CHAPTER 23 Degradation of Cellular Components*
An individual cell can live for weeks, months, years, or even the entire lifetime of the organism, but the cell’s constituent proteins, lipids, and RNA turn over continuously. This process of molecular degradation and replacement serves three functions. Constitutive turnover is a “housekeeping” function that ensures regular replacement of older molecules with newly synthesized ones or that removes misfolded, mislocalized, or otherwise damaged molecules so that they do not hinder the function of native molecules. Regulated or induced turnover results in rapid degradation of specific target molecules and functions in signal transduction, regulation of the cell cycle, and remodeling of cells and tissues during development. Finally, macroautophagy, a more global mechanism for degradation of cellular proteins or lipids, can be triggered under conditions of starvation, when the cell perceives a shortage of specific raw materials, such as amino acids. This chapter focuses primarily on the mechanisms that govern protein degradation and turnover, as these are the best studied; lipid turnover is also discussed. Chapter 16 covers RNA turnover.
Characteristics of Constitutive Protein Turnover
The turnover of cellular constituents at steady state arises from a careful balance of the rates of synthesis and degradation. Turnover can be massive. For example, approximately 40% of total cellular protein in rat liver is degraded every day. The rates of transcription and translation determine the rate of synthesis of each protein. Once synthesized, each type of protein turns over at a characteristic rate. Degradation is random, so older molecules are not selected over younger ones. The time course of degradation of the population of any specific protein follows a single exponential—strong evidence that chance, rather than aging, determines which copies of the protein are degraded (see Fig. 4-1 for first-order reactions). The rate is usually expressed as a half-life, the time for half of the molecules to be degraded.
The intrinsic rate of degradation of a given protein is determined by many factors, including its size, overall charge, thermal instability, flexibility, hydrophobicity, folding, and assembly with other protein subunits (if it is multimeric). Long-lived proteins have half-lives measured in days, while short-lived proteins have half-lives of hours or minutes. Small, basic proteins tend to have longer half-lives than do large, acidic proteins, and key enzymes of metabolic pathways often have very short half-lives. A protein may have specific sequences or structural motifs that are recognized by the proteolytic machinery. Phosphorylation marks some proteins for destruction. The rate at which a protein is degraded can be altered either by increasing the activity of its degradative pathway or by exposure or creation of “degradation motifs” on the protein to initiate destruction.
Degradation in Lysosomes
Lysosomes, the major digestive organelles, contain at least 60 distinct hydrolytic enzymes, including proteases, lipases, phospholipases, glycosidases, and nucleases. Lysosomal hydrolases are tagged in the Golgi apparatus with mannose-6-phosphate groups on their N-linked oligosaccharides. Mannose-6-phosphate re-ceptors in the trans-Golgi network bind lysosomal hydrolases for diversion to endosomes and lysosomes (see Chapter 22). Most lysosomal hydrolases are synthesized as inactive precursors and are activated by proteolysis on arrival in lysosomes. The low pH of lysosomes, maintained by the vacuolar adenosine triphosphatase (ATPase) proton pump (see Fig. 8-5C), is essential for efficient degradation. Most lysosomal enzymes have maximal hydrolytic activity at pH 4 to 5 rather than at the cytoplasmic pH of 6.5 to 7.0. Moreover, the low pH helps to denature many proteins, increasing their susceptibility to degradation. It is important to note that the hydrolases themselves are more resistant than are most macromolecules to the harsh environment. Lysosomal enzymes degrade proteins, lipids, and nucleic acids to fragments that are small enough to be transported either actively or passively across the lysosomal membrane to the cytoplasm, where they are reused to synthesize new macromolecules.
Lysosomes degrade substrates that originate both outside and inside the cell. Extracellular substrates taken into the cell by endocytosis are delivered to lysosomes via the endocytic pathway (see Chapter 22). Lysosomes also degrade cellular constituents, accounting for 50% to 70% of cellular protein turnover. Substrates are delivered to lysosomes by endocytosis and by direct translocation from the cytoplasm. Degradation of intracellular substrates is generally termed autophagy, which occurs by three distinct mechanisms called crinophagy, macroautophagy, and microautophagy, which are described later.
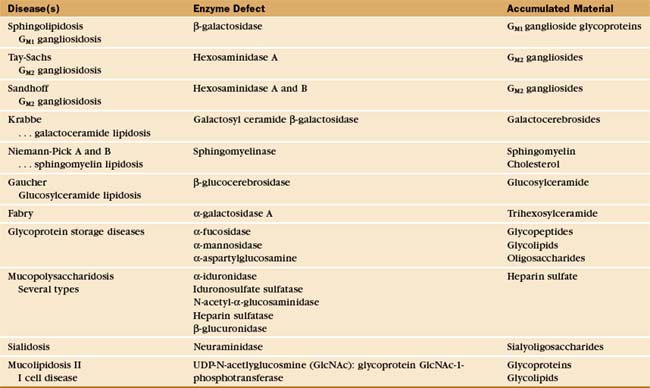
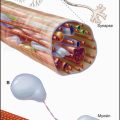
The essential role of lysosomes as the primary site for constitutive degradation is revealed by the more than 30 distinct human lysosomal storage diseases (Appendix 23-1). Patients with these diseases lack the function of one or more lysosomal hydrolases. Consequently, undigested material accumulates in lysosomes and causes them to swell (Fig. 23-1), ultimately killing the cell.
