310 |
Hypersensitivity Pneumonitis and Pulmonary Infiltrates with Eosinophilia |
HYPERSENSITIVITY PNEUMONITIS
INTRODUCTION AND DEFINITION
Hypersensitivity pneumonitis (HP), also referred to as extrinsic allergic alveolitis, is a pulmonary disease that occurs due to inhalational exposure to a variety of antigens leading to an inflammatory response of the alveoli and small airways. Systemic manifestations such as fever and fatigue can accompany respiratory symptoms. Although sensitization to an inhaled antigen as manifested by specific circulating IgG antibodies is necessary for the development of HP, sensitization alone is not sufficient as a defining characteristic, because many sensitized individuals do not develop HP. The incidence and prevalence of HP are variable, depending on geography, occupation, avocation, and environment of the cohort being studied. As yet unexplained is the decreased risk of developing HP in smokers.
OFFENDING ANTIGENS
HP can be caused by any of a large list of potential offending inhaled antigens (Table 310-1). The various antigens and environmental conditions described to be associated with HP give rise to an expansive list of monikers given to specific forms of HP. Antigens derived from fungal, bacterial, mycobacterial, bird-derived, and chemical sources have all been implicated in causing HP.
|
EXAMPLES OF HYPERSENSITIVITY PNEUMONITIS |
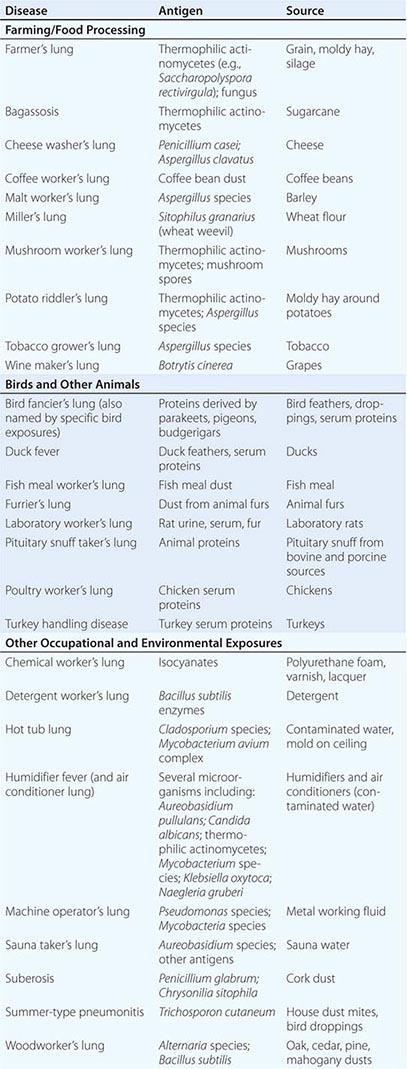
Categories of individuals at particular risk in the United States include farmers, bird owners, industrial workers, and hot tub users. Farmer’s lung occurs as a result of exposure to one of several possible sources of bacterial or fungal antigens such as grain, moldy hay, or silage. Potential offending antigens include thermophilic actinomycetes or Aspergillus species. Bird fancier’s lung (also referred to by names corresponding to specific birds) must be considered in patients who give a history of keeping birds in their home and is precipitated by exposure to antigens derived from feathers, droppings, and serum proteins. Occupational exposure to birds may also cause HP, as is seen in poultry worker’s lung. Chemical worker’s lung is provoked by exposure to occupational chemical antigens such as diphenylmethane diisocyanate and toluene diisocyanate. Mycobacteria may cause HP rather than frank infection, a phenomenon observed in hot tub lung and in HP due to metalworking fluid.
PATHOPHYSIOLOGY
The pathophysiology of HP has not been characterized in depth on an immunologic level, although it has been established that HP is an immune-mediated condition that occurs in response to inhaled antigens that are small enough to deposit in distal airways and alveoli. From a lymphocyte perspective, HP has been categorized as a condition with a TH1 inflammatory pattern. However, emerging evidence suggests that TH17 lymphocyte subsets may be involved in the pathogenesis of the disease as well. Although the presence of precipitating IgG antibodies against specific antigens in HP suggests a prominent role for adaptive immunity in the pathophysiology of HP, innate immune mechanisms may also make an important contribution. This is highlighted by the observation that Toll-like receptors and downstream signaling proteins such as MyD88 are activated in HP. Although no clear genetic basis for HP has been established, in specific cohorts, polymorphisms in genes involved in antigen processing and presentation, including TAP1 and major histocompatibility complex type II, have been observed.
CLINICAL PRESENTATION
Given the heterogeneity among patients, variability in offending antigens, and differences in the intensity and duration of exposure to antigen, the presentation of HP is accordingly variable. Although these categories are not fully satisfactory in capturing this variability, HP has been traditionally categorized as having acute, subacute, and chronic forms. Acute HP usually manifests itself 4–8 h following exposure to the inciting antigen, often intense in nature. Systemic symptoms, including fevers, chills, and malaise, are prominent and are accompanied by dyspnea. Symptoms resolve within hours to days if no further exposure to the offending antigen occurs. In subacute HP resulting from ongoing antigen exposure, the onset of respiratory and systemic symptoms is typically more gradual over the course of weeks. A similar presentation may occur as a culmination of intermittent episodes of acute HP. Although respiratory impairment may be quite severe, antigen avoidance generally results in resolution of the symptoms, although with a slower time course, on the order of weeks to months, than that seen with acute HP. Chronic HP can present with an even more gradual onset of symptoms than subacute HP, with progressive dyspnea, cough, fatigue, weight loss, and clubbing of the digits. The insidious onset of symptoms and frequent lack of an anteceding episode of acute HP make diagnosing chronic HP a challenge. Unlike with the other forms of HP, there can be an irreversible component to the respiratory impairment that is not responsive to removal of the responsible antigen from the patient’s environment. The disease progression to hypoxemic respiratory failure can mirror that seen in idiopathic pulmonary fibrosis (IPF). Fibrotic lung disease is a potential feature of chronic HP due to exposure to bird antigens, whereas an emphysematous phenotype may be seen in farmer’s lung.
The categories of acute, subacute, and chronic HP are not completely sufficient in classifying HP. The HP Study Group found on cluster analysis that a cohort of HP patients is best described in bipartite fashion, with one group featuring recurrent systemic signs and symptoms and the other featuring more severe respiratory findings.
Concordant with the variability in the presentation of HP is the observed variability in outcome. HP that has not progressed to chronic lung disease has a more favorable outcome with likely resolution if antigen avoidance can be achieved. However, chronic HP resulting in lung fibrosis has a poorer prognosis, with patients with chronic pigeon breeder’s lung having demonstrated a similar mortality as seen in IPF.
DIAGNOSIS
Although there is no set of universally accepted criteria for arriving at a diagnosis of HP, diagnosis depends foremost on establishing a history of exposure to an offending antigen that correlates with respiratory and systemic symptoms. A careful occupational and home exposure history should be taken and may be supplemented if necessary by a clinician visit to the work or home environment. Specific inquiries will be influenced by geography and the occupation of the patient. When HP is suspected by history, the additional workup is aimed at establishing an immunologic and physiologic response to inhalational antigen exposure with chest imaging, pulmonary function testing, serologic studies, bronchoscopy, and, on occasion, lung biopsy.
Chest Imaging Chest x-ray findings in HP are nonspecific and can even lack any discernible abnormalities. In cases of acute and subacute HP, findings may be transient and can include ill-defined micronodular opacities or hazy ground-glass airspace opacities. Findings on chest x-ray will often resolve with removal from the offending antigen, although the time course of resolution may vary. With chronic HP, the abnormalities seen on the chest radiograph are frequently more fibrotic in nature and may be difficult to distinguish from IPF.
With the wide availability of high-resolution computed tomography (HRCT), this modality has become a common component in the diagnostic workup for HP. Although the HRCT may be normal in acute forms of HP, this may be due to lack of temporal correlation between exposure to the offending antigen and obtaining the imaging. Additionally, because of the transient nature of acute HP, HRCT is not always performed. In subacute forms of the disease, ground-glass airspace opacities are characteristic, as is the presence of centrilobular nodules. Expiratory images may show areas of air trapping that are likely caused by involvement of the small airways (Fig. 310-1). Reticular changes and traction bronchiectasis can be observed in chronic HP. Subpleural honeycombing similar to that seen in IPF may be present in advanced cases, although unlike in IPF, the lung bases are frequently spared.
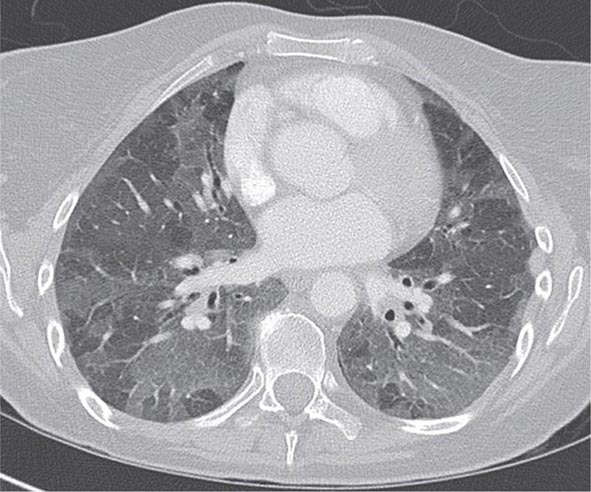
FIGURE 310-1 Chest computed tomography scan of a patient with subacute hypersensitivity pneumonitis in which scattered regions of ground-glass infiltrates in a mosaic pattern consistent with air trapping are seen bilaterally. This patient had bird fancier’s lung. (Courtesy of TJ Gross; with permission.)
Pulmonary Function Testing (PFT) Either restrictive or obstructive PFTs can be present in HP, so the pattern of PFT change is not useful in establishing the diagnosis of HP. However, obtaining PFTs is of use in characterizing the physiologic impairment of an individual patient and in gauging the response to antigen avoidance and/or corticosteroid therapy. Diffusion capacity for carbon monoxide may be significantly impaired, particularly in cases of chronic HP with fibrotic pulmonary parenchymal changes.
Serum Precipitins Assaying for precipitating IgG antibodies against specific antigens can be a useful adjunct in the diagnosis of HP. However, the presence of an immunologic response alone is not sufficient for establishing the diagnosis, because many asymptomatic individuals with high levels of exposure to antigen may display serum precipitins, as has been observed in farmers and in pigeon breeders. It should also be noted that panels that test for several specific serum precipitins often provide false-negative results, because they represent an extremely limited proportion of the universe of potential offending environmental antigens.
Bronchoscopy Bronchoscopy with bronchoalveolar lavage (BAL) may be used in the evaluation of HP. Although not a specific finding, BAL lymphocytosis is characteristic of HP. However, in active smokers, a lower threshold should be used to establish BAL lymphocytosis, because smoking will result in lower lymphocyte percentages. Most cases of HP have a CD4+/CD8+ lymphocyte ratio of less than 1, but again, this is not a specific finding and has limited utility in the diagnosis of HP.
Lung Biopsy Tissue samples may be obtained by a bronchoscopic approach using transbronchial biopsy, or more architecturally preserved specimens may be obtained by a surgical approach (video-assisted thoracoscopy or open approach). As is the case with BAL, histologic specimens are not absolutely necessary to establish the diagnosis of HP, but they can be useful in the correct clinical context. A common histologic feature in HP is the presence of noncaseating granulomas in the vicinity of small airways (Fig. 310-2). As opposed to pulmonary sarcoidosis, in which noncaseating granulomas are well defined, the granulomas seen in HP are loose and poorly defined in nature. Within the alveolar spaces and in the interstitium, a mixed cellular infiltrate with a lymphocytic predominance is observed that is frequently patchy in distribution. Bronchiolitis with the presence of organizing exudate is also often observed. Fibrosis may be present as well, particularly as the disease progresses to its chronic form. Fibrotic changes may be focal but can be diffuse and severe with honeycombing in advanced cases, similar to findings in IPF.
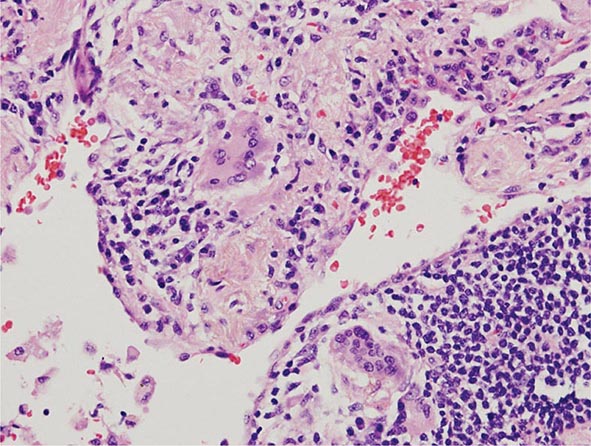
FIGURE 310-2 Open-lung biopsy from a patient with subacute hypersensitivity pneumonitis demonstrating a loose, nonnecrotizing granuloma made up of histiocytes and multinucleated giant cells. Peribronchial inflammatory infiltrate made up of lymphocytes and plasma cells is also seen. (Courtesy of TJ Gross; with permission.)
Clinical Prediction Rule Although not meant as a set of validated diagnostic criteria, a clinical prediction rule for predicting the presence of HP has been published by the HP Study Group. They identified six statistically significant predictors for HP, the strongest of which was exposure to an antigen known to cause HP. Other predictive criteria were the presence of serum precipitins, recurrent symptoms, symptoms occurring 4–8 h after antigen exposure, crackles on inspiration, and weight loss.
DIFFERENTIAL DIAGNOSIS
Differentiating HP from other conditions that cause a similar constellation of respiratory and systemic symptoms requires an increased index of suspicion based on obtaining a history of possible exposure to an offending antigen. Presentations of acute or subacute HP can be mistaken for respiratory infection. In cases of chronic disease, HP must be differentiated from interstitial lung disease, such as IPF or nonspecific interstitial pneumonitis (NSIP); this can be a difficult task even with lung biopsy. Given the presence of pulmonary infiltrates and noncaseating granulomas on biopsy, sarcoidosis is also a consideration in the differential diagnosis of HP. Unlike in HP, however, hilar adenopathy may be prominent on chest x-ray, organs other than the lung may be involved, and noncaseating granulomas in pathologic specimens tend to be well formed. Other inhalational syndromes, such as organic toxic dust syndrome (OTDS), can be misdiagnosed as HP. OTDS occurs with exposure to organic dusts, including those produced by grains or mold silage, but neither requires prior antigen sensitization nor is characterized by positive serum precipitins.
GLOBAL CONSIDERATIONS
![]() As the ever-expanding list of antigens and exposures associated with the development of HP suggests, populations at risk for HP will vary globally based on specifics of local occupational, avocational, and environmental factors. Specific examples of geographically limited HP include summer-type pneumonitis seen in Japan and suberosis seen in cork workers in Portugal and Spain.
As the ever-expanding list of antigens and exposures associated with the development of HP suggests, populations at risk for HP will vary globally based on specifics of local occupational, avocational, and environmental factors. Specific examples of geographically limited HP include summer-type pneumonitis seen in Japan and suberosis seen in cork workers in Portugal and Spain.
PULMONARY INFILTRATES WITH EOSINOPHILIA
Although eosinophils are normal constituents of the lungs, there are several pulmonary eosinophilic syndromes that are characterized by pulmonary infiltrates on imaging along with an increased number of eosinophils in lung tissue, in sputum, and/or in BAL fluid, with resultant increased respiratory symptoms and the potential for systemic manifestations. Because the eosinophil plays such an important role in each of these syndromes, it is often difficult to distinguish between them, but there are important clinical and pathologic differences as well as differences in prognosis and treatment paradigms.
CLASSIFYING PULMONARY INFILTRATES WITH EOSINOPHILIA AND GENERAL APPROACH
Because there are so many different diagnoses associated with pulmonary infiltrates with eosinophilia, the first step in classifying pulmonary eosinophilic syndromes is distinguishing between primary pulmonary eosinophilic lung disorders and those with eosinophilia that are secondary to a specific cause such as a drug reaction, an infection, a malignancy, or another pulmonary condition such as asthma. Table 310-2 lists primary and secondary pulmonary eosinophilic disorders.
|
PULMONARY INFILTRATES WITH EOSINOPHILIA |
For each patient, a detailed history is of utmost importance and can help elucidate what the underlying disease is. Details regarding onset, timing, and precipitants of specific symptoms can help discern one diagnosis from another. History regarding pharmacologic, occupational, and environmental exposures is instructive, and family and travel history are crucial. In addition to details about the sinuses and lungs, it is important to inquire about systemic manifestations and assess for physical findings of cardiac, gastrointestinal (GI), neurologic, dermatologic, and genitourinary involvement, all of which may give clues to specific diagnoses. Once the details from history and physical are teased out, laboratory testing (including measurements of blood eosinophils, cultures, and markers of inflammation), spirometry and radiographic imaging can help distinguish between different diseases. Often, however, BAL, transbronchial, or open lung biopsies are required. In many cases, biopsies or noninvasive diagnostic studies of other organs (e.g., echocardiogram, electromyogram, or bone marrow biopsy) can be helpful.
PATHOPHYSIOLOGY
Pathologically, the pulmonary eosinophilic syndromes are characterized by tissue infiltration by eosinophils (Fig. 310-2). In eosinophilic granulomatosis with polyangiitis (EGPA), extravascular granulomas and necrotizing vasculitis may occur in the lungs, as well as in the heart, skin, muscle, liver, spleen, and kidneys, and may be associated with fibrinoid necrosis and thrombosis.
The exact etiology of the various pulmonary eosinophilic syndromes is unknown; however, it is felt that these syndromes result from dysregulated eosinophilopoiesis or an autoimmune process because of the prominence of allergic features and the presence of immune complexes, heightened T cell immunity, and altered humoral immunity as evidenced by elevated IgE and rheumatoid factor. Because of its integral involvement in eosinophilopoiesis, interleukin 5 (IL-5) has been hypothesized to play an etiologic role, and efforts to block this cytokine are being investigated. Antineutrophil cytoplasmic antibodies (ANCAs) are present in about half of patents with EGPA; binding of ANCAs to vascular walls likely contributes to vascular inflammation and injury as well as chemotaxis of inflammatory cells.
