Chapter 4 Coronary Physiology and Atherosclerosis
ANATOMY AND PHYSIOLOGY OF BLOOD VESSELS
Normal Artery Wall
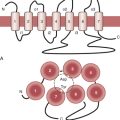
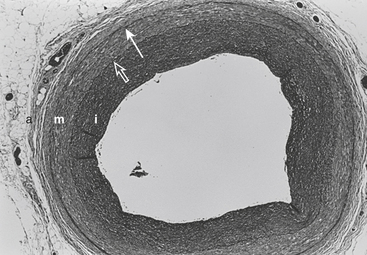
The arterial lumen is lined by a monolayer of endothelial cells that overlies smooth muscle cells (Fig. 4-1). The inner layer of smooth muscle cells, known as the intima, is circumscribed by the internal elastic lamina. Between the internal elastic lamina and external elastic lamina is another layer of smooth muscle cells, the media. Outside the external elastic lamina is an adventitia that is sparsely populated by cells and microvessels of the vasa vasorum.
Endothelium
Endothelium-Derived Relaxing Factors
The first vasoactive endothelial substance to be discovered was prostacyclin (PGI2), a product of the cyclooxygenase pathway of arachidonic acid metabolism (Box 4-1). The production of PGI2 is activated by shear stress, pulsatility of flow, hypoxia, and a variety of vasoactive mediators. Upon production it leaves the endothelial cell and acts in the local environment to cause relaxation of the underlying smooth muscle or to inhibit platelet aggregation. Both actions are mediated by the stimulation of adenylyl cyclase in the target cell to produce cyclic adenosine monophosphate (cAMP).
BOX 4-1 Endothelium-Derived Relaxing and Contracting Factors
Healthy endothelial cells have an important role in modulating coronary tone by producing:
It has been shown that many physiologic stimuli cause vasodilation by stimulating the release of a labile, diffusible, nonprostanoid molecule termed endothelium-derived relaxing factor (EDRF), now known to be nitric oxide (NO). NO is the basis of a widespread paracrine signal transduction mechanism whereby one cell type can modulate the behavior of adjacent cells of a different type.1,2 NO is a very small lipophilic molecule that can readily diffuse across biologic membranes and into the cytosol of nearby cells. The half-life of the molecule is less than 5 seconds so that only the local environment can be affected. NO is synthesized from the amino acid L-arginine by NO synthase (NOS). When NO diffuses into the cytosol of the target cell, it binds with the heme group of soluble guanylate cyclase, resulting in a 50- to 200-fold increase in production of cyclic guanosine monophosphate (cGMP), its second messenger. If the target cells are vascular smooth muscle cells, vasodilation occurs; if the target cells are platelets, adhesion and aggregation are inhibited.
It is likely that NO is the final common effector molecule of nitrovasodilators (including sodium nitroprusside and organic nitrates such as nitroglycerin). The cardiovascular system is in a constant state of active vasodilation that is dependent on the generation of NO. The molecule is more important in controlling vascular tone in veins and arteries compared with arterioles. Abnormalities in the ability of the endothelium to produce NO likely play a role in diseases such as diabetes, atherosclerosis, and hypertension. The venous circulation of humans seems to have a lower basal release of NO and an increased sensitivity to nitrovasodilators compared with the arterial side of the circulation.3
Endothelium-Derived Contracting Factors
Contracting factors produced by the endothelium include prostaglandin H2, thromboxane A2 (via cyclooxygenase), and the peptide endothelin. Endothelin is a potent vasoconstrictor peptide (100-fold more potent than norepinephrine).4
Endothelial Inhibition of Platelets
A primary function of endothelium is to maintain the fluidity of blood. This is achieved by the synthesis and release of anticoagulant (e.g., thrombomodulin, protein C), fibrinolytic (e.g., tissue-type plasminogen activator), and platelet inhibitory (e.g., PGI2, NO) substances (Box 4-2). Mediators released from aggregating platelets stimulate the release of NO and PGI2 from intact endothelium, which act together to increase blood flow and decrease platelet adhesion and aggregation, thereby flushing away microthrombi and maintaining the patency of the vessel.
