[level-membership-for-pediatrics-category]
Chapter 79 Defects in Metabolism of Amino Acids
79.1 Phenylalanine
Phenylalanine is an essential amino acid. Dietary phenylalanine not utilized for protein synthesis is normally degraded by way of the tyrosine pathway (Fig. 79-1). Deficiency of the enzyme phenylalanine hydroxylase (PAH) or of its cofactor tetrahydrobiopterin (BH4) causes accumulation of phenylalanine in body fluids and in the brain. The severity of hyperphenylalaninemia depends on the degree of enzyme deficiency and may vary from very high plasma concentrations (>20 mg/dL or >1,200 µmole/L, classic phenylketonuria [PKU]) to mildly elevated levels (2-6 mg/dL or 120-360 µmole/L). In affected infants with plasma concentrations >20 mg/dL, excess phenylalanine is metabolized to phenylketones (phenylpyruvate and phenylacetate; see Fig. 79-1) that are excreted in the urine, giving rise to the term phenylketonuria (PKU). These metabolites have no role in pathogenesis of central nervous system (CNS) damage in patients with PKU; their presence in the body fluids simply signifies the severity of the condition. The term hyperphenylalaninemia implies lower plasma levels (<20 mg/dL) of phenylalanine; these patients may or may not need dietary therapy based on their blood phenylalanine level. The brain is the main organ affected by hyperphenylalaninemia. The CNS damage in affected patients is caused by the elevated concentration of phenylalanine in brain tissue. The high blood levels of phenylalanine in PKU saturate the transport system across the blood-brain barrier causing inhibition of the cerebral uptake of other large neutral amino acids such as tyrosine and tryptophan. The exact mechanism of damage caused by elevated levels of intracerebral phenylalanine remains elusive. There are a few adults with classic PKU and normal intelligence who have never been treated with a phenylalanine-restricted diet. Phenylalanine content of the brain in these individuals was found to be close to that of normal subjects when studied by magnetic resonance spectroscopy (MRS) and imaging (MRI) techniques.
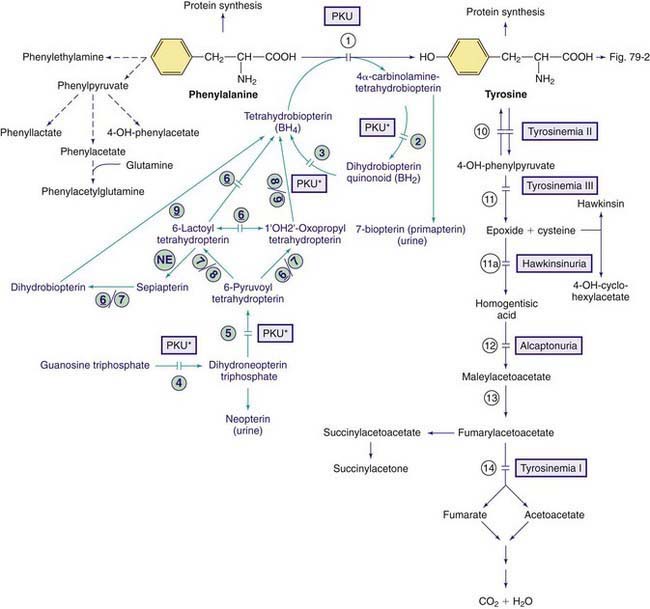
Figure 79-1 Pathways of phenylalanine and tyrosine metabolism. Enzyme defects causing genetic conditions are depicted as horizontal bars crossing the reaction arrow(s). Pathways for synthesis of cofactor BH4 are shown in purple. PKU* refers to defects of BH4 metabolism that affect the phenylalanine, tyrosine, and tryptophan hydroxylases (see Figs. 79-2 and 79-5). Enzymes: (1) phenylalanine hydroxylase (PAH), (2) pterin-carbinolamine dehydratase (PCD), (3) dihydrobiopterin reductase, (4) guanosine triphosphate (GTP) cyclohydrolase, (5) 6-pyruvoyltetrahydropterin synthase (6-PTS), (6) seriapterin reductase, (7) carbonyl reductase, (8) aldolase reductase, (9) dihydrofolate reductase, (10) tyrosine aminotransferase, (11a) intramolecular rearrangement, (11) 4-hydroxyphenylpyruvate dioxygenase, (12) homogentisic acid dioxygenase, (13) maleylacetoacetate isomerase, (14) fumarylacetoacetate hydroxylase, (NE) nonenzymatic.
Milder Forms of Hyperphenylalaninemia, Non-PKU Hyperphenylalaninemias
Hyperphenylalaninemia due to Deficiency of the Cofactor BH4
In 1-3% of infants with hyperphenylalaninemia, the defect resides in 1 of the enzymes necessary for production or recycling of the cofactor BH4 (Fig. 79-2). If these infants are misdiagnosed as having PKU, they may deteriorate neurologically despite adequate control of plasma phenylalanine. BH4 is synthesized from guanosine triphosphate (GTP) through several enzymatic reactions (see Fig. 79-1). In addition to acting as a cofactor for PAH, BH4 is also a cofactor for tyrosine hydroxylase and tryptophan hydroxylase, which are involved in the biosynthesis of dopamine (see Fig. 79-2) and serotonin (see Fig 79-5), respectively. Therefore, patients with hyperphenylalaninemia due to BH4 deficiency also manifest neurologic findings related to deficiencies of the neurotransmitters dopamine and serotonin. Four enzyme deficiencies leading to defective BH4 formation cause hyperphenylalaninemia and deficiencies of dopamine and serotonin. These include autosomal recessive GTP cyclohydrolase deficiency, pterin-carbinolamine dehydratase (PCD) deficiency, dihydropteridine reductase (DHPR) deficiency, and 6-pyruvoyltetrahydropterin synthase (PTPS or 6-PTS) deficiency. More than half of the reported patients have had a deficiency of 6-pyruvoyltetrahydropterin synthase. Autosomal dominant form of GTP deficiency and sepiapterin reductase deficiency result in deficiencies of neurotransmitters without hyperphenylalaninemia (Chapter 79.11 and Fig. 79-1).
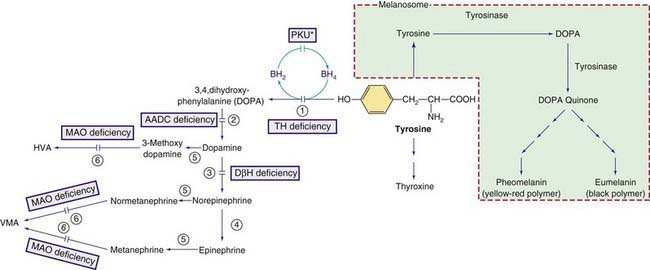
Figure 79-2 Other pathways involving tyrosine metabolism. PKU* indicates hyperphenylalanemia due to tetrahydrobiopterin (BH4) deficiency (see Fig. 79-1). HVA, homovanillic acid; VMA, vanillymandelic acid. Enzymes: (1) tyrosine hydroxylase (TH), (2) aromatic L-amino acid decarboxylase (AADC), (3) dopamine hydroxylase, (4) phenylethanolamine-N-methyltransferase (PNMT), (5) catechol O-methyltransferase (COMT), (6) Monoamine oxidase (MAO).
Diagnosis
BH4 deficiency and the responsible enzyme defect may be diagnosed by the following studies:
Blau N. Defining tetrahydrobiopterin (BH4)-responsiveness in PKU. J Inherit Metab Dis. 2008;31:2-3.
Blau N, Bélanger-Quintana A, Demirkol M, et al. Optimizing the use of sapropterin (BH4) in the management of phenylketonuria. Mol Genet Metab. 2009;96:158-163.
Blau N, Bélanger-Quintana A, Demirkol M, et al. Management of phenylketonuria in Europe: survey results from 19 countries. Mol Genet Metab. 2010;99:109-115.
Blau N, van Spronsen FJ, Levy HL. Phenylketonuria. Lancet. 2010;376:1417-1427.
Burton BK, Adams DJ, Grange DK, et al. Tetrahydrobiopterin therapy for phenylketonuria in infants and young children. J Pediatr. 2011;158:410-415.
Cederbaum S. Tetrahydrobiopterin and PKU: into the future. J Pediatr. 2011;158:351-353.
Committee on Genetics. Maternal phenylketonuria. Pediatrics. 2008;122:445-449.
Feillet F, van Spronsen FJ, MacDonald A, et al. Challenges and pitfalls in the management of phenylketonuria. Pediatrics. 2010;126:333-341.
Giovannini M, Verduci E, Salvatici E, et al. Phenylketonuria: dietary and therapeutic challenges. J Inherit Metab Dis. 2007;30:145-152.
Mitchell JJ, Scriver CR. Phenylalanine hydroxylase deficiency, 2010. GeneReviews at GeneTests: Medical Information Resource. University of Washington, Seattle, 1997-2010. www.genetests.org. Accessed May 2010
Scriver CR, Levy H, Donlon J. Hyperphenylalaninemia: phenylalanine hydroxylase deficiency. In: Scriver CR, Beaudet AL, Sly WS, et al, editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill, 2008.
Waisbren SE, Noel K, Fahrbach K, et al. Phenylalanine blood levels and clinical outcomes in phenylketonuria: a systematic literature review and meta-analysis. Mol Genet Metab. 2007;92:63-70.
Waisbren SE, White D, van Spronsen F. Phenylketonuria, psychology and the brain. Mol Genet Metab. 2010;99(Suppl):S1-S108.
Zurfluh MR, Zschocke J, Lindner M, et al. Molecular genetics of tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency. Hum Mutat. 2008;29:167-175.
79.2 Tyrosine
Tyrosine is derived from ingested proteins or is synthesized endogenously from phenylalanine. It is used for protein synthesis and is a precursor of dopamine, norepinephrine, epinephrine, melanin, and thyroxine. Excess tyrosine is metabolized to carbon dioxide and water (see Fig. 79-1). Hereditary causes of hypertyrosinemia include deficiencies of tyrosine aminotransferase, 4-hydroxyphenylpyruvate dioxygenase (4-HPPD), or fumarylacetoacetate hydrolase (FAH). Acquired hypertyrosinemia may occur in severe hepatocellular dysfunction (liver failure), scurvy (vitamin C is the cofactor for the enzyme 4-HPPD), and hyperthyroidism. Hypertyrosinemia is common in blood samples obtained soon after eating.
Tyrosinemia Type I (Tyrosinosis, Hereditary Tyrosinemia, Hepatorenal Tyrosinemia)
Laboratory Findings
The presence of elevated levels of succinylacetone in serum and urine is diagnostic for tyrosinemia type I (see Fig. 79-1). In untreated patients, routinely available tests have a characteristic pattern. α-Fetoprotein level is increased, often markedly, and liver-synthesized coagulation factors are decreased in most patients; serum levels of transaminases are often increased, with marked increases being possible during acute hepatic episodes. Serum concentration of bilirubin is usually normal but can be increased with liver failure. Increased levels of α-fetoprotein are present in the cord blood of affected infants, indicating intrauterine liver damage. Plasma tyrosine level is usually elevated at diagnosis but this is a nonspecific finding and is dependent on dietary intake. Other amino acids, particularly methionine, may also be elevated in patients with liver damage. Hyperphosphaturia, hypophosphatemia, and generalized aminoaciduria may occur. The urinary level of 5-aminolevulinic acid is elevated (due to inhibition of 5-aminolevulinic hydratase by succinylacetone).
Diagnosis is usually established by demonstration of elevated levels of succinylacetone in urine or blood. Neonatal screening for hypertyrosinemia detects only a minority of patients with tyrosinemia type I. Succinylacetone, which is now assayed by most screening programs, has higher sensitivity and specificity than tyrosine and is the preferable metabolite for screening. Tyrosinemia type I should be differentiated from other causes of hepatitis and hepatic failure in infants, including galactosemia, hereditary fructose intolerance, neonatal iron storage disease, giant cell hepatitis, and citrullinemia type II (Chapter 79.11).
Treatment and Outcome
A diet low in phenylalanine and tyrosine can slow but does not halt the progression of the condition. The treatment of choice is nitisinone (NTBC), which inhibits tyrosine degradation at 4-HPPD (see Fig. 79-1). This treatment prevents acute hepatic and neurologic crises. Although nitisinone stops or greatly slows disease progression, some pretreatment liver damage is not reversible. Therefore, patients must be followed for development of cirrhosis or hepatocellular carcinoma. On imaging, the presence of even a single liver nodule usually indicates underlying cirrhosis. Most liver nodules in tyrosinemic patients are benign but current imaging techniques do not accurately distinguish all malignant nodules. Liver transplantation is an effective therapy for tyrosinemia type I and alleviates the risk of hepatocellular carcinoma. The impact of nitisinone treatment on the need for liver transplantation is still under study but the greatest effect is in patients treated early, such as children detected by neonatal screening, prior to the development of clinical symptoms. Rarely, nitisinone-treated patients develop corneal crystals, presumably of tyrosine, which are reversible by strict dietary compliance. This finding, combined with observations of developmental delay in some patients with chronically elevated tyrosine such as tyrosinemia type II, suggests that a diet low in phenylalanine and tyrosine should be continued in patients treated with nitisinone.
Tyrosinemia Type II (Richner-Hanhart Syndrome, Oculocutaneous Tyrosinemia)
This rare autosomal recessive disorder is caused by deficiency of tyrosine aminotransferase, resulting in palmar and plantar hyperkeratosis, herpetiform corneal ulcers, and mental retardation (see Fig. 79-1). Ocular manifestations include excessive tearing, redness, pain, and photophobia and often occur before skin lesions. Corneal lesions are presumed to be due to tyrosine deposition. In contrast to herpetic ulcers, corneal lesions in tyrosinemia type II stain poorly with fluorescein and often are bilateral. Skin lesions, which may develop later in life, include painful, nonpruritic hyperkeratotic plaques on the soles, palms and fingertips. Mental retardation, which occurs in <50% of patients, is usually mild to moderate.
The principal laboratory finding in untreated patients is marked hypertyrosinemia (20-50 mg/dL; 1,100-2,750 µmole/L). Surprisingly, 4-hydroxyphenylpyruvic acid and its metabolites are also elevated in urine despite being downstream from the metabolic block (see Fig. 79-1). This is hypothesized to occur via the action of other transaminases in the presence of high tyrosine concentrations, producing 4HPP in cellular compartments like the mitochondrion in which it is not further degraded. In contrast to tyrosinemia type I, liver and kidney function are normal, as are serum concentrations of other amino acids and succinylacetone. Tyrosinemia type II is due to TAT gene mutations, causing deficiency of cytosolic tyrosine aminotransferase activity in liver.
Transient Tyrosinemia of the Newborn
In a small number of newborn infants, plasma tyrosine may be as high as 60 mg/dL (3,300 µmole/L) during the 1st 2 weeks of life. Most affected infants are premature and are receiving high-protein diets. Transient tyrosinemia is felt to result from delayed maturation of 4-HPPD (see Fig. 79-1). Lethargy, poor feeding, and decreased motor activity are noted in some patients. Most are asymptomatic and are identified by a high blood phenylalanine or tyrosine level on screening. Laboratory findings include marked elevation of plasma tyrosine with a moderate increase in plasma phenylalanine. The finding of hypertyrosinemia differentiates this condition from PKU. 4-Hydroxyphenylpyruvic acid and its metabolites are present in the urine. Hypertyrosinemia usually resolves spontaneously in the 1st mo of life. It can be corrected promptly by reducing dietary protein to below 2 g/kg/24 hr and by administering vitamin C (200-400 mg/24 hr). Mild intellectual deficits have been reported in some infants that had this condition, but the causal relationship to hypertyrosinemia is not conclusively established.
Hawkinsinuria
This rare autosomal dominant condition is caused by a mutant 4-HPPD enzyme that catalyzes a partial reaction, releasing an intermediate compound used for diagnosis (see Fig. 79-1). This intermediate is either reduced to form 4-hydroxycyclohexylacetic acid (4-HCAA) or reacts with glutathione to form the unusual organic acid hawkinsin (2-L-cysteine-S-yl-1-4-dihydroxycyclohex-5-en-1-yl-acetic acid); secondary glutathione deficiency may occur.
Alcaptonuria
This rare (with an incidence of ≈1/250,000) autosomal recessive disorder is due to a deficiency of homogentisic acid oxidase, which causes large amounts of homogentisic acid to accumulate in the body and then to be excreted in the urine (see Fig. 79-1).
Albinism
Albinism is due to deficiency of melanin, the main pigment of the skin and eye (Table 79-1). Melanin is synthesized by melanocytes from tyrosine in a membrane-bound intracellular organelle, the melanosome. Melanocytes originate from the embryonic neural crest and migrate to the skin, eyes (choroid and iris), hair follicles, and inner ear. The melanin in the eye is confined to the retinal pigment epithelium, whereas in skin and hair follicles, it is secreted into the epidermis and hair shaft. Albinism can be caused by deficiencies of melanin synthesis, by some hereditary defects of melanosomes, or by disorders of melanocyte migration. Although albinism is a classical example of a biochemical genetic disease, neither the biosynthetic pathway of melanin nor many facets of melanocyte cell biology are completely elucidated (see Fig. 79-2). The end products are 2 pigments: pheomelanin, which is a yellow-red pigment; and eumelanin, a brown-black pigment.
| TYPE | GENE | CHROMOSOME |
|---|---|---|
| OCULOCUTANEOUS ALBINISM (OCA) | ||
| OCA1 (tyrosinase deficient) | TYR | 11q |
| OCA1A (severe deficiency) | TYR | 11q |
| OCA1B (mild deficiency)* | TYR | 11q |
| OCA2 (tyrosinase positive)† | P (pink-eyed dilution) | 15q |
| OCA3 (Rufous, red OCA) | TYRP1‡ | 9p |
| OCA4 | MATP | 5p13.3 |
| Hermansky-Pudlak syndrome | HPS1 | 10q |
| Chédiak-Higashi syndrome | LYST | 1q |
| OCULAR ALBINISM | ||
| OA1 (Nettleship-Falls type) | OA | xp |
| LOCALIZED ALBINISM | ||
| Piebaldism | KIT | 4q |
| Waardenburg syndrome I & III | PAX3 | 2q |
| Waardenburg syndrome II | MITF | 3p |
* This includes Amish, minimal pigment, yellow albinism, and platinum and temperature-sensitive variants.
‡ Tyrosinase related protein 1.
Many clinical forms of albinism have been identified. Some of the seemingly distinct clinical forms are caused by different mutations of the same gene. Several genes located on different chromosomes are shown to be involved in melanogenesis (see Table 79-1). Attempts to differentiate types of albinism based on the mode of inheritance, tyrosinase activity, or the extent of hypopigmentation have failed to yield a comprehensive classification. The following classification is based on the distribution of albinism in the body and the type of mutated gene.
Mutation detection is clinically available for most albinism genes (see Table 79-1). Molecular diagnosis is of little use therapeutically in isolated albinism but can be helpful for precise genetic counseling of families.
Oculocutaneous (Generalized) Albinism (OCA)
OCA2 (Tyrosinase-Positive OCA)
This is the most common form of generalized OCA, particularly in African blacks. Clinically, patients demonstrate some pigmentation of the skin and eyes at birth and continue to accumulate pigment throughout their lives. The hair is yellow at birth and may darken with age. They have pigmented nevi and freckles but do not tan. They may be clinically indistinguishable from OCA1 B. These individuals have normal tyrosinase activity in hair bulbs. The defect is in the OCA2 gene on chromosome 15q, orthologous to the p (pink-eyed dilution) gene in the mouse. This gene produces the P protein, a melanosome membrane protein. Patients with Prader-Willi and Angelman syndromes with microdeletion in chromosome 15q12 lack 1 copy of the OCA2 gene and have mild pigmentary dilution (Chapter 76).
Syndromic Forms of Generalized Albinism
Chédiak-Higashi Syndrome
Patients with this rare autosomal recessive condition (Chapter 124) have albinism of variable severity and susceptibility to infection. Bacterial infections of skin and upper respiratory tract are common. Giant peroxidase-positive lysosomal granules can be seen in granulocytes in a blood smear. Patients have a reduced number of melanosomes, which are abnormally large (macromelanosomes). The bleeding tendency is typically mild. The major, life-threatening complication is macrophage activation with hemophagocytic lymphohistiocytosis, manifested by fever, lymphadenopathy, hepatosplenomegaly, cytopenias, and elevated plasma ferritin level. Patients surviving childhood may develop cerebellar atrophy, peripheral neuropathy, and cognitive delay. Mutations in the LYST gene on chromosome 1q are the only known cause of this syndrome.