ACUTE EOSINOPHILIC PNEUMONIA
Acute eosinophilic pneumonia is a syndrome characterized by fevers, acute respiratory failure that often requires mechanical ventilation, diffuse pulmonary infiltrates, and pulmonary eosinophilia in a previously healthy individual (Table 310-3).
|
DIAGNOSTIC CRITERIA OF ACUTE EOSINOPHILIC PNEUMONIA |
Clinical Features and Etiology At presentation, acute eosinophilic pneumonia is often mistaken for acute lung injury or acute respiratory distress syndrome (ARDS), until a BAL is performed and reveals >25% eosinophils. Although the predominant symptoms of acute eosinophilic pneumonia are cough, dyspnea, malaise, myalgias, night sweats, and pleuritic chest pain, physical exam findings include high fevers, basilar rales, and rhonchi on forced expiration. Acute eosinophilic pneumonia most often affects males between age 20 and 40 with no history of asthma. Although no clear etiology has been identified, several case reports have linked acute eosinophilic pneumonia to recent initiation of tobacco smoking or exposure to other environmental stimuli including dust from indoor renovations.
In addition to a suggestive history, the key to establishing a diagnosis of acute eosinophilic pneumonia is the presence of >25% eosinophilia on BAL fluid. While lung biopsies show eosinophilic infiltration with acute and organizing diffuse alveolar damage, it is generally not necessary to proceed to biopsy to establish a diagnosis. Although patients present with an elevated white blood cell count, in contrast to other pulmonary eosinophilic syndromes, acute eosinophilic pneumonia is often not associated with peripheral eosinophilia upon presentation. However, between 7 and 30 days of disease onset, peripheral eosinophilia often occurs with mean eosinophil counts of 1700. Erythrocyte sedimentation rate (ESR), C-reactive protein, and IgE levels are high but nonspecific, whereas HRCT is always abnormal with bilateral random patchy ground-glass or reticular opacities, and small pleural effusions in as many as two-thirds of patients. Pleural fluid is characterized by a high pH with marked eosinophilia.
Clinical Course and Response to Therapy Although some patients improve spontaneously, most patients require admission to an intensive care unit and respiratory support with either invasive (intubation) or noninvasive mechanical ventilation. However, what distinguishes acute eosinophilic pneumonia from both other cases of acute lung injury as well as some of the other pulmonary eosinophilic syndromes is the absence of organ dysfunction or multisystem organ failure other than respiratory failure. One of the characteristic features of acute eosinophilic pneumonia is the high degree of corticosteroid responsiveness and the excellent prognosis. Another distinguishing feature of acute eosinophilic pneumonia is that complete clinical and radiographic recovery without recurrence or residual sequelae occurs in almost all patients within several weeks of initiation of therapy.
CHRONIC EOSINOPHILIC PNEUMONIA
In contrast to acute eosinophilic pneumonia, chronic eosinophilic pneumonia is a more indolent syndrome that is characterized by pulmonary infiltrates and eosinophilia in both the tissue and blood. Most patients are female nonsmokers with a mean age of 45, and patients do not usually develop the acute respiratory failure and significant hypoxemia appreciated in acute eosinophilic pneumonia. Similar to EGPA, a majority have asthma, with many having a history of allergies.
Patients present with a subacute illness over weeks to months, with cough, low-grade fevers, progressive dyspnea, weight loss, wheezing, malaise, and night sweats, and a chest x-ray with migratory bilateral peripheral or pleural-based opacities. Although this “photographic negative pulmonary edema” appearance on chest x-ray and chest CT is pathognomonic of chronic eosinophilic pneumonia, less than 25% of patients present with this finding. Other radiographic findings include atelectasis, pleural effusions, lymphadenopathy, and septal line thickening.
Almost 90% of patients have peripheral eosinophilia, with mean eosinophil counts of over 30% of total white blood cell count. BAL eosinophilia is also an important distinguishing feature with mean BAL eosinophil counts of close to 60%. Both peripheral and BAL eosinophilia are very responsive to treatment with corticosteroids. Other laboratory features of chronic eosinophilic pneumonia include increased ESR, C-reactive protein, platelets, and IgE. Lung biopsy is also often not required to establish a diagnosis, but may show accumulation of eosinophils and histiocytes in the lung parenchyma and interstitium, as well as cryptogenic organizing pneumonia, but with minimal fibrosis. Nonrespiratory manifestations are uncommon, but arthralgias, neuropathy, and skin and GI symptoms have all been reported; their presence may suggest EGPA or hypereosinophilic syndrome. Another similarity is the rapid response to corticosteroids with quick resolution of peripheral and BAL eosinophilia and improvement in symptoms. In contrast to acute eosinophilic pneumonia, though, over 50% of patients relapse, and many require prolonged courses of corticosteroids for months to years.
EOSINOPHILIC GRANULOMATOSIS WITH POLYANGIITIS (EGPA)
Previously known as allergic angiitis granulomatosis or Churg-Strauss syndrome, this complex syndrome is characterized by eosinophilic vasculitis that may involve multiple organ systems including the lungs, heart, skin, GI tract, and nervous system. Although EGPA is characterized by peripheral and pulmonary eosinophilia with infiltrates on chest x-ray, the primary features that distinguish EGPA from other pulmonary eosinophilic syndromes are the presence of eosinophilic vasculitis in the setting of asthma and involvement of multiple end organs (a feature it shares with hypereosinophilic syndrome). Although perceived to be quite rare, in the last few years, there has appeared to be an increased incidence of this disease, particularly in association with various asthma therapies.
The primary features of EGPA include asthma, peripheral eosinophilia, neuropathy, pulmonary infiltrates, paranasal sinus abnormality, and presence of eosinophilic vasculitis. It typically occurs in several phases. The prodromal phase is characterized by asthma and allergic rhinitis, and usually begins when the individual is in his or her twenties or thirties, typically persisting for many years. The eosinophilic infiltrative phase is characterized by peripheral eosinophilia and eosinophilic tissue infiltration of various organs including the lungs and GI tract. The third phase is the vasculitic phase and may be associated with constitutional signs and symptoms including fever, weight loss, malaise, and fatigue. The mean age at diagnosis is 48 years, with a range of 14 to 74 years; the average length of time between diagnosis of asthma and vasculitis is 9 years.
Similar to other pulmonary eosinophilic syndromes, constitutional symptoms are very common in EGPA and include weight loss of 10–20 lb, fevers, and diffuse myalgias and migratory polyarthralgias. Myositis may be present with evidence of vasculitis on muscle biopsies. In contrast to the eosinophilic pneumonias, EGPA involves many organ systems including the lungs, skin, nerves, heart, GI tract, and kidneys.
Symptoms and Clinical Manifestations • RESPIRATORY Most EGPA patients have asthma that arises later in life and in individuals who have no family history of atopy. The asthma can often be severe, and oral corticosteroids are often required to control symptoms but may lead to suppression of vasculitic symptoms. In addition to the more common symptoms of cough, dyspnea, sinusitis, and allergic rhinitis, alveolar hemorrhage and hemoptysis may also occur.
NEUROLOGIC Over three-fourths of EGPA patients have neurologic manifestations. Mononeuritis multiplex most commonly involves the peroneal nerve, but also involves the ulnar, radial, internal popliteal, and occasionally, cranial nerves. Cerebral hemorrhage and infarction may also occur and are important causes of death. Despite treatment, neurologic sequelae often do not completely resolve.
DERMATOLOGIC Approximately half of EGPA patients develop dermatologic manifestations. These include palpable purpura, skin nodules, urticarial rashes, and livedo.
CARDIOVASCULAR Granulomas, vasculitis, and widespread myocardial damage may be found on biopsy or at autopsy, and cardiomyopathy and heart failure may be seen in up to half of all patients but are often at least partially reversible. Acute pericarditis, constrictive pericarditis, myocardial infarction, and other electrocardiographic changes all may occur. The heart is a primary target organ in EGPA, and cardiac involvement often portends a worse prognosis.
GI GI symptoms are common in EGPA and likely represent an eosinophilic gastroenteritis characterized by abdominal pain, diarrhea, GI bleeding, and colitis. Ischemic bowel, pancreatitis, and cholecystitis have also been reported in association with EGPA and usually portend a worse prognosis.
RENAL Renal involvement is more common than once thought, and approximately 25% of patients have some degree of renal involvement. This may include proteinuria, glomerulonephritis, renal insufficiency, and rarely, renal infarct.
Lab Abnormalities Systemic eosinophilia is the hallmark laboratory finding in patients with EGPA and reflects the likely pathogenic role that the eosinophil plays in this disease. Eosinophilia greater than 10% is one of the defining features of this illness and may be as high as 75% of the peripheral white blood cell count. It is present at the time of diagnosis in over 80% of patients but may respond quickly (often within 24 h) to initiation of systemic corticosteroid therapy. Even in the absence of systemic eosinophilia, tissue eosinophilia may be present.
Although not specific to EGPA, ANCAs are present in up to two-thirds of patients, mostly with a perinuclear staining pattern. Nonspecific lab abnormalities that may be present in patients with EGPA include a marked elevation in ESR, a normochromic normocytic anemia, an elevated IgE, hypergammaglobulinemia, and positive rheumatoid factor and antinuclear antibodies (ANA). Although BAL often reveals significant eosinophilia, this may be seen in other eosinophilic lung diseases. Similarly, PFT often reveals an obstructive defect similar to asthma.
Radiographic Features Chest x-ray abnormalities are extremely common in EGPA and consist of bilateral, nonsegmental, patchy infiltrates that often migrate and may be interstitial or alveolar in appearance. Reticulonodular and nodular disease without cavitation can be seen, as can pleural effusions and hilar adenopathy. The most common CT findings include bilateral ground-glass opacity and airspace consolidation that is predominantly subpleural. Other CT findings include bronchial wall thickening, hyperinflation, interlobular septal thickening, lymph node enlargement, and pericardial and pleural effusions. Angiography may be used diagnostically and may show signs of vasculitis in the coronary, central nervous system, and peripheral vasculature.
Treatment and Prognosis of EGPA Most patients diagnosed with EGPA have previously been diagnosed with asthma, rhinitis, and sinusitis, and have received treatment with inhaled or systemic corticosteroids. Because these agents are also the initial treatment of choice for EGPA patients, institution of these therapies in patients with EGPA who are perceived to have severe asthma may delay the diagnosis of EGPA because signs of vasculitis may be masked. Corticosteroids dramatically alter the course of EGPA: up to 50% of those who are untreated die within 3 months of diagnosis, whereas treated patients have a 6-year survival of over 70%. Common causes of death include heart failure, cerebral hemorrhage, renal failure, and GI bleeding. Recent data suggest that clinical remission may be obtained in over 90% of patients treated; approximately 25% of those patients may relapse, often due to corticosteroid tapering, with a rising eosinophil count heralding the relapse. Myocardial, GI, and renal involvement most often portend a poor prognosis. In such cases, treatment with higher doses of corticosteroids or the addition of cytotoxic agents such as cyclophosphamide is often warranted. Although survival does not differ between those treated or untreated with cyclophosphamide, cyclophosphamide is associated with a reduced incidence of relapse and an improved clinical response to treatment. Other therapies that have been used successfully in the management of EGPA include azathioprine, methotrexate, intravenous gamma globulin, and interferon α. Plasma exchange has not been shown to provide any additional benefit. Recent studies examining the efficacy of anti-IL-5 therapy have shown promise.
HYPEREOSINOPHILIC SYNDROMES
Hypereosinophilic syndromes (HES) constitute a heterogeneous group of disease entities manifest by persistent eosinophilia >1500 eosinophils/μL in association with end organ damage or dysfunction, in the absence of secondary causes of eosinophilia. In addition to familial, undefined, and overlap syndromes with incomplete criteria, the predominant HES subtypes are the myeloproliferative and lymphocytic variants. The myeloproliferative variant may be divided into three subgroups: (1) chronic eosinophilic leukemia with demonstrable cytogenetic abnormalities and/or blasts on peripheral smear; (2) the platelet-derived growth factor receptor α (PDGFRα)–associated HES, attributed to a constitutively activated tyrosine kinase fusion protein (Fip1L1-PDGFRα) due to a chromosomal deletion on 4q12; this variant is often responsive to imatinib; and (3) the FIP1-negative variant associated with clonal eosinophilia and at least four of the following: dysplastic peripheral eosinophils, increased serum vitamin B12, increased tryptase, anemia, thrombocytopenia, splenomegaly, bone marrow cellularity >80%, spindle-shaped mast cells, and myelofibrosis.
Extrapulmonary Manifestations of HES More common in men than in women, HES occurs between the ages of 20 and 50 and is characterized by significant extrapulmonary involvement, including infiltration of the heart, GI tract, kidney, liver, joints, and skin. Cardiac involvement includes myocarditis and/or endomyocardial fibrosis, as well as a restrictive cardiomyopathy.
Pulmonary Manifestations of HES Similar to the other pulmonary eosinophilic syndromes, these HES are manifest by high levels of blood, BAL, and tissue eosinophilia. Lung involvement occurs in 40% of these patients and is characterized by cough and dyspnea, as well as pulmonary infiltrates. Although it is often difficult to discern the pulmonary infiltrates and effusions seen on chest x-ray from pulmonary edema resulting from cardiac involvement, CT scan findings include interstitial infiltrates, ground-glass opacities, and small nodules. HES are typically not associated with ANCA or elevated IgE.
Course and Response to Therapy Unlike the other pulmonary eosinophilic syndromes, less than half of patients with these HES respond to corticosteroids as first-line therapy. Although other treatment options include hydroxyurea, cyclosporine, and interferon, the tyrosine kinase inhibitor imatinib has emerged as an important therapeutic option for patients with the myeloproliferative variant. Anti-IL-5 therapy with mepolizumab also holds promise for these patients and is currently being investigated.
ALLERGIC BRONCHOPULMONARY ASPERGILLOSIS
Allergic bronchopulmonary aspergillosis (ABPA) is an eosinophilic pulmonary disorder that occurs in response to allergic sensitization to antigens from Aspergillus species fungi. The predominant clinical presentation of ABPA is an asthmatic phenotype, often accompanied by cough with production of brownish plugs of mucus. ABPA has also been well described as a complication of cystic fibrosis. A workup for ABPA may be beneficial in patients who carry a diagnosis of asthma but have proven refractory to usual therapy. ABPA is a distinct diagnosis from simple asthma, characterized by prominent peripheral eosinophilia and elevated circulating levels of IgE (>417 IU/mL). Establishing a diagnosis of ABPA also requires establishing sensitivity to Aspergillus antigens by skin test reactivity, positive serum precipitins for Aspergillus, and/or direct measurement of circulating specific IgG and IgE to Aspergillus. Central bronchiectasis is described as a classic finding on chest imaging in ABPA but is not necessary for making a diagnosis. Other possible findings on chest imaging include patchy infiltrates and evidence of mucus impaction.
Systemic glucocorticoids may be used in the treatment of ABPA that is persistently symptomatic despite the use of inhaled therapies for asthma. Courses of glucocorticoids should be tapered over 3–6 months, and their use must be balanced against the risks of prolonged steroid therapy. Antifungal agents such as fluconazole and voriconazole given over a 4-month course reduce the antigenic stimulus in ABPA and may therefore modulate disease activity in selected patients. The use of monoclonal antibody against IgE (omalizumab) has been described in treating severe ABPA, particularly in individuals with ABPA as a complication of cystic fibrosis.
ABPA-like syndromes have been reported as a result to sensitization to several non-Aspergillus species fungi. However, these conditions are substantially rarer than ABPA, which may be present in a significant proportion of patients with refractory asthma.
INFECTIOUS PROCESSES
Infectious etiologies of pulmonary eosinophilia are largely due to helminths and are of particular importance in the evaluation of pulmonary eosinophilia in tropical environments and in the developing world (Table 310-4). These infectious conditions may also be considered in recent travelers to endemic regions. Loffler syndrome refers to transient pulmonary infiltrates with eosinophilia that occurs in response to passage of helminthic larvae through the lungs, most commonly larvae of Ascaris species (roundworm). Symptoms are generally self-limited and may include dyspnea, cough, wheeze, and hemoptysis. Loffler syndrome may also occur in response to hookworm infection with Ancylostoma duodenale or Necator americanus. Chronic Strongyloides stercoralis infection can lead to recurrent respiratory symptoms with peripheral eosinophilia between flares. In immunocompromised hosts, including patients on glucocorticoids, a severe, potentially fatal, hyperinfection syndrome can result from Strongyloides infection. Paragonimiasis, filariasis, and visceral larval migrans can all cause pulmonary eosinophilia as well.
|
INFECTIOUS CAUSES OF PULMONARY EOSINOPHILIA |
Source: Adapted from P Akuthota, PF Weller: Clin Microbiol Rev 25:649, 2012.
DRUGS AND TOXINS
A host of medications are associated with the development of pulmonary infiltrates with peripheral eosinophilia. Therefore, drug reaction must always be included in the differential diagnosis of pulmonary eosinophilia. Although the list of medications associated with pulmonary eosinophilia is ever expanding, common culprits include nonsteroidal anti-inflammatory medications and systemic antibiotics, most specifically nitrofurantoin. Additionally, various and diverse environmental exposures such as particulate metals, scorpion stings, and inhalational drugs of abuse may also cause pulmonary eosinophilia. Radiation therapy for breast cancer has been linked with eosinophilic pulmonary infiltration as well. The mainstay of treatment is removal of the offending exposure, although glucocorticoids may be necessary if respiratory symptoms are severe.
GLOBAL CONSIDERATIONS
![]() In the United States, drug-induced eosinophilic pneumonias are the most common cause of eosinophilic pulmonary infiltrates. A travel history or evidence of recent immigration should prompt the consideration of parasite-associated disorders. Tropical eosinophilia is usually caused by filarial infection; however, eosinophilic pneumonias also occur with other parasites such as Ascaris spp., Ancylostoma spp., Toxocara spp., and Strongyloides stercoralis. Tropical eosinophilia due to Wuchereria bancrofti or Wuchereria malayi occurs most commonly in southern Asia, Africa, and South America and is treated successfully with diethylcarbamazine. In the United States, Strongyloides is endemic to the southeastern and Appalachian regions.
