Chapter 15 Congenital Heart Disease in Adults
Since the first surgical ligation of a patent ductus arteriosus by Gross in 1938, enormous advances have been made in the repair and palliation of congenital heart disease. Excluding bicuspid aortic valve, approximately 0.9% of infants are born with congenital heart disease; the incidences of the major types of defect are listed in Table 15-1. As a result of improvements in medical and surgical treatment, the population of adults with congenital heart disease has grown such that it now exceeds the population of children with congenital heart disease in many parts of the developed world.1,2
Table 15-1 Incidence of Congenital Heart Disease
| Lesion | Incidence/1000 Live Births |
|---|---|
| Ventricular septal defect | 3.57±2.9 |
| Patent ductus arteriosus | 0.80±1.4 |
| Atrial septal defect | 0.94±1.0 |
| Atrioventricular septal defects | 0.35±0.16 |
| Pulmonary stenosis | 0.73±0.73 |
| Aortic stenosis | 0.40±0.54 |
| Coarctation of the aorta | 0.41±0.25 |
| Tetralogy of Fallot | 0.42±0.19 |
| d-Transposition of the heart arteries | 0.32±0.12 |
| Hypoplastic right heart | 0.22±0.20 |
| Tricuspid atresia | 0.08±0.05 |
| Ebstein anomaly | 0.11±0.14 |
| Pulmonary atresia | 0.13±0.12 |
| Hypoplastic left heart | 0.27±0.22 |
| Truncus arteriosus | 0.11±0.07 |
| Double-outlet right ventricle | 0.16±0.10 |
| Bicuspid aortic valve | 13.56±13.05 |
| All congenital heart disease (excluding bicuspid aortic valve) | 9.60±7.40 |
Adapted from Hoffman JI, Kaplan S: The incidence of congenital heart disease. J Am Coll Cardiol 39:1890-1900, 2002.
CLASSIFICATION OF CONGENITAL HEART DISEASE
Surgically corrected or palliated congenital heart disease may be classified on the basis of whether there is a two-ventricle or a single-ventricle circulation (Table 15-2). With a two-ventricle circulation, the right ventricle is usually the pulmonary pump and the left ventricle the systemic pump, but in certain situations (e.g., atrial baffle repairs for transposition of the great arteries), the right ventricle is the systemic pump. For some defects a two-ventricle repair is not possible, and a staged palliation is performed; it results in a functionally single-ventricle circulation. The final stage of palliation is a Fontan-type operation, as explained later.
Table 15-2 Congenital Cardiac Defects in Which a Functionally Normal Circulation May Be Obtained vs. Defects Managed by Fontan-type Palliation
| Biventricular Repair |
| Ventricular septal defect |
| Atrial septal defect |
| Atrioventricular canal defect |
| Tetralogy of Fallot |
| Transposition of the great arteries |
| Truncus arteriosis |
| Anomalous pulmonary venous drainage |
| Valvular/subvalvular/supravalvular aortic stenosis |
| Interrupted aortic arch |
| Coarctation of the aorta |
| Ebstein anomaly of the tricuspid valve |
| Fontan Operation |
| Tricuspid atresia |
| Hypoplastic left heart |
| Pulmonary atresia with intact ventricular septum |
| Double-outlet left ventricle |
| Heterotaxy syndromes |
Multisystem Disease
Children who have undergone repair or palliation for congenital heart disease have, on average, lower IQs than their peers and are more likely to have developmental disabilities.3,4 Many adults with congenital heart disease were cyanotic for long periods of time during their childhoods; they may have experienced periods of circulatory compromise and may have undergone cardiopulmonary bypass during the formative years of that technology. In addition, some patients have syndromes in which congenital heart disease and mental disability are associated (e.g., Down syndrome). Anxiety and depression are also relatively common and may be exacerbated by admission to the ICU.
