Chapter 93 Congenital Myopathies
The congenital myopathies are a group of disorders characterized by their histopathologic findings on muscle biopsy. A prevalence of 3.5–5/10,000 children has been reported for all congenital myopathies [Chung et al., 2003; Sharma et al., 2009]. The discovery of this heterogeneous group of disorders grew out of the technical ability to investigate abnormal muscle by histochemistry, introduced in the 1950s and 1960s [Jungbluth et al., 2003]. Since then, electron microscopy, proteomics, and genomics have expanded the understanding of these conditions, as well as complicated physicians’ ability to create a simple classification schema.
The majority of these conditions manifest at or shortly after birth with hypotonia, static or nonprogressive muscle weakness, normal to decreased deep tendon reflexes, and delays in reaching milestones [Riggs et al., 2003; Taratuto, 2002]. They can also become apparent in late childhood or adulthood. Patients may have a mildly progressive course or may even be asymptomatic when presenting later in life [Riggs et al., 2003]. The serum creatine kinase level is typically normal or mildly elevated, and electromyography often reveals a myopathic pattern: namely, short, small-action potentials with rapid recruitment. A muscle biopsy is usually necessary to classify the condition and improve the clinician’s ability to prognosticate for the patient and family. The findings commonly seen in the biopsy include type I fiber predominance or hypotrophy, or both, with the absence of fiber necrosis and other evidence of chronic degeneration, such as endomysial fibrosis and fatty infiltration, findings typically seen in muscular dystrophies. Protein aggregation, often of defective proteins, then allows for final classification. Overall, there are no cures for these conditions, but supportive care is important for quality of life and longevity.
Central Core Disease
Central core disease was the first congenital myopathy described by Magee and Shy [1956]. It was named for the round delineated areas, typically within type I muscle fibers, that are devoid of oxidative enzyme activity [Jungbluth et al., 2003; Quinlivan et al., 2003; Taratuto, 2002].
Clinical Features
In addition to the hypotonia, muscle weakness (which typically affects the pelvic girdle), and developmental delay, children often present with skeletal deformities, such as scoliosis, congenital hip dislocation, and pes cavus [Jungbluth et al., 2003; Quinlivan et al., 2003; Taratuto, 2002]. Facial weakness can also be present, is usually mild, and may be seen only in the inability to bury the eyelashes completely [De Cauwer et al., 2002; Jungbluth et al., 2003]. It mainly manifests in infancy or early childhood, although it can appear later in life or even never. The weakness tends to be static or slowly progressive. Many patients are able to walk eventually, and some families may report muscle cramping [Jungbluth et al., 2003]. Central core disease is allelic with malignant hyperthermia [Quinlivan et al., 2003]. The serum creatine kinase level tends to be normal to mildly elevated.
Pathology
Muscle biopsy specimens from patients with central core disease demonstrate type I fiber predominance, with cores seen in the center of many type 1 fibers that extend throughout a large part of the fiber’s length [Jungbluth et al., 2003; Taratuto, 2002]. The cores are seen most easily when stained with the nicotinamide adenine dinucleotide-tetrazolium reductase technique [Riggs et al., 2003].
The 1995 diagnostic criteria created by the European Neuromuscular Centre include the presence of cores, which are central, although they may be eccentric or multiple, and well demarcated, visible with oxidative stains, and affecting only type 1 fibers [De Cauwer et al., 2002]. Histologic diagnosis also requires distinctive electron microscopy and a type 1 fiber predominance. Corelike lesions can also be seen with denervation and are called targetoid lesions or targets if they are trilayered [Goebel, 2003]. Various proteins have been found to accumulate within cores (Box 93-1), and this finding often helps differentiate central core disease from mini-core disease, which is discussed later.























Genetics
Central core disease is most commonly an autosomal-dominant condition with variable penetrance; however, sporadic cases have been reported [Jungbluth et al., 2003; Riggs et al., 2003; Taratuto, 2002]. It has been mapped to chromosome region 19q12–p13.1 [Fananapazir et al., 1993; Haan et al., 1990; Monnier et al., 2000; Tilgen et al., 2001], which is also associated with malignant hyperthermia. This locus is linked to a ryanodine receptor (RYR1), which is a ligand-gated release channel for calcium [McCarthy et al., 2000], mediating calcium release after sarcolemma depolarization [Taratuto, 2002]. Mutations in this gene can give rise to malignant hyperthermia, central core disease, or both, and account for almost 80 percent of cases of central core disease [Taratuto, 2002]. There are three regions on the RYR1 gene where mutations have been found. The majority of lesions are found in regions 1 and 2, which reside in the myoplasmic foot domain [Tilgen et al., 2001]. Mutations have been reported more recently in region 3, which is located in the highly conserved transmembrane C-terminal region. A mutation in this region has been found in a French family with a severe form of the disease, characterized by malignant hyperthermia and cores and rods in muscle fibers on biopsy [Monnier et al., 2000]. Central core disease has also been reported with hypertrophic cardiomyopathy, but not malignant hyperthermia, and found to result from a mutation in β-myosin heavy gene [Fananapazir et al., 1993].