In the United States, drug-induced eosinophilic pneumonias are the most common cause of eosinophilic pulmonary infiltrates. A travel history or evidence of recent immigration should prompt the consideration of parasite-associated disorders. Tropical eosinophilia is usually caused by filarial infection; however, eosinophilic pneumonias also occur with other parasites such as Ascaris spp., Ancylostoma spp., Toxocara spp., and Strongyloides stercoralis. Tropical eosinophilia due to Wuchereria bancrofti or Wuchereria malayi occurs most commonly in southern Asia, Africa, and South America and is treated successfully with diethylcarbamazine. In the United States, Strongyloides is endemic to the southeastern and Appalachian regions.
ACKNOWLEDGMENTS
We acknowledge the contributions of Dr. Alicia K. Gerke and Dr. Gary W. Hunninghake to the previous edition of this chapter.
311 |
Occupational and Environmental Lung Disease |
Occupational and environmental lung diseases are difficult to distinguish from those of nonenvironmental origin. Virtually all major categories of pulmonary disease can be caused by environmental agents, and environmentally related disease usually presents clinically in a manner indistinguishable from that of disease not caused by such agents. In addition, the etiology of many diseases may be multifactorial; occupational and environmental factors may interact with other factors (such as smoking and genetic risk). It is often only after a careful exposure history is taken that the underlying workplace or general environmental exposure is uncovered.
Why is knowledge of occupational or environmental etiology so important? Patient management and prognosis are affected significantly by such knowledge. For example, patients with occupational asthma or hypersensitivity pneumonitis often cannot be managed adequately without cessation of exposure to the offending agent. Establishment of cause may have significant legal and financial implications for a patient who no longer can work in his or her usual job. Other exposed people may be identified as having the disease or prevented from getting it. In addition, new associations between exposure and disease may be identified (e.g., nylon flock worker’s lung disease and diacetyl-induced bronchiolitis obliterans).
Although the exact proportion of lung disease due to occupational and environmental factors is unknown, a large number of individuals are at risk. For example, 15–20% of the burden of adult asthma and chronic obstructive pulmonary disease (COPD) has been estimated to be due to occupational factors.
HISTORY AND EXPOSURE ASSESSMENT
The patient’s history is of paramount importance in assessing any potential occupational or environmental exposure. Inquiry into specific work practices should include questions about the specific contaminants involved, the presence of visible dusts, chemical odors, the size and ventilation of workspaces, the use of respiratory protective equipment, and whether co-workers have similar complaints. The temporal association of exposure at work and symptoms may provide clues to occupation-related disease. In addition, the patient must be questioned about alternative sources of exposure to potentially toxic agents, including hobbies, home characteristics, exposure to secondhand smoke, and proximity to traffic or industrial facilities. Short-term and long-term exposures to potential toxic agents in the distant past also must be considered.
Workers in the United States have the right to know about potential hazards in their workplaces under federal Occupational Safety and Health Administration (OSHA) regulations. Employers must provide specific information about potential hazardous agents in products being used through Material Safety Data Sheets as well as training in personal protective equipment and environmental control procedures. However, the introduction of new processes and/or new chemical compounds may change exposure significantly, and often only the employee on the production line is aware of the change. For the physician caring for a patient with a suspected work-related illness, a visit to the work site can be very instructive. Alternatively, an affected worker can request an inspection by OSHA. If reliable environmental sampling data are available, that information should be used in assessing a patient’s exposure. Because many of the chronic diseases result from exposure over many years, current environmental measurements should be combined with work histories to arrive at estimates of past exposure.
PULMONARY FUNCTION TESTS AND CHEST IMAGING
Exposures to inorganic and organic dusts can cause interstitial lung disease that presents with a restrictive pattern and a decreased diffusing capacity (Chap. 306e). Similarly, exposures to a number of organic dusts or chemical agents may result in occupational asthma or COPD that is characterized by airway obstruction. Measurement of change in forced expiratory volume (FEV1) before and after a working shift can be used to detect an acute bronchoconstrictive response.
The chest radiograph is useful in detecting and monitoring the pulmonary response to mineral dusts, certain metals, and organic dusts capable of inducing hypersensitivity pneumonitis. The International Labour Organisation (ILO) International Classification of Radiographs of Pneumoconioses classifies chest radiographs by the nature and size of opacities seen and the extent of involvement of the parenchyma. In general, small rounded opacities are seen in silicosis or coal worker’s pneumoconiosis, and small linear opacities are seen in asbestosis. Although useful for epidemiologic studies and screening large numbers of workers, the ILO system can be problematic when applied to an individual worker’s chest radiograph. With dusts causing rounded opacities, the degree of involvement on the chest radiograph may be extensive, whereas pulmonary function may be only minimally impaired. In contrast, in pneumoconiosis causing linear, irregular opacities like those seen in asbestosis, the radiograph may lead to underestimation of the severity of the impairment until relatively late in the disease. For patients with a history of asbestos exposure, conventional computed tomography (CT) is more sensitive for the detection of pleural thickening, and high-resolution CT (HRCT) improves the detection of asbestosis.
Other procedures that may be of use in identifying the role of environmental exposures in causing lung disease include skin prick testing or specific IgE antibody titers for evidence of immediate hypersensitivity to agents capable of inducing occupational asthma (flour antigens in bakers), specific IgG precipitating antibody titers for agents capable of causing hypersensitivity pneumonitis (pigeon antigen in bird handlers), and assays for specific cell-mediated immune responses (beryllium lymphocyte proliferation testing in nuclear workers or tuberculin skin testing in health care workers). Sometimes a bronchoscopy to obtain transbronchial biopsies of lung tissue may be required for histologic diagnosis (chronic beryllium disease). Rarely, video-assisted thoracoscopic surgery to obtain a larger sample of lung tissue may be required to determine the specific diagnosis of environmentally induced lung disease (hypersensitivity pneumonitis or giant cell interstitial pneumonitis due to cobalt exposure).
DETERMINANTS OF INHALATIONAL EXPOSURE
The chemical and physical characteristics of inhaled agents affect both the dose and the site of deposition in the respiratory tract. Water-soluble gases such as ammonia and sulfur dioxide are absorbed in the lining fluid of the upper and proximal airways and thus tend to produce irritative and bronchoconstrictive responses. In contrast, nitrogen dioxide and phosgene, which are less soluble, may penetrate to the bronchioles and alveoli in sufficient quantities to produce acute chemical pneumonitis.
Particle size of air contaminants must also be considered. Because of their settling velocities in air, particles >10–15 μm in diameter do not penetrate beyond the nose and throat. Particles <10 μm in size are deposited below the larynx. These particles are divided into three size fractions on the basis of their size characteristics and sources. Particles ~2.5–10 μm (coarse-mode fraction) contain crustal elements such as silica, aluminum, and iron. These particles mostly deposit relatively high in the tracheobronchial tree. Although the total mass of an ambient sample is dominated by these larger respirable particles, the number of particles, and therefore the surface area on which potential toxic agents can deposit and be carried to the lower airways, is dominated by particles <2.5 μm (fine-mode fraction). These fine particles are created primarily by the burning of fossil fuels or high-temperature industrial processes resulting in condensation products from gases, fumes, or vapors. The smallest particles, those <0.1 μm in size, represent the ultrafine fraction and make up the largest number of particles; they tend to remain in the airstream and deposit in the lung only on a random basis as they come into contact with the alveolar walls. If they do deposit, however, particles of this size range may penetrate into the circulation and be carried to extrapulmonary sites. New technologies create particles of this size (“nanoparticles”) for use in many commercial applications. Besides the size characteristics of particles and the solubility of gases, the actual chemical composition, mechanical properties, and immunogenicity or infectivity of inhaled material determine in large part the nature of the diseases found among exposed persons.
OCCUPATIONAL EXPOSURES AND PULMONARY DISEASE
Table 311-1 provides broad categories of exposure in the workplace and diseases associated with chronic exposure in those industries.
|
CATEGORIES OF OCCUPATIONAL EXPOSURE AND ASSOCIATED RESPIRATORY CONDITIONS |
ASBESTOS-RELATED DISEASES
Asbestos is a generic term for several different mineral silicates, including chrysolite, amosite, anthophyllite, and crocidolite. In addition to workers involved in the production of asbestos products (mining, milling, and manufacturing), many workers in the shipbuilding and construction trades, including pipe fitters and boilermakers, were occupationally exposed because asbestos was widely used during the twentieth century for its thermal and electrical insulation properties. Asbestos also was used in the manufacture of fire-resistant textiles, in cement and floor tiles, and in friction materials such as brake and clutch linings.
Exposure to asbestos is not limited to persons who directly handle the material. Cases of asbestos-related diseases have been encountered in individuals with only bystander exposure, such as painters and electricians who worked alongside insulation workers in a shipyard. Community exposure resulted from the use of asbestos-containing mine and mill tailings as landfill, road surface, and playground material (e.g., Libby, MT, the site of a vermiculite mine in which the ore was contaminated with asbestos). Finally, exposure can occur from the disturbance of naturally occurring asbestos (e.g., from increasing residential development in the foothills of the Sierra Mountains in California).
Asbestos has largely been replaced in the developed world with synthetic mineral fibers such as fiberglass and refractory ceramic fibers, but it continues to be used in the developing world. The major health effects from exposure to asbestos are pleural and pulmonary fibrosis, cancers of the respiratory tract, and pleural and peritoneal mesothelioma.
Asbestosis is a diffuse interstitial fibrosing disease of the lung that is directly related to the intensity and duration of exposure. The disease resembles other forms of diffuse interstitial fibrosis (Chap. 315). Usually, exposure has taken place for at least 10 years before the disease becomes manifest. The mechanisms by which asbestos fibers induce lung fibrosis are not completely understood but are known to involve oxidative injury due to the generation of reactive oxygen species by the transition metals on the surface of the fibers as well as from cells engaged in phagocytosis.
Past exposure to asbestos is specifically indicated by pleural plaques on chest radiographs, which are characterized by either thickening or calcification along the parietal pleura, particularly along the lower lung fields, the diaphragm, and the cardiac border. Without additional manifestations, pleural plaques imply only exposure, not pulmonary impairment. Benign pleural effusions also may occur. The fluid is typically a serous or bloody exudate. The effusion may be slowly progressive or may resolve spontaneously.
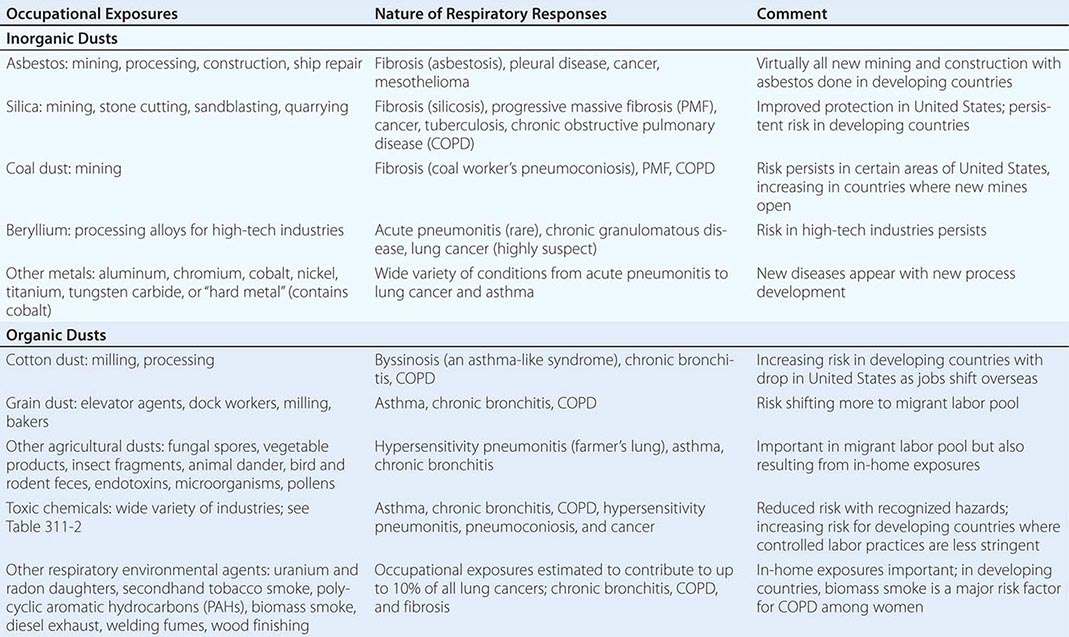
Irregular or linear opacities that usually are first noted in the lower lung fields are the chest radiographic hallmark of asbestosis. An indistinct heart border or a “ground-glass” appearance in the lung fields may be seen. HRCT may show distinct changes of subpleural curvilinear lines 5–10 mm in length that appear to be parallel to the pleural surface (Fig. 311-1).
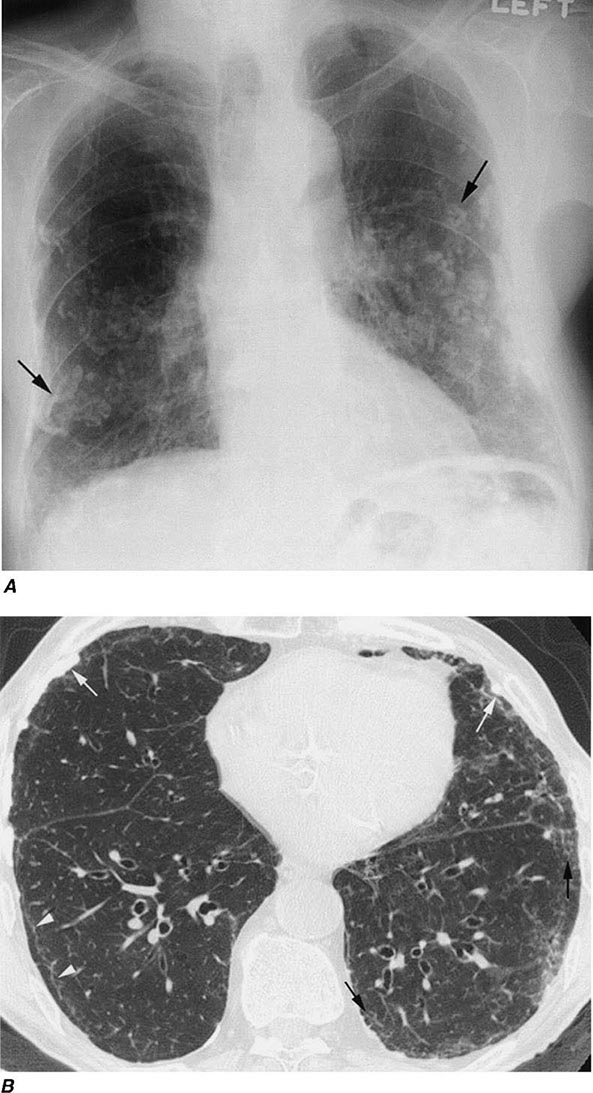
FIGURE 311-1 Asbestosis. A. Frontal chest radiograph shows bilateral calcified pleural plaques consistent with asbestos-related pleural disease. Poorly defined linear and reticular abnormalities are seen in the lower lobes bilaterally. B. Axial high-resolution computed tomography of the thorax obtained through the lung bases shows bilateral, subpleural reticulation (black arrows), representing fibrotic lung disease due to asbestosis. Subpleural lines are also present (arrowheads), characteristic of, though not specific for, asbestosis. Calcified pleural plaques representing asbestos-related pleural disease (white arrows) are also evident.
Pulmonary function testing in asbestosis reveals a restrictive pattern with a decrease in both lung volumes and diffusing capacity. There may also be evidence of mild airflow obstruction (due to peribronchiolar fibrosis).
Because no specific therapy is available for asbestosis, supportive care is the same as that given to any patient with diffuse interstitial fibrosis of any cause. In general, newly diagnosed cases will have resulted from exposures that occurred many years before.
Lung cancer (Chap. 107) is the most common cancer associated with asbestos exposure. The excess frequency of lung cancer (all histologic types) in asbestos workers is associated with a minimum latency of 15–19 years between first exposure and development of the disease. Persons with more exposure are at greater risk of disease. In addition, there is a significant interactive effect of smoking and asbestos exposure that results in greater risk than what would be expected from the additive effect of each factor.
Mesotheliomas (Chap. 316), both pleural and peritoneal, are also associated with asbestos exposure. In contrast to lung cancers, these tumors do not appear to be associated with smoking. Relatively short-term asbestos exposures of ≤1–2 years, occurring up to 40 years in the past, have been associated with the development of mesotheliomas (an observation that emphasizes the importance of obtaining a complete environmental exposure history). Although the risk of mesothelioma is much less than that of lung cancer among asbestos-exposed workers, over 2000 cases were reported in the United States per year at the start of the twenty-first century.
Because epidemiologic studies have shown that >80% of mesotheliomas may be associated with asbestos exposure, documented mesothelioma in a patient with occupational or environmental exposure to asbestos may be compensable.
SILICOSIS
Despite being one of the oldest known occupational pulmonary hazards, free silica (SiO2), or crystalline quartz, is still a major cause of disease. The major occupational exposures include mining; stonecutting; sand blasting; glass and cement manufacturing; foundry work; packing of silica flour; and quarrying, particularly of granite. Most often, pulmonary fibrosis due to silica exposure (silicosis) occurs in a dose-response fashion after many years of exposure.
Workers heavily exposed through sandblasting in confined spaces, tunneling through rock with a high quartz content (15–25%), or the manufacture of abrasive soaps may develop acute silicosis with as little as 10 months of exposure. The clinical and pathologic features of acute silicosis are similar to those of pulmonary alveolar proteinosis (Chap. 315). The chest radiograph may show profuse miliary infiltration or consolidation, and there is a characteristic HRCT pattern known as “crazy paving” (Fig. 311-2). The disease may be quite severe and progressive despite the discontinuation of exposure. Whole-lung lavage may provide symptomatic relief and slow the progression.
