Chapter 41 Degenerative Disorders Primarily of Gray Matter
Rett’s Syndrome
Rett’s syndrome is an X-linked disease that primarily affects females. It is the second leading cause of mental retardation in females, with an incidence of 1 case per 10,000–22,000 females [Hagberg et al., 1985; Kozinetz et al., 1993]. All ethnicities are equally affected. The hallmarks of this syndrome are a period of normal development followed by regression of speech and development of stereotypical hand gestures. The genes that cause this syndrome are MECP2, which maps to the Xq28 locus, CDKL5 (cyclin-dependent kinase-like 5) gene (previously known as STK9) located in Xp22 [Scala et al., 2005; Kalscheue et al., 2003; Evans et al., 2005], Netrin G1 gene, located on chromosome 1 [Borg et al., 2005], and FOXG1 gene, located in 14q12 [Ariani et al., 2008]. The MECP2 protein is thought to be necessary for the maintenance of neurons during the later stages of development and after neuronal maturation is complete. The structure and function of the MECP2 protein continue to be the focus of intense scrutiny. Although this syndrome has many severe manifestations, approximately 50 percent of affected individuals live into the third decade of life. Despite the advances in knowledge about the cause and defects of Rett’s syndrome during the past decade, treatment remains primarily supportive.
History
Rett’s syndrome was first reported in 1966 by Dr. Andreas Rett [Rett, 1966]. This initial case report was followed in 1978 with a publication about Japanese female patients with a particular pattern of symptoms, including mental retardation and stereotypical hand-wringing [Ishikawa et al., 1978]. It was not until 1983, when Hagberg et al. [1983] published a case report of 35 female patients, that Rett’s syndrome gained international attention. Intense investigation of Rett’s syndrome over the past 20 years led to identification of the genetic defect in 1999 [Amir et al., 1999], 2005 [Borg et al., 2005; Scala et al., 2005; Kalscheue et al., 2003; Evans et al., 2005], and 2008 [Ariani et al., 2008].
Clinical Description
The diagnosis of Rett’s syndrome is based on a set of clinical observations accompanied by changes in various laboratory test results. The clinical criteria for classic Rett’s syndrome were established in the 1980s [Hagberg et al., 1985; Trevathan and Moser, 1988], and include loss of speech, seizures, mental retardation, and classic motor (specifically hand) movements. Criteria for atypical Rett’s syndrome were reported in 1993 [Hagberg and Gillberg, 1993]. More than 75 percent of patients have classic Rett’s syndrome, whereas 25 percent have atypical Rett’s syndrome variants [Hagberg, 2002].
In classic Rett’s syndrome (Table 41-1), the newborn initially appears developmentally normal. This period is followed by deceleration of head growth, loss of purposeful hand movements, development of stereotypic hand movements, and gait dyspraxia. These five criteria must be met for the diagnosis of classic Rett’s syndrome [Hagberg, 1995]. The chronology of these symptoms is critical for the diagnosis. Normal development is typical for the first 3–6 months of life. Deceleration in the rate of head growth occurs between 3 months and 4 years. Patients lose the ability to use their hands in a purposeful manner between 9 months and 2.5 years. Stereotypic hand movements appear between 1 and 3 years of age, and a dyspraxic gait manifests between 2 and 4 years of age if the patient is ambulatory.
Table 41-1 Obligatory Criteria for the Diagnosis of Rett’s Syndrome
| Manifestation | Age | Comments |
|---|---|---|
| Period of normal neonatal development | 0–6 mo | Prenatal or perinatal period into the first 6 months of life, sometimes longer |
| Stagnation of rate of head circumference growth | 3 mo–4 yr | Normal at birth, then decelerates |
| Loss of purposeful hand skills | 9 mo–2.5 yr | Communicative dysfunction, social withdrawal, mental deficiency, loss of speech or babbling |
| Classic stereotypic hand movements | 1–3 yr | Hand-washing or hand-wringing and variants, including clapping and tapping, are common |
| Gait or posture dyspraxia Absence of organomegaly, optic atrophy, retinal changes, or delayed intrauterine growth |
2–4 yr | Truncal “ataxia” |
(Data from Hagberg B. Clinical manifestations and stages of Rett syndrome. Ment Retard Dev Disabil 2002;8:61–65, and from Percy AK. Clinical trials and treatment prospects. Ment Retard Dev Disabil 2002;8:106–111.)
Clinical manifestations of classic Rett’s syndrome are grouped into four stages: early onset (3–6 months of age), regression (1–4 years of age), stabilization, and late motor impairment (after the age of 3 years) [Jellinger, 2003]. The early-onset stage is characterized by developmental delay, deceleration of head growth [Neul and Zoghbi, 2004; Schultz et al., 1993], onset of autistic-like behavior, and classic hand-wringing [Jellinger, 2003]. Weight and height percentiles for age also decrease; the median values fall below the fifth percentile by age 7 years [Percy, 2002]. Although Rett’s syndrome can manifest earlier, clear signs of a central nervous system abnormality are usually not evident until 6 months of age [Akbarian, 2003]. The second stage of the syndrome is characterized by cognitive decline and regression [Hagberg, 2002]. Loss of speech and purposeful hand movements, emergence of stereotypic movements, seizures, breathing irregularities, other signs of autonomic instability, inattentive behavior, and hypotonia appear [Hagberg, 2002; Jellinger, 2003; Kerr et al., 2001]. The third stage consists of stabilization of symptoms; this stage differentiates Rett’s syndrome clinically from other pediatric neurodegenerative disorders. Sometimes there is a return of communication skills, with preservation of remaining ambulatory skills. This stage is also known as the pseudostationary stage because slow neuromotor regression continues [Hagberg, 1995]. In patients older than 3 years, bradykinesia and rigidity set in [Fitzgerald et al., 1990]. Stabilization can last years to decades. Late motor impairment begins when ambulation ceases; this signals the end of the stabilization or pseudostationary stage. This final stage of Rett’s syndrome is characterized by nonambulation and severe disability. The length of late motor impairment is variable and can last decades.
Common clinical manifestations of classic Rett’s syndrome include stereotypic hand movements, intense staring, breathing irregularities, bruxism, sleep disturbances and night laughter, scoliosis, lower limb spasticity and dystonia, seizures, swallowing dysfunction, constipation, gastroesophageal reflux, and small, bluish or red feet. The stereotypic hand movements occur while the individual is awake. These gestures are individualized, but they typically include continuous and repetitive twisting, wringing, knitting, and clapping motions. The intense eye communication may be compensatory for the loss of speech. This eye pointing has been observed in many individuals with Rett’s syndrome. The breathing irregularities are of two types: hyperventilation and breath-holding. Typically, they occur only while the individual is awake, but can also occur during sleep [d’Orsi et al., 2009a].
Seizures are reported in 30–80 percent of individuals with Rett’s syndrome. The electroencephalogram (EEG) is always abnormal after the age of 2 years. Early-onset seizures were reported in patients with CDKL5 [Artuso et al., 2010] and Netrin G1 gene mutations [Borg et al., 2005]. Prevalence of drug-resistant epilepsy in RTT patients with MeCP2 mutations was 16 percent. No significant relationship was found between clinical severity of drug-resistant epilepsy and quantitative or qualitative EEG scores. In addition, no significant relationship was found between the drug-resistant epilepsy and the RTT genotype category, or a specific MECP2 genotype [Buoni et al., 2008]. Myoclonic status had been misdiagnosed as a movement disorder of gait impairment [Pelc and Dan, 2009; d’Orsi et al., 2009b]. Infrequent clinical manifestations of classic Rett’s syndrome include bloating, violent screaming, abnormal nociception, pain insensitivity [Devarakonda et al., 2009], hyperkalemic distal renal tubular acidosis [Assadi et al., 2006], and cardiac arrhythmias [Acampa and Guideri, 2006]. Bloating or air swallowing is generally mild, but 5–10 percent of individuals with Rett’s syndrome demonstrate severe bloating. Massive gastric dilatation, with total necrosis and perforation due to bloating, has been reported [Baldassarrea et al., 2006]. The gastrointestinal disturbances are attributed to changes within the autonomic nervous system. Screaming typically is encountered in teenage patients. The screaming may be associated with ill-defined pain, but no known pathology can be found. Occasionally, patients have abnormally prolonged responses or insensitivity to pain. Children and adults with Rett’s syndrome are at substantially increased risk of fracture. The lower limbs, especially the femur, are particularly susceptible and patients with the R270X and R168X mutations genotype are especially vulnerable. The presence of epilepsy also increased fracture risk [Downs et al., 2008]. Although a decreased life span is characteristic for this syndrome, many patients survive into adulthood [Sekul and Percy, 1992], with 50 percent remaining alive in their 30s [Akbarian, 2003].
The atypical Rett’s syndrome variants include a forme fruste variant, early seizure type variant, late childhood regression variant, preserved speech variant, and congenital Rett’s syndrome. Diagnosis of atypical Rett’s syndrome is complex. The criteria for variants of classic Rett’s syndrome, as outlined by Hagberg and Skjeldal [1994], are especially helpful (Box 41-1). Forme fruste is the most common atypical variant, accounting for about 80 percent of nonclassic Rett’s syndrome. There is a wide variability of function in forme fruste; it is a milder variant. It is seldom diagnosed before 8–10 years of age, and it is usually suspected in older individuals who are just beginning to develop symptoms of Rett’s syndrome. The early-onset seizure variant is linked to mutations in CDKL5 and Netrin G1 gene and manifests with early epilepsy onset between the first week and 5 months, hand stereotypies, severely impaired psychomotor development, and severe hypotonia [Artuso et al., 2010; Borg et al., 2005]. The late childhood regression form is characterized by a normal head circumference and by a more gradual and later onset (late childhood) of regression of language and motor skills. The preserved speech variant was first described in 1992 [Zappella, 1992]. It is characterized by the preservation of speech, but preserved head size and obesity are also common features [Zappella, 1992; Zappella et al., 2001]. There is some debate about whether the preserved speech variant is part of the autistic spectrum disorders, as well as the Rett’s syndrome spectrum [Percy et al., 1990]. Congenital Rett’s syndrome is rare. It differs from classic Rett’s syndrome because of the absence of the 3- to 6-month period of normal development [Hagberg and Skjeldal, 1994]. It is linked to mutations in FOXG1 gene [Ariani et al., 2008].




Clinical Diagnostic Tests
Routine Laboratory Tests
Levels of lactate, pyruvate, and glutamate are increased in cerebrospinal fluid [Budden et al., 1990; Lappalainen et al., 1997a]. Cerebrospinal fluid testing yields decreased levels of β-phenylalanine, substance P, and gangliosides [Lekman et al., 1991; Matsuishi et al., 1997; Satoi et al., 2000]. There are increased levels of biogenic amines and creatine in the urine [Lekman et al., 1990]. Plasma levels of levels of β-endorphin and prolactin are decreased [Fanchetti et al., 1986]. The increased levels of lactate and pyruvate in the cerebrospinal fluid may result from hyperventilation [Budden et al., 1990], whereas the decreased levels of cerebrospinal fluid β-phenylalanine are caused by dysregulation of the dopaminergic pathways in patients with Rett’s syndrome [Satoi et al., 2000]. The levels of IgA and IgG antibodies to gluten and gliadin proteins found in grains and to casein found in milk are significantly increased in girls with Rett’s syndrome [Reichelt and Skjeldal, 2006].
Neurophysiologic Tests
The EEG is abnormal in Rett’s syndrome. Initial abnormalities are noticed in the rapid eye movement stage of sleep [Kudo et al., 2003]. During the stabilization stage of Rett’s syndrome, a slow spike-wave pattern resembling that in Lennox–Gastaut syndrome is observed [Glaze, 1987]. After 3 years of age, there is a decrease in alpha activity with a subsequent increase in theta activity [Bashina et al., 1994]. Evoked potential studies indicate intact visual and auditory peripheral pathways and dysfunction of central cortical pathways involved in processing and integration of sensory information in young girls with Rett’s syndrome. Somatosensory-evoked potentials can be characterized by “giant” responses, suggesting cortical hyperexcitability [Glaze, 2005]. There is a prolongation of somatosensory-evoked responses in older patients, suggesting involvement of the upper spinal cord and spinothalamic tracts [Bader et al., 1989]. Results of nerve conduction studies are consistent with an axonopathy and denervation indicative of lower motor dysfunction [Jellinger et al., 1990]. Impairment of the autonomic nervous system in Rett’s syndrome is suggested by an increased incidence of long QT intervals during electrocardiographic recordings and diminished heart rate variability [Glaze, 2005].
Neuroimaging Studies
Initial cranial computed tomographic (CT) scans and magnetic resonance imaging (MRI) are normal. As the patient ages and neurologic symptoms develop, generalized atrophy of the cerebral hemispheres and decreased volume of the caudate nucleus become apparent [Reiss et al., 1993]. Imaging of the basal ganglia reveals decreased volume of the caudate head [Dunn et al., 2002]. Commonly, there is a decrease in gray and white matter volumes, specifically within the frontal and temporal regions, and of the midbrain and cerebellum [Subramaniam et al., 1997]. Hypoperfusion of the prefrontal and temporoparietal regions is also reported [Lappalainen et al., 1997b]. Although imaging studies can help in making the diagnosis, there is no correlation between spectroscopic changes and clinical status [Gokcay et al., 2002]. One study reported an association between the level of hypoperfusion and early-onset Rett’s syndrome [Lappalainen et al., 1997b]. In more recent MR spectroscopy studies of RTT patients with MeCP2 mutations, NAA/Cr ratios decreased and myoinositol/Cr ratios increased with age. The mean glutamate and glutamine/Cr ratio was increased. The mean NAA/Cr ratio decreased in RTT patients with seizures and with increasing clinical severity score. Compared to patients with T158X, R255X, and R294X mutations, and C-terminal deletions, patients with the R168X mutation tended to have the greatest severity score and the lowest NAA/Cr ratio. Decreasing NAA/Cr and increasing myoinositol/Cr with age are suggestive of progressive axonal damage and astrocytosis in RTT, respectively, whereas increased glutamate and glutamine/Cr ratio may be secondary to increasing glutamate/glutamine cycling at the synaptic level [Horska et al., 2009].
Pathology
Brain
Gross findings include generalized atrophy of the frontal and temporal regions, the cerebellum, and especially the vermis. The corpus callosum decreases in size by as much as 30 percent [Oldfors et al., 1990; Reiss et al., 1993]. The brain is the only organ that is decreased in size compared with height [Armstrong et al., 1999]. Cerebellar volume is reported to remain relatively normal. The average weight of a brain from a patient with Rett’s syndrome is about 950 g, equivalent to the weight of a brain from a developmentally normal 1-year-old child [Armstrong, 2000]. More importantly, the brain weight does not continue to decrease with age, because Rett’s syndrome is not a progressive neurodevelopmental disorder in the classic sense.
There are many microscopic findings in brain tissue from Rett’s syndrome patients. Neuronal size is decreased, but cell density in the cerebral cortex, thalamus, basal ganglia, amygdala, and hippocampus is increased [Bauman et al., 1995]. The previous findings contrast with a report of an overall decrease in the number of neurons in the frontal cortex, the temporal cortex, and the cholinergic nucleus basalis of Meynert [Belichenko et al., 1994; Kitt and Wilcox, 1995]. Decreases in dendritic branching and dendritic number are found in the frontal, motor, and subicular areas [Armstrong, 1997; Armstrong et al., 1995; Cornford, 1994]. In addition to decreases in dendritic number and branches, shortening of the apical and basilar dendritic branches within these same regions of the brain has been reported [Armstrong et al., 1998]. Afferent neurons have decreased synaptic contacts. The striatum and internal pallidum exhibit hypochromia, whereas hypomyelination is observed in the substantia nigra pars compacta [Jellinger et al., 1988]. The neocortex has decreased expression of microtubule-associated protein 2, and disruption of the cytoskeleton within the neocortex is apparent [Kaufmann et al., 1995]. The caudate nucleus and putamen exhibit reduced levels of dopamine transporter protein [Wong et al., 1998]. Degenerative changes of the substantia nigra, caudate nucleus, and putamen have been demonstrated in neuropathological and neurochemical studies of RTT brains. Stereotypies and other movement disorders present in RTT could be interpreted as signs of dysfunction of the nigrostriatal-dopaminergic pathway [Kitt and Wilcox, 1995; Wenk, 1995].
There are conflicting results regarding the expression of nerve growth factor in Rett’s syndrome patients. One study documented no reduction in the cortical levels of nerve growth factor [Wenk and Hauss-Wgrzyniak, 1999], and others demonstrated large decreases in the expression of nerve growth factor and the neurotrophic tyrosine kinase type receptor, which binds to nerve growth factor with high affinity [Lipani et al., 2000]. Adults with Rett’s syndrome also have axonal degeneration, loss of motor neurons, loss of spinal ganglion cells, and decreased glutamate and gamma-aminobutyric acid type B (GABA B) receptor density [Oldfors et al., 1988; Blue et al., 1999]. Blue et al. [1999] reported age-specific alterations in amino acid neurotransmitter receptors within the basal ganglia of adults.
Electron microscopy of neurons depicts distinct abnormalities [Papadimitriou et al., 1988]. These changes include abnormal neurites that are filled with lysosomes and laminate bodies. Axonal swellings, large mitochondria, and membranous multilamellar bodies are seen. Although electron microscopy reveals intraneuronal inclusion bodies that contain lipofuscin-like material, there are no other characteristics of a lipid storage disorder.
Muscle
Type I and type II fiber atrophy is sometimes seen on muscle biopsy [Wakia et al., 1990]. Decreased cytochrome c oxidase and succinate cytochrome c reductase activities in muscle biopsies have also been reported [Coker and Melnyk, 1991]. The myocardium has no gross abnormalities. The atrioventricular node has an abnormal or immature rearrangement of muscle fibers within the conduction system [Armstrong, 1997]. Electron microscopy of muscle biopsy specimens reveals dumbbell-shaped mitochondria with foamy vacuoles [Ruch et al., 1989].
Genetics
Rett’s syndrome is an X-linked dominant disorder that has been mapped to the Xq28 locus [Ellison et al., 1992; Sirianni et al., 1998]. Although most cases of Rett’s syndrome are sporadic, genetic mapping was possible because familial inheritance does occur, and there is concordance in monozygotic twins [Jellinger, 2003]. Mutations within the methyl-CpG binding protein 2 gene (MECP2) cause 70–80 percent of reported cases of Rett’s syndrome in females [Auranen et al., 2001; Van den Veyver and Zoghbi, 2002]. This gene was identified in 1999 [Amir et al., 1999]. Most mutations in males lead to fetal demise. Although DNA mitochondrial mutations are found in some cases of Rett’s syndrome, there is no indication that mitochondrial DNA plays a part in the development of this syndrome [Nielson et al., 1993; Colantuoni et al., 2001]. Mutations within MECP2 have also been linked to childhood-onset schizophrenia, Angelman’s syndrome, and mild mental retardation [Watson et al., 2001].
The MECP2 protein has three known functional domains: an amino-terminal methyl-CpG binding domain [Lewis et al., 1992], a transcriptional repressor domain, and a carboxyl-terminal domain [Chandler et al., 1999]. The MECP2 protein binds to methylated CpG dinucleotides by the methyl-CpG binding domain [Nan et al., 1993]. The transcriptional repressor domain interacts with various co-repressor complexes and disrupts transcription [Nan et al., 1996, 1997]. The nuclear localization signal (NLS), consisting of amino acid residues 265–271, is contained within the transcriptional repressor domain. The biochemical function of the carboxyl-terminal region is unknown [Kriaucionis and Bird, 2003]. Seventy percent of the mutations within MECP2 are in eight hotspots affecting translation of the following amino acids: R106, R133, T158, R168, R255, R270, R294, and R306. Seven of these eight mutation hotspots affect arginine, which contains a CpG in its codon. These mutations may result from unrepaired deamination of 5-methylcytosine. This mechanism is thought to cause one-third of all point mutations that lead to human genetic disease [Cooper and Youssoufian, 1988]. Eighty percent of females with classic Rett’s syndrome have nonsense or frameshift mutations within the MECP2 gene [Van den Veyver and Zoghbi, 2002].
Genotype–Phenotype Correlation
Genotype–phenotype correlation has been attempted, but it is complicated by MECP2 gene X-chromosome inactivation. This inactivity allows a mother with a mutation of MECP2 to have a normal phenotype because of skewing of X-chromosome inactivation. If this mother has a daughter with the mutation of MECP2 but balanced X-chromosome inactivation, the daughter will have Rett’s syndrome [Amir et al., 2000]. Despite the problems with X-chromosome inactivation, many studies of genotype–phenotype correlations exist. It is reported that truncated mutations of MECP2 are more severe than missense mutations [Chae et al., 2002; Cheadle et al., 2000; Monros et al., 2001]. The location of the truncation generally does not affect the phenotype [Bienvenu et al., 2000; Giunti et al., 2001; Huppke et al., 2000; Satoi et al., 2000; Amir et al., 2000] reported that truncation mutations led to increased levels of homovanillic acid in cerebrospinal fluid and to increased respiratory problems. The same study reported an increased incidence of scoliosis in cases of missense mutations. Huppke et al. [2002] examined mutations from 123 patients with Rett’s syndrome. They determined that mutations affecting the NLS caused the most severe phenotype. They also reported that deletions within the carboxyl terminus caused the least severe clinical presentation. Truncations result in more severe disease than missense mutations, except when the truncation affects the carboxyl terminus. Single-amino acid mutations cause less severe phenotypes, presumably because they lead to mild impairment of protein function [Laccone et al., 2002].
Rett’s Syndrome Variants
Most of the mutations within MECP2 cause classic Rett’s syndrome. Twenty-nine cases of the preserved speech variant of Rett’s syndrome have mutations within the MECP2 gene [Conforti et al., 2003; Hoffbuhr et al., 2001; Huppke et al., 2000; Neul and Zoghbi, 2004; Nielsen et al., 2001; Obata et al., 2000; Weaving et al., 2003; Yamashita et al., 2001; Zappella et al., 2001]. These mutations were evenly distributed among the three known functional domains of the MECP2 gene: the methyl-CpG binding domain, the transcriptional repressor domain, and the carboxyl-terminal domain. Mutations resulting in less severe phenotypes (i.e., mutations within the carboxyl terminus or a truncation after the NLS motif) were common in patients with the preserved speech variant of Rett’s syndrome. Patients with the preserved speech variant who had mutations normally associated with severe disease had skewed X-chromosome inactivation (92:8 in one case), explaining the less severe phenotypes [Hoffbuhr et al., 2001; Zappella et al., 2001].
