[level-membership-for-neurology-category]
4 Cranial Nerve II
Optic Nerve and Visual System
Intraocular Optic Nerve
Clinical Vignette
The optic nerve is not a peripheral nerve but rather a central nervous system (CNS) tract containing central myelin formed by oligodendrocytes. It is composed of long axons, whose cell bodies comprise the ganglion cell layer of the inner retina (Figs. 4-1 and 4-2). The axons run in the retina’s nerve fiber layer to gather at the optic disk.
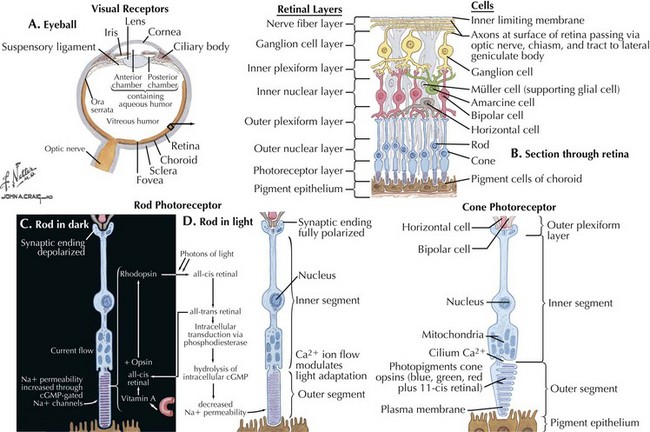
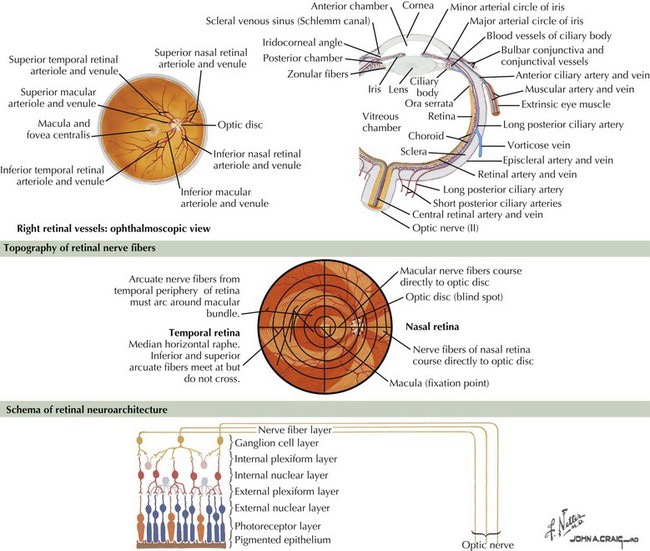
The optic nerve nominally begins when the axons of the ganglion cells (the nerve fiber layer of the retina) turn 90°, changing orientation from horizontal along the inner retinal surface to vertical, passing through the outer retina via the scleral canal (Fig. 4-3). The gathering of axons at the canal forms the optic disk (also, optic nerve head) of the fundus. Myelin is usually absent from the nerve fiber layer where the nerve exits the globe.
