[level-membership-for-cardiovascular-category]
Chapter 10 Congenital Heart Disease
Over one million adults have congenital heart disease (CHD) in the United States. For the first time, there are now more adults living with CHD than children. The reported incidence of CHD varies widely because of differences in counting minor lesions at birth (such as bicuspid aortic valve, small ventricular septal defects, silent patent ductus arteriosus, and anomalous pulmonary veins), and increasingly better diagnostic methods. The incidence of severe CHD requiring surgery is about 3 per 1000 live births. More accurate diagnosis at presentation and improved medical, surgical, and postoperative care have greatly improved survival so that 90% of children born today with CHD will reach adulthood.
SEGMENTAL ANALYSIS OF CARDIAC MALFORMATIONS
The steps for the segmental diagnostic approach to cardiac malformations are:
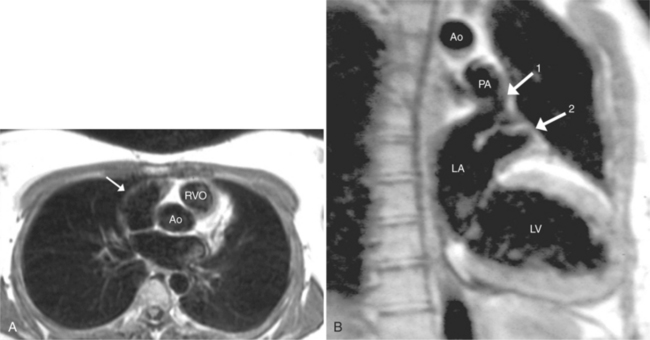
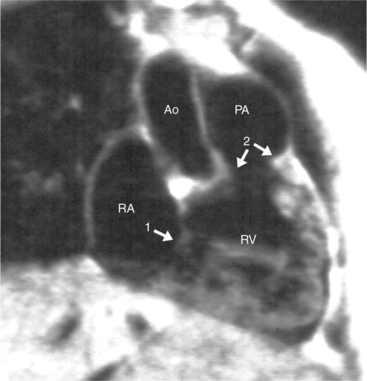
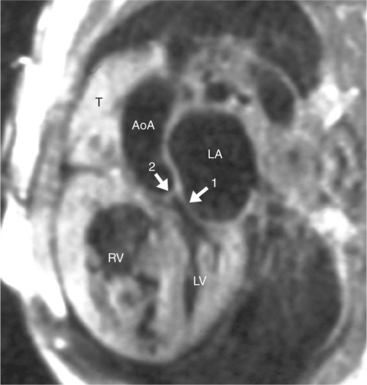
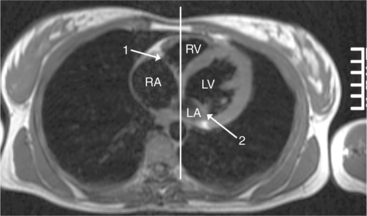
Cardiac Axis and Visceral Situs
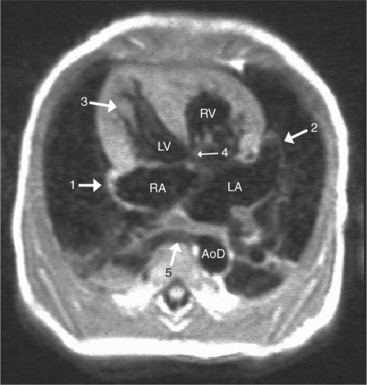
The situs is determined by chest and abdominal radiographs. Situs solitus and situs inversus are recognized by the asymmetry of the tracheobronchial tree and by the positions of the abdominal organs. Symmetrically lobed lungs (Fig. 10-10), midline liver, gastrointestinal malrotations, asplenia, and polysplenia denote the heterotaxy syndrome (Fig. 10-11), which can also be recognized by isomerism of the atria. Isomerism means that both atria have features of the right atrium or of the left atrium. The visceral-atrial rule is that the right and left atria develop on the same side as the thoracic and abdominal viscera do. In situs solitus, the right atrium is on the right side of the mediastinum and the left atrium is on the left side. In situs inversus, the morphologic right atrium is on the left side and the left atrium lies on the right side. In situs ambiguus, right and left sides cannot be determined because the lungs and abdomen are symmetric. For example, in asplenia there are two right (trilobed) lungs and both atria are morphologically right atria. In polysplenia there are two (bilobed) left lungs and both atria are morphologic left atria.
In most people, the major portion of the heart lies slightly to the left of midline. The cardiac apex denotes the location of the heart within the thorax. Dextrocardia (Fig. 10-12), levocardia (Fig. 10-13), and mesocardia then indicate the possible positions of the heart. Using this terminology, a cardiac malposition is any heart that does not have a leftward cardiac axis in situs solitus. A malposition includes dextrocardia in situs solitus and levocardia in situs inversus (Fig. 10-14), as well as dextrocardia in situs inversus. All these positions represent deviation from normal embryologic development without necessarily implying any hemodynamic or morphologic derangement.
In primary dextrocardia, the main defect is in the heart. There are two types of primary dextrocardia: (1) dextroversion, in which the heart is rotated or pivoted so that its apex lies on the right side with the atria as a fulcrum; and (2) mirror-image dextrocardia. In secondary dextrocardia, the heart is normal but the mediastinum is shifted to the right because of extracardiac abnormalities that involve the lungs, pleura, or skeleton (Box 10-1). Examples of the latter include pneumothorax, congenital herniation of the gastrointestinal tract into the thorax, and thoracolumbar scoliosis (Fig. 10-15).
Atrial Morphology
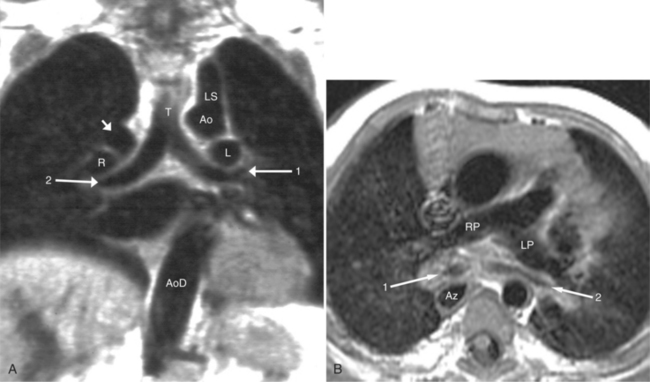
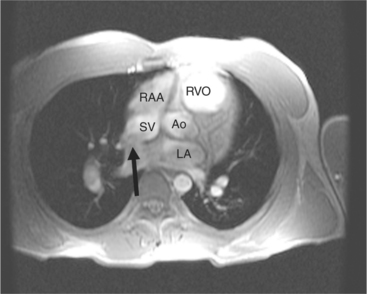
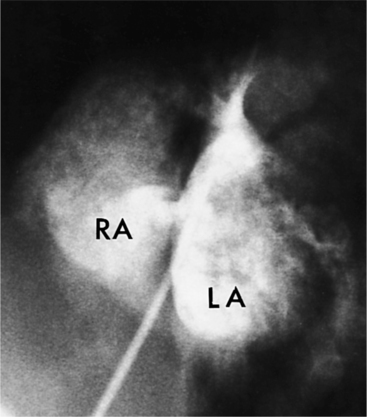
With rare exceptions, the morphology of the atria corresponds closely with the situs of the tracheobronchial tree and the abdominal viscera. Of the various criteria for distinguishing between right and left atria, the most reliable are the shape of the atrial appendage (see Figure 10-1) and the connection to the inferior vena cava (Box 10-2). The right atrial appendage is broad and pyramidal, whereas the left atrial appendage is thin with a narrow neck. The inferior vena cava almost always connects with the right atrium. This is true even in the “absence” of the inferior vena cava and azygos continuation. In this entity, there is no intrahepatic portion of the cava but the hepatic veins connect to the subdiaphragmatic portion of the inferior vena cava, which joins the right atrium.
Ventricular Morphology
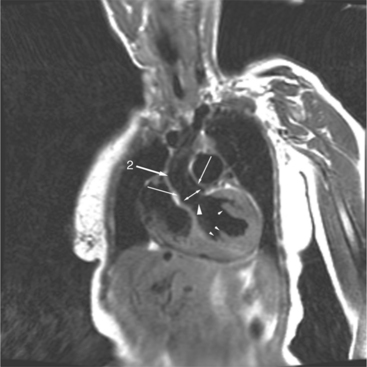
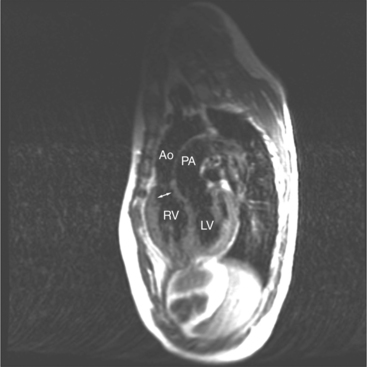
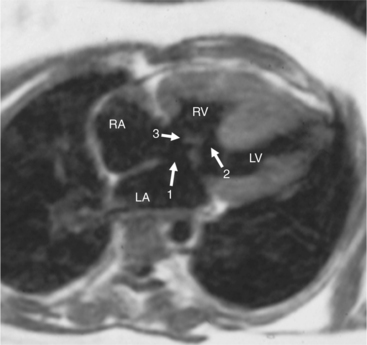
When the right and left ventricles are normal, identification of the two ventricles is relatively simple (Box 10-3). The normal right ventricle has coarse, trabeculated walls when compared with the smooth-walled left ventricle. The right ventricle has a contractile muscle called the conus or infundibulum between the tricuspid and pulmonary valves (see Figure 10-2), whereas the left ventricle has mitral-aortic continuity with no intervening muscle (Fig. 10-16). The right ventricle has trabeculations and papillary muscles on its septum, whereas in the left ventricle these structures are not present on the septum. A bicuspid (mitral) atrioventricular valve is a part of the left ventricle, whereas a tricuspid atrioventricular valve is part of the right ventricle, although either of these valves may have a cleft or be absent.
Box 10-3 Normal ventricles
| Right Ventricle | Left Ventricle |
|---|---|
| Coarse, trabeculated walls | Smooth walls |
| Contractile muscle (conus, infundibulum between tricuspid and pulmonary valves) | Mitral aortic continuity with no intervening muscle |
| Trabeculation and papillary muscles on septum | Septum free from trabeculations and papillary muscles |
| Tricuspid atrioventricular valve | Bicuspid (mitral) atrioventricular valve |
| Complex triangular shape | Spheroidal shape |
There is general agreement that an inflow tract must be present for a chamber to be considered a ventricle. The trabecular portion determines whether the chamber is of the right or left ventricular type. In these instances, the single ventricle consists of one large chamber that receives both atrioventricular valves. (Note that this definition excludes mitral or tricuspid atresia.) If only the trabecular and outflow segments are present, this structure is called an outlet chamber. Examples of such hearts are the univentricular heart of the left ventricular type, with or without a rudimentary outflow chamber.
Atrioventricular Connections
The atrioventricular connections are called concordant when the right atrium connects to the right ventricle (see Figure 10-4) and the left atrium connects to the left ventricle. When the right atrium connects to the left ventricle and the left atrium connects to the right ventricle, the ventricles are discordant in relation to the atria (see Figure 10-5). In the heterotaxy syndrome, in which either two right atria or two left atria may exist, the atrioventricular connection is ambiguous. This schema is less clear when either atresia of one of the atrioventricular valves exists or when one of the atrioventricular valves straddles the interventricular septum. When there is a double-inlet or a straddling atrioventricular valve, the tensor apparatus (the attachments of the chordae tendineae) may connect to either side of the interventricular septum.
Relations of the Great Arteries
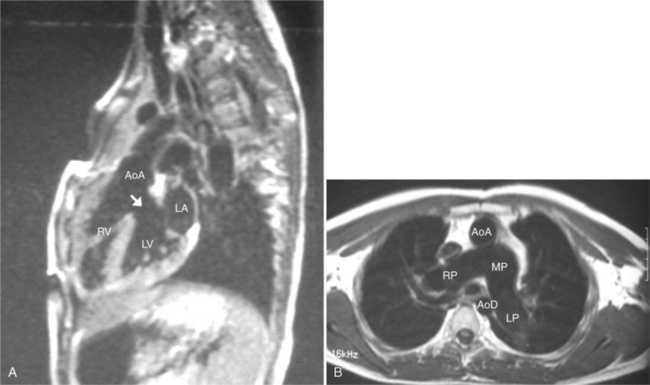
The position of the aorta is described relative to the pulmonary artery in both the anteroposterior and lateral planes (Fig. 10-17). In the normal heart, the aorta is to the right of and posterior to the pulmonary artery (see Figure 10-6). An anterior aorta to the right of the pulmonary artery is common in transposition of the great arteries (TGA; see Figures 10-7, 10-9). An aorta to the left of and anterior to the pulmonary artery is typical in, but not diagnostic of, corrected transposition (Fig. 10-18). Then there is a characteristic leftward convexity of the aorta (Fig. 10-19).
Ventriculoarterial Connections
The pulmonary valve is part of the pulmonary artery (not part of the right ventricle) and the aortic valve is part of the aorta. When the pulmonary artery or the aorta is related to, or overrides more than 50% of, a particular ventricle, it is defined as being connected to that ventricle. This association is particularly strong when there is a continuity between an atrioventricular valve and the semilunar valve. Concordant connections exist when the left ventricle is connected to the aorta and the right ventricle to the pulmonary artery (see Figures 10-2, 10-8). Discordant connections result when the left ventricle is connected to the pulmonary artery and the right ventricle to the aorta (see Figure 10-9). This latter connection is also called transposition. When both great arteries arise predominantly from one ventricle, there is a double-outlet right ventricle or double-outlet left ventricle. The final type of arterial connection is a single-outlet heart, of which there are three varieties:
The ventricular outflow tracts may be one of four distinct types:
Certain types of conus are associated with particular malformations:
Associated Malformations
Cardiotyping
At this point in the segmental analysis of the heart, the atrial and ventricular situs, the atrioventricular connections, the ventriculoarterial connections, and the position of the aorta are known. This data can be expressed in a notation developed by Van Praagh to categorize all possible types of hearts (Box 10-4). The situs of the atria, as indicated by the position of the abdominal viscera and the trachea and lungs, is designated as solitus (S), inversus (I), or ambiguus (A). The ventricular situs is characterized as a D-loop (D), L-loop (L), or undiagnosed loop (X). The position of the aorta in relation to the pulmonary valve is to the right (D), to the left (L), or directly anterior (A). These three letters are written in sequence (atrial situs, ventricular situs, and aortic situs). Examples are a normal heart (S,D,D), a dextrotransposition of the great arteries (S,D,D), and a congenitally corrected TGA or levotransposition (S,L,L) in which the D- and L- indicate the aortic position.
Box 10-4 Cardiotypes
DETERMINE ATRIAL SITUS BY ANALYZING ABDOMEN AND TRACHEOBRONCHIAL TREE
| S | solitus |
| I | inversus |
| A | ambiguus |
DETERMINE VENTRICULAR SITUS
| D | D-loop or solitus |
| L | L-loop or inverted |
| X | X-loop or undiagnosed |
DETERMINE AORTIC SITUS IN RELATIONS TO PULMONARY VALVE
| D | Normally related great arteries with aorta to the right of the pulmonary artery |
| L | Inverted great arteries with aorta to left of pulmonary artery |
| A | Aortic valve is directly anterior to pulmonary valve |
NOTATION (ATRIA, VENTRICLES, GREAT ARTERIES)
| Normal | (S,D,D) |
| D-transportation of great arteries | (S,D,D) |
| L-transportation of great arteries | (S,L,L) |
| Situs inversus | (I,L,L) |
| Asplenia, dextrocardia, transposition of great arteries | (A,L,L) |
MALPOSITIONS AND ABNORMAL CONNECTIONS
Cardiac Connections and Positions
Dextrocardia
Dextrocardia signifies that the apex of the heart is directed toward the right. Primary dextrocardia exists because of an embryologic abnormality. This type of dextrocardia can exist with any type of situs position (Fig. 10-21). When dextrocardia exists with situs inversus, the atrial and ventricular relations are a mirror image of their positions in the usual situs solitus. When the dextrocardia exists in situs solitus, the term isolated dextrocardia is frequently applied. It is clear then that dextrocardia can occur in situs solitus, inversus, and ambiguus. Many associated cardiac anomalies exist in primary dextrocardia. Frequent conditions include ventricular septal defect, TGA, corrected TGA, double-outlet right ventricle, and juxtaposition of the atrial appendages.
The goal of echocardiography, magnetic resonance imaging (MRI), and angiography is to define the position and location of each chamber of the heart and their connections and relations with one another and with the great arteries. In those malpositioned hearts in which the location of the interventricular septum is not known before angiography, posteroanterior and lateral projections serve as initial guidelines. Frequently, the projections can be reversed for a malposition; that is, those structures that are normally best seen in the left anterior oblique projection in the normal heart would be studied in the right anterior oblique projection in dextrocardia. As a rule, the dextrocardia itself does not cause clinical problems but rather the associated malformations mandate medical or surgical alleviation.