Multi-Mini-Core Disease
Multi-mini-core disease is histologically similar to central core disease; however, there are many significant differences. It was first described by Engel and associates in 1971 and is characterized by multiple small areas of sarcomeric disorganization lacking oxidative activity [Ferreiro and Fardeau, 2002].
Clinical Features
There are four phenotypic groups identified, all of which are characterized by approximately normal creatine kinase levels, myopathic findings on electromyography, and no cardiac involvement [Ferreiro and Fardeau, 2002]. Patients typically present at birth or within the first 18 months of life. The classic form, or first group, manifests with severe neonatal hypotonia; predominant axial muscle weakness, especially in neck flexors; delayed motor development; severe scoliosis; and significant respiratory involvement. Its characteristics are similar to those of congenital muscular dystrophy with early rigidity of the spine (RSMD1) [Jungbluth et al., 2003]. Limb joint hyperlaxity and myopia have been found in many of the patients [Ferreiro and Fardeau, 2002; Taratuto, 2002]. The second group, or ophthalmoplegia form, consists of the typical findings plus variable ophthalmoplegia and often severe facial weakness. The third group manifests with early onset and arthrogryposis, and the fourth group is characterized by slow progression and hand amyotrophy.
Pathology
There are several pathologic differences between central core and mini-core disease. In multi-mini-core disease, the lesions tend to have poorly defined boundaries and are short; thus, longitudinal muscle sections must be evaluated to help differentiate the two conditions. Mini-cores are found in both fiber types, as opposed to just type 1 fibers in central core disease [Ferreiro and Fardeau, 2002], and contain different proteins (see Box 93-1) [Goebel, 2003].
Genetics
These disorders are mostly autosomal-recessive, but many sporadic cases have been reported [Jungbluth et al., 2003]. Recessive mutations have been found in the RYR1 gene in patients with group 3 disease [Jungbluth et al., 2002] and in one family with distal weakness and amyotrophy [Ferreiro et al., 2002a]. Patients with the classic form have been found to have recessive mutations in the selenoprotein N gene [Ferreiro et al., 2002b] and in the RSMD1 gene [Moghadaszadeh et al., 2001]. Selenoprotein N is a glycoprotein found in the endoplasmic reticulum, expressed mostly in fetal tissues and in dividing cells [Petit et al., 2003].
Nemaline Myopathy
Nemaline myopathy was the second congenital myopathy to be reported. Its classification system has undergone a dramatic change as a result of advances in molecular genetics, and six different clinical forms can be identified [Goebel, 2003].
Clinical Features
The severe congenital form manifests with no spontaneous movements or respirations at birth, and may be associated with contractures and congenital fractures [Wallgren-Pettersson and Laing, 2000]. The fetal akinesia sequence has been reported to be associated with intrauterine onset [Lammens et al., 1997]. High-arched palate [Taratuto, 2002], cardiomyopathy, and ophthalmoplegia have also been associated with this form [Wallgren-Pettersson and Laing, 2000]. Children affected with the intermediate congenital type are able to breathe and move at birth, but during early childhood they become unable to breathe independently and typically cannot sit or walk [Wallgren-Pettersson and Laing, 2000]. Contractures tend to develop early. The typical form manifests in early childhood with proximal weakness, especially in neck flexors, in association with facial, bulbar, and respiratory weakness [Wallgren-Pettersson and Laing, 2000]. Distal involvement can occur later, but affected children are able to reach their milestones, albeit delayed, and have a slowly progressive or even nonprogressive course [Wallgren-Pettersson and Laing, 2000]. The mild childhood or juvenile form is similar to the classic form without facial weakness [Goebel, 2003]. There is also an adult-onset form and other, less common forms that may manifest with an unusual distribution of weakness [Wallgren-Pettersson and Laing, 2000].
Pathology
Nemaline myopathy is histopathologically characterized through the use of the Gomori trichrome technique by red-staining “rods” that are predominantly subsarcolemmal [Jungbluth et al., 2003], although they can be intermyofibrillar or intranuclear and are reactive to α-actin [Taratuto, 2002]. They are associated with the Z-disk and often are in continuity with the Z-lines [Jungbluth et al., 2003; Riggs et al., 2003]. They have a similar lattice structure and are composed of filaments. There is no correlation between the amount of rods seen and the severity of the condition [Jungbluth et al., 2003; Taratuto, 2002]. The rods can be seen solely in type I fibers or in both fiber types, and measure 2–7 mm in length [Riggs et al., 2003]. These findings are typically in conjunction with type 1 fiber predominance or type 1 fiber hypotrophy, or both.
