Delivery of biopharmaceuticals
Ijeoma F. Uchegbu and Andreas G. Schätzlein
Chapter contents
Key points
Introduction
Biopharmaceuticals, also known as biologicals or biologics, are medicines in which the active is derived from a biological (usually non-plant) source (Table 46.1). Vaccines are also considered here, even though they are constituted from whole attenuated/inactivated organisms as well as antigen subunits. The biopharmaceutical class of drugs includes proteins and peptides, nucleic acids and carbohydrates, all chemical components that exist in nature (Fig. 46.1). There are few carbohydrate drugs derived from biological sources, with heparin being the most well known, and while carbohydrate therapeutics are now being studied more intensely, they will not be treated in detail here because of the paucity of commercial products and information available. Other biopharmaceuticals include cell types such as the emerging area of transplanted stem cells and engineered tissues.
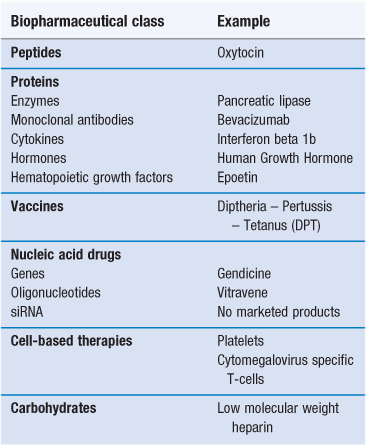
Since the 1990s, there has been huge growth in the area of biopharmaceuticals and this growth is predicted to continue, largely fuelled by the difficulty in identifying small molecule candidates for the diseases that still endure less than optimal treatment regimens or no treatments at all. The biopharmaceuticals market is dominated by three classes of drugs: protein and peptide hormones, e.g. insulin; monoclonal antibodies, e.g. trastuzumab and vaccines, e.g. the influenza and the diphtheria – pertussis – tetanus (DPT) vaccines. Other biopharmaceuticals include haematopoietic factors such as erythropoietin (epoetin); cytokines, e.g. the interferons; and enzymes such as the pancreatic enzyme, pancrealipase. Gene therapeutics currently constitute only a tiny fraction of the market, in essence limited to three authorized products – Gendicine, marketed in China, Rexin G, marketed in the Philippines and Glybera, recently approved in Europe. Ribonucleic acid gene silencing agents, commonly known as small interfering RNAs (siRNAs), while an area of active scientific endeavour, are yet to be marketed.
One aspect that unites these therapeutic molecules is that they are still administered mainly using a syringe and needle, despite the extensive efforts that have been made to deliver these compounds by non-parenteral means. Biopharmaceuticals are largely high molecular weight molecules (>5,000 daltons), with the exception of the short peptides and they suffer from instability problems, either on storage or after being administered. These properties make the administration of these compounds by non-parenteral means problematic at best and frequently impossible.
This chapter will serve as an introduction to the delivery issues associated with key members of this class of drugs, as well as an introduction to the established and emerging delivery solutions.
Protein and peptide drugs
Introduction
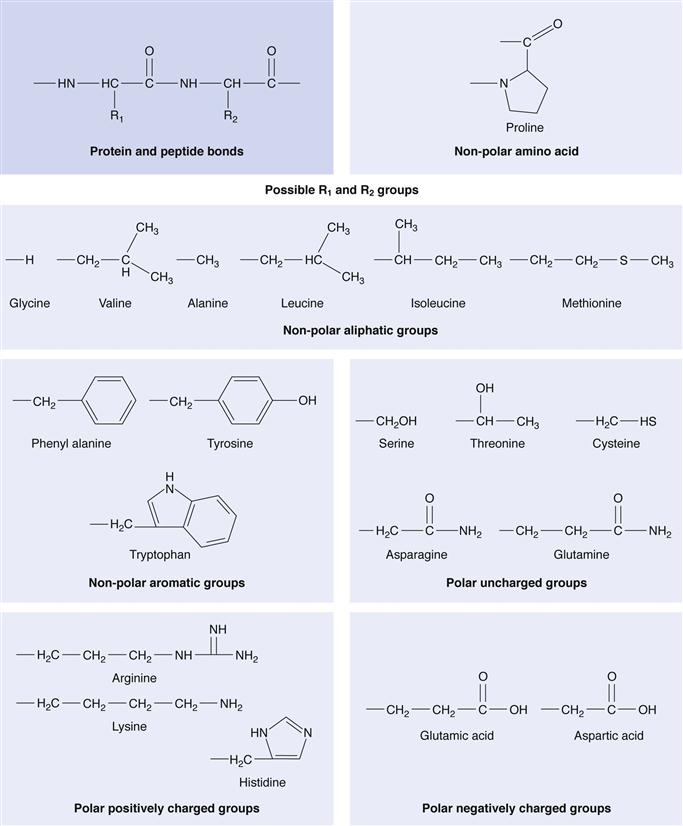
Proteins are composed of individual amino acids linked by amide bonds. The 20 known amino acids (Fig. 46.1) are the constituent parts of proteins. Amino acids are chiral compounds and amino acids of biological origin are L-amino acids. Peptides differ from proteins mainly in the number of amino acids contained within each molecule. The amino acid residue distinction between peptides and proteins is not exact: peptides are generally defined as having less than 50 amino acids, while proteins usually contain hundreds of amino acids and have a tertiary (folded) structure. However, insulin with 51 amino acids is defined as a peptide.
