Chapter 136 Congenital Hypertrophy of the Retinal Pigment Epithelium
Introduction
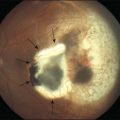
Congenital hypertrophy of the retinal pigment epithelium (CHRPE) is generally an asymptomatic congenital hamartoma that occurs in three variant forms: either as solitary, or grouped or multiple pigmented fundus lesions. Lesions are usually observed during routine ophthalmoscopy.1 Multiple CHRPE may be associated with familial adenomatous polyposis (FAP), an autosomal dominant disease with numerous adenomatous polyps of the colon and rectum. FAP with prominent extracolonic manifestations has been termed Gardner syndrome (GS) or another variant as Turcot syndrome for FAP with brain tumors lesions (Fig. 136.1). The observation of CHRPE in a premature child provides evidence for the congenital nature of this lesion.2
Epidemiology/demographics
The reported prevalence of CHRPE is as low as 3 described cases among 2400 ophthalmic examinations (1.25%). The prevalence of FAP varies from 1 in 7000 to 1 in 22 000 individuals. Approximately 70–90% of FAP patients have pigmented fundus lesions.3
Clinical findings and classification
Solitary CHRPE
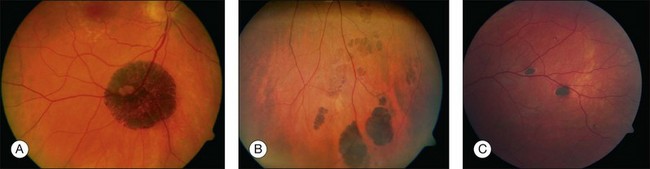
Solitary CHRPE is typically a flat, round, hyperpigmented lesion with smooth or scalloped margins that is well-demarcated from normal-appearing retinal pigment epithelium (RPE) (Fig. 136.2A). The color may vary from light gray, brown to black and is independent of the patient’s race.1 A marginal halo of depigmentation may surround the lesion and “punched-out” inner lacunae with hypopigmentation may be present in larger lesions. Both depigmented zones show the tendency to progress gradually over time, and eventually, may involve the entire lesion.4–7 The overlying retina and its vasculature appear normal in most cases, however discrete focal intraretinal pigmentation may become visible near the margin on biomicroscopy.4 Some lesions may be associated with retinal vascular abnormalities including capillary and large-vessel obliteration, microaneurysmal changes, chorioretinal anastomoses, and neovascularization. The size may vary from 100 µm to several disc diameters, occasionally occupying an entire quadrant. Lesions may be found anywhere in the fundus, with predominance in the superotemporal and equatorial region while the macula is rarely involved. Solitary CHRPE enlarge in 46–83% of cases over three or more years of follow-up. Extension into the fovea may result in reduced visual acuity.8 Some heavily pigmented nodular lesions may develop within CHRPE.9 Histopathologically nodular lesions proved to be adenocarcinomas in two reports.10,11 Untreated nodular lesions may enlarge to form pedunculated tumors of >7 mm thickness with serous retinal detachment.11
Grouped CHRPE
When several lesions of varying size are arranged in a cluster, resembling the footprint of an animal (‘bear tracks’), they are named “grouped CHRPE” (233800, OMIM). Grouped lesions are flat, well-demarcated round oval or geographic black spots with increasing size towards the fundus periphery. Each cluster includes approximately 3–30 lesions, which may vary in size from 100 to 300 µm lesions (Fig. 136.2B). Etiology and development of grouped CHRPE remain unknown. The presence of grouped CHRPE and additional ipsilateral sectorial pigmented skin lesions following the “cutaneous lines of Blaschko” gave evidence for possible pigmentary mosaicism in both the eye and skin.12 Therefore, grouped CHRPE are considered as a cluster of atypical hyperpigmented RPE cells, that derive from the edge of the optic nerve and migrate along the stream of embryologic tissue lines, analogous to the “cutaneous lines of Blaschko.” The sectorial pattern of grouped CHRPE obviously reflects the stream, outgrowth and migration path of RPE cells during embryogenesis.13 Bilateral grouped CHRPE has rarely been observed. There are no systemic associations in patients with solitary and grouped CHRPE.