Brilliant MH. Oculocutaneous albinism type 4, 2007. GeneReviews at GeneTests: Medical Genetics Information Resources. University of Washington, Seattle, 1997-2010. www.genetests.org. Accessed June 2010
Charfeddine C, Monastiri K, Mokni M, et al. Clinical and mutational investigations of tyrosinemia type II in Northern Tunisia: identification and structural characterization of two novel TAT mutations. Mol Genet Metab. 2006;88:184-191.
El-Karaksy H, Rashed M, El-Sayed R, et al. Clinical practice. NTBC therapy for tyrosinemia type 1: how much is enough? Eur J Pediatr. 2010;169:689-693.
Gahl WA. Hermansky-Pudlak syndrome, 2010. GeneReviews at GeneTests: Medical Genetics Information Resources. University of Washington, Seattle, 1997-2010. www.genetests.org. Accessed June 2010
Introne WJ, Westbroek W, Golas GA, et al. Chédiak-Higashi syndrome, 2009. GeneReviews at GeneTests: Medical Genetics Information Resources. University of Washington, Seattle, 1997-2010. www.genetests.org. Accessed May 2010
King RA, Oetting W. Oculocutaneous albinism type 2, 2007. GeneReviews at GeneTests: Medical Genetics Information Resources. University of Washington, Seattle, 1997-2010. www.genetests.org. Accessed June 2010
Masurel-Paulet A, Poggi-Bach J, Rolland MO, et al. NTBC treatment in tyrosinaemia type I: long-term outcome in French patients. J Inherit Metab Dis. 2008;31:81-87.
Pingault V, Ente D, Dastot-LeMoal F, et al. Review and update of mutations causing Waardenburg syndrome. Hum Mutat. 2010;31:391-406.
Rezvani I. 50 years ago in Journal of Pediatrics: Waardenburg’s syndrome. J Pediatr. 2010;157:732.
Scott CR. The genetic tyrosinemias. Am J Med Genet C Semin Med Genet. 2006;142C:121-126.
79.3 Methionine
The normal pathway for catabolism of methionine, an essential amino acid, produces S-adenosylmethionine, which serves as a methyl group donor for methylation of a variety of compounds in the body, and cysteine, which is formed through a series of reactions collectively called trans-sulfuration (Fig. 79-3).
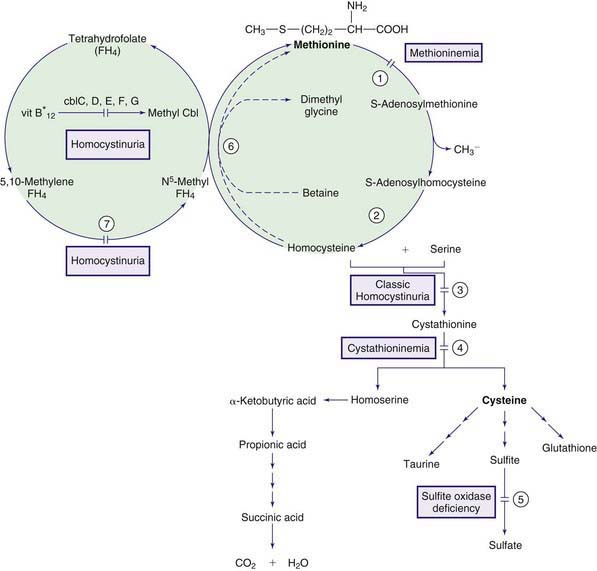
Homocystinuria (Homocystinemia)
Normally, most homocysteine, an intermediate compound of methionine degradation, is remethylated to methionine. This methionine-sparing reaction is catalyzed by the enzyme methionine synthase, which requires a metabolite of folic acid (5-methyltetrahydrofolate) as a methyl donor and a metabolite of vitamin B12 (methylcobalamin) as a cofactor (see Fig. 79-3). Only 20-30% of total homocysteine (and its dimer homocystine) is in free form in the plasma of normal individuals. The rest is bound to proteins as mixed disulfides. Three major forms of homocystinemia and homocystinuria have been identified.
Homocystinuria Due to Cystathionine β-Synthase (CBS) Deficiency (Classic Homocystinuria)
Infants with this disorder are normal at birth. Clinical manifestations during infancy are nonspecific and may include failure to thrive and developmental delay. The diagnosis is usually made after 3 yr of age, when subluxation of the ocular lens (ectopia lentis) occurs. This causes severe myopia and iridodonesis (quivering of the iris). Astigmatism, glaucoma, staphyloma, cataracts, retinal detachment, and optic atrophy may develop later in life. Progressive mental retardation is common. Normal intelligence has been reported. In an international survey of >600 patients, IQ scores ranged from 10 to 135. Higher IQ scores are seen in vitamin B6 responsive patients. Psychiatric and behavioral disorders have been observed in >50% of affected patients. Convulsions occur in about 20% of patients. Affected individuals with homocystinuria manifest skeletal abnormalities resembling those of Marfan syndrome (Chapter 693); they are usually tall and thin, with elongated limbs and arachnodactyly. Scoliosis, pectus excavatum or carinatum, genu valgum, pes cavus, high arched palate, and crowding of the teeth are commonly seen. These children usually have fair complexions, blue eyes, and a peculiar malar flush. Generalized osteoporosis, especially of the spine, is the main roentgenographic finding. Thromboembolic episodes involving both large and small vessels, especially those of the brain, are common and may occur at any age. Optic atrophy, paralysis, cor pulmonale, and severe hypertension (due to renal infarcts) are among the serious consequences of thromboembolism, which is caused by changes in the vascular walls and increased platelet adhesiveness secondary to elevated homocystine levels. The risk of thromboembolism increases after surgical procedures. Spontaneous pneumothorax and acute pancreatitis are rare complications.
Treatment with high doses of vitamin B6 (200-1,000 mg/24 hr) causes dramatic improvement in most patients who are responsive to this therapy. The degree of response to vitamin B6 treatment may be different in different families. Some patients may not respond because of folate depletion; a patient should not be considered unresponsive to vitamin B6 until folic acid (1-5 mg/24 hr) has been added to the treatment regimen. Restriction of methionine intake in conjunction with cysteine supplementation is recommended for patients who are unresponsive to vitamin B6. The need for dietary restriction and its extent remains controversial in patients with vitamin B6 responsive form. In some patients with this form, addition of betaine may obviate the need for any dietary restriction. Betaine (trimethylglycine, 6-9 g/24 hr for adults or 200-250 mg/kg/day for children) lowers homocysteine levels in body fluids by remethylating homocysteine to methionine (see Fig. 79-3); this may result in further elevation of plasma methionine levels. This treatment has produced clinical improvement (preventing vascular events) in patients who are unresponsive to vitamin B6 therapy. Cerebral edema has occurred in a patient with vitamin B6 nonresponsive homocystinuria and dietary noncompliance during betaine therapy. Administration of large doses of vitamin C (1 g/day) has improved the endothelial function; long-term clinical efficacy is not known.
Homocystinuria Due to Defects in Methylcobalamin Formation
Methylcobalamin is the cofactor for the enzyme methionine synthase, which catalyzes remethylation of homocysteine to methionine. There are at least 5 distinct defects in the intracellular metabolism of cobalamin that may interfere with the formation of methylcobalamin. To better understand the metabolism of cobalamin, see methylmalonic acidemia (Fig. 79-4; Chapter 79.6 and Fig. 79-3). The 5 defects are designated as cblC, cblD (including cblD variant 1), cblE (methionine synthase reductase), cblG (methionine synthase), and cblF. Patients with cblC, cblD (not those with cblD variant 2), and cblF defects have methylmalonic acidemia in addition to homocystinuria because formation of both adenosylcobalamin and methylcobalamin is impaired (Chapter 79.6).
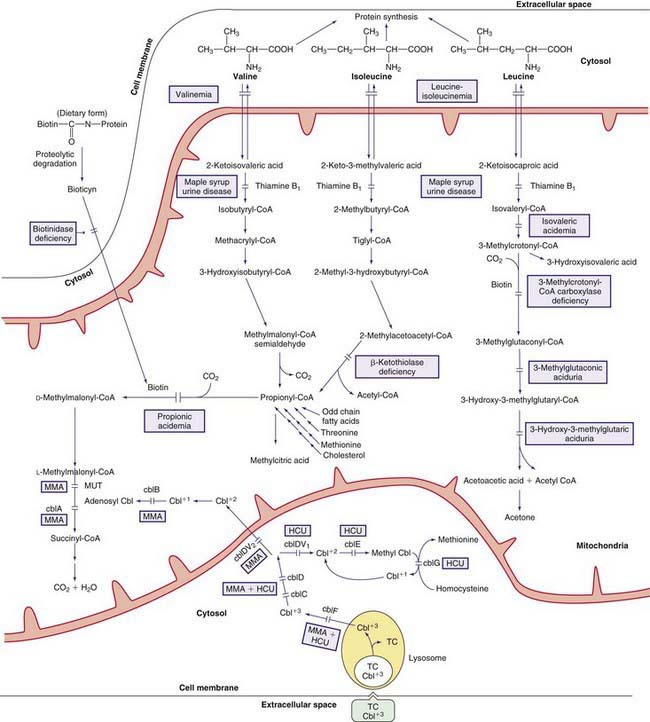
Patients with cblE and cblG defects are unable to form methylcobalamin and develop homocystinuria without methylmalonic acidemia (see Fig. 79-4); fewer than 40 patients are known with each of these diseases.
Hypermethioninemia
Secondary hypermethioninemia occurs in liver disease, tyrosinemia type I, and classic homocystinuria. Hypermethioninemia has also been found in premature and some full-term infants receiving high-protein diets, in whom it may represent delayed maturation of the enzyme methionine adenosyltransferase. Lowering the protein intake usually resolves the abnormality. Primary hypermethioninemia caused by the deficiency of hepatic methionine adenosyltransferase (MAT I/III; MAT II, which is present in other tissues, is not affected; see Fig. 79-3) has been reported in approximately 60 patients. The majority of these patients have been diagnosed in the neonatal period through screening for homocystinuria. Affected individuals with residual enzyme activity remain asymptomatic throughout life despite persistent hypermethioninemia. Some complain of unusual odor to their breath (boiled cabbage). A few patients with complete enzyme deficiency have had neurologic abnormalities related to demyelination (mental retardation, dystonia, dyspraxia). Normal pregnancies producing normal offspring have been reported in mothers with methionine adenoslytransferase deficiency. The condition is inherited as an autosomal recessive trait. The gene for hepatic methionine adenosyltransferase is on chromosome 10q22 and several disease-causing mutations have been identified. A novel defect, glycine N-methyltransferase deficiency, also causes isolated hypermethioninemia.
Adams D, Venditti CP. Disorders of intracellular cobalamin metabolism. GeneReviews at GeneTests: Medical Genetics Information Resources. University of Washington, Seattle, 1997-2010. www.genetests.org. Accessed May 2010
Allen NC, Bagade S, McQueen MB, et al. Systematic meta-analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nat Genet. 2008;30:827-834.
Forges T, Chery C, Audonnet S, et al. Life-threatening methylenetetrahydrofolate reductase (MTHFR) deficiency with extremely early onset: characterization of two novel mutations in compound heterozygous patients. Mol Genet Metab. 2010;100:143-148.
Holm PK, Hustad S, Ueland PM, et al. Modulation of the homocysteine-betaine relationship by methylenetetrahydrofolate reductase 677C-T genotypes and B-vitamin status in a large-scale epidemiologic study. J Clin Endocr Metab. 2007;92:1535-1541.
Lawson-Yuen A, Levy HL. The use of betaine in the treatment of elevated homocysteine. J Mol Genet Metab. 2006;88:201-207.
Lin HJ, Neidich JA, Salazar D, et al. Asymptomatic maternal combined homocystinuria and methylmalonic aciduria (cbIC) detected through low carnitine levels on newborn screening. J Pediatr. 2009;155:924-927.
Picker JD, Levy HL. Homocystinuria caused by cystathionine beta-synthase deficiency, 2006. GeneReviews at GeneTests: Medical Information Resource. University of Washington, Seattle, 1997-2010. www.genetests.org. Accessed May 2010
Schiff M, Benoist JF, Tilea B, et al. Isolated remethylation disorders: do our treatments benefit patients? J Inherit Metab Dis. 2011;34:137-145.
Skovby F, Gaustadnes M, Mudd SH. A revisit to the natural history of homocystinuria due to cystathionine beta-synthase deficiency. Mol Genet Metab. 2010;99:1-3.
Strauss KA, Morton DH, Puffenberger EG, et al. Prevention of brain disease from severe 5,10-methylenetetrahydrofolate reductase deficiency. Mol Genet Metab. 2007;91:165-175.
Testai FD, Gorelick PB. Inherited metabolic disorders and stroke part 2: homocystinuria, organic acidurias, and urea cycle disorders. Arch Neurol. 2010;67:148-153.
79.4 Cysteine/Cystine
Cysteine is a sulfur-containing nonessential amino acid that is synthesized from methionine (see Fig. 79-3). In the presence of oxygen, 2 molecules of cysteine are oxidized to form cystine. The most common disorders of cysteine/cystine metabolism, cystinuria (Chapter 541) and cystinosis (Chapter 523.3).
Sulfite Oxidase Deficiency (Molybdenum Cofactor Deficiency)
At the last step in cysteine metabolism, sulfite is oxidized to sulfate by sulfite oxidase, and the sulfate is excreted in the urine (see Fig. 79-3). This enzyme requires a molybdenum-pterin complex named molybdenum cofactor. This cofactor is also necessary for the function of 2 other enzymes in humans: xanthine dehydrogenase (which oxidizes xanthine and hypoxanthine to uric acid) and aldehyde oxidase. Three enzymes, encoded by 3 different genes, are involved in the synthesis of the cofactor. The genes are mapped to chromosomes 14q24, 6p21.3, and 5q11. Deficiency of any of the 3 enzymes causes cofactor deficiency with identical phenotype. Most patients who were originally diagnosed as having sulfite oxidase deficiency have been proven to have molybdenum cofactor deficiency. Both conditions are inherited as autosomal recessive traits. The gene for sulfite oxidase is on chromosome 12.
Kugler S, Hahnewald R, Garrido M, et al. Long-term rescue of a lethal inherited disease by adeno-associated virus-mediated gene transfer in a mouse model of molybdenum-cofactor deficiency. Am J Hum Genet. 2007;80:291-297.
Schwarz G, Mendel RR, Ribbe MW. Molybdenum cofactors, enzymes and pathways. Nature. 2009;460:839-847.
Tan WH, Eickler FS, Hoda S, et al. Isolated sulfite oxidase deficiency: a case report with a novel mutation and review of the literature. Pediatrics. 2005;116:757-766.
Veldman A, Santamaria-Araujo JA, Sollazzo S, et al. Successful treatment of molybdenum cofactor deficiency type A with cPMP. Pediatrics. 2010;125:e1249-e1254.
79.5 Tryptophan
Tryptophan is an essential amino acid and a precursor for nicotinic acid (niacin) and serotonin (Fig. 79-5). The genetic disorders of metabolism of serotonin, 1 of the major neurotransmitters, are discussed in Chapter 79.11.
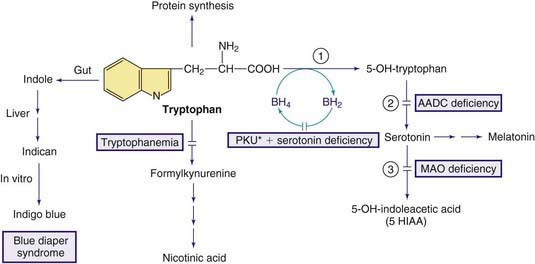
Figure 79-5 Pathways in the metabolism of tryptophan. PKU* indicates hyperphenylalanemia due to tetrahydrobiopterin deficiency (see Fig. 79-1). Enzymes: (1) tryptophan hydroxylase, (2) aromatic L-amino acid decarboxylase (AADC), (3) monoamine oxidase (MAO).
79.6 Valine, Leucine, Isoleucine, and Related Organic Acidemias
The early steps in the degradation of these 3 essential amino acids, the branched-chain amino acids, are similar (see Fig. 79-4). The intermediate metabolites are all organic acids, and deficiency of any of the degradative enzymes, except for the transaminases, causes acidosis; in such instances, the organic acids before the enzymatic block accumulate in body fluids and are excreted in the urine. These disorders commonly cause metabolic acidosis, which usually occurs in the 1st few days of life. Although most of the clinical findings are nonspecific, some manifestations may provide important clues to the nature of the enzyme deficiency. An approach to infants suspected of having an organic acidemia is presented in Figure 79-6. Definitive diagnosis is usually established by identifying and measuring specific organic acids in body fluids (blood, urine), by the enzyme assay, and by identification of the mutant gene.
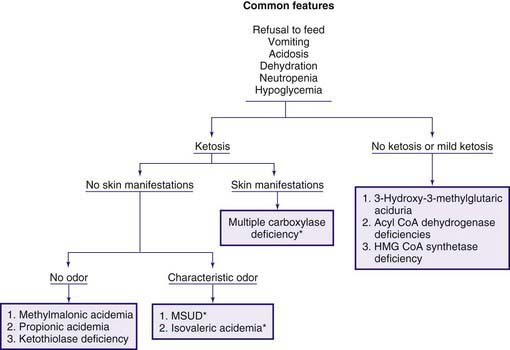
Figure 79-6 Clinical approach to infants with organic acidemia. Asterisks indicate disorders in which patients have a characteristic odor (see text and Table 79-2). MSUD, maple syrup urine disease.
Organic acidemias are not limited to defects in the catabolic pathways of branched-chain amino acids. Disorders causing accumulation of other organic acids include those derived from lysine (Chapter 79.14), those associated with lactic acid (Chapter 81), and dicarboxylic acidemias associated with defective fatty acid degradation (Chapter 80.1).
Maple Syrup Urine Disease (MSUD)
Decarboxylation of leucine, isoleucine, and valine is accomplished by a complex enzyme system (branched-chain α-ketoacid dehydrogenase) using thiamine pyrophosphate (vitamin B1) as a coenzyme. This mitochondrial enzyme consists of 4 subunits: E1α, E1β, E2, and E3. The E3 subunit is shared with 2 other dehydrogenases in the body, namely pyruvate dehydrogenase and α-ketoglutarate dehydrogenase. Deficiency of this enzyme system causes MSUD (see Fig. 79-4), named after the sweet odor of maple syrup found in body fluids, especially urine. Based on clinical findings and response to thiamine administration, 5 phenotypes of MSUD have been identified.
Classic MSUD
Diagnosis is often suspected because of the peculiar odor of maple syrup found in urine, sweat, and cerumen (see Fig. 79-6). It is usually confirmed by amino acid analysis showing marked elevations in plasma levels of leucine, isoleucine, valine, and alloisoleucine (a stereoisomer of isoleucine not normally found in blood) and depression of alanine. Leucine levels are usually higher than those of the other 3 amino acids. Urine contains high levels of leucine, isoleucine, and valine and their respective ketoacids. These ketoacids may be detected qualitatively by adding a few drops of 2,4-dinitrophenylhydrazine reagent (0.1% in 0.1 N HCl) to the urine; a yellow precipitate of 2,4-dinitrophenylhydrazone is formed in a positive test. Neuroimaging during the acute state may show cerebral edema, which is most prominent in the cerebellum, dorsal brainstem, cerebral peduncle, and internal capsule. After recovery from the acute state and with advancing age, hypomyelination and cerebral atrophy may be seen in neuroimaging of the brain. The enzyme activity can be measured in leukocytes and cultured fibroblasts.
Isovaleric Acidemia
This rare condition is due to the deficiency of isovaleryl coenzyme A (CoA) dehydrogenase (see Fig. 79-5).