Clinical Presentations
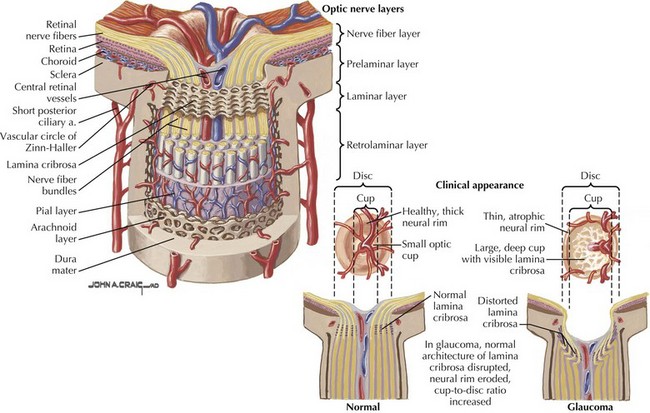
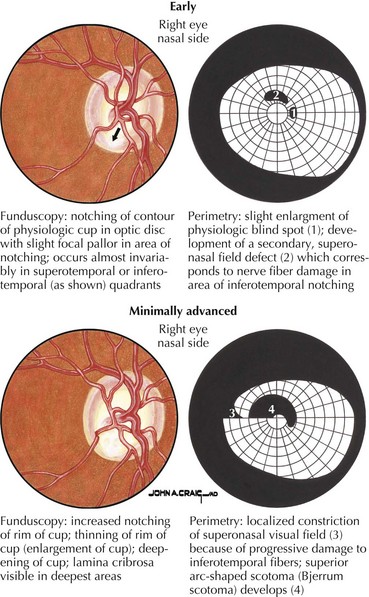
Primary open-angle glaucoma (POAG) is a chronic, progressive, degenerative disease of the optic nerve. Its usual hallmark is high intraocular pressure (IOP; above 21 mm Hg), but glaucoma without high IOP (normal pressure or low-tension glaucoma) is occasionally seen, especially in the elderly. The typical optic nerve finding is cupping atrophy (i.e., enlargement of the disk’s central cup as nerve fibers are lost), coupled by progressive visual field loss that often starts nasally, progresses superiorly and inferiorly, and finally extinguishes the central and temporal fields (Fig. 4-4). POAG is usually bilateral and asymmetric and the visual loss is permanent. The time course is measured in years, and because of the slow pace and the late involvement of the central field, patients may remain asymptomatic until the disease is quite advanced. It is essential that all standard eye examinations include screening IOP measurements and optic disk inspection.
CRAO, BRAO, and TMVL may also serve as a warning sign of impending hemispheric stroke. Identification and treatment of the embolic source, if one can be identified, becomes the main focus of therapy after the window for acute treatment of the involved eye has passed. CRAO is often a sign of carotid stenosis, the appropriate management of which will significantly reduce long-term stroke risk (see Chapter 55, “Ischemic Stroke”). Heart embolism is another cause and a full stroke investigation is usually required. Nevertheless, up to 40% of cases remain without a definite identifiable cause with the presumed mechanism relating to intrinsic narrowing of the retinal artery due to atherosclerosis or, less commonly, other arteritides or compression.
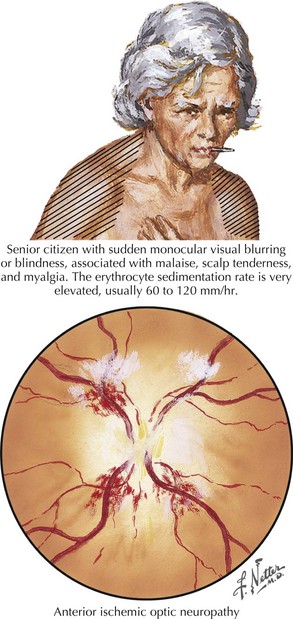
Anterior ischemic optic neuropathy can be divided into nonarteritic and arteritic (associated with temporal arteritis [TA]) and is caused by loss of blood flow in the short posterior ciliary arteries. Patients usually experience sudden and severe painless monocular visual loss, often on awakening. Examination classically reveals an altitudinal (superior or inferior) visual field loss, with a unilaterally swollen, hemorrhagic disk (Fig. 4-5). The disk loses its swelling and becomes pale within weeks. The visual loss in most cases does not change following the event but 20% may show measurable change for better or worse over days. In contrast to retinal artery occlusions, embolic AION is extremely rare. In most cases, AION occurs in middle-aged individuals who have a congenitally small, elevated (“crowded”) optic disk, or in those with one or more vascular disease risk factors, such as diabetes, hypertension, or sleep apnea. In these cases, a transient fall in blood pressure causes hypoperfusion of the posterior ciliary circulation and subsequent ischemic damage to the optic nerve head.
Papilledema (see Fig. 1-6) is bilateral optic nerve elevation and expansion due to high intracranial pressure (ICP). In mild cases, patients may have no visual symptoms. Moderate papilledema is typically accompanied by transient binocular visual obscurations, either spontaneously or during coughing, straining, or abrupt postural change. Other symptoms of high ICP may be present and include headaches (worse with recumbency) and diplopia (resulting from nonlocalizing abducens palsy; see Chapter 5). When visual loss occurs, it starts with blind spot enlargement (see Fig. 1-6), a nonspecific and often reversible change. Visual field loss resembling that of glaucoma can ensue, often over a period of many weeks. However, papilledema due to very high ICP can progress rapidly, with severe permanent visual loss within days.