Cyanotic Heart Disease
The presence of right-to-left intracardiac shunting increases the risk for cerebral embolic events. Until recently, it was accepted that polycythemia itself was a risk factor for stroke. This belief led to the widespread use of phlebotomy to control hematocrit, typically to less than 55%. More recently, the detrimental effects of phlebotomy on oxygen delivery and iron stores have been appreciated,5 and many centers now reserve phlebotomy for patients with symptomatic polycythemia. The increased red cell turnover associated with polycythemia predisposes to the development of gallstones and gout. The risk for gout is increased by concomitant renal dysfunction and the use of diuretics.
General Perioperative Considerations
Most surgeries performed for congenital heart disease in adults are reoperations, and in many cases cardiopulmonary bypass times are long. Conduits from previous operations may be located directly behind the sternum and can be damaged during sternotomy. Postoperative problems involving excessive bleeding, coagulopathy, myocardial stunning, and pulmonary hypertension are not uncommon. Chronic volume and pressure overload of the cardiac chambers, along with scarring from previous surgeries, predispose to the development of ventricular dysfunction and arrhythmias. Poor nutritional state, chronic hypoxemia, and low cardiac output contribute to poor wound healing and the occurrence of nosocomial infection. Adults with congenital heart disease are at increased risk for developing endocarditis (see Table 10-7) and require antibiotic prophylaxis prior to certain invasive procedures (see Endocarditis Prophylaxis in Chapter 10). If these patients become febrile it is essential to draw blood cultures prior to commencing antibiotic treatment.
SPECIFIC CONGENITAL HEART DISEASES
Tetralogy of Fallot
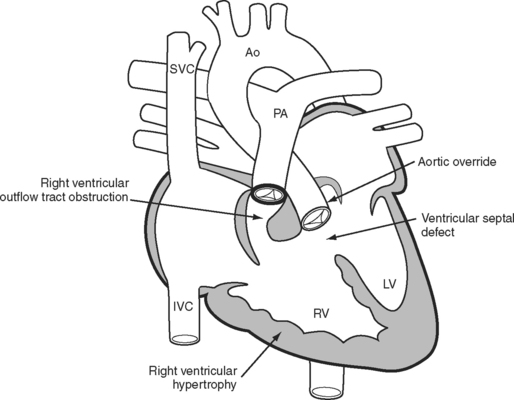
The essence of the tetralogy of Fallot (Fig. 15-1) is anterior displacement of the conal septum. This results in obstruction of the right ventricular outflow tract (RVOT) and a ventricular septal defect (VSD) and causes the aorta to override the crest of the ventricular septum. RVOT obstruction and right ventricular volume loading cause right ventricular hypertrophy, which is the fourth feature of the tetralogy. RVOT obstruction in the presence of a VSD causes a variable degree of right-to-left shunting and hypoxemia. Patients usually present in infancy with a murmur. If right ventricular outflow obstruction is severe, patients become cyanotic and may experience hypercyanotic “spells.”
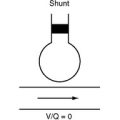
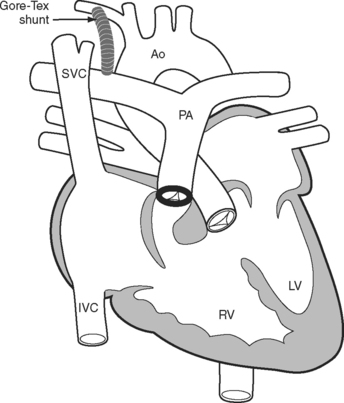
A tetralogy of Fallot was first palliated in 1944 by the classical Blalock-Taussig shunt. In this procedure, the subclavian artery is transected and anastomosed directly onto the pulmonary artery. Additional palliative strategies have included the Waterston and Potts shunts, in which the aorta (ascending or descending, respectively) is anastomosed side-to-side to a branch pulmonary artery. A modified version of the Blalock-Taussig shunt, in which a tube graft is used to connect the right subclavian artery to the right pulmonary artery (Fig. 15-2), is still performed to palliate lesions in which pulmonary blood flow is inadequate.