Levocardia
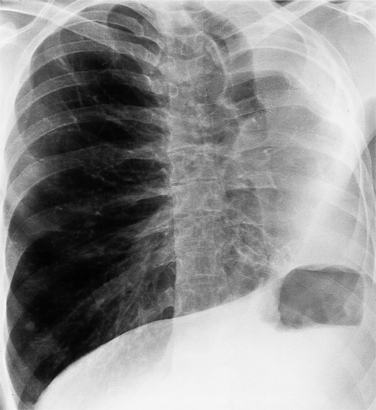
Strictly speaking, levocardia means that the cardiac apex is left sided. Isolated levocardias are those hearts that are left sided when situs inversus is present. This anomaly occurs in less than 1% of all patients with congenital cardiac malformation compared with a 2% incidence of dextrocardia in patients with CHD. With levocardia, the position of the thoracic and abdominal organs ranges from partial to complete situs inversus and also to heterotaxy (Fig. 10-22). Severe malformations are always associated with levocardia and frequently include ventricular septal defect, complete atrioventricular canal defects, and pulmonary stenosis or atresia. Isolated levocardia may be suspected on the chest film with a right-sided stomach bubble and left-sided liver shadow and a left cardiac apex. In contrast, in extrinsic levocardia the heart is intrinsically normal but the mediastinum is shifted from skeletal or pulmonary abnormalities (Fig. 10-23).
Heterotaxy and the Syndromes of Asplenia and Polysplenia
Heterotaxy is the failure of the developing embryo to establish normal left-right asymmetry. Asplenia and polysplenia are part of this spectrum. In these patients the thoracic and abdominal contents have a degree of symmetry, unlike those in situs solitus or inversus in which right- and left-sided organs exist together. In the thorax, both lungs may be trilobed with bilateral epiarterial bronchi, or both lungs may be bilobed with bilateral hypoarterial bronchi. In the abdomen, the asymmetry is also frequently lost. The liver may be midline. The attachment of the mesentery, which usually runs from the left upper quadrant to the right lower quadrant, may have a midline attachment. The spleen may be absent (asplenia), bilobed with multiple accessory spleens, or multiple small spleens (Fig. 10-24) may be found throughout the mesentery (polysplenia). Situs ambiguus exists either when the right and left sides of the lungs, heart, and abdomen are similar or where a right-left relationship is difficult to identify. Boxes 10-5 and 10-6 summarize the characteristics of asplenia and polysplenia.)
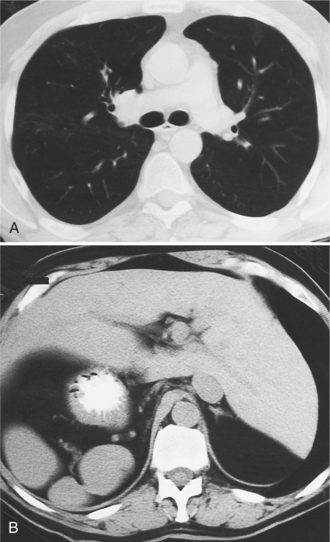
Splenic anomalies with malpositions and malformations in multiple organ systems have been recognized since 1826 when Martin and later Ivemark described the absence of the spleen in cyanotic CHD. Complex cardiac malformations are typical when the type of thoracic and abdominal situs abnormality is uncertain or has features of both situs solitus and situs inversus (Table 10-1).
TABLE 10-1 Cardiovascular abnormalities in asplenia and polysplenia
| Abnormality | Asplenia (%) | Polysplenia (%) |
|---|---|---|
| SUPERIOR VENA CAVA | ||
| Bilateral | 53 | 33 |
| Right | 34 | 33 |
| Left | 10 | 33 |
| Uncertain | 3 | — |
| INFERIOR VENA CAVA | ||
| Right sided | 60 | — |
| Left sided | 28 | — |
| Uncertain | 12 | — |
| Azygos continuation | — | 84 |
| Anomalous pulmonary veins | 84 | 50 |
| Total anomalous connection | 72 | — |
| Partial anomalous connection | 12 | — |
| CARDIAC APEX | ||
| Left | 56 | 58 |
| Right | 41 | 42 |
| Uncertain | 3 | — |
| AORTIC ARCH | ||
| Left | 56 | 33 |
| Right | 38 | 67 |
| Unknown | 6 | — |
| Great vessels | ||
| Normally related | 19 | 84 |
| Transposition of great arteries | 72 | 8 |
| Double-outlet right ventricle | 9 | 8 |
| PULMONARY VALVE | ||
| Normal | 22 | 58 |
| Stenosis | 34 | 33 |
| Atresia | 44 | 9 |
| Patent ductus arteriosus | 56 | 50 |
| Absent coronary sinus | 85 | 42 |
| Single ventricle | 44 | 8 |
| Ventricular septal defects | 90* | 67 |
* Of the ventricular septal defects in asplenia, 84% were of the atrioventricular canal type.
Modified from Rose V, Izukawa T, Moes CAF: Syndromes of asplenia and polysplenia; a review of cardiac and non-cardiac malformations in 60 cases with special reference to diagnosis and prognosis. Br Heart J 37:840-852, 1975.
Segmental analysis of the defects in hearts associated with asplenia begins with the atria and the atrial septum. On the chest film, the external contours of the heart frequently do not conform to the expected heart chambers (Fig. 10-25). Almost all these hearts show a common atrioventricular valve, frequently associated with separate, large atrial septal defects in the primum and secundum location. The size and location of these atrial defects are such that the malformation is called a common atrium. The ventricles also almost invariably have major malformations. About one fourth of the ventricles are inverted (as seen in corrected transposition), and half of the hearts have a univentricular chamber with a rudimentary outflow tract.
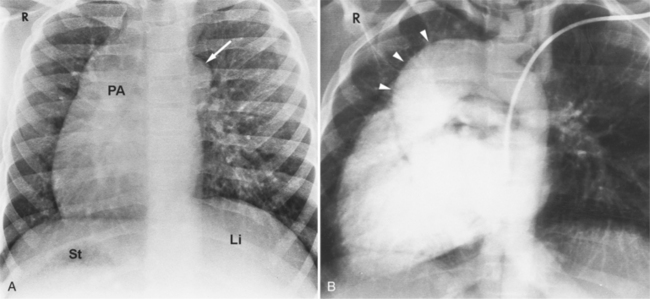
Anomalies of the great vessels, including TGA and double-outlet right ventricle, have an incidence of 3% to 30%. Angiographically, the posteroanterior and lateral projections are best to allow identification of their right-left relationships. Anomalies in the ventricular septum and semilunar valve stenosis are common, so that filming is also done with the x-ray beam parallel to the interventricular septum with cranial angulation. Because two thirds of persons with asplenia have anomalous systemic venous or pulmonary venous connections, or both, these malformations frequently complicate catheterization.
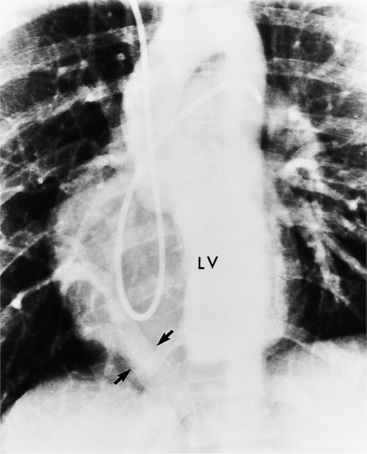
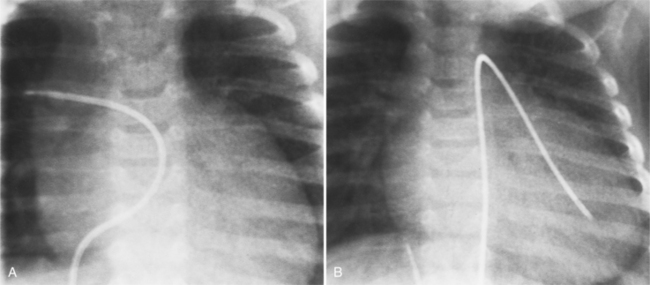
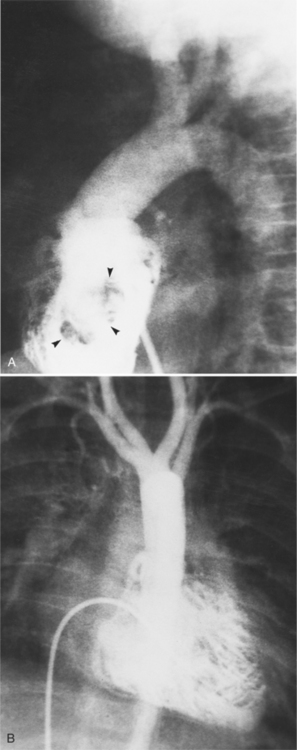
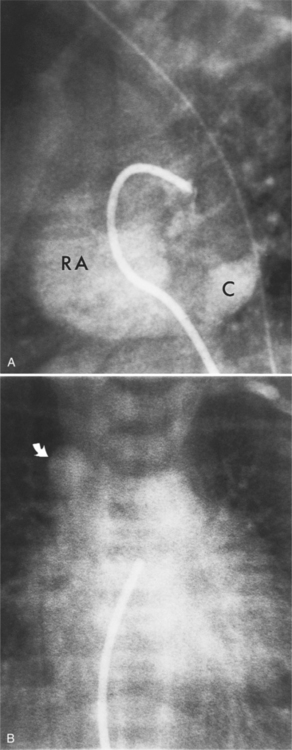
The features of the heart in polysplenia are quite variable, and in fact, there is occasionally no cardiac malformation. With a femoral vein approach, you can recognize azygos continuation by the course of the catheter around the azygos arch (Figures 10-10A, 10-26). You should not make the diagnosis of tricuspid atresia if the catheter tip fails to pass leftward through the heart above the diaphragm but instead should continue advancing the catheter superiorly until it goes around the azygos arch.
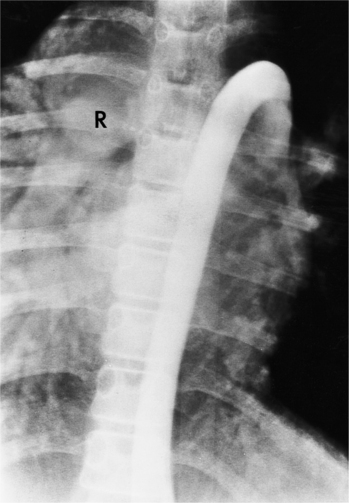
Atrioventricular Discordance
In the schema of segmental cardiac analysis, after recognition of the atria and ventricles, the next step is to determine whether the connections between them are concordant or discordant. Atrioventricular discordance means that the right atrium is connected to the left ventricle and the left atrium is connected to the right ventricle (see Figures 10-4, 10-5). Implicit in this definition is the presence of two atria, two atrioventricular valves, and two ventricles. This diagnosis is not appropriate when atrial identification is indeterminate in situs ambiguus or when there is a single common atrioventricular valve. Similarly, distinct right and left ventricles are necessary for this definition, although a ventricular septal defect may exist.
Congenitally Corrected Transposition of the Great Vessels (Levotransposition of the Great Arteries)
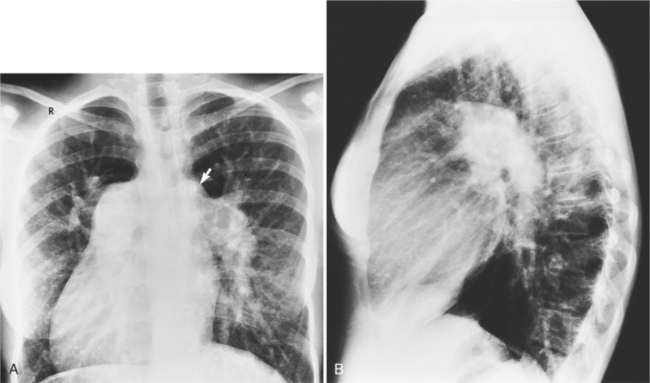
In 1875 Rokitansky reported a form of transposition in which blood passed in normal serial fashion through the pulmonary and systemic circuits. The right atrium was connected to the left ventricle, which was connected to the pulmonary artery. On the oxygenated side of the lungs, the left atrium was connected to the right ventricle, which was connected to the aorta. The atrioventricular valves always correspond with their ventricles, even when there is atrioventricular discordance. That is, the mitral valve is a left ventricular structure, and the tricuspid valve is a right ventricular structure. In congenitally corrected transposition of the great vessels, the aorta lies to the left of and anterior to the pulmonary artery, whereas the pulmonary valve lies to the right and posterior. The aortic valve is usually somewhat anterior to the pulmonary valve, although the two great vessels may be exactly lateral to each other. The ascending aorta frequently has an unusual course, passing in a direction toward the left shoulder so that occasionally a distinctive contour in the left side of the mediastinum is visible on the chest film.
If there are no other defects, this malformation causes no hemodynamic problems and may go undetected during a normal life span. Unfortunately, associated malformations are the rule, and their site and severity determine the clinical course. Ventricular septal defects are frequent (Figures 10-27, 10-28) and may be large enough to cause pulmonary arterial hypertension. These defects are usually in the membranous septum adjacent to the pulmonary valve; muscular defects and supracristal defects are less common. Generally, the left-sided atrioventricular valve (i.e., the valve between the left atrium and the right ventricle) is displaced slightly into the ventricle in a manner resembling Ebstein anomaly. If the displacement is more than a few millimeters (because the tricuspid valve is usually displaced to the apex by that amount), the diagnosis of Ebstein anomaly is quite likely. Pulmonary stenosis is frequently associated with ventricular septal defect and may be caused by a malformed valve, a subpulmonary membrane, aneurysms of the membranous ventricular septum, or rarely, accessory tissue in the atrioventricular valve or a muscular bar in the subpulmonary region.
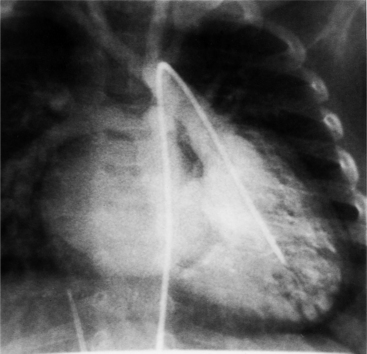
Imaging establishes the atrioventricular connections, the morphology of the ventricles, and the position of the aorta and pulmonary artery. The position of the venous and arterial catheters frequently give the first clue to a corrected transposition (Fig. 10-29). In situs solitus and levocardia, the venous catheter passes through the heart in the midline to reach the pulmonary arteries. The catheter in the pulmonary artery is posterior to its usual location, which is where the aorta should be in normal hearts. The retrograde arterial catheter has a distinctive curve in the ascending aorta as its course becomes convex medially and to the left before entering the heart (see Figure 10-18). On the lateral view, the aortic catheter is anterior and superior to the venous catheter. The venous and arterial catheters indicate the fundamental relationship between the aorta and the pulmonary artery in corrected transposition with situs solitus and levocardia; the pulmonary artery lies to the right and posterior, whereas the aorta is anterior and to the left.
In the sagittal view, the left ventricle appears to “stand on its apex” with a conical shape whose apex is in the diaphragmatic-sternal angle. The anterior wall of the left ventricle extends superiorly into a distinctive pouch that is characteristic of inverted ventricles, namely the anteriorly placed left ventricle (Fig. 10-30). This recess is separate from both the mitral and pulmonary valves and is the most anterior and superior structure of either ventricle. The outflow portion of the left ventricle in the lateral projection is posterior and connects to a pulmonary artery, which is beside or posterior to the aorta. The posterior wall of the left ventricle beneath the pulmonary valve is the membranous septum, and the anterior wall forms a neck above the blind recess and below the pulmonary valve.
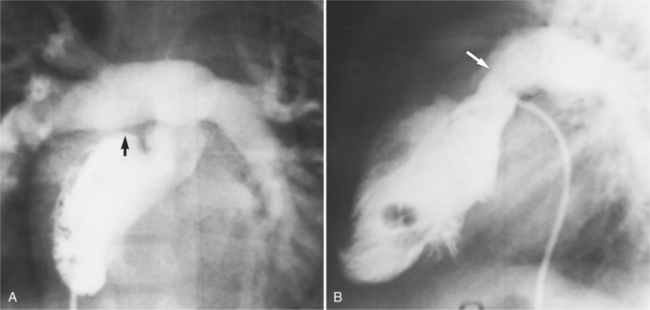
In the frontal view, the right ventricle is to the left of and slightly superior to the left ventricle (Fig. 10-31). In this position, the right ventricle has an oval to triangular shape and the usual coarse trabeculations. The tricuspid annulus is in the posteroanterior plane separated from the aortic valve by the muscular infundibulum. This morphologic right ventricle connects with the left atrium. The crista supraventricularis in this projection is the medial wall of the infundibulum above the tricuspid valve. In the lateral projection, the crista is the posterior wall of the infundibulum and separates the tricuspid from the aortic valve.