Endogenous proteins are synthesized in the cell in an amino acid sequence that is defined by a specific nucleotide base pair sequence. Following the synthesis of the protein, there is post translational modification, including glycosylation and protein folding to give the functional three-dimensional structure. Endogenous bioactive peptides are also synthesized within the cell and are normally the result of cleavage of larger proteins to give the peptide active. There are a number of therapeutic classes of proteins on the market (Table 46.1). The peptide therapeutic classes mostly comprise peptide hormones, such as insulin and calcitonin as well as endogenous peptide analogues, such as goserelin.
Production
Protein drugs are produced in mammalian cells, e.g. the Chinese Hamster Ovary (CHO) cell line; bacteria, e.g. Escherichia coli (E. coli) or yeast cells (Fig. 46.2). The gene of interest is transfected into the cells and the cells are grown in a bioreactor. The protein product is isolated by cell lysis and centrifugation/filtration and the protein is purified using chromatographic techniques. Protein yields are an important determinant of the efficiency of the process, with yields in CHO cells of about 5 g L−1. In an effort to improve yields, new cell lines have been introduced, e.g. the PER. C6 cell line, created by the transfection of human retinal cells with the Adenovirus 5 E1A gene. This cell line gives high cell densities (160 X 106 cells mL−1) and antibody yields as high as 25 g L−1.
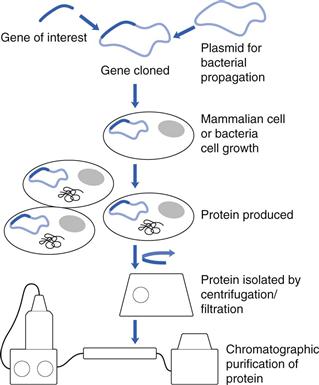
Fig. 46.2 The production of biopharmaceuticals.
Whilst the usual sources of proteins are mammalian and yeast cells, others such as animal sources are also being investigated. For example, transgenic animals have been used to produce human antithrombin in goats’ milk. Cheap plant sources of proteins could revolutionize the biotechnology industry, lowering production costs, reducing drug prices and would have a positive effect on patient access to these therapeutic agents. The feasibility of producing high value protein products in plant species has been recently demonstrated with the production of trastuzumab in the Nicotiana benthamiana specie using viral gene expression systems.
Peptides, such as insulin, are produced using the above described recombinant techniques, whereas shorter peptide chains (less than 50 amino acids) are synthesized using relatively expensive and laborious solid phase synthetic techniques.
Once proteins are produced using recombinant means, the product is characterized to establish its identity and purity using the analytical techniques outlined in Table 46.2.
Table 46.2
| Analytical technique | Physical/chemical basis of the technique | Protein characterization |
| Sodium dodecyl sulphate (SDS) PAGE analysis – a qualitative evaluation of molecular weight against molecular weight markers | Separates proteins according to their electrophoretic mobility and molecular weight | Provides a qualitative evaluation of protein molecular weight and detects proteolysis and dimerization impurities |
| Isoelectric focusing | Separates proteins according to their charge, as a function of pH | Characterizes the homogeneity of the protein product |
| Capillary zone electrophoresis | Separates proteins according to hydrodynamic radius, friction and charge | Characterization of glycosylation patterns |
| Size exclusion chromatography plus light scattering | Separates proteins according to hydrodynamic radius and measures molecular weight | Determines protein molecular weight |
| High performance liquid chromatography | Separates molecules according to polarity and quantifies protein levels | Demonstrates protein purity and used to analyse impurity levels |
| Spectroscopic methods Nuclear magnetic resonance (NMR) spectrometery Ultraviolet absorption spectroscopy Circular dichroism infrared (CDIR) spectroscopy |
Records electronic and molecular transitions in response to electromagnetic energy | Primary structure from NMR spectroscopy Reference material characteristics (e.g. from the IR fingerprint region) Secondary structure from CD and IR spectroscopy |
| Liquid chromatography – mass spectrometry (LC-MS) | Separation according to hydrophobic – hydrophilic balance and the detection of mass fragments | Peptide mapping |
| Differential scanning calorimetry | Measures the enthalpy change of protein thermal transitions (e.g. protein folding) | Data on protein stability |
| Epitope mapping | Identifies antibody binding sites on a protein – these may be linear peptide epitopes or conformational epitopes arising from protein folding Requires synthesis of the epitope followed by binding studies |
Measures efficacy and immunogenicity |
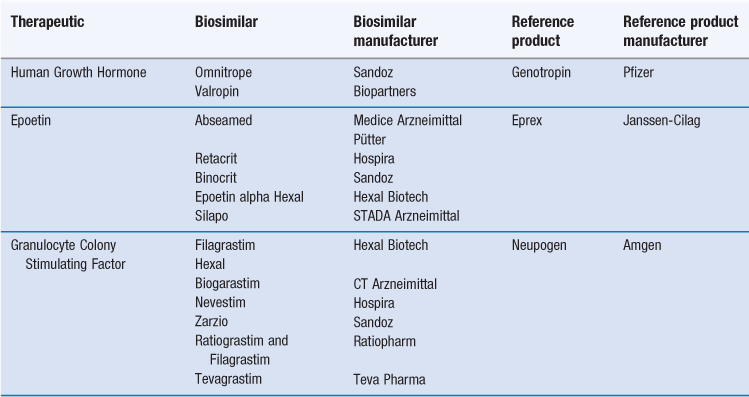
Production processes are normally proprietary and as a consequence, it is impossible, once the protecting patents have expired, for other companies to produce exactly the same therapeutic protein with identical glycosylation patterns without access to the original bioreactor procedures. This realization has led to a new category of medicine – the ‘follow-on biologic’ made by a separate manufacturer after patent expiry. Such follow-on products are also known as ‘biosimilars’ or ‘biobetters’. They must be comparable with respect to a number of key indicators in order to be considered biosimilars of reference marketed products. A number of biosimilars have been introduced and the biosimlars currently available in Europe are listed in Table 46.3.
