Chapter 640 Cutaneous Defects
Skin Dimples
Sacral dimples are common and usually are isolated findings. They may be seen in multiple syndromes or in association with spina bifida occulta and diastomyelia. Association with a mass or other cutaneous stigma (hair, aplasia cutis, hemangioma) should increase concern for underlying spinal dysraphism (Chapter 585). Ultrasonography during the first 3 mo of life, before ossification of the posterior elements of the lower spine, may provide a cost-effective, noninvasive method of assessing any associated lumbosacral spine abnormalities.
Amniotic Constriction Bands
Adhesive bands involve the craniofacial area and are associated with severe defects such as encephalocele and facial clefts. Adhesive bands result from broad fusion between disrupted fetal parts and an intact amniotic membrane. The craniofacial defects appear not to be caused by constrictive amniotic bands but to result from a vascular disruption sequence with or without cephaloamniotic adhesion (Chapter 102).
Accessory Tragi
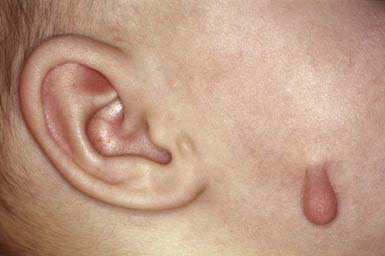
An accessory tragus typically appears as a single pedunculated, flesh-colored papule in the preauricular region anterior to the tragus. Less commonly, accessory tragi are multiple or bilateral, and may be located in the preauricular area, on the cheek along the line of the mandible (Fig. 640-1), or on the lateral aspect of the neck anterior to the sternocleidomastoid muscle. In contrast to the rest of the pinna, which develops from the 2nd branchial arch, the tragus and accessory tragi derive from the 1st branchial arch. Accessory tragi may occur as isolated defects or in chromosomal 1st branchial arch syndromes that include anomalies of the ears and face, such as cleft lip, cleft palate, and mandibular hypoplasia. An accessory tragus is consistently found in oculo-auriculo-vertebral syndrome (Goldenhar syndrome). Surgical excision is appropriate.
Branchial Cleft and Thyroglossal Cysts and Sinuses
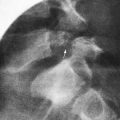
Thyroglossal cysts and fistulas are similar defects located in or near the midline of the neck; they may extend to the base of the tongue. A pathognomonic sign is vertical motion of the mass with swallowing and tongue protrusion. In nearly 50% of affected children, the cyst or fistula manifests as an infected midline upper neck mass. Cysts in the tongue base may be differentiated from an undescended lingual thyroid by radionuclide scanning. Unlike branchial cysts, a thyroglossal duct cyst often appears after an upper respiratory infection (Chapter 557).
Supernumerary Nipples
Solitary or multiple accessory nipples may occur in a unilateral or bilateral distribution along a line from the anterior axillary fold to the inguinal area. They are more common among black (3.5%) than white (0.6%) children. Accessory nipples may or may not have areolae and may be mistaken for congenital nevi. They may be excised for cosmetic reasons. Renal or urinary tract anomalies and hematologic abnormalities may occur in children with this finding (Chapter 545).
Aplasia Cutis Congenita (Congenital Absence of Skin)
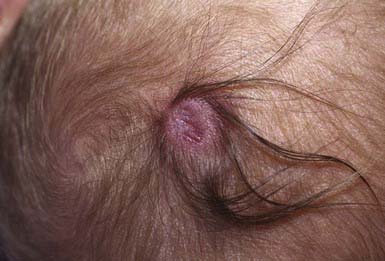
Developmental absence of skin is usually noted on the scalp as multiple or solitary (70%), noninflammatory, well-demarcated, oval or circular 1- to 2-cm ulcers. The appearance of lesions varies, depending on when they occurred during intrauterine development. Those that form early in gestation may heal before delivery and appear as atrophic, fibrotic scars with associated alopecia, whereas more recent defects may manifest as ulcerations. Most occur at the vertex just lateral to the midline, but similar defects may also occur on the face, trunk, and limbs, where they are often symmetric. The depth of the ulcer varies. Only the epidermis and upper dermis may be involved, resulting in minimal scarring or hair loss, or the defect may extend to the deep dermis, to the subcutaneous tissue, and, rarely, to the periosteum, skull, and dura. Lesions may be surrounded by a collar of hair (Fig. 640-2).
Focal Facial Ectodermal Dysplasia (Bitemporal Aplasia Cutis Congenita, Ectodermal Dysplasia of the Face)
Dyskeratosis Congenita (Zinsser-Engman-Cole Syndrome)
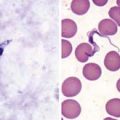
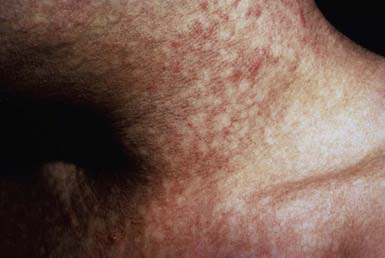
Dyskeratosis congenita, a rare familial syndrome, consists classically of the triad of reticulated hyperpigmentation of the skin (Fig. 640-3), dystrophic nails, and mucous membrane leukoplakia in association with immunologic and hematologic abnormalities. Patients with dyskeratosis congenita also show signs of premature aging and increased occurrence of cancer, especially squamous cell carcinoma. Dyskeratosis congenita may be X-linked recessive (DKC-1 gene), autosomal dominant (hTERC and TINF2 genes), or autosomal recessive (NOLA3 gene). Onset occurs in childhood, most commonly as nail dystrophy. The nails become atrophic and ridged longitudinally with progression to pterygia and complete nail loss. Skin changes usually appear after onset of nail changes and consist of reticulated gray-brown pigmentation, atrophy, and telangiectasia, especially on the neck, face, and chest. Hyperhidrosis and hyperkeratosis of the palms and soles, sparse scalp hair, and easy blistering of the hands and feet are also characteristic. Blepharitis, ectropion, and excessive tearing due to atresia of the lacrimal ducts are occasional manifestations. Oral leukokeratosis may give rise to squamous cell carcinoma. Other mucous membranes, including conjunctival, urethral, and genital, may be involved. Infection, malignancy, and bone marrow failure are common, and death before age 40 yr is typical (Chapter 462).
Belliard C, Guillet G, Vabres P, et al. Bi-acromial dimples: a series of seven cases. Pediatr Dermatol. 2005;22:412-414.
Ferrara P, Giorgio V, Vitelli O, et al. Still a marker of urinary tract anomalies in children? Scand J Urol Nephrol. 2008;3:1-4.
Kirwan M, Dokal I. Dyskeratosis congenita: a genetic disorder of many faces. Clin Genet. 2008;73:103-112.
Krathen MS, Rosenback M, Yan AC, et al. Focal preauricular dermal dysplasia: report of 2 cases and a review of the literature. Pediatr Dermatol. 2008;25:344-348.
Turkyilmaz Z, Karabulut R, Bayazit YA, et al. Congenital neck masses in children and their embryologic and clinical features. B-ENT. 2008;4:7-18.