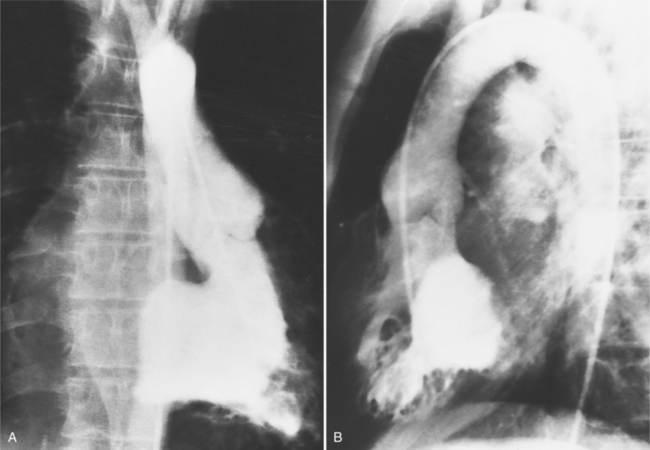
About one third of patients with corrected transposition have tricuspid regurgitation (i.e., from the right ventricle connected to the left atrium). The apically displaced tricuspid leaflets of Ebstein anomaly are usually the cause of the regurgitation, but there are occasionally other leaflet abnormalities. When there is severe regurgitation in infants, the details of the leaflets and the origin of their insertion are frequently difficult to identify. If technical factors such as arrhythmia and catheter position can be excluded, it should be presumed that severe regurgitation into the left atrium is associated with a “left-sided” Ebstein anomaly (Fig. 10-32). The tricuspid annulus is adjacent to the right coronary artery, which may be opacified during the ventriculogram.
Coronary Artery Patterns
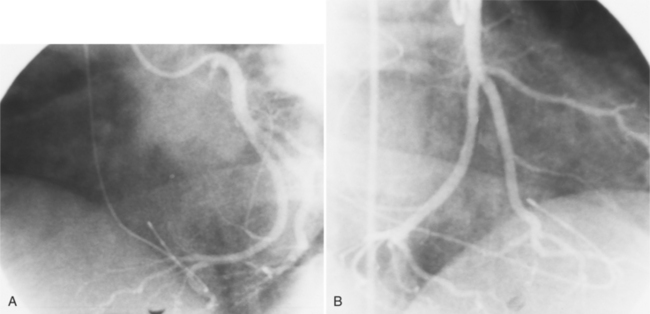
The coronary anatomy in congenitally corrected transposition of the great vessels is unique to inverted ventricles. The right coronary artery supplies the morphologic right ventricle and the left coronary artery provides an anterior descending branch in the interventricular sulcus and a variable circumflex branch over the morphologic left ventricle (Fig. 10-33). In congenitally corrected TGA, the right coronary artery passes to the left and inferior in the atrioventricular groove between the left atrium and right ventricle. Distally, this artery branches into the atrioventricular nodal branch, the posterior descending artery, and a variable set of branches to the inferior portion of the left ventricle (Fig. 10-34). The marginal branches over the right ventricular epicardial surface tend to be large with numerous branches. In contrast, the left coronary artery lies anterior and to the right of the right coronary artery. The left main coronary artery continues mainly as the anterior descending branch, which has numerous septal and diagonal branches. The circumflex artery in the atrioventricular groove between the right atrium and left ventricle tends to be vestigial. The position of the coronary arteries within the thorax may be different because of dextrocardia or other relative rotations, but the coronary distribution corresponds uniquely to the respective ventricle. When confusing ventricular morphology does not allow identification of the right or left ventricle, visualization of the coronary arteries permits accurate identification of the ventricles.
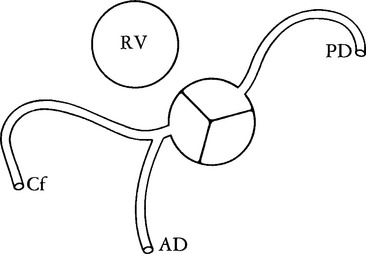
Isolated Ventricular Inversion
Atrioventricular discordance with ventriculoarterial concordance is termed isolated ventricular inversion. The segmental connections for the venous side of the heart are right atrium to left ventricle to aorta, and for the systemic side left atrium to right ventricle to pulmonary artery. As originally reported by the Van Praaghs (1966), the pulmonary artery arises anteriorly and to the left of the aorta. A large subaortic ventricular septal defect is adjacent to the septal leaf of the tricuspid valve. The aorta may also originate anterior to the pulmonary artery.
Ventriculoarterial Discordance
Complete Dextrotransposition of the Great Arteries
In 1797 Baillie described the heart of an infant in which the aorta connected to the right ventricle and the pulmonary artery to the left ventricle. The term transposition of the aorta and pulmonary artery is ascribed to Farre in 1814. Since that time, there has been controversy about whether it should be defined by the abnormal anteroposterior position of the great arteries or by the abnormal connections to the ventricles. TGA is a ventriculoarterial abnormality in which the aorta originates above the right ventricle and the pulmonary artery originates over the left ventricle.
Ventricular septal defects occur in about one third of babies with transposition (see Figure 10-7A), and when present, may result in congestive heart failure from the large blood flow. Extracardiac shunts may occur, as in patent ductus arteriosus or with bronchopulmonary connections to the pulmonary vascular bed. The ductus arteriosus remains patent in one fourth to one half of infants who do not receive prostaglandin E1 and allows blood to flow from the pulmonary artery to the aorta if the pulmonary vascular resistance is high and from the aorta to the pulmonary artery when the high fetal pulmonary artery pressures fall below the systemic blood pressure.
Besides the ventricular septal defect, the other major associated malformation is obstruction to blood entering the pulmonary arteries. About one fourth of those with TGA have some form of pulmonary stenosis (Fig. 10-35). The site of obstruction is usually in the subpulmonary region and it has a variety of causes:
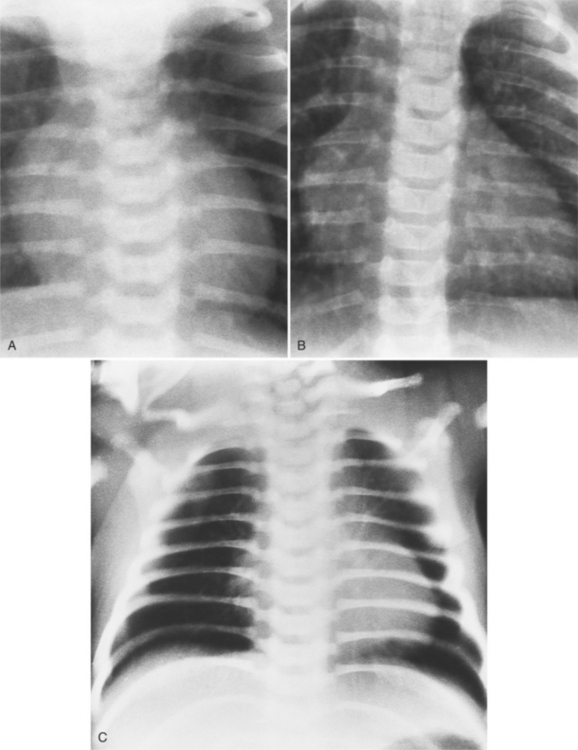
The typical chest radiograph in the first few days of life of a child with TGA shows mild cardiomegaly, a narrow superior mediastinum, and increased size of the pulmonary vessels that is consistent with a left-to-right shunt. All these signs are variable, and in fact, the chest radiograph may appear normal. The size of the pulmonary vessels depends on the amount of blood carried by them: If the degree of mixing between the pulmonary and systemic circuits is small or if there is some type of pulmonary stenosis, the pulmonary vessels will be small; if there are large intracardiac or extracardiac shunts, the pulmonary vessels are large (Fig. 10-36).
Imaging Anatomy
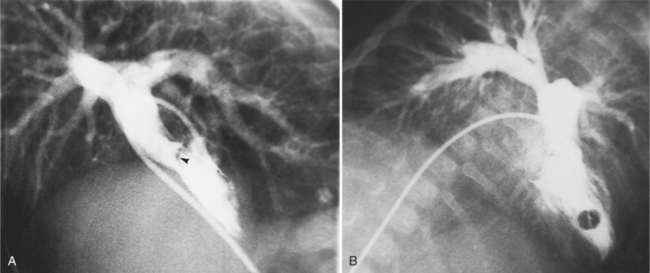
Complete TGA is usually diagnosed by echocardiography (Chapter 2). If angiography is performed, standard right and left ventriculography demonstrates the typical coarsely trabeculated body and cylindrical infundibulum, which connects to the aortic valve and aorta. A catheter passed through the right atrium and right ventricle and around the aortic arch has a distinctive shape (Fig. 10-37). The right ventriculogram demonstrates that the aorta is medial and anterior to the pulmonary artery (Fig. 10-38). In contrast to the normal heart, the plane of the aortic valve is parallel to the long axis of the body. The subaortic conus elevates the position of the aortic valve superior to the pulmonary valve and creates the “untucked” aorta.
In complete transposition, left ventriculography locates a posterior left ventricle, which connects directly to the pulmonary valve and main pulmonary artery (Figures 10-9, 10-39). There is no subpulmonary conus, that is, the mitral valve lies adjacent to the pulmonary valve. The interventricular septum usually lies parallel to the anterior chest wall. In hearts with normal pulmonary artery pressures, the interventricular septum may appear concave to the left ventricle, reflecting the systemic pressures in the right ventricle. In the cranial left anterior oblique projection, ventricular septal defects are visible with streaming of contrast material into the right ventricle.
Development of left ventricular outflow tract obstruction with or without ventricular septal defects is best evaluated with oblique cranially angled views. The upper aspect of the ventricular septum may protrude dynamically into the subpulmonary region during systole. If the mitral leaflets are drawn toward the septum during systole, similar to the same motion seen in idiopathic hypertrophic subaortic stenosis, subpulmonary obstruction develops with an increase in the left ventricular afterload (Fig. 10-40). A fixed form of subpulmonary stenosis is the fibromuscular ridge or membrane, visible as an irregular, radiolucent line on the interventricular septum. This ridge may be more visible during systole when the subpulmonary region is contracted.
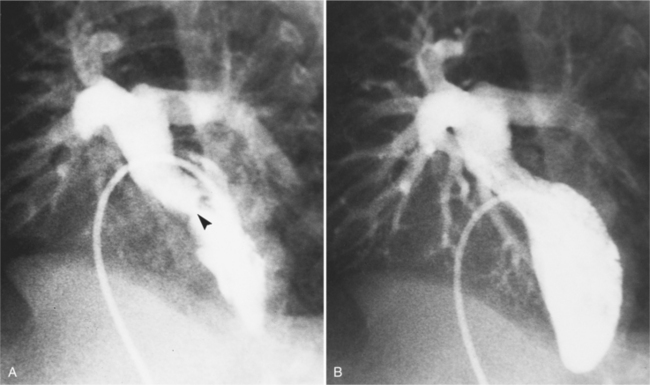
Associated malformations, such as coarctation, other aortic arch anomalies, and patent ductus arteriosus, are frequently detected in this way when they are not recognized by the hemodynamic measurements (Fig. 10-41).
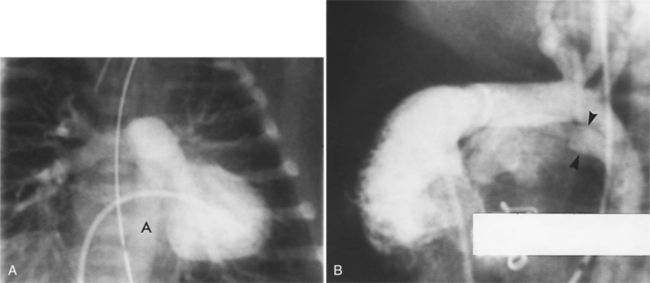
D-Transposition of the Great Arteries
In D-transposition of the great arteries (D-TGA), the aorta arises from the right ventricle and the pulmonary artery arises from the left ventricle. The blood circulates through two parallel circuits, and mixing of blood is necessary to get oxygenated blood to the systemic circulation. Up to 45% of patients can have a ventricular septal defect for mixing. When the ventricular septum is intact, mixing must occur through a patent foramen ovale or a PDA, and if these communications are inadequate, an emergent balloon atrial septostomy is performed. There are four different approaches to the repair of D-TGA which either correct the circulation physiologically (Mustard and Senning operations) or anatomically (Jatene and Rastelli operations; Table 10-2).
TABLE 10-2 Surgical repair of D-transposition of the great arteries
| Procedure | Description |
|---|---|
| PHYSIOLOGIC CORRECTION OR ATRIAL SWITCH | Creation of intracardiac baffle to “switch” the flow of blood at the inflow level |
| • Senning | Baffle created from right atrial wall and septal tissue |
| • Mustard | Atrial septum excised and baffle created from pericardium or synthetic material |
| ANATOMIC CORRECTION OR ARTERIAL SWITCH | |
| • Jatene | Switch great arteries (Lecompte maneuver), transfer coronaries and close ventricular septal defect |
| • Rastelli | Used for patients with pulmonary (left ventricle) outflow tract obstruction and ventricular septal defect |
| Intracardiac baffle from left ventricle to aorta via ventricular septal defect | |
| Right-ventricle-to-pulmonary-artery conduit | |
Beginning in the 1960s, the Mustard and Senning operations were the first definitive approaches for repair of TGA. Both operations correct the abnormal blood flow of the transposed vessels by switching the blood at the inflow (atrial) level. An intraatrial baffle composed of atrial tissue (Senning; Fig. 10-42) or pericardium or synthetic tissue (Mustard) directs the pulmonary venous return into the right ventricle and the systemic venous return into the left ventricle. Although this does correct the physiologic abnormality of TGA with deoxygenated blood now flowing to the pulmonary circulation and oxygenated blood flowing to the systemic circulation, the left ventricle remains as the pulmonary ventricle and the right ventricle as the systemic ventricle. The Senning and Mustard operations were initially well tolerated with relatively few complications. Late follow-up has shown increased incidence of complications requiring reoperation, exercise intolerance, arrhythmias, and sudden death. There continues to be great concern about the ability of the anatomic right ventricle to sustain the systemic circulation in the long term, and many patients have moderate to severe right ventricular dysfunction 25 years after repair.
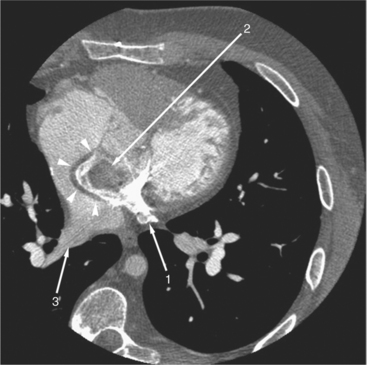
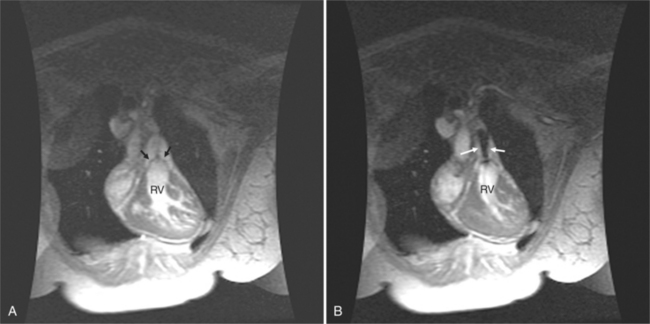
Cardiac magnetic resonance (MR) imaging is now used as the primary imaging for follow-up of patients after the atrial switch operation (Fig. 10-43). In addition to assessing right ventricular volumes and function, detection of baffle leaks and quantification of tricuspid regurgitation is performed. These are important volume-loading lesions that contribute to impaired right ventricular function and can be treated with transcatheter or surgical intervention. Cardiac MR can detect obstruction to either the systemic or pulmonary venous pathways with better accuracy as compared with echocardiography.
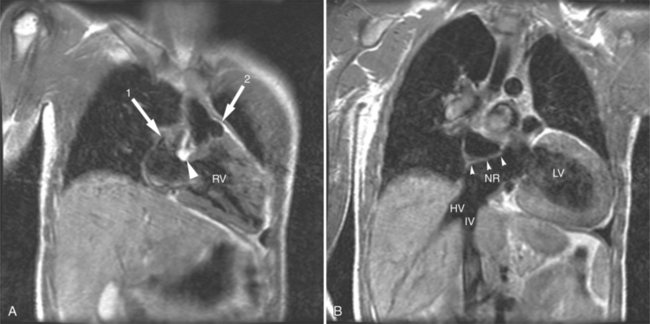
When patients with D-TGA have a ventricular septal defect and left ventricular outflow tract obstruction (usually from a sub pulmonary conus), the Rastelli operation is performed. An intracardiac baffle is created to divert blood flow from the left ventricle via the ventricular septal defect to the aorta (Fig. 10-44). The pulmonary valve is over sewn, and a right-ventricle-topulmonary-artery conduit is placed. Although it is advantageous to have the left ventricle as the systemic ventricle, patients will usually require future operations for replacement of the right-ventricle-to-pulmonary-artery conduit.
