[level-membership-for-anesthesiology-category]
20 Congenital Heart Disease in Adults
Advances in perioperative care for children with congenital heart disease (CHD) over the past several decades has resulted in an ever-increasing number of these children reaching adulthood with their cardiac lesions palliated or repaired. The first article on adult CHD was published in 19731; the field has grown such that there are now several texts devoted to it, and even a specialty society dedicated to it, the International Society for Adult Congenital Cardiac Disease (http: //www.isaccd.org). There are estimated to be about 32,000 new cases of CHD each year in the United States and 1.5 million worldwide.2 More than 85% of infants born with CHD are expected to grow to adulthood. It is estimated that there are more than 1,000,000 adults in the United States with CHD,3 and this population is growing at approximately 5% per year; 55% of these adults remain at moderate-to-high risk, and more than 115,000 have complex disease.4 The increasing survival of children with complex disease has shifted the spectrum of adults with CHD. Where once it was thought that adults represent milder degrees of disease, this is now changing.3 Put another way, there are more adults than children with CHD and, in fact, as many adults as children have congenital cardiac defects considered severe.5 As an example to support the increased life expectancy of this patient group, the leading cause of mortality in adults with CHD in the United States is currently coronary artery disease.6 These patients can be seen by anesthesiologists for primary cardiac repair, repair after a prior palliation, revision of repair because of failure or lack of growth of prosthetic material, or conversion of a suboptimal repair to a more modern operation (Box 20-1). In addition, these adults with CHD will be seen for all the other ailments of aging and trauma that require surgical intervention. Lastly, women of child-bearing age with CHD may become pregnant. They must cope with the added physiologic demands of pregnancy and will require analgesia for labor and anesthesia for Cesarean delivery. Although it has been suggested that teenagers and adults can have repair of congenital cardiac defects with morbidity and mortality approaching that of surgery done during childhood, these data are limited and may reflect only a relatively young and acyanotic sampling.7 Other data suggest that, in general, adults older than 50 years represent an excessive proportion of the early postoperative mortality encountered, and the number of previous operations and cyanosis are both risk factors.8 Risk factors for noncardiac surgery include heart failure, pulmonary hypertension, and cyanosis.9
Despite the fact that congenital cardiac disease carries implications for lifelong medical problems, a significant number of patients, even those with lesions deemed severe, will not have continuing cardiology follow-up, despite ongoing general medical care.10 These patients bring with them anatomic and physiologic complexities of which physicians accustomed to caring for adults may be unaware, as well as medical problems associated with aging or pregnancy that might not be familiar to physicians used to caring for children. This problem has led to the establishment of the growing subspecialty of adult CHD. There have been two Bethesda conferences devoted to the issue, most recently in 200111; and the American College of Cardiology reviewed the available evidence and published superb guidelines for the care of these patients in 2008.12 It is important to note that most of the current recommendations are based on Level C evidence (only consensus opinion or case studies) because prospective studies and large registries of patient outcomes are rare. An informed anesthesiologist is a critical member of the team required to care optimally for these patients. A specific recommendation of that conference was that noncardiac surgery on adult patients with moderate-to-complex CHD be done at an adult congenital heart center (regional centers) with the consultation of an anesthesiologist experienced with adult CHD.11,13 In fact, one of the founding fathers of the subspecialty wrote: “A cardiac anesthesiologist with experience in CHD is pivotal.…The cardiac anesthesiologist and the attending cardiologist are more important than the noncardiac surgeon.”2 High-risk patients include, but are not limited to, those with Fontan physiology, cyanotic disease, severe pulmonary arterial hypertension, complex disease with residua such as heart failure, valve disease or the need for anticoagulation, or the potential for malignant arrhythmias (Table 20-1). Centers may find it helpful to delegate one attending anesthesiologist to be the liaison with the cardiology service to centralize preoperative evaluations and triage of patients to an anesthesiologist with specific expertise in managing patients with CHD, rather than random consultations with generalist anesthesiologists.
TABLE 20-1 Rhythm Disturbances in Adults with Congenital Heart Disease
| Rhythm Disturbance | Associated Lesions |
|---|---|
| Tachycardias | |
| Wolff-Parkinson-White syndrome | Ebstein’s anomaly |
| Congenitally corrected transposition | |
| Intra-atrial reentrant tachycardia (atrial flutter) | Postoperative Mustard |
| Postoperative Senning | |
| Postoperative Fontan | |
| Tetralogy of Fallot | |
| Other | |
| Atrial fibrillation | Mitral valve disease |
| Aortic stenosis | |
| Tetralogy of Fallot | |
| Palliated single ventricle | |
| Ventricular tachycardia | Tetralogy of Fallot |
| Aortic stenosis | |
| Other | |
| Bradycardias | |
| Sinus node dysfunction | Postoperative Mustard |
| Postoperative Senning | |
| Postoperative Fontan | |
| Sinus venosus ASD | |
| Heterotaxy syndrome | |
| Spontaneous AV block | AV septal defects |
| Congenitally corrected transposition | |
| Surgically induced AV block | VSD closure |
| Subaortic stenosis relief | |
| AV valve replacement |
ASD, atrial septal defect; AV, atrioventricular; VSD, ventricular.
(Reprinted from ACC/AHA 2008 guidelines for the management of adults with congenital heart disease: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Develop Guidelines on the Management of Adults With Congenital Heart Disease). Developed in Collaboration with the American Society of Echocardiography, Heart Rhythm Society, International Society for Adult Congenital Heart Disease, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J Am Coll Cardiol 52:e143–e263, 2008.)
General noncardiac issues with longstanding congenital heart disease
A variety of organ systems can be affected by longstanding CHD; these are summarized in Table 20-2 and Box 20-2. Because congenital cardiac disease can be one manifestation of a multiorgan genetic or dysmorphic syndrome, all patients require a full review of systems and examination.
TABLE 20-2 Potential Noncardiac Organ Involvement in Patients with Congenital Heart Disease
| Potential Respiratory Implications |
| Decreased compliance (with increased pulmonary blood flow or impediment to pulmonary venous drainage) |
| Compression of airways by large, hypertensive pulmonary arteries |
| Compression of bronchioles |
| Scoliosis |
| Hemoptysis (with end-stage Eisenmenger syndrome) |
| Phrenic nerve injury (prior thoracic surgery) |
| Recurrent laryngeal nerve injury (prior thoracic surgery; rarely from encroachment of cardiac structures) |
| Blunted ventilatory response to hypoxemia (with cyanosis) |
| Underestimation of Paco2 by capnometry in cyanotic patients |
| Potential Hematologic Implications |
| Symptomatic hyperviscosity |
| Bleeding diathesis |
| Abnormal von Willebrand factor |
| Artifactually increased prothrombin/partial thromboplastin times with erythrocytic blood |
| Artifactual thrombocytopenia with erythrocytic blood |
| Gallstones |
| Potential Renal Implication |
| Hyperuricemia and arthralgias (with cyanosis) |
| Potential Neurologic Implications |
| Paradoxical emboli |
| Brain abscess (with right-to-left shunts) |
| Seizure (from old brain abscess focus) |
| Intrathoracic nerve injury (iatrogenic phrenic, recurrent laryngeal, or sympathetic trunk injury) |
BOX 20-2 Noncardiac Organ Systems with Potential Involvement By Longstanding Congenital Heart Disease

Pulmonary
Any lesion that results in either increased pulmonary blood flow or pulmonary venous obstruction can cause increased pulmonary interstitial fluid with decreased pulmonary compliance and increased work of breathing.14 Patients with cyanotic heart disease have increased minute ventilation and maintain normocarbia.15 These patients have a normal ventilatory response to hypercapnia but a blunted response to hypoxemia that normalizes after corrective surgery and the establishment of normoxia.16–18 End-tidal CO2 underestimates arterial Paco2 in cyanotic patients with decreased, normal, or even increased pulmonary blood flow.19
Although enlarged hypertensive pulmonary arteries or an enlarged left atrium can impinge on bronchi in children, this is rare in adults. Late-stage Eisenmenger syndrome can result in hemoptysis, and patients with Eisenmenger physiology and erythrocytosis can develop thrombosis of upper lobe pulmonary arteries.20 Prior thoracic surgery could have injured the phrenic nerve with resultant diaphragmatic paresis or paralysis.
Scoliosis can occur in up to 19% of CHD patients, most commonly in cyanotic patients. It can also develop in adolescence, years after surgical resolution of cyanosis.21 The relative contributions of cyanosis and congenital cardiovascular defects versus early lateral thoracotomy remain unclear: Scoliosis occurs more commonly with open surgical versus interventional techniques but is nevertheless increased over the general population in children with coarctation or patent ductus who had percutaneous rather than open interventions.22 Scoliosis is rarely severe enough to impact respiratory function.

Hematologic
Hematologic manifestations of chronic CHD are primarily a consequence of longstanding cyanosis and incorporate abnormalities of both hemostasis and red cell regulation. Longstanding hypoxemia causes increased erythropoietin production in the kidney and resultant increased red cell mass. Because solely red cell production is affected, these patients are correctly referred to as erythrocytotic rather than polycythemic. There is, however, a fairly poor relation among oxygen saturation, red cell mass, and 2,3-diphosphoglycerate.23 The oxygen-hemoglobin dissociation curve is normal or minimally shifted to the right. Most patients have established an equilibrium state at which they have a stable hematocrit and are iron replete. Some patients, however, develop excessive hematocrits and are iron deficient, causing a hyperviscous state. Iron-deficient red cells are less deformable and have been said to cause increased viscosity for the same hematocrit.24 However, recently there is some conflicting evidence.25 Symptoms of hyperviscosity are uncommon and typically develop only at hematocrits exceeding 65% provided the patient is iron replete. Iron deficiency also shifts the oxygen-hemoglobin dissociation curve to the right, decreasing oxygen affinity in the lungs.26 Symptoms of hyperviscosity are listed in Box 20-3. Iron deficiency can be the result of misguided attempts to reduce the hematocrit by means of repeated phlebotomies.27 The red cells in these patients may be hypochromic and microcytic despite the high hematocrit. Assessment of iron status is best done by measures of serum ferritin and transferrin rather than inferences from red cell indices.27 Treatment with oral iron needs to be undertaken with care because rapid increases in hematocrit can result.
Symptomatic hyperviscosity is the indication for treatment to temporarily relieve symptoms. It is not indicated to treat otherwise asymptomatic increased hematocrits (generally hemoglobin > 20 and hematocrit > 65). Treatment is by means of a partial isovolumic exchange transfusion, and it is assumed that the increased hematocrit is not related to dehydration. Partial isovolumic exchange transfusion usually results in regression of symptoms within 24 hours. It is rare to require exchange of more than 1 unit of blood. Before surgery, phlebotomized blood can be banked for autologous perioperative retransfusion if required. Elective preoperative isovolumic exchange transfusion has decreased the incidence of hemorrhagic complications of surgery.28,29
Hyperviscosity and erythrocytosis can cause cerebral venous thrombosis in younger children, but it is not a problem in adults, regardless of the hematocrit.20 Protracted preoperative fasts need to be avoided in erythrocytotic patients because they can be accompanied by rapid increases in the hematocrit.
Bleeding dyscrasias have been described in up to 20% of patients. A variety of clotting abnormalities have been described in association with cyanotic CHD, although none uniformly.30 Bleeding dyscrasias are uncommon until the hematocrit exceeds 65%, although excessive surgical bleeding can occur at lower hematocrits. Generally, higher hematocrits are associated with a greater bleeding diathesis. Abnormalities of a variety of factors in both the intrinsic and extrinsic coagulation pathways have been described inconsistently. Both cyanotic and acyanotic patients have been reported with deficiencies of the largest von Willebrand factor multimers, which have normalized after reparative surgery.31 Fibrinolytic pathways are normal.32

Platelet counts are typically normal or occasionally low, but bleeding is not due to thrombocytopenia. Platelets are reported per milliliter of blood, not milliliter of plasma. When corrected for the decreased plasma fraction in erythrocytic blood, the total plasma platelet count is closer to normal. That said, abnormalities in platelet function and life span have on occasion been reported.33,34 Patients with low-pressure conduits (Fontan pathway) or synthetic vascular anastomoses are often maintained on antiplatelet drugs.
Cyanotic erythrocytotic patients have excessive hemoglobin turnover, and adults have an increased incidence of calcium bilirubinate gallstones. Biliary colic can develop years after cyanosis has been resolved by cardiac surgery.20
A variety of mechanical factors can also affect excessive surgical bleeding in cyanotic CHD patients. These factors include increased tissue capillary density, increased systemic venous pressure, aortopulmonary and transpleural collaterals that have developed to increase pulmonary blood flow, and prior thoracic surgery. Aprotinin and ε-aminocaproic acid improve postoperative hemostasis in patients with cyanotic CHD.35 The results with tranexamic acid have been mixed.36

Renal
Some degree of renal insufficiency is not uncommon in adults with CHD, and the severity is a predictor of mortality. Moderate or severe renal dysfunction (estimated glomerular filtration rate < 60 mL/min/m2) carry a fivefold increased risk for death at 6-year follow-up compared with patients with normal glomerular filtration rate and a threefold increase over those with mild increases in glomerular filtration rate.37 Renal dysfunction is particularly prevalent in cyanotic patients and those with poor cardiac function. Adult patients with cyanotic CHD can develop abnormal renal histology with hypercellular glomeruli and basement membrane thickening, focal interstitial fibrosis, tubular atrophy, and hyalinized afferent and efferent arterioles.38 Cyanotic CHD is often accompanied by increases in plasma uric acid levels that are due to inappropriately low fractional uric acid excretion.39 Decreased urate reabsorption is thought to result from renal hypoperfusion with a high filtration fraction. Despite the increased uric acid levels, urate stones and urate nephropathy are rare.40 Although arthralgias are common, true gouty arthritis is less frequent than would be expected from the degree of hyperuricemia.39 There appears to be an increased incidence of postcardiopulmonary bypass renal dysfunction in adults with longstanding cyanosis.41

Neurologic
Adults with persistent or potential intracardiac shunts remain at risk for paradoxic embolism. Paradoxical emboli can occur even through shunts that are predominantly left to right because during the cardiac cycle there can be small transient reversals of the shunt direction. It has been said that, unlike in children, adults with cyanotic CHD are not at risk for the development of cerebral thrombosis despite the hematocrit.20,42 However, this assertion has been challenged by Ammash and Warnes,43 who suggested an association of stroke not with red cell mass, but with iron deficiency and repeated venesection. Adults do, however, remain at risk for the development of brain abscess. A healed childhood brain abscess can provide the nidus for the development of seizures throughout life.
Prior thoracic surgery can result in permanent peripheral nerve damage. Surgery at the apices of the lungs is particularly associated with the risk for nerve damage. These operations would include Blalock–Taussig shunts, ligation of patent ductus arteriosus (PDA), banding of the pulmonary artery, and repair of aortic coarctation. Nerves that are susceptible to injury include the recurrent laryngeal nerve, the phrenic nerve, and the sympathetic chain. The incidence of migraine headaches is greater in adults with CHD compared with a control group with acquired heart disease (45% vs. 11%) and is increased in left-to-right, right-to-left, and no-shunt groups.44

Vasculature
Vessel abnormalities can be congenital or iatrogenic. They can affect the suitability of vessels for cannulation by the anesthesiologist or measurement of correct pressures. These abnormalities are described in Table 20-3.
| Vessel | Possible Problem |
|---|---|
| Femoral vein(s) | May have been ligated if cardiac catheterization was done by cutdown; large therapeutic catheters in infants often thrombose femoral veins |
| Inferior vena cava | Some lesions, particularly when associated with heterotaxy (polysplenia), have discontinuity of the inferior vena cava; will not be able to pass a catheter from the groin to the right atrium |
| Left subclavian and pedal arteries | Distal blood pressure will be low in the presence of coarctation of the aorta or after subclavian flap repair (subclavian artery only), and variably so if postoperative recoarctation; pulses can be absent or palpable with abnormal blood pressure |
| Subclavian artery | Blood pressure low with classic Blalock-Taussig shunt on that side, and variably so with modified Blalock-Taussig shunt |
| Right subclavian artery | Blood pressure artifactually high with supravalvular aortic stenosis (Coanda effect) |
| Superior vena cava | Risk for catheter-related thrombosis with Glenn operation |

Pregnancy
The physiologic changes of pregnancy, labor, and delivery can significantly alter the physiologic status of women with CHD. Several texts are available that specifically discuss issues of the pregnant woman with CHD in more detail than is possible here,45–47 and there has been a recent review of generic cardiac surgery in the parturient.48 Management and clinical outcomes during pregnancy and delivery for several cardiac lesions are included under the later discussions of those lesions.
Although cardiac complications, spontaneous abortions, premature delivery, thrombotic complications, peripartum endocarditis, and poor fetal outcomes can occur,49 successful pregnancy to term with vaginal delivery is possible for most patients with congenital defects.50 High-risk factors for mother and fetus are shown in Box 20-4. Eisenmenger physiology is a particular risk factor. Up to 47% of cyanotic women have worsening of functional capacity during pregnancy.51 Hematocrits greater than 44% are associated with birth weights in less than the 50th percentile, and fetal death is about 90% or more with hemoglobin levels greater than 18 gm/dL or oxygen saturation less than 85%, with most losses in the first trimester. The increases in stroke volume and cardiac output during pregnancy can stress an already pressure-overloaded ventricle. The decrease in systemic vascular resistance that accompanies pregnancy is better tolerated by women with regurgitant lesions and typically offsets the added insult of pregnancy-related hypervolemia. The decrease in systemic vascular resistance can, however, increase right-to-left shunting. Hypervolemia can be problematic in patients with poor ventricular function. Maternal cyanosis is associated with increased incidences of prematurity and intrauterine growth retardation. Profound cyanosis is associated with a high rate of spontaneous abortion.52 Endocarditis prophylaxis is not currently recommended for vaginal deliveries.53 The recurrence risk rate of any congenital cardiac defect in a newborn is 2.3% with one affected older sibling (any defect), 7.3% with two, 6.7% if the mother has a congenital cardiac defect, but only 2.1% if the father is affected.2 However, it has become apparent that recurrence risk can be specific to the type of maternal defect and the underlying genetic basis. If possible, pregnancies in mothers with CHD should be managed in a high-risk obstetric center with cardiologists experienced with the care of adult CHD, and with early consultation with the obstetric anesthesia service. Women on long-term anticoagulation will likely need peripartum modifications, and postpartum thromboembolism is a potential significant problem.
Some mothers will be taking medications for their cardiac condition, including antiarrhythmics. In general, these are safe for the infant.54 Exceptions include β-blockers that can interfere with fetal growth and the response of the fetus to the stress of labor, as well as amiodarone that can affect fetal thyroid function. Maternal cardioversion would appear to be safe for the fetus at all stages, because of the low intensity of the electrical field at the uterus. However, the fetus should be monitored throughout the procedure. Women with implanted internal defibrillators have carried successfully to term.55 Should cardiopulmonary bypass (CPB) be required during pregnancy, it carries with it increased fetal risk, particularly if hypothermia is used.

