[level-membership-for-neurology-category]
18 Dementia
Mild Cognitive Impairment, Alzheimer Disease, Lewy Body Dementia, Frontotemporal Lobar Dementia, Vascular Dementia
Mild Cognitive Impairment
The utility of these tests in detecting MCI is less reliable. Indeed, most patients with MCI score within the normal range on the MMSE. Interview-based dementia assessments, such as the Clinical Dementia Rating scale (CDR), provide a more sensitive means for reliable detection of MCI but may require considerably more time to administer. Another brief screening tool, the Montreal Cognitive Assessment Test (www.mocatest.org), may provide greater sensitivity in detecting MCI. The definitive diagnosis of MCI requires formal neuropsychological assessment. However, neuropsychological test batteries take several hours to administer and interpret. Therefore, they are not practical as screening tools. In the hands of an experienced neuropsychologist, formal neuropsychological tests provide the most sensitive means of detecting cognitive impairment. They may also provide greater specificity in identifying the underlying cause, although there may be significant variability among neuropsychologists’ interpretations.
Alzheimer Disease
Pathogenesis
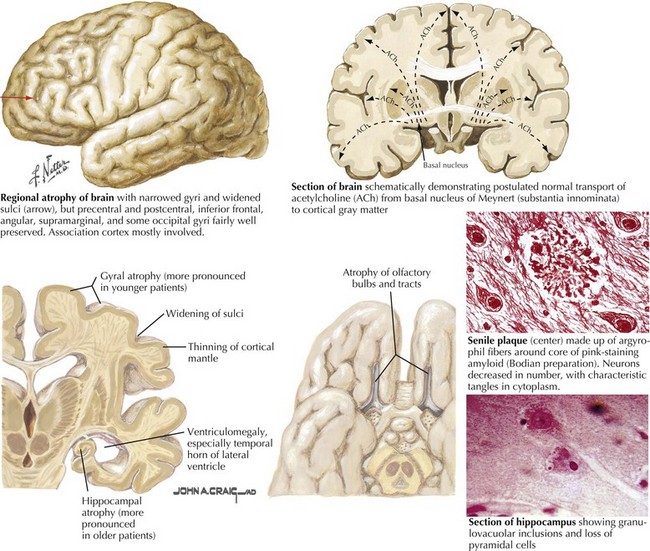
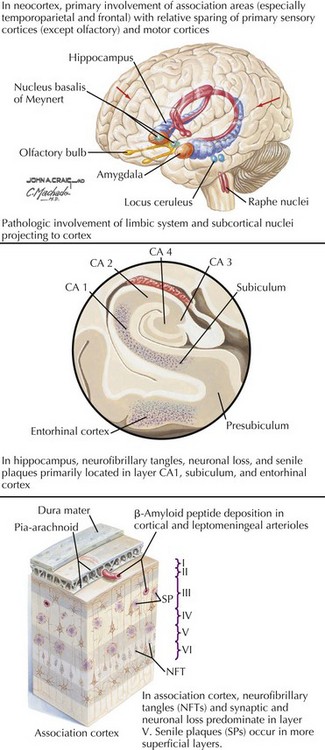
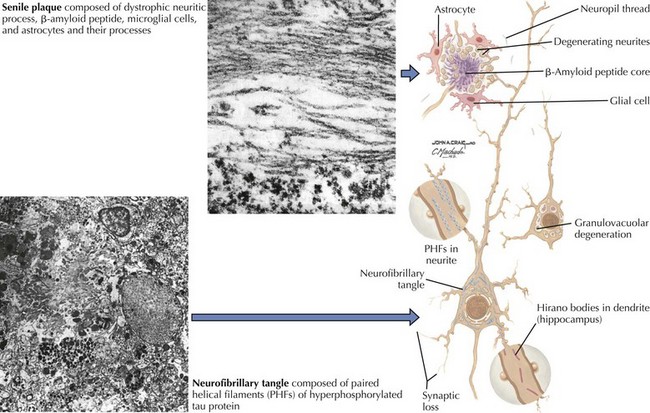
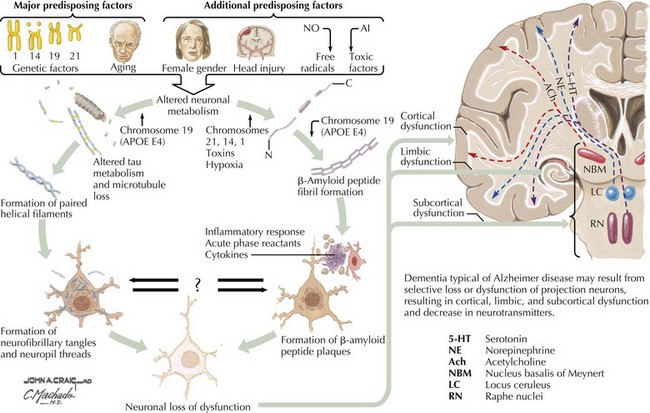
There is pronounced gross cerebral atrophy clearly evident on both imaging studies and post mortem. Typically, the dementia of AD preferentially affects the frontal, temporal, and parietal cortex. This is particularly evident in the temporoparietal and frontal association areas as well as the olfactory cortex. In contrast, other primary sensory cortical areas are unaffected. Additionally, the limbic system as well as subcortical nuclei as well as the nucleus basalis of Meynert are preferentially affected. Microscopically, there is clear loss of both neurons and neuropil. The classic findings include senile plaques and neurofibrillary tangles (Figs. 18-1 and 18-2). The white matter sometimes demonstrates a secondary demyelination.