MECP2 Mutations in Males
Three outcomes occur in males: Rett’s syndrome, severe encephalopathy with neonatal fatality, and mild neuropsychiatric phenotypes [Geerdink et al., 2002; Villard et al., 2000; Wan et al., 1999; Zeev et al., 2002]. The classic form of Rett’s syndrome can occur in males [Jan et al., 1999]. Although similar mutations are seen in males and females with this disease, there is a report of a unique mutation within MECP2 that causes Rett’s syndrome in males [Ravn et al., 2003]. Male siblings of female Rett’s syndrome patients with identical MECP2 mutations develop a severe encephalopathy and die by 1–2 years of age. These mutations typically affect the methyl-CpG binding domain or the NLS portion of the MECP2 protein.
Rett’s syndrome is produced as a result of somatic mosaicism, meaning there is a mixed population of cells with the wild type of MECP2 and mutated MECP2 [Armstrong et al., 2001; Clayton-Smith et al., 2000; Topcu et al., 2002]. Males with Rett’s syndrome have a unique genetic composition. Klinefelter’s syndrome (46,XXY) allows phenotypic males to replicate the somatic mosaicism achieved by females and avoid neonatal fatality [Leonard et al., 2003; Schwartzman et al., 2001]. There are case reports of Rett’s syndrome occurring in a phenotypic male, in whom the SRY region of the Y chromosome that produces “maleness” is translocated on to an X chromosome, so that a phenotypic male is genotypically a female (46,XX) [Maiwald et al., 2002]. Mutations in the MECP2 gene also occur in males with mental retardation and no other symptoms of Rett’s syndrome. These mutations generally affect the carboxyl terminus of the methyl-CpG binding domain region of the MECP2 protein [Couvert et al., 2001; Kleefstra et al., 2002; Yntema et al., 2002a]. Whether these mutations contribute to the phenotypes observed, or are normal polymorphisms, is being explored [Laccone et al., 2002; Yntema et al., 2002b].
Cell Biology
Because Rett’s syndrome is caused primarily by mutations within the MECP2 gene, it is necessary to understand the function and interactions of the MECP2 protein. The MECP2 gene encodes a protein with three known functional domains: the methyl-CpG binding domain, the transcriptional repressor domain, and the carboxyl terminus. Human MECP2 has 48 amino acids (about 80 kDa) [Akbarian, 2003]. The methyl-CpG binding domain contains 85 amino acids [Nan et al., 1993] and binds to single- and double-methylated CpG dinucleotides [Bird and Wolfe, 1999; Lewis et al., 1992]. There is a correlation between the capability of the methyl-CpG binding domain to bind to pericentromeric heterochromatic regions of DNA and the ability of the protein to repress methylated promoters [Kudo et al., 2003]. Residues R111, R133C, and R134C within the methyl-CpG binding domain are thought to come into contact with methylated cystines. Mutations affecting R111 cause the MECP2 protein to lose its binding ability to heterochromatic DNA and its capability to repress transcription. Mutations resulting in R133C or R134C affect neither of the aforementioned protein properties. The MECP2 protein associates with chromatin remodeling complexes and aids in the regulation of the structure and function of chromatin.
The transcriptional repressor domain is 100 base pairs (bp) long, and it interacts with various co-repressor complexes [Nan et al., 1997]. One of these complexes is the Sin3A co-repressor complex. This complex contains histone deacetylases 1 and 2, which remove acetyl groups from histones and create a compressed form of chromatin that then inhibits or represses gene expression [Nan et al., 1998]. The action of the transcriptional repressor domain is partially reversed by trichostatin A, a histone deacetylase inhibitor. This finding suggests that repression by means of the transcriptional repressor domain is caused by histone deacetylation. MECP2 recruits these histone deacetylases and other chromatin remodeling complexes to methylated CpG dinucleotides. This leads to chromatin condensation that interferes with the binding of transcription complexes [Akbarian, 2003]. The transcriptional repressor domain has also been seen to bind to TFIIB (also designated GTF2B), SKI (a proto-oncogene), DNMT1 (which codes for a DNA methyltransferase), and SUV39H1 (which codes for a histone methyltransferase), although the importance of these interactions is unknown [Fuks et al., 2003; Kaludov and Wolffe, 2000; Kimura and Shiota, 2003]. Repression can be mediated in other ways, because the MECP2 protein binds to general transcription factors and interferes with the binding of transcription complexes. The transcriptional repressor domain is able to repress transcription when bound as far as 2000 bp from the transcription initiation site. The carboxyl terminus is thought to be involved in the binding of MECP2 to naked and nucleosomal DNA. Specifically, the carboxyl-terminal region of the MECP2 protein binds to DNA that is coiled around histone octamers [Chandler et al., 1999].
The MECP2 protein is mostly located within the nucleus of cells, and a small portion is seen within the perikarya [Kaufmann et al., 1995]. Although MECP2 binds throughout chromosomes, binding is most dense around pericentromeric heterochromatic regions of DNA. Forty percent of methyl-CpGs (i.e., binding sites for the methyl-CpG binding domain) are found within pericentromeric heterochromatic DNA. The immunoreactivity of MECP2 is increased around centromeric and perinucleolar heterochromatin. MECP2 does not associate with ribosomal DNA, despite its many methylations. MECP2 distribution is regulated by unknown factors and does not simply distribute to where methylated CpGs are found. The MECP2 protein is thought to play a role in the maintenance of neuronal nuclei in the later stages of development and within the mature brain [Akbarian, 2003]. It is hypothesized that MECP2 makes chromatin more stable and less accessible to transcription factors by anchoring chromatin fibers into the nuclear matrix.
There are reduced levels of dopamine, serotonin, and their metabolites, homovanillic acid and 5-hydroxy-indoleacetic acid, in Rett’s syndrome [Lekman et al., 1990]. Some researchers have noticed a decreased density of postsynaptic D2 receptors in older patients with Rett’s syndrome [Dunn, 2001], whereas others describe increased specific binding at D2 receptors. The latter finding implies that the decreased levels of dopamine are causing increased levels or density of postsynaptic receptors [Chiron et al., 1993; Dunn et al., 2002]. The D1 receptor density is unchanged [Wenk, 1995]. Jellinger et al. [1990] proposed that the different densities of postsynaptic receptors within the dopaminergic pathways might be age-specific. Increased choline concentrations and decreased choline acetyltransferase levels are thought to result from problems within the cholinergic system in the forebrain [Gokcay et al., 2002]. The frontal cortex and striatum have decreased levels of ferritin [Sofic et al., 1987]. Decreased levels of binding protein for the benzodiazepine receptor in the frontotemporal, parietal, and occipital regions of the brain also have been reported [Yamashita et al., 1998]. Significantly increased oxidative stress markers (intraerythrocyte non-protein-bound iron, plasma non-protein-bound iron, free F2-isoprostanes, esterified F2-isoprostanes, total F2-isoprostanes, and protein carbonyl concentrations) were evident in Rett’s syndrome subjects and associated with reduced arterial oxygen levels compared to controls. Biochemical evidence of oxidative stress was related to clinical phenotype severity and lower peripheral and arterial oxygen levels. Pulmonary 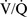 mismatch was found in the majority of the Rett’s syndrome population. These data identify hypoxia-induced oxidative stress as a key factor in the pathogenesis of classic Rett’s syndrome [De Felice et al., 2009].
mismatch was found in the majority of the Rett’s syndrome population. These data identify hypoxia-induced oxidative stress as a key factor in the pathogenesis of classic Rett’s syndrome [De Felice et al., 2009].
Animal Models
In a model of MECP2-null mice, males and females were affected. Homozygous female mice and heterozygous male mice were developmentally normal for the first several weeks of life, but they died soon after neurologic symptoms appeared [Chen et al., 2001; Guy et al., 2001]. This model replicates the genetic component of Rett’s syndrome, but it was difficult to study because of the rapid deterioration and early death of the animals after symptoms appeared. Shahbazian et al. [2002] developed a mouse model that has a truncation mutation within the MECP2 gene with a less severe phenotype than the MECP2-null mice. The truncated protein mimics a commonly observed human mutation, and it is partially functional, containing the methyl-CpG binding domain and transcriptional repressor domain. This group observed that the mice appeared developmentally normal up to 6 weeks after birth. After this period of normal development, the mice developed neurologic symptoms, including tremors, motor impairment, hypoactivity, seizures, kyphosis, and the classic forearm movements associated with human Rett’s syndrome [Shahbazian et al., 2002b]. Random X-inactivation causes a variety of phenotypes due to a single genotype in females, which makes analysis of the mouse model difficult [Young and Zoghbi, 2004]. One solution is to use male mice, because they are not subject to random X-inactivation.
Luikenhuis et al. [2004] overexpressed MECP2 in postmitotic neurons of homozygous MECP2-null mice. These mice did not display any neurologic symptoms, and they were developmentally equivalent to the control population. In normal mice, MECP2-encoded RNA is not expressed until about 10 days after conception, and it reaches adult levels by 16 days after conception. It has been postulated that the defect in Rett’s syndrome involves neuronal maintenance and maturation, and therefore affects developmental stability. Ballas et al. found that the loss of MeCP2 occurs not only in neurons but also in glial cells of Rett brains, and that mutant astrocytes from a Rett mouse model, and their conditioned medium, failed to support normal dendritic morphology of either wild-type or mutant hippocampal neurons [Ballas et al., 2009]. This suggests that astrocytes in the Rett brain carrying MeCP2 mutations have a non-cell autonomous effect on neuronal properties, probably as a result of aberrant secretion of soluble factor(s).
Pathogenesis
The timeline of MECP2 expression suggests that it is needed in the later phase of cortical development in neonates and after maturation in adults. MECP2 is expressed in normal fetal brains until 20 weeks’ gestation, after which it disappears from the cerebellum. It disappears from the brainstem after the perinatal period. MECP2 does not reappear in the brain until after adolescence. Lack of MECP2 during any of these developmental periods may lead to synaptic and neuronal dysfunction of the catecholaminergic neurons in patients with Rett’s syndrome [Itoh and Takashima, 2002]. Mice with MECP2 overexpressed in postmitotic neurons are rescued from the Rett’s syndrome phenotype. A decrease in MECP2 in postmitotic neurons during the later stages of development is sufficient to cause Rett’s syndrome [Chen et al., 2001], but MECP2 deficiency in neuronal precursors is probably not a major contributor to the pathogenesis in Rett’s syndrome.
MECP2 causes transcriptional repression, and loss of function of this protein may cause an imbalance between transcription and gene silencing, leading to dysregulated gene expression and pathologic changes. Some groups have found no evidence for MECP2 transcriptional repression in neurons or glia. Gene expression studies found an increase in glial transcription, in contrast to the predicted decrease. Levels of presynaptic proteins, however, were decreased. MECP2 may be causing perturbations within the presynaptic signal transduction pathway [Colantuoni et al., 2001]. The critics of this theory point out that these studies were performed on postmortem brains, suggesting that relevant time points in gene expression may have been missed. Additional evidence argues against MECP2 deficiency unsilencing transcription and causing Rett’s syndrome. Affymetrix GeneChip analysis of MECP2-deficient human fibroblasts did not demonstrate any large-scale dysregulation of gene expression. There were only small differences in gene expression in presymptomatic, early symptomatic, and late symptomatic MECP2-deficient mouse brains [Tudor et al., 2002].
If MECP2 does not act as a transcriptional repressor, it may play a maintenance role during development. MECP2-encoded mRNA is undetectable in the mouse forebrain during mid-gestation [Coy et al., 1999]. Immunohistochemical studies in nonhuman primates and mice demonstrate MECP2 expression in neuronal nuclei correlates with neuronal maturity, and levels of expression are highest in the adult cerebral cortex [Akbarian et al., 2001]. In human cerebral cortex, MECP2 is seen only in Cajal–Retzius cells, the earliest maturing neurons [Marin-Padilla, 1998], at 14 weeks’ gestation. At 26 weeks’ gestation, MECP2-immunoreactive neurons are seen in the deeper, more differentiated cortical layers. MECP2 neuronal immunoreactivity increases with age. Only about 10 percent of cells are immunoreactive during the third trimester of pregnancy, but approximately 80 percent of neurons demonstrate immunoreactivity for MECP2 at 10 years of age [Shahbazian et al., 2002a]. These findings are also seen in rodents and nonhuman primates. The observation that MECP2 expression is decreased in immature neurons and elevated in mature neurons suggests a role for MECP2 in neuronal maintenance. MECP2 expression studies show high levels of the protein in mature neurons but not in glia or astrocytes.
MECP2 may also play a role in cell division. Removing pericentromeric heterochromatin or disrupting heterochromatin silencing by inhibition of histone deacetylases in Drosophila and yeast reduced chromosome transmission during cell division [Henikoff, 2001]. Because MECP2 associates with pericentromeric heterochromatin, it may influence cell division.
Treatment
Rett’s syndrome has no cure. The treatments available have been empirically derived and are designed to combat specific symptoms. Antiepileptic agents include carbamazepine, valproic acid, and lamotrigine. The use of l-DOPA and dopamine agonists to increase motor ability in Rett’s syndrome patients is controversial [Zappella, 1990]. A study of the treatment of Rett’s syndrome with folate and betaine did not find any objective evidence of improvement [Glaze et al., 2009]. Zinc sulfate, lithium, and antidepressants have been demonstrated to increase central brain-derived neurotropic factor (BDNF) levels or signaling in human as well as animal studies. Thus, it is proposed that these agents could have therapeutic potential for RTT subjects [Tsai, 2006]. The breathing irregularities associated with Rett’s syndrome can be treated with naltrexone, an opiate antagonist. Naltrexone (1–3 mg/kg/day) can reduce disorganized breathing and increases oxygen saturation levels [Percy, 2002]. Use of high-fat, high-calorie diets is recommended in the late stages of the disease, and it has been suggested that individuals with Rett’s syndrome require a higher protein intake [Motil et al., 1999]. Feeding by means of a gastrostomy tube is sometimes indicated [Jellinger, 2003]. Constipation in Rett’s syndrome has been managed with high-fiber foods, enemas, mineral oils, milk of magnesia, and polyethylene glycol or MiraLax, with varying degrees of success. Orthopedists and physical therapists routinely see individuals with Rett’s syndrome. The goals are to improve balance, enhance flexibility, and strengthen atrophying muscles. Bracing for scoliosis is necessary when a 25° curvature exists, and surgery is advised when the curvature exceeds 40°. Speech and occupational therapy are occasionally used to improve communication.
Small-scale clinical trials using l-carnitine and the ketogenic diet have been completed. l-Carnitine has been reported to improve the respiratory features of Rett’s syndrome [Ellaway et al., 2001], and the ketogenic diet may reduce seizure frequency during the first 3 months, but no long-term clinical trials have been reported. A comprehensive, life-span approach to the management of scoliosis in Rett’s syndrome is recommended that takes into account factors such as physical activity, posture, and nutritional and bone health needs. Surgery should be considered when the Cobb angle is approximately 40–50° and must be supported by specialist management of anesthesia, pain control, seizures, and early mobilization. Evidence- and consensus-based guidelines were successfully created and have the potential to improve care of a complex comorbidity in a rare condition and stimulate research to improve the current limited [Downs et al., 2009].
Menkes’ Disease
History
The story of the discovery of Menkes’ disease begins in the 1930s, when veterinary physicians in Australia noticed the importance of copper for the normal development of sheep [Bennetts and Chapman, 1937]. They observed that mothers that grazed in copper-deficient pastures had offspring with cerebral demyelination, ataxia, and porencephaly. They concluded that copper deficiency in sheep was associated with ataxia and a demyelinating disease. The disease was described in 1962 by John Menkes [Menkes et al., 1962]. He described five males of English–Irish heritage who had “peculiar” hair, failure to thrive, and a neurodegenerative disorder. The syndrome was initially called Menkes’ kinky hair syndrome in reference to the unique appearance of the hair of these patients. The basis for Menkes’ disease was not known until the association was made with the illness in sheep [Danks et al., 1972, 1973]. The distinctive hair found in the copper-deficient sheep and in patients provided the necessary link. Serum testing revealed that patients who had this distinctive hair also had decreased serum copper and ceruloplasmin levels. Danks et al. [1972, 1973] then concluded that Menkes’ disease was a human example of a neurodevelopmental disorder caused by copper deficiency. In 1993, three groups used positional cloning to discover the ATP7A gene [Chelly et al., 1993; Mercer et al., 1993].
Clinical Description
Menkes’ disease typically occurs in males. Its occipital horn syndrome variant is also known as X-linked cutis laxa [Danks, 1995]. In classic Menkes’ disease, there is a period of normal development that typically lasts for 2–3 months [Kaler, 1994]. Developmental regression follows, with seizures, hypotonia, and failure to thrive [Kaler, 1998]. Patients exhibit severe mental retardation with symptoms of neurodegeneration [Mercer, 1998]. Most individuals with classic Menkes’ disease die between the ages of 7 months and 4 years [Bankier, 1995]. Occipital horn syndrome is characterized by a less severe genetic mutation that results in a connective tissue disorder.
The characteristic findings in classic Menkes’ disease are related to the patient’s hair. Commonly described as steel-woolish, the hair on the scalp and eyebrows is short, sparse, coarse, and twisted. The amount of hair is decreased, and it is generally shorter on the sides of the head. The color is often light, with white, silver, and gray being common. Light microscopy of hair reveals three characteristic findings [Kaler, 1998; Moore and Howell, 1985]:
Hypopigmentation is common but not the rule, and is caused by a deficiency of catechol oxidase. Patients typically have large jowls, sagging cheeks, large ears, and a high-arched palate. The skin appears loose at the nape of the neck, in the axillae, and on the trunk. Delayed tooth eruption and pectus excavatum are common, and the incidence of umbilical hernias is increased. Nephrocalcinosis and chronic renal failure have also been reported [Balestracci et al., 2009].
Classic Menkes’ disease has several distinctive neurologic findings. These patients exhibit truncal hypotonia with poor head control, hyperactive deep tendon reflexes, impaired visual fixation or tracking, and cortical adducted thumbs. There is increased appendicular tone, and asymmetric growth failure that appears shortly after neurodegeneration begins. EEGs are moderately or severely abnormal, and hypsarrhythmia occurs frequently [Venta-Sobero et al., 2004]. Ophthalmologic findings common in Menkes’ disease include myopia, strabismus, and problems with visual fixation and tracking.
Connective tissue disorders are common in the three variants. Pelvic ultrasound and cystograms reveal diverticula of the urinary bladder [Daly and Rabinovitch, 1981; Harke et al., 1977]. Associated vascular disorders include lumbar and iliac artery aneurysms [Adaletli et al., 2005]. Skull and skeletal radiographs are notable for Wormian bones in the skull, metaphyseal spurring of the long bones, and anterior flaring or multiple fractures of the ribs [Adams et al., 1974; Capesius et al., 1977; Koslowski and McCrossin, 1979; Stanley et al., 1976]. Congenital skull fracture, related to global osteopenia, subdural hematoma, intrauterine growth delay, and lethal outcome, is reported in neonatal Menkes’ disease [Veit-Sauca et al., 2009]. MRI demonstrates white matter changes and impaired myelination, cerebral blood vessel tortuosity, ventriculomegaly, and diffuse cerebral atrophy [Faerber et al., 1989; Johnsen et al., 1991]. White matter lesions localized in the deep periventricular white matter in the absence of diffuse cortical atrophy [Lee et al., 2007], and transient temporal lobe lesions related to vasogenic and cytotoxic edema [Ito et al., 2008] were reported.
Patients with occipital horn syndrome have hyperelastic skin and may develop other connective tissue disorders, including aortic aneurysms, hernias, bladder diverticula, and skeletal abnormalities, which most likely result from lysyl oxidase deficiency. The characteristic occipital horns are symmetric exostoses protruding from the occipital bone and pointing down. These may be present around 1–2 years of age, but are usually detected only around 5–10 years of age and continue to grow up to early adulthood [Tümer and Moller, 2009]. These individuals also have mild mental retardation and autonomic dysfunction that manifests as syncope, hypothermia, and diarrhea [Byers et al., 1980]. The severity of mild Menkes’ disease falls between that in the classic form and the much milder occipital horn syndrome [Procopis et al., 1981]. It is important for patients with milder variations of Menkes’ disease to have frequent vision examinations because ophthalmologic problems can greatly impair functioning of affected individuals.
Clinical and Biochemical Diagnoses
The clinical diagnosis of Menkes’ disease is supported by specific laboratory findings [Poulsen et al., 2002]. Early diagnosis of affected newborns is necessary for the institution of appropriate therapy and survival. A high index of suspicion based on clinical grounds is essential because supportive laboratory findings may be problematic in the first few months of life.
Initially, few or no neurologic manifestations occur [Gunn et al., 1984]. Kinky hair with light hair pigmentation is most suggestive of Menkes’ disease. The pili torti pathognomonic for Menkes’ disease are usually seen only in older patients. The diagnosis should be considered for a neonate born after premature labor and delivery with large cephalohematomas, unexplained hypothermia or hypoglycemia, jaundice requiring phototherapy, pectus excavatum, and inguinal or umbilical hernias [Kaler, 1998].
The classic biochemical markers of Menkes’ disease are low serum levels of copper and ceruloplasmin. Decreased intestinal copper absorption leads to low levels of copper in the plasma, liver, and brain. In contrast, copper stores are increased in the duodenum, kidney, spleen, pancreas, and skeletal muscle [Heydorn et al., 1975; Horn, 1984; Williams and Atkins, 1981].
