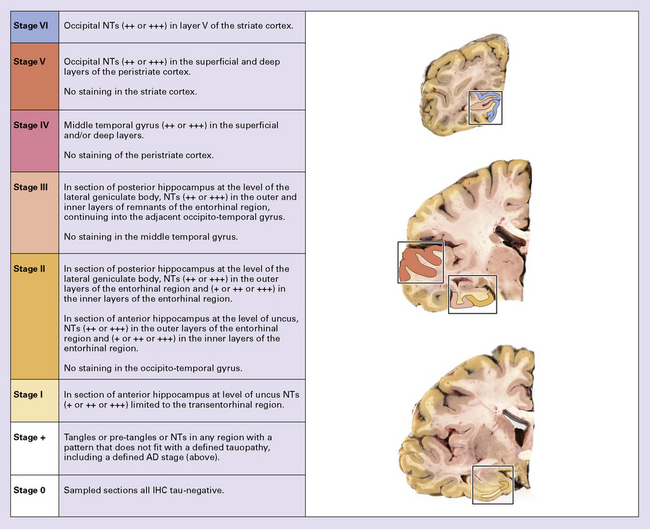
NFT stages correlated well with the severity of dementia (Fig. 31.25). The Braak staging of neurofibrillary degeneration is incorporated in the 2012 National Institute on Aging–Alzheimer’s Association (NIA-AA) guidelines for the neuropathologic assessment of Alzheimer’s disease neuropathologic criteria for diagnosis of AD (p. 625). The BrainNet Europe Consortium has validated a scheme incorporating immunohistochemistry for hyperphosphorylated tau instead of silver staining for staging of neurofibrillary changes in AD (Fig. 31.26).
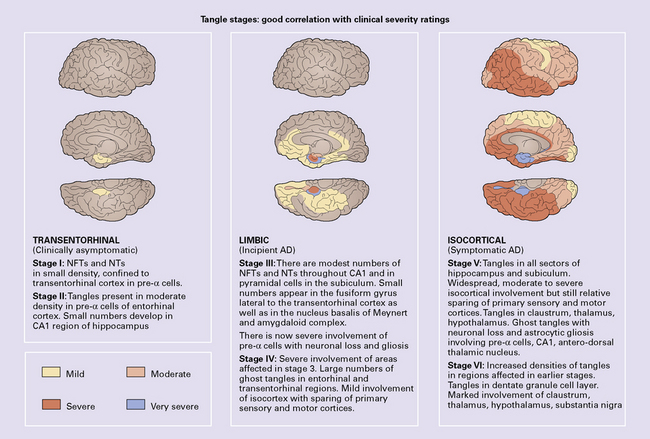
AD pathology in the cognitively normal elderly
The histologic changes that affect AD patients may be found in a restricted distribution or low density in cognitively normal elderly individuals. Plaques occur in the cortex with increased frequency in aging. In normal aging, the plaques are mainly diffuse. There may be small numbers of neuritic plaques, most associated with ubiquitin- and chromogranin-immunoreactive neurites, that do not contain tau protein. NFTs may be seen in small numbers in the hippocampus and entorhinal cortex in the cognitively normal elderly. This corresponds to Braak stages I–III (Fig. 31.25).
Pathologic diagnostic criteria for AD
Several different criteria have been proposed for the pathologic diagnosis for AD:
 The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) guidelines for the diagnosis of AD have been widely used and are based on semi-quantitative assessment of neuritic plaque density by comparison with standard reference illustrations (Tables 31.7–31.9, Figs 31.24, 31.25). This has been shown to have good reproducibility between different laboratories. The patient’s age and the clinical history of dementia are taken into account in determining the diagnostic category for each case. The CERAD scheme specifies silver staining or fluorescent dye staining, which has prompted workers to evaluate equivalent immunohistochemical staining methods for evaluation of cases.
The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) guidelines for the diagnosis of AD have been widely used and are based on semi-quantitative assessment of neuritic plaque density by comparison with standard reference illustrations (Tables 31.7–31.9, Figs 31.24, 31.25). This has been shown to have good reproducibility between different laboratories. The patient’s age and the clinical history of dementia are taken into account in determining the diagnostic category for each case. The CERAD scheme specifies silver staining or fluorescent dye staining, which has prompted workers to evaluate equivalent immunohistochemical staining methods for evaluation of cases.
Table 31.7
CERAD protocol for diagnosis of Alzheimer’s disease
Macroscopic appearance
The following features are noted:
brain weight
regional neocortical atrophy and ventricular enlargement (rated semiquantitatively as none, mild, moderate, or severe)
atrophy of the hippocampus and entorhinal cortex (present or absent)
pallor of the substantia nigra and locus ceruleus (present or absent)
atherosclerosis, significant obstruction or aneurysms of cerebral blood vessels (present or absent)
lacunar infarcts, regional infarcts, hemorrhages (number, size, frequency, distribution, and laterality recorded)
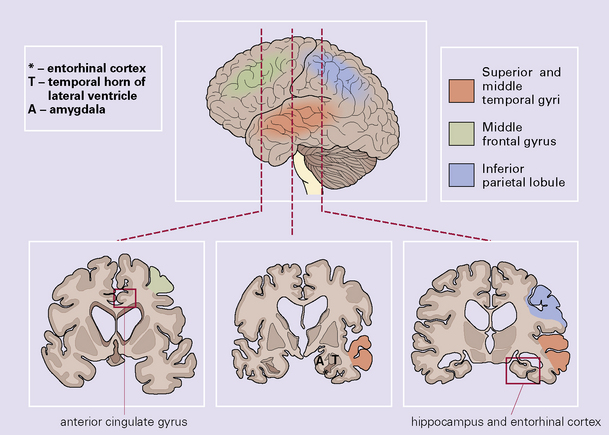
Histologic sampling and staining
A minimum of six anatomic regions is designated for histologic examination (Fig. 31.26):
middle frontal gyrus
superior and middle temporal gyri
anterior cingulate gyrus
inferior parietal lobule
hippocampus and entorhinal cortex
midbrain including the substantia nigra
Paraffin-embedded sections are cut at a thickness of 6–8 μm and stained with:
Hematoxylin and eosin (H&E)
A silver stain, such as the modified Bielschowsky impregnation, for the detection of neuritic plaques and neurofibrillary tangles
Thioflavin-S stained sections viewed under UV light can be used to assess plaques, tangles, and vascular amyloid
A Congo red stain can be used for evaluating vascular amyloid
Diagnostic classification
The CERAD classification is performed in three steps:
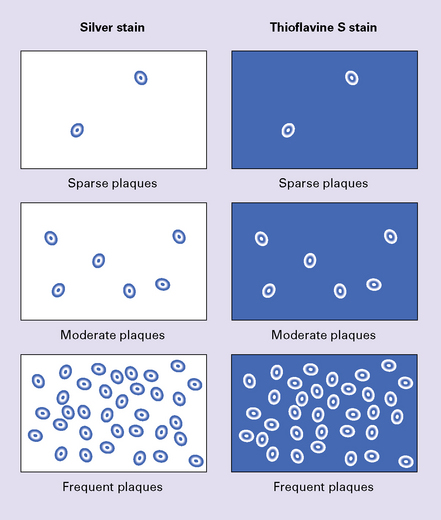
A semiquantitative assessment is made of the density of neuritic plaques (i.e. that include thickened, silver-impregnated neurites) in the sections of the neocortex. The density is scored by comparison with reference photomicrographs and diagrams as none, sparse, moderate, or frequent (Fig. 31.27). The density of tangles is also estimated but this does not contribute to the diagnostic classification in the CERAD protocol
An age-related plaque score is obtained by relating the maximum plaque density in sections of frontal, temporal, or parietal cortex, to the age of the patient at death (in the ranges <50, 50–75, or >75 years) (Table 31.8)
The age-related plaque score is then integrated with the clinical presence or absence of dementia to allow cases to be categorized as normal with respect to AD, probable AD, or definite AD (Table 31.9)

Table 31.9
Normal (with respect to Alzheimer’s disease or other dementing processes) if:
Either
No histologic evidence of Alzheimer’s disease (0 score), and no clinical history of dementia, and absence of other neuropathologic lesions likely to cause dementia
Or
An A age-related plaque score and no clinical history of dementia
CERAD NP definite Alzheimer’s disease
C age-related plaque score, and clinical history of dementia, and presence or absence of other neuropathologic lesions likely to cause dementia
CERAD NP probable Alzheimer’s disease
B age-related plaque score, and clinical history of dementia, and presence or absence of other neuropathologic lesions likely to cause dementia
CERAD NP possible Alzheimer’s disease if:
Either
A age-related plaque score, and clinical history of dementia, and presence or absence of other neuropathologic lesions likely to cause dementia
Or
B or C age-related plaque score and absence of clinical manifestations of dementia


 Aβ plaques are staged according to the Thal Phase scheme (A).
Aβ plaques are staged according to the Thal Phase scheme (A).
 NFT stage is determined according to the Braak criteria (B).
NFT stage is determined according to the Braak criteria (B).
 Neuritic plaques are scored according to the CERAD scheme (C) (Fig. 31.27, Table 31.10).
Neuritic plaques are scored according to the CERAD scheme (C) (Fig. 31.27, Table 31.10).
Table 31.10
ABC scoring scheme for AD neuropathologic change
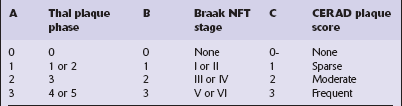
Modified from National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease, 2012.
Combining these three scores (‘ABC scoring’) allows the pathologist to allocate a probability that AD-associated abnormalities accounted for the patient’s dementia in life. AD lesions seen in the post-mortem brain from cognitively normal elderly people are considered pathologic rather than a part of a normal aging process. The likelihood that clinical dementia has been caused by AD lesions in the brain is stratified on the basis of the post-mortem neuropathologic findings, as follows (Table 31.11):
Table 31.11
NIA-AA ABC scoring for Alzheimer neuropathologic change
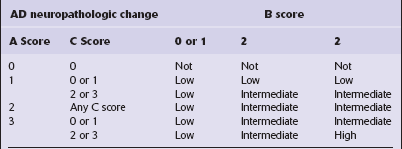
(Modified from National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimer’s Dement 2012 8(1):1–13)
 High probability that dementia was due to AD lesions, if the brain has both neuritic plaques and neurofibrillary tangles in the neocortex (CERAD frequent neuritic plaque score 3, Thal plaques score 3 and Braak and Braak stage V/VI score 3).
High probability that dementia was due to AD lesions, if the brain has both neuritic plaques and neurofibrillary tangles in the neocortex (CERAD frequent neuritic plaque score 3, Thal plaques score 3 and Braak and Braak stage V/VI score 3).


In applying earlier NIA–Reagan criteria, it became apparent that many laboratories had moved away from use of silver-stained preparations in evaluating AD pathology and that alternate approaches were desirable. The BrainNet Europe group published staging criteria based on evaluation of tau-labeled histologic sections from four brain regions (Figs 31.27–31.30).
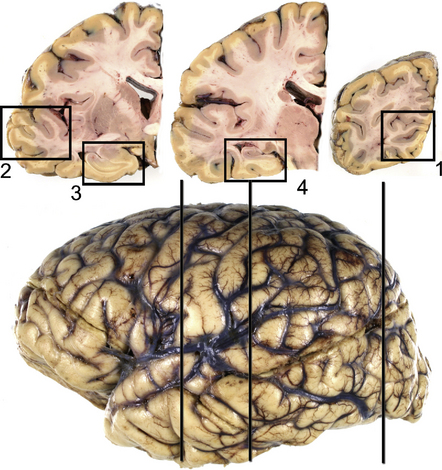
31.28 Cortical sampling for immunohistochemical staging of neurofibrillary changes (BrainNet Europe).
Section 1: visual cortex including the calcarine fissure to include the primary visual cortex with band of Gennari (involved in stage VI) and parastriate/peristriate region (Brodmann area 18/19, the six-layered cortex (involved in stage V). Section 2: middle and superior temporal gyrus (involved in stage IV). Section 3: anterior hippocampus and/or amygdala at the level of uncus (involved in stages I–III). Section 4: posterior hippocampus at level of lateral geniculate body (involved in stages II and III).
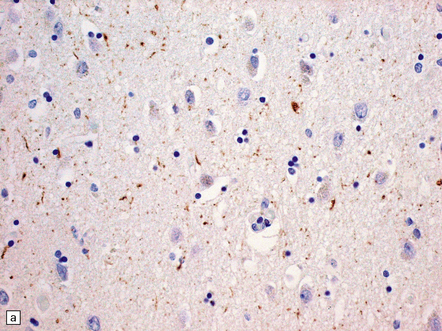
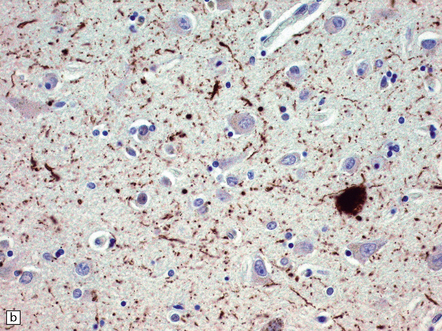
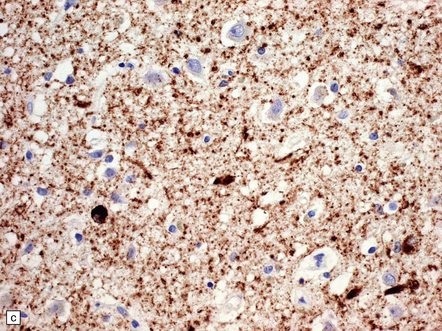
31.29 Semiquantitative assessment of immunohistochemical labeling of neurofibrillary changes, with antibody to hyperphosphorylated tau protein.
0, No labeling (not illustrated); +, labeling barely detectable (a); ++, labeling readily detectable (b); +++, dense labeling that can be seen by macroscopic inspection of the slide (c).
DIFFICULTIES IN DIAGNOSING AD
 Specific tauopathies such as PSP (see Chapter 28) and CBD (see Chapter 28, and later in this chapter) may present as a dementia syndrome or a syndrome of parkinsonism and dementia. A search for the characteristic abnormalities should be made of subcortical structures known to be affected in these disorders. Cortical regions should be assessed for swollen neurons characteristic of CBD, and both cortex and white matter should be examined for tau-immunopositive glial inclusions.
Specific tauopathies such as PSP (see Chapter 28) and CBD (see Chapter 28, and later in this chapter) may present as a dementia syndrome or a syndrome of parkinsonism and dementia. A search for the characteristic abnormalities should be made of subcortical structures known to be affected in these disorders. Cortical regions should be assessed for swollen neurons characteristic of CBD, and both cortex and white matter should be examined for tau-immunopositive glial inclusions.
 Tangle-only dementia (see below) is an uncommon but increasingly recognized disorder.
Tangle-only dementia (see below) is an uncommon but increasingly recognized disorder.
Some patients have an abundance of plaques but very few NFTs.
 A careful search for cortical Lewy bodies should be made in cortical and subcortical regions, with a view to making a diagnosis of dementia with Lewy bodies (DLB), which accounts for many cases previously regarded as ‘plaque-only’ AD.
A careful search for cortical Lewy bodies should be made in cortical and subcortical regions, with a view to making a diagnosis of dementia with Lewy bodies (DLB), which accounts for many cases previously regarded as ‘plaque-only’ AD.

 Limbic AD, characterized by clinical dementia and large numbers of NFTs restricted to the amygdala and hippocampus but with large numbers of neocortical plaques.
Limbic AD, characterized by clinical dementia and large numbers of NFTs restricted to the amygdala and hippocampus but with large numbers of neocortical plaques.




 Swollen neurons in AD. In a few cases swollen cortical neurons are a feature of disease that would otherwise be pathologically typical of AD. Care should be taken to ensure that the case does not meet criteria for CBD (see Chapter 28, and below) and that grain pathology is not present (p. 639).
Swollen neurons in AD. In a few cases swollen cortical neurons are a feature of disease that would otherwise be pathologically typical of AD. Care should be taken to ensure that the case does not meet criteria for CBD (see Chapter 28, and below) and that grain pathology is not present (p. 639).

