[level-membership-for-pediatrics-category]
Chapter 395 Cystic Fibrosis
CF is responsible for most cases of exocrine pancreatic insufficiency in early life and is the major cause of severe chronic lung disease in children. It is also responsible for many cases of salt depletion, nasal polyposis, pansinusitis, rectal prolapse, pancreatitis, cholelithiasis, and insulin-dependent hyperglycemia. CF may manifest as failure to thrive and, occasionally, as cirrhosis or other forms of hepatic dysfunction. Therefore, this disorder enters into the differential diagnosis of many pediatric conditions (Table 395-1).
Table 395-1 COMPLICATIONS OF CYSTIC FIBROSIS
RESPIRATORY
GASTROINTESTINAL
OTHER
Adapted from Silverman FN, Kuhn JP: Essentials of Caffrey’s pediatric x-ray diagnosis, 1990, Chicago, Year Book, p 649.
Genetics
CF occurs most frequently in white populations of northern Europe, North America, and Australia/New Zealand. The prevalence in these populations varies but approximates 1/3,500 live births (1/9,200 individuals of Hispanic descent and 1/15,000 in African Americans). Although less frequent in African, Hispanic, Middle Eastern, South Asian, and eastern Asian populations, the disorder does exist in these populations as well (Fig. 395-1).
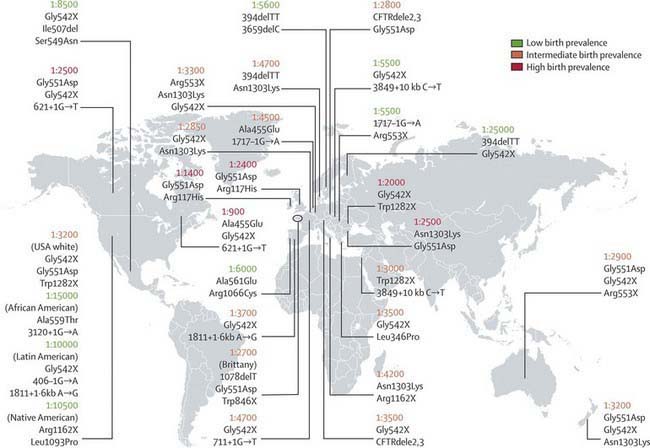
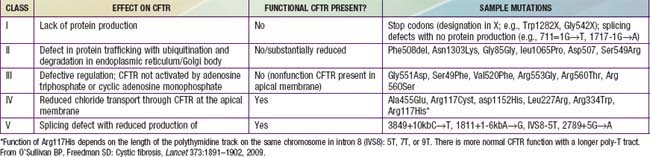
CF is inherited as an autosomal recessive trait. The CF gene codes for the CFTR protein, which is 1,480 amino acids. CFTR is expressed largely in epithelial cells of airways, the gastrointestinal tract (including the pancreas and biliary system), the sweat glands, and the genitourinary system. CFTR is a member of the adenosine triphosphate (ATP)–binding cassette superfamily of proteins. It functions as a chloride channel and has other regulatory functions that are perturbed variably by the different mutations. More than 1,500 CFTR polymorphisms grouped into 5 main classes of mutations that affect protein function are associated with the CF syndrome (Table 395-2). The most prevalent mutation of CFTR is the deletion of a single phenylalanine residue at amino acid 508 (ΔF508). This mutation is responsible for the high incidence of CF in northern European populations and is considerably less frequent in other populations, such as those of southern Europe and Israel. Approximately 50% of individuals with CF who are of northern European ancestry are homozygous for ΔF508, and >80% carry at least one ΔF508 gene. The remainder of patients has an extensive array of mutations, none of which has a prevalence of more than several percentage points, except in circumscribed populations; the W1282X mutation occurs in 60% of Ashkenazi Jews with CF. The relationship between CFTR genotype and clinical phenotype is highly complex and is not predictable for individual patients. Mutations categorized as “severe” are associated almost uniformly with pancreatic insufficiency but only in general with more rapid progression of lung disease. Modifier gene polymorphisms appear to be responsible for much of the variation in the progression of lung disease. The most compelling association with more severe disease is with a single nucleotide change in the transforming growth factor-β1(TGF-β1) gene. Variant alleles of the mannose-binding lectin, a key factor in systemic innate immunity, are associated with more serious lung infections and reduced survival. Polymorphism in the IRFD1 gene, a transcriptional co-regulator that is essential for neutrophil differentiation, has been associated with a more serious CF pulmonary phenotype. Several mutations, such as 3849 + 10kbC→T, are found in patients with normal sweat chloride concentrations. Some individuals with polymorphisms of both CFTR genes have few or no CF manifestations until adolescence or adulthood, when they present with pancreatitis, sinusitis, diffuse bronchiectasis, or male infertility. Whereas CFTR mutations are a sine qua non for CF, two mutations of CFTR can cause disorders that do not meet diagnostic criteria for CF and, occasionally, do not cause discernible clinical problems.
Pathogenesis
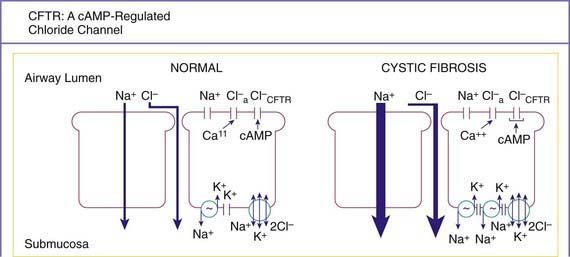
A number of long-standing observations of CF are of fundamental pathophysiologic importance; they include failure to clear mucous secretions, a paucity of water in mucous secretions, an elevated salt content of sweat and other serous secretions, and chronic infection limited to the respiratory tract. Additionally, there is a greater negative potential difference across the respiratory epithelia of patients with CF than across the respiratory epithelia of control subjects. Aberrant electrical properties are also demonstrated for CF sweat gland duct and rectal epithelia. The membranes of CF epithelial cells are unable to secrete chloride ions in response to cyclic adenosine monophosphate (cAMP)–mediated signals, and at least in the respiratory tract, excessive amounts of sodium are absorbed through these membranes (Fig. 395-2). These defects can be traced to a dysfunction of CFTR (Figs. 395-3 and 395-4).