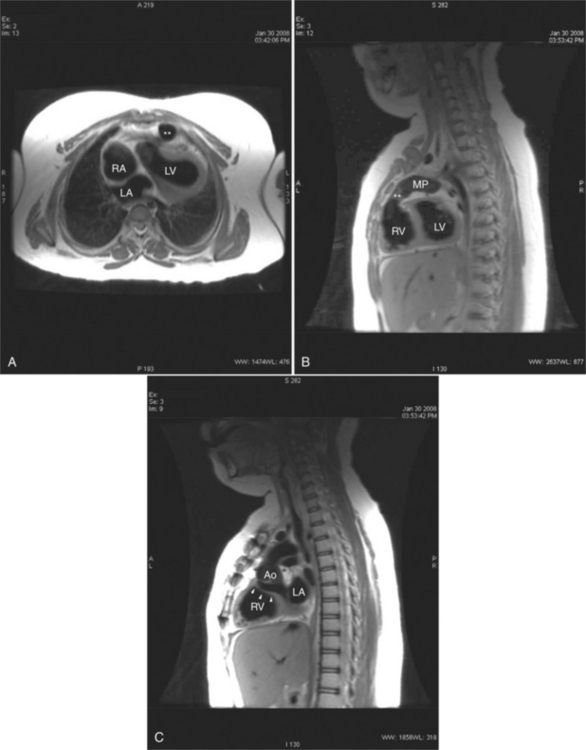
Following the Rastelli operation for D-TGA, there are long-term complications similar to those occurring after the atrial switch operations, including left and right ventricular dysfunction, arrhythmias, and sudden death (Table 10-3). In addition to assessing ventricular function, the cardiac MRI examination should include evaluation for intracardiac baffle obstruction or leak, and right-ventricle-to-pulmonary-artery conduit obstruction and regurgitation.
TABLE 10-3 Complications after repair of D-transposition of the great arteries
| ATRIAL SWITCH OPERATION |
| Pulmonary or systemic venous pathway obstruction |
| Intraatrial baffle leaks |
| Tricuspid valve regurgitation |
| Right ventricular systolic and diastolic dysfunction |
| RASTELLI OPERATION |
| Intracardiac baffle (left ventricular outflow tract) obstruction |
| Intracardiac baffle leak |
| Aortic regurgitation |
| Right-ventricle-to-pulmonary-artery conduit stenosis and regurgitation |
| ARTERIAL SWITCH OPERATION |
| Anastomotic stenosis main pulmonary artery |
| Anastomotic stenosis ascending aorta |
| Neoaortic regurgitation |
| Branch pulmonary artery stenosis |
| Coronary artery stenosis |
In 1975, Jatene described the first successful arterial switch operation (ASO) for D-TGA, which has now replaced the atrial switch procedures. The operation includes transection of the aorta and pulmonary artery above the level of the valve sinuses and transfer of the coronary arteries to the neoaortic root. The main pulmonary artery is then moved anterior to the aorta (the Lecompte maneuver) and the great arteries are sutured in place. The postoperative appearance of the great arteries is characteristic, with the main pulmonary artery located anteriorly and the pulmonary arteries “draped” to either side of the ascending aorta (Fig. 10-45). One of the most important aspects of the preoperative evaluation before the ASO is identification of the coronary artery anatomy by echocardiography. Although the overall perioperative mortality is low, there are certain coronary artery patterns that are associated with early mortality and need for early repair.
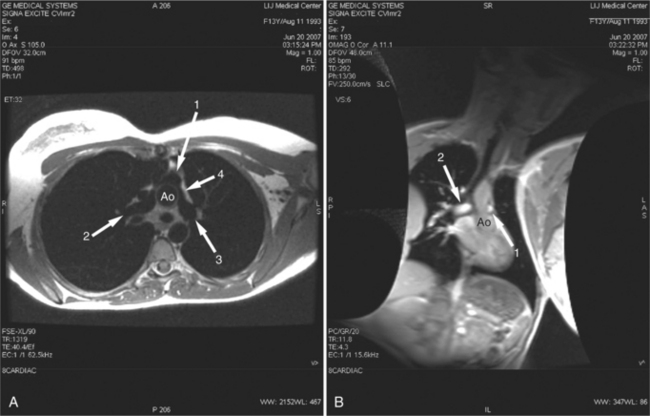
Partial Transposition of the Great Arteries
Double-Outlet Right Ventricle
Double-outlet right ventricle is a congenital cardiac defect in which both great arteries originate exclusively from the morphologic right ventricle. The only outlet for the left ventricle is through the ventricular septal defect, which is usually large but may be restrictive. The malformation occurs in two varieties, depending on the location of the ventricular septal defect in relation to the aorta and pulmonary artery. In the more common type, the ventricular septal defect is adjacent to the aorta with the pulmonary outflow situated on the far side of the right ventricle. In the less common type, the Taussig-Bing heart (Fig. 10-46), the ventricular septal defect is adjacent to the pulmonary outflow, whereas the aorta arises on the far side of the right ventricle. The aorta is usually to the right of the pulmonary artery, either slightly to the front or directly to the side, but it may arise in front of the pulmonary artery. Although there are rare exceptions, bilateral conus is a major criterion for distinguishing double-outlet right ventricle from tetralogy of Fallot or complete transposition of the great arteries. Bilateral conus is recognizable by muscle between each atrioventricular valve and the semilunar valves. These features place the aortic and pulmonary valves on approximately the same level in the transverse plane.
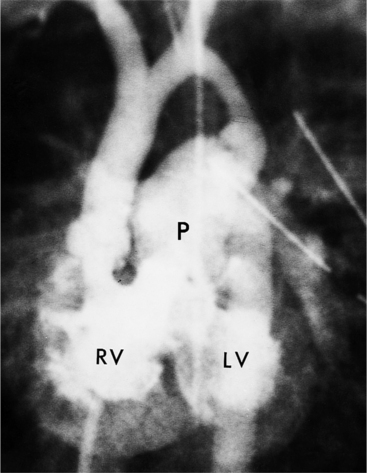
There are many angiographic features characteristic of double-outlet right ventricle in situs solitus (Fig. 10-47). In the frontal view, the heavily trabeculated right ventricle partially overlies the posterior left ventricle. Both semilunar valves have the same height and are separated from the rest of the heart by a bilateral conus. The central lucency between the aortic and pulmonary outflow tracts is the crista supraventricularis. On the lateral view, there is discontinuity between the atrioventricular and semilunar valves. Behind the great arteries and the mitral valve there is a notch that represents the ventriculoinfundibular recess. The conus under both great arteries generally overlaps completely so that, unless one artery fills before the other, the relation of the conus to the ventricle may be difficult to recognize.
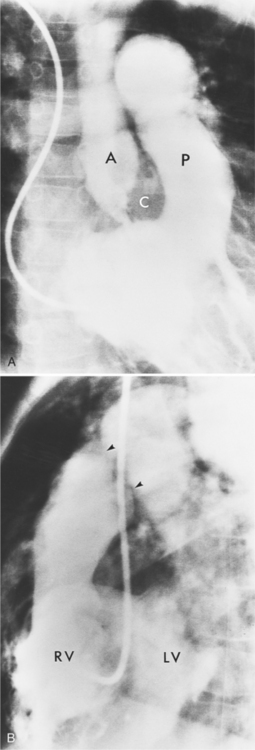
Pulmonary stenosis (Fig. 10-48), both the subvalvular (Fig. 10-49) and valvular varieties (see Figure 10-35), exists in about half of patients with double-outlet right ventricle. Conversely, subaortic stenosis occurs in about 20% of patients with this defect. Because the semilunar valves tend to lie on a transaxial plane, nonangled films in the frontal plane project the annulus of both the aortic and pulmonary valves in tangent. An injection in the outflow tract or directly beneath the crista supraventricularis may aid in making these outflow obstructions visible. (Table 10-4 illustrates the differential diagnosis of double-outlet right ventricle.)
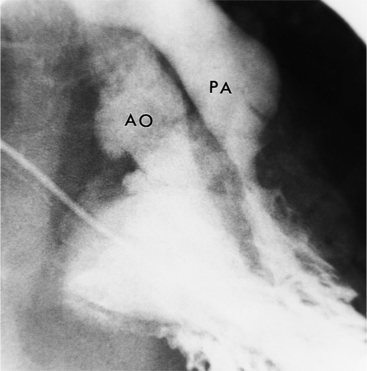
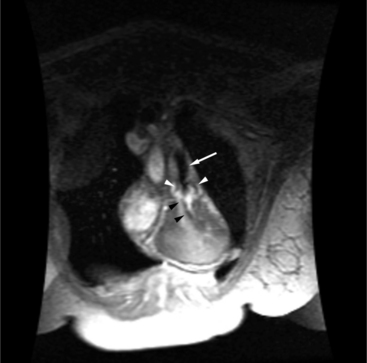
TABLE 10-4 Differential diagnosis of double-outlet right ventricle
| Findings in Double-Outlet Right Ventricle | Alternative Findings | Alternative Diagnosis |
|---|---|---|
| Bilateral conus | Pulmonary mitral continuity | Transposition of great arteries |
| Aortic–mitral continuity | Tetralogy of Fallot |
Double-Outlet Left Ventricle
The purpose of imaging in double-outlet left ventricle is to identify the associated anomalies and to locate the position of the great arteries. Because the only outlet for the right ventricle is through the ventricular septal defect, a right ventricular injection alone may allow misinterpretation of this defect as another form of transposition or even as tetralogy of Fallot. To recognize double-outlet left ventricle you must be able to visualize both great arteries over the left ventricle or one artery over the ventricular septal defect and the other over the left ventricle (Fig. 10-50). With this definition there is usually, but not always, continuity between the mitral valve and either the aortic or pulmonary valves. For example, in complete TGA with a ventricular septal defect, the pulmonary and mitral valves are adjacent and the aorta may override the ventricular septal defect; if the aorta is malpositioned so that more than 50% of its annulus corresponds to the left ventricle, then there is double-outlet left ventricle rather than complete transposition.
Anomalies of Aortopulmonary Septation
Truncus Arteriosus
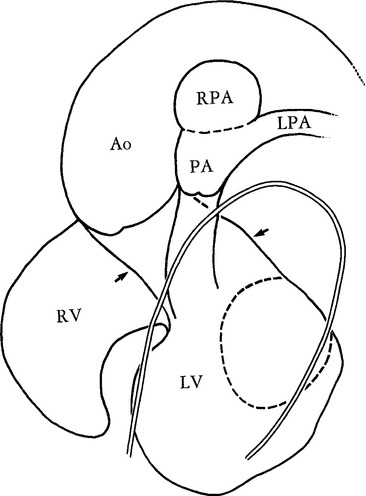
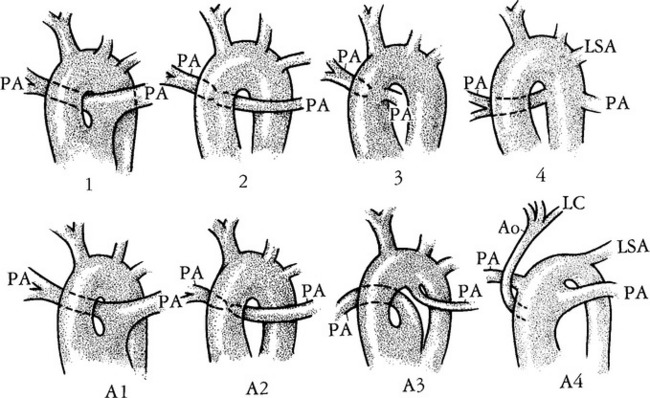
Truncus arteriosus is a congenital malformation in which only one great artery arises from the base of the heart and gives origin to the systemic, pulmonary, and coronary arteries proximal to the aortic arch (Fig. 10-51). Buchanan first described this in 1864 from an autopsy specimen. In 1949 Collett and Edwards described five types of truncus arteriosus based on the connections of the pulmonary artery and the aorta (Fig. 10-52):
The coronary circulation is quite variable in its origin. There is frequently an ectopic high origin of one or both coronary arteries. There is a single coronary artery in 13%, usually arising from the posterior cusp. The left coronary artery tends to arise from a more posterior level than in normal hearts.
Chest Film Abnormalities
The chest film usually shows substantial cardiac enlargement (Fig. 10-53) and an engorged pulmonary vasculature. Both ventricles are enlarged because of the central shunting across the ventricular septal defect. In the neonate, the thymic shadow hides the pulmonary arteries and aortic arch but in older children there may be a concave pulmonary artery segment. The shape of this segment reflects not only the absence of the right ventricular origin of the pulmonary artery but also the posterior position of the pulmonary arteries behind the truncus and ascending aorta. Also, in type A2, the left pulmonary artery originates above the right pulmonary artery. In some patients, the pulmonary arteries may appear to be elevated away from the cardiac silhouette. The size of the peripheral pulmonary arteries reflects the amount of blood flowing through them; they are generally large except when there is pulmonary artery stenosis.
Imaging Features
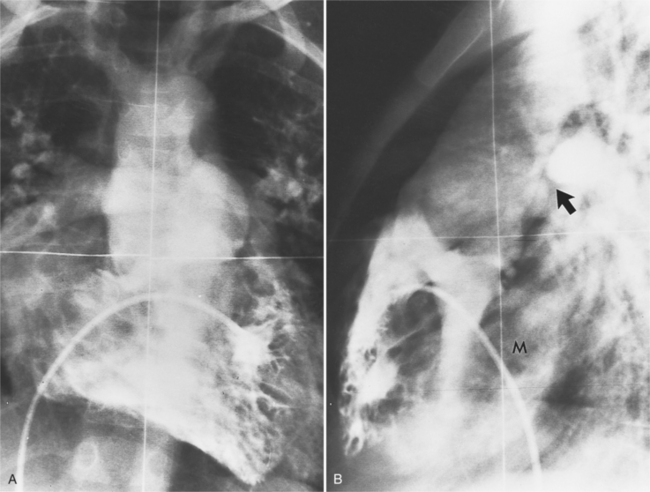
In addition to establishing the diagnosis of truncus arteriosus, it is essential to image the pulmonary arteries to see if there is stenosis. Imaging shows truncal-mitral continuity and the relation of the tricuspid valve to the truncal valve (Fig. 10-54). Establishing the degree of truncal regurgitation and the presence of arch interruption completes the examination (Fig. 10-55).
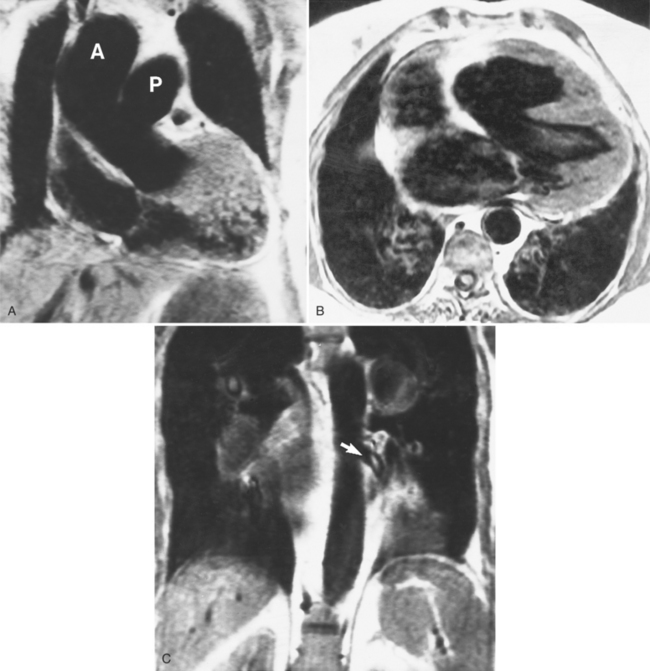
MRI can provide a valuable adjunct to angiography and echocardiography by noninvasively locating the mediastinal pulmonary arteries (Fig. 10-56). Spin echo images are obtained in the coronal and axial planes for the origins of the pulmonary arteries. Small arteries near the lung border are better identified with flow sequences such as cine gradient-recalled techniques or phase reconstruction.
Truncus Arteriosus
The surgical repair of truncus arteriosus with confluent or near confluent pulmonary arteries consists of separating the pulmonary arteries from the arterial trunk and connecting them to the right ventricle via a conduit or homograft and closure of the ventricular septal defect (see Figure 10-44). When the aortic arch is interrupted or has a significant coarctation, the repair also consists of reconstruction of the aortic arch with the arterial trunk.