Clinical manifestations in the acute form include vomiting and severe acidosis in the 1st few days of life. Lethargy, convulsions, and coma may ensue, and death may occur if proper therapy is not initiated. The vomiting may be severe enough to suggest pyloric stenosis. The characteristic odor of “sweaty feet” may be present (see Fig. 79-6). Infants who survive this acute episode will go on to have the chronic intermittent form later on in life. A milder form of the disease (chronic intermittent form) also exists; in this, the 1st clinical manifestation (vomiting, lethargy, acidosis or coma) may not appear until the child is a few months or a few years old. In both forms, acute episodes of metabolic decompensations may occur during a catabolic state such as an infection. Sensitive methods for newborn screening have identified yet a milder and potentially asymptomatic phenotype of the condition; a few older siblings of these affected newborns were found to have identical genotype and biochemical abnormalities without any clinical manifestations.
Laboratory findings during the acute attacks include ketoacidosis, neutropenia, thrombocytopenia, and occasionally pancytopenia. Hypocalcemia, hyperglycemia, and moderate to severe hyperammonemia may be present in some patients. Increases in plasma ammonia may suggest a defect in the urea cycle. In urea cycle defects the infant is not acidotic (see Fig. 79-6).
Treatment of the acute attack is aimed at hydration, reversal of the catabolic state (by providing adequate calories orally or intravenously), correction of metabolic acidosis (by infusing sodium bicarbonate), and removal of the excess isovaleric acid. Because isovalerylglycine has a high urinary clearance, administration of glycine (250 mg/kg/24 hr) is recommended to enhance formation of isovalerylglycine. L-carnitine (100 mg/kg/24 hr orally) also increases removal of isovaleric acid by forming isovalerylcarnitine, which is excreted in the urine. In patients with significant hyperammonemia (blood ammonia >200 µM), measures that reduce blood ammonia should be employed (Chapter 79.12). Exchange transfusion and peritoneal dialysis may be needed if the just described measures fail to induce significant clinical and biochemical improvement. After recovery from the acute attack, the patient should receive a low-protein diet (1.0-1.5 g/kg/24 hr) and should be given glycine and carnitine supplements. Pancreatitis (acute or recurrent forms) has been reported in survivors. Normal development can be achieved with early and proper treatment.
Multiple Carboxylase Deficiencies (Defects in Utilization of Biotin)
Biotin is a water-soluble vitamin that is a cofactor for all 4 carboxylase enzymes in humans: pyruvate carboxylase, acetyl CoA carboxylase, propionyl CoA carboxylase, and 3-methylcrotonyl CoA carboxylase. The latter 2 are involved in the metabolic pathways of leucine, isoleucine, and valine (see Fig. 79-4).
Dietary biotin is bound to proteins; free biotin is generated in the intestine by the action of digestive enzymes, by intestinal bacteria, and perhaps by biotinidase. The latter enzyme, which is found in serum and most tissues in the body, is also essential for the recycling of biotin in the body by releasing it from the apoenzymes (carboxylases, see Fig. 79-4). Free biotin must form a covalent peptide bond with the apoprotein of the 4 carboxylases to activate them (holocarboxylase). This binding is catalyzed by holocarboxylase synthetase. Deficiencies in this enzyme or in biotinidase result in malfunction of all the carboxylases and in organic acidemia.
Holocarboxylase Synthetase Deficiency (Multiple Carboxylase Deficiency—Infantile or Early Form)
Infants with this rare autosomal recessive disorder become symptomatic in the 1st few weeks of life. Symptoms may appear as early as a few hours after birth to 21 mo of age. Clinically, the affected infants who seem normal at birth develop breathing difficulties (tachypnea and apnea) shortly after birth. Feeding problems, vomiting, and hypotonia are also commonly present. If the condition remains untreated, generalized erythematous rash with exfoliation and alopecia (partial or total), failure to thrive, irritability, seizures, lethargy, and even coma may occur. Developmental delay is common. Immune deficiency manifests with susceptibility to infection. The urine may have a peculiar odor, which has been described as similar to tomcat urine. The rash, when present, differentiates this condition from other organic acidemias (see Fig. 79-6).
Isolated 3-Methylcrotonyl CoA Carboxylase Deficiency
This enzyme is 1 of 4 carboxylase enzymes in the body that require biotin as a cofactor (see Fig. 79-4). An isolated deficiency of this enzyme must be differentiated from disorders of biotin metabolism (multiple carboxylase deficiency), which cause diminished activity of all 4 carboxylases. 3-Methylcrotonyl CoA carboxylase is a heteromeric enzyme consisting of α (biotin containing) and β subunits.
3-Methylglutaconic Aciduria
At least 3 inherited conditions are known to be associated with excessive excretion of 3-methylglutaconic acid in the urine. Deficiency of the enzyme 3-methylglutaconyl CoA hydratase (see Fig. 79-4) has been documented in only 1 condition (type I). In the other 2 conditions, the enzyme activity is normal despite a modest 3-methylglutaconic aciduria.
3-Methylglutaconic Aciduria Type II (X-Linked Cardiomyopathy, Neutropenia, Growth Retardation, and 3-Methylglutaconic Aciduria With Normal 3-Methylglutaconyl CoA Hydratase, Barth Syndrome)
β-Ketothiolase (3-Oxothiolase) Deficiency (Mitochondrial Acetoacetyl CoA Thiolase [T2] Deficiency)
This reversible mitochondrial enzyme cleaves 2-methylacetoacetyl CoA (see Fig. 79-4) or acetoacetyl CoA in 1 direction and synthesizes these compounds in a reverse action (Fig. 79-7).
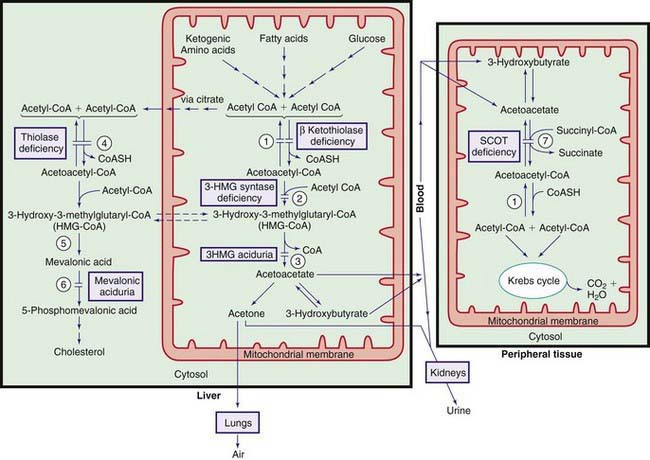
The pathogenesis of ketosis in this condition is not adequately explained because, in this enzyme deficiency, one expects impaired ketone formation (see Fig. 79-7). It is postulated that excess acetoacetyl CoA produced from other sources is used as a substrate for 3-hydroxy-3-methylglutaryl (HMG) CoA synthesis in the liver.
Cytosolic Acetoacetyl CoA Thiolase (ACAT2) Deficiency
This enzyme catalyzes the cytosolic production of acetoacetyl CoA from 2 moles of acetyl CoA (see Fig. 79-7). Cytosolic acetoacetyl CoA is the precursor of hepatic cholesterol synthesis. Cytosolic acetoacetyl CoA thiolase is a completely different enzyme from that of mitochondrial thiolase (see earlier and Fig. 79-4). Clinical manifestations in patients with this rare enzyme deficiency are similar to those in patients with mevalonic acidemia (see later). Severe progressive developmental delay, hypotonia, and choreoathetoid movements develop in the 1st few months of life. Laboratory findings are nonspecific; elevated levels of lactate, pyruvate, acetoacetate, and 3-hydroxybutyrate may be found in blood and urine. One patient had normal levels of acetoacetate and 3-hydroxybutyrate. Diagnosis can be established by demonstrating a deficiency in cytosolic thiolase activity in liver biopsy or in cultured fibroblasts or by DNA analysis. No effective treatment is available. The gene for this condition is mapped to chromosome 6q25.3-q26.
Mitochondrial 3-Hydroxy-3-Methylglutaryl (HMG) CoA Synthase Deficiency
This enzyme catalyzes synthesis of HMG-CoA from acetoacetyl CoA in the mitochondria. This is a critical step in ketone body synthesis in the liver (see Fig. 79-7). A few patients with deficiency of this enzyme have been reported. All patients have had similar presentations and outcomes. Signs and symptoms of acute hypoglycemia have occurred after an acute illness (gastroenteritis). Age at presentation has ranged from 18 mo to 6 yr. All children were asymptomatic before the episodes and remained normal after the recovery (except for mild hepatomegaly with fatty infiltration). None of the patients has had a 2nd episode, perhaps as a result of preventive measures to avoid prolonged fasting during ensuing intercurrent illnesses. Hepatomegaly was a consistent physical finding in all patients. Laboratory findings included hypoglycemia, acidosis with mild or no ketosis, elevation of liver function tests, and massive dicarboxylic aciduria. The clinical and laboratory findings may be confused with those of patients with defects in fatty acid metabolism (see Chapter 80.1). In contrast to the latter, blood concentrations of acylcarnitine conjugates are normal in patients with HMG-CoA synthase deficiency. Fasting of these patients has produced the abovementioned clinical and biochemical abnormalities.
3-Hydroxy-3-Methylglutaric Aciduria
This condition is due to a deficiency of HMG-CoA lyase (see Fig. 79-4). This enzyme catalyzes the conversion of HMG-CoA to acetoacetate and is a rate-limiting enzyme for ketogenesis (see Fig. 79-7). Clinically, >60% of patients become symptomatic between 3 and 11 mo of age, whereas about 30% develop symptoms in the 1st few days of life. One child remained asymptomatic until 15 yr of age. Episodes of vomiting, severe hypoglycemia, hypotonia, acidosis with mild or no ketosis, and dehydration may rapidly lead to lethargy, ataxia, and coma. These episodes often occur during a catabolic state such as fasting or an intercurrent infection. Hepatomegaly is common. These manifestations may be mistaken for Reye syndrome or medium-chain acyl CoA dehydrogenase (MCAD) deficiency. Patients are usually clinically asymptomatic between the attacks; 1 patient died of acute cardiomyopathy at age 7 mo during a febrile illness. Development is usually normal, but mental retardation and seizure with abnormalities of white matter (shown by MRI) have been observed in patients with prolonged episodes of hypoglycemia.
Laboratory findings include hypoglycemia, moderate to severe hyperammonemia, and acidosis. There is mild or no ketosis (see Fig. 79-7). Urinary excretion of 3-hydroxy-3-methylglutaric acid and other proximal intermediate metabolites of leucine catabolism (3-methylglutaconic acid and 3-hydroxyisovaleric acid) is markedly increased causing the urine to smell like cat urine. These organic acids are excreted in the urine as carnitine conjugates, resulting in secondary carnitine deficiency. Glutaric and adipic acids may also be increased in urine during acute attacks. Diagnosis may be confirmed by enzyme assay in cultured fibroblasts, leukocytes, or liver specimens or by identification of the mutant gene. Prenatal diagnosis is possible by the assay of the enzyme in cultured amniocytes or a chorionic villi biopsy or by DNA analysis.
Treatment of acute episodes includes hydration, infusion of glucose to control hypoglycemia, provision of adequate calories, and administration of bicarbonate to correct acidosis. Hyperammonemia should be treated promptly (Chapter 79.12). Exchange transfusion and peritoneal dialysis may be required in patients with severe hyperammonemia. Restriction of protein and fat intake is recommended for long-term management. Oral administration of L-carnitine (50-100 mg/kg/24 hr) prevents secondary carnitine deficiency. Prolonged fasting should be avoided. One child died after routine immunization. The condition is inherited as an autosomal recessive trait. The gene for HMG-CoA lyase resides on chromosome 1pter-p33 and several disease-causing mutations have been identified in different families. The gene defect appears to be more common in the Arabic population, especially in Saudi Arabia.
Succinyl CoA:3-Ketoacid CoA Transferase (SCOT) Deficiency
This enzyme is necessary for the metabolism of ketone bodies (acetoacetate and 3-hydroxybutyrate) in peripheral tissue (see Fig. 79-7). A deficiency of this enzyme results in the underutilization and accumulation of ketone bodies and ketoacidosis. Only a few patients with SCOT deficiency have been reported to date; the condition may not be rare because many cases are undiagnosed.
Mevalonic Aciduria
Mevalonic acid, an intermediate metabolite of cholesterol synthesis, is converted to 5-phosphomevalonic acid by the action of the enzyme mevalonate kinase (MVK) (see Fig. 79-7). Based on clinical manifestations, 2 forms of this condition have been recognized.
Periodic Fever With Hyperimmunoglobulinemia D (Mevalonic Aciduria, Mild Form)
Some mutations of mevalonic kinase gene (MVK) cause mild deficiencies of the enzyme and produce the clinical picture of periodic fever with hyperimmunogobulinemia D (Chapter 157). These patients have periodic bouts of fever associated with abdominal pain, vomiting, diarrhea, arthralgia, arthritis, hepatosplenomegaly, lymphadenopathy, and morbilliform rash (even petechia and purpura), which usually start before 1 yr of age. The attacks can be produced by vaccination, minor trauma, or stress; usually occur every 1-2 mo and last 2-7 days. Patients are free of symptoms between acute attacks. The diagnostic laboratory finding is elevation of serum immunoglobulin gamma D (IgD); IgA is also elevated in 80% of patients. During acute attacks, leukocytosis, increased C-reactive protein, and mild mevalonic aciduria may be present. High concentration of serum IgD differentiates this condition from familial Mediterranean fever.
Propionic Acidemia (Propionyl CoA Carboxylase Deficiency)
Propionic acid is an intermediate metabolite of isoleucine, valine, threonine, methionine, odd-chain fatty acids, and cholesterol catabolism. It is normally carboxylated to methylmalonic acid by the mitochondrial enzyme propionyl CoA carboxylase, which requires biotin as a cofactor (see Fig. 79-4). The enzyme is composed of 2 nonidentical subunits, α and β. Biotin is bound to the α subunit.
The diagnosis of propionic acidemia should be differentiated from multiple carboxylase deficiencies (see earlier and Fig. 79-6). Infants with the latter condition may have skin manifestations and excrete large amounts of lactic acid, 3-methylcrotonic acid, and 3-hydroxyisovaleric acid in addition to propionic acid. The presence of hyperammonemia may suggest a genetic defect in the urea cycle enzymes. Infants with defects in the urea cycle are usually not acidotic (see Fig. 79-1). Definitive diagnosis of propionic acidemia can be established by measuring the enzyme activity in leukocytes or cultured fibroblasts.
Treatment of acute attacks includes hydration, correction of acidosis, and amelioration of the catabolic state by provision of adequate calories through parenteral hyperalimentation. Minimal amounts of protein (0.25 g/kg/24 hr), preferably a protein deficient in propionate precursors, should be provided in the hyperalimentation fluid very early in the course of treatment. To curtail the possible production of propionic acid by intestinal bacteria, sterilization of the intestinal tract flora by antibiotics (oral neomycin, or metronidazole) should be promptly initiated. Constipation should also be treated. Patients with propionic acidemia may develop carnitine deficiency, presumably as a result of urinary loss of propionylcarnitine formed from the accumulated organic acid. Administration of L-carnitine (50-100 mg/kg/24 hr orally or 10 mg/kg/24 hr intravenously) normalizes fatty acid oxidation and improves acidosis. In patients with concomitant hyperammonemia, measures to reduce blood ammonia should be employed (Chapter 79.12). Very ill patients with severe acidosis and hyperammonemia require peritoneal dialysis or hemodialysis to remove ammonia and other toxic compounds efficiently. Although infants with true propionic acidemia are rarely responsive to biotin, this compound should be administered (10 mg/24 hr orally) to all infants during the 1st attack and until the diagnosis is established.
Methylmalonic Acidemia
Methylmalonic acid, a structural isomer of succinic acid, is normally derived from propionic acid as part of the catabolic pathways of isoleucine, valine, threonine, methionine, cholesterol, and odd-chain fatty acids. Two enzymes are involved in the conversion of D-methylmalonic acid to succinic acid: methylmalonyl CoA racemase, which forms the L-isomer; and methylmalonyl CoA mutase, which converts the L-methylmalonic acid to succinic acid (see Fig. 79-4). The latter enzyme requires adenosylcobalamin, a metabolite of vitamin B12, as a coenzyme. Deficiency of either the mutase or its coenzyme causes the accumulation of methylmalonic acid and its precursors in body fluids. A deficiency of the racemase can be associated with mild elevations of methylmalonic acid, but the clinical consequence of racemase deficiency is not known.
Defects in Metabolism of Vitamin B12 (Cobalamin, Cbl)
Dietary vitamin B12 requires intrinsic factor (IF), a glycoprotein secreted by the gastric parietal cells, for absorption in the terminal ileum. It is transported in the blood by haptocorrin (TCI) and transcobalamin II (TC). The complex of transcobalamin II-cobalamin (TC-Cbl) is recognized by a specific receptor on the cell membrane and enters the cell by endocytosis. The TC-Cbl complex is hydrolyzed in the lysosome, and free cobalamin is released into the cytosol (see Fig. 79-4). The cobalt of the molecule is reduced in the cytosol from 3 valences (cob[III]alamin) to 2 (cob[II]alamin) before it enters the mitochondria, where further reduction to cob(I)alamin occurs. The latter compound reacts with adenosine to form adenosylcobalamin (coenzyme for methylmalonyl CoA mutase). The free cobalamin in the cytosol may also undergo a series of poorly understood enzymatic steps to form methylcobalamin (coenzyme for methionine synthase, see Fig. 79-3).
Laboratory findings include ketosis, acidosis, anemia, neutropenia, thrombocytopenia, hyperglycinemia, hyperammonemia, hypoglycemia, and the presence of large quantities of methylmalonic acid in body fluids (see Fig. 79-6). Propionic acid and its metabolites 3-hydroxypropionate and methylcitrate are also found in the urine. Hyperammonemia may suggest the presence of genetic defects in the urea cycle enzymes; patients with defects in urea cycle enzymes are not acidotic (see Fig. 79-12). The reason for hyperammonemia is not well understood.
Successful pregnancy with normal outcomes for both the mother and the baby has been reported.
Combined Methylmalonic Aciduria and Homocystinuria (cblC, cblD, and cblF Defects)
Over 300 patients with methylmalonic acidemia and homocystinuria due to cblC have been reported. Indeed with the advent of expanded newborn screening, cblC may be as common as mutase deficiency. The cblD, and cblF defects are much rarer with less than a dozen patients known with each (see Figs. 79-3 and 79-4). Neurologic findings are prominent in patients with cblC and cblD defects. Most patients with the cblC defect present in the 1st yr of life because of failure to thrive, lethargy, poor feeding, mental retardation, and seizures. However, late-onset defects with sudden development of dementia and myelopathy have been reported, even with presentation is adulthood. Megaloblastic anemia was a common finding in patients with cblC defect. Mild to moderate increases in concentrations of methylmalonic acid and homocysteine were found in body fluids. Unlike patients with classic homocystinuria, plasma levels of methionine are low to normal in these defects. Neither hyperammonemia nor hyperglycinemia is present in these patients. The clinical findings in the cblF defect are quite variable; the 1st 2 patients had poor feeding, growth and developmental delay, and persistent stomatitis manifesting in the 1st 3 wk of life. One patient was not diagnosed until age 10 yr and had findings suggestive of rheumatoid arthritis, a pigmented skin abnormality, and encephalopathy. Vitamin B12 malabsorption has been noted in patients with cblF defect.
Patients with cblD variant 1, cblE and cblG defects do not have methylmalonic acidemia (Chapter 79.3).
Bailey LM, Ivanov RA, Jitrapakdee S, et al. Reduced half-life of holocarboxylase synthetase from patients with severe multiple carboxylase deficiency. Hum Mutat. 2008;29:e47-e57.
Coelho D, Suormala T, Stucki M, et al. Gene identification for the cbID defect of vitamin B12 metabolism. N Engl J Med. 2008;358:1454-1564.
Eminoglu FT, Ozcelik AA, Okur I, et al. 3-Methylcrotonyl-CoA carboxylase deficiency: phenotypic variability in a family. J Child Neurol. 2009;24:478-481.
Goldsmith DP. Periodic fever syndromes. Pediatr Rev. 2009;30:e34-e41.
