Cushing’s Syndrome
Harvey Cushing1,2 was the first to codify the symptom complex of obesity, diabetes, hirsutism, and adrenal hyperplasia, and to postulate that the basophilic adenomas found at autopsy in six of eight patients caused the disease that now bears his name. Shortly thereafter, Walters and colleagues3 identified the etiologic contribution of adrenal tumors and the therapeutic role of adrenalectomy. Over the ensuing century, our understanding of the pathogenesis of Cushing’s syndrome has expanded to include ectopic production of adrenocorticotropic hormone (ACTH)4 and corticotropin-releasing hormone (CRH),5 and recognition of bilateral adrenal stimulation by factors other than ACTH.6–9 Because florid Cushing’s syndrome is ultimately fatal, early diagnosis and treatment have always been important. A plethora of tests have been developed over the years to improve the diagnostic yield. Similarly, the treatment options for Cushing’s syndrome have increased to include medical agents that decrease the secretion or block the activity of circulating cortisol and surgical resection of eutopic and ectopic ACTH-producing tumors. Despite all these advances, Cushing’s syndrome continues to tax endocrinologists and is likely to continue to do so. This chapter reviews the manifestations, causes, approaches to diagnosis, and treatments for this complicated and multifaceted syndrome.
Etiology and Pathophysiology
The causes of Cushing’s syndrome can be divided into those that are ACTH dependent and those that are ACTH independent (Table 8-1). The ACTH-dependent forms are characterized by excessive ACTH production from a corticotroph adenoma (known as pituitary-dependent Cushing’s syndrome or Cushing’s disease), from an ectopic tumoral source (ectopic ACTH syndrome), or (rarely) from normal corticotrophs under the influence of excessive CRH production (ectopic CRH secretion). ACTH stimulates all three layers of the adrenal cortex to grow and secrete steroids. When excessive, this results in histologic hyperplasia and increased adrenal weight. Micronodules and macronodules (>1 cm) may be seen. Circulating glucocorticoids are increased, often in association with some increase in adrenal androgens.
Cushing’s Disease
Cushing’s disease is almost always caused by a solitary (probably monoclonal) corticotroph adenoma.10 Although nodular corticotroph hyperplasia without evidence of a CRH-producing neoplasm does occur, it represents 2% or less of large surgical series,11,12 and some doubt its existence. Most tumors are intrasellar microadenomas (<1 cm in diameter), although macroadenomas account for approximately 5% to 10% of tumors, and extrasellar extension or invasion may occur. The cause of Cushing’s disease remains unknown, despite much work on the molecular characterization of these tumors. Traditionally, whether the development of pituitary adenomas is due to abnormal hypothalamic hormonal stimulation or feedback regulation or an intrinsic pituitary defect has been the subject of debate, although most data support a primary pituitary abnormality or a series of abnormalities. More recently, a model has been proposed that encompasses both theories. Here, tumors can arise either as a clonal expansion from a primary intrinsic pituitary defect or as excessive hormonal stimulation/abnormal feedback leading to hyperplasia, which in turn predisposes the cells to mutate, with subsequent clonal expansion.13 Analysis of the primary corticotroph stimulatory and negative feedback pathways has not revealed a common defect.14,15 Similarly, the common oncogenes and tumor suppressor genes implicated in other cancers do not seem to be commonly involved in the pathogenesis of corticotropinomas. Studies of knockout mice and analysis of human pituitary tumor samples have implicated the cyclin-dependent kinase inhibitor p27 (Kip1) in corticotroph tumorigenesis. Overall, reduced p27 protein levels in corticotropinomas and a high phosphorylated p27/p27 ratio suggest increased inactivation of this negative cell-cycle regulator, although the cause of this change remains to be elucidated.16 Cytogenetic studies have revealed a surprising number of gross chromosomal changes in benign pituitary adenomas, and although the number of corticotroph tumors studied has been small, gain of chromosome 6p and loss of chromosomes 2, 15q, and 22 seem to be the most common abnormalities.17–19 Perhaps improvement in molecular biologic techniques, particularly microarray analysis, will lead to the implication of new genes in the pathogenesis of these tumors that then will require further study.20
Ectopic Adrenocorticotropic Hormone Syndrome
The syndrome of ectopic hormone secretion was first codified by Liddle and colleagues, who defined it as “any hormone produced by a neoplasm which is derived from tissue not normally engaged in the production of the hormone in question.”4 ACTH and other pro-opiomelanocortin (POMC) products were subsequently identified in many noncorticotroph tumors, although not all were associated with increased circulating levels or the development of Cushing’s syndrome.4,21
Although small cell lung cancer is probably the most common cause of ectopic ACTH syndrome, it is not the most common seen in larger series of generally less obvious tumors investigated at endocrine centers, as discussed later (Table 8-2). An intrathoracic neoplasm (carcinoma of the lung or carcinoid of the bronchus or thymus) accounts for approximately 60% of ectopic ACTH secretion, followed by pancreatic tumors (islet cell or carcinoid), pheochromocytoma (≈5% to 10%), and medullary carcinoma of the thyroid (<5%).
Table 8-2
Percentage Incidence of Tumor Types Causing Ectopic Adrenocorticotropic Hormone Syndrome in Four Large Series from 1969 to 2003

*Other tumors reported to uncommonly secrete adrenocorticotropic hormone include appendix, breast, cloacogenic carcinoma of the anal canal, colon, esophagus, gallbladder, gastric carcinoid, kidney, melanoma, mesothelioma, myeloblastic leukemia, ovary, prostate, salivary glands, and testes.
The mechanism whereby the POMC gene becomes derepressed in noncorticotroph tumors is not understood. One hypothesis is that these cells are derived from a common multipotential progenitor cell capable of producing peptide hormones, such that ACTH production is a reversion to a less differentiated state.22 The speculation that many ACTH-producing tumors are derived from neural crest amine precursor uptake and decarboxylation (APUD) cells may support this view,23 although this embryological hypothesis is not supported by the most recent data. However, because endodermally derived tumors also produce ACTH, the acquisition of APUD characteristics may be but one manifestation of dedifferentiation and may not represent the cause of ectopic ACTH production.
Although the mechanism of gene derepression is not understood, the regulation of POMC production and processing has been investigated. POMC, corticotropin-like intermediate lobe protein, and larger forms of ACTH (“big” or pro-ACTH) that are not usually secreted may circulate, and the intracellular ratio of the POMC products may be abnormal.24,25 Investigation of cell lines of small cell carcinoma of the lung that synthesize POMC and pro-ACTH showed that only ACTH precursors were secreted, suggesting that processing to ACTH is defective.26 The pattern of POMC mRNA species in ACTH-producing tumors has been characterized. A 1200 bp transcript similar to that of a corticotroph adenoma,27 a shorter than normal 800 bp mRNA lacking a signal sequence for secretion,27,28 and a larger 1400 to 1500 bp POMC transcript have been identified. The larger species appears to originate upstream of the usual pituitary promoter, with preservation of the normal translation start site.29,30 It is possible that the promoters that initiate this transcription are not regulated by glucocorticoids, and this may explain in part the lack of responsiveness to glucocorticoid suppression noted clinically in these patients. In vitro investigation of human small cell cancer cell lines and pancreatic islet cell tumors with normal glucocorticoid receptor binding has found, for the most part, no regulation of POMC, tyrosine aminotransferase, or the glucocorticoid receptor mRNA at doses of hydrocortisone that would normally suppress pituitary production.31–33 However, clinical observation of suppression of ACTH production by some bronchial carcinoids during glucocorticoid administration suggests retention of a functional glucocorticoid response element that regulates POMC production, at least in some ectopic tumors.34
Ectopic Corticotropin-Releasing Hormone Secretion
Tumor secretion of CRH with or without ACTH secretion is a rare cause of Cushing’s syndrome. Although many tumors immunostain for CRH, its secretion is less common, and most patients do not develop cushingoid features.35 Thus, the diagnosis primarily rests on the demonstration of elevated plasma CRH levels (requiring an assay that is not readily available). The literature includes fewer than 20 patients who fit this criterion. Tumors may have negative immunostaining for ACTH, but this may be related to reduced storage and rapid secretion. In cases such as these, a CRH and ACTH gradient across the tumor bed can be suggestive that, in fact, the tumor secretes both peptides.36 Tumors include bronchial and thymic carcinoids, small cell lung cancer, medullary thyroid carcinoma, pheochromocytoma, gangliocytoma, prostate carcinoma, and ganglioneuroblastoma.37,38 The biochemical responses to diagnostic tests can be similar to those seen in ectopic ACTH secretion or in pituitary ACTH-dependent disease.38 It is important to note that many, if not all, ectopic secretors of CRH causing Cushing’s syndrome are also ectopic ACTH secretors.
Primary Adrenal Disease
The primary adrenal forms of Cushing’s syndrome do not share a common cause. Although the cause of adrenocortical neoplasia is not known, some events important in the development of adrenal cancer have been identified. Paternal isodisomy at 11p15.5 with overexpression of insulin-like growth factor-2 (IGF-2) and reduced expression of CDKN1C (a G1 cyclin-dependent kinase inhibitor) and H19 (a putative growth suppressor) seems to be a key event. Mutations of p53 may be involved in a small subset of carcinomas, and mutations of β-catenin may be an early event. Other genes important in pathogenesis remain to be elucidated, although potential loci have been identified at chromosomes 17p, 1p, 2p16, and 11q13 for tumor suppressor genes, and at chromosomes 4, 5, and 12 for oncogenes.39 Adenomas and carcinomas tend to be monoclonal, although the nodular hyperplasias are often polyclonal.40 Adrenal adenomas are encapsulated benign tumors, usually less than 40 g in weight. Adrenal carcinomas usually are encapsulated, generally weigh more than 100 g, and may lack classic histologic features of malignancy, although nuclear pleomorphism, necrosis, mitotic figures, and vascular or lymphatic invasion suggest the diagnosis.41 The adjacent adrenal tissue is atrophic in both conditions.
Primary pigmented nodular adrenal disease (PPNAD), also known as micronodular adrenal disease, is a rare form of Cushing’s syndrome characterized histologically by small to normal-size glands (combined weight <12 g) with cortical micronodules (average 2 to 3 mm) that may be dark or black in color. The intervening cortex is usually atrophic.42 Most cases of PPNAD occur as part of the Carney complex in association with a variety of other abnormalities, including myxomatous masses of the heart, skin, or breast; blue nevi or lentigines; and other endocrine disorders (sexual precocity; Sertoli cell, Leydig cell, or adrenal rest tumors; acromegaly). The Carney complex is inherited as an autosomal dominant condition, and Cushing’s syndrome occurs in approximately 30% of cases. The tumor suppressor gene PRKAR1A, coding for the type 1A regulatory subunit of protein kinase A, has been shown to be mutated in approximately one half of patients with Carney complex. Mutations in this gene and also the phosphodiesterase 11A (PDE11A) gene have been shown to be associated with an isolated distinct form of PPNAD.43
Cushing’s syndrome resulting from bilateral nodular adrenal disease is an uncommon feature of the McCune-Albright syndrome,44 which is characterized by fibrous dysplasia of bone, café-au-lait skin pigmentation, and endocrine dysfunction (usually precocious puberty). In this disease, an activating mutation at codon 201 of the α subunit of the G protein that stimulates cyclic adenosine monophosphate formation occurs in a mosaic pattern in early embryogenesis.45 If this affects some adrenal cells, constitutive activation of adenylate cyclase and the steroidogenic cascade leads to nodule formation and glucocorticoid excess. The internodular adrenal cortex, where the mutation is not present, becomes atrophic.46
A missense mutation of the ACTH receptor, resulting in its constitutive activation and ACTH-independent Cushing’s syndrome, also has been reported.47
ACTH-independent bilateral macronodular adrenal hyperplasia (AIMAH) is a rare form (<1%) of Cushing’s syndrome that involves large or even huge adrenal glands, usually with definite nodules on imaging. Most cases are sporadic, but a few familial cases have been reported.48 Although the cause remains unclear in most cases, some nodules express increased numbers of receptors normally found on the adrenal gland, or ectopic receptors for circulating ligands that then can stimulate cortisol production. Perhaps the best known example of this phenomenon is food-dependent Cushing’s syndrome. The normal postprandial increase in gastric inhibitory peptide (GIP) appeared to cause Cushing’s syndrome in two middle-aged women with bilateral multinodular adrenal enlargement, mildly elevated urinary free cortisol (UFC) values, and undetectable plasma ACTH values. Fasting morning serum cortisol values were low or normal. Cortisol values increased dramatically after meals and after in vivo or in vitro exposure to GIP.7,8 In one patient, curative bilateral adrenalectomy revealed multinodular adrenal glands weighing 20 and 35 g.8 In the other, treatment with octreotide ameliorated the syndrome.7 Ectopic expression of GIP receptors was found in these patients. Aberrant expression of vasopressin, β-adrenergic luteinizing hormone/human chorionic gonadotropin, serotonin, angiotensin, leptin, glucagon, interleukin (IL)-1, and thyroid-stimulating hormone (TSH) has been described as functionally linked to cortisol production.49 However, it is possible that this apparent ectopic induction of receptors on the adrenal is a response to the adrenal hyperplasia rather than its cause.
Adrenal rest tissue in the liver, in the adrenal beds, or in association with the gonads may rarely cause Cushing’s syndrome, usually in the setting of ACTH-dependent disease after adrenalectomy.50–53 Ectopic cortisol production by an ovarian carcinoma has been reported.54
PSEUDO-CUSHING’S STATES
A pseudo-Cushing’s state may be defined as one in which some or all of the clinical features that resemble true Cushing’s syndrome, and some evidence of hypercortisolism, are present but disappear after resolution of the underlying condition.55 The pathophysiology of these states has not been established. One hypothesis is that these stressful conditions increase the activity of the CRH neuron, resulting in excessive ACTH secretion, adrenal hyperplasia, and increased cortisol production.56 The model predicts only intermittent and modest hypercortisolism because of appropriate corticotroph reduction in ACTH secretion in response to negative feedback by cortisol (Fig. 8-1). This construct presumes also that hypertrophied adrenal glands produce excessive glucocorticoids in response to normal ACTH levels, an assumption that is supported by the blunted ACTH, but not cortisol, response to exogenous CRH in anorexia nervosa,57 depression,58 and obligate athleticism.59
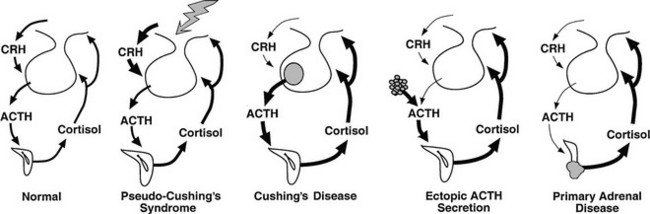
FIGURE 8-1 Physiology of the hypothalamic-pituitary-adrenal axis in normal individuals and hypercortisolemic states. Corticotropin-releasing hormone (CRH) secretion from the hypothalamus normally stimulates adrenocorticotropic hormone (ACTH) secretion from the pituitary gland. This in turn results in increased cortisol production from the adrenal glands. The system is modulated by negative feedback inhibition by cortisol of both CRH and ACTH secretion. In pseudo-Cushing’s syndrome, the CRH neuron is activated by central input (large shaded arrow), resulting in increased CRH output that eventuates in hypercortisolism. Increased cortisol production restrains corticotroph activation but does not completely reverse the activation of the CRH neuron, so that mild to moderate hypercortisolism may persist. In Cushing’s disease, a corticotroph adenoma secretes ACTH in excess and is inhibited only partially by rising cortisol levels. In this setting and that of ectopic ACTH secretion and primary adrenal disease, the CRH neuron is suppressed by hypercortisolism. In ectopic ACTH secretion, excessive secretion of ACTH from a nonpituitary tumor is not inhibited by glucocorticoid feedback. In this setting and that of autonomous production of cortisol by the adrenal gland, ACTH secretion by normal corticotrophs is suppressed by hypercortisolism.