Delivery issues
The delivery issues surrounding protein and peptide drugs are divided into two main categories: a) maintaining stability on storage and b) the optimization of in vivo efficacy. Storage stability issues may be classified according to the chemical and physical origins of protein instability. Of the chemical origins of protein instability – deamination (Fig. 46.3) is arguably the most intensively studied and occurs with the most frequency. Deamination is the result of base catalysed hydrolysis of asparagine (usually) and glutamine side chain amides to give aspartic acid and glutamic acid respectively. The reaction mechanism, which proceeds via the 5-membered cyclic ring, is the more prevalent mechanism, and the loss of ammonia on cyclization effectively makes the process irreversible. On deamination of asparagines, both aspartic acid and L-isoaspartic acid are formed.
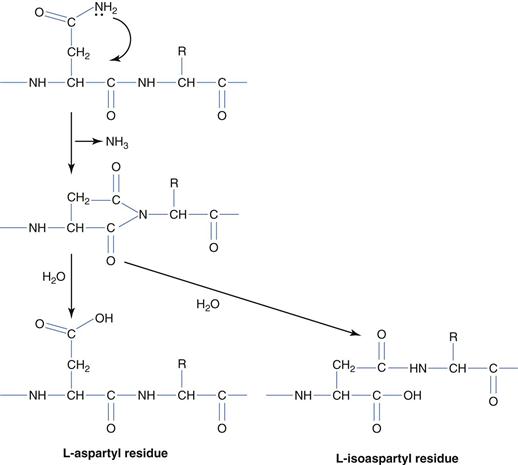
Peptide bond hydrolysis is also a source of instability and this occurs at aspartic acid and tryptophan sites and the hinge region of antibodies. Racemization of amino acids to convert from the L-form to the non-natural D-form also occurs at aspartic acid residues. Additionally, base-catalysed nucleophilic attack mediated amine terminal cyclization, to give diketopiperazine groups; occasionally with the loss of the first two amino acids or an amine terminal attack on the side chain of glutamic acid groups to produce a cyclic pyroglutamic acid residue, are both promoted at basic pH. Oxidation of proteins by reactive oxygen species occurs at histidine, methionine, cysteine, tyrosine and tryptophan residues and disulphide bond formation between cysteine residues is also a source of protein chemical instability. Base-catalysed protein degradation may be controlled by storage at acid pH (pH = 3–6).
Care must be taken when selecting excipients for protein formulations as glycation of proteins may occur if proteins are placed in contact with reducing sugars; the net result being the reaction of a side chain lysine with the reducing sugar to form a Schiff’s base – the Maillard reaction. Hence, protein stabilization with sugars should not be attempted using reducing sugars.
Proteins are also susceptible to physical instabilities. From a physical instability perspective, proteins are prone to denature (alter their native secondary or tertiary structure) on exposure to heat, extremes of pH or organic solvents. One of the most important challenges associated with protein formulation is to prevent protein aggregation in solution. Protein aggregation to form non-native aggregates, while not completely understood, is known to be mediated by either hydrophobic attraction between the less polar parts of the protein molecule or covalent bonding between two protein molecules. Partial unfolding to reveal hydrophobic faces has been implicated in protein aggregation and hence methods, which stabilize the protein in its native state, prevent protein aggregation. The administration of non-native aggregates is undesirable, since protein aggregates are known, specifically, to provoke an unwanted immune response as well as to compromise the efficacy of the drug.
When optimizing in vivo efficacy, the source of poor in vivo efficacy must first be thoroughly understood. One major source of poor efficacy is their chemical structure. Protein and peptide drugs are composed of hydrolysable bonds (Fig. 46.1) and are generally large molecules. They are highly water soluble and the combination of a high molecular weight, high propensity for degradation and hydrophilic character means that these molecules have difficulty in traversing biological, lipid rich membranes (Fig. 46.4), such as those found in the gastrointestinal tract and brain capillary endothelial cells. There are certain delivery routes that are precluded as a result of the fact that proteins are easily hydrolysed, and as such the oral delivery of proteins is not currently a possibility.