 AD with vascular disease. There may be ischemic or hemorrhagic disease due to the cerebral and cerebellar amyloid angiopathy in AD (see Chapter 10), and AD is often associated with atherosclerotic and/or arteriosclerotic vascular disease of the brain; a diagnosis of mixed AD and vascular dementia is appropriate in some cases.
AD with vascular disease. There may be ischemic or hemorrhagic disease due to the cerebral and cerebellar amyloid angiopathy in AD (see Chapter 10), and AD is often associated with atherosclerotic and/or arteriosclerotic vascular disease of the brain; a diagnosis of mixed AD and vascular dementia is appropriate in some cases.
LEWY BODIES IN DEMENTIA SYNDROMES
 Some patients have a primary clinical dementia syndrome, characterized by fluctuation, early visual hallucinations, extrapyramidal tremor, which meets clinical criteria for dementia with Lewy bodies (DLB). These patients typically have Lewy bodies in brain stem and midbrain nuclei as well as widespread involvement of neocortex and limbic cortex by Lewy bodies. Many have AD-type pathology.
Some patients have a primary clinical dementia syndrome, characterized by fluctuation, early visual hallucinations, extrapyramidal tremor, which meets clinical criteria for dementia with Lewy bodies (DLB). These patients typically have Lewy bodies in brain stem and midbrain nuclei as well as widespread involvement of neocortex and limbic cortex by Lewy bodies. Many have AD-type pathology.


 GENETIC FACTORS IN ALZHEIMER’S DISEASE
GENETIC FACTORS IN ALZHEIMER’S DISEASE
Down syndrome and Alzheimer’s disease
Patients with Down syndrome (trisomy 21) develop pathologic lesions of Alzheimer’s neuropathology with age. Clinical dementia typically develops by 40 years. This is believed to be related to triplication of the APP gene on Chr 21 and aligns strongly with the amyloid cascade hypothesis. The earliest changes are deposition of Aβ as diffuse plaques with later development of tau pathology as neuritic plaques and neurofibrillary tangles.
Genetic risk factors for late onset sporadic Alzheimer’s disease
Genome-wide association studies as well as earlier linkage studies have identified several genes that increase the risk of development of AD and which now account for about 50% of the genetic risk in late-onset sporadic AD. Four main pathways can now be suggested as being linked to AD risk by a small set of genes. There is probably significant interplay between each pathway.
 Aβ metabolism – CLU, APOE, ABCA7, PICALM.
Aβ metabolism – CLU, APOE, ABCA7, PICALM.
 Immune system function – CLU, CR1, ABCA7, MS4A cluster, CD33, and EPHA1.
Immune system function – CLU, CR1, ABCA7, MS4A cluster, CD33, and EPHA1.
 Cholesterol metabolism – APOE, CLU and ABCA7.
Cholesterol metabolism – APOE, CLU and ABCA7.
 Synaptic function and cell membrane processing – PICALM, BIN1, CD33, CD2AP and EPHA1.
Synaptic function and cell membrane processing – PICALM, BIN1, CD33, CD2AP and EPHA1.
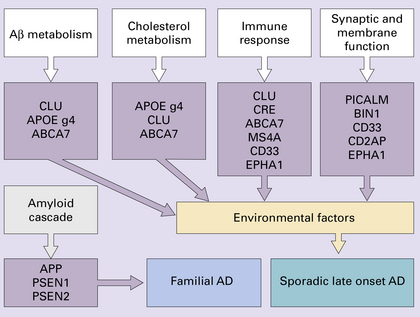
(Adapted from Morgan K. The three new pathways leading to Alzheimer’s disease. Neuropathol Appl Neurobiol 2011; 37(4):353–357.
DEMENTIA WITH LEWY BODIES (DLB)
 CLINICAL DIAGNOSIS OF DLB
CLINICAL DIAGNOSIS OF DLB The central requirement is progressive cognitive decline of sufficient magnitude to interfere with normal social or occupational function. Prominent or persistent memory impairment may not necessarily occur in the early stages, but is usually evident with progression. Deficits may be prominent on testing of attention and frontal subcortical skills, and visuospatial ability.
The central requirement is progressive cognitive decline of sufficient magnitude to interfere with normal social or occupational function. Prominent or persistent memory impairment may not necessarily occur in the early stages, but is usually evident with progression. Deficits may be prominent on testing of attention and frontal subcortical skills, and visuospatial ability.



MACROSCOPIC APPEARANCES
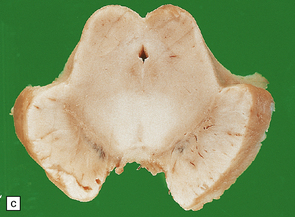
The macroscopic appearance is similar to that in AD, but atrophy of the frontal, temporal, and parietal cortex is typically only mild to moderate (Fig. 31.31a) and the occipital lobe is spared. Atrophy of limbic structures is usually moderate to severe (Fig. 31.31b). There is pallor of the substantia nigra and locus ceruleus (Fig. 31.31c).
31.31 Macroscopic appearances of DLB.
(a) Cortical atrophy over the cerebral convexities is typically less severe than that in a patient with AD of similar clinical severity. (b) There is typically significant atrophy involving the limbic system, as seen here on the medial surface of the cerebral hemisphere. (c) The substantia nigra is abnormally pale in patients with DLB, due to loss of pigmented neurons.
MICROSCOPIC APPEARANCES
The defining feature of DLB is the presence of Lewy bodies in several brain regions (Table 31.12). As in idiopathic PD, there is almost invariably a significant loss of neurons from the substantia nigra and locus ceruleus, but many of the residual neurons contain classic Lewy bodies (Fig. 31.32). There are also Lewy bodies in the cerebral cortex. Cortical Lewy bodies can be seen in sections stained with H&E (Fig. 31.33a–c) but are better demonstrated by immunochemistry for α-synuclein, P62 or ubiquitin (Fig. 31.33d).
Table 31.12
Pathologic features of dementia with Lewy bodies
Essential for diagnosis of DLB
Lewy bodies
Associated but not essential
Lewy-related neurites
Plaques (all morphological types)
Neurofibrillary tangles
Regional neuronal loss – especially brain stem (substantia nigra and locus ceruleus) and nucleus basalis of Meynert
Microvacuolation (spongiform change) and synapse loss
Neurochemical abnormalities and neurotransmitter deficits.
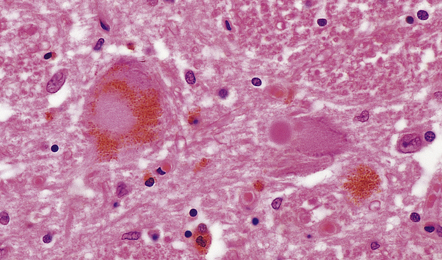
31.32 Lewy bodies in substantia nigra in DLB.
Lewy bodies and pale bodies are seen in residual neurons of the substantia nigra. The appearances are identical to those in Parkinson’s disease.
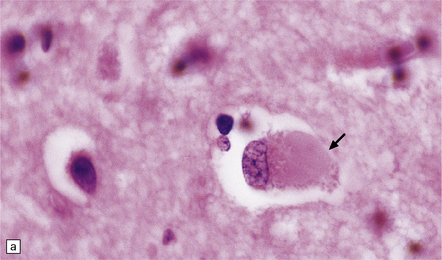
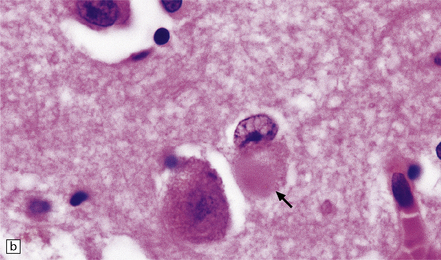
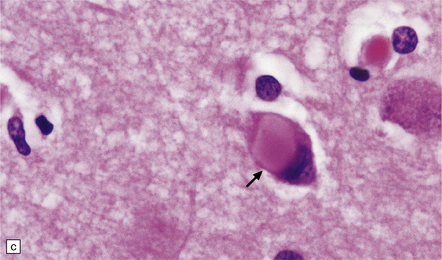
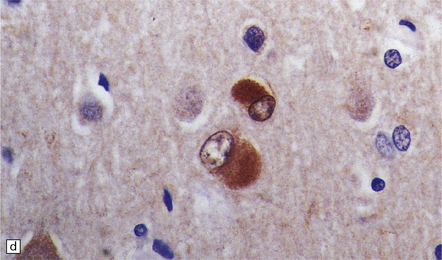
31.33 Cortical Lewy bodies.
Cortical Lewy bodies (arrows) have a variety of morphological appearances (a–c). Because the inclusions stain quite homogeneously and often have a poorly defined margin, cortical neurons that contain Lewy bodies may, on cursory examination, be confused with reactive astrocytes. Ubiquitin immunohistochemistry can be used to detect cortical Lewy bodies (d) as can staining for P62 or α-synuclein, which produces a similar appearance.
Lewy-related neurites demonstrable by immunostaining for α-synuclein or ubiquitin but not generally for tau may be present in the CA2–3 region of the hippocampus (Fig. 31.34) and in the subcortical nuclei affected by cell loss. Transcortical microvacuolation resembling that in prion disease is seen in the mesial temporal cortex in a small proportion of patients (Fig. 31.35).
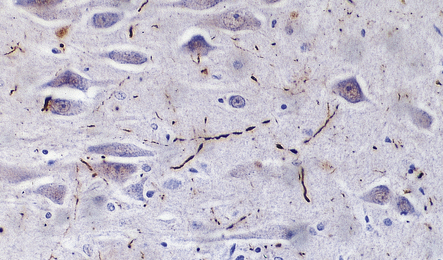
31.34 Lewy-related neurites in the hippocampus.
Lewy-related neurites are not visible on hematoxylin and eosin staining but can be stained with antibody to ubiquitin, as shown here. They are also immunopositive for α-synuclein.
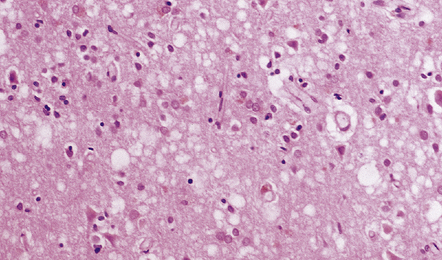
31.35 Cortical microvacuolation in DLB.
Fine transcortical vacuolation closely resembling that in prion disease may be present, but the distribution is largely restricted to the medial temporal neocortex and amygdala.
Overlap with the pathology of AD is as follows:
 Approximately 80% of affected patients with DLB have numerous diffuse plaques and smaller numbers of neuritic plaques (Fig. 31.36).
Approximately 80% of affected patients with DLB have numerous diffuse plaques and smaller numbers of neuritic plaques (Fig. 31.36).
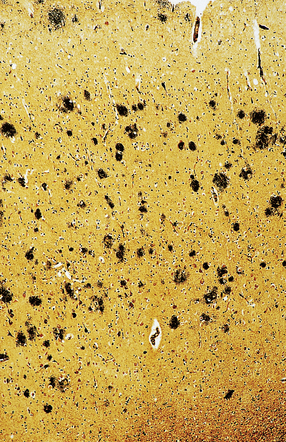


A small proportion of DLB patients have cortical and brain stem Lewy body pathology in the complete absence of AD changes. This pattern is sometimes referred to as the pure form of DLB, while the more frequent combination of cortical and brain stem Lewy bodies in the presence of AD changes is referred to as the common form of DLB. The pathologic assessment of DLB is summarized in Figure 31.37.
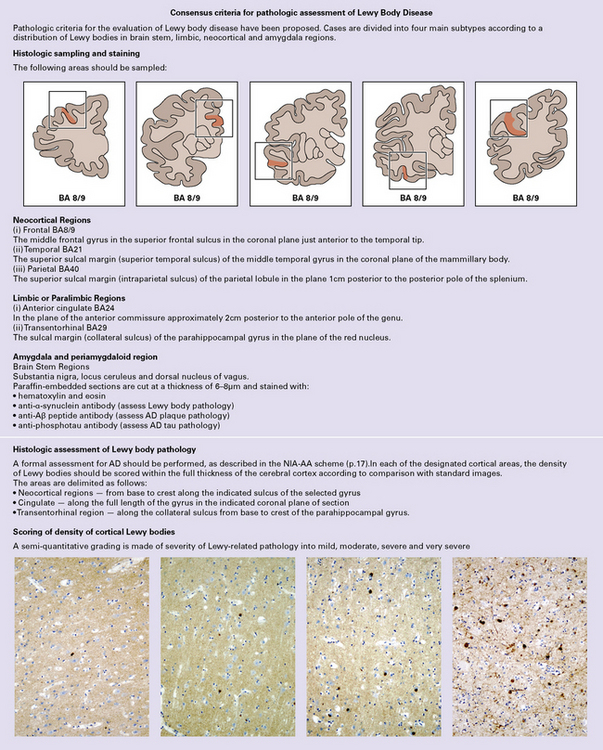
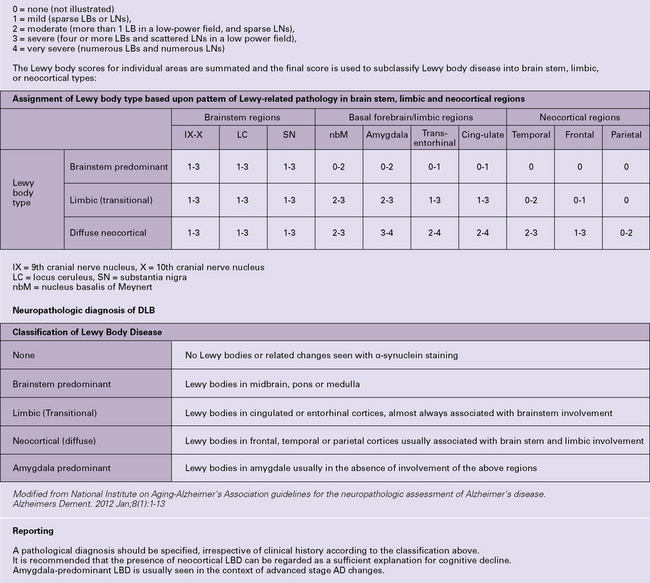
31.37 Diagnosis of Lewy body disease.
A scheme for the pathologic categorization of Lewy Body Disease into the brain stem predominant, limbic, neocortical and amygdala predominant subtypes. Reporting: (1) A pathologic diagnosis should be specified, irrespective of clinical history according to the classification above. (2) It is recommended that the presence of neocortical LBD can be regarded as a sufficient explanation for cognitive decline. (3) Amygdala-predominant LBD is usually seen in the context of advanced stage AD changes. (Modified from National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimer’s Dement 2012; 8(1):1–13)
 NEUROCHEMICAL PATHOLOGY OF DLB
NEUROCHEMICAL PATHOLOGY OF DLB

 GENETICS OF DLB
GENETICS OF DLB The APOE ε4 allele is over-represented in patients with DLB at a level intermediate between that of populations with pure AD and non-demented populations. The ε4 allele frequency is probably normal in the pure form of DLB (as it is in PD).
The APOE ε4 allele is over-represented in patients with DLB at a level intermediate between that of populations with pure AD and non-demented populations. The ε4 allele frequency is probably normal in the pure form of DLB (as it is in PD).


 Heterozygous mutations in the glucocerebrosidase gene, GBA, elevate the risk of PD and DLB.
Heterozygous mutations in the glucocerebrosidase gene, GBA, elevate the risk of PD and DLB.

FRONTOTEMPORAL LOBAR DEGENERATIONS, INCLUDING TAUOPATHIES
 CLINICAL FEATURES OF FTLD
CLINICAL FEATURES OF FTLD There are three main clinical syndromes associated with FTLD:
There are three main clinical syndromes associated with FTLD:
1. Behavioral variant of frontotemporal dementia (bvFTD) is associated with cognitive decline leading to changes in social and personal conduct associated with difficulty in regulating behavior. Patients may present with neglect of personal hygiene, ‘uncaring’ incontinence, impaired judgment, disinhibition, and stereotyped behavior. Patients typically lack insight into their cognitive decline. Imaging commonly shows that prefrontal and anterior temporal regions are atrophic.
2. Progressive nonfluent aphasia (PNFA) – patients present with problems in word retrieval (expression) but have preservation of comprehension. This pattern is typically associated with atrophy of peri-Sylvian regions in the dominant hemisphere. Imaging commonly shows that the dominant frontotemporal region is atrophic.
3. Semantic dementia – patients have impairment of that realm of memory that relates to the meaning of verbal or visual inputs (semantic memory) but have preservation of episodic memory. This pattern is typically associated with anterior temporal atrophy, particularly in the dominant hemisphere. Imaging commonly shows that temporal regions are atrophic.
 Patients may have a movement disorder, usually parkinsonism or motor neuron disease.
Patients may have a movement disorder, usually parkinsonism or motor neuron disease.