Psychosocial
Teenagers with CHD are certainly no different from other teenagers in that issues of denial, a sense of immortality, and risk-taking behavior can affect optimal care for these youngsters. Bodies that carry scars from prior surgery and physical limitations can complicate the body-conscious teenage years. Although most adolescents and adults with CHD function well, adults with CHD are less likely to be married or cohabitating and are more likely to be living with their parents.56 There are several series of adolescent and adult patients and psychosocial outcome, but there are no well-done controlled studies.57–64 It has been suggested that depression is common and can exacerbate the clinical consequences of the cardiac defect.
Adolescent CHD patients have higher medical care expenses than the general population, and they can have difficulty in obtaining life and health insurance after they can no longer be covered under their parents’ policies.65–67 Life insurance is somewhat more available to adult CHD patients than in the past; however, policies vary widely among insurers.68
Cardiac issues
The basic hemodynamic effects of an anatomic cardiac lesion can be modified by time and by the superimposed effects of chronic cyanosis, pulmonary disease, or the effects of aging. Although surgical cure is the goal, true universal cure, without residua, sequelae, or complications, is uncommon on a population-wide basis. Exceptions include closure of a nonpulmonary hypertensive PDA or atrial septal defect (ASD), probably in childhood. Although there have been reports of series of surgeries on adults with CHD, the wide variety of defects and sequelae from prior surgery make generalizations difficult, if not impossible. Poor myocardial function can be inherent in the CHD but can also be affected by longstanding cyanosis or superimposed surgical injury, including inadequate intraoperative myocardial protection.68,69 This is particularly true of adults who had their cardiac repair several decades ago when myocardial protection may not have been as good and when repair was undertaken at an older age. Postoperative arrhythmias are common, particularly when surgery entails long atrial suture lines, and the incidence of atrial arrhythmias increases with time, either as a primary sequela or as an indicator of diminished cardiac function.70 Thrombi can be found in these atria precluding immediate cardioversion.71 Bradyarrhythmias can be secondary to surgical injury to the sinus node or conducting tissue or can be a component of the cardiac defect.
The number of cardiac lesions and subtypes, together with the large number of contemporary and obsolescent palliative and corrective surgical procedures, make a complete discussion of all CHD impossible. The reader is referred to one of the current texts on pediatric cardiac anesthesia for more detailed descriptions of these lesions, the available surgical repairs, and the anesthetic implications during primary repair.72,73 Some general perioperative guidelines to caring for these patients are offered in Table 20-4. This chapter provides a discussion of the more common and physiologically important defects that will be encountered in an adult CHD population. Defining outcome after CHD surgery is like trying to hit a continuously moving target. Both short-term and long-term results from older series can differ significantly from contemporary results.
TABLE 20-4 General Approach to Anesthesia for Patients with Congenital Heart Disease
| General |
| The best care for both cardiac and noncardiac surgery in adult patients with CHD is afforded in a center with a multidisciplinary team experienced in the care of adults with CHD, and knowledgeable about both the anatomy and physiology of CHD and the manifestations and considerations specific to adults with CHD |
| Preoperative |
| Review most recent laboratory data, catheterization, and echocardiogram, and other imaging data; the most recent office letter from the cardiologist is often most helpful; obtain and review these in advance |
| Drawing a diagram of the heart with saturations, pressures, and direction of blood flow often clarifies complex and superficially unfamiliar anatomy and physiology |
| Avoid prolonged fast if patient is erythrocytotic to avoid hemoconcentration |
| No generalized contraindication to preoperative sedation |
| Intraoperative |
| Large-bore intravenous access for redo sternotomy and cyanotic patients (see Table 20-3 for vascular access considerations) |
| Avoid air bubbles in all intravenous catheters; there can be transient right-to-left shunting even in lesions with predominant left-to-right shunting (filters are available but will severely restrict ability to give volume and blood) |
| Apply external defibrillator pads for redo sternotomies and patients with poor cardiac function |
| Appropriate endocarditis prophylaxis (orally or intravenously before skin incision)40 |
| Consider antifibrinolytic therapy, especially for patients with prior sternotomy |
| Transesophageal echocardiography for cardiac operations |
| Modulate pulmonary and systemic vascular resistances as appropriate pharmacologically and by modifications in ventilation |
| Postoperative |
| Appropriate pain control (cyanotic patients have normal ventilatory response to hypercarbia and narcotics) |
| Maintain hematocrit appropriate for arterial saturation |
| Maintain central venous and left atrial pressures appropriate for altered ventricular diastolic compliance or presence of beneficial atrial level shunting |
| Pao2 may not increase significantly with the application of supplemental oxygen in the face of right-to-left shunting; similarly, neither will it decrease much with the withdrawal of oxygen (in the absence of lung pathology) |

Aortic Stenosis
Most aortic stenosis in adults is due to a congenitally bicuspid valve that does not become problematic until late middle age or later, although endocarditis risk is lifelong. Congenital aortic stenosis can, on some occasions, however, become severe enough to warrant surgical correction in adolescence or young adulthood, in addition to those severely affected valves that present in infancy. Once symptoms (angina, syncope, near-syncope, heart failure) develop, survival is markedly shortened. Median survival is 5 years after the development of angina, 3 years after syncope, and 2 years after heart failure.74 Anesthetic management of aortic stenosis does not vary whether the stenosis is congenital (most common) or acquired (see Chapter 19).

Aortopulmonary Shunts
Depending on their age, adult patients may have had one or more of several aortopulmonary shunts to palliate cyanosis during childhood. These are shown in Figure 20-1. Although life saving, there were considerable shortcomings of these shunts in the long term. All were inherently inefficient, as some of the oxygenated blood returning through the pulmonary veins to the left atrium and ventricle would then return to the lungs through the shunt, thus volume loading the ventricle. It was difficult to quantify the size of the earlier shunts such as the Waterston (side-to-side ascending aorta to right pulmonary artery) and Potts (side-to-side descending aorta to left pulmonary artery). If too small, the patient was left excessively cyanotic; if too large, there was pulmonary overcirculation with the risk for development of pulmonary vascular disease. The Waterston, in fact, could, on occasion, stream blood flow unequally, resulting in a hyperperfused, hypertensive ipsilateral (right) pulmonary artery and a hypoperfused contralateral (left) pulmonary artery. There were also surgical issues when complete repair became possible. Takedown of Waterston shunts often required a pulmonary arterioplasty to correct deformity of the pulmonary artery at the site of the anastomosis, and the posteriorly located Potts anastomoses could not be taken down from a median sternotomy. Patients with a classic Blalock–Taussig shunt almost always lack palpable pulses on the side of the shunt. Even if there is a palpable pulse (from collateral flow around the shoulder), blood pressure obtained from that arm will be artifactually low. Even after a modified Blalock–Taussig shunt (using a piece of Gore-Tex tubing instead of an end-to-side anastomosis of the subclavian and pulmonary arteries), there can be a blood pressure disparity between the arms. Preoperative blood pressure should be measured in both arms to ensure a valid measurement (Box 20-5).
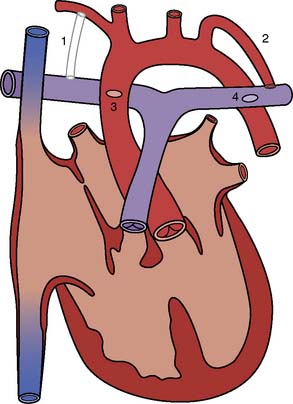
Figure 20-1 Various aortopulmonary anastomoses.
(From Baum VC: The adult with congenital heart disease. J Cardiothorac Vasc Anesth 10:261, 1996.)
BOX 20-5 Aortopulmonary Shunts
| Waterston | Ascending aorta→right pulmonary artery | No longer done |
| Potts | Descending aorta→left pulmonary artery | No longer done |
| Classic Blalock–Taussig | Subclavian artery→ipsilateral pulmonary artery | No longer done |
| Modified Blalock–Taussig | Gore-Tex tube subclavian artery→ipsilateral pulmonary artery | Current |
| Central shunt | Gore-Tex tube ascending aorta→main pulmonary artery | Current |

Atrial Septal Defect and Partial Anomalous Pulmonary Venous Return
There are several anatomic types of ASD. The most common type, and if otherwise undefined the presumptive type, is the secundum type located in the midseptum. The primum type at the lower end of the atrial septum is a component of endocardial cushion defects, the most primitive of which is the common atrioventricular canal (see later). The sinus venosus type, high in the septum near the entry of the superior vena cava (SVC), is almost always associated with partial anomalous pulmonary venous return, most frequently drainage of the right upper pulmonary vein to the low SVC. An uncommon atrial septal–type defect is when blood passes from the left atrium to the right via an unroofed coronary sinus. For purposes of this section, only secundum defects are considered, although the natural histories of all of the defects are similar (Box 20-6).
Both the natural history and the postoperative outcome of ASDs and the physiologically related partial anomalous venous return are similar.75–77 Because the symptoms and clinical findings of an ASD can be quite subtle and patients often remain asymptomatic until adulthood, ASDs represent approximately one third of all CHD discovered in adults. Although asymptomatic survival to adulthood is common, significant shunts (p/s > 1.5:1) will probably cause symptoms over time, and paradoxical emboli can occur through defects with smaller shunts. Surgical repair of restrictive lesions 5 mm or smaller in size does not impact the natural history. Thus, surgical closure of small lesions is not indicated in the absence of paradoxical emboli. Effort dyspnea occurs in 30% by the third decade, and atrial flutter or fibrillation in about 10% by age 40.75 The avoidance of complications developing in adulthood provides the rationale for surgical repair of asymptomatic children. The mortality for a patient with an uncorrected ASD is 6% per year after 40 years of age, and essentially all patients older than 60 years are symptomatic.75–77 Large, unrepaired defects can cause death from atrial tachyarrhythmias or right ventricular failure in 30- to 40-year-old patients.78 With the decreased left ventricular diastolic compliance accompanying the systemic hypertension or coronary artery disease that is common with aging, left-to-right shunting increases with age. Pulmonary vascular disease typically does not develop until after the age of 40, unlike ventricular or ductal level shunts, which can lead to it in early childhood. Mitral insufficiency can be found in adult patients and is significant in about 15% of adult patients.79 Paradoxical emboli remain a lifelong risk.
Late closure of the defect, after 5 years of age, has been associated with incomplete resolution of right ventricular dilation.80 Left ventricular dysfunction has been reported in some patients having defect closure in adulthood, and closure, particularly in middle age, may not prevent the development of atrial tachyarrhythmias or stroke.81–84 Survival of patients without pulmonary vascular disease has been reported to be best if operated on before 24 years of age, intermediate if operated on between 25 and 41 years of age, and worst if operated on thereafter.84 However, more recent series have shown that even at ages older than 40, surgical repair provides an overall survival and complication-free benefit compared with medical management.85 Surgical morbidity in these patients is primarily atrial fibrillation, atrial flutter, or junctional rhythm.82 Current practice is to close these defects in adults in the catheterization laboratory via transvascular devices if anatomically practical (Figure 20-2). For example, there needs to be an adequate rim of septum around the defect to which the device can attach. Device closure is inappropriate if the defect is associated with anomalous pulmonary venous drainage. The indications for closure with a transvascular device are the same as for surgical closure.86
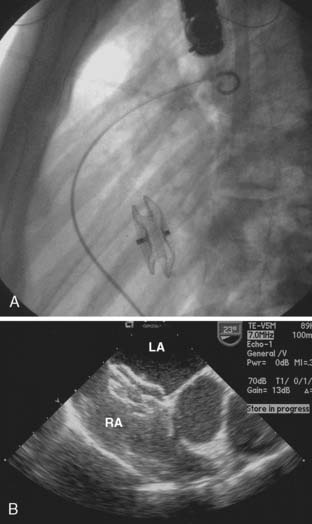
An otherwise uncomplicated secundum ASD, unlike most congenital cardiac defects, is not associated with an increased endocarditis risk.87 Presumably, this is because the shunt, although potentially large, is low pressure and unassociated with jet lesions of the endocardium.

Coarctation of the Aorta
Unrepaired coarctation of the aorta in the adult brings with it significant morbidity and mortality. Mortality rate is 25% by age 20, 50% by age 30, 75% by age 50, and 90% by age 60.78,88–90 Left ventricular aneurysm, rupture of cerebral aneurysms, and dissection of a postcoarctation aneurysm all contribute to the excessive mortality. Left ventricular failure can occur in patients older than 40 with unrepaired lesions. If repair is not undertaken early, there is incremental risk for the development of premature coronary atherosclerosis. Even with surgery, coronary artery disease remains the leading cause of death 11 to 25 years after surgery.91 Coarctation is accompanied by a bicuspid aortic valve in the majority of patients. Although endocarditis of this abnormal valve is a lifelong risk, these valves often do not become stenotic until middle age or later. Coarctation can also be associated with mitral valve abnormalities (Box 20-7).
Aneurysms at the site of coarctation repair can develop years later (Figure 20-3), and restenosis as well can develop in adolescence or adulthood. Repair includes resection of the coarctation and end-to-end anastomosis. Because this sometimes resulted in recoarctation when done in infancy, for many years a common repair was the Waldhausen or subclavian flap operation, in which the left subclavian artery is ligated and the proximal segment opened and rotated as a flap to open the area of the coarctation. Aneurysms in the area of repair are a particular concern in adolescents and adults after coarctectomy. Persistent systemic hypertension is common after coarctation repair.92 The risk for hypertension parallels the duration of unrepaired coarctation. Adult patients require continued periodic follow-up for hypertension. A pressure gradient of 20 mm Hg or more (less in the presence of extensive collaterals) is an indication for treatment.93 Recoarctation can be treated surgically or by balloon angioplasty with stenting.94 Surgical repair of recoarctation or aneurysm in adults is associated with increased mortality and can be associated with significant intraoperative bleeding because of previous scar or extensive collateral vessels. It requires lung isolation for optimal surgical exposure and placement of an arterial catheter in the right arm. Endovascular repair by ballooning/stenting has proved useful for these patients.95,96
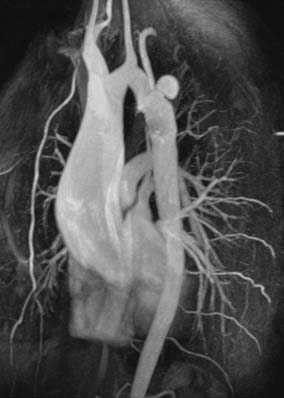
Half of patients operated on after age 40 have persistent hypertension, and many of the remainder have an abnormal hypertensive response to surgery. Long-term survival is worse for patients having repair later in life. Patients older than 40 undergoing repair have a 15-year survival rate of only 50%.91

Congenitally Corrected Transposition of the Great Vessels (l-Transposition, Ventricular Inversion)
“Transposition” in this context refers solely to the fact that the aorta arises anterior to the pulmonary artery. It bears no reference to the origin of blood in the aorta or pulmonary artery, or to the ventricle of origin of those vessels. In l-transposition, as a consequence of the embryonic heart tube rotating to the left rather than to the right, the flow of blood is through normal vena cavae to the right atrium, through a mitral valve to a right-sided anatomic left ventricle, to the pulmonary artery, through the pulmonary circulation, to the left atrium, through a tricuspid valve to a left-sided anatomic right ventricle, and thence to the aorta (Figure 20-4) Although anatomically altered, the physiologic flow of blood is appropriate and there are no associated shunts. l-Transposition of the great arteries (L-TGA) is frequently associated with other cardiac lesions, most commonly a ventricular septal defect (VSD), subpulmonic stenosis, heart block, or systemic atrioventricular (tricuspid) valve regurgitation. In the absence of any of these associated cardiac defects, L-TGA will usually be asymptomatic through infancy and childhood. When l-transposition is an isolated lesion, most patients maintain normal biventricular function through early adulthood, and can attain a normal life span.
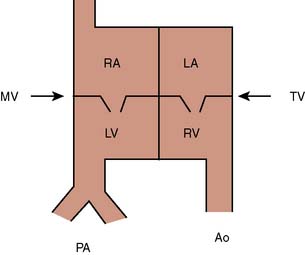
Systemic atrioventricular (tricuspid) valve insufficiency may not develop until later in life, resulting in approximately 60% of patients being diagnosed as adults.97 Dysfunction of the right or systemic ventricle can develop with aging, and asymptomatic aging is relatively uncommon.98 By age 45, heart failure will be present in 67% of those patients with associated lesions and 25% of those without.99 These patients can be born with congenital heart block, which can progress in degree. Second- or third-degree heart block occurs at a rate of about 2% per year. More than 75% of patients develop some degree of heart block, although the intrinsic pacemaker remains above the His bundle with a narrow QRS complex. l-Transposition can be associated with an Ebstein-like deformity of the systemic atrioventricular (tricuspid) valve, and there can be a bundle of Kent causing the Wolff-Parkinson-White syndrome associated with that abnormal valve. There is a significant incidence of tricuspid valve insufficiency in the systemic ventricle, and this is greater still in patients with an Ebstein’s deformity of the valve.100 Anesthetic management depends on the presence of any associated lesions and the adequacy of right (systemic) ventricular function.
Although women generally do well with pregnancy,101 the physiologic stresses of pregnancy and delivery can result in ventricular or valvular dysfunction, particularly with baseline dysfunction and/or an insufficient systemic atrioventricular valve; but even if these develop, pregnancy can be successfully managed.101–104 The decrease in systemic vascular resistance associated with both pregnancy and neuraxial analgesia might be advantageous to women with tricuspid (systemic) valve insufficiency. The acute autotransfusion associated with delivery could potentially cause problems for women with existing diminished systemic ventricle function.

Ebstein’s Anomaly of the Tricuspid Valve
Ebstein’s anomaly, one of arrested or incomplete delamination of the embryonic tricuspid valve from the right ventricular myocardium, resulting in apically displaced valve tissue, is the most common cause of congenital tricuspid insufficiency. The septal leaflet tends to be the most dysplastic. The anterior leaflet tends to be large and redundant. The defect is associated with a patent foramen ovale or a secundum ASD. There is “atrialization” of part of the right ventricle. The displacement of the tricuspid valve toward the right ventricular apex results in a portion of the right ventricle being above the tricuspid valve and becoming functionally part of the right atrium. Apicalization of the tricuspid valve results in a portion of the heart above the valve having a ventricular intracardiac electrogram (it is ventricular myocardium) but atrial pressures (it lies above the tricuspid valve). The right atrium can be massively enlarged (Figure 20-5), and the right ventricle, lacking the inflow portion that is now part of the right atrium, is smaller than usual with varying degrees of pulmonic stenosis. Patients with l-transposition (see earlier) can have Ebstein’s anomaly of a left-sided tricuspid valve.
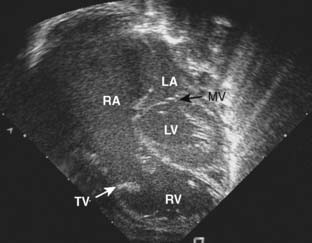
Figure 20-5 Ebstein’s anomaly of the tricuspid valve.
(From Baum VC: Abnormalities of the atrioventricular valves. In Lake CL, Booker P [eds]: Pediatric cardiac anesthesia, 4th ed. Philadelphia, 2004: Lippincott Williams & Wilkins.)
Symptoms will vary based on the amount of displacement of the valve and the size of the smaller than normal right ventricle. Cyanosis from atrial-level right-to-left shunting can be a neonatal phenomenon, resolving with the normal postnatal decrease in pulmonary vascular resistance, only to recur in adolescence or adulthood. Very mild disease is quite compatible with asymptomatic survival into adulthood, although overall earlier reports suggested a mean age at death of about 20 years, with about one third dying before 10 years of age and only 15% survival rate by 60 years.105–107 About half of patients experience development of arrhythmias, typically supraventricular tachyarrhythmias. Once symptoms develop, disability rapidly progresses.
Valve repair is currently the approach of choice, and only uncommonly will tricuspid valve replacement be required.108 Additional intervention may be required for progressive insufficiency, stenosis, prosthetic valve failure, or valve replacement with growth. After valve replacement, up to 25% of patients experience development of high-grade atrioventricular block. Ebstein’s valves are often associated with a right-sided bypass tract, causing Wolff-Parkinson-White syndrome. This is of concern because 25% to 30% of patients experience development of supraventricular tachyarrhythmias. The dilated right atrium is ready substrate for the development of atrial fibrillation. In addition to a decrease in cardiac performance, atrial fibrillation is also potentially dangerous in patients with underlying Wolff-Parkinson-White syndrome because very high atrial rates can be conducted to the ventricle through the bypass tract.
In the absence of marked cyanosis, pregnancy and delivery are generally well tolerated. There is, however, an increased incidence of prematurity and fetal loss, and birth weights are lower in infants of cyanotic mothers,109 as well as an increased incidence of CHD in the offspring.