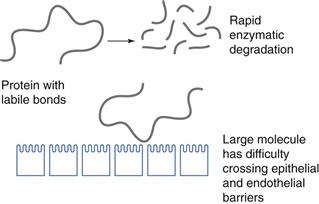
While there has been a huge effort aimed at making peptides orally active, there are only two oral peptide drugs on the market and both are cyclic peptides: ciclosporin and desmopressin. Cyclization undoubtedly makes these peptides less susceptible to gut amino and carboxy peptidases. Delivery of proteins to the brain by intravenous administration is also not currently a clinical reality. Other delivery routes that have been attempted include the nasal route, which is useful experimentally for delivering peptides to the brain and has been used commercially for the systemic (not specifically brain targeted) delivery of calcitonin, a 32 amino acid peptide used to treat osteoporosis. However, dose volumes are limited via this route to about 150 µL and a dose of about 25 mg is the upper limit (see Chapter 38).
Even when injected parenterally, e.g. intravenously, therapeutic proteins are cleared rapidly from the blood (Table 46.4) and this rapid clearance adversely affects their therapeutic outcomes. A further delivery challenge for therapeutic proteins, in particular, is their immunogenicity, where proteins generate neutralising antibodies, which make subsequent doses of the drug ineffective.
Table 46.4
| Protein | Plasma half-life following intravenous administration (h) |
| Arginine deaminase | 2.8 |
| GCSF | 1.8 |
| Human growth hormone | 0.34 |
| Interferon alpha 2a | 0.7 |
| Interferon beta 1a | 0.98 |
| Interferon beta 1b | 1.1 |
| Interleukin 6 | 0.05 |
| Tumour necrosis factor alpha | 0.07 |
(Adapted from Veronese, 2009.)
Delivery systems
Protein stabilization
The formulation of proteins and peptides involves preventing chemical degradation and enhancing in vivo activity. To prevent chemical degradation, a low pH is preferred (pH = 3–6) as this limits the reactivity of nucleophiles. Nucleophilic attack leads to deamination or amine terminal cyclization. Furthermore, drying, especially freeze-drying may be used to stabilize proteins against a variety of degradative influences (e.g. hydrolytic peptide bond cleavage and protein unfolding); although on freeze-drying lyoprotectants/cryoprotectants such as trehalose and sucrose, must be added to the protein formulation to replace the hydrogen bonding of the protein that is lost on removal of water. To prevent oxidative damage, limiting access to oxygen is an obvious step and this is achieved by reducing the head space in the final vial. Additionally, metal chelating agents, e.g. ethylene diamine tetraacetic acid (EDTA) may be added to prevent metal catalysed oxidation. Protein aggregation may also be prevented by the inclusion of various sugars, glycerol, arginine and urea; and although the mechanism of action of these stabilizers is not entirely clear, they appear to prevent the inter-protein interaction of protein hydrophobic faces. Such hydrophobic faces may be exposed on protein unfolding and there is evidence that polyols, such as glycerol, preferentially bind to the exposed hydrophobic faces, preventing inter-protein binding.
Protein delivery
Protein therapeutic products are formulated with a number of excipients. As an illustrative example, the anti-angiogenic monoclonal antibody, bevacizumab is formulated with trehalose (cryoprotectant and prevention of aggregation), phosphate buffer (for pH maintenance) and polysorbate 20 (to stabilize against interaction with surfaces, unfolding and aggregation).
Protein therapeutics are administered parenterally: mainly intravenously and sometimes subcutaneously or intramuscularly. Other non-parenteral routes, such as the nasal and pulmonary route, have also been attempted. There have been reports of the experimental administration of human growth hormone via the nasal route and interferon alpha via the pulmonary route; proteins are not administered via the oral route. Following intravenous administration, pharmaceutical proteins are generally rapidly cleared from the blood with short plasma half-lives (Table 46.4).
One method of prolonging the circulation time of proteins is by the use of poly(ethylene glycol) (PEG) conjugation. Conjugation of PEG to the therapeutic protein typically reduces the activity of the protein but markedly extends the biological half-life of the protein and in essence extends its duration of action, leading to longer dosing intervals. For example, the activity of PEGylated human growth hormone is reduced by 75% when compared to the native hormone but its intravenous half-life is increased 30 fold from 20 minutes to 10 hours. It is widely accepted that PEG acts by increasing the protein’s molecular volume above the glomerular filtration threshold of approximately 122 nm3 or 40 kDa. For example, the conjugation of haemoglobin to two chains of intermediate molecular weight PEG (10 kDa) or two chains of higher molecular weight PEG (20 kDa) results in molecular volumes of 712 and 1436 nm3, respectively. PEG conjugation also reduces the immunogenicity of therapeutic proteins, such as interferon beta 1b and recombinant human erythropoietin (epoetin), and masks sensitive degradation sites, preventing premature degradation. A number of PEG conjugates are available commercially including: PEG-asparaginase, PEG-Intron (interferon alpha) and Pegasys (interferon alpha).