β-Amyloid
β-Amyloid is a short fragment of the APP, typically 40–42 amino acids in length, which accumulates outside the cell during APP processing (Figs. 18-3 and 18-4). The tertiary structure of the 42–amino acid fragment is a β-pleated sheet that renders it insoluble. Consequently, it accumulates slowly, over many years, in the extracellular space and within synapses. In vitro studies confirm that β-amyloid is toxic to surrounding synapses and neurons, causing synaptic membrane destruction and eventual cell death. Transgenic mouse models show a clear association between accumulation of β-amyloid fragments, formation of amyloid plaques, and development of cognitive impairment.
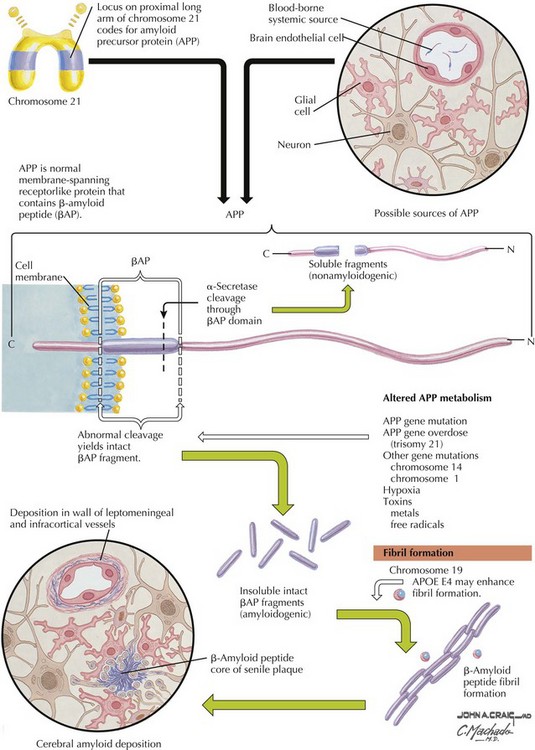
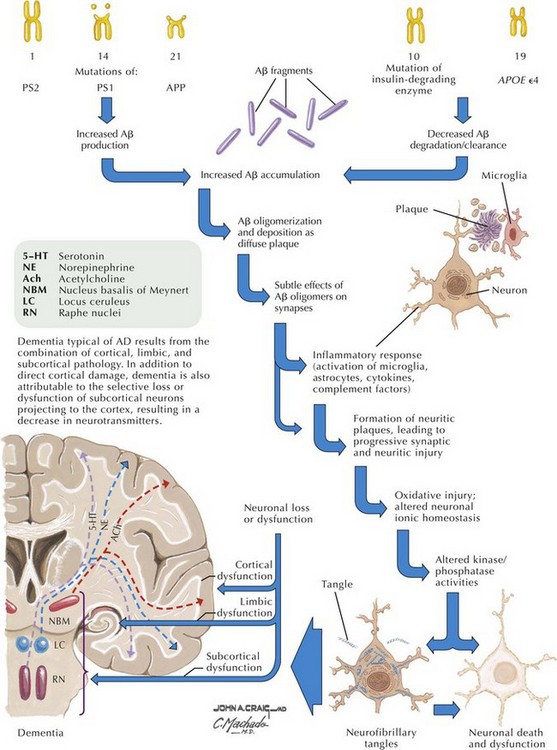
In vivo, β-amyloid fragments coalesce to form “diffuse” or immature plaques, best seen with silver-staining techniques. Diffuse plaques, however, are not sufficient to produce dementia; many nondemented elderly patients have substantial depositions of diffuse plaques throughout the cortex, a condition termed pathologic aging. It is when these plaques mature into “senile” or neuritic plaques that dementia becomes more likely (Fig. 18-5, top). Senile plaques consist of other substances in addition to β-amyloid, including synaptic proteins, inflammatory proteins, neuritic threads, activated glial cells, and other components. Unlike diffuse plaques, senile plaques are composed of a central core of β-amyloid surrounded by a myriad of proteins and cellular debris. Senile plaques are distributed diffusely in the cortex, typically starting in the hippocampus and the basal forebrain. Senile plaque formation correlates with increasing loss of synapses, which correlates with the earliest clinical sign, namely, short-term memory loss. The anatomic pattern of progression gradually spreads to neocortical and subcortical gray matter of the temporal, parietal, frontal, and, eventually, occipital cortex. Subcortical nuclei become involved relatively late in the process.