The cardiac MR protocol for evaluating patients following repair of truncus arteriosus is essentially the same as the protocol for tetralogy of Fallot because the complications are similar with respect to conduit stenosis and regurgitation and branch pulmonary artery stenosis and their inherent effects on right ventricular volumes and function (Table 10-5). In addition, there is a significant incidence of progressive truncal root dilatation and valvular regurgitation postoperatively that can be quantified and followed serially by cardiac MR for possible root or valve replacement.
TABLE 10-5 Complications after repair of tetralogy of Fallot
| Complication | Description |
|---|---|
| Residual right ventricular outflow tract obstruction | Infundibulum, pulmonary valve, main and branch pulmonary arteries |
| Residual pulmonary regurgitation | Well tolerated if mild to moderate, can lead to impaired ventricular function if severe, especially with concurrent pulmonary artery stenosis |
| Right and left ventricular dysfunction | Associated with longstanding right ventricular outflow tract obstruction, chronic volume overload resulting from palliative shunts, or residual ventricular septal defect (left ventricle) or pulmonary regurgitation (right ventricle) |
| Aortic root dilatation | Damage to the aortic valve resulting from ventricular septal defect closure, or intrinsic aortic root abnormality |
| Exercise intolerance | Related to pulmonary regurgitation and right ventricular dysfunction |
Hemitruncus Arteriosus
In hemitruncus one of the pulmonary arteries originates from the aorta and the other from the right ventricle. There are separate aortic and pulmonary valves that distinguish hemitruncus from truncus type A3. The lung connected to the aorta is subject to the systemic pressure and has aneurysmal hilar branches and serpentine peripheral vessels. The lung supplied by the right ventricle has normal vasculature. The aortic arch tends to be large and may displace the trachea away from the side of the arch. It is right sided in 20% to 30% of cases. When the thymus does not obscure the mediastinum, the right boundary is the edge of the large ascending aorta.
Aortopulmonary Septation
Separate aortic and pulmonary valves and a defect between the ascending aorta and the main or right pulmonary artery are characteristic of aortopulmonary septation (Fig. 10-57). This rare malformation is also called aortopulmonary septal defect and aortic pulmonary window. The defect is usually in the left lateral wall of the ascending aorta and connects with the right lateral wall of the pulmonary trunk. The defect can extend from the annulus of the two semilunar valves to involve the ascending aorta to varying degrees. About half have aortic arch interruption or a preductile coarctation.
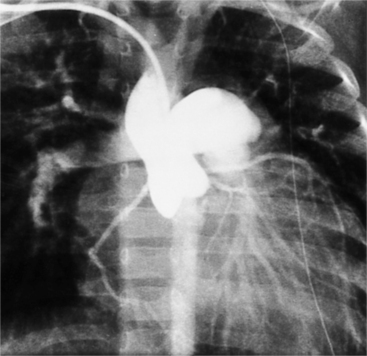
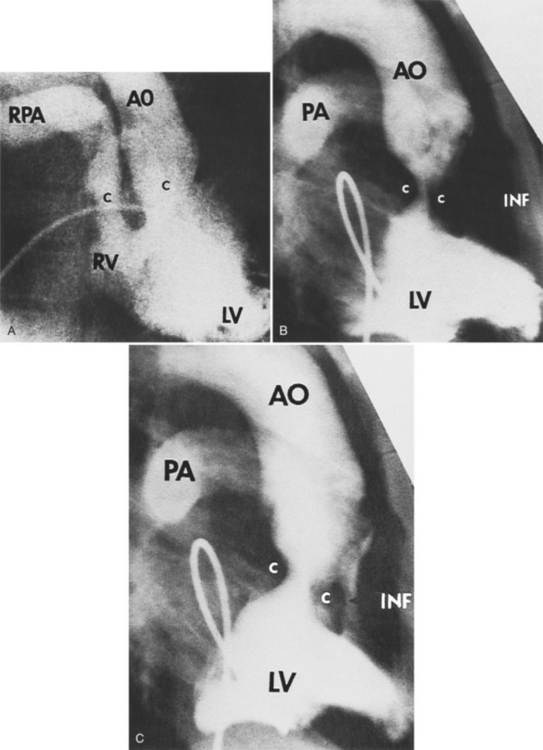
Anatomically Corrected Malposition of the Great Arteries
In this malformation, the left ventricle ejects into the aorta and the right ventricle into the pulmonary artery. However, the great arteries are abnormally related to one another. In situs solitus, the aorta is to the left of the pulmonary artery. The ventriculoarterial connections are normal but their relations are malpositioned (Fig. 10-58). The levotransposed aorta is characteristic of corrected transposition of the great arteries, and if the loop rule applies, indicates ventricular inversion and L-looping of the ventricles. Levotransposed aorta rarely occurs in other types of malformations, including those that defy the loop rule and have noninverted ventricles.
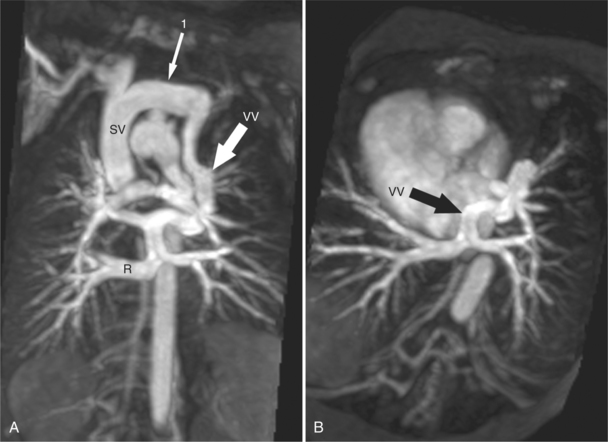
Abnormal Connections of the Pulmonary Veins
Total Anomalous Pulmonary Venous Connection
All pulmonary veins may connect anomalously in three ways:
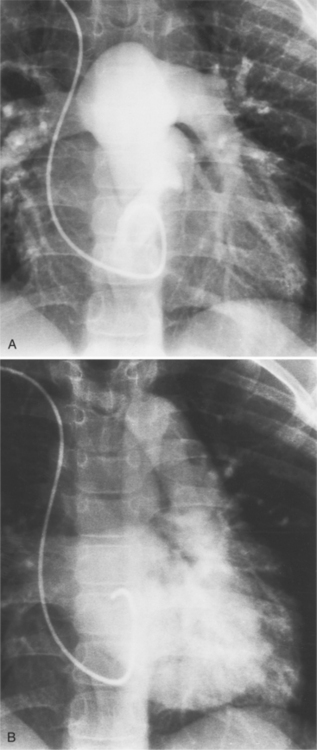
The most common anomaly is connection of all pulmonary veins to a systemic vein in the thorax (Fig. 10-59). The one or two veins draining each lung merge to form a common confluence behind the left atrium but without connecting to it. The outflow of this common vein may go into the left innominate vein, the right superior vena cava, or to the azygos or hemiazygos system of veins. When the connection is to a vein on the left side of the mediastinum, this vascular structure may be either a left superior vena cava or an anomalous vertical vein. Most vertical veins in the supracardiac type of total anomalous pulmonary venous drainage do not connect to the coronary sinus, hemiazygos, or left atrium as a left superior vena cava would. Therefore, it is actually an anomalous vein between the common pulmonary vein and the left innominate vein. This anomalous vertical vein may pass posterior to the pulmonary artery or may pass between the left main stem bronchus and the left pulmonary artery. In the latter instance, an obstruction to flow may occur by compression of these adjacent structures on to the anomalous vein.
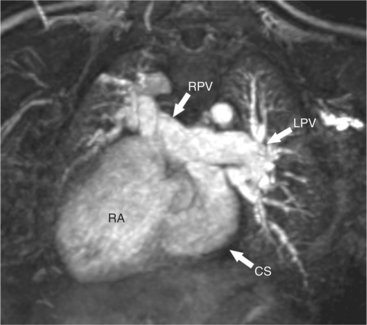
In the intracardiac type of total anomalous pulmonary venous connection (TAPVC), the common pulmonary vein connects directly to the right atrium or coronary sinus (Fig. 10-60), either as one vessel or with separate connections of two or more of the pulmonary veins. The intracardiac type of anomaly is frequently accompanied by polysplenia or asplenia with the associated bilateral superior vena cava and azygos continuation. The pulmonary veins may also attach to the coronary sinus, in which case this structure is considerably enlarged.
The infracardiac type of anomalous pulmonary connection always demonstrates pulmonary vascular obstruction. In this type, a venous channel from the common pulmonary vein passes inferiorly through the diaphragm at the esophageal hiatus to connect with the portal vein or its tributaries or the ductus venosus. In the extreme form, which represents atresia of the common pulmonary vein, all egress from the common pulmonary vein is absent except into minute venous channels adjacent to the esophagus.
Chest Film Findings
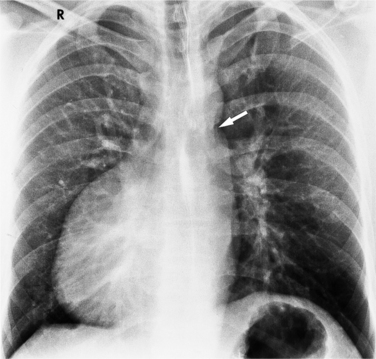
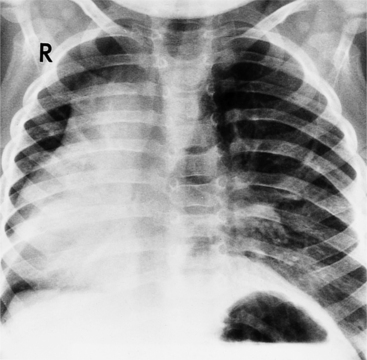
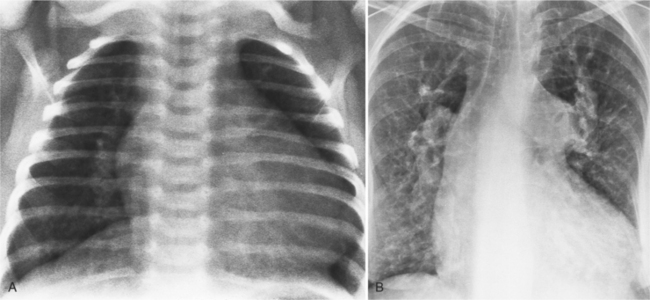
The findings on the chest film depend on the same variables that control the blood flow to the heart and lungs. When there is no pulmonary venous obstruction, typically in the supracardiac and intracardiac types, the chest film generally demonstrates cardiomegaly and large pulmonary vessels. The right atrial and ventricular contours appear prominent, and there is no evidence of left atrial enlargement, even on the barium esophagogram. The right superior vena cava is dilated when it is part of the circuit that receives pulmonary venous drainage. The only sign pathognomonic of the supracardiac variety that is seen on the chest film is the “snowman” configuration (Fig. 10-61). The anomalous left vertical vein forms the convex left superior mediastinal border as it joins the dilated left innominate vein. The enlarged right superior vena cava protrudes into the right lung, completing the head of the snowman.
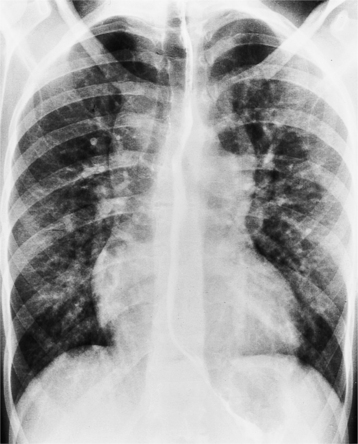
In those cases that are obstructive, the chest film is quite different (Fig. 10-62). Generally, the heart is of normal size without prominence of any chamber. The striking abnormality occurs in the lungs, which show evidence of pulmonary edema. An interstitial pattern is invariably present in the infracardiac type. Interstitial and alveolar pulmonary edema, and occasionally, Kerley B lines are frequently mistaken for noncardiac causes of pulmonary consolidation, such as neonatal pneumonia, aspiration, respiratory distress syndrome, and transient tachypnea of the newborn.
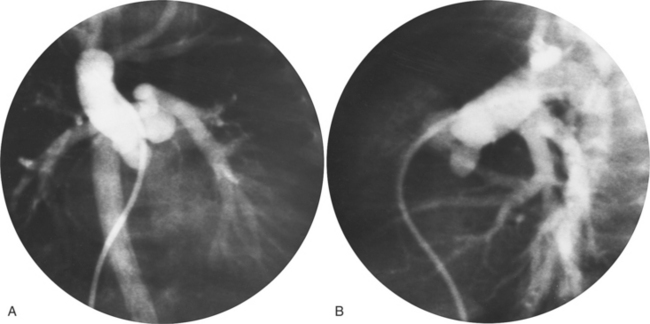
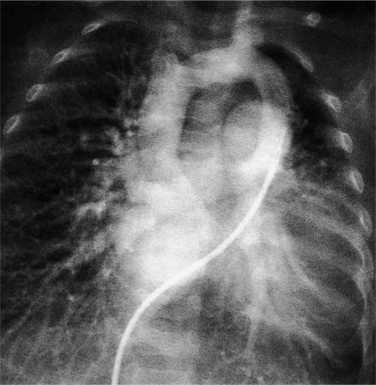
The angiographic technique for identifying the course and location of the anomalous veins begins with pulmonary arteriography. In the supracardiac variety with the anomalous left vertical vein, the vein itself may be injected directly. In the newborn, this structure may be fragile so that indirect opacification from a more peripheral site is desirable. A main pulmonary artery angiogram in the posteroanterior projection demonstrates the pulmonary veins on the levophase (Fig. 10-63). In the lateral view, the confluence of veins appears behind the usual position of the left atrium before it ascends into the anomalous vertical vein to join the left innominate vein. If other cardiac defects are discovered, additional injections will clarify their morphology.
In the intracardiac type, the location of the anomalous connection may be difficult to identify because of overlapping cardiac chambers. Pulmonary angiography with late filming for the levophase structures shows opacification of the coronary sinus or right atrium before the left atrium (Fig. 10-64).
The infracardiac variety is distinctive and has considerably reduced blood flow (Fig. 10-65). For this reason, the levophase of the pulmonary angiogram should extend for 10 to 12 seconds until you can see the long venous channel extending below the diaphragm to enter the portal vein.
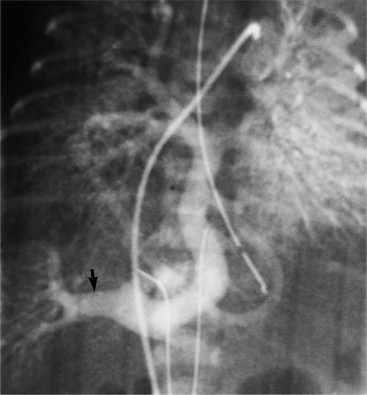
Partial Anomalous Pulmonary Venous Connection
Partial anomalous pulmonary venous connection (PAPVC) is present when one or more, but not all, of the pulmonary veins connect to a systemic vein (Fig. 10-66). The veins of the right lung have 2 to 10 times as many anomalous connections as those of the left lung. When an anomalous vein exists, it usually connects to the nearest adjacent systemic vein or to the right atrium (Fig. 10-67). For example, the right superior pulmonary vein usually connects to the right superior vena cava (Fig. 10-68) or azygos vein. The right inferior vein may connect with the inferior or superior vena cava, a hepatic vein, or occasionally the azygos vein. The left lung veins connect to an anomalous vertical vein draining into the left innominate vein, the coronary sinus, or the hemiazygos vein (Fig. 10-69). Although these examples are typical, there are many variations in number, size, and connections of the pulmonary veins. The atrial septum is usually intact. Conversely, if an atrial septal defect is present, about 10% of these patients will have a pulmonary venous anomaly.