Delivery to Lysosomes via the Endocytic Pathway
Lysosomal degradation of plasma membrane proteins internalized by endocytosis plays an important role in remodeling the plasma membrane in response to cell stimuli. For example, the half-life of the receptor for the epidermal growth factor (EGF) is normally approximately 10 hours. However, when circulating EGF binds, the activated receptor is more efficiently internalized and degraded in lysosomes with a half-time of less than 1 hour. This downregulates the biological response (see Fig. 27-6).
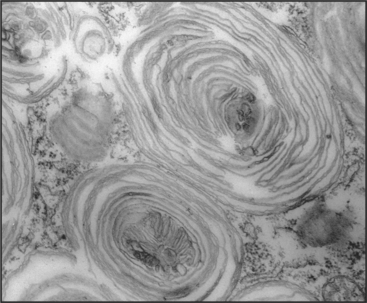
The degradation of lipids and membrane proteins in lysosomes poses a topologic problem, since fusion of lysosomes with other membrane compartments would simply merge the two membranes. This problem is solved by segregating the components to be degraded into regions of membrane that bud into endosomes, forming the intraluminal vesicles or tubules of multivesicular bodies (see Fig. 22-16). Fusion of a multivesicular body with a lysosome delivers the intraluminal vesicles to the lumen of the lysosome for digestion (Fig. 23-2). Resident lysosomal membrane proteins remain in the limiting membrane.
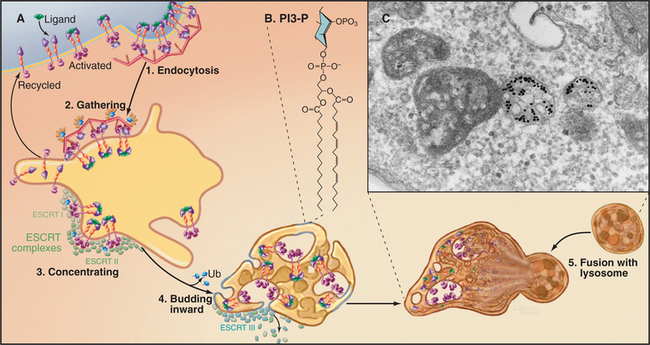
The formation of multivesicular bodies starts with a sorting process in endosomes (see Chapter 22). Proteins that are destined for degradation are tagged with single ubiquitin molecules, a process that is distinct from the polyubiquitination reaction required for targeting to proteasomes (see later). These monoubiquitinated proteins are gathered together in endosomes by ubiquitin-binding proteins that are localized there by interaction with phosphatidylinositol-3-phosphate (PI3-P), formed by PI3-kinase (see Fig. 26-7). A sequence of three multiprotein ESCRT complexes (endosomal com-plex required for transport [Fig. 22-16]) then further concentrates the monoubiquitinated proteins on regions of membrane that bud into the vesicle interior by an unknown mechanism. Components of the ESCRT complexes initially bind monoubiquitin but later recruit enzymes that remove and recycle the ubiquitin. Interestingly, budding of a number of viruses, including HIV-1, the virus that causes AIDS, resembles the process of multivesicular body formation. In fact, HIV hijacks two of the ESCRT complexes for this purpose.
Autophagy
Microautophagy and macroautophagy both describe the consumption of cytoplasmic constituents within lysosomes. Microautophagy occurs as a by-product of the formation of multivesicular bodies (Fig. 23-2; also see Fig. 22-16). Small volumes of cytoplasm are captured in the intraluminal vesicles and tubules that invaginate within endosomal or lysosomal membranes. The cytoplasmic components are degraded as the vesicles are consumed. However, studies in yeast suggest the exist-ence of alternative pathways for the selective packaging and delivery of cytosolic proteins to lysosomes. When starved for glucose, Saccharomyces cerevisiae expresses several cytosolic enzymes and membrane transporters that are required to process more complex sugars. When glucose becomes available, the yeast switches metabolic pathways and degrades the enzymes it no longer needs. Transporters in the plasma membrane are internalized and delivered to the vacuole (the yeast lysosome equivalent) through the formation of multivesicular bodies and microautophagy. Unneeded cytoplasmic enzymes are selectively packaged into small vesicles that deliver their contents to the vacuole by fusion. Genetic studies of yeast are unraveling the mechanisms of these rapidly induced and selective degradation pathways.