Genetics
Currently, five filament encoding genes have been found to carry mutations. The rods have been found to be morphologically the same among all types [Goebel, 2003]. The most common mutations have been found in the α-actin and nebulin genes. Alpha-actin gene mutations, which account for 10–20 percent of cases [Taratuto, 2002], are frequently reported in the severe cases [Wallgren-Pettersson and Laing, 2001], although the presentation in patients with these mutations is heterogeneous [Agrawal et al., 2004]. Autosomal-dominant and recessive forms have also been described [Nowak et al., 1999]. The α-actin gene is at chromosome 1q42.1 [Goebel, 2003; Taratuto, 2002], and most cases are sporadic [Agrawal et al., 2004]. Nebulin gene mutations have also been reported in the severe congenital form [Wallgren-Pettersson et al., 2002]. The nebulin gene is located on chromosome region 2q21.2–q22 [Pelin et al., 1999]. Mutations are usually recessive and are associated with the typical form [Jungbluth et al., 2003; Wallgren-Pettersson and Laing, 2003].
The first genetic locus to be reported in nemaline myopathy is in the slow α-tropomyosin gene, found on chromosome region 1q21–q23 [Laing et al., 1992, 1995]. It, along with β-tropomyosin, is an actin-related skeletal muscle fiber protein [Goebel, 2003]. Alpha-tropomyosin gene mutations [Laing et al., 1995] and, less frequently, β-tropomyosin gene mutations are located on 9p13.2–p13.1 [Donner et al., 2002], and have been reported in the mild form. The often-fatal infantile autosomal-recessive type has been associated with a mutation in the troponin T1 gene on chromosome 19q13.4 in an Amish population [Johnston et al., 2000]. Troponins are a component of the actin filaments [Goebel, 2003]. Finally, an autosomal-dominant phenotype, consisting of muscle slowness and proximal muscle weakness with associated corelike lesions on muscle biopsy, was found in a Dutch family [Gommans et al., 2002] and has been linked to chromosome region 15q21–q23 [Gommans et al., 2003]. However, no disease-associated mutations in the α-tropomyosin-1 gene, which is located within the critical region, could be found.
Management
Nemaline myopathy is the only myopathy in this group with a possible treatment apart from supportive care. l-Tyrosine has been reported to improve certain clinical aspects of patients with this condition [Wallgren-Pettersson and Laing, 2003]. Tyrosine treatment appears to diminish drooling and increase appetite and physical activity level. These symptoms returned on cessation of the supplement and were again controlled with reinstituting the treatment. Tyrosine is a nonessential amino acid required in the synthesis of catecholamines, which possibly helps explain the benefits seen from supplementation. Several trials evaluating the benefits of tyrosine in patients with nemaline myopathy are currently in progress.
Centronuclear/Myotubular Myopathy
The group of disorders known as centronuclear myopathies (CNM) has recently expanded to include X-linked myotubular myopathy, which accounts for the majority of cases and is the most severe [Taratuto, 2002], as well as CNM of childhood and CNM of adulthood [Sharma et al., 2009]. Pathologically, there are numerous centrally located nuclei in muscle fibers, which appear similar to the myotube, the immature stage of the muscle fiber [Goebel, 2003]. It is this appearance that led to the name myotubular myopathy [Spiro et al., 1966], and later centronuclear myopathy [Sher et al., 1967].
Clinical Features
Respiratory failure typically leads to death within the first year of life [Taratuto, 2002], and often necessitates ventilatory dependence [Bertini et al., 2004]. Milder forms, with the development of spontaneous breathing, have been reported [Barth and Dubowitz, 1998]. Associated conditions include polyhydramnios, reduced to absent fetal movements, severe hypotonia, and limited extraocular movements [Jungbluth et al., 2003; Taratuto, 2002]. It appears to be a nonprogressive condition; however, affected infants do not exhibit progress in motor developmental milestones [Jungbluth et al., 2003]. Female carriers are usually asymptomatic [Taratuto, 2002], although they can develop early limb-girdle weakness with secondary kyphoscoliosis. Facial weakness and dysarthria can develop. These findings are more likely caused by an abnormal expression or distribution of myotubularin, the abnormal protein leading to this condition, rather than by X-inactivation [Sutton et al., 2001]. Other clinical courses have been described. Patients with onset during late infancy or childhood present with weakness that includes the extraocular muscles, hypotonia, and delayed achievement of milestones [Riggs et al., 2003]. Other patients present in adulthood with mild to moderate lower-extremity weakness.