Gunay-Aygun M, Gahl WA, Anikster Y. 3-Methylglutaconic aciduria type 3, 2009. GeneReviews at GeneTests: Medical Information Resource. University of Washington, Seattle, 1997-2010. www.genetests.org. Accessed May 2010
Hörster F, Baumgartner MR, Viardot C, et al. Long-term outcome in methylmalonic acidurias is influenced by the underlying defect (mut0, mut-, cbIA, cbIB). Pediatr Res. 2007;62:225-230.
Kayer MA. Disorders of ketone production and utilization. Mol Genet Metab. 2006;87:281-283.
Korman SH. Inborn errors of isoleucine degradation: a review. Mol Genet Metab. 2006;89:289-299.
Lerner-Ellis JP, Anastasio N, Liu J, et al. Spectrum of mutations in MMACHC, allelic expression, and evidence for genotype-phenotype correlations. Hum Mutat. 2009;30:1072-1081.
Lerner-Ellis JP, Tirone JC, Pawelek PD, et al. Identification of the gene responsible for methylmalonic aciduria and homocystinuria, cbIC type. Nat Genet. 2006;38:93-100.
Pié J, López-Viñas E, Puisac B, et al. Molecular genetics of HMG-CoA lyase deficiency. Mol Genet Metab. 2007;92:198-209.
Rela M, Battula N, Madanur M, et al. Auxiliary liver transplantation for propionic acidemia: a 10-year follow-up. Am J Transplant. 2007;7:2200-2203.
Romano S, Valayannopoulos V, Touati G, et al. Cardiomyopathies in propionic aciduria are reversible after liver transplantation. J Pediatr. 2010;156:128-134.
Rutsch F, Gailus S, Miousse IR, et al. Identification of a putative lysosomal cobalamin exporter mutated in the cbIF inborn error of vitamin B12 metabolism. Nat Genet. 2009;41:234-239.
Scholl-Bürgi S, Haberlandt E, Gotwald T, et al. Stroke-like episodes in propionic academia caused by central focal metabolic decompensation. Neuropediatrics. 2009;40:76-81.
Shafqat N, Turnbull A, Zschocke J, et al. Crystal structures of human HMG-CoA synthase isoforms provide insights into inherited ketogenesis disorders and inhibitor design. J Mol Biol. 2010;398:497-506.
Simon E, Flaschker N, Schadewaldt P, et al. Variant maple syrup urine disease (MSUD)—the entire spectrum. J Inherit Metab Dis. 2006;29:716-724.
Simon E, Schwarz M, Wendel U. Social outcome in adults with maple syrup urine disease (MSUD). J Inherit Metab Dis. 2007;30:264.
Skvorak KJ. Animal models of maple syrup urine disease. J Inherit Metab Dis. 2009;32:229-246.
Strauss KA, Mazariegos GV, Sindhi R, et al. Elective liver transplantation for the treatment of classical maple syrup urine disease. Am J Transplant. 2006;6:557-564.
Strauss KA, Wardley B, Robinson D, et al. Classical maple syrup urine disease and brain development: principles of management and formula design. Mol Genet Metab. 2010;99:333-345.
Tilbrook LK, Slater J, Agarwal A, et al. An unusual cause of interference in a salicylate assay caused by mitochondrial acetoacetyl-CoA thiolase deficiency. Ann Clin Biochem. 2008;45(Pt 5):524-526.
van der Hilst JC, Bodar EJ, Barron KS, et al. Long-term follow-up, clinical features, and quality of life in a series of 103 patients with hyperimmunoglobulinemia D syndrome. Medicine (Baltimore). 2008;87:301-310.
Vockley J, Ensenauer R. Isovaleric acidemia: new aspects of genetic and phenotypic heterogeneity. Am J Med Genet C Semin Med Genet. 2006;142C:95-103.
Watkins D, Whitehead VM, Rosenblatt DS. Megaloblastic anemia. In: Orkin SH, Ginsburg D, Nathan DA, et al, editors. Nathan and Oski’s hematology of infancy and childhood. ed 7. Philadelphia: Elsevier; 2009:467-520.
Whitehead VM. Acquired and inherited disorders of cobalamin and folate in children. Br J Haematol. 2006;134:125-136.
Wolf B. Clinical issues and frequent questions about biotinidase deficiency. Mol Genet Metab. 2010;100:6-13.
Zinnanti WJ, Lazovic J, Griffin K, et al. Dual mechanism of brain injury and novel treatment strategy in maple syrup urine disease. Brain. 2009;132:903-918.
79.7 Glycine
Glycine is a nonessential amino acid synthesized mainly from serine and threonine. Structurally, it is the simplest amino acid. It is involved in many reactions in the body, especially in the nervous system where it functions as a neurotransmitter (excitatory in the cortex, inhibitory in the brainstem and the spinal cord; Chapter 79.11). Its main catabolic pathway requires the complex glycine cleavage enzyme to cleave the first carbon of glycine and convert it to carbon dioxide (Fig. 79-8). The glycine cleavage protein, a mitochondrial multienzyme, is composed of 4 proteins: P protein (glycine decarboxylase), H protein, T protein, and L protein, which are encoded by 4 different genes.
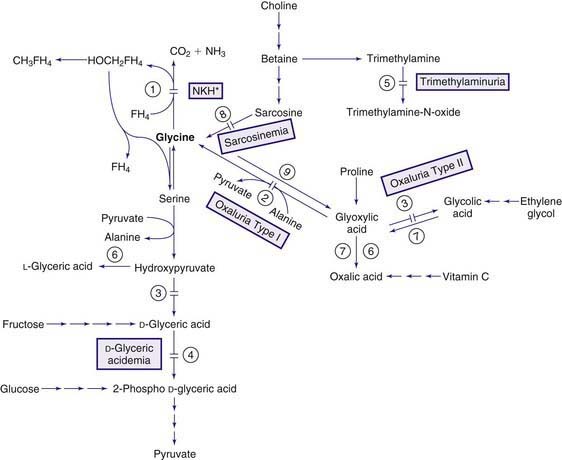
Hypoglycinemia
Defects in the biosynthetic pathway of serine (Chapter 79.8) cause deficiency of glycine in addition to that of serine in body fluids, especially in the CSF. Isolated primary deficiency of glycine has not been reported.
Hyperglycinemia
Elevated levels of glycine in body fluids occur in propionic acidemia and methylmalonic acidemia that are collectively referred to as ketotic hyperglycinemia because episodes of severe acidosis and ketosis occur. The pathogenesis of hyperglycinemia in these disorders is not fully understood, but inhibition of the glycine cleavage enzyme system by the various organic acids has been shown to occur in some of the patients. The term nonketotic hyperglycinemia (NKH) is reserved for the clinical condition caused by the genetic deficiency of the glycine cleavage enzyme system (see Fig. 79-8). In this condition, hyperglycinemia is present without ketosis.
Nonketotic Hyperglycinemia, Glycine Encephalopathy
Four forms of this condition have been identified: neonatal, infantile, late onset, and transient.
Infantile NKH
Laboratory findings in these patients are identical to those seen in the neonatal form.
Late-Onset NKH, Mild Episodic Form
Laboratory findings are similar to but not as pronounced as in the neonatal form.
Sarcosinemia
Increased concentrations of sarcosine (N-methylglycine) are observed in both blood and urine, but no consistent clinical picture has been attributed to this metabolic defect. This is a recessively inherited metabolic condition caused by a defect in sarcosine dehydrogenase, the enzyme that converts sarcosine to glycine (see Fig. 79-8). The gene for this enzyme is on chromosome 9q33-q34.
D-Glyceric Aciduria
D-Glyceric acid is an intermediate metabolite of serine and fructose metabolism (see Fig. 79-8). In this rare condition, clinical manifestations of severe encephalopathy (hypotonia, seizures, and mental and motor deficits) and the laboratory findings of hyperglycinemia and hyperglycinuria were suggestive of NKH. These patients excreted large quantities of D-glyceric acid. This compound is not normally detectable in urine. Enzyme studies indicated a deficiency of glycerate kinase in 1 patient and decreased activity of D-glyceric dehydrogenase in another.
Trimethylaminuria
Trimethylamine is normally produced in the intestine from the breakdown of dietary choline and trimethylamine oxide by bacteria. Egg yolk and liver are the main sources of choline, and fish is the major source of trimethylamine oxide. Trimethylamine is absorbed and oxidized in the liver by trimethylamine oxidase (flavin-containing monooxygenases) to trimethylamine oxide, which is odorless and excreted in the urine (see Fig. 79-8). Deficiency of this enzyme results in massive excretion of trimethylamine in urine. Several asymptomatic patients with trimethylaminuria have been reported; there is a foul body odor that resembles that of a rotten fish, which may have significant social and psychosocial ramifications. Restriction of fish, eggs, liver, and other sources of choline (such as nuts and grains) in the diet significantly reduce the odor. Treatment with short courses of oral metronidazole, neomycin or lactulose cause temporary reduction in the body odor. The gene for trimethylamine oxidase has been mapped to chromosome 1q23-q25.
Hyperoxaluria and Oxalosis
Normally, oxalic acid is derived mostly from oxidation of glyoxylic acid, and to a lesser degree, from oxidation of ascorbic acid (see Fig. 79-8). Glyoxylic acid is formed from the oxidation of glycolic acid and the deamination of glycine in the peroxisomes. The source of glycolic acid is poorly understood. Foods containing oxalic acid, such as spinach and rhubarb, are the main exogenous sources of this compound. Oxalic acid cannot be further metabolized in humans and is excreted in the urine as oxalates. Calcium oxalate is relatively insoluble in water and precipitates in tissues (kidneys and joints) if its concentration increases in the body.
Secondary hyperoxaluria has been observed in pyridoxine deficiency (cofactor for alanine-glyoxylate aminotransferase; see Fig. 79-8) after ingestion of ethylene glycol or high doses of vitamin C, after administration of the anesthetic agent methoxyflurane (which oxidizes directly to oxalic acid), and in patients with inflammatory bowel disease or extensive resection of the bowel (enteric hyperoxaluria). Acute, fatal hyperoxaluria may develop after ingestion of plants with a high oxalic acid content such as sorrel. Precipitation of calcium oxalate in tissues causes hypocalcemia, liver necrosis, renal failure, cardiac arrhythmia, and death. The lethal dose of oxalic acid is estimated to be between 5 and 30 g.
Primary Hyperoxaluria Type I
This rare condition is the most common form of primary hyperoxaluria. It is due to a deficiency of the peroxisomal enzyme alanine-glyoxylate aminotransferase, which is expressed only in the liver peroxisomes and requires pyridoxine (vitamin B6) as its cofactor. In the absence of this enzyme, glyoxylic acid, which cannot be converted to glycine, is transferred to the cytosol, where it is oxidized to oxalic acid (see Fig. 79-8).
Primary Hyperoxaluria Type II (L-Glyceric Aciduria)
This rare condition is due to a deficiency of D-glycerate dehydrogenase (hydroxypyruvate reductase)/glyoxylate reductase enzyme complex (see Fig. 79-8). A deficiency in the activity of this enzyme results in an accumulation of 2 intermediate metabolites, hydroxypyruvate (the ketoacid of serine) and glyoxylic acid. Both these compounds are further metabolized by lactate dehydrogenase (LDH) to L-glyceric acid and oxalic acid, respectively. About 30% of reported patients are from the Saulteaux-Ojibway Indians of Manitoba.
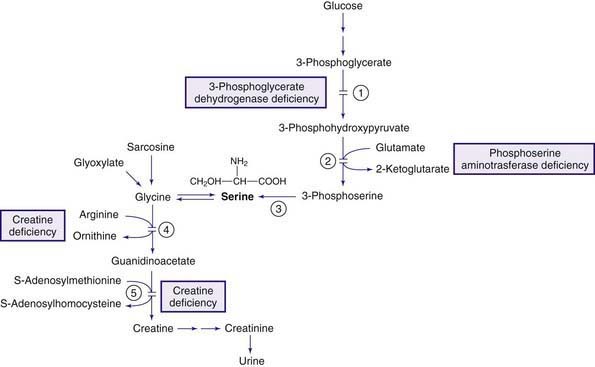
Creatine Deficiency
Creatine is synthesized in the liver, pancreas, and kidneys from arginine and glycine (Fig. 79-9) and is transported to muscles and the brain, in which there is high activity of the enzyme creatine kinase. Phosphorylation and dephosphorylation of creatine in conjunction with adenosine triphosphate and diphosphate (ATP and ADP), respectively, provide high-energy phosphate transfer reactions in these organs. Creatine is nonenzymatically metabolized to creatinine at a constant daily rate and is excreted in the urine. Three genetic conditions are known to cause creatine deficiency in tissues. Two are due to deficiency of the enzymes involved in the biosynthesis of creatine. The enzymes are arginine:glycine amidinotransferase (AGAT) and guanidinoacetate methyltransferase (GAMT) (see Fig. 79-9). Both conditions respond well to creatine supplementation. The third condition is caused by the defect in the creatinine transporter (CRTR) and is not responsive to creatine administration.
Barger AV, Capeau NG, Port JD, et al. MRS is the test of choice for detecting and monitoring disorders of creatine metabolism. Pediatr Neurol. 2009;40:408-410.
Boneh A, Allan S, Mendelson D, et al. Clinical, ethical and legal considerations in the treatment of newborns with non-ketotic hyperglycinaemia. Mol Genet Metab. 2008;94:143-147.
Chiong MA, Procopis P, Carpenter K, et al. Late-onset nonketotic hyperglycinemia with leukodystrophy and an unusual clinical course. Pediatr Neurol. 2007;37:283-286.
Coulter-Mackie M, White C, Hurley RM, et al. Primary hyperoxaluria type 1, 2009. GeneReviews at GeneTests: Medical Information Resource. University of Washington, Seattle, 1997-2010. www.genetests.org. Accessed June 2010
Dhar SU, Scaglia F, Li FY, et al. Expanded clinical and molecular spectrum of guanidinoacetate methyltransferase (GAMT) deficiency. Mol Genet Metab. 2009;96:38-43.
Hamosh A, Scharer G. Glycine encephalopathy, 2009. GeneReviews at GeneTests: Medical Information Resource. University of Washington, Seattle, 1997-2010. www.genetests.org. Accessed May 2010
Mercimek-Mahmutoglu S, Stockler-Ipsiroglu S. Creatine deficiency syndrome, 2009. GeneReviews at GeneTests: Medical Information Resource. University of Washington, Seattle, 1997-2010. www.genetests.org. Accessed May 2010
Nasrallah F, Feki M, Kaabachi N. Creatine and creatine deficiency syndromes: biochemical and clinical aspects. Pediatr Neurol. 2010;42:163-171.
Philips IR, Shepard EAR. Trimethylaminuria, 2008. GeneReviews at GeneTests: Medical Information Resource. University of Washington, Seattle, 1997-2010. www.genetests.org. Accessed May 2010
Rossi S, Saniele I, Bastrenta P, et al. Early myoclonic encephalopathy and nonketotic hyperglycinemia. Pediatr Neurol. 2009;41:371-374.
Rumsby G. Primary hyperoxaluria type 2, 2008. GeneReviews at GeneTests: Medical Information Resource. University of Washington, Seattle, 1997-2010. www.genetests.org. Accessed June 2010
79.8 Serine
Serine is a nonessential amino acid supplied through dietary sources and through its endogenous synthesis, mainly from glucose and glycine (see Fig. 79-9). The endogenous production of serine comprises an important portion of the daily requirement of this amino acid, especially in the synaptic junctions where it functions as a neurotransmitter (Chapter 79.11). Deficiencies of the enzymes involved in the biosynthesis of serine, therefore, cause neurologic manifestations. Affected patients respond favorably to oral supplementation with serine and glycine provided that the treatment is initiated very early in life. The catabolic pathway of serine is shown in Figure 79-8.
Phosphoserine Aminotransferase Deficiency
This enzyme catalyzes conversion of 3-phosphohydroxypyruvate to 3-phosphoserine (see Fig. 79-9). Deficiency of this enzyme has been reported in 2 siblings from an English family. Poor feeding, cyanotic episodes, and jerky movements developed shortly after birth in the 1st affected infant and progressed to intractable seizures by 9 wk of age. The infant was microcephalic. EEG was consistent with multifocal seizures. Neuroimaging showed generalized cerebral and cerebellar atrophies. Laboratory studies were all within normal limits except for mild decrease in plasma levels of serine and glycine with pronounced deficiencies of these 2 amino acids in the CSF. Treatment with serine (500 mg/kg/day) and glycine (200 mg/kg/day) was started at 11 wk of age but resulted in only marginal clinical improvement; the child died at 7 mo of age. The younger affected sibling, who was treated with serine and glycine within a few hours after birth, has remained asymptomatic at 3 yr of age.
de Koning TJ. Treatment with amino acids in serine deficiency disorders. J Inherit Metab Dis. 2006;29:347-351.
Hart CE, Race V, Achouri Y, et al. Phosphoserine aminotransferase deficiency: a novel disorder of the serine biosynthesis pathway. Am J Hum Genet. 2007;80:931-937.
Tabatabaie L, Klomp LW, Berger R, et al. L-serine synthesis in the central nervous system: a review on serine deficiency disorders. Mol Genet Metab. 2010;99:256-262.
79.9 Proline
Proline is a nonessential amino acid synthesized endogenously from glutamic acid, ornithine, and arginine (Fig. 79-10). Proline and hydroxyproline are found in high concentrations in collagen. Neither of these amino acids is normally found in urine in the free form except in early infancy. Excretion of proline and hydroxyproline as iminopeptides (dipeptides and tripeptides containing proline or hydroxyproline) reflects collagen turnover and is increased in disorders of accelerated collagen turnover, such as rickets or hyperparathyroidism. Proline is also found at synaptic junctions and functions as a neurotransmitter (Chapter 79.11).
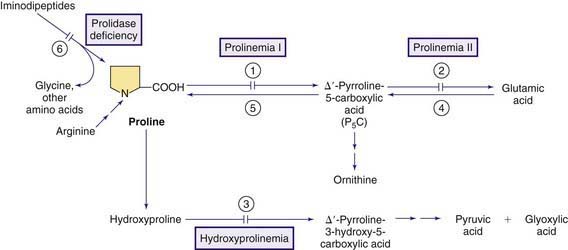
Hyperprolinemia
Two types of primary hyperprolinemia have been described.
Hyperprolinemia Type I
This rare autosomal recessive condition is due to deficiency of proline oxidase (proline dehydrogenase, see Fig. 79-10). Clinical manifestations are variable; some affected individuals are asymptomatic but patients with severe psychomotor retardation and seizures have been reported. Schizophrenia is a common finding in these patients. The gene for proline oxidase is located on chromosome 22q11.2 and several disease-causing mutations have been identified. Microdeletions involving this region of chromosome 22 cause velocardiofacial (DiGeorge, Shprintzen) syndrome; approximately 50% of patients with this syndrome have been reported to have hyperprolinemia type I. Therefore, all patients with hyperprolinemia type I should be screened (by FISH analysis) for presence of DiGeorge syndrome.
Hyperprolinemia Type II
This is a rare autosomal recessive condition caused by the deficiency of pyrroline-carboxylic acid dehydrogenase (aldehyde dehydrogenase 4, ALDH4; see Fig. 79-10). Psychomotor retardation (modest to severe) and seizures (usually precipitated by an intercurrent infection) have been reported in most affected children, but asymptomatic patients have also been reported.
Laboratory studies reveal increased concentrations of proline and Δ1-pyrroline-5-carboxylic acid (P5C) in blood, urine, and the CSF. Increased excretion of xanthurenic acid has also been reported in this condition. The presence of P5C differentiates this condition from hyperprolinemia type I (see earlier). Increased levels of P5C in body fluids, especially in the CNS, cause inactivation of vitamin B6 and generate a state of vitamin B6 dependency (Chapter 79.14). Deficiency of vitamin B6 is perhaps the main cause of neurologic findings in this condition and may explain the variability in clinical manifestations in different patients. Treatment with high doses of vitamin B6 in conjunction with a diet low in proline is recommended but the experience remains very limited because of paucity of patients. The gene for P5C dehydrogenase (ALDH4) is on chromosome 1p36.