Macroautophagy involves the engulfment of large regions of cytoplasm—that might include glycogen granules, ribosomes, and organelles, such as mitochondria and peroxisomes—into an autophagic vacuole. Autophagic vacuoles begin to form when a flattened membrane cisterna encircles a region of cytosol and closes into a vesicle with two membranes (Fig. 23-3). The origin of the smooth membrane cisternae is uncertain and could be either the smooth endoplasmic reticulum (ER), the trans-Golgi apparatus, or the endosomal system. The trans-Golgi apparatus has the advantage of having targeting information for fusion with lysosomes. Fusion of a nascent autophagic vacuole with late endosomes and lysosomes forms an autolysosome with acid hydrolases in the lumen to degrade the contents (Figs. 23-3 and 23-4). The end stage of an autolysosome is typically a residual body with a dense core of undegraded material. The process of formation and degradation of autophagic vacuoles in the liver requires less than 15 minutes.
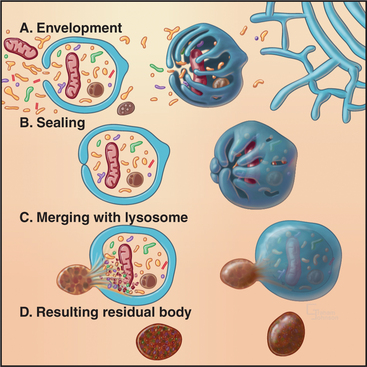
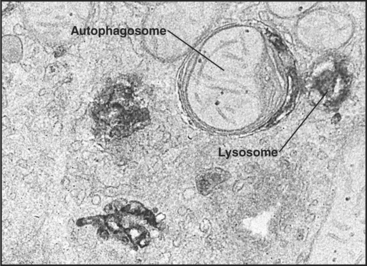
Because large volumes of cytoplasm and entire organelles are destroyed, macroautophagy must be regulated precisely and directly. The intracellular signal that triggers macroautophagy is thought to be tied to the intracellular levels of particular amino acids, which are, in turn, related to the extracellular concentrations of these amino acids. Amino acids are potent inhibitors of autophagy. Circulating peptide hormones also regulate autophagy by binding receptors and activating signaling cascades that involve protein phosphorylation. For example, starvation increases the circulating levels of the hormone glucagon, which stimulates autophagy in liver cells. Feeding produces the opposite reaction by increasing the levels of insulin, which reduce auto-phagy. Diurnal feeding rhythms cause the numbers of autophagic vacuoles to vary along with the fluctuation of essential amino acids in the blood.
Budding yeast use autophagy as a cell survival pathway during starvation, but autophagy can kill some cells. For example, autophagy can lead to death when a cell receives a lethal insult under conditions in which apoptotic pathways (see Chapter 46) are not functional. Whether autophagy is a physiological cell death pathway in addition to apoptosis remains controversial. Autophagy has also been implicated in the developmental programs of a number of higher organisms and in the destruction of intracellular protein aggregates.
Degradation by Proteasomes
The proteasome is the second major cellular compartment for proteolysis. Proteasomes are multisubunit structures about half the size of a ribosome that are located in both the cytoplasm and nucleoplasm (Fig. 23-5). They are abundant, often accounting for up to 1% of total cellular protein. Proteasomes contain an array of proteolytic active sites arrayed on the interior wall of a cylindrical chamber. They degrade abnormal and misfolded proteins as well as selected normal proteins down to the level of small peptides (Fig. 23-6). Proteasomes degrade key substrates in response to signaling cascades or at key transitions of the cell cycle. One class of proteasomes processes intracellular antigens for presentation by the immune system.
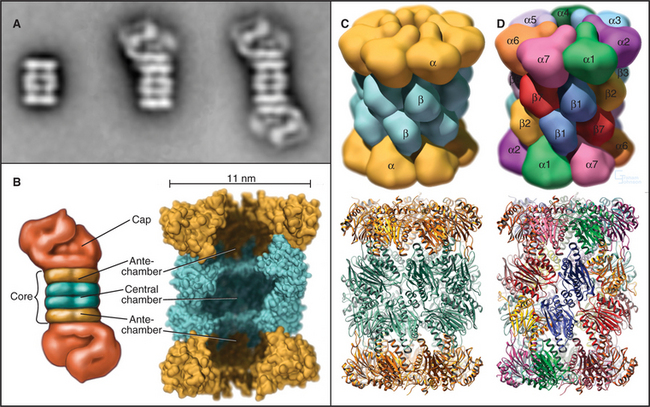
The proteasome has two major structural components: the core and the cap. The core, referred to as the 20S proteasome (named according to its sedimentation coefficient; see Chapter 6) is structurally conserved from bacteria to mammals, although the subunit composition varies. In mammals, the cylindrical 20S core is assembled from two copies each of 14 different 25- to 35-kD subunits arranged into four seven-membered rings: α-type subunits form the top and bottom rings flanking the two rings of β-type subunits. Four constrictions divide the interior into three cavities: a central chamber surrounded by the β-subunits and two antechambers at either end of the cylinder (Fig. 23-5B). The proteolytic active sites on the β-subunits face the central chamber. An N-terminal threonine residue on the β-subunits is exposed by autocatalytic proteolysis and serves as the key active site residue for proteolysis. The antibiotic lactacystin reacts covalently and selectively with these threonine residues to inactivate the proteasome.