Peptide delivery
Peptides, containing 2–50 amino acid residues are largely administered parenterally although there are a few marketed products in which peptides are administered via the nasal route (e.g. calcitonin nasal spray – 32 amino acids).
For the past three decades, there has been a huge focus on alternative forms of insulin delivery, presumably as a result of the Type II diabetes epidemic, which emerged at the turn of the century. It is estimated that there are 366 million diabetes sufferers worldwide, and all of the insulin-dependent diabetics administer or are administered their insulin via parenteral routes. There have been hundreds of studies examining the feasibility of delivering insulin via the oral route, but at the present time there are no commercially available insulin oral dosage forms.
Insulin is administered subcutaneously in fast acting and long acting forms. Insulin is composed of A and B chains. Rapidly acting insulin (e.g. Novolog) contains a mutation in the B28 proline, which is substituted with an aspartic acid residue; this prevents insulin from forming hexamers due to charge repulsion, allowing the monomers to be rapidly absorbed. Long acting insulin (e.g. Levemir) is prepared by decreasing the solubility of insulin such that insulin forms a depot. With Levemir, this is achieved by the conjugation of a C14 fatty acid chain to the B29 amino acid residue and the omission of threonine; the net result is that the molecules associate with albumin and have prolonged activity. An alternative method of reducing insulin’s solubility involves the substitution of amino acids, such that insulin achieves its isolectric point at neutral pH and forms an insoluble depot when injected subcutaneously, e.g. insulin glargine (Lantus) where the A21 glycine is substituted for asparagine and a further two arginines are added to the carboxy terminal of the B chain.
Long-acting peptides are required in certain disease states, such as for the treatment of prostate cancer, via chemical castration. In this therapeutic regimen, a luteinizing hormone releasing hormone (LHRH) agonist goserelin needs to be administered for several weeks to achieve its therapeutic effect. Initial doses of goserelin lead to an increase in plasma testosterone levels. This elevated testosterone ultimately creates a negative feedback loop within 14–21 days and eventually diminished levels of testosterone are achieved, a process termed chemical castration. The mechanism of action of goserelin is best suited to a long acting formulation. Goserelin is hence formulated within poly(DL-lactide–co-glycolide) microspheres (Chapter 45) as a one-month or three-month depot, known commercially as Zoladex. The one month formulation contains 3.6 mg goserelin in a poly(DL-lactide-co-glycolide) matrix consisting of 50% lactide and 50% glycolide, while the three-month depot contains 10.8 mg goserelin in a poly(DL-lactide-co-glycolide) matrix consisting of 95% lactide and 5% glycolide. The higher level of the less soluble lactide in the three-month depot formulation ensures that matrix erosion/degradation and drug release occur at a slower rate.
Vaccines
Introduction
Vaccines are administered prophylactically to patients to protect against infectious diseases. Mass immunization programmes at the start of the 20th century, coupled with access to clean water and the invention of antibiotics have had the most profound effect on human health ever witnessed. United Kingdom death rates from infectious diseases fell from a high of 300 deaths per 100 000 of the population in 1917 to a low of 4 deaths per 100 000 of the population in 2010. Smallpox has been eradicated by vaccination and the disease burden for a number of infectious diseases has been significantly reduced in the United States (US). For example, US cases of diptheria, pertussis, tetanus, measles, rubella and mumps were all reduced in the 21st century from their peak levels in the early 20th century by 99.95%, 98.2%, 98.34%, 99.99%, 99.97% and 99.86% respectively. Additionally, polio is all but eradicated in the Western Hemisphere. Vaccination is thus undeniably a success story.
Vaccines consist of antigens, which activate the immune system, produce antibodies against the antigen and induce immunological memory, enabling the immune system to recognize and destroy specific pathogens if exposed to the pathogenic molecules a second time (Fig. 46.5). There are three types of vaccines: live organisms, which have been attenuated to ensure that they do not cause disease; inactivated vaccines (inactivated by heat or chemical means) and subunit vaccines.
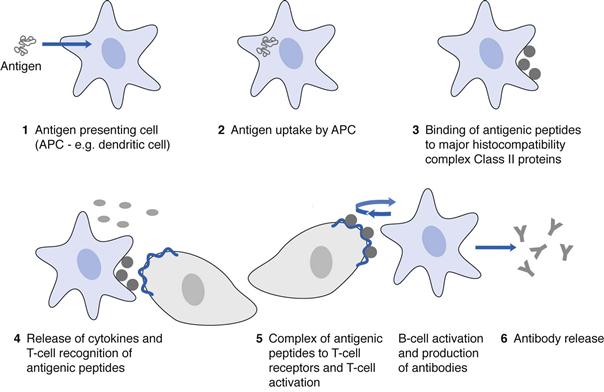
Prophylaxis against disease stems from innate and adaptive immune responses. The innate immune response is rapid and non-specific, and is a response to pathogen-associated molecular patterns (PAMPs) on a vaccine or pathogen via pattern recognition receptors (PRRs) on antigen presenting cells (APCs). The most studied PRRs are the toll-like receptors. The innate immune response involves an activation of the immune system, the removal of foreign cells and proteins and an activation of the adaptive immune response. This early immune response provides a rapid and generic defence to threats and is vital for the initiation of the adaptive and longer lasting immune response, which leads to immunological memory and to the antigen specific removal of the pathogen. On application, vaccines are taken up by APCs, the most abundant of which are the dendritic cells. The antigenic peptides are then presented on the major histocompatibility complex, Class II proteins of the APCs and there is recognition of this complex by the T-cells bearing T-cell receptors, which bind the complex. The result is T-cell activation and expansion. The release of co-stimulatory molecules and cytokines as part of the innate immune response also contributes to the activation of T-cells. T-cell activation is followed by B-cell activation and expansion and the release of antibodies specific for the antigen. The mechanism by which vaccines stimulate an immune response has largely informed the development of vaccine delivery systems.