79.10 Glutamic Acid
Glutamic acid and its aminated derivative glutamine have a wide range of functions in the body. One of the major products of glutamic acid is glutathione (γ-glutamylcysteinylglycine). This ubiquitous tripeptide, with its function as the major antioxidant in the body, is synthesized and degraded through a complex cycle called the γ-glutamyl cycle (Fig. 79-11). Because of its free sulfhydryl (-SH) group and its abundance in the cell, glutathione protects other sulfhydryl-containing compounds (such as enzymes and CoA) from oxidation. It is also involved in the detoxification of peroxides, including hydrogen peroxide, and in keeping the intracellular milieu in a reduced state. The common consequence of glutathione deficiency is hemolytic anemia. Glutathione also participates in amino acid transport across the cell membrane through the γ-glutamyl cycle. Glutamic acid is also the precursor of γ-aminobutyric acid (GABA), a major neurotransmitter in the nervous system (Chapter 79.11).
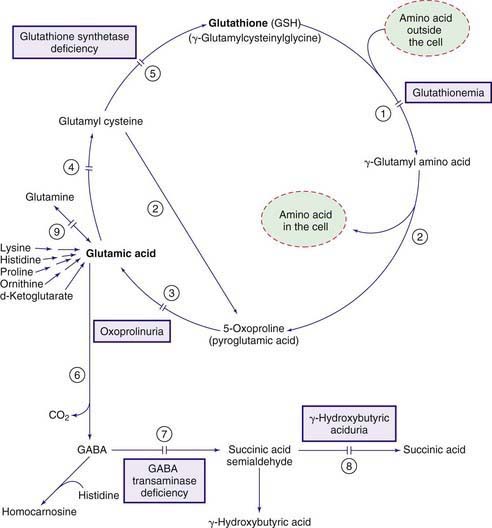
Glutathione Synthetase Deficiency (See Fig. 79-11)
Glutathione Synthetase Deficiency, Severe Form (Pyroglutamic Acidemia, Severe 5-Oxoprolinuria) and Moderate Form
Laboratory findings include metabolic acidosis, mild to moderate degrees of hemolytic anemia, and 5-oxoprolinuria. High concentrations of 5-oxoproline are also found in blood. The glutathione content of erythrocytes is markedly decreased. Increased synthesis of 5-oxoproline in this disorder is believed to be due to the conversion of γ-glutamylcysteine to 5-oxoproline by the enzyme γ-glutamyl cyclotransferase (see Fig. 79-11). γ-Glutamylcysteine production increases greatly because the normal inhibitory effect of glutathione on the γ-glutamylcysteine synthetase enzyme is removed. A deficiency of glutathione synthetase has been demonstrated in a variety of cells including erythrocytes.
γ-Glutamylcysteine Synthetase Deficiency
Only a few patients with this enzyme deficiency have been reported. The most consistent clinical manifestation has been mild chronic hemolytic anemia. Acute attacks of hemolysis have occurred after exposure to sulfonamides. Peripheral neuropathy and progressive spinocerebellar degeneration have been noted in 2 siblings in adulthood. Laboratory findings of chronic hemolytic anemia were present in all patients. Generalized aminoaciduria is also present because the γ-glutamyl cycle is involved in amino acid transport in cells (see Fig. 79-11). Treatment is that of hemolytic anemia and avoidance of drugs and oxidants that may trigger the hemolytic process. The condition is inherited as an autosomal recessive trait.
Glutathionemia (γ-Glutamyl Transpeptidase [GGT] Deficiency)
Laboratory findings include marked elevations in urinary concentrations of glutathione (up to 1 g/day), γ-glutamylcysteine, and cysteine. None of the reported patients has had generalized aminoaciduria, a finding that would have been expected to occur in this enzyme deficiency (see Fig. 79-11).
Häberle J, Görg B, Toutain A, et al. Inborn error of amino acid synthesis: human glutamine synthetase deficiency. J Inherit Metab Dis. 2006;29:352-358.
Njålsson R, Norgren S. Physiological and pathological aspects of GSH metabolism. Acta Paediatr. 2005;94:132-137.
Njålsson R, Ristoff E, Carlsson K, et al. Genotype, enzyme activity, glutathione level, and clinical phenotype in patients with glutathione synthetase deficiency. Hum Genet. 2005;116:384-389.
79.11 Genetic Disorders of Neurotransmitters
Neurotransmitters are released from the axonal end of excited neurons at the synaptic junctions and cause propagation and amplification or inhibition of neural impulses. A number of amino acids and their metabolites comprise the bulk of neurotransmitters. Mutations in genes responsible for the synthesis or degradation of these substances may cause conditions that are usually manifested by neurologic and/or psychiatric abnormalities (Table 79-2). Previously, affected children have been diagnosed with cerebral palsy, seizure disorder, parkinsonism, or dystonia. Correct diagnosis is important because most of these conditions respond favorably to therapy. Diagnosis requires specialized laboratory studies of the CSF in most cases because some of the neurotransmitters generated in the CNS (dopamine and serotonin) do not cross the blood-brain barrier and are therefore not detected in the serum or urine. An ever-growing number of these conditions are being identified; diseases that were once thought to be very rare are now diagnosed with increasing frequency.
Table 79-2 GENETIC DISORDERS OF NEUROTRANSMITTERS IN CHILDREN
| TRANSMITTER | SYNTHESIS DEFECTS | DEGRADATION DEFECTS |
|---|---|---|
| MONOAMINES | ||
| Dopamine | TH deficiency | MAO deficiency |
| Serotonin and dopamine | AADC deficiency | MAO deficiency |
| BH4 deficiency | ||
| With hyperphe | ||
| Without hyperphe | ||
| Norepinephrine | DβH deficiency | MAO deficiency |
| GABA | GAD deficiency? | GABA transaminase deficiency GHB aciduria |
| Histamine | HDC deficiency | ? |
| AMINO ACIDS | ||
| Proline | ? | Hyperprolinemia |
| Serine | 3-PGD, PSAT deficiencies | ? |
| Glycine | 3-PDG, PSAT deficiencies | NKH |
TH, tyrosine hydroxylase; MAO, monoamine oxidase; AADC, aromatic L-amino acid decarboxylase; BH4, tetrahydrobiopterin; Hyperphe, hyperphenylalaninemia; DβH, dopamine β-hydroxylase; GABA, γ-aminobutyric acid; GAD, glutamic acid decarboxylase; GHB, γ-hydroxybutyric; HDC, histidine decarboxylase; 3-PGD, 3-phosphoglycerate dehydrogenase; PSAT, phosphoserine aminotransferase; NKH, nonketotic hyperglycinemia.
Tyrosine Hydroxylase Deficiency (Infantile Parkinsonism, Autosomal Recessive Dopa-Responsive Dystonia)
Tyrosine hydroxylase catalyzes the formation of L-dopa from tyrosine (see Fig. 79-2). Deficiency of this enzyme has been reported in a few children with dystonia and parkinsonism. The clinical picture resembles that of autosomal dominant dystonia due to GTP cyclohydrolase deficiency (see later). The spectrum of the condition is not fully appreciated.
Aromatic L-Amino Acid Decarboxylase Deficiency
Aromatic L-amino acid decarboxylase (AADC) enzyme catalyzes decarboxylation of both 5-hydroxytryptophan (to form serotonin, see Fig. 79-5) and L-dopa (to generate dopamine, see Fig. 79-2). Clinical manifestations of this relatively rare enzyme deficiency are related to underproduction of both dopamine and serotonin. Poor feeding, lethargy, hypotension, hypothermia, eye rolling (oculogyric crises), and ptosis have been observed in affected neonates. Clinical findings in infants and older children include developmental delay, truncal hypotonia with hypertonia of limbs, oculogyric crises, extrapyramidal movements (choreoathetosis, dystonia, myoclonus), and autonomic abnormalities (sweating, salivation, irritability, temperature instability, hypotension). Laboratory findings include decreased concentrations of dopamine and serotonin and their metabolites (homovanillic acid, 5-hydroxyindoleacetic acid, vanillylmandelic acid and norepinephrine), and increased levels of 5-hydroxytryptophan, L-dopa and its metabolite (3-O-methyldopa) in body fluids, especially in CSF. Elevated serum concentrations of prolactin (due to dopamine deficiency) have also been observed. The electroencephalogram and imaging of the brain are usually normal but progressive cerebral atrophy may be present in older patients. Patients with AADC enzyme deficiency may be misdiagnosed as having cerebral palsy, epilepsy, mitochondrial cytopathy, myasthenia gravis or dystonia. The condition should also be differentiated from other neurotransmitter disorders such as tyrosine hydroxylase deficiency and Segawa disease. Treatment with neurotransmitter precursors has produced limited clinical improvement. Dopamine and serotonin have no therapeutic value because of their inability to cross the blood-brain barrier. Dopamine agonists (L-dopa/carbidopa, bromocriptine), monoamine oxidase (MAO) inhibitors (tranylcypromine), serotonergic agents and high doses of pyridoxine (cofactor for AADC enzyme) have been tried. No treatment of choice has yet emerged because of the paucity of patients. The condition is inherited as an autosomal recessive trait and several disease-producing mutations have been identified in different families. The condition seems to be more severe in females. The gene for the enzyme is mapped to chromosome 7p11.
Tetrahydrobiopterin (BH4) Deficiency (Chapter 79.1)
Tetrahydrobiopterin is the cofactor for PAH (see Fig. 79-1), tyrosine hydroxylase (see Fig. 79-2), tryptophan hydroxylase (see Fig. 79-5), and nitric oxide synthase. It is synthesized from GTP in many tissues of the body (see Fig. 79-1). Deficiencies of enzymes involved in the biosynthesis of BH4 result in inadequate production of this cofactor which in turn causes deficiencies of the neurotransmitters serotonin and dopamine with or without concomitant hyperphenylalaninemia.
BH4 Deficiency Without Hyperphenylalaninemia
Hereditary progressive dystonia, autosomal dominant dopa-responsive dystonia, Segawa disease (Chapter 590.3).
Laboratory findings show no hyperphenylalaninemia, but reduced levels of BH4 and neopterin are found in the CSF. Dopamine and its metabolite (homovanillic acid) may also be reduced in the spinal fluid. The serotonergic pathway is less affected by this enzyme deficiency; thus, concentrations of serotonin and its metabolites are usually normal. Plasma phenylalanine is normal but an oral phenylalanine loading test (100 mg/kg) produces an abnormally high plasma phenylalanine level with a high ratio of phenylalanine to tyrosine. It is believed that the enzyme deficiency in this condition is less severe than that of the autosomal recessive form of GTP cyclohydrolase I deficiency, which is associated with hyperphenylalaninemia (Chapter 79.1). The existence of asymptomatic carriers indicates that other factors or genes may play a role in the pathogenesis of the phenotype. The asymptomatic carrier may be identified by the phenylalanine loading test (see earlier).
Diagnosis may be confirmed by reduced levels of BH4 and neopterin in the spinal fluid, by measurement of the enzyme activity, and by identification of the gene defect (Chapter 79.1). Clinically, the condition should be differentiated from other causes of dystonias and childhood parkinsonism, especially tyrosine hydroxylase, sepiapterin reductase, and aromatic amino acid decarboxylase deficiencies.
Sepiapterin Reductase Deficiency
Sepiapterin reductase is 1 of the enzymes that is involved in conversion of 6-pyruvoyl-tetrahydropterin to tetrahydrobiopterin (BH4). It also participates in the salvage pathway of tetrahydrobiopterin synthesis (see Fig. 79-1). Deficiency of this enzyme results in accumulation of 6-lactoyl-tetrahydropterin, which is converted to sepiapterin nonenzymatically. Most of the sepiapterin is metabolized to tetrahydrobiopterin through the salvage pathway in peripheral tissues (see Fig. 79-1), but because of the low enzyme activity of dihydrofolate reductase (DHFR) in the human brain, the amount of BH4 remains inadequate for proper synthesis of dopamine and serotonin in the CNS. This explains the lack of hyperphenylalaninemia in this condition. Fewer than 40 patients with this disorder have been identified but the condition may be underdiagnosed since the diagnosis requires highly specialized assays of the CSF.
Monoamine Oxidase (MAO) Deficiency
There are 2 monoamine oxidase isoenzymes, MAO A and MAO B. Both enzymes catalyze oxidative deamination of most biogenic amines in the body including serotonin (see Fig 79-5), norepinephrine, epinephrine, and dopamine (see Fig. 79-2). The genes for both isoenzymes are on chromosome X (Xp11.23). The deficiencies of these enzymes, therefore, are expected to be of clinical significance mainly in hemizygous males. Deficiency of MAO A has been reported in a large Dutch kindred. All affected males showed mild mental retardation with aggressive, violent behavior. MAO B deficiency has been found in patients with Norrie disease (Chapter 614); the importance of the enzyme deficiency in the pathogenesis of this condition is not known. Isolated MAO B deficiency has not been reported. Etiologic contribution of the deficiency of these enzymes to psychiatric diseases has been postulated but has not been supported by clinical studies. Diagnosis is established by elevated levels of norepinephrine, dopamine, serotonin, in conjunction with low levels of their metabolites in body fluids. No effective therapy has yet emerged.
γ-Aminobutyric Acid (GABA)
γ-Aminobutyric acid (GABA) is the main inhibitory neurotransmitter, which is synthesized in the synapses through decarboxylation of glutamic acid by glutamic acid decarboxylase (GAD). The same pathway is responsible for production of GABA in other organs, especially the kidneys and the β cells of the pancreas. GABA is metabolized to succinic acid by 2 enzymes, GABA transaminase and succinic semialdehyde dehydrogenase (SSADH) (see Fig. 79-11).
γ-Hydroxybutyric Aciduria (Succinic Semialdehyde Dehydrogenase Deficiency)
This is the most common genetic disorder of neurotransmitters. More than 350 patients with this enzyme deficiency (see Fig. 79-11) have been identified. Clinical manifestations, which usually begin in early infancy, include mild to moderate mental retardation, delayed speech, marked hypotonia, neuropsychiatric symptoms (sleep disturbances, anxiety, inattention, hyperactivity), nonprogressive ataxia, and seizures. Other associated findings are autistic features, hallucinations, and aggressive behavior.
The role of γ-hydroxybutyric acid in the pathogenesis of this condition remains unclear, since administration of this compound to humans and animals has produced variably conflicting effects. γ-Hydroxybutyrate (GHB) has been used illicitly as a recreational drug with anesthetic effect and is 1 of the date-rape drugs (Chapter 108).
Hyperprolinemia
Psychomotor retardation and seizures are common findings in most patients with hyperprolinemia type I and type II. Patients with type I hyperprolinemia also have an increased risk of developing schizophrenia. The contribution of increased concentration of proline to the pathogenesis of these conditions, however, remains unclear. The neurologic abnormalities observed in hyperprolinemia type II is now believed to be mainly due to development of vitamin B6 dependency in this condition (Chapter 79.9).
Acosta MT, Munasinghe J, Pearl PL, et al. Cerebellar atrophy in human and murine succinic semialdehyde dehydrogenase deficiency. J Child Neurol. 2010;25:1457-1461.
Dósa Z, Nieto-Gonzalez JL, Korghoej AR, et al. Effect of gene dosage on single-cell hippocampal and electrophysiology in a murine model of SSADH deficiency (gamma-hydroxybutyric aciduria). Epilepsy Res. 2010;90:39-46.
Ercan-Sencicek AG, Stillman AA, Ghosh AK, et al. L-histidine decarboxylase and Tourette’s syndrome. N Engl J Med. 2010;362:1901-1908.
Hyland K, Gibson KM, Sharma R, et al. Neurotransmitter disorders. In: Sarafoglu K, Hoffmann GF, Roth K, editors. Pediatric endrocrinology and inborn errors of metabolism. New York: McGraw Hill; 2009:789-820.
Knerr I, Gibson KM, Murdoch G, et al. Neuropathology in succinic semialdehyde dehydrogenase deficiency. Pediatr Neurol. 2010;42:255-258.
Pearl PL, Capp PK, Novotny EJ, et al. Inherited disorders of neurotransmitters in children and adults. Clin Biochem. 2005;38:1051-1058.
Pearl PL, Gibson KM, Cortez MA, et al. Succinic semialdehyde dehydrogenase deficiency: lessons from mice and men. J Inherit Metab Dis. 2009;32:343-352.
Pearl PL, Gibson KM, Quezado Z, et al. Decreased GABA-A binding on FMZ-PET in succinic semialdehyde dehydrogenase deficiency. Neurology. 2009;73:423-429.
Pearl PL, Jakobs C, Gibson KM. Disorders of β and Y amino acids in free and peptide linked forms. In: Scriver CR, Beaudet AL, Sly WS, et al, editors. The metabolic and molecular bases of inherited disease. New York: McGraw Hill, 2008.
Pearl PL, Taylor JL, Trzcinski S, et al. The pediatric neurotransmitter disorders. J Child Neurol. 2007;22:606-616.
Tsuji M, Aida N, Obata T, et al. A new case of GABA transaminase deficiency facilitated by proton MR spectroscopy. J Inherit Metab Dis. 2010;33:85-90.
79.12 Urea Cycle and Hyperammonemia (Arginine, Citrulline, Ornithine)
Catabolism of amino acids results in the production of free ammonia, which, in high concentration, is extremely toxic to the CNS. In mammals ammonia is detoxified to urea via the urea cycle (Fig. 79-12). Five enzymes are involved in the synthesis of urea: carbamyl phosphate synthetase (CPS), ornithine transcarbamylase (OTC), argininosuccinate synthetase (AS), argininosuccinate lyase (AL), and arginase. A 6th enzyme, N-acetylglutamate synthetase, is also required for synthesis of N-acetylglutamate, which is an activator (effector) of the CPS enzyme. Individual deficiencies of these enzymes have been observed and, with an overall estimated prevalence of 1/30,000 live births, they are the most common genetic causes of hyperammonemia in infants.
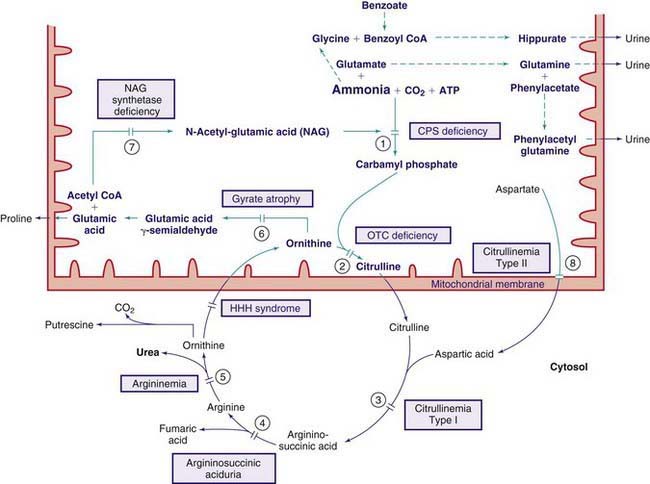
Genetic Causes of Hyperammonemia
In addition to genetic defects of the urea cycle enzymes, a marked increase in plasma level of ammonia is observed in other genetic disorders of metabolism (Table 79-3); the pathogenesis for hyperammonemia in some of these conditions is not fully understood.
Diagnosis
The main criterion for diagnosis is hyperammonemia. Each clinical laboratory should establish its own normal values for blood ammonia; some variation is common. In the older child and adult, the normal limit is typically <35 µmole/L. Blood concentrations in healthy newborn infants often are higher (as high as 100 µmole/L in a full-term baby and 150 µmole/L in a premature infant). An approach to the differential diagnosis of hyperammonemia in the newborn infant is illustrated in Figure 79-13. Plasma amino acids reveal abnormalities that may help the diagnosis. In patients with deficiencies of either CPS, OTC, or N-acetylglutamate (NAG) synthetase frequent findings are elevations in plasma glutamine and alanine with concurrent decrements in citrulline and arginine. These disorders cannot be differentiated from one another by the plasma amino acid levels alone. A marked increase in urinary orotic acid in patients with OTC deficiency differentiates this defect from CPS deficiency. Differentiation between the CPS deficiency and the NAG synthetase deficiency may require an assay of the respective enzymes. Clinical improvement occurring after oral administration of carbamylglutamate, however, may suggest NAG synthetase deficiency. Patients with deficiencies of AS, AL, or arginase have marked increases in the plasma levels of citrulline, argininosuccinic acid, or arginine, respectively. Indeed, the combination of hyperammonemia and marked hypercitrullinemia or argininosuccinic acidemia is virtually pathognomonic for these disorders.