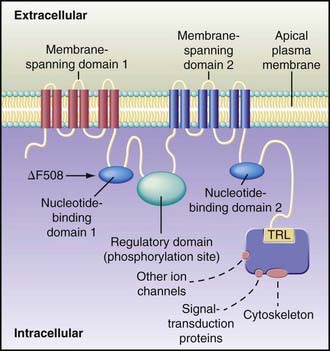
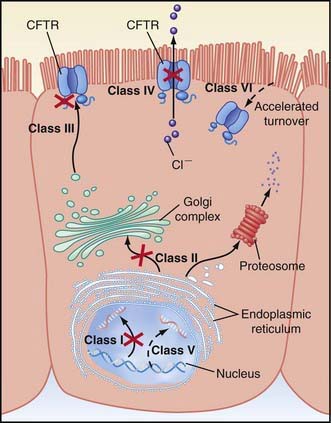
Cyclic AMP–stimulated protein kinase A (PKA) regulation of chloride conductance is the primary function of CFTR; this function is absent in epithelial cells with many different mutations of the CFTR gene. CFTR mutations fall into 6 classes in another classification system, albeit with some overlap (see Fig. 395-4). Individuals with class I, II, and III mutations, on average, have shorter survival than those with “mild” genotypes (class IV or V). The clinical importance of these functional categories is limited because they do not uniformly correlate with specific clinical features or their severity. Rather, clinical features correlate with the residual CFTR activity.
Chronic infection in CF is limited to the airways. A likely explanation for infection is a sequence of events starting with failure to clear inhaled bacteria promptly and then proceeding to persistent colonization and an inflammatory response in airway walls. In addition, it has been proposed that abnormal CFTR creates a proinflammatory state or amplifies the inflammatory response to initial infections (viral or bacterial). Some investigators have identified primary differences in CF-affected immune cells and have suggested that these alterations contribute to this proinflammatory state. It appears that inflammatory events occur first in small airways, perhaps because clearance of altered secretions and microorganisms from these regions is more difficult. Chronic bronchiolitis and bronchitis are the initial lung manifestations (Chapter 383), but after months to years, structural changes in airway walls produce bronchiolectasis and bronchiectasis.
A finding that is not readily explained by CFTR dysfunction is the high prevalence in patients with CF of airway colonization with Staphylococcus aureus (Chapter 174.1), Pseudomonas aeruginosa (Chapter 197.1), and Burkholderia cepacia (Chapter 197.2), organisms that rarely infect the lungs of other individuals. It has been postulated that the CF airway epithelial cells or surface liquids may provide a favorable environment for harboring these organisms. CF airway epithelium may be compromised in its innate defenses against these organisms, through either acquired or genetic alterations. Antimicrobial activity is diminished in CF secretions; this diminution may be related to hyperacidified surface liquids or other effects on innate immunity. Another puzzle is the propensity for P. aeruginosa to undergo mucoid transformation in the CF airways. The complex polysaccharide produced by these organisms generates a biofilm that provides a hypoxic environment and thereby protects Pseudomonas against antimicrobial agents.
Pathology
The earliest pathologic lesion in the lung is that of bronchiolitis (mucous plugging and an inflammatory response in the walls of the small airways); with time, mucus accumulation and inflammation extend to the larger airways (bronchitis) (Chapter 383). Goblet cell hyperplasia and submucosal gland hypertrophy become prominent pathologic findings, which is most likely a response to chronic airway infection. Organisms appear to be confined to the endobronchial space; invasive bacterial infection is not characteristic. With long-standing disease, evidence of airway destruction such as bronchiolar obliteration, bronchiolectasis, and bronchiectasis (Chapter 393) becomes prominent. Imaging modalities demonstrate both increased airway wall thickness and luminal cross-sectional area relatively early in lung disease evaluation. Bronchiectatic cysts and emphysematous bullae or subpleural blebs are frequent with advanced lung disease, the upper lobes being most commonly involved. These enlarged air spaces may rupture and cause pneumothorax. Interstitial disease is not a prominent feature, although areas of fibrosis appear eventually. Bronchial arteries are enlarged and tortuous, contributing to a propensity for hemoptysis in bronchiectatic airways. Small pulmonary arteries eventually display medial hypertrophy, which would be expected in secondary pulmonary hypertension.
The paranasal sinuses are uniformly filled with secretions containing inflammatory products, and the epithelial lining displays hyperplastic and hypertrophied secretory elements (Chapter 372). Polypoid lesions within the sinuses and erosion of bone have been reported. The nasal mucosa may form large or multiple polyps, usually from a base surrounding the ostia of the maxillary and ethmoidal sinuses.
Clinical Manifestations
Mutational heterogeneity and environmental factors appear responsible for highly variable involvement of the lungs, pancreas, and other organs. A list of presenting manifestations is lengthy, although pulmonary and gastrointestinal presentations predominate (Fig. 395-5). With inclusion of CF newborn screening panels, an increasing proportion of children are diagnosed before symptoms appear (Table 395-3).
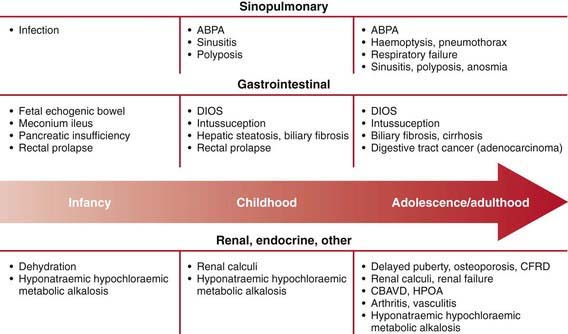
Table 395-3 PRESENTING FEATURES OF MORE THAN 25,000 PATIENTS WITH CYSTIC FIBROSIS IN THE USA
| FEATURE | % | % of Patients Presenting in 2007 |
|---|---|---|
| Acute or persistent respiratory symptoms | 45.6 | 31.2 |
| Failure to thrive, malnutrition | 37.5 | 18.7 |
| Abnormal stools | 28.8 | 13.8 |
| Meconium ileus, intestinal obstruction | 19.9 | 14 |
| Family history | 16.0 | 12 |
| Newborn Screening | 6.4 | 30.7 |
| Electrolyte, acid-base abnormality | 4.2 | 1.1 |
| Rectal prolapse | 3.3 | 3.3 |
| Nasal polyps, sinus disease | 3.3 | 4.6 |
| Hepatobiliary disease | 1.2 | 1.4 |
| Other* | 3-4 | 6.7 |
* Includes pseudotumor cerebri, azoospermia, acrodermatitis-like rash, vitamin deficiency states, hypoproteinemic edema, hypoprothrombinemia with bleeding, and meconium plug syndrome.