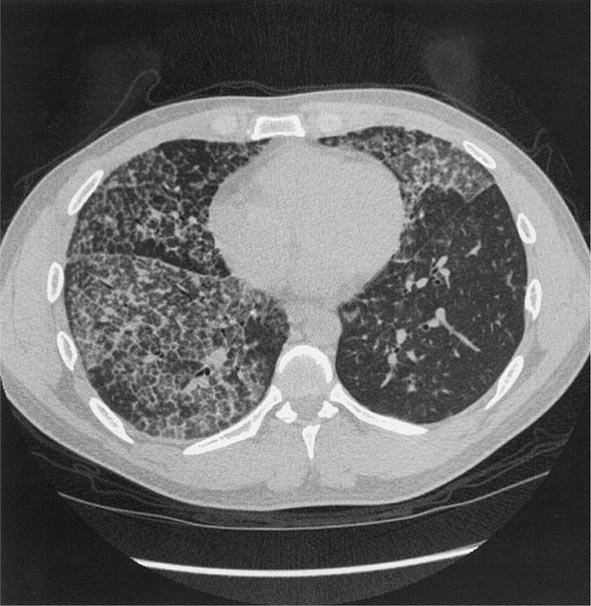
FIGURE 311-2 Acute silicosis. This high-resolution computed tomography scan shows multiple small nodules consistent with silicosis but also diffuse ground-glass densities with thickened intralobular and interlobular septa producing polygonal shapes. This has been referred to as “crazy paving.”
With long-term, less intense exposure, small rounded opacities in the upper lobes may appear on the chest radiograph after 15–20 years of exposure, usually without associated impairment of lung function (simple silicosis). Calcification of hilar nodes may occur in as many as 20% of cases and produces a characteristic “eggshell” pattern. Silicotic nodules may be identified more readily by HRCT (Fig. 311-3). The nodular fibrosis may be progressive in the absence of further exposure, with coalescence and formation of nonsegmental conglomerates of irregular masses >1 cm in diameter (complicated silicosis). These masses can become quite large, and when this occurs, the term progressive massive fibrosis (PMF) is applied. Significant functional impairment with both restrictive and obstructive components may be associated with PMF.
FIGURE 311-3 Chronic silicosis. A. Frontal chest radiograph in a patient with silicosis shows variably sized, poorly defined nodules (arrows) predominating in the upper lobes. B. Axial thoracic computed tomography image through the lung apices shows numerous small nodules, more pronounced in the right upper lobe. A number of the nodules are subpleural in location (arrows).
Because silica is cytotoxic to alveolar macrophages, patients with silicosis are at greater risk of acquiring lung infections that involve these cells as a primary defense (Mycobacterium tuberculosis, atypical mycobacteria and fungi). Because of the increased risk of active tuberculosis, the recommended treatment of latent tuberculosis in these patients is longer. Another potential clinical complication of silicosis is autoimmune connective tissue disorders such as rheumatoid arthritis and scleroderma. In addition, there are sufficient epidemiologic data that the International Agency for Research on Cancer lists silica as a probable lung carcinogen.
Other, less hazardous silicates include fuller’s earth, kaolin, mica, diatomaceous earths, silica gel, soapstone, carbonate dusts, and cement dusts. The production of fibrosis in workers exposed to these agents is believed to be related either to the free silica content of these dusts or, for substances that contain no free silica, to the potentially large dust loads to which these workers may be exposed. Some silicates, including talc and vermiculite, may be contaminated with asbestos. Fibrosis of lung or pleura, lung cancer, and mesothelioma have been associated with chronic exposure to talc and vermiculite dusts.
COAL WORKER’S PNEUMOCONIOSIS (CWP)
Occupational exposure to coal dust can lead to CWP, which has enormous social, economic, and medical significance in every nation in which coal mining is an important industry. Simple radiographically identified CWP is seen in ~10% of all coal miners and in as many as 50% of anthracite miners with more than 20 years of work on the coal face. The prevalence of disease is lower in workers in bituminous coal mines.
With prolonged exposure to coal dust (i.e., 15–20 years), small, rounded opacities similar to those of silicosis may develop. As in silicosis, the presence of these nodules (simple CWP) usually is not associated with pulmonary impairment. In addition to CWP, coal dust can cause chronic bronchitis and COPD (Chap. 314). The effects of coal dust are additive to those of cigarette smoking.
Complicated CWP is manifested by the appearance on the chest radiograph of nodules ≥1 cm in diameter generally confined to the upper half of the lungs. As in silicosis, this condition can progress to PMF that is accompanied by severe lung function deficits and associated with premature mortality. Despite improvements in technology to protect coal miners, cases of PMF still occur in the United States at a disturbing rate.
Caplan syndrome (Chap. 380), first described in coal miners but subsequently in patients with silicosis, is the combination of pneumoconiotic nodules and seropositive rheumatoid arthritis. Silica has immunoadjuvant properties and is often present in anthracitic coal dust.
CHRONIC BERYLLIUM DISEASE
Beryllium is a lightweight metal with tensile strength, good electrical conductivity, and value in the control of nuclear reactions through its ability to quench neutrons. Although beryllium may produce an acute pneumonitis, it is far more commonly associated with a chronic granulomatous inflammatory disease that is similar to sarcoidosis (Chap. 390). Unless one inquires specifically about occupational exposures to beryllium in the manufacture of alloys, ceramics, or high-technology electronics in a patient with sarcoidosis, one may miss entirely the etiologic relationship to the occupational exposure. What distinguishes chronic beryllium disease (CBD) from sarcoidosis is evidence of a specific cell-mediated immune response (i.e., delayed hypersensitivity) to beryllium.
The test that usually provides this evidence is the beryllium lymphocyte proliferation test (BeLPT). The BeLPT compares the in vitro proliferation of lymphocytes from blood or bronchoalveolar lavage in the presence of beryllium salts with that of unstimulated cells. Proliferation is usually measured by lymphocyte uptake of radiolabeled thymidine.
Chest imaging findings are similar to those of sarcoidosis (nodules along septal lines) except that hilar adenopathy is somewhat less common. As with sarcoidosis, pulmonary function test results may show restrictive and/or obstructive ventilatory deficits and decreased diffusing capacity. With early disease, both chest imaging studies and pulmonary function tests may be normal. Fiberoptic bronchoscopy with transbronchial lung biopsy usually is required to make the diagnosis of CBD. In a beryllium-sensitized individual, the presence of noncaseating granulomas or monocytic infiltration in lung tissue establishes the diagnosis. Accumulation of beryllium-specific CD4+ T cells occurs in the granulomatous inflammation seen on lung biopsy. Susceptibility to CBD is highly associated with human leukocyte antigen DP (HLA-DP) alleles that have a glutamic acid in position 69 of the β chain.
OTHER METALS
Aluminum and titanium dioxide have been rarely associated with a sarcoid-like reaction in lung tissue. Exposure to dust containing tungsten carbide, also known as “hard metal,” may produce giant cell interstitial pneumonitis. Cobalt is a constituent of tungsten carbide and is the likely etiologic agent of both the interstitial pneumonitis and the occupational asthma that may occur. The most common exposures to tungsten carbide occur in tool and dye, saw blade, and drill bit manufacture. Diamond polishing may also involve exposure to cobalt dust. In patients with interstitial lung disease, one should always inquire about exposure to metal fumes and/or dusts. Especially when sarcoidosis appears to be the diagnosis, one should always consider possible CBD.
OTHER INORGANIC DUSTS
Most of the inorganic dusts discussed thus far are associated with the production of either dust macules or interstitial fibrotic changes in the lung. Other inorganic and organic dusts (see categories in Table 311-1), along with some of the dusts previously discussed, are associated with chronic mucus hypersecretion (chronic bronchitis), with or without reduction of expiratory flow rates. Cigarette smoking is the major cause of these conditions, and any effort to attribute some component of the disease to occupational and environmental exposures must take cigarette smoking into account. Most studies suggest an additive effect of dust exposure and smoking. The pattern of the irritant dust effect is similar to that of cigarette smoking, suggesting that small airway inflammation may be the initial site of pathologic response in those cases and continued exposure may lead to chronic bronchitis and COPD.
ORGANIC DUSTS
Some of the specific diseases associated with organic dusts are discussed in detail in the chapters on asthma (Chap. 309) and hypersensitivity pneumonitis (Chap. 310). Many of these diseases are named for the specific setting in which they are found, e.g., farmer’s lung, malt worker’s disease, and mushroom worker’s disease. Often the temporal relation of symptoms to exposure furnishes the best evidence for the diagnosis. Three occupational exposures are singled out for discussion here because they affect the largest proportions of workers.
Cotton Dust (Byssinosis) Workers occupationally exposed to cotton dust (but also to flax, hemp, or jute dust) in the production of yarns for textiles and rope making are at risk for an asthma-like syndrome known as byssinosis. Exposure occurs throughout the manufacturing process but is most pronounced in the portions of the factory involved with the treatment of the cotton before spinning, i.e., blowing, mixing, and carding (straightening of fibers). The risk of byssinosis is associated with both cotton dust and endotoxin levels in the workplace environment.
Byssinosis is characterized clinically as occasional (early-stage) and then regular (late-stage) chest tightness toward the end of the first day of the workweek (“Monday chest tightness”). Exposed workers may show a significant drop in FEV1 over the course of a Monday workshift. Initially the symptoms do not recur on subsequent days of the week. However, in 10–25% of workers, the disease may be progressive, with chest tightness recurring or persisting throughout the workweek. After >10 years of exposure, workers with recurrent symptoms are more likely to have an obstructive pattern on pulmonary function testing. The highest grades of impairment generally are seen in smokers.
Dust exposure can be reduced by the use of exhaust hoods, general increases in ventilation, and wetting procedures, but respiratory protective equipment may be required during certain operations. Regular surveillance of pulmonary function in cotton dust–exposed workers using spirometry before and after the workshift is required by OSHA. All workers with persistent symptoms or significantly reduced levels of pulmonary function should be moved to areas of lower risk of exposure.
Grain Dust Worldwide, many farmers and workers in grain storage facilities are exposed to grain dust. The presentation of obstructive airway disease in grain dust–exposed workers is virtually identical to the characteristic findings in cigarette smokers, i.e., persistent cough, mucus hypersecretion, wheeze and dyspnea on exertion, and reduced FEV1 and FEV1/FVC (forced vital capacity) ratio (Chap. 306e).
Dust concentrations in grain elevators vary greatly but can be >10,000 μg/m3 with many particles in the respirable size range. The effect of grain dust exposure is additive to that of cigarette smoking, with ~50% of workers who smoke having symptoms. Smoking grain dust–exposed workers are more likely to have obstructive ventilatory deficits on pulmonary function testing. As in byssinosis, endotoxin may play a role in grain dust–induced chronic bronchitis and COPD.
Farmer’s Lung This condition results from exposure to moldy hay containing spores of thermophilic actinomycetes that produce a hypersensitivity pneumonitis (Chap. 310). A patient with acute farmer’s lung presents 4–8 h after exposure with fever, chills, malaise, cough, and dyspnea without wheezing. The history of exposure is obviously essential to distinguish this disease from influenza or pneumonia with similar symptoms. In the chronic form of the disease, the history of repeated attacks after similar exposure is important in differentiating this syndrome from other causes of patchy fibrosis (e.g., sarcoidosis).
A wide variety of other organic dusts are associated with the occurrence of hypersensitivity pneumonitis (Chap. 310). For patients who present with hypersensitivity pneumonitis, specific and careful inquiry about occupations, hobbies, and other home environmental exposures is necessary to uncover the source of the etiologic agent.
TOXIC CHEMICALS
Exposure to toxic chemicals affecting the lung generally involves gases and vapors. A common accident is one in which the victim is trapped in a confined space where the chemicals have accumulated to harmful levels. In addition to the specific toxic effects of the chemical, the victim often sustains considerable anoxia, which can play a dominant role in determining whether the individual survives.
Table 311-2 lists a variety of toxic agents that can produce acute and sometimes life-threatening reactions in the lung. All these agents in sufficient concentrations have been demonstrated, at least in animal studies, to affect the lower airways and disrupt alveolar architecture, either acutely or as a result of chronic exposure. Some of these agents may be generated acutely in the environment (see below).
|
SELECTED COMMON TOXIC CHEMICAL AGENTS THAT AFFECT THE LUNG |
Firefighters and fire victims are at risk of smoke inhalation, an important cause of acute cardiorespiratory failure. Smoke inhalation kills more fire victims than does thermal injury. Carbon monoxide poisoning with resulting significant hypoxemia can be life-threatening (Chap. 473e). Synthetic materials (plastic, polyurethanes), when burned, may release a variety of other toxic agents (such as cyanide and hydrochloric acid), and this must be considered in evaluating smoke inhalation victims. Exposed victims may have some degree of lower respiratory tract inflammation and/or pulmonary edema.
Exposure to certain highly reactive, low-molecular-weight agents used in the manufacture of synthetic polymers, paints, and coatings (diisocyanates in polyurethanes, aromatic amines and acid anhydrides in epoxies) is associated with a high risk of occupational asthma. Although this occupational asthma manifests clinically as if sensitization has occurred, an IgE antibody–mediated mechanism is not necessarily involved. Hypersensitivity pneumonitis–like reactions also have been described in diisocyanate and acid anhydride–exposed workers.
Fluoropolymers such as Teflon, which at normal temperatures produce no reaction, become volatilized upon heating. The inhaled agents cause a characteristic syndrome of fever, chills, malaise, and occasionally mild wheezing, leading to the diagnosis of polymer fume fever. A similar self-limited, influenza-like syndrome—metal fume fever—results from acute exposure to fumes containing zinc oxide, typically from welding of galvanized steel. These inhalational fever syndromes may begin several hours after work and resolve within 24 h, only to return on repeated exposure.
Two other agents have been associated with potentially severe lung disease. Occupational exposure to nylon flock has been shown to induce a lymphocytic bronchiolitis, and workers exposed to diacetyl, which is used to provide “butter” flavor in the manufacture of microwave popcorn and other foods, have developed bronchiolitis obliterans (Chap. 315).
World Trade Center Disaster A consequence of the attack on the World Trade Center (WTC) on September 11, 2001, was relatively heavy exposure of a large number of firefighters and other rescue workers to the dust generated by the collapse of the buildings. Environmental monitoring and chemical characterization of WTC dust has revealed a wide variety of potentially toxic constituents, although much of the dust was pulverized cement. Possibly because of the high alkalinity of WTC dust, significant cough, wheeze, and phlegm production occurred among firefighters and cleanup crews. New cough and wheeze syndromes also occurred among local residents. Heavier exposure to WTC dust among New York City firefighters was associated with accelerated decline of lung function over the first year after the disaster. More recently, concerns have been raised about risk of interstitial lung disease, especially of a granulomatous nature.
OCCUPATIONAL RESPIRATORY CARCINOGENS
Exposures at work have been estimated to contribute to 10% of all lung cancer cases. In addition to asbestos, other agents either proven or suspected to be respiratory carcinogens include acrylonitrile, arsenic compounds, beryllium, bis(chloromethyl) ether, chromium (hexavalent), formaldehyde (nasal), isopropanol (nasal sinuses), mustard gas, nickel carbonyl (nickel smelting), polycyclic aromatic hydrocarbons (coke oven emissions and diesel exhaust), secondhand tobacco smoke, silica (both mining and processing), talc (possible asbestos contamination in both mining and milling), vinyl chloride (sarcomas), wood (nasal cancer only), and uranium. Workers at risk of radiation-related lung cancer include not only those involved in mining or processing uranium but also those exposed in underground mining operations of other ores where radon daughters may be emitted from rock formations.
ASSESSMENT OF DISABILITY
Disability is the term used to describe the decreased ability to work due to the effects of a medical condition. Physicians are generally able to assess physiologic dysfunction, or impairment, but the rating of disability for compensation of loss of income also involves nonmedical factors such as the education and employability of the individual. The disability rating scheme differs with the compensation-granting agency. For example, the U.S. Social Security Administration requires that an individual be unable to do any work (i.e., total disability) before he or she will receive income replacement payments. Many state workers’ compensation systems allow for payments for partial disability. In the Social Security scheme, no determination of cause is done, whereas work-relatedness must be established in workers’ compensation systems.
For respiratory impairment rating, resting pulmonary function tests (spirometry and diffusing capacity) are used as the initial assessment tool, with cardiopulmonary exercise testing (to assess maximal oxygen consumption) used if the results of the resting tests do not correlate with the patient’s symptoms. Methacholine challenge (to assess airway reactivity) can also be useful in patients with asthma who have normal spirometry when evaluated. Some compensation agencies (e.g., Social Security) have proscribed disability classification schemes based on pulmonary function test results. When no specific scheme is proscribed, the Guidelines of the American Medical Association should be used.
GENERAL ENVIRONMENTAL EXPOSURES
OUTDOOR AIR POLLUTION
In 1971, the U.S. government established national air quality standards for several pollutants believed to be responsible for excess cardiorespiratory diseases. Primary standards regulated by the U.S. Environmental Protection Agency (EPA) designed to protect the public health with an adequate margin of safety exist for sulfur dioxide, particulates matter, nitrogen dioxide, ozone, lead, and carbon monoxide. Standards for each of these pollutants are updated regularly through an extensive review process conducted by the EPA. (For details on current standards, go to http://www.epa.gov/air/criteria.html.)
Pollutants are generated from both stationary sources (power plants and industrial complexes) and mobile sources (motor vehicles), and none of the regulated pollutants occurs in isolation. Furthermore, pollutants may be changed by chemical reactions after being emitted. For example, sulfur dioxide and particulate matter emissions from a coal-fired power plant may react in air to produce acid sulfates and aerosols, which can be transported long distances in the atmosphere. Oxides of nitrogen and volatile organic compounds from automobile exhaust react with sunlight to produce ozone. Although originally thought to be confined to Los Angeles, photochemically derived pollution (“smog”) is now known to be a problem throughout the United States and in many other countries. Both acute and chronic effects of these exposures have been documented in large population studies.