Production
Bacterial vaccines are grown in bioreactors, while viral vaccines are produced in fertilized chicken eggs, although there has recently been a move towards the manufacture of viral vaccines in mammalian cells, such as MDCK cell lines. When viral vaccines are grown in MDCK cells, there is initially cell multiplication, followed by viral infection. The virus is taken up by the cell and the viral genomic material enters the nucleus where multiple copies of the virus are produced; the virus then infects other cells and when viral titres are sufficient the viruses are harvested. Once the viral or bacterial vaccine has been harvested it is inactivated using chemical methods or heat, prior to formulation. Alternatively, vaccine recombinant subunits are grown in host cells, by inserting the gene for the antigen into bacterial, yeast or mammalian cells and growing multiple copies of the antigen. Subunit vaccines, e.g. the hepatitis B vaccine or the human papilloma virus vaccine (which consists of the viral capsid devoid of genetic material), are usually composed of the viral antigenic coat proteins. The antigen is isolated from the cells using ultracentrifugation and chromatographic means.
Delivery issues
The foremost delivery challenge surrounding vaccines is the maintenance of the cold chain to prevent antigen degradation and ensure potency. It is also desirable to prevent unwanted bacterial growth and to ensure a sufficiently high and prolonged immune response as this will allow for fewer vaccination events. With subunit vaccines, it is necessary to present the antigenic proteins in particulate form to enable efficient uptake by antigen presenting cells.
Delivery systems
Most multiple use vaccines contain a preservative, such as the mercury compound, thiomersal. Vaccines may also contain antibiotics to prevent unwanted bacterial contamination and stabilizers such as 2-phenoxy esters. Furthermore, as stated above, all vaccines must be stored in a continuous cold chain to maintain antigen potency. This requirement for refrigeration contributes significantly to the costs of vaccine distribution and prevents low resource communities from accessing vaccines. In order to circumvent the considerable expense associated with the maintenance of a cold chain, researchers have prepared vaccine-sugar glasses in which a vaccine is mixed with trehalose and sucrose and dried on a membrane to be hydrated when required.
To produce vaccines with a prolonged and enhanced immune response, huge efforts have been put into designing vaccine formulations. Vaccine formulation excipients may work in one or both of two ways: as delivery systems which present the antigen to antigen presenting cells, and as adjuvants or immunopotentiators which stimulate innate immunity and ensure T-cell activation. Particulate delivery systems such as emulsions, liposomes and virosomes enable the subunit vaccines to be presented in a particle as it would be if it was still part of an infectious organism, allowing the antigen to then be taken up by antigen presenting cells (APCs). Immunopotentiators include the imidazoquinolones, which have been shown to enhance the immune response in preclinical studies and the potassium aluminium salts (e.g. KAl(SO4)2.12H2O, commonly known as alum) are adjuvants which stimulate dendritic cells to release cytokines. Alum is licensed for human use as an adjuvant, as are the microemulsions MS59 and AS03, which both contain squalene as the oil and have been used in influenza and avian influenza vaccines respectively. Both of these microemulsions potentiate the immune response.
There are various innovative approaches to vaccine delivery: one of which is the dry vaccine formulation mentioned above. A second innovation is intradermal vaccination using microneedles; this offers the two advantages of painless delivery and the ability to vaccinate large populations rapidly. Microneedles, which are 500–750 µm long, deliver their cargo into the epidermis or just below it and do not penetrate to the nerve endings, making the injection painless. The pores formed on insertion of the microneedles into the epidermis rapidly and painlessly reseal on withdrawal of the microneedles. Microneedles may be fabricated from solid, hollow or dissolvable materials and dissolvable microneedles have been fabricated from maltose and amylopectin. These devices, if adopted could really change the way in which populations are vaccinated as they may be used for mass vaccination in the event of a pandemic, with patients vaccinating themselves in their own homes, having received the vaccines by post.