Pathology
Muscle nuclei, which normally are peripherally placed, are found displaced centrally in a large number of muscle fibers in patients with this disorder. Perinuclear halos – areas with absence of myofilaments – often surround these central nuclei [Jungbluth et al., 2003]. As with the other myopathies in this category, type 1 fiber predominance is usually seen with type 1 fiber hypotrophy. Central nuclei can be seen in just type 1 fibers or in both fiber types [Riggs et al., 2003]. Although the disorder has been named after the similar appearance that muscle fibers seen on biopsy samples have to fetal myotubes, studies have demonstrated the presence of mature myofibrillar proteins [Jungbluth et al., 2003].
Genetics
The severe form of this disorder tends to arise from mutations in the myotubularin gene on chromosome Xq28 [Laporte et al., 1996], and likely accounts for up to 80 percent of X-linked cases [Laporte et al., 2000]. Myotubularin is a tyrosine phosphatase [Laporte et al., 1996] that dephosphorylates phosphatidylinositol-3-phosphate, a second messenger, and exerts its effects during myogenesis by regulating cellular levels of this lipid [Taylor et al., 2000]. Most mutations lead to absence or inactivation of myotubularin [Bertini et al., 2004]. Eight other forms of myotubularin have been reported [Goebel, 2003], and mutations in one have been found in patients with one of the demyelinating forms of Charcot–Marie–Tooth disease [Bolino et al., 2000; Houlden et al., 2001].
Autosomal-dominant forms, which tend to be milder and manifest later in life, and autosomal-recessive forms, with variable manifestation from mild generalized weakness with external ophthalmoplegia to early onset with severe proximal muscle weakness, have also been reported [Jungbluth et al., 2003]. Mutations in myogenic factor-6 (MYF6) [Kerst et al., 2000], as well as dynamin 2 (DNM2) [Bitoun et al., 2009], have been associated with dominant forms of the condition. DNM2 is a large GTPase involved in the release of nascent vesicles during endocytosis and intracellular membrane trafficking. In CNM, mutations affecting the middle domain (MD) have been associated with mild late-onset disease, while mutations in the pleckstrin homology domain (PH) tend to cause a more severe phenotype [Bitoun et al., 2009]. Interestingly, mutations in DNM2 have been reported in the intermediate [Zuchner et al., 2005] and axonal forms of Charcot–Marie–Tooth disease (CMT) [Bitoun et al., 2008]. An autosomal-recessive form of CNM has been associated with a mutation in amphiphysin 2 (BIN 1) [Nicot et al., 2007], a protein that interacts with dynamin 2.
Fiber Type Disproportion Myopathy
Case reports of patients with presentations similar to the congenital myopathies and the finding of type 1 fibers at least 12 percent smaller than type 2 fibers were published in the 1960s and 1970s [Clarke and North, 2003]. The term congenital fiber type disproportion was then coined by Brooke [1973]. However, since then, these pathologic findings have been reported in the other congenital myopathies and in myotonic dystrophy, other myopathies, neuropathies, and central nervous system disorders [Clarke and North, 2003; Imoto and Nonaka, 2001; Taratuto, 2002] (Box 93-2). This condition should therefore be diagnosed only through exclusion, if at all.
Box 93-2 More Common Causes of Fiber Size Disproportion










(Adapted from Clarke NF, North KN. Congenital fiber type disproportion – 30 years on. J Neuropathol Exp Neurol 2003;62:977.)
Clinical Features
Overall, children with a diagnosis of this condition tend to have proximal and limb-girdle static weakness in association with myopathic facies and high-arched palate [Clarke and North, 2003]. Most cases have a mild to moderate course, although severe cases have been reported. There is often a positive family history of this condition, but the disease can also be sporadic.
Other Structural Congenital Myopathies
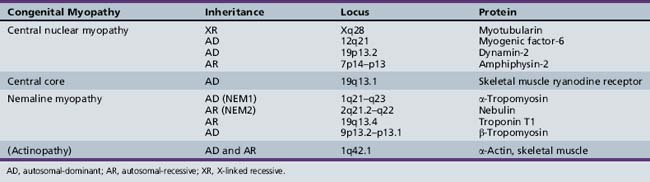
Many other forms of congenital myopathy have been reported over the years as resulting from abnormal structural arrangements seen on biopsy (Table 93-1). Some have emerged as a result of the presence of intracytoplasmic inclusions or aggregates of filaments, whereas others have been characterized by vacuolated areas. As knowledge of the proteins involved in muscle structure and function increases, investigators have been able to determine the underlying etiology for some of these conditions. Unusual inclusions described in cases of congenital myopathy include zebra bodies, which are a nonspecific finding in muscle at the myotendinous junction and in extraocular muscles; cylindric spirals; caps; and reducing bodies, which also have been observed in a congenital myopathy with rigid spine [Taratuto, 2002]. Some of the less common myopathies that contain myocyte inclusions are described as follows.