Eisenmenger Syndrome
Eisenmenger described a particular type of large VSD with dextroposition of the aorta.110 In a general way, the term Eisenmenger syndrome has come to describe the clinical setting in which a large left-to-right cardiac shunt results in the development of pulmonary vascular disease and has been the subject of recent reviews.111 Although early on the pulmonary vasculature remains reactive, with continued insult, pulmonary hypertension becomes fixed and does not respond to pulmonary vasodilators. Ultimately, the level of pulmonary vascular resistance is so high that the shunt reverses and becomes right to left. Clinically, patients who are cyanotic from intracardiac right-to-left shunting are deemed to have Eisenmenger physiology even though their PVR may not yet truly be “fixed.” This is the intermediate phase of the disease before progression to a truly fixed PVR. That is, at baseline they shunt right to left but may still retain some pulmonary vascular reactivity in the presence of vasodilating agents such as oxygen or nitric oxide. The degree of reactivity can be determined in the catheterization laboratory by measuring the pulmonary blood flow on room air, pure oxygen, and pure oxygen with nitric oxide added. The development of pulmonary vascular disease is dependent on shear rate. Lesions with high shear rates, such as a large VSD or a large PDA, can result in pulmonary hypertension in early childhood. Lesions such as an ASD with high pulmonary blood flow but low pressure may not result in pulmonary vascular disease until late middle age. Pulmonary vascular disease progression is also accelerated in patients living at high altitudes.
The most common presenting symptom is dyspnea on exertion. Additional symptoms include palpitations, edema, hemoptysis, syncope, hyperpnea, and of course, increasing cyanosis. Hepatic synthetic function can be altered from the increased central venous pressure. There can be CNS symptoms from increased blood viscosity from the erythrocytosis associated with cyanosis. Right ventricular ischemia is a possibility. Patients may be on chronic therapy with drugs such as intravenous prostacyclin, an oral phosphodiesterase-5 inhibitor such as sildenafil (e.g., Revatio), or an oral endothelin receptor antagonist such as bosentan (e.g., Tracleer). Because of the risk for pulmonary thromboses,112 patients may be on chronic anticoagulants.
Eisenmenger physiology is compatible with survival into adulthood.113,114 However, reported rates of survival after diagnosis vary, probably based on the relatively long life expectancy and variability in the time of diagnosis. Cantor et al115 reported median survival to 53 years but with wide variation. Saha et al116 reported a survival rate of 80% at 10 years after diagnosis and 42% at 25 years. Oya et al,117 however, reported survival rate of 77% at 5 years and 58% at 10 years. Syncope, increased central venous pressure, and arterial desaturation to less than 85% are all associated with poor short-term outcome.113 Other factors associated with mortality include syncope, age at presentation, functional status, supraventricular arrhythmias, increased right atrial pressure, renal insufficiency, severe right ventricular dysfunction, and trisomy 21. Most deaths are sudden cardiac deaths. Other causes of death include heart failure, hemoptysis, brain abscess, thromboembolism, and complications of pregnancy and noncardiac surgery.118 These patients face potentially significant perioperative risks. Findings of Eisenmenger syndrome are summarized in Table 20-5.
Surgical closure of cardiac defects with fixed pulmonary vascular hypertension is associated with very high mortality. Lung or heart-lung transplantation is a surgical alternative.119 Although there are several surgical series reporting survival after heart-lung or single- or double-lung transplantation performed for primary pulmonary hypertension, it is unclear whether this cohort of patients is similar to patients with Eisenmenger physiology.
When noncardiac surgery is deemed essential and time permits, a preoperative cardiac catheterization may be helpful to determine the presence of pulmonary reactivity to oxygen or nitric oxide. Fixed pulmonary vascular resistance precludes rapid adaptation to perioperative hemodynamic changes. Changes in systemic vascular resistance are mirrored by changes in intracardiac shunting. A decrease in systemic vascular resistance is accompanied by increased right-to-left shunting and a decrease in systemic oxygen saturation. Systemic vasodilators, including regional anesthesia, need to be used with caution, and close assessment of intravascular volume is important. Epidural analgesia has been used successfully in patients with Eisenmenger physiology, but the local anesthetic needs to be delivered slowly and incrementally with close observation of blood pressure and oxygen saturation.120 Postoperative postural hypotension can also increase the degree of right-to-left shunting, and these patients should change position slowly. All intravenous catheters need to be maintained free of air bubbles.
Placement of pulmonary artery catheters in these patients is problematic for a variety of reasons, and they are of less utility than might be expected. Pulmonary arterial hypertension is a risk factor for pulmonary artery rupture from a pulmonary artery catheter. Rupture is particularly worrisome in these cyanotic patients who can also have hemostatic deficits associated with erythrocytosis.121 Abnormal intracardiac anatomy and right-to-left shunting can make successful passage into the pulmonary artery difficult without fluoroscopy. Relative resistances of the pulmonary and systemic beds are reflected in the systemic oxygen saturation, readily measured by pulse oximetry, so measures of pulmonary artery pressure are not required. In addition, in the presence of right-to-left shunting, thermodilution cardiac outputs do not accurately reflect systemic output. Thus, the value of pulmonary artery catheters in these patients is minimal at best, and they essentially are never indicated. The one potential exception is the patient with an ASD who is at risk for development of right ventricular failure if suprasystemic right ventricular pressure develops.122
Pregnancy carries a very high mortality risk and risk for premature delivery. From 20% to 30% of pregnancies result in spontaneous abortions, and premature delivery occurs in about half.123 At least half of newborns have intrauterine growth retardation. From 30% to 45% of all pregnancies end in maternal death intrapartum or during the first postpartum week, and successful first pregnancy does not preclude maternal death during a subsequent pregnancy.124 The hemodynamic changes of both pregnancy and delivery increase maternal risk. Pulmonary microembolism and macroembolism have caused peripartum maternal deaths, even days after delivery. Factors that influence mortality include thromboembolism (44%), hypovolemia (26%), and preeclampsia (18%).123,124 Mortality is similar with Cesarean section or vaginal delivery, and both are significantly greater than the mortality rate for spontaneous abortions. Pregnancy should be discouraged in these women. Women who do become pregnant should be closely monitored with arterial catheters during delivery. Epidural analgesia, delivered slowly and incrementally, can moderate many of the deleterious hemodynamic changes of active labor. Pulmonary arterial catheters are of little to no use during delivery. Prompt treatment of blood loss and hypotension during delivery is absolutely required. Postpartum observation should be in an intensive care setting. Pulmonary hypertension in pregnancy was reviewed in detail.125,126

Endocardial Cushion Defects (Atrioventricular Canal Defects)
There is much subtlety required to construct two separate functional atrioventricular valves without residual stenosis or insufficiency of either component. Postoperative or residual mitral insufficiency is not uncommon, and 10% to 30% require repeat surgery.127 The strongest predictor of postoperative mitral insufficiency is the degree of preoperative regurgitation of the common atrioventricular valve. Anesthesia for these patients depends on the degree of shunt, valve insufficiency, and pulmonary vascular disease.

Fontan Physiology
In 1968, performing the operation that now bears his name, Fontan and Baudet128 proved that it was possible to deliver the entire systemic venous return to the lungs without the benefit of a ventricular pump. The Fontan operation was a landmark development in CHD because it established a “normal” series circulation in patients with a single ventricle. The price to be paid for a series circulation is the unique physiologic demand of passive pulmonary blood flow. Complications never envisioned at the time of the original operation have occurred, necessitating significant changes in operative technique. Fontan’s original operation (Figure 20-6) was soon modified to an atriopulmonary connection129 (Figure 20-7). The original strict eligibility criteria130 have been liberalized, but patients meeting as many of the criteria as possible still have the best prognosis for good long-term survival. By the mid-1980s, it became clear that the success of Fontan circulation was based on an unobstructed pathway from systemic veins to pulmonary artery, a pulmonary vasculature that was free from anatomic distortion (from previous Blalock-Taussig shunt, for example), low PVR, and good ventricular function without significant atrioventricular valve regurgitation. The incorporation of the atrium in the Fontan pathway proved disappointing. The atrium lost its contractile function, providing no assistance to pulmonary blood flow and causing serious complications. Understanding these complications and how the Fontan operation has evolved is the key to managing these challenging patients whose complex CHD has been palliated, not cured.
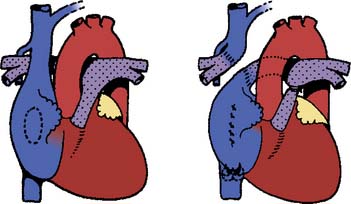
Figure 20-6 The original Fontan operation.
(From Fontan F, Baudet E: Surgical repair of tricuspid atresia. Thorax 26:240, 1971.)
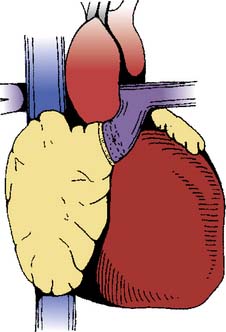
Figure 20-7 The atriopulmonary modification of the Fontan operation.
(From Kreutzer G, Galindez E, Bono H, et al: An operation for the correction of tricuspid atresia. J Thorac Cardiovasc Surg 66:613, 1973.)
Complications
The dilated, noncontractile atrium serves as a reservoir of blood and a ready source of thrombus.131–133 Pulmonary embolization will impair the passive blood flow necessary for successful Fontan circulation. Atrial thrombus could embolize paradoxically through residual right-to-left shunts. Patients are also at risk for arterial thrombosis, secondary to a mild hypercoagulable state.134,135 Given the morbidity associated with thromboembolism, it seems reasonable to prescribe aspirin therapy to all Fontan patients.136 Those who display further potential for thrombosis such as low cardiac output state, atrial arrhythmia with significant atrial dilation, or marked venous hypertension may benefit from warfarin (Coumadin). Box 20-8 lists complications of the Fontan procedure.
Fontan patients show a steady increase in atrial tachyarrhythmias with an incidence of more than 50% at 20 years.137 Changes in surgical technique evolved, in part, to decrease the rate of atrial arrhythmias. Although initial results were promising,138 unfortunately, much of this benefit is lost with longer term follow-up.139 Fontan surgical patients tolerate tachycardia poorly, and acute episodes usually require urgent treatment using medical therapy to control ventricular rate or cardioversion. Late-onset atrial tachyarrhythmias usually occur between 6 and 10 years after Fontan completion.140 The most common tachyarrhythmia is right intra-atrial reentrant tachycardia. Over time, episodic attacks of tachycardia become more frequent. Frequently, atrial fibrillation occurs and the loss of atrioventricular synchrony results in decreased effort tolerance. The onset of atrial tachyarrhythmias mandates an evaluation of the Fontan pathway with attention turned to relieving any significant obstructions. In the setting of passive pulmonary blood flow, even small gradients can be hemodynamically significant.141 Therapies for chronic atrial arrhythmias consist of medication, catheter ablation, and surgery. Given the complex anatomy, dilated atrium and atrial scar with suture lines from prior surgeries, it is not surprising that atrial arrhythmias can become refractory to standard treatment in many patients. Catheter ablation typically has high initial success rates that are not maintained.141
Bradyarrhythmias, caused by sinus node ischemia, are common. In a large cohort of patients with atriopulmonary connections, the incidence rate of bradyarrhythmias requiring pacemakers was 13%.142 Progressive fibrosis and scar around the sinus node, caused by prior surgical dissection, eventually leads to ischemia and clinical sinus node dysfunction. If accompanied by premature atrial contractions, sinus or junctional bradycardia can precipitate an intra-atrial reentry tachycardia. Thus, sinus node dysfunction also serves as a risk factor for the development of atrial tachyarrhythmias. Clinically significant bradyarrhythmias require pacing. Pacemakers pose special problems in the Fontan patient because the altered anatomy precludes transvenous placement. Thus, Fontan patients who require pacing end up with epicardial leads placed via repeat sternotomy with all the risks that entails. Even though atrioventricular synchrony can be achieved with pacing, it is still not as good as intrinsic sinus rhythm. The incidence of sinus node dysfunction is less with a cavopulmonary versus an atriopulmonary connection.143 However, a clear benefit from an extracardiac connection (see later) when compared with the lateral tunnel approach has been difficult to prove.144,145
The last major complication of Fontan physiology is protein-losing enteropathy (PLE), a condition as confounding as it is serious. The incidence rate is quoted as high as 15%, but a large international multicenter study found a rate of 3.7%.146 Clinically, there is an edematous state with ascites and pleural-pericardial effusions. Serum albumin is low and the diagnosis is confirmed by finding enteric protein loss with increased levels of stool α1 antitrypsin. Most ominously, PLE is accompanied by a 50% mortality rate 5 years from diagnosis despite treatment. It was believed that PLE constituted a straightforward situation of increased portal pressures secondary to central venous hypertension. Increased portal pressures lead to vascular congestion, lymphatic obstruction, and enteric protein loss from the gut. Unfortunately, there is not a good correlation between central venous pressures and PLE.147 This has led to a broader understanding of PLE as a multifactorial phenomenon caused by reduced mesenteric perfusion,147 chronic inflammation,148 and enterocyte dysfunction.149 Patients who present with PLE should have a complete hemodynamic evaluation. This is vital because interventions that improve cardiac output have proved successful in PLE. Any Fontan pathway obstruction should be treated and cardiac output optimized with medical therapy, fenestration, or pacing. In the absence of correctable obstructions, PLE portends a poor prognosis, despite surgery or cardiac transplantation.
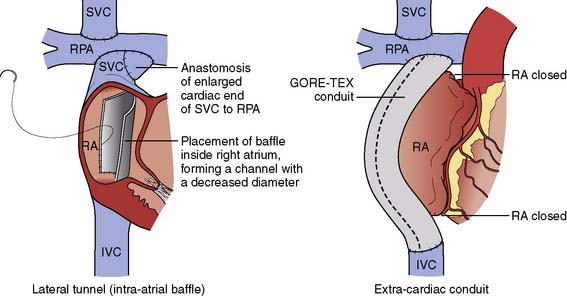
The Modern Fontan Operation
The atriopulmonary connection proved an inefficient method of pulmonary blood flow. Colliding streams of blood from the SVC and inferior vena cava (IVC) resulted in energy loss and turbulence within the atrium.150 The energy required to propel blood forward into the pulmonary vasculature was lost as blood swirled sluggishly in the dilated atrium (Figure 20-8). The modern Fontan operation is a total cavopulmonary connection (Figure 20-9). The “lateral tunnel Fontan” improved pulmonary blood flow, and only the lateral wall of the atrium was exposed to central venous hypertension. There was no dilated atrium to serve as a source of thrombus. The extensive atrial suture lines, however, remained a risk for arrhythmia. The “extracardiac Fontan” is a further modification of the total cavopulmonary connection. The “extracardiac Fontan” greatly reduces the number of atrial incisions and hopefully the long-term development of atrial arrhythmias. Has the modern Fontan improved outcomes? Reductions in arrhythmia and improvements in overall survival have been noted.151 Results for the extracardiac Fontan are even better than the lateral tunnel Fontan but are limited by the shorter duration of follow-up. It is not yet certain whether the development of long-term complications has been truly reduced or only delayed.
Preoperative Assessment
Patients with Fontan physiology are presenting in larger numbers for the entire array of noncardiac surgery including obstetric procedures. Preoperative assessment begins with a directed history, concentrating on functional status and the presence of major complications. Heightened suspicion is clearly needed for patients with atriopulmonary connections, as well as those with a systemic right ventricle. Patients with Fontan circulation have a low cardiac output state. This low output state exists despite the presence of good ventricular function, minimal atrioventricular valve regurgitation, and low PVR. A cohort of patients with an atriopulmonary Fontan performed at an older age showed striking reductions in anaerobic threshold (< 50% of control subjects),  O2 max (< 33% of control subjects), and systemic ejection fraction both at rest and exercise.152 Further compounding the issue is that patients’ self-assessment grossly overestimates their objective exercise capacity.153 This places the anesthesiologist in a considerable dilemma when faced with a Fontan patient who rates his or her functional status as “good.” The authors believe that transthoracic echocardiography should be the initial preoperative investigation and is mandatory except in cases of very minor surgery. Further testing is guided by the results of the echocardiogram and in consultation with a cardiologist experienced in caring for adults with CHD. Normal ventricular function on an echocardiogram would stratify the patient as “low risk” only in the context of patients with Fontan circulation.
O2 max (< 33% of control subjects), and systemic ejection fraction both at rest and exercise.152 Further compounding the issue is that patients’ self-assessment grossly overestimates their objective exercise capacity.153 This places the anesthesiologist in a considerable dilemma when faced with a Fontan patient who rates his or her functional status as “good.” The authors believe that transthoracic echocardiography should be the initial preoperative investigation and is mandatory except in cases of very minor surgery. Further testing is guided by the results of the echocardiogram and in consultation with a cardiologist experienced in caring for adults with CHD. Normal ventricular function on an echocardiogram would stratify the patient as “low risk” only in the context of patients with Fontan circulation.
A term that should immediately get the attention of the anesthesiologist is “failing Fontan.” The specific reason for failing may be different, but the common denominator in these patients is a marked limitation of functional status. They will manifest some combination of refractory arrhythmias, PLE, liver dysfunction, hypoxemia, or congestive heart failure. Although PLE always signifies a failing Fontan, the converse is not always true. That is, patients can have severely limited function with increased central venous pressures and even evidence of cirrhosis on liver biopsy without demonstrating PLE.154 Patients with a “failing Fontan” require a search for correctable lesions.155 First, any obstructions within the Fontan pathway should be treated, preferably with percutaneous techniques of dilation and stenting. Second, loss of sinus rhythm should be treated with pacing. If loss of sinus rhythm is accompanied by severe tachyarrhythmias, Fontan conversion surgery is indicated. Third, some patients develop collateral vessels. Aortopulmonary collaterals result in a progressive volume load on the single ventricle. Collaterals from the venous system to the systemic atrium or ventricle cause hypoxemia. In both cases, large collaterals should be coil occluded in the catheterization laboratory. Another option is the creation of a fenestration, which can improve cardiac output and lower central venous pressures but at the expense of a right-to-left shunt. Unfortunately, not all of these therapeutic options are indicated or successful in every patient. At this point, if no realistic hope of further improvement exists, the only option is cardiac transplantation.
The functional state of Fontan patients exists across a spectrum but generally falls into two groups. The first and largest group is those who report New York Heart Association Class I-II level of function but have been shown to possess much less cardiorespiratory reserve than age-matched two-ventricle control subjects. These patients will tolerate most surgical procedures with an acceptably low risk. The second group is smaller but consists of those patients who have manifested one of more of the “failing Fontan” criteria. Surgery in these patients carries much greater risk and should only be undertaken after careful consultation with physicians experienced in adult CHD. When it comes to a discussion of anesthetic technique, the same lessons learned in caring for patients with acquired coronary artery disease apply. That is, there is no right drug for these patients, nor is there a single “best” anesthetic technique. Rather, the critical issue is to gain a clear and comprehensive understanding of the patient’s pathophysiology. It is not the drugs used, but rather how they are used. Certain principles for patients with Fontan physiology are important and need to be stressed (Box 20-9).
BOX 20-9 Management Principles for Patients with Fontan Physiology
Ventilatory Management
In an effort to minimize pulmonary vascular resistance, functional residual capacity should be maintained by the application of small amounts of positive end-expiratory pressure or continuous positive airway pressure, and excessive lung volumes should be avoided. Positive end-expiratory pressure or continuous positive airway pressure will not significantly impede cardiac output if less than 6 cm H2O. Spontaneous ventilation has been assumed to be optimal for these patients to minimize intrathoracic pressure and encourage forward flow into the pulmonary circulation; but as discussed by Steven and McGowan,156 hard evidence for this approach is mostly lacking.
Pregnancy
It was inevitable that as some of the female Fontan patients reached childbearing age they would become pregnant. Case reports first began appearing in 1989.157,158 Unfortunately, the body of knowledge on this important subject consists primarily of more case reports with no large registry documenting outcomes. The physiologic changes of pregnancy are well-known and described in standard texts. Can a Fontan patient cope with the increased cardiovascular demands of pregnancy? The dilemma facing physicians caring for these patients is that Fontan patients are known to have decreased cardiac reserve, even those who report good functional status. Because pregnancy is a “stress test,” who will pass this test and who will fail? The literature provides conflicting data. One series of 33 pregnancies found women tolerated pregnancy, labor, and delivery well, but there was an increased risk for spontaneous abortion.50 More recently, a smaller series found live birth pregnancies complicated by high rates of New York Heart Association class deterioration, atrial arrhythmia, prematurity, and intrauterine growth retardation.49 What can be made of these reports? They suffer from the usual problems of retrospective review and self-reporting. However, they do provide clinicians with some reassuring information. First, pregnancy is usually undertaken only in those patients with relatively good functional status, thereby removing the highest-risk patients. Undoubtedly, most adult congenital cardiologists would counsel against pregnancy in any patient with evidence of a failing Fontan circulation. In patients with good functional status, pregnancy can be successfully carried to term, albeit with increased risk for miscarriage and premature delivery. A review of the case reports in the anesthetic literature shows that epidural analgesia is well tolerated and indeed recommended for the first stage of labor. The Caesarian section rate approaches 50%.50 Neuraxial anesthesia for Caesarian section, in addition to its usual benefits, preserves spontaneous ventilation, which is desirable in Fontan patients. However, no increased risk from general anesthesia was identified. Perioperative complications are low, and peripartum cardiac decompensation is rare.
Fontan Conversion Surgery
There is now a large cohort of patients with atriopulmonary connections suffering from some degree of thrombosis, arrhythmia, PLE, or ventricular dysfunction. These patients are candidates for Fontan conversion surgery, which is the most commonly performed high-risk operation in the adult CHD population. Case reports and small case series began to appear in the literature in the mid-1990s. At that time, interest focused on the best indications for this major surgery, outcome predictors, and optimizing the surgical technique. There are now some answers to these important questions. It was believed that conversion of an atriopulmonary Fontan to the improved hemodynamics of the modern Fontan would relieve severe atrial arrhythmias. The profile of the early patient undergoing Fontan conversion surgery was one of refractory atrial arrhythmias and poor functional state.159 Two general trends have been identified. First, in this very high risk group of patients, perioperative mortality was low. Second, arrhythmia control was much better in the group that underwent extracardiac connection with arrhythmia surgery. Conversion to extracardiac Fontan without an ablative procedure resulted in a high rate of arrhythmia recurrence. The largest experience came from Mavroudis,160 whose preferred technique was conversion to an extracardiac Fontan with intraoperative electrophysiologic mapping, arrhythmia ablation, and pacemaker placement. The risk factors for death or transplantation were right or ambiguous ventricular morphology, PLE, atrioventricular valve regurgitation graded moderate or worse, and long CPB time.