Epidemiology
Iatrogenic causes account for most cases of Cushing’s syndrome because of the common therapeutic use of high-dose glucocorticoids. Large series have reported the distribution of endogenous cases as follows: Cushing’s disease (68%), adrenal adenomas (8% to 19%), adrenal carcinoma (6% to 7%), ectopic ACTH syndrome (6% to 15%), and nodular adrenal hyperplasia (2%).55,60 However, a paucity of information is available on the true incidence of these causes. Perhaps the best data come from a population-based study covering the whole of Denmark (population of 5.3 million), which used stringent methods of data collection, aided by the small number of centers treating the disorder.61 The incidences of Cushing’s disease, adrenal adenoma, and adrenal carcinoma were 1.2 per million per year, 0.6 per million per year, and 0.2 per million per year, respectively. The reported incidence of ectopic ACTH syndrome was extremely low (0.1 per million per year). This is probably due (as the authors concede) to the fact that many cases were never recognized, but may be explained in part by a group of patients with ACTH-dependent Cushing’s syndrome (0.5 per million per year) with presumed but unproven pituitary disease. Some of these may well have had ectopic ACTH syndrome. The incidence of ectopic ACTH syndrome most certainly is underestimated in the endocrine literature because most cases reaching endocrinologists are those caused by occult tumors as opposed to those caused by overt malignancy. However, given that Cushing’s syndrome will be present in 3% to 12% of cases of small cell lung cancer,62,63 and that the recent incidence of small cell lung cancer in Europe is approximately 120 per million per year in men and 40 per million per year in women,64 this is by far the most common cause. Other epidemiologic studies have looked at just the incidence of Cushing’s disease and have found rates between 0.7 per million per year in northern Italy65 and 2.4 per million per year in northern Spain.66
Gender and age distribution varies with the cause of Cushing’s syndrome. Adrenal adenomas and Cushing’s disease present much more commonly in women than in men, and adrenal carcinoma is approximately 1.5 times as common as in men.55,60 Nodular adrenal hyperplasia has an approximately equal gender ratio.
Ectopic ACTH syndrome is the only cause of the syndrome that is more common in men (other than Cushing’s disease in prepubertal children), although this may change as more women are developing small cell lung cancer. Lung cancer is more common after age 40, and this accounts for the increased mean age of patients with ectopic ACTH syndrome compared with Cushing’s disease, which occurs between 25 and 40 years of age.67 The other major cause of ectopic ACTH secretion, intrathoracic carcinoids, has a peak incidence around 40 years and only a slightly increased male-to-female ratio.68 The age distribution of adrenal cancer is bimodal, with peaks in childhood and adolescence and late in life, although adrenal adenoma occurs most often around 35 years of age.
Clinical Features
Excessive cortisol production has widespread systemic effects67,69–72 (Table 8-3). Although the full-blown cushingoid phenotype is unmistakable, the clinical diagnosis may be equivocal for patients with few of the typical characteristics (Fig. 8-2). Some nonspecific features consistent with the diagnosis of Cushing’s syndrome, such as obesity, hypertension, and menstrual irregularity, are common in the general population and may provoke unwarranted and costly screening tests for patients not likely to be affected.
Table 8-3
Percentage Frequency of Clinical Signs and Symptoms of Cushing’s Syndrome as Described in Six Large Studies from 1952 to 2003
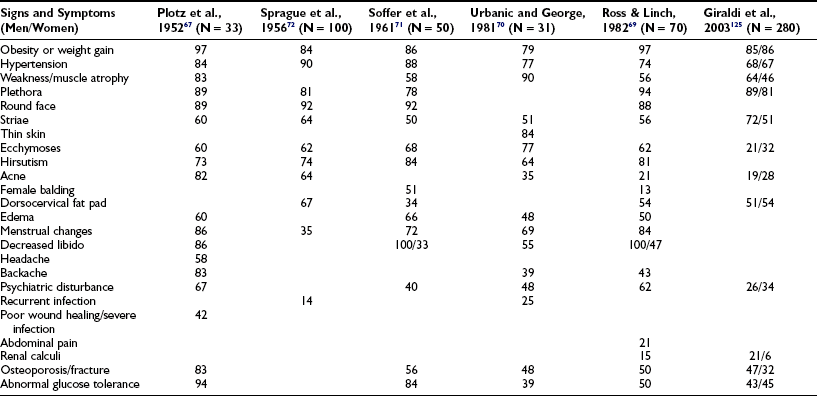
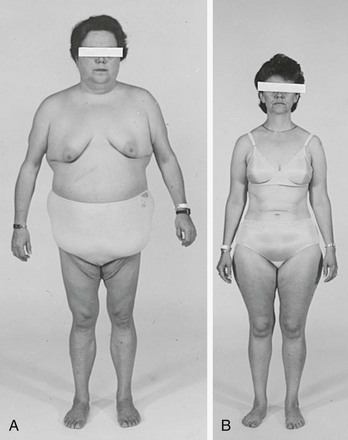
FIGURE 8-2 Body habitus of two patients with proven Cushing’s syndrome. Features typical of the syndrome—central obesity, round face, and supraclavicular fat pads—are present in the patient in A, but not in the patient in B, illustrating that the diagnosis is not always apparent from the initial physical examination.
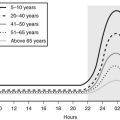
One useful strategy when the diagnosis of Cushing’s syndrome is considered, is to look for evidence of progressive physical changes by examination of serial photographs, especially of individuals photographed at annual events such as holidays, birthdays, or school milestones (Fig. 8-3). Another approach relies on identification of signs and symptoms that correctly classify patients suspected of having the disorder. Truncal obesity, ecchymoses, plethora, proximal muscle weakness, and osteopenia are useful discriminant indices for Cushing’s syndrome, with osteoporosis, ecchymoses, and muscle weakness being the most reliable.69,73,74
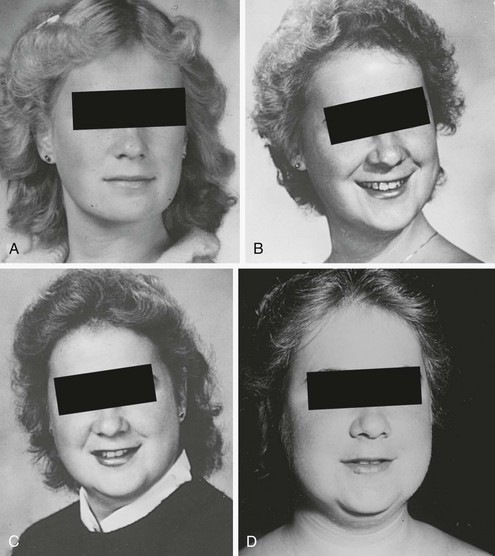
FIGURE 8-3 Progression of cushingoid features as shown in photographs taken at 1-year intervals (A through D, progress from earliest to latest).
Increased deposition of fat, one of the earliest signs, occurs in almost all patients and is reported as increasing weight or difficulty in maintaining weight. The distribution of fat is altered in both men and women, with increased amounts in the visceral compartments75 and subcutaneous sites on the face and neck. Increased intra-abdominal fat results in the truncal obesity described by Cushing in approximately 50% of patients. Increased fat in the face (moon facies), the supraclavicular or temporal fossae, and the dorsocervical area (“buffalo hump”) is uncommon in normal people. When extreme, the supraclavicular fat may present as a “collar” rising above the clavicles (Fig. 8-4); filling of the temporal fossae may prevent eyeglass frames from seating properly. Abnormal fat deposition may occur in the epidural space. Spinal epidural lipomatosis causing neurologic deficit, a rare complication of long-term exogenous steroid use, has been reported in a few patients with endogenous Cushing’s syndrome.76,77 Lumbosacral findings were seen in both men and women, whereas thoracic obstruction was restricted to men. The condition can be diagnosed by magnetic resonance imaging (MRI).78
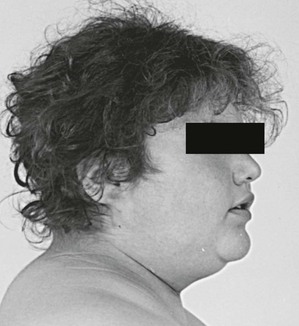
FIGURE 8-4 Fat may fill or, in this case, rise above the supraclavicular fossa of patients with Cushing’s syndrome.
Loss of subcutaneous tissue results in a variety of skin abnormalities that are unusual in the general population and suggest hypercortisolism. Ecchymoses, often after minimal trauma, and cutaneous atrophy, seen as a fine “cigarette paper” wrinkling or tenting over the dorsum of the hand and elbows, are typical. Cutaneous atrophy is influenced by gender and age, with men and the young having greater skin thickness. Two maxims follow: First, it is useful to compare the patient’s skin with that of a near age- and gender-matched healthy person; and second, skin thickness is relatively preserved in cushingoid women with increased androgen production or preservation of ovarian function (Fig. 8-5).

Purple striae more than 1 cm in diameter are virtually pathognomonic for Cushing’s syndrome (Fig. 8-6). Although the silvery, healed striae that are typical postpartum are not caused by active Cushing’s syndrome, other pink, less pigmented, and thinner striae may be seen. Although most common over the abdomen, striae occur also over the hips, buttocks, thighs, breasts, and upper arms. The tear in the subcutaneous tissue may be best appreciated by indirect (side) lighting, which throws the striae into relief, or by light stroking of the skin. The violaceous hue is not dependent on ACTH-dependent pigmentation and may be seen in Cushing’s syndrome in association with primary adrenal causes.
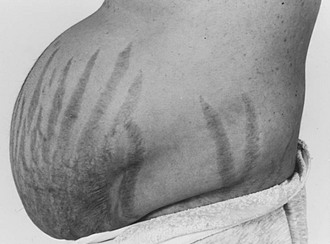
FIGURE 8-6 Typical abdominal striae of a patient with hypercortisolism. These are greater than 1 cm in width and are violaceous.
Proximal muscle weakness with preservation of distal strength is a hallmark of Cushing’s syndrome. Histologically, this is reflected in profound atrophy of fibers without necrosis.79–81 Weakness is best assessed historically by questions related to the use of these muscles: Is there difficulty or weakness in climbing stairs, getting up from a chair or bed without using hand propulsion, or performing activities using the shoulders (e.g., brushing hair, reaching objects in overhead cabinets, changing ceiling light bulbs)? Formal muscle testing is useful. Assess the strength of the hip flexors by asking the patient to get out of a chair without using his or her arms. If this can be done, the patient is asked to rise from a squat. Inability to perform either task, in the absence of hip or lower extremity arthropathy or other myopathic processes, is suggestive of Cushing’s syndrome. Leg extension while seated is a quantifiable test of proximal muscle strength. The number of seconds for which this position is held can be used to judge deterioration or progress after treatment.
Osteopenia is common. A history of fractures, typically of the feet, ribs, or vertebrae, may be one of the only signs of Cushing’s syndrome, especially in men.70,71,82 Avascular necrosis of bone, a rare complication of endogenous hypercortisolism, is more common in iatrogenic hypercortisolism.83,84 It usually occurs in the hips, but we have also seen it in the knees.
Vellous hypertrichosis of the forehead or upper cheeks distinguishes Cushing’s syndrome from the more common causes of hirsutism and may be appreciated only by careful visual and tactile inspection (Fig. 8-7). Excessive terminal hair on the face and body, and acne—pustular, reflecting increased androgens, or papular, reflecting pure glucocorticoid excess—may be present.85 Severe hirsutism and virilization are uncommon and suggest adrenal carcinoma.
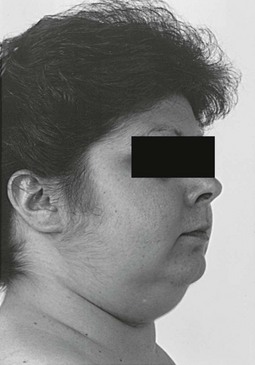
FIGURE 8-7 Vellous hirsutism, especially on the cheeks, is often present in women with Cushing’s syndrome.
Most patients experience emotional and cognitive changes (including increased fatigue, irritability, crying, and restlessness, depressed mood, decreased libido, insomnia, anxiety, impaired memory, concentration, and verbal communication) and changes in appetite. These changes correlate with the degree of hypercortisolism.86 Irritability, characterized as a decreased threshold for uncontrollable verbal outbursts, may be one of the earliest symptoms. Global impairment in neuropsychological function correlates well with the performance of seven serial subtractions and recall of the names of three cities—bedside tests that can be used by the clinician to quantify this symptom complex.87 Approximately 80% of patients meet strict criteria for a major affective disorder—50% with unipolar depression and 30% with bipolar illness.88,89 Although the quality of the depressed mood ranges from suicide attempts to sadness, the time course is characteristically intermittent, rarely lasting longer than 3 days, in contrast to the constant dysphoria reported by depressed patients without Cushing’s syndrome.86 A minority of patients are manic. The improvement in neuropsychiatric findings after treatment of Cushing’s syndrome, coupled with similar features in patients treated with exogenous steroids, and the association of hypercortisolism with poor cognitive performance in depressed patients suggest glucocorticoid excess as a cause.90,91
Hypertension is present in approximately 80% of patients, and although hypertension is also common in the general population, its presence in patients younger than 40 years of age, especially if difficult to control, may alert one to the syndrome. Hypertension usually resolves with treatment of the Cushing’s syndrome but may persist, possibly as the result of microvessel remodeling and/or underlying essential hypertension.92
Patients with marked hypercortisolism (plasma cortisol >43 µg/dL [1200 nmol/L], UFC >2000 µg/day [5520 nmol/day]) are at risk for two potentially catastrophic events: perforation of the viscera and severe infection, either bacterial or opportunistic, such as Pneumocystis carinii, aspergillosis, nocardiosis, cryptococcosis, histoplasmosis, and Candida.93–95 Classic clinical signs, such as loss of bowel sounds and fever, may be absent in peritonitis, and the typical leukocytosis of hypercortisolism may not increase further. Thus, the threshold of suspicion for opportunistic infection and a surgical abdomen must be low in patients with severe hypercortisolism.