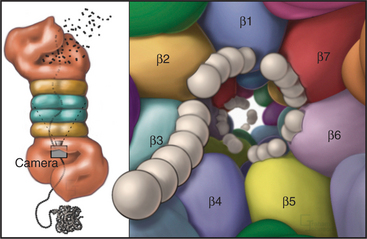
Eukaryotic proteasomes have multiple types of hydrolytic activities that can be ascribed to distinct β-type subunits (Fig. 23-6). In yeast and probably in mammalian proteasomes, the caspase-like activity of the b1-subunit cleaves after acidic residues, the trypsin-like activity of the b2-subunit cleaves after basic residues, and the chymotrypsin-like activity of the β5-subunit cleaves after hydrophobic residues (see Fig. 46-10 for a description of caspases). These three activities combine to give the proteasome broad specificity, allowing it to cleave diverse substrates into short peptides. Other β-type subunits in eukaryotic proteasomes are apparently not posttranslationally processed to mature, catalytically active enzymes. It was initially thought that the restricted length (seven to nine residues) of the final products of proteolysis reflected an “intrinsic molecular ruler,” the length of which corresponded to the distance between active sites. However, mutagenesis to selectively inactivate individual β-subunits has no effect on product length, as would be predicted. Instead, the length of the peptides that are produced might be related to the kinetics of translocation of the unfolded substrate polypeptide through the pro-teasome.
The proteasomes of eukaryotes and Archaea are “capped” on one or both ends of the 20S barrel with regulatory complexes to form the 26S proteasome. The type of regulatory complex varies depending on the function of the proteasome. One such complex, called the 19S regulator or PA700, has a mass of approximately 700 kD (see Fig. 23-5A). Six of its subunits are members of the AAA ATPase family (see Box 36-1) and are believed to play a role in the dissociation of target protein oligomers, protein unfolding, and translocation into the central channels. Other subunits are involved in recognition of ubiquitinated proteins and in recycling ubiquitin from proteins destined for degradation (see the next section for more about ubiquitin).
A second regulator complex, the 11S cap or PA28 regulator, is associated with a subpopulation of 20S proteasome cores only in cells of higher vertebrates. This 20S/11S proteasome is induced by the cytokine γ-interferon as part of the immune response. This specialized “immunoproteasome” participates in ubiquitin-independent cleavage of intracellular antigens, such as those derived from an infecting virus, into peptides of uniform length for presentation on the cell surface of antigen-presenting cells (see Fig. 27-8). The 11S cap does not recognize ubiquitinated protein substrates and may be an adapter for the interaction of molecular chaperones with the immunoproteasome. Specialized catalytic β-subunits in the 20S core of the immunoproteasome generate somewhat longer peptides that are better suited for antigen presentation. The immunoproteasome is physically and functionally coupled to an ABC transporter (see Fig. 8-9) called the TAP (transporter associated with antigen presentation) that translocates peptides generated by the proteasome into the ER. Another integral membrane protein of the ER directly loads translocated peptides onto class I major histocompatibility antigen I (MHC) molecules for transport to the cell surface. On the cell surface, the MHC-peptide complex stimulates T-cells (see Fig. 27-8). To avoid detection by the immune system, some viruses commandeer this pathway and force the translocation of MHC molecules backward through TAP, out of the ER, and into the waiting jaws of the proteasome.
Ubiquitination Targets Proteins to Proteasomes
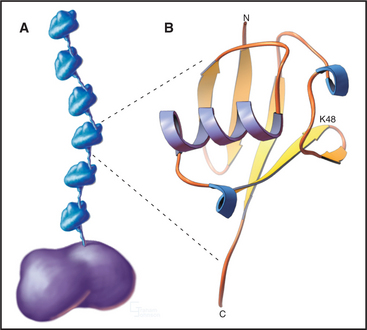
The key to regulating degradation by proteasomes is controlling access of molecules into the central proteolytic chamber. The best-characterized targeting mechanism involves the reversible, covalent linkage of a small protein, ubiquitin, onto the target protein. Ubiquitin is a very abundant and highly conserved protein of 76 residues. The C-terminal four amino acids extend from the compact globular structure, and its C-terminus is linked to target proteins (Fig. 23-7).