Multiple CHRPE
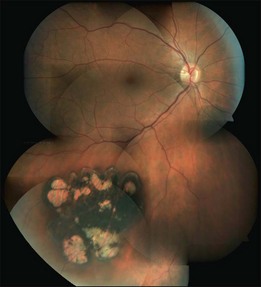
Multiple CHRPE lesions in FAP are generally smaller (50–100 µm in diameter) compared with solitary CHRPE. They are black, brown, or light gray (Fig. 136.2C). Larger lesions may be surrounded by a depigmented halo, mottled RPE,14 window-defect-type changes adjacent to retinal vessels,15 may contain depigmented lacunae, and can be accompanied by small pigmented satellite lesions. Retinal invasion and glial, capillary and pigment epithelial proliferation and hypertrophy are typical. More than four widely spaced lesions per eye or bilateral involvement are suggestive of FAP.16 Traboulsi et al. examined 16 GS families for multiple CHRPE. Of 41 GS patients, 37 (90%) had multiple CHRPE lesions.14 Lesions were bilateral in 32 patients (78%). The presence of bilateral lesions or multiple unilateral lesions (>4) appeared to be a useful clinical marker (specificity, 0.95; sensitivity, 0.78) for GS. In view of the multiple, bilateral character of retinal lesions observed in FAP, they can be considered a clinical disease marker. However, the absence of CHRPE has no predictive value for absence of GS or FAP.
Differential diagnosis
Often CHRPE has been misdiagnosed as choroidal malignant melanoma. The latter is almost always elevated, less homogeneously pigmented and less sharply demarcated compared with CHRPE, and usually exhibits growth in all three dimensions. Choroidal nevi are flat and located below the RPE. Their color may vary from light to dark brown. The borders are less well demarcated and often are feathery because nevus cells extend along larger choroidal vessels. Frequently, drusen and pigmentary mottling are present on the surface of nevi. Melanocytomas of the choroid have a similar appearance to CHRPE, except for their homogeneously black color. Black sunburst lesions in sickle cell retinopathy may appear as dark gray to brown convex-shaped lesions predominantly in the midperiphery. True hyperplasia of the RPE has ill-defined borders and invades the retina, often leading to retinal distortion. Focal pigmentation caused by injury, inflammation, or drug toxicity may resemble CHRPE but can be differentiated on the basis of a more irregular shape, widespread distribution, and associated clues suggesting acquired disease.17
Associated extraocular findings
Solitary, unilateral lesions and grouped CHRPE are restricted to the RPE with no other ocular or extracellular findings.18 Multiple or bilateral CHRPE may be associated with autosomal dominant FAP or GS (175100, OMIM)). Gardner syndrome is characterized by FAP of the large and small intestine, hamartomas of the skeleton and various soft-tissue tumefactions.19,20 Intestinal polyps generally appear during the third decade of life and invariably progress to adenocarcinomas by the fifth decade. Skeletal hamartomas, most commonly benign osteomas of mandible skull, orbits and long bones, become apparent in the third decade of life, as well as soft-tissue abnormalities including epidermoid and sebaceous cysts, dermal fibromas, lipomas, leiomyomas, desmoid, and mesenteric fibromatosis. Further extracolonic manifestations of GS include tumors of the thyroid, adrenal, and bladder, as well as sarcomas and hepatoblastomas. Turcot syndrome (233800, OMIM) is associated with FAP and brain tumors, e.g., astrocytomas, medulloblastomas, and ependymomas.
Pathophysiology/histopathology
Histopathologic studies demonstrated that most solitary1,4 and grouped CHRPE lesions are a monocellular layer of hypertrophied RPE cells, densely packed with large round pigment granules instead of the wedge-shaped melanin granules frequently found in normal RPE cell lesions (Fig. 136.3A). These large round granules represent primarily macromelanosomes.21 The underlying Bruch’s membrane may be thickened1,4,22,23 due to widening of the basement membrane of the hypertrophied RPE cells. The remaining layers of Bruch’s membrane, the choriocapillaris, and choroid appear normal. The subjacent photoreceptor layer degenerates progressively with increasing age. The inner retinal layers, including the retinal vasculature, remain normal. In the area of depigmented lacunae, both the hypertrophied RPE and photoreceptor cell layers are absent and replaced by glial cells.4 The transition from the margin of CHRPE lesions to normal RPE lesions occurs abruptly. A surrounding hypopigmented halo corresponds to areas of less pigmented hypertrophied RPE cells. All lesions remain flat.