Desminopathies
Desmin is a 52-kDa chief intermediate filament of skeletal and cardiac muscle [Dalakas et al., 2000] that has been found to accumulate in a group of myopathies known as myofibrillar myopathies [Taratuto, 2002]. These aggregates appear as cytoplasmic, sarcoplasmic, and spheroid bodies, and can also be seen as granulofilamentous material [Goebel, 2003; Taratuto, 2002]. Several structural desmin-related myopathies have also been discovered and include αB-crystallinopathy, hereditary inclusion body myopathies, and possibly hyaline body myopathy [Goebel and Fardeau, 2002].
Clinical Features
Desminopathies typically manifest with distal muscle weakness. Cardiac conduction defects may be present, as may a cardiomyopathy [Taratuto, 2002]. When a cardiomyopathy is present, granulofilamentous material is seen on muscle biopsy [Goebel, 2003]. Peripheral neuropathy can accompany these features as well, and giant axons resulting from neurofilament accumulation are seen on nerve biopsy [Goebel, 2003]. The protein aggregates of desmin, which have been found to be hyperphosphorylated [Goebel, 2003], are typically surrounded by areas lacking oxidative enzymes [Taratuto, 2002]. Autosomal-dominant and recessive types have been reported [Goebel and Fardeau, 2002; Taratuto, 2002]. The desmin gene maps to chromosome 2q35 [Viegas-Pequignot et al., 1989]. A desmin knockout mouse develops normally but becomes afflicted with an early skeletal and cardiac myopathy, accompanied by vascular disease [Goebel, 2003].
Alpha B-Crystallinopathy
A French family with a clinical picture suggestive of a desminopathy and accompanied by small lens opacities was found to have a mutation on chromosome 11 in a gene encoding αB-crystallin [Goebel and Fardeau, 2002], a protein that co-aggregates with desmin [Goebel, 2003]. Alpha B-crystallin is a small heat shock protein that acts as a chaperone protein, and participates in the breakdown of intracellular proteins outside of the lysosome [Goebel, 2003]. Therefore, mutated αB-crystallin would lead naturally to protein accumulation. Alpha B-crystallin has been found in many tissues, including skeletal muscle and lens tissue, and in cardiocytes [Goebel, 2003].
Hyaline Body Myopathy
Hyaline body myopathy, which was previously known as myofibrillar lysis myopathy [Goebel and Fardeau, 2002], is characterized by non-membrane-bound subsarcolemmal aggregation of dense, disorganized filaments in continuity with myosin staining, also known as hyaline bodies [Goebel and Anderson, 1999; Taratuto, 2002]. These bodies are usually seen in 10–30 percent of type 1 fibers, exhibit myosin immunoreactivity, and stain intensely with acid myosin adenosine triphosphatase [Goebel and Anderson, 1999]. Children affected with this disorder typically present with mild, nonprogressive scapuloperoneal weakness and atrophy early in life [Goebel and Anderson, 1999; Taratuto, 2002]. Most cases are sporadic, but autosomal-dominant and recessive forms have been described [Goebel and Fardeau, 2002; Taratuto, 2002].
Myosin-Related Myopathy
Two disorders so far have been linked to mutations in the genes encoding myosin heavy chains. The first is in a Swedish family with a progressive myopathy and rimmed vacuoles, tubulofilamentous aggregates, and mini-core-like lesions. This condition was once called hereditary inclusion body myopathy type 3 and now is known as type 1c [Goebel, 2003]. The gene is on chromosome 17 and encodes for myosin heavy chain IIa. Hereditary cardiomyopathies have also been associated with mutations in cardiac myosin heavy chains.
Fingerprint Body Myopathy
Fingerprint body myopathy was first described by Engel and colleagues in 1972, and manifests with early onset, limb and trunk weakness, and delayed motor development [Riggs et al., 2003]. Fingerprint bodies are identified on electron microscopy as subsarcolemmal inclusions of osmiophilic lamellae that resemble fingerprints [Goebel and Anderson, 1999; Riggs et al., 2003]. There is one case report of a pair of siblings with proximal muscle weakness and susceptibility to malignant hyperthermia, who were found to have central cores in conjunction with fingerprint bodies [Stojkovic et al., 2001]. No mutations in the RYR1 gene were found. Fingerprint bodies have been reported in other muscle disorders, such as myotonic dystrophy [Tomé and Fardeau, 1973], a patient with Marfan’s syndrome and slowly progressive weakness [Jadro-Santel et al., 1980], and oculopharyngeal muscular dystrophy, as well as other myopathies and neurodegenerative disorders [Riggs et al., 2003; Stojkovic et al., 2001]. They have also been found in fetal muscle, which suggests that these bodies may be transient structures of developmental significance, and could account for their infrequent occurrence in these conditions [Ambler et al., 1987].