Many pathophysiological mechanisms are associated with papilledema, including CNS tumor with mass effect or edema, obstructive hydrocephalus, meningitis, certain medications (e.g., tetracycline or vitamin A), and intracranial venous thrombosis or obstruction. Papilledema is occasionally seen without explanation in obese women of childbearing age and is then termed idiopathic intracranial hypertension (IIH; also, pseudotumor cerebri; see Chapter 11). Treatment involves only weight loss if the condition is mild and there is no evidence of progressive visual loss or debilitating headache. In progressive IIH, in addition to weight loss, carbonic anhydrase inhibitors such as acetazolamide (typically 1–2 g/day in divided doses) are used to reduce cerebrospinal fluid (CSF) production and optic nerve edema. When medical treatment fails, two surgical options exist: optic nerve sheath fenestration or CSF shunting either with lumboperitoneal or ventriculoperitoneal shunts.
Orbital and Intracanalicular Optic Nerve
Clinical Vignette
Multiple sclerosis (see Chapter 46), however, remains the chief cause of orbital optic nerve disease and is the initial manifestation in approximately 20% of patients. An additional 20% will eventually experience it throughout the course of the disease. It is estimated that more than 90% of patients suffering “isolated” optic neuritis will eventually receive a diagnosis of MS. Diagnostic testing in optic neuritis naturally mirrors that for MS, with brain MRI and CSF analysis being the primary tools.
Clinical Presentations
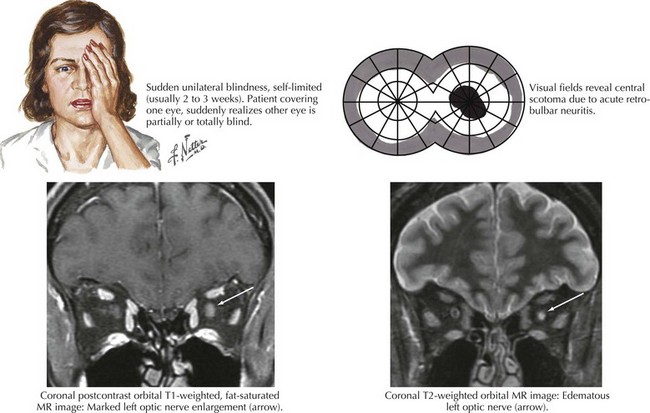
Optic neuritis is the clinical syndrome of subacute painful, monocular visual loss. The pain often precedes visual loss by a day or more and is a periorbital ache made worse with eye movements. Ensuing visual loss is often sudden and severe, with perceived worsening over several days. The degree of visual field loss varies, but a central scotoma is the classic finding (Fig. 4-6). Examination may also demonstrate loss of central acuity, contrast sensitivity, and color perception in the affected eye.
Optic Chiasm
Clinical Vignette
The optic chiasm is the intersection of the optic nerves from each eye and is located above the pituitary body that lies within the sella turcica of the sphenoid bone, and covered by the diaphragm sellae (Fig. 4-7). The chiasmatic cistern is located between the chiasm and the diaphragm sella. Superior to the chiasm is the third ventricle. The internal carotid arteries flank the optic chiasm laterally and then bifurcate into the anterior and middle cerebral arteries. The anterior cerebral arteries and the anterior communicating artery are anterior to the optic chiasm.
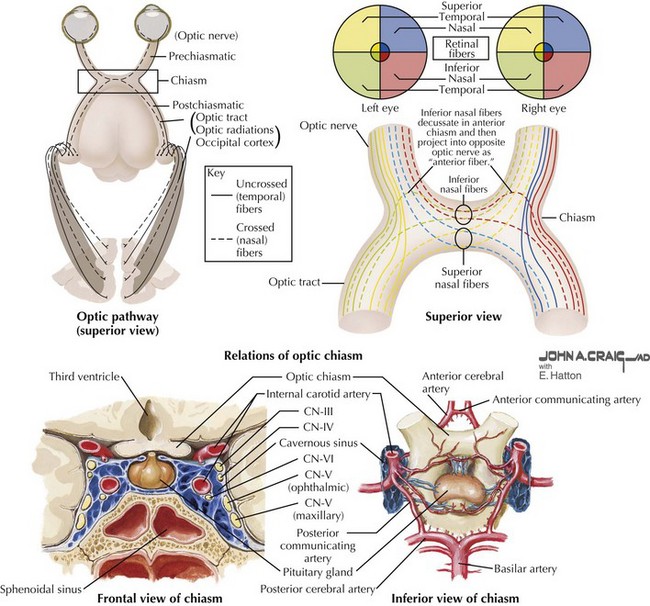
Within the chiasm, axons from the temporal retina (nasal field) comprise its lateral aspect and remain ipsilateral as they pass through the chiasm to the optic tract. In contrast, the nasal retinal fibers decussate, carrying temporal visual field information to the contralateral side. Inferior nasal fibers decussate within the chiasm more anteriorly than superior ones. As the inferior nasal retinal fibers approach the posterior aspect of the chiasm, the fibers shift to occupy the lateral aspect of the contralateral optic tract (see Fig. 4-7).