Early laboratory diagnosis of neonates is complicated by the fact that copper and ceruloplasmin concentrations are normally low in healthy newborns, and these values can overlap with those typically found for older patients with Menkes’ disease [Lockitch et al., 1986, 1988]. Copper egress assay in cultured fibroblasts is the definitive diagnostic study at this age, but the test is lengthy and requires several weeks of cell culture. Rapid diagnosis of Menkes’ disease can be achieved by measurement of plasma catecholamines or polymerase chain reaction (PCR) detection of known deletions or point mutations in the ATP7A gene. The copper deficiency in Menkes’ disease affects the function of many enzymes requiring copper. Dopamine mono-oxygenase is one of the cuproenzymes that is dysfunctional in Menkes’ disease, resulting in abnormal plasma catechol concentrations in newborns and fetuses [Kaler et al., 1993a, 1993b, 1993c]. The plasma catecholamine profile is considered to be the most rapid and reliable way to diagnose Menkes’ disease during the neonatal period. Patients with Menkes’ disease have high plasma dopamine and low norepinephrine levels. Considered alone, neither dopamine nor norepinephrine levels have perfect sensitivity, whereas the ratio of dopamine to norepinephrine is high in all affected patients. Analogously, levels of the dopamine metabolite, dihydroxyphenylacetic acid, and the norepinephrine metabolite, dihydroxyphenylglycol, were imperfectly sensitive, whereas the dihydroxyphenylacetic acid to dihydroxyphenylglycol ratio is high in all patients. Plasma dihydroxyphenylalanine and the ratio of epinephrine to norepinephrine levels are high in affected neonates [Goldstein et al., 2009]. Increased urine ratios of homovanillic acid/vanillylmandelic acid (HVA/VMA) have been proposed as a screening tool for Menkes’ disease also [Matsuo et al., 2005]. PCR methods are helpful in the diagnosis of partial deletions and point mutations when they are already identified for a specific family. The DNA-based technologies used for screening ATP7A for point mutations are chemical cleavage mismatch detection [Das et al., 1994], single-strand conformational polymorphism analysis [Tumer et al., 1997], and dideoxy fingerprinting [Moller et al., 2000]. PCR-based methods for screening ATP7A for large partial deletions include multiplex PCR, genomic PCR, and reverse transcriptase PCR [Poulsen et al., 2002]. These PCR methods are useful in neonatal and prenatal diagnosis, and they are helpful for carrier screening.
Prenatal diagnosis and identification of carrier status are important for families that are at risk for Menkes’ disease. Assays looking for increased levels of copper in cultured fibroblasts also can be used for prenatal diagnosis and for testing of potential carriers [Goka et al., 1976; Poulsen et al., 2002; Tümer et al., 2003], but random X-inactivation renders carrier testing using this technique uninformative when negative. The only definitive tests that can exclude this disease are DNA-based assays.
An important component of all variants of Menkes’ disease is connective tissue involvement. Deoxypyridinoline is a cross-linking residue of type I collagen and is a good marker for lysyl oxidase activity, the cuproenzyme deficiency that is responsible for the connective tissue disorders observed in these patients. Deoxypyridinoline has been proposed as a marker for the presence of connective tissue disorders associated with Menkes’ disease [Kodama et al., 2003].
Genetics
Menkes’ disease is an X-linked disease, and one-third of cases are thought to represent new mutations [Haldane, 1935]. The gene responsible for Menkes’ disease, ATP7A, was discovered in 1993 [Chelly et al., 1993; Mercer et al., 1993; Vulpe et al., 1993]. The gene encodes the copper transporting ATPase known as ATP7A [Mercer, 1998]. ATP7A is part of a highly conserved family of cation-transporting ATPase proteins [Odermatt et al., 1993; Pederson and Carafoli, 1987]. This protein family includes the protein WD that is defective in Wilson’s disease. The ATP7A gene (formerly known as MNK) is located at Xq13.2–q13.3 [Tümer et al., 2003]. It has 23 exons and is about 140 kb long [Dierick et al., 1995]. ATP7A is 8.5 kDa. About 357 different mutations have been identified and are distributed as follows: nonsense mutations 18 percent, splice-site mutations 16 percent, missense mutations 17 percent, partial insertions 4 percent, minus mutations 5 percent, partial deletions 17 percent, chromosomal mutations 1 percent, and small indels 22 percent [Møller et al., 2009].
Occipital horn syndrome results from point mutations (75 percent), chromosomal rearrangements (about 1 percent), and large or partial deletions (about 15 percent) in the ATP7A gene [Liu et al., 1999; Tümer et al., 1999]. Severe classic Menkes’ disease RNA contains low levels of active ATP7A mRNA, whereas mild variants of Menkes’ disease have higher levels of active ATP7A mRNA. Occipital horn syndrome has 20–35 percent residual ATP7A mRNA in cultured cells, whereas classic Menkes’ disease has lower levels of mRNA in the cells [Kaler, 1994, 1998]. Decreased mRNA levels result from premature stop codons, missense mutations, frame shifts, and other deletions or mutations that affect RNA splicing [Das et al., 1995]. Patients with the identical deletion or mutation may have different clinical outcomes [Tümer et al., 2003]. This finding suggests that other modifying genes or proteins must play a role in the pathogenesis of this disease. Alternatively, other pathways for copper transport may exist at the cellular level.
Biochemistry
The genetic mutation of the ATP7A gene leads to problems with copper transport within the body. Normally, ATP7A allows efflux of copper from gut epithelium into the portal circulation, transport of copper across the blood–brain barrier, and transfer of copper that is reabsorbed by the kidney back into circulation. ATP7A protein deficiency results in an inability of the body to absorb copper from the gastrointestinal tract in amounts required to satisfy nutritional needs, as well as in impaired use and handling of the copper that is absorbed from the intestine. Copper is mobilized from the cytoplasm of cells so that it can be incorporated into secretory pathways [Tümer et al., 2003]. The mutation of a copper-transporting ATPase causes impaired cellular copper efflux, leading to increased intracellular copper concentrations. Patients with Menkes’ disease have high concentrations of copper in gut epithelial cells, and they absorb little copper from their diet. Copper accumulates in kidney tubules. At high levels, copper causes lipid peroxidation, protein cleavage, enzyme inhibition, and DNA damage. Normally, basal intracellular stores are maintained at low levels [Rae et al., 1999; Voskoboinik and Camakaris, 2002]. The disease is caused by a decreased amount of the ATP7A protein, or it can result from alterations to the protein that impair its ability to transport copper.
ATP7A has 6–8 transmembrane domains, and it transports Cu2+ ions using energy from adenosine triphosphate (ATP) hydrolysis [Vulpe et al., 1993]. The protein has several known motifs or domains within it, such as the ATP binding domain. The phosphorylation motif (DKTG) contains an aspartic acid that becomes phosphorylated during the protein’s cycle (present in all P-type ATPases). The cation transduction motif (CPC) features a conserved proline, which plays a role in the conformation changes that occur with cation transport [Silver et al., 1989]. There are six metal binding sites (MBSs) in the amino-terminal region of ATP7A. The consensus sequence of the MBS is GMxCxxC. Although human ATP7A has six different forms of MBS, microbial cells show that only one or two are necessary for a functional protein [Odermatt et al., 1993; Solioz et al., 1994]. Human ATP7A does not need an MBS, demonstrated by the fact that mutations in all six MBSs do not stop ATP7A from functioning [Forbes et al., 1999; Payne and Gitlin, 1998; Tsivkovskii et al., 2002]. The MBS may act as a sensor for intracellular copper levels, and it may play a regulatory role when concentrations are low [Goodyer et al., 1999; Strausak et al., 1999; Voskoboinik and Camakaris, 2002]. ATP7A also features a magnesium-binding motif (TGE), the “hinge” domain of the protein, and a phosphorylation motif (DKTG).
ATP7A-encoded mRNA is found is most cell types, but it is missing in liver. ATP7B, the protein mutated in Wilson’s disease (Table 41-2), transports copper in the liver [Vulpe et al., 1993; Paynter et al., 1994]. ATP7A and ATP7B are members of the P-type ATPases group IB family, as are bacterial heavy metal transporters [Tsivkovskii et al., 2002; Voskoboinik et al., 2001]. ATP7A has been localized to the trans-Golgi compartment under basal conditions [Petris et al., 1996]. This is consistent with the ability of ATP7A to supply cuproenzymes (i.e., lysyl oxidase) that are in secretory pathways. There is continuous recycling of ATP7A between the plasma membrane and the trans-Golgi compartment [Petris and Mercer, 1999]. ATP7A traffics to the plasma membrane with increased extracellular copper concentrations and is endocytosed back to the trans-Golgi compartment after extracellular levels decrease to normal. Copper-dependent vesicular trafficking moves ATP7A from the plasma membrane to the trans-Golgi network and back. Lower organisms have two copper ATPases, but this is unnecessary in humans because ATP7A traffics between the two areas where such ATPases are needed [Yuan et al., 1997]. Cu(I) may be the type of copper used by ATP7A as its substrate. It is unknown whether Cu(I) becomes Cu(II) before or after it is released in the Golgi lumen.
Table 41-2 Comparison of Menkes’ Disease and Wilson’s Disease
| Characteristic | Menkes’ Disease | Wilson’s Disease |
|---|---|---|
| Inheritance pattern | X-linked recessive | Autosomal-recessive |
| Location | Xq13.3 | 13q14.3 |
| Incidence | 1:300,000 | 1:100,000 |
| Clinical manifestations | Onset at birth Cerebral degeneration Global delay Kinky hair Pili torti Abnormal facies Hypopigmentation Arterial rupture or thrombosis Bone changes or cutis laxa |
Dysarthria Kayser–Fleischer rings |
| Laboratory test findings | ↓Serum Cu ↓Serum ceruloplasmin ↑Intestinal or kidney Cu ↓ Liver Cu |
↓ Serum Cu ↓ Serum ceruloplasmin ↑ Urinary Cu ↑ Liver Cu |
| Prognosis | Lethal in classic cases Death <3 yr |
Can be treated effectively with chelating agents |
| Gene product | 1500-amino acid copper-binding ATPase | 1411-amino acid copper-binding ATPase |
| Location, expression | All tissues except liver | Liver, kidney, and placenta |
| Mutation | Partial deletions in 15 percent; most others are point mutations | Point mutations and small rearrangements |
Menkes’ disease is caused by mutations that result in a substitution of highly conserved amino acids or those in highly conserved motifs. Any mutation that affects the structure and function of the ATP7A gene will lead to disease [Guy et al., 2001]. Mutations that induce Menkes’ disease include those that abolish the Mg2+ binding domain [Seidel et al., 2001] and those that change the cation transduction motif. Classic Menkes’ disease is usually caused by a premature truncation, typically occurring before the first transmembrane domain and resulting in loss of all catalytic activity. Occipital horn syndrome and mild Menkes’ disease usually result from missense or splice mutations. It is unknown how much catalytic activity is needed to result in a milder phenotype (i.e., ATP7A still able to absorb sufficient amounts of intestinal copper and enable its delivery to the requisite enzymes) [Tümer et al., 1997]. A case study of a patient with occipital horn syndrome showed that the splice mutation allowed 2–5 percent of ATP7A transcripts to be produced. This amount of protein was sufficient to allow partial absorption of copper from the gut epithelium and partial transport across the blood–brain barrier [Møller et al., 2000]. There was, however, too little protein for lysyl oxidase to function correctly. Between 2 and 5 percent of ATP7A activity is the proposed amount of protein necessary to decrease the severity of the Menkes’ disease phenotype.
Dopamine mono-oxygenase, also known as dopamine β-hydroxylase, is part of the catecholamine biosynthetic pathway. In Menkes’ disease, there is complete or partial deficiency of dopamine β-hydroxylase. This deficiency causes abnormal plasma and cerebrospinal fluid patterns. The degree of deficiency can be evaluated by looking at norepinephrine concentrations or the ratio of dihydroxyphenylalanine to dihydroxyphenylglycol. Cases of Menkes’ disease with deficient dopamine β-hydroxylase exist that have normal plasma and cerebrospinal fluid concentrations of norepinephrine. It is unknown why compensatory mechanisms are active in some patients but not in others. In the mouse model of Menkes’ disease, normal concentrations of norepinephrine are observed in certain areas of the brain [Prohaska and Bailey, 1994]. The dopamine β-hydroxylase deficiency causes temperature instability, hypoglycemia, eyelid ptosis, and loss of sympathetic adrenergic function [Biaggioni et al., 1990; Robertson et al., 1986].
Peptidylglycine mono-oxygenase is a cuproenzyme necessary for the removal of the carboxyl-terminal glycine residue from neuroendocrine precursors, including gastrin, cholecystokinin, vasoactive intestinal peptide, corticotropin-releasing factor, calcitonin, vasopressin, and thyrotropin-releasing hormone [Eipper et al., 1983, 1992]. When this enzyme is deficient, these neuroendocrine factors have 100- to 1000-fold decreased bioactivity. Affected animal models of Menkes’ disease have decreased peptidylglycine mono-oxygenase activity within their brains [Prohaska and Bailey, 1995]. The decreased activity of these neuroendocrine factors contributes to the phenotype of Menkes’ disease.
Cytochrome c oxidase is a copper-dependent enzyme that is deficient in Menkes’ disease. The decreased cytochrome c oxidase activity causes a subacute necrotizing encephalomyelitis without the severe lactic acidemia that is generally associated with complex IV defects [DiMauro et al., 1990; Robinson, 1989; Robinson et al., 1987]. Peripheral hypotonia and muscle weakness in patients with Menkes’ disease is partially caused by decreased activity of this enzyme.
The normal function of lysyl oxidase (protein lysine 6-oxidase) is to deaminate lysine and hydroxylysine during the first step of collagen cross-link formation [Siegel, 1979]. ATP7A may be needed to transport copper into the trans-Golgi for use by lysyl oxidase. Deficiency of lysyl oxidase decreases the strength of connective tissue in certain tissues or organs and leads to a host of connective tissue disorders that usually are associated with Menkes’ disease, including vascular tortuosity [Royce and Steinmann, 1990], bladder diverticula [Daly and Rabinovitch, 1981; Harke et al., 1977], and gastric polyps. ATP7A deficiency affects lysyl oxidase more profoundly than any of the other cuproenzymes [Gacheru et al., 1993]. It is possible that other cuproenzymes can acquire copper from cytoplasmic carriers without using ATP7A (an intermediate carrier), and this may explain why some mutations of ATP7A lead to the less severe occipital horn syndrome.
Cu/Zn SOD is also deficient in this disease [Rohmer et al., 1977]. Lowered levels of Cu/Zn SOD can increase susceptibility to damage by oxygen free radicals. It is not known whether decreased Cu/Zn SOD causes any developmental regression, because animal models without Cu/Zn SOD have normal development [Reaume et al., 1996] Postmortem studies of patients with Menkes’ disease have found increased levels of manganese SOD, which may be a compensatory change for the decreased levels of Cu/Zn SOD [Shibata et al., 1995]. It is unknown whether the decrease in this enzyme contributes to the phenotype observed in Menkes’ disease. Tyrosinase and sulfhydryl oxidase act on pigment formation and cross-linking of keratin, respectively; hence the manifestations of hypopigmentation, abnormal hair, and dry skin [Horn and Tümer 2002].
Pathology
Kidney
Copper accumulation within the kidneys leads to problems with renal reabsorption. Animal models of Menkes’ disease demonstrate an accumulation of copper within the proximal tubules of the kidney [Kodama and Murata, 1999]. Beta2-microglobulin is absorbed by the renal proximal tubules. Urinary β2-microglobulin levels rise with increasing age of Menkes’ disease patients, regardless of whether they undergo treatment [Ozawa et al., 2003].
Brain
Microscopy of tissues from animal models of Menkes’ disease reveals an increased number of apoptotic cells within the neocortex and the hippocampus [Rossi et al., 2001]. Postmortem cerebral cortex and cerebellum analysis showed downregulation of genes involved in myelination, energy metabolism, and translation, with the cerebellum being more sensitive to copper deficiency [Liu et al., 2005]. Brain CT scans demonstrate atrophy that is generalized or diffuse. Brain MRI reveals infarcts that result from tortuous arteries [Venta-Sobero et al., 2004]. MRI also demonstrates white matter disturbances, ventriculomegaly, and diffuse atrophy [Faerber et al., 1989].
Pathogenesis
Defective copper trafficking explains most changes seen in Menkes’ disease. The body’s nutritional needs for copper are crucial in the first 12 months of life. Brain growth and motor development are most rapid during this phase. Neurodevelopment and motor development are processes that require copper [Berg, 1994]. Expression of BCL2, an anti-apoptotic protein, is decreased in children with Menkes’ disease. Decreased levels of copper lead to mitochondrial damage that causes decreased levels of BCL2 [Rossi et al., 2001]. These decreased levels of BCL2 may explain the increased number of apoptotic cells seen in the brain of the animal model, as well as the mechanism behind neurodegeneration in Menkes’ disease.
Animal Models
Complete deficiency of the Menkes’ protein in mice causes lethality before birth, whereas humans can survive for a limited time without ATP7A [Mercer, 1998]. The murine homolog of ATP7A is Atp7a [Levinson et al., 1994; Mercer et al., 1994]. Just as in humans, there are a variety of observed phenotypes, with most of them reflecting their human counterparts. The severity of the murine phenotypes depends on the amount of ATP7A-encoded mRNA. Four murine genotypes are used to study Menkes’ disease:
Treatment
Treatment of Menkes’ disease is limited to supportive therapy and supplementation with parenteral copper histidine. Unlike nutritional copper deficiency, Menkes’ disease cannot be cured by copper replacement. Copper histidine is more effective in patients with milder phenotypes. A small subset of patients can achieve normal neurodevelopment. Early detection and intervention are critical for the copper histidine treatment to have an effect. Daily copper injections may improve the outcome in Menkes’ disease if started within days after birth [Kaler et al., 2008]. Many individuals with Menkes’ disease fare poorly, with little or no developmental improvement despite early intervention. Older patients with neurologic signs of Menkes’ disease demonstrate little improvement with copper histidine replacement. Therapy may, however, reduce irritability and allow for calmer sleeping patterns and minor improvements in personal and social development [Kaler, 1998]. These minor responses to therapy may help lessen the burden placed on caretakers. Copper histidine therapy helps the neurologic signs and symptoms of Menkes’ disease, but it has no effect on connective tissue disorders associated with this disease.
Future Directions
Improved therapeutic strategies are critical for better outcomes. Biochemical approaches to bypass the block in copper absorption from the intestine and allow requisite copper absorption are needed. Affected individuals must be identified early to initiate treatment before manifestations of neurologic signs and symptoms occur. This is the period when treatment has the most potential. ATP7A is necessary for the transport of copper through the blood–brain barrier. To prevent neurologic developmental regression, other ways of delivering copper stores to the brain must be identified. The symptoms of Menkes’ disease result from the dysfunction of multiple cuproenzymes, and copper must be made available to these enzymes to reduce the non-neurologic symptoms experienced by these patients [Kaler, 1998]. Although advances have been made in early detection of affected individuals, there has been little progress in finding ways to deliver copper to the necessary cuproenzymes or through the blood–brain barrier. Because classic Menkes’ disease is the result of the partial or complete absence of the traditional copper transport pathway, identification of alternate routes may allow the delivery essential for normal development.
Alpers’ Disease
Alpers’ disease is a fatal, progressive disease that affects the gray matter of the brain [Simonati et al., 2003]. This disease was initially described formally in 1931 [Alpers, 1931]. Alpers’ disease is known by other names: diffuse progressive degeneration of the gray matter, poliodystrophia cerebri progressiva, degeneration of the cerebral gray matter of Alpers’ [Ford et al., 1951], diffuse cerebral degeneration of infancy [Blackwood et al., 1963], progressive poliodystrophy [Dreifuss and Netsky, 1964], spongy glioneuronal dystrophy [Klein and Dichgans, 1969], spongy degeneration of the gray matter [Janota, 1974], Alpers–Huttenlocher syndrome [Huttenlocher et al., 1976], and progressive neuronal degeneration of childhood with liver disease [Harding et al., 1986]. Characteristic manifestations include neurologic deterioration, intractable seizures, and liver failure [Harding, 1990]. There is no known treatment. The clinical manifestations of Alpers’ disease are caused by myriad genetic defects with various forms of inheritance. Most of the genetic mutations that lead to Alpers’ disease are still unknown.
History
While visiting a colleague’s laboratory in Hamburg, American neuropathologist Bernard Alpers conducted a detailed study of a 4-month-old girl who died after a month of intractable seizures. The patient had a normal birth and normal development until age 4 months. There was necrosis of the deep gray matter nuclei and diffuse loss of ganglion cells in the third layer of the cortex. Alpers believed that the degeneration had a toxic cause. Many similar case reports have been made since then. Huttenlocher et al. [1976] were first to report that the neuronal degeneration was associated with liver failure. The pattern of inheritance has been postulated to be autosomal-recessive [Sandbank and Lerman, 1972]. A mitochondrial defect leading to Alpers’ disease has also been hypothesized [Naviaux et al., 1999]. The disease most likely has several causes, accounting for subtypes such as hepatocerebral or myopathic Alpers’ disease.
Clinical Description
Alpers’ disease is a clinical diagnosis that is documented on MRI but confirmed on postmortem examination. The disease is a diagnosis of exclusion, but specific clinical findings suggest its presence. Characteristic manifestations are liver failure, refractory seizures, and psychomotor retardation [Wefring and Lamvik, 1967]. Patients with Alpers’ disease are developmentally normal initially. Onset can occur between 1 month and 25 years of age [Harding et al., 1995]. Onset is more common during a patient’s infancy and adolescence, with most cases showing initial symptoms in infancy, usually before the age of 5. Death occurs between 3 months and 12 years of age, with most patients dying before the age of 3 years. The course of Alpers’ disease is variable, alternating between periods of development, degeneration, and mild recovery or stasis. Rare cases of Alpers’ disease have been reported in individuals as old as 25 years. Because of the variable age of onset, Alpers’ disease has been categorized as having juvenile, infantile, and prenatal forms [Frydman et al., 1993; Harding et al., 1995; Montine et al., 1995; Simonati et al., 2003; Worle et al., 1998]. Prenatal Alpers’ disease has been described in one family, and it is characterized by microcephaly, intrauterine growth retardation, retrognathia, joint limitations, and chest deformity. The infantile form manifests with early onset, a slowly progressive course, and late-occurring severe signs and symptoms. The juvenile form of Alpers’ disease is identical to the infantile form, but it is notable for a peripheral ataxia resulting from central and peripheral sensory axonopathy.
In addition to the classic triad of symptoms (i.e., psychomotor retardation, intractable seizures, and liver failure), patients with Alpers’ disease may have other manifestations. Hypotonia may occur initially [Egger et al., 1987]. Ataxia, febrile illness, and cortical blindness are less common [Naviaux and Nguyen, 2004]. A progressive ataxia that involves the sensory pathways has been described. This occurrence is similar to other sensory neuropathies caused by mitochondrial disorders [Fadic et al., 1997].