Neurofibrillary Tangles
The second pathologic hallmark of AD is the neurofibrillary tangle (Fig. 18-5, bottom). These lesions develop and conform to an anatomic pattern that correlates with the clinical syndrome; the number and distribution of tangles are directly related to the severity and clinical features of the dementia. Neurofibrillary tangles form intracellularly, consisting of a microtubule-associated protein, tau, which has a vital role in the maintenance of neuronal cytoskeleton structure and function. Tau is hyperphosphorylated in AD, causing it to dissociate from the cytoskeleton and accumulate, forming a paired helical filament protein structure. The cytoskeleton is compromised structurally and functionally, disrupting normal cell function. The most commonly used pathologic criteria for definitive AD diagnosis at autopsy require the presence of senile plaques and neurofibrillary tangles. Other lesions, such as Hirano bodies, are also seen in AD but have little diagnostic specificity.
Neurotransmitters
In addition to neuronal and synaptic loss, there is a gradual loss of various neurotransmitters. Acetylcholine synthesis is the earliest and most prominently affected. Most acetylcholinergic neurons arise within the nucleus basalis of Meynert in the basal forebrain (see Fig. 18-2). This nucleus is affected relatively early in the process; acetylcholine levels within the brain and spinal fluid of patients with AD quickly decline with disease progression. This observation supported the cholinergic hypothesis—that acetylcholine depletion results in the cognitive decline observed in patients with AD—eventually leading to the first symptomatic treatment of AD.
Risk Factors
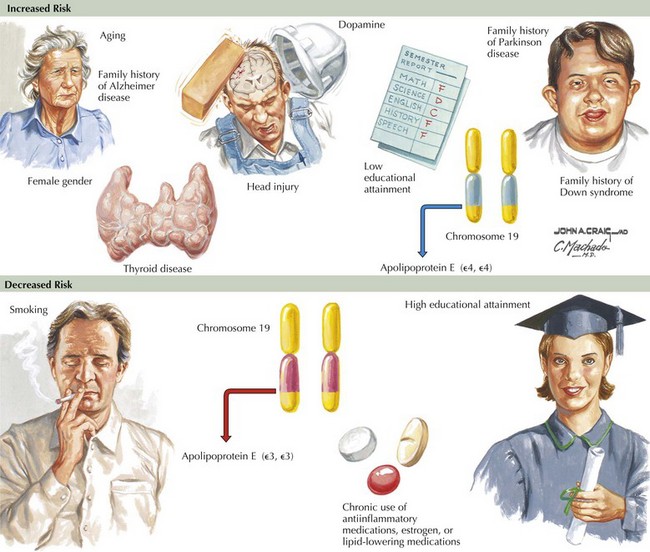
Epidemiologic studies identify several potential risk factors for AD. The most consistent risk factors include advanced age, family history (especially in first-degree relatives), and ApoE genotype. Other risk factors include hypertension, stroke, and fasting homocysteine levels (Fig. 18-6). Because vascular risk factors are modifiable, they may affect risk reduction and treatment for patients with AD and those at risk for development of AD.
Clinical Presentation
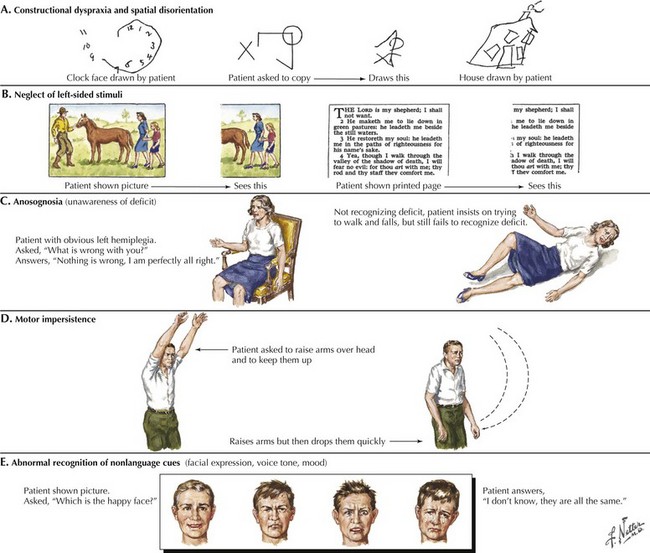
The early signs of AD may be subtle (Fig. 18-8). In the initial stages of AD, memory losses can be clinically distinguished from normal aging, although formal memory testing is often required to confirm suspicion of early dementia. The early signs of Alzheimer begin insidiously, progress slowly, and are often covered up by patients. Detection may be challenging even for close family members. The physician may observe changes in patient’s pattern of behavior, such as missing appointments or poor compliance with medications. It is important to discuss such issues openly with family members given the patient often cannot recall examples of memory problems. Indeed, it is common for patients to have limited insight into their deficit, and for family members to initiate an evaluation for memory loss. In these early stages, patients maintain their social graces. It is not uncommon during mental status testing to discover the significant cognitive problems concealed by a patient’s friendly and sociable affect. “Very pleasant” patients sometimes fool even seasoned geriatricians. The Alzheimer’s Association lists 10 key warning signs of AD.