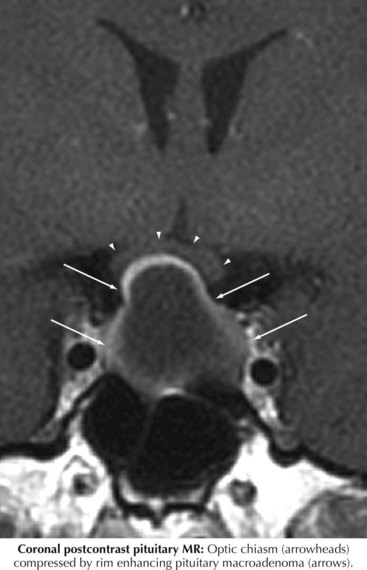
The location of the chiasm renders it vulnerable to compression from vascular structures (e.g., aneurysm near the origin of the anterior communicating artery or the ophthalmic artery), from tumors of the meninges, from sphenoid sinus masses, and most important from the pituitary (Fig. 4-8).
Clinical Presentations
Central chiasmatic lesions most commonly produce a bitemporal hemianopia (Fig. 4-9A) that ensues when the optic chiasm is compressed or damaged midsagittally at its decussation. Such lesions preferentially affect the crossing nasal retinal fibers responsible for temporal vision, as in the vignette in this chapter.
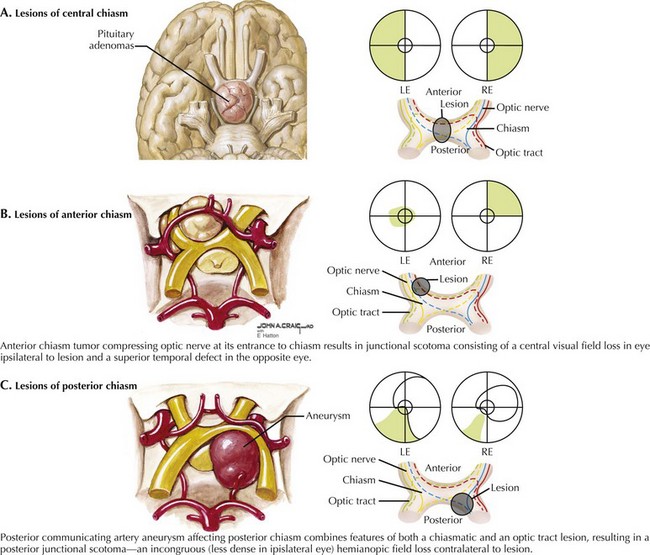
Variants on the classic bitemporal hemianopia are seen with compression of the optic nerve at its entrance to the anterior chiasm, resulting in a junctional scotoma, with central visual loss in the ipsilateral eye and a superotemporal defect in the other. The field loss in the contralateral eye reflects involvement of the opposite inferior nasal optic nerve fibers that swing forward into the ipsilateral anterior chiasm (Willebrand knee) before decussating to the optic tract (Fig. 4-9B).
Posterior optic chiasm lesions lead to a posterior junctional scotoma, which displays the features of chiasmatic and optic tract lesions. The classic finding is incongruous (less dense in the ipsilateral eye) hemianopic visual field loss contralateral to the lesion from involvement of the anterior optic tract and an inferotemporal visual field loss in the ipsilateral eye—from pressure on the posterior chiasm affecting the late-crossing superotemporal retinal fibers (Fig. 4-9C). Such defects occur in lesions located near the anterior aspect of the third ventricle that approach the chiasm posteromedially. The incongruous nature of the hemianopsia is caused by the incomplete intermixing of the decussating fibers entering the optic tract with their corresponding uncrossed fibers from the contralateral eye.