 Neuroimaging shows variable cerebral atrophy. Patterns of atrophy have some correlation with underlying pathology and molecular cause of disease (Fig. 31.38).
Neuroimaging shows variable cerebral atrophy. Patterns of atrophy have some correlation with underlying pathology and molecular cause of disease (Fig. 31.38).
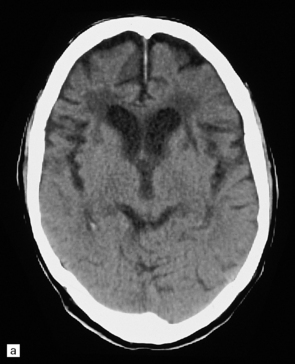
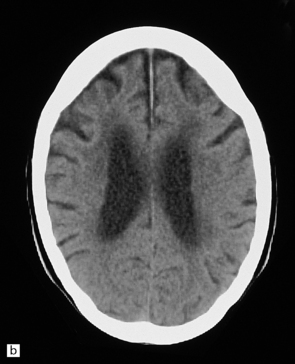
31.38 Imaging in severe frontotemporal dementia.
(a,b) CT scans from a patient with advanced frontotemporal dementia show marked atrophy of the frontal lobes, with ventricular dilatation, and low signal attenuation in the frontal white matter. (a) Horizontal scan at mid-thalamic level. (b) Horizontal scan at higher level, through body of lateral ventricles.
PATHOLOGIC SUBTYPES OF FTLD
There are five main pathologic groups in the frontotemporal lobar degenerations (Table 31.13).
Table 31.13
Tauopathies (FTLD-tau) (sporadic or inherited) – 50% cases
Frontotemporal lobar degeneration with TDP-43 pathology (FTLD-TDP) (sporadic or inherited) 45%
Frontotemporal lobar degeneration with FUS pathology (FTLD-FUS) 5%
Frontotemporal lobar degeneration with ubiquitin pathology (FTLD-UPS) <1%
Frontotemporal lobar degeneration with no inclusions seen (FTLD-ni) <1%
 GENETICS OF FTLD
GENETICS OF FTLD

There are two genes located on chromosome 17 which are linked to different forms of FTLD:
 Autosomal dominant inclusion body myopathy associated with Paget’s disease of the bone and frontotemporal dementia (IBMPFD) is related to mutations in the gene for valosin-containing protein, VCP, on chromosome 9p13–p12. This is an AAA (ATPases Associated with diverse cellular Activities) protein involved in several cellular processes including a function as a ubiquitin-protein conjugate chaperone with a role in degradation of protein complexes. Mutations are linked to a pattern of FTLD-TDP.
Autosomal dominant inclusion body myopathy associated with Paget’s disease of the bone and frontotemporal dementia (IBMPFD) is related to mutations in the gene for valosin-containing protein, VCP, on chromosome 9p13–p12. This is an AAA (ATPases Associated with diverse cellular Activities) protein involved in several cellular processes including a function as a ubiquitin-protein conjugate chaperone with a role in degradation of protein complexes. Mutations are linked to a pattern of FTLD-TDP.

THE TAUOPATHIES INCLUDING FTLD-TAU
The tauopathies can be divided into two main groups:
 The majority of patients with a tauopathy do not have a family history and have one of the conditions that are regarded as sporadic tauopathies.
The majority of patients with a tauopathy do not have a family history and have one of the conditions that are regarded as sporadic tauopathies.
 In a small number of kindreds the tauopathy is inherited, associated with mutation in the tau gene, MAPT (Tables 31.14, 31.15).
In a small number of kindreds the tauopathy is inherited, associated with mutation in the tau gene, MAPT (Tables 31.14, 31.15).
Table 31.14
Sporadic and inherited tauopathies
Sporadic tauopathy
Alzheimer’s disease
Parkinsonism-dementia complex of Guam
Postencephalitic parkinsonism
Dementia pugilistica
Familial British dementia
Progressive supranuclear palsy (PSP)
Corticobasal degeneration (CBD)
Argyrophilic grain disease (AGD)
Pick’s disease
Inherited tauopathy
Frontotemporal lobar degeneration with Parkinsonism linked to chromosome 17 tau (FTDP-17tau)
FTLD-tau pattern
Progressive supranuclear palsy pattern
Corticobasal degeneration pattern
Pick’s disease pattern
Table 31.15
Tau accumulation in different conditions
| Western blot of insoluble brain extracts | Predominant tau isoform | Diseases |
| Tau triplet 60, 64 & 68 kDa |
4R & 3R | Alzheimer’s disease Parkinsonism-dementia Complex of Guam Postencephalitic parkinsonism Dementia pugilistica Familial British dementia FTDP-17tau |
| Tau doublet 64 & 69 kDa |
4R | Progressive supranuclear palsy Corticobasal degeneration Argyrophilic grain disease FTDP-17tau |
| Tau doublet 60 & 64 kDa |
3R | Pick’s disease FTDP-17tau |
This section will consider both familial and sporadic tauopathies.
 BIOLOGY AND BIOCHEMISTRY OF TAU
BIOLOGY AND BIOCHEMISTRY OF TAU
 Tau protein is one of a family of microtubule-associated proteins (MAPs). Tau is abundant in axons, where it functions to stabilize microtubules.
Tau protein is one of a family of microtubule-associated proteins (MAPs). Tau is abundant in axons, where it functions to stabilize microtubules.
 The tau gene, MAPT, is located on chromosome 17. Alternate splicing of transcripts of MAPT leads to the synthesis of six main isoforms of tau, all containing tandem repeat regions that are encoded by exons 9–12 and serve as microtubule-binding domains. Three of the isoforms include exon 10 and have four microtubule-binding repeat domains (4Rtau). Three of the tau isoforms lack exon 10 and have three microtubule-binding repeat domains (3Rtau). The tau isoforms range from 352 to 441 amino acids in length, with molecular weights of 45–65 kDa (Table 31.15).
The tau gene, MAPT, is located on chromosome 17. Alternate splicing of transcripts of MAPT leads to the synthesis of six main isoforms of tau, all containing tandem repeat regions that are encoded by exons 9–12 and serve as microtubule-binding domains. Three of the isoforms include exon 10 and have four microtubule-binding repeat domains (4Rtau). Three of the tau isoforms lack exon 10 and have three microtubule-binding repeat domains (3Rtau). The tau isoforms range from 352 to 441 amino acids in length, with molecular weights of 45–65 kDa (Table 31.15).

PICK’S DISEASE: CLINICAL FEATURES
MACROSCOPIC APPEARANCES
In Pick’s disease atrophy of the frontal and temporal lobes is typically very severe, in some cases producing ‘blade-like’ or ‘knife-edge’ gyri (Fig. 31.39). The posterior part of the superior temporal gyrus is usually spared. In some cases of Pick’s disease, atrophy is only moderate.
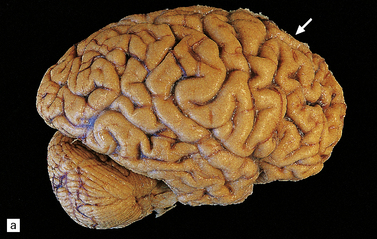
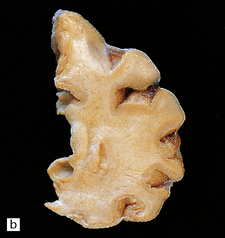
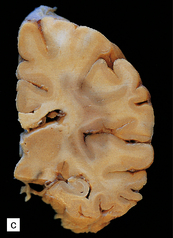
31.39 Pick’s disease macroscopic features.
(a) Lateral view of brain. There is sharp demarcation (arrow) between the atrophic frontal lobe and the posterior part of the cerebrum. Temporal atrophy is moderate in this case. (b) A coronal slice through the frontal lobe from the same brain reveals marked cortical atrophy, especially inferiorly and superomedially. The white matter has a gelatinous gray appearance. (c) There is good preservation of the cerebrum in the posterior frontal/anterior parietal region. The inferomedial temporal neocortex and subjacent white matter are atrophic. No macroscopic abnormalities are discernible in the hippocampus.
MICROSCOPIC APPEARANCES
The cardinal histologic abnormality is the presence of Pick bodies. These are spherical inclusions in neuronal cell bodies. In contrast to Lewy bodies, Pick bodies are slightly basophilic and have a crisp margin (Fig. 31.40a). They are strongly argyrophilic (Fig. 31.40b,c) and may be seen in pyramidal neurons and dentate granule cells in the hippocampus and in affected regions of neocortex. They may be present in low density in subcortical nuclei (Fig. 31.40d).
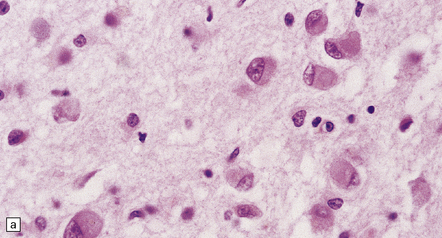
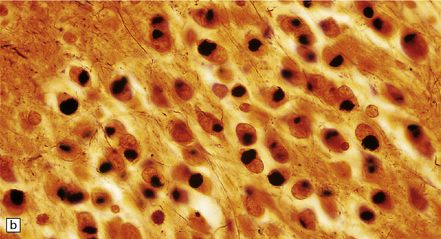
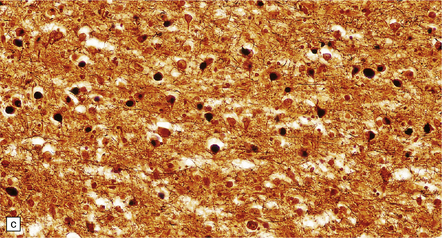
31.40 Pick bodies.
(a) In sections stained with hematoxylin and eosin, Pick bodies appear as well demarcated round, slightly basophilic inclusions in the neuronal cytoplasm. They are shown here in the amygdala. (b) Bielschowsky silver impregnation of Pick bodies in neurons of the hippocampal dentate gyrus. (c) Silver impregnation of Pick bodies in the superficial frontal cortex. (d) Pick body in a large neuron in the putamen.
Electron microscopy of Pick bodies shows that they contain 15 nm straight filaments, and some 22–24 nm twisted filaments appearing similar to the PHF in AD. Entrapped vesicular structures are also present. Immunohistochemistry shows reactivity for phosphorylated tau protein (Fig. 31.41), ubiquitin, tubulin, and chromogranin-A. The tau protein in Pick’s disease differs from that in other tau disorders in that only 3Rtau isoforms are present, from transcripts lacking exon 10.
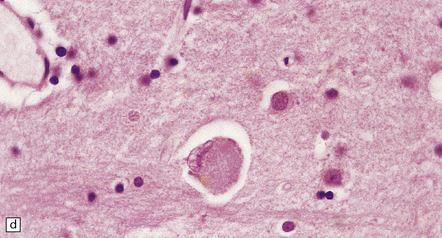
31.41 Tau immunostaining of Pick bodies.
(a) At low magnification one can see that Pick bodies are numerous in laminae II and III but are also present in the deeper parts of the cortex. (b) High magnification view of the immunostained spherical neuronal inclusions. Fine tau-immunoreactive neurites may also be present.
Swollen neurons are typical of Pick’s disease, when they are termed Pick cells, but vary in number in relation to the severity of the neuronal loss. Swollen neurons are argyrophilic and can be stained with antisera to phosphorylated neurofilament protein or αB-crystallin (Fig. 31.42). Tau immunoreactivity is often also present in swollen neurons.
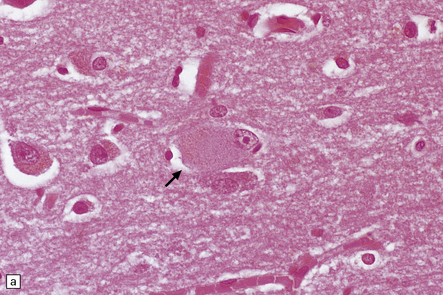
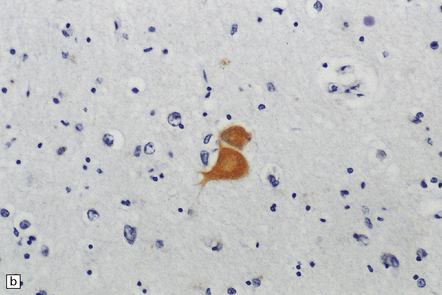
31.42 Swollen neurons in Pick’s disease called Pick cells.
(a) Swollen neuron (arrow) with eosinophilic cytoplasm and eccentrically displaced nucleus. (b) αB-crystallin immunolabeling.
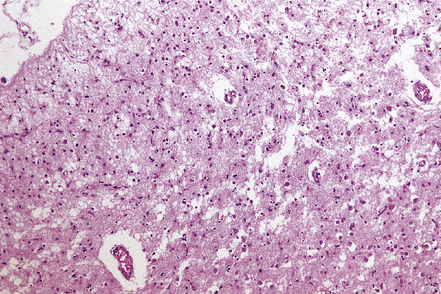
Neuronal loss relates to the degree of cortical atrophy present. In severe cases neuronal loss in affected regions of cortex is virtually complete, resulting in status spongiosus (Fig. 31.43). In cases with moderate cortical atrophy there is microvacuolation in Layer II of the cortex and restricted neuronal loss. Astrocytic gliosis is present in areas of cortical neuronal loss and in underlying white matter. Granulovacuolar degeneration is commonly seen in remaining neurons.
FRONTOTEMPORAL DEGENERATION AND PARKINSONISM LINKED TO CHROMOSOME 17 TAU (FTDP-17TAU)
MICROSCOPIC APPEARANCES
Immunohistochemical staining is the key to establishing a diagnosis, and reveals extensive accumulation of phosphorylated tau in neurons and glial cells, both astrocytes and oligodendrocytes (Fig. 31.44). Diffuse tau-immunoreactivity within neurons in the form of so-called pretangles is the dominant component of neuronal pathology. Tau-immunoreactive glial lesions are frequently seen and include tufted astrocytes and astrocytic plaques. Oligodendroglial coiled bodies are common. Myelin loss and astrocytic gliosis of white matter are seen in some cases.
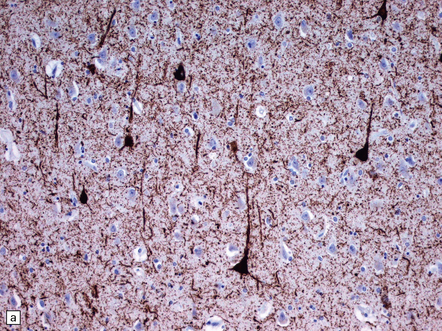
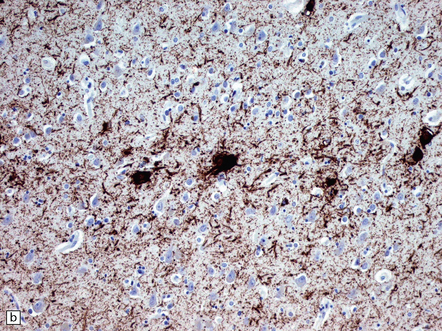
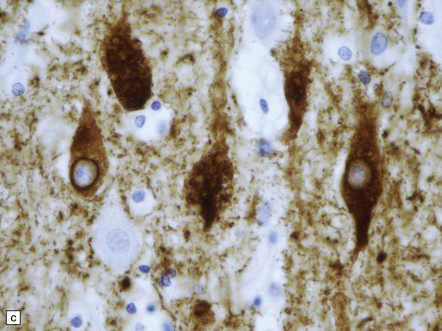
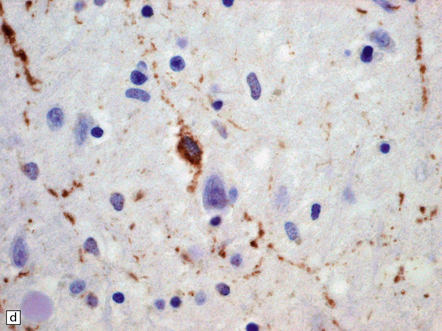
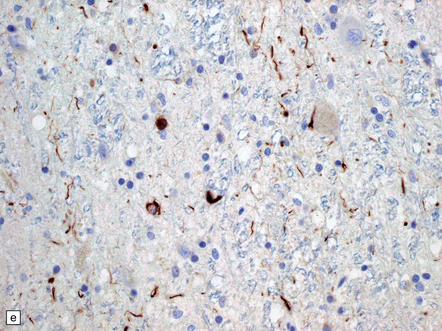
31.44 Tau pathology in FTDP-17tau.
Immunostaining for phosphorylated tau typically shows extensive neuronal and glial pathology. Affected cerebral cortex shows neuronal pre-tangles as well as widespread neuritic staining (a). In some cases tau-positive swollen neurons are present (b). Much of the tau staining is as pretangles. A ring-shaped pattern of tau around the nucleus is sometime seen (c). Phosphorylated tau accumulates in both astrocytes and oligodendroglia, including in some cases in the form of white matter threads (d,e).
The tau pathology shows some correlation with the type of underlying MAPT mutation:
 Missense mutations outside exon 10 (e.g. G272V, V337M, R406W) cause tau to accumulate predominantly in neurons, and to consist of straight filaments composed of all six isoforms (both 4R and 3R tau). These filaments are similar to the paired helical filaments (PHFs) and straight filaments of AD. In some cases the tau pathology resembles that in AD (with the exception of the plaque-related tau), in others it resembles that in PSP and CBD (p. 627).
Missense mutations outside exon 10 (e.g. G272V, V337M, R406W) cause tau to accumulate predominantly in neurons, and to consist of straight filaments composed of all six isoforms (both 4R and 3R tau). These filaments are similar to the paired helical filaments (PHFs) and straight filaments of AD. In some cases the tau pathology resembles that in AD (with the exception of the plaque-related tau), in others it resembles that in PSP and CBD (p. 627).