DETERMINANTS OF CORONARY BLOOD FLOW
Perfusion Pressure and Myocardial Compression
Coronary blood flow is proportional to the pressure gradient across the coronary circulation (Box 4-3). This gradient is calculated by subtracting downstream coronary pressure from the pressure in the root of the aorta.
Myocardial Metabolism
Myocardial blood flow, like flow in the brain and skeletal muscle, is primarily under metabolic control. Even when the heart is cut off from external control mechanisms (neural and humoral factors), its ability to match blood flow to its metabolic requirements is almost unaffected. Because coronary venous oxygen tension is normally 15 to 20 mm Hg, there is only a small amount of oxygen available through increased extraction. A major increase in cardiac oxygen consumption (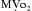 ), beyond the normal resting value of 80 to 100 mL O2/100 g of myocardium, can occur only if oxygen delivery is increased by augmentation of coronary blood flow. Normally, flow and metabolism are closely matched so that over a wide range of oxygen consumption coronary sinus oxygen saturation changes little.5
), beyond the normal resting value of 80 to 100 mL O2/100 g of myocardium, can occur only if oxygen delivery is increased by augmentation of coronary blood flow. Normally, flow and metabolism are closely matched so that over a wide range of oxygen consumption coronary sinus oxygen saturation changes little.5
Hypotheses of metabolic control propose that vascular tone is linked either to a substrate that is depleted, such as oxygen or adenosine triphosphate (ATP), or to the accumulation of a metabolite such as carbon dioxide (CO2) or hydrogen ion (Box 4-4). Adenosine has been proposed in both categories.
Neural and Humoral Control
 causes a metabolically-mediated coronary vasoconstriction. The direct effect of activation of cholinergic receptors on coronary vessels is vasodilation. These direct effects can be abolished by atropine.
causes a metabolically-mediated coronary vasoconstriction. The direct effect of activation of cholinergic receptors on coronary vessels is vasodilation. These direct effects can be abolished by atropine.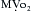 is fixed, coronary blood flow remains relatively constant between mean arterial pressures of 60 to 140 mm Hg.
is fixed, coronary blood flow remains relatively constant between mean arterial pressures of 60 to 140 mm Hg.Coronary Reserve
Myocardial ischemia causes intense coronary vasodilation. Following a 10- to 30-second coronary occlusion, restoration of perfusion pressure is accompanied by a marked increase in coronary flow. This large increase in flow, which can be five or six times resting flow in the dog, is termed reactive hyperemia. The repayment volume is greater than the debt volume. There is, however, no overpayment of the oxygen debt because oxygen extraction falls during the hyperemia. The presence of high coronary flows when coronary venous oxygen content is high suggests that mediators other than oxygen are responsible for this metabolically-induced vasodilation. The difference between resting coronary blood flow and peak flow during reactive hyperemia represents the autoregulatory coronary flow reserve: the further capacity of the arteriolar bed to dilate in response to ischemia.6
Transmural Blood Flow
It is well known that when coronary perfusion pressure is inadequate, the inner one third to one fourth of the left ventricular wall is the first region to become ischemic or necrotic.7 This increased vulnerability of the subendocardium may be due to an increased demand for perfusion or a decreased supply, compared with the outer layers.
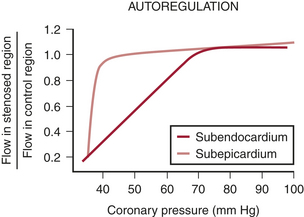
If coronary pressure is gradually reduced, autoregulation is exhausted and flow decreases in the inner layers of the left ventricle before it begins to decrease in the outer layers (Fig. 4-2). This indicates that there is less flow reserve in the subendocardium than in the subepicardium.