The symptoms and diseases associated with air pollution are the same as conditions commonly associated with cigarette smoking. In addition, decreased growth of lung function and asthma have been associated with chronic exposure to only modestly elevated levels of traffic-related gases and respirable particles. Multiple population-based time-series studies within cities have demonstrated excess health care utilization for asthma and other cardiopulmonary conditions as well as increased mortality rates. Cohort studies comparing cities that have relatively high levels of particulate exposures with less polluted communities suggest excess morbidity and mortality rates from cardiopulmonary conditions in long-term residents of the former. The strong epidemiologic evidence that fine particulate matter is a risk factor for cardiovascular morbidity and mortality has prompted toxicologic investigations into the underlying mechanisms. The inhalation of fine particles from combustion sources probably generates oxidative stress followed by local injury and inflammation in the lungs that in turn lead to autonomic and systemic inflammatory responses that can induce endothelial dysfunction and/or injury. Recent research findings on the health effects of air pollutants has led to stricter U.S. ambient air quality standards for ozone, oxides of nitrogen, and particulate matter as well as greater emphasis on publicizing pollution alerts to encourage individuals with significant cardiopulmonary impairment to stay indoors during high-pollution episodes.
INDOOR EXPOSURES
Secondhand tobacco smoke (Chap. 470), radon gas, wood smoke, and other biologic agents generated indoors must be considered. Several studies have shown that the respirable particulate load in any household is directly proportional to the number of cigarette smokers living in that home. Increases in prevalence of respiratory illnesses, especially asthma, and reduced levels of pulmonary function measured with simple spirometry have been found in the children of smoking parents in a number of studies. Recent meta-analyses for lung cancer and cardiopulmonary diseases, combining data from multiple secondhand tobacco smoke epidemiologic studies, suggest an ~25% increase in relative risk for each condition, even after adjustment for major potential confounders.
Exposure to radon gas in homes is a risk factor for lung cancer. The main radon product (radon-222) is a gas that results from the decay series of uranium-238, with the immediate precursor being radium-226. The amount of radium in earth materials determines how much radon gas will be emitted. Levels associated with excess lung cancer risk may be present in as many as 10% of the houses in the United States. When smokers reside in the home, the problem is potentially greater, because the molecular size of radon particles allows them to attach readily to smoke particles that are inhaled. Fortunately, technology is available for assessing and reducing the level of exposure.
Other indoor exposures of concern are bioaerosols that contain antigenic material (fungi, cockroaches, dust mites, and pet danders) associated with an increased risk of atopy and asthma. Indoor chemical agents include strong cleaning agents (bleach, ammonia), formaldehyde, perfumes, pesticides, and oxides of nitrogen from gas appliances. Nonspecific responses associated with “tight-building syndrome,” perhaps better termed “building-associated illness,” in which no particular agent has been implicated, have included a wide variety of complaints, among them respiratory symptoms that are relieved only by avoiding exposure in the building in question. The degree to which “smells” and other sensory stimuli are involved in the triggering of potentially incapacitating psychological or physical responses has yet to be determined, and the long-term consequences of such environmental exposures are unknown.
GLOBAL CONSIDERATIONS
![]() Indoor exposure to biomass smoke (wood, dung, crop residues, charcoal) is estimated to be responsible for >4% of worldwide disability-adjusted life-years (DALYs) lost, due to acute lower respiratory infections in children, COPD and lung cancer in women, and cardiovascular disease among men. This burden of disease places indoor exposure to biomass smoke as the leading environmental hazard for poor health and the third most important risk factor overall.
Indoor exposure to biomass smoke (wood, dung, crop residues, charcoal) is estimated to be responsible for >4% of worldwide disability-adjusted life-years (DALYs) lost, due to acute lower respiratory infections in children, COPD and lung cancer in women, and cardiovascular disease among men. This burden of disease places indoor exposure to biomass smoke as the leading environmental hazard for poor health and the third most important risk factor overall.
Almost one-half of the world’s population uses biomass fuel for cooking, heating, or baking. This occurs predominantly in the rural areas of developing countries. Because many families burn biomass fuels in open stoves, which are highly inefficient, and inside homes with poor ventilation, women and young children are exposed on a daily basis to high levels of smoke. In these homes, 24-h mean levels of fine particulate matter, a component of biomass smoke, have been reported to be 2–30 times higher than the National Ambient Air Quality Standards set by the U.S. EPA.
Epidemiologic studies have consistently shown associations between exposure to biomass smoke and both chronic bronchitis and COPD, with odds ratios ranging between 3 and 10 and increasing with longer exposures. In addition to the common occupational exposure to biomass smoke of women in developing countries, men from such countries may be occupationally exposed. Because of increased migration to the United States from developing countries, clinicians need to be aware of the chronic respiratory effects of exposure to biomass smoke, which can include interstitial lung disease (Fig. 311-4). Evidence is beginning to emerge that improved stoves with chimneys can reduce biomass smoke–induced respiratory illness in both children and women.
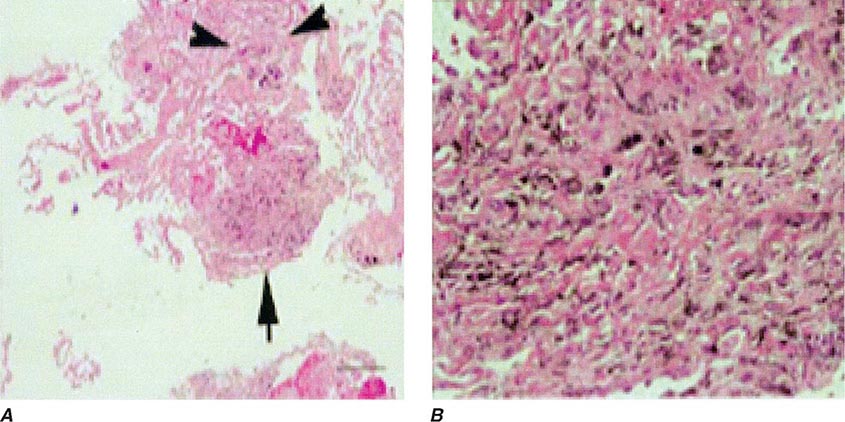
FIGURE 311-4 Histopathologic features of biomass smoke–induced interstitial lung disease. A. Anthracitic pigment is seen accumulating along alveolar septae (arrowheads) and within a pigmented dust macule (single arrow). B. A high-power photomicrograph contains a mixture of fibroblasts and carbon-laden macrophages.
312 |
Bronchiectasis |
Bronchiectasis refers to an irreversible airway dilation that involves the lung in either a focal or a diffuse manner and that classically has been categorized as cylindrical or tubular (the most common form), varicose, or cystic.
ETIOLOGY
Bronchiectasis can arise from infectious or noninfectious causes (Table 312-1). Clues to the underlying etiology are often provided by the pattern of lung involvement. Focal bronchiectasis refers to bronchiectatic changes in a localized area of the lung and can be a consequence of obstruction of the airway—either extrinsic (e.g., due to compression by adjacent lymphadenopathy or parenchymal tumor mass) or intrinsic (e.g., due to an airway tumor or aspirated foreign body, a scarred/stenotic airway, or bronchial atresia from congenital underdevelopment of the airway). Diffuse bronchiectasis is characterized by widespread bronchiectatic changes throughout the lung and often arises from an underlying systemic or infectious disease process.
|
MAJOR ETIOLOGIES OF BRONCHIECTASIS AND PROPOSED WORKUP |

More pronounced involvement of the upper lung fields is most common in cystic fibrosis (CF) and is also observed in postradiation fibrosis, corresponding to the lung region encompassed by the radiation port. Bronchiectasis with predominant involvement of the lower lung fields usually has its source in chronic recurrent aspiration (e.g., due to esophageal motility disorders like those in scleroderma), end-stage fibrotic lung disease (e.g., traction bronchiectasis from idiopathic pulmonary fibrosis), or recurrent immunodeficiency-associated infections (e.g., hypogammaglobulinemia). Bronchiectasis resulting from infection by nontuberculous mycobacteria (NTM), most commonly the Mycobacterium avium-intracellulare complex (MAC), often preferentially affects the midlung fields. Congenital causes of bronchiectasis with predominant midlung field involvement include the dyskinetic/immotile cilia syndrome. Finally, predominant involvement of the central airways is reported in association with allergic bronchopulmonary aspergillosis (ABPA), in which an immune-mediated reaction to Aspergillus damages the bronchial wall. Congenital causes of central airway–predominant bronchiectasis resulting from cartilage deficiency include tracheobronchomegaly (Mounier-Kuhn syndrome) and Williams-Campbell syndrome.
In many cases, the etiology of bronchiectasis is not determined. In case series, as many as 25–50% of patients referred for bronchiectasis have idiopathic disease.
EPIDEMIOLOGY
The overall reported prevalence of bronchiectasis in the United States has recently increased, but the epidemiology of bronchiectasis varies greatly with the underlying etiology. For example, patients born with CF often develop significant clinical bronchiectasis in late adolescence or early adulthood, although atypical presentations of CF in adults in their thirties and forties are also possible. In contrast, bronchiectasis resulting from MAC infection classically affects nonsmoking women >50 years of age. In general, the incidence of bronchiectasis increases with age. Bronchiectasis is more common among women than among men.
![]() In areas where tuberculosis is prevalent, bronchiectasis more frequently occurs as a sequela of granulomatous infection. Focal bronchiectasis can arise from extrinsic compression of the airway by enlarged granulomatous lymph nodes and/or from development of intrinsic obstruction as a result of erosion of a calcified lymph node through the airway wall (e.g., broncholithiasis). Especially in reactivated tuberculosis, parenchymal destruction from infection can result in areas of more diffuse bronchiectasis. Apart from cases associated with tuberculosis, an increased incidence of non-CF bronchiectasis with an unclear underlying mechanism has been reported as a significant problem in developing nations. It has been suggested that the high incidence of malnutrition in certain areas may predispose to immune dysfunction and development of bronchiectasis.
In areas where tuberculosis is prevalent, bronchiectasis more frequently occurs as a sequela of granulomatous infection. Focal bronchiectasis can arise from extrinsic compression of the airway by enlarged granulomatous lymph nodes and/or from development of intrinsic obstruction as a result of erosion of a calcified lymph node through the airway wall (e.g., broncholithiasis). Especially in reactivated tuberculosis, parenchymal destruction from infection can result in areas of more diffuse bronchiectasis. Apart from cases associated with tuberculosis, an increased incidence of non-CF bronchiectasis with an unclear underlying mechanism has been reported as a significant problem in developing nations. It has been suggested that the high incidence of malnutrition in certain areas may predispose to immune dysfunction and development of bronchiectasis.
PATHOGENESIS AND PATHOLOGY
The most widely cited mechanism of infectious bronchiectasis is the “vicious cycle hypothesis,” in which susceptibility to infection and poor mucociliary clearance result in microbial colonization of the bronchial tree. Some organisms, such as Pseudomonas aeruginosa, exhibit a particular propensity for colonizing damaged airways and evading host defense mechanisms. Impaired mucociliary clearance can result from inherited conditions such as CF or dyskinetic cilia syndrome, and it has been proposed that a single severe infection (e.g., pneumonia caused by Bordetella pertussis or Mycoplasma pneumoniae) can result in significant airway damage and poor secretion clearance. The presence of the microbes incites continued chronic inflammation, with consequent damage to the airway wall, continued impairment of secretion and microbial clearance, and ongoing propagation of the infectious/inflammatory cycle. Moreover, it has been proposed that mediators released directly from bacteria can interfere with mucociliary clearance.
Classic studies of the pathology of bronchiectasis from the 1950s demonstrated significant small-airway wall inflammation and larger-airway wall destruction as well as dilation, with loss of elastin, smooth muscle, and cartilage. It has been proposed that inflammatory cells in the small airways release proteases and other mediators, such as reactive oxygen species and proinflammatory cytokines, that damage the larger-airway walls. Furthermore, the ongoing inflammatory process in the smaller airways results in airflow obstruction. It is thought that antiproteases, such as α1 antitrypsin, play an important role in neutralizing the damaging effects of neutrophil elastase and in enhancing bacterial killing. Bronchiectasis and emphysema have been observed in patients with α1 antitrypsin deficiency.
Proposed mechanisms for noninfectious bronchiectasis include immune-mediated reactions that damage the bronchial wall (e.g., those associated with systemic autoimmune conditions such as Sjögren’s syndrome and rheumatoid arthritis). Traction bronchiectasis refers to dilated airways arising from parenchymal distortion as a result of lung fibrosis (e.g., postradiation fibrosis or idiopathic pulmonary fibrosis).
CLINICAL MANIFESTATIONS
The most common clinical presentation is a persistent productive cough with ongoing production of thick, tenacious sputum. Physical findings often include crackles and wheezing on lung auscultation, and some patients with bronchiectasis exhibit clubbing of the digits. Mild to moderate airflow obstruction is often detected on pulmonary function tests, overlapping with that seen at presentation with other conditions, such as chronic obstructive pulmonary disease (COPD). Acute exacerbations of bronchiectasis are usually characterized by changes in the nature of sputum production, with increased volume and purulence. However, typical signs and symptoms of lung infection, such as fever and new infiltrates, may not be present.
DIAGNOSIS
The diagnosis is usually based on presentation with a persistent chronic cough and sputum production accompanied by consistent radiographic features. Although chest radiographs lack sensitivity, the presence of “tram tracks” indicating dilated airways is consistent with bronchiectasis. Chest computed tomography (CT) is more specific for bronchiectasis and is the imaging modality of choice for confirming the diagnosis. CT findings include airway dilation (detected as parallel “tram tracks” or as the “signet-ring sign”—a cross-sectional area of the airway with a diameter at least 1.5 times that of the adjacent vessel), lack of bronchial tapering (including the presence of tubular structures within 1 cm from the pleural surface), bronchial wall thickening in dilated airways, inspissated secretions (e.g., the “tree-in-bud” pattern), or cysts emanating from the bronchial wall (especially pronounced in cystic bronchiectasis; Fig. 312-1).
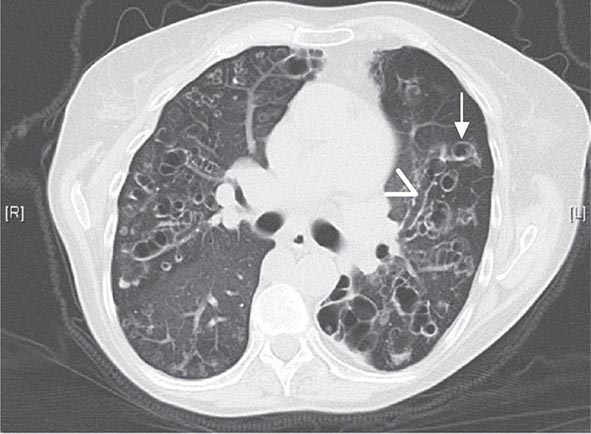
FIGURE 312-1 Representative chest computed tomography (CT) image of severe bronchiectasis. This patient’s CT demonstrates many severely dilated airways, seen both longitudinally (arrowhead) and in cross-section (arrow).
COMPLICATIONS
In more severe cases of infectious bronchiectasis, recurrent infections and repeated courses of antibiotics can lead to microbial resistance to antibiotics. In certain cases, combinations of antibiotics that have their own independent toxicity profiles may be necessary to treat resistant organisms.
Recurrent infections can result in injury to superficial mucosal vessels, with bleeding and, in severe cases, life-threatening hemoptysis. Management of massive hemoptysis usually requires intubation to stabilize the patient, identification of the source of bleeding, and protection of the nonbleeding lung. Control of bleeding often necessitates bronchial artery embolization and, in severe cases, surgery.
PROGNOSIS
Outcomes of bronchiectasis can vary widely with the underlying etiology and may also be influenced by the frequency of exacerbations and (in infectious cases) the specific pathogens involved. In one study, the decline of lung function in patients with non-CF bronchiectasis was similar to that in patients with COPD, with the forced expiratory volume in 1 s (FEV1) declining by 50–55 mL per year as opposed to 20–30 mL per year for healthy controls.
PREVENTION
Reversal of an underlying immunodeficient state (e.g., by administration of gamma globulin for immunoglobulin-deficient patients) and vaccination of patients with chronic respiratory conditions (e.g., influenza and pneumococcal vaccines) can decrease the risk of recurrent infections. Patients who smoke should be counseled about smoking cessation.
After resolution of an acute infection in patients with recurrences (e.g., ≥3 episodes per year), the use of suppressive antibiotics to minimize the microbial load and reduce the frequency of exacerbations has been proposed, although there is less consensus with regard to this approach in non-CF-associated bronchiectasis than in patients with CF-related bronchiectasis. Possible suppressive treatments include (1) administration of an oral antibiotic (e.g., ciprofloxacin) daily for 1–2 weeks per month; (2) use of a rotating schedule of oral antibiotics (to minimize the risk of development of drug resistance); (3) administration of a macrolide antibiotic (see below) daily or three times per week (with mechanisms of possible benefit related to non-antimicrobial properties, such as anti-inflammatory effects and reduction of gram-negative bacillary biofilms); (4) inhalation of aerosolized antibiotics (e.g., tobramycin inhalation solution) by select patients on a rotating schedule (e.g., 30 days on, 30 days off), with the goal of decreasing the microbial load without eliciting the side effects of systemic drug administration; and (5) intermittent administration of IV antibiotics (e.g., “clean-outs”) for patients with more severe bronchiectasis and/or resistant pathogens. In relation to macrolide therapy (point 3 above), a number of double-blind, placebo-controlled, randomized trials have recently been published in non-CF bronchiectasis and support a benefit of long-term macrolides (6–12 months of azithromycin or erythromycin) in decreasing rates of bronchiectasis exacerbation, mucus production, and decline in lung function. However, two of these studies also reported increased macrolide resistance in commensal pathogens, dampening enthusiasm for universal use of macrolides in this setting and raising the question of whether there might be select non-CF bronchiectasis patients with higher morbidity for whom benefits of long-term macrolides might outweigh the risks of emergence of antibiotic resistance. In particular, development of macrolide-resistant NTM is a significant concern, making treatment of that pathogen much more difficult. Therefore, it is advised to rule out NTM infection before chronic macrolide therapy is considered.
In addition, ongoing consistent attention to bronchial hygiene can promote secretion clearance and decrease the microbial load in the airways.