Ubiquitination of protein substrates proceeds through a tightly regulated multistep pathway, which has been elucidated through biochemical purification of mammalian components and in vitro reconstitution of partial reactions. The overall scheme can be subdivided into three stages (Fig. 23-8):
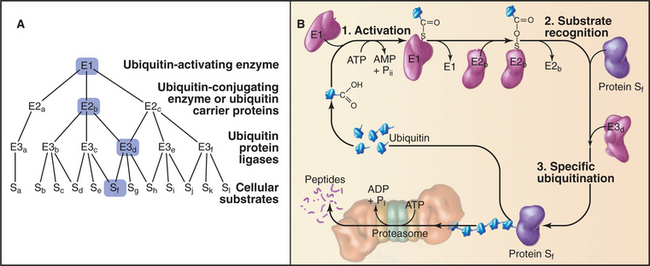
Humans have more than 500 E3 enzymes that confer specificity to the ubiquitination reaction. Many E3 enzymes contain a protein structural motif of 40 to 60 amino acids called a RING finger. This is a specialized type of Zn2+ finger (see Fig. 15-17) that mediates protein-protein interactions.
Additional ubiquitin molecules are then conjugated by an as yet unknown mechanism onto lysine 48 of the preceding ubiquitin to create a polyubiquitin chain (Figs. 23-7 and 23-8). In general, chains of four or more ubiquitins are sufficient for targeting to the proteasome. Subunits of the proteasome cap complete the cycle by deubiquitinating the substrates as they are fed into the proteolytic central chamber. The released ubiquitin is reutilized.
If polyubiquitin chains are assembled by linking ubiquitins between the C-terminal and lysine 63, rather than lysine 48, the modified protein is not targeted for destruction. Instead, this form of ubiquitination modulates other aspects of protein behavior. For example, such lysine 63–linked ubiquitin chains regulate the dynamic behavior of the chromosomal passenger protein Survivin at centromeres during mitosis (see Fig. 44-10) and are essential for normal chromosome segregation. Lysine 63–linked ubiquitin chains have also been implicated in signaling pathways, DNA repair, and vesicle trafficking.
In addition to ubiquitin, at least eight other highly conserved small proteins can be conjugated to e-amino groups of lysine on target proteins. They are generally not involved in protein degradation and instead serve a variety of functions. The best known of these is called SUMO (small ubiquitin-like modifier). SUMO can be conjugated to the same residues on target proteins as ubiquitin, but it typically stabilizes the target protein rather than promoting degradation as ubiquitin does. The ubiquitin-like proteins are ligated to their targets by E1, E2, and E3 enzymes, which are distinct from those involved in conjugation of ubiquitin.
Motifs That Specify Ubiquitination
Tight regulation of ubiquitination pathways ensures that only the appropriate target proteins are recognized, ubiquitinated, and consequently degraded. The primary responsibility for substrate selectivity lies with the E3 family of enzymes. These ubiquitin ligases can bind directly to protein substrates or indirectly through adapter molecules. Although the 40 or more E2 enzymes can interact directly with substrate, in general, they recognize the E3-substrate complex. One E2 enzyme can cooperate with several different E3 enzymes in the ubiquitination reaction (Fig. 23-8). E2s accept substrates from a single E1 in yeast.
Particular E3 enzymes recognize different determinants on protein substrates, but only a few of the rules or motifs governing E3-substrate recognition signals have been identified (Table 23-1). The first and simplest signal to be identified is described by the “N-end rule.” Through studies of chimeric and fusion proteins in yeast, researchers found that specific destabilizing amino acids at the N-terminus of a protein are recognized for ubiquitination by a specific E3 and subsequent destruction. Both the destabilizing N-terminal amino acid and a mobile lysine on the substrate are needed for ubiquitination and rapid degradation. The extent to which this simple rule governs proteolysis in vivo remains uncertain, although an increasing number of potential substrates that encode destabilizing amino acids at their N-terminus are being identified. One physiological substrate of the N-end rule is the C-terminal fragment of the Scc1 chromosome cohesin molecule that is produced by specific proteolysis at the outset of anaphase (see Fig. 44-16). Interference with N-end rule degradation of this protein is lethal to the cell, presumably because chromosome segregation is disrupted during mitosis. The first known N-end rule substrate in metazoans was an IAP inhibitor of apoptosis in Drosophila (see Chapter 46). Cleavage of this protein by a caspase (see Fig. 46-10) exposes an N-terminal destabilizing residue. Subsequent destruction of the protein is essential for its function as an apoptosis inhibitor.
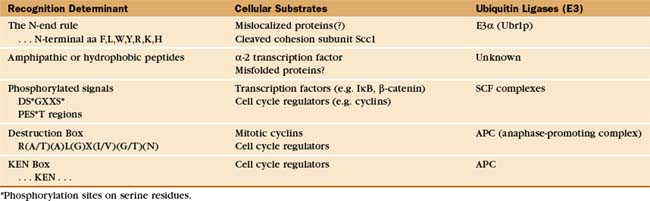
Regulated proteolysis is key in controlling cell cycle progression and transcription activation. In these cases, targeting signals for degradation can be generated by specific phosphorylation events. For example, phosphorylation of a conserved sequence near the N-terminus of several transcription factors or their regulatory subunits generates a determinant recognized by the SCF complex (a family of modular E3 enzymes named for their three core components: skp1, cdc53/cullin, and an F-box-containing protein; see Fig. 40-17). This phosphorylation is often performed by cyclin-dependent kinases (Cdks [see Fig. 40-14]) and is thereby tightly linked to the cell cycle. Phosphorylation of cell cycle proteins containing internal sequences that are rich in proline, glutamic acid, serine, and threonine (called PEST sequences) can also target these proteins via SCF family E3 enzymes for rapid degradation.