DEMENTIA WITH CHANGES OF CBD OR PSP
Some patients who develop a clinical frontotemporal dementia syndrome have pathologic changes of a tauopathy with the pattern of either CBD or PSP. These conditions have been discussed in Chapter 28 as examples of extrapyramidal movement disorders. Review has shown a proportion of such cases to have a mutation in MAPT and therefore to fall within the spectrum of FTDP-17tau.
Progressive supranuclear palsy
PSP usually presents with an extrapyramidal movement disorder (described in Chapter 28) but cognitive impairment is common and in some patients can be the presenting feature. Tauopathy with features of PSP has also been seen underlying clinical primary progressive aphasia and corticobasal syndrome. Histologic features are those of PSP.
Corticobasal degeneration
The corticobasal clinical syndrome involves both movement disorder due to nigral and basal ganglia disease as well as cortical features leading to cognitive symptoms including apraxia, sensory deficits and the “alien hand” phenomenon. Some patients have a clinical presentation of frontotemporal dementia or progressive aphasia, hence its inclusion as part of the spectrum of FTLD-tau. (Macroscopic and histologic features are as described in Chapter 28 on page 579 and Figure 31.37.) Some patients who present with a corticobasal syndrome have a non-tau form of FTLD (FTLD-TDP).
TANGLE-ONLY DEMENTIA
MACROSCOPIC AND MICROSCOPIC APPEARANCES
Numerous 3R and 4R tau-immunoreactive NFTs and NTs are present in the hippocampus, entorhinal cortex, and amygdala, corresponding to Braak stage III, with many tangles being ‘ghost tangles’ (Fig. 31.45). In about 60% of reported cases plaques have been absent. In others there have been only a few diffuse Aβ deposits, consistent with aging. The substantia nigra typically shows neuronal loss and NFT formation. In making this diagnosis, a search should be made to exclude diagnostic features in other defined tauopathies such as PSP, and argyrophillic grain disease (AGD).
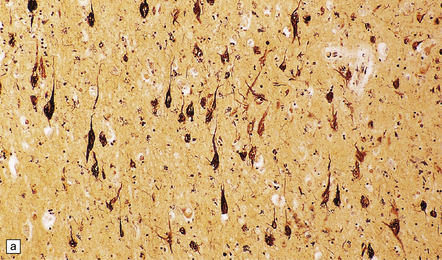
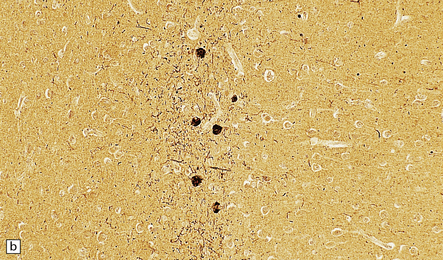
31.45 Tangle-only dementia.
(a) In tangle-only dementia the temporal cortex contains abundant NFTs, demonstrated here by silver impregnation, in the absence of plaques. (b) Tau-immunostaining sometimes shows a band of involved neurons in the entorhinal cortex, with abundant surrounding neuropil threads.
ARGYROPHILIC GRAIN DEMENTIA (AGD)
MACROSCOPIC AND MICROSCOPIC APPEARANCES
Small, spindle-shaped, argyrophilic grains and coiled bodies or filaments are found in the hippocampus, entorhinal cortex and surrounding regions, and in some subcortical nuclei (Fig. 31.46). These argyrophilic structures are composed of tau-immunoreactive straight filaments with a diameter of about 9 nm. Many pyramidal neurons in these same regions show coarse granular accumulation of phosphorylated tau within the cell body and dendrites, but NFTs and NTs are relatively sparse. Tau reactive astrocytes, oligodendroglial coil bodies and occasional interfascicular threads may be seen, mainly in the parahippocampal white matter. There may also be occasional astrocytic tau plaques of the type seen in CBD but restricted to the anterior temporal cortex. Aβ plaques are often present but tend to be predominantly diffuse. Ballooned neurons may be seen in nearby temporal cortex.
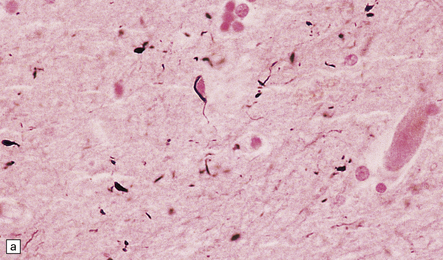
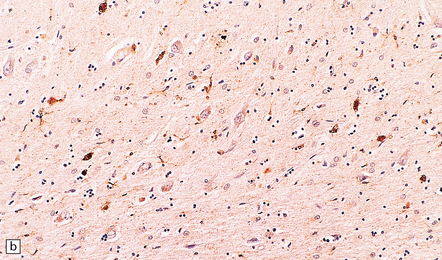
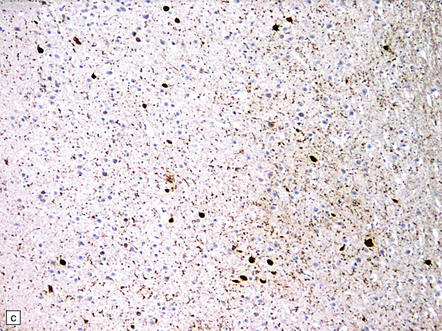
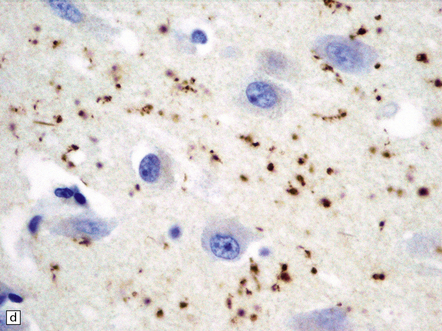
31.46 Argyrophilic grain dementia (AGD).
(a) Gallyas silver impregnation of coils and grains. (b) Tau immunostaining also highlights the coils and grains with larger structures being neurons containing tangles. (c) P62 staining shows grains with larger structures being neurons containing tangles. (d) P62 stain to show morphology of grains.
FRONTOTEMPORAL LOBAR DEGENERATION WITH TDP-43 PATHOLOGY (FTLD-TDP)
MACROSCOPIC APPEARANCES
Frontal and temporal atrophy can vary from mild/moderate to very severe (Fig. 31.47). The basal ganglia may show mild to moderate atrophy, and the substantia nigra moderate pallor.
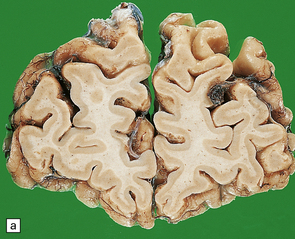
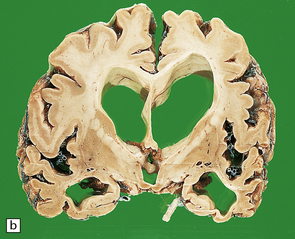
31.47 Macroscopic features of FTLD-TDP.
These coronal slices are from a patient with clinical features of frontotemporal dementia. Neuropathologic assessment revealed ubiquitin-positive neuronal inclusions and neurites which were TDP-43 immunoreactive. (a) There is marked frontal lobe atrophy. (b) Marked ventricular enlargement, with atrophy of the temporal lobe and moderate atrophy of the basal ganglia. Some cases show hippocampal sclerosis.
MICROSCOPIC APPEARANCES
In early disease, neuronal loss and astrocytic gliosis are restricted to the superficial neocortical laminae, which show microvacuolation (Fig. 31.48). Variable numbers of swollen neurons may be present. Severe disease is characterized by virtually complete transcortical loss of neurons from affected regions, with status spongiosus (Fig. 31.49). In patients with associated amyotrophic lateral sclerosis/motor neuron disease, loss of upper or lower motor neurons can be seen together with variable loss of corticospinal tracts. There is usually rarefaction and gliosis of white matter in the affected regions. Neuronal loss and astrocytic gliosis may involve the basal ganglia and substantia nigra.
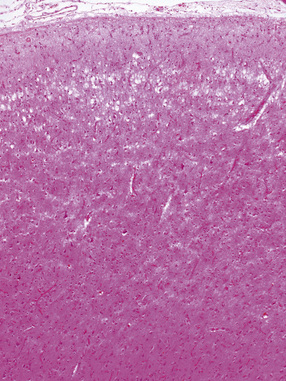
31.48 Cortical microvacuolization.
This image shows characteristic Layer 2 microvacuolation and neuronal loss in a case of FTLD.
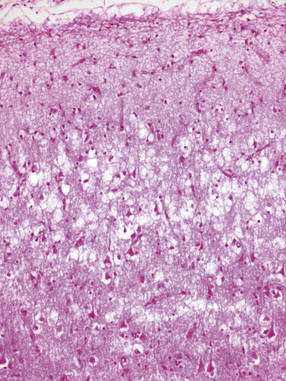
31.49 Cortical neuronal loss in FTLD.
This image shows more severe neuronal loss in FTLD. In severe cases, this can be through the full cortical thickness and associated with astrocytic gliosis.
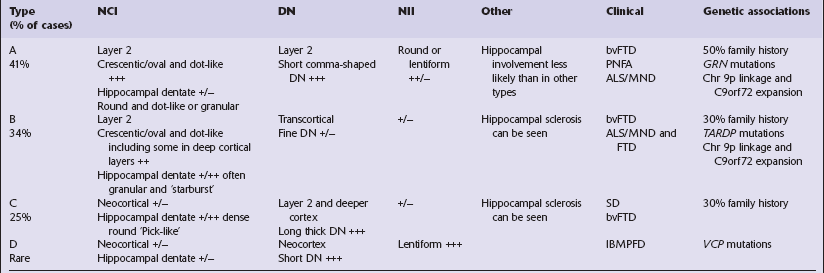
Three main histologic abnormalities are seen on immunohistochemistry for ubiquitin, P62, and TDP-43. Several authors have proposed conceptually similar classification systems for FTLD-TDP cases based on these pathologic findings, which have been reconciled into four groups, A–D, in a consensus proposal (Table 31.16).
 Neuronal cytoplasmic inclusions (NCI) can be seen in neocortical neurons and in dentate granule cells of the hippocampus. A range of morphological types of NCI have been described. Hippocampal involvement varies in particular types of FTLD-TDP (Figs 31.50–31.52, Table 31.16).
Neuronal cytoplasmic inclusions (NCI) can be seen in neocortical neurons and in dentate granule cells of the hippocampus. A range of morphological types of NCI have been described. Hippocampal involvement varies in particular types of FTLD-TDP (Figs 31.50–31.52, Table 31.16).
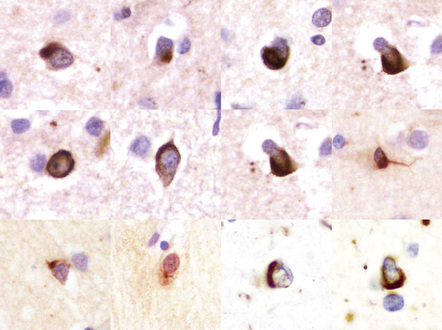
31.50 Cortical NCI in FTLD-TDP.
Inclusions can be detected with antibodies to TDP-43, ubiquitin or P62. They range from small paranuclear granular deposits, through crescent-shaped and ring-shaped structures, to small spherical paranuclear inclusions.
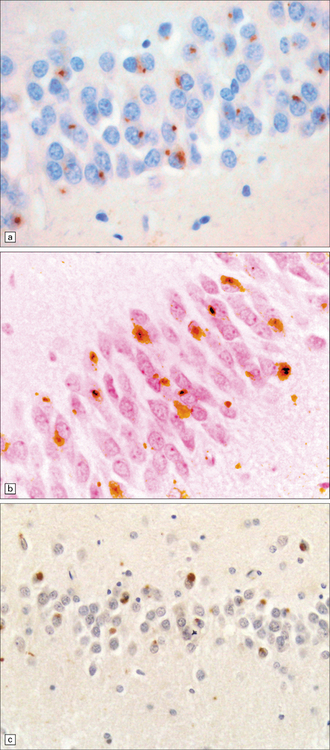
31.51 Hippocampal NCI in dentate granule cells FTLD-TDP.
The morphology of hippocampal neuronal cytoplasmic inclusions varies between different types of FTLD-TDP. Some are dot-like (a). Others have a more dispersed granular appearance (b). In some cases inclusions appear as larger structures which are Pick body-like or bean-shaped (c).
31.52 Neuronal cytoplasmic inclusions in hippocampal dentate granule cells labeled with antibody to TDP-43.
TDP-43 is normally detectable in the nucleus but not the cytoplasm. Neurons with cytoplasmic inclusions have unlabeled (blue) nuclei. Neurons that do not have inclusions show normal nuclear labeling of TDP-43.
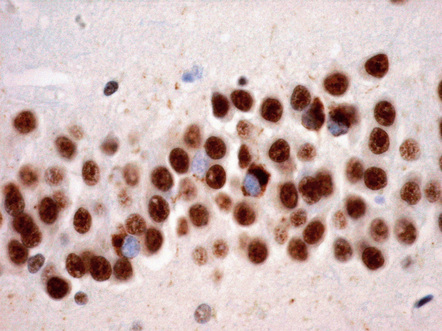
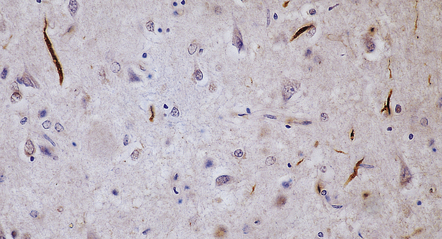
 Dystrophic neurites (DN) seem to be of two qualitatively different types and most prominent in regions of cortical microvacuolation. Long, linear neurites are mainly in outer cortical layers while shorter, comma-shaped neurites occur in all cortical layers. The different types of neurite are associated with different types of disease (Figs 31.53, 31.54, Table 31.16).
Dystrophic neurites (DN) seem to be of two qualitatively different types and most prominent in regions of cortical microvacuolation. Long, linear neurites are mainly in outer cortical layers while shorter, comma-shaped neurites occur in all cortical layers. The different types of neurite are associated with different types of disease (Figs 31.53, 31.54, Table 31.16).
31.53 Long dystrophic neurites in FTLD-TDP.
These structures are mainly seen in outer cortical layers and typically run perpendicular to the cortical surface. They can be detected with antibodies to TDP-43, P62 or ubiquitin.
31.54 Short cortical dystrophic neurites in FTLD-TDP.
This low magnification image of a section labeled with anti-P62 shows layer 2, which includes numerous short, twisted and comma-like DN. Many small neurons also contain NCI.
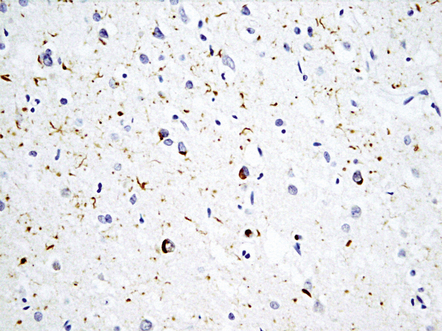
 Neuronal intranuclear inclusions (NII) are seen in a proportion of cases. These are typically elongated and lenticular in shape. There are particularly prominent in patients with VCP mutations but may be seen in lower density in other types of FTLD-TDP (Fig. 31.55 and Table 31.16).
Neuronal intranuclear inclusions (NII) are seen in a proportion of cases. These are typically elongated and lenticular in shape. There are particularly prominent in patients with VCP mutations but may be seen in lower density in other types of FTLD-TDP (Fig. 31.55 and Table 31.16).
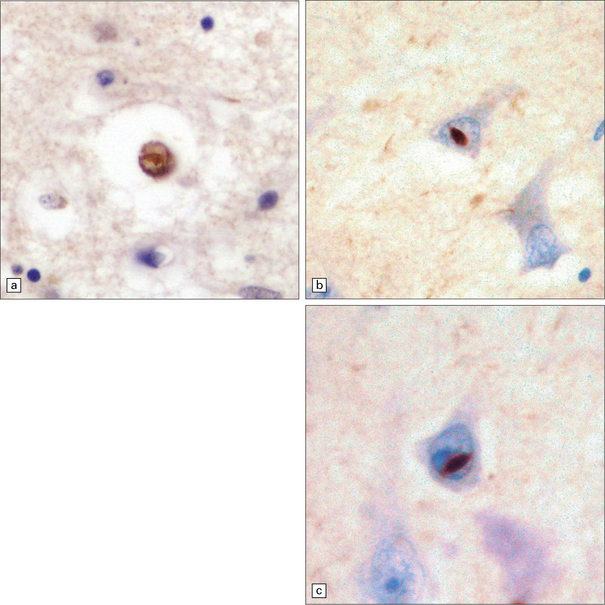
Hippocampal sclerosis is seen in some cases, as detailed in Table 31.16. It is now recognized that certain forms of hippocampal sclerosis previously classified as representing an isolated (‘pure’) abnormality are in reality cases of TDP-43 proteinopathy.
 BIOLOGY AND SIGNIFICANCE OF TDP-43 IMMUNOSTAINING
BIOLOGY AND SIGNIFICANCE OF TDP-43 IMMUNOSTAINING
TDP-43 in inclusions undergoes truncation, phosphorylation, and ubiquitination.
TDP-43 inclusions have been reported in about 3% of elderly neurologically normal patients.
Inclusion body myopathy with Paget’s disease of the bone and frontotemporal dementia (IBMPFD)
This is an autosomal dominant multi-system disease linked to mutations in VCP. About a third of patients develop a frontotemporal dementia associated with brain atrophy. In addition, patients develop a myopathy with pathologic features similar to inclusion body myositis, as well as Paget’s disease of bone. This condition is associated with a type D form of TDP-43 proteinopathy (Table 31.16). Neuropathologic assessment has shown neuronal loss and astrocytic gliosis in neocortical areas, with superficial cortical microvacuolation similar to that shown in Figure 31.48.
C9orf72 and chromosome 9-linked FTLD (C9FTD/ALS)
 p62 positive NCIs and NIIs in the pyramidal cell layer of the hippocampus.
p62 positive NCIs and NIIs in the pyramidal cell layer of the hippocampus.
 p62 positive NCIs w in the cerebellar granular and molecular layers.
p62 positive NCIs w in the cerebellar granular and molecular layers.
 FRONTOTEMPORAL DEMENTIA WITH MOTOR NEURON DISEASE
FRONTOTEMPORAL DEMENTIA WITH MOTOR NEURON DISEASE
A proportion of patients with MND develop a frontotemporal dementia syndrome. In a reverse situation, a proportion of patients who present with bvFTD, PNFA, SD or CBS may develop motor neuron signs. This condition has been referred to as FTD-MND to describe the clinical association, however understanding of the disease pathogenesis in such patients would put the majority of cases into the group of FTLD-TDP. Motor neuron disorders (MND) are discussed in Chapter 27. In patients with pure MND it is possible to see extramotor pathology of the type described in FTLD.
Frontotemporal lobar degenerations with FUS pathology (FTLD-FUS)