313 |
Cystic Fibrosis |
CLINICAL FEATURES
Cystic fibrosis (CF) is an autosomal recessive exocrinopathy affecting multiple epithelial tissues. The gene product responsible for CF (the cystic fibrosis transmembrane conductance regulator [CFTR]) serves as an anion channel in the apical (luminal) plasma membranes of epithelial cells and regulates volume and composition of exocrine secretion. An increasingly sophisticated understanding of CFTR molecular genetics and membrane protein biochemistry has facilitated CF drug discovery, with a number of new agents advancing through the clinical testing phase.
Respiratory Manifestations The major morbidity and mortality associated with CF is attributable to respiratory compromise, characterized by copious hyperviscous and adherent pulmonary secretions that obstruct small and medium-sized airways. CF airway secretions are exceedingly difficult to clear, and a complex bacterial flora that includes Staphylococcus aureus, Haemophilus influenzae, and Pseudomonas aeruginosa (among other pathogens) is routinely cultured from CF sputum. Robust pulmonary inflammation in the setting of inspissated mucus and chronic bacterial infection leads to collateral tissue injury and further aggravates respiratory decline. Organisms such as P. aeruginosa exhibit a stereotypic mode of pathogenesis; a sentinel and early colonization event often engenders lifelong pulmonary infection by the same genetic strain. Over the course of many years, P. aeruginosa evolves in CF lungs to adopt a mucoid phenotype (attributable to release of alginate exoproduct) that confers selective advantage for the pathogen and poor prognosis for the host. Infection with other bacterial organisms such as Burkholderia cepacia also indicates a less favorable pulmonary outlook. Strategies to eradicate organisms such as P. aeruginosa early in the pathogenesis cascade have been successful and are thought to improve prognosis significantly if sustained.
Pancreatic Findings The complete name of the disease, cystic fibrosis of the pancreas, refers to profound tissue destruction of the exocrine pancreas, with fibrotic scarring and/or fatty replacement, cyst proliferation, loss of acinar tissue, and ablation of normal pancreatic architecture. As in the lung, tenacious exocrine secretions (sometimes termed concretions) obstruct pancreatic ducts and impair production and flow of digestive enzymes to the duodenum. The sequelae of exocrine pancreatic insufficiency include chronic malabsorption, poor growth, fat-soluble vitamin insufficiency, high levels of serum immunoreactive trypsinogen (a diagnostic test used in newborn screening), and loss of pancreatic islet cell mass. CF-related diabetes mellitus is a manifestation in over 30% of adults with the disease and is likely multifactorial in nature (attributable to progressive destruction of the endocrine pancreas, insulin resistance due to stress hormones, and other factors).
Other Organ System Damage As in CF lung and pancreas, thick and tenacious secretions compromise numerous other exocrine tissues. Obstruction of intrahepatic bile ducts and parenchymal fibrosis are commonly observed in pathologic specimens, with multilobular cirrhosis in 4–15% of patients with CF and significant hepatic insufficiency as a resulting manifestation among adults. Contents of the intestinal lumen are often difficult to excrete, leading to meconium ileus (a presentation in approximately 10–20% of newborns with CF) or distal intestinal obstructive syndrome in older individuals. Men typically exhibit complete involution of the vas deferens and infertility (despite functioning spermatogenesis), and approximately 99% of males with CF are infertile. The etiology of this dramatic anatomic defect in the male genitourinary system is not understood but may represent a developmental abnormality secondary to secretory obstruction of the vas. Abnormalities of female reproductive tract secretions are likely contributors to an increased incidence of infertility among women with CF. Radiographic evidence of sinusitis occurs in most CF patients and is associated with pathogens similar to those recovered from lower airways, suggesting that the sinus may serve as a reservoir for bacterial seeding.
PATHOGENESIS
Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) CFTR is an integral membrane protein that functions as an epithelial anion channel. The ~1480-amino-acid molecule encodes a passive conduit for chloride and bicarbonate transport across plasma membranes of epithelial tissues, with direction of ion flow dependent on the electrochemical driving force. Gating of CFTR involves conformational cycling between an open and closed configuration and is augmented by hydrolysis of adenosine triphosphate (ATP). Anion flux mediated by CFTR does not involve active transport against a concentration gradient but utilizes the energy provided from ATP hydrolysis as a central feature of ion channel mechanochemistry and gating.
CFTR is situated in the apical plasma membranes of acinar and other epithelial cells where it regulates the amount and composition of secretion by exocrine glands. In numerous epithelia, chloride and bicarbonate release is followed passively by the flow of water, allowing for mobilization and clearance of exocrine products. Along respiratory mucosa, CFTR is necessary to provide sufficient depth of the periciliary fluid layer (PCL), allowing normal ciliary extension and mucociliary transport. CFTR-deficient airway cells exhibit depleted PCL, causing ciliary collapse and failure to clear overlying mucus (Video 313-1). In airway submucosal glands, CFTR is highly expressed in acini and may participate both in the formation of mucus and extrusion of glandular secretion onto the airway surface (Fig. 313-1). In other exocrine glands characterized by abrogated mucus transport (e.g., pancreatic acini and ducts, bile canaliculi, intestinal lumen), similar pathogenic mechanisms have been implicated. In these tissues, a driving force for apical chloride and/or bicarbonate secretion is believed to promote CFTR-mediated fluid and electrolyte release into the lumen, which confers proper rheology of mucins and other exocrine products. Failure of this mechanism disrupts normal hydration and transport of glandular secretion and is widely viewed as a proximate cause of ductular obstruction, with concomitant tissue injury.
FIGURE 313-1 Extrusion of mucus secretion onto the epithelial surface of airways in cystic fibrosis. A. Schematic of the surface epithelium and supporting glandular structure of the human airway. B. The submucosal glands of a patient with cystic fibrosis are filled with mucus, and mucopurulent debris overlies the airway surfaces, essentially burying the epithelium. C. A higher magnification view of a mucus plug tightly adhering to the airway surface, with arrows indicating the interface between infected and inflamed secretions and the underlying epithelium to which the secretions adhere. (Both B and C were stained with hematoxylin and eosin, with the colors modified to highlight structures.) Infected secretions obstruct airways and, over time, dramatically disrupt the normal architecture of the lung. D. CFTR is expressed in surface epithelium and serous cells at the base of submucosal glands in a porcine lung sample, as shown by the dark staining, signifying binding by CFTR antibodies to epithelial structures (aminoethylcarbazole detection of horseradish peroxidase with hematoxylin counterstain). (From SM Rowe, S Miller, EJ Sorscher: N Engl J Med 352:1992, 2005.)
Pulmonary Inflammation and Remodeling The CF airway is characterized by an aggressive, unrelenting, neutrophilic inflammatory response with release of proteases and oxidants leading to airway remodeling and bronchiectasis. Intense pulmonary inflammation is largely driven by chronic respiratory infection. Macrophages resident in CF lungs augment elaboration of proinflammatory cytokines, which contribute to innate and adaptive immune reactivity. CFTR-dependent abnormalities of airway surface fluid composition (e.g., pH) have been reported as contributors to impaired bacterial killing in CF lungs. The role of CFTR as a direct mediator of inflammatory responsiveness and/or pulmonary remodeling represents an important and topical area of investigation.
MOLECULAR GENETICS
DNA sequencing of CFTR from patients (and others) worldwide has revealed almost 2000 allelic variants; however, only about 10% of these have been well-characterized as disease-causing mutations. Distinguishing the single nucleotide transversions or other polymorphisms with causal relevance often presents a significant challenge. The CFTR2 resource (www.cftr2.org/) delineates gene variants with a clear etiologic role.
CFTR defects known to elicit disease are often categorized based on molecular mechanism. For example, the common F508del mutation (nomenclature denotes omission of a single phenylalanine residue [F] at CFTR position 508) leads to a folding abnormality recognized by cellular quality control pathways. CFTR encoding F508del retains partial ion channel function, but protein maturation is arrested in the endoplasmic reticulum, and CFTR fails to arrive at the plasma membrane. Instead, F508del CFTR is misrouted and undergoes endoplasmic reticulum–associated degradation via the proteasome. CFTR mutations that disrupt protein maturation are termed class II defects and are by far the most common genetic abnormalities. F508del alone accounts for ~70% of defective CFTR alleles in the United States, where approximately 90% of individuals with CF carry at least one F508del mutation.
Other gene defects include CFTR ion channels properly trafficked to the apical cell surface but unable to open and/or gate. Such channel proteins include G551D (a glycine to aspartic acid replacement at CFTR position 551), which leads to an inability to transport Cl– or HCO3– in the presence of ATP (a class III abnormality). Individuals with at least one G551D allele represent 4–5% of CF patients in North America. CFTR nonsense alleles such as G542X, R553X, and W1282X (premature termination codon replaces glycine, arginine, or tryptophan at positions 542, 553, or 1282, respectively) are among the common class I defects, in addition to large deletions or other major disruptions of the gene. The W1282X mutation, for example, is prevalent among individuals of Ashkenazi descent and is a predominant CF genotype in Israel. Additional categories of CFTR mutation include defects in the ion channel pore (class IV), RNA splicing (class V), and increased plasma membrane turnover (class VI) (Fig. 313-2).
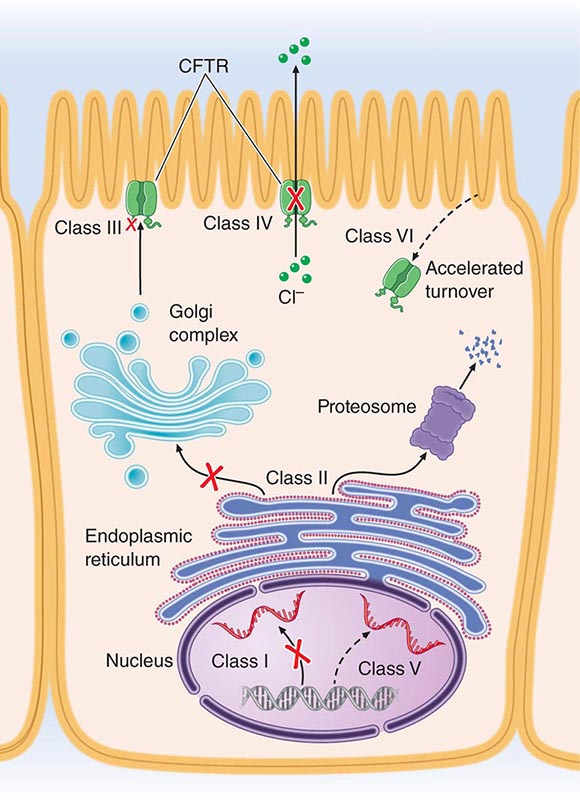
FIGURE 313-2 Categories of CFTR mutations. Classes of defects in the CFTR gene include the absence of synthesis (class I); defective protein maturation and premature degradation (class II); disordered gating/regulation, such as diminished adenosine triphosphate (ATP) binding and hydrolysis (class III); defective conductance through the ion channel pore (class IV); a reduced number of CFTR transcripts due to a promoter or splicing abnormality (class V); and accelerated turnover from the cell surface (class VI). (From SM Rowe, S Miller, EJ Sorscher: N Engl J Med 352:1992, 2005.)
DIAGNOSIS
The diagnosis of CF is based in part on clinical symptoms, family history, or positive newborn screening. CFTR mutation analysis together with sweat electrolyte measurements represent cardinal diagnostic tests. DNA-based evaluation typically surveys numerous disease-associated mutations; panels that identify 20–80 gene defects are available through commercial sources. For difficult cases, complete CFTR exonic sequencing together with analysis of splice junctions and key regulatory elements can be obtained. Sweat electrolytes following pilocarpine iontophoresis comprise an invaluable diagnostic measurement, with levels of chloride markedly elevated in CF compared to non-CF individuals. The sweat test result is highly specific and served as the mainstay of diagnosis for many decades prior to availability of CFTR genotyping. Notably, hyperviscosity of eccrine sweat is not a clinical feature of the disease. Sweat ducts function to reabsorb chloride from a primary sweat secretion produced by the glandular coil. Malfunction of CFTR leads to diminished chloride uptake from the ductular lumen, and sweat emerges on the skin with markedly elevated levels of chloride. For the unusual situation in which both CFTR genotype and sweat electrolytes are inconclusive, in vivo measurement of ion transport across the nasal airways can serve as a specific test for CF and is used by a number of referral centers. For example, elevated (sodium-dependent) transepithelial charge separation across airway epithelial tissue and failure of isoproterenol-dependent chloride secretion (via CFTR) represent bioelectric findings highly specific for the disease. Measurements of CFTR activity in excised rectal mucosal biopsies can also be obtained.
COMPLEXITY OF A CF PHENOTYPE
CF classically presents in childhood with chronic productive cough, malabsorption including steatorrhea, and failure to thrive. The disease is most common among whites (~1 in 3300 live births) and much less frequent among African-American (~1 in 15,000) or Asian populations (~1 in 33,000). Several “severe” defects that impair CFTR activity (including F508del, G551D, and truncation alleles) are predictive of pancreatic insufficiency, which is clinically evident in 80–90% of individuals with CF. These few specific genotype-phenotype correlations notwithstanding, genotype is, in general, a poor predictor of overall respiratory prognosis.
A spectrum of CFTR-related diseases with features resembling classic CF has been well described. In addition to multiorgan involvement, forme frustes, such as isolated congenital bilateral absence of the vas deferens or pancreatitis (without other organ system findings), are strongly associated with CFTR mutations in at least one allele. Although CF is a classic monogenic disease, the importance of non-CFTR gene modifiers and proteins that regulate ion flux, inflammatory pathways, and airway remodeling has been increasingly appreciated as influencing clinical course. For example, the magnitude of transepithelial sodium reabsorption in CF airways, which helps control periciliary fluid depth and composition, is strongly influenced by CFTR and represents a molecular target for disease intervention.
THERAPEUTICS DIRECTED TOWARD CF SEQUELAE
Standard care for outpatients with CF is intensive, with regimens that include exogenous pancreatic enzymes taken with meals, nutritional supplementation, anti-inflammatory medication, bronchodilators, and chronic or periodic administration of oral or aerosolized antibiotics (e.g., as maintenance therapy for patients with P. aeruginosa). Recombinant DNAse aerosols (degraded DNA strands that contribute to mucus viscosity) and nebulized hypertonic saline (serves to augment PCL depth, activate mucociliary clearance, and mobilize inspissated airway secretions) are administered routinely. Chest physiotherapy several times each day is a standard means to promote clearance of airway mucus. Among older individuals with CF, malabsorption, chronic inflammation, and endocrine abnormalities can lead to poor bone mineralization, requiring treatment with vitamin D, calcium, and other measures. The time, complexity, and expense of home care are considerable and take a significant toll on patients and their families.
Severe respiratory exacerbation is commonly managed by hospital admission for frequent chest physiotherapy and parenteral antibiotics directed against serious (and often multiply resistant) bacterial pathogens. Aggressive intervention in this setting can restore a large component of lung function, but ongoing and cumulative loss of pulmonary reserve reflects the natural history of the disease. Poor prognostic indicators such as sputum culture containing B. cepacia, mucoid P. aeruginosa, or atypical mycobacteria are rigorously monitored in the CF patient population. An increasing incidence of methicillin-resistant S. aureus has also been observed, although the clinical significance of this finding has not been fully elucidated. Typical inpatient antibiotic coverage includes combination drug therapy with an aminoglycoside and β-lactam for up to 14 days. Maximal improvement in lung function is often achieved by 8–10 days in this setting. Many families elect parenteral antibiotic treatment at home, and additional studies are needed to evaluate specific drug combinations, duration of therapy, and home versus inpatient management. Other CF respiratory sequelae that may require hospitalization include hemoptysis and pneumothorax. Hypersensitivity to Aspergillus (allergic bronchopulmonary aspergillosis) occurs in approximately 5% of individuals with the disease and should be suspected in the absence of a response to conventional treatment.
Lung transplantation remains a viable therapeutic option in the setting of end-stage CF pulmonary failure, with 5-year postoperative survival rates on the order of 50–60%. Determining the optimal timing for surgery presents a substantial challenge, particularly because overall prognosis for individuals with severe lung disease is sometimes difficult to predict, and mortality associated with transplantation is significant (1-year survival rates of approximately 80%). Forced expiratory volume in 1 s (FEV1) measurements less than 30% predicted, together with an assortment of other clinical features, are often used as thresholds for entry onto transplantation lists, although waiting periods for healthy donor lungs can be quite protracted. Based on clinical outcome and limited access to healthy donor lungs, many CF patients and their families do not pursue this option.
CFTR MODULATION
Potentiation of Mutant CFTR Gating A massive effort directed toward high-throughput drug analysis of large compound libraries (containing millions of individual agents) has identified novel and promising approaches to CF therapy. The approved compound ivacaftor, for example, robustly potentiates CFTR channel opening and stimulates ion transport. Ivacaftor overcomes the G551D CFTR gating defect, and individuals carrying this mutation exhibit dramatic improvement in lung function, weight gain, and other clinical parameters after only a few weeks of oral therapy. Remarkably, sweat chloride values are significantly improved with this treatment in patients with G551D CFTR. No clinical intervention of any sort has previously been shown to normalize the CF sweat chloride abnormality. Long-term studies of the drug in patients with G551D CFTR are ongoing. Ivacaftor has been viewed as the harbinger of a new era for CF therapeutics directed at treating the most fundamental causes of the disease.
Correction of the F508del Processing Abnormality Advancement of new drugs that address specific CFTR defects in protein folding and maturation has been bolstered by clinical studies of F508del rescue in combination with ivacaftor. So-called “corrector” molecules (as distinct from CFTR gating “potentiators” such as ivacaftor) discovered through compound library screening are suitable for promoting cell surface localization of the F508del protein. Significant improvement in pulmonary function of F508del homozygous individuals has been achieved with potentiator/corrector combination therapy in early clinical trials, and several candidate molecules are under evaluation.