Data from the Patient Registry, Cystic Fibrosis Foundation, Bethesda, MD.
Intestinal Tract
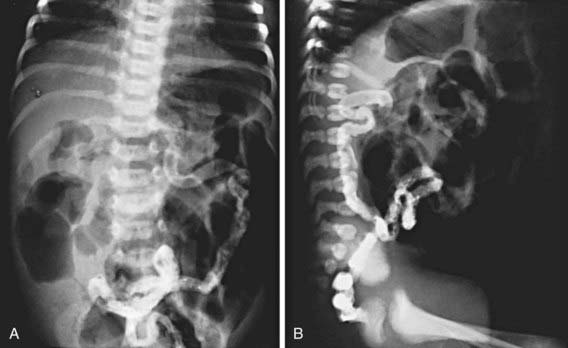
In 15-20% of newborn infants with CF, the ileum is completely obstructed by meconium (meconium ileus). The frequency is greater (≈30%) among siblings born subsequent to a child with meconium ileus and is particularly striking in monozygotic twins, reflecting a genetic contribution from one or more modifying genes. Abdominal distention, emesis, and failure to pass meconium appear in the 1st 24-48 hr of life (Chapters 96.1 and 322.2). Abdominal radiographs (Fig. 395-6) show dilated loops of bowel with air-fluid levels and, frequently, a collection of granular, “ground-glass” material in the lower central abdomen. Rarely, meconium peritonitis results from intrauterine rupture of the bowel wall and can be detected radiographically as the presence of peritoneal or scrotal calcifications. Meconium plug syndrome occurs with increased frequency in infants with CF but is less specific than meconium ileus. Ileal obstruction with fecal material (distal intestinal obstruction syndrome [DIOS]) occurs in older patients, causing cramping abdominal pain and abdominal distention.
Diagnosis and Assessment
The diagnosis of CF has been based on a positive quantitative sweat test (Cl− ≥ 60 mEq/L) in conjunction with 1 or more of the following features: typical chronic obstructive pulmonary disease, documented exocrine pancreatic insufficiency, and a positive family history. With newborn screening, diagnosis is often made prior to obvious clinical manifestations such as failure to thrive and chronic cough (Table 395-3). Diagnostic criteria have been recommended to include additional testing procedures (Table 395-4).
Table 395-4 DIAGNOSTIC CRITERIA FOR CYSTIC FIBROSIS (CF)
Presence of typical clinical features (respiratory, gastrointestinal, or genitourinary)
OR
A history of CF in a sibling
OR
A positive newborn screening test
PLUS
Laboratory evidence for CFTR (CF transmembrane regulator) dysfunction:
Two elevated sweat chloride concentrations obtained on separate days
OR
Identification of two CF mutations
OR
An abnormal nasal potential difference measurement
Sweat Testing
More than 60 mEq/L of chloride in sweat is diagnostic of CF when 1 or more other criteria are present. Threshold levels of 30-40 mEq/L for infants have been suggested. Borderline (or intermediate) values of 40 to 60 mEq/L have been reported in patients of all ages who have CF with atypical involvement and require further testing. Chloride concentrations in sweat are somewhat lower in individuals who retain exocrine pancreatic function but usually remain within the diagnostic range. Conditions associated with false-negative and false-positive results are noted in Table 395-5.
Table 395-5 CONDITIONS ASSOCIATED WITHFALSE-POSITIVE AND FALSE-NEGATIVE SWEAT TEST RESULTS
WITH FALSE-POSITIVE RESULTS
WITH FALSE-NEGATIVE RESULTS
Radiology
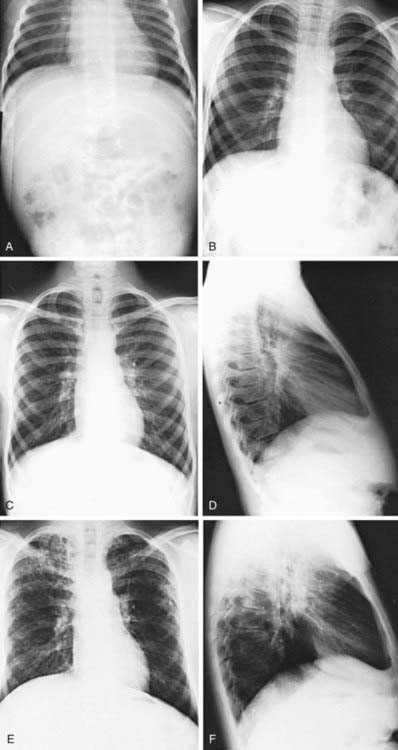
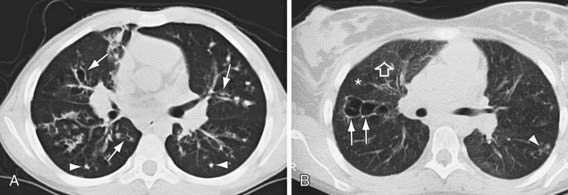
Pulmonary radiologic findings suggest the diagnosis but are not specific. Hyperinflation of lungs occurs early and may be overlooked in the absence of infiltrates or streaky densities. Bronchial thickening and plugging and ring shadows suggesting bronchiectasis usually appear first in the upper lobes. Nodular densities, patchy atelectasis, and confluent infiltrate follow. Hilar lymph nodes may be prominent. With advanced disease, impressive hyperinflation with markedly depressed diaphragms, anterior bowing of the sternum, and a narrow cardiac shadow are noted. Cyst formation, extensive bronchiectasis, dilated pulmonary artery segments, and segmental or lobar atelectasis are often apparent with advanced disease. Typical progression of lung disease is seen in Figure 395-7. Most CF centers obtain chest radiographs (posteroanterior [PA] and lateral) at least annually. Standardized scoring of roentgenographic changes has been used to follow progression of lung disease. CT of the chest can detect and localize thickening of bronchial airway walls, mucous plugging, focal hyperinflation, and early bronchiectasis (Fig. 395-8); it is generally not used for routine evaluation of chest disease. Many children with normal lung function have bronchiectasis on CT, indicating that this imaging modality is sensitive to early lung changes.