Differential Diagnosis
The absence of motor deficits early in AD differentiates it from most other dementias. Other dementias lacking motor signs include amnestic syndrome (Korsakoff encephalopathy), Pick disease, vascular dementia, and HIV dementia complex. Depression can also produce dementia-like symptoms without motor deficits. Poor concentration and short-term memory impairment result from lack of effort, disinterest, or distractibility. “Pseudo dementia” due to depression is usually not progressive, and functional loss is often disproportionately severe relative to cognitive impairment (Fig. 18-9).
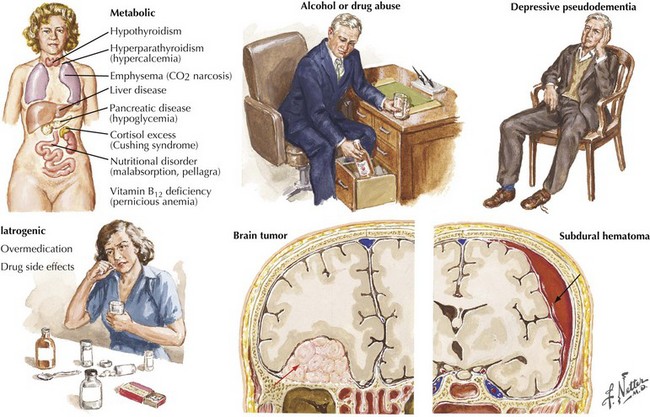
Reversible causes of dementia without motor signs include toxic and metabolic causes of chronic delirium. Chronic use of medication with anticholinergic side effects (e.g., antihistamines and tricyclic antidepressants) is a possible cause of chronic delirium that may mimic AD. β-Blockers, digoxin, H2 blockers, and various antibiotics may also contribute to chronic delirium. Chronic mass effect, caused by a slow-growing tumor (Fig. 18-9), may also produce reversible cognitive impairment.
Normal pressure hydrocephalus is a relatively uncommon condition that late in its course may be characterized by a significant dementia (see Fig. 32-2). Typically, these individuals present with a broad-based magnetic gait as if their feet were partially glued to the ground. Eventually these patients may become unwittingly incontinent, unaware of their loss of sphincter control as the dementing process evolves. Although these patients most often have no identifiable cause on occasion, they have previously sustained a subarachnoid hemorrhage or meningitis leading to poor cerebrospinal fluid (CSF) reabsorption. This leads to the characteristic hydrocephalus without an associated loss of cortical mantel. A CSF shunt may lead to a remarkable improvement.
Diagnosis
Mental Status Exam
The mental status examination (see Chapter 2) should assess all major cognitive domains, including Memory, Attention, Language, Construction, Orientation, Praxis, and Executive function (MALCOPE). Standardized global measures of cognitive function such as the MMSE are of limited diagnostic value. The widely used MMSE is relatively insensitive to the milder stages of AD. Other tests, such as the Montreal Cognitive Assessment test, include test items more suited to detecting earlier stages of cognitive impairment allowing improved sensitivity for detecting MCI. Another measure, the Alzheimer Disease 8 (AD8) is an informant-based tool that may shorten screening considerably. It must be emphasized that such instruments are not diagnostic tests, and interpretation of results must take into consideration level of education, native language, and physical or sensory impairment that might affect performance.
Clock drawing is useful when testing construction and executive function (Fig. 18-10). Patients draw a clock indicating 1:45, for example, on a blank sheet of paper. Their performance is observed from beginning to end, including the shape and size, number order and placement, hand size and placement, etc. The strategy (or lack thereof) used to draw the clock manifests itself readily, indicating impaired executive function in following a set of rules, or organizing and executing a multistep task. When patients finish, they should try copying a clock that the examiner draws in front of them. The numbers 12, 6, 3, and 9 are placed first, and the hands drawn accurately. If construction problems exist, patients have difficulty with the copy task as well as with the command task. If the copy is good, construction problems may not be a factor in cognitive impairment. There are many standardized, brief mental status tests like these available to clinicians. Routine use of such tests allows for longitudinal assessment and staging of dementia severity.
Additional Testing
Brain Imaging
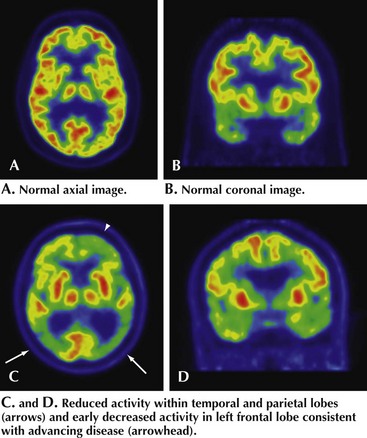
All patients with evidence of cognitive impairment should undergo structural brain imaging with MRI or when not available, CT. This may show findings of non-AD-related changes such as stroke, subdural hematoma, tumor, or hydrocephalus (Fig. 18-9).