Most commonly, chiasmatic compression results from a benign pituitary adenoma (see Chapter 52). These are common brain tumors, and with high-resolution MRI imaging are detected in 10% of patients. Tumors smaller than 10 mm, termed microadenomas, are generally too small to place significant pressure on the optic chiasm. and are usually discovered because of the effects of excess pituitary hormone (e.g., prolactin) secretion. Small nonsecreting adenomas can be found as an incidental finding on brain MRI obtained for other reasons. Once a tumor grows sufficiently and obliterates the 10 mm distance from diaphragm sella to the chiasm, the potential for visual loss exists. Typically, the chiasm can accommodate slowly growing tumors, so that chiasmatic impingement or displacement by such tumors may be seen without any field defect. When the macroadenoma, however, reaches 20–25 mm, field defects are likely. The usual indications for surgical excision are continued tumor growth or the presence of visual compromise. Prolactinomas can often be treated medically using bromocriptine or cabergoline to shrink the tumor. Similarly, mitotane has been used for adrenocorticotropic hormone–secreting tumors, and somatostatin analogues for tumors secreting human growth hormone. Failure of medical therapy leaves the options of transsphenoidal surgical excision, or perhaps precision radiotherapy (e.g., gamma-knife).
Posterior Visual Afferent System: Optic Tracts, Lateral Geniculate Nucleus, Optic Radiations
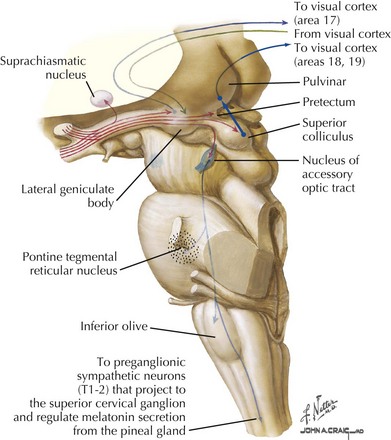
The axons comprising the optic tract are still those emanating from the retinal ganglion cells, which have yet to synapse. Nevertheless, after they leave the chiasm for the optic tract, they nominally become part of the “posterior visual pathway” (Fig. 4-10). Axons of the optic tract course via the anterior limb of the internal capsule, between the tuber cinereum and the anterior perforated substance, then continue posteriorly as a band of flattened fibers around the cerebral peduncles to synapse in the lateral geniculate nucleus (LGN) within the thalamus. The LGN is a thalamic relay nucleus that serves as the synapse point of the retinal ganglion cells. It comprises six gray matter layers separated by five white matter layers. The layers are folded over, forming a bend or small knee. Each layer has a retinotopic organization, creating a map of the contralateral hemifield (Fig. 4-11). The ratio of geniculate cells to retinal axons is approximately 1 : 1. Retinal input to the LGN comprises only one fifth of its afferent fibers. The remainder comes from the mesencephalic reticular formation, posterior parietal cortex, occipital cortex, and other thalamic nuclei. The LGN may use these nonretinal elements to “screen” the visual input, gating certain inputs to the visual cortex while blocking other signals, depending on the relevance of the inputs.
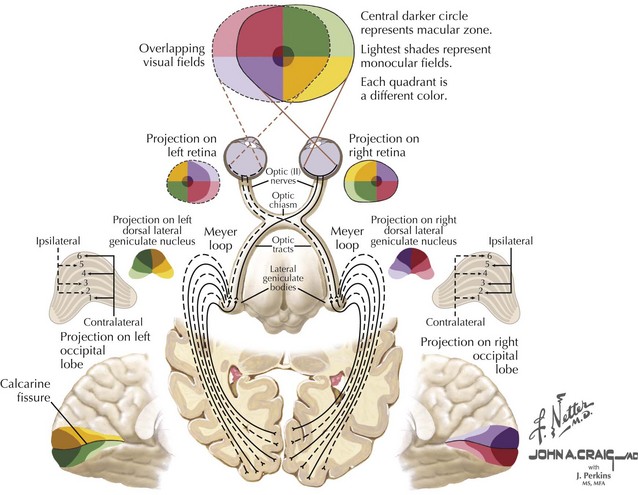
The optic radiations are myelinated axons emanating from LGN and course to the primary visual cortex. After they leave the LGN, they continue through the posterior limb of the internal capsule. Most fibers take a fairly direct path to the calcarine cortex, following the curve of the corona radiata through the parietal lobe to the occipital lobe. However, the most inferior axons (Meyer loop) that carry visual information from the opposite superior field detour laterally around the lateral ventricles and through the posterior temporal lobe (Fig. 4-11). Therefore, stroke or injury confined to this portion of the temporal lobe affects only this portion of the optic radiations. Meyer loop fibers rejoin the rest of the optic radiations after their detour.