A second multisubunit class of E3 enzymes, designated the APC/C (anaphase-promoting complex/cyclosome), recognizes a partially conserved, nine-residue “destruction box” sequence near the N-terminus of several cell cycle regulatory proteins that targets these proteins for degradation (see Chapter 40). The short amino acid sequence lysine, glutamic acid, asparagine (KEN), the “KEN-box,” can also serve as a destruction signal for the APC/C. Deletion of the destruction box or the KEN box stabilizes the protein, whereas their transfer to a normally stable protein often results in cell cycle–dependent ubiquitination and rapid degradation. Destruction or KEN boxes are not themselves regulated, but APC/C activity and specificity are regulated by Cdk phosphorylation (see Chapter 40).
Role of Proteasomes in Elimination of Misfolded Proteins from the Endoplasmic Reticulum
Integral membrane proteins and secretory proteins fold and assemble in the lipid bilayer or lumen of the endoplasmic reticulum (ER) (see Fig. 20-7). Proteins that fail to fold or assemble are retrieved from the ER and degraded by the proteasome in a pathway known as ERAD (ER-associated protein degradation). The ERAD pathway also regulates levels of a number of ER resident proteins. Interestingly, the E3 ligases responsible for ubiquitinating ERAD target proteins are localized in the cytoplasm, so a poorly understood mechanism must expose target proteins to the cytoplasm for ubiquitin tagging. After ubiquitination in the cytoplasm, association with ubiquitin-binding proteins appears to ensure the extraction of these proteins from the ER by AAα-ATPases, and their subsequent targeting to proteasomes.
The ERAD pathway is of considerable medical interest. Defects in ubiquitination of particular proteins are associated with the pathology of Parkinson’s disease. Furthermore, the most common form of cystic fibrosis results from ERAD-mediated degradation of a slow-folding (but catalytically competent) variant of the CFTR ABC transporter (see Fig. 11-4).
Other Regulated Intracellular Proteolysis
Another form of regulated intracellular proteolysis is activation of inactive proenzymes or transcription factors (see Fig. 15-22 for NFkB) by proteolytic cleavage. An important example of activation by proteolytic cleavage is provided by caspases. Extracellular or intracellular signals trigger the cleavage of procaspases, turning on their proteolytic activity and initiating a cascade that leads to apoptosis (see Fig. 46-10). In all cases, intracellular proteolysis is tightly regulated through a combination of triggered activation of the protease, specific substrate recognition, and compartmentalization.
Lipid Turnover and Degradation
Distinct pathways exist for the turnover of the three classes of cellular lipids: phosphoglycerides, glycolipids, and cholesterol. Glycolipids, which are restricted to the extracellular leaflet of a lipid bilayer, are degraded primarily in lysosomes, as evidenced by their abnormal accumulation in lysosomal storage diseases (Appendix 23-1). Sphingomyelin and gangliosides are delivered to lysosomes via vesicular transport and degraded to the level of ceramide, sugars, and fatty acids by a series of lysosomal hydrolases. Recent evidence suggests that the specialized lipid composition of lysosomal membranes, including the phospholipid species lysobisphosphatidic acid (see Fig. 22-15), which is enriched in intraluminal vesicles of multivesicular bodies and lysosomes, may play a role in activating sphingomyelinases and restricting their hydrolytic activity to the intraluminal side of the membranes. Cell surface sphingomyelinases also exist, and their activation triggers production of ceramide, which can function in signal transduction pathways as a second messenger (see Fig. 26-11).
The turnover of phosphoglycerides is much more varied in mechanism and location. Some phosphoglycerides, particularly those found in the outer leaflet of the plasma membrane (and in topologically equivalent surfaces), are degraded in lysosomes to their fatty acids, head group, and glycerol constituents. More frequently, phosphoglyceride degradation is only partial, and the degradative products (e.g., fatty acids, lysophospholipids, and diacylglycerol) are salvaged and reutilized in “short-circuit pathways.” In this way, “old” phospholipids are “remodeled,” forming new ones with altered properties. These phospholipid-remodeling reactions are catalyzed by a variety of phospholipases that cleave distinct bonds in the phospholipid to generate distinct products (see Fig. 26-4). Localized lipid remodeling can generate specialized lipid subdomains that are required for vesicle fusion or fission or for the selective recruitment of proteins to the membrane. In addition, molecules released from partial degradation of phosphoglycerides, fatty acids, diacylglycerol, and some head groups function as second messengers in signaling cascades (see Fig. 26-4).