Personalized Molecular Therapies The advent of modulators with robust clinical impact has engendered new optimism regarding care of patients with CF. It is clear that future interventions will be tailored to specific genotypic abnormalities. Drug screening campaigns and other research programs have identified agents capable of suppressing CFTR nonsense alleles, augmenting potentiator activity, and promoting F508del correction. Efforts to apply these compounds in a fashion that will benefit CF subjects carrying a single copy of F508del (i.e., with a distinct or unusual CFTR mutation on the second allele) comprise an essential priority for the future. Progress in CF drug discovery is emblematic of what might be accomplished in other refractory genetic diseases using an approach grounded in molecular mechanism and unbiased compound library screening.
CF QUALITY IMPROVEMENT, INCLUDING ASPECTS OF GLOBAL HEALTH
As a direct result of advances in basic research, new therapies have transformed CF from a disease typically leading to death in early childhood to a condition with frequent survival well into the fourth decade of life. It has also become increasingly clear that specified approaches to patient management can have an impact on overall prognosis. For example, standardization of clinical intervention throughout the United States has led to remarkable benefit among the CF population. Well-defined measures for outpatient care are now established, including thresholds for hospital admission, antibiotic regimens, nutritional guidelines, periodicity of diagnostic tests, and other clinical parameters. These therapeutic recommendations have become standardized throughout approximately 110 specialized CF care centers and 55 affiliated programs. The initiative has improved endpoints such as weight gain, body mass index, and pulmonary function. Information regarding standardized protocols for CF therapy can be accessed at (www.cff.org/treatments/cfcareguidelines/) or through a number of excellent reviews.
Newborn screening for CF is now universal throughout the United States, most of the Canadian provinces, Australia, New Zealand, and much of Europe, and will facilitate early CF intervention. Based on data indicating that early nutritional and other therapies can be beneficial, newborn diagnosis is expected to significantly promote health in the CF population. Export of quality control measures and novel therapeutics worldwide has become an increasing imperative. For example, median survival of individuals with CF is less than 20 years in much of Central and South America (compared to ~40 years in the United States and Canada), and efforts to apply state-of-the-art management to underdiagnosed and underserved CF patient populations are expected to improve outcome and mitigate CF health disparities in the future.
VIDEO 313-1 Initial video sequences describe establishment of the normal periciliary fluid layer bathing the surface airway epithelium, with spheres representing chloride and bicarbonate ions secreted through CFTR and across the apical (mucosal) respiratory surface. Later video sequences depict failure of CFTR anion transport and resulting depletion of the periciliary layer, “plastering” of cilia against the mucosal surface, and accumulation of mucus in the airway with resulting bacterial infection. (Video courtesy of the Cystic Fibrosis Foundation.)
314 |
Chronic Obstructive Pulmonary Disease |
Chronic obstructive pulmonary disease (COPD) is defined as a disease state characterized by airflow limitation that is not fully reversible (http://www.goldcopd.com/). COPD includes emphysema, an anatomically defined condition characterized by destruction and enlargement of the lung alveoli; chronic bronchitis, a clinically defined condition with chronic cough and phlegm; and small airways disease, a condition in which small bronchioles are narrowed. COPD is present only if chronic airflow obstruction occurs; chronic bronchitis without chronic airflow obstruction is not included within COPD.
COPD is the third leading cause of death and affects >10 million persons in the United States. COPD is also a disease of increasing public health importance around the world. Estimates suggest that COPD will rise from the sixth to the third most common cause of death worldwide by 2020.
PATHOGENESIS
Airflow limitation, the major physiologic change in COPD, can result from both small airway obstruction and emphysema. As described below, small airways may become narrowed by cells (hyperplasia and accumulation), mucus, and fibrosis. Of note, activation of transforming growth factor β (TGF-β) contributes to airway fibrosis, while lack of TGF-β may contribute to parenchymal inflammation and emphysema. Largely due to greater similarity of animal air spaces than airways to humans, we know more about mechanisms involved in emphysema than small airway obstruction.
The dominant paradigm of the pathogenesis of emphysema comprises four interrelated events (Fig. 314-1): (1) Chronic exposure to cigarette smoke leads to inflammatory and immune cell recruitment within the terminal air spaces of the lung. (2) These inflammatory cells release elastolytic and other proteinases that damage the extracellular matrix of the lung. (3) Structural cell death (endothelial and epithelial cells) occurs directly through oxidant-induced cigarette smoke damage and senescence as well as indirectly via proteolytic loss of matrix attachment. (4) Ineffective repair of elastin and other extracellular matrix components result in air space enlargement that defines pulmonary emphysema.
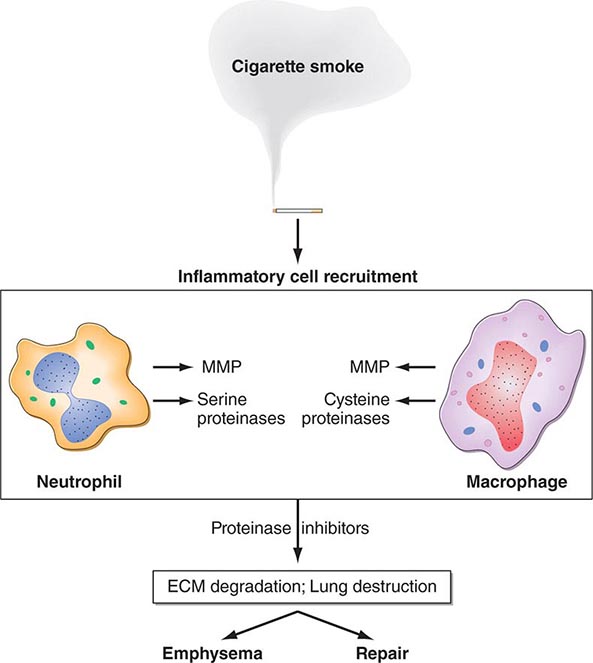
FIGURE 314-1 Pathogenesis of emphysema. Upon long-term exposure to cigarette smoke, inflammatory cells are recruited to the lung; they release proteinases in excess of inhibitors, and if repair is abnormal, this leads to air space destruction and enlargement or emphysema. ECM, extracellular matrix; MMP, matrix metalloproteinase.
THE ELASTASE:ANTIELASTASE HYPOTHESIS
Elastin, the principal component of elastic fibers, is a highly stable component of the extracellular matrix that is critical to the integrity of the lung. The elastase:antielastase hypothesis proposed in the mid-1960s states that the balance of elastin-degrading enzymes and their inhibitors determines the susceptibility of the lung to destruction resulting in air space enlargement. This hypothesis was based on the clinical observation that patients with genetic deficiency in α1 antitrypsin (α1AT), the inhibitor of the serine proteinase neutrophil elastase, were at increased risk of emphysema, and that instillation of elastases, including neutrophil elastase, into experimental animals results in emphysema. The elastase:antielastase hypothesis remains a prevailing mechanism for the development of emphysema. However, a complex network of immune and inflammatory cells and additional proteinases that contribute to emphysema have subsequently been identified.
INFLAMMATION AND EXTRACELLULAR MATRIX PROTEOLYSIS
Upon exposure to oxidants from cigarette smoke, macrophages and epithelial cells become activated, producing proteinases and chemokines that attract other inflammatory and immune cells. One mechanism of macrophage activation occurs via oxidant-induced inactivation of histone deacetylase-2, shifting the balance toward acetylated or loose chromatin, exposing nuclear factor-κB sites, and resulting in transcription of matrix metalloproteinases, proinflammatory cytokines such as interleukin 8 (IL-8), and tumor necrosis factor α (TNF-α); this leads to neutrophil recruitment. CD8+ T cells are also recruited in response to cigarette smoke and release interferon-inducible protein-10 (IP-10, CXCL-7), which in turn leads to macrophage production of macrophage elastase (matrix metalloproteinase-12 [MMP-12]). Matrix metalloproteinases and serine proteinases, most notably neutrophil elastase, work together by degrading the inhibitor of the other, leading to lung destruction. Proteolytic cleavage products of elastin also serve as a macrophage chemokine, fueling this destructive positive feedback loop.
Autoimmune mechanisms may promote the progression of disease. Increased B cells and lymphoid follicles are present in patients, particularly those with advanced disease. Antibodies have been found against elastin fragments as well; IgG autoantibodies with avidity for pulmonary epithelium and the potential to mediate cytotoxicity have been detected.
Concomitant cigarette smoke–induced loss of cilia in the airway epithelium and impaired macrophage phagocytosis predispose to bacterial infection with neutrophilia. In end-stage lung disease, long after smoking cessation, there remains an exuberant inflammatory response, suggesting that mechanisms of cigarette smoke–induced inflammation that initiate the disease differ from mechanisms that sustain inflammation after smoking cessation.
Cell Death Cigarette smoke oxidant-mediated structural cell death occurs via a variety of mechanisms including rt801 inhibition of mammalian target of rapamycin (mTOR), leading to cell death as well as inflammation and proteolysis. Involvement of mTOR and other senescence markers has led to the recent concept that emphysema resembles premature aging of the lung. Uptake of apoptotic cells by macrophages results in production of growth factors and dampens inflammation, promoting lung repair. Cigarette smoke impairs macrophage uptake of apoptotic cells, limiting repair.
Ineffective Repair The ability of the adult lung to repair damaged alveoli appears limited. It is unlikely that the process of septation that is responsible for alveologenesis during lung development can be reinitiated. The capacity of stem cells to repopulate the lung is under active investigation. It appears difficult for an adult human to completely restore an appropriate extracellular matrix, particularly functional elastic fibers.
PATHOLOGY
Cigarette smoke exposure may affect the large airways, small airways (≤2 mm diameter), and alveoli. Changes in large airways cause cough and sputum, while changes in small airways and alveoli are responsible for physiologic alterations. Emphysema and small airway pathology are both present in most persons with COPD; however, they do not appear to be mechanistically related to each other, and their relative contributions to obstruction vary from one person to another.
LARGE AIRWAY
Cigarette smoking often results in mucus gland enlargement and goblet cell hyperplasia, leading to cough and mucus production that define chronic bronchitis, but these abnormalities are not related to airflow limitation. Goblet cells not only increase in number but in extent through the bronchial tree. Bronchi also undergo squamous metaplasia, predisposing to carcinogenesis and disrupting mucociliary clearance. Although not as prominent as in asthma, patients may have smooth-muscle hypertrophy and bronchial hyperreactivity leading to airflow limitation. Neutrophil influx has been associated with purulent sputum of upper respiratory tract infections. Independent of its proteolytic activity, neutrophil elastase is among the most potent secretagogues identified.
SMALL AIRWAYS
The major site of increased resistance in most individuals with COPD is in airways ≤2 mm diameter. Characteristic cellular changes include goblet cell metaplasia, with these mucus-secreting cells replacing surfactant-secreting Clara cells. Smooth-muscle hypertrophy may also be present. These abnormalities may cause luminal narrowing by fibrosis, excess mucus, edema, and cellular infiltration. Reduced surfactant may increase surface tension at the air-tissue interface, predisposing to airway narrowing or collapse. Respiratory bronchiolitis with mononuclear inflammatory cells collecting in distal airway tissues may cause proteolytic destruction of elastic fibers in the respiratory bronchioles and alveolar ducts where the fibers are concentrated as rings around alveolar entrances. Narrowing and drop-out of small airways precede the onset of emphysematous destruction.
LUNG PARENCHYMA
Emphysema is characterized by destruction of gas-exchanging air spaces, i.e., the respiratory bronchioles, alveolar ducts, and alveoli. Their walls become perforated and later obliterated with coalescence of small distinct air spaces into abnormal and much larger air spaces. Macrophages accumulate in respiratory bronchioles of essentially all young smokers. Bronchoalveolar lavage fluid from such individuals contains roughly five times as many macrophages as lavage from nonsmokers. In smokers’ lavage fluid, macrophages comprise >95% of the total cell count, and neutrophils, nearly absent in nonsmokers’ lavage, account for 1–2% of the cells. T lymphocytes, particularly CD8+ cells, are also increased in the alveolar space of smokers.
Emphysema is classified into distinct pathologic types, the most important being centriacinar and panacinar. Centriacinar emphysema, the type most frequently associated with cigarette smoking, is characterized by enlarged air spaces found (initially) in association with respiratory bronchioles. Centriacinar emphysema is usually most prominent in the upper lobes and superior segments of lower lobes and is often quite focal. Panacinar emphysema refers to abnormally large air spaces evenly distributed within and across acinar units. Panacinar emphysema is usually observed in patients with α1AT deficiency, which has a predilection for the lower lobes.
PATHOPHYSIOLOGY
Persistent reduction in forced expiratory flow rates is the most typical finding in COPD. Increases in the residual volume and the residual volume/total lung capacity ratio, nonuniform distribution of ventilation, and ventilation-perfusion mismatching also occur.
AIRFLOW OBSTRUCTION
Airflow limitation, also known as airflow obstruction, is typically determined by spirometry, which involves forced expiratory maneuvers after the subject has inhaled to total lung capacity. Key parameters obtained from spirometry include the volume of air exhaled within the first second of the forced expiratory maneuver (FEV1) and the total volume of air exhaled during the entire spirometric maneuver (forced vital capacity [FVC]). Patients with airflow obstruction related to COPD have a chronically reduced ratio of FEV1/FVC. In contrast to asthma, the reduced FEV1 in COPD seldom shows large responses to inhaled bronchodilators, although improvements up to 15% are common. Asthma patients can also develop chronic (not fully reversible) airflow obstruction.
Airflow during forced exhalation is the result of the balance between the elastic recoil of the lungs promoting flow and the resistance of the airways limiting flow. In normal lungs, as well as in lungs affected by COPD, maximal expiratory flow diminishes as the lungs empty because the lung parenchyma provides progressively less elastic recoil and because the cross-sectional area of the airways falls, raising the resistance to airflow. The decrease in flow coincident with decreased lung volume is readily apparent on the expiratory limb of a flow-volume curve. In the early stages of COPD, the abnormality in airflow is only evident at lung volumes at or below the functional residual capacity (closer to residual volume), appearing as a scooped-out lower part of the descending limb of the flow-volume curve. In more advanced disease, the entire curve has decreased expiratory flow compared to normal.
HYPERINFLATION
Lung volumes are also routinely assessed in pulmonary function testing. In COPD there is often “air trapping” (increased residual volume and increased ratio of residual volume to total lung capacity) and progressive hyperinflation (increased total lung capacity) late in the disease. Hyperinflation of the thorax during tidal breathing preserves maximum expiratory airflow, because as lung volume increases, elastic recoil pressure increases, and airways enlarge so that airway resistance decreases.
Despite compensating for airway obstruction, hyperinflation can push the diaphragm into a flattened position with a number of adverse effects. First, by decreasing the zone of apposition between the diaphragm and the abdominal wall, positive abdominal pressure during inspiration is not applied as effectively to the chest wall, hindering rib cage movement and impairing inspiration. Second, because the muscle fibers of the flattened diaphragm are shorter than those of a more normally curved diaphragm, they are less capable of generating inspiratory pressures than normal. Third, the flattened diaphragm (with increased radius of curvature, r) must generate greater tension (t) to develop the transpulmonary pressure (p) required to produce tidal breathing. This follows from Laplace’s law, p = 2t/r. Also, because the thoracic cage is distended beyond its normal resting volume, during tidal breathing the inspiratory muscles must do work to overcome the resistance of the thoracic cage to further inflation instead of gaining the normal assistance from the chest wall recoiling outward toward its resting volume.
GAS EXCHANGE
Although there is considerable variability in the relationships between the FEV1 and other physiologic abnormalities in COPD, certain generalizations may be made. The partial pressure of oxygen in arterial blood PaO2 usually remains near normal until the FEV1 is decreased to ~50% of predicted, and even much lower FEV1 values can be associated with a normal PaO2, at least at rest. An elevation of arterial level of carbon dioxide (PaCO2) is not expected until the FEV1 is <25% of predicted and even then may not occur. Pulmonary hypertension severe enough to cause cor pulmonale and right ventricular failure due to COPD typically occurs in individuals who have marked decreases in FEV1 (<25% of predicted) and chronic hypoxemia (PaO2 <55 mmHg); however, recent evidence suggests that some patients will develop significant pulmonary hypertension independent of COPD severity (Chap. 304).
Nonuniform ventilation and ventilation-perfusion mismatching are characteristic of COPD, reflecting the heterogeneous nature of the disease process within the airways and lung parenchyma. Physiologic studies are consistent with multiple parenchymal compartments having different rates of ventilation due to regional differences in compliance and airway resistance. Ventilation-perfusion mismatching accounts for essentially all of the reduction in PaO2 that occurs in COPD; shunting is minimal. This finding explains the effectiveness of modest elevations of inspired oxygen in treating hypoxemia due to COPD and therefore the need to consider problems other than COPD when hypoxemia is difficult to correct with modest levels of supplemental oxygen.
RISK FACTORS
CIGARETTE SMOKING
By 1964, the Advisory Committee to the Surgeon General of the United States had concluded that cigarette smoking was a major risk factor for mortality from chronic bronchitis and emphysema. Subsequent longitudinal studies have shown accelerated decline in FEV1 in a dose-response relationship to the intensity of cigarette smoking, which is typically expressed as pack-years (average number of packs of cigarettes smoked per day multiplied by the total number of years of smoking). This dose-response relationship between reduced pulmonary function and cigarette smoking intensity accounts for the higher prevalence rates of COPD with increasing age. The historically higher rate of smoking among males is the likely explanation for the higher prevalence of COPD among males; however, the prevalence of COPD among females is increasing as the gender gap in smoking rates has diminished in the past 50 years.
Although the causal relationship between cigarette smoking and the development of COPD has been absolutely proved, there is considerable variability in the response to smoking. Although pack-years of cigarette smoking is the most highly significant predictor of FEV1 (Fig. 314-2), only 15% of the variability in FEV1 is explained by pack-years. This finding suggests that additional environmental and/or genetic factors contribute to the impact of smoking on the development of airflow obstruction.