Liver failure is a complication that manifests late in the course of the disease [Smith et al., 1996], and it is usually the cause of death. Most cases of liver failure in these patients were attributed to hepatotoxicity caused by antiepileptic drugs, specifically valproic acid. However, some cases of liver failure and identical hepatic histology have occurred in patients who did not receive antiepileptic drugs. Valproic acid may cause hepatotoxicity in some cases of Alpers’ disease, but it cannot explain all cases. Orthotopic liver transplantation has not been helpful in cases with liver failure. Liver transplantation in patients with Alpers’ disease has been associated with neurologic deterioration [Delarue et al., 2000; Kayihan et al., 2000]. Complications of neuronal degeneration can lead to death from respiratory failure or primary hypoventilation.
Clinical Diagnostic Tests
Initial laboratory test results can be normal. Liver function test results can be elevated initially, although this finding is rare [Egger et al., 1987]. Values are elevated in the later stages of the disease because of liver cirrhosis. There are no specific in vivo serum or cerebrospinal fluid markers for Alpers’ disease, and the diagnosis must rely on neuropathologic evaluation. Lactate levels may be raised in the blood, and the protein and lactate levels may be elevated in the cerebrospinal fluid [Worle et al., 1998]. Positive cerebrospinal fluid oligoclonal bands, and very high immunoglobulin (Ig) G synthesis rate and IgG index were also reported [Bao et al., 2008]. Elevated cerebrospinal fluid neopterin, interleukin (IL)-6, IL-8, interferon (IFN)-c, reduced cerebrospinal fluid 5-methyltetrahydrofolate (5-MTHF), and increased serum, as well as cerebrospinal fluid folate receptor blocking autoantibodies, are present [Hasselmann et al., 2009].
Cranial CT scans demonstrate progressive atrophy and low densities in the occipital and temporal lobes. There is involvement of the cortex and the white matter [Kendall et al., 1987; Flemming et al., 2002]. Generalized atrophy is common throughout the brain in the later stages of the disease. Multiple findings are apparent on conventional MRI scans. These include diminished white matter and cortical thinning of the frontal, posterotemporal, and occipital lobes [Barkovich et al., 1993]. Lesions of the thalamus also have been reported. Occipital lobe atrophy is widespread. Proton MR spectroscopy reveals increased cerebrospinal fluid levels of lactate [Charles et al., 1994] and a reduced N-acetylaspartate to creatine ratio. The increase in lactate marks the switch from oxidative to anaerobic metabolism. N-acetylaspartate is an indicator of neuronal viability [Neumann-Haefelin et al., 2000].
EEG findings correlate with the lesions seen on MRI that are most apparent in the occipital lobes. The EEG of Alpers’ disease is described as slow (<1 Hz) with high-amplitude activity (0.2–1.0 mV). Lower-amplitude polyspikes are also recognized on the EEG. This pattern is seen in 75 percent of patients, but it may be present only transiently in periodic bursts [Martinez-Mena et al., 1998]. These bursts increase in number and duration as the disease progresses. Unilateral occipital rhythmic high-amplitude delta with superimposed (poly)spikes (RHADS) was described in convulsive status epilepticus in Alpers’ patients [Wolf et al., 2009]. Mild axonal sensory neuropathy was reported, as was central-peripheral sensory axonopathy in a juvenile case of the syndrome [Simonati et al., 2003]. There can be a loss of visual-evoked potentials [Martinez-Mena et al., 1998]. The triad of requisite clinical symptoms and MRI and EEG findings suggests the diagnosis of Alpers’ disease.
Pathology
Alpers originally described the neuronal pathology as cortical lesions with reactive gliosis, demyelination, nerve cell loss, spongy degeneration, and accumulation of neutral lipids. The occipital cortices are usually involved, and involvement can be symmetric or asymmetric. Patchy cerebral cortical destruction usually is worst within the striate cortex. Striate cortex involvement is a hallmark of Alpers’ disease [Harding, 1990; Harding et al., 1986]. Because of the destruction of the visual cortices, patients often have cortical blindness [Charles et al., 1994; Dietrich et al., 2001; Parsons et al., 2000]. Multiple case studies have described the cortical destruction as neuronal loss, spongiosis, astrocytosis, and gliosis. Milder changes are seen in the parietal cortices [Montine et al., 1995]. Necroses in the hippocampi, lateral geniculate nuclei, amygdala, substantia nigra, and dorsal columns may be evident. In patients with severe liver failure in the late stages of Alpers’ disease, Alzheimer’s disease type II astrocytes are seen. The white matter is only minimally affected.
Premortem liver biopsies reveal lobular disarray, microvesicular steatosis, and inflammation with acute and chronic hepatocyte necrosis [Narkewicz et al., 1991]. Common hepatic findings at autopsy are fibrosis, regenerative nodules, hepatocyte dropout, bile duct proliferation, fatty changes, and bile stasis. Pancreatitis is infrequently observed in these patients. Muscle biopsies may contain ragged red fibers and cytochrome c oxidase-negative fibers when Alpers’ disease is caused by a mitochondrial defect. A subset of patients with Alpers’ disease lacks any detectable energy metabolism defect and has normal hepatic histology [Frydman et al., 1993].
Biochemistry
The clinical manifestations of Alpers’ disease have several causes. Abnormalities within the citric acid cycle of leukocytes, cultured fibroblasts, and hepatocytes due to a defect in pyruvate metabolism or mitochondria have been suggested [Gabreels et al., 1984; Prick et al., 1981]. These defects result in deficiencies of cytochrome c oxidase, pyruvate cocarboxylase, and mitochondrial electron transport chain complex I. Mitochondrial DNA (mtDNA) polymerase gamma activity is less than 5 percent of normal in muscle and liver cells [Naviaux and Nguyen, 2004]. Immunoreactive subunits of the mtDNA polymerase are still present in patients. The deletion that causes the reduced amount of mtDNA polymerase is small because detectable levels of protein are still present. Southern blot analysis of two infants with Alpers’ disease revealed decreased mtDNA in muscle, liver, brain, and fibroblasts. The loss of mtDNA may be tissue-specific [Tesarova et al., 2004].
Genetics
The mode of transmission of Alpers’ disease is understood, and it is consistent between families. Case studies and biochemical evidence support autosomal-recessive inheritance and maternal or mitochondrial inheritance patterns. Cases with autosomal-recessive inheritance patterns have no mitochondrial deficiencies [Harding, 1990]. Simonati et al. [2003] observed that, although the disease affects both genders, there appears to be a mild male predominance. The same group also observed that children with juvenile Alpers’ disease do not have depletion or significant mutations in mtDNA.
Two mutations within the polymerase gamma gene (POLG) are associated with Alpers’ disease: G2899T and G1681A. G2899T causes a premature stop codon (Glu873Stop). Some Alpers’ disease patients are heterozygous for the POLG mutation Glu873Stop. It has been theorized that this stop codon is incomplete and allows a small number of ribosomes to be read. The number of ribosomes that are able to bypass the premature stop codon may be regulated in an age- and tissue-dependent manner, allowing Alpers’ disease to manifest at different ages and in different tissue types. In Drosophila melanogaster, proteins are regulated in a tissue-specific manner by changing the ratio of short to long forms of the protein [Robinson and Cooley, 1997]. The premature stop codon in POLG may change the ratio and cause tissue-dependent regulation.
The G1681A causes an Ala167Thr substitution. This corresponds to the linker region of the POLG protein. POLG is located on chromosome 15q24–26 [Zullo et al., 1997]. No other polymerase can be substituted for decreased POLG activity. POLG is the only polymerase of the 15 known DNA polymerases that has a mitochondrial import signal. Mutations within POLG are found in other mitochondrial diseases: progressive external ophthalmoplegia (autosomal-recessive and dominant forms) [Van Goethem et al., 2002, 2003], ophthalmoparesis, sensory ataxia, neuropathy, dysarthria, and male infertility. Children homozygous for the G2899T mutation are affected with Alpers’ disease, whereas control groups consisting of patients with neuromuscular diseases or patients with other mitochondrial diseases do not have this mutation. Many patients with Alpers’ disease are heterozygous for G1681A, but the mutation is not specific. Heterozygous G1681A mutations are also seen in ophthalmoparesis, both types of progressive external ophthalmoplegia, and in the asymptomatic parents of children with Alpers’ disease.
Pathogenesis
Alpers’ disease was originally believed to be a complication of concomitant hypoxia and epilepsy. Comparisons with status epilepticus cases are not valid because the cortical lesions in the two diseases differ significantly. Others have proposed that the underlying defect is metabolic. Alpers’ disease most likely can be caused by multiple metabolic defects. The cerebral destruction of the striate cortex in Alpers’ disease may result from a novel form of hepatocerebral toxicity, which is different from that seen in Wilsonian hepatocerebral degeneration. The possibility of a viral infection has been raised after brain tissue from a patient who died from Alpers’ disease was injected into hamsters, and caused a spongiform encephalopathy [Rasmussen et al., 2000].
Management
There is no known treatment for Alpers’ disease. It is important to recognize this disease early and avoid the use of valproic acid, because this increases the incidence of hepatotoxicity in these patients. Plasma alanine aminotransferase (AAT) can be used as an index of liver cell damage. Vigabatrin, which suppresses AAT activity, is better avoided in Alpers’ patients [Williams et al., 1998]. Although some patients may respond to antiepileptic medications early in their disease course, the seizures become refractory to treatment. Treatment with oral leucovorin (5-formyl-tetrahydrofolate) in a patient with increased serum as well as cerebrospinal fluid folate receptor-blocking autoantibodies was initiated at 0.25 mg/kg b.i.d., and later increased to 4 mg/kg twice daily; this resulted in improvement of seizure frequency and communicative abilities [Hasselmann et al., 2009]. Ketogenic diet is reported to cause clinical and EEG improvement [Joshi et al., 2009].
Neuronal Ceroid-Lipofuscinosis: Batten’s Disease
The neuronal ceroid-lipofuscinoses, or Batten’s disease, comprise a group of inherited neurodegenerative diseases of childhood caused by defects in different genes and proteins. Their unifying clinical hallmarks are seizures, blindness, cognitive and motor decline, and early death. Eight genes and their protein products have been identified to account for 8 of the 10 or more described clinical entities: CLN1/protein palmitoyl thioesterase (PPT1), CLN2/tripeptidyl peptidase (TTP1), CLN3/battenin, CLN5, CLN6, CLN7/MFSD8 (major facilitator superfamily), CLN8/EPMR, and CLN10/CTSD (cathepsin D) [Siintola et al., 2007]. The clinical types are classic infantile (INCL/CLN1); classic late infantile (LINCL/CLN2); variant late infantile Finnish (CLN5); variant late infantile, also known as Costa Rican or Portuguese but not limited to these populations (CLN6); classic juvenile (JNCL/CLN3); Scottish juvenile (CLN1); epilepsy with mental retardation (EPMR/CLN8); Turkish variant infantile, previously classified as CLN7 (with defects in the CLN8 gene); a second type of Turkish variant, infantile due to CLN7/MFSD8 defects; CLN9 variant (CLN9; gene to be identified); and a number of adult-onset variants that are dominantly or recessively inherited (CLN4, gene to be identified) [Boustany, 1996; Goebel et al., 1999; Mole, 2004] (Table 41-3). Naturally occurring mouse models with defects in either Ctsf (cathepsin F) [Tang, 2006], Clcn-3 [Yoshikawa et al., 2002], or Clcn-7 [Kasper et al., 2005] result in an NCL-like phenotype, but mutations within these genes have not yet been reported in humans.
A plethora of novel information has emerged over the past decade regarding the genetics, molecular and cell biology, and biochemistry of this group of disorders. [Gao et al., 2002; Persaud-Sawin et al., 2004; Puranam et al., 1997; Ranta et al., 1999; Schulz et al., 2004; Sleat et al., 1997]. Three of the proteins identified, protein palmitoyl thioesterase, tripeptidyl peptidase, and cathepsin D, are soluble lysosomal proteins, although INCL, LINCL, and congenital NCL are not typical lysosomal storage diseases. Two other proteins, CLN6 and CLN8, are resident proteins in the endoplasmic reticulum, and CLN3 is a protein that traffics between Golgi, early recycling endosomes, and lipid rafts in the plasma membrane. CLN3, CLN6, and CLN8 are hydrophobic membrane proteins. CLN5 is characterized as a lysosomal membrane glycoprotein. There are many naturally occurring animal models. Two of these, the nclf mouse and the New Zealand Southhampshire sheep, are models for CLN6. The mnd mouse is a model for CLN8. Transgenic models for INCL, LINCL, and CLN6-deficient variant LINCL and CLN10 also are available.
Treatment options are beginning to expand beyond anticonvulsants and supportive nutritional and physical measures. Targeted therapies have been used in patients with known biochemical and cell biologic processes, such as cysteamine in INCL; flupirtine in INCL, LINCL, variant forms of LINCL, JNCL, and CLN6- and CLN9-deficient variants [Batten and Mayou, 1915; Dhar et al., 2002; Mayou, 1904]; and mycophenolate mofetil in JNCL. Gene- and protein-based delivery systems are being developed for the classic late infantile and infantile types with defects in soluble proteins, and they are being tested in transgenic mouse models. Stem cell replacement is being explored in animal models as potential therapy for these terminal diseases, as well as in terminal CLN2 and CLN1 human cases.
A clinical rating scale, the Unified Batten Disease Rating Scale (UBDRS), was developed to assess motor, behavioral, and functional capability in JNCL [Marshall et al., 2005]. This scale should be of assistance in evaluating novel treatment strategies mentioned.
History and Terminology
The first clinical description of neuronal ceroid-lipofuscinosis was that of the juvenile form by Stengel [1826]. This was soon followed by clinical and pathologic descriptions by Batten, Mayou, Spielmeyer, Vogt, and Sjögren [Batten and Mayou, 1915; Mayou, 1904; Spielmeyer, 1923; Vogt, 1909]. The CLN3 gene responsible for the juvenile form (JNCL) was discovered in 1995 [Lerner et al., 1995]. The late infantile form (LINCL) was described by Jansky and Bielchowski in 1908 and 1913 [Jansky, 1908]. These two variants were previously referred to as Batten’s disease, a term that now refers to all variants. The adult form, or Kufs’ disease, an early-onset dementia with seizures and absence of visual findings, was described in 1925 [Dom et al., 1979]. The adult form (ANCL) has been described in sporadic cases and familial cases, with some families suggesting a dominant pattern of inheritance. Chromosomal location of the CLN4 gene or genes responsible for the adult disease remains unknown.
The term neuronal ceroid-lipofuscinosis was introduced by Zeman and Dyken in 1969 as a descriptive term referring to the autofluorescent, waxy, dusky lipid accumulating in neuronal endosomes, reminiscent of lipofuscin, the aging pigment [Zeman et al., 1970]. The infantile form (INCL) was described by Hagberg and then by Haltia and Santavuori in 1973 [Hagberg et al., 1968; Haltia et al., 1973a, 1973b; Santavuori et al., 1973]. The CLN1 gene was identified in 1995, followed by the CLN2 gene responsible for LINCL [Sleat et al., 1997]. Other types have been described since then, including variant late infantile forms and early juvenile forms due to defects in the CLN5, CLN6, and CLN8 genes [Gao et al., 2002; Ranta et al., 2001; Savukoski et al., 1998; Wheeler et al., 2002]. A CLN9 form is also described that is clinically similar to the juvenile form [Lin et al., 2001; Schulz et al., 2004]. The CLN9 gene remains to be characterized.
The terminology is confusing because it was established before many variants or clinical forms were defined and before any of the genes were identified. The terms INCL, LINCL, JNCL, and ANCL were initially chosen to separate the forms according to age of onset. This holds true for the main classic variants described. Since discovery of the various genes, many atypical cases have been described with variable ages of onset. It has, therefore, been decided to refer to these diseases by a unified nomenclature, illustrated by the following example: CLN1 disease infantile; CLN1 disease late infantile; and so on, as referred to in Table 41-3 [Mole et al., 2010].
Major Neuronal Ceroid-Lipofuscinosis Clinical Types or Syndromes
The clinical features, laboratory tests, pathology, biochemistry, and genetics are summarized for each of the major types (see Table 41-3).
Infantile Neuronal Ceroid-Lipofuscinosis
INCL (i.e., Haltia–Santavuori variant, CLN1-defective, PPT1-deficient form) is caused by a deficiency in palmitoyl protein thioesterase. The function of this lysosomal thioesterase is to remove fatty acids attached in thioester linkages to cysteine residues in proteins [Schriner et al., 1996]. The first description of this disease was in 1968 by Hagberg et al. [1968]. A comprehensive clinical and pathologic characterization of this autosomal-recessive disorder was provided later from Finland [Haltia et al., 1973b; Santavuori et al., 1973].
Clinical description
The Finnish cases represent the most severe form of the disease. Development is normal until 10–18 months of age. Deceleration of head growth can start as early as 5 months of age. Developmental arrest, hypotonia, and ataxia ensue. All children become microcephalic. Visual impairment is apparent at 1 year of age, and children are blind by the age of 2 years. Optic atrophy, thinned retinal vessels, and a discolored, brownish macula are seen funduscopically. Frequent myoclonic jerks begin after year 1, and many affected children develop generalized seizures. Hand-knitting movements reminiscent of Rett’s syndrome are observed early but disappear by age 2 years. By the third year of life, patients are bedridden, hypotonic, irritable, and spastic. At age 5 years, severe flexion contractures, acne, hirsutism, and rarely, precocious puberty are observed. Children with INCL have an increased risk of hypothermia and bradycardia late in the course, especially during anesthesia due to impaired thermoregulation. These children should be carefully monitored and warming interventions applied during operative procedures [Miao et al., 2009]. Most children die between the ages of 7 and 13 years. There are rare cases in which the age of onset may be as late as 4 years. A distinct adolescent phenotype reminiscent of the classic juvenile variant has been described in some patients of Scottish descent [Mitchison et al., 1998].
Clinical diagnostic tests
The single best diagnostic test is measurement of PPT1 enzyme activity in leukocytes [Das et al., 1998]. Enzyme activity can be measured from a dried blood spot on filter paper or from cultured fibroblasts. In the proper clinical setting, an enzyme activity less than 5 percent of normal is diagnostic for INCL. Salivary PPT1 measurement also is reported as a reliable method of diagnosis [Kohan et al., 2005]. DNA diagnostics are also widely available. Before the availability of the enzyme assay or DNA diagnostics for this disease, the diagnosis was based on the clinical presentation and electron microscopic examination of skin or other available tissue. The characteristic finding is membrane-bound GRODs, which typically are seen in endothelial, periepithelial, and autonomic nerve cells of the submucosal myenteric nerve plexus, but they also have been reported in other cell types. The EEG may initially be normal but then reveals lack of sleep spindles and absence of the attenuation in amplitude seen with eye opening by the ages of 16–24 months. There is gradual loss of amplitude, and the EEG becomes isoelectric by age 3 years. The ERGs, visual-evoked responses, and somatosensory-evoked responses are also abnormal but tedious to demonstrate in young children, and they are not needed for the diagnosis. The ERG is abnormal, with cone function affected before rod function. CT and MRI findings are present early and include signal loss in the thalami and cerebral atrophy with high-signal-intensity, thinned periventricular rims. Postmortem T2-weighted MRI scans reveal a remarkable hypointensity of the gray matter with respect to the white matter. Prenatal diagnosis has been performed on chorionic villus samples as early as 11 weeks, and it can be achieved by examining amniocytes at a later stage (16–18 weeks). Initially, electron microscopic or ultrastructural studies were performed to look for GRODs, but the PPT1 enzyme assay and a DNA analysis can be performed instead. Prenatal diagnosis using allele specific primer extension (ASPE) is also reported [Zhong et al., 2005]. There remains a role for electron microscopy when enzymatic diagnosis is not available, when enzyme activities and DNA analysis are not clear-cut, or when identification of the existing mutation is absent. Ideally, all three diagnostic methods should be used because of the importance of the decision, based on these results, to be taken by the family, treating obstetrician, and geneticist. The diagnosis of a normal or carrier fetus should be confirmed at birth by analysis of cord blood.
Pathology
At the time of death, brain weight is drastically reduced to 250–400 g. Serial pathologic studies in Finland revealed progressive cortical neuronal loss beginning at 1 year of age, with a subtotal loss of neurons by 4 years. Giant Betz cells and neurons of the hippocampal CA1 and CA4 sectors are relatively preserved. Reactive astrocytes become more prominent with time. GRODs are apparent in neurons and macrophages as early as 8 weeks’ gestation. Ultimately, cerebellar Purkinje cells and granule cells are destroyed and are replaced by a rim of Bergman glia. The white matter appears gliotic. The brainstem and basal ganglia are also involved. The spinal cord is relatively preserved with anterior horn cells that demonstrate storage only. Storage granules can be seen in many different tissues of the body, but tissue destruction is reserved for the brain and retina [Goebel and Wisniewski, 2004; Wisniewski et al., 2004b].
Biochemistry, cell biology, and pathophysiology
There is loss of function of PPT1, which removes long-chain fatty acids attached in thioester linkage to the cysteine residues of proteins. Proteins containing the fatty acylated cysteine residues are usually found at the inner plasma membrane leaflet. Normally, reversible acylation and deacylation of these may have impact on protein–protein and protein–lipid membrane interactions. S-acylated proteins are degraded in lysosomes, and this function also may be impaired in INCL. INCL, like other neuronal ceroid-lipofuscinosis disorders, differs from other storage diseases in that the material that accumulates in the cell has no demonstrable link to the actual defect and may be a secondary occurrence. PPT1 is located in the lysosome and taken up in a mannose-6 phosphate-dependent manner, but it is also activated at neutral and basic pH, and likely functions in the lysosome and elsewhere in the cell. PPT1 co-localizes with synaptophysin to presynaptic vesicles in neurons. Sphingolipid activator proteins A and D accumulate in storage cytosomes, probably as a secondary phenomenon [Tyynela et al., 1993]. There are reported abnormalities in brain sphingomyelin and other phospholipids, levels of which are decreased in the INCL brain. There are reports of increased rates of apoptosis in lymphocytes, cultured lymphoblasts, and fibroblasts from patients, as well as neurons rendered deficient in PPT1 [Cho et al., 2001].