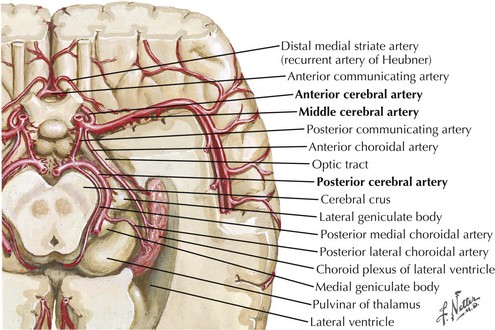
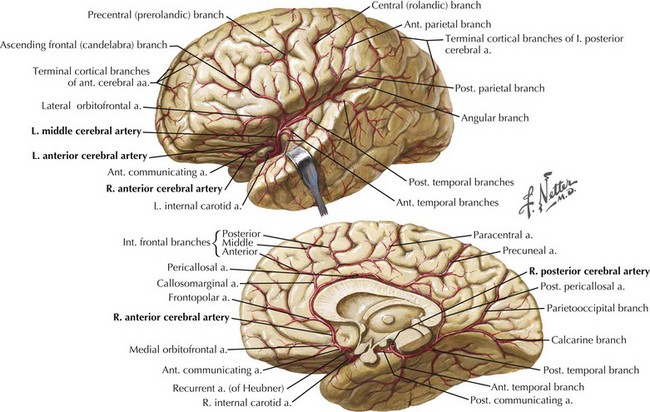
Five primary arteries supply blood to the optic radiation: the anterior and posterior choroidal arteries, the middle and posterior cerebral arteries, and the calcarine artery (Fig. 4-12). The anterior choroidal artery supplies the anterior portion of the optic radiations, the optic tract, and the lateral geniculate body. The anterior optic radiations are also fed by a meshwork of branches from the posterior choroidal arteries. The middle portion of the optic radiations, however, is fed via the deep optic branch of the middle cerebral artery, which lies lateral to the ventricle. The posterior portion of the optic radiation is fed by the posterior cerebral artery and one of its branches, the calcarine artery.
Differential and Diagnostic Approach
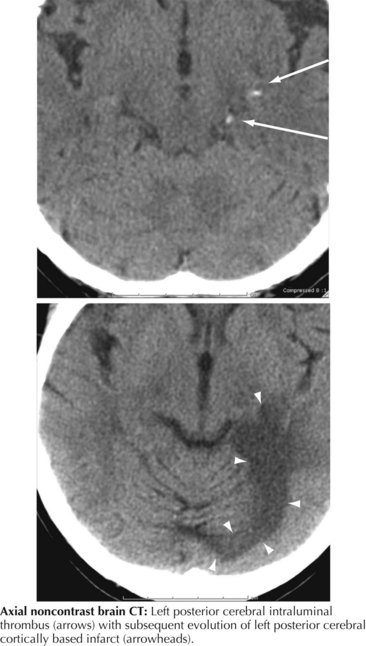
As with any visual loss, the diagnostic course for hemianopic visual loss includes complete eye examination, visual fields with attention to assessment of central acuity, pupillary reactions, pursuit movements, and funduscopy. The presence of additional neurologic or systemic symptoms and the speed of onset may help both localize the lesion and suggest an etiology. Review of past medical history may reveal if the patient has known risks for some etiologies, such as stroke, demyelination, or metastasis. The most important ancillary test is diagnostic imaging. Typically, MRI examination is recommended as best able to detect, and distinguish among, the potential pathologies. Diffusion-weighted images can be particularly useful in defining recent stroke (see Chapter 55). Management and therapy are dictated by the etiology.