Patent Ductus Arteriosus
Beyond the neonatal period, spontaneous closure of a PDA is uncommon. The risk for a longstanding moderate-to-large PDA is volume overloading of the left atrium and left ventricle with the risk for development of pulmonary vascular disease. The progression of pulmonary vascular disease is relatively accelerated when compared with patients with other types of right-to-left shunts. The development of pulmonary vascular disease is dependent on the volume and pressure of the right-to-left shunt. A PDA delivers blood at high shear stress (i.e., arterial pressure) to the pulmonary vasculature, and flow occurs continuously throughout the cardiac cycle. With time, the ductus can become calcified or aneurysmally dilated with a risk for rupture. Ductal calcification or aneurysm increases the risk for surgery, which rarely requires CPB.161 Unrepaired, the natural history is for one third of patients to die of heart failure, pulmonary hypertension, or endocarditis by 40 years of age and two thirds by age 60.162 Although small PDAs are of no hemodynamic consequence, even small PDAs carry relatively high endocarditis risk. Surgical closure should be considered for all adults with PDA, and transvascular closure by means of one of several devices is possible.161 With calcification and friability of the ductus, if device closure is not practicable, it is possible to do a patch closure from inside the aorta or pulmonary artery.

Pulmonary Valve Stenosis
Long-term asymptomatic survival is typical of patients, with the exception of neonates with critical stenosis.163 There is a 94% survival rate 20 years after diagnosis, and adults generally do not require surgical intervention.164 With aging, however, right ventricular fibrosis and failure can develop, and this is the most common cause of death, usually in the fourth decade of life. Almost all patients who have relief of stenosis either surgically or by balloon valvuloplasty have normal right ventricular function after surgery. However, abnormal ventricular function may not resolve after late surgical correction. The development of isolated pulmonary valvular stenosis, even of a severe degree, is usually well tolerated during pregnancy, even in the face of the volume overload that accompanies it.165

Single Ventricle
See the Fontan Physiology section earlier in this chapter for a detailed discussion.

Tetralogy of Fallot
As with many things in medicine, tetralogy of Fallot was first described by someone else—probably in 1673 by Stenson. The classic description of tetralogy of Fallot includes: (1) a large, nonrestrictive malaligned VSD, with (2) an overriding aorta, (3) infundibular pulmonic stenosis, and (4) consequent right ventricular hypertrophy, all derived from an embryonic anterocephalad deviation of the outlet septum. However, there is a spectrum of disease with more severe defects including stenosis of the pulmonary valve, stenosis of the pulmonary valve annulus, or stenosis and hypoplasia of the pulmonary arteries in the most severe cases. Pentalogy of Fallot refers to the addition of an ASD. With advances in genetics, up to one third or more of cases of tetralogy have been ascribed to one of several genetic abnormalities, including trisomy 21, the 22q11 microdeletion, the genes NKX 2.5 and FOG 2.4, and others. Tetralogy of Fallot is the most common cyanotic lesion encountered in the adult population. Unrepaired or nonpalliated, approximately 25% of patients survive to adolescence, after which the mortality rate is 6.6% per year. Only 3% survive to age 40.166 Unlike children, teenagers and adults with tetralogy do not develop “tet spells.” Long-term survival with a good quality of life is expected after repair. The 32- to 36-year survival rate has been reported to be 85% to 86%, although symptoms, primarily arrhythmias and decreased exercise tolerance, occur in 10% to 15% at 20 years after the primary repair167–170 (Box 20-10). In the past, most children with tetralogy were managed with a preliminary palliation with an aortopulmonary shunt such as a Blalock-Taussig, followed by complete correction. Essentially all patients would eventually have come for complete repair. Currently, most children are managed with a complete repair in infancy, without preceding palliation.
It is uncommon to encounter an adolescent or adult with unrepaired tetralogy. However, it can be encountered in immigrants or in patients whose anatomic variation was considered to be inoperable when they were children. In tetralogy, the right ventricle “sees” the obstruction from the pulmonic stenosis. Pulmonary vascular resistance is typically normal to low. Right-to-left shunting is caused by obstruction at the level of the right ventricular outflow tract and is unaffected by attempts at modulating pulmonary vascular resistance. Shunting is minimized, however, by pharmacologically increasing systemic vascular resistance. Increases in the inotropic state of the heart increase the dynamic obstruction at the right ventricular infundibulum and worsen right-to-left shunting. β-Blockers are often used to decrease inotropy. Although halothane was the historic anesthetic of choice in children with tetralogy because of its myocardial depressant effects and ability to maintain systemic vascular resistance, current practice is to use sevoflurane, without undue consequence from a reduction in systemic vascular resistance.171 Anesthetic induction in adults can easily be achieved with any of the available agents, keeping in mind the principles of maintenance of systemic blood pressure, avoidance of hypovolemia, and preventing increases in inotropy.
Patients require closure of the VSD and resolution of the pulmonic stenosis. Although current practice is to repair the VSD through the right atrium in an effort to maintain competence of the pulmonary valve and limit any ventriculotomy, older patients will likely have had repair via a right ventriculotomy. A large right ventriculotomy increases the risks for arrhythmias and sudden death.172 Patients who have had a right ventriculotomy will have an obligate right bundle branch block pattern on the electrocardiogram. However, unlike the more usual bundle branch block in adults, this represents disruption of the His-Purkinje system only in the right ventricular outflow, in the area of the right ventricular incision. Because most His-Purkinje conduction is intact, it does not carry increased risk for the development of complete heart block. These patients can have an abnormal response to exercise.
Some patients require repair of pulmonic stenosis by placement of a transannular patch, with obligate residual pulmonary insufficiency. Isolated mild-to-moderate pulmonary insufficiency is generally well tolerated, but in the long term, it can contribute to right ventricular dysfunction with a risk for ventricular tachycardia and sudden death. Patients requiring pulmonary valve replacement in their late teens or early 20s after a transannular patch in early childhood are a growing proportion of the adult CHD population. Atrial tachyarrhythmias occur in about one third of adults late after repair and can contribute to late morbidity.173,174 The development of atrial flutter or atrial reentrant tachycardia is often a harbinger of hemodynamic compromise. The substrate is usually an atrial surgical scar and the trigger is atrial dilation, such as from tricuspid insufficiency with right ventricular dysfunction. The mechanism for the development of ventricular arrhythmias is presumably the same, namely, dilation superimposed on surgical scar.
In some cases, the right ventricular outflow tract patch needs to be extended onto the branch pulmonary arteries to relieve obstruction. Patients with abnormal coronary arteries may have required repair using a right ventricle-to-pulmonary artery conduit to avoid doing a right ventriculotomy in the area of the coronary artery. Repair at a younger age (< 12 years) results in better postoperative right ventricular function.175 Because there is an unrestrictive VSD, in the unrepaired adult, systemic hypertension developing in adult life imposes an additional load on both ventricles, not just the left. The increase in systemic vascular resistance decreases right-to-left shunting and diminishes cyanosis but at the expense of right ventricular or biventricular failure.
Sudden death or ventricular tachycardia requiring treatment can occur in up to 5.5% of postoperative patients older than 30 years, often years after surgery.168,169,176 The foci for these arrhythmias are typically in the right ventricular outflow tract in the area that has had surgery, and they can be ablated in the catheterization laboratory. Older age at repair, severe left ventricular dysfunction, postoperative right ventricular hypertension from residual or recurrent outflow tract obstruction, wide-open pulmonary insufficiency, and prolongation of the QRS (to > 180 milliseconds) are all predictors of sudden death.172,177 Premature ventricular contractions and even nonsustained ventricular tachycardia are not rare but do not seem to be associated with sudden death, making appropriate treatment options difficult.178 QRS prolongation to longer than 180 milliseconds, although highly sensitive, has a low positive predictive value.179 The impact of this risk factor in the current group of younger patients who have not had ventriculotomies is unclear because their initial postoperative QRS durations are shorter than in patients who had a right ventriculotomy.
Although for many years it was thought that moderate-to-severe pulmonary insufficiency in these patients was well tolerated, it has become apparent from a number of series that right ventricular dysfunction and both atrial and ventricular arrhythmias can be common long-term sequelae. For this reason, patients with symptomatic pulmonary insufficiency from a transannular patch or aneurysm formation at the site of a right ventricular outflow tract patch can require reoperation to replace a widely incompetent pulmonary valve with a bioprosthetic valve with or without a tricuspid annuloplasty.180 Interestingly, the incidence of atrial arrhythmias may not be diminished when adult patients have a pulmonary valve placed, although the incidence of ventricular arrhythmias is decreased. Right ventricular dysfunction improves in a variable number of adults, suggesting that pulmonary valve placement be done sooner rather than later. The development of a pulmonary valve that can be delivered via a vascular catheter holds much promise.181
Most adult patients require reoperation to repair the right ventricular outflow tract or to insert or replace a valve in the pulmonic position. Other reasons for reoperation include repair of an outflow tract aneurysm at the site of a patch, repair of a residual VSD, or repair of an incompetent tricuspid valve.170 These patients often have diminished right ventricular diastolic compliance and require higher than normal central venous pressure. Postoperative management includes minimizing pulmonary vascular resistance and maintaining central venous pressure. Patients often require treatment postbypass with an inotrope and afterload reduction.182
Women with good surgical results without residual defects should tolerate pregnancy and delivery well with outcomes approximating those of the general population.183 Women with uncorrected tetralogy, particularly those with significant cyanosis, have a high incidence of fetal loss (80% with hematocrit > 65%). The decline in systemic vascular resistance that accompanies pregnancy and delivery can worsen cyanosis, and the physiologic volume loading of pregnancy can exaggerate failure of both ventricles.

Transposition of the Great Arteries (d-Transposition)
Atrial repairs function by redirecting systemic venous blood to the left ventricle (and thence to the transposed pulmonary artery) and pulmonary venous blood to the right ventricle (and thence to the aorta). The Mustard operation uses an intra-atrial conduit of native pericardium (Figure 20-10), whereas the Senning operation uses native atrial tissue to fashion the conduit. The arterial switch operation transposes transected aorta and pulmonary artery such that they now arise above the appropriate ventricle. This operation also requires transposing the coronary arteries from the aorta to the pulmonary root, which, after the procedure, becomes the aortic root. The Rastelli procedure closes the VSD on a bias such that the left ventricle empties into the aorta and connects the right ventricle to the pulmonary artery by means of a valved conduit.
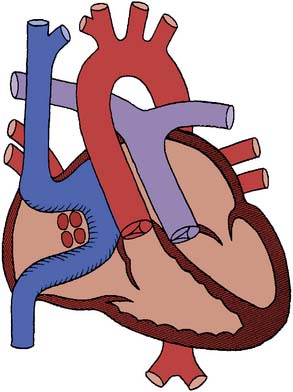
Figure 20-10 The Mustard operation.
(From Mullins C, Mayer D: Congenital heart disease. A diagrammatic atlas. New York: Wiley-Liss, 1988, by permission of Wiley-Liss, Inc., a subsidiary of John Wiley & Sons, Inc.)
Atrial repairs result in a systemic right ventricle, and these patients consistently have abnormal right ventricular function that can be progressive with a right ventricular ejection fraction of about 40%.184 Mild tricuspid insufficiency is common, but severe tricuspid insufficiency suggests the development of severe right ventricular dysfunction. There is an 85% to 90% 10-year survival with these operations, but by 20 years, survival is less than 80%.185–187 Over 25 years, about half experience development of moderate right ventricular dysfunction and one third experience development of severe tricuspid insufficiency.185,186,188–190 Although it always remains abnormal, it has been suggested that earlier surgery minimizes right ventricular dysfunction.191 Because of the incidence of right ventricular dysfunction, some patients with atrial repairs have been converted to an arterial switch, after preparation of the left ventricle by a pulmonary artery band to prepare it to tolerate systemic arterial pressure.192
Atrial repairs bring an incidence of late electrophysiologic sequelae including sinus node dysfunction (bradycardia), junctional escape rhythms, atrioventricular block, and supraventricular arrhythmias. Atrial flutter occurs in 20% of patients by age 20, with half having progressive sinus node dysfunction by that time.185,189 On occasion, these tachyarrhythmias can result in sudden death, presumably from 1:1 conduction producing ventricular fibrillation.185,193 The loss of sinus rhythm in the face of right ventricular (the systemic ventricle) dysfunction can also contribute to late sudden death. The risk for late death after an atrial repair is almost three times greater if there is an associated VSD. The incidence of tachyarrhythmias does decrease, however, after the tenth postoperative year.
An arterial switch operation can be done after a failed atrial repair in adults, but the outcome is generally poor. It is suggested that younger patients do better.194 Survival after an arterial switch operation, even early in the experience with this operation, is approximately 90% at 10 years.195 Very-long-term outcome after the arterial switch procedure is still not known. It does appear that there is essentially no mortality after 5 years after surgery, and late surgical reintervention is mostly because of supravalvular pulmonic stenosis.196 Although many of these children have abnormal resting myocardial perfusion, up to 9% can have evidence of exercise-induced myocardial ischemia.197 The implication for the development of premature coronary artery disease in adulthood is not known, and there is also some concern about the ultimate function of the neoaortic valve. Patients who have had a Rastelli repair will require episodic reoperation for replacement of the prosthetic conduit valve.

Truncus Arteriosus
Because of the very high risk for congestive heart failure followed by pulmonary vascular disease in childhood from high pulmonary blood flow from the aorta, essentially all patients who survive to adolescence have had surgical repair or will have inoperable pulmonary vascular disease. The rare exception is the patient with stenosis near the origin of the pulmonary arteries from the aorta. Patients with valved conduits placed in infancy and early childhood have requisite reoperations to replace the conduit with patient growth, even in the face of adequate valve function. Conduits placed in later childhood can suffice until adulthood. There can be ongoing problems with incompetence and stenosis of the truncal valve (postoperatively functioning as the aortic valve), and eventual dysfunction from stenosis and/or incompetence of the homograft conduit is routinely encountered, requiring replacement.198,199 Because these patients require multiple sternotomies and the valved conduit tends to lie immediately behind and in close proximity to the sternum, sternotomy carries with it significant potential risk for laceration of the conduit. On occasion, the femoral vessels are cannulated for bypass before sternotomy.