Genetics
All cases of INCL in Finland are caused by an identical common missense mutation (R122W, arginine to tryptophan) that leads to an unstable protein that is degraded in the endoplasmic reticulum [Mole et al., 1999]. At 1 in 70, the carrier frequency is high in Finland, and the incidence of the disease is 1 case per 20,000 individuals. The incidence in the United States has never been accurately computed, but it probably accounts for about 20 percent of all diagnosed cases of Batten’s disease. A juvenile-onset variant of this disorder was first described in a person from Scotland; it has a threonine to proline substitution at position 75 [Mitchison et al., 1998]. This mutation and another with a premature stop codon at arginine 151 account for most alleles from patients in the United States, all of whom have Irish or Scottish ancestry. The former is found in juvenile cases and the latter in infantile cases. At least 40 mutations in the CLN1 gene have been described.
Management and treatment
In a mouse model for PPT1 deficiency, virally mediated CLN1 gene delivery has cleared storage and improved the clinical condition of these neurologically impaired mice. This therapy is being developed for use in humans. Enzyme replacement is a theoretical possibility, although protein delivery to the central nervous system is not trivial. Under development are methods to chemically open the blood–brain barrier or chemically camouflage proteins to enable them to cross it selectively. Bone marrow replacement has failed, but stem cell therapies are being explored. Gene therapy using intravitreal injection of AAV2-PPT1 increased enzyme levels in the eye and correlated with improvements in the histological abnormalities and mixed rod/cone and pure cone functions in murine models. In addition, PPT1 activity was detected in the brain following intravitreal injection [Griffey et al., 2005]. A clinical trial is using phosphocysteamine, a safe oral drug that deacylates proteins and is anti-apoptotic, is on-going. Clinical efficacy is unknown, but the drug clears storage material and decreases apoptosis in vitro [Zhang et al., 2001]. Flupirtine is an oral anti-apoptotic drug with analgesic, antispasmodic, and weak antiepileptic properties that protects PPT1-deficient cells from apoptosis. It is approved for use as an analgesic and antispasmodic in Europe, but it has not been approved by the U.S. Food and Drug Administration (FDA). Its clinical efficacy in INCL remains unknown.
Late Infantile Neuronal Ceroid-Lipofuscinosis or Late Infantile Batten’s Disease
LINCL (i.e., Jansky–Bielchowski, CLN2-defective, TPP1-deficient form) results from a deficiency of lysosomal tripeptidyl peptidase. The defect was discovered by comparing mannose-6-phosphate-modified lysosomal proteins from a normal and an LINCL-affected brain [Sleat et al., 1997]. It was first described by Jansky in 1908 and then by Bielchowski in 1913. It is the most pan-ethnic of neuronal ceroid-lipofuscinosis disorders, having been described in European, Middle Eastern, Chinese, Pakistani, and Indian patients. It is the second most common form of Batten’s disease in the United States, although the total number of cases at any time is less than 500, making it an orphan disease according to the FDA. It is thought that a large number of cases originate from Europe.
Clinical description
LINCL manifests with a generalized seizure disorder and ataxia due to unrecognized, frequent absence seizures occurring between the ages of 2.5 and 3.5 years. Within 6 months, vision, motor, and cognitive skills deteriorate rapidly. Affected children are blind by age 4 years because of tapetoretinal degeneration. Most children are nonambulatory and mute by age 5 years, and they require gavage feeding. In addition to prolonged, generalized tonic-clonic seizures, all have frequent myoclonic jerks, which are most prominent in the face but are also observed in the trunk and extremities. An early hypotonia gives way to severe spasticity with flexion contractures. Autoregulation of vascular tone is lost, resulting in mottled, cold hands and feet. Hypothalamic involvement leads to temperature instability vacillating between hyperthermia and hypothermia. Hyperthermia often leads to unnecessary fever evaluations. Copious secretions and shallow breathing resulting from poor chest wall excursions often cause pneumonias. Sepsis and uncontrollable seizures are frequently the cause of death at the end of the first decade or in the early teens [Boustany, 1996; Zhong et al., 2000]. Some atypical cases have a later onset and protracted course [Wisniewski et al., 2004c].
Clinical diagnostic tests
The most definitive test is measurement of TPP1 enzyme activity when the clinical history and course fit the description. Typically, enzyme activity less than 5 percent of normal is diagnostic for LINCL. This test can be performed on leukocytes, cultured fibroblasts, or amniocytes, and on dried blood from a filter paper. Salivary PPT1 measurement is also reported as a reliable method of diagnosis [Kohan et al., 2005]. DNA-based diagnosis is also available, but it is more tedious, particularly if the specific family mutation or mutations are not known. In this instance, the diagnostic laboratory excludes the most commonly reported mutations first. Before availability of enzyme or DNA diagnosis, ultrastructural study of a skin biopsy provided objective proof for the disease. The appearance of curvilinear bodies enclosed within unilamellar endosomes in multiple cell types (i.e., endothelial cell, pericyte, Schwann cell, and others) is the most characteristic feature. Rarely, few fingerprint profiles may be seen. Electron microscopy continues to be a valuable diagnostic tool when other forms of diagnosis are not available and in evaluating atypical cases. It has sometimes led to identification of novel neuronal ceroid-lipofuscinosis variant. The ERG reveals reduced amplitudes early in the course of this illness, even before changes of thinned vessels and pale discs become apparent. The process is extinguished within a few months of presentation. Characteristic giant occipital polyspike-spike discharges are seen on the EEG in response to a single flash of light or to low-frequency, repetitive stimulation. These discharges represent the early phase of an exaggerated visual-evoked response. Wave amplitudes of visual-evoked and somatosensory-evoked responses are also high. These tests are seldom used diagnostically. Neuroimaging studies often help to confirm the diagnosis. An initial CT or MRI scan may be normal, but usually within 6 months of onset and before the age of 4 years, cerebral and cerebellar atrophy is prominent. Within 2 years of onset, there is a 40 percent loss of volume of the cerebellum and a fivefold increase in lateral ventricle to hemisphere volume ratio. Caudate and thalamic volumes are markedly reduced compared with age-matched controls, and there is relative preservation of brainstem volume early in the course [Boustany and Filipek, 1993]. Prenatal diagnosis may be possible using ASPE [Zhong et al., 2005].
Pathology
The small number of remaining neurons has distended cell bodies and granular cytoplasm. This material is positive for periodic acid–Schiff (PAS), Luxol fast blue, and Sudan black B in light microscopy sections. White matter appears relatively intact, strongly speaking against a primary inflammatory component in LINCL. Condensed chromatin identified by electron microscopy, upregulation of BCL2 protein, and positive terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) stains all provide evidence for the occurrence of apoptosis, a feature common to multiple neurodegenerative diseases [Puranam et al., 1995, 1997]. There is strong reactivity with an antibody to subunit C of mitochondrial ATP synthase [Johnson et al., 1995]. The reason for this is still unknown, but it may represent a form of apoptosis observed in neurodegenerative illnesses called mitopsis.
Ultrastructurally, neurons and many other cells contain curvilinear inclusions enclosed within a single membrane. Frequently, these inclusions are admixed with fingerprint profiles. This is more commonly observed in cells outside the central nervous system, such as smooth muscle cells, eccrine sweat glands, endothelial cells, and pericytes. Before the availability of enzymatic diagnosis, prenatal diagnosis was determined by analyzing the ultrastructure of amniocytes obtained at 16–17 weeks’ gestation [Wisniewski et al., 2004a].
Genetics
LINCL is an autosomal-recessive disorder that appears to be pan-ethnic, with cases diagnosed from many countries on all continents except Africa. There is a notable lack of African and Jewish cases. It is the second most common form of Batten’s disease in the United States, accounting for one-third of diagnosed cases. More than 53 mutations have been described. In the United States, two common mutations account for 65 percent of diagnosed cases. One is a nonsense mutation, Arg208X, and the other affects a splice junction site, IVS5-1G>C. The few cases with a milder course and later onset carry one of these two mutations on one allele and an Arg447His on the other. A mouse model for this disease has been developed [Sleat et al., 2004].
Management and treatment
Treatment is primarily supportive. Areas requiring attention include seizures that become uncontrollable with antiepileptic drug monotherapy. Single or combined use of valproic acid, clonazepam, and clorazepate is helpful, particularly in the early stages. There is also a role for phenobarbital, zonisamide, and levetiracetam. Gavage feeding becomes a necessity by age 5–7 years, when frequent pneumonias imply difficulty in swallowing and aspiration. Attention should be given to development of contractures. Bone marrow transplantation has been tried and has failed [Lake et al., 1997]. Gene and enzyme replacement strategies are being developed and soon may be tried in a developed mouse model. Gene replacement achieved by a number of strategically placed burr holes in three patients with advanced disease is being evaluated. Stem cell therapy also is being developed as a treatment option. It has been proposed that oral use of an anti-apoptotic drug such as flupirtine may slow the progression of this disease. The safety of this drug and its analgesic, antispasmodic, and weak antiepileptic effects make it particularly attractive [Dhar et al., 2002]. Its efficacy in LINCL remains unproven.
Variant Late Infantile Forms
Three genes have been described. The gene for the variant Finnish type, CLN5, was the first to be identified. This rare variant is mostly restricted to a region in Finland and has been found in 16 families, with one Swedish and one Dutch case also reported. Northern epilepsy and its gene, CLN8, were identified in cases from the northeast part of Finland. A subset of Turkish cases with variant LINCL are also caused by mutations in the CLN8 gene. The variant LINCL type referred to as Costa Rican/Portuguese or CLN6-deficient has been identified in patients of Venezuelan, Pakistani, and Indian descent, as well as in a case from the United States. It had been previously described as an early juvenile form, and it is also referred to as the Lake Cavanaugh variant [Gao et al., 2002; Savukoski et al., 1998; Wheeler et al., 2002].
Clinical description
For northern epilepsy or EPMR (i.e., CLN8-deficient form) [Ranta et al., 1999], the first stage of disease occurs from age 5 to puberty, and is characterized by frequent but short generalized tonic-clonic convulsions and complex partial seizures, as well as by cognitive decline to a low average level. After puberty, the second stage is notable for slowness of movement and a slowing of the rate of cognitive decline. In the final stage of the illness, seizures diminish in frequency, but mental dullness and cognitive decline lead to moderate mental retardation by the end of the third decade. This stage is also notable for clumsiness, ataxia, and impaired vision. Age at death varies from 17 years to late middle age.
For the Costa Rican/Portuguese vLINCL/Lake Cavanaugh variant (i.e., CLN6-deficient form) [Teixeira et al., 2003], the initial presenting symptoms at the age of 4 years are ataxia and speech difficulties after a period of normal development. Visual failure due to retinitis pigmentosa, myoclonic jerks, and seizures are accompanied by ataxia and intellectual decline. Death occurs in the early to middle teens.
Pathology
In cases of Finnish variant LINCL (i.e., CLN5-deficient form), the brain weighs about 500 g at postmortem examination. Most notable is the severe cerebellar atrophy. Findings are otherwise quite similar to those for classic LINCL. There is strong immunoreactivity to subunit C or 9 of mitochondrial ATP synthase and weak immunoreactivity to the saposins. Electron microscopic findings are notable for the presence of rectilinear profiles and curvilinear and fingerprint bodies [Goebel and Wisniewski, 2004; Goebel et al., 1999; Topcu et al., 2004; Wisniewski et al., 2004b].
For Turkish vLINCL or tLINCL (i.e., CLN8-deficient form), no pathology reports are available in the literature. The ultrastructure of skin fibroblasts reveals the presence of curvilinear, rectilinear, and fingerprint profiles [Topcu et al., 2004].
Genetics
For the Costa Rican/Portuguese vLINCL/Lake Cavanaugh variant (i.e., CLN6-deficient form), more than 19 mutations are recognized. The most prevalent are the nonsense mutation (c.214G>T) reported in 20 Costa Rican families and the 3-bp deletion (I154del) in 6 Portuguese families [Teixeira et al., 2003]. Other mutations have been described in those of Venezuelan, Indian, Pakistani, Greek, U.S., Trinidadian, or East Indian ancestry. The nclf mouse is a naturally occurring model for CLN6-deficient vLINCL [Mole, 2004].
For northern epilepsy or EPMR (i.e., CLN8-deficient form) and variant Turkish tLINCL, one mutation, R24G, accounts for all patients with northern epilepsy. The four other mutations result in the more severe phenotype of tLINCL. Two missense mutations (R204C and W263C) occur in exon 3, and two others, L16M and T170M, occur in exon 2 and are also missense mutations. R204C occurs in the conserved TLC lipid-sensing domain and predicts a potential role for CLN8 in the sphingolipid synthetic pathways. The mnd mouse is a naturally occurring mouse model for CLN8-deficient forms of neuronal ceroid-lipofuscinosis [Ranta et al., 2004].
Biochemistry and cell biology
CLN6 and CLN8 are transmembrane proteins that reside in the endoplasmic reticulum. The CLN5 protein has been reported as being a transmembrane protein in some papers and a secreted lysosomal glycoprotein in others [Holmberg et al., 2004; Savukoski et al., 1998]. CLN5 has been reported to co-immunoprecipitate with CLN2 protein and with CLN3 protein [Vesa et al., 2002]. This interaction implies that these are dynamic proteins that most likely exist in many subcellular locations and that they may functionally interact, forming a complex whereby one protein may substitute for the other. This finding can have great implications for therapy, because some of these proteins, such as CLN2, are soluble and amenable to protein or gene replacement therapy, whereas transmembrane proteins, such as CLN3, are not.
MFSD8/CLN7
Siintola et al. identified the novel gene, major facilitator superfamily (MFS) domain containing 8 (MFSD8), for Turkish vLINCL in 2007. The gene was identified after homozygosity mapping of 10 families, for which known human NCL loci and homologous genes (CLCN3 and CLCN7) causing NCL-like phenotypes in animal models were excluded by haplotype analysis [Siintola et al., 2007]. CLN7/MFSD8 mutations are now reported from India, the Netherlands, Italy, Czech Republic, Albania, and Greece [Kousi et al., 2009].
Clinical description
The age of presentation varies between 3 and 8 years. Patients present with delayed speech development, stereotypic hand movements, and sleep disorders. Later, myoclonic, clonic, and nocturnal seizures develop. These respond to medical therapy, then become refractory. In some patients, the presenting feature is epilepsy. Status epilepticus has been also reported. Progressive neurological decline and psychological symptoms of agitation and aggressiveness occur in late childhood. Axial akinesia, hypertonia, palilalia, and blindness ensue. Patients become disabled, cannot walk without support, and become wheelchair-bound at the end stages. Death usually occurs in late childhood but some patients survive till the second and third decade. Patients with missense mutation in exon 5 (c.362a>g /p.Tyr121Cys) were reported to have unaffected vision [Siintola et al., 2007; Stogmann et al., 2009].
Clinical diagnostic tests
Brain MRI reflects cerebellar and cerebral atrophy and increased signal intensity in white matter, with evidence of delayed white matter maturation [Siintola et al., 2007]. EEG documents slow background activity, multifocal epileptic discharges [Siintola et al., 2007], and occipital epileptiform activity [Aiello et al., 2009]. Eye ground examination reveals retinopathy and optic atrophy [Siintola et al., 2007].
Pathology
Peripheral blood lymphocytes show vacuoles. Skin biopsy reveals vacuoles with fingerprint profiles. Rectal biopsy contains neuronal curvilinear bodies [Siintola et al., 2007]. Electron microscopy performed in skin fibroblasts details fingerprint profiles, curvilinear bodies, and granular osmiophilic deposits [Aiello et al., 2009].
Biochemistry
The two identified missense mutations (p.Gly310Asp and p.Gly429Asp) change the highly conserved glycines that reside in the predicted eight and tenth transmembrane domains, respectively, to aspartic acids. Two of the mutations (c.697ArG and c.1102GrC) may change highly conserved amino acids (p.Arg233Gly and p.Asp368His, respectively), and may result in changes in the protein structure and/or function. Alternatively, they may affect splicing of the transcript, because they change sequences at the exon–intron junctions in the 3′ ends of exons 7 and 11, and thus may lead to production of abnormal mRNAs and/or proteins [Siintola et al., 2007].
Genetics
Analysis of genome-wide single nucleotide polymorphism (SNP) data in families with vLINCL revealed three regions on chromosomes 4, 8, and 15, with heterozygosity log of odds (HLOD) scores >2. Ninety known putative genes were identified. After excluding TRAM1L1 and TRPC3 by sequencing, six homozygous mutations in MFSD were identified. Homozygous nonsense mutation c.894TrG, in exon 10, creates a premature stop codon (p.Tyr298X) predicted to truncate the protein by 221 aa. Homozygous missense mutations c.929GrA (p.Gly310Asp) in exon 10 and c.1286GrA (p.Gly429Asp) in exon 12, respectively, affect amino acids that are conserved across vertebrates. Homozygous nucleotide changes at the exon–intron junctions may either change an amino acid or affect the splicing of the transcript. An arginine to glycine transition at the second-to-last nucleotide of exon 7 (c.697Arg; p.Arg233Gly) and a transversion of G to C at the last nucleotide of exon 11 (c.1102GrC; p.Asp368His) are two examples. Both Arg233 and Asp368 are conserved in vertebrates. Intronic mutation (c.754_2TrA) was identified in intron 8 [Siintola et al., 2007]. Other mutations reported are a missense mutation in exon 5 (c.362a>g/p.Y121C) [Stogmann et al., 2009], nonsense mutations (p.Arg35Stop, p.Glu381Stop, p.Arg482Stop), missense mutations (p.Met1Thr, p.Gly52Arg, p.Thr294Lys, p.Pro447Leu), splice site mutations (c.863+3_4insT, c.863+1G>C), 17-bp deletion predicting a frameshift and premature protein truncation (c.627_643del17/p.Met209IlefsX3) [Aiello et al., 2009], missense mutation in exon 12 (c.1398C>T) [Aldahmesh et al., 2009], and other mutations in Indian and European families [Kousi et al., 2009].
Juvenile Neuronal Ceroid-Lipofuscinosis or Juvenile Batten’s Disease
Although cases of JNCL (i.e., JNCL, Spielmeyer–Vogt–Batten–Mayou, CLN3-defective or deficient form) from all over the world have been described, there is a preponderance of cases with Northern European ancestry (i.e., Finland, Iceland, Norway, Sweden, Denmark, Germany, and Holland). There is a notable absence of African or Jewish cases. Japanese, Portuguese, Polish, British, Turkish, Moroccan, and Lebanese cases and cases from other countries have been described. It is the most prevalent type of neuronal ceroid-lipofuscinosis in the United States [Boustany, 1996]. JNCL was the first Batten variant to be recognized, and the gene responsible for it, CLN3, was the first to be cloned [Lerner et al., 1995]. Description of the first juvenile cases is credited to a Danish physician, Otto Christian Stengel [Stengel, 1826]. The genetic nature of the illness was established in the Norwegian family he described, who had four affected siblings. Because they were raised in different geographic areas by different family members, an environmental cause for the illness was eliminated.
Clinical description
Early development is normal. The first sign of trouble is decreased central vision caused by retinitis pigmentosa. This sets in between 4 and 6 years of age. These children are followed by ophthalmologists as normal children with retinitis pigmentosa. They ultimately are enrolled in schools for the visually impaired. Patients become completely blind between the ages of 10 and 14 years, but sometimes even later. Retinal findings include macular retinal pigment epithelium atrophy, pigment stippling, epiretinal membrane, bull’s eye maculopathy, retinitis with the appearance of peripheral bone spicules, and variable disk pallor [Hainsworth et al., 2009]. Complete blindness is accompanied by a disturbed sleep–wake cycle and insomnia. Retrospectively, a subset of affected children may manifest difficult behavior between the ages of 7 and 9 years. By age 10 years, cognitive decline sets in. The diagnosis is first suspected by teachers, who may be familiar with this condition in the pediatric visually impaired population. Seizures make their appearance as early as age 12 years, but they often do not occur until 14 years of age. Early-onset seizures that are difficult to control often foretell a more rapidly declining course. Speech becomes echolalic. Perseveration of speech and actions becomes routine. A cogwheel rigidity of the limbs sets in. Patients walk with a stooped, shuffling gait reminiscent of patients with Parkinson’s disease. An intention tremor of variable severity is often observed.
Clinical diagnostic tests
When clinical suspicion is strong, DNA-based CLN3 gene tests can confirm the diagnosis (see “Genetics”). The EEG is abnormal from age 9 years onward. Large-amplitude spike and slow-wave complexes are observed. CT and MRI scans may initially be normal and can remain so until age 12 years. Ultimately, cerebral atrophy with gaping sulci and large ventricles is the norm. Cerebellar atrophy is often present. Morphometric MRI measurements indicate loss of hemispheric, caudate, thalamic, and lenticular volumes [Boustany and Filipek, 1993]. There is a low signal in the white matter seen in T2-weighted images. Positron emission tomography (PET) has demonstrated decreased glucose use that starts in the calcarine area and progresses to involve all gray matter structures. The latter two techniques are not done routinely on patients, but when carried out, they can help to understand disease progression better. The ERG is often abnormal, even before the patients complain of decreased vision. Visual-evoked potentials reveal reduced-amplitude potentials, and somatosensory-evoked potentials are enhanced, but these are not particularly useful tests. The ultrastructure of the skin biopsy sample is often helpful, particularly if the common 1-kb deletion is absent from one or both alleles. Schwann cells, endothelial cells, pericytes, neurons, macrophages, and eccrine sweat glands all contain inclusions. Fingerprint-like inclusions enclosed by a unit membrane are typical. Curvilinear inclusions are frequently seen, sometimes within the same cell. Vacuolated lymphocytes are a hallmark of JNCL, but these have to be processed swiftly and correctly, otherwise the number of false-positive results becomes high. Unfortunately, very few diagnostic laboratories can accurately evaluate vacuolated lymphocytes. Skin fibroblast electron microscopy is a more robust test that has proved extremely helpful over the years.
Biochemistry, cell biology, and pathophysiology
An initial observation was that ceramide, the pro-apoptotic lipid second messenger, was elevated in JNCL brains [Puranam et al., 1999]. This elevation correlated with the identification of apoptosis in JNCL brains and anti-apoptotic amino acid stretches within the CLN3 protein [Persaud-Sawin et al., 2002]. In addition to ceramide, galactosyl-ceramide, glucosylceramide, ceramide trihexoside, and sphingomyelin levels are elevated, pointing to sphingolipid overproduction [Persaud-Sawin et al., 2004]. The CLN3 protein is upregulated in a number of human and mouse cancer cell lines and in solid colon cancer specimens [Rylova et al., 2002]. The CLN3 protein localizes to the Golgi, early recycling endosomes, and lipid rafts in plasma membranes. The VYFAE motif within the CLN3 protein is embedded in a larger galactosylceramide lipid raft binding domain. In CLN3-deficient cells, mutant CLN3 incorrectly localizes to late endosomes and lysosomes, and mutant CLN3 protein and galactosylceramide, an important component of lipid rafts, remain stuck in the Golgi, never reaching the plasma membrane and lipid rafts. Reversal of this after restoring CLN3 to the deficient cells suggests that CLN3 normally functions as a galactosylceramide transporter from the Golgi to lipid rafts by recycling endosomes. This may explain the increase in apoptosis that is often initiated from lipid rafts and the increased production of sphingolipids in an attempt to rectify the galactosylceramide deficiency in lipid rafts [Rusyn et al., 2008].