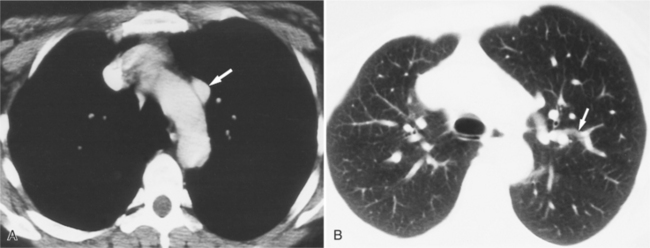
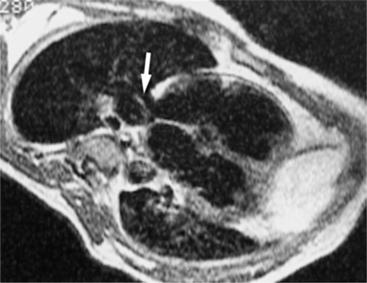
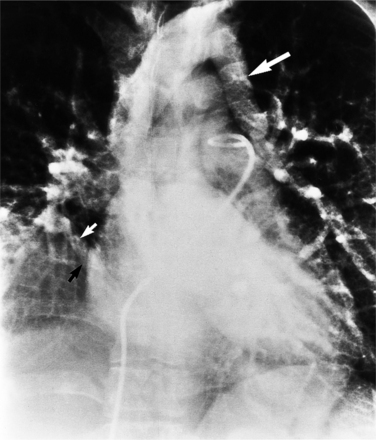
The chest radiograph is usually normal because the pulmonary-to-systemic flow ratio is generally less than 2:1. When an entire lung is drained by an anomalous connection, the main pulmonary arteries and peripheral branches are enlarged similarly to the situation with a left-to-right shunt. In this case, the right ventricle is enlarged, whereas the other cardiac chambers and the vena cava are equivocally dilated. The isolated lower lobe veins that connect to the inferior vena cava present the most striking radiographic abnormalities. The anomalous vein is almost always seen on the right side but may occasionally be a left-sided structure. Curving medially and inferiorly through the lung, the vein has been likened to a scimitar because, unlike a normal pulmonary artery, it increases in diameter as it goes toward the base of the lung (Fig. 10-70).
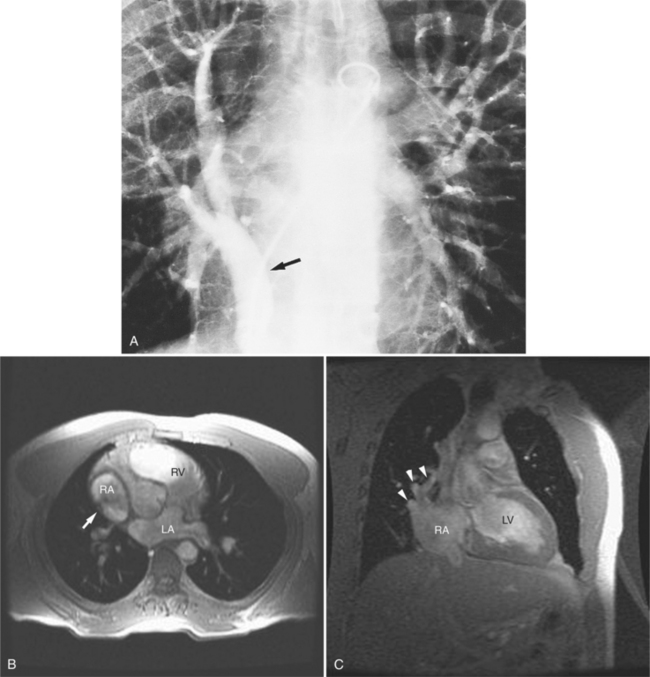
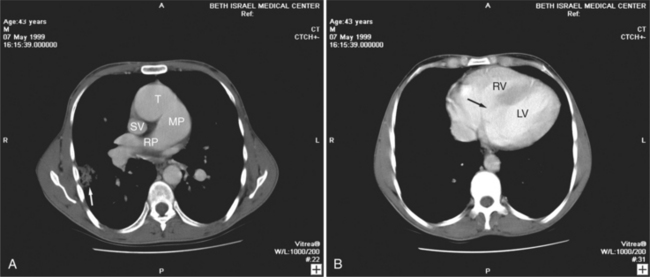
Felson named it the venolobar syndrome. Fraser preferred the hypogenetic lung syndrome. The syndrome is a rare congenital anomaly that consists of an anomalous pulmonary venous connection of the right lung to the inferior vena cava; it is associated with hypoplasia of the right lung and its bronchus and pulmonary artery. The heart is secondarily rotated toward the right hemithorax because of the small right lung. The right lung may have abnormal lobation, frequently having two rather than three lobes. This condition is also called the pulmonary venolobar syndrome.
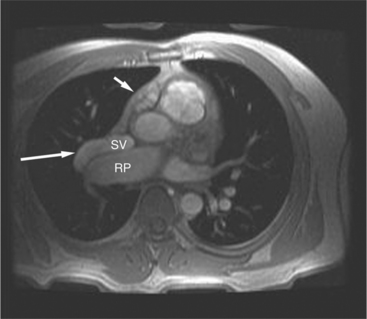
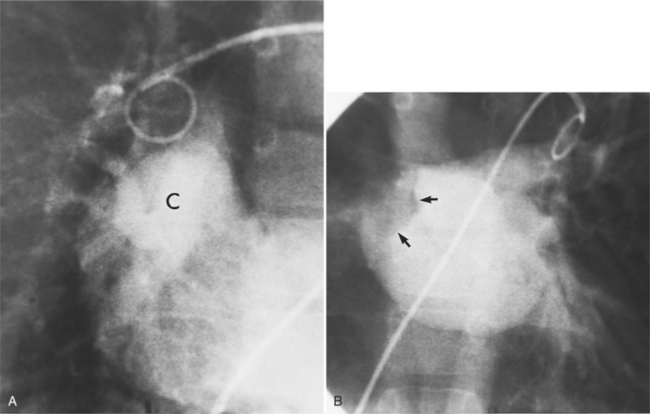
Pulmonary angiography is a common examination for locating all the pulmonary veins. If the right atrium is visible during the levophase, an anomalous pulmonary vein, an atrial septal defect, or both must be present (Fig. 10-71). When a PAPVC to the superior vena cava or right atrium exists, an atrial septal defect occurs in 90% of patients, whereas when the anomalous vein connects to the inferior vena cava, only 15% have an atrial septal defect. A technique of localizing the anomalous pulmonary vein with respect to the atrial septal defect involves placing a catheter into the right pulmonary vein from an inferior vena caval approach. Using cranial angulation with the hepatoclavicular or “four-chamber” view, the atrial septum is projected in profile so that the connection of the vein to the atrium is seen during a 10-ml injection.
SEPTAL DEFECTS, HYPOPLASIAS, AND ATRESIAS
Atrial Septal Defects
Ostium Secundum and Primum Defects
An ostium secundum atrial septal defect is an absence or deficiency of tissue in the region of the fossa ovalis (Fig. 10-72). An ostium primum atrial septal defect is medial and adjacent to both atrioventricular valves. This abnormality is associated with developmental defects in the endocardial cushions that contribute to the inferior atrial septum (Fig. 10-73). If all endocardial cushions are deficient, associated defects occur in the superior ventricular septum and in the leaflets and chordal attachments of both the mitral and tricuspid valves.
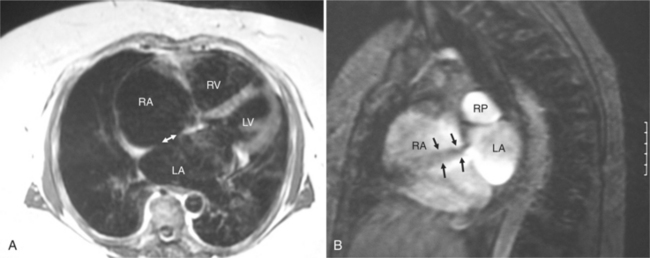
Sinus Venosus Defects
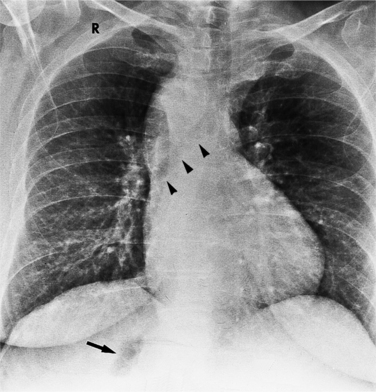
A sinus venosus atrial septal defect occurs in the superior and lateral portion of the atrial septum adjacent to the connection with the right pulmonary veins (Fig. 10-74). Because of this location, anomalies of pulmonary venous connection are commonly found in the triangle defined by the superior vena cava, the interatrial septum, and the right superior pulmonary vein.
Less Common Defects
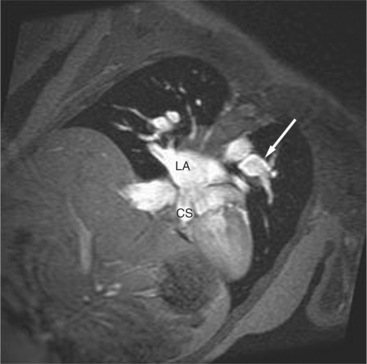
Less common atrial defects are those between the fossa ovalis and the inferior vena cava and in the region of the coronary sinus (Fig. 10-75). An absent coronary sinus, connection of the left superior vena cava to the left atrium, and a large atrial septal defect make up this unusual developmental complex.
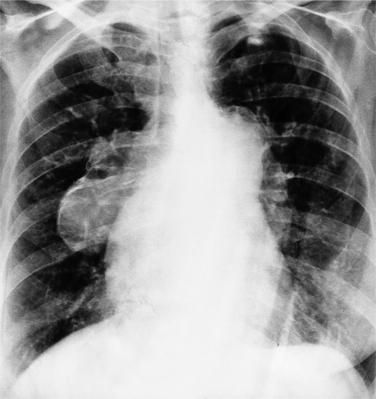
Chest Film Features
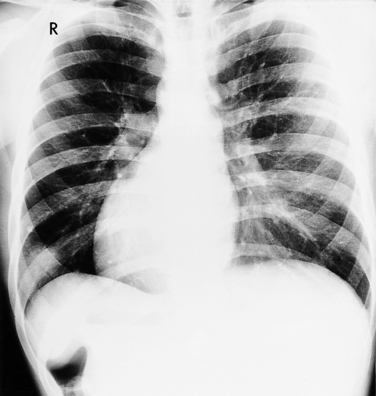
The classic chest film features of an atrial septal defect (Fig. 10-76) are enlargement of the right atrium, the right ventricle, and all segments of the pulmonary arteries (“shunt vascularity”). Because the chest film does not accurately reflect the size of the right atrium and ventricle, the heart size may appear normal. These signs of enlargement are apparent when the flow through the pulmonary artery is at least twice that through the aorta. The large size of the main pulmonary artery is usually striking when compared with the normal size of the aortic arch.
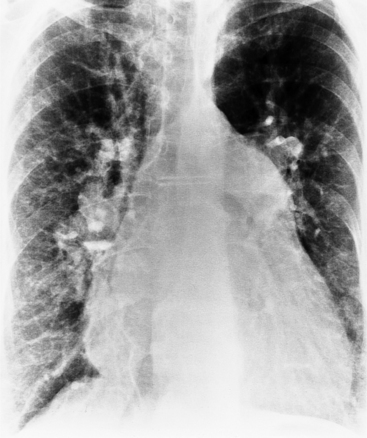
There is no enlargement of the left heart in a simple atrial septal defect unless there are other complications. For example, when the right ventricle is enlarged as a result of increased volume, there is a distortion of the geometry of the left ventricle so that mitral prolapse and occasionally mitral regurgitation ensue. Similarly, in an atrioventricular canal defect, the left atrium and ventricle are enlarged when either a ventricular septal defect or mitral regurgitation causes volume overload in these chambers.
When cyanosis results from pulmonary arterial hypertension (Eisenmenger syndrome), the pulmonary arteries become quite large. However, evaluating the size of the hilar pulmonary vessels is not a reliable way of differentiating pulmonary arterial hypertension with a small left-to-right shunt from normal pulmonary artery pressures with a torrential shunt. When an Eisenmenger syndrome causes blood to flow in a reverse direction (from the right to the left atrium), the heart becomes smaller. Radiographic signs of pulmonary arterial hypertension at this stage include calcification of the pulmonary arteries (Fig. 10-77), aneurysmal dilatation of the hilar branches, and a serpentine course of the lower lobe pulmonary arteries.
Imaging Techniques and Features
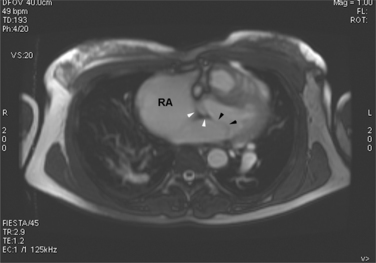
Atrial septal defects are routinely imaged by echocardiography. In the four-chamber view (Fig. 10-78), the entire atrial septum can be seen: superiorly from the entrance of the right upper pulmonary vein and inferiorly to the tricuspid valve. With a catheter in the right upper pulmonary vein, contrast material streams along the left atrial side of the septum and then through the defect to opacify the right atrium. In these projections, if the interatrial septum is divided into thirds, a sinus venosus defect will occur in the upper outer third, a secundum defect will occur in the middle third, and a primum defect will occur in the central third.
Atrioventricular Septal Defects
Complete Atrioventricular Canal Defects
In the complete form of atrioventricular septal defect, the mitral and tricuspid valves may be formed into a single large common valve, or they may have separate annuli. The mitral valve has a deficiency or cleft in the anterior leaflet, while the tricuspid valve has a cleft in its septal leaflet. This division results in five leaflets, two of which bridge from the left to the right ventricle. A posterior leaflet extends from the posterior papillary muscle of the left ventricle to the right posterior papillary muscle of the right ventricle. The anterior leaflet extends from the medial papillary muscle of the right ventricle to the anterolateral papillary muscle of the left ventricle. The anterior leaflet may be divided with chordae tendineae going to the septum and to the papillary muscles, or it may be undivided with no attachments to the septum. The combination of defects makes the size of the mitral orifice appear two or three times as large as that of the aortic valve.
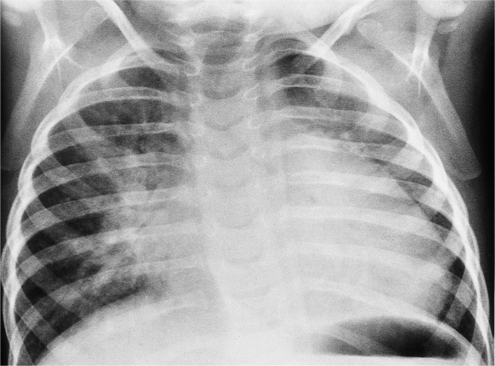
The complete form includes unequal inlet and outlet lengths of the ventricular septum, a malorientation of the aortic valve relative to the atrioventricular valves, atrial and ventricular septal defects, and a deficiency and malattachment of part of the right and left atrioventricular valves. The complete form of an atrioventricular canal defect, which constitutes about 20% of all types of canal defects, has an atrial septal defect that communicates with a ventricular septal defect. The atrioventricular valve may be common or may be partitioned by the tricuspid and mitral annuli. Among patients with a complete atrioventricular canal defect, 50% to 80% have Down syndrome. Of patients with Down syndrome, 40% have an atrioventricular septal defect (Fig. 10-79).
Partial or Incomplete Atrioventricular Canal Defects
A partial or incomplete atrioventricular canal defect has separate mitral and tricuspid orifices, and may have atrial or ventricular septal defects, or both, and clefts in the mitral and tricuspid valve leaflets (see Figure 10-73). About two thirds of all patients with an atrioventricular canal defect have the partial form. The most common partial form is an ostium primum atrial septal defect and a cleft in the anterior (septal) leaflet of the mitral valve.
Segmental Analysis
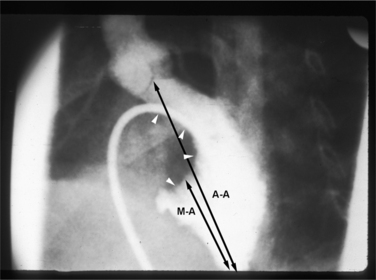
The mitral valve is displaced toward the apex of the left ventricle, as compared with the aortic valve. The papillary muscles are in front of one another rather than in the anterolateral and posteromedial locations. In the normal heart, the distance from the aortic valve to the apex is the same as that from the mitral valve to the apex. The ratio of the length of the left ventricular inflow tract to the outflow tract may vary from 0.5 to almost 1.0 (in the normal heart, this ratio is 1.0). In the complete form of atrioventricular septal defect, the ratio tends to be between 0.5 and 0.7, and in the partial form is between 0.7 and 0.9 (Fig. 10-80). The mitral leaflets are quite redundant and scalloped. Rarely, this extra valve tissue can appear as a tumor mass and obstruct the outflow tract. Because of the cleft, the mitral regurgitation may go directly into the right atrium through the atrial septal defect rather than into the left atrium.
Chest Film Abnormalities
The appearance of the heart and lungs on the chest film reflects the abnormal blood flow (see Figure 10-79). Because there is a left-to-right shunt across an atrial or ventricular septal defect, or across both, all segments of the pulmonary arteries are enlarged. All four chambers of the heart are large, reflecting not only the flow through the septal defect but also mitral and tricuspid regurgitation. When a combination of high cardiac output and mitral regurgitation coexist, interstitial and alveolar pulmonary edema may be visible, particularly in the hilar regions, even though the left atrial pressure is normal.