Ventricular Septal Defects
The natural history of VSDs has been reviewed in detail.163 More than 75% of small and moderate VSDs close spontaneously during childhood by a gradual ingrowth of surrounding septum. Of those that close spontaneously, almost all have closed by 10 years of age. Other mechanisms for natural closure include closure by tricuspid valve tissue, closure by prolapsed aortic leaflet, and closure by endocarditis. Some VSDs result in the development of aortic insufficiency in adults from prolapse of the aortic valve into the defect.200 Although the risk for endocarditis is ongoing, there is no hemodynamic risk for a small VSD in the adult. If pulmonary vascular disease is present, it can progress if closure of a large VSD is delayed.
Although some studies have reported possible ventricular dysfunction years after surgical repair, these are older reports and patients were operated on later than by current standards.201–203 It does appear, though, that the ventricle successfully remodels from chronic volume overload if surgical correction is done by 5 years of age and perhaps up to 10 to 12 years of age. Iatrogenic heart block is a possible surgical complication, but was much more common in the earlier days of cardiac surgery. Percutaneous closure devices for use with certain VSDs are available, but not currently for widespread commercial use.
Echo cases
 Case Study 1
Case Study 1Atrial Septal Defect
Framing
The different types of ASDs arise from problems that occur in the various embryologic structures that combine to form the atrial septum (Figure 20-11). The echocardiographer is required to know the different types of ASD and any associated cardiac defects that accompany each lesion. Secundum ASDs are by far the most common type and are rarely associated with other cardiac defects. Those lesions requiring intervention are usually treated with percutaneous device closure providing there is an adequate rim of surrounding atrial septum for the device to “grab on to.” The echocardiographer is vital in helping the interventional cardiologist to “see” the atrium in three dimensions and guide placement of the device.
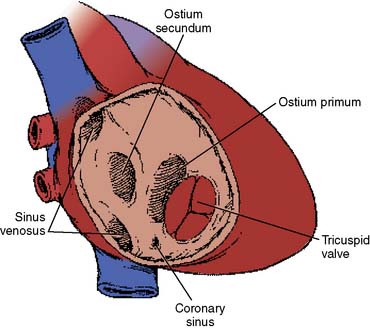
Data Collection and Interpretation
The intraoperative TEE examination is focused on confirming the presumptive diagnosis of ASD and determining the presence of any other associated defects. The Society of Cardiovascular Anesthesiologists has published guidelines with 20 views that constitute a comprehensive intraoperative TEE examination.204 When a more detailed examination of a specific cardiac structure is sought, we recommend focusing on the structure by using the zoom feature. The specific structure can then be carefully assessed by using the multiplane function to advance 15 to 30 degrees at a time until the structure has been visualized in multiple views from 0 to 180 degrees. In this example, the echocardiographer would focus on the atrial septum in the midesophageal four-chamber view at 0 degree and then slowly multiplane forward to 180 degrees, providing a comprehensive examination of the structure. The pulmonary veins can be difficult to identify, even for experienced echocardiographers. In the midesophageal four-chamber view with the multiplane angle at 0 degree, the ultrasound image is focused on the left atrium. The probe is then gently turned to the patient’s left to identify the left-sided pulmonary veins and then to the right to identify the right-sided pulmonary veins. Visualizing two separate pulmonary veins from each side is challenging because they arrive at a confluence as they enter the left atrium. The use of color Doppler is often helpful to identify blood flowing within the pulmonary veins.
Decision Making and Interpretation
A patient was scheduled for surgical resection of an ASD. Preoperative transthoracic echocardiography confirmed a large secundum ASD with inferior extension toward the IVC (Figure 20-12). The size and inferior extension of the ASD precluded percutaneous device closure. The TEE examination confirmed the preoperative findings and demonstrated four pulmonary veins returning to the left atrium. The volume of left-to-right shunting was large (Figure 20-13). Right ventricular volume overload was present with significant right ventricular dilation (Figure 20-14). After closure of the ASD and separation from CPB, a residual ASD was seen at the inferior aspect of the ASD patch (Figure 20-15). Comparing Figures 20-13 and 20-15 demonstrates the echocardiographic difference in appearance between a high-volume, low-velocity shunt and a more restrictive, higher velocity shunt. The configuration of the TEE machine in the color Doppler mode identifies blood flow toward the transducer in red and blood flow away from the transducer in blue. There is also a threshold of velocity set, such that when blood flow exceeds the velocity threshold, speckling occurs. In Figure 20-13, there is a uniform blue color demonstrating a significant volume shunt. The absence of color speckling confirms low-velocity blood flow. Figure 20-15, taken after attempted repair of the ASD, shows a smaller blue area with orange speckling. The speckling confirms higher velocity blood flow, which is to be expected because the residual defect is much smaller than the unrepaired ASD. Velocity increases as blood flows through a narrower orifice.
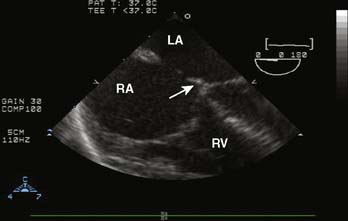
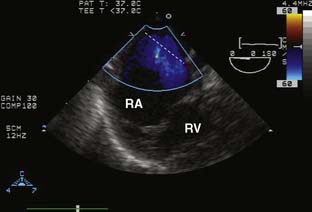
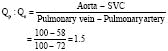
After the second attempt at repair, the patient separated easily from CPB. The ASD patch appeared intact on TEE examination, but there was an unusual color-flow Doppler jet seen originating from the junction of the IVC and right atrium. What are the possible explanations? Could this simply be turbulence in the area of the ASD repair? The echocardiographer must now rely on knowledge of the lesion and the potential surgical complications associated with its repair. Figure 20-11 clearly demonstrates the close proximity of an inferior sinus venosus ASD to the IVC. A known, albeit rare, complication of inferior sinus venosus ASD repair is to suture the ASD patch from the IVC to the ASD, thus creating a path from the IVC to left atrium. This suspicion was bolstered by the fact the peripheral pulse oximeter reading varied between 85% and 88%. This was confirmed with an arterial blood gas. The presence of a right-to-left shunt demanded correction. The patient returned to CPB for a third time for takedown of the patch and closure of the ASD. Based on the two previous attempted repairs, the surgeon realized the secundum ASD might extend even farther into the sinus venosus region of the septum than previously imagined. The IVC venous cannula was repositioned more inferiorly, allowing greater exposure of the defect. After repair, the patient once again separated easily from CPB. The TEE examination confirmed the success of the repair with no residual shunting. This case well demonstrates the need for the intraoperative echocardiographer to be more than a “technician” and integrate supporting physiologic information with the TEE images.
 Case Study 2
Case Study 2Anomalous Left Main Coronary Artery
Framing
Anomalous coronary arterial lesions comprise a spectrum of defects. In one variant, the left main coronary artery (LMCA) originates from the pulmonary artery. The clinical presentation is heart failure secondary to left ventricular ischemia and generally occurs in the first few months of life. A more insidious but also potentially lethal form of anomalous coronary arterial lesion is both coronary arteries arising from the aorta but from abnormal locations. Most well described is an anomalous LMCA that originates from the right coronary sinus. The LMCA may originate from a separate ostium or may share the same ostium as the right coronary artery (RCA) (Figure 20-16). As Figure 20-16 demonstrates, the path of the LMCA is abnormal. First, the LMCA travels within the wall of the aorta for a short distance before exiting onto the epicardial surface of the heart. This is known as an intramural coronary artery. The path of LMCA then follows a path between the aorta and pulmonary artery. The abnormal path of the anomalous LMCA explains why the clinical presentation is sudden death, most often during exercise. The increased blood flow caused by exercise results in dilation of both the aorta and pulmonary artery. This dilation can compress the LMCA between the aorta and pulmonary artery. Alternatively, if the LMCA is intramural, it may be compressed within the wall of the aorta. Either case leads to ischemia in the entire territory of the LMCA with sudden cardiac death. Tragically, an anomalous LMCA is often a postmortem finding after an unexplained sudden death in an otherwise healthy young person. For reasons that are unclear, patients rarely, if ever, develop exertional chest pain. Also, the amount of exertion that precipitates a cardiac event is not predictable. That is, the patient may have vigorously exercised in the past without a problem but suffers a cardiac event during more modest exertion. For these reasons, if a diagnosis of anomalous LMCA is made, surgery is indicated for the prevention of sudden cardiac death.
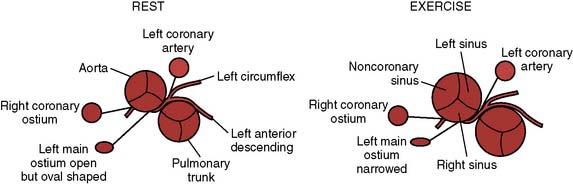
Decision Making and Interpretation
During investigations for nonspecific chest pains, an otherwise healthy patient was discovered to have an anomalous LMCA and was scheduled for coronary reimplantation. After induction of anesthesia, the TEE probe was inserted that demonstrated normal left ventricular function and the LMCA originating from the right coronary cusp, sharing a common ostium with the RCA (Figure 20-17). After the aortic cross-clamping was applied and cardioplegia delivered, the heart was slow to arrest. The cardioplegia circuit was checked and found to be working properly. However, throughout the period of aortic cross-clamping, the heart frequently recovered electrical and mechanical activity before the next scheduled dose of cardioplegia. At the conclusion of the coronary reimplantation, the surgeon believed the myocardial preservation had been poor and the patient would likely need inotropic support. On attempted separation from CPB, the patient was hypotensive with poor myocardial function. What is the differential diagnosis of ventricular dysfunction after CPB? The electrocardiogram showed sinus tachycardia with nonspecific ST- changes in lead II and V5. This was confirmed in other electrocardiogram leads. The question to be answered was whether ventricular dysfunction was caused by possible poor myocardial preservation or whether the LMCA reimplantation was unsuccessful. By visual inspection, the right ventricle was contracting well. There was no ST- elevation in lead II to suggest air embolus to the RCA. What is the preferred TEE view to assess ventricular function? The transgastric short-axis view at the midpapillary level is used routinely to assess preload, contractility, and regional wall motion abnormalities. Knowing the coronary artery that corresponds to the various left ventricular segments allows the echocardiographer to identify regional wall motions abnormalities caused by ischemia (Figure 20-18). The transgastric image showed marked left ventricular dilation with akinesis of the septal, anterior, lateral, and posterior walls. There was minimal decrease in left ventricular cavity size during systole (see Video 20-1, available online). The reimplanted LMCA could not be seen originating from the left coronary sinus. Global ventricular dysfunction secondary to poor myocardial preservation was unlikely because right ventricular function was good. The inferior region of left ventricle, which is supplied by the RCA, had normal function. The findings of severe regional wall motion abnormalities in the distribution of the LMCA confirmed the suspicion of unsuccessful LMCA reimplantation. The extensive wall motion abnormalities suggested the patient had a left dominant coronary circulation. By inspection, the reimplanted LMCA appeared to be free of tension or kinking. After consultation with the surgeon and a description of the TEE findings, it was decided that a second attempt at LMCA reimplantation was unlikely to be successful.
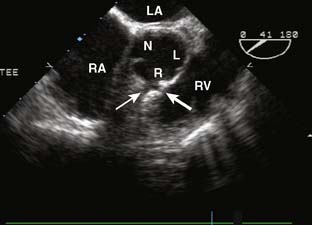
1 Perloff J.K. Pediatric congenital cardiac patient becomes a postoperative adult. The changing population of congenital heart disease. Circulation. 1973;47:606-619.
2 Perloff J.K., Warnes C.A. Challenges posed by adults with repaired congenital heart disease. Circulation. 2001;103:2637-2643.
3 Williams R.G., Pearson G.D., Barst R.J., et al. Report of the National Heart, Lung, and Blood Institute Working Group on research in adult congenital heart disease. J Am Coll Cardiol. 2006;47:701-707.
4 Warnes C.A., Liberthson R., Danielson G.K., et al. Task force 1: The changing profile of congenital heart disease in adult life. J Am Coll Cardiol. 2001;37:1170-1175.
5 Marelli A.J., Mackie A.S., Ionescu-Ittu R., et al. Congenital heart disease in the general population: Changing prevalence and age distribution. Circulation. 2007;115:163-172.
6 Pillutla P., Shetty K.D., Foster E. Mortality associated with adult congenital heart disease: Trends in the US population from 1979 to 2005. Am Heart J. 2009;158:874-879.
7 Andropoulos D.B., Stayer S.A., Skjonsby B.S., et al. Anesthetic and perioperative outcome of teenagers and adults with congenital heart disease. J Cardiothorac Vasc Anesth. 2002;16:731-736.
8 Dore A., Glancy D.L., Stone S., et al. Cardiac surgery for grown-up congenital heart patients: Survey of 307 consecutive operations from 1991 to 1994. Am J Cardiol. 1997;80:906-913.
9 Warnes C.A. The adult with congenital heart disease: Born to be bad? J Am Coll Cardiol. 2005;46:1-8.
10 Mackie A.S., Ionescu-Ittu R., Therrien J., et al. Children and adults with congenital heart disease lost to follow-up: Who and when? Circulation. 2009;120:302-309.
11 Webb G.D., Williams R.G. Care of the adult with congenital heart disease: Introduction. J Am Coll Cardiol. 2001;37:1166.
12 Warnes C.A., Williams R.G., Bashore T.M., et al. ACC/AHA 2008 guidelines for the management of adults with congenital heart disease: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Develop Guidelines on the Management of Adults with Congenital Heart Disease). Developed in Collaboration with the American Society of Echocardiography, Heart Rhythm Society, International Society for Adult Congenital Heart Disease, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J Am Coll Cardiol. 2008;52:e143-e263.
13 Landzberg M.J., Murphy D.J.Jr, Davidson W.R.Jr, et al. Task force 4: Organization of delivery systems for adults with congenital heart disease. J Am Coll Cardiol. 2001;37:1187-1193.
14 Bancalari E., Jesse M.J., Gelband H., et al. Lung mechanics in congenital heart disease with increased and decreased pulmonary blood flow. J Pediatr. 1977;90:192-195.
15 Sietsema K.E., Perloff J.K. Cyanotic congenital heart disease: Dynamics of oxygen uptake and control of ventilation during exercise. In: Perloff J.K., Child J.S., editors. Congenital heart disease in adults. Philadelphia: WB Saunders; 1991:104-110.
16 Blesa M.I., Lahiri S., Rashkind W.J., et al. Normalization of the blunted ventilatory response to acute hypoxia in congenital cyanotic heart disease. N Engl J Med. 1977;296:237-241.
17 Edelmann N.H., Lahiri S., Braudo L., et al. The ventilatory response to hypoxia in cyanotic congenital heart disease. N Engl J Med. 1970;282:405-411.
18 Sorensen S.C., Severinghaus J.W. Respiratory insensitivity to acute hypoxia persisting after correction of tetralogy of Fallot. J Appl Physiol. 1968;25:221-223.
19 Burrows F.A. Physiologic dead space, venous admixture, and the arterial to end-tidal carbon dioxide difference in infants and children undergoing cardiac surgery. Anesthesiology. 1989;70:219-225.
20 Perloff J.K., Rosove M.H., Child J.S., et al. Adults with cyanotic congenital heart disease: Hematologic management. Ann Intern Med. 1988;109:406-413.
21 Kawakami N., Mimatsu K., Deguchi M., et al. Scoliosis and congenital heart disease. Spine. 1995;20:1252-1255.
22 Roclawski M., Sabiniewicz R., Potaz P., et al. Scoliosis in patients with aortic coarctation and patent ductus arteriosus: Does standard posterolateral thoracotomy play a role in the development of the lateral curve of the spine? Pediatr Cardiol. 2009;30:941-945.
23 Berman W.J., Wood S.C., Yabek S.M., et al. Systemic oxygen transport in patients with congenital heart disease. Circulation. 1987;75:360-368.
24 Linderkamp O., Klose H.J., Betke K., et al. Increased blood viscosity in patients with cyanotic congenital heart disease and iron deficiency. J Pediatr. 1979;95:567-569.
25 Broberg C.S., Bax B.E., Okonko D.O., et al. Blood viscosity and its relationship to iron deficiency, symptoms, and exercise capacity in adults with cyanotic congenital heart disease. J Am Coll Cardiol. 2006;48:356-365.
26 Gidding S.S., Stockman J.A.3rd. Effect of iron deficiency on tissue oxygen delivery in cyanotic congenital heart disease. Am J Cardiol. 1988;61:605-607.
27 Spence M.S., Balaratnam M.S., Gatzoulis M.A. Clinical update: Cyanotic adult congenital heart disease. Lancet. 2007;370:1530-1532.
28 Maurer H.M., McCue C.M., Robertson L.W., et al. Correction of platelet dysfunction and bleeding in cyanotic congenital heart disease by simple red cell volume reduction. Am J Cardiol. 1975;35:831-835.
29 Wedemeyer A.L., Lewis J.H. Improvement in hemostasis following phlebotomy in cyanotic patients with heart disease. J Pediatr. 1973;83:46-50.
30 Tempe D.K., Virmani S. Coagulation abnormalities in patients with cyanotic congenital heart disease. J Cardiothorac Vasc Anesth. 2002;16:752-765.
31 Weinstein M., Ware J.A., Troll J., et al. Changes in von Willebrand factor during cardiac surgery: Effect of desmopressin acetate. Blood. 1988;71:1648-1655.
32 Rosove M.H., Hocking W.G., Harwig S.S., et al. Studies of beta-thromboglobulin, platelet factor 4, and fibrinopeptide A in erythrocytosis due to cyanotic congenital heart disease. Thromb Res. 1983;29:225-235.
33 Waldman J.D., Czapek E.E., Paul M.H., et al. Shortened platelet survival in cyanotic heart disease. J Pediat. 1975;87:77-79.
34 Ware J.A., Reaves W.H., Horak J.K., et al. Defective platelet aggregation in patients undergoing surgical repair of cyanotic congenital heart disease. Ann Thorac Surg. 1983;36:289-294.
35 Chauhan S., Kumar B.A., Rao B.H., et al. Efficacy of aprotinin, epsilon aminocaproic acid, or combination in cyanotic heart disease. Ann Thorac Surg. 2000;70:1308-1312.
36 Levin E., Wu J., Devine D.V., et al. Hemostatic parameters and platelet activation marker expression in cyanotic and acyanotic pediatric patients undergoing cardiac surgery in the presence of tranexamic acid. Thromb Haemost. 2000;83:54-59.
37 Dimopoulos K., Diller G.P., Koltsida E., et al. Prevalence, predictors, and prognostic value of renal dysfunction in adults with congenital heart disease. Circulation. 2008;117:2320-2328.
38 Spear G.S. The glomerular lesion of cyanotic congenital heart disease. Johns Hopkins Med J. 1977;140:185-188.
39 Ross E.A., Perloff J.K., Danovitch G.M., et al. Renal function and urate metabolism in late survivors with cyanotic congenital heart disease. Circulation. 1986;73:396-400.
40 Young D. Hyperuricemia in cyanotic congenital heart disease. Am J Dis Child. 1980;134:902-903.
41 Dittrich S., Kurschat K., Dahnert I., et al. Renal function after cardiopulmonary bypass surgery in cyanotic congenital heart disease. Int J Cardiol. 2000;73:173-179.
42 Perloff J.K., Marelli A.J., Miner P.D. Risk of stroke in adults with cyanotic congenital heart disease. Circulation. 1993;87:1954-1959.
43 Ammash N., Warnes C.A. Cerebrovascular events in adult patients with cyanotic congenital heart disease. J Am Coll Cardiol. 1996;28:768-772.
44 Truong T., Slavin L., Kashani R., et al. Prevalence of migraine headaches in patients with congenital heart disease. Am J Cardiol. 2008;101:396-400.
45 Colman J.M., Sermer M., Seaward G., et al. Congenital heart disease: Pathophysiology, clinical recognition, diagnosis, and management. In: Wilansky S., Willerson J.T., editors. Heart disease in women. New York: Churchill Livingstone; 2002:443-455.
46 Gatzoulis M., Webb G.D., Daubeney P.E.F. Diagnosis and management of adult congenital heart disease. Philadelphia: Churchill Livingstone, 2003.
47 Warnes C.A., Elkayam U. Congenital heart disease and pregnancy. In: Elkayam U., Gleicher N., editors. Cardiac problems in pregnancy. ed 3. New York: Wiley-Liss; 1998:39-53.
48 Chandrasekhar S., Cook C.R., Collard C.D. Cardiac surgery in the parturient. Anesth Analg. 2009;108:777-785.
49 Drenthen W., Pieper P.G., Roos-Hesselink J.W., et al. Pregnancy and delivery in women after Fontan palliation. Heart. 2006;92:1290-1294.
50 Canobbio M.M., Mair D.D., van der Velde M., et al. Pregnancy outcomes after the Fontan repair. J Am Coll Cardiol. 1996;28:763-767.
51 Shime J., Mocarski E.J., Hastings D., et al. Congenital heart disease in pregnancy: Short- and long-term implications. Am J Obstet Gynecol. 1987;156:313-322.
52 Presbitero P., Somerville J., Stone S., et al. Pregnancy in cyanotic congenital heart disease. Outcome of mother and fetus. Circulation. 1994;89:2673-2676.
53 Wilson W., Taubert K.A., Gewitz M., et al. Prevention of infective endocarditis: Guidelines from the American Heart Association: A guideline from the American Heart Association Rheumatic Fever, Endocarditis, and Kawasaki Disease Committee, Council on Cardiovascular Disease in the Young, and the Council on Clinical Cardiology, Council on Cardiovascular Surgery and Anesthesia, and the Quality of Care and Outcomes Research Interdisciplinary Working Group. Circulation. 2007;116:1736-1754.
54 Qasqas S.A., McPherson C., Frishman W.H., et al. Cardiovascular pharmacotherapeutic considerations during pregnancy and lactation. Cardiol Rev. 2004;12:240-261.
55 Natale A., Davidson T., Geiger M.J., et al. Implantable cardioverter-defibrillators and pregnancy: A safe combination? Circulation. 1997;96:2808-2812.
56 Foster E., Graham T.P.Jr, Driscoll D.J., et al. Task force 2: Special health care needs of adults with congenital heart disease. J Am Coll Cardiol. 2001;37:1176-1183.
57 Bromberg J.I., Beasley P.J., D’Angelo E.J., et al. Depression and anxiety in adults with congenital heart disease: A pilot study. Heart Lung. 2003;32:105-110.
58 Cox D., Lewis G., Stuart G., et al. A cross-sectional study of the prevalence of psychopathology in adults with congenital heart disease. J Psychosomat Res. 2002;52:65-68.
59 Kamphuis M., Ottenkamp J., Vliegen H.W., et al. Health related quality of life and health status in adult survivors with previously operated complex congenital heart disease. Heart. 2002;87:356-362.
60 Kamphuis M., Vogels T., Ottenkamp J., et al. Employment in adults with congenital heart disease. Arch Pediatr Adolesc Med. 2002;156:1143-1148.
61 Lane D.A., Lip G.Y., Millane T.A. Quality of life in adults with congenital heart disease. Heart. 2002;88:71-75.