Management and treatment
JNCL is the most challenging of the clinical types to manage. Although initially seizure control is easily achieved with one drug, as the disease advances, some patients progress to having over 100 seizures per day despite use of a multitude of antiepileptic medications. The seizures are of mixed type, including generalized, myoclonic, and partial complex seizures. The emotional and psychiatric aspects of this disorder present a therapeutic dilemma. Many patients require antipsychotic drugs and mood stabilizers, which lower seizure threshold and aggravate parkinsonian symptoms. Insomnia is a problem that should be addressed with the use of benzodiazepines and other drugs. Weight loss becomes an issue in the final few years. It requires gastrostomy tubes for adequate provision of calories, liquids, and anticonvulsants. Anecdotal reports of the use of the anti-apoptotic medication flupirtine suggests improved seizure control and sleep patterns. Of those patients that develop bradycardia, only a handful have needed pacemakers placed. Nonconventional therapies based on findings in a small number of patients positive for GAD-65 and anti α-fetoprotein antibodies [Castaneda and Pearce, 2008] includes a trial of modulators of the immune system by a variety of ways, including oral prednisone and mycophenolate mofetil (Cellcept). The latter has been FDA-approved for human clinical trials [personal communication to R-MB, NCL Resource, UK]. Eight patients with JNCL positive for GAD65 antibodies were treated with oral prednisolone 0.75 mg/kg/day, maximum dose of 40 mg, for 10 days each month. Two had a significant increase in verbal IQ, alertness, and ability to move on days 3–4 of treatment. This effect lasted just a few days beyond therapy, only to reappear during the next month with treatment [Aberg et al., 2008].
CLN9-Deficient Juvenile-like Variant
Two German brothers and two American sisters of Serbian descent had been clinically diagnosed with the JNCL variant before identification of the CLN3 gene [Lin et al., 2001; Schulz et al., 2004]. When DNA from these cases was examined, it was determined that they had no defects in the CLN3 gene, and they had normal levels of CLN3 mRNA. Analysis of cDNA from CLN3-, CLN1-, CLN2-, and CLN6-deficient variants, together with cDNA from these unknown cases, using Affymetrix GeneChips, revealed a distinctive gene profile that grouped them together as a separate variant (Figure 41-1). Results of enzyme assays and molecular tests for all other known neuronal ceroid-lipofuscinosis variants were normal.
Pathology
The brain weight of the older brother at the time of death was 1140 g. Neurons were ballooned with fine, granular material. Large neurons remaining in the cerebral cortex and the deep gray structures were ballooned. The substantia nigra and nuclear thalami had moderate to severe astrogliosis, as did the spinal cord. The storage material was stained gray with Sudan black, and it had a yellow autofluorescence. The ultrastructure of neurons reveals GRODs and curvilinear bodies (Figure 41-2). Neurons also demonstrated immunoreactivity to subunit C or 9 of mitochondrial ATP synthase.
Management and treatment
Treatment is similar to that outlined for the JNCL variant. Flupirtine used empirically in the two sisters has led to stabilization of the course and better control of seizures. After the entire sphingolipid pathway and its perturbations are better defined for this variant, it will become possible to use drugs to correct defects in the pathway. CLN9 protein may be a regulator of dihydroceramide synthase. 4-hydroxyphenyl retinamide (4-HPR), an activator of the enzyme, could be developed as a treatment for CLN9-deficient patients [Schulz et al., 2006].
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Aberg L., Talling M., Härkönen T., et al. Intermittent prednisolone and autoantibodies to GAD65 in juvenile neuronal ceroid lipofuscinosis. Neurology. 2008;70(14):1218-1220.
Acampa M., Guideri F. Cardiac disease and Rett syndrome. Arch Dis Child. 2006;91:440-443.
Adaletli I., Omeroglu A., Kurugoglu S., et al. Lumbar and iliac artery aneurysms in Menkes’ disease: endovascular cover stent treatment of the lumbar artery aneurysm. Pediatr Radiol. 2005;35:1006-1009.
Adams P., Strand R., Bresnan M.J., et al. Kinky hair syndrome: Serial study of radiological findings with emphasis on the similarity to the battered child syndrome. Radiology. 1974;112:401-407.
Aiello C., Terracciano A., Simonati A., et al. Mutations in MFSD8/CLN7 are a frequent cause of variant-late infantile neuronal ceroid lipofuscinosis. Hum Mutat. 2009;30:E530-E540.
Akbarian S. The neurobiology of Rett syndrome. Neuroscientist. 2003;9:57-63.
Akbarian S., Chen R.Z., Gribnau J., et al. Expression pattern of the Rett syndrome gene MeCP2 in primate prefrontal cortex. Neurobiol Dis. 2001;87:84-91.
Aldahmesh M.A., Al-Hassnan Z.N., Aldosari M., et al. Neuronal ceroid lipofuscinosis caused by MFSD8 mutations: a common theme emerging. Neurogenetics. 2009;10:307-311.
Alpers B. Diffuse progressive degeneration of the gray matter of the cerebrum. Arch Neurol Psychiatry. 1931;25:469-505.
Amir R.E., Van den Veyver I.B., et al. Influence of mutation type and X chromosome inactivation on Rett syndrome phenotypes. Ann Neurol. 2000;47:670-679.
Amir R.E., Van den Veyver I.B., et al. Rett syndrome is caused by mutations in X-linked MeCP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185-188.
Ariani F., Hayek G., Rondinella D., et al. FOXG1 is responsible for the congenital variant of Rett syndrome. Am J Hum Genet. 2008;83:89-93.
Armstrong D., Dunn K., et al. Decreased dendritic branching in frontal, motor and limbic cortex in Rett syndrome compared with trisomy 21. J Neuropathol Exp Neurol. 1998;57:1013-1017.
Armstrong D.D. Rett syndrome: Neuropathology review. Brain Dev. 2000;23(Suppl 1):S72-S76.
Armstrong D.D. Review of Rett syndrome. J Neuropathol Exp Neurol. 1997;56:843-849.
Armstrong D.D., Dunn J.K., et al. Organ growth in Rett syndrome, a post mortem examination analysis. Pediatr Neurol. 1999;20:125-129.
Armstrong D.D., Dunn J.K., et al. Selective dendritic alterations in the cortex of Rett syndrome. J Neuropathol Exp Neurol. 1995;54:195-201.
Armstrong J., Pineda M., et al. Classic Rett syndrome in a boy as a result of a somatic mosaicism for a MECP2 mutation. Ann Neurol. 2001;50:692.
Artuso R., Mencarelli M.A., Polli R., et al. Early-onset seizure variant of Rett syndrome: Definition of the clinical diagnostic criteria. Brain Dev. 2010;32(1):17-24.
Assadi F., Crowe C., Rouhi O. Hyperkalemic distal renal tubular acidosis associated with Rett syndrome. Pediatr Nephrol. 2006;21:588-590.
Auranen M., Vanhala R., et al. MeCP2 gene analysis in classical Rett syndrome and in patients with Rett-like features. Neurology. 2001;56:611-617.
Bader G.G., Witt-Engerstrom I., et al. Neurophysiological findings in the Rett syndrome. II. Visual and auditory brainstem, middle and late evoked responses. Brain Dev. 1989;11:110-114.
Baldassarrea E., Capuanob G., Valentib G., et al. A case of massive gastric necrosis in a young girl with Rett Syndrome. Brain Dev. 2006;28:49-51.
Balestracci A., Caletti M.G., Missoni M. A case of Menkes’ disease with nephrocalcinosis and chronic renal failure. Pediatr Nephrol. 2009;24:1255-1256.
Ballas N., Lioy D.T., Grunseich C., et al. Non-cell autonomous influence of MeCP2-deficient glia on neuronal dendritic morphology. Nat Neurosci. 2009;12(3):311-317.
Bankier A. Menkes disease. J Med Genet. 1995;32:213-215.
Bao X., Wu Y., Wong L.J., et al. Alpers syndrome with prominent white matter changes. Brain Dev. 2008;30(4):295-300.
Barkovich A., Good W., et al. Mitochondrial disorders: Analysis of their clinical and imaging characteristics. Am J Neuroradiol. 1993;14:1119-1137.
Bashina V.M., Gorbachevskaya N.L., et al. Clinical, neurophysiological and differential diagnostic aspects of severe early childhood autism. J Neuropathol. 1994;4:68-71.
Batten F.E., Mayou M.S. Family cerebral degeneration with macular changes. Proc R Soc Med. 1915;8:70-90.
Bauman J.L., Kemper T.L., et al. Pervasive neuroanatomic abnormalities of the brain in three cases of Rett syndrome. Neurology. 1995;45:1581-1586.
Belichenko P.V., Oldfors A., et al. Rett syndrome: 3-D confocal microscopy of cortical pyramidal dendrites and afferents. Neuroreport. 1994;5:1509-1513.
Bennetts H., Chapman F. Copper deficiency in sheep in western Australia: A preliminary account of the aetiology of enzootic ataxia of lambs and anaemia of ewes. Aust Vet J. 1937;13:138-149.
Berg B. The neurologic examination. In: Berg B., editor. Child neurology: A clinical manual. Philadelphia: JB Lippincott; 1994:1-26.
Biaggioni I., Goldstein D., et al. Dopamine-beta-hydroxylase deficiency in humans. Neurology. 1990;40:370-373.
Bienvenu T., Carrie A., et al. MECP2 mutations account for most cases of typical forms of Rett syndrome. Hum Mol Genet. 2000;9:1377-1384.
Bird A.P., Wolfe A.P. Methylation-induced repression-belts, braces, and chromatin. Cell. 1999;99:451-454.
Blackwood W., Buxton P., et al. Diffuse cerebral degeneration in infancy (Alpers’ disease). Arch Dis Child. 1963;38:193-204.
Blue M.E., Naidu S., et al. Altered development of glutamate and GABA receptors in the basal ganglia of girls with Rett syndrome. Exp Neurol. 1999;156:345-352.
Borg I., Freude K., Kubart S., et al. Disruption of Netrin G1 by a balanced chromosome translocation in a girl with Rett syndrome. Eur J Hum Genet. 2005;13:921-927.
Boustany R.M. Batten disease or neuronal ceroid lipofuscinosis. In: Handbook of clinical neurology: Neurodystrophies and neurolipidoses. New York: Elsevier; 1996:671-900.
Boustany R.M., Filipek P. Seizures, depression and dementia in teenagers with Batten disease. J Inherit Metab Dis. 1993;16:252-255.
Budden S.S., Myer E.C., et al. Cerebrospinal fluid studies in the Rett syndrome: Biogenic amines and P-endorphins. Brain Dev. 1990;12:81-84.
Buoni S., Zannolli R., Felice C.D., et al. Drug-resistant epilepsy and epileptic phenotype-EEG association in MECP2 mutated Rett syndrome. Clin Neurophysiol. 2008;119(11):2455-2458.
Byers P., Siegel R., et al. X-linked cutis laxa: Defective cross-link formation in collagen due to decreased lysyl oxidase activity. N Engl J Med. 1980;303:61-65.
Capesius P., Van Damme W., et al. The metaphyseal lesions of the Menkes’ syndrome – a case report. J Belge Radiol. 1977;60:345-350.
Castaneda J.A., Pearce D.A. Identification of alpha-fetoprotein as an autoantigen in juvenile Batten disease. Neurobiol Dis. 2008;29:92-102.
Chae J.H., Hwang Y.S., et al. Mutation analysis of MECP2 and clinical characterization in Korean patients with Rett syndrome. J Child Neurol. 2002;17:33-36.
Chandler S.P., Guschin D., Landsberger N., et al. The methyl-CpG binding transcriptional repressor MeCP2 stably associates with nucleosomal DNA. Biochemistry. 1999;38:7008-7018.
Charles H., Lazeyras F., et al. Proton spectroscopy of human brain: Effects of age and sex. Prog Neuropsychopharmacol Biol Psychiatry. 1994;18:995-1004.
Cheadle J., Gill H., Fleming N., et al. Long-read sequence analysis of the MECP2 gene in Rett syndrome patients: Correlation of disease severity with mutation type and location. Hum Mol Genet. 2000;9:1119-1129.
Chelly J., Tumer Z., et al. Isolation of a candidate gene for Menkes disease that encodes a potential heavy mental binding protein. Nat Genet. 1993;3:14-19.
Chen R.Z., Akbaraian S., et al. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 2001;27:327-331.
Chiron C., Bulteau C., et al. Dopaminergic D2 receptor SPECT imaging in Rett syndrome: Increase of specific binding in striatum. J Nucl Med. 1993;34:1717-1721.
Cho S., Dawson P.E., et al. Role of palmitoyl-protein thioesterase in cell death: Implications for infantile neuronal ceroid lipofuscinosis. Eur J Paediatr Neurol. 2001;5(Suppl A):53-55.
Clayton-Smith J., Watson P., et al. Somatic mutation in MECP2 as a non-fatal neurodevelopment disorder in males. Lancet. 2000;356:830-832.
Coker S.B., Melnyk A.R. Rett syndrome and mitochondrial enzyme deficiencies. J Child Neurol. 1991;6:164-166.
Colantuoni C., Jeon O.H., Hyder K., et al. Gene expression profiling in postmortem Rett syndrome brain: Differential gene expression and patient classification. Neurobiol Dis. 2001;8:847-865.
Conforti F.L., Mazzei R., Magariello A., et al. Mutation analysis of the MECP2 gene in patients with Rett syndrome. Am J Med Genet A. 2003;117:184-187.
Cooper D.N., Youssoufian H. The CpG dinucleotide and human genetic disease. Hum Genet. 1988;78:151-155.
Cornford M.E. Neuropatholgoy of Rett syndrome: Case report with neuronal and mitochondrial abnormalities in the brain. J Child Neurol. 1994;9:424-431.
Couvert P., Bienvenu T., et al. MECP2 is highly mutated in X-liked mental retardation. Hum Mol Genet. 2001;10:941-946.
Coy J., Sedlacek Z., et al. A complex pattern of evolutionary conservation and alternative polyadenylation within the long 3-untranslated region of the methyl-CpG-binding protein 2 gene (MeCP2) suggests a regulatory role in gene expression. Hum Mol Genet. 1999;8:1253-1262.
d’Orsi G., Demaio V., Scarpelli F., et al. Central sleep apnoea in Rett syndrome. Neurol Sci. 2009;30:389-391.
d’Orsi G., Demaio V., Minervini M.G. Myoclonic status misdiagnosed as movement disorders in Rett syndrome: a video-polygraphic study. Epilepsy Behav. 2009;15(2):260-262.
Daly W., Rabinovitch H. Urologic abnormalities in Menkes’ syndrome. J Utol. 1981;126:262-264.
Danks D. Disorders of copper transport. In: Scriver C., Beaudet A., Sly W., et al, editors. The metabolic and molecular basis of inherited disease. New York: McGraw-Hill; 1995:2211-2235.
Danks D., Cartwright E., et al. Menkes’ kinky hair disease: Further definition of the defect in copper transport. Science. 1973;179:1140-1142.
Danks D., Stevens, et al. Menkes’ kinky hair syndrome. Lancet. 1972;1:1100-1102.
Das A.K., Becerra C.H.R., et al. Molecular genetics of palmitoyl-protein thioesterase deficiency in the U.S. J Clin Invest. 1998;102:361.
Das S., Levinson B., et al. Diverse mutations in patients with Menkes disease often leads to exon skipping. Am J Hum Genet. 1994;55:883-889.
Das S., Levinson B., et al. Similar splicing mutations of the Menkes/mottled copper-transporting ATPase gene in occipital horn syndrome and the blotchy mouse. Am J Hum Gen. 1995;56:570-576.
De Felice C., Ciccoli L., Leoncini S., et al. Systemic oxidative stress in classic Rett syndrome. Radic Biol Med. 2009;47(4):440-448.
Delarue A., Paut O., Guys J.M., et al. Inappropriate liver transplantation in a child with Alpers-Huttenlocher syndrome misdiagnosed as valproate-induced acute liver failure. Pediatr Transplant. 2000;4:67-71.
Devarakonda K.M., Lowthian D., Raghavendra T. A case of Rett syndrome with reduced pain sensitivity. Paediatr Anaesth. 2009;19(6):625-627.
Dhar S., Bitting R.L., et al. Flupirtine blocks apoptosis in Batten patient lymphoblasts and in human postmitotic CLN3- and CLN2-deficient neurons. Ann Neurol. 2002;51:448-466.
Dierick H., Ambrosini L., et al. Molecular structure of the Menkes disease gene (ATP7A). Genomics. 1995;28:462-469.
Dietrich O., Heiland S., et al. Noise correction for exact determination of apparent diffusion coefficients at low SNR. Magn Reson Med. 2001;45:448-453.
DiMauro S., Lombes, et al. Cytochrome c oxidase deficiency. Pediatr Res. 1990;28:536-541.
Dom R., Brucher J.M., et al. Adult ceroid-lipofuscinosis (Kufs disease) in two brothers: Retinal and visceral storage in one; diagnostic muscle biopsy in the other. Acta Neuropathol. 1979;45:67-72.
Downs J., Bebbington A., Woodhead H., et al. Early determinants of fractures in Rett syndrome. Pediatrics. 2008;121(3):540-546.
Downs J., Bergman A., Carter P., et al. Guidelines for management of scoliosis in Rett syndrome patients based on expert consensus and clinical evidence. Spine. 2009;34(17):E607-E617. (Phila Pa 1976)
Dreifuss F., Netsky M. Progressive poliodystrophy. Am J Dis Child. 1964;107:649-656.
Dunn H.G. Rett syndrome: Review of biological abnormalities. Can J Neurol Sci. 2001;28:16-29.
Dunn H.G., Stoessl A.J., et al. Rett syndrome: Investigation of nine patients, including PET scan. Can J Neurol Sci. 2002;29:345-357.
Egger J., Harding B., et al. Progressive neuronal degeneration of childhood (PNDC) with liver disease. Clin Pediatr. 1987;26:167-173.
Eipper B., Mains R., Glembotski C.C. Identification in pituitary tissue of a peptide alpha-amidation activity that acts on glycine extended peptides and requires molecular oxygen, copper, and ascorbic acid. Proc Natl Acad Sci USA. 1983;80:5144-8514.
Eipper B., Stoffers D., et al. The biosynthesis of neuropeptides. Peptide alpha-amidation. Annu Rev Neurosci. 1992;15:57-85.
Ellaway C., Peat J., et al. Medium-term open label trial of l-carnitine in Rett syndrome. Brain Dev. 2001;23(Suppl):S85-S89.
Ellison K.A., Fill C.P., et al. Examination of X chromosome markers in Rett syndrome: Exclusion mapping with a novel variation on multilocus linkage analysis. Am J Hum Genet. 1992;50:278-287.
Evans J.C., Archer H.L., Colley J.P., et al. Early onset seizures and Rett-like features associated with mutations in CDKL5. Eur J Hum Genet. 2005;13:1113-1120.
Fadic R., Russell J., et al. Sensory ataxic neuropathy as the presenting feature of a novel mitochondrial disease. Neurology. 1997;49:239-245.
Faerber E., Grover W., et al. Cerebral MR of Menkes’ kinky hair disease. Am J Neuroradiol. 1989;10:190-192.
Fanchetti E., Zappela M., et al. Plasma endorphins in Rett syndrome. Preliminary data. Am J Med Genet. 1986;24:331-338.
Fitzgerald P.M., Jankovic J., et al. Rett syndrome and associated movement disorders. Mov Disord. 1990;5:195-202.
Flemming K., Ulmer S., et al. MR spectroscopic findings in a case of Alpers-Huttenlocher syndrome. Am J Neuroradiol. 2002;23:1421-1423.
Forbes J., His G., et al. Role of the copper-binding domain in the copper transport function of ATP7B, the P-type ATPase defective in Wilson disease. J Biol Chem. 1999;274:12408-12413.
Ford F., Livingston S., et al. Familial degeneration of the cerebral gray matter in childhood, with convulsions, myoclonus, spasticity, cerebellar ataxia, choreoathetosis, dementia, and death in status epilepticus: Differentiation of infantile and juvenile types. J Pediatr. 1951;39:33-43.
Frydman M., Jager-Roman E., et al. Alpers progressive infantile neuronal poliodystrophy: An acute neonatal form with findings of the fetal akinesia syndrome. Am J Med Genet. 1993;47:31-36.
Fujii T., Ito M., et al. Biochemical study on the critical period for treatment of the mottled brindled mouse. J Neurol. 1990;55:885-889.
Fuks F., Hurd P., et al. The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J Biol Chem. 2003;278:4035-4040.
Gabreels F., Prick M., et al. Defects in citric acid cycle and the electron transport chain in progressive poliodystrophy. Acta Neurol Scand. 1984;70:145-154.
Gacheru S., McGee C., et al. Expression and accumulation of lysyl oxidase, elastin, and type 1 procollagen in human Menkes and mottle mouse fibroblasts. Arch Biochem Biophys. 1993;301:325-329.
Gao H., Boustany R.M., et al. Mutations in a novel CLN6 encoded transmembrane protein cause variant neuronal ceroid lipofuscinosis in mouse and man. Am J Hum Genet. 2002;70:324-335.
Geerdink N., Rotteveel J.J., et al. MECP2 mutation in a boy with severe neonatal encephalopathy: Clinical, neuropathological and molecular findings. Neuropediatrics. 2002;33:33-36.
Giunti L., Pelagatti S., et al. Spectrum and distribution of MECP2 mutations in 64 Italian Rett syndrome girls: Tentative genotype/ phenotype correlation. Brain Dev. 2001;23(Suppl 1):S242-S245.
Glaze D.G. Rett syndrome: Characterization of respiratory patterns and sleep. Ann Neurol. 1987;21:377-382.
Glaze D.G. Neurophysiology of Rett syndrome. J Child Neurol. 2005;2(9):740-746.