 MOLECULAR PATHOLOGY AND SIGNIFICANCE OF FUS
MOLECULAR PATHOLOGY AND SIGNIFICANCE OF FUSAtypical FTLD with ubiquitin-only immunoreactive changes
Histologically, there are ubiquitin- or P62-positive, tau/TDP-43-negative neuronal cytoplasmic inclusions (NCI) which are strongly immunoreactive for FUS. The majority of NCI appear as small, compact, round or oval inclusions, which sit next to the nucleus and are rarely larger than it. These bean-shaped inclusions are commonly seen in outer layers of the cerebral cortex (Fig. 31.56), hippocampal dentate fascia (Fig. 31.57) and periaqueductal gray matter. In addition to bean-shaped FUS inclusions, crescentic NCI can also be seen in the striatum. A coarse granular pattern of FUS accumulation is demonstrable in pyramidal neurons in some cases. Neuritic pathology is not evident although rare fine FUS-positive filaments may be seen. A small proportion of lower motor neurons contain inclusions, some of which may have a skein appearance.
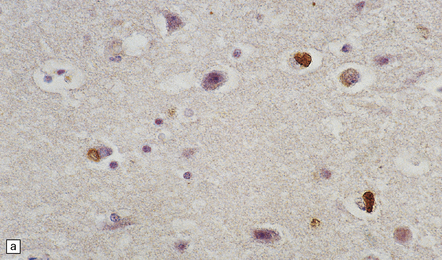
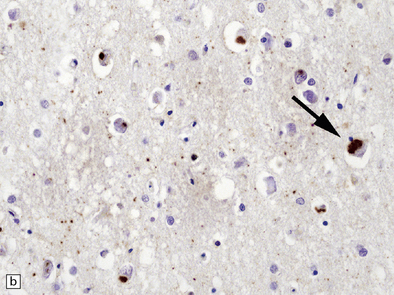
31.56 aFTLD-U NCI P62 and FUS staining in cerebral cortex.
Inclusions are typically dense-stained oval and ‘bean shaped’ roughly the same size as the neuronal nucleus. They can be seen with P62 (a) or FUS (b) staining (arrow). The section illustrated in (b) was immersed for a few days in formalin prior to immunohistochemistry, to minimize the normal nuclear immunopositivity for FUS.
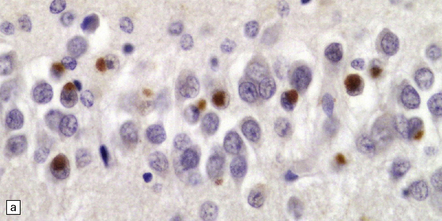
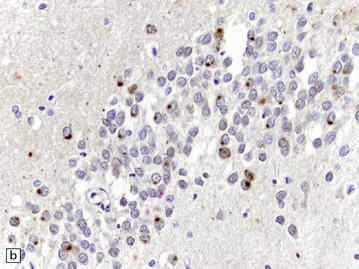
31.57 aFTLD-U NCI ubiquitin and FUS staining hippocampus.
Hippocampal dentate granule cells contain compact round inclusions that can be labeled for ubiquitin (a), P62, or FUS (b).
Another prominent abnormality is the presence of neuronal intranuclear inclusions (NII) which are long and thin, and may either appear to run straight across the nucleus as a rod or to be curved around the nuclear margin (vermiform NII). These are present in the hippocampus, especially in dentate granule cells (Fig. 31.58).
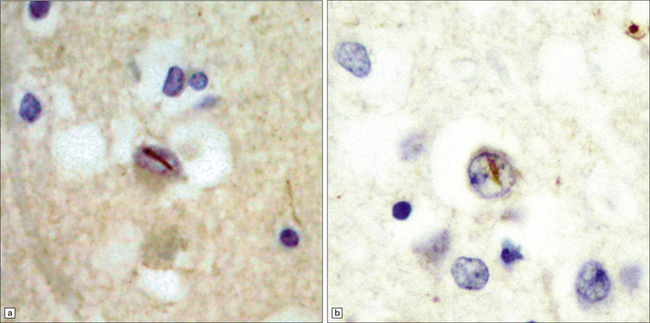
NEURONAL INTERMEDIATE FILAMENT INCLUSION DISEASE (NIFID)
Histologically, affected cortical regions show neuronal loss, superficial microvacuolation and astrocytic gliosis. Neuronal cytoplasmic inclusions can be seen in neocortex, hippocampus and basal ganglia. Some of the inclusions are usually visible on H&E staining (Fig. 31.59a,b) but these represent but a small subset of those that are demonstrable immunohistochemically. It is important to note that one cannot rely on ubiquitin IHC to screen for these inclusions as they typically show only weak labeling (Fig. 31.59f).
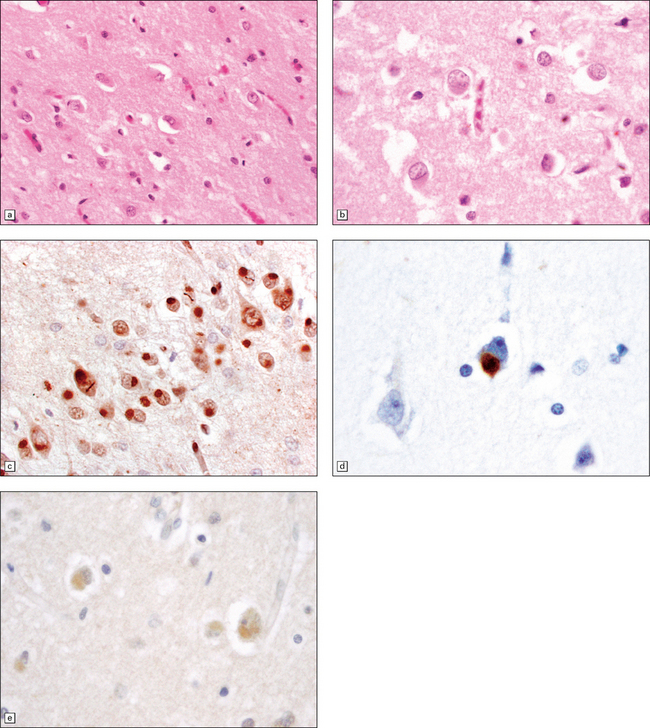
31.59 NIFID inclusions in neocortex.
H&E staining reveals small ovoid cytoplasmic inclusions, about the same size as the neuronal nucleus. Some have a more strongly eosinophilic core (a,b). FUS staining shows the inclusions to have varied morphologies (c). A proportion of inclusions is immunoreactive for neurofilament protein (d) but only very weakly for ubiquitin (e).
Immunohistochemistry for FUS shows many more inclusions than are seen in H&E sections. Neocortical NCI are of varied morphology: crescent shaped, annular, flame-shaped, or forming small, round Pick-like inclusions. Rod-shaped and curved vermiform neuronal intranuclear inclusions are present (Fig. 31.59c,d). A subset of cortical inclusions is also immunoreactive for neurofilament protein (NF) (Fig. 31.59e) and/or α-internexin. Hippocampal dentate granule cells contain FUS-positive rounded NCI as well as vermiform or annular intranuclear inclusions. Occasional FUS-positive oligodendroglial inclusions have been noted.
BASOPHILIC INCLUSION BODY DISEASE (BIBD)
This neurodegenerative condition was originally characterized pathologically by the presence of neuronal cytoplasmic inclusions which were well defined and basophilic on H&E staining: basophilic inclusions (BI) (Fig. 31.60). BIBD may be associated with sporadic ALS/MND (including, in particular, juvenile ALS), ALS/MND with dementia and a form of pure frontotemporal dementia (FTD). Since the discovery that these inclusion bodies contain FUS as well as related proteins, BIBD is now classed as one of the TDP-FUS group.
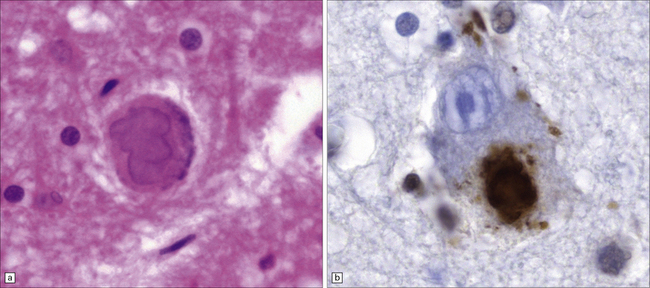
31.60 BIBD.
On H&E staining, inclusions can be seen in a proportion of neurons as basophilic areas in the cytoplasm, typically with crisply-defined, more deeply basophilic margins (a). FUS staining is the most sensitive way to detect the inclusions (b). (Courtesy of Dr Olaf Ansorge, Oxford.)
On immunostaining for FUS, cytoplasmic inclusions can be seen in neocortical neurons, hippocampal pyramidal cells and dentate granule cells, neurons in the globus pallidus and thalamus. Inclusions are also present in the midbrain, pons and medulla (including the hypoglossal nuclei) and in LMN of the spinal cord. Neuronal cytoplasmic inclusions appear as one of three main types (Fig. 31.37):
 Compact, round inclusions in the cerebral cortex.
Compact, round inclusions in the cerebral cortex.
 Crescentic and annular inclusions in the cerebral cortex and striatum.
Crescentic and annular inclusions in the cerebral cortex and striatum.
 Large round inclusions or complex pleomorphic inclusions in brain stem and deep gray matter nuclei.
Large round inclusions or complex pleomorphic inclusions in brain stem and deep gray matter nuclei.
HIPPOCAMPAL SCLEROSIS
Macroscopically, the hippocampal region shows atrophy. Histologically, loss of hippocampal pyramidal cells is seen in CA1 and the subiculum (Fig. 31.61)
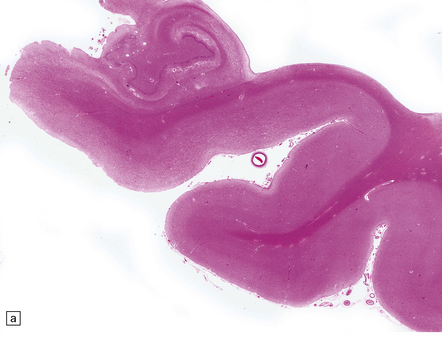
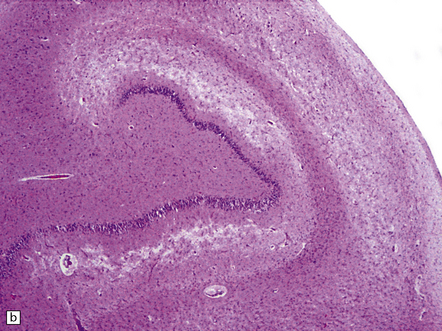
FAMILIAL BRITISH DEMENTIA AND FAMILIAL DANISH DEMENTIA
MICROSCOPIC APPEARANCES
The characteristic findings are of severe widespread amyloid angiopathy with parenchymal amyloid plaques. The amyloid is composed of a 24-amino-acid peptide (ABRI) and antibodies to this peptide can be used in diagnosis (Fig. 31.62). Immunohistochemistry for phosphorylated tau reveals NFTs and NTs in a distribution corresponding approximately to Braak stage II but also clustered around amyloid-laden parenchymal blood vessels.
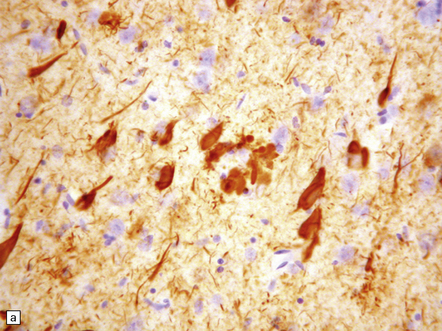
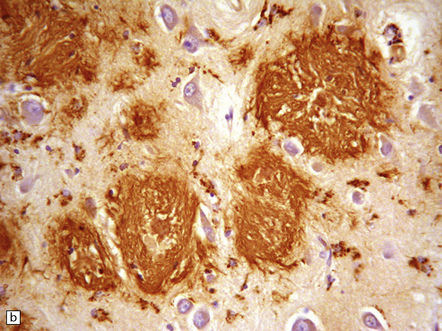
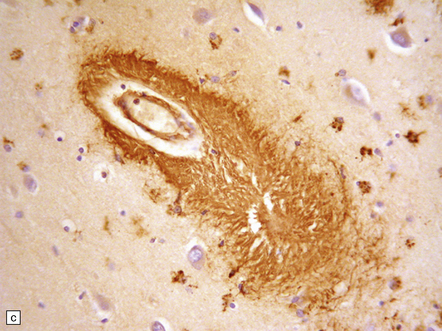
31.62 Familial British dementia.
Neurofibrillary tangles are a feature of disease, shown here by immunohistochemistry for phosphorylated tau (a). Immunostaining for ABri shows parenchymal plaques (b) as well as abundant vascular staining with adjacent perivascular parenchymal amyloid (c). Tau-immunopositive neuronal processes are present in the regions of perivascular amyloid. (Courtesy of Professor T. Revesz, Institute of Neurology, London, UK.)
DEMENTIA ASSOCIATED WITH OTHER DEGENERATIVE DISEASES
Several diseases typically present as early onset degenerative diseases of the nervous system but can present with a dementia syndrome in adult life (Table 31.17).
Table 31.17
Degenerative diseases that cause dementia and usually present in childhood
Mitochondrial cytopathy
Neuronal intranuclear inclusion disease
Lafora body disease
Neuronal ceroid lipofuscinosis
Wilson’s disease
Fabry’s disease
Krabbe’s disease
Metachromatic leukodystrophy
Alexander’s disease
Cerebrotendinous xanthomatosis
Nasu–Hakola disease
Adrenoleukodystrophy
GM1 gangliosidosis type III
GM2 gangliosidosis
Gaucher’s disease
Niemann–Pick disease type C
Mucopolysaccharidosis type IIIB
THALAMIC DEMENTIAS
Severe disturbance of memory and other cognitive functions amounting to a dementia syndrome may result from thalamic pathology. Neuronal loss and astrocytic gliosis affect the thalamus in a range of neurodegenerative disorders, including rare conditions regarded as primary thalamic degenerations (see Chapter 33).
VASCULAR DEMENTIA
 Primary vascular dementia, in which cerebrovascular disease is the dominant pathologic cause of cognitive decline.
Primary vascular dementia, in which cerebrovascular disease is the dominant pathologic cause of cognitive decline.