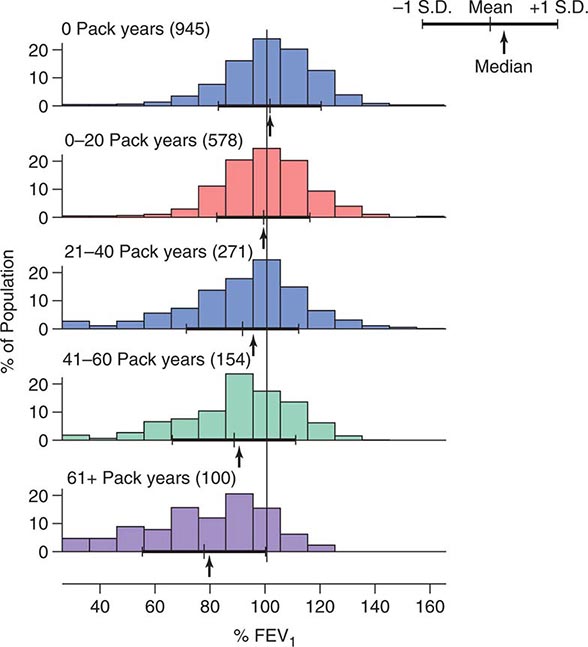
FIGURE 314-2 Distributions of forced expiratory volume in 1 s (FEV1) values in a general population sample, stratified by pack-years of smoking. Means, medians, and ±1 standard deviation of percent predicted FEV1 are shown for each smoking group. Although a dose-response relationship between smoking intensity and FEV1 was found, marked variability in pulmonary function was observed among subjects with similar smoking histories. (From B Burrows et al: Am Rev Respir Dis 115:95, 1977; with permission.)
Although cigar and pipe smoking may also be associated with the development of COPD, the evidence supporting such associations is less compelling, likely related to the lower dose of inhaled tobacco by-products during cigar and pipe smoking.
AIRWAY RESPONSIVENESS AND COPD
A tendency for increased bronchoconstriction in response to a variety of exogenous stimuli, including methacholine and histamine, is one of the defining features of asthma (Chap. 309). However, many patients with COPD also share this feature of airway hyperresponsiveness. The considerable overlap between persons with asthma and those with COPD in airway responsiveness, airflow obstruction, and pulmonary symptoms led to the formulation of the Dutch hypothesis. This suggests that asthma, chronic bronchitis, and emphysema are variations of the same basic disease, which is modulated by environmental and genetic factors to produce these pathologically distinct entities. The alternative British hypothesis contends that asthma and COPD are fundamentally different diseases: Asthma is viewed as largely an allergic phenomenon, whereas COPD results from smoking-related inflammation and damage. Determination of the validity of the Dutch hypothesis versus the British hypothesis awaits identification of all of the genetic predisposing factors for asthma and/or COPD, as well as the interactions between these postulated genetic factors and environmental risk factors.
Longitudinal studies that compared airway responsiveness at the beginning of the study to subsequent decline in pulmonary function have demonstrated that increased airway responsiveness is clearly a significant predictor of subsequent decline in pulmonary function. Thus, airway hyperresponsiveness is a risk factor for COPD.
RESPIRATORY INFECTIONS
The impact of adult respiratory infections on decline in pulmonary function is controversial, but significant long-term reductions in pulmonary function are not typically seen following an episode of bronchitis or pneumonia. The impact of the effects of childhood respiratory illnesses on the subsequent development of COPD has been difficult to assess due to a lack of adequate longitudinal data. Thus, although respiratory infections are important causes of exacerbations of COPD, the association of both adult and childhood respiratory infections with the development and progression of COPD remains to be proven.
OCCUPATIONAL EXPOSURES
Increased respiratory symptoms and airflow obstruction have been suggested to result from exposure to dust and fumes at work. Several specific occupational exposures, including coal mining, gold mining, and cotton textile dust, have been suggested as risk factors for chronic airflow obstruction. Although nonsmokers in these occupations can develop some reductions in FEV1, the importance of dust exposure as a risk factor for COPD, independent of cigarette smoking, is not certain for most of these exposures. However, among coal miners, coal mine dust exposure was a significant risk factor for emphysema in both smokers and nonsmokers. In most cases, the magnitude of these occupational exposures on COPD risk is likely substantially less important than the effect of cigarette smoking.
AMBIENT AIR POLLUTION
Some investigators have reported increased respiratory symptoms in those living in urban compared to rural areas, which may relate to increased pollution in the urban settings. However, the relationship of air pollution to chronic airflow obstruction remains unproved. Prolonged exposure to smoke produced by biomass combustion—a common mode of cooking in some countries—also appears to be a significant risk factor for COPD among women in those countries. However, in most populations, ambient air pollution is a much less important risk factor for COPD than cigarette smoking.
PASSIVE, OR SECOND-HAND, SMOKING EXPOSURE
Exposure of children to maternal smoking results in significantly reduced lung growth. In utero, tobacco smoke exposure also contributes to significant reductions in postnatal pulmonary function. Although passive smoke exposure has been associated with reductions in pulmonary function, the importance of this risk factor in the development of the severe pulmonary function reductions in COPD remains uncertain.
GENETIC CONSIDERATIONS
![]() Although cigarette smoking is the major environmental risk factor for the development of COPD, the development of airflow obstruction in smokers is highly variable. Severe α1AT deficiency is a proven genetic risk factor for COPD; there is increasing evidence that other genetic determinants also exist.
Although cigarette smoking is the major environmental risk factor for the development of COPD, the development of airflow obstruction in smokers is highly variable. Severe α1AT deficiency is a proven genetic risk factor for COPD; there is increasing evidence that other genetic determinants also exist.
α1 Antitrypsin Deficiency Many variants of the protease inhibitor (PI or SERPINA1) locus that encodes α1AT have been described. The common M allele is associated with normal α1AT levels. The S allele, associated with slightly reduced α1AT levels, and the Z allele, associated with markedly reduced α1AT levels, also occur with frequencies of >1% in most white populations. Rare individuals inherit null alleles, which lead to the absence of any α1AT production through a heterogeneous collection of mutations. Individuals with two Z alleles or one Z and one null allele are referred to as PiZ, which is the most common form of severe α1AT deficiency.
Although only approximately 1% of COPD patients are found to have severe α1AT deficiency as a contributing cause of COPD, these patients demonstrate that genetic factors can have a profound influence on the susceptibility for developing COPD. PiZ individuals often develop early-onset COPD, but the ascertainment bias in the published series of PiZ individuals—which have usually included many PiZ subjects who were tested for α1AT deficiency because they had COPD—means that the fraction of PiZ individuals who will develop COPD and the age-of-onset distribution for the development of COPD in PiZ subjects remain unknown. Approximately 1 in 3000 individuals in the United States inherits severe α1AT deficiency, but only a small minority of these individuals has been identified. The clinical laboratory test used most frequently to screen for α1AT deficiency is measurement of the immunologic level of α1AT in serum (see “Laboratory Findings”).
A significant percentage of the variability in pulmonary function among PiZ individuals is explained by cigarette smoking; cigarette smokers with severe α1AT deficiency are more likely to develop COPD at early ages. However, the development of COPD in PiZ subjects, even among current or ex-smokers, is not absolute. Among PiZ nonsmokers, impressive variability has been noted in the development of airflow obstruction. Asthma and male gender also appear to increase the risk of COPD in PiZ subjects. Other genetic and/or environmental factors likely contribute to this variability.
Specific treatment in the form of α1AT augmentation therapy is available for severe α1AT deficiency as a weekly IV infusion (see “Treatment,” below).
The risk of lung disease in heterozygous PiMZ individuals, who have intermediate serum levels of α1AT (~60% of PiMM levels), is controversial. Several recent large studies have suggested that PiMZ subjects are at slightly increased risk for the development of airflow obstruction, but it remains unclear if all PiMZ subjects are at slightly increased risk for COPD or if a subset of PiMZ subjects are at substantially increased risk for COPD due to other genetic or environmental factors.
Other Genetic Risk Factors Studies of pulmonary function measurements performed in general population samples have suggested that genetic factors other than PI type influence variation in pulmonary function. Familial aggregation of airflow obstruction within families of COPD patients has also been demonstrated.
Association studies have compared the distribution of variants in candidate genes hypothesized to be involved in the development of COPD in COPD patients and control subjects. However, the results have been quite inconsistent, often due to underpowered studies. However, a well-powered association study comprising 8300 patients and 7 separate cohorts found that a minor allele single nucleotide polymorphism (SNP) of MMP12 (rs2276109) associated with decreased MMP12 expression has a positive effect on lung function in children with asthma and in adult smokers. Recent genome-wide association studies have identified several COPD susceptibility loci, including a region near the hedgehog interacting protein (HHIP) gene on chromosome 4, a cluster of genes on chromosome 15 (including components of the nicotinic acetylcholine receptor), and a region within a gene of unknown function (FAM13A). A regulatory SNP upstream from the HHIP gene has been identified as one potential functional variant; the specific genetic determinants in the other genomic regions have yet to be definitively identified.
NATURAL HISTORY
The effects of cigarette smoking on pulmonary function appear to depend on the intensity of smoking exposure, the timing of smoking exposure during growth, and the baseline lung function of the individual; other environmental factors may have similar effects. Most individuals follow a steady trajectory of increasing pulmonary function with growth during childhood and adolescence, followed by a gradual decline with aging. Individuals appear to track in their quantile of pulmonary function based on environmental and genetic factors that put them on different tracks. The risk of eventual mortality from COPD is closely associated with reduced levels of FEV1. A graphic depiction of the natural history of COPD is shown as a function of the influences on tracking curves of FEV1 in Fig. 314-3. Death or disability from COPD can result from a normal rate of decline after a reduced growth phase (curve C), an early initiation of pulmonary function decline after normal growth (curve B), or an accelerated decline after normal growth (curve D). The rate of decline in pulmonary function can be modified by changing environmental exposures (i.e., quitting smoking), with smoking cessation at an earlier age providing a more beneficial effect than smoking cessation after marked reductions in pulmonary function have already developed. Genetic factors likely contribute to the level of pulmonary function achieved during growth and to the rate of decline in response to smoking and potentially to other environmental factors as well.
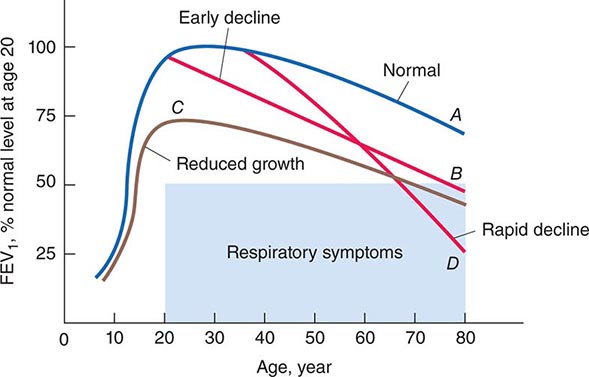
FIGURE 314-3 Hypothetical tracking curves of forced expiratory volume in 1 s (FEV1) for individuals throughout their life spans. The normal pattern of growth and decline with age is shown by curve A. Significantly reduced FEV1 (<65% of predicted value at age 20) can develop from a normal rate of decline after a reduced pulmonary function growth phase (curve C), early initiation of pulmonary function decline after normal growth (curve B), or accelerated decline after normal growth (curve D). (From B Rijcken: Doctoral dissertation, p 133, University of Groningen, 1991; with permission.)
CLINICAL PRESENTATION
HISTORY
The three most common symptoms in COPD are cough, sputum production, and exertional dyspnea. Many patients have such symptoms for months or years before seeking medical attention. Although the development of airflow obstruction is a gradual process, many patients date the onset of their disease to an acute illness or exacerbation. A careful history, however, usually reveals the presence of symptoms prior to the acute exacerbation. The development of exertional dyspnea, often described as increased effort to breathe, heaviness, air hunger, or gasping, can be insidious. It is best elicited by a careful history focused on typical physical activities and how the patient’s ability to perform them has changed. Activities involving significant arm work, particularly at or above shoulder level, are particularly difficult for patients with COPD. Conversely, activities that allow the patient to brace the arms and use accessory muscles of respiration are better tolerated. Examples of such activities include pushing a shopping cart or walking on a treadmill. As COPD advances, the principal feature is worsening dyspnea on exertion with increasing intrusion on the ability to perform vocational or avocational activities. In the most advanced stages, patients are breathless doing simple activities of daily living.
Accompanying worsening airflow obstruction is an increased frequency of exacerbations (described below). Patients may also develop resting hypoxemia and require institution of supplemental oxygen.
PHYSICAL FINDINGS
In the early stages of COPD, patients usually have an entirely normal physical examination. Current smokers may have signs of active smoking, including an odor of smoke or nicotine staining of fingernails. In patients with more severe disease, the physical examination is notable for a prolonged expiratory phase and may include expiratory wheezing. In addition, signs of hyperinflation include a barrel chest and enlarged lung volumes with poor diaphragmatic excursion as assessed by percussion. Patients with severe airflow obstruction may also exhibit use of accessory muscles of respiration, sitting in the characteristic “tripod” position to facilitate the actions of the sternocleidomastoid, scalene, and intercostal muscles. Patients may develop cyanosis, visible in the lips and nail beds.
Although traditional teaching is that patients with predominant emphysema, termed “pink puffers,” are thin and noncyanotic at rest and have prominent use of accessory muscles, and patients with chronic bronchitis are more likely to be heavy and cyanotic (“blue bloaters”), current evidence demonstrates that most patients have elements of both bronchitis and emphysema and that the physical examination does not reliably differentiate the two entities.
Advanced disease may be accompanied by cachexia, with significant weight loss, bitemporal wasting, and diffuse loss of subcutaneous adipose tissue. This syndrome has been associated with both inadequate oral intake and elevated levels of inflammatory cytokines (TNF-α). Such wasting is an independent poor prognostic factor in COPD. Some patients with advanced disease have paradoxical inward movement of the rib cage with inspiration (Hoover’s sign), the result of alteration of the vector of diaphragmatic contraction on the rib cage as a result of chronic hyperinflation.
Signs of overt right heart failure, termed cor pulmonale, are relatively infrequent since the advent of supplemental oxygen therapy.
Clubbing of the digits is not a sign of COPD, and its presence should alert the clinician to initiate an investigation for causes of clubbing. In this population, the development of lung cancer is the most likely explanation for newly developed clubbing.
LABORATORY FINDINGS
The hallmark of COPD is airflow obstruction (discussed above). Pulmonary function testing shows airflow obstruction with a reduction in FEV1 and FEV1/FVC (Chap. 306e). With worsening disease severity, lung volumes may increase, resulting in an increase in total lung capacity, functional residual capacity, and residual volume. In patients with emphysema, the diffusing capacity may be reduced, reflecting the lung parenchymal destruction characteristic of the disease. The degree of airflow obstruction is an important prognostic factor in COPD and is the basis for the Global Initiative for Lung Disease (GOLD) severity classification (Table 314-1). More recently it has been shown that a multifactorial index incorporating airflow obstruction, exercise performance, dyspnea, and body mass index is a better predictor of mortality rate than pulmonary function alone. In 2011, the GOLD added an additional classification system incorporating symptoms and exacerbation history; the utility of this system remains to be defined.
|
GOLD CRITERIA FOR SEVERITY OF AIRFLOW OBSTRUCTION IN COPD |
Arterial blood gases and oximetry may demonstrate resting or exertional hypoxemia. Arterial blood gases provide additional information about alveolar ventilation and acid-base status by measuring arterial PCO2 and pH. The change in pH with PCO2 is 0.08 units/10 mmHg acutely and 0.03 units/10 mmHg in the chronic state. Knowledge of the arterial pH therefore allows the classification of ventilatory failure, defined as PCO2 >45 mmHg, into acute or chronic conditions. The arterial blood gas is an important component of the evaluation of patients presenting with symptoms of an exacerbation. An elevated hematocrit suggests the presence of chronic hypoxemia, as does the presence of signs of right ventricular hypertrophy.
Radiographic studies may assist in the classification of the type of COPD. Obvious bullae, paucity of parenchymal markings, or hyperlucency suggests the presence of emphysema. Increased lung volumes and flattening of the diaphragm suggest hyperinflation but do not provide information about chronicity of the changes. Computed tomography (CT) scan is the current definitive test for establishing the presence or absence of emphysema in living subjects (Fig. 314-4). From a practical perspective, the CT scan currently does little to influence therapy of COPD except in individuals considering surgical therapy for their disease (described below) and as screening for lung cancer.
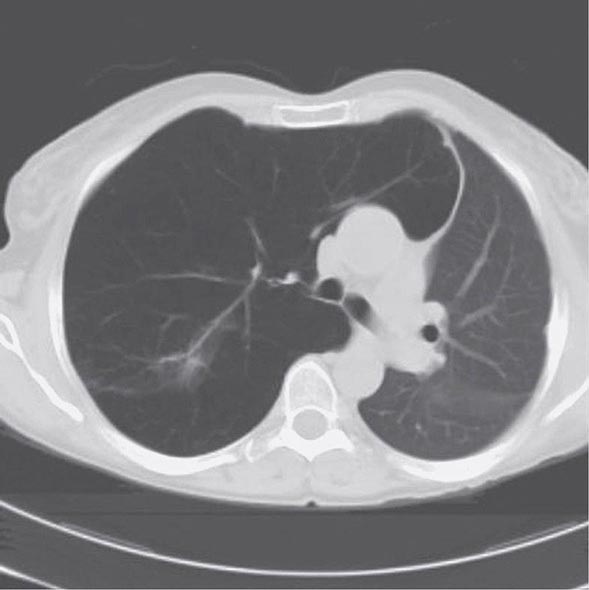
FIGURE 314-4 Chest computed tomography scan of a patient with chronic obstructive pulmonary disease who underwent a left single-lung transplant. Note the reduced parenchymal markings in the right lung (left side of figure) as compared to the left lung, representing emphysematous destruction of the lung, and mediastinal shift to the left, indicative of hyperinflation.
Recent guidelines have suggested testing for α1AT deficiency in all subjects with COPD or asthma with chronic airflow obstruction. Measurement of the serum α1AT level is a reasonable initial test. For subjects with low α1AT levels, the definitive diagnosis of α1AT deficiency requires protease inhibitor (PI) type determination. This is typically performed by isoelectric focusing of serum, which reflects the genotype at the PI locus for the common alleles and many of the rare PI alleles as well. Molecular genotyping of DNA can be performed for the common PI alleles (M, S, and Z).