Primary Visual Cortex and Visual Association Cortices
Clinical Vignette
Axons of the optic radiations synapse with the primary visual cortex. A unique white stripe or stria (stripe or line of Gennari for the discovering anatomist) represents a myelin-rich cortical layer; it is easily seen in gross sections through the cortex and bespeaks the layered, highly structured organization of V1 (also known as the primary visual cortex, the striate cortex, or Brodmann area 17). Primarily located on the mesial surface of the occipital lobe within and surrounding the calcarine fissure, the most posterior aspect of V1 typically wraps around the posterior (occipital) pole for a short distance (Fig. 4-13).
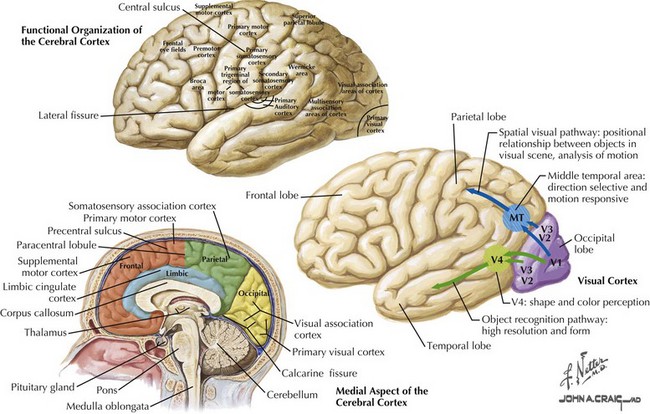
The blood supply of the striate cortex primarily derives from the calcarine artery, a branch of the posterior cerebral artery, and sometimes the middle cerebral artery, or anastomoses from it (Fig. 4-14).The calcarine artery is a major supply to the visual area; however, in 75% of cases, other arteries contribute as well: the posterior temporal or parietooccipital arteries, and, occasionally, anastomotic connections from the middle cerebral artery.
Clinical Presentations
Features of homonymous hemianopias that suggest occipital lobe origin include extremely congruous partial defects between eyes, macular sparing, central homonymous defects, keyhole defects, as well as temporal crescent defects. Because of the specialized nature of V1, lesions in it affect only the vision, without other neurologic dysfunction (except, occasionally, headache). In addition to the above, striate cortex lesions produce no signs of anterior visual pathway involvement such as optic pallor or relative afferent papillary defect. Typically, central acuity in the preserve field is normal (Fig. 4-15).
Future Directions
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
4 Cranial Nerve II
Optic Nerve and Visual System
Intraocular Optic Nerve
Clinical Vignette
The optic nerve is not a peripheral nerve but rather a central nervous system (CNS) tract containing central myelin formed by oligodendrocytes. It is composed of long axons, whose cell bodies comprise the ganglion cell layer of the inner retina (Figs. 4-1 and 4-2). The axons run in the retina’s nerve fiber layer to gather at the optic disk.
The optic nerve nominally begins when the axons of the ganglion cells (the nerve fiber layer of the retina) turn 90°, changing orientation from horizontal along the inner retinal surface to vertical, passing through the outer retina via the scleral canal (Fig. 4-3). The gathering of axons at the canal forms the optic disk (also, optic nerve head) of the fundus. Myelin is usually absent from the nerve fiber layer where the nerve exits the globe.
Clinical Presentations
Primary open-angle glaucoma (POAG) is a chronic, progressive, degenerative disease of the optic nerve. Its usual hallmark is high intraocular pressure (IOP; above 21 mm Hg), but glaucoma without high IOP (normal pressure or low-tension glaucoma) is occasionally seen, especially in the elderly. The typical optic nerve finding is cupping atrophy (i.e., enlargement of the disk’s central cup as nerve fibers are lost), coupled by progressive visual field loss that often starts nasally, progresses superiorly and inferiorly, and finally extinguishes the central and temporal fields (Fig. 4-4). POAG is usually bilateral and asymmetric and the visual loss is permanent. The time course is measured in years, and because of the slow pace and the late involvement of the central field, patients may remain asymptomatic until the disease is quite advanced. It is essential that all standard eye examinations include screening IOP measurements and optic disk inspection.