Glaze D.G., Percy A.K., Motil K.J., et al. A study of the treatment of Rett syndrome with folate and betaine. J Child Neurol. 2009;24(5):551-556.
Goebel H.H., Mole S.E., et al, editors. The neuronal ceroid lipofuscinoses (Batten disease). Burke, VA: IOS Press, 1999.
Goebel H.H., Wisniewski K.E. Current state of clinical and morphological features in human NCL. Brain Pathol. 2004;14:61-69.
Goka T., Stevenson R., et al. Menkes disease: A biochemical abnormality in cultured human fibroblasts. Proc Natl Acad Sci USA. 1976;73:604-606.
Gokcay A., Kitis O., et al. Proton MR spectroscopy in Rett syndrome. Comput Med Imaging Graph. 2002;26:271-275.
Goldstein D.S., Holmes C.S., Kaler S.G. Relative efficiencies of plasma catechol levels and ratios for neonatal diagnosis of Menkes dsease. Neurochem Res. 2009;34:1464-1468.
Goodyer I., Jones E., et al. Characterization of the Menkes protein copper-binding domains and their role in copper-induced protein relocalization. Hum Mol Genet. 1999;8:1473-1478.
Griffey M., Macauley S.L., Ogilvie J.M., et al. AAV2-mediated ocular gene therapy for infantile neuronal ceroid lipofuscinosis. Mol Ther. 2005;12:413-421.
Gunn T., McFarlane S., et al. Difficulties in the neonatal diagnosis of Menkes’ kinky hair syndrome-trichopoliodystrophy. Clin Pediatr. 1984;23:514-516.
Guy J., Hendrich B., et al. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet. 2001;27:322-326.
Hagberg B. Clinical manifestations and stages of Rett syndrome. Ment Retard Dev Disabil. 2002;8:61-65.
Hagberg B. Rett syndrome: Clinical peculiarities and biologic mysteries. Acta Paediatr. 1995;84:971-976.
Hagberg B., Aicardi J., et al. A progressive syndrome of autism, dementia, ataxia and loss of purposeful hand use in girls: Rett syndrome. Report of 35 cases. Ann Neurol. 1983;14:471-479.
Hagberg B., Gillberg C. Rett variants – retoid phenotypes. Clin Dev Med. 1993;127:40-60.
Hagberg B., Goutieres F., et al. Rett syndrome: Criteria for inclusion and exclusion. Brain Dev. 1985;7:372-373.
Hagberg B., Skjeldal O.H. Rett variants: A suggested model for inclusion criteria. Pediatr Neurol. 1994;11:5-11.
Hagberg B., Sourander P., et al. Late Infantile progressive encephalopathy with disturbed polyunsaturated fat metabolism. Acta Paediatr Scand. 1968;57:495-499.
Hainsworth D.P., Liu G.T., Hamm C.W., et al. Funduscopic and angiographic appearance in the neuronal ceroid lipofuscinoses. Retina. 2009;29:657-668.
Haldane J. The rate of spontaneous mutation of a human gene. J Genet. 1935;31:317-326.
Haltia M., Rapola J., et al. Infantile type of so-called neuronal ceroid lipofuscinosis. Part II. Histological and electron microscopic studies. Acta Neuropathol. 1973;26:157.
Haltia M., Rapola J., et al. Infantile type of so-called neuronal ceroid-lipofuscinosis. Part 2. Morphological and biochemical studies. J Neurol Sci. 1973;18:269.
Harding B. Progressive neuronal degeneration of childhood with liver disease (Alpers-Huttenlocher syndrome): A personal review. J Child Neurol. 1990;5:273-287.
Harding B., Egger J., et al. Progressive neuronal degeneration of childhood with liver disease. Brain. 1986;109:181-206.
Harding B.N., Alsanjari N., Smith S.J.M., et al. Progressive neuronal degeneration of childhood with liver disease (Alpers disease) presenting in young adults. J Neurol Neurosurg Psychiatry. 1995;58:320-325.
Harke H.J., Capitanio M., et al. Bladder diverticula and Menkes’ syndrome. Radiology. 1977;124:459-461.
Hasselmann Oa, Blau N., Ramaekers V.T., et al. Cerebral folate deficiency and CNS inflammatory markers in Alpers disease. Mol Genet Metab. 2009. Aug 22
Henikoff S. Chromosomes on the move. Trends Genet. 2001;17:689-690.
Heydorn K., Damsgaard E., et al. Extrahepatic storage of copper. A male foetus suspected of Menkes’ disease. Hum Genet. 1975;29:171-175.
Hoffbuhr K., Devaney J.M., et al. MeCP2 mutations in children with and without the phenotype of Rett syndrome. Neurology. 2001;56:1486-1495.
Holmberg V., Jalanko A., Isosomppi J., et al. The mouse ortholog of the neuronal ceroid lipofuscinosis CLN5 gene encodes a soluble lysosomal glycoprotein expressed in the developing brain. Neurobiol Dis. 2004;16:29-40.
Horn N.. Copper metabolism in Menkes’ disease. Rennert O., Chan W.Y., editors. Metabolism of trace elements in man. Genetic implications. Boca Raton, FL: CRC Press; 1984:25-51. vol II
Horn N., Tümer Z. Menkes disease and the occipital horn syndrome. In: Royce P.M., Steinmann B., editors. Connective Tissue and its Heritable Disorders: Molecular, Genetic, and Medical Aspects. ed 2. New York: John Wiley and Sons Inc; 2002:651-685.
Horská A., Farage L., Bibat G., et al. Brain metabolism in Rett syndrome: age, clinical, and genotype correlations. Ann Neurol. 2009;65(1):90-97.
Huppke P., Held M., Hanefield F., et al. Influence of mutation type and location on phenotype in 123 patients with Rett syndrome. Neuropediatrics. 2002;33:63-68.
Huppke P., Laccone F., et al. Rett syndrome: Analysis of MECP2 and clinical characterization of 31 patients. Hum Mol Genet. 2000;9:1369-1375.
Huttenlocher P., Solitare G., et al. Infantile diffuse cerebral degeneration with hepatic cirrhosis. Arch Neurol. 1976;33:186-192.
Ishikawa A., Goto T., et al. A new syndrome (?) of progressive psychomotor deterioration with peculiar stereotype movement and autistic tendency: A report of three cases. Brain Dev. 1978;3:258.
Ito H., Mori K., Sakata M., et al. Pathophysiology of the transient temporal lobe lesion in a patient with Menkes disease. Pediatr Int. 2008(6):825-827.
Itoh M., Takashima S. Neuropathology and immunohistochemistry of brains with Rett syndrome. No To Hattatsu. 2002;34:211-216.
Jan M.M., Dooley J.M., et al. Male Rett syndrome variant: Application of diagnostic criteria. Pediatr Neurol. 1999;20:238-240.
Janota I. Spongy degeneration of grey matter in three children: Neuropathological report. Arch Dis Child. 1974;49:571-575.
Jansky J. Dosud nepopsany pripad familiarni amazuroticke idiotie komplikovanem a hypoplasii mozeckovou. Sborn Lek. 1908;13:165-196.
Jellinger K., Armstrong D., et al. Neuropathology of Rett syndrome. Acta Neuropathol. 1988;76:142-158.
Jellinger K., Grisold W., et al. Peripheral nerve involvement in the Rett syndrome. Brain Dev. 1990;12:109-114.
Jellinger K.A. Rett syndrome – an update. J Neural Transm. 2003;110:681-701.
Johnsen D., Coleman L., et al. MR of progressive neurodegenerative change in treated Menkes’ kinky hair disease. Neuroradiology. 1991;33:181-182.
Johnson D.W., Speier S., et al. Role of subunit-9 of mitochondrial ATP synthase in Batten disease. Am J Med Genet. 1995;57:350-360.
Joshi C.N., Greenberg C.R., Mhanni A.A., et al. Ketogenic diet in Alpers-Huttenlocher syndrome. Pediatr Neurol. 2009;40:314-316.
Kaler S. Diagnosis and therapy of Menkes syndrome, a genetic form of copper deficiency. Am J Clin Nutr. 1998;67(Suppl):1029S-1034S.
Kaler S. Menkes disease. Adv Pediatr. 1994;41:262-303.
Kaler S., Goldstein D., et al. Plasma and cerebrospinal fluid neurochemical pattern in Menkes disease. Ann Neurol. 1993;33:171-175.
Kaler S., Miller R., et al. In utero treatment of Menkes disease [Abstract]. Pediatr Res. 1993;33:192A.
Kaler S., Westman J., et al. Gastrointestinal hemorrhage associated with gastric polyps in Menkes disease. J Pediatr. 1993;122:93-95.
Kaler S.G., Holmes C.S., Goldstein D.S., et al. Neonatal diagnosis and treatment of Menkes disease. N Engl J Med. 2008;358:605-614.
Kaludov N., Wolffe A. MeCP2 driven transcriptional repression in vitro: Selectivity for methylated DNA, action at a distance and contacts with the basal transcription machinery. Nucleic Acids Res. 2000;28:1921-1928.
Kasper D., et al. Loss of the chloride channel CLC-7 leads to lysosomal storage disease and neurodegeneration. EMBO J. 2005;24:1079-1091.
Kaufmann W.E., Naidu S., et al. Abnormal expression of microtubule-associated protein 2 (MAP-2) in neocortex in Rett syndrome. Neuropediatrics. 1995;26:109-113.
Kayihan N., Nennesomo I., et al. Fatal deterioration of neurological disease after orthotopic liver transplantation for valproic acid-induced liver damage. Pediatr Transplant. 2000;4:211-214.
Kendall B., Boyd S., et al. Progressive neuronal degeneration of childhood with liver disease. Computed tomographic features. Neuroradiology. 1987;29:174-180.
Kerr A.M., Nomura Y., et al. Guidelines for reporting clinical features in cases with MECP2 mutations. Brain Dev. 2001;23:208-211.
Kimura H., Shiota K. Methyl-CpG-binding protein, MeCP2, is a target molecule for maintenance DNA methyltransferase, Dnmt1. J Biol Chem. 2003;278:4806-4812.
Kitt C.A., Wilcox B. Preliminary evidence for neurodegenerative changes in the substantia nigra of Rett syndrome. Neuropediatrics. 1995;26:114-118.
Kleefstra T., Yntema H.G., et al. De novo MECP2 frameshift mutation in a boy with moderate retardation, obesity and gynaecomastia. Clin Genet. 2002;61:359-362.
Klein H., Dichgans J. Familiare juvenile glio-neurale Dystrophie: Akut beginnende progressive Encephalopathie mit rechtsseitigen occipito-parietalen Herdsymptomen und Status epilepticus. Arch Psychiatr Nervenkr. 1969;212:400-422.
Kodama H., Murata Y. Molecular genetics and pathophysiology of Menkes disease. Pediatr Int. 1999;41:430-435.
Kodama H., Sato E., et al. Biochemical indicator for evaluation of connective tissue abnormalities in Menkes’ disease. J Pediatr. 2003;142:726-728.
Kohan R., de Halac I.N., Tapia Anzolini V., et al. Palmitoyl Protein Thioesterase1 (PPT1) and Tripeptidyl Peptidase-I (TPP-I) are expressed in the human saliva. A reliable and non-invasive source for the diagnosis of infantile (CLN1) and late infantile (CLN2) neuronal ceroid lipofuscinoses. Clin Biochem. 2005;38:492-494.
Koslowski K., McCrossin R. Early osseous abnormalities in Menkes kinky hair syndrome. Pediatr Radiol. 1979;8:191-194.
Kousi M., Siintola E., Dvorakova L., et al. Mutations in CLN7/MFSD8 are a common cause of variant late-infantile neuronal ceroid lipofuscinosis. Brain. 2009;132:810-819.
Kozinetz C.A., Skender M.L., et al. Epidemiology of Rett syndrome: A population-based registry. Pediatrics. 1993;91:445-450.
Kriaucionis S., Bird A. DNA methylation and Rett syndrome. Hum Mol Genet. 2003;12:R221-R227.
Kudo S., Nomura Y., et al. Heterogeneity in residual function of MeCP2 carrying missense mutations in the methyl CpG binding domain. J Med Genet. 2003;40(7):487-493.
Laccone F., Zoll B., et al. MECP2 gene nucleotide changes and their pathogenicity in males: Proceed with caution. J Med Genet. 2002;39:586-588.
Lake B.D., Steward C.G., Oakhill A., et al. Bone marrow transplantation in late infantile Batten disease and juvenile Batten disease. Neuropediatrics. 1997;28:80-81.
Lappalainen R., Lindholm D., et al. Low levels of nerve growth factor in cerebrospinal fluid of children with Rett syndrome. J Child Neurol. 1997;11:296-300.
Lappalainen R., Liewendahl K., et al. Brain perfusion SPECT and EEG findings in Rett syndrome. Acta Neurol Scand. 1997;95:44-50.
Lee E.S., Ryoo J.W., Choi D.S., et al. Diffusion-weighted MR imaging of unusual white matter lesion in a patient with Menkes disease. Korean J Radiol. 2007;8:82-85.
Lekman A., Hagberg B., et al. Membrane cerebral lipids in Rett syndrome. Pediatr Neurol. 1991;7:186-190.
Lekman A., Witt-Engerstrom I., et al. CSF and urine biogenic amine metabolites in Rett syndrome. Clin Genet. 1990;37:173-178.
Leonard H., Colvin L., Christodoulou J., et al. Patients with the R133C mutation: Is their phenotype different from patients with Rett syndrome with other mutations? J Med Genet. 2003;40:E52.
Lerner T.J., Boustant R.M., et al. Isolation of a novel gene underlying Batten disease, CLN3. Cell. 1995;82:949-957.
Levinson B., Vulpe C., et al. The mottled gene is the mouse homologue of the Menkes disease gene. Nat Genet. 1994;6:369-373.
Lewis J.D., Meehan R.R., et al. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell. 1992;69:843-849.
Lin S.M., Dhar S., et al. Extracting knowledge from gene expression data: A case study of Batten Disease. In: Proceedings of Data Mining in Bioinformatics. San Francisco, CA: Association for Computing Machinery–Knowledge Discovery and Data Mining (ACM-KDD); 2001.
Lipani J.D., Bhattacharjee M.B., et al. Reduced nerve growth factor in Rett syndrome postmortem brain tissue. J Neuropathol Exp Neurol. 2000;59:889-895.
Liu P.C., Chen Y.W., Centeno J.A., et al. Downregulation of myelination, energy, and translational genes in Menkes disease brain. Mol Genet Metab. 2005;85:291-300.
Liu Y., Wada R., et al. A genetic model of substrate deprivation therapy for a glycosphingolipid storage disorder. J Clin Invest. 1999;103:439-440.
Lockitch G., Halstead A., et al. Age- and sex-specific pediatric reference intervals and correlations for zinc, copper, selenium, iron, vitamins A and E and related proteins. Clin Chem. 1988;34:1625-1628.
Lockitch G., McCallum C., et al. Reference intervals for eight plasma proteins in preterm and term newborns by laser nephelometry [Letter]. Clin Chem. 1986;32:2106.
Luikenhuis S., Giacometti E., et al. Expression of MeCP2 in postmitotic neurons rescues Rett syndrome in mice. Proc Natl Acad Sci USA. 2004;101:6033-6038.
Maiwald R., Bonte A., et al. De novo MECP2 mutation in a 46, XX male patient with Rett syndrome. Neurogenetics. 2002;4:107-108.
Marin-Padilla M. Cajal-Retzius cells and the development of the neocortex. Trends Neurosci. 1998;21:64-71.
Marshall F.J., de Blieck E.A., Mink J.W., et al. A clinical rating scale for Batten disease: reliable and relevant for clinical trials. Neurology. 2005;65:275-279.
Martinez-Mena J., Manquillo A., Saez J, et al. Neurophysiological study in Alpers syndrome [in Spanish]. Rev Neurol. 1998;26:70-77.
Matsuishi T., Nagamitsu S., et al. Decreased cerebrospinal fluid levels of substance P in patients with Rett syndrome. Ann Neurol. 1997;42:978-981.
Matsuo M., Tasaki R., Kodama H., et al. Screening for Menkes disease using the urine HVA/VMA ratio. J Inherit Metab Dis. 2005;28:89-93.
Mayou M.S. Cerebral degeneration with symmetrical changes in the maculae in three members of a family. Trans Ophthalmol Soc UK. 1904;24:142-145.
Menkes J., Alter M., et al. A sex-linked recessive disorder with retardation of growth, peculiar hair and focal cerebellar degeneration. Pediatrics. 1962;29:764-769.
Mercer J. Menkes syndrome and animal models. Am J Clin Nutr. 1998;67(Suppl):1022S-1028S.
Mercer J., Grimers A., et al. Mutations in the murine homologue of the Menkes disease gene in dappled and blotchy mice. Nat Genet. 1994;6:374-378.
Mercer J., Livingston J., et al. Isolation of a partial candidate gene for Menkes disease by positional cloning. Nat Genet. 1993;3:20-25.
Miao N., Levin S.W., Baker E.H., et al. Children with infantile neuronal ceroid lipofuscinosis have an increased risk of hypothermia and bradycardia during anesthesia. Anesth Analg. 2009;109:372-378.
Mitchison H.M., Hofmann S.L., Becerra C.H., et al. Mutations in the palmitoyl-protein thioesterase gene (PPT; CLN1) causing juvenile neuronal ceroid lipofuscinosis with granular osmiophilic deposits. Hum Mol Genet. 1998;7:291-297.
Mole S.E. The genetic spectrum of human neuronal ceroid-lipofuscinoses. Brain Pathol. 2004;14:70-76.
Mole S.E., Mitchison H.M., et al. Molecular basis of the neuronal ceroid lipofuscinoses: Mutations in CLN1, CLN2, CLN3, and CLN5. Hum Mutat. 1999;14:199-215.
Mole S.E., Williams R.E., Goebel H.H., editors. The Neuronal Ceroid Lipofuscinoses (Batten Disease), ed 2, Oxford University Press, 2010. In preparation
Møller L.B., Mogensen M., Horn N. Molecular diagnosis of Menkes disease: Genotype–phenotype correlation. Biochimie. 2009;91:1273-1277.
Møller L., Tumer Z., et al. Similar splice-site mutations of the ATP7A gene lead to different phenotypes: Classical Menkes disease or occipital horn syndrome. Am J Hum Genet. 2000;66:1211-1220.
Monros E., Armstrong J., et al. Rett syndrome in Spain: Mutation analysis and clinical correlations. Brain Dev. 2001;23(Suppl 1):S251-S253.
Montine T., Powers J., et al. Alpers’ syndrome presenting with seizures and multiple stroke-like episodes in a 17-year-old male. Clin Neuropathol. 1995;14:322-326.
Moore C., Howell R. Ectodermal manifestation in Menkes’ disease. Clin Genet. 1985;28:532-537.
Motil K., Schultz R., et al. Oropharyngeal dysfunction and gastroesophageal dysmotility are present in girls and women with Rett syndrome. J Pediatr Gastroenterol Nutr. 1999;29:31-37.
Nan X., Campoy F.J., et al. McCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell. 1997;88:471-481.
Nan X., Meehan R.R., et al. Dissection of the methyl-CpG binding domain from the chromosomal protein McCP2. Nucleic Acids Res. 1993;21:4886-4892.
Nan X., Ng H., et al. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386-389.
Nan X., Tate P., et al. DNA methylation specifies chromosomal localization of MeCP2. Mol Cell Biol. 1996;16:414-421.
Narkewicz M., Sokol R., et al. Liver involvement in Alpers disease. J Pediatr. 1991;119:260-267.
Naviaux R., Nguyen K. POLG mutations associated with Alpers’ syndrome and mitochondrial DNA depletion. Ann Neurol. 2004;55:706-712.
Naviaux R., Nyhan W., et al. Mitochondrial DNA polymerase gamma deficiency and mtDNA depletion in a child with Alpers’ syndrome. Ann Neurol. 1999;45:54-58.
Neul J.L., Zoghbi Y. Rett syndrome: A prototypical neurodevelopmental disorder. Neuroscientist. 2004;10:118-128.
Neumann-Haefelin T., Wittask H., et al. Diffusion- and perfusion-weighted MRI: Influence of severe carotid artery stenosis on the DWI/ PWI mismatch in acute stroke. Stroke. 2000;31:1311-1317.
Nielsen J.B., Henriksen K.F., et al. MECP2 mutations in Danish patients with Rett syndrome: High frequency of mutations but no consistent correlations with clinical severity or with the X chromosome inactivation pattern. Eur J Hum Genet. 2001;9:178-184.
Nielson J.B., Toft P.B., et al. Cerebral magnetic resonance spectroscopy in Rett syndrome. Failure to detect mitochondrial disorder. Brain Dev. 1993;15:107-112.
Obata K., Matsuishi T., et al. Mutation analysis of the methyl-CpG biding protein 2 gene (MECP2) in patients with Rett syndrome. J Med Genet. 2000;37:608-610.
Odermatt A., Suter H., Krapf R., et al. Primary structure of two P-type ATPases involved in copper homeostasis in Enterococcus hirae. J Biol Chem. 1993;268:12775-12779.
Oldfors A., Hagberg B., et al. Rett syndrome: Spinal cord pathology. Pediatr Neurol. 1988;4:172-174.
Oldfors A., Sourander P., et al. Rett syndrome: Cerebellar pathology. Pediatr Neurol. 1990;6:310-331.
Ozawa H., Kodama H., Kawaguchi H., et al. Renal function in patients with Menkes disease. Eur J Pediatr. 2003;162:51-52.
Papadimitriou J.M., Hockey A., Tan N., et al. Rett syndrome: Abnormal membrane-bound lamellated inclusions in neurons and oligodendroglia. Am J Med Genet. 1988;29:365-368.
Parsons M., Li T., et al. Combined (1)H MR spectroscopy and diffusion-weighted MRI improves the prediction of stroke outcome. Neurology. 2000;55:498-505.
Payne A., Gitlin J. Functional expression of the Menkes disease protein reveals common biochemical mechanisms among the copper-transporting P-type ATPases. J Biol Chem. 1998;273:3765-3770.
Paynter J., Grimers A., et al. Expression of the Menkes gene homologue in mouse tissue: Lack of effect of copper on the mRNA levels. FEBS Lett. 1994;351:186-190.
Pederson P., Carafoli E. Ion motive ATPases. I. Ubiquity, properties, and significance to the cell function. Trends Biochem Sci. 1987;12:146-150.