ATHEROSCLEROSIS
The atherosclerotic lesion consists of an excessive accumulation of smooth muscle cells in the intima, with quantitative and qualitative changes in the noncellular connective tissue components of the artery wall and intracellular and extracellular deposition of lipoproteins and mineral components (e.g., calcium). By definition, atherosclerosis is a combination of “atherosis” and “sclerosis.” The latter term, sclerosis, refers to the hard collagenous material that accumulates in lesions and is usually more voluminous than the pultaceous “gruel” of the atheroma (Fig. 4-3).
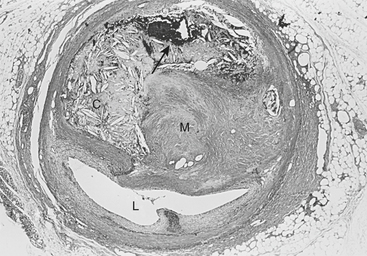
Stary noted that the earliest detectable change in the evolution of coronary atherosclerosis in young people was the accumulation of intracellular lipid in the subendothelial region, giving rise to lipid-filled macrophages or “foam cells.”8 Grossly, a collection of foam cells may give the artery wall the appearance of a “fatty streak.” In general, fatty streaks are covered by a layer of intact endothelium and are not characterized by excessive smooth muscle cell accumulation. At later stages of atherogenesis, extracellular lipoproteins accumulate in the musculoelastic layer of the intima, eventually forming an avascular core of lipid-rich debris that is separated from the central arterial lumen by a fibrous cap of collagenous material. Foam cells are not usually seen deep within the atheromatous core but are frequently found at the periphery of the lipid core.
Arterial Wall Inflammation
A number of studies have demonstrated the presence of monocytes/macrophages and T lymphocytes in the arteries of not only advanced lesions but also early atherosclerotic lesions of young adults.9 Moreover, in experimental atherosclerosis, leukocyte infiltration into the vascular wall is known to precede smooth muscle cell hyperplasia. Once inside the artery wall, mononuclear cells may play several important roles in lesion development. For example, monocytes may transform into macrophages and become involved in the local oxidation of low-density lipoproteins (LDLs) and accumulation of oxidized LDLs. Alternatively, macrophages in the artery wall may act as a rich source of factors that, for example, promote cell proliferation, migration, or the breakdown of local tissue barriers. The latter process of local tissue degradation may be very important for the initiation of acute coronary artery syndromes because loss of arterial wall integrity may lead to plaque fissuring or rupture.
Role of Lipoproteins in Lesion Formation
One of the major consequences of cholesterol accumulation in the artery wall is thought to be the impairment of endothelial function. The endothelium is more than a physical barrier between the bloodstream and the artery wall. Under normal conditions, the endothelium is capable of modulating vascular tone (e.g., via NO), thrombogenicity, fibrinolysis, platelet function, and inflammation. In the presence of traditional risk factors, particularly dyslipidemias, these protective endothelial functions are reduced or lost. A number of clinical studies demonstrate dramatic improvements in endothelial function, as well as cardiovascular morbidity and mortality, with the use of inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, or “statins.”10
PATHOPHYSIOLOGY OF CORONARY BLOOD FLOW
Coronary Artery Stenoses and Plaque Rupture
Coronary atherosclerosis is a chronic disease that develops over decades, remaining clinically silent for prolonged periods of time (Box 4-5). Clinical manifestations of CAD occur when the atherosclerotic plaque mass encroaches on the vessel lumen and obstructs coronary blood flow, causing angina. Alternatively, cracks or fissures may develop in the atherosclerotic lesions and result in acute thromboses that cause unstable angina or myocardial infarction.