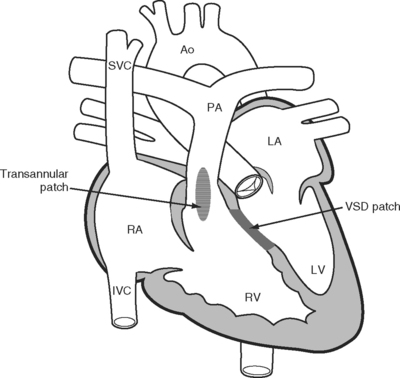
In the 1950s a more complete repair of the tetralogy of Fallot was developed. Repair involves patch closure of the VSD and reconstruction of the obstructed RVOT (Fig. 15-3); the latter is commonly achieved by placing a patch across the annulus of the pulmonary valve, rendering it incompetent. Patients usually undergo surgical correction in early childhood; depending on institutional practice and the presence and severity of cyanosis, it may have been preceded by a Blalock-Taussig shunt in infancy. Long-term survival rates are excellent. However, free pulmonary regurgitation causes right ventricular overload, which can eventually lead to right ventricular failure and ventricular arrhythmias. Patients who have undergone repair of a tetralogy of Fallot are at increased risk for sudden death, and those with ventricular tachyarrhythmias should be considered for internal cardiac defibrillators. Pulmonary valve replacement can prevent these problems, although there is disagreement about the optimal timing of surgery. Most centers use a combination of symptoms, measured exercise tolerance, and quantitative assessment of right ventricular function (usually by means of a magnetic resonance imaging scan) to establish the need for pulmonary valve replacement.
Preoperative Assessment for Pulmonary Valve Replacement
Patients typically have signs of pulmonary regurgitation and right ventricular dysfunction, including a diastolic murmur at the left upper sternal border, raised jugular venous pressure, hepatic congestion, and systemic edema. There may be systolic murmurs suggestive of residual RVOT obstruction or a patch margin VSD. Clinical examination and imaging should also assess any stenoses of branch pulmonary arteries, which are present in many adult patients with repaired tetralogy of Fallot, as consequences of a previous shunt or of the hemodynamic state created at the time of the childhood repair.
Postoperative Care Following Pulmonary Valve Replacement
Most patients have uneventful postoperative courses. Problems, if they do occur, usually relate to bleeding, arrhythmias, and right ventricular dysfunction. Arrhythmias include complete heart block, junctional ectopic tachycardia characterized by atrioventricular (AV) dissociation and rapid junctional rates, and ventricular ectopy. If the right atrium is dilated, atrial flutter and fibrillation may occur. Prophylactic amiodarone may be considered if there is a history of arrhythmias or if ventricular function is impaired. Preoperative antiarrhythmic medications should be reinstituted as early as possible postoperatively. Right ventricular dysfunction is manifested as low cardiac output or hypotension in association with elevated central venous pressure. There may be large V waves on the central venous pressure trace; they are indicative of tricuspid regurgitation. If right ventricular dysfunction is suspected, an echocardiogram should be obtained to confirm the diagnosis and rule out other causes of hemodynamic instability, particularly cardiac tamponade. The treatment of right ventricular dysfunction is described in Chapter 20.