Nucleic acid drugs
Introduction
Nucleic acid-based drugs fall into three classes: anti-sense oligonucleotides, small interfering ribonucleic acids (siRNAs) and genes. Deoxyribonucleic acid (DNA) is the constituent material of genes and ribonucleic acid (RNA) is the constituent material of messenger and transfer RNA. These nucleic acids consist of double stranded chains of nucleotides (Figure 46.6). Genes are an information repository for the cell and organism, containing genetic information which is established at conception. Genes are usually faithfully copied during the billions of cell division events that occur throughout a lifetime. However, a number of diseases may be traced to the mutation of various genes and thus have a genetic basis. Mutations may be present at conception or occur at some later time, e.g. as a result of environmental pollutants, such as those contained in tobacco smoke, or following excessive exposure to the sun’s ultraviolet radiation. Genes give rise to the cell’s proteins via transcription (messenger RNA synthesis) and translation – protein synthesis (Fig. 46.7); proteins are the functional components of the cell. Gene mutations will thus alter the resulting protein and such alterations may lead to disease. Disease linked gene mutations, as in cystic fibrosis, result in a non-functioning protein – the non-functioning cystic fibrosis transmembrane regulator (CFTR). The mutated CFTR gives rise to poor ion and fluid transport across membranes and specifically the poor quality lung function observed in cystic fibrosis patients. Theoretically, gene therapy may be used to replace a mutated gene and thus achieve a functioning protein. However, gene replacement therapy is not straightforward, as delivery vectors are required to achieve the gene therapy goal. Proof of this therapeutic concept has been demonstrated in humans by the licensed gene medicine, Gendicine. Gendicine contains wild type p53 to replace mutated p53 in cancer cells and is delivered in an adenoviral vector.
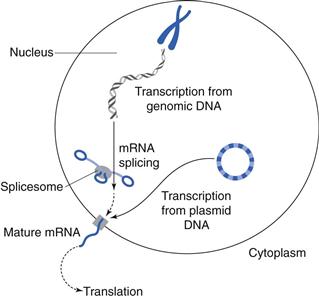
Oligoucleotides, e.g. vitravene are single stranded chains of nucleotides which inhibit translation and in the case of vitravene, inhibit translation of viral messenger RNA, thus providing a treatment for cytomegalovirus ocular infections. Small interfering RNA (siRNA) sequences inhibit translation by combining with the RNA induced silencing complex (RISC) to selectively and specifically cause the hydrolytic degradation of messenger RNA. The RISC precursor complex (~250 kDa) is transformed in the presence of adenosine triphosphate (ATP) into the active 100 kDa complex that cleaves the substrate mRNA in regions homologous to the siRNA template. siRNA therapies are still largely experimental and there are no siRNA therapeutics on the market at present.
Production
For genes to be used as therapeutic agents there is an absolute requirement for a delivery system. The two marketed gene therapeutics (Gendicine and Rexin G) are delivered using viral vectors. Gendicine, which is delivered in an adenovirus vector, is produced as a viral particle. The Gendicine adenovirus consists of an E1 gene deleted adenoviral vector, with E1 gene deletion necessary to prevent replication. The adenovirus is produced in SBN-Cells, which supply the E1 proteins, necessary for replication. As there are some limitations associated with viral vectors for gene therapy, notable of which is their limited use as systemic therapeutics, there have been a number of studies looking at the development of synthetic vectors. If synthetic (chemical compound) vectors are to be used for gene therapy, the gene product is delivered as a bacterial plasmid. Plasmids containing the gene of interest are grown in E. coli cell lines and purified by cell lysis, filtration, chromatographic separation and centrifugation.
siRNA and oligonucleotides are synthesized chemically with many companies offering custom synthesis for particular siRNA or oligonucleotide sequences.
Delivery issues
The main delivery issues surrounding nucleic acid drugs are: a) poor plasma stability of DNA, siRNA and oligonucleotides and b) the inability of a large polar (negatively charged at physiological pH) molecule such as DNA, or even double stranded siRNA to cross the lipid rich plasma cell membrane. A further issue surrounding DNA delivery is the fact that therapeutic DNA must gain entry to the cell nucleus in order to produce its therapeutic product, the functional protein. Entry to the nucleus is limited largely to cell division events. These delivery issues mean that gene and siRNA therapeutics have an absolute requirement for a delivery system.
Delivery systems
The delivery of genes in commercial gene therapies has so far been achieved using viruses, despite the fact that one third of the over 2000 gene therapy trials conducted to date use synthetic (non-viral) vectors or no vectors at all. Gendicine, the world’s first gene therapeutic, and currently only licensed for use in China, comprises the wild type p53 gene within an E1 deleted gene adenovirus for the treatment of head and neck cancers. This replication incompetent virus performs a mutation compensation function and is administered intratumourally. The need for intratumoural injections is a severe limitation of the therapy as it may not be easily used to treat metastatic cancers. Patients receive one injection per week for 4–8 weeks and each injection consists of 1012 viral particles in 1mL of water for injection containing glycerol (to maintain tonicity).
Normally p53 is upregulated in cancer cells and causes cell apoptosis, anti-angiogensis, an activation of an anti-tumour immune response and a down regulation of the expression of the multi-drug resistance genes. However p53 is mutated in 50 – 70% of human tumours. On intratumoural administration of Gendicine, the adenovirus enters the cancer cell via the coxsakie adenovirus receptor and the cell then begins to over express wild type p53, causing apoptosis. Gendicine is only effective when administered alongside radiotherapy.