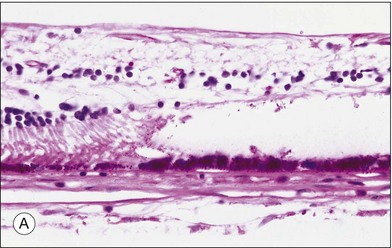
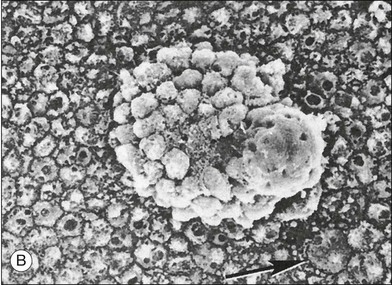
Multiple lesions associated with FAP show hypertrophy and hyperplasia of plump RPE cells, retinal invasion as well as retinal partially vascular changes. Hyperplastic lesions with multiple layers of hypertrophied RPE cells are resembling “hamartomas,” causing an elevated appearance and present as clusters of enlarged RPE cells on scanning electron microscopy (Fig. 136.3B).21,22 In another lesion, hypertrophied RPE cells traversed the full thickness of the retina. These histopathologic characteristics allow the classification of the pigmented ocular lesions in FAP as hamartomas of the RPE.24 To distinguish these lesions nosologically the terms “polyposis-associated congenital hamartoma of the RPE” and “multiple RPE hamartomas (MRPEH)” from the classic lesion of CHRPE have been proposed.25 In grossly normal-appearing areas of the fundus, individual and small clusters of enlarged RPE cells contain the same large spherical melanin pigment granules found in the pigmented lesions.
Clinical examination/ancillary testing
The vast majority of pigmented fundus lesions are easily recognized by indirect ophthalmoscopy or three-mirror contact lens examination after dilatation of the pupil. Color fundus photography is conventionally used to document the location, color, size and appearance of the lesions (Fig. 136.4) and novel ultra wide-field scanning-laser ophthalmoscopy has been recommended as a screening tool.2,26 However, absence of CHRPE lesions in a family with FAP does not indicate absence of the gene, since variable phenotype expression may exist or tiny lesions can be overlooked.
As small or lightly pigmented lesions may easily be missed, fundus autofluorescence (FAF) may be helpful to identify these lesions. Since CHRPE contains predominantly melanin and only small amounts of lipofuscin these lesions typically demonstrate hypoautofluorescence. Only areas with lacunae or nonpigmented halos show a trace to moderate autofluorescence (Fig. 136.5A).27,28
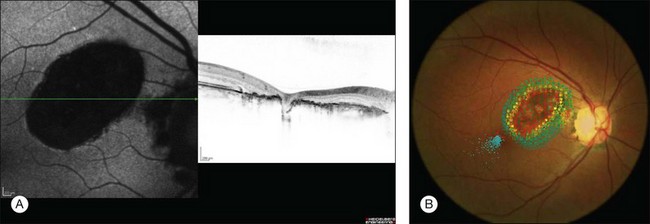
Spectral domain optical coherence tomography (SD-OCT) highlights the cross-sectional retinal anatomy of CHRPE lesions. A general thinning of the neural retina with a predominant photoreceptor loss becomes apparent over the CHRPE lesion (Fig. 136.5A). Darkly pigmented CHRPE shadows the underlying choroidal structures. Areas of lacunae show absence of RPE and increased transmission of light. Retinal thinning and photoreceptor loss over CHRPE account directly for related visual field loss. Patients with CHRPE rarely complain of related symptoms, except when the fovea is involved. Microperimetry may reveal a relative or absolute scotomata corresponding in size and location to the lesions. Microperimetric defects tend to progress from relative to absolute scotoma and expand slightly in size with aging (Fig. 136.5B).