Pulmonary Function
Standard pulmonary function studies are not obtained until patients are 4-6 yr of age, by which time many patients show the typical pattern of obstructive pulmonary involvement (Chapter 376). Decrease in the midmaximal flow rate is an early functional change, reflecting small airway obstruction. This lesion also affects the distribution of ventilation and increases the alveolar-arterial oxygen difference. The findings of obstructive airway disease and modest responses to a bronchodilator are consistent with the diagnosis of CF at all ages. Residual volume and functional residual capacity are increased early in the course of lung disease. Restrictive changes, characterized by declining total lung capacity and vital capacity, correlate with extensive lung injury and fibrosis and are a late finding. Testing at each clinic visit is recommended to evaluate the course of the pulmonary involvement and allow for early intervention when substantial decrements are documented. Increasing numbers of CF centers are equipped to measure airflow patterns of sedated infants (infant pulmonary function tests). Some patients reach adolescent or adult life with normal pulmonary function and without evidence of overinflation.
Treatment
General Approach to Care
Initial efforts after diagnosis should be intensive and should include baseline assessment, initiation of treatment, clearing of pulmonary involvement, and education of the patient and parents. Follow-up evaluations are scheduled every 1-3 mo, depending on the age at diagnosis, because many aspects of the condition require careful monitoring. An interval history and physical examination should be obtained at each visit. A sputum sample or, if that is not available, a lower pharyngeal swab taken during or after a forced cough is obtained for culture and antibiotic susceptibility studies. Because irreversible loss of pulmonary function from low-grade infection can occur gradually and without acute symptoms, emphasis is placed on a thorough pulmonary history. Table 395-6 lists symptoms and signs that suggest the need for more intensive antibiotic and physical therapy. Protection against exposure to methicillin-resistant S. aureus, P. aeruginosa, B. cepacia, and other resistant gram-negative organisms is essential, including isolation procedures and careful attention to sterilization of inhalation therapy equipment. A nurse, respiratory therapist, social worker, dietitian, and psychologist, as members of the multidisciplinary care team, should evaluate children regularly and contribute to the development of a comprehensive daily care plan. Considerable education and programs to empower families and older children to take responsibility for care are likely to result in the best adherence to daily care programs. Standardization of practice, on the part of both caregivers and families, as well as close monitoring and early intervention for new or increasing symptoms appears to result in the best long-term outcomes.
Table 395-6 SYMPTOMS AND SIGNS ASSOCIATED WITH EXACERBATION OF PULMONARY INFECTION IN PATIENTS WITH CYSTIC FIBROSIS
SYMPTOMS
SIGNS
From Ramsey B: Management of pulmonary disease in patients with cystic fibrosis, N Engl J Med 335:179, 1996.
The basic daily care program varies according to the age of the child, the degree of pulmonary involvement, other system involvement, and the time available for therapy. The major components of this care are pulmonary and nutritional therapies. Because therapy is medication-intensive, iatrogenic problems frequently arise. Monitoring for these complications is also an important part of management (Table 395-7).
| COMPLICATION | AGENT |
|---|---|
| Gastrointestinal bleeding | Ibuprofen |
| Hyperglycemia | Corticosteroids (systemic) |
| Growth retardation | Corticosteroids (systemic, inhaled) |
| Renal dysfunction: | |
| Tubular | Aminoglycosides |
| Interstitial nephritis | Semisynthetic penicillins, nonsteroidal anti-inflammatory drugs |
| Hearing loss, vestibular dysfunction | Aminoglycosides |
| Peripheral neuropathy or optic atrophy | Chloramphenicol (prolonged course) |
| Hypomagnesemia | Aminoglycosides |
| Hyperuricemia, colonic stricture | Pancreatic extracts (very large doses) |
| Goiter | Iodine-containing expectorants |
| Gynecomastia | Spironolactone |
| Enamel hypoplasia or staining | Tetracyclines (used in 1st 8 yr of life) |
* Note: Common hypersensitivity reactions to drugs are not included.
Pulmonary Therapy
Inhalation Therapy
Aerosolized antibiotics are often used when the airways are colonized with Pseudomonas as part of daily therapy. Aerosolized tobramycin, TOBI, used as a suppressive therapy (on 1 month, off 1 month) may reduce symptoms, improve pulmonary function, and alleviate the need for hospitalization (see Aerosolized Antibiotic Therapy).
Oral Antibiotic Therapy
Indications for oral antibiotic therapy in a patient with CF include the presence of respiratory tract symptoms and identification of pathogenic organisms in respiratory tract cultures. Whenever possible, the choice of antibiotics should be guided by in vitro sensitivity testing. Common organisms, include S. aureus, nontypable Haemophilus influenzae, P. aeruginosa; B. cepacia and other gram-negative rods, are encountered with increasing frequency. The first 2 can be eradicated from the respiratory tract in CF with use of oral antibiotics, but Pseudomonas is more difficult to treat. The usual course of therapy is ≥2 wk, and maximal doses are recommended. Useful oral antibiotics are listed in Table 395-8. The quinolones are the only broadly effective oral antibiotics for Pseudomonas infection, but resistance against these agents emerges rapidly. Infection with mycoplasmal or chlamydial organisms has been documented, providing a rationale for the use of macrolides on an empirical basis for flare of symptoms. Macrolides may reduce the virulence properties of P. aeruginosa, such as biofilm production, and contribute anti-inflammatory effects. Long-term therapy with azithromycin times a week has been shown to improve lung function in patients with chronic P. aeruginosa infection.