Libido is decreased uniformly in men and to a lesser extent (44%) in women,70 in whom increased libido may indicate excess androgen production by an adrenocortical carcinoma. Menstrual irregularities, amenorrhea, and infertility are common and may be the presenting complaints.96 Impotence is common.
Pathology
The cardinal laboratory findings in endogenous Cushing’s syndrome reflect overproduction of glucocorticoids. Although morning plasma cortisol values may be normal, an increased nighttime nadir blunts or obliterates the normal diurnal rhythm.97–99 This increase in mean 24-hour plasma values is reflected in increased levels of free, or unbound, cortisol in urine100 and saliva.101 The capacity of corticosteroid-binding globulin for cortisol is exceeded at a serum cortisol value of approximately 20 µg/dL (≈600 nmol/L). At this point, the excretion of free cortisol increases dramatically in direct proportion to the increased unbound circulating cortisol values.
Hypokalemic metabolic alkalosis usually is observed when daily urine cortisol excretion is greater than 1500 µg (4100 nmol), and thus mainly in cases of ectopic ACTH syndrome.102 This probably represents a mineralocorticoid action of cortisol at the renal tubule due to saturation of the enzyme 11β-hydroxysteroid dehydrogenase type 2, which inactivates cortisol to cortisone.103 However, although a common feature of ectopic ACTH secretion, it also may occur in approximately 10% of patients with Cushing’s disease. Serum albumin is inversely correlated with cortisol levels, but this is of clinical significance only at very high cortisol levels, and it reverses with treatment for Cushing’s syndrome.104 Drastic reductions in serum albumin should alert the physician to the possibility of concomitant pathology such as infection. Circulating elevated glucocorticoids increase clotting factors, including factor VIII, fibrinogen, and von Willebrand factor, and reduce fibrinolytic activity, resulting in a fourfold risk of thrombotic events.105–107 Lipid abnormalities show increases in very–low density lipoprotein, low-density lipoprotein, high-density lipoprotein, and consequently total cholesterol and triglycerides. These changes probably are caused by a direct cortisol effect of increased hepatic synthesis of very low–density lipoprotein without altered clearance.108,109
Cushing’s syndrome is characterized by insulin resistance and hyperinsulinemia, with frank diabetes mellitus occurring in 30% to 40% of patients, and glucose intolerance in a further 20% to 30%.110,111 A recent study has suggested that as many as 2% of overweight, poorly controlled patients with diabetes may have occult Cushing’s syndrome if fully investigated.112 In the absence of clinical suspicion, the yield is probably lower.113
Patients with Cushing’s disease show accelerated cardiovascular disease, including increased carotid artery intima-media thickness and atherosclerotic plaques on Doppler ultrasonography.114 This increased risk is maintained even as long as 5 years after cure of the hypercortisolemia is attained.115 It also is likely that glucocorticoids have a direct pathogenic effect on the myocardium.
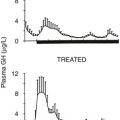
Hypercortisolism suppresses the thyroidal, gonadal, and growth hormone axes. Thyrotropin-releasing hormone and thyroid-stimulating hormone release is disturbed, and particularly the nocturnal surge of thyroid-stimulating hormone is lost, resulting in reduced total thyroxine, total triiodothyronine, and free triiodothyronine levels compared with controls.116 Others have found no differences in free thyroxine or free triiodothyronine levels but have shown a significantly increased prevalence of autoimmune thyroid disease in patients treated for Cushing’s syndrome.117,118 In both men and women, low levels of luteinizing hormone, follicle-stimulating hormone, and gonadal steroids consistent with hypogonadotropic hypogonadism are common and correlate with the degree of hypercortisolemia.119,120 In addition, the coexistence of polycystic ovarian syndrome in Cushing’s syndrome may be more common than was previously thought.96 Hypercortisolemia causes reduced growth hormone (GH) secretion during sleep and blunted GH response to stimulation tests.121
The prevalence of osteoporosis as assessed by dual-energy x-ray absorptiometry is approximately 50% in adult Cushing’s syndrome.122 It appears more common in adrenal Cushing’s syndrome than in Cushing’s disease, and this may relate to the protective effect of increased adrenal androgens in the latter.123
The accentuated visceral fat distribution characteristic of Cushing’s syndrome can be marked when visualized by computed tomography (CT),75 and the liver frequently (20%) is steatotic on imaging.124
Clinical Spectrum
The typical patient with Cushing’s disease presents at midlife complaining of the gradual development of symptoms, although males tend to present at an earlier age and with more severe clinical consequences.125 Hypokalemia, virilization, and extremely high cortisol excretion (>10-fold normal) are distinctly uncommon and should alert the physician to an alternative cause. The clinical presentation of pituitary corticotroph macroadenomas, apart from visual field changes caused by suprasellar expansion, is not unique. By contrast, invasive pituitary adenomas present at a slightly younger age; cavernous sinus and dural involvement may result in cranial neuropathies and facial neuralgia.126,127 Only a few case reports attest to cerebrospinal or extracranial metastasis of ACTH-producing pituitary tumors.128
An abrupt onset of severe Cushing’s syndrome should prompt an evaluation for ectopic ACTH secretion. This variant of ectopic ACTH secretion classically presents as a paraneoplastic syndrome in the context of a known malignancy. The features were captured in the initial formulation of Liddle4: weight loss, hypokalemia, weakness, and diabetes. However, Cushing’s syndrome caused by less obvious ectopic ACTH secretion often presents in the more classic way with weight gain and striae and can be difficult to differentiate clinically from Cushing’s disease. It is patients with this syndrome who most often present a diagnostic dilemma. They tend to have UFC excretion in the range seen in pituitary disease and may not show hypokalemia, hyperpigmentation, or the other findings typical of severe classical ectopic ACTH secretion.
Diagnosis and Differential Diagnosis
The diagnosis of Cushing’s syndrome rests on the demonstration of both physical and biochemical features of glucocorticoid excess. Thus, the diagnosis is unequivocal in a typical patient, with many of the physical features discussed earlier in the setting of UFC levels more than fourfold above normal.129 However, many of the signs of hypercortisolism, such as obesity, hypertension, glucose intolerance, mood changes, menstrual irregularity, and hirsutism, are common in the general population. Similarly, mild glucocorticoid excess is seen in affective disorders,130 strenuous exercise,59 alcoholism and alcohol withdrawal states,131 renal failure,132 and hypoglycemia. Diagnostic strategies for distinguishing between these pseudo-Cushing’s states and true Cushing’s syndrome are discussed later.
Glucocorticoid resistance is characterized by an abnormal glucocorticoid receptor number or binding, which causes compensatory increases in ACTH and excessive glucocorticoid production to maintain normal glucocorticoid-mediated effects at the target tissues. The diagnosis should be considered in the hypokalemic, hypertensive, hypercortisolemic patient without typical glucocorticoid-mediated signs of Cushing’s syndrome.133
ESTABLISHING THE DIAGNOSIS OF CUSHING’S SYNDROME
When a careful history and physical examination reveal clinical features that could be consistent with the syndrome, exogenous glucocorticoid use must be excluded (Table 8-4). In addition to inquiring about the use of oral, rectal, inhaled, injected, or topical glucocorticoid administration, it is important to evaluate the use of “tonics,” herbs, and skin bleaching creams, which may contain glucocorticoids. In the absence of exogenous glucocorticoids, biochemical confirmation of the diagnosis of Cushing’s syndrome is needed. It is important to remember that the urgency for diagnosis and treatment of Cushing’s syndrome is greatest when the symptoms are severe. In milder cases, the patient may be best served by waiting until the diagnosis is clear. Periodic reevaluation with urine screening tests and documentation of body habitus with photographs may reveal progression.
Table 8-4
Evaluation of Suspected Cushing’s Syndrome
History
Growth retardation in children
Impotence/irregular or no menses
Emotional, cognitive, mood changes (fatigue, irritability, anxiety, insomnia, depression, impaired memory and concentration)
Examination
Fat distribution (centripetal obesity; rounded face; dorsocervical, supraclavicular, temporal fat pads)
Laboratory Findings
First-Line Screening Tests
Elevated 24-hour urinary free cortisol (three collections)
Additional Screening Tests (if required)
CRH, Corticotropin-releasing hormone; LDDST, low-dose dexamethasone suppression test.
Initial Screening Tests
Hypercortisolemia, demonstrated by loss of the normal circadian rhythm of cortisol secretion, and disturbed feedback of the hypothalamic-pituitary-adrenal (HPA) axis are the cardinal biochemical features of Cushing’s syndrome. Tests to confirm the diagnosis are based on these principles. To screen for Cushing’s syndrome, tests of high sensitivity should be used initially to avoid missing milder cases. All of these screening tests may miss identification of mild cases of hypercortisolemia, and multiple samples or a combination of tests may be needed. A recent guideline suggests that two abnormal first-line test results should be required for the diagnosis of Cushing’s syndrome.134
Urinary Free Cortisol
Under normal conditions, 10% of plasma cortisol is free or unbound and physiologically active. Unbound cortisol is filtered by the kidney, with most being reabsorbed in the tubules and the remainder excreted unchanged. Thus, 24-hour UFC collection produces an integrated measure of serum cortisol, smoothing out variations in cortisol during the day. UFC determinations first became clinically available in 1968135 and have superseded the historical measurement of urinary metabolites of glucocorticoids and androgens (17-hydroxycorticosteroids [17-OHCS], 17-ketosteroids, and 17-ketogenic steroids).
The major drawback of the test is the potential for overcollection or undercollection of the 24-hour specimen, and written instructions must be given to the patient. In addition, creatinine excretion in the collection can be measured to assess completeness and should equal approximately 1 g per 24 hours in a 70 kg patient (variations depend on muscle mass). This value should not vary by more than 10% between collections in the same individual.136 It cannot be used to correct for incomplete collection, however, because rates of cortisol and creatinine excretion are not parallel over the 24-hour period. Various groups have tried to overcome the collection issue by proposing shorter collection periods, usually at night, when the loss of circadian rhythm differs most from normal controls,137,138 but this approach has not been widely accepted. High-performance liquid chromatography and tandem mass spectrometry are now used to measure UFC, which overcomes the previous problem of cross-reactivity of some exogenous glucocorticoids and other structurally similar steroids with conventional radioimmunoassay.139 Occasionally, substances such as carbamazepine, digoxin, and fenofibrate can coelute with cortisol during high-performance liquid chromatography, causing falsely elevated results.140,141
If the previous caveats have been satisfied, the UFC measurement can be interpreted. In large series, measurement of an elevated UFC above the normal range has a high sensitivity for the diagnosis of Cushing’s syndrome (≈95% to 100%).100,142 However, it should be noted that in the latter study, 11% of 146 patients with proven Cushing’s syndrome had at least one of four UFC collections within the normal range, which confirms the need for multiple collections. Values greater than fourfold normal are rare except in Cushing’s syndrome. Values between this and down to the upper limit of normal are compatible with Cushing’s syndrome or pseudo-Cushing’s states, so that one must exclude the latter diagnosis. In summary, UFC measurements have a high sensitivity if collected correctly, and several completely normal collections make the diagnosis of Cushing’s syndrome very unlikely. However, when biochemical evidence of Cushing’s syndrome is not obtained in the setting of clinical features that suggest the diagnosis, repeated measurement of urine cortisol may demonstrate cyclicity or progression. The specificity is somewhat lower, thus patients with marginally elevated levels require further investigation.55
Late-Night Salivary Cortisol
Salivary cortisol measurement offers an excellent reflection of the plasma free cortisol concentration in health and disease because it circumvents the changes in total cortisol due to corticosteroid-binding globulin alterations.143,144 Salivary cortisol is stable for some days at room temperature, and the simple noninvasive collection procedure means that it can be performed conveniently at home and delivered via mail. Thus, it offers a number of attractive advantages over blood collection, particularly in children. Analysis is performed using a modification of the plasma cortisol radioimmunoassay, enzyme-linked immunosorbent assay, or liquid chromatography/tandem mass spectrometry, and commercial kits are internationally available for this.145 The diagnostic value cutoff varies between studies (0.13 µg/dL [3.6 nmol/L] to 0.55 µg/dL [15.2 nmol/L]) because of different assays and comparison groups studied.146–153 Normal values also differ between adult and pediatric populations, and this may be affected by other comorbidities such as diabetes and hypertension.154 However, from these studies, the sensitivity and specificity of this test appear to be relatively consistent at different centers, ranging from 92% to 100%, and from 93% to 100% respectively. It does not appear to make a difference if sampling is done at bedtime (≈23.00 hr) or at midnight, although it should be determined that the patient has a normal sleep pattern. Positive or negative results should be confirmed by repeat sampling. In summary, therefore, although late-night salivary cortisol appears to be a useful and convenient additional screening test for Cushing’s syndrome, particularly in the outpatient setting, local normal ranges should be validated based on the assay used and the population studied.