Fontan Procedures
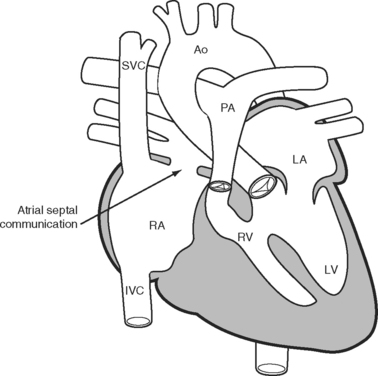
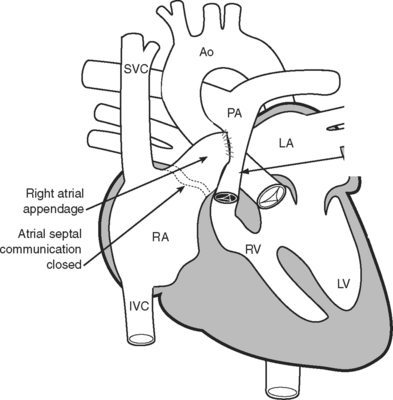
The classical Fontan operation, first described for tricuspid atresia (Fig. 15-4), involved connecting the right atrial appendage directly to the pulmonary artery and closing the interatrial communication (Fig. 15-5). This operation provides good initial palliation, but over time, right atrial dilatation develops, resulting in persistent atrial arrhythmias.6–8 Patients are also at risk for developing right atrial thrombi and pulmonary emboli which, in a circulation that relies on passive blood flow through the pulmonary circulation, are very poorly tolerated. Additionally, right atrial dilatation can cause compression of the right-sided pulmonary venous return, contributing to impaired cardiac function.
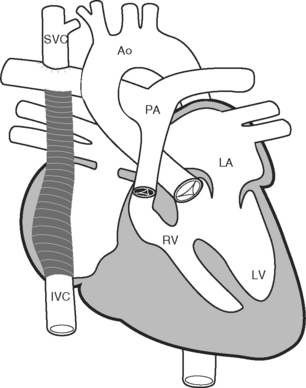
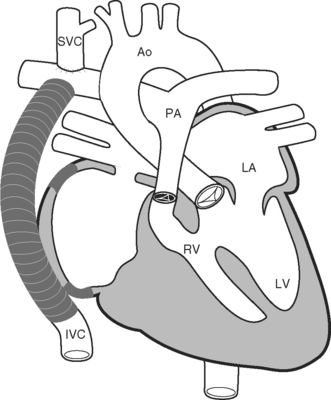
Because of these complications, a number of modifications to the classical Fontan operation have been introduced, including the lateral tunnel (Fig. 15-6) and extracardiac (Fig. 15-7) techniques, with the aim of streamlining venous return to the lung and minimizing atrial scarring. These modifications, in particular the extracardiac Fontan, have become the procedures of choice for children currently undergoing single-ventricle palliation.
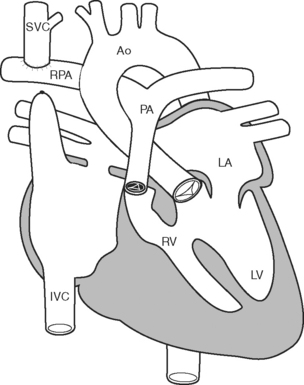
After this initial operation, most children then undergo a bidirectional Glenn (Fig. 15-8) or hemi-Fontan procedure at between 3 and 9 months of age. These procedures direct superior vena caval blood directly to the pulmonary arteries. Patients remain cyanotic following this procedure, with oxygen saturations usually in the mid 80s. The Fontan operation itself is usually performed in the preschool years.
Postoperative Issues
The vulnerabilities of single ventricle physiology may be exposed in the postoperative period. Without a pulmonary pumping chamber, pulmonary blood flow and left heart filling are critically dependent on adequate systemic venous pressure and on low pulmonary vascular resistance. Central venous pressure is a reflection of both systemic venous pressure and pulmonary artery pressure. The goal is to optimize cardiac output at the lowest possible central venous pressure.
Central venous and left atrial catheters are often put in place before the completion of surgery so as to enable postoperative monitoring. In the early postoperative period, central venous pressure is normally 14 to 18 mmHg, gradually settling to 12 to 16 mmHg over the first few days as pulmonary vascular resistance falls with recovery from surgery. The normal range for left atrial pressure is 6 to 10 mmHg. A central venous pressure greater than 18 to 20 mmHg or a transpulmonary gradient (see Chapter 1) more than 10 mmHg is never normal, and both readings demand further investigation.