 RISK FACTORS FOR VASCULAR DEMENTIA
RISK FACTORS FOR VASCULAR DEMENTIA
Risk factors for vascular dementia can be divided into five main groups: demographic, atherosclerotic, stroke-related, poor cerebral perfusion, and genetic (Table 31.18).
Table 31.18
Risk factors for vascular dementia
| Risk category | Risk factor |
| Demographic | Age, male sex, lower educational level |
| Atherosclerotic | Hypertension, cigarette smoking, myocardial infarction, diabetes mellitus, hyperlipidemia |
| Stroke-related | Large cerebral infarcts, bilateral cerebral infarcts, ischemic white matter disease, strategic infarcts (thalamic, angular gyrus, or subcortical frontal infarction) |
| Poor cerebral perfusion or oxygenation | Obstructive sleep apnea, congestive cardiac failure, cardiac arrhythmias, major surgery, orthostatic hypotension |
| Genetic | Familial vascular encephalopathies (CADASIL) |
MACROSCOPIC AND MICROSCOPIC APPEARANCES
The neuropathologic abnormalities which are potentially associated with vascular cognitive impairment can be classified according to the cause of the ischemia: large vessel disease, small vessel disease and hypoperfusion (see Chapter 9). Rarely there are specific vasculopathies leading to disease. (Table 31.19).
Table 31.19
Pathology of vascular dementia
Large vessel disease
Large volume infarcts or bilateral infarcts
Strategically placed infarcts
Small vessel disease
Lacunar infarcts
Ischemic white matter damage (leukoencephalopathy)
Cribriform change
Cortical microinfarcts
Hypoperfusion lesions
Hippocampal sclerosis
Border zone infarcts
Cortical laminar necrosis
Rare local vascular disorders
CADASIL
Cerebral amyloidosis
Cerebral vasculitis (including secondary to autoimmune disorders)
Antiphospholipid antibody syndrome
 Ischemic white matter degeneration. Macroscopically, the brain is usually of normal or slightly reduced weight with little external evidence of atrophy. The major cerebral arteries are usually atherosclerotic. The lateral and third ventricles are moderately dilated. The cerebral white matter often feels soft on sectioning. The cut surface appears slightly granular and may be pitted with small depressions (Fig. 31.63). When these changes are seen in life on imaging it is termed ‘leukoaraiosis’. Typically, these changes are most pronounced in the deep frontal and temporal white matter. Coexistent lacunar infarcts are usually present in the basal ganglia, thalamus, pons, and, less often, the hemispheric white matter (Fig. 31.64). Histology reveals arteriosclerosis and arteriolosclerosis – hyalinization of small arteries and arterioles with loss of smooth muscle and replacement by collagen (Fig. 31.65). The white matter shows patchy pallor on myelin staining, axonal loss, reduction in the number of oligodendrocytes, and mild astrocytic gliosis (Fig. 31.66). There is often an associated loss of axons in these areas. Dementia with these changes is now clinically termed subcortical vascular dementia (SVaD) but has also been referred to in the past as lacunar dementia and Binswanger’s disease. Note that if histologic sampling is restricted to small blocks that include only 1–2 cm of subcortical white matter, significant deep white matter disease may well be missed.
Ischemic white matter degeneration. Macroscopically, the brain is usually of normal or slightly reduced weight with little external evidence of atrophy. The major cerebral arteries are usually atherosclerotic. The lateral and third ventricles are moderately dilated. The cerebral white matter often feels soft on sectioning. The cut surface appears slightly granular and may be pitted with small depressions (Fig. 31.63). When these changes are seen in life on imaging it is termed ‘leukoaraiosis’. Typically, these changes are most pronounced in the deep frontal and temporal white matter. Coexistent lacunar infarcts are usually present in the basal ganglia, thalamus, pons, and, less often, the hemispheric white matter (Fig. 31.64). Histology reveals arteriosclerosis and arteriolosclerosis – hyalinization of small arteries and arterioles with loss of smooth muscle and replacement by collagen (Fig. 31.65). The white matter shows patchy pallor on myelin staining, axonal loss, reduction in the number of oligodendrocytes, and mild astrocytic gliosis (Fig. 31.66). There is often an associated loss of axons in these areas. Dementia with these changes is now clinically termed subcortical vascular dementia (SVaD) but has also been referred to in the past as lacunar dementia and Binswanger’s disease. Note that if histologic sampling is restricted to small blocks that include only 1–2 cm of subcortical white matter, significant deep white matter disease may well be missed.
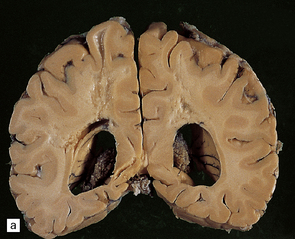

31.63 Ischemic white matter degeneration.
(a) Coronal slices show the parieto-occipital white matter to contain foci of granular gray discoloration that have sunken away from the cut surface. The sunken appearance and discoloration are seen at higher magnification in (b), which also demonstrates the ‘pin-hole’ effect caused by the dilatation of perivascular spaces.
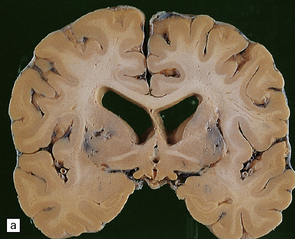
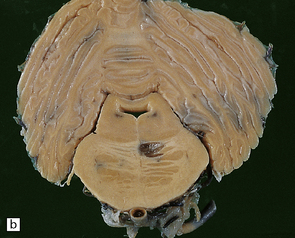
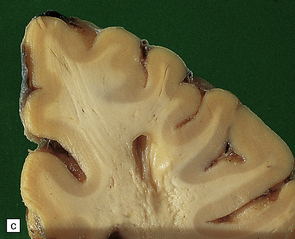
31.64 Lacunar infarcts.
These are seen in association with small vessel disease of the brain. (a) Bilateral lacunar infarcts in basal ganglia. (b) Lacuna in pons. (c) Large lacuna in white matter.
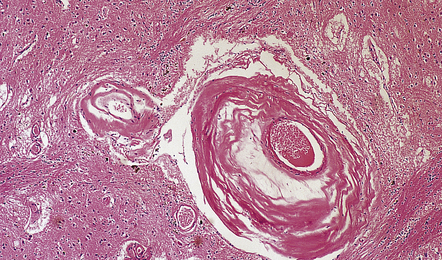
31.65 Arteriosclerosis and arteriolosclerosis.
The tunica media of small penetrating arteries and arterioles has been replaced by many layers of hyaline collagenous connective tissue.
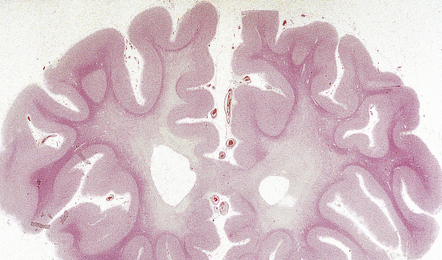
31.66 Ischemic white matter degeneration.
Whole mount of frontal lobes, showing severe rarefaction of deep white matter with subcortical white matter preservation.
 Lacunar infarction. Lacunar infarcts are seen as very small cavities and may be present in both gray and white matter. They are most commonly found in the basal ganglia, thalamus and pons (Fig. 31.64). Clinicopathologic studies have shown a relationship with cognitive abnormality and the pattern of disease termed subcortical vascular dementia (SVaD).
Lacunar infarction. Lacunar infarcts are seen as very small cavities and may be present in both gray and white matter. They are most commonly found in the basal ganglia, thalamus and pons (Fig. 31.64). Clinicopathologic studies have shown a relationship with cognitive abnormality and the pattern of disease termed subcortical vascular dementia (SVaD).
 Cribriform atrophy of white matter. This appears as myriad fine pin-size holes in the white matter that are due to dilatation of perivascular spaces and most numerous in the anterior temporal and frontal regions. Histologically, hyalinized vessels are surrounded by a dilated perivascular space with surrounding myelin pallor and astrocytic gliosis that extend a small distance from the vessel (Fig. 31.67). The significance of this change and relationships with clinical features are in need of further clarification.
Cribriform atrophy of white matter. This appears as myriad fine pin-size holes in the white matter that are due to dilatation of perivascular spaces and most numerous in the anterior temporal and frontal regions. Histologically, hyalinized vessels are surrounded by a dilated perivascular space with surrounding myelin pallor and astrocytic gliosis that extend a small distance from the vessel (Fig. 31.67). The significance of this change and relationships with clinical features are in need of further clarification.
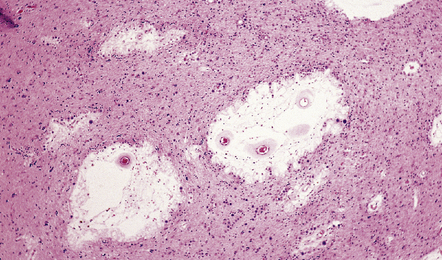
31.67 Perivascular changes in vascular dementia.
A feature of vascular dementia is the prominent hyaline arteriosclerosis and dilatation of perivascular spaces in the cerebral white matter. There is astrocytic gliosis of the surrounding white matter.
 Granular cortical atrophy. This is a relatively uncommon finding and its relationship with cognitive decline is in need of further clarification. Macroscopically, affected regions of cerebral cortex appear pitted by depressions 1–2 mm in diameter (Fig. 31.68). Histology shows these to correspond to numerous cortical microinfarcts, usually associated with hyaline arteriosclerosis of small vessels but sometimes associated with amyloid angiopathy. Other diseases that involve small vessels can cause this pattern of lesions, the most notable example being the microvascular thrombosis associated with the antiphospholipid antibody syndrome.
Granular cortical atrophy. This is a relatively uncommon finding and its relationship with cognitive decline is in need of further clarification. Macroscopically, affected regions of cerebral cortex appear pitted by depressions 1–2 mm in diameter (Fig. 31.68). Histology shows these to correspond to numerous cortical microinfarcts, usually associated with hyaline arteriosclerosis of small vessels but sometimes associated with amyloid angiopathy. Other diseases that involve small vessels can cause this pattern of lesions, the most notable example being the microvascular thrombosis associated with the antiphospholipid antibody syndrome.
Large vessel disease: Large regional cerebral infarcts rarely contribute significantly to global cognitive decline in the setting of a clinical dementia. The term ‘multi-infarct dementia’ has been used for this clinical association. Rarely, severe cognitive dysfunction is caused by bilateral small infarcts in critical sites such as the medial thalamus or hippocampal region (Table 31.20). Other reasons for the association of large vessel disease or regional cerebral infarcts with dementia are:
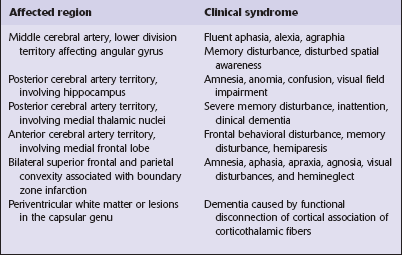



The causes and consequences of various forms of large vessel disease, including atherosclerotic and embolic disease, vasculitis, and CADASIL are considered in Chapter 9.
Global cerebral hypoperfusion: Conditions leading to cerebral hypoperfusion can cause ischemic injury to the hippocampus and watershed/boundary zone regions (see Chapters 8 and 9). Hippocampal sclerosis with clinical dementia is seen in some patients after a defined episode of hypotension complicating cardiovascular disease (Fig. 31.69). Boundary zone infarcts in the frontal and parietal cortex can also lead to dementia.
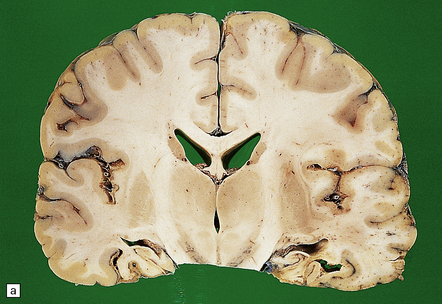

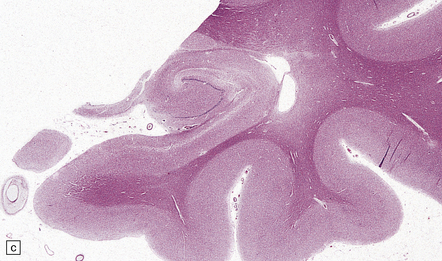
31.69 Hippocampal sclerosis due to hypoperfusion
(a) Macroscopic view showing bilateral hippocampal sclerosis in a patient following a period of circulatory arrest. (b) The marked shrinkage of the hippocampus can be appreciated in this close-up view. (c) Histology shows extensive neuronal loss from the hippocampus, with associated astrocytic gliosis. Where there is no history of hypoperfusion injury a search for a neurodegenerative cause, for example a FTLD, should be performed.
 CLINICAL FEATURES OF VASCULAR DEMENTIA
CLINICAL FEATURES OF VASCULAR DEMENTIA





 CLINICAL FEATURES OF VASCULAR COGNITIVE IMPAIRMENT (VCI)
CLINICAL FEATURES OF VASCULAR COGNITIVE IMPAIRMENT (VCI)
 The term VCI encompasses all forms of cognitive deficits from VaD to mild cognitive impairment (MCI) of vascular origin.
The term VCI encompasses all forms of cognitive deficits from VaD to mild cognitive impairment (MCI) of vascular origin.





 There is cognitive impairment and imaging evidence of cerebrovascular disease and:
There is cognitive impairment and imaging evidence of cerebrovascular disease and:

There is cognitive impairment and imaging evidence of cerebrovascular disease but:







 There is cognitive impairment and imaging evidence of cerebrovascular disease and:
There is cognitive impairment and imaging evidence of cerebrovascular disease and:

There is cognitive impairment and imaging evidence of cerebrovascular disease but:




(Adapted from AHA/ASA Scientific Statement – Vascular Contributions to Cognitive Impairment and Dementia. Stroke 2011 42(9):2672–2713.)
NEUROPATHOLOGIC ASSESSMENT IN VASCULAR DEMENTIA
An approach to neuropathologic diagnostic evaluation of VaD has been published (see below) alongside a classification for the neuropathologic diagnosis of vascular dementia into six subtypes (Table 31.21). A diagnosis of probable VaD is appropriate when cerebrovascular pathology meets criteria for one of the subtypes of VaD and other causes of dementia have been excluded. A diagnosis of possible VaD is appropriate when neuropathologic examination shows a vascular pathology which does not meets criteria for one of the subtypes of VaD.
Table 31.21
| Lesions | Diagnostic group | Subtype |
| Large infarct or several infarcts (>50 mL loss of tissue) | Multi-infarct dementia | I |
| Multiple small or microinfarcts (>3 with minimum diameter 5 mm; involving >3 coronal levels; vascular hyalinization, CAA, lacunar infarcts, perivascular changes) | Small vessel vascular dementia | II |
| Infarcts in critical areas (e.g. thalamus, hippocampus, basal forebrain) | Strategic infarct dementia | III |
| Incomplete or diffuse infarction (hippocampal sclerosis, ischemic–anoxic damage, cortical laminar necrosis, border zone infarcts involving brain tissue in at least three different coronal planes) | Vascular dementia due to cerebral hypoperfusion | IV |
| Multiple cerebral hemorrhages (lobar, intracerebral or subarachnoid) | Vascular dementia due to cerebral hemorrhages | V |
| Any of the above cerebrovascular changes with concurrent AD-type pathology (Braak stage >III) | Mixed dementia | VI |
Adapted from Kalaria et al. (2004).
INTERMITTENTLY RAISED PRESSURE HYDROCEPHALUS
 PATHOLOGIC CHARACTERIZATION OF LESIONS IN SUSPECTED VASCULAR DEMENTIA
PATHOLOGIC CHARACTERIZATION OF LESIONS IN SUSPECTED VASCULAR DEMENTIA
 Characterize lesions as ischemic or hemorrhagic infarcts.
Characterize lesions as ischemic or hemorrhagic infarcts.
 Note presence of lacunes and lacunar infarcts.
Note presence of lacunes and lacunar infarcts.
 Note location of infarcts: cortex, white matter, basal ganglia, brain stem, cerebellum.
Note location of infarcts: cortex, white matter, basal ganglia, brain stem, cerebellum.