Low-Dose Dexamethasone Suppression Tests
In normal individuals, administration of the potent synthetic glucocorticoid dexamethasone results in suppression of the HPA axis, whereas patients with Cushing’s syndrome are resistant, at least partially, to this negative feedback. The original low-dose dexamethasone test (LDDST), as described by Liddle in 1960, measured urinary 17-OHCS before and during 48 hours of 0.5 mg dexamethasone every 6 hours, and an excretion of greater than 4 mg/day on the second day of dexamethasone treatment was considered to indicate Cushing’s syndrome.155 Dexamethasone does not cross-react with modern cortisol immunoassays, and the simpler measurement of a single plasma cortisol post dexamethasone has been validated in various series and gives the test a sensitivity of between 97% and 100% for the diagnosis of Cushing’s syndrome.156–159 The simpler overnight LDDST was proposed by Nugent and colleagues in 1965; this measured a 9:00 a.m. plasma cortisol after a single dose of 1 mg dexamethasone taken at midnight.160 Since then, various other doses, between 0.5 and 2 mg, have been proposed for the overnight test, and various diagnostic cutoffs have been applied.161–163 There appears to be no difference in discrimination between single doses of 1, 1.5, and 2 mg.164 Higher doses significantly decrease the sensitivity of the test.165 In a comprehensive review of the LDDST, both the original 2 day test and the 1 mg overnight protocol appear to have comparably high sensitivities (98% to 100%), provided a conservative postdexamethasone serum cortisol cutoff of 1.8 µg/dL (50 nmol/L) is applied. However, the specificity of the overnight test (88%) is lower compared with the 2 day test, particularly if serum cortisol is measured at both 24 and 48 hours (97% to 100%), with potential misclassification of patients with pseudo-Cushing’s states and acute or chronic illnesses. Many endocrinologists use the overnight test because of its greater simplicity and lower cost, although some centers still advocate the 48-hour test because of its high sensitivity and specificity, and the information it can provide in the differential diagnosis of ACTH-dependent Cushing’s syndrome (see later).159 Written instructions should be given to the patient if the latter is to be performed on an outpatient basis. Salivary rather than serum cortisol has been evaluated as the end point for the LDDST. This offers potential benefit in terms of convenience but requires further evaluation.149,166
Factors such as variable absorption and increased or decreased dexamethasone metabolism due to other compounds (Table 8-5) can influence any oral dexamethasone test.167 Therefore, a history of symptoms of malabsorption and a careful drug history should be taken before the test is used in a patient. Measurement of plasma dexamethasone is available in some centers and can be useful in patients of concern. One solution to overcome demonstrated malabsorption is to use one of the published intravenous dexamethasone suppression tests, recognizing that criteria for response have not been standardized.168,169 Pregnancy and other causes of increased or decreased corticosteroid-binding globulin (such as exogenous estrogens and the nephrotic syndrome) also should be excluded because these are likely to result in false-positive and false-negative tests.170
Second-Line Tests
The Dexamethasone-CRH Test: In 1993, a combined dexamethasone-CRH (Dex-CRH) test was introduced for the difficult scenario of the differentiation of pseudo-Cushing’s states from true Cushing’s syndrome in patients with only mild hypercortisolemia and equivocal physical findings.171 Dexamethasone 0.5 mg every 6 hours was given for eight doses, ending 2 hours before administration of ovine CRH (1 µg/kg intravenously) to 58 adults with UFC less than 360 µg/day (<1000 nmol/day). Subsequent evaluation proved that 39 had Cushing’s syndrome and 19 had a pseudo-Cushing’s state. The plasma cortisol value 15 minutes after CRH was less than 1.4 µg/dL (38 nmol/L) in all patients with pseudo-Cushing’s states and was greater in all patients with Cushing’s syndrome. A prospective follow-up study by the same group in 98 patients continued to show that the test had an impressive sensitivity and specificity of 99% and 96%, respectively.172 However, results from a number of other smaller studies have challenged the diagnostic utility of this test over the standard LDDST.173–175 Overall, in these reports, the specificity of the LDDST in 92 patients without Cushing’s syndrome was 79%, versus 70% for the Dex-CRH. Test sensitivity in 59 patients with Cushing’s syndrome was 96% for LDDST, versus 98% for the Dex-CRH group. It perhaps is not surprising that the diagnostic utility of the Dex-CRH has altered with additional studies at a greater number of centers. This might be the case for a number of reasons, including variable dexamethasone metabolism in individuals, different definitions of patients with pseudo-Cushing’s, different protocols and assays, and variable diagnostic thresholds.176 Of note, the original cortisol criteria performed poorly at these other centers, and this may have happened because many cortisol assays do not reliably measure levels <1.8 µg/dL (50 nmol/L). It does highlight that as a clinician one must be confident in the assay that is to be used for a particular test, and diagnostic criteria should be chosen that are appropriate for that assay. The Dex-CRH test remains a test that should be considered in patients with equivocal results.
Plasma Cortisol Circadian Rhythm: The normal diurnal rhythm of plasma cortisol is blunted or absent in Cushing’s syndrome, with normal or increased morning values and an increase in the nighttime nadir. Although less convenient than salivary cortisol, midnight plasma cortisol levels may be useful to obtain in patients admitted for investigation. Samples are best obtained around midnight, through an indwelling line for awake patients or by direct venipuncture within 5 to 10 minutes of waking of sleeping patients. In one study, 20 normal sleeping subjects had values less than 1.8 µg/dL (50 nmol/L), whereas all 150 patients with Cushing’s syndrome had midnight plasma cortisol concentrations greater than this.158 The suggested cutoff criterion in awake patients is higher, 7.5 to 8.3 µg/dL (207 to 229 nmol/L) and less discriminatory (sensitivity 92% to 94%, and specificity 96% to 100%).177,178 This difference probably reflects a different comparison group—patients suspected to have Cushing’s but in whom it was excluded. Patients with severe medical illness, depression, and mania may have cortisol values one to three times normal.130,164 Therefore, a sleeping midnight cortisol value less than 1.8 µg/dL (50 nmol/L) effectively excludes active Cushing’s syndrome, but higher values, unless very high, are less specific for Cushing’s syndrome.
Other Second-Line Tests
The insulin tolerance test has been used to distinguish Cushing’s syndrome from pseudo-Cushing’s states. Serum cortisol values increase in normal people after acute hypoglycemia, presumably because of central stimulation of CRH and vasopressin. The sustained hypercortisolism of Cushing’s syndrome suppresses CRH and vasopressin secretion and so blunts this response. The CRH/vasopressin neurons are presumed to be overactive in pseudo-Cushing’s states, particularly those that are depression associated, so a normal response to hypoglycemia (<40 mg/dL; <2.2 nmol/L) usually is maintained. Unfortunately, approximately 18% of patients with Cushing’s syndrome, especially those with minimal hypercortisolism, show a normal response to adequate hypoglycemia.164 Additionally, criteria for interpretation of results have not been established. If used, a dose of insulin of 0.3 U/kg should be used to overcome insulin resistance in these patients.130
The opiate agonist loperamide (16 mg orally) has been shown to inhibit CRH and thus ACTH and cortisol levels in most normal individuals, but not in patients with Cushing’s syndrome. This test has not been used widely but has been evaluated in one center, revealing a sensitivity of 100% and a specificity of 95%.179,180 However, it is unclear as to how well this test may exclude pseudo-Cushing’s states because a significant proportion of patients with depression also fail to suppress the HPA axis.181 It does not appear to be affected by drugs that affect the metabolism of dexamethasone and could potentially be useful in assessing patients on such treatment.180
DIFFERENTIAL DIAGNOSIS OF CUSHING’S SYNDROME
Once the diagnosis of Cushing’s syndrome is made, its cause must be determined. The strategy for the differential diagnosis of Cushing’s syndrome (Fig. 8-8) begins with measurement of plasma ACTH to distinguish between ACTH-dependent and ACTH-independent causes. Modern two-site immunoradiometric assays are more sensitive than the older radioimmunoassays and therefore provide the best discrimination. Only assays that can reliably detect values to below 10 ng/L should be used, and appropriate collection and processing of the sample are essential, because ACTH is susceptible to degradation by peptidases; therefore the sample must be kept in an ice water bath and centrifuged, aliquoted, and frozen within a few hours to avoid a spuriously low result. Repeated measurements are usually necessary because patients with ACTH-dependent Cushing’s disease have been shown to have on occasion ACTH levels less than 10 ng/L (2 pmol/L) on conventional radioimmunoassay,182 but consistent ACTH measurements of less than 10 ng/L (2 pmol/L) at 9:00 am with concomitant hypercortisolemia essentially confirm ACTH-independent Cushing’s syndrome. When the basal ACTH level is indeterminate (10 to 20 g/L [2 to 4 pmol/L]), the response to CRH may be useful in this setting. Patients with primary adrenal disease rarely show maximal ACTH values greater than 20 ng/L (4 pmol/L), although patients with Cushing’s disease usually exceed this value.
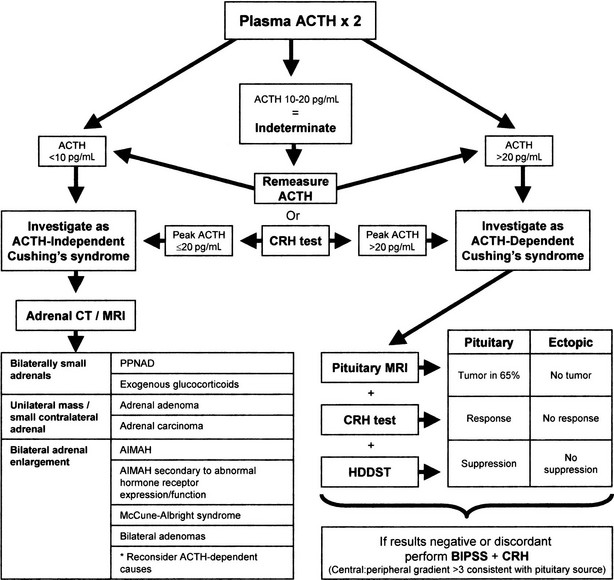
FIGURE 8-8 Suggested strategy for the differential diagnosis of Cushing’s syndrome. ACTH, Adrenocorticotropic hormone; AIMAH, ACTH-independent bilateral macronodular adrenal hyperplasia; BIPSS, bilateral inferior petrosal sinus sampling; CRH, corticotropin-releasing hormone; CT, computed tomography; HDDST, high-dose dexamethasone suppression test; MRI, magnetic resonance imaging; PPNAD, primary pigmented nodular adrenal disease.
Investigating Adrenocorticotropic Hormone–Independent Cushing’s Syndrome
Radiologic tests are the mainstay in differentiating between the various types of ACTH-independent Cushing’s syndrome. High-resolution CT scanning of the adrenal glands has excellent diagnostic accuracy for masses greater than 1 cm and allows evaluation of the contralateral gland.183 MRI may be useful for the differential diagnosis of adrenal masses; the T2-weighted signal is progressively brighter in normal tissue, adenoma, carcinoma, and finally pheochromocytoma.184 With this approach, adrenal tumors appear as a unilateral mass with an atrophic or less commonly a normal-size contralateral gland.185 If the lesion is greater than 5 cm in diameter, it should be considered to be malignant until proven otherwise, and imaging characteristics should not be relied upon. Very rarely, bilateral adenomas may be present.186 The adrenal glands in PPNAD appear normal or slightly lumpy from multiple small nodules but generally are not enlarged.187 AIMAH is characterized by bilaterally huge (>5 cm) nodular or hyperplastic glands.188 Exogenous administration of glucocorticoids results in adrenal atrophy; very small glands may provide a clue as to this entity.
The CT appearance of the adrenals in AIMAH may be similar to that of ACTH-dependent forms of Cushing’s syndrome, in which adrenal enlargement is present in 70% of cases.189 However, in our experience, the adrenal glands in Cushing’s disease are smaller and usually are symmetrically enlarged with an occasional nodule, as opposed to large or huge glands with definite nodules in AIMAH. In addition, the two can usually be differentiated by the ACTH level, although some patients with the macronodular subset of Cushing’s disease can develop a degree of adrenal autonomy that can cause biochemical confusion.190 Occasionally, confusion also may arise with apparent unilateral adrenal lesions, when the biochemistry is consistent with an ACTH-dependent cause; we generally would rely on the biochemistry in this situation and would examine the contralateral gland to see whether it is hyperplastic.
Differentiating Between Adrenocorticotropic Hormone–Dependent Causes of Cushing’s Syndrome
Although some patients with ectopic ACTH secretion, usually those with overt tumors, have extremely elevated values of plasma ACTH (>100 ng/L [>20 pmol/L]), complete overlap is seen between values in occult ectopic ACTH secretion and in Cushing’s disease.191 Therefore, ACTH values alone cannot differentiate reliably the ACTH-dependent forms of Cushing’s syndrome.
The ACTH-dependent forms of Cushing’s syndrome present the greatest diagnostic challenge. Cushing’s disease accounts for by far the majority of cases of ACTH-dependent Cushing’s syndrome—overall approximately 80% to 90% in most series. This percentage is gender dependent and is higher in women than in men,192 although in prepubertal childhood, an anomalous 80% male preponderance is noted. Therefore, even before one starts further investigation, the pretest probability that the patient has Cushing’s disease is very high, and any investigation must improve on this. The specificity of any test should be as close to 100% as possible for the diagnosis of Cushing’s disease, to avoid inappropriate pituitary surgery in patients with ectopic ACTH production. A variety of functional tests of the HPA axis have been developed to take advantage of the differences in pathophysiology between ACTH-dependent causes of Cushing’s syndrome. Some of these investigations have evolved, and others have fallen by the wayside.
Bilateral Inferior Petrosal Sinus Sampling: Bilateral inferior petrosal sinus sampling (BIPSS) is the best test for distinguishing ACTH-dependent forms of Cushing’s syndrome, as long as the patient has active hypercortisolemia, which should be confirmed at the time of the procedure.193,194 The test exploits the normal venous drainage of each half of the pituitary gland via the cavernous sinus into the corresponding petrosal sinus. Each petrosal sinus is catheterized separately via a femoral approach, and blood for measurement of ACTH is obtained simultaneously from each sinus and a peripheral vein at two timepoints before and at 3 to 5 minutes and possibly also 10 minutes after the administration of ovine or human CRH (Ferring) (1 µg/kg or 100 µg intravenously)195 (Fig. 8-9). Where CRH is unavailable for whatever reason, recent data suggest that 10 µg desmopessin may be a suitable alternative.
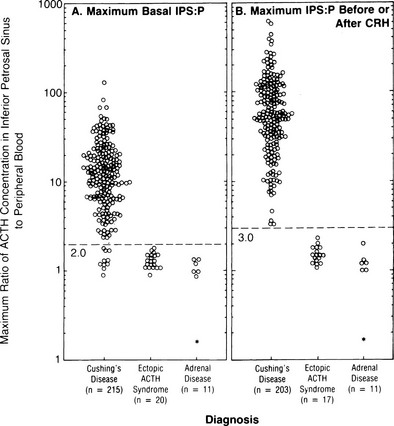
FIGURE 8-9 Maximal ratio of adrenocorticotropic hormone (ACTH) concentration in the inferior petrosal sinus to peripheral blood in patients with confirmed Cushing’s disease, ectopic ACTH syndrome, or adrenal disease before corticotropin-releasing hormone (CRH) (A) or at any time before or after CRH administration (B). A ratio of 3.0 had 100% sensitivity and specificity. (Data from Oldfield EH, Doppman JL, Nieman LK, et al: Petrosal sinus sampling with and without corticotropin-releasing hormone for the differential diagnosis of Cushing’s syndrome. N Engl J Med 325:897–905, 1991.)
ACTH concentrations are greater in the central samples in Cushing’s disease and increase after CRH administration, reflecting ACTH secretion by the corticotroph adenoma. In contrast, ACTH values in the central and peripheral specimens are similar in ectopic ACTH secretion and do not increase after CRH. A ratio of central (i.e., petrosal) to peripheral ACTH values is calculated. In earlier series, pre-CRH ratios greater than 2 or post-CRH ratios greater than 3 were 100% specific for Cushing’s disease,196,197 but a small number of false positives have been reported in later series.198,199 The sensitivity of the test is improved after CRH; however, false negatives still occur in about 6% of patients with Cushing’s disease. False positives in ectopic ACTH are extremely rare.