Coarctation of the Aorta
Coarctation of the aorta most commonly occurs in proximity to the point of entry of the ductus arteriosus to the descending aorta (Fig. 15-9). The narrowing can be discrete or associated with aortic hypoplasia. Coarctation of the aorta can occur as an isolated abnormality or in association with congenital heart disease, most commonly bicuspid aortic valve. Severe coarctation usually presents in infancy but milder degrees can present for the first time in adulthood, usually with systemic hypertension. Patients can also develop recurrent coarctation at the site of a previous repair.
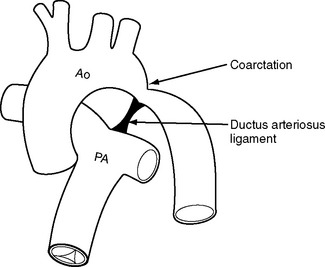
In infancy and childhood, most coarctations are repaired by end-to-end anastomosis. An alternative technique is the use of a subclavian flap, in which the subclavian artery is divided and turned down adjacent to the descending aorta to be used as a patch to bridge the narrowed segment. Patients who have undergone this procedure will have weak or absent pulses in the left arm. The use of Dacron interposition grafts or patch aortoplasty for repair in children is associated with the development of aortic aneurysms in later life. Aortic coarctation that presents in adulthood can usually be managed by percutaneous balloon dilation and stent placement, but occasionally open surgical repair is required.
Postoperative Care
Common problems after surgery include:
Secundum Atrial Septal Defects
Postoperative Care
Postoperative management is determined by the degree of right (and occasionally left) ventricular dysfunction and the severity of pulmonary hypertension. Many younger patients without these features can be extubated in the operating room or shortly after arriving in the ICU. Older patients may have considerable right ventricular dysfunction and are prone to atrial arrhythmias. Right ventricular volume overload can also cause left ventricular diastolic dysfunction (see Chapter 20). Following closure of the defect (and loss of left-to-right atrial shunting), left atrial pressure may transiently rise, causing pulmonary edema. Arrhythmias may occur due to chronic right atrial dilatation. Temporary or permanent sinus node dysfunction may occur, resulting in a nodal bradycardia. Pacing may be required. The likelihood of perioperative mortality is increased considerably by the presence of pulmonary hypertension. Treatments for right ventricular dysfunction and pulmonary hypertension are discussed in Chapters 20 and Chapter 24, respectively.
Transposition of the Great Arteries
Transposition of the great arteries is a condition in which the connections of the great arteries are reversed so that the aorta arises from the right ventricle and the pulmonary artery from the left ventricle. The current approach to surgical correction is the arterial switch procedure, which results in anatomically normal circulation. However, most adult patients with transposition have previously undergone palliative atrial switch procedures. They include the Senning and Mustard procedures (Fig. 15-10), in which baffles are inserted within the atria to redirect the pulmonary and systemic venous return. The systemic venous return is redirected to the anatomic left ventricle and then to the pulmonary artery; the pulmonary venous return is redirected to the anatomic right ventricle and then to the aorta. In this way, a functionally normal circulation is obtained, but the anatomic right ventricle remains as the systemic ventricle. These operations provide excellent initial palliation. However, over the longer term two important problems typically develop. First, the systemic right ventricle gradually fails, resulting in congestive cardiac failure; and second, atrial arrhythmias become a growing problem.