On chest film it is often impossible to distinguish a secundum atrial septal defect or an isolated membranous ventricular septal defect from either the complete or partial form of atrioventricular canal defect. There are, however, several clues on the chest film that may suggest the latter (Table 10-6). First, the age of the patient is helpful in that it is unusual in a person less than a few months old for a secundum atrial septal defect to produce visible large pulmonary vessels and signs of congestive heart failure, whereas canal defects may be associated with signs of heart failure within the first few weeks of life. Second, the left atrium is not visibly enlarged in a secundum atrial septal defect; this feature, however, is typical of an atrioventricular septal defect with mitral regurgitation. Third, an enlarged right atrium is not a feature of a simple ventricular septal defect but is present in an atrioventricular septal defect when there is either an atrial septal defect or a left ventricular-right atrial defect. These findings, coupled with left axis deviation of the QRS complex on the electrocardiogram (ECG), are likely to represent some type of atrioventricular septal defect.
| Shunt Lesions | Chamber Enlargement | Dilated Aorta |
|---|---|---|
| Atrial septal defect | Right atrium | No |
| Right ventricle | ||
| Ventricular septal defect | Left atrium | No |
| Left ventricle | ||
| Right ventricle | ||
| Patent ductus arteriosus | Left atrium | Yes |
| Left ventricle |
From Van Praagh R, Weinberg PM, Smith SD, et al.: Malpositions of the heart. In: Emmanoulides GC, Riemenschneider TA, Allen HD, et al., eds.: Moss and Adams heart disease in infants, children and adolescents including the fetus and young adults, ed 5, Baltimore, 1995, Williams & Wilkins.
Angiographic Features
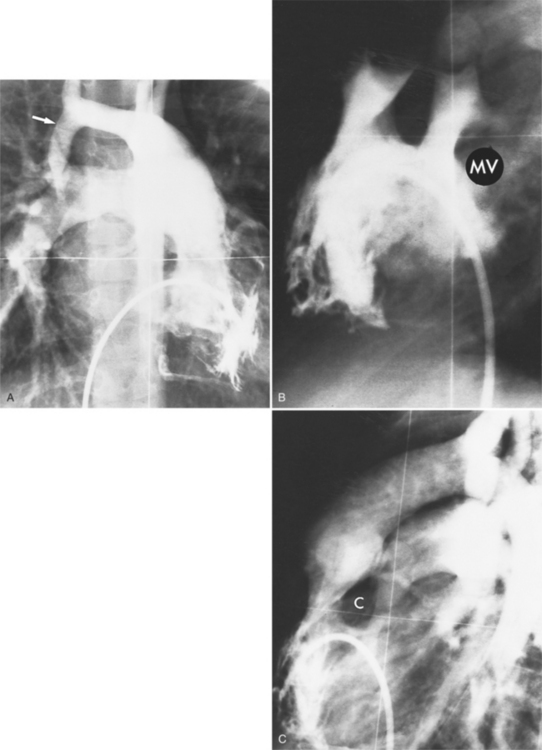
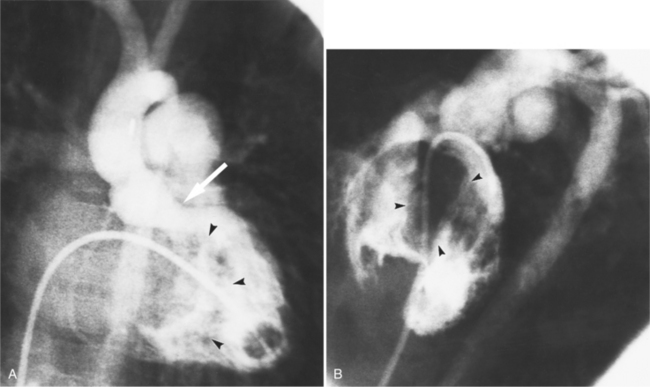
The angiographic features of atrioventricular canal defects were first recognized in a classic paper by Baron and colleagues in 1964. They described the diagnostic pattern in the left ventriculogram as a “gooseneck” deformity and explained how this resulted from a deficiency in the inferior interventricular septum. The characteristic deformity in the left ventricle is visible in the right anterior oblique projection and is accentuated in diastole. The left ventricular inflow is concave and not straight as it is in the normal heart. This concave edge represents in part the apically displaced mitral valve; it has a serrated edge, which is the redundant leaflet of tissue pulled by the chordae tendineae. A radiolucent horizontal line in the mitral valve represents the cleft of the anterior leaflet. These features are present with or without a ventricular septal defect and also persist after surgical closure of the atrial and ventricular septal defects (Fig. 10-81).
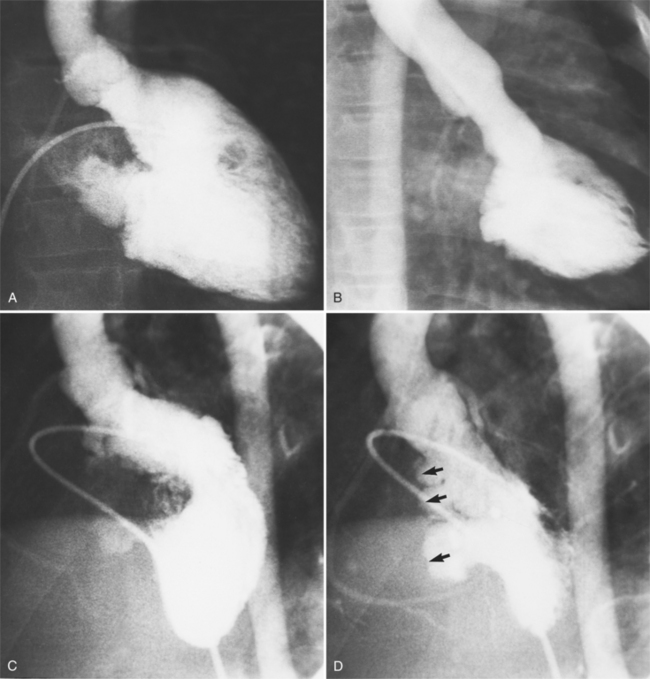
When separate mitral and tricuspid valves exist, the septal leaflet forms the right border of the left ventricular outflow tract. When there is a common atrioventricular valve, contrast material passes from the left ventricle into the right ventricle beneath the leaflets of the common valve (Fig. 10-82). The leaflets may be attached to the septum or may form a continuous bridge from the superior rim of the common valve. The chordal attachments of the common leaflet may extend to their adjacent ventricular papillary muscles or the lip of the ventricular septal defect or may cross into the opposite ventricle. The partial form of this malformation, in which the chordae attach on opposite sides of the septum, is the straddling atrioventricular valve.
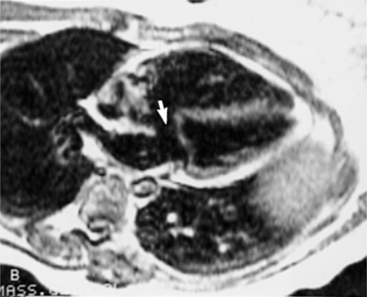
Magnetic Resonance Imaging Features
In the four-chamber view perpendicular to the interventricular septum, spin echo sequences will show defects at the junction of the interatrial and interventricular septum (Fig. 10-83). White blood sequences, which are sensitive to movement, are needed to assess valve regurgitation and shunt flow.
Ventricular Septal Defects
Classifications
Several surgical and pathologic classifications have divided these defects into supracristal and infracristal varieties. However, the crista supraventricularis is not always identified in its entirety by imaging. In addition, the ventricular septum is curvilinear so that other landmarks, such as the mitral and aortic valves, are more readily recognizable. To overcome these problems, Soto devised a classification in 1980 that divides the left ventricle into the inlet part near the mitral valve, the muscular portion, and the outflow tract portion below the aortic valve. Using this way of dividing the ventricle, defects in the posterior ventricular septum are those associated with atrioventricular septal defects. Muscular defects have slight overlap with defects in the other categories but are mainly holes of varying sizes between the trabeculations of the two ventricles. Ventricular septal defects in the left ventricular outflow region mainly occur in the membranous part of the septum. In complex defects, such as tetralogy of Fallot, the defect extends into the crista and occasionally into the muscular septum. Isolated membranous defects, viewed from the right ventricular side, are infracristal in location. The least common defect is in the supracristal location below the pulmonary valve. Also called conoventricular defects, these malformations are frequently associated with malalignment between the ventricular septum and the conal septum that separates the aortic from the pulmonary valves.
Chest Film Findings
In all types of ventricular septal defect, the appearance of the heart and lungs on the chest radiograph depends on the flow and pressure in the pulmonary artery. In small shunts that represent less than a 2:1 shunt-to-pulmonary-artery ratio, the chest film is usually normal. In larger shunts, cardiac size increases roughly in proportion to the amount of the shunt. The left ventricular apex projects to the left and inferiorly and posteriorly. The left atrium is enlarged and can be seen as a double density behind the right atrium. The main pulmonary artery and all pulmonary vessels increase in size depending on the degree of the blood flow (Fig. 10-84). If pulmonary vascular obstruction increases so that blood flow reverses and becomes a right-to-left shunt (Eisenmenger physiology), the heart size becomes smaller. At this point, the central pulmonary arteries appear as aneurysms, and rarely, may be calcified. The basilar pulmonary arteries tend to elongate when pulmonary artery hypertension occurs, resulting in a serpentine course. The right ventricle, seen mainly on the lateral chest film, also enlarges as the size of the shunt increases. Both ventricles have a corresponding increase in diastolic volume and a similar ejection fraction.
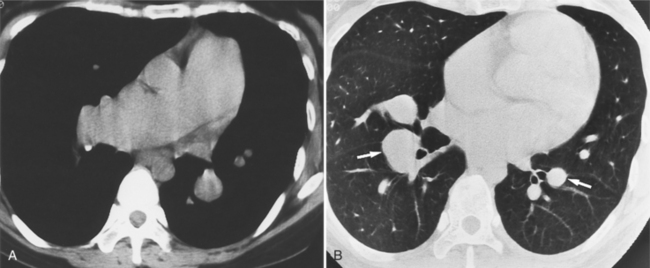
Imaging Features
Muscular ventricular septal defects typically appear on MR cine angiography as thin, small jets of contrast material that pass directly into the right ventricle. These defects are usually located in the apical half of the ventricular septum but may exist adjacent to the membranous septum at its trabeculated edge (Fig. 10-85). Although these defects usually range from a few millimeters to a centimeter in size, most of the muscular septum may be absent, making a common ventricle. Small muscular defects, which are seen as thin streams into the right ventricle, are visible throughout most of systole. If the defects are tiny, the contracting trabeculations may close these holes so that they are visible on ventriculography only during the early part of systole.
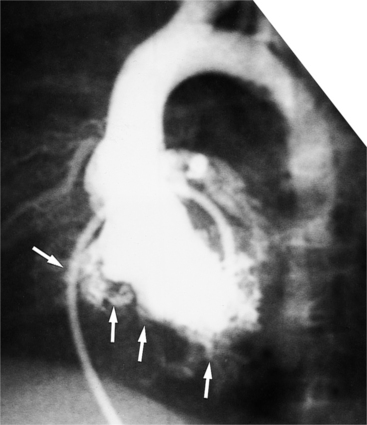
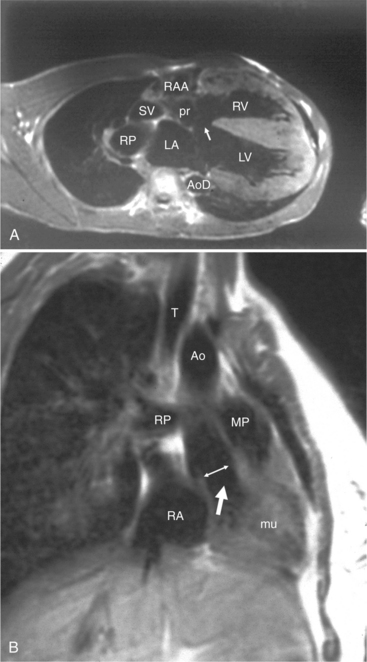
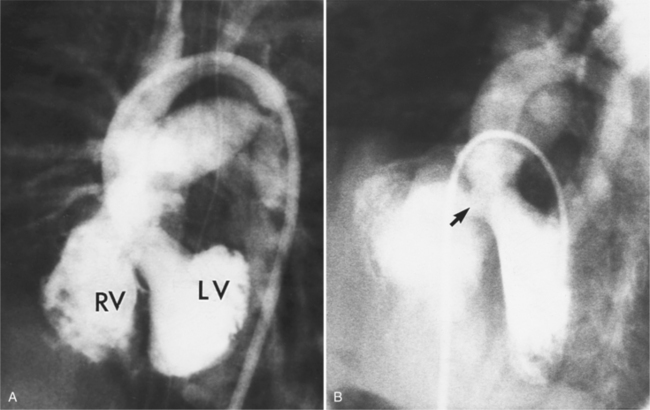
Defects in the membranous septum are seen in the outflow tract inferior to the aortic valve and medial to the mitral valve (Figures 10-86, 10-87). From the right ventricular side, the septum is deficient below the crista and behind and adjacent to the sepal leaflet of the tricuspid valve. During a left ventriculogram the path of contrast material is seen going through the septum, then apically down the right ventricular side of the septum (Fig. 10-88). This “waterfall” motion is typical of a membranous defect and opacifies the entire right ventricle when the size of the defect is moderate. When the pulmonary artery pressures are normal, left-to-right shunting is visible during the left ventriculogram in both systole and diastole, although the major portion of contrast media crosses the ventricular septal defect in systole. If the peak right ventricular pressure is within 30 mm Hg of that in the left ventricle, transient right-to-left shunting occurs during isovolumetric relaxation. When pulmonary vascular obstruction ensues with systemic pulmonary artery pressures and cyanosis (Eisenmenger syndrome), most of the blood flow is from the right ventricle into the left ventricle; the degree of opacification depends less on the size of the defect than on the pressure gradient between the ventricles.
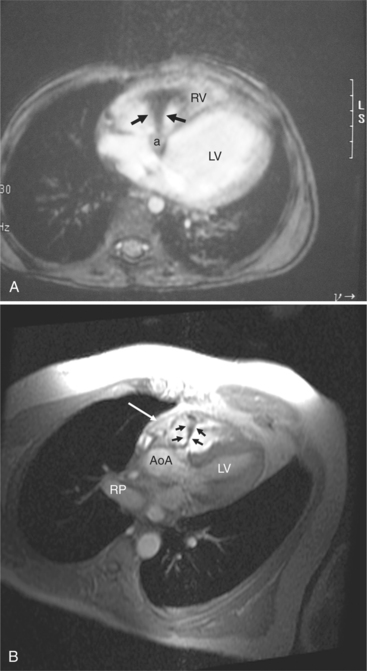
Aneurysms of the membranous septum look like a windsock (Fig. 10-89) and project into the ventricle adjacent to the septal leaflet of the tricuspid valve. These aneurysms, which appear to represent closing membranous defects, are frequently round and smooth-walled but may have a scalloped and irregular margin. The aneurysm may connect to the right ventricle through its center, or it may be completely closed allowing no communication between the ventricles. Rarely, the aneurysms extend into the right atrium. Common lesions associated with a membranous septal aneurysm are aortic insufficiency with prolapse of a cusp into a portion of this aneurysm, bacterial endocarditis, and (rarely) right ventricular outflow obstruction and tricuspid regurgitation. In views other than the left anterior oblique projection, the aneurysms may appear as masses adjacent to either the aortic or mitral valve. Masses in the right atrium and right ventricle represent the other side of the sac.
Patent Ductus Arteriosus
Characteristics
The altered hemodynamics that arise from a persistent ductus arteriosus depend on the size of the ductus and the ratio of the pulmonary-to-systemic vascular resistance. In fetal life, the shunt created by the ductus is from right to left because of the high pulmonary vascular resistance. Shortly after birth, the complex changes that happen in the first few minutes in the lungs result in a lowered pulmonary arterial pressure of one fourth to one sixth of the aortic pressure. A left-to-right shunt is thereby created through the patent ductus. This results in varying degrees of overcirculation to the lungs, left atrium, left ventricle, and the ascending and arch portions of the aorta. If the ductus does not close and an Eisenmenger reaction to the increased pulmonary flow results, the pulmonary vascular resistance again exceeds the systemic resistance and causes a right-to-left shunt across the ductus arteriosus.
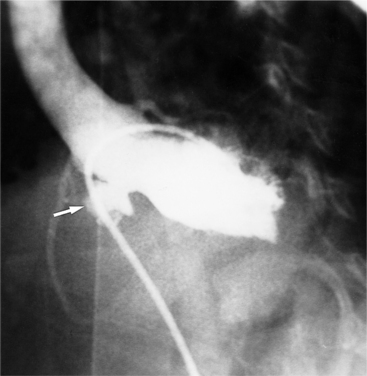
The diameter and length of the ductus arteriosus varies considerably depending on whether partial closure has occurred (Fig. 10-90). In the newborn infant, the ductus is nearly the size of the descending aorta and may be mistaken for the aortic arch because of the size similarity. The length of the ductus is also quite variable and ranges from several centimeters to an aortic-pulmonary fistula without any intervening neck.