Familial adenomatous polyposis prognosis and management options
The presence of extracolonic symptoms and the severity of disease correlate with the location of the mutation on the APC gene. Attenuated FAP (AFAP, <100 colorectal adenomas) is caused by mutations localized after codon 1595, before codon 157 and in the spliced region of exon 9. Severe FAP (>1000 adenomas) is seen in patients with mutations between codons 1250 and 1464. Mutations in the remaining APC gene lead to intermediate FAP (100–1000 adenomas) (Fig. 136.6).29–31
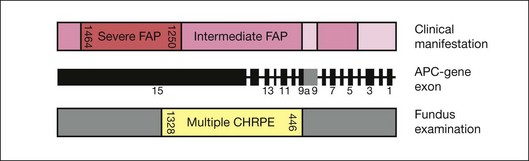
Phenotypic expression of CHRPE correlates with the localization of mutations. FAP patients with mutations between codon 446 and codon 1338 regularly present with CHRPE lesions, whereas FAP patients with mutations between 1445–1578 lack multiple CHRPE at the retinal fundus. This relationship between CHRPE manifestations and the site of the APC mutation points out a specific role of the APC protein in development of retinal tissue. Moreover, the CHRPE status adds important information about the location of the genetic mutation and plays a critical role in the detection of the corresponding APC mutations. Recent well-defined assessment criteria necessitate the presence of at least four CHRPE lesions whatever their size or at least two lesions, of which one is large, to support the diagnosis of FAP.32,33
1 Kurz GH, Zimmerman LE. Vagaries of the retinal pigment epithelium. Int Ophthalmol Clin. 1962;2:441–464.
2 Aiello LP, Traboulsi EI. Pigmented fundus lesions in a preterm infant with familial adenomatous polyposis. Arch Ophthalmol. 1980;90:661–667.
3 Coleman P, Barnard NA. Congenital hypertrophy of the retinal pigment epithelium: prevalence and ocular features in the optometric population. Ophthalmic Physiol Opt. 2007;27:547–555.
4 Buettner H. Congenital hypertrophy of the retinal pigment epithelium. Am J Ophthalmol. 1975;79:177–189.
5 Shields CL, Mashayekhi A, Ho T, et al. Solitary congenital hypertrophy of the retinal pigment epithelium. Clinical features and frequency of enlargement in 330 patients. Ophthalmology. 2003;110:1968–1976.
6 Shields CL, Mashayekhi A, Ho T, et al. Solitary congenital hypertrophy of the retinal pigment epithelium. Ophthalmology. 2003;110:1968–1976.
7 Augsburger JJ, Henson GL, Hershberger VS, et al. Topographical distribution of typical unifocal congenital hypertrophy of retinal pigment epithelium. Graefes Arch Clin Exp Ophthalmol. 2006;244:1412–1414.
8 Van der Toren K, Luyten GP. Progression of papillomacular congenital hypertrophy of the retinal pigment epithelium associated with impaired visual function. Arch Ophthalmol. 1998;116:256–257.
9 Shields JA, Shields CL, Eagle RC, Jr., et al. Adenocarcinoma arising from congenital hypertrophy of retinal pigment epithelium. Arch Ophthalmol. 2001;119:597–602.
10 Shields JA, Eagle RC, Jr., Shields CL, et al. Malignant transformation of congenital hypertrophy of the retinal pigment epithelium. Ophthalmology. 2009;116:2213–2216.
11 Trichopoulos N, Augsburger JJ, Schneider S. Adenocarcinoma arising from congenital hypertrophy of the retinal pigment epithelium. Graefes Arch Clin Exp Ophthalmol. 2006;244:125–128.
12 Meyer CH, Freyschmidt-Paul P, Happle R, et al. Unilateral linear hyperpigmentation of the skin with ipsilateral sectorial hyperpigmentation of the retina. Am J Med Genet A. 2004;126A:89–92.
13 Meyer CH, Rodrigues EB, Mennel S, et al. Grouped congenital hypertrophy of the retinal pigment epithelium follows developmental patterns of pigmentary mosaicism. Ophthalmology. 2005;112:841–847.