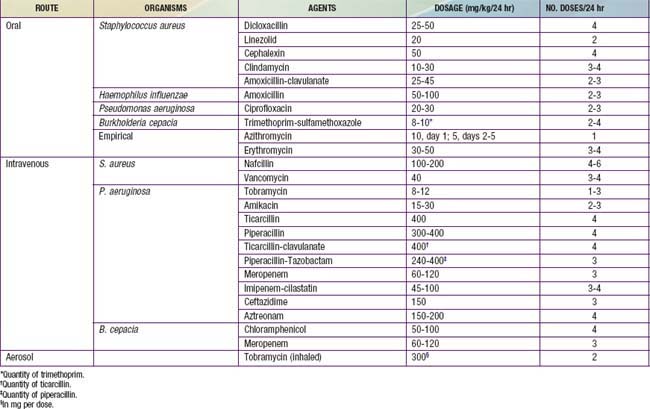
Intravenous Antibiotic Therapy
Commonly used intravenous antibiotics are listed in Table 395-8. In general, treatment of Pseudomonas infection requires 2-drug therapy. A 3rd agent may be required for optimal coverage of S. aureus or other organisms. The aminoglycosides have a relatively short half-life in many patients with CF. The initial parenteral dose, noted in Table 395-8, is generally given every 8 hr. After blood levels have been determined, the total daily dose should be adjusted. Peak levels of 10-15 mg/L are desirable, and trough levels should be kept at <2 mg/L to minimize the risk of ototoxicity and nephrotoxicity. Once- or twice-daily aminoglycoside dosing may have advantages over dosing every 8 hr. Changes in therapy should be guided by lack of improvement and by culture results. If patients do not show improvement, complications such as heart failure and reactive airways or infection with viruses, Aspergillus fumigatus (Chapter 229), nontuberculous mycobacteria (Chapter 209), or other unusual organisms should be considered. B. cepacia is the most frequent of a growing list of gram-negative rods that may be particularly refractory to antimicrobial therapy.
Anti-Inflammatory Agents
Corticosteroids are useful for the treatment of allergic bronchopulmonary aspergillosis and severe reactive airway disease occasionally encountered in children with CF. Prolonged treatment of standard CF lung disease using an alternate-day regimen initially appeared to improve pulmonary function and diminish hospitalization rates. However, a 4-yr double-blind, multicenter study of this regimen for patients with mild to moderate lung disease found only modest efficacy and prohibitive side effects, including growth retardation, cataracts, and abnormalities of glucose tolerance at a dose of 2 mg/kg and growth retardation at 1 mg/kg. Inhaled corticosteroids have theoretical appeal, but there are few data documenting their efficacy and safety; it appears that discontinuing inhaled corticosteroids in patients with CF had no effect on lung function, antibiotic use, or bronchodilator use. Ibuprofen, given long term (dose adjusted to achieve a peak serum concentration of 50-100 µg/mL) for 4 yr, is associated with a slowing of disease progression, particularly in younger patients with mild lung disease. Side effects of nonsteroidal anti-inflammatory drugs have been encountered (see Table 395-7); therefore, this therapy has not gained broad acceptance even though ibuprofen is the only anti-inflammatory agent with documented efficacy in the patient population.
Treatment of Pulmonary Complications
Atelectasis
Lobar atelectasis occurs relatively infrequently; it may be asymptomatic and noted only at the time of a routine chest radiograph. Aggressive intravenous therapy with antibiotics and increased chest PT directed at the affected lobe may be effective. If there is no improvement in 5-7 days, bronchoscopic examination of the airways may be indicated. If the atelectasis does not resolve, continued intensive home therapy is indicated, because atelectasis may resolve during a period of weeks or months. Persistent atelectasis may be asymptomatic. Lobectomy should be considered only if expansion is not achieved and the patient has progressive difficulty from fever, anorexia, and unrelenting cough (Chapter 402).
Pneumothorax
Pneumothorax (Chapter 405) is encountered in <1% of children and teenagers with CF, although it is more frequently encountered in older patients and may be life-threatening. The episode may be asymptomatic but is often attended by chest and shoulder pain, shortness of breath, or hemoptysis. A small air collection that does not grow can be observed closely. Chest tube placement with or without pleurodesis is often the initial therapy. Intravenous antibiotics are also begun on admission. An open thoracotomy or video-assisted thoracoscopy (VATS) with plication of blebs, apical pleural stripping, and basal pleural abrasion should be considered if the air leak persists. Surgical intervention is usually well tolerated even in cases of advanced lung disease. The thoracotomy tube is removed as soon as possible, usually on the 2nd or 3rd postoperative day. The patient can then be mobilized, and full postural drainage therapy resumed. Previous pneumothorax with or without pleurodesis is not a contraindication to subsequent lung transplantation.
Allergic Aspergillosis
Allergic aspergillosis occurs in 5-10% of patients with CF and may manifest as wheezing, increased cough, shortness of breath, and marked hyperinflation (Chapters 229 and 391). In some patients, a chest radiograph shows new, focal infiltrates. The presence of rust-colored sputum, the recovery of Aspergillus organisms from the sputum, the demonstration of serum precipitating and specific immunoglobulin E (IgE) and IgG antibodies against A. fumigatus, or the presence of eosinophils in a fresh sputum sample supports the diagnosis. The serum IgE level is usually high. Treatment is directed at controlling the inflammatory reaction with oral corticosteroids and preventing central bronchiectasis. For refractory cases, oral antifungals may be required.
Acute Respiratory Failure
Acute respiratory failure (Chapter 65) rarely occurs in patients with mild to moderate lung disease and is usually the result of a severe viral or other infectious illness. Because patients with this complication can regain their previous status, intensive therapy is indicated. In addition to aerosol, postural drainage, and intravenous antibiotic treatment, oxygen is required to raise the arterial PaO2. An increasing PCO2 may require ventilatory assistance. Endotracheal or bronchoscopic suction may be necessary to clear airway inspissated secretions and can be repeated daily. Right-sided heart failure should be treated vigorously. Recovery is often slow. Intensive intravenous antibiotic therapy and postural drainage should be continued for 1-2 wk after the patient has regained baseline status.