Patients with stable angina typically have lesions with smooth borders on angiography. Only a minority of coronary lesions are concentric, with most having a complex geometry varying in shape over their length. Eccentric stenoses, with a remaining pliable, musculoelastic arc of normal wall, can vary in diameter and resistance in response to changes in vasomotor tone or intraluminal pressure. The majority of human coronary stenoses are compliant. The intima of the normal portion of the vessel wall is often thickened, making endothelial dysfunction probable. In contrast, patients with unstable angina usually have lesions characterized by overhanging edges, scalloped or irregular borders, or multiple irregularities. These complicated stenoses likely represent ruptured plaque or partially occlusive thrombus or both.11 Superficial intimal injury (plaque erosions) and intimal tears of variable depth (plaque fissures) with overlying microscopic mural thrombosis are commonly found in atherosclerotic plaques. In the absence of obstructive luminal thrombosis, these intimal injuries do not cause clinical events. However, disruption of the fibrous cap, or plaque rupture, is a more serious event that typically results in the formation of clinically significant arterial thromboses. From autopsy studies it is known that rupture-prone plaques tend to have a thin, friable fibrous cap. The site of plaque rupture is thought to be the shoulder of the plaque, where substantial numbers of mononuclear inflammatory cells are commonly found.12 The mechanisms responsible for the local accumulation of these cells at this location in the plaque are unknown; presumably, monocyte chemotactic factors, the expression of leukocyte cell adhesion molecules, and specific cytokines are involved. Moreover, macrophages in plaques have been shown to express factors such as stromelysin, which promote the breakdown of the extracellular matrix and thereby weaken the structural integrity of the plaque.
Coronary Collateral Vessels
Coronary collateral vessels are anastomotic connections, without an intervening capillary bed, between different coronary arteries or between branches of the same artery. In the normal human heart, these vessels are small and have little or no functional role. In patients with CAD, well-developed coronary collateral vessels may play a critical role in preventing death and myocardial infarction. Individual differences in the capability of developing a sufficient collateral circulation is a determinant of the vulnerability of the myocardium to coronary occlusive disease.13
It has been estimated that, in humans, perfusion via collateral vessels can equal perfusion via a vessel with a 90% diameter obstruction. Although coronary collateral flow can be sufficient to preserve structure and resting myocardial function, muscle dependent on collateral flow usually becomes ischemic when oxygen demand rises above resting levels. It is possible that evidence from patients with angina underestimates collateral function of the population of all patients with CAD.14
Determinants of Myocardial Oxygen Supply/Demand Ratio
An increase in myocardial oxygen requirement beyond the capacity of the coronary circulation to deliver oxygen results in myocardial ischemia (Box 4-6). This is the most common mechanism leading to ischemic episodes in chronic stable angina and during exercise testing. Intraoperatively, the anesthesiologist must measure and control the determinants of myocardial oxygen consumption and protect the patient from “demand” ischemia. The major determinants of myocardial oxygen consumption are heart rate, myocardial contractility, and wall stress (chamber pressure × radius/wall thickness).
Dynamic Stenosis
Patients with CAD can have variable exercise tolerance during the day and between days. Ambulatory monitoring of the ECG has demonstrated that ST-segment changes indicative of myocardial ischemia, in the absence of changes in oxygen demand, are common.15 These findings are explained by variations over time in the severity of the obstruction to blood flow imposed by coronary stenoses.
Although the term hardening of the arteries suggests rigid, narrowed vessels, in fact most stenoses are eccentric and have a remaining arc of compliant tissue. A modest amount (10%) of shortening of the muscle in the compliant region of the vessel can cause dramatic changes in lumen caliber. This was part of Prinzmetal’s original proposal to explain coronary spasm. Maseri and associates16 suggest that the term spasm be reserved for “situations where coronary constriction is both focal, is sufficiently profound to cause transient coronary occlusion, and is responsible for reversible attacks of angina at rest” (i.e., variant angina).
Coronary Steal
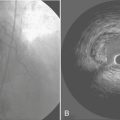
Steal occurs when the perfusion pressure for a vasodilated vascular bed (in which flow is pressure dependent) is lowered by vasodilation in a parallel vascular bed, both beds usually being distal to a stenosis.17 Two kinds of coronary steal are illustrated: collateral and transmural (Fig. 4-4).