Treatment
Treatment of Alzheimer Disease
General Approach
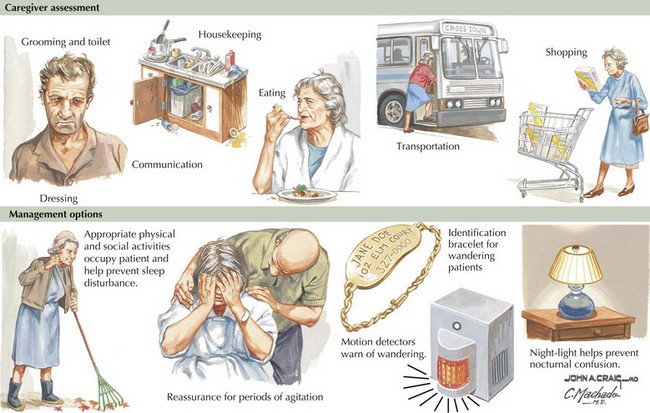
Much of the management of the patient with AD revolves around family interactions. Caregivers should provide patients with a comforting and respectful living environment. As their cognitive abilities slip away, it is important to provide a setting that preserves patient dignity. AD patients particularly benefit from a structured simple approach to daily life, maintaining a routine of social and physical activities (Fig. 18-12).
Cholinesterase Inhibitors
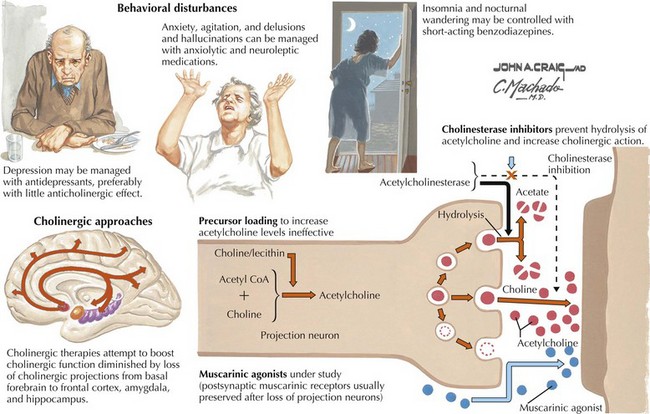
The various agents available include donepezil, rivastigmine, and galantamine. Several studies show these medications reduce the decline on standardized tests better than placebo when used for 6–12 months, but do not slow the degenerative process. These drugs may provide some benefit if taken consistently over time. When initiating therapy patients need to increase the dose gradually. Additionally, the eventual maximum required doses of these medications is not predictable. Cholinesterase inhibitors are generally utilized in patients with mild to moderate AD (Fig. 18-13).
Dementia with Lewy Bodies
Pathogenesis
Lewy bodies are intracytoplasmic inclusion bodies. They are the hallmark histopathologic lesions of primary PD where these occur within neurons of the substantia nigra and other brainstem nuclei. A spherical shape and eosinophilic staining properties characterize LBs morphologically (Fig. 18-14). The center stains densely, and a pale halo surrounds it. In cases of PD with dementia, LBs occur in cortical neurons and other gray matter regions. Cortical LBs are characterized by irregular shapes and do not have the characteristic pale halo seen with PD. Hence, cortical LBs can easily be missed with routine neuropathologic staining techniques. Moreover, LBs do not stain with silver-based stains often used to identify neuropathologic lesions in AD. A synaptic protein called alpha synuclein is the major LB component. Specific immunohistochemical stains for α-synuclein greatly improve LB detection throughout the brain. Ubiquitin staining also detects these lesions well. The function of α-synuclein is not completely understood. It may have a role in regulating presynaptic, nerve-terminal vesicular function. Mutations in the α-synuclein gene produce a mixed phenotype within members of affected kindred. Symptoms are predominantly PD-like, with cases of dementia occurring less frequently. α-Synuclein appears to be the main pathologic substrate in multiple systems atrophy (MSA) as well.
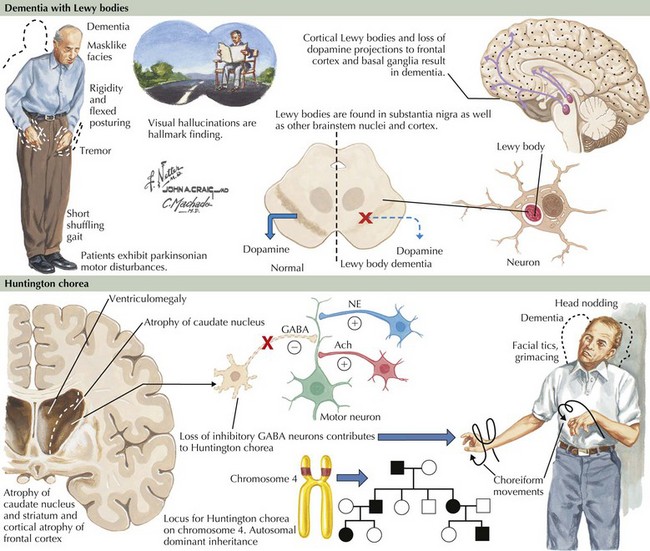
Clinical Presentation and Differential Diagnosis
Prominent psychotic features, including hallucinations and delusions, also develop in patients with DLB, although such symptoms are not typically seen early in the disease course. In DLB, psychosis can be an early and severely disabling feature, sometimes heralding the onset of dementia. Recurrent vivid and detailed visual hallucinations are particularly prevalent in DLB. The emotional response to these hallucinations ranges from relative indifference to severe agitation and combativeness. Agitation typically occurs when the patient has little insight or the hallucinations are perceived as threatening. Hallucinations having other sensory (i.e., nonvisual) also occur but are less specific for DLB. Delusions are frequently bizarre, complex, and unrelated to cognitive impairment. In contrast, delusions in patients with AD often occur from misinterpretation secondary to forgetfulness. For example, patients with AD may become suspicious of others when they cannot find things they misplaced. Other behavioral problems such as depression and anxiety also occur frequently but are not unique to DLB (Table 18-1).