 Note laterality: right or left.
Note laterality: right or left.

 Presence and location of small vessel disease: lipohyalinosis, fibroid necrosis, CAA.
Presence and location of small vessel disease: lipohyalinosis, fibroid necrosis, CAA.
 Presence of white matter disease: rarefaction or incomplete infarction.
Presence of white matter disease: rarefaction or incomplete infarction.
 Presence of hippocampal sclerosis.
Presence of hippocampal sclerosis.


Adapted from Kalaria et al. (2004).
MACROSCOPIC AND MICROSCOPIC APPEARANCES
Pathology confirms these imaging features (Fig. 31.70). There is symmetric dilatation of the lateral and third ventricles and good preservation of the cerebral cortex. The cerebral white matter appears macroscopically normal, but histology may reveal mild to moderate periventricular rarefaction and gliosis. Vascular dementia and primary neurodegenerative disease should be excluded by histologic examination. If hydrocephalus is diagnosed in life a ventricular shunt is usually inserted. A frequent complication of this procedure in the elderly is the development of large subdural hematomas.
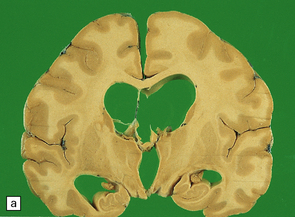
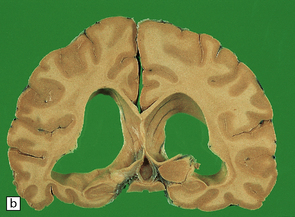
A STAGED APPROACH TO PATHOLOGIC DIAGNOSIS
Strategies for the staged examination of the post-mortem brain have been published using conventional, immunohistochemical and molecular techniques. The schematic diagram in Figure 31.71 allows a staged examination to achieve a diagnosis with appropriate use of resources.
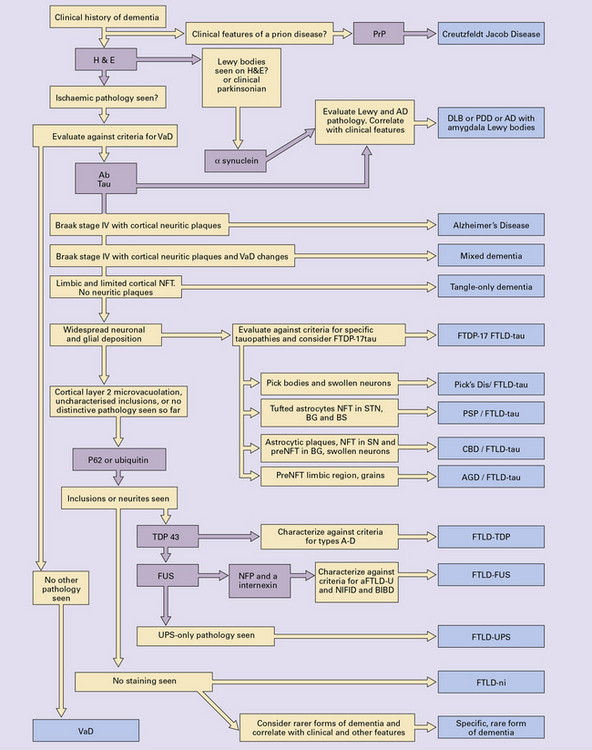
31.71 Staged approach to diagnostic evaluation in dementia.
Strategic sampling of the brain should be performed as described in the evaluation of Alzheimer’s disease (p. 612). If a prion disease is suspected then appropriate containment or material should be undertaken. AGD, argyrophilic grain disease; CBD, corticobasal degeneration; DLB, dementia with Lewy bodies; FTDP-17tau, frontotemporal degeneration and parkinsonism linked to chromosome 17 tau; FTLD-tau, frontotemporal lobar degeneration-tau; FTDP-FUS, frontotemporal lobar degeneration FUS; FTLD-TDP, frontotemporal lobar degeneration TDP; FTLD-UPS, frontotemporal lobar degeneration-ubiquitin proteasome system; FTLD-ni, frontotemporal lobar degeneration with no inclusions; PrP, prion protein; NFT, neurofibrillary tangle; NFP, neurofilament protein; Aβ, amyloid-β beta peptide; H&E, hematoxylin and eosin; PDD, Parkinson’s disease dementia; VaD, vascular dementia; PSP, progressive supranuclear palsy; NIFID, neuronal intermediate filament inclusion disease; BIBD, basophilic inclusion body disease.
REFERENCES
Alafuzoff, I., Thal, D.R., Arzberger, T., et al. Assessment of beta-amyloid deposits in human brain: a study of the BrainNet Europe Consortium. Acta Neuropathol (Berl). 2009;117(3):309–320.
Armstrong, R.A. The molecular biology of senile plaques and neurofibrillary tangles in Alzheimer’s disease. Folia Neuropathol. 2009;47(4):289–299.
Ballard, C., Gauthier, S., Corbett, A., et al. Alzheimer’s disease. Lancet. 2011;377(9770):1019–1031.
Bekris, L.M., Yu, C.E., Bird, T.D., et al. Genetics of Alzheimer disease. J Geriatr Psychiatry Neurol. 2010;23(4):213–227.
Braak, B. Neuropathologic staging of Alzheimer related changes. Acta Neuropathol (Berl). 1991;82:239–259.
Braak, H., Alafuzoff, I., Arzberger, T., et al. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol (Berl). 2006;112(4):389–404.
Chow, V.W., Mattson, M.P., Wong, P.C., et al. An overview of APP processing enzymes and products. Neuromolecular Med. 2010;12(1):1–12.
Crews, L., Masliah, E. Molecular mechanisms of neurodegeneration in Alzheimer’s disease. Hum Mol Genet. 2010;19(R1):R12–R20.
Duyckaerts, C., Delatour, B., Potier, M.C. Classification and basic pathology of Alzheimer disease. Acta Neuropathol (Berl). 2009;118(1):5–36.
Gearing, M., Mirra, S.S., Hedreen, J.C., The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part X. Neuropathology confirmation of the clinical diagnosis of Alzheimer’s disease. Neurology. 1995;45:461–466.
Goedert, M., Klug, A., Crowther, R.A. Tau protein, the paired helical filament and Alzheimer’s disease. J Alzheimers Dis. 2006;9(3 suppl):195–207.
Hyman, B.T., Phelps, C.H., Beach, T.G., et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimer’s Dement. 2012;8(1):1–13.
Montine, T.J., Phelps, C.H., Beach, T.G., et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol (Berl). 2012;123(1):1–11.
Iqbal, K., Liu, F., Gong, C.X., et al. Tau in Alzheimer disease and related tauopathies. Curr Alzheimer Res.. 2010;7(8):656–664.
Iqbal, K., Wang, X., Blanchard, J., et al. Alzheimer’s disease neurofibrillary degeneration: pivotal and multifactorial. Biochem Soc Trans. 2010;38(4):962–966.
Ittner, L.M., Götz, J. Amyloid-β and tau–a toxic pas de deux in Alzheimer’s disease. Nat Rev Neurosci. 2011;12(2):65–72.
LaFerla, F.M. Pathways linking Abeta and tau pathologies. Biochem Soc Trans. 2010;38(4):993–995.
Markesbery, W.R. Neuropathologic alterations in mild cognitive impairment: a review. J Alzheimer’s Dis. 2010;19(1):221–228.
Metsaars, W.P., Hauw, J.J., van Welsem, M.E., et al. A grading system of Alzheimer disease lesions in neocortical areas. Neurobiol Aging. 2003;24(4):563–572.
Mirra, S.S., Heyman, A., McKeel, D., The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41:479–486.
Morgan, K. The three new pathways leading to Alzheimer’s disease. Neuropathol Appl Neurobiol. 2011;37(4):353–357.
Munoz, D.G., Wang, D. Tangle-associated neuritic clusters. A new lesion in Alzheimer’s disease and aging suggests that aggregates of dystrophic neurites are not necessarily associated with beta/A4. Am J Pathol. 1992;140(5):1167–1178.
Murray, M.E., Graff-Radford, N.R., Ross, O.A., et al. Neuropathologically defined subtypes of Alzheimer’s disease with distinct clinical characteristics: a retrospective study. Lancet Neurol. 2011;10(9):785–796.
National Institute on Aging and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease. Consensus recommendations for the postmortem diagnosis of Alzheimer’s disease. Neurobiol Aging. 1997;18(4 suppl):S1–S2.
Perl, D.P. Neuropathology of Alzheimer’s disease. Mt Sinai J Med. 2010;77(1):32–42.
Probst, A., Herzig, M.C., Mistl, C., et al. Perisomatic granules (non-plaque dystrophic dendrites) of hippocampal CA1 neurons in Alzheimer’s disease and Pick’s disease: a lesion distinct from granulovacuolar degeneration. Acta Neuropathol (Berl). 2001;102(6):636–644.
Reitz, C., Brayne, C., Mayeux, R. Epidemiology of Alzheimer disease. Nat Rev Neurol. 2011;7(3):137–152.
Thal, D.R., Rüb, U., Orantes, M., et al. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58(12):1791–1800.
Zhang, Y.W., Thompson, R., Zhang, H., et al. APP processing in Alzheimer’s disease. Mol Brain. 2011;4:3.
Dementia with Lewy bodies and Parkinson’s disease dementia
Aarsland, D., Kurz, M.W. The epidemiology of dementia associated with Parkinson’s disease. Brain Pathol. 2010;20(3):633–639.
Ballard, C., Ziabreva, I., Perry, R., et al. Differences in neuropathologic characteristics across the Lewy body dementia spectrum. Neurology. 2006;67(11):1931–1934.
Bedford, L., Paine, S., Rezvani, N., et al. The UPS and autophagy in chronic neurodegenerative disease: six of one and half a dozen of the other – or not? Autophagy. 2009;5(2):224–227.
Beyer, K., Ariza, A. Protein aggregation mechanisms in synucleinopathies: commonalities and differences. J Neuropathol Exp Neurol. 2007;66(11):965–974.
Dickson, D.W., Fujishiro, H., Orr, C., et al. Neuropathology of non-motor features of Parkinson disease. Parkinsonism Relat Disord. 2009;15(suppl 3):S1–S5.
Goetz, C.G., Emre, M., Dubois, B. Parkinson’s disease dementia: definitions, guidelines, and research perspectives in diagnosis. Ann Neurol. 2008;64(suppl 2):S81–S92.
Halliday, G.M., McCann, H. The progression of pathology in Parkinson’s disease. Ann N Y Acad Sci. 2010;1184:188–195.
Hyman, B.T., Phelps, C.H., Beach, T.G., et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimer’s Dement. 2012;8(1):1–13.
Iwatsubo, T. Pathological biochemistry of alpha-synucleinopathy. Neuropathology. 2007;27(5):474–478.
Jellinger, K.A., Wenning, G.K., Seppi, K. Predictors of survival in dementia with Lewy bodies and Parkinson dementia. Neurodegener Dis. 2007;4(6):428–430.
Jellinger, K.A. The neuropathologic substrate of Parkinson disease dementia. Acta Neuropathol (Berl). 2010;119(1):151–153.
Jellinger, K.A. Formation and development of Lewy pathology: a critical update. J Neurol. 2009;256(suppl 3):270–279.
Jellinger, K.A. Significance of brain lesions in Parkinson disease dementia and Lewy body dementia. Front Neurol Neurosci. 2009;24:114–125.
Kalaitzakis, M.E., Pearce, R.K. The morbid anatomy of dementia in Parkinson’s disease. Acta Neuropathol (Berl). 2009;118(5):587–598.
Kovacs, G.G., Alafuzoff, I., Al-Sarraj, S., et al. Mixed brain pathologies in dementia: the BrainNet Europe consortium experience. Dement Geriatr Cogn Disord. 2008;26(4):343–350.
Lippa, C.F., Duda, J.E., Grossman, M., et al. DLB and PDD boundary issues: diagnosis, treatment, molecular pathology. Neurology. 2007;68(11):812–819.
Lowe, J. Neuropathology of dementia with Lewy bodies. Handb Clin Neurol. 2008;89:321–330.
McKeith, I.G., Dickson, D.W., Lowe, J., et al. Consortium on DLB. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65(12):1863–1872.
Uchikado, H., Lin, W.L., DeLucia, M.W., et al. Alzheimer disease with amygdala Lewy bodies: a distinct form of alpha-synucleinopathy. J Neuropathol Exp Neurol. 2006;65(7):685–697.
Waxman, E.A., Giasson, B.I. Molecular mechanisms of alpha-synuclein neurodegeneration. Biochim Biophys Acta. 2009;1792(7):616–624.
Frontotemporal lobar degenerations (FTLD): Terminology and general approaches
Cairns, N.J., Bigio, E.H., Mackenzie, I.R., Consortium for Frontotemporal Lobar Degeneration. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol (Berl). 2007;114(1):5–22.
Dormann, D., Haass, C. TDP-43 and FUS: a nuclear affair. Trends Neurosci. 2011. [PMID: 21700347 [Epub ahead of print]].
Ferrari, R., Hardy, J., Momeni, P. Frontotemporal Dementia: From Mendelian Genetics Towards Genome Wide Association Studies. J Mol Neurosci. 2011;45(3):500–515.
Fiesel, F.C., Kahle, P.J. TDP-43 and FUS/TLS: cellular functions and implications for neurodegeneration. FEBS J. 2011;278(19):3550–3568.
Goldman, J.S., Rademakers, R., Huey, E.D., et al. An algorithm for genetic testing of frontotemporal lobar degeneration. Neurology. 2011;76(5):475–483.
Knopman, D.S., Roberts, R.O. Estimating the number of persons with frontotemporal lobar degeneration in the US population. J Mol Neurosci. 2011;45(3):330–335.
Kril, J.J., Halliday, G.M. Pathological staging of frontotemporal lobar degeneration. J Mol Neurosci. 2011;45(3):379–383.
Mackenzie, I.R., Neumann, M., Baborie, A., et al. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol (Berl). 2011;122(1):111–113.
Mackenzie, I.R., Neumann, M., Bigio, E.H., et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol (Berl). 2010;119(1):1–4.
Mackenzie, I.R., Neumann, M., Bigio, E.H., et al. Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: consensus recommendations. Acta Neuropathol (Berl). 2009;117(1):15–18.
Mackenzie, I.R., Neumann, M., Cairns, N.J., et al. Novel types of frontotemporal lobar degeneration: beyond tau and TDP-43. J Mol Neurosci. 2011;45(3):402–408.
Mackenzie, I.R., Rademakers, R., Neumann, M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 2010;9(10):995–1007.
Piguet, O., Hornberger, M., Mioshi, E., et al. Behavioral-variant frontotemporal dementia: diagnosis, clinical staging, and management. Lancet Neurol. 2011;10(2):162–172.
Rohrer, J.D. Behavioral Variant frontotemporal dementia – defining genetic and pathological subtypes. J Mol Neurosci. 2011;45(3):583–588.
Strong, M.J., Yang, W. The frontotemporal syndromes of ALS. Clinicopathological correlates. J Mol Neurosci. 2011;45(3):648–655.
Dickson, D.W. Neuropathology of Pick’s disease. Neurology. 2001;56(11 suppl 4):S16–S20.
Dickson, D.W. Neuropathology of non-Alzheimer degenerative disorders. Int J Clin Exp Pathol. 2009;3(1):1–23.
Dickson, D.W., Ahmed, Z., Algom, A.A., et al. Neuropathology of variants of progressive supranuclear palsy. Curr Opin Neurol. 2010;23(4):394–400.
Dickson, D.W., Bergeron, C., Chin, S.S., Office of Rare Diseases of the National Institutes of Health. Office of rare diseases neuropathologic criteria for corticobasal degeneration. J Neuropathol Exp Neurol. 2002;61(11):935–946.
Dickson, D.W., Kouri, N., Murray, M.E., et al. Neuropathology of frontotemporal lobar degeneration-tau (FTLD-tau). J Mol Neurosci. 2011;45(3):384–389.
Ferrer, I., Santpere, G., van Leeuwen, F.W. Argyrophilic grain disease. Brain.. 2008;131(Pt 6):1416–1432.
Goedert, M., Spillantini, M.G. Pathogenesis of the tauopathies. J Mol Neurosci. 2011;45(3):425–431.
Jellinger, K.A., Attems, J. Neurofibrillary tangle-predominant dementia: comparison with classical Alzheimer disease. Acta Neuropathol (Berl). 2007;113(2):107–117.