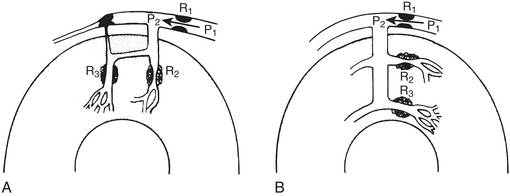
Collateral steal in which one vascular bed (R3), distal to an occluded vessel, is dependent on collateral flow from a vascular bed (R2) supplied by a stenotic artery is diagrammed in Figure 4-4A. Because collateral resistance is high, the R3 arterioles are dilated to maintain flow in the resting condition (autoregulation). Dilation of the R2 arterioles increases flow across the stenosis R1 and decreases pressure P2. If R3 resistance cannot further decrease sufficiently, flow there decreases, producing or worsening ischemia in the collateral-dependent bed.
Transmural steal is illustrated in Figure 4-4B. Normally, vasodilator reserve is less in the subendocardium. In the presence of a stenosis, flow may become pressure dependent in the subendocardium while autoregulation is maintained in the subepicardium.
SUMMARY
1. Ignarro L.J. Nitric oxide: A novel signal transduction mechanism for transcellular communication. Hypertension. 1990;16:477.
2. Lincoln T.M., Dey N., Sellak H. Invited review: cGMP-dependent protein kinase signaling mechanisms in smooth muscle: From the regulation of tone to gene expression. J Appl Physiol. 2001;91:1421.
3. Harrison D.G., Cai H. Endothelial control of vasomotion and nitric oxide production. Cardiol Clin. 2003;21:289.
4. Goodwin A.T., Yacoub M.H. Role of endogenous endothelin on coronary flow in health and disease. Coron Artery Dis. 2001;12:517.
5. Feigl E.O. Coronary physiology. Physiol Rev. 1983;63:1.
6. Kern M.J. Coronary physiology revisited: Practical insights from the cardiac catheterization laboratory. Circulation. 2000;101:1344.
7. Hoffman J.I.E. Transmural myocardial perfusion. Prog Cardiovasc Dis. 1987;29:429.
8. Stary H.C. Evolution and progression of atherosclerotic lesions in coronary arteries of children and young adults. Arteriosclerosis. 1989;9(suppl 1):I-19.
9. Katsuda S., Boyd H.C., Fligner C., et al. Human atherosclerosis: Immunocytochemical analysis of the cell composition of lesions of young adults. Am J Pathol. 1992;140:907.
10. Treasure C.B., Klein J.L., Weintraub W.S. Beneficial effects of cholesterol-lowering therapy on the coronary endothelium in patients with coronary artery disease. N Engl J Med. 1995;332:481.
11. Pasterkamp G., de Kleijn D., Borst C. Arterial remodeling in atherosclerosis, restenosis and after alteration of blood flow: Potential mechanisms and clinical implications. Cardiovasc Res. 2000;45:843.
12. Van der Wal A.C., Becker A.E., van der Loos C.M., et al. Site of intimal rupture or erosion of thrombosed coronary atherosclerotic plaque is characterized by an inflammatory process irrespective of the dominant plaque morphology. Circulation. 1994;89:36.
13. Koerselman J., van der Graf Y., De Jaegere P.P., et al. Coronary collaterals: An important and underexposed aspect of coronary artery disease. Circulation. 2003;107:2507.
14. Fujita M., Tambara K. Recent insights into human coronary collateral development. Heart. 2004;90:246.
15. Stone P.H. Mechanisms of silent myocardial ischemia: Implications for selection of optimal therapy. Adv Cardiol. 1990;37:328.
16. Maseri A., Newman C., Davies G. Coronary vasomotor tone: A heterogeneous entity. Eur Heart J. 1989;10(suppl F):2.
17. Konidala S., Gutterman D.D. Coronary vasospasm and the regulation of coronary blood flow. Prog Cardiovasc Dis. 2004;46:349.