| Manifestation | DLB | AD |
|---|---|---|
| Memory loss | Less pronounced, poor retrieval | Characteristic, poor encoding |
| Visuospatial and construction skills | Severely impaired early | Mildly impaired early |
| Executive function | Impaired earlier | Impaired later |
| Fluctuating mental status | Pronounced | Less pronounced |
| Psychotic features | Can be prominent early | Not typical early |
| Delusions | Bizarre, unrelated to impaired cognitive function | Often related to memory loss |
| Depression and anxiety | Common | Common |
| Parkinsonism | Within 1–2 years of dementia | Later in disease course |
AD, Alzheimer disease; DLB, dementia with Lewy bodies.
Frontotemporal Lobar Dementia
Clinical Presentation
Behavior Subtype of Frontotemporal Dementia (bvFTD)
The distinguishing clinical features of bvFTD and Pick disease are striking behavioral and personality changes. Most patients are unaware of their problem. There is often a major breakdown in social behavior, personal hygiene, and affect. Mental processes become concrete and perseverative (Fig. 18-15). Three major behavioral subtypes include disinhibition, apathy, and stereotypic behavior.
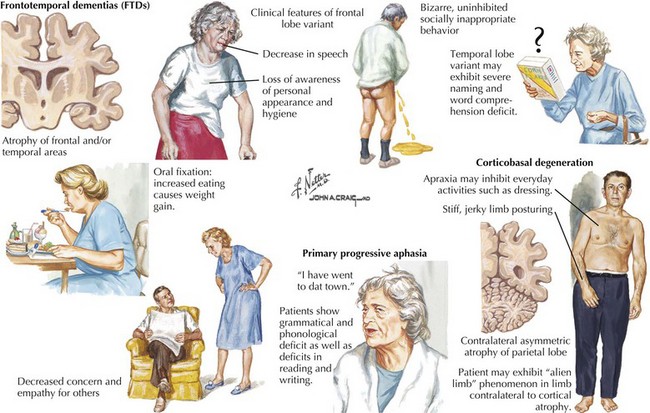
Vascular Cognitive Impairment
Clinical Presentation and Differential Diagnosis
Almost all standard diagnostic criteria for VCI require imaging studies demonstrating evidence of stroke (Fig. 18-16). However, there is no specific characteristic appearance on imaging studies that provide a diagnosis of VCI per se. The absence of specific cerebrovascular lesions of course mitigates this diagnosis. MRI is more sensitive than CT in showing subcortical and periventricular white matter changes consistent with small-vessel disease, and smaller infarcts. Therefore, a VCI diagnosis requires recognition of various syndromic features to correlate with findings on imaging studies.
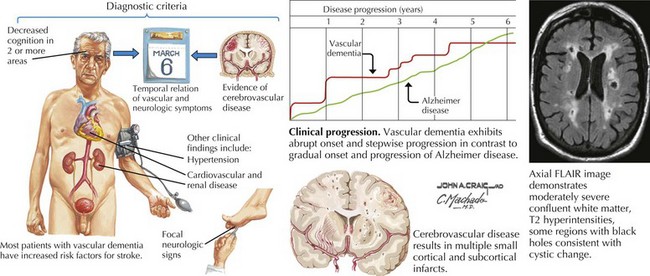
Moreover, small-vessel pathology is often seen in the context of “normal” aging, where the smallest branches become increasingly tortuous, producing twists and loops along paths deep in the brain. Morphologic changes are amplified by hypertension and diabetes. This results in diffuse myelin loss within deep vascular territories such as periventricular and subcortical white matter regions. The clinical correlates of small-vessel disease may include executive dysfunction, apathy, inattentiveness, and personality changes typical of frontal lobe syndromes as occur with hydrocephalus and frontotemporal dementia (see Fig. 18-15). Involvement of specific circuits correlates with recognized clinical manifestations.
Alzheimer’s Association. www.alz.org.
Cummings JL. Alzheimer’s disease. N Engl J Med. 2004 Jul 1;351(1):56-67. Review
Dubinsky RM, Stein AC, Lyons K. Practice parameter: Risk of driving and Alzheimer’s disease (an evidence-based review): Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2000 Jun;54:2205-2211.