Cholesterol Homeostasis
Cholesterol metabolism in mammals involves multiple organs. Approximately 90% of the free cholesterol in animal cells is in the plasma membrane. Cholesterol is the precursor for steroid hormones, which are synthesized in specialized cells but utilized throughout the body for a myriad of essential functions. Cholesterol is also the precursor for bile acids, which are synthesized by the liver and transported to the gut, where they aid in the digestion of dietary fat. Unlike the case with virtually all other intracellular molecules, individual cells cannot degrade cholesterol. Instead, cellular levels of cholesterol are regulated by a complex balance of endogenous synthesis, uptake of extracellular cholesterol, and efflux of intracellular cholesterol to vascular fluids (Fig. 23-9). When present in excess, cholesterol accumulates as plaques in the walls of major arteries, contributing to atherosclerosis.
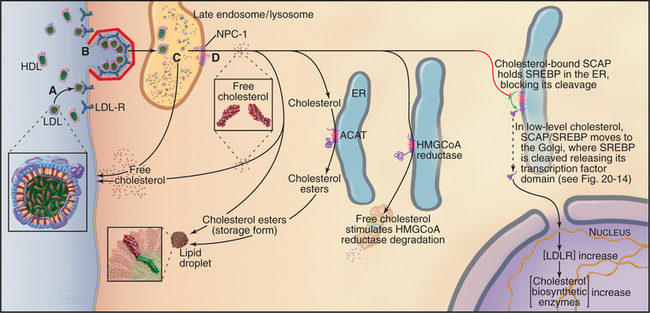
Figure 23-9 the intracellular processing and regulation of cholesterol biosynthesis. A, Dietary cholesterol is delivered to cells in LDL particles. B, LDL particles are taken up by clathrin-mediated endocytosis. C, Free cholesterol is released in late endosomes/lysosomes and transported to the cell surface or internal membranes, depending, in part, on the activity of NPC-1 (D), an integral membrane protein. Excess cholesterol can be acylated by ACAT activity and stored in cytoplasmic lipid droplets as cholesterol esters. ACAT activity is increased by high intracellular cholesterol levels. At the same time, high cholesterol in the membrane decreases new cholesterol synthesis by triggering the proteasome-dependent degradation of the enzyme HMG-CoA reductase. Finally, high cellular cholesterol decreases the uptake of LDL particles and dietary cholesterol by blocking the proteolytic processing of the transcription factor, SREBP, required for LDL-receptor expression (see Fig. 20-14). Genetic defects that perturb steps A to D required for maintaining the delicate balance of cholesterol homeostasis cause several human diseases. Familial hypercholesterolemia is caused either by a lack of LDL receptor (LDL-R) (A) or by LDL-R that is defective in endocytic activity (B). Wolman disease is a lysosomal storage disease that is caused by defective lysosomal cholesterol esterase activity; Niemann-Pick disease type C, another lysosomal storage disease, results in defective trafficking of cholesterol out of late endosomes and lysosomes caused by mutations in NPC-1 (D).
Cholesterol is insoluble and is transported through the body as cholesterol esters packaged with other lipids and proteins. The intestine assembles dietary cholesterol into particles called chylomicrons, which are transported through the blood and eventually taken up by the liver, the major site of cholesterol synthesis in mammals. The liver packages dietary and de novo–synthesized cholesterol into low-density lipoproteins (LDLs), which are secreted into the blood for transport to other tissues. Other cells take up LDL particles via receptor-mediated endocytosis and deliver them along the endocytic pathway to lysosomes (Fig. 23-9). Within the lysosome, cholesterol esters are hydrolyzed, and the bulk of free LDL-derived cholesterol is transported by a yet-to-be identified cytoplasmic carrier protein back to the plasma membrane. Importantly, a small portion of cholesterol is also transported to the ER, where the cholesterol level controls the activity of transcription factors that regulate genes involved with cholesterol metabolism.
Two key enzymes in the endoplasmic reticulum have sterol-sensing domains that allow them to respond to the cholesterol content of the membrane and control intracellular free cholesterol levels (Fig. 23-10). Accumulation of LDL-derived cholesterol in the ER activates acyl CoA:cholesterol acyltransferase (ACAT), the enzyme that converts free cholesterol to cholesterol esters for storage. Substantial increases in the levels of free cholesterol (or an oxygenated metabolite of it) trigger the destruction of the enzyme that catalyzes the first step in cholesterol biosynthesis, HMG-CoA reductase (Fig. 23-9). Cholesterol triggers the degradation of HMG-CoA reductase through a ubiquitin- and proteasome-dependent pathway. A third protein with a sterol-sensing domain—SCAP—traps the membrane-bound transcription activator, SREBP, in the ER, thereby limiting the expression of the genes for both HMG-CoA reductase and the LDL receptor. When ER cholesterol is low, SCAP/SREBP escapes to the Golgi apparatus, where proteolytic cleavage liberates the SREBP activation domain (see Fig. 20-14). This then travels to the nucleus to drive the expression of LDL receptor, HMG-CoA reductase, and other proteins involved with cholesterol metabolism. This negative feedback mechanism reduces cholesterol input both from de novo synthesis and from extracellular sources (Fig. 23-9).