CRAO, BRAO, and TMVL may also serve as a warning sign of impending hemispheric stroke. Identification and treatment of the embolic source, if one can be identified, becomes the main focus of therapy after the window for acute treatment of the involved eye has passed. CRAO is often a sign of carotid stenosis, the appropriate management of which will significantly reduce long-term stroke risk (see Chapter 55, “Ischemic Stroke”). Heart embolism is another cause and a full stroke investigation is usually required. Nevertheless, up to 40% of cases remain without a definite identifiable cause with the presumed mechanism relating to intrinsic narrowing of the retinal artery due to atherosclerosis or, less commonly, other arteritides or compression.
Anterior ischemic optic neuropathy can be divided into nonarteritic and arteritic (associated with temporal arteritis [TA]) and is caused by loss of blood flow in the short posterior ciliary arteries. Patients usually experience sudden and severe painless monocular visual loss, often on awakening. Examination classically reveals an altitudinal (superior or inferior) visual field loss, with a unilaterally swollen, hemorrhagic disk (Fig. 4-5). The disk loses its swelling and becomes pale within weeks. The visual loss in most cases does not change following the event but 20% may show measurable change for better or worse over days. In contrast to retinal artery occlusions, embolic AION is extremely rare. In most cases, AION occurs in middle-aged individuals who have a congenitally small, elevated (“crowded”) optic disk, or in those with one or more vascular disease risk factors, such as diabetes, hypertension, or sleep apnea. In these cases, a transient fall in blood pressure causes hypoperfusion of the posterior ciliary circulation and subsequent ischemic damage to the optic nerve head.
Papilledema (see Fig. 1-6) is bilateral optic nerve elevation and expansion due to high intracranial pressure (ICP). In mild cases, patients may have no visual symptoms. Moderate papilledema is typically accompanied by transient binocular visual obscurations, either spontaneously or during coughing, straining, or abrupt postural change. Other symptoms of high ICP may be present and include headaches (worse with recumbency) and diplopia (resulting from nonlocalizing abducens palsy; see Chapter 5). When visual loss occurs, it starts with blind spot enlargement (see Fig. 1-6), a nonspecific and often reversible change. Visual field loss resembling that of glaucoma can ensue, often over a period of many weeks. However, papilledema due to very high ICP can progress rapidly, with severe permanent visual loss within days.
Many pathophysiological mechanisms are associated with papilledema, including CNS tumor with mass effect or edema, obstructive hydrocephalus, meningitis, certain medications (e.g., tetracycline or vitamin A), and intracranial venous thrombosis or obstruction. Papilledema is occasionally seen without explanation in obese women of childbearing age and is then termed idiopathic intracranial hypertension (IIH; also, pseudotumor cerebri; see Chapter 11). Treatment involves only weight loss if the condition is mild and there is no evidence of progressive visual loss or debilitating headache. In progressive IIH, in addition to weight loss, carbonic anhydrase inhibitors such as acetazolamide (typically 1–2 g/day in divided doses) are used to reduce cerebrospinal fluid (CSF) production and optic nerve edema. When medical treatment fails, two surgical options exist: optic nerve sheath fenestration or CSF shunting either with lumboperitoneal or ventriculoperitoneal shunts.
Orbital and Intracanalicular Optic Nerve
Clinical Vignette
Multiple sclerosis (see Chapter 46), however, remains the chief cause of orbital optic nerve disease and is the initial manifestation in approximately 20% of patients. An additional 20% will eventually experience it throughout the course of the disease. It is estimated that more than 90% of patients suffering “isolated” optic neuritis will eventually receive a diagnosis of MS. Diagnostic testing in optic neuritis naturally mirrors that for MS, with brain MRI and CSF analysis being the primary tools.
Clinical Presentations
Optic neuritis is the clinical syndrome of subacute painful, monocular visual loss. The pain often precedes visual loss by a day or more and is a periorbital ache made worse with eye movements. Ensuing visual loss is often sudden and severe, with perceived worsening over several days. The degree of visual field loss varies, but a central scotoma is the classic finding (Fig. 4-6). Examination may also demonstrate loss of central acuity, contrast sensitivity, and color perception in the affected eye.
Indirect traumatic optic neuropathy
[/not-level-membership-for-neurology-category]