Tubular Aggregate Myopathies
Tubular aggregates are basophilic deposits made up of fascicles of parallel tubules, possibly derived from the sarcoplasmic reticulum [Riggs et al., 2003]. They have been reported as being seen in up to 98 percent of muscle fibers on biopsy in a family with slowly progressive weakness of the iliopsoas, deltoid, triceps, and sternocleidomastoid muscles. This condition is inherited in an autosomal-dominant fashion [Goebel and Anderson, 1999]. However, like fingerprint bodies, tubular aggregates have been reported in many other neurologic conditions, such as periodic paralyses; myalgic syndromes, in which the aggregates were in type 2 fibers; and familial myopathies, in which they were seen in both fiber types [Riggs et al., 2003].
Vacuolar Myopathy
Families have been reported with what appears to be X-linked mental retardation, cardiomyopathy, and a vacuolar myopathy [Goebel and Anderson, 1999; Muntoni et al., 1994]. Affected males developed proximal and axial muscle weakness in their teens and a cardiomyopathy in their 20s. They all had moderate to severe cognitive impairment. Creatine kinase values were moderately elevated, and muscle biopsy specimens revealed severe vacuolar changes in association with many fibers having internal nuclei.
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Agrawal P.B., Strickland C.D., Midgett C., et al. Heterogeneity of nemaline myopathy cases with skeletal muscle alpha-actin gene mutations. Ann Neurol. 2004;56:86.
Ambler M.W., Neave C., Entwistle R. Fingerprint inclusions in normal fetal muscle. Acta Neuropathol (Berl). 1987;73:185.
Barth P.G., Dubowitz V. X-linked myotubular myopathy – A long-term follow-up study. Eur J Paediatr Neurol. 1998;2:49.
Bertini E., Biancalana V., Bolino A., et al. 118th ENMC International Workshop on Advances in Myotubular Myopathy. 26–28 September 2003, Naarden, The Netherlands. (5th Workshop of the International Consortium on Myotubular Myopathy). Neuromuscul Disord. 2004;14:387.
Bitoun M., Bevilacqua J.A., Eymard B., et al. A new centronuclear myopathy phenotype due to a novel dynamin 2 mutation. Neurology. 2009;72:93.
Bitoun M., Stojkovic T., Prudhon B., et al. A novel mutation in the dynamin 2 gene in a Charcot-Marie-Tooth type 2 patient: clinical and pathological findings. Neuromuscul Disord. 2008;18:334.
Bolino A., Muglia M., Conforti F.L., et al. Charcot-Marie-Tooth type 4B is caused by mutations in the gene encoding myotubularin-related protein-2. Nat Genet. 2000;25:17.
Brooke M.H. Congenital fiber type disproportion. In: Kakulas B.A., editor. 2nd International Congress on Muscle Disease. Perth, Australia: Excerpta Medica; 1973:147. November 22–29, 1971
Chung B., Wong V., Ip P. Prevalence of neuromuscular diseases in Chinese children: a study in southern China. J Child Neurol. 2003;18:217.
Clarke N.F., North K.N. Congenital fiber type disproportion – 30 years on. J Neuropathol Exp Neurol. 2003;62:977.
Dalakas M.C., Park K.Y., Semino-Mora C., et al. Desmin myopathy, a skeletal myopathy with cardiomyopathy caused by mutations in the desmin gene. N Engl J Med. 2000;342:770.
De Cauwer H., Heytens L., Martin J.J. Workshop report of the 89th ENMC International Workshop: Central Core Disease, 19th-20th January 2001, Hilversum, The Netherlands. Neuromuscul Disord. 2002;12:588.
Donner K., Ollikainen M., Ridanpaa M., et al. Mutations in the beta-tropomyosin (TPM2) gene – A rare cause of nemaline myopathy. Neuromuscul Disord. 2002;12:151.
Engel A.G., Angelini C., Gomez M.R. Fingerprint body myopathy, a newly recognized congenital muscle disease. Mayo Clin Proc. 1972;47:377.
Engel A.G., Gomez M.R., Groover R.V. Multicore disease. A recently recognized congenital myopathy associated with multifocal degeneration of muscle fibers. Mayo Clin Proc. 1971;46:666.
Fananapazir L., Dalakas M.C., Cyran F., et al. Missense mutations in the beta-myosin heavy-chain gene cause central core disease in hypertrophic cardiomyopathy. Proc Natl Acad Sci USA. 1993;90:3993.