14 Traboulsi EI, Krush AJ, Gardner EJ, et al. Prevalence and importance of pigmented ocular fundus lesions in Gardner’s syndrome. N Engl J Med. 1987;316:661–667.
15 Chamot L, Zografos L, Klainguti G. Fundus changes associated with congenital hypertrophy of the retinal pigment epithelium. Am J Ophthalmol. 1993;115:154–161.
16 Romania A, Zakov ZN, McGannon E, et al. Congenital hypertrophy of the retinal pigment epithelium in familial adenomatous polyposis. Ophthalmology. 1989;96:879–884.
17 Meyer CH, Becker R, Schmidt JC, et al. When is congenital hypertrophy of the retinal pigment epithelium (CHRPE) associated with the Gardner’s syndrome? An overview with clinical examples. Klin Monbl Augenheilkd. 2002;219:644–648.
18 Shields JA, Shields CL, Pankajkumar GS, et al. Lack of association among typical congenital hypertrophy of the retinal pigment epithelium, adenomatous polyposis, and Gardner syndrome. Ophthalmology. 1992;99:1709–1713.
19 Gardner EJ, Richards RC. Multiple cutaneous and subcutaneous lesions occurring simultaneously with hereditary polyposis and osteomatosis. Am J Hum Genet. 1953;5:139–148.
20 Buettner H. Congenital hypertrophy of the retinal pigment epithelium in familial polyposis coli. Int Ophthalmol. 1987;10:109–110.
21 Lloyd WC, III., Eagle RC, Jr., Shields JA, et al. Congenital hypertrophy of the retinal pigment epithelium: electron microscopic and morphometric observations. Ophthalmology. 1990;97:1052–1060.
22 Roseman RL, Gass JDM. Solitary hypopigmented nevus of the retinal pigment epithelium in the macula. Arch Ophthalmol. 1992;110:1358–1359.
23 Kasner L, Traboulsi EI, Delacruz Z, et al. A histopathologic study of the pigmented fundus lesions in familial adenomatous polyposis. Retina. 1992;12:35–42.
24 Traboulsi EI, Murphy SF, de la Cruz ZC, et al. A clinicopathologic study of the eyes in familial adenomatous polyposis with extracolonic manifestations (Gardner’s syndrome). Am J Ophthalmol. 1990;110:550–561.
25 Hennesy MP, Collins F. The distinction between multiple retinal pigment epithelial hamartomata (MRPHE) in familial adenomatous polyposis (FAP) and congenital hypertrophy of the retinal pigment epithelium (CHRPE). Aust NZ J Ophthalmol. 1993;21:275–276.
26 Meyer CH, Holz FG. Documentation of congenital hypertrophy of the retinal pigment epithelium with wide-field funduscopy. Semin Ophthalmol. 2009;24:251–253.
27 Shields CL, Pirondini C, Bianciotto C, et al. Autofluorescence of congenital hypertrophy of the retinal pigment epithelium. Retina. 2007;27:1097–1111.
28 Shields CL, Materin MA, Walker C, et al. Photoreceptor loss overlying congenital hypertrophy of the retinal pigment epithelium by optical coherence tomography. Ophthalmology. 2006;113:661–665.
29 Nieuwenhuis MH, Vasen HF. Correlations between mutation site in APC and phenotype of familial adenomatous polyposis (FAP): a review of the literature. Crit Rev Oncol Hematol. 2007;61:153–161.
30 Friedl W, Caspari R, Sengteller M, et al. Can APC mutation analysis contribute to therapeutic decisions in familial adenomatous polyposis? Experience from 680 FAP families. Gut. 2001;48:515–521.
31 Caspari R, Olschwang S, Friedl W, et al. Familial adenomatous polyposis: desmoid tumours and lack of ophthalmic lesions (CHRPE) associated with APC mutations beyond codon 1444. Hum Mol Genet. 1995;4:337–340.
32 Tiret A, Taiel-Sartral M, Tiret E, et al. Diagnostic value of fundus examination in familial adenomatous polyposis. Br J Ophthalmol. 1997;81:755–758.
33 Gebert JF, Dupon C, Kadmon M, et al. Combined molecular and clinical approaches for the identification of families with familial adenomatous polyposis coli. Ann Surg. 1999;229:350–361.