It should be remembered that the technique is highly specialized, and allied with this are a number of important points. First, both petrosal sinuses must be cannulated adequately and catheter placement confirmed before and after sampling.200 Second, the radiologist must confirm the venous anatomy because anomalous venous drainage can give false-negative results. Preliminary data have suggested that simultaneous measurement of prolactin can be used as an index of pituitary venous drainage, and prolactin should be measured when results indicate a noncentral source of ACTH.201 Third, the procedure carries a small risk of complications. Transient ear discomfort or pain can occur, as can local groin hematomas. More serious transient and permanent neurologic sequelae, including brain stem infarction, have been reported, although these are rare (<1%), and most have been related to the particular type of catheter used202,203; if any early warning signs of such events are observed, the procedure should be halted immediately. Patients should be given heparin during sampling to prevent thrombotic events.129 CRH itself generally is tolerated well, although patients may experience brief facial flushing and a metallic taste in the mouth. One case of CRH induced pituitary apoplexy in a patient with Cushing’s disease has been reported.204
Another potential advantage of BIPSS involves lateralizing microadenomas within the pituitary using the inferior petrosal sinus ACTH gradient, with a basal or post-CRH intersinus ratio of at least 1.4 being the criterion used for lateralization in all large studies.196,197,205,206 In these studies, the diagnostic accuracy of localization as assessed by operative outcome varied between 59% and 83%. This is improved if venous drainage is assessed to be symmetric.207 Some discrepancy has been noted between studies as to whether CRH improves the predictive value of the test.208 If a reversal of lateralization is seen pre- and post-CRH, the test cannot be relied upon.209 Sampling of the internal jugular veins is a simpler procedure but is not as sensitive as BIPSS.210 However, it may be a useful technique in less experienced centers, with the caveat that patients with negative results then are referred for BIPSS.211 In our opinion, sampling from the cavernous sinus itself offers no great advantage.
High-Dose Dexamethasone Suppression Test: The original high-dose dexamethasone suppression test (HDDST) was described in the same paper as the 48-hour LDDST; 2 mg dexamethasone is used in place of 0.5 mg, with a 50% reduction in urinary 17-OHCS shown to differentiate 96% of patients with Cushing’s disease rather than adrenal tumors.155 The role of the HDDST in the differential diagnosis of ACTH-dependent Cushing’s syndrome is based on the same premise, that is, that most pituitary corticotroph tumors retain some responsiveness (albeit reduced) to negative glucocorticoid feedback on ACTH secretion, whereas ectopic ACTH-secreting tumors, like adrenal tumors, typically do not. Measurement of UFC or plasma/serum cortisol has superseded that of urinary 17-OHCS, and an overnight test has been advocated, with a single dose of 8 mg dexamethasone given at 11:00 pm, and with the criterion of a 50% reduction in plasma cortisol levels on the morning after administration.212 Despite evidence that only about 80% of patients with Cushing’s disease will show suppression of plasma cortisol to less than 50% of the basal value, and large numbers of patients with ectopic Cushing’s syndrome have false-positive results (≈30%),60,213 the HDDST is still used widely. Some data suggest that that suppression to HDDST can be inferred by a greater than 30% suppression of serum cortisol to the 2 day LDDST (Fig. 8-10); therefore, in centers that use this form of the LDDST, the HDDST may not confer any extra information.159 It should not be forgotten that patients are receiving large doses of glucocorticoids, in addition to their high endogenous cortisol production, and one should be alert for the precipitation of psychosis and/or worsening of glycemic control or other complications.
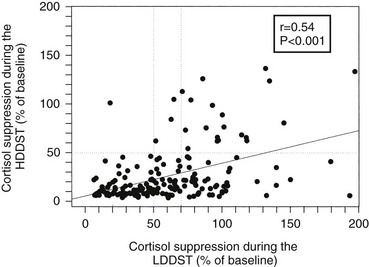
FIGURE 8-10 Correlation between the degree of suppression during the low-dose dexamethasone suppression test (LDDST) and during the high-dose dexamethasone suppression test (HDDST) in 185 patients with Cushing’s disease. (Reproduced with permission from Isidori AM, Kaltsas GA, Mohammed S, et al: Discriminatory value of the low-dose dexamethasone suppression test in establishing the diagnosis and differential diagnosis of Cushing’s syndrome. J Clin Endocrinol Metab 88:5299–5306, 2003. Copyright © 2003, The Endocrine Society.)
Corticotropin-Releasing Hormone Stimulation Test: The use of CRH stimulation for the differential diagnosis of ACTH-dependent Cushing’s syndrome is based on two assumptions: (1) that corticotropinomas retain responsivity to CRH, whereas noncorticotroph tumors lack CRH receptors and cannot respond to the agent; and (2) that hypercortisolism has been sufficient to inhibit the normal corticotroph response. Indeed, most patients with Cushing’s disease respond to CRH, either 1 µg/kg or 100 µg intravenous synthetic ovine or human sequence CRH, with increases in plasma ACTH or cortisol, and patients with ectopic ACTH secretion typically do not.214–216 Human sequence CRH has qualitatively similar properties to ovine CRH, although it is shorter acting with a slightly smaller increase in plasma cortisol and ACTH in normal and obese patients and in those with Cushing’s disease217; this may be related to the more rapid clearance of the human sequence by endogenous CRH-binding protein.218 Availability differs worldwide, with ovine CRH predominant in North America but human CRH elsewhere.
Because different centers have used differing protocols, including different types of CRH and different sampling timepoints, little consensus has been reached on a universal criterion for interpreting the test. However, where the test has been validated in experienced centers, the diagnostic utility appears similar. For instance, in the largest published series of the use of ovine CRH in ACTH-dependent Cushing’s syndrome, an increase in ACTH of at least 35% from a mean basal (5 minutes and 1 minute) to a mean of 15 and 30 minutes after ovine CRH in 100 patients with Cushing’s disease and in 16 patients with ectopic ACTH syndrome (Fig. 8-11) gave the test a sensitivity of 93% for diagnosing Cushing’s disease and 100% specificity. The best cortisol criterion was an increase of at least 20% at a mean of 30 and 45 minutes, revealing a sensitivity of 91% and a specificity of 88%.219 Similarly, in the largest series involving use of the human CRH test in 101 patients with Cushing’s disease and in 14 with ectopic ACTH syndrome, the best criterion used to differentiate Cushing’s disease from ectopic ACTH syndrome was an increase in cortisol of at least 14% from a mean basal (15 and 0 minutes) to a mean of 15 and 30 minutes, yielding a sensitivity of 85% and a specificity of 100% (Fig. 8-12). In contrast, the best ACTH response was a maximal increase of at least 105%, indicating 70% sensitivity and 100% specificity.192 The CRH test is a useful discriminator between causes of ACTH-dependent Cushing’s syndrome; however, which cutoff to use must be evaluated at individual centers, and caution should be exercised because undoubtedly there will be patients with ectopic ACTH syndrome who respond outside these cutoffs. However, an increase in cortisol outside the normal range can differentiate Cushing’s disease from normality, albeit in only approximately 50% of cases, and, as noted previously, the measurement of plasma ACTH in the test can help to discriminate ACTH-dependent from ACTH-independent causes of Cushing’s syndrome when basal levels of ACTH are equivocal. We therefore believe that this test plays a useful role in the investigation of patients with Cushing’s syndrome.
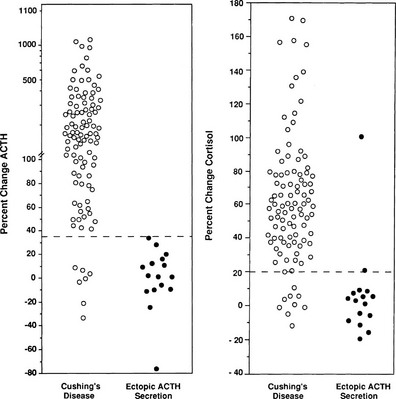
FIGURE 8-11 Response of adrenocorticotropic hormone (ACTH) and cortisol to ovine corticotropin-releasing hormone in patients with Cushing’s disease and ectopic ACTH secretion. ACTH responses are expressed as the percentage of change in mean concentration 15 and 30 minutes after ovine corticotropin-releasing hormone from the mean basal value 1 and 5 minutes before the injection. The dashed line indicates a response of 35%, representing a diagnostic criterion with 100% specificity and 93% sensitivity. Cortisol responses are expressed as percentage of change in mean cortisol concentration 30 and 45 minutes after ovine corticotropin-releasing hormone from the mean basal value 1 and 5 minutes before the injection. The dashed line indicates a response of 20%, representing a diagnostic criterion with 88% specificity and 91% sensitivity. (Data from Nieman LK, Oldfield EH, Wesley R, et al: A simplified morning ovine corticotropin-releasing hormone stimulation test for the differential diagnosis of ACTH-dependent Cushing syndrome. J Clin Endocrinol Metab 77:1308–1312, 1993.)
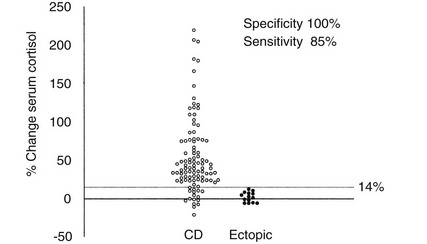
FIGURE 8-12 Percentage change in serum cortisol from a mean basal at 15 and 0 minutes to a mean value calculated from the levels at 15 and 30 minutes after the administration of human corticotropin-releasing hormone (100 mg intravenously) in 100 patients with Cushing’s disease (CD) and 14 patients with ectopic adrenocorticotropic hormone syndrome. (Reproduced with permission from Newll-Price J, Morris DG, Drake WM, et al: Optimal response criteria for the human CRH test in the differential diagnosis of ACTH-dependent Cushing’s syndrome. J Clin Endocrinol Metab 87:1640–1645, 2002.)
Other Stimulation Tests: Vasopressin and desmopressin (a synthetic long-acting vasopressin analogue without V1-mediated pressor effects) are thought to stimulate ACTH release in Cushing’s disease through the corticotroph-specific V3 (or V1b) receptor. Hexarelin (a growth hormone secretagogue) also stimulates ACTH release to a sevenfold greater extent than human CRH; although the mechanism has not been entirely elucidated, this probably occurs through stimulation of vasopressin release in normal subjects,220 and through stimulation of aberrant growth hormone secretagogue receptors in patients with corticotroph tumors.221 These peptides all have been used in a similar manner to CRH, to try to improve the differentiation of ACTH-dependent Cushing’s syndrome, but they generally have proved inferior or of no advantage.222–225 However, desmopressin may be useful in centers where CRH is unavailable. A combined desmopressin (10 µg) and human CRH (100 µg) test initially looked extremely promising.226 However, a later study of this combined test in 26 patients with Cushing’s disease and 5 patients with ectopic ACTH syndrome showed significant overlap in responses.227 The disappointing discriminatory outcome of these stimulants is undoubtedly due to the expression of both vasopressin and growth hormone secretagogue receptors by some ectopic ACTH-secreting tumors.129,228
Combined Test Strategies
Because none of the noninvasive tests have 100% diagnostic accuracy, a number of investigators have evaluated the utility of combined test strategies. The CRH and the HDDST have been paired in this way, and combined, they have a diagnostic accuracy greater than that of either test alone, yielding 98% to 100% sensitivity and 88% to 100% specificity.229–231 Similar high accuracy has been obtained by combining the results of the LDDST and the CRH test.159
Imaging of the Adrenocorticotropin Hormone Source: Pituitary MRI imaging before and after gadolinium enhancement should be performed in all patients with ACTH-dependent Cushing’s syndrome via T1-weighted spin echo and/or spoiled gradient recalled acquisition (SPGR) techniques. These will identify an adenoma in up to 80% of patients with Cushing’s disease,60,232,233 and in approximately 10% of normal individuals.234 Most adenomas (95%) exhibit a hypointense signal with no postgadolinium enhancement, and the remaining 5% show an isointense signal post gadolinium enhancement.235 CT imaging typically shows a hypodense lesion that fails to enhance post contrast but is less sensitive than MRI in detecting small (<5 mm) adenomas and is not recommended for this reason.60,236
Imaging is the most helpful way to identify the source of ectopic ACTH production. Given the likely sites of tumors, CT and/or MRI of the neck, chest, and abdomen should be obtained. The most common source is a bronchial carcinoid tumor, but small (<1 cm) lesions often can prove difficult to locate. Fine-cut high-resolution CT scanning with both supine and prone images can help differentiate between tumors and vascular shadows.55 MRI can identify chest lesions that are not evident on CT scanning and that characteristically show a high signal on T2-weighted and short-inversion time inversion recovery (STIR) images.237 Additionally, pheochromocytomas are bright on T2-weighted MRI. CT-guided aspiration of masses for measurement of ACTH may provide useful functional information.238
Because most ectopic ACTH-secreting tumors are of neuroendocrine origin and therefore may express somatostatin receptor subtypes, radiolabeled somatostatin analogue (111In-pentetreotide) scintigraphy may be useful to show functionality of identified tumors, and sporadic reports have indicated that it identifies lesions not apparent on conventional imaging.239–241 However, in most patients, including a recent series of 35 patients with ectopic ACTH secretion, 111In-pentetreotide scintigraphy was not able to detect tumors when the MRI or CT scan was negative, and significant numbers of false-positive scans resulted.242 Thus, CT and MRI represent the best initial screening examinations, but scintigraphy may be a useful adjunctive imaging modality to help confirm abnormalities seen on CT or MRI. 18-Flurodeoxyglucose positron emission tomography (PET) generally does not offer any advantage over conventional CT or MRI unless the tumors are metabolically active,243 which is not usually the case.244 However, 67Ga-octreotate PET scintigraphy may show advantages over 111In-pentetreotide scintigraphy.245
Strategy for Diagnosis and Differential Diagnosis of Cushing’s Syndrome
After an international workshop in 2002, a consensus statement was published for the diagnosis and differential diagnosis of Cushing’s syndrome.129 Most of its recommendations are still valid. We would advocate that three 24-hour UFC, late-night salivary cortisols and/or the LDDST should be used as first-line screening tests. (This approach also was advocated in a recent guidelines statement issued by the Endocrine Society.134) False-positive results will be common, and second-line tests should be used as necessary for confirmation. Once the diagnosis of Cushing’s syndrome is unequivocal, ACTH levels, the CRH test, and a dexamethasone suppression test, together with appropriate imaging, are the most useful noninvasive investigations to determine the cause. Bilateral inferior petrosal sinus sampling is recommended in cases of ACTH-dependent Cushing’s syndrome in which the clinical, biochemical, or radiologic results are discordant or equivocal, although others would recommend its use in almost all cases of ACTH-dependent Cushing’s syndrome.