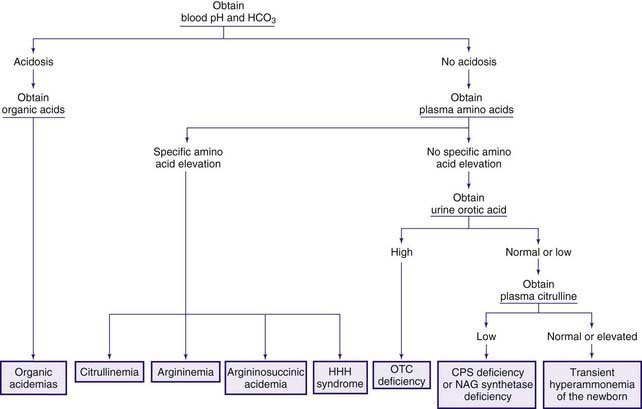
Treatment of Acute Hyperammonemia
The outcome for patients with hyperammonemic episodes depends mainly on the severity and duration of the hyperammonemia. Serious neurologic sequelae are highly likely in newborn infants with severe elevations in blood ammonia (>300 µmole/L) for more than 12 hr. Thus, hyperammonemia should be treated promptly and vigorously. The goal of therapy is to lower the concentration of ammonia in the body. This is accomplished in two ways: (1) by removal of ammonia from the body in a form other than urea, and (2) by provision of adequate calories and essential amino acids to minimize the endogenous protein degradation and favor protein synthesis (Table 79-4). Fluid, electrolytes, glucose (5-15%), and lipids (1-2 g/kg/24 hr) should be infused intravenously together with a minimal amount of protein (0.25 g/kg/24 hr), preferably in the form of essential amino acids. As soon as the clinical condition of the patient allows, oral feeding with a low-protein formula (0.5-1.0 g/kg/24 hr) through a nasogastric tube should be started.
Table 79-4 TREATMENT OF ACUTE HYPERAMMONEMIA IN AN INFANT
* The concentration of sodium chloride should be calculated to be 0.45% to 0.9% including the amount of the sodium in the drugs.
† These compounds are usually prepared as a 1-2% solution for intravenous use. Sodium from these drugs should be included as part of the daily sodium requirement.
‡ The higher dose is recommended in the treatment of patients with citrullinemia and argininosuccinic aciduria. Arginine is not recommended in patients with arginase deficiency and in those whose hyperammonemia is secondary to organic acidemia.
An important advance in treatment of hyperammonemia has been the advent of acylation therapy in which exogenously administered organic acids form acyl adducts with endogenous nonessential amino acids. These adducts are nontoxic compounds with high renal clearances. The main organic acids used for this purpose are sodium salts of benzoic acid and phenylacetic acid. Benzoate forms hippuric acid with endogenous glycine in the liver (see Fig. 79-12). Each mole of benzoate removes 1 mole of ammonia as glycine. Phenylacetate conjugates with glutamine to form phenylacetylglutamine, which is readily excreted in the urine. One mole of phenylacetate removes 2 moles of ammonia as glutamine from the body (see Fig. 79-12).
Arginine administration is effective in the treatment of hyperammonemia that is due to most defects of the urea cycle because it supplies the urea cycle with ornithine and NAG (see Fig. 79-12). In patients with citrullinemia, 1 mole of arginine reacts with 1 mole of ammonia (as carbamyl phosphate) to form citrulline. In patients with argininosuccinic acidemia, 2 moles of ammonia (as carbamyl phosphate and aspartate) react with arginine to form argininosuccinic acid. Citrulline and argininosuccinic acid are far less toxic and more readily excreted by the kidneys than ammonia. In patients with CPS or OTC deficiency, arginine administration is indicated because arginine becomes an essential amino acid in these disorders. Patients with OTC deficiency benefit from supplementation with citrulline (200 mg/kg/24 hr), which reacts with 1 mole of ammonia (as aspartic acid) to form arginine. Administration of arginine or citrulline is contraindicated in patients with arginase deficiency, a rare condition in which the presenting clinical picture is one of spastic diplegia rather than hyperammonemia (see later). Furthermore, arginine therapy is of no benefit if hyperammonemia is secondary to an organic acidemia. However, in a newborn infant with a 1st attack of hyperammonemia, arginine should be used until the diagnosis is established.
Benzoate, phenylacetate, and arginine may be administered together for maximal therapeutic effect. A priming dose of these compounds is followed by continuous infusion until recovery from the acute state occurs (see Table 79-4). Both benzoate and phenylacetate are usually supplied as concentrated solutions and should be properly diluted (1-2% solution) for intravenous use. The recommended therapeutic doses of both compounds deliver a substantial amount of sodium to the patient that should be calculated as part of the daily sodium requirement. A commercial preparation of sodium benzoate plus sodium phenylacetate is available for intravenous use (Ammonul; www.ammonul.com). Benzoate and phenylacetate should be used with caution in newborn infants with hyperbilirubinemia because they may displace bilirubin from albumin. However, despite this theoretical risk, no documented case of kernicterus (Chapter 96.4) has yet been reported in neonates with hyperammonemia who have received such therapies.
Long-Term Therapy
Once the infant is alert, therapy should be tailored to the underlying cause of the hyperammonemia. In general, all patients, regardless of the enzymatic defect, require some degree of protein restriction (1-2 g/kg/24 hr). In patients with defects in the urea cycle, chronic administration of benzoate (250-500 mg/kg/24 hr), phenylacetate (250-500 mg/kg/24 hr), and arginine (200-400 mg/kg/24 hr) or citrulline (in patients with OTC deficiency, 200-400 mg/kg/24 hr) is effective in maintaining blood ammonia levels within the normal range. Phenylbutyrate may be used in place of phenylacetate, because the patient and the family may not accept the latter owing to its offensive odor. A commercial preparation of the compound is available for oral use (Buphenyl; www.buphenyl.com).
Carbamyl Phosphate Synthetase (CPS) and N-Acetylglutamate (NAG) Synthetase Deficiencies (See Fig. 79-12)
Laboratory findings include hyperammonemia. The plasma aminogram commonly shows a marked increase of glutamine and alanine with a relatively low levels of citrulline and arginine. Urinary orotic acid is usually low or may be absent (see Fig. 79-13).
Treatment of acute hyperammonemic attacks and the long-term therapy of the condition is outlined earlier (see Table 79-4). Patients with NAG synthetase deficiency benefit from oral administration of carbamylglutamate. It is therefore important to differentiate between CPS and NAG synthetase deficiencies by assay of the enzyme activities in liver biopsy specimens. Deficiency of NAG synthetase is rare in North America.
Ornithine Transcarbamylase (OTC) Deficiency (See Fig. 79-12)
The major laboratory finding during the acute attack is hyperammonemia accompanied by marked elevations of plasma concentrations of glutamine and alanine with low levels of citrulline and arginine. Blood level of urea (BUN) is usually low. A marked increase in the urinary excretion of orotic acid differentiates this condition from CPS deficiency (see Fig. 79-13). Orotates may precipitate in urine as pink gravel. In the mild form, these laboratory abnormalities may revert to normal between attacks. This form should be differentiated from all the episodic conditions of childhood. In particular, patients with lysinuric protein intolerance (Chapter 79.13) may demonstrate some features of OTC deficiency, but the former can be differentiated by increased urinary excretion of lysine, ornithine, and arginine and elevated blood concentrations of citrulline.
Argininosuccinate Synthetase (AS) Deficiency (Citrullinemia) (See Fig. 79-12)
Citrullinemia Type I (Classic Citrullinemia, CTLN 1)
This condition is caused by the deficiency of AS (see Fig. 79-12) and has variable clinical manifestation depending on the degree of the enzyme deficiency. Two major forms of the condition have been identified. The severe or neonatal form, which is most common, appears in the 1st few days of life with signs and symptoms of hyperammonemia (see earlier). In the subacute or mild form, clinical findings such as failure to thrive, frequent vomiting, developmental delay, and dry, brittle hair appear gradually after 1 yr of age. Acute hyperammonemia, triggered by an intercurrent catabolic state, may bring the diagnosis to light.
Laboratory findings are similar to those found in patients with OTC deficiency except that the plasma citrulline concentration is markedly elevated (50-100 times normal) in citrullinemia type I (see Fig. 79-13). Urinary excretion of orotic acid is moderately increased; crystalluria due to precipitation of orotates may also occur. The diagnosis is confirmed by enzyme assay in cultured fibroblasts or by DNA analysis. Prenatal diagnosis is feasible with the assay of the enzyme activity in cultured amniotic cells or by DNA analysis of chorionic villi biopsy.
Citrullinemia Due to Citrin Deficiency (Citrullinemia Type II, CLTN 2)
Citrin (aspartate-glutamate carrier, AGC2) is a mitochondrial transport protein encoded by a gene (SLC25A13) on chromosome 7q21.3. The major function of this protein is to transport aspartate from mitochondria to cytoplasm; aspartate is required for converting citrulline to argininosuccinic acid (see Fig. 79-12). If aspartate is unavailable to the cytoplasmic component of the urea cycle, urea will not be formed at a normal rate and citrulline will accumulate in the body. AS activity is deficient in the liver of these patients, but no mutation in the gene for AS has been found. It is postulated that citrin deficiency or its mutated gene interferes with translation of mRNA for AS enzyme in the liver. The condition initially was reported among Japanese individuals but a few non-Japanese patients have also been identified. Two forms of citrin deficiency have been described.
Neonatal Intrahepatic Cholestasis (Citrullinemia Type II-Neonatal Form)
Clinical and laboratory manifestations, which usually start before 1 yr of age, include cholestatic jaundice with mild to moderate direct (conjugated) hyperbilirubinemia, marked hypoproteinemia, clotting dysfunction (increased prothrombin time and partial thromboplastin times), and increased serum γ-glutamyltranspeptidase (GGTP) and alkaline phosphatase activities; liver transaminases are usually normal. Plasma concentrations of ammonia and citrulline are usually normal, but moderate elevations are reported. There may be increases in plasma concentrations of methionine, tyrosine, alanine, and threonine. Elevated levels of serum galactose may occur, but all enzymes involved in galactose metabolism are normal. The reason for hypergalactosemia is not known. Marked elevation in serum level of α-fetoprotein is also present. These findings resemble those of tyrosinemia type I, but unlike the latter condition, urinary excretion of succinylacetone is not elevated (Chapter 79.2). Liver biopsy shows fatty infiltration, cholestasis with dilated canaliculi, and a moderate degree of fibrosis. The condition is usually self-limiting and the majority of infants recover spontaneously by 1 yr of age with only supportive and symptomatic treatment. Hyperammonemia and hypercitrullinemia, if present, should be treated with low-protein diet and other appropriate measures (see earlier). Hepatic failure requiring liver transplantation has occurred in a few cases. The diagnosis should be considered in cases of unexplained neonatal hepatitis with cholestasis. Data on the long-term prognosis and the natural history of the condition are limited; development into the adult form of the condition after several years of seemingly asymptomatic hiatus has been observed.
Argininosuccinate Lyase (AL) Deficiency (Argininosuccinic Aciduria) (See Fig. 79-12)
Laboratory findings include hyperammonemia, moderate elevations in liver enzymes, nonspecific increases in plasma levels of glutamine and alanine, moderate increase in plasma levels of citrulline (less than that seen in citrullinemia), and marked increase in plasma levels of argininosuccinic acid (see Fig. 79-13). In most amino acid analyzers, argininosuccinic acid appears as series of anhydrides within the isoleucine or methionine region, which may cause confusion in the diagnosis. Argininosuccinic acid can also be found in large amounts in urine and spinal fluid. The levels in the spinal fluid are usually higher than those in plasma. The enzyme is normally present in erythrocytes, the liver, and cultured fibroblasts. Prenatal diagnosis is possible by measurement of the enzyme activity in cultured amniotic cells or by identification of the mutant gene. Argininosuccinic acid is also elevated in the amniotic fluid of affected fetuses.
Arginase Deficiency (Hyperargininemia) (See Fig. 79-12)
Laboratory findings include marked elevations of arginine in plasma and CSF (see Fig. 79-13). Urinary orotic acid is moderately increased. Plasma ammonia levels may be normal or mildly elevated. Urinary excretions of arginine, lysine, cystine, and ornithine are usually increased, but normal levels have also been noted. Therefore, determination of amino acids in plasma is a critical step in the diagnosis of argininemia. The guanidino compounds (α-keto-guanidinovaleric acid, argininic acid) are markedly increased in urine. The diagnosis is confirmed by assaying arginase activity in erythrocytes.
Ornithine
Ornithine is 1 of the intermediate metabolites of the urea cycle that is not incorporated into natural proteins. Rather, it is generated in the cytosol from arginine and must be transported into the mitochondria where it is used as a substrate for the enzyme OTC to form citrulline. Excess ornithine is catabolized by 2 enzymes, ornithine 5-aminotransferase, which is a mitochondrial enzyme and converts ornithine to a proline precursor, and ornithine decarboxylase, which resides in the cytosol and converts ornithine to putrescine (see Fig. 79-12). Two genetic disorders result in hyperornithinemia: gyrate atrophy of the retina and hyperammonemia-hyperornithinemia-homocitrullinemia syndrome.
Gyrate Atrophy of the Retina And Choroid
This is a rare autosomal recessively inherited disorder caused by the deficiency of the enzyme ornithine 5-aminotransferase (see Fig. 79-12). About 30% of the reported cases are from Finland. Clinical manifestations are limited to the eyes and include night blindness, myopia, loss of peripheral vision, and posterior subcapsular cataracts. These eye changes start between 5 and 10 yr of age and progress to complete blindness by the 4th decade of life. Atrophic lesions in the retina resemble cerebral gyri. These patients usually have normal intelligence. There is a 10- to 20-fold increase in plasma levels of ornithine (400-1,400 µmole/L). There are no hyperammonemia and no increases in any other amino acids; plasma levels of glutamate, glutamine, lysine, creatine, and creatinine are moderately decreased. Some patients respond partially to high doses of pyridoxine (500-1,000 mg/24 hr). Arginine-restricted diet in conjunction with supplemental lysine, proline, and creatine has been successful in reducing plasma ornithine concentration and has produced some clinical improvements. The gene for ornithine 5-aminotransferase is on chromosome 10q26. Many (at least 60) disease-causing mutations have been identified in different families.
Hyperammonemia-Hyperornithinemia-Homocitrullinemia (HHH) Syndrome
In this rare autosomal recessively inherited disorder, the defect is in the transport system of ornithine from the cytosol into the mitochondria, resulting in accumulation of ornithine in the cytosol and its deficiency in the mitochondria. The former causes hyperornithinemia and the latter results in disruption of the urea cycle and hyperammonemia (see Fig. 79-12). Homocitrulline is presumably formed from the reaction of mitochondrial carbamyl phosphate with lysine, which occurs because of the intramitochondrial deficiency of ornithine. Clinical manifestations of hyperammonemia may develop shortly after birth or may be delayed until adulthood. Acute episodes of hyperammonemia manifest as refusal to feed, vomiting, and lethargy; coma may occur during infancy. Progressive neurologic signs, such as lower limb weakness, increased deep tendon reflexes, spasticity, clonus, seizures, and varying degrees of psychomotor retardation may develop if the condition remains undiagnosed. No clinical ocular findings have been observed in these patients.
Cederbaum S, Grombez EA. Arginase deficiency, 2009. GeneReviews at GeneTests: Medical Information Resource. University of Washington, Seattle, 1997-2010. www.genetests.org. Accessed June 2010
Cederbaum S, LeMons C, Lee B. New developments and future directions for urea cycle disorders. Mol Genet Metab. 2010;100(Suppl 1):1-106.
Enns GM. Neurologic damage and neurocognitive dysfunction in urea cycle disorders. Semin Pediatr Neurol. 2008;15:132-139.
Ficicioglu C, Mandell R, Shih VE. Argininosuccinate lyase deficiency: long-term outcome of 13 patients detected by newborn screening. Mol Genet Metab. 2009;98:273-277.
Kimura A, Kage M, Nagata I, et al. Histological findings in the livers of patients with neonatal intrahepatic cholestasis caused by citrin deficiency. Hepatol Res. 2010;40:295-303.
Saheki T, Inoue K, Tushima A, et al. Citrin deficiency and current treatment concepts. Mol Genet Metab. 2010;100(Suppl 1):S59-S64.
Summar ML, Dobbelaere D, Brusilow S, et al. Diagnosis, symptoms, frequency and mortality of 260 patients with urea cycle disorders from a 21-year, multicentre study of acute hyperammonaemic episodes. Acta Paediatr. 2008;97:1420-1425.
Thoene JG. Citrullinemia type I, 2009. GeneReviews at GeneTests: Medical Information Resource. University of Washington, Seattle, 1997-2010. www.genetests.org. Accessed June 2010
Tuchman M, Lee B, Lichter-Konecki U, et alfor the Urea Cycle Disorders Consortium of the Rare Diseases Clinical Research Network. Cross-sectional multicenter study of patients with urea cycle disorders in the United States. Mol Genet Metab. 2008;94:397-402.
Tessa A, Fiermonte G, Dionisi-Vici C, et al. Identification of novel mutations in the SLC25A15 gene in hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome: a clinical, molecular, and functional study. Hum Mutat. 2009;20:741-748.
Valayannopoulos V, Boddaert N, Mention K, et al. Secondary creatine deficiency in ornithine delta-aminotransferase deficiency. Mol Genet Metab. 2009;97:109-113.
Yudkoff M, Mew NA, Daikhin Y, et al. Measuring in vivo ureagenesis with stable isotopes. Mol Genet Metab. 2010;100(Suppl 1):S37-S41.
79.13 Histidine
Decarboxylation of histidine by histidine decarboxylase produces histamine. Deficiency of this enzyme has been shown to be the cause of familial form of Tourette’s syndrome (Chapter 79.11).
79.14 Lysine
Lysine is catabolized through 2 pathways. In the first pathway, lysine is condensed with α-ketoglutaric acid to form saccharopine. Saccharopine is then metabolized to α-aminoadipic acid semialdehyde and glutamic acid. These 1st 2 steps are catalyzed by α-aminoadipic semialdehyde synthase, which has 2 activities: lysine-ketoglutarate reductase, and saccharopine dehydrogenase (Fig. 79-14). In the second pathway, lysine is first transaminated and then condensed to its cyclic forms, pipecolic acid and piperidine-6-carboxylic acid. The latter compound and its linear form, α-aminoadipic acid semialdehyde, are oxidized to α-aminoadipic acid by the enzyme antiquitin. This is the major pathway for D-lysine in the body and for the L-lysine in the brain (see Fig. 79-14).
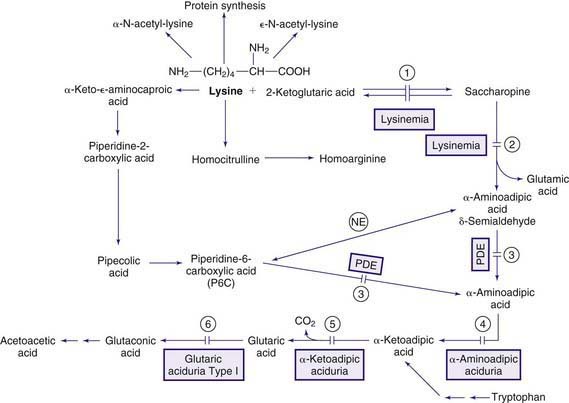
Pyridoxine (Vitamin B6)-Dependent Epilepsy
Antiquitin (α-Aminoadipic Semialdehyde Dehydrogenase) Deficiency
This is the most common cause of pyridoxine-dependent epilepsy. Deficiency of antiquitin (named because of preservation of its structure from garden pea to human) results in accumulation of Δ1-piperidine-6-carboxylic acid (P6C) in brain tissue (see Fig. 79-14); P6C reacts with pyridoxal 5′-phosphate and renders it inactive. Large doses of pyridoxine are, therefore, needed to overcome this inactivation.
Hyperprolinemia Type II
In this condition, accumulation of Δ1-pyrroline-5-carboxylic acid (P5C) in brain tissue causes inactivation of pyridoxal 5′-phosphate and hence pyridoxine dependency (Chapter 79.9 and Fig. 79-10).