Pelc K., Dan B. Postural cortical myoclonus during gait in Rett syndrome. Epilepsy Behav. 2009;16(1):188.
Percy A.K. Clinical trials and treatment prospects. Ment Retard Dev Disabil. 2002;8:106-111.
Percy A.K., Gillberg C., et al. Rett syndrome and the autistic disorders. Neurol Clin. 1990;8:659-676.
Persaud-Sawin D.A., McNamara J.2nd, Rylova S., et al. A galactosylceramide binding motif is involved in trafficking of CLN3 from Golgi to rafts via recycling endosomes. Pediatr Res. 2004;56:449-463.
Persaud-Sawin D.A., VanDongen A., et al. Motifs within the CLN3 protein: Modulation of cell growth rates and apoptosis. Hum Mol Genet. 2002;11:2129-2142.
Petris M., Mercer J. The Menkes protein (ATP7A; MNK) cycles via the plasma membrane both in basal and elevated extracellular copper using a C-terminal di-leucine endocytic signal. Hum Mol Genet. 1999;8:2107-2115.
Petris M., Mercer J., et al. Ligand-regulated transport of the Menkes copper P-type ATPase efflux pump from the Golgi apparatus to the plasma membrane: A novel mechanism of regulated trafficking. EMBO J. 1996;15:6084-6095.
Poulsen L., Horn N., et al. X-linked recessive Menkes disease: Identification of a partial gene deletions in affected males. Clin Genet. 2002;62:449-457.
Prick M., Gabreels F., et al. Pyruvate dehydrogenase deficiency restricted to the brain. Neurology. 1981;31:398-404.
Procopis P., Camakaris J., et al. A mild form of Menkes syndrome. J Pediatr. 1981;98:97-100.
Prohaska J., Bailey W. Alterations of peptidylglycine alpha-amidating monooxygenase and other cuproenzyme activities following perinatal copper deficiency. Proc Soc Exp Biol Med. 1995;210:107-116.
Prohaska J., Bailey W. Regional specificity in alterations of rat brain copper and catecholamines following perinatal copper deficiency. J Neurochem. 1994;63:1551-1557.
Puranam K., Lane S.C., et al. Overexpression of Bcl–2 is associated with apoptosis in neuronal death. J Cell Biochem. 1995;19(Suppl B):318.
Puranam K., Qian W.H., et al. Upregulation of Bcl-2 and elevation of ceramide in Batten disease. Neuropediatrics. 1997;28:37-41.
Puranam K.L., Guo W.X., et al. CLN3 defines a novel antiapoptotic pathway operative in neurodegeneration and mediated by ceramide. Mol Genet Metab. 1999;66:294-308.
Rae T., Schmidt P., et al. Undetectable intracellular free copper: The requirement of a copper chaperone for superoxide dismutase. Science. 1999;184:805-808.
Ranta S., Savukoski M., et al. Studies of homogenous populations: CLN5 and CLN8. Adv Genet. 2001;45:123-140.
Ranta S., Topcu M., et al. Variant late infantile neuronal ceroid lipofuscinosis in a subset of Turkish patients is allelic to Northern epilepsy. Hum Mutat. 2004;23:300-3005.
Ranta S., Zhang Y., et al. The neuronal ceroid lipofuscinoses in human EPMR and mnd mutant mice are associated with mutations in CLN8. Nat Genet. 1999;23:233-236.
Rasmussen M., Sanengen T., Skullerud K., et al. Evidence that Alpers-Huttenlocher syndrome could be a mitochondrial disease. J Child Neurol. 2000;15:473-477.
Ravn K., Nielsen J.B., Uldall P., et al. No correlation between phenotype and genotype in boys with a truncating MECP2 mutation. J Med Genet. 2003;40:E5.
Reaume A., Elliott J., Hoffman E.K., et al. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Genet. 1996;13:43-47.
Reichelt K.L., Skjeldal O. IgA antibodies in Rett syndrome. Autism. 2006;10(2):189-197.
Reiss A.L., Farugue F., et al. Neuroanatomy of Rett syndrome: A volumetric imaging study. Ann Neurol. 1993;34:227-234.
Rett A. Uber ein eigenartiges hirnatrophisches Syndrom bei Hyperammonamie im Kindesalter. Wien Klin Wochenschr. 1966;116:723-725.
Robertson D., Goldberg M., et al. Isolated failure of autonomic noradrenergic neurotransmission. N Engl J Med. 1986;314:843-849.
Robinson B. Lactic academia. In: Scriver C., Beaudet A., Sly W., et al, editors. The metabolic and molecular basis of inherited disease. New York: McGraw-Hill; 1989:879-882.
Robinson B., De Meirleir L., et al. Clinical presentation of patients with mitochondrial respiratory chain defects in NADH-coenzyme Q reductase and cytochrome oxidase: Clues to the pathogenesis of Leigh disease. J Pediatr. 1987;110:216-222.
Robinson D., Cooley L. Genetic analysis of the actin cytoskeleton in the Drosophila ovary. Annu Rev Cell Biol. 1997;13:147-170.
Rohmer A., Krug J., et al. Maladie de Menkes: Etude de deux enzymes cupro-dependants. Pediatrie. 1977;32:447-456.
Rossi L., DeMartino A., et al. Neurodegeneration in the animal model of Menkes’ disease involves Bcl-2-linked apoptosis. Neuroscience. 2001;103:181-188.
Royce P., Steinmann B. Markedly reduced activity of lysyl oxidase in skin and aorta from a patient with Menkes disease showing unusually severe connective tissue manifestations. Pediatr Res. 1990;28:137-141.
Ruch A., Kurczynski T.W., et al. Mitochondrial alterations in Rett syndrome. Pediatr Neurol. 1989;5:320-323.
Rusyn E., Mousallem T., Persaud-Sawin D., et al. CLN3p impacts Galactosylceramide Transport, Raft Morphology and Lipid Content. Pediatr Res. 2008;63(6):625-631.
Rylova S.N., Amalfitano A., et al. The CLN3 gene is a novel molecular target for cancer drug discovery. Cancer Res. 2002;62:801-808.
Sandbank U., Lerman P. Progressive cerebral poliodystrophy—Alpers’ disease. Disorganized giant neuronal mitochondria on electron microscopy. J Neurol Neurosurg Psychiatry. 1972;35:749-755.
Santavuori P., Haltia M., et al. Infantile type of so-called neuronal ceroid-lipofuscinosis: Part 1, a clinical study of 15 patients. J Neurol Sci. 1973;18:257-267.
Satoi M., Matsuishi T., et al. Decreased cerebrospinal fluid levels of beta-phenylethylamine in patients with Rett syndrome. Ann Neurol. 2000;47:801-803.
Savukoski M., Klockars T., et al. CLN5, a novel gene encoding a putative transmembrane protein mutated in Finnish variant late infantile neuronal ceroid lipofuscinosis. Nat Genet. 1998;19:286-288.
Scala E., Ariani F., Mari F., et al. CDKL5/STK9 is mutated in Rett syndrome variant with infantile spasms. J Med Genet. 2005;42:103-107.
Schriner J.E., Yi W., et al. cDNA and genomic cloning of human palmitoyl-protein thioesterase (PPT), the enzyme defective in infantile neuronal ceroid lipofuscinosis [published erratum appears in Genomics 1996;38:458]. Genomics. 1996;34:317.
Schultz R.J., Glaze D.G., et al. The pattern of growth failure in Rett syndrome. Am J Dis Child. 1993;147:663-667.
Schulz A., Dhar S., et al. Impaired cell adhesion and apoptosis in the novel CLN9 Batten Disease variant. Ann Neurol. 2004;56:342-350.
Schulz A., Mousallem T., Venkataramani M., et al. The CLN9 protein, a regulator of dihydroceramide synthase. J Biol Chem. 2006;281:2784-2794.
Schwartzman J.S., Bernardino A., et al. Rett syndrome in a boy with a 47,XXY karyotype confirmed by a rare mutation in the MECP2 gene. Neuropediatrics. 2001;32:164.
Seidel J., Moller L., et al. Disturbed copper transport in humans. Part 1. Mutations of the ATP7A gene lead to Menkes disease and occipital horn syndrome. Cell Mol Biol. 2001;47:141-148.
Sekul E.A., Percy A.K. Rett syndrome: Clinical features, genetic considerations, and the search for a biological marker. Curr Neurol. 1992;12:173-200.
Shahbazian M., Sun Y., et al. Balanced X chromosome inactivation patterns in the Rett syndrome brain. Am J Med Genet. 2002;111:164-168.
Shahbazian M., Young J., Yuva-Paylor L., et al. Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron. 2002;35:243-254.
Shibata N., Hirano A., et al. Cerebellar superoxide dismutase expression in Menkes kinky hair disease: An immunohistochemical investigation. Acta Neuropathol. 1995;90:198-202.
Siegel R.C. Lysyl oxidase. Int Rev Connect Tissue Res. 1979;8:73-118.
Siintola E., Topcu M., Aula N., et al. The novel neuronal ceroid lipofuscinosis gene MFSD8 encodes a putative lysosomal transporter. Am J Hum Genet. 2007;81:136-146.
Silver S., Nucifora G., et al. Bacterial resistance ATPases: Primary pumps for exporting toxic cations and anions. Trends Biochem Sci. 1989;14:76-80.
Simonati A., Filostro M., Tomelleri G., et al. Central-peripheral sensory axonopathy in a juvenile case of Alpers-Huttenlocher disease. J Neurol. 2003;250:702-706.
Sirianni N., Naidu S., et al. Rett syndrome: Confirmation of X-linked dominant inheritance, and localization of the gene to Xq28. Am J Hum Gen. 1998;63:1552-1558.
Sleat D., Wiseman J., et al. A mouse model of classical late-infantile neuronal ceroid lipofuscinosis based on targeted disruption of the CLN2 gene results in a loss of tripeptidyl-peptidase I activity and progressive neurodegeneration. J Neurosci. 2004;24:9117-9126.
Sleat D.E., Donnelly R.J., et al. Associations of mutations in a lysosomal protein with classical late-infantile neuronal ceroid lipofuscinosis. Science. 1997;277:1802-1805.
Smith J., Mah J., et al. Brain MR imaging findings in two patients with Alpers’ syndrome. Clin Imaging. 1996;20:235-237.
Sofic E., Riederer P., Killian W., et al. Reduced concentration of ascorbic acid and glutathione in a single case of Rett syndrome: A postmortem brain study. Brain Dev. 1987;9:529-532.
Solioz M., Odermatt A., et al. Copper pumping ATPases: Common concepts in bacteria and man. FEBS Lett. 1994;346:44-47.
Spielmeyer W. Familiare amaurotische Idiotie. Zentralbl Gesamte Ophthalmol. 1923;10:161-208.
Stanley P., Gwinn J., et al. The osseous abnormalities in Menkes syndrome. Ann Radiol. 1976;19:167-172.
Stengel E. Beretning om et maerkeligt Sygdomstilfoelde hos fire Sodskende i Naerheden af Roraas. Eyr Med Tidskr. 1826;1:347-352.
Stogmann E., El Tawil S., Wagenstaller J., et al. A novel mutation in the MFSD8 gene in late infantile neuronal ceroid lipofuscinosis. Neurogenetics. 2009;10:73-77.
Strausak D., La Fontaine S., Hill J., et al. The role of GMXCXXC metal binding sites in the copper-induced redistribution of the Menkes protein. J Biol Chem. 1999;274:11170-11177.
Subramaniam B., Naidu S., et al. Neuroanatomy in Rett syndrome: Cerebral cortex and posterior fossa. Neurology. 1997;48:399-407.
Tang. Murine cathepsin F deficiency causes neuronal lipofuscinosis and late-onset neurological disease. Mol Cell Biol. 2006;26:2309-2316.
Teixeira C.A., Espinola J., et al. Novel mutations in the CLN6 gene causing a variant late infantile neuronal ceroid lipofuscinosis. Hum Mutat. 2003;21:502-508.
Tesarova M., Mayr J., et al. Mitochondrial DNA depletion in Alpers syndrome. Neuropediatrics. 2004;35:217-223.
Topcu M., Akyerli C., et al. Somatic mosaicism for a MECP2 mutation associated with classic Rett syndrome in a boy. Eur J Hum Genet. 2002;10:77-81.
Topcu M., Tan H., et al. Evaluation of 36 patients from Turkey with neuronal ceroid lipofuscinosis: Clinical, neurophysiological, neuroradiological and histopathologic studies. Turk J Pediatr. 2004;46:1-10.
Trevathan E., Moser H.W. Diagnostic criteria for Rett syndrome. The Rett Syndrome Diagnostic Criteria Work Group. Ann Neurol. 1988;23:425-428.
Tsai S. Lithium and antidepressants: Potential agents for the treatment of Rett syndrome. Med Hypotheses. 2006;67:626-629.
Tsivkovskii R., Eisses J., et al. Functional properties of the copper-transporting ATPase ATP7B (the Wilson’s disease protein) expressed in insect cells. J Biol Chem. 2002;277:976-983.
Tudor M., Akbarian S., et al. Transcriptional profiling of a mouse model for Rett syndrome reveals subtle transcriptional changes in the brain. Proc Natl Acad Sci USA. 2002;99:15536-15541.
Tumer Z., Lund C., et al. Identification of point mutations in 41 unrelated patients affected with Menkes disease. Am J Hum Genet. 1997;60:63-71.
Tumer Z., Moller L., et al. Mutation spectrum of ATP7A, the gene defective in Menkes disease. Adv Exp Med Biol. 1999;448:83-95.
Tumer Z., Moller L., et al. Screening of 383 unrelated patients affected with Menkes disease and findings of 57 gross deletions in ATP7A. Hum Mutat. 2003;22:457-464.
Tümer Z., Møller L.B. Menkes disease. Eur J Hum Genet. 2009:1-8.
Tyynela J., Palmer D.N., et al. Storage of saposins A and D in infantile neuronal ceroid-lipofuscinosis. Fed Eur Biochem Soc. 1993;330(1):8-12.
Van den Veyver I.B., Zoghbi H.Y. Genetic basis of Rett syndrome. Ment Retard Dev Disabil Res Rev. 2002;8:82-86.
Van Goethem G., Martin J., et al. Progressive external ophthalmoplegia and multiple mitochondrial DNA deletions. Acta Neurol Belg. 2002;102:39-42.
Van Goethem G., Martin J., Van Broeckhoven C. Progressive external ophthalmoplegia characterized by multiple deletions of mitochondrial DNA: Unraveling the pathogenesis of human mitochondrial DNA instability and the initiation of a genetic classification. Neuromol Med. 2003;3:129-146.
Veit-Sauca B., Cambonie G., Salloum R., et al. A moderate intrauterine growth delay with lethal outcome: neonatal Menkes disease. Arch Pediatr. 2009;16(1):41-45. Epub 2008 Nov 28
Venta-Sobero J.A., Porras-Kattz E., et al. West syndrome as an epileptic presentation in Menkes’ disease. Rev Neurol. 2004;39:133-136.
Vesa J., Chin M.H., et al. Neuronal ceroid lipofuscinoses are connected at molecular level: Interaction of CLN5 protein with CLN2 and CLN3. Mol Biol Cell. 2002;13:2410-2420.
Villard L., Kpebe A., et al. Two affected boys in a Rett syndrome family: Clinical and molecular findings. Neurology. 2000;55:1188-1193.
Vogt H. Familiare amaurotische Idiotie, histologische und histopathologische Studien. Arch Kinderheilkd. 1909;51:1-35.
Voskoboinik I., Camakaris J. Menkes copper-translocating P-type ATPases (ATP7A): Biochemical and cell biology properties, and role in Menkes disease. J Bioenerg Biomembr. 2002;34:363-371.
Voskoboinik I., Greenough M., et al. Functional studies on the Wilson copper P-type ATPase and toxic milk mouse mutant. Biochem Biophys Res Commun. 2001;281:966-970.
Vulpe C., Levinson B., et al. Isolation of a candidate gene for Menkes disease and evidence that it encodes a copper-transporting ATPase. Nat Genet. 1993;3:7-13.
Wakia S., Kameda I., et al. Rett syndrome: Findings suggesting axonopathy and mitochondrial abnormalities. Pediatr Neurol. 1990;6:339-343.
Wan M., Lee S.S., et al. Rett syndrome and beyond: Recurrent spontaneous and familial MECP2 mutations at CpG hotspots. Am J Hum Genet. 1999;65:1520-1529.
Watson P., Black G., et al. Angelman syndrome phenotype associated with mutations in MECP2, a gene encoding a methyl CpG binding protein. J Med Genet. 2001;38:224-228.
Weaving L.S., Williamson S.L., Bennetts B., et al. Effects of MECP2 mutation type, location and X-inactivation in modulating Rett syndrome phenotype. Am J Med Genet A. 2003;118:103-114.
Wefring K., Lamvik J. Familial progressive poliodystrophy with cirrhosis of the liver. Acta Paediatr Scand. 1967;56:295-300.
Wenk G.L. Alterations in dopaminergic function in Rett syndrome. Neuropediatrics. 1995;26:123-125.
Wenk G.L., Hauss-Wgrzyniak B. Altered cholinergic function in the basal forebrain of girls with Rett syndrome. Neuropediatrics. 1999;30:125-129.
Wheeler R.B., Sharp J.D., Schultz R.A., et al. The gene mutated in variant late-infantile neuronal ceroid lipofuscinosis (CLN6) and in nclf mutant mice encodes a novel predicted transmembrane protein. Am J Hum Genet. 2002;70:537-542.
Williams A., Sekaninova S., Coakley J. Suppression of elevated alanine aminotransferase activity in liver disease by vigabatrin. J Paediatr Child Health. 1998;34:395-397.
Williams D., Atkins C. Tissue copper concentrations of patients with Menkes’ kinky hair disease. Am J Dis Child. 1981;135:375-376.
Wisniewski K., Golabek A., et al. Classic late-infantile NCL with tripeptidyl-peptidase I deficiency (CLN2). Pathology and genetics. In: Golden J., Harding B., editors. Developmental neuropathology. Basel: ISN Neuropath Press; 2004:274-276.
Wisniewski K., Golabek A., et al. Palmitoyl-protein thioesterase 1 deficiency with granular osmiophilic deposits (CLN1). Pathology and genetics. In: Golden J., Harding B., editors. Developmental neuropathology. Basel: ISN Neuropath Press; 2004:271-273.
Wisniewski K., Golabek A., et al. Rare forms of neuronal ceroid lipofuscinoses. Pathology and genetics. In: Golden J., Harding B., editors. Developmental neuropathology. Basel: ISN Neuropath Press; 2004:280-282.
Wolf N.I., Rahman S., Schmitt B., et al. Status epilepticus in children with Alpers’ disease caused by POLG1 mutations: EEG and MRI features. Epilepsia. 2009;50(6):1596-1607.
Wong D.F., Ricaurte G., et al. Dopamine transporter changes in neuropsychiatric disorders. Adv Pharmacol. 1998;42:219-223.
Worle H., Köhler B., Schlote W., et al. Progressive cerebral degeneration of childhood with liver disease (Alpers-Huttenlocher disease) with cytochrome oxidase deficiency presenting with epilepsia partialis continua as a first clinical manifestation. Clin Neuropathol. 1998;17:63-68.
Yamashita Y., Kondo I., et al. Mutation analysis of the methyl-CpG–binding protein 2 gene (MECP2) in Rett patients with preserved speech. Brain Dev. 2001;23(Suppl 1):S157-S1560.
Yamashita Y., Matsuishi T., et al. Decrease in benzodiazepine receptor binding in the brains of adult patients with Rett syndrome. J Neurol Sci. 1998;154:146-150.
Yntema H.G., Kleefstra T., et al. Low frequency of MECP2 mutations in mentally retarded males. Eur J Hum Genet. 2002;10:487-490.
Yntema H.G., Oudakker A.R., et al. In-frame deletion in MECP2 causes mild nonspecific mental retardation. Am J Med Genet. 2002;107:81-83.
Yoshikawa M., Uchida S., Ezaki J., et al. CLC-3 deficiency leads to phenotypes similar to human neuronal ceroid lipofuscinosis. Genes to Cells. 2002;7:597-605.
Young J.I., Zoghbi H.Y. X-chromosome inactivation patterns are unbalanced and affect the phenotypic outcome in a mouse model of Rett syndrome. Am J Hum Genet. 2004;74:511-520.
Yuan D., Dancis A., et al. Restriction of copper export in Saccharomyces cerevisiae to a late Golgi or post-Golgi compartment in the secretory pathway. J Biol Chem. 1997;272:25787-25793.
Zappella M. A double blind trial of bromocriptine in the Rett syndrome. Brain Dev. 1990;12:148-150.
Zappella M. The Rett girls with preserved speech. Brain Dev. 1992;14:98-101.
Zappella M., Meloni I., et al. Preserved speech variants of the Rett syndrome: Molecular and clinical analysis. Am J Med Genet. 2001;104:14-22.
Zeev B.B., Yaron Y., et al. Rett syndrome: Clinical manifestations in males with MECP2 mutations. J Child Neurol. 2002;17:20-24.
Zeman W., Donahue P., et al. The neuronal ceroid-lipofuscinoses (Batten-Vogt syndrome). In: Vinken P.J., Bruyn G.W., editors. Handbook of clinical neurology: Leukodystrophies and poliodystrophies. Amsterdam: North-Holland Publishing; 1970:588-687.
Zhang Z., Butler J.D., Levin S.W., et al. Lysosomal ceroid depletion by drugs: therapeutic implications for a hereditary neurodegenerative disease of childhood. Nat Med. 2001;7(4):478-484.
Zhong N., Ju W., Moroziewicz D., et al. Prenatal diagnostic testing for infantile and late-infantile neuronal ceroid lipofusinoses (NCL) using allele specific primer extension (ASPE). Beijing Da Xue Xue Bao. 2005;37:20-25.
Zhong N., Moroziewicz D.N., et al. Heterogeneity of late-infantile neuronal ceroid lipofuscinosis. Genet Med. 2000;2:312-318.
Zullo S.J., Butler L., Zahorchak R.J., et al. Localization by fluorescence in situ hybridization (FISH) of human mitochondrial polymerase gamma (POLG) to human chromosome band 15q24→q26, and of mouse mitochondrial polymerase gamma (Polg) to mouse chromosome band 7E, with confirmation by direct sequence analysis of bacterial artificial chromosomes (BACs). Cytogenet Cell Genet. 1997;78:281-284.