Ferreiro A., Fardeau M. 80th ENMC International Workshop on Multi-Minicore Disease: 1st International MmD Workshop. 12–13th May, 2000, Soestduinen, The Netherlands. Neuromuscul Disord. 2002;12:60.
Ferreiro A., Monnier N., Romero N.B., et al. A recessive form of central core disease, transiently presenting as multi-minicore disease, is associated with a homozygous mutation in the ryanodine receptor type 1 gene. Ann Neurol. 2002;51:750.
Ferreiro A., Quijano-Roy S., Pichereau C., et al. Mutations of the selenoprotein N gene, which is implicated in rigid spine muscular dystrophy, cause the classical phenotype of multiminicore disease: Reassessing the nosology of early-onset myopathies. Am J Hum Genet. 2002;71:739.
Goebel H.H. Congenital myopathies at their molecular dawning. Muscle Nerve. 2003;27:527.
Goebel H.H., Anderson J.R. Structural congenital myopathies (excluding nemaline myopathy, myotubular myopathy and desminopathies): 56th European Neuromuscular Centre (ENMC) sponsored International Workshop. December 12–14, 1997, Naarden, The Netherlands. Neuromuscul Disord. 1999;9:50.
Goebel H.H., Fardeau M. Desmin – Protein surplus myopathies, 96th European Neuromuscular Centre (ENMC)-sponsored International Workshop held 14–16 September 2001, Naarden, The Netherlands. Neuromuscul Disord. 2002;12:687.
Gommans I.M., Davis M., Saar K., et al. A locus on chromosome 15q for a dominantly inherited nemaline myopathy with core-like lesions. Brain. 2003;126(Pt 7):1545.
Gommans I.M., van Engelen B.G., ter Laak H.J., et al. A new phenotype of autosomal dominant nemaline myopathy. Neuromuscul Disord. 2002;12:13.
Haan E.A., Freemantle C.J., McCure J.A., et al. Assignment of the gene for central core disease to chromosome 19. Hum Genet. 1990;86:187.
Houlden H., King R.H., Wood N.W., et al. Mutations in the 5 region of the myotubularin-related protein 2 (MTMR2) gene in autosomal recessive hereditary neuropathy with focally folded myelin. Brain. 2001;124(Pt 5):907.
Imoto C., Nonaka I. The significance of type 1 fiber atrophy (hypotrophy) in childhood neuromuscular disorders. Brain and Development. 2001;23:298.
Jadro-Santel D., Grcevic N., Dogan S., et al. Centronuclear myopathy with type I fibre hypotrophy and “fingerprint” inclusions associated with Marfan’s syndrome. J Neurol Sci. 1980;45:43.
Johnston J.J., Kelley R.I., Crawford T.O., et al. A novel nemaline myopathy in the Amish caused by a mutation in troponin T1. Am J Hum Genet. 2000;67:814.
Jungbluth H., Muller C.R., Halliger-Keller B., et al. Autosomal recessive inheritance of RYR1 mutations in a congenital myopathy with cores. Neurology. 2002;59:284.
Jungbluth H., Sewry C.A., Muntoni F. What’s new in neuromuscular disorders? The congenital myopathies. Eur J Paediatr Neurol. 2003;7:23.
Kerst B., Mennerich D., Schuelke M., et al. Heterozygous myogenic factor 6 mutation associated with myopathy and severe course of Becker muscular dystrophy. Neuromuscul Disord. 2000;10:572.
Laing N.G., Majda B.T., Akkari P.A., et al. Assignment of a gene (NEMI) for autosomal dominant nemaline myopathy to chromosome I. Am J Hum Genet. 1992;50:576.
Laing N.G., Wilton S.D., Akkari P.A., et al. A mutation in the alpha tropomyosin gene TPM3 associated with autosomal dominant nemaline myopathy NEM1. Nat Genet. 1995;10:249.
Lammens M., Moerman P., Fryns J.P., et al. Fetal akinesia sequence caused by nemaline myopathy. Neuropediatrics. 1997;28:116.
Laporte J., Biancalana V., Tanner S.M., et al. MTM1 mutations in X-linked myotubular myopathy. Hum Mutat. 2000;15:393.
Laporte J., Hu L.J., Kretz C., et al. A gene mutated in X-linked myotubular myopathy defines a new putative tyrosine phosphatase family conserved in yeast. Nat Genet. 1996;13:175.
Magee K.R., Shy G.M. A new congenital non-progressive myopathy. Brain. 1956;79:610.
McCarthy T.V., Quane K.A., Lynch P.J. Ryanodine receptor mutations in malignant hyperthermia and central core disease. Hum Mutat. 2000;15:410.
Moghadaszadeh B., Petit N., Jaillard C., et al. Mutations in SEPN1 cause congenital muscular dystrophy with spinal rigidity and restrictive respiratory syndrome. Nat Genet. 2001;29:17.