62 Lip G.Y., Lane D.A., Millane T.A., et al. Psychological interventions for depression in adolescent and adult congenital heart disease. Cochrane Database Syst Rev. 2003. CD004394
63 van Rijen E.H., Utens E.M., Roos-Hesselink J.W., et al. Psychosocial functioning of the adult with congenital heart disease: A 20-33 years follow-up. Eur Heart J. 2003;24:673-683.
64 Kovacs A.H., Saidi A.S., Kuhl E.A., et al. Depression and anxiety in adult congenital heart disease: Predictors and prevalence. Int J Cardiol. 2009;137:158-164.
65 Moons P., Siebens K., De Geest S., et al. A pilot study of expenditures on, and utilization of resources in, health care in adults with congenital heart disease. Cardiol Young. 2001;11:301-313.
66 Skorton D.J., Garson A.Jr, Allen H.D., et al. Task force 5: Adults with congenital heart disease: Access to care. J Am Coll Cardiol. 2001;37:1193-1198.
67 Vonder Muhll I., Cumming G., Gatzoulis M.A. Risky business: Insuring adults with congenital heart disease. Eur Heart J. 2003;24:1595-1600.
68 Truesdell S.C., Clark E.B. Health insurance status in a cohort of children and young adults with congenital cardiac diagnoses. Circulation. 1991;84(Suppl 2):II-386.
69 Graham T.P.Jr, Cordell G.D., Bender H.W. Ventricular function following surgery. In: Kidd B.S., Rowe R.D., editors. The child with congenital heart disease after surgery. Mt. Kisco, NY: Futura Publishing Co, 1995.
70 Bouchardy J., Therrien J., Pilote L., et al. Atrial arrhythmias in adults with congenital heart disease. Circulation. 2009;120:1679-1686.
71 Feltes T.F., Friedman R.A. Transesophageal echocardiographic detection of atrial thrombi in patients with nonfibrillation atrial tachyarrhythmias and congenital heart disease. J Am Coll Cardiol. 1994;24:1365-1370.
72 Andropoulos D.B., Stayer S.A., Russell I.A. Anesthesia for congenital heart disease. Armonk, NY: Futura, 2004.
73 Lake C.L., Booker P.D. Pediatric cardiac anesthesia. Philadelphia: Lippincott-Williams & Wilkins, 2004.
74 Carabello B.A., Carawford F.A.J. Valvular heart disease. N Engl J Med. 1997;337:32-41.
75 Craig R.J., Selzer A. Natural history and prognosis of atrial septal defect. Circulation. 1968;37:805-815.
76 Markman P., Howitt G., Wade E.G. Atrial septal defect in the middle-aged and elderly. Q J Med. 1965;34:409-426.
77 Mattila S., Merikallio E., Tala P. ASD in patients over 40 years of age. Scand J Thorac Cardiovasc Surg. 1979;13:21-24.
78 Campbell M. Natural history of coarctation of the aorta. Br Heart J. 1970;32:633-640.
79 Boucher C.A., Liberthson R.R., Buckley M.J. Secundum atrial septal defect and significant mitral regurgitation: Incidence, management and morphologic basis. Chest. 1979;75:697-702.
80 Liberthson R.R., Boucher C.A., Strauss H.W., et al. Right ventricular function in adult atrial septal defect. Preoperative and postoperative assessment and clinical implications. Am J Cardiol. 1981;47:56-60.
81 Davies H., Oliver G.C., Rappoport W.J., et al. Abnormal left heart function after operation for atrial septal defect. Br Heart J. 1970;32:747-753.
82 Gatzoulis M.A., Freeman M.A., Siu S.C., et al. Atrial arrhythmia after surgical closure of atrial septal defects in adults. N Engl J Med. 1999;340:839-846.
83 Konstantinides S., Geibel A., Olschewski M., et al. A comparison of surgical and medical therapy for atrial septal defect in adults. N Engl J Med. 1995;333:469-473.
84 Murphy J.G., Gersh B.J., McGoon M.D., et al. Long-term outcome after surgical repair of isolated atrial septal defect. Follow-up at 27 to 32 years. N Engl J Med. 1990;323:1645-1650.
85 Attie F., Rosas M., Granados N., et al. Surgical treatment for secundum atrial septal defects in patients > 40 years old. A randomized clinical trial. J Am Coll Cardiol. 2001;38:2035-2042.
86 Du Z.D., Hijazi Z.M., Kleinman C.S., et al. Comparison between transcatheter and surgical closure of secundum atrial septal defect in children and adults: Results of a multicenter nonrandomized trial. J Am Coll Cardiol. 2002;39:1836-1844.
87 Dajani A.S., Taubert K.A., Wilson W., et al. Prevention of bacterial endocarditis. Recommendations by the American Heart Association. JAMA. 1997;277:1794-1801.
88 Abbott M.E. Coarctation of the aorta of adult type: II. A statistical study and historical retrospect of 200 recorded cases with autopsy, of stenosis or obliteration of the descending arch in subjects above the age of two years. Am Heart J. 1928;3:392-421.
89 Mitchell S.C., Korones S.B., Berendes H.W. Congenital heart disease in 56,109 births. Incidence and natural history. Circulation. 1971;43:323-332.
90 Reifenstein G.H., Levine S.A., Gross R.E. Coarctation of the aorta: A review of 104 autopsied cases of the “adult type,” 2 years of age or older. Am Heart J. 1947;33:146-168.
91 Maron B.J., Humphries J.O., Rowe R.D., et al. Prognosis of surgically corrected coarctation of the aorta. A 20-year postoperative appraisal. Circulation. 1973;47:119-126.
92 Clarkson P.M., Nicholson M.R., Barratt-Boyes B.G., et al. Results after repair of coarctation of the aorta beyond infancy: A 10 to 28 year follow-up with particular reference to late systemic hypertension. Am J Cardiol. 1983;51:1481-1488.
93 Therrien J., Gatzoulis M., Graham T., et al. Canadian Cardiovascular Society Consensus Conference 2001 update: Recommendations for the Management of Adults with Congenital Heart Disease—Part II. Can J Cardiol. 2001;17:1029-1050.
94 Hamdan M.A., Maheshwari S., Fahey J.T., et al. Endovascular stents for coarctation of the aorta: Initial results and intermediate-term follow-up. J Am Coll Cardiol. 2001;38:1518-1523.
95 Golden A.B., Hellenbrand W.E. Coarctation of the aorta: Stenting in children and adults. Catheter Cardiovasc Interv. 2007;69:289-299.
96 Kutty S., Greenberg R.K., Fletcher S., et al. Endovascular stent grafts for large thoracic aneurysms after coarctation repair. Ann Thorac Surg. 2008;85:1332-1338.
97 Beauchesne L.M., Warnes C.A., Connolly H.M., et al. Outcome of the unoperated adult who presents with congenitally corrected transposition of the great arteries. J Am Coll Cardiol. 2002;40:285-290.
98 Graham T.P.Jr, Parrish M.D., Boucek R.J.J., et al. Assessment of ventricular size and function in congenitally corrected transposition of the great arteries. Am J Cardiol. 1983;51:244-251.
99 Graham T.P.Jr, Bernard Y.D., Mellen B.G., et al. Long-term outcome in congenitally corrected transposition of the great arteries: A multi-institutional study. J Am Coll Cardiol. 2000;36:255-261.
100 Connelly M.S., Robertson P., Liu P., et al. Congenitally corrected transposition of the great arteries in adults: Natural history. Circulation. 1994;90:I-51.
101 Connolly H.M., Grogan M., Warnes C.A. Pregnancy among women with congenitally corrected transposition of great arteries. J Am Coll Cardiol. 1999;33:1692-1695.
102 Arendt K.W., Connolly H.M., Warnes C.A., et al. Anesthetic management of parturients with congenitally corrected transposition of the great arteries: Three cases and a review of the literature. Anesth Analg. 2008;107:1973-1977.
103 Cordone M., Wolfson A., Wolfson N., et al. Anesthetic management of labor in a patient with congenitally corrected transposition of the great arteries. Int J Obstet Anesth. 2008;17:57-60.
104 Therrien J., Barnes I., Somerville J. Outcome of pregnancy in patients with congenitally corrected transposition of the great arteries. Am J Cardiol. 1999;84:820-824.
105 Celermajer D.S., Cullen S., Sullivan I.D., et al. Outcome in neonates with Ebstein’s anomaly. J Am Coll Cardiol. 1992;19:1041-1046.
106 Kumar A.E., Fyler D.C., Miettinen O.S., et al. Ebstein’s anomaly. Clinical profile and natural history. Am J Cardiol. 1971;28:84-95.
107 Spitaels S.E. Ebstein’s anomaly of the tricuspid valve complexities and strategies. Cardiol Clin. 2002;20:431-439. vii
108 Stulak J.M., Dearani J.A., Danielson G.K. Surgical management of Ebstein’s anomaly. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu. 2007:105-111.
109 Connolly H.M., Warnes C.A. Ebstein’s anomaly: Outcome of pregnancy. J Am Coll Cardiol. 1994;23:1194-1198.
110 Eisenmenger V. Die angeborenen Defects des Kammerscheidewand des Herzen. Z Klin Med. 1897;32(Suppl):1-28.
111 Diller G.P., Gatzoulis M.A. Pulmonary vascular disease in adults with congenital heart disease. Circulation. 2007;115:1039-1050.
112 Silversides C.K., Granton J.T., Konen E., et al. Pulmonary thrombosis in adults with Eisenmenger syndrome. J Am Coll Cardiol. 2003;42:1982-1987.
113 Vongpatanasin W., Brickner M.E., Hillis L.D., et al. The Eisenmenger syndrome in adults. Ann Intern Med. 1998;128:745-755.
114 Diller G.P., Dimopoulos K., Broberg C.S., et al. Presentation, survival prospects, and predictors of death in Eisenmenger syndrome: A combined retrospective and case-control study. Eur Heart J. 2006;27:1737-1742.
115 Cantor W.J., Harrison D.A., Moussadji J.S., et al. Determinants of survival and length of survival in adults with Eisenmenger syndrome. Am J Cardiol. 1999;84:677-681.
116 Saha A., Balakrishnan K.G., Jaiswal P.K., et al. Prognosis for patients with Eisenmenger syndrome of various aetiology. Int J Cardiol. 1994;45:199-207.
117 Oya H., Nagaya N., Uematsu M., et al. Poor prognosis and related factors in adults with Eisenmenger syndrome. Am Heart J. 2002;143:739-744.
118 Daliento L., Somerville J., Presbitero P., et al. Eisenmenger syndrome. Factors relating to deterioration and death. Eur Heart J. 1998;19:1845-1855.
119 Bando K., Armitage J.M., Paradis I.L., et al. Indications for and results of single, bilateral, and heart-lung transplantation for pulmonary hypertension. J Thorac Cardiovasc Surg. 1994;108:1056-1065.
120 Holzman R.S., Nargozian C.D., Marnach R., et al. Epidural anesthesia in patients with palliated cyanotic congenital heart disease. J Cardiothorac Vasc Anesth. 1992;6:340-343.
121 Devitt J.H., Noble W.H., Byrick R.J. A Swan-Ganz catheter related complication in a patient with Eisenmenger’s syndrome. Anesthesiology. 1982;57:335-337.
122 Perloff J.K. The clinical recognition of congenital heart disease, ed 3. Philadelphia: WB Saunders, 1987.
123 Avila W.S., Grinberg M., Snitcowsky R., et al. Maternal and fetal outcome in pregnant women with Eisenmenger’s syndrome. Eur Heart J. 1995;16:460-464.
124 Gleicher N., Midwall J., Hochberger D., et al. Eisenmenger’s syndrome and pregnancy. Obstet Gynecol Surv. 1979;34:721-741.
125 Weiss B.M., Hess O.M. Pulmonary vascular disease and pregnancy: Current controversies, management strategies, and perspectives. Eur Heart J. 2000;21:104-115.
126 Weiss B.M., Zemp L., Seifert B., et al. Outcome of pulmonary vascular disease in pregnancy: A systematic overview from 1978 through 1996. J Am Coll Cardiol. 1998;31:1650-1657.
127 Masuda M., Kado H., Kajihara N., et al. Early and late results of total correction of congenital cardiac anomalies in infancy. Jpn J Thorac Cardiovasc Surg. 2001;49:497-503.
128 Fontan F., Baudet E. Surgical repair of tricuspid atresia. Thorax. 1971;26:240-248.
129 Kreutzer G., Galindez E., Bono H., et al. An operation for the correction of tricuspid atresia. J Thorac Cardiovasc Surg. 1973;66:613-621.
130 Choussat, Fontan A., Besse P. Selection criteria for Fontan’s procedure. In: Anderson R.H., Shinebourne E.A., editors. Pediatric cardiology. Edinburgh: Churchill Livingstone, 1978.
131 Coon P.D., Rychik J., Novello R.T., et al. Thrombus formation after the Fontan operation. Ann Thorac Surg. 2001;71:1990-1994.
132 Monagle P., Karl T.R. Thromboembolic problems after the Fontan operation. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu. 2002;5:36-47.
133 Varma C., Warr M.R., Hendler A.L., et al. Prevalence of “silent” pulmonary emboli in adults after the Fontan operation. J Am Coll Cardiol. 2003;41:2252-2258.
134 Cromme-Dijkhuis A.H., Henkens C.M., Bijleveld C.M., et al. Coagulation factor abnormalities as possible thrombotic risk factors after Fontan operations. Lancet. 1990;336:1087-1090.
135 Odegard K., McGowan F.X.J., Zurakowski D., et al. Procoagulant and anticoagulant factor abnormalities following the fontan procedure: Increased factor VIII may predispose to thrombosis. J Thorac Cardiovasc Surg. 2003;125:1260-1267.
136 Jacobs M.L., Pourmoghadam K.K., Geary E.M., et al. Fontan’s operation: Is aspirin enough? Is coumadin too much? Ann Thorac Surg. 2002;73:64-68.
137 Weipert J., Noebauer C., Schreiber C., et al. Occurrence and management of atrial arrhythmia after long-term Fontan circulation. J Thorac Cardiovasc Surg. 2004;127:457-464.
138 Gelatt M., Hamilton R.M., McCrindle B.W., et al. Risk factors for atrial tachyarrhythmias after the Fontan operation. J Am Coll Cardiol. 1994;24:1735-1741.
139 Durongpisitkul K., Porter C.J., Cetta F., et al. Predictors of early- and late-onset supraventricular tachyarrhythmias after Fontan operation. Circulation. 1998;98:1099-1107.
140 Kirsh J.A., Walsh E.P., Triedman J.K. Prevalence of and risk factors for atrial fibrillation and intra-atrial reentrant tachycardia among patients with congenital heart disease. Am J Cardiol. 2002;90:338-340.
141 Deal B.J., Mavroudis C., Backer C.L. Arrhythmia management in the Fontan patient. Pediatr Cardiol. 2007;28:448-456.
142 Driscoll D.J., Offord K.P., Feldt R.H., et al. Five- to fifteen-year follow-up after Fontan operation. Circulation. 1992;85:469-496.
143 Balaji S., Gewillig M., Bull C., et al. Arrhythmias after the Fontan procedure. Comparison of total cavopulmonary connection and atriopulmonary connection. Circulation. 1991;84:III162-III167.
144 Cohen M.I., Bridges N.D., Gaynor J.W., et al. Modifications to the cavopulmonary anastomosis do not eliminate early sinus node dysfunction. J Thorac Cardiovasc Surg. 2000;120:891-900.
145 Giannico S., Hammad F., Amodeo A., et al. Clinical outcome of 193 extracardiac Fontan patients: The first 15 years. J Am Coll Cardiol. 2006;47:2065-2073.
146 Mertens L., Hagler D.J., Sauer U., et al. Protein-losing enteropathy after the Fontan operation: An international multicenter study. PLE study group. J Thorac Cardiovasc Surg. 1998;115:1063-1073.
147 Ostrow A., Freeze H., Rychik J. Protein-losing enteropathy after Fontan operation: Investigations into possible pathophysiologic mechanisms. Ann Thorac Surg. 2006;82:695-701.
148 Rychik J., Piccoli D.A., Barber G. Usefulness of corticosteroid therapy for protein-losing enteropathy after the Fontan procedure. Am J Cardiol. 1991;68:819-821.
149 Donnelly J.P., Rosenthal A., Castle V.P., et al. Reversal of protein-losing enteropathy with heparin therapy in three patients with univentricular hearts and Fontan palliation. J Pediatr. 1997;130:474-478.
150 de Leval M.R., Kilner P., Gewillig M., et al. Total cavopulmonary connection: A logical alternative to atriopulmonary connection for complex Fontan operations. Experimental studies and early clinical experience. J Thorac Cardiovasc Surg. 1988;96:682-695.
151 d’Udekem Y., Iyengar A.J., Cochrane A.D., et al. The Fontan procedure: Contemporary techniques have improved long-term outcomes. Circulation. 2007;116:I157-I164.
152 Harrison D.A., Liu P., Walters J.E., et al. Cardiopulmonary function in adult patients late after Fontan repair. J Am Coll Cardiol. 1995;26:1016-1021.
153 Gratz A., Hess J., Hager A. Self-estimated physical functioning poorly predicts actual exercise capacity in adolescents and adults with congenital heart disease. Eur Heart J. 2009;30:497-504.
154 Kiesewetter C.H., Sheron N., Vettukattill J.J., et al. Hepatic changes in the failing Fontan circulation. Heart. 2007;93:579-584.
155 Ghanayem N.S., Berger S., Tweddell J.S. Medical management of the failing Fontan. Pediatr Cardiol. 2007;28:465-471.
156 Steven J.M., McGowan F.X. Neuraxial blockade for pediatric cardiac surgery: Lessons yet to be learned. Anesth Analg. 2000;90:1011-1013.
157 Carp H., Jayaram A., Vadhera R., et al. Epidural anesthesia for cesarean delivery and vaginal birth after maternal Fontan repair: Report of two cases. Anesth Analg. 1994;78:1190-1192.
158 Fyfe D.A., Gillette P.C., Jones J.S., et al. Successful pregnancy following modified Fontan procedure in a patient with tricuspid atresia and recurrent atrial flutter. Am Heart J. 1989;117:1387-1388.
159 Sheikh A.M., Tang A.T., Roman K., et al. The failing Fontan circulation: Successful conversion of atriopulmonary connections. J Thorac Cardiovasc Surg. 2004;128:60-66.
160 Mavroudis C., Deal B.J., Backer C.L., et al. J. Maxwell Chamberlain Memorial Paper for congenital heart surgery. 111 Fontan conversions with arrhythmia surgery: Surgical lessons and outcomes. Ann Thorac Surg. 2007;84:1457-1465. discussion 65–66
161 Fisher R.G., Moodie D.S., Sterba R., et al. Patent ductus arteriosus in adults—long-term follow-up: Nonsurgical versus surgical treatment. J Am Coll Cardiol. 1986;8:280-284.
162 Campbell M. Natural history of patent ductus arteriosus. Br Heart J. 1968;30:4-13.
163 O’Fallon W.M., Weidman W.H. Long-term follow-up of congenital aortic stenosis, pulmonary stenosis and ventricular septal defect. Circulation. 1993;87(Suppl I):I1-I126.
164 Hayes C.J., Gersony W.M., Driscoll D.J., et al. Second natural history study of congenital heart defects. Results of treatment of patients with pulmonary valvar stenosis. Circulation. 1993;87:I28-I37.
165 Hameed A.B., Goodwin T.M., Elkayam U. Effect of pulmonary stenosis on pregnancy outcomes—a case-control study. Am Heart J. 2007;154:852-854.
166 Bertranou E.G., Blackstone E.H., Hazelrig J.B., et al. Life expectancy without surgery in tetralogy of Fallot. Am J Cardiol. 1978;42:458-466.
167 Harrison D.A., Harris L., Siu S.C., et al. Sustained ventricular tachycardia in adult patients late after repair of tetralogy of Fallot. J Am Coll Cardiol. 1997;30:1368-1373.
168 Murphy J.G., Gersh B.J., Mair D.D., et al. Long-term outcome in patients undergoing surgical repair of tetralogy of Fallot. N Engl J Med. 1993;329:593-599.
169 Nollert G., Fischlein T., Bouterwek S., et al. Long-term survival in patients with repair of tetralogy of Fallot: 36-year follow-up of 490 survivors of the first year after surgical repair. J Am Coll Cardiol. 1997;30:1374-1383.
170 Oechslin E.N., Harrison D.A., Harris L., et al. Reoperation in adults with repair of tetralogy of Fallot: Indications and outcomes. J Thorac Cardiovasc Surg. 1999;118:245-251.
171 Russell I.A., Miller Hance W.C., Gregory G., et al. The safety and efficacy of sevoflurane anesthesia in infants and children with congenital heart disease. Anesth Analg. 2001;92:1152-1158.
172 Gatzoulis M.A., Balaji S., Webber S.A., et al. Risk factors for arrhythmia and sudden cardiac death late after repair of tetralogy of Fallot: A multicentre study. Lancet. 2000;356:975-981.
173 Harrison D.A., Siu S.C., Hussain F., et al. Sustained atrial arrhythmias in adults late after repair of tetralogy of Fallot. Am J Cardiol. 2001;87:584-588.
174 Roos-Hesselink J., Perlroth M.G., McGhie J., et al. Atrial arrhythmias in adults after repair of tetralogy of Fallot. Correlations with clinical, exercise, and echocardiographic findings. Circulation. 1995;91:2214-2219.
175 Rammohan M., Airan B., Bhan A., et al. Total correction of tetralogy of Fallot in adults—surgical experience. Int J Cardiol. 1998;63:121-128.
176 Kavey R.E., Blackman M.S., Sondheimer H.M. Incidence and severity of chronic ventricular dysrhythmias after repair of tetralogy of Fallot. Am Heart J. 1982;103:342-350.
177 Abd El Rahman M.Y., Abdul-Khaliq H., Vogel M., et al. Relation between right ventricular enlargement, QRS duration, and right ventricular function in patients with tetralogy of Fallot and pulmonary regurgitation after surgical repair. Heart. 2000;84:416-420.
178 Cullen S., Celermajer D.S., Franklin R.C., et al. Prognostic significance of ventricular arrhythmia after repair of tetralogy of Fallot: A 12-year prospective study. J Am Coll Cardiol. 1994;23:1151-1155.
179 Gatzoulis M.A., Till J.A., Somerville J., et al. Mechanoelectrical interaction in tetralogy of Fallot. QRS prolongation relates to right ventricular size and predicts malignant ventricular arrhythmias and sudden death. Circulation. 1995;92:231-237.
180 Discigil B., Dearani J.A., Puga F.J., et al. Late pulmonary valve replacement after repair of tetralogy of Fallot. J Thorac Cardiovasc Surg. 2001;121:344.
181 Momenah T.S., El Oakley R., Al Najashi K., et al. Extended application of percutaneous pulmonary valve implantation. J Am Coll Cardiol. 2009;53:1859-1863.
182 Heggie J., Poirer N., Williams R.G., et al. Anesthetic considerations for adult cardiac surgery patients with congenital heart disease. Semin Cardiothorac Vasc Anesth. 2003;7:141-152.
183 Veldtman G.R., Connolly H.M., Grogan M., et al. Outcomes of pregnancy in women with tetralogy of Fallot. J Am Coll Cardiol. 2004;44:174-180.
184 Graham T.P.Jr. Ventricular performance in congenital heart disease. Circulation. 1991;84:2259-2274.
185 Gelatt M., Hamilton R.M., McCrindle B.W., et al. Arrhythmia and mortality after the Mustard procedure: A 30-year single-center experience. J Am Coll Cardiol. 1997;29:194-201.
186 Wilson N.J., Clarkson P.M., Barratt-Boyes B.G., et al. Long-term outcome after the mustard repair for simple transposition of the great arteries. 28-year follow-up. J Am Coll Cardiol. 1998;32:758-765.
187 Oechslin E., Jenni R. 40 years after the first atrial switch procedure in patients with transposition of the great arteries: Long-term results in Toronto and Zurich. J Thorac Cardiovasc Surg. 2000;48:233-237.
188 Myridakis D.J., Ehlers K.H., Engle M.A. Late follow-up after venous switch operation (Mustard procedure) for simple and complex transposition of the great arteries. Am J Cardiol. 1994;74:1030-1036.
189 Puley G., Siu S., Connelly M., et al. Arrhythmia and survival in patients > 18 years of age after the mustard procedure for complete transposition of the great arteries. Am J Cardiol. 1999;83:1080-1084.
190 Warnes C.A., Somerville J. Transposition of the great arteries: Late results in adolescents and adults after the Mustard procedure. Br Heart J. 1987;58:148-155.
191 Graham T.P.Jr, Burger J., Bender H.W., et al. Improved right ventricular function after intra-atrial repair of transposition of the great arteries. Circulation. 1985;72:II45-II51.
192 Cochrane A.D., Karl T.R., Mee R.B. Staged conversion to arterial switch for late failure of the systemic right ventricle. Ann Thorac Surg. 1993;56:854-861.
193 Garson A.J. The emerging adult with arrhythmias after congenital heart disease: Management and financial health care policy. Pacing Clin Electrophysiol. 1990;13:951-954.
194 Mavroudis C., Backer C.L. Arterial switch after failed atrial baffle procedures for transposition of the great arteries. Ann Thorac Surg. 2000;69:851-857.
195 Hutter P.A., Kreb D.L., Mantel S.F., et al. Twenty-five years’ experience with the arterial switch operation. J Thorac Cardiovasc Surg. 2002;124:790-797.
196 Losay J., Touchot A., Serraf A., et al. Late outcome after arterial switch operation for transposition of the great arteries. Circulation. 2001;104:I121-I126.
197 Mahle W.T., McBride M.G., Paridon S.M. Exercise performance after the arterial switch operation for D-transposition of the great arteries. Am J Cardiol. 2001;87:753-758.
198 Rajasinghe H.A., McElhinney D.B., Reddy V.M., et al. Long-term follow-up of truncus arteriosus repaired in infancy: A twenty-year experience. J Thorac Cardiovasc Surg. 1997;113:869-878.