A further approved gene therapeutic, available in the Philippines for the treatment of metastatic lesions is Rexin G: a replication incompetent retroviral (murine leukemia virus) particle, bearing the human Cyclin G1 gene. The Cyclin G1 gene causes apoptosis and necrosis in dividing tumour cells and in the cells of the tumour vasculature.
The use of viral delivery systems for gene therapy has had a problematic history with the high profile death of an otherwise healthy patient in an adenovirus gene therapy trial and patients developing leukaemia, stemming from insertional mutagenesis, in a trial involving ex-vivo retroviral gene therapy for the treatment of Severe Combined Immunodeficiency Syndrome. As a result, a number of synthetic gene vectors have been explored, using poly(propylenimine) dendrimers, liposomes and naked DNA. Good preclinical efficacy (tumour regression) data has been obtained with the poly(propylenimine) dendrimer gene therapy system.
The delivery of siRNA has been accomplished clinically using a cyclodextrin based nanoparticle carrier covered with PEG. Gene silencing of the M2 subunit of ribonuclease reductase was achieved in human melanoma tissue in this pivotal human study.
Summary
Biopharmaceuticals (peptides, proteins, vaccines and nucleic acid medicines) are varied in chemical structure but have in common the presence of labile covalent bonds that are prone to hydrolysis, either on storage or administration. A further factor confounding the long term stability of these medicines is the functional tertiary structure of the protein class of molecules, a structure that is prone to unfolding at extremes of temperature, pH or in the presence of various chemical agents such as metal ions. Stabilization of the functional protein entity on storage requires a fundamental understanding of the instability profile (e.g. the underlying mechanisms of protein unfolding and protein aggregations), and extreme care during production, processing and storage. A complete understanding of the mechanisms underpinning protein unfolding and aggregation is yet to be realized. This knowledge gap prevents formulators from adopting a rational set of steps towards protein formulation.
The most important factors to consider when formulating protein, peptide and gene biopharmaceuticals are the in vivo delivery challenges, i.e. the rapid clearance and degradation of the active agent in vivo and the active agent’s difficulty in traversing cell membranes and cells. Solutions to these formulation/delivery difficulties include the attachment of PEG chains to proteins to reduce protein clearance and degradation and the fabrication of poly(lactide-co-glycolide) matrices for the controlled and prolonged release of peptides. To enable genes to cross cell membranes and arrive at the cell nucleus, the commercial gene medicines are delivered using adenoviral and retroviral vectors.
From the foregoing it is clear that, if at all possible, the formulation of new biopharmaceuticals should be approached systematically by considering the nature of the source of the active agent’s instability both on storage and on administration and a desire to match the drug to the disease, without introducing unwanted toxic effects.
References
1. Dranitsaris G, Amir E, Dorward K. Biosimilars of biological drug therapies, regulatory, clinical and commercial considerations. Drugs. 2011;71:1527–1536.
2. Veronese F, ed. PEGylated Protein Drugs: Basic Science and Clincial Applications. Berlin: Birkhäuser; 2009.
Bibliography
1. Dufes C, Keith WN, Bilsland A, Proutski I, Uchegbu IF, Schätzlein AG. Synthetic anticancer gene medicine exploits intrinsic antitumor activity of cationic vector to cure established tumours. Cancer Research. 2005;65:8079–8084.
2. Falconer RJ, Jackson-Matthews D, Mahler SM. Analytical strategies for assessing the comparability of biosimilars. Journal of Chemical Technology and Biotechnology. 2011;86:915–922.
3. Hedge NR, Srivnas VK, Bayry J. Recent advances in the administration of vaccines for infectious diseases: microneedles as painless delivery devices for mass vaccination. Drug Discovery Today. 2011;16:1061–1067.
4. Hou JJC, Codamo J, Pilborough W, Hughes B, Gray PP, Munro TP. New frontiers in cell line development: challenges for biosimilars. Journal of Chemical Technology and Biotechnology. 2011;86:895–904.
5. Illum L. Nasal drug delivery – recent developments and future prospects. Journal of Controlled Release. 2012;161:254–263.
6. Manning MC, Chou DK, Murphy BM, Payne RW, Katayama DS. Stability of pharmaceutical proteins. Pharmaceutical Research. 2010;27:544–575.
7. Nelson DL, Cox MM. Lehninger Principles of Biochemistry. 4th edn New York: W.H. Freeman and Company; 2005.
8. O’Hagen DT, Valiante NM. Recent advances in the discovery and delivery of vaccine adjuvants. Nature Reviews Drug Discovery. 2003;2:727–735.
9. Peng Z. Current status of Gendicine in China – Recombinant Ad-p53 agent for treatment of cancers. Human Gene Therapy. 2005;16:1016–1027.
10. Shukla D, Schneider CP, Trout BL. Molecular level insight into intra-solvent interaction effects on protein stability and aggregation. Advanced Drug Delivery Reviews. 2011;63:1074–1085.
11. Uchegbu IF, Schätzlein AG, eds. Polymers in Drug Delivery. Boca Raton: Taylor and Francis; 2006.