Hypophosphatasia
Pyridoxal 5′-phosphate is the main circulating form of pyridoxine. Alkaline phosphatase is required for dephosphorylation of pyridoxal 5′-phosphate to generate free pyridoxine which is the only form of vitamin B6 that can cross the blood-brain barrier and enter the brain cells. Pyridoxine is rephosphorylated intracellularly to form pyridoxal 5′-phosphate. In the infantile form of hypophosphatasia, marked deficiency of tissue nonspecific alkaline phosphatase causes intracellular deficiency of pyridoxine and, therefore, pyridoxine-dependent epilepsy (Chapter 696).
Glutaric Aciduria Type I
Glutaric acid is an intermediate in the degradation of lysine (see Fig. 79-14), hydroxylysine, and tryptophan. Glutaric aciduria type I, a disorder caused by a deficiency of glutaryl CoA dehydrogenase, should be differentiated from glutaric aciduria type II, a distinct clinical and biochemical disorder caused by defects in the electron transport system (Chapter 80.1).
Lysinuric Protein Intolerance (Familial Protein Intolerance)
Mild anemia and increased serum levels of ferritin, lactic dehydrogenase (LDH), and thyroxine-binding globulin have also been observed in these patients. This condition should be differentiated from hyperammonemia due to urea cycle defects (Chapter 79.11), especially in heterozygous females with OTC deficiency. Increased urinary excretion of lysine, ornithine, and arginine and elevated blood levels of citrulline are not seen in patients with OTC deficiency.
Treatment with a low-protein diet (1.0-1.5 g/kg/24 hr) supplemented with citrulline (3-8 g/day) has produced biochemical and clinical improvements. Episodes of hyperammonemia should be treated promptly (Chapter 79.12). Supplementation with lysine is not useful because it is poorly absorbed and tends to produce diarrhea and abdominal pain. Treatment with high doses of prednisone and bronchoalveolar lavage has been effective in the management of acute pulmonary complications.
Basura GJ, Hagland SP, Wiltse AM, et al. Clinical features and the management of pyridoxine-dependent and pyridoxine-responsive seizures: review of 63 North American cases submitted to a patient registry. Eur J Pediatr. 2009;168:697-704.
Clayton PT. B6-responsive disorders: a model of vitamin dependency. J Inherit Metab Dis. 2006;29:317-326.
Gospe SM. Pyridoxine-dependent seizures, 2009. GeneReviews at GeneTests: Medical Information Resource. University of Washington, Seattle, 1997-2010. www.genetests.org. Accessed June 2010
Kölker S, Christensen E, Leonard JV, et al. Guideline for the diagnosis and management of glutaryl-CoA dehydrogenase deficiency (glutaric aciduria type I). J Inherit Metab Dis. 2007;30:5-22.
Pearl PL. New treatment paradigms in neonatal metabolic epilepsies. J Inherit Metab Dis. 2009;32:204-213.
Strauss KA, Donnelly P, Wintermark M. Cerebral haemodynamics in patients with glutaryl-coenzyme A dehydrogenase deficiency. Brain. 2010;133(Pt 1):76-92.
Zinnanti WJ, Lazovic J. Mouse model of encephalopathy and novel treatment strategies with substrate competition in glutaric aciduria type I. Mol Genet Metab. 2010;100(Suppl 1):S88-S91.
79.15 Aspartic Acid (Canavan Disease)
Canavan Disease
Diagnosis
In a typical patient with Canavan disease, CT scan and MRI reveal diffuse white matter degeneration, primarily in the cerebral hemispheres, with less involvement of the cerebellum and brainstem (Fig. 79-15). Repeated evaluations may be required. MRS performed at the time MRI is done can show the high peak of N-acetylaspartic acid, suggesting Canavan disease. The differential diagnosis of Canavan disease should include Alexander disease, which is another leukodystrophy associated with macrocephaly. Progression is usually slower in Alexander disease; hypotonia is not as pronounced as it is in Canavan disease. Brain biopsies of patients with Canavan disease show spongy degeneration of the myelin fibers, astrocytic swelling, and elongated mitochondria. Definitive diagnosis can be established by finding elevated amounts of N-acetylaspartic acid in the urine or blood. A deficiency of aspartoacylase can be found in cultured skin fibroblasts. The biochemical method is the preferred choice for diagnosis. N-acetylaspartic acid is found only in trace amounts (24 ± 16 µmol/mmol creatinine) in the urine of unaffected individuals, whereas in patients with Canavan disease its concentration is in the range of 1,440 ± 873 µmol/mmol creatinine. High levels of N-acetylaspartic acid can also be detected in plasma, CSF, and brain tissue. The activity of aspartoacylase in the fibroblasts of obligate carriers is less than or equal to half of the activity found in normal individuals.
Leone P, Janson CG, Bilianuk L, et al. Aspartoacylase gene transfer to the mammalian central nervous system with therapeutic implications for Canavan disease. Ann Neurol. 2000;48:27-38.
Matalon R, Michals K. Molecular basis of Canavan disease. Eur J Paediatr Neurol. 1998;2:69-76.
Matalon R, Michals-Matalon K. Spongy degeneration of the brain, Canavan disease: biochemical and molecular findings. Front Biosci. 2000;5:307-311.
Surendran S, Bamforth FJ, Chan A, et al. Mild elevation of N-acetylaspartic acid and macrocephaly: diagnostic problem. J Child Neurol. 2003;18:809-812.
Tacke U, Olbrich H, Sass JO, et al. Possible genotype-phenotype correlations in children with mild clinical course of Canavan disease. Neuropediatrics. 2005;36:252-255.
Topcu M, Erdem G, Saatsi I, et al. Clinical and magnetic resonance imaging features of L-2-hydroxyglutaric aciduria: report of three cases in comparison with Canavan disease. J Child Neurol. 1996;11:373-377.
Yalcinkaya C, Benbir G, Salomons GS, et al. Atypical MRI findings in Canavan disease: a patient with a mild course. Neuropediatrics. 2005;36:336-339.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 79 Defects in Metabolism of Amino Acids
79.1 Phenylalanine
Phenylalanine is an essential amino acid. Dietary phenylalanine not utilized for protein synthesis is normally degraded by way of the tyrosine pathway (Fig. 79-1). Deficiency of the enzyme phenylalanine hydroxylase (PAH) or of its cofactor tetrahydrobiopterin (BH4) causes accumulation of phenylalanine in body fluids and in the brain. The severity of hyperphenylalaninemia depends on the degree of enzyme deficiency and may vary from very high plasma concentrations (>20 mg/dL or >1,200 µmole/L, classic phenylketonuria [PKU]) to mildly elevated levels (2-6 mg/dL or 120-360 µmole/L). In affected infants with plasma concentrations >20 mg/dL, excess phenylalanine is metabolized to phenylketones (phenylpyruvate and phenylacetate; see Fig. 79-1) that are excreted in the urine, giving rise to the term phenylketonuria (PKU). These metabolites have no role in pathogenesis of central nervous system (CNS) damage in patients with PKU; their presence in the body fluids simply signifies the severity of the condition. The term hyperphenylalaninemia implies lower plasma levels (<20 mg/dL) of phenylalanine; these patients may or may not need dietary therapy based on their blood phenylalanine level. The brain is the main organ affected by hyperphenylalaninemia. The CNS damage in affected patients is caused by the elevated concentration of phenylalanine in brain tissue. The high blood levels of phenylalanine in PKU saturate the transport system across the blood-brain barrier causing inhibition of the cerebral uptake of other large neutral amino acids such as tyrosine and tryptophan. The exact mechanism of damage caused by elevated levels of intracerebral phenylalanine remains elusive. There are a few adults with classic PKU and normal intelligence who have never been treated with a phenylalanine-restricted diet. Phenylalanine content of the brain in these individuals was found to be close to that of normal subjects when studied by magnetic resonance spectroscopy (MRS) and imaging (MRI) techniques.
Figure 79-1 Pathways of phenylalanine and tyrosine metabolism. Enzyme defects causing genetic conditions are depicted as horizontal bars crossing the reaction arrow(s). Pathways for synthesis of cofactor BH4 are shown in purple. PKU* refers to defects of BH4 metabolism that affect the phenylalanine, tyrosine, and tryptophan hydroxylases (see Figs. 79-2 and 79-5). Enzymes: (1) phenylalanine hydroxylase (PAH), (2) pterin-carbinolamine dehydratase (PCD), (3) dihydrobiopterin reductase, (4) guanosine triphosphate (GTP) cyclohydrolase, (5) 6-pyruvoyltetrahydropterin synthase (6-PTS), (6) seriapterin reductase, (7) carbonyl reductase, (8) aldolase reductase, (9) dihydrofolate reductase, (10) tyrosine aminotransferase, (11a) intramolecular rearrangement, (11) 4-hydroxyphenylpyruvate dioxygenase, (12) homogentisic acid dioxygenase, (13) maleylacetoacetate isomerase, (14) fumarylacetoacetate hydroxylase, (NE) nonenzymatic.
Milder Forms of Hyperphenylalaninemia, Non-PKU Hyperphenylalaninemias
Hyperphenylalaninemia due to Deficiency of the Cofactor BH4
In 1-3% of infants with hyperphenylalaninemia, the defect resides in 1 of the enzymes necessary for production or recycling of the cofactor BH4 (Fig. 79-2). If these infants are misdiagnosed as having PKU, they may deteriorate neurologically despite adequate control of plasma phenylalanine. BH4 is synthesized from guanosine triphosphate (GTP) through several enzymatic reactions (see Fig. 79-1). In addition to acting as a cofactor for PAH, BH4 is also a cofactor for tyrosine hydroxylase and tryptophan hydroxylase, which are involved in the biosynthesis of dopamine (see Fig. 79-2) and serotonin (see Fig 79-5), respectively. Therefore, patients with hyperphenylalaninemia due to BH4 deficiency also manifest neurologic findings related to deficiencies of the neurotransmitters dopamine and serotonin. Four enzyme deficiencies leading to defective BH4 formation cause hyperphenylalaninemia and deficiencies of dopamine and serotonin. These include autosomal recessive GTP cyclohydrolase deficiency, pterin-carbinolamine dehydratase (PCD) deficiency, dihydropteridine reductase (DHPR) deficiency, and 6-pyruvoyltetrahydropterin synthase (PTPS or 6-PTS) deficiency. More than half of the reported patients have had a deficiency of 6-pyruvoyltetrahydropterin synthase. Autosomal dominant form of GTP deficiency and sepiapterin reductase deficiency result in deficiencies of neurotransmitters without hyperphenylalaninemia (Chapter 79.11 and Fig. 79-1).
Figure 79-2 Other pathways involving tyrosine metabolism. PKU* indicates hyperphenylalanemia due to tetrahydrobiopterin (BH4) deficiency (see Fig. 79-1). HVA, homovanillic acid; VMA, vanillymandelic acid. Enzymes: (1) tyrosine hydroxylase (TH), (2) aromatic L-amino acid decarboxylase (AADC), (3) dopamine hydroxylase, (4) phenylethanolamine-N-methyltransferase (PNMT), (5) catechol O-methyltransferase (COMT), (6) Monoamine oxidase (MAO).
Diagnosis
BH4 deficiency and the responsible enzyme defect may be diagnosed by the following studies:
Blau N. Defining tetrahydrobiopterin (BH4)-responsiveness in PKU. J Inherit Metab Dis. 2008;31:2-3.
Blau N, Bélanger-Quintana A, Demirkol M, et al. Optimizing the use of sapropterin (BH4) in the management of phenylketonuria. Mol Genet Metab. 2009;96:158-163.
Blau N, Bélanger-Quintana A, Demirkol M, et al. Management of phenylketonuria in Europe: survey results from 19 countries. Mol Genet Metab. 2010;99:109-115.
Blau N, van Spronsen FJ, Levy HL. Phenylketonuria. Lancet. 2010;376:1417-1427.
Burton BK, Adams DJ, Grange DK, et al. Tetrahydrobiopterin therapy for phenylketonuria in infants and young children. J Pediatr. 2011;158:410-415.
Cederbaum S. Tetrahydrobiopterin and PKU: into the future. J Pediatr. 2011;158:351-353.
Committee on Genetics. Maternal phenylketonuria. Pediatrics. 2008;122:445-449.
Feillet F, van Spronsen FJ, MacDonald A, et al. Challenges and pitfalls in the management of phenylketonuria. Pediatrics. 2010;126:333-341.
Giovannini M, Verduci E, Salvatici E, et al. Phenylketonuria: dietary and therapeutic challenges. J Inherit Metab Dis. 2007;30:145-152.
Mitchell JJ, Scriver CR. Phenylalanine hydroxylase deficiency, 2010. GeneReviews at GeneTests: Medical Information Resource. University of Washington, Seattle, 1997-2010. www.genetests.org. Accessed May 2010
Scriver CR, Levy H, Donlon J. Hyperphenylalaninemia: phenylalanine hydroxylase deficiency. In: Scriver CR, Beaudet AL, Sly WS, et al, editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill, 2008.
Waisbren SE, Noel K, Fahrbach K, et al. Phenylalanine blood levels and clinical outcomes in phenylketonuria: a systematic literature review and meta-analysis. Mol Genet Metab. 2007;92:63-70.
Waisbren SE, White D, van Spronsen F. Phenylketonuria, psychology and the brain. Mol Genet Metab. 2010;99(Suppl):S1-S108.
Zurfluh MR, Zschocke J, Lindner M, et al. Molecular genetics of tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency. Hum Mutat. 2008;29:167-175.
79.2 Tyrosine
Tyrosine is derived from ingested proteins or is synthesized endogenously from phenylalanine. It is used for protein synthesis and is a precursor of dopamine, norepinephrine, epinephrine, melanin, and thyroxine. Excess tyrosine is metabolized to carbon dioxide and water (see Fig. 79-1). Hereditary causes of hypertyrosinemia include deficiencies of tyrosine aminotransferase, 4-hydroxyphenylpyruvate dioxygenase (4-HPPD), or fumarylacetoacetate hydrolase (FAH). Acquired hypertyrosinemia may occur in severe hepatocellular dysfunction (liver failure), scurvy (vitamin C is the cofactor for the enzyme 4-HPPD), and hyperthyroidism. Hypertyrosinemia is common in blood samples obtained soon after eating.
Tyrosinemia Type I (Tyrosinosis, Hereditary Tyrosinemia, Hepatorenal Tyrosinemia)
Laboratory Findings
The presence of elevated levels of succinylacetone in serum and urine is diagnostic for tyrosinemia type I (see Fig. 79-1). In untreated patients, routinely available tests have a characteristic pattern. α-Fetoprotein level is increased, often markedly, and liver-synthesized coagulation factors are decreased in most patients; serum levels of transaminases are often increased, with marked increases being possible during acute hepatic episodes. Serum concentration of bilirubin is usually normal but can be increased with liver failure. Increased levels of α-fetoprotein are present in the cord blood of affected infants, indicating intrauterine liver damage. Plasma tyrosine level is usually elevated at diagnosis but this is a nonspecific finding and is dependent on dietary intake. Other amino acids, particularly methionine, may also be elevated in patients with liver damage. Hyperphosphaturia, hypophosphatemia, and generalized aminoaciduria may occur. The urinary level of 5-aminolevulinic acid is elevated (due to inhibition of 5-aminolevulinic hydratase by succinylacetone).
Diagnosis is usually established by demonstration of elevated levels of succinylacetone in urine or blood. Neonatal screening for hypertyrosinemia detects only a minority of patients with tyrosinemia type I. Succinylacetone, which is now assayed by most screening programs, has higher sensitivity and specificity than tyrosine and is the preferable metabolite for screening. Tyrosinemia type I should be differentiated from other causes of hepatitis and hepatic failure in infants, including galactosemia, hereditary fructose intolerance, neonatal iron storage disease, giant cell hepatitis, and citrullinemia type II (Chapter 79.11).
Treatment and Outcome
A diet low in phenylalanine and tyrosine can slow but does not halt the progression of the condition. The treatment of choice is nitisinone (NTBC), which inhibits tyrosine degradation at 4-HPPD (see Fig. 79-1). This treatment prevents acute hepatic and neurologic crises. Although nitisinone stops or greatly slows disease progression, some pretreatment liver damage is not reversible. Therefore, patients must be followed for development of cirrhosis or hepatocellular carcinoma. On imaging, the presence of even a single liver nodule usually indicates underlying cirrhosis. Most liver nodules in tyrosinemic patients are benign but current imaging techniques do not accurately distinguish all malignant nodules. Liver transplantation is an effective therapy for tyrosinemia type I and alleviates the risk of hepatocellular carcinoma. The impact of nitisinone treatment on the need for liver transplantation is still under study but the greatest effect is in patients treated early, such as children detected by neonatal screening, prior to the development of clinical symptoms. Rarely, nitisinone-treated patients develop corneal crystals, presumably of tyrosine, which are reversible by strict dietary compliance. This finding, combined with observations of developmental delay in some patients with chronically elevated tyrosine such as tyrosinemia type II, suggests that a diet low in phenylalanine and tyrosine should be continued in patients treated with nitisinone.
Tyrosinemia Type II (Richner-Hanhart Syndrome, Oculocutaneous Tyrosinemia)
This rare autosomal recessive disorder is caused by deficiency of tyrosine aminotransferase, resulting in palmar and plantar hyperkeratosis, herpetiform corneal ulcers, and mental retardation (see Fig. 79-1). Ocular manifestations include excessive tearing, redness, pain, and photophobia and often occur before skin lesions. Corneal lesions are presumed to be due to tyrosine deposition. In contrast to herpetic ulcers, corneal lesions in tyrosinemia type II stain poorly with fluorescein and often are bilateral. Skin lesions, which may develop later in life, include painful, nonpruritic hyperkeratotic plaques on the soles, palms and fingertips. Mental retardation, which occurs in <50% of patients, is usually mild to moderate.
The principal laboratory finding in untreated patients is marked hypertyrosinemia (20-50 mg/dL; 1,100-2,750 µmole/L). Surprisingly, 4-hydroxyphenylpyruvic acid and its metabolites are also elevated in urine despite being downstream from the metabolic block (see Fig. 79-1). This is hypothesized to occur via the action of other transaminases in the presence of high tyrosine concentrations, producing 4HPP in cellular compartments like the mitochondrion in which it is not further degraded. In contrast to tyrosinemia type I, liver and kidney function are normal, as are serum concentrations of other amino acids and succinylacetone. Tyrosinemia type II is due to TAT gene mutations, causing deficiency of cytosolic tyrosine aminotransferase activity in liver.
Transient Tyrosinemia of the Newborn
In a small number of newborn infants, plasma tyrosine may be as high as 60 mg/dL (3,300 µmole/L) during the 1st 2 weeks of life. Most affected infants are premature and are receiving high-protein diets. Transient tyrosinemia is felt to result from delayed maturation of 4-HPPD (see Fig. 79-1). Lethargy, poor feeding, and decreased motor activity are noted in some patients. Most are asymptomatic and are identified by a high blood phenylalanine or tyrosine level on screening. Laboratory findings include marked elevation of plasma tyrosine with a moderate increase in plasma phenylalanine. The finding of hypertyrosinemia differentiates this condition from PKU. 4-Hydroxyphenylpyruvic acid and its metabolites are present in the urine. Hypertyrosinemia usually resolves spontaneously in the 1st mo of life. It can be corrected promptly by reducing dietary protein to below 2 g/kg/24 hr and by administering vitamin C (200-400 mg/24 hr). Mild intellectual deficits have been reported in some infants that had this condition, but the causal relationship to hypertyrosinemia is not conclusively established.
Hawkinsinuria
This rare autosomal dominant condition is caused by a mutant 4-HPPD enzyme that catalyzes a partial reaction, releasing an intermediate compound used for diagnosis (see Fig. 79-1). This intermediate is either reduced to form 4-hydroxycyclohexylacetic acid (4-HCAA) or reacts with glutathione to form the unusual organic acid hawkinsin (2-L-cysteine-S-yl-1-4-dihydroxycyclohex-5-en-1-yl-acetic acid); secondary glutathione deficiency may occur.