199 Schreiber C., Eicken A., Balling G., et al. Single centre experience on primary correction of common arterial trunk: Overall survival and freedom from reoperation after more than 15 years. Eur J Cardiothorac Surg. 2000;18:68-73.
200 Wilson N.J., Neutze J.M. Adult congenital heart disease: Principles and management guidelines: Part II. Aust NZ J Med. 1993;23:697-705.
201 Jarmakani J.M., Graham T.P.Jr, Canent R.V.Jr, et al. The effect of corrective surgery on left heart volume and mass in children with ventricular septal defect. Am J Cardiol. 1971;27:254-258.
202 Jarmakani J.M., Graham T.P.J., Canent R.V.J. Left ventricular contractile state in children with successfully corrected ventricular septal defect. Circulation. 1972;45(Suppl 1):102-110.
203 Maron B.J., Redwood D.R., Hirshfeld J.W.J., et al. Postoperative assessment of patients with ventricular septal defect and pulmonary hypertension. Response to intense upright exercise. Circulation. 1973;48:864-874.
204 Shanewise J.S., Cheung A.T., Aronson S., et al. ASE/SCA guidelines for performing a comprehensive intraoperative multiplane transesophageal echocardiography examination: Recommendations of the American Society of Echocardiography Council for Intraoperative Echocardiography and the Society of Cardiovascular Anesthesiologists Task Force for Certification in Perioperative Transesophageal Echocardiography. Anesth Analg. 1999;89:870-874.
[/level-membership-for-anesthesiology-category][not-level-membership-for-anesthesiology-category]
20 Congenital Heart Disease in Adults
Advances in perioperative care for children with congenital heart disease (CHD) over the past several decades has resulted in an ever-increasing number of these children reaching adulthood with their cardiac lesions palliated or repaired. The first article on adult CHD was published in 19731; the field has grown such that there are now several texts devoted to it, and even a specialty society dedicated to it, the International Society for Adult Congenital Cardiac Disease (http: //www.isaccd.org). There are estimated to be about 32,000 new cases of CHD each year in the United States and 1.5 million worldwide.2 More than 85% of infants born with CHD are expected to grow to adulthood. It is estimated that there are more than 1,000,000 adults in the United States with CHD,3 and this population is growing at approximately 5% per year; 55% of these adults remain at moderate-to-high risk, and more than 115,000 have complex disease.4 The increasing survival of children with complex disease has shifted the spectrum of adults with CHD. Where once it was thought that adults represent milder degrees of disease, this is now changing.3 Put another way, there are more adults than children with CHD and, in fact, as many adults as children have congenital cardiac defects considered severe.5 As an example to support the increased life expectancy of this patient group, the leading cause of mortality in adults with CHD in the United States is currently coronary artery disease.6 These patients can be seen by anesthesiologists for primary cardiac repair, repair after a prior palliation, revision of repair because of failure or lack of growth of prosthetic material, or conversion of a suboptimal repair to a more modern operation (Box 20-1). In addition, these adults with CHD will be seen for all the other ailments of aging and trauma that require surgical intervention. Lastly, women of child-bearing age with CHD may become pregnant. They must cope with the added physiologic demands of pregnancy and will require analgesia for labor and anesthesia for Cesarean delivery. Although it has been suggested that teenagers and adults can have repair of congenital cardiac defects with morbidity and mortality approaching that of surgery done during childhood, these data are limited and may reflect only a relatively young and acyanotic sampling.7 Other data suggest that, in general, adults older than 50 years represent an excessive proportion of the early postoperative mortality encountered, and the number of previous operations and cyanosis are both risk factors.8 Risk factors for noncardiac surgery include heart failure, pulmonary hypertension, and cyanosis.9
Despite the fact that congenital cardiac disease carries implications for lifelong medical problems, a significant number of patients, even those with lesions deemed severe, will not have continuing cardiology follow-up, despite ongoing general medical care.10 These patients bring with them anatomic and physiologic complexities of which physicians accustomed to caring for adults may be unaware, as well as medical problems associated with aging or pregnancy that might not be familiar to physicians used to caring for children. This problem has led to the establishment of the growing subspecialty of adult CHD. There have been two Bethesda conferences devoted to the issue, most recently in 200111; and the American College of Cardiology reviewed the available evidence and published superb guidelines for the care of these patients in 2008.12 It is important to note that most of the current recommendations are based on Level C evidence (only consensus opinion or case studies) because prospective studies and large registries of patient outcomes are rare. An informed anesthesiologist is a critical member of the team required to care optimally for these patients. A specific recommendation of that conference was that noncardiac surgery on adult patients with moderate-to-complex CHD be done at an adult congenital heart center (regional centers) with the consultation of an anesthesiologist experienced with adult CHD.11,13 In fact, one of the founding fathers of the subspecialty wrote: “A cardiac anesthesiologist with experience in CHD is pivotal.…The cardiac anesthesiologist and the attending cardiologist are more important than the noncardiac surgeon.”2 High-risk patients include, but are not limited to, those with Fontan physiology, cyanotic disease, severe pulmonary arterial hypertension, complex disease with residua such as heart failure, valve disease or the need for anticoagulation, or the potential for malignant arrhythmias (Table 20-1). Centers may find it helpful to delegate one attending anesthesiologist to be the liaison with the cardiology service to centralize preoperative evaluations and triage of patients to an anesthesiologist with specific expertise in managing patients with CHD, rather than random consultations with generalist anesthesiologists.
TABLE 20-1 Rhythm Disturbances in Adults with Congenital Heart Disease
| Rhythm Disturbance | Associated Lesions |
|---|---|
| Tachycardias | |
| Wolff-Parkinson-White syndrome | Ebstein’s anomaly |
| Congenitally corrected transposition | |
| Intra-atrial reentrant tachycardia (atrial flutter) | Postoperative Mustard |
| Postoperative Senning | |
| Postoperative Fontan | |
| Tetralogy of Fallot | |
| Other | |
| Atrial fibrillation | Mitral valve disease |
| Aortic stenosis | |
| Tetralogy of Fallot | |
| Palliated single ventricle | |
| Ventricular tachycardia | Tetralogy of Fallot |
| Aortic stenosis | |
| Other | |
| Bradycardias | |
| Sinus node dysfunction | Postoperative Mustard |
| Postoperative Senning | |
| Postoperative Fontan | |
| Sinus venosus ASD | |
| Heterotaxy syndrome | |
| Spontaneous AV block | AV septal defects |
| Congenitally corrected transposition | |
| Surgically induced AV block | VSD closure |
| Subaortic stenosis relief | |
| AV valve replacement |
ASD, atrial septal defect; AV, atrioventricular; VSD, ventricular.
(Reprinted from ACC/AHA 2008 guidelines for the management of adults with congenital heart disease: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Develop Guidelines on the Management of Adults With Congenital Heart Disease). Developed in Collaboration with the American Society of Echocardiography, Heart Rhythm Society, International Society for Adult Congenital Heart Disease, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J Am Coll Cardiol 52:e143–e263, 2008.)
General noncardiac issues with longstanding congenital heart disease
A variety of organ systems can be affected by longstanding CHD; these are summarized in Table 20-2 and Box 20-2. Because congenital cardiac disease can be one manifestation of a multiorgan genetic or dysmorphic syndrome, all patients require a full review of systems and examination.
TABLE 20-2 Potential Noncardiac Organ Involvement in Patients with Congenital Heart Disease
| Potential Respiratory Implications |
| Decreased compliance (with increased pulmonary blood flow or impediment to pulmonary venous drainage) |
| Compression of airways by large, hypertensive pulmonary arteries |
| Compression of bronchioles |
| Scoliosis |
| Hemoptysis (with end-stage Eisenmenger syndrome) |
| Phrenic nerve injury (prior thoracic surgery) |
| Recurrent laryngeal nerve injury (prior thoracic surgery; rarely from encroachment of cardiac structures) |
| Blunted ventilatory response to hypoxemia (with cyanosis) |
| Underestimation of Paco2 by capnometry in cyanotic patients |
| Potential Hematologic Implications |
| Symptomatic hyperviscosity |
| Bleeding diathesis |
| Abnormal von Willebrand factor |
| Artifactually increased prothrombin/partial thromboplastin times with erythrocytic blood |
| Artifactual thrombocytopenia with erythrocytic blood |
| Gallstones |
| Potential Renal Implication |
| Hyperuricemia and arthralgias (with cyanosis) |
| Potential Neurologic Implications |
| Paradoxical emboli |
| Brain abscess (with right-to-left shunts) |
| Seizure (from old brain abscess focus) |
| Intrathoracic nerve injury (iatrogenic phrenic, recurrent laryngeal, or sympathetic trunk injury) |
BOX 20-2 Noncardiac Organ Systems with Potential Involvement By Longstanding Congenital Heart Disease
Pulmonary
Any lesion that results in either increased pulmonary blood flow or pulmonary venous obstruction can cause increased pulmonary interstitial fluid with decreased pulmonary compliance and increased work of breathing.14 Patients with cyanotic heart disease have increased minute ventilation and maintain normocarbia.15 These patients have a normal ventilatory response to hypercapnia but a blunted response to hypoxemia that normalizes after corrective surgery and the establishment of normoxia.16–18 End-tidal CO2 underestimates arterial Paco2 in cyanotic patients with decreased, normal, or even increased pulmonary blood flow.19
Although enlarged hypertensive pulmonary arteries or an enlarged left atrium can impinge on bronchi in children, this is rare in adults. Late-stage Eisenmenger syndrome can result in hemoptysis, and patients with Eisenmenger physiology and erythrocytosis can develop thrombosis of upper lobe pulmonary arteries.20 Prior thoracic surgery could have injured the phrenic nerve with resultant diaphragmatic paresis or paralysis.
Scoliosis can occur in up to 19% of CHD patients, most commonly in cyanotic patients. It can also develop in adolescence, years after surgical resolution of cyanosis.21 The relative contributions of cyanosis and congenital cardiovascular defects versus early lateral thoracotomy remain unclear: Scoliosis occurs more commonly with open surgical versus interventional techniques but is nevertheless increased over the general population in children with coarctation or patent ductus who had percutaneous rather than open interventions.22 Scoliosis is rarely severe enough to impact respiratory function.
Hematologic
Hematologic manifestations of chronic CHD are primarily a consequence of longstanding cyanosis and incorporate abnormalities of both hemostasis and red cell regulation. Longstanding hypoxemia causes increased erythropoietin production in the kidney and resultant increased red cell mass. Because solely red cell production is affected, these patients are correctly referred to as erythrocytotic rather than polycythemic. There is, however, a fairly poor relation among oxygen saturation, red cell mass, and 2,3-diphosphoglycerate.23 The oxygen-hemoglobin dissociation curve is normal or minimally shifted to the right. Most patients have established an equilibrium state at which they have a stable hematocrit and are iron replete. Some patients, however, develop excessive hematocrits and are iron deficient, causing a hyperviscous state. Iron-deficient red cells are less deformable and have been said to cause increased viscosity for the same hematocrit.24 However, recently there is some conflicting evidence.25 Symptoms of hyperviscosity are uncommon and typically develop only at hematocrits exceeding 65% provided the patient is iron replete. Iron deficiency also shifts the oxygen-hemoglobin dissociation curve to the right, decreasing oxygen affinity in the lungs.26 Symptoms of hyperviscosity are listed in Box 20-3. Iron deficiency can be the result of misguided attempts to reduce the hematocrit by means of repeated phlebotomies.27 The red cells in these patients may be hypochromic and microcytic despite the high hematocrit. Assessment of iron status is best done by measures of serum ferritin and transferrin rather than inferences from red cell indices.27 Treatment with oral iron needs to be undertaken with care because rapid increases in hematocrit can result.
Symptomatic hyperviscosity is the indication for treatment to temporarily relieve symptoms. It is not indicated to treat otherwise asymptomatic increased hematocrits (generally hemoglobin > 20 and hematocrit > 65). Treatment is by means of a partial isovolumic exchange transfusion, and it is assumed that the increased hematocrit is not related to dehydration. Partial isovolumic exchange transfusion usually results in regression of symptoms within 24 hours. It is rare to require exchange of more than 1 unit of blood. Before surgery, phlebotomized blood can be banked for autologous perioperative retransfusion if required. Elective preoperative isovolumic exchange transfusion has decreased the incidence of hemorrhagic complications of surgery.28,29
Hyperviscosity and erythrocytosis can cause cerebral venous thrombosis in younger children, but it is not a problem in adults, regardless of the hematocrit.20 Protracted preoperative fasts need to be avoided in erythrocytotic patients because they can be accompanied by rapid increases in the hematocrit.
Bleeding dyscrasias have been described in up to 20% of patients. A variety of clotting abnormalities have been described in association with cyanotic CHD, although none uniformly.30 Bleeding dyscrasias are uncommon until the hematocrit exceeds 65%, although excessive surgical bleeding can occur at lower hematocrits. Generally, higher hematocrits are associated with a greater bleeding diathesis. Abnormalities of a variety of factors in both the intrinsic and extrinsic coagulation pathways have been described inconsistently. Both cyanotic and acyanotic patients have been reported with deficiencies of the largest von Willebrand factor multimers, which have normalized after reparative surgery.31 Fibrinolytic pathways are normal.32
Platelet counts are typically normal or occasionally low, but bleeding is not due to thrombocytopenia. Platelets are reported per milliliter of blood, not milliliter of plasma. When corrected for the decreased plasma fraction in erythrocytic blood, the total plasma platelet count is closer to normal. That said, abnormalities in platelet function and life span have on occasion been reported.33,34 Patients with low-pressure conduits (Fontan pathway) or synthetic vascular anastomoses are often maintained on antiplatelet drugs.
Cyanotic erythrocytotic patients have excessive hemoglobin turnover, and adults have an increased incidence of calcium bilirubinate gallstones. Biliary colic can develop years after cyanosis has been resolved by cardiac surgery.20
A variety of mechanical factors can also affect excessive surgical bleeding in cyanotic CHD patients. These factors include increased tissue capillary density, increased systemic venous pressure, aortopulmonary and transpleural collaterals that have developed to increase pulmonary blood flow, and prior thoracic surgery. Aprotinin and ε-aminocaproic acid improve postoperative hemostasis in patients with cyanotic CHD.35 The results with tranexamic acid have been mixed.36
Renal
Some degree of renal insufficiency is not uncommon in adults with CHD, and the severity is a predictor of mortality. Moderate or severe renal dysfunction (estimated glomerular filtration rate < 60 mL/min/m2) carry a fivefold increased risk for death at 6-year follow-up compared with patients with normal glomerular filtration rate and a threefold increase over those with mild increases in glomerular filtration rate.37 Renal dysfunction is particularly prevalent in cyanotic patients and those with poor cardiac function. Adult patients with cyanotic CHD can develop abnormal renal histology with hypercellular glomeruli and basement membrane thickening, focal interstitial fibrosis, tubular atrophy, and hyalinized afferent and efferent arterioles.38 Cyanotic CHD is often accompanied by increases in plasma uric acid levels that are due to inappropriately low fractional uric acid excretion.39 Decreased urate reabsorption is thought to result from renal hypoperfusion with a high filtration fraction. Despite the increased uric acid levels, urate stones and urate nephropathy are rare.40 Although arthralgias are common, true gouty arthritis is less frequent than would be expected from the degree of hyperuricemia.39 There appears to be an increased incidence of postcardiopulmonary bypass renal dysfunction in adults with longstanding cyanosis.41
Neurologic
Adults with persistent or potential intracardiac shunts remain at risk for paradoxic embolism. Paradoxical emboli can occur even through shunts that are predominantly left to right because during the cardiac cycle there can be small transient reversals of the shunt direction. It has been said that, unlike in children, adults with cyanotic CHD are not at risk for the development of cerebral thrombosis despite the hematocrit.20,42 However, this assertion has been challenged by Ammash and Warnes,43 who suggested an association of stroke not with red cell mass, but with iron deficiency and repeated venesection. Adults do, however, remain at risk for the development of brain abscess. A healed childhood brain abscess can provide the nidus for the development of seizures throughout life.
Prior thoracic surgery can result in permanent peripheral nerve damage. Surgery at the apices of the lungs is particularly associated with the risk for nerve damage. These operations would include Blalock–Taussig shunts, ligation of patent ductus arteriosus (PDA), banding of the pulmonary artery, and repair of aortic coarctation. Nerves that are susceptible to injury include the recurrent laryngeal nerve, the phrenic nerve, and the sympathetic chain. The incidence of migraine headaches is greater in adults with CHD compared with a control group with acquired heart disease (45% vs. 11%) and is increased in left-to-right, right-to-left, and no-shunt groups.44
Vasculature
Vessel abnormalities can be congenital or iatrogenic. They can affect the suitability of vessels for cannulation by the anesthesiologist or measurement of correct pressures. These abnormalities are described in Table 20-3.
| Vessel | Possible Problem |
|---|---|
| Femoral vein(s) | May have been ligated if cardiac catheterization was done by cutdown; large therapeutic catheters in infants often thrombose femoral veins |
| Inferior vena cava | Some lesions, particularly when associated with heterotaxy (polysplenia), have discontinuity of the inferior vena cava; will not be able to pass a catheter from the groin to the right atrium |
| Left subclavian and pedal arteries | Distal blood pressure will be low in the presence of coarctation of the aorta or after subclavian flap repair (subclavian artery only), and variably so if postoperative recoarctation; pulses can be absent or palpable with abnormal blood pressure |
| Subclavian artery | Blood pressure low with classic Blalock-Taussig shunt on that side, and variably so with modified Blalock-Taussig shunt |
| Right subclavian artery | Blood pressure artifactually high with supravalvular aortic stenosis (Coanda effect) |
| Superior vena cava | Risk for catheter-related thrombosis with Glenn operation |
Pregnancy
The physiologic changes of pregnancy, labor, and delivery can significantly alter the physiologic status of women with CHD. Several texts are available that specifically discuss issues of the pregnant woman with CHD in more detail than is possible here,45–47 and there has been a recent review of generic cardiac surgery in the parturient.48 Management and clinical outcomes during pregnancy and delivery for several cardiac lesions are included under the later discussions of those lesions.
Although cardiac complications, spontaneous abortions, premature delivery, thrombotic complications, peripartum endocarditis, and poor fetal outcomes can occur,49 successful pregnancy to term with vaginal delivery is possible for most patients with congenital defects.50 High-risk factors for mother and fetus are shown in Box 20-4. Eisenmenger physiology is a particular risk factor. Up to 47% of cyanotic women have worsening of functional capacity during pregnancy.51 Hematocrits greater than 44% are associated with birth weights in less than the 50th percentile, and fetal death is about 90% or more with hemoglobin levels greater than 18 gm/dL or oxygen saturation less than 85%, with most losses in the first trimester. The increases in stroke volume and cardiac output during pregnancy can stress an already pressure-overloaded ventricle. The decrease in systemic vascular resistance that accompanies pregnancy is better tolerated by women with regurgitant lesions and typically offsets the added insult of pregnancy-related hypervolemia. The decrease in systemic vascular resistance can, however, increase right-to-left shunting. Hypervolemia can be problematic in patients with poor ventricular function. Maternal cyanosis is associated with increased incidences of prematurity and intrauterine growth retardation. Profound cyanosis is associated with a high rate of spontaneous abortion.52 Endocarditis prophylaxis is not currently recommended for vaginal deliveries.53 The recurrence risk rate of any congenital cardiac defect in a newborn is 2.3% with one affected older sibling (any defect), 7.3% with two, 6.7% if the mother has a congenital cardiac defect, but only 2.1% if the father is affected.2 However, it has become apparent that recurrence risk can be specific to the type of maternal defect and the underlying genetic basis. If possible, pregnancies in mothers with CHD should be managed in a high-risk obstetric center with cardiologists experienced with the care of adult CHD, and with early consultation with the obstetric anesthesia service. Women on long-term anticoagulation will likely need peripartum modifications, and postpartum thromboembolism is a potential significant problem.