Chronic Respiratory Failure
Lung transplantation is an option for end-stage lung disease (Chapter 437) but a topic of vigorous debate. Criteria for referral continue to be a subject of investigation and ideally include estimates of longevity with and without transplant based on lung function and exercise tolerance data. Because of bronchiolitis obliterans (Chapter 386.1) and other complications, transplanted lungs cannot be expected to function for the lifetime of a recipient, and repeat transplantation is increasingly common. The demand for donor lungs exceeds the supply, and waiting lists as well as duration of waits continue to grow. The protocol for matching donor organs with lung transplant recipients has been revised to account for the severity of the patients’ lung disease. In a review of lung transplantation in children with CF between 1992 and 2002, pre-transplantation colonization with B. cepacia, diabetes, and older age decreased post-transplantation survival. The review suggests that transplantation is often associated with many complications and may not prolong life nor significantly improve its quality.
Heart Failure
Some patients experience reversible right-sided heart failure (Chapter 436) as the result of an acute event such as a viral infection or pneumothorax. Individuals with long-standing, advanced pulmonary disease, especially those with severe hypoxemia (PaO2 <50 mm Hg), often acquire chronic right-sided heart failure. The mechanisms include hypoxemic pulmonary arterial constriction and loss of the pulmonary vascular bed. Pulmonary arterial wall changes contribute to increased vascular resistance with time. Evidence for concomitant left ventricular dysfunction is often found. Cyanosis, increased shortness of breath, increased liver size with a tender margin, ankle edema, jugular venous distention, an unusual weight gain, increased heart size seen on chest radiograph, or evidence for right-sided heart enlargement on electrocardiogram or echocardiogram helps to confirm the diagnosis. Diuresis induced by furosemide (1 mg/kg administered intravenously) confirms the suspicion of fluid retention. Repeated doses are often required at 24- to 48-hr intervals to reduce fluid accumulation and accompanying symptoms. Concomitant use of spironolactone may protect against potassium depletion and facilitate long-term diuresis. Hypochloremic alkalosis complicates the long-term use of loop diuretics. Digitalis is not effective in pure right-sided failure but may be useful when there is an associated left-sided dysfunction. The arterial PO2 should be maintained at >50 mm Hg if possible. Intensive pulmonary therapy, including intravenous antibiotics, is most important. Initially, the salt intake should be limited. Volume overload and antibiotics with high sodium content should be avoided. No clear-cut long-term benefit from pulmonary vasodilators has been demonstrated. The prognosis for heart failure is poor, but a number of patients survive for ≥5 yr after the appearance of heart failure. Heart-lung transplantation may be an option (see preceding section).
Treatment of Intestinal Complications
Meconium Ileus
When meconium ileus (Chapter 96.1) is suspected, a nasogastric tube is placed for suction and the newborn is hydrated. In many cases, diatrizoate (Gastrografin) enemas with reflux of contrast material into the ileum not only confirm the diagnosis but have also resulted in the passage of a meconium plug and clearing of the obstruction. Use of this hypertonic solution requires careful correction of water losses into the bowel. Children in whom this procedure fails require operative intervention. Children who are successfully treated generally have a prognosis similar to that of other patients with severe CF mutations. Infants with meconium ileus should be treated as if they have CF until adequate diagnostic testing can be carried out.
Distal Intestinal Obstruction Syndrome (Meconium Ileus Equivalent) and Other Causes of Abdominal Symptoms
Despite appropriate pancreatic enzyme replacement, 2-5% of patients accumulate fecal material in the terminal portion of the ileum and in the cecum, which may result in partial or complete obstruction. For intermittent symptoms, pancreatic enzyme replacement should be continued or even increased, and stool softeners (polyethylene glycol [MiraLAX] or docusate sodium [Colace]) given. Increased fluid intake is also recommended. Failure to relieve symptoms signals the need for large-volume bowel lavage with a balanced salt solution containing polyethylene glycol taken by mouth or by nasogastric tube. When there is complete obstruction, a diatrizoate enema, accompanied by large amounts of intravenous fluids, can be therapeutic. Intussusception (Chapter 325.3) and volvulus (Chapter 321.4) must also be considered in the differential diagnosis. Intussusception, usually ileocolic, occurs at any age and often follows a 1- to 2-day history of “constipation.” It can often be diagnosed and reduced via a diatrizoate enema. If a nonreducible intussusception or a volvulus is present, laparotomy is required. Repeated episodes of intussusception may be an indication for cecectomy.
Hepatobiliary Disease
Liver function abnormalities associated with biliary cirrhosis can be improved by treatment with ursodeoxycholic acid. The ability of bile acids to prevent progression of cirrhosis has not been clearly documented. Portal hypertension with esophageal varices, hypersplenism, or ascites occurs in ≤8% of children with CF (Chapter 359). The acute management of bleeding esophageal varices includes nasogastric suction and cold saline lavage. Sclerotherapy is recommended after an initial hemorrhage. In the past, significant bleeding has also been treated successfully with portosystemic shunting. Splenorenal anastomosis has been the most effective treatment. Pronounced hypersplenism may require splenectomy. Cholelithiasis should prompt surgical consultation. The management of ascites is discussed in Chapter 362.
Obstructive jaundice in newborns with CF needs no specific therapy. Hepatomegaly with steatosis requires careful attention to nutrition and may respond to carnitine repletion. Rarely, biliary cirrhosis proceeds to hepatocellular failure, which should be treated as in patients without CF (Chapters 356 and 359). End-stage liver disease is an indication for liver transplantation in children with CF, especially if pulmonary function is good (Chapter 360).
Pancreatitis
Pancreatitis can be precipitated by fatty meals, alcohol ingestion, or tetracycline therapy. Serum amylase and lipase values may remain elevated for long periods. Treatment of this disorder is discussed in Chapter 343.
Other Therapy
Nasal Polyps
Nasal polyps (Chapter 370) occur in 15-20% of patients with CF and are most prevalent in the 2nd decade of life. Local corticosteroids and nasal decongestants occasionally provide some relief. When the polyps completely obstruct the nasal airway, rhinorrhea becomes constant, or widening of the nasal bridge is noticed, surgical removal of the polyps is indicated; polyps may recur promptly or after a symptom-free interval of months to years. Polyps inexplicably stop developing in many adults.
Aaron SD, Vandemheen KL, Ferris W, et al. Combination antibiotic susceptibility testing to treat exacerbations of cystic fibrosis associated with multiresistant bacteria: a randomized, double-blind, controlled clinical trial. Lancet. 2005;366:463-471.