Klafki HW, Staufenbiel M, Kornhuber J, Wiltfang J. Therapeutic approaches to Alzheimer’s disease. Brain. 2006 Nov;129(Pt.11):2840-2855. Epub 2006 Oct 3. Review
Knopman DS, Dekosky ST, Cummings JL, et al. The American Academy of Neurology. Practice Parameter: Diagnosis of Dementia (an evidence-based review—Report of the Quality Standards Subcommittee of the American Academy of Neurology). Neurology. 2001;56:1143-1153.
Lesser JM, Hughes S. Psychosis-related disturbances. Psychosis, agitation, and disinhibition in Alzheimer’s disease: definitions and treatment options. Geriatrics. 2006 Dec;61(12):14-20. Review
Raina P, Santaguida P, Ismaila A, et al. Effectiveness of cholinesterase inhibitors and memantine for treating dementia: evidence review for a clinical practice guideline. Ann Intern Med. 2008 Mar 4;148(5):379-397. Review
Small BJ, Gagnon E, Robinson B. Early identification of cognitive deficits: preclinical Alzheimer’s disease and mild cognitive impairment. Geriatrics. 2007 Apr;62(4):19-23. Review
Fantini ML, Ferini-Strambi L, Montplaisir J. Idiopathic REM sleep behavior disorder: toward a better nosologic definition. Neurology. 2005 Mar 8;64(5):780-786. Review
Huey ED, Putnam KT, Grafman J. A systematic review of neurotransmitter deficits and treatments in frontotemporal dementia. Neurology. 2006 Jan 10;66(1):17-22. Review
McKeith IG, Dickson DW, Lowe J. Consortium on DLB. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005 Dec 27;65(12):1863-1872. Review. Erratum in: Neurology. 2005
Murphy J, Henry R, Lomen-Hoerth C. Establishing subtypes of the continuum of frontal lobe impairment in amyotrophic lateral sclerosis. Arch Neurol. 2007;64:330-334. Review
Sleegers K, Kumar-Singh S, Cruts M, Van Broeckhoven C. Molecular pathogenesis of frontotemporal lobar degeneration: basic science seminar in neurology. Arch Neurol. 2008 Jun;65(6):700-704. Review
van der Zee J, Sleegers K, Van Broeckhoven C. Invited article: the Alzheimer disease–frontotemporal lobar degeneration spectrum. Neurology. 2008 Oct 7;71:1191-1197. Review
Josephs KA. Frontotemporal dementia and related disorders: deciphering the enigma. Annals of Neurology. 2008;64:4-14.
Bronge L, Wahlund LO. White matter changes in dementia: does radiology matter? Br J Radiol. 2007 Dec;80(Spec No 2):S115-S120. Review
Farlow MR. Use of antidementia agents in vascular dementia: beyond Alzheimer disease. Mayo Clin Proc. 2006 Oct;81(10):1350-1358. Review
Knopman DS. Cerebrovascular disease and dementia. Br J Radiol. 2007 Dec;80(Spec No 2):S121-S127. Review
Papademetriou V. Hypertension and cognitive function. Blood pressure regulation and cognitive function: a review of the literature. Geriatrics. 2005 Jan;60(1):20-22. 24. Review
Viswanathan A, Rocca WA, Tzourio C. Vascular risk factors and dementia: how to move forward? Neurology. 2009 Jan 27;72(4):368-374. Review
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
18 Dementia
Mild Cognitive Impairment, Alzheimer Disease, Lewy Body Dementia, Frontotemporal Lobar Dementia, Vascular Dementia
Mild Cognitive Impairment
The utility of these tests in detecting MCI is less reliable. Indeed, most patients with MCI score within the normal range on the MMSE. Interview-based dementia assessments, such as the Clinical Dementia Rating scale (CDR), provide a more sensitive means for reliable detection of MCI but may require considerably more time to administer. Another brief screening tool, the Montreal Cognitive Assessment Test (www.mocatest.org), may provide greater sensitivity in detecting MCI. The definitive diagnosis of MCI requires formal neuropsychological assessment. However, neuropsychological test batteries take several hours to administer and interpret. Therefore, they are not practical as screening tools. In the hands of an experienced neuropsychologist, formal neuropsychological tests provide the most sensitive means of detecting cognitive impairment. They may also provide greater specificity in identifying the underlying cause, although there may be significant variability among neuropsychologists’ interpretations.
Alzheimer Disease
Pathogenesis
There is pronounced gross cerebral atrophy clearly evident on both imaging studies and post mortem. Typically, the dementia of AD preferentially affects the frontal, temporal, and parietal cortex. This is particularly evident in the temporoparietal and frontal association areas as well as the olfactory cortex. In contrast, other primary sensory cortical areas are unaffected. Additionally, the limbic system as well as subcortical nuclei as well as the nucleus basalis of Meynert are preferentially affected. Microscopically, there is clear loss of both neurons and neuropil. The classic findings include senile plaques and neurofibrillary tangles (Figs. 18-1 and 18-2). The white matter sometimes demonstrates a secondary demyelination.