Kouri, N., Whitwell, J.L., Josephs, K.A., et al. Corticobasal degeneration: a pathologically distinct 4R tauopathy. Nat Rev Neurol. 2011;7(5):263–272.
Piguet, O., Halliday, G.M., Reid, W.G., et al. Clinical phenotypes in autopsy-confirmed Pick disease. Neurology. 2011;76(3):253–259.
Tolnay, M., Probst, A. Argyrophilic grain disease. Handb Clin Neurol. 2008;89:553–563.
van Swieten, J., Spillantini, M.G. Hereditary frontotemporal dementia caused by Tau gene mutations. Brain Pathol. 2007;17(1):63–73.
Yamada, M., Itoh, Y., Sodeyama, N., et al. Senile dementia of the neurofibrillary tangle type: a comparison with Alzheimer’s disease. Dement Geriatr Cogn Disord. 2001;12(2):117–126.
Amador-Ortiz, C., Lin, W.L., Ahmed, Z., et al. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann Neurol. 2007;61(5):435–445.
Benajiba, L., Le Ber, I., Camuzat, A., French Clinical and Genetic Research Network on Frontotemporal Lobar Degeneration/Frontotemporal Lobar Degeneration with Motoneuron Disease. TARDBP mutations in motoneuron disease with frontotemporal lobar degeneration. Ann Neurol. 2009;65(4):470–473.
Bigio, E.H. TDP-43 variants of frontotemporal lobar degeneration. J Mol Neurosci. 2011;45(3):390–401.
Bigio, E.H. TAR DNA-binding protein-43 in amyotrophic lateral sclerosis, frontotemporal lobar degeneration, and Alzheimer disease. Acta Neuropathol (Berl). 2008;116(2):135–140.
Borroni, B., Bonvicini, C., Alberici, A., et al. Mutation within TARDBP leads to frontotemporal dementia without motor neuron disease. Hum Mutat. 2009;30(11):E974–E983.
Chen-Plotkin, A.S., Martinez-Lage, M., Sleiman, P.M., et al. Genetic and clinical features of progranulin-associated frontotemporal lobar degeneration. Arch Neurol. 2011;68(4):488–497.
Cohen, T.J., Lee, V.M., Trojanowski, J.Q. TDP-43 functions and pathogenic mechanisms implicated in TDP-43 proteinopathies. Trends Mol Med. 2011;17(11):659–667.
Forman, M.S., Mackenzie, I.R., Cairns, N.J., et al. Novel ubiquitin neuropathology in frontotemporal dementia with valosin-containing protein gene mutations. J Neuropathol Exp Neurol. 2006;65(6):571–581.
Geser, F., Martinez-Lage, M., Robinson, J., et al. Clinical and pathological continuum of multisystem TDP-43 proteinopathies. Arch Neurol. 2009;66(2):180–189.
Hasegawa, M., Nonaka, T., Tsuji, H., et al. Molecular Dissection of TDP-43 Proteinopathies. J Mol Neurosci. 2011;45(3):480–485.
Josephs, K.A., Hodges, J.R., Snowden, J.S., et al. Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathol (Berl). 2011;122(2):137–153.
Ju, J.S., Weihl, C.C. Inclusion body myopathy, Paget’s disease of the bone and fronto-temporal dementia: a disorder of autophagy. Hum Mol Genet. 2010;19(R1):R38–R45.
Mackenzie, I.R., Neumann, M., Baborie, A., et al. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol (Berl). 2011;122(1):111–113.
Neumann, M., Mackenzie, I.R., Cairns, N.J., et al. TDP-43 in the ubiquitin pathology of frontotemporal dementia with VCP gene mutations. J Neuropathol Exp Neurol. 2007;66(2):152–157.
Rohrer, J.D., Geser, F., Zhou, J., et al. TDP-43 subtypes are associated with distinct atrophy patterns in frontotemporal dementia. Neurology. 2010;75(24):2204–2211.
Strong, M.J., Yang, W. The frontotemporal syndromes of ALS. Clinicopathological Correlates. J Mol Neurosci. 2011;45(3):648–655.
Tartaglia, M.C., Sidhu, M., Laluz, V., et al. Sporadic corticobasal syndrome due to FTLD-TDP. Acta Neuropathol (Berl). 2010;119(3):365–374.
Watts, G.D., Wymer, J., Kovach, M.J., et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet. 2004;36(4):377–381.
Weihl, C.C. Valosin containing protein associated fronto-temporal lobar degeneration: clinical presentation, pathologic features and pathogenesis. Curr Alzheimer Res. 2011;8(3):252–260.
Weihl, C.C., Pestronk, A., Kimonis, V.E. Valosin-containing protein disease: inclusion body myopathy with Paget’s disease of the bone and fronto-temporal dementia. Neuromuscul Disord. 2009;19(5):308–315.
Whitwell, J.L., Jack, C.R., Jr., Parisi, J.E., et al. Does TDP-43 type confer a distinct pattern of atrophy in frontotemporal lobar degeneration? Neurology. 2010;75(24):2212–2220.
Wilson, A.C., Dugger, B.N., Dickson, D.W., et al. TDP-43 in aging and Alzheimer’s disease – a review. Int J Clin Exp Pathol. 2011;4(2):147–155.
Yu, C.E., Bird, T.D., Bekris, L.M., et al. The spectrum of mutations in progranulin: a collaborative study screening 545 cases of neurodegeneration. Arch Neurol. 2010;67(2):161–170.
Al-Sarraj, S., King, A., Troakes, C., et al. p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol (Berl). 2011;122(6):691–702.
Simón-Sánchez, J., Dopper, E.G., Cohn-Hokke, P.E., et al. The clinical and pathological phenotype of C9ORF72 hexanucleotide repeat expansions. Brain. 2012;135:723–735.
Snowden, J.S., Rollinson, S., Thompson, J.C., et al. Distinct clinical and pathological characteristics of frontotemporal dementia associated with C9ORF72 mutations. Brain. 2012;135:693–708.
Troakes, C., Maekawa, S., Wijesekera, L., et al. An MND/ALS phenotype associated with C9orf72 repeat expansion: Abundant p62-positive, TDP-43-negative inclusions in cerebral cortex, hippocampus and cerebellum but without associated cognitive decline. Neuropathology. 2011. [PMID: 22181065 [Epub ahead of print]].
Brelstaff, J., Lashley, T., Holton, J.L., et al. Transportin1: a marker of FTLD-FUS. Acta Neuropathol (Berl). 2011;122(5):591–600.
Broustal, O., Camuzat, A., Guillot-Noël, L., et al. French clinical and genetic research network on FTD/FTD-MND. FUS mutations in frontotemporal lobar degeneration with amyotrophic lateral sclerosis. J Alzheimers Dis. 2010;22(3):765–769.
Huang, E.J., Zhang, J., Geser, F., et al. Extensive FUS-immunoreactive pathology in juvenile amyotrophic lateral sclerosis with basophilic inclusions. Brain Pathol. 2010;20(6):1069–1076.
Ito, H., Fujita, K., Nakamura, M., et al. Optineurin is co-localized with FUS in basophilic inclusions of ALS with FUS mutation and in basophilic inclusion body disease. Acta Neuropathol (Berl). 2011;121(4):555–557.
Josephs, K.A., Holton, J.L., Rossor, M.N., et al. Neurofilament inclusion body disease: a new proteinopathy? Brain. 2003;126(Pt 10):2291–2303.
Josephs, K.A., Whitwell, J.L., Parisi, J.E., et al. Caudate atrophy on MRI is a characteristic feature of FTLD-FUS. Eur J Neurol. 2010;17(7):969–975.
Jekyll Dr, K.H., Hyde, M.r. The Two Faces of the FUS/EWS/TAF15 Protein Family. Sarcoma. 2011, 2011. [837474].
Loy, C.T., McCusker, E., Kril, J.J., et al. Very early-onset frontotemporal dementia with no family history predicts underlying fused in sarcoma pathology. Brain. 2010;133(Pt 12):e158. [author reply e159].
Mackenzie, I.R., Ansorge, O., Strong, M., et al. Pathological heterogeneity in amyotrophic lateral sclerosis with FUS mutations: two distinct patterns correlating with disease severity and mutation. Acta Neuropathol (Berl). 2011;122(1):87–98.
Mackenzie, I.R., Munoz, D.G., Kusaka, H., et al. Distinct pathological subtypes of FTLD-FUS. Acta Neuropathol (Berl). 2011;121(2):207–218.
Matsuoka, T., Fujii, N., Kondo, A., et al. An autopsied case of sporadic adult-onset amyotrophic lateral sclerosis with FUS-positive basophilic inclusions. Neuropathology. 2011;31(1):71–76.
Menon, R., Baborie, A., Jaros, E., et al. What’s in a name? Neuronal intermediate filament inclusion disease (NIFID), frontotemporal lobar degeneration-intermediate filament (FTLD-IF) or frontotemporal lobar degeneration-fused in sarcoma (FTLD-FUS)? J Neurol Neurosurg Psychiatry. 2010;82(12):1412–1414.
Munoz, D.G., Neumann, M., Kusaka, H., et al. FUS pathology in basophilic inclusion body disease. Acta Neuropathol (Berl). 2009;118(5):617–627.
Neumann, M., Rademakers, R., Roeber, S., et al. A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain. 2009;132(Pt 11):2922–2931.
Rohrer, J.D., Lashley, T., Holton, J., et al. The clinical and neuroanatomical phenotype of FUS associated frontotemporal lobar degeneration. J Neurol Neurosurg Psychiatry. 2010;82(12):1405–1407.
Snowden, J.S., Hu, Q., Rollinson, S., et al. The most common type of FTLD-FUS (aFTLD-U) is associated with a distinct clinical form of frontotemporal dementia but is not related to mutations in the FUS gene. Acta Neuropathol (Berl). 2011;122(1):99–110.
Urwin, H., Josephs, K.A., Rohrer, J.D., et al. FUS pathology defines the majority of tau- and TDP-43-negative frontotemporal lobar degeneration. Acta Neuropathol (Berl). 2010;120(1):33–41.
Yokota, O., Tsuchiya, K., Terada, S., et al. Basophilic inclusion body disease and neuronal intermediate filament inclusion disease: a comparative clinicopathological study. Acta Neuropathol (Berl). 2008;115(5):561–575.
Isaacs, A.M., Johannsen, P., Holm, I., the FReJA consortium. Frontotemporal Dementia Caused by CHMP2B Mutations. Curr Alzheimer Res. 2011;8(3):246–251.
Amador-Ortiz, C., Ahmed, Z., Zehr, C., et al. Hippocampal sclerosis dementia differs from hippocampal sclerosis in frontal lobe degeneration. Acta Neuropathol (Berl). 2007;113(3):245–252.
Josephs, K.A., Dickson, D.W. Hippocampal sclerosis in tau-negative frontotemporal lobar degeneration. Neurobiol Aging. 2007;28(11):1718–1722.
Lippa, C.F., Dickson, D.W. Hippocampal sclerosis dementia: expanding the phenotypes of frontotemporal dementias? Neurology. 2004;63(3):414–415.
Nelson, P.T., Schmitt, F.A., Lin, Y., et al. Hippocampal sclerosis in advanced age: clinical and pathological features. Brain. 2011;134(Pt 5):1506–1518.
Probst, A., Taylor, K.I., Tolnay, M. Hippocampal sclerosis dementia: a reappraisal. Acta Neuropathol (Berl). 2007;114(4):335–345.
Ghiso, J., Rostagno, A., Tomidokoro, Y., et al. Genetic alterations of the BRI2 gene: familial British and Danish dementias. Brain Pathol. 2006;16(1):71–79.
Holton, J.L., Ghiso, J., Lashley, T., et al. Regional distribution of amyloid-Bri deposition and its association with neurofibrillary degeneration in familial British dementia. Am J Pathol. 2001;158(2):515–526.
Revesz, T., Ghiso, J., Lashley, T., et al. Cerebral amyloid angiopathies: a pathologic, biochemical, and genetic view. J Neuropathol Exp Neurol. 2003;62(9):885–898.
Black, S.E. Vascular cognitive impairment: epidemiology, subtypes, diagnosis and management. J R Coll Physicians Edinb. 2011;41(1):49–56.
Caplan, L.R., Gomes, J.A. Binswanger disease – an update. J Neurol Sci. 2010;299(1–2):9–10.
Debette, S., Seshadri, S. Vascular risk factors and dementia revisited. J Neurol Neurosurg Psychiatry. 2009;80(11):1183–1184.
Enciu, A.M., Constantinescu, S.N., Popescu, L.M., et al. Neurobiology of vascular dementia. J Aging Res.. 2011;2011:401604.
Gorelick, P.B., Scuteri, A., Black, S.E., on behalf of the American Heart Association Stroke Council, Council on Epidemiology and Prevention, Council on Cardiovascular Nursing, Council on Cardiovascular Radiology and Intervention, Council on Cardiovascular Surgery and Anesthesia. Vascular Contributions to Cognitive Impairment and Dementia: A Statement for Healthcare Professionals From the American Heart Association/American Stroke Association. Stroke. 2011;42(9):2672–2713.
Jellinger, K.A. Morphologic diagnosis of “vascular dementia” – a critical update. J Neurol Sci. 2008;270(1–2):1–12.
Jellinger, K.A. The enigma of vascular cognitive disorder and vascular dementia. Acta Neuropathol (Berl). 2007;113(4):349–388.
Kalaria, R.N., Kenny, R.A., Ballard, C.G., et al. Towards defining the neuropathological substrates of vascular dementia. J Neurol Sci. 2004;226(1–2):75–80.
Matthews, F.E., Brayne, C., Lowe, J., et al. Epidemiological pathology of dementia: attributable-risks at death in the Medical Research Council Cognitive Function and Ageing Study. PLoS Med. 2009;6(11):e1000180.
Menon, U., Kelley, R.E. Subcortical ischemic cerebrovascular dementia. Int Rev Neurobiol. 2009;84:21–33.
Pantoni, L., Sarti, C., Alafuzoff, I., et al. Postmortem examination of vascular lesions in cognitive impairment: a survey among neuropathological services. Stroke. 2006;37(4):1005–1009.
Tomimoto, H. Subcortical vascular dementia. Neurosci Res. 2011;71(3):193–199.
Coker, S.B. The diagnosis of childhood neurodegenerative disorders presenting as dementia in adults. Neurology. 1991;41:794–798.
Davis, R.L., Shrimpton, A.E., Holohan, P.D., et al. Familial dementia caused by polymerization of mutant neuroserpin. Nature. 1999;401:376–379.
Davis, R.L., Holohan, P.D., Shrimpton, A.E., et al. Familial encephalopathy with neuroserpin inclusion bodies. Am J Pathol. 1999;155:1901–1913.
Kosaka, K. Diffuse neurofibrillary tangles with calcification: a new presenile dementia. J Neurol Neurosurg Psychiatry. 1994;57:594–596.
Leinonen, V., Koivisto, A.M., Savolainen, S., et al. Post-mortem findings in 10 patients with presumed normal pressure hydrocephalus and review of the literature. Neuropathol Appl Neurobiol. 2011;38(1):72–86.
Schott, J.M., Reiniger, L., Thom, M., et al. Brain biopsy in dementia: clinical indications and diagnostic approach. Acta Neuropathol (Berl). 2010;120(3):327–341.