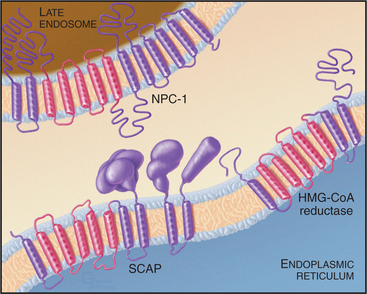
Cholesterol homeostasis is critical to human health, as is evidenced by the number of genetic diseases that result from defects in cholesterol metabolism. Defects in the LDL receptor reduce or eliminate LDL uptake, and LDL builds up in the blood, leading to cholesterol deposition in the walls of arteries and arteriosclerosis. Rare defects in the enzyme that hydrolyzes cholesterol esters in lysosomes lead to Wolman disease, which causes death within the first year of life. The devastating neurodegenerative disease called Niemann-Pick type C disease results from mutations in a multitransmembrane domain protein (designated NPC-1) that is required for transport of LDL-derived cholesterol from late endosomes to both the plasma membrane and ER. Cholesterol accumulates in lysosomes of diseased patients, and cholesterol homeostasis is impaired. Interestingly, NPC-1 has a “sterol-sensing domain” found in enzymes that are regulated by the cholesterol composition of membranes (Fig. 23-10; also see Fig. 20-14).
Abele R, Tampe R. Modulation of the antigen transport machinery TAP by friends and enemies. FEBS Lett. 2006;580:1156-1163.
Babst M. A protein’s final ESCRT. Traffic. 2005;6:2-9.
Bonifacino JS, Weissman AM. Ubiquitin and the control of protein fate in the secretory and endocytic pathways. Annu Rev Cell Dev Biol. 1998;14:9-58.
Brown MS, Goldstein JL. A proteolytic pathway that controls the cholesterol content of membranes, cells, and blood. Proc Natl Acad Sci U S A. 1999;96:11041-11048.
Ciechanover A. Proteolysis: From the lysosome to ubiquitin and the proteasome. Nat Rev Mol Cell Biol. 2005;6:79-86.
Cuervo AM. Autophagy: In sickness and in health. Trends Cell Biol. 2004;14:70-77.
D’Azzo A, Bongiovanni A, Nastasi T. E3 ubiquitin ligases as regulators of membrane protein trafficking and degradation. Traffic. 2005;6:429-441.
Deshaies RJ. SCF and Cullin/Ring H2-based ubiquitin ligases. Annu Rev Cell Dev Biol. 1999;15:435-468.
Earnshaw WC, Martins LM, Kaufmann SH. Mammalian caspases: Structure, activation, substrates, and functions during apoptosis. Annu Rev Biochem. 1999;68:383-442.
Fineschi B, Miller J. Endosomal proteases and antigen processing. Trends Biochem Sci. 1997;22:377-382.
Hicke L, Dunn R. Regulation of membrane protein transport by ubiquitin and ubiquitin-binding proteins. Annu Rev Cell Dev Biol. 2003;19:141-172.
Klionsky DJ. The molecular machinery of autophagy: Unanswered questions. J Cell Sci. 2005;118:7-18.
McGrath ME. Lysosomal cysteine proteases. Annu Rev Biophys Biomol Struct. 1999;28:181-204.
Meusser B, Hirsch C, Jarosch E, Sommer T. ERAD: The long road to destruction. Nat Cell Biol. 2005;7:766-772.
Mizushima N, Yoshimori T, Ohsumi Y. Role of the Apg12 conjugation system in mammalian autophagy. Int J Biochem Cell Biol. 2003;35:553-561.
Pickart CM. Mechanisms underlying ubiquitination. Annu Rev Biochem. 2001;70:503-534.
Pines J. Mitosis: A matter of getting rid of the right protein at the right time. Trends Cell Biol. 2006;16:55-63.
Römisch K. Endoplasmic reticulum-associated degradation. Annu Rev Cell Dev Biol. 2005;21:435-456.
Schwartz DC, Hochstrasser M. A superfamily of protein tags: Ubiquitin, SUMO and related modifiers. Trends Biochem Sci. 2003;28:321-328.
Simons K, Ikonen E. How cells handle cholesterol. Science. 2000;290:1721-1726.
Sun L, Chen ZJ. The novel functions of ubiquitination in signaling. Curr Opin Cell Biol. 2004;16:119-126.
Varshavsky A. The ubiquitin system. Trends Biochem Sci. 1997;22:383-387.
Wolf DH, Hilt W. The proteasome: A proteolytic nanomachine of cell regulation and waste disposal. Biochim Biophys Acta. 2004;1695:19-31.