Monnier N., Romero N.B., Lerale J., et al. An autosomal dominant congenital myopathy with cores and rods is associated with a neomutation in the RYR1 gene encoding the skeletal muscle ryanodine receptor. Hum Mol Genet. 2000;9:2599.
Muntoni F., Catani G., Mateddu A., et al. Familial cardiomyopathy, mental retardation and myopathy associated with desmin-type intermediate filaments. Neuromuscul Disord. 1994;4:233.
Nicot A.S., Toussaint A., Tosch V., et al. Mutations in amphiphysin 2 (BIN1) disrupt interaction with dynamin 2 and cause autosomal recessive centronuclear myopathy. Nat Genet. 2007;39:1134.
Nowak K.J., Wattanasirichaigoon D., Goebel H.H., et al. Mutations in the skeletal muscle alpha-actin gene in patients with actin myopathy and nemaline myopathy. Nat Genet. 1999;23:208.
Pelin K., Hilpela P., Donner K., et al. Mutations in the nebulin gene associated with autosomal recessive nemaline myopathy. Proc Natl Acad Sci USA. 1999;96:2305.
Petit N., Lescure A., Rederstorff M., et al. Selenoprotein N: An endoplasmic reticulum glycoprotein with an early developmental expression pattern. Hum Mol Genet. 2003;12:1045.
Quinlivan R.M., Muller C.R., Davis M., et al. Central core disease: Clinical, pathological, and genetic features. Arch Dis Child. 2003;88:1051.
Riggs J.E., Bodensteiner J.B., Schochet S.S.Jr. Congenital myopathies/dystrophies. Neurol Clin. 2003;21:779.
Sharma M.C., Jain D., Sarkar C., et al. Congenital myopathies – a comprehensive update of recent advancements. Acta Neurol Scand. 2009;119:281.
Sher J.H., Rimalovski A.B., Athanassiades T.J., et al. Familial centronuclear myopathy: A clinical and pathological study. Neurology. 1967;17(8 Pt 1):727.
Spiro A.J., Shy G.M., Gonatas N.K. Myotubular myopathy. Persistence of fetal muscle in an adolescent boy. Arch Neurol. 1966;14:1.
Stojkovic T., Maurage C.A., Moerman A., et al. Congenital myopathy with central cores and fingerprint bodies in association with malignant hyperthermia susceptibility. Neuromuscul Disord. 2001;11:538.
Sutton I.J., Winer J.B., Norman A.N., et al. Limb girdle and facial weakness in female carriers of X-linked myotubular myopathy mutations. Neurology. 2001;57:900.
Taratuto A.L. Congenital myopathies and related disorders. Curr Opin Neurol. 2002;15:553.
Taylor G.S., Maehama T., Dixon J.E. Inaugural article: Myotubularin, a protein tyrosine phosphatase mutated in myotubular myopathy, dephosphorylates the lipid second messenger, phosphatidylinositol 3-phosphate. Proc Natl Acad Sci USA. 2000;97:8910.
Tilgen N., Zorzato F., Halliger-Keller B., et al. Identification of four novel mutations in the C-terminal membrane spanning domain of the ryanodine receptor 1: Association with central core disease and alteration of calcium homeostasis. Hum Mol Genet. 2001;10:2879.
Tomé F.M., Fardeau M. “Fingerprint inclusions” in muscle fibres in dystrophia myotonica. Acta Neuropathol (Berl). 1973;24:62.
Viegas-Pequignot E., Li Z.L., Dutrillaux B., et al. Assignment of human desmin gene to band 2q35 by nonradioactive in situ hybridization. Hum Genet. 1989;83:33.
Wallgren-Pettersson C., Donner K., Sewry C., et al. Mutations in the nebulin gene can cause severe congenital nemaline myopathy. Neuromuscul Disord. 2002;12:674.
Wallgren-Pettersson C., Laing N.G. 109th ENMC International Workshop: 5th workshop on nemaline myopathy, 11th-13th October 2002, Naarden, The Netherlands. Neuromuscul Disord. 2003;13:501.
Wallgren-Pettersson C., Laing N.G. Report of the 70th ENMC International Workshop: Nemaline myopathy, 11–13 June 1999, Naarden, The Netherlands. Neuromuscul Disord. 2000;10:299.
Wallgren-Pettersson C., Laing N.G. Report of the 83rd ENMC International Workshop: 4th Workshop on Nemaline Myopathy, 22–24 September 2000, Naarden, The Netherlands. Neuromuscul Disord. 2001;11:589.
Zuchner S., Noureddine M., Kennerson M., et al. Mutations in the pleckstrin homology domain of dynamin 2 cause dominant intermediate Charcot-Marie-Tooth disease. Nat Genet. 2005;37:289.