Treatment
Optimal treatment for Cushing’s syndrome renders the patient eucortisolemic with minimal morbidity and mortality. With the advent of synthetic glucocorticoid therapy, adrenalectomy became the treatment of choice because it conferred rapid and, in most cases, permanent resolution of Cushing’s syndrome.246 Improvements in neurosurgical techniques and appreciation of the sources of ectopic ACTH secretion have changed the therapeutic approach to Cushing’s syndrome, so that surgery now is directed toward resection of abnormal tissue, whether ACTH or cortisol producing. In patients with only mild hypercortisolemia and subclinical disease, the benefits of surgical treatment have not been proved.247 The optimal surgical approach cannot be realized if the patient is unable to safely undergo surgery, or if the tumor is occult or metastatic. Other second-line therapies that are less specific and that may have greater morbidity must be chosen in these settings. In 2007, an international meeting of endocrinologists outlined a consensus statement on the treatment of ACTH-dependent Cushing’s syndrome.248
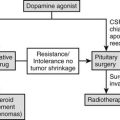
Surgery
Preoperative Evaluation and Treatment
Some centers use routine preoperative medical adrenal blockade to attain a period of eucortisolemia for 4 to 6 weeks before surgery. The aim is to allow reversal of some of the metabolic and catabolic effects of the hypercortisolemia that may inhibit wound healing and cause other complications in the perioperative period. The disadvantage of this strategy is that the normal corticotrope may be disinhibited by the time of surgery, so that expected hypocortisolism after successful tumor resection does not occur, and this index of remission is not reliable. This rationale is only empirical, and a randomized trial undertaken to see whether this approach improves outcome would be welcomed.249
Because Cushing’s syndrome is a prothrombotic state, anticoagulant prophylaxis should be considered perioperatively.250 The lipid abnormalities, hypertension, and diabetes common in Cushing’s syndrome predispose these patients to atherosclerotic cardiac disease and should be treated by conventional approaches.
Transsphenoidal Resection of Corticotropinoma
Transsphenoidal resection is regarded as the treatment of choice in Cushing’s disease.248 The procedure usually is performed via a gingival approach, as originally devised by Kanavel and Halsted and later popularized by Cushing.251 The development of the operating microscope led to the introduction of transsphenoidal resection of pituitary microadenomas by Hardy in 1968. Additional advances have been made over the past decade with the use of rigid endoscopes and more recently flexible endoscopes. The goal of surgery is a selective adenomectomy and thus preservation of as much normal pituitary tissue as possible; the procedure should be performed by a neurosurgeon who is experienced in transsphenoidal surgery. Most large series report immediate remission rates of approximately 70% to 90%,252–259 with lower rates noted for macroadenomas.254,260 These reports use variable remission criteria that include normal postoperative cortisol levels, normal UFC levels, and/or a normal response to an LDDST, associated with clinical resolution of the disease. Data are insufficient regarding whether endoscopic surgery offers any benefit.261 However, a recent report suggests that resection achieved through a technique that enucleates microadenomas via their pseudocapsule may improve remission rates to 98%.262
Long-term recurrence rates are as high as 20% by 10 years after surgery. Thus, in reality, transsphenoidal surgery achieves long-term cure even in the best of hands in only approximately 60% to 80% of adult patients, and therefore is somewhat disappointing; the data emphasize the need for long-term endocrinologic follow-up of these patients.263 Favorable indicators of long-term remission are patients older than 25 years, a microadenoma detected by MRI, lack of invasion of the dura or cavernous sinus, histologic confirmation of an ACTH-secreting tumor, low postoperative cortisol levels, and long-lasting adrenal insufficiency.248 If a tumor cannot be identified at surgery, hemihypophysectomy on the side of the gland with an ACTH gradient on petrosal sinus sampling is usually the best way to proceed, and in some series, this yields better results than those attained by selective microadenomectomy.256,264
The mortality associated with transsphenoidal surgery is approximately 1% to 2%.11,258 Transient diabetes insipidus is probably the most common complication, being reported in as many as 28% of patients.264 Other perioperative complications, including cerebrospinal fluid leak, meningitis, and profuse bleeding, occur in less than 10% of patients. Permanent complications such as persistent diabetes insipidus and injury to the optic nerve or nerves of the cavernous sinus (causing ptosis or diplopia) occur much less frequently.11,258 However, hypopituitarism, particularly growth hormone deficiency, is common (53% to 59%), with other anterior hormone deficits occurring in approximately 35% to 45% of patients.264,265 Such complications are more common after resection of larger tumors or with the presence of larger amounts of normal pituitary tissue (or stalk), or after repeat surgery.
Postoperative Evaluation and Management
Patients typically receive supraphysiologic doses of glucocorticoids to cover transsphenoidal surgery at initial daily doses of as high as 400 mg hydrocortisone (4 mg dexamethasone), tapering off within 1 to 3 days. Morning (9:00 am) serum cortisol measurements then are obtained for 3 days, starting 24 hours after the last glucocorticoid administration, during which time the patient should be observed for development of signs of adrenal insufficiency. This approach allows prompt classification of likely cure, normocortisolemia, or persistent hypercortisolism. Where close perioperative supervision is possible, such glucocorticoid “cover” may be omitted, permitting a very early postoperative assessment of cure. What defines apparent cure or remission after transsphenoidal surgery is still debated. Postoperative hypocortisolemia (<1.8 µg/dL [50 nmol/L] at 9:00 am) is probably the best indicator of the likelihood of long-term remission. However, detectable cortisol levels of <5 µg/dL (140 nmol/L) are also compatible with sustained remission.266,267 Higher postoperative cortisol levels are more likely to be associated with failed surgery; however, cortisol levels sometimes may decline gradually over 4 to 6 weeks, reflecting gradual infarction of remnant tumor or some degree of adrenal semiautonomy. Persistent cortisol levels >5 µg/dL (140 nmol/L) 6 weeks after surgery require further investigation.
Dynamic tests have been used to predict long-term remission. The cortisol and ACTH response to CRH in the early postoperative period may provide a useful index of the risk for recurrence of Cushing’s disease, the rationale being that responsiveness may indicate residual tumor.60,268 A study of postoperative responses to CRH in 221 patients suggested that a cortisol response greater than 5 µg/dL (138 nmol/L) at 60 minutes had a positive predictive value of 42% and a negative predictive value of 94% for recurrence of disease.269 Because patients with partial recovery of the axis would be expected to have a normal response, regardless of the risk for recurrence, the CRH test cannot be interpreted and should not be used in this setting. Some evidence suggests that persistence of the ACTH response to desmopressin postoperatively probably is linked to a higher rate of relapse.270,271
Patients who are hypocortisolemic should be started on glucocorticoid replacement, and 15 to 30 mg hydrocortisone (12 to 15 mg/m2) in two to three divided doses is the preferred choice. The first dose (usually half to two thirds of the total dose) should be taken before getting out of bed, and the last dose should be taken no later than 6:00 pm, because later administration of glucocorticoids may result in disordered sleep. In this situation, the lowest possible dose of hydrocortisone should be used to avoid long-term suppression of the HPA axis. All patients receiving long-term glucocorticoid replacement therapy should be instructed that they are “dependent” on taking glucocorticoids as prescribed, and that failure to take or absorb the medication will lead to adrenal crisis and possibly death. They should be prescribed a 100 mg hydrocortisone (or other high-dose glucocorticoid) intramuscular injection pack for emergency use. They also should obtain a medical information bracelet or necklace that identifies this requirement (Medic Alert Foundation). Education should stress the effects of glucocorticoid withdrawal272; the need for compliance with the daily dose of glucocorticoid; the need to double the oral dose for nausea, diarrhea, and fever; and the need for parenteral administration and medical evaluation during emesis, trauma, or severe medical stress.
The patient should be told to expect desquamation of the skin and flu-like symptoms (malaise, joint aching, anorexia, and nausea) during the postoperative months, and that these are signs that indicate remission: some of these symptoms have been related to high levels of circulating interleukin-6.273 Most patients tolerate these symptoms of glucocorticoid withdrawal much better if they are forewarned and alerted to their positive nature. Physicians should not increase the glucocorticoid dose in the absence of intercurrent illness based on these symptoms alone but should seek signs of adrenal insufficiency, such as vomiting, electrolyte abnormalities, and postural hypotension.274 The affective and cognitive changes associated with Cushing’s syndrome are particularly slow to resolve and may not normalize. Evidence of persisting physical and emotional dysfunction has been found, even after prolonged duration of cure.275 Postoperatively, assessment for deficiencies of other pituitary hormones should be sought, and the appropriate replacement regimen initiated as necessary.
Diuresis is common after transsphenoidal surgery and may result from intraoperative or glucocorticoid-induced fluid overload or may be due to diabetes insipidus. For these reasons, assessment of paired serum and urine osmolality and the serum sodium concentration is essential. It is advisable to withhold specific therapy unless the serum osmolality is greater than 295 mOsm/kg, the serum sodium is greater than 145 mmol/L, and the urine output is greater than 200 mL/hr, with an inappropriately low urine osmolality. Desmopressin (DDAVP, Ferring) 1 µg given subcutaneously, will provide adequate vasopressin replacement for 12 hours or longer. Hyponatremia may occur in as many as 20% of patients within 10 days of surgery. This may be due to injudicious fluid replacement or inappropriate antidiuretic hormone secretion, as is frequently seen after extensive gland exploration, and fluid intake should be restricted.276 A small minority of patients proceed to (apparently) permanent diabetes insipidus, requiring long-term treatment with a vasopressin analogue. A dose and schedule of administration should be chosen to provide unbroken sleep but allowing a period of “breakthrough” urination each day. This goal is often achieved when 10 to 20 µg desmopressin is given intranasally (or an equivalent oral dose) in the evening.
Some glucocorticoid-induced abnormalities, including hypokalemia, hypertension, and glucose intolerance, may be normalized during the postoperative period, so that preoperative treatments for these need to reassessed. Some evidence indicates that deficits in bone mass may be partially reversed after treatment of hypercortisolemia.277,278 Bisphosphonate treatment may induce a more rapid improvement in bone mineral density279 and should be considered (along with calcium and vitamin D supplements) in patients with osteoporosis.
Treatment options for patients with persistent Cushing’s disease include repeat surgery, radiation therapy, and adrenalectomy. If immediate surgical remission is not achieved at the first exploration, early repeat transsphenoidal surgery may be worthwhile in a significant proportion of patients, at the expense of increased likelihood of hypopituitarism.280,281 The likelihood of remission after repeat surgery is greatest when some or all of the following outcome parameters are present: The diagnosis is correct, as evidenced by previous curative surgery with pathologic confirmation of an ACTH-staining adenoma; the initial exposure or resection was incomplete; or residual tumor is seen on MRI scan without evidence of cavernous sinus invasion. Repeat sellar exploration is less likely to be helpful in patients with empty sella syndrome or very little pituitary tissue on MRI scans. Patients with cavernous sinus or dural invasion identified at the initial procedure are not candidates for repeat surgery to treat hypercortisolism and should receive radiation therapy.
Recovery of the HPA axis can be monitored by measurement of 9:00 am serum cortisol after omission of hydrocortisone replacement. Because recovery after transsphenoidal surgery rarely occurs before 3 to 6 months and is common at 1 year, initial testing at 6 to 9 months is cost-effective.282 If the cortisol is undetectable on 2 consecutive days, then recovery of the axis has not occurred, and glucocorticoid replacement can be restarted. If the cortisol is measurable, adequate reserve of the HPA axis can be assessed with the insulin tolerance test,283 with a peak cortisol value of >18 µg/dL (500 nmol/L), indicating adequate reserve on modern assays.284 Many centers use the cortisol response to 250 µg synthetic (1-24) ACTH as an alternative means of assessing HPA reserve,285,286 but some controversy regarding its reliability in this situation has been ongoing.287,288 If it is used instead of the insulin tolerance test, a 30 minute cortisol of 22 µg/dL (600 nmol/L) is probably more reliable than the traditional cutoff of 18 µg/dL (500 nmol).284 Glucocorticoid replacement can be discontinued abruptly if the cortisol response is shown to be normal.
Two late but unrelated conundrums may arise: the questions of recurrence and permanent lack of recovery of the axis. Patients who articulate that Cushing’s syndrome has returned are often correct, even before physical and biochemical evidence is unequivocal. Assessment is warranted in a patient with these complaints or with recurrent physical signs characteristic of the hypercortisolemic phase. If this is to be done on an outpatient basis, UFC can be measured initially on dexamethasone 0.5 mg/day, if not yet weaned from glucocorticoids. Measurement of late-night salivary cortisol after omission of the afternoon dose of hydrocortisone also may be useful. However, ideally, assessment of a cortisol circadian rhythm can be done on an inpatient basis after the hydrocortisone has been stopped completely. If the UFC result is increased, evaluation of hypercortisolism should proceed. If recurrent Cushing’s disease is diagnosed, the therapeutic options are the same as for persistent disease. It should be remembered when investigating recurrence that long-standing ACTH stimulation by a pituitary adenoma causing macronodular adrenal hyperplasia subsequently may involve autonomous cortisol production.289
Adrenalectomy
Resection of the affected adrenal gland(s) is the treatment of choice only for non–ACTH-dependent hypercortisolism of adrenal origin, or when a specific surgical approach to ACTH-dependent causes is not feasible. In adrenal adenomas, the cure rate is 100% when performed by experienced adrenal surgeons.290 Surgery is the mainstay of treatment for adrenal cancer; more aggressive surgical approaches probably account for the increase in life span reported in this disease.85,291 This approach may require multiple operations to resect primary lesions, local recurrences, and hepatic, thoracic, and, occasionally, intracranial metastases. Adjuvant medical treatment with mitotane and other chemotherapeutic agents is discussed later.
The mortality and morbidity of traditional open adrenalectomy via an anterior or posterior incision range from 1% to 20% in various series, probably reflecting differences in the severity of Cushing’s syndrome and the presence of associated conditions, such as cardiovascular disease.184,246 Apart from resection of suspected carcinoma, these approaches have been supplanted by laparoscopic resection, which has low mortality and morbidity when done by an experienced surgeon.292 Glands as large as 7.5 cm may be removed through this approach.293 Serum cortisol levels become undetectable after successful adrenalectomy. Early failure to achieve hypocortisolism usually is related to incomplete resection of the gland(s). Recurrence, especially in the ACTH-dependent forms of Cushing’s syndrome, may be related to regrowth of adrenal cells in the surgical bed, or to growth of adrenal rest tissue.