Aaron SD, Vandemheen KL, Ramotar K, et al. Infection with transmissible strains of Pseudomonas aeruginosa and clinical outcomes in adults with cystic fibrosis. JAMA. 2010;304(19):2145-2152.
Accurso FJ, Rowe SM, Clancy JP, et al. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N Engl J Med. 2010;363(21):1991-2003.
Aldámiz-Echevarria L, Preito JA, Andrade F, et al. Persistence of essential fatty acid deficiency in cystic fibrosis despite nutritional therapy. Pediatr Res. 2009;66:585-589.
Bartlett JR, Friedman KJ, Ling SC, et al. Genetic modifiers of liver disease in cystic fibrosis. JAMA. 2009;302:1076-1082.
Boyle MP. Adult cystic fibrosis. JAMA. 2007;298:1787-1793.
Buranawuti K, Boyle MP, Cheng S, et al. Variants in mannose-binding lectin and tumor necrosis factor α affect survival in cystic fibrosis. J Med Genet. 2007;44:209-214.
Casaulta C, Stirnimann A, Schoeni MH, et al. Sweat test in patients with glucose-6-phosphate-1-dehydrogenase deficiency. Arch Dis Child. 2008:93:878-879.
Colombo C, Battezzati A. Growth failure in cystic fibrosis: a true need for anabolic agents? J Pediatr. 2005;146:303-305.
Collaco J, Cutting G. Update on gene modifiers in cystic fibrosis. Curr Opin Pulmon Med. 2008;14:559-566.
Collaco JM, Blackman SM, McGready J, et al. Quantification of the relative contribution of environmental and genetic factors to variation in cystic fibrosis lung function. J Pediatr. 2010;157:802-807.
Collaco JM, Vanscoy L, Bremer L, et al. Interactions between secondhand smoke and genes that affect cystic fibrosis lung disease. JAMA. 2008;299:417-424.
Corey M, Edwards L, Levison H, et al. Longitudinal analysis of pulmonary function decline in patients with cystic fibrosis. J Pediatr. 1997;131:809-814.
Dasenbrook EC, Checkley W, Merlo CA, et al. Association between respiratory tract methicillin-resistant Staphylococcus aureus and survival in cystic fibrosis. JAMA. 2010;303:2386-2392.
Davies JC, Alton EWFW, Bush A. Cystic fibrosis. BMJ. 2007;335:1255-1259.
Donaldson SH, Bennett WD, Zeman KL, et al. Mucus clearance and lung function in cystic fibrosis with hypertonic saline. N Engl J Med. 2006;354:241-250.
Drumm ML, Konstan MW, Schluchter MD, et al. Genetic modifiers of lung disease in cystic fibrosis. N Engl J Med. 2005;353:1443-1453.
Elkins MR, Robinson M, Rose BR, et al. A controlled trial of long-term inhaled hypertonic saline on patients with cystic fibrosis. N Engl J Med. 2006;354:229-240.
Emerson J, Rosenfeld M, McNamara S, et al. Pseudomonas aeruginosa and other predictors of mortality and morbidity in young children with cystic fibrosis. Pediatr Pulmonol. 2002;34:91-100.
Ferkol T, Rosenfeld M, Milla CE. Cystic fibrosis pulmonary exacerbations. J Pediatr. 2006;148:259-264.
FitzSimmons SC, Burkhart GA, Borowitz D, et al. High-dose pancreatic-enzyme supplements and fibrosing colonopathy in children with cystic fibrosis. N Engl J Med. 1997;336:1283-1289.
Fuchs HJ, Borowitz DS, Christiansen DH, et al. Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. The Pulmozyme Study Group. N Engl J Med. 1994;331:637-642.
Flume PA, Robinson KA, O’Sullivan BP, et al. Cystic fibrosis pulmonary guidelines: airway clearance therapies. Respir Care. 2009;54:522-537.
Flume PA, Robinson KA, O’Sullivan BP, et al. Cystic fibrosis pulmonary guidelines: chronic medications for maintenance of lung health. Am J Respir Crit Care Med. 2007;176:957-969.
Garcon-Michel N, Roguedas-Contios AM, Rault G, et al. Frequency of aquagenic palmoplantar keratoderma in cystic fibrosis: a new sign of cystic fibrosis? Br J Dermatol. 2010;163:162-166.
Gild R, Clay CD, Morey S. Aquagenic wrinkling of the palms in cystic fibrosis and the cystic fibrosis carrier state: a case-control study. Br J Dermatol. 2010;163:1082-1084.
Grangeia A, Sa R, Carvalho F, et al. Molecular characterization of the cystic fibrosis transmembrane conductance regulator gene in congenital absence of the vas deferens. Genet Med. 2007;9:163-172.
Grasemann H, Stehling F, Brunar H, et al. Inhalation of Moli1901 in patients with cystic fibrosis. Chest. 2007;131:1461-1466.
Green D, Carson K, Leonard A, et al. Current treatment recommendations for correcting vitamin D deficiency in pediatric patients with cystic fibrosis are inadequate. J Pediatr. 2008;153:554-559.
Groman JD, Karczeki B, Sheridan M, et al. Phenotypic and genetic characterization of patients with features of nonclassic forms of cystic fibrosis. J Pediatr. 2005;146:675-680.
Grosse SD, Rosenfeld M, Devine OJ, et al. Potential impact of newborn screening for cystic fibrosis on child survival: a systematic review and analysis. J Pediatr. 2006;149:362-366.
Gu Y, Harley ITW, Henderson LB, et al. Identification of IFRD1 as a modifier gene for cystic fibrosis lung disease. Nature. 2009;458:1039-1042.
Hardin DS, Rice J, Ahn C, et al. Growth hormone treatment enhances nutrition and growth in children with cystic fibrosis receiving enteral nutrition. J Pediatr. 2005;146:324-328.
Harrison AN, Regelmann WE, Zirbes JM, et al. Longitudinal assessment of lung function from infancy to childhood in patients with cystic fibrosis. Pediatr Pulmonol. 2009;44:330-339.
Hewer SCL, Tyrrell K. Cystic fibrosis and the transition to adult health services. Arch Dis Child. 2008;93:817-820.
Kappler M, Griese M. Nutritional supplements in cystic fibrosis. BMJ. 2006;332:618-619.