Alcaptonuria
This rare (with an incidence of ≈1/250,000) autosomal recessive disorder is due to a deficiency of homogentisic acid oxidase, which causes large amounts of homogentisic acid to accumulate in the body and then to be excreted in the urine (see Fig. 79-1).
Albinism
Albinism is due to deficiency of melanin, the main pigment of the skin and eye (Table 79-1). Melanin is synthesized by melanocytes from tyrosine in a membrane-bound intracellular organelle, the melanosome. Melanocytes originate from the embryonic neural crest and migrate to the skin, eyes (choroid and iris), hair follicles, and inner ear. The melanin in the eye is confined to the retinal pigment epithelium, whereas in skin and hair follicles, it is secreted into the epidermis and hair shaft. Albinism can be caused by deficiencies of melanin synthesis, by some hereditary defects of melanosomes, or by disorders of melanocyte migration. Although albinism is a classical example of a biochemical genetic disease, neither the biosynthetic pathway of melanin nor many facets of melanocyte cell biology are completely elucidated (see Fig. 79-2). The end products are 2 pigments: pheomelanin, which is a yellow-red pigment; and eumelanin, a brown-black pigment.
| TYPE | GENE | CHROMOSOME |
|---|---|---|
| OCULOCUTANEOUS ALBINISM (OCA) | ||
| OCA1 (tyrosinase deficient) | TYR | 11q |
| OCA1A (severe deficiency) | TYR | 11q |
| OCA1B (mild deficiency)* | TYR | 11q |
| OCA2 (tyrosinase positive)† | P (pink-eyed dilution) | 15q |
| OCA3 (Rufous, red OCA) | TYRP1‡ | 9p |
| OCA4 | MATP | 5p13.3 |
| Hermansky-Pudlak syndrome | HPS1 | 10q |
| Chédiak-Higashi syndrome | LYST | 1q |
| OCULAR ALBINISM | ||
| OA1 (Nettleship-Falls type) | OA | xp |
| LOCALIZED ALBINISM | ||
| Piebaldism | KIT | 4q |
| Waardenburg syndrome I & III | PAX3 | 2q |
| Waardenburg syndrome II | MITF | 3p |
* This includes Amish, minimal pigment, yellow albinism, and platinum and temperature-sensitive variants.
‡ Tyrosinase related protein 1.
Many clinical forms of albinism have been identified. Some of the seemingly distinct clinical forms are caused by different mutations of the same gene. Several genes located on different chromosomes are shown to be involved in melanogenesis (see Table 79-1). Attempts to differentiate types of albinism based on the mode of inheritance, tyrosinase activity, or the extent of hypopigmentation have failed to yield a comprehensive classification. The following classification is based on the distribution of albinism in the body and the type of mutated gene.
Mutation detection is clinically available for most albinism genes (see Table 79-1). Molecular diagnosis is of little use therapeutically in isolated albinism but can be helpful for precise genetic counseling of families.
Oculocutaneous (Generalized) Albinism (OCA)
OCA2 (Tyrosinase-Positive OCA)
This is the most common form of generalized OCA, particularly in African blacks. Clinically, patients demonstrate some pigmentation of the skin and eyes at birth and continue to accumulate pigment throughout their lives. The hair is yellow at birth and may darken with age. They have pigmented nevi and freckles but do not tan. They may be clinically indistinguishable from OCA1 B. These individuals have normal tyrosinase activity in hair bulbs. The defect is in the OCA2 gene on chromosome 15q, orthologous to the p (pink-eyed dilution) gene in the mouse. This gene produces the P protein, a melanosome membrane protein. Patients with Prader-Willi and Angelman syndromes with microdeletion in chromosome 15q12 lack 1 copy of the OCA2 gene and have mild pigmentary dilution (Chapter 76).
Syndromic Forms of Generalized Albinism
Chédiak-Higashi Syndrome
Patients with this rare autosomal recessive condition (Chapter 124) have albinism of variable severity and susceptibility to infection. Bacterial infections of skin and upper respiratory tract are common. Giant peroxidase-positive lysosomal granules can be seen in granulocytes in a blood smear. Patients have a reduced number of melanosomes, which are abnormally large (macromelanosomes). The bleeding tendency is typically mild. The major, life-threatening complication is macrophage activation with hemophagocytic lymphohistiocytosis, manifested by fever, lymphadenopathy, hepatosplenomegaly, cytopenias, and elevated plasma ferritin level. Patients surviving childhood may develop cerebellar atrophy, peripheral neuropathy, and cognitive delay. Mutations in the LYST gene on chromosome 1q are the only known cause of this syndrome.
Brilliant MH. Oculocutaneous albinism type 4, 2007. GeneReviews at GeneTests: Medical Genetics Information Resources. University of Washington, Seattle, 1997-2010. www.genetests.org. Accessed June 2010
Charfeddine C, Monastiri K, Mokni M, et al. Clinical and mutational investigations of tyrosinemia type II in Northern Tunisia: identification and structural characterization of two novel TAT mutations. Mol Genet Metab. 2006;88:184-191.
El-Karaksy H, Rashed M, El-Sayed R, et al. Clinical practice. NTBC therapy for tyrosinemia type 1: how much is enough? Eur J Pediatr. 2010;169:689-693.
Gahl WA. Hermansky-Pudlak syndrome, 2010. GeneReviews at GeneTests: Medical Genetics Information Resources. University of Washington, Seattle, 1997-2010. www.genetests.org. Accessed June 2010
Introne WJ, Westbroek W, Golas GA, et al. Chédiak-Higashi syndrome, 2009. GeneReviews at GeneTests: Medical Genetics Information Resources. University of Washington, Seattle, 1997-2010. www.genetests.org. Accessed May 2010
King RA, Oetting W. Oculocutaneous albinism type 2, 2007. GeneReviews at GeneTests: Medical Genetics Information Resources. University of Washington, Seattle, 1997-2010. www.genetests.org. Accessed June 2010
Masurel-Paulet A, Poggi-Bach J, Rolland MO, et al. NTBC treatment in tyrosinaemia type I: long-term outcome in French patients. J Inherit Metab Dis. 2008;31:81-87.
Pingault V, Ente D, Dastot-LeMoal F, et al. Review and update of mutations causing Waardenburg syndrome. Hum Mutat. 2010;31:391-406.
Rezvani I. 50 years ago in Journal of Pediatrics: Waardenburg’s syndrome. J Pediatr. 2010;157:732.
Scott CR. The genetic tyrosinemias. Am J Med Genet C Semin Med Genet. 2006;142C:121-126.
79.3 Methionine
The normal pathway for catabolism of methionine, an essential amino acid, produces S-adenosylmethionine, which serves as a methyl group donor for methylation of a variety of compounds in the body, and cysteine, which is formed through a series of reactions collectively called trans-sulfuration (Fig. 79-3).
Homocystinuria (Homocystinemia)
Normally, most homocysteine, an intermediate compound of methionine degradation, is remethylated to methionine. This methionine-sparing reaction is catalyzed by the enzyme methionine synthase, which requires a metabolite of folic acid (5-methyltetrahydrofolate) as a methyl donor and a metabolite of vitamin B12 (methylcobalamin) as a cofactor (see Fig. 79-3). Only 20-30% of total homocysteine (and its dimer homocystine) is in free form in the plasma of normal individuals. The rest is bound to proteins as mixed disulfides. Three major forms of homocystinemia and homocystinuria have been identified.
Homocystinuria Due to Cystathionine β-Synthase (CBS) Deficiency (Classic Homocystinuria)
Infants with this disorder are normal at birth. Clinical manifestations during infancy are nonspecific and may include failure to thrive and developmental delay. The diagnosis is usually made after 3 yr of age, when subluxation of the ocular lens (ectopia lentis) occurs. This causes severe myopia and iridodonesis (quivering of the iris). Astigmatism, glaucoma, staphyloma, cataracts, retinal detachment, and optic atrophy may develop later in life. Progressive mental retardation is common. Normal intelligence has been reported. In an international survey of >600 patients, IQ scores ranged from 10 to 135. Higher IQ scores are seen in vitamin B6 responsive patients. Psychiatric and behavioral disorders have been observed in >50% of affected patients. Convulsions occur in about 20% of patients. Affected individuals with homocystinuria manifest skeletal abnormalities resembling those of Marfan syndrome (Chapter 693); they are usually tall and thin, with elongated limbs and arachnodactyly. Scoliosis, pectus excavatum or carinatum, genu valgum, pes cavus, high arched palate, and crowding of the teeth are commonly seen. These children usually have fair complexions, blue eyes, and a peculiar malar flush. Generalized osteoporosis, especially of the spine, is the main roentgenographic finding. Thromboembolic episodes involving both large and small vessels, especially those of the brain, are common and may occur at any age. Optic atrophy, paralysis, cor pulmonale, and severe hypertension (due to renal infarcts) are among the serious consequences of thromboembolism, which is caused by changes in the vascular walls and increased platelet adhesiveness secondary to elevated homocystine levels. The risk of thromboembolism increases after surgical procedures. Spontaneous pneumothorax and acute pancreatitis are rare complications.
Treatment with high doses of vitamin B6 (200-1,000 mg/24 hr) causes dramatic improvement in most patients who are responsive to this therapy. The degree of response to vitamin B6 treatment may be different in different families. Some patients may not respond because of folate depletion; a patient should not be considered unresponsive to vitamin B6 until folic acid (1-5 mg/24 hr) has been added to the treatment regimen. Restriction of methionine intake in conjunction with cysteine supplementation is recommended for patients who are unresponsive to vitamin B6. The need for dietary restriction and its extent remains controversial in patients with vitamin B6 responsive form. In some patients with this form, addition of betaine may obviate the need for any dietary restriction. Betaine (trimethylglycine, 6-9 g/24 hr for adults or 200-250 mg/kg/day for children) lowers homocysteine levels in body fluids by remethylating homocysteine to methionine (see Fig. 79-3); this may result in further elevation of plasma methionine levels. This treatment has produced clinical improvement (preventing vascular events) in patients who are unresponsive to vitamin B6 therapy. Cerebral edema has occurred in a patient with vitamin B6 nonresponsive homocystinuria and dietary noncompliance during betaine therapy. Administration of large doses of vitamin C (1 g/day) has improved the endothelial function; long-term clinical efficacy is not known.
Homocystinuria Due to Defects in Methylcobalamin Formation
Methylcobalamin is the cofactor for the enzyme methionine synthase, which catalyzes remethylation of homocysteine to methionine. There are at least 5 distinct defects in the intracellular metabolism of cobalamin that may interfere with the formation of methylcobalamin. To better understand the metabolism of cobalamin, see methylmalonic acidemia (Fig. 79-4; Chapter 79.6 and Fig. 79-3). The 5 defects are designated as cblC, cblD (including cblD variant 1), cblE (methionine synthase reductase), cblG (methionine synthase), and cblF. Patients with cblC, cblD (not those with cblD variant 2), and cblF defects have methylmalonic acidemia in addition to homocystinuria because formation of both adenosylcobalamin and methylcobalamin is impaired (Chapter 79.6).
Patients with cblE and cblG defects are unable to form methylcobalamin and develop homocystinuria without methylmalonic acidemia (see Fig. 79-4); fewer than 40 patients are known with each of these diseases.
Hypermethioninemia
Secondary hypermethioninemia occurs in liver disease, tyrosinemia type I, and classic homocystinuria. Hypermethioninemia has also been found in premature and some full-term infants receiving high-protein diets, in whom it may represent delayed maturation of the enzyme methionine adenosyltransferase. Lowering the protein intake usually resolves the abnormality. Primary hypermethioninemia caused by the deficiency of hepatic methionine adenosyltransferase (MAT I/III; MAT II, which is present in other tissues, is not affected; see Fig. 79-3) has been reported in approximately 60 patients. The majority of these patients have been diagnosed in the neonatal period through screening for homocystinuria. Affected individuals with residual enzyme activity remain asymptomatic throughout life despite persistent hypermethioninemia. Some complain of unusual odor to their breath (boiled cabbage). A few patients with complete enzyme deficiency have had neurologic abnormalities related to demyelination (mental retardation, dystonia, dyspraxia). Normal pregnancies producing normal offspring have been reported in mothers with methionine adenoslytransferase deficiency. The condition is inherited as an autosomal recessive trait. The gene for hepatic methionine adenosyltransferase is on chromosome 10q22 and several disease-causing mutations have been identified. A novel defect, glycine N-methyltransferase deficiency, also causes isolated hypermethioninemia.
Adams D, Venditti CP. Disorders of intracellular cobalamin metabolism. GeneReviews at GeneTests: Medical Genetics Information Resources. University of Washington, Seattle, 1997-2010. www.genetests.org. Accessed May 2010
Allen NC, Bagade S, McQueen MB, et al. Systematic meta-analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nat Genet. 2008;30:827-834.
Forges T, Chery C, Audonnet S, et al. Life-threatening methylenetetrahydrofolate reductase (MTHFR) deficiency with extremely early onset: characterization of two novel mutations in compound heterozygous patients. Mol Genet Metab. 2010;100:143-148.
Holm PK, Hustad S, Ueland PM, et al. Modulation of the homocysteine-betaine relationship by methylenetetrahydrofolate reductase 677C-T genotypes and B-vitamin status in a large-scale epidemiologic study. J Clin Endocr Metab. 2007;92:1535-1541.
Lawson-Yuen A, Levy HL. The use of betaine in the treatment of elevated homocysteine. J Mol Genet Metab. 2006;88:201-207.
Lin HJ, Neidich JA, Salazar D, et al. Asymptomatic maternal combined homocystinuria and methylmalonic aciduria (cbIC) detected through low carnitine levels on newborn screening. J Pediatr. 2009;155:924-927.
Picker JD, Levy HL. Homocystinuria caused by cystathionine beta-synthase deficiency, 2006. GeneReviews at GeneTests: Medical Information Resource. University of Washington, Seattle, 1997-2010. www.genetests.org. Accessed May 2010
Schiff M, Benoist JF, Tilea B, et al. Isolated remethylation disorders: do our treatments benefit patients? J Inherit Metab Dis. 2011;34:137-145.
Skovby F, Gaustadnes M, Mudd SH. A revisit to the natural history of homocystinuria due to cystathionine beta-synthase deficiency. Mol Genet Metab. 2010;99:1-3.
Strauss KA, Morton DH, Puffenberger EG, et al. Prevention of brain disease from severe 5,10-methylenetetrahydrofolate reductase deficiency. Mol Genet Metab. 2007;91:165-175.
Testai FD, Gorelick PB. Inherited metabolic disorders and stroke part 2: homocystinuria, organic acidurias, and urea cycle disorders. Arch Neurol. 2010;67:148-153.
79.4 Cysteine/Cystine
Cysteine is a sulfur-containing nonessential amino acid that is synthesized from methionine (see Fig. 79-3). In the presence of oxygen, 2 molecules of cysteine are oxidized to form cystine. The most common disorders of cysteine/cystine metabolism, cystinuria (Chapter 541) and cystinosis (Chapter 523.3).
Sulfite Oxidase Deficiency (Molybdenum Cofactor Deficiency)
At the last step in cysteine metabolism, sulfite is oxidized to sulfate by sulfite oxidase, and the sulfate is excreted in the urine (see Fig. 79-3). This enzyme requires a molybdenum-pterin complex named molybdenum cofactor. This cofactor is also necessary for the function of 2 other enzymes in humans: xanthine dehydrogenase (which oxidizes xanthine and hypoxanthine to uric acid) and aldehyde oxidase. Three enzymes, encoded by 3 different genes, are involved in the synthesis of the cofactor. The genes are mapped to chromosomes 14q24, 6p21.3, and 5q11. Deficiency of any of the 3 enzymes causes cofactor deficiency with identical phenotype. Most patients who were originally diagnosed as having sulfite oxidase deficiency have been proven to have molybdenum cofactor deficiency. Both conditions are inherited as autosomal recessive traits. The gene for sulfite oxidase is on chromosome 12.
Kugler S, Hahnewald R, Garrido M, et al. Long-term rescue of a lethal inherited disease by adeno-associated virus-mediated gene transfer in a mouse model of molybdenum-cofactor deficiency. Am J Hum Genet. 2007;80:291-297.
Schwarz G, Mendel RR, Ribbe MW. Molybdenum cofactors, enzymes and pathways. Nature. 2009;460:839-847.
Tan WH, Eickler FS, Hoda S, et al. Isolated sulfite oxidase deficiency: a case report with a novel mutation and review of the literature. Pediatrics. 2005;116:757-766.
Veldman A, Santamaria-Araujo JA, Sollazzo S, et al. Successful treatment of molybdenum cofactor deficiency type A with cPMP. Pediatrics. 2010;125:e1249-e1254.
79.5 Tryptophan
Tryptophan is an essential amino acid and a precursor for nicotinic acid (niacin) and serotonin (Fig. 79-5). The genetic disorders of metabolism of serotonin, 1 of the major neurotransmitters, are discussed in Chapter 79.11.
Figure 79-5 Pathways in the metabolism of tryptophan. PKU* indicates hyperphenylalanemia due to tetrahydrobiopterin deficiency (see Fig. 79-1). Enzymes: (1) tryptophan hydroxylase, (2) aromatic L-amino acid decarboxylase (AADC), (3) monoamine oxidase (MAO).
79.6 Valine, Leucine, Isoleucine, and Related Organic Acidemias
The early steps in the degradation of these 3 essential amino acids, the branched-chain amino acids, are similar (see Fig. 79-4). The intermediate metabolites are all organic acids, and deficiency of any of the degradative enzymes, except for the transaminases, causes acidosis; in such instances, the organic acids before the enzymatic block accumulate in body fluids and are excreted in the urine. These disorders commonly cause metabolic acidosis, which usually occurs in the 1st few days of life. Although most of the clinical findings are nonspecific, some manifestations may provide important clues to the nature of the enzyme deficiency. An approach to infants suspected of having an organic acidemia is presented in Figure 79-6. Definitive diagnosis is usually established by identifying and measuring specific organic acids in body fluids (blood, urine), by the enzyme assay, and by identification of the mutant gene.
Figure 79-6 Clinical approach to infants with organic acidemia. Asterisks indicate disorders in which patients have a characteristic odor (see text and Table 79-2). MSUD, maple syrup urine disease.
Organic acidemias are not limited to defects in the catabolic pathways of branched-chain amino acids. Disorders causing accumulation of other organic acids include those derived from lysine (Chapter 79.14), those associated with lactic acid (Chapter 81), and dicarboxylic acidemias associated with defective fatty acid degradation (Chapter 80.1).
Maple Syrup Urine Disease (MSUD)
Decarboxylation of leucine, isoleucine, and valine is accomplished by a complex enzyme system (branched-chain α-ketoacid dehydrogenase) using thiamine pyrophosphate (vitamin B1) as a coenzyme. This mitochondrial enzyme consists of 4 subunits: E1α, E1β, E2, and E3. The E3 subunit is shared with 2 other dehydrogenases in the body, namely pyruvate dehydrogenase and α-ketoglutarate dehydrogenase. Deficiency of this enzyme system causes MSUD (see Fig. 79-4), named after the sweet odor of maple syrup found in body fluids, especially urine. Based on clinical findings and response to thiamine administration, 5 phenotypes of MSUD have been identified.
Classic MSUD
Diagnosis is often suspected because of the peculiar odor of maple syrup found in urine, sweat, and cerumen (see Fig. 79-6). It is usually confirmed by amino acid analysis showing marked elevations in plasma levels of leucine, isoleucine, valine, and alloisoleucine (a stereoisomer of isoleucine not normally found in blood) and depression of alanine. Leucine levels are usually higher than those of the other 3 amino acids. Urine contains high levels of leucine, isoleucine, and valine and their respective ketoacids. These ketoacids may be detected qualitatively by adding a few drops of 2,4-dinitrophenylhydrazine reagent (0.1% in 0.1 N HCl) to the urine; a yellow precipitate of 2,4-dinitrophenylhydrazone is formed in a positive test. Neuroimaging during the acute state may show cerebral edema, which is most prominent in the cerebellum, dorsal brainstem, cerebral peduncle, and internal capsule. After recovery from the acute state and with advancing age, hypomyelination and cerebral atrophy may be seen in neuroimaging of the brain. The enzyme activity can be measured in leukocytes and cultured fibroblasts.