Bilateral adrenalectomy as a second line of treatment for Cushing’s disease has the advantage of providing rapid resolution of hypercortisolism and has no risk of hypopituitarism, in contrast to radiation therapy. Adrenalectomy may be chosen over radiation therapy by young patients who desire fertility, who have concerns about radiation-induced hypopituitarism and loss of reproductive function. Its disadvantages include perioperative morbidity and mortality and the life-long requirement of glucocorticoid and mineralocorticoid replacement therapy. In addition, patients with Cushing’s disease have a risk for developing Nelson’s syndrome, which may occur more frequently if adrenalectomy is performed at a younger age, and if a pituitary adenoma is confirmed at previous pituitary surgery.259,294 Regular pituitary MRI and monitoring of ACTH levels are mandatory in these patients.295 Prophylactic pituitary radiotherapy appears to reduce the risk for developing Nelson’s syndrome from 50% to 25%,296 but no consensus has been reached on this topic.
After unilateral adrenalectomy, the components of the HPA axis gradually recover after surgical cure of Cushing’s syndrome. The time to recovery may be as short as 3 months and as long as 2 years.268,282 The duration of recovery may be shorter in patients with mild hypercortisolism and in those with recurrence, and longer after resection of an adrenal adenoma.
Radiotherapy
Conventional Radiotherapy
Conventional pituitary radiotherapy is delivered at a total dose of 4500 to 5000 cGy (rad) in 25 fractional doses over 35 days through a three-field (opposed lateral fields and vertex field) technique. Nowadays, this usually is based on stereotactic conformal field planning to optimize the tumor dose and to minimize radiation to other areas. This approach ensures that the daily dose to neural tissue does not exceed 180 cGy and avoids the complications of optic neuritis and cortical necrosis associated with larger total and fractional doses.297 The latent onset of action of radiotherapy means that adjunctive medical therapy usually is instituted before or at the time of treatment. The therapeutic response is assessed at least with annual monitoring, with weaning of medical treatment if possible. When conventional radiotherapy is used as primary treatment for Cushing’s disease, remission is achieved in only 40% to 60% of adult patients.259,298–300 The response is even worse if a lower dose of radiation (2000 cGy) is used.301 More usually, conventional radiotherapy is used in the setting of failure to cure after transsphenoidal surgery. In this regard, it performs rather better, with reported remission rates as high as 83%.299,302 After conventional radiotherapy is given, remission usually starts by 9 months after treatment, and most patients are in remission within 2 years, although this can take much longer.303
Hypopituitarism is the most common side effect of pituitary radiotherapy, with growth hormone deficiency occurring in 36% to 68% of treated adults.265,302 Gonadotropin and thyroid-stimulating hormone deficiency is seen less commonly. The risk for optic neuropathy is low and probably is less than 1% as long as low-dose fractions are used304 (200 cGy). Similarly, the occurrence of brain necrosis is exceedingly rare.305 The issue of secondary tumors remains contentious; although meningiomas and gliomas have been reported after pituitary radiotherapy, it is not clear whether the incidence is significantly greater than the background risk for developing such tumors in patients who already have one intracranial tumor and have undergone careful surveilllance.304
Stereotactic Radiosurgery
In stereotactic radiosurgery, concentrated beams of high doses of radiation are aimed precisely at the mapped discrete lesion, delivering very high doses to the tumor and relatively low doses to normal surrounding tissue. A number of techniques can be used to do this: using narrow beams from multiple gamma cobalt sources (gamma knife), using heavy charged particles (proton or helium beams), or using a single beam from a linear accelerator that is arced around the target (X-knife, SMART, Linac). The advent of high-resolution MRI for mapping has facilitated this therapy. Usually, only a single therapy dose is required, and this is biologically more effective than delivery of the same dose in fractions.306 This technique also may be used to deliver radiation to the entire pituitary gland when a specific target is not known after surgery or MRI examination.
Probably the technique most widely used currently for Cushing’s syndrome is the gamma knife. As adjunctive therapy after failed transsphenoidal surgery, it probably is no better than conventional radiotherapy.307 Radiosurgery of the pituitary gland using proton beams has similar efficacy.308 Linear accelerator radiotherapy for Cushing’s disease is less well described, but success has been reported in small numbers of patients.309
As with conventional radiotherapy, the main side effect is hypopituitarism, and although the perceived risk of damage to adjacent structures such as the optic chiasm is less, additional studies and longer follow-up are needed. Some centers use it mainly for salvage of difficult recurrent tumors, for example, in the cavernous sinus or in Nelson’s syndrome, whereas others have suggested it as an alternative to conventional radiotherapy in adenomas smaller than 30 mm with a minimal distance from the optic chiasm of 2 to 3 mm.305
Interstitial Radiotherapy
Two centers have used interstitial irradiation (yttrium-90 or gold-198 implants) as primary therapy for Cushing’s disease.310,311 Remission rates in these cohorts were high (75% to 77%), although some required a second implant. The principal side effect was hypopituitarism.
Other Tumors
No significant evidence suggests that radiotherapy improves overall survival in adrenocortical carcinoma, although sporadic reports indicate that it may be helpful adjuvant treatment to radical surgery in selected cases.312,313 Local radiotherapy after surgical resection of an ectopic ACTH-secreting source may be beneficial, particularly in nonmetastatic thoracic carcinoid tumors.314,315
Medical Treatment
Agents Inhibiting Steroidogenesis
The oral inhibitors of adrenal steroidogenesis are the most commonly used medical agents as treatment for Cushing’s syndrome; of these, metyrapone, ketoconazole, and mitotane are the most effective. Aminoglutethimide316 is no longer available, and trilostane317 is now rarely used. Etomidate is the only available agent that can be given parenterally. We usually would recommend partial inhibition of cortisol production (adjusted adrenal blockade) with frequent monitoring to identify a dosing regimen that maintains eucortisolism while avoiding adrenal insufficiency or excess. However, in some patients, particularly those with variable cortisol production, it can be difficult to achieve this. In these cases, full adrenal blockade with glucocorticoid replacement to avoid symptoms of adrenal insufficiency (a block-and-replace regimen) may be advocated. In all forms of treatment, patients and their physicians must be alert to the signs and symptoms of adrenal insufficiency.
Metyrapone: Metyrapone acts primarily to inhibit the enzyme 11β-hydroxylase,318 and the subsequent elevation of 11-deoxycortisol can be monitored in the serum of patients treated with metyrapone. The decrease in cortisol is rapid, with trough levels at 2 hours post dose, and a test dose of 750 mg with hourly cortisol estimation for 4 hours can be used to predict response: A rapid and sustained decrease in cortisol to less than approximately 7 µg/dL (≈200 nmol/L), as is often seen with ectopic ACTH and adrenal tumors, suggests that a smaller dose of metyrapone may be appropriate, whereas a decrease 10 to 12 µg/dL (≈300 nmol/L), as often is seen in Cushing’s disease, would indicate a higher dose requirement.319 Therapy is started at 0.75 to 1.5 g/day in three to four divided doses daily. The usual requirement is approximately 2 g/day, although higher doses (as high as 6 g/day) may be needed in ectopic ACTH syndrome.319 Metyrapone is useful in treating patients with Cushing’s syndrome from adrenal tumors, ectopic ACTH syndrome, and Cushing’s disease.319,320 The principal side effects are hirsutism and acne (as predicted by the increase in adrenal androgens), dizziness, and gastrointestinal upset. The androgenic effects can be particularly problematic and may preclude its use in some younger female patients. Hypokalemia, edema, and hypertension due to increased mineralocorticoids are infrequent319 but may require cessation of therapy. Our experience would suggest that the only major problems are associated with the increase in adrenal androgens; careful monitoring of treatment to avoid hypoadrenalism and education of the patient are required. Although not previously reported, we have seen a case of hemolysis in a patient with glucose-6-phosphate dehydrogenase deficiency.
Ketoconazole and Other Antifungals: Ketoconazole is an imidazole derivative whose primary indication is as an oral antifungal agent. However, reports of gynecomastia in some ketoconazole-treated patients led to the realization that it is an inhibitor of cytochrome P-450 enzymes, including side-chain cleavage, C17,20-lyase, 11β-hydroxylase, and 17β-hydroxylase.321,322 It has also been reported to have a direct effect on ectopic ACTH secretion from a thymic carcinoid tumor.323 Treatment for Cushing’s syndrome usually is started at a dose of 200 mg twice daily, and its onset of action is slower than metyrapone. It has been used successfully to lower cortisol levels in patients with Cushing’s syndrome of various causes, including adrenal carcinoma, ectopic ACTH syndrome, and invasive ACTH-producing pituitary carcinoma, with doses between 200 and 1200 mg/day required in as many as four divided daily doses.324–326 The principal side effect of ketoconazole is hepatotoxicity. Reversible elevation of hepatic serum transaminases occurs in approximately 5% to 10% of patients and need not result in discontinuation of the agent if levels remain below twofold to threefold the upper normal range. The incidence of serious hepatic injury is approximately 1 in 15,000 patients,327 and it can be fatal or may require liver transplantation.328,329 The hepatotoxicity appears to be idiosyncratic and has been reported within 7 days of the start of treatment in a patient with Cushing’s syndrome.330 Other adverse reactions of ketoconazole include skin rashes and gastrointestinal upset, but these occur in less than 15%,324 and one must always be wary of causing adrenal insufficiency.331 Because of its C17-20 lyase inhibition and consequent antiandrogenic properties, ketoconazole is particularly useful in female patients in whom hirsutism, which may be worsened with metyrapone, is an issue. Conversely, gynecomastia and reduced libido in male patients may be unacceptable and may require alternative agents. One further advantage of ketoconazole is its inhibition of cholesterol synthesis, particularly low-density lipoprotein cholesterol.332 We have found it to be particularly useful in combination with low doses of metyrapone. Fluconazole, another oral imidazole, is less well studied but has been shown to be effective, and it provides the advantage of less toxicity.333
Mitotane: Mitotane, or o,p′DDD, is derived from the family of insecticides that includes DDT. It inhibits adrenal steroidogenesis catalyzed by cholesterol desmolase,334 11- and 18-hydroxylase, and 3β-hydroxysteroid dehydrogenase.335 It also has marked direct cytotoxic effects on the zona fasciculata and the zona reticulosa, which led to its original use in high doses (5 to 20 g/day) as treatment for inoperable adrenocortical carcinoma.336,337 In this condition, it now is used more commonly as adjunctive treatment to surgery, and it does appear to prolong recurrence-free survival with lower doses of between 1 and 5 g/day.338,339 At such doses, it may take a longer time to reach therapeutic levels, and where possible, monitoring of serum levels should be undertaken. It is suggested that target levels should be between 15 and 20 µg/mL.338,340 Combined treatment of mitotane with standard chemotherapeutic agents such as cisplatin, etoposide, and doxorubicin has been used in a number of small studies in advanced adrenal carcinoma with variable benefit.341–343
In Cushing’s disease, mitotane alone in high doses (4 to 12 g/day) can achieve remission in as many as 83% of patients, but more commonly, it is used in lower doses (0.5 to 4 g/day), sometimes in combination with radiation therapy, with clinical and biochemical remission achieved in approximately 80%.344,345 At these doses, the onset of effect can take approximately 6 to 8 weeks, and additional adjunctive medical treatment may be needed in this interim. Similarly, the agent has a long half-life (18 to 159 days), due in part to its lipophilic properties, and it effects can last for weeks or months after discontinuation of therapy. Mitotane, alone or in combination with metyrapone or aminoglutethimide, also has proved useful as treatment for hypercortisolism associated with ectopic secretion of ACTH.346
The utility of mitotane is limited by its gastrointestinal and neurologic toxicity. Nausea and anorexia are common at doses as high as 4 g/day and are ubiquitous at more than 4 g/day.347 These side effects may be avoided by beginning at a dose of 0.5 to 1.0 g/day and increasing gradually, by 0.5 to 1.0 g every 1 to 4 weeks. Doses should be taken with meals or at bedtime with food. If significant adverse effects do occur, the drug should be discontinued for 3 to 5 days and then restarted at a lower dose.345 At higher doses and serum levels >20 µg/mL,340 neurologic side effects are common and include drowsiness, gait disturbances, dizziness or vertigo, confusion, and problems with language. Other adverse effects at any dose include fatigue (perhaps due to decreased cortisol levels), gynecomastia, skin rash, hypouricemia, elevated liver enzymes, and abnormal platelet function.337,348,349 The hypercholesterolemia, which is common even at low doses, can be reversed with 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors such as simvastatin.350 Mitotane is relatively contraindicated in women desiring fertility within 2 to 5 years. It may induce spontaneous abortion and may act as a teratogen, effects that may persist for a number of years after discontinuation because of deposition in fat.351 Mitotane increases hormone-binding proteins (cortisol-binding globulin, sex hormone binding–globulin352 and thyroxine-binding globulin). Therefore, total serum cortisol cannot be relied on to monitor therapy, and UFC should be used instead. The value of serum-free or salivary cortisol remains to be determined. In addition, mitotane increases the metabolic clearance of exogenously administered steroids,353 and replacement doses of glucocorticoid must be increased by approximately one third.
Etomidate: Etomidate, an imidazole-derived anesthetic agent, was reported in 1983 to have adrenolytic effects.354 Compared with the other imidazole derivative ketoconazole, etomidate more potently inhibits adrenocortical 11β-hydroxylase and shows similar inhibition of 17-hydroxylase, but it has less of an effect on C17-20 lyase.355 At higher concentrations, it also appears to have an effect on cholesterol side-chain cleavage.356 Short-term etomidate infusions can reduce severe hypercortisolemia in patients with Cushing’s syndrome of various causes at doses as high as 0.3 mg/kg/hr,357 although such high doses tend to be sedative.358 Most case reports of long-term therapy therefore have described the use of lower nonhypnotic total doses of between 1.2 and 8.3 mg/hr to good effect.359–362 It may be more difficult to normalize cortisol levels in patients with Cushing’s disease, which probably reflects increased ACTH drive from the pituitary, as opposed to relatively fixed production from an ectopic source.360 Clearly, etomidate is an effective adrenolytic agent that acts rapidly but is limited in its use by the fact that it has to be given parenterally. However, in this situation, it may be lifesaving. It is important to recognize that the etomidate preparations available in Europe are dissolved in an alcohol-based vehicle, although the currently available preparation in the United States uses propylene glycol, which may have potential side effects such as nephrotoxicity.361
Glucocorticoid Receptor Antagonists
Mifepristone (RU 486), the antiprogesterone antagonist that is marketed as an abortifacient, is also a competitive antagonist of glucocorticoid and androgen receptors. The major drawback is the lack of biochemical markers to monitor overtreatment, and its long half-life and minimal agonist activity leave the patient open to hypoadrenalism.363
Agents That Modulate ACTH Release
A number of agents, including the serotonin antagonists cyproheptadine364 and ritanserin,365 the dopamine agonists bromocriptine366 and cabergoline,367 and sodium valproate,368 have been examined in Cushing’s disease. The precise mechanism of action of many of these agents is incompletely understood, although most seem to reduce ACTH secretion through an effect on the hypothalamic-pituitary axis. Their efficacy in Cushing’s disease seems to be variable between individual patients; therefore, we do not recommend their routine use. However, more recent data on the use of cabergoline suggest that this drug may be more effective than was previously realized.