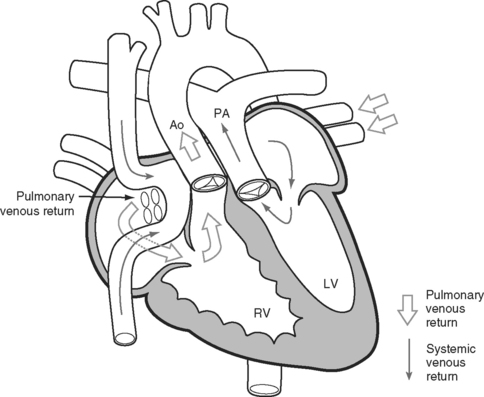
Some centers have performed late arterial switch, usually in the first decade or early teenage years, in patients who have undergone Senning or Mustard procedures.9 The main problem limiting a more general application of this approach is that the anatomic left ventricle, having been the pulmonary pump since birth, may be unable to function as the systemic ventricle following anatomic correction. Sequential pulmonary artery banding usually precedes arterial switch in an attempt to “train” the anatomic left ventricle to tolerate an increased afterload. The major postoperative problem is left ventricular failure.
Atrioventricular Canal Defects
Defects of the central portion of the heart are referred to as AV canal defects. They consist of a spectrum of diseases and may be categorized as partial, intermediate, or complete. Partial defects consist of an ostium primum ASD and a mitral cleft. Intermediate defects consist of an ostium primum ASD, a VSD, and separate, but abnormal, AV valves. Complete AV canal defects consist of an ostium primum ASD, a VSD, and a common AV valve. There is left-to-right shunting and AV valve regurgitation, which cause a variable degree of pulmonary hypertension and congestive heart failure. AV canal defects are usually repaired in childhood. Failure to achieve early repair of defects with a significant ventricular component can result in severe pulmonary hypertension and, potentially, Eisenmenger syndrome. Patients may present in late childhood or adulthood for AV valve repair or replacement and can have significant pulmonary hypertension and ventricular dysfunction.
Eisenmenger Syndrome
Patients with Eisenmenger syndrome occasionally require noncardiac surgery or intensive care treatment. The principles of management include avoiding acute increases in pulmonary vascular resistance and decreases in systemic vascular resistance and supporting right ventricular function. Abrupt increases in pulmonary vascular resistance, for instance due to hypercarbia or acidosis, cause profound hypoxemia and may precipitate acute right heart failure. Support of right ventricular function and control of pulmonary vascular resistance are discussed in Chapters 20 and Chapter 24, respectively.
1 Williams WG, Webb GD. The emerging adult population with congenital heart disease. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu. 2000;3:227-233.
2 Webb GD, Williams RG. Care of the adult with congenital heart disease: introduction. J Am Coll Cardiol. 2001;37:1166.
3 Mahle WT, Wernovsky G. Long-term developmental outcome of children with complex congenital heart disease. Clin Perinatol. 2001;28:235-247.
4 Bellinger DC, Wypij D, du Duplessis AJ, et al. Neurodevelopmental status at eight years in children with dextro-transposition of the great arteries: the Boston Circulatory Arrest Trial. J Thorac Cardiovasc Surg. 2003;126:1385-1396.
5 Thorne SA. Management of polycythaemia in adults with cyanotic congenital heart disease. Heart. 1998;79:315-316.
6 Gelatt M, Hamilton RM, McCrindle BW, et al. Risk factors for atrial tachyarrhythmias after the Fontan operation. J Am Coll Cardiol. 1994;24:1735-1741.
7 Fishberger SB, Wernovsky G, Gentles TL, et al. Factors that influence the development of atrial flutter after the Fontan operation. J Thorac Cardiovasc Surg. 1997;113:80-86.
8 Durongpisitkul K, Porter CJ, Cetta F, et al. Predictors of early- and late-onset supraventricular tachyarrhythmias after Fontan operation. Circulation. 1998;98:1099-1107.
9 Poirier NC, Yu JH, Brizard CP, et al. Long-term results of left ventricular reconditioning and anatomic correction for systemic right ventricular dysfunction after atrial switch procedures. J Thorac Cardiovasc Surg. 2004;127:975-981.
10 Hoffman JI, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol. 2002;39:1890-1900.