Some mothers will be taking medications for their cardiac condition, including antiarrhythmics. In general, these are safe for the infant.54 Exceptions include β-blockers that can interfere with fetal growth and the response of the fetus to the stress of labor, as well as amiodarone that can affect fetal thyroid function. Maternal cardioversion would appear to be safe for the fetus at all stages, because of the low intensity of the electrical field at the uterus. However, the fetus should be monitored throughout the procedure. Women with implanted internal defibrillators have carried successfully to term.55 Should cardiopulmonary bypass (CPB) be required during pregnancy, it carries with it increased fetal risk, particularly if hypothermia is used.
Psychosocial
Teenagers with CHD are certainly no different from other teenagers in that issues of denial, a sense of immortality, and risk-taking behavior can affect optimal care for these youngsters. Bodies that carry scars from prior surgery and physical limitations can complicate the body-conscious teenage years. Although most adolescents and adults with CHD function well, adults with CHD are less likely to be married or cohabitating and are more likely to be living with their parents.56 There are several series of adolescent and adult patients and psychosocial outcome, but there are no well-done controlled studies.57–64 It has been suggested that depression is common and can exacerbate the clinical consequences of the cardiac defect.
Adolescent CHD patients have higher medical care expenses than the general population, and they can have difficulty in obtaining life and health insurance after they can no longer be covered under their parents’ policies.65–67 Life insurance is somewhat more available to adult CHD patients than in the past; however, policies vary widely among insurers.68
Cardiac issues
The basic hemodynamic effects of an anatomic cardiac lesion can be modified by time and by the superimposed effects of chronic cyanosis, pulmonary disease, or the effects of aging. Although surgical cure is the goal, true universal cure, without residua, sequelae, or complications, is uncommon on a population-wide basis. Exceptions include closure of a nonpulmonary hypertensive PDA or atrial septal defect (ASD), probably in childhood. Although there have been reports of series of surgeries on adults with CHD, the wide variety of defects and sequelae from prior surgery make generalizations difficult, if not impossible. Poor myocardial function can be inherent in the CHD but can also be affected by longstanding cyanosis or superimposed surgical injury, including inadequate intraoperative myocardial protection.68,69 This is particularly true of adults who had their cardiac repair several decades ago when myocardial protection may not have been as good and when repair was undertaken at an older age. Postoperative arrhythmias are common, particularly when surgery entails long atrial suture lines, and the incidence of atrial arrhythmias increases with time, either as a primary sequela or as an indicator of diminished cardiac function.70 Thrombi can be found in these atria precluding immediate cardioversion.71 Bradyarrhythmias can be secondary to surgical injury to the sinus node or conducting tissue or can be a component of the cardiac defect.
The number of cardiac lesions and subtypes, together with the large number of contemporary and obsolescent palliative and corrective surgical procedures, make a complete discussion of all CHD impossible. The reader is referred to one of the current texts on pediatric cardiac anesthesia for more detailed descriptions of these lesions, the available surgical repairs, and the anesthetic implications during primary repair.72,73 Some general perioperative guidelines to caring for these patients are offered in Table 20-4. This chapter provides a discussion of the more common and physiologically important defects that will be encountered in an adult CHD population. Defining outcome after CHD surgery is like trying to hit a continuously moving target. Both short-term and long-term results from older series can differ significantly from contemporary results.
TABLE 20-4 General Approach to Anesthesia for Patients with Congenital Heart Disease
| General |
| The best care for both cardiac and noncardiac surgery in adult patients with CHD is afforded in a center with a multidisciplinary team experienced in the care of adults with CHD, and knowledgeable about both the anatomy and physiology of CHD and the manifestations and considerations specific to adults with CHD |
| Preoperative |
| Review most recent laboratory data, catheterization, and echocardiogram, and other imaging data; the most recent office letter from the cardiologist is often most helpful; obtain and review these in advance |
| Drawing a diagram of the heart with saturations, pressures, and direction of blood flow often clarifies complex and superficially unfamiliar anatomy and physiology |
| Avoid prolonged fast if patient is erythrocytotic to avoid hemoconcentration |
| No generalized contraindication to preoperative sedation |
| Intraoperative |
| Large-bore intravenous access for redo sternotomy and cyanotic patients (see Table 20-3 for vascular access considerations) |
| Avoid air bubbles in all intravenous catheters; there can be transient right-to-left shunting even in lesions with predominant left-to-right shunting (filters are available but will severely restrict ability to give volume and blood) |
| Apply external defibrillator pads for redo sternotomies and patients with poor cardiac function |
| Appropriate endocarditis prophylaxis (orally or intravenously before skin incision)40 |
| Consider antifibrinolytic therapy, especially for patients with prior sternotomy |
| Transesophageal echocardiography for cardiac operations |
| Modulate pulmonary and systemic vascular resistances as appropriate pharmacologically and by modifications in ventilation |
| Postoperative |
| Appropriate pain control (cyanotic patients have normal ventilatory response to hypercarbia and narcotics) |
| Maintain hematocrit appropriate for arterial saturation |
| Maintain central venous and left atrial pressures appropriate for altered ventricular diastolic compliance or presence of beneficial atrial level shunting |
| Pao2 may not increase significantly with the application of supplemental oxygen in the face of right-to-left shunting; similarly, neither will it decrease much with the withdrawal of oxygen (in the absence of lung pathology) |
Aortic Stenosis
Most aortic stenosis in adults is due to a congenitally bicuspid valve that does not become problematic until late middle age or later, although endocarditis risk is lifelong. Congenital aortic stenosis can, on some occasions, however, become severe enough to warrant surgical correction in adolescence or young adulthood, in addition to those severely affected valves that present in infancy. Once symptoms (angina, syncope, near-syncope, heart failure) develop, survival is markedly shortened. Median survival is 5 years after the development of angina, 3 years after syncope, and 2 years after heart failure.74 Anesthetic management of aortic stenosis does not vary whether the stenosis is congenital (most common) or acquired (see Chapter 19).
Aortopulmonary Shunts
Depending on their age, adult patients may have had one or more of several aortopulmonary shunts to palliate cyanosis during childhood. These are shown in Figure 20-1. Although life saving, there were considerable shortcomings of these shunts in the long term. All were inherently inefficient, as some of the oxygenated blood returning through the pulmonary veins to the left atrium and ventricle would then return to the lungs through the shunt, thus volume loading the ventricle. It was difficult to quantify the size of the earlier shunts such as the Waterston (side-to-side ascending aorta to right pulmonary artery) and Potts (side-to-side descending aorta to left pulmonary artery). If too small, the patient was left excessively cyanotic; if too large, there was pulmonary overcirculation with the risk for development of pulmonary vascular disease. The Waterston, in fact, could, on occasion, stream blood flow unequally, resulting in a hyperperfused, hypertensive ipsilateral (right) pulmonary artery and a hypoperfused contralateral (left) pulmonary artery. There were also surgical issues when complete repair became possible. Takedown of Waterston shunts often required a pulmonary arterioplasty to correct deformity of the pulmonary artery at the site of the anastomosis, and the posteriorly located Potts anastomoses could not be taken down from a median sternotomy. Patients with a classic Blalock–Taussig shunt almost always lack palpable pulses on the side of the shunt. Even if there is a palpable pulse (from collateral flow around the shoulder), blood pressure obtained from that arm will be artifactually low. Even after a modified Blalock–Taussig shunt (using a piece of Gore-Tex tubing instead of an end-to-side anastomosis of the subclavian and pulmonary arteries), there can be a blood pressure disparity between the arms. Preoperative blood pressure should be measured in both arms to ensure a valid measurement (Box 20-5).
Figure 20-1 Various aortopulmonary anastomoses.
(From Baum VC: The adult with congenital heart disease. J Cardiothorac Vasc Anesth 10:261, 1996.)
BOX 20-5 Aortopulmonary Shunts
| Waterston | Ascending aorta→right pulmonary artery | No longer done |
| Potts | Descending aorta→left pulmonary artery | No longer done |
| Classic Blalock–Taussig | Subclavian artery→ipsilateral pulmonary artery | No longer done |
| Modified Blalock–Taussig | Gore-Tex tube subclavian artery→ipsilateral pulmonary artery | Current |
| Central shunt | Gore-Tex tube ascending aorta→main pulmonary artery | Current |
Atrial Septal Defect and Partial Anomalous Pulmonary Venous Return
There are several anatomic types of ASD. The most common type, and if otherwise undefined the presumptive type, is the secundum type located in the midseptum. The primum type at the lower end of the atrial septum is a component of endocardial cushion defects, the most primitive of which is the common atrioventricular canal (see later). The sinus venosus type, high in the septum near the entry of the superior vena cava (SVC), is almost always associated with partial anomalous pulmonary venous return, most frequently drainage of the right upper pulmonary vein to the low SVC. An uncommon atrial septal–type defect is when blood passes from the left atrium to the right via an unroofed coronary sinus. For purposes of this section, only secundum defects are considered, although the natural histories of all of the defects are similar (Box 20-6).
Both the natural history and the postoperative outcome of ASDs and the physiologically related partial anomalous venous return are similar.75–77 Because the symptoms and clinical findings of an ASD can be quite subtle and patients often remain asymptomatic until adulthood, ASDs represent approximately one third of all CHD discovered in adults. Although asymptomatic survival to adulthood is common, significant shunts (p/s > 1.5:1) will probably cause symptoms over time, and paradoxical emboli can occur through defects with smaller shunts. Surgical repair of restrictive lesions 5 mm or smaller in size does not impact the natural history. Thus, surgical closure of small lesions is not indicated in the absence of paradoxical emboli. Effort dyspnea occurs in 30% by the third decade, and atrial flutter or fibrillation in about 10% by age 40.75 The avoidance of complications developing in adulthood provides the rationale for surgical repair of asymptomatic children. The mortality for a patient with an uncorrected ASD is 6% per year after 40 years of age, and essentially all patients older than 60 years are symptomatic.75–77 Large, unrepaired defects can cause death from atrial tachyarrhythmias or right ventricular failure in 30- to 40-year-old patients.78 With the decreased left ventricular diastolic compliance accompanying the systemic hypertension or coronary artery disease that is common with aging, left-to-right shunting increases with age. Pulmonary vascular disease typically does not develop until after the age of 40, unlike ventricular or ductal level shunts, which can lead to it in early childhood. Mitral insufficiency can be found in adult patients and is significant in about 15% of adult patients.79 Paradoxical emboli remain a lifelong risk.
Late closure of the defect, after 5 years of age, has been associated with incomplete resolution of right ventricular dilation.80 Left ventricular dysfunction has been reported in some patients having defect closure in adulthood, and closure, particularly in middle age, may not prevent the development of atrial tachyarrhythmias or stroke.81–84 Survival of patients without pulmonary vascular disease has been reported to be best if operated on before 24 years of age, intermediate if operated on between 25 and 41 years of age, and worst if operated on thereafter.84 However, more recent series have shown that even at ages older than 40, surgical repair provides an overall survival and complication-free benefit compared with medical management.85 Surgical morbidity in these patients is primarily atrial fibrillation, atrial flutter, or junctional rhythm.82 Current practice is to close these defects in adults in the catheterization laboratory via transvascular devices if anatomically practical (Figure 20-2). For example, there needs to be an adequate rim of septum around the defect to which the device can attach. Device closure is inappropriate if the defect is associated with anomalous pulmonary venous drainage. The indications for closure with a transvascular device are the same as for surgical closure.86
An otherwise uncomplicated secundum ASD, unlike most congenital cardiac defects, is not associated with an increased endocarditis risk.87 Presumably, this is because the shunt, although potentially large, is low pressure and unassociated with jet lesions of the endocardium.
Coarctation of the Aorta
Unrepaired coarctation of the aorta in the adult brings with it significant morbidity and mortality. Mortality rate is 25% by age 20, 50% by age 30, 75% by age 50, and 90% by age 60.78,88–90 Left ventricular aneurysm, rupture of cerebral aneurysms, and dissection of a postcoarctation aneurysm all contribute to the excessive mortality. Left ventricular failure can occur in patients older than 40 with unrepaired lesions. If repair is not undertaken early, there is incremental risk for the development of premature coronary atherosclerosis. Even with surgery, coronary artery disease remains the leading cause of death 11 to 25 years after surgery.91 Coarctation is accompanied by a bicuspid aortic valve in the majority of patients. Although endocarditis of this abnormal valve is a lifelong risk, these valves often do not become stenotic until middle age or later. Coarctation can also be associated with mitral valve abnormalities (Box 20-7).
Aneurysms at the site of coarctation repair can develop years later (Figure 20-3), and restenosis as well can develop in adolescence or adulthood. Repair includes resection of the coarctation and end-to-end anastomosis. Because this sometimes resulted in recoarctation when done in infancy, for many years a common repair was the Waldhausen or subclavian flap operation, in which the left subclavian artery is ligated and the proximal segment opened and rotated as a flap to open the area of the coarctation. Aneurysms in the area of repair are a particular concern in adolescents and adults after coarctectomy. Persistent systemic hypertension is common after coarctation repair.92 The risk for hypertension parallels the duration of unrepaired coarctation. Adult patients require continued periodic follow-up for hypertension. A pressure gradient of 20 mm Hg or more (less in the presence of extensive collaterals) is an indication for treatment.93 Recoarctation can be treated surgically or by balloon angioplasty with stenting.94 Surgical repair of recoarctation or aneurysm in adults is associated with increased mortality and can be associated with significant intraoperative bleeding because of previous scar or extensive collateral vessels. It requires lung isolation for optimal surgical exposure and placement of an arterial catheter in the right arm. Endovascular repair by ballooning/stenting has proved useful for these patients.95,96
Half of patients operated on after age 40 have persistent hypertension, and many of the remainder have an abnormal hypertensive response to surgery. Long-term survival is worse for patients having repair later in life. Patients older than 40 undergoing repair have a 15-year survival rate of only 50%.91
Congenitally Corrected Transposition of the Great Vessels (l-Transposition, Ventricular Inversion)
“Transposition” in this context refers solely to the fact that the aorta arises anterior to the pulmonary artery. It bears no reference to the origin of blood in the aorta or pulmonary artery, or to the ventricle of origin of those vessels. In l-transposition, as a consequence of the embryonic heart tube rotating to the left rather than to the right, the flow of blood is through normal vena cavae to the right atrium, through a mitral valve to a right-sided anatomic left ventricle, to the pulmonary artery, through the pulmonary circulation, to the left atrium, through a tricuspid valve to a left-sided anatomic right ventricle, and thence to the aorta (Figure 20-4) Although anatomically altered, the physiologic flow of blood is appropriate and there are no associated shunts. l-Transposition of the great arteries (L-TGA) is frequently associated with other cardiac lesions, most commonly a ventricular septal defect (VSD), subpulmonic stenosis, heart block, or systemic atrioventricular (tricuspid) valve regurgitation. In the absence of any of these associated cardiac defects, L-TGA will usually be asymptomatic through infancy and childhood. When l-transposition is an isolated lesion, most patients maintain normal biventricular function through early adulthood, and can attain a normal life span.
Systemic atrioventricular (tricuspid) valve insufficiency may not develop until later in life, resulting in approximately 60% of patients being diagnosed as adults.97 Dysfunction of the right or systemic ventricle can develop with aging, and asymptomatic aging is relatively uncommon.98 By age 45, heart failure will be present in 67% of those patients with associated lesions and 25% of those without.99 These patients can be born with congenital heart block, which can progress in degree. Second- or third-degree heart block occurs at a rate of about 2% per year. More than 75% of patients develop some degree of heart block, although the intrinsic pacemaker remains above the His bundle with a narrow QRS complex. l-Transposition can be associated with an Ebstein-like deformity of the systemic atrioventricular (tricuspid) valve, and there can be a bundle of Kent causing the Wolff-Parkinson-White syndrome associated with that abnormal valve. There is a significant incidence of tricuspid valve insufficiency in the systemic ventricle, and this is greater still in patients with an Ebstein’s deformity of the valve.100 Anesthetic management depends on the presence of any associated lesions and the adequacy of right (systemic) ventricular function.
Although women generally do well with pregnancy,101 the physiologic stresses of pregnancy and delivery can result in ventricular or valvular dysfunction, particularly with baseline dysfunction and/or an insufficient systemic atrioventricular valve; but even if these develop, pregnancy can be successfully managed.101–104 The decrease in systemic vascular resistance associated with both pregnancy and neuraxial analgesia might be advantageous to women with tricuspid (systemic) valve insufficiency. The acute autotransfusion associated with delivery could potentially cause problems for women with existing diminished systemic ventricle function.
Ebstein’s Anomaly of the Tricuspid Valve
Ebstein’s anomaly, one of arrested or incomplete delamination of the embryonic tricuspid valve from the right ventricular myocardium, resulting in apically displaced valve tissue, is the most common cause of congenital tricuspid insufficiency. The septal leaflet tends to be the most dysplastic. The anterior leaflet tends to be large and redundant. The defect is associated with a patent foramen ovale or a secundum ASD. There is “atrialization” of part of the right ventricle. The displacement of the tricuspid valve toward the right ventricular apex results in a portion of the right ventricle being above the tricuspid valve and becoming functionally part of the right atrium. Apicalization of the tricuspid valve results in a portion of the heart above the valve having a ventricular intracardiac electrogram (it is ventricular myocardium) but atrial pressures (it lies above the tricuspid valve). The right atrium can be massively enlarged (Figure 20-5), and the right ventricle, lacking the inflow portion that is now part of the right atrium, is smaller than usual with varying degrees of pulmonic stenosis. Patients with l-transposition (see earlier) can have Ebstein’s anomaly of a left-sided tricuspid valve.
Figure 20-5 Ebstein’s anomaly of the tricuspid valve.
(From Baum VC: Abnormalities of the atrioventricular valves. In Lake CL, Booker P [eds]: Pediatric cardiac anesthesia, 4th ed. Philadelphia, 2004: Lippincott Williams & Wilkins.)
Symptoms will vary based on the amount of displacement of the valve and the size of the smaller than normal right ventricle. Cyanosis from atrial-level right-to-left shunting can be a neonatal phenomenon, resolving with the normal postnatal decrease in pulmonary vascular resistance, only to recur in adolescence or adulthood. Very mild disease is quite compatible with asymptomatic survival into adulthood, although overall earlier reports suggested a mean age at death of about 20 years, with about one third dying before 10 years of age and only 15% survival rate by 60 years.105–107 About half of patients experience development of arrhythmias, typically supraventricular tachyarrhythmias. Once symptoms develop, disability rapidly progresses.
Valve repair is currently the approach of choice, and only uncommonly will tricuspid valve replacement be required.108 Additional intervention may be required for progressive insufficiency, stenosis, prosthetic valve failure, or valve replacement with growth. After valve replacement, up to 25% of patients experience development of high-grade atrioventricular block. Ebstein’s valves are often associated with a right-sided bypass tract, causing Wolff-Parkinson-White syndrome. This is of concern because 25% to 30% of patients experience development of supraventricular tachyarrhythmias. The dilated right atrium is ready substrate for the development of atrial fibrillation. In addition to a decrease in cardiac performance, atrial fibrillation is also potentially dangerous in patients with underlying Wolff-Parkinson-White syndrome because very high atrial rates can be conducted to the ventricle through the bypass tract.
[/not-level-membership-for-anesthesiology-category]