Somatostatin Analogues
Long-acting somatostatin analogues such as octreotide and lanreotide are used widely in the therapy of various neuroendocrine tumors, and somatostatin receptors have been demonstrated on corticotroph adenomas, in addition to ectopic sources of ACTH.369 Octreotide appears to inhibit ACTH release in Nelson’s syndrome but rarely in patients with Cushing’s disease, and this has been postulated to be due to somatostatin receptor downregulation from the circulating hypercortisolemia.370 In ectopic ACTH-secreting tumors, octreotide has produced a prolonged (>3 months) reduction in ACTH and hypercortisolemia in approximately 70% of published cases, but this high rate of response may be due to selective reporting. Preoperative assessment with pentetreotide scintigraphy may help predict which tumors might respond to treatment. Octreotide treatment produces a temporary response in GIP-dependent Cushing’s syndrome but is unhelpful in other causes of ACTH-independent Cushing’s syndrome.371 So far, little experience has been reported with use of the sustained-release preparations in treating Cushing’s syndrome. Much interest surrounds somatostatin analogues with a broader spectrum of activity for somatostatin receptor subtypes. Such an agent, pasireotide (SOM230; Novartis, Basel, Switzerland), has been shown in vitro to reduce human corticotroph proliferation and ACTH secretion,372 and early results in vivo are encouraging.373,374 Additional clinical studies are ongoing.
Thiazolidinediones
Rosiglitazone, a peroxisome-proliferator–activated receptor-γ (PPAR-γ) agonist, at suprapharmacologic doses has been shown in vitro and in animal models to suppress ACTH secretion and corticotroph tumor size.375 However, a number of clinical trials of the thiazolidinediones rosiglitazone and pioglitazone in patients with Cushing’s syndrome have generally been disappointing, with only short-term benefit seen in occasional patients even at high doses.376–380
Potential Novel Medical Agents
In the rare cases of Cushing’s syndrome due to AIMAH and aberrant receptor expression, specific receptor antagonists have proved useful at least in the short term, and further investigation is needed.381 Retinoic acid has been found to inhibit ACTH secretion and cell proliferation both in vitro in ACTH-producing tumor cell lines and cultured human corticotroph adenomas and in vivo in nude mice382 and dogs.383 The potential antisecretory and antiproliferative activities of this agent in Cushing’s syndrome have not to our knowledge been investigated further in man.
Special Clinical Scenarios
Most patients with Cushing’s syndrome demonstrate consistently elevated glucocorticoid values. A small subset show significant variability in glucocorticoid secretion, alternating normal and elevated values on a regular or irregular basis.384 The few cases of spontaneous remission of Cushing’s syndrome, including Cushing’s first patient, may fit into this category.2,385,386 The clinical course of patients with this type of intermittent, cyclic, or periodic Cushing’s syndrome may be invariant, usually with mild signs and cushingoid symptoms, or it may parallel the biochemical abnormalities, with exacerbation of cushingoid features that parallel increased glucocorticoid production. The etiologic distribution is altered; in a recent review, Cushing’s disease, ectopic ACTH secretion, and primary adrenal causes accounted for 54%, 26%, and 11%, respectively, of 65 cases, with the remainder being unclassified.387
Patients with periodic Cushing’s syndrome often show conflicting or “inappropriate” responses to standard diagnostic tests, particularly dexamethasone suppression.388 Discrepant urine tests have been reported in these patients, with elevated 17-OHCS and normal UFC excretion.389 If studied during a quiescent period, patients with nonpituitary disease may be misclassified as having Cushing’s disease, and those with “normal” responses to the LDDST may be incorrectly diagnosed as not having Cushing’s syndrome.390 Dynamic testing should be performed only during a sustained period of hypercortisolism, as documented by failure to suppress on an LDDST, concurrent increased evening salivary or plasma cortisol, and/or elevated UFC excretion.
Iatrogenic and Factitious Cushing’s Syndrome
Appropriate therapeutic but supraphysiologic doses of glucocorticoids given for a medical condition cause most cases of iatrogenic Cushing’s syndrome, which usually is an expected, unavoidable adverse effect of therapy. Exogenous hypercortisolism may result also when a prescribed dose of glucocorticoid is increased inappropriately by the patient. Although most common with oral agents, Cushing’s syndrome may result from glucocorticoids administered to the nasal or rectal mucosa, the tracheobronchial tree, or the skin.391–393 The use of all prescription drugs, over-the-counter medications, and herbal remedies,394 including nasal drops, inhalants, and topical agents, should be assessed in all cushingoid patients. Agents not given for their glucocorticoid activity, such as fludrocortisone acetate and megestrol,395 also may produce cushingoid features on occasion. Agents sold for cosmetic skin whitening may contain potent glucocorticoids.
Factitious Cushing’s syndrome, which may be a form of Münchausen’s syndrome, is rare. The typical suppression of plasma ACTH and dehydroepiandrosterone sulfate may lead to a mistaken diagnosis of primary adrenal disease.396 Plasma and urine cortisol values vary, depending on the route, schedule, and type of glucocorticoid ingested. For example, intravenous injection of hydrocortisone may suppress ACTH values and increase UFC levels without increasing single random plasma cortisol values.397 If basal urine or plasma cortisol values are low, it may be useful to screen the urine for synthetic glucocorticoids.396
Chronic Renal Failure
Cushing’s syndrome has been described in the setting of chronic renal failure only rarely.398–400 Plasma levels of cortisol are normal in chronic renal failure when assessed with radioimmunoassays using an organic extraction procedure,401,402 but they may be increased if other assay techniques are used.403 ACTH levels are increased.401 Glomerular filtration rates of less than 30 mL/min result in decreased cortisol excretion, and the UFC may be normal despite excessive cortisol production.404 ACTH and cortisol responses to ovine CRH may be suppressed in patients with renal failure, except for those undergoing continuous ambulatory peritoneal dialysis.402 The metabolism of dexamethasone is normal in chronic renal failure, but oral absorption can be altered in some patients, which may necessitate measurement of plasma dexamethasone levels.405,406 The reduced degree of suppression of cortisol by dexamethasone suggests a prolonged half-life of cortisol. Normal suppression of the overnight 1 mg LDDST is uncommon, and the 2 day LDDST does better in this regard.132,406,407 The cortisol response to insulin-induced hypoglycemia is normal or absent.405,408
Pediatric Cushing’s Syndrome
The most common presentation of Cushing’s syndrome in children is growth retardation, often with a decrease in height percentile over time as the weight percentile increases.409–411 However, hypercortisolemic patients with virilizing adrenal tumors may show growth acceleration; thus, the absence of growth failure does not exclude the diagnosis of Cushing’s syndrome.412 Other virilizing signs such as acne and hirsutism are seen in approximately 50% of patients regardless of etiology.411 Hypertension and striae are seen in approximately 50% of cases.413 Muscle weakness may be less common in the pediatric patient.70 This may reflect the effect of exercise rather than age because older patients who follow an exercise program tend to maintain strength. In addition to the spectrum of psychiatric and cognitive changes seen in adults, which can affect school performance, children may show “compulsive diligence” and actually do well academically.409 Depression is less common in children than in adults. Headache and fatigue are common.411 Cushing’s disease accounts for between 75% and 80% of Cushing’s syndrome in children and adolescents, but before the age of 10 years, ACTH-independent causes of Cushing’s syndrome are more common. Cushing’s disease has a male predominance in prepubertal children. Two primary adrenal causes of Cushing’s syndrome, McCune-Albright syndrome and PPNAD, are typically diseases of childhood or young adulthood. Signs of virilization or feminization in the very young (<4 years) suggest adrenal carcinoma. Ectopic secretion of ACTH occurs rarely in the pediatric population and usually is due to bronchial or thymic carcinoids.414
As mentioned previously, late-night salivary cortisol measurement has particular logistical benefits in children. Study of its utility in the pediatric population has been limited, and diagnostic criteria are not clear, although a cutoff of 0.27 µg/dL (7.5 nmol/L) has been suggested if the sample is taken around midnight.166,415 Serum cortisol measurement in inpatients has high sensitivity.416 In children, UFC should be corrected for body surface area (×1.72 m2).417 The standard 2 day LDDST adult protocol can be used in children weighing 40 kg or more, otherwise the dexamethasone dose is adjusted to 30 µg/kg/day.410 As in adults, there is good correlation between cortisol suppression on the LDDST and on the HDDST for the differential diagnosis; thus the latter is not necessary.418 Although it can be argued that the ectopic ACTH syndrome is so rare in children that the CRH test or BIPSS is not necessary, they do add reassurance in those with a negative pituitary MRI, which is the case in more than 50% of cases. In addition, BIPSS has arguably better accuracy in lateralization of the pituitary tumor.414 MRI is at least as useful as CT in the evaluation of adrenal causes.419
Adrenalectomy, either unilateral or bilateral depending on the cause, is first-line therapy in ACTH-independent Cushing’s syndrome. Transsphenoidal surgery is the treatment of choice in children with Cushing’s disease, with similar rates of remission as in adults in expert hands.420 Conventional radiotherapy used in the setting of failure to cure after transsphenoidal surgery performs even better than in adults with reported remission rates as high as 100%, with cure occurring within 12 months.421 Following pituitary surgery, plus or minus radiotherapy, the incidence of growth hormone deficiency is high, but prompt diagnosis and treatment with human growth hormone ensure acceptable growth acceleration and catch-up growth, although an abnormal body composition often persists.422 Similarly, normalization of reduced bone mineral density can be achieved.423
Cushing’s Syndrome in Pregnancy
The pregnant woman with possible Cushing’s syndrome presents a diagnostic challenge to the physician because of the physical and biochemical changes that are common to both conditions, including weight gain, fatigue, striae, hypertension, and glucose intolerance. The investigative screening process has to be based on the recognition of physiologic changes in pregnancy.424 Total serum cortisol levels increase in pregnancy, beginning in the first trimester and peaking at 6 months, with a decrease only after delivery, probably reflecting increased induction of hepatic corticosteroid-binding globulin production by estrogen. UFC excretion is normal in the first trimester and then rises up to threefold by term. Thus, only UFC values greater than threefold normal are diagnostic in the last two trimesters. Suppression to dexamethasone is blunted, but not because of reduced bioavailability of dexamethasone. The cortisol diurnal rhythm is maintained in pregnancy but with a higher nadir, and appropriate diagnostic cutoffs in pregnancy have yet to be established. Adrenal adenomas are frequently the cause of Cushing’s syndrome in pregnancy (40% to 50%), but it is interesting to note that ACTH levels may not be suppressed in these patients, possibly because of placental CRH stimulation of the pituitary corticotrophs or placental ACTH secretion. The HDDST may be useful in distinguishing these patients from those with Cushing’s disease. The CRH test also has been used to identify patients with Cushing’s disease, and no evidence of harm has been found in animal studies and in the small number of pregnant patients studied with this drug. MRI without gadolinium enhancement is considered safe in the third trimester, and its use in combination with the noninvasive tests discussed earlier should resolve most diagnostic issues. IPSS with appropriate additional radiation protection for the fetus should be reserved only for cases in which diagnostic uncertainty remains.
Although Cushing’s syndrome is rare in pregnancy, maternal hypercortisolism is associated with poor outcomes, both maternal and fetal. Definitive surgical treatment for adrenal or pituitary disease is recommended to achieve eucortisolemia, although adverse fetal outcomes may persist: The second trimester is probably the safest time for operative intervention. Medical treatment carries potential risk for the fetus and should be considered only as second-line therapy when the benefit outweighs the risk, and then generally only as an interim measure. Metyrapone is probably the adrenolytic agent of choice, although its association with preeclampsia has been reported. Ketoconazole has been utilized successfully in a small number of patients but is teratogenic in animals and therefore should be used cautiously.425
Prognosis
The life expectancy of patients with nonmalignant causes of Cushing’s syndrome, at one time a uniformly fatal illness, has improved dramatically with effective surgical and medical treatments and the availability of antibiotics, antihypertensive agents, and glucocorticoids. In a 1952 review, Plotz67 reported a 5-year mortality rate of 50% in actively hypercortisolemic patients, with 46% caused by bacterial infection and 40% due to cardiovascular complications (cardiac failure, cardiovascular accidents, or renal insufficiency). In 1961, the mortality rate was similar, but the causes had changed: Two thirds were due to postoperative adrenal crisis before cortisone was available or from metastatic adrenal cancer. Cardiovascular events related to hypertension (stroke, heart failure, renal failure, myocardial infarction) led to death in approximately 20%; infectious causes decreased to approximately 15%.71 Ten years later, in 1971, 30% of patients with benign causes of Cushing’s syndrome died within 5 years of diagnosis, most from cardiovascular disease or infection, despite decreased postoperative mortality.426 In 1979, a lower incidence of death (6%) was noted within 2 to 10 years of radiation therapy, mitotane, or combination treatment for Cushing’s disease.344 This improvement may reflect earlier detection of Cushing’s syndrome and better treatment for hypercortisolism and associated medical complications, such as hypertension, or lower perioperative mortality. Three studies on long-term survival in Cushing’s disease treated in the era of transsphenoidal surgery have been completed; two were epidemiologic studies from northern Spain66 and Denmark,61 and the third was a series from a single neurosurgical center where all patients had undergone transsphenoidal surgery.255 Investigators report varying standardized mortality ratios of 3.8, 1.7, and 0.98, respectively. The discrepancy in findings between the three series is not completely clear, but it is difficult to make absolute comparisons, not least because the study by Swearingen and colleagues undoubtedly is affected by selection bias. However, the latter two studies do appear to show that after curative transsphenoidal surgery, long-term mortality is not significantly different from that in the general population. This is perhaps surprising because increased cardiovascular risk markers and evidence of atherosclerotic disease persist when measured 5 years after remission of Cushing’s disease.115 The outcome of pediatric Cushing’s disease is excellent if treated at centers with appropriate experience.413
Cushing’s syndrome results in significant impairment in quality of life. Unfortunately, in the long term, this is improved only partially with treatment.275,427
The prognosis of the potentially malignant causes of Cushing’s syndrome is variable. Adrenal cancer, as reviewed earlier, has an extremely poor prognosis. Tumors that produce ACTH ectopically tend to have a poor prognosis, particularly when compared with tumors from the same tissue that do not produce ACTH. Small cell lung cancer, islet cell tumors, and thymic carcinoids428 illustrate this phenomenon. As many as 82% of patients with small cell lung cancer and Cushing’s syndrome die within 2 weeks from the start of chemotherapy.63 Among the causes of ectopic ACTH syndrome, pheochromocytoma and bronchial carcinoid appear to offer the best prognosis after tumor resection, but this is not universal.