Kerem E, Hirawat S, Armoni S, et al. Effectiveness of PTC124 treatment of cystic fibrosis caused by nonsense mutations: a prospective phase 2 trial. Lancet. 2008;372:719-727.
LeGrys VA, Yankaskas JR, Quittell LM, et al. Diagnostic sweat testing: the cystic fibrosis foundation guidelines. J Pediatr. 2007;151:85-89.
Lenaerts C, Lapierre C, Patriqui H, et al. Surveillance for cystic fibrosis-associated hepatobiliary disease: early ultrasound changes and predisposing factors. J Pediatr. 2003;143:343-350.
Levy H, Kalish LA, Cannon CL, et al. Predictors of mucoid Pseudomonas colonization in cystic fibrosis patients. Pediatr Pulmonol. 2008;43:463-467.
Li Z, Kosorok MR, Farrell PM, et al. Longitudinal development of mucoid Pseudomonas aeruginosa infection and lung disease progression in children with cystic fibrosis. JAMA. 2005;293:581-588.
Liou TG, Adler FR, Cox DR, Cahill TGI. Lung transplantation and survival in children with cystic fibrosis. N Engl J Med. 2007;357:2143-2152.
Liou TG, Rubenstein RC. Carrier screening, incidence of cystic fibrosis, and difficult decisions. JAMA. 2009;302:2595-2596.
McColley SA. Cystic fibrosis lung disease: when does it start, and how can it be prevented? J Pediatr. 2004;145:6-7.
Mishra A, Greaves R, Smith K, et al. Diagnosis of cystic fibrosis by sweat testing: age-specific reference intervals. J Pediatr. 2008;153:758-763.
Moran A, Hardin D, Rodman D, et al. Diagnosis, screening and management of cystic fibrosis related diabetes mellitus: a consensus conference report. Diabetes Res Clin Pract. 1999;45:61-73.
Moss RB. Infection, inflammation, and the downward spiral of cystic fibrosis lung disease. J Pediatr. 2009;154:162-163.
Moyer K, Balistreri W. Hepatobiliary disease in patients with cystic fibrosis. Curr Opin Gastroenterol. 2009;25:272-278.
Munck A, Houssin E, Rousey M. The importance of sweat testing for older siblings of patients with cystic fibrosis identified by newborn screening. J Pediatr. 2009;155:928-930.
O’Sullivan BP, Freedman SD. Cystic fibrosis. Lancet. 2009;373:1891-1902.
Poustie VJ, Russell JE, Watling, et al. Oral protein energy supplements for children with cystic fibrosis: CALICO multicentre randomized controlled trial. BMJ. 2006;332:632-635.
Ravzi S, Saiman L. Nontuberculous mycobacteria in cystic fibrosis. Pediatr Infect Dis J. 2007;26:263-264.
Ren CL, Pasta DJ, Rasouliyan L, et al. Relationship between inhaled corticosteroid therapy and rate of lung function decline in children with cystic fibrosis. J Pediatr. 2008;153:746-751.
Retsch-Bogart GZ, Quittner AL, Gibson RL, et al. Efficacy and safety of inhaled aztreonam lysine for airway pseudomonas in cystic fibrosis. Chest. 2009;135:1223-1232.
Ross LF. Newborn screening for cystic fibrosis: a lesson in public health disparities. J Pediatr. 2008;153:308-313.
Saiman L, Marshall BC, Mayer-Hamblett N, et al. Azithromycin in patients with cystic fibrosis chronically infected with Pseudomonas aeruginosa: a randomized controlled trial. JAMA. 2003;290:1749-1756.
Sims EJ, McCormick J, Mehta G, et al. Newborn screening for cystic fibrosis is associated with reduced treatment intensity. J Pediatr. 2005;147:306-311.
Smyth A, Tan KHV, Hyman-Taylor P, et al. Once versus three-times daily regimens of tobramycin treatment for pulmonary exacerbations of cystic fibrosis—the TOPIC study: a randomized controlled trial. Lancet. 2005;365:573-578.
Smyth RL. Daily activity and disease status in cystic fibrosis: an important area for research. J Pediatr. 2005;147:281-283.
Smyth RL. Diagnosis and management of cystic fibrosis. Arch Dis Child. 2005;90:ep1-ep6.
Southern KW, Mérelle MME, Dankert-Roelse JE, et al: Newborn screening for cystic fibrosis (review), Cochrane Database System Rev (1):CD001402, 2009.
Spultan ZN, Foster MM, Newman NB, et al. Sweat chloride testing in infants identified as heterozygote carriers by newborn screening. J Pediatr. 2008;13:857-859.
Sterescu AE, Rhodes B, Jackson R, et al. Natural history of glucose intolerance in patients with cystic fibrosis: ten-year prospective observation program. J Pediatr. 2010;156:613-617.
Stick SM, Brennan S, Murray C, et al. Bronchiectasis in infants and preschool children diagnosed with cystic fibrosis after newborn screening. J Pediatr. 2009;155:623-628.
Sugarman EA, Rohlfs EM, Silverman LM, et al. CFTR mutation distribution among U.S. Hispanic and African American individuals: evaluation in cystic fibrosis patient and carrier screening populations. Genet Med. 2004;6:392-399.
Texereau J, Marullo S, Hubert D, et al. Nitric oxide synthase 1 as a potential modifier gene of decline in lung function in patients with cystic fibrosis. Thorax. 2004;59:156-158.
Tuchman LK, Schwartz LA, Sawcki GS, et al. Cystic fibrosis and transition to adult medical care. Pediatrics. 2010;125:566-573.
Vandenbussche HL, Klepser ME. Single daily tobramycin dosing in cystic fibrosis: is it better for the patients or the bugs? Lancet. 2005;365:547-548.
Welsch MJ. Targeting the basic defect in cystic fibrosis. N Engl J Med. 2010;363(21):2056-2057.
Wilcken B. Cystic fibrosis: refining the approach to newborn screening. J Pediatr. 2009;155:605-606.
Wilschanski M, Yahav Y, Yaacov Y, et al. Gentamicin-induced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutations. N Engl J Med. 2003;349:1433-1441.
Zeitlin P. Emerging drug treatments for cystic fibrosis. Expert Opin Emerg Drugs. 2007;12:329-336.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]<
[/not-level-membership-for-pediatrics-category]