β-Amyloid
β-Amyloid is a short fragment of the APP, typically 40–42 amino acids in length, which accumulates outside the cell during APP processing (Figs. 18-3 and 18-4). The tertiary structure of the 42–amino acid fragment is a β-pleated sheet that renders it insoluble. Consequently, it accumulates slowly, over many years, in the extracellular space and within synapses. In vitro studies confirm that β-amyloid is toxic to surrounding synapses and neurons, causing synaptic membrane destruction and eventual cell death. Transgenic mouse models show a clear association between accumulation of β-amyloid fragments, formation of amyloid plaques, and development of cognitive impairment.
In vivo, β-amyloid fragments coalesce to form “diffuse” or immature plaques, best seen with silver-staining techniques. Diffuse plaques, however, are not sufficient to produce dementia; many nondemented elderly patients have substantial depositions of diffuse plaques throughout the cortex, a condition termed pathologic aging. It is when these plaques mature into “senile” or neuritic plaques that dementia becomes more likely (Fig. 18-5, top). Senile plaques consist of other substances in addition to β-amyloid, including synaptic proteins, inflammatory proteins, neuritic threads, activated glial cells, and other components. Unlike diffuse plaques, senile plaques are composed of a central core of β-amyloid surrounded by a myriad of proteins and cellular debris. Senile plaques are distributed diffusely in the cortex, typically starting in the hippocampus and the basal forebrain. Senile plaque formation correlates with increasing loss of synapses, which correlates with the earliest clinical sign, namely, short-term memory loss. The anatomic pattern of progression gradually spreads to neocortical and subcortical gray matter of the temporal, parietal, frontal, and, eventually, occipital cortex. Subcortical nuclei become involved relatively late in the process.
Neurofibrillary Tangles
The second pathologic hallmark of AD is the neurofibrillary tangle (Fig. 18-5, bottom). These lesions develop and conform to an anatomic pattern that correlates with the clinical syndrome; the number and distribution of tangles are directly related to the severity and clinical features of the dementia. Neurofibrillary tangles form intracellularly, consisting of a microtubule-associated protein, tau, which has a vital role in the maintenance of neuronal cytoskeleton structure and function. Tau is hyperphosphorylated in AD, causing it to dissociate from the cytoskeleton and accumulate, forming a paired helical filament protein structure. The cytoskeleton is compromised structurally and functionally, disrupting normal cell function. The most commonly used pathologic criteria for definitive AD diagnosis at autopsy require the presence of senile plaques and neurofibrillary tangles. Other lesions, such as Hirano bodies, are also seen in AD but have little diagnostic specificity.
Neurotransmitters
In addition to neuronal and synaptic loss, there is a gradual loss of various neurotransmitters. Acetylcholine synthesis is the earliest and most prominently affected. Most acetylcholinergic neurons arise within the nucleus basalis of Meynert in the basal forebrain (see Fig. 18-2). This nucleus is affected relatively early in the process; acetylcholine levels within the brain and spinal fluid of patients with AD quickly decline with disease progression. This observation supported the cholinergic hypothesis—that acetylcholine depletion results in the cognitive decline observed in patients with AD—eventually leading to the first symptomatic treatment of AD.
Risk Factors
Epidemiologic studies identify several potential risk factors for AD. The most consistent risk factors include advanced age, family history (especially in first-degree relatives), and ApoE genotype. Other risk factors include hypertension, stroke, and fasting homocysteine levels (Fig. 18-6). Because vascular risk factors are modifiable, they may affect risk reduction and treatment for patients with AD and those at risk for development of AD.
Clinical Presentation
The early signs of AD may be subtle (Fig. 18-8). In the initial stages of AD, memory losses can be clinically distinguished from normal aging, although formal memory testing is often required to confirm suspicion of early dementia. The early signs of Alzheimer begin insidiously, progress slowly, and are often covered up by patients. Detection may be challenging even for close family members. The physician may observe changes in patient’s pattern of behavior, such as missing appointments or poor compliance with medications. It is important to discuss such issues openly with family members given the patient often cannot recall examples of memory problems. Indeed, it is common for patients to have limited insight into their deficit, and for family members to initiate an evaluation for memory loss. In these early stages, patients maintain their social graces. It is not uncommon during mental status testing to discover the significant cognitive problems concealed by a patient’s friendly and sociable affect. “Very pleasant” patients sometimes fool even seasoned geriatricians. The Alzheimer’s Association lists 10 key warning signs of AD.
Differential Diagnosis
The absence of motor deficits early in AD differentiates it from most other dementias. Other dementias lacking motor signs include amnestic syndrome (Korsakoff encephalopathy), Pick disease, vascular dementia, and HIV dementia complex. Depression can also produce dementia-like symptoms without motor deficits. Poor concentration and short-term memory impairment result from lack of effort, disinterest, or distractibility. “Pseudo dementia” due to depression is usually not progressive, and functional loss is often disproportionately severe relative to cognitive impairment (Fig. 18-9).
[/not-level-membership-for-neurology-category]