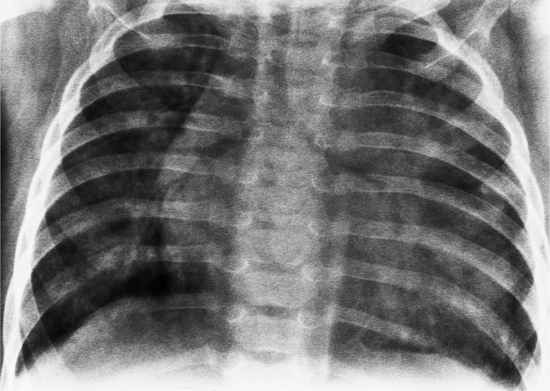
Infant Chest Film Abnormalities
The radiographic signs of a persistent patent ductus have a rough correlation with the degree of pulmonary circulation. In the infant and young child, the hilar and segmental pulmonary arteries are enlarged, when not obscured by the thymus (Fig. 10-91). When the shunt is greater than 2:1, perihilar pulmonary edema may begin to develop from the high cardiac output in the infant and may progress to generalized alveolar edema. Concomitant cardiac changes include an enlarged cardiac silhouette with cardiothoracic ratios greater than 0.55. In the infant, the differentiation of individual enlarged chambers is frequently difficult; however, on the lateral view, left atrial enlargement can be detected by the posterior displacement of the left mainstem bronchus. Other noninvasive tests, such as the echocardiogram, show more accurately that the left atrium is enlarged and has an anteroposterior diameter exceeding that of the aorta. In the normal newborn infant, the anteroposterior left atrial dimension is less than that of the adjacent ascending aorta.
Adult Chest Film Abnormalities
Because the mediastinum is not obscured by the thymus in the adult, other radiologic signs may point to the existence of a PDA. The “ductus bump” is a localized dilatation adjacent to the descending aorta next to its arch at the entrance of the ductus. Embryologically, this dilatation is thought to be distinct from the involuted end of the right aortic arch. The large ascending aorta and arch may elongate so that the ascending aorta and innominate artery are visible on the right side of the mediastinum, whereas the left subclavian artery projects into the left lung apex. Assuming an isolated left-to-right shunt is present, the large aortic arch indicates that the shunt is extracardiac; for example, a PDA is present rather than an atrial or ventricular septal defect. An aortopulmonary septal defect (a connection between the ascending aorta and the main pulmonary artery) produces similar findings, but the amount of the shunt usually is larger. Rarely, a linear or circular calcification exists in the region of a ductus arteriosus (Fig. 10-92). The significance of this deposition of calcium usually is unclear; it may be a result of either an episode of aortitis or atherosclerosis at the aortic end of the duct. A calcified ligamentum arteriosus implies a closed ductus.
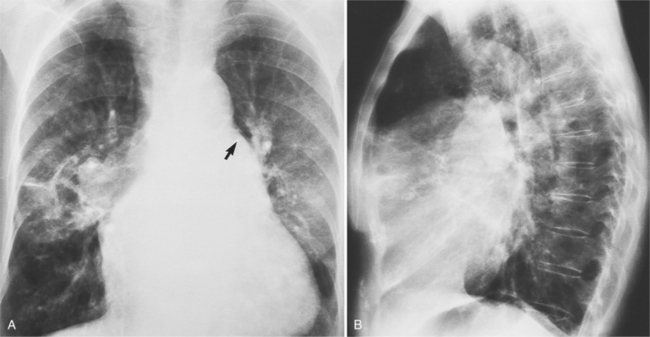
Angiographic Examination
In the cardiac catheterization laboratory, a PDA can be evaluated by cineangiography in several ways. A catheter advanced up the inferior vena cava can usually be manipulated out of the pulmonary artery, through the duct, and into the descending aorta. In the frontal projection, the shape of the catheter looks like a musical treble clef (Fig. 10-93).
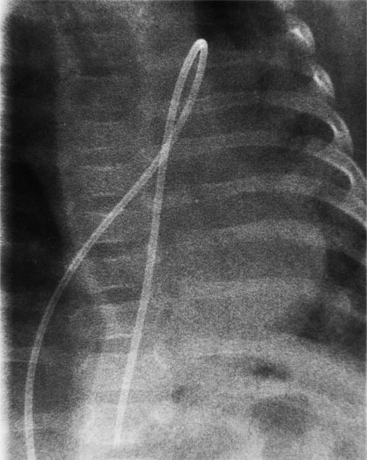
When the flow through the ductus arteriosus is left to right, if the duct is not seen with ultrasound, aortography provides the best visualization of the ductus (Fig. 10-94) and of associated juxtaductal abnormalities, such as aortic coarctation. Similarly, when the flow is reversed, a pulmonary artery injection defines the ductal anatomy, but the proximal aortic arch is usually not visualized. The flow through the ductus recorded by angiography depends on the force of the injection and the natural direction of shunting. In addition, most shunts have some degree of bidirectional flow so that contrast material occasionally seems to oscillate from aorta to pulmonary artery and back.
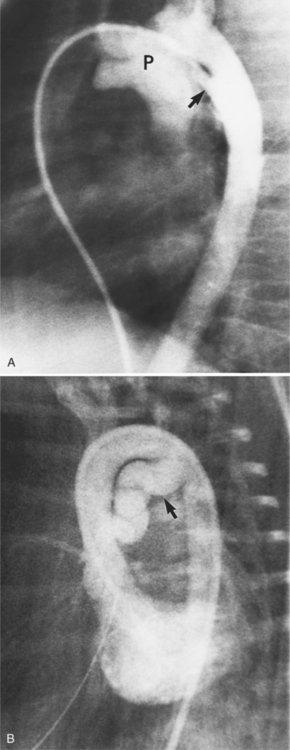
A PDA may exist with other more complex cardiac defects. When a ventricular septal defect exists, a left ventriculogram is inadequate for identifying a PDA. In this case, the mixing through the ventricular septal defect will opacify the arch of the aorta and the main right and left pulmonary arteries nearly simultaneously, thereby obscuring the ductus. In this situation and in other connections between the aorta and pulmonary artery, such as truncus arteriosus, aortic-pulmonary window, or other aortic-pulmonary fistulas, an injection must be made in the descending aorta at the mouth of the suspected ductus. In a similar fashion, the rare anomaly of a bilateral ductus arteriosus, which forms a vascular ring around the trachea and esophagus, necessitates an injection in the proximal descending aorta with 30 to 40 degrees of cranial angulation to project both ductus from their respective aortic arches in the right and left anterior oblique projection.
Single Ventricle
Definition
A common ventricle is not a single ventricle because the atrioventricular valves enter separately on each side of the rudimentary interventricular septum. The interventricular septum has either single or multiple large defects with the remaining ventricular muscle having recognizable elements of right ventricular morphology on one side and of left ventricular morphology on the other.
Chest Film Features
An inverted ventricle is the only distinctive sign of single ventricle that separates it from all other types of CHD on a chest film with a large heart and shunt vascularity. The trabecular pouch or rudimentary outflow chamber is anterior, superior, and to the left. In this position, the outflow chamber has a convexity separate from the other cardiac chambers, projecting where the left atrial appendage would normally be (Fig. 10-95). This bulge extends along the lateral border of the cardiac silhouette as far as and frequently further than does the usual enlarged left atrial appendage. The pulmonary vasculature has a shunt pattern unless there is severe pulmonary stenosis. The hilar arteries are quite large, particularly those on the right side, which are not hidden behind other mediastinal structures.
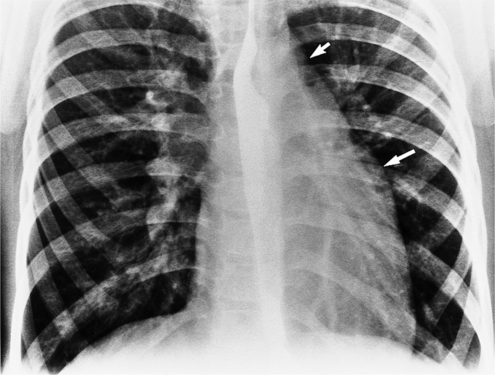
Imaging Examination
The cineangiographic evaluation starts with a biplane ventriculogram in the frontal and lateral projections (Fig. 10-96). However, the cranially angled views in the left anterior oblique projection better delineate the straddling atrioventricular valves and the trabecular pouch (Fig. 10-97). Selective injections in the subaortic and subpulmonary regions or in the trabeculated pouch help to determine whether the great arteries originate from an outlet chamber.
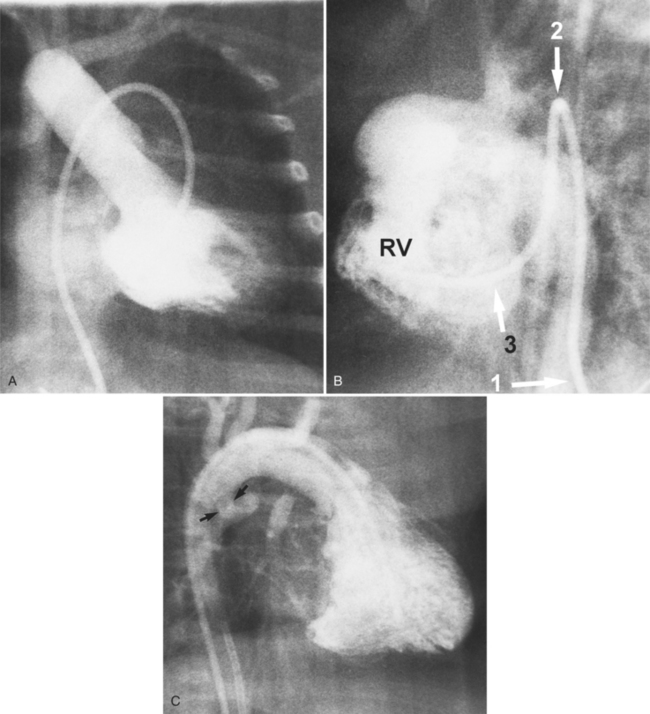
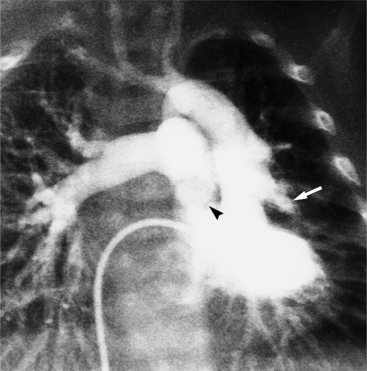
Tricuspid Atresia
Characteristics
Tricuspid atresia is characterized by congenital absence of the morphologic tricuspid valve. This anomaly, which results in cyanosis, is always associated with a group of other malformations. There is an interatrial communication and enlargement of the left ventricle and mitral valve. The right ventricle is always hypoplastic (Fig. 10-98). Variable features include a ventricular septal defect, malposition of the great arteries, abnormalities in the ventriculoarterial connections, and obstruction to pulmonary blood flow.
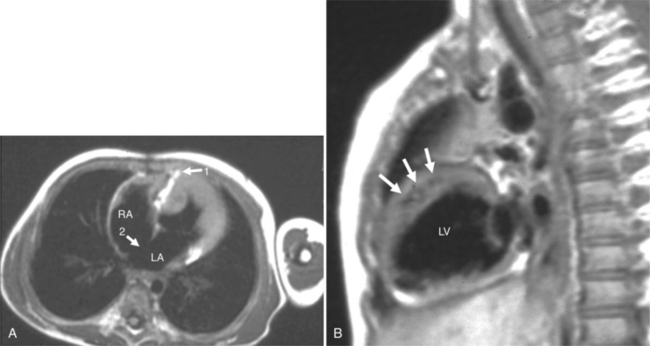
Anatomy
Both the left ventricle and the mitral valve are relatively large compared with the remainder of the heart. The left ventricle is both dilated and hypertrophied and occupies the diaphragmatic and anterolateral positions in the mediastinum that are usually taken by the right ventricle. In addition, the remainder of the left ventricle lies in its usual lateral and posterior location. The right ventricle is always hypoplastic, ranging from a moderate-sized chamber in some hearts to a thin sinus only a few millimeters long in the most extreme cases. A ventricular septal defect may be present, and when it exists, frequently serves as another location to obstruct pulmonary blood flow. In the absence of a ventricular septal defect, the lungs are supplied by a patent ductus arteriosus and other bronchial collaterals.
Chest Film Abnormalities
The chest film in tricuspid atresia illustrates that phrases such as “left (or right) ventricular enlargement,” which are appropriate for acquired diseases, are inappropriate in discussing certain types of CHD. The position, size, and relation of the chambers in tricuspid atresia are not those of a normal adult heart. Yet the cardiac silhouette sometimes has an apex that points inferiorly and to the left, which in acquired heart disease resembles left ventricular enlargement. Other cases of tricuspid atresia have a cardiac silhouette that resembles the coeur en sabot appearance of right ventricular enlargement in tetralogy of Fallot. Obviously, because the right ventricle is never enlarged in tricuspid atresia, this latter interpretation is fallacious. Similarly, when the ventricles are inverted or TGA is present, the usual position of the heart and mediastinal structures is altered so that their position in the cardiomediastinal silhouette may be unknown. In these instances, reference to a particular chamber of the heart on the chest film may be incorrect.
The size of the heart on the frontal radiograph is typically normal to slightly enlarged in tricuspid atresia (Fig. 10-99). The right atrial segment has slight rounding at the superior vena caval border, indicating atrial enlargement. The inferior half of the right atrial segment has a straight or sometimes concave appearance, the latter suggesting juxtaposition of the atrial appendages. When there is decreased pulmonary flow, the pulmonary artery segment is flat or concave. In this situation, the heart size tends to be normal, and the cardiac silhouette may appear to have an uplifted apex. This coeur en sabot shape is identical to that seen in tetralogy of Fallot. When pulmonary artery flow is increased, indicating no obstruction to pulmonary flow, the overall heart size may become quite large, and transposition of the great arteries is quite likely.
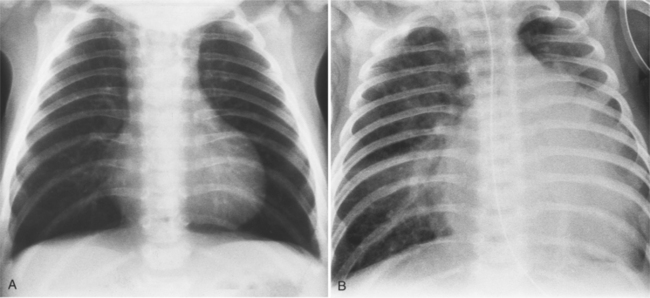
Angiographic Findings
Right atrial angiography in the frontal projection delineates most of the features of tricuspid atresia (Fig. 10-100). The flow of contrast material is from right atrium to left atrium to left ventricle; the right ventricle opacifies later than the left ventricle. A triangular lucency representing the right ventricle exists inferiorly between the medial border of the right atrium and the opacified left ventricle. This defect is typical of tricuspid atresia but may rarely be seen in congenital tricuspid stenosis (Fig. 10-101).
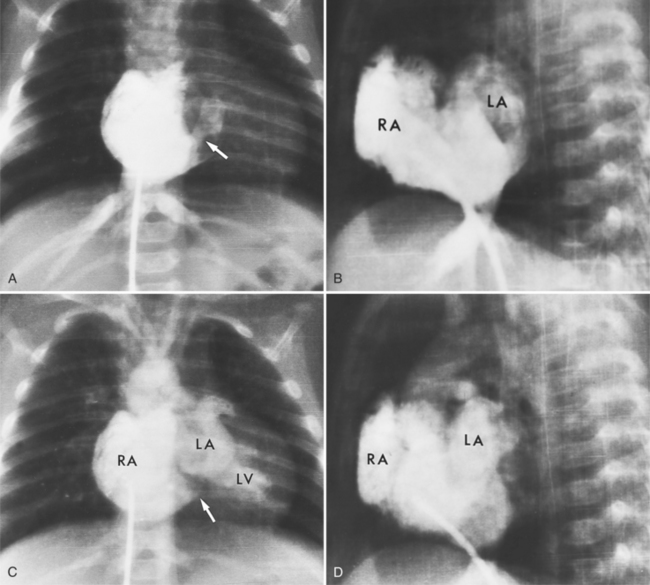
In tricuspid atresia, the left ventriculogram shows a large chamber and mitral valve. The ventricular septal defect, if present, is usually between the membranous septum and the lower muscular septum (Fig. 10-102). In the lateral view, origin of the aorta anterior to the pulmonary artery indicates transposition. The pulmonary valve and subpulmonary region may be clearer on a cranially angled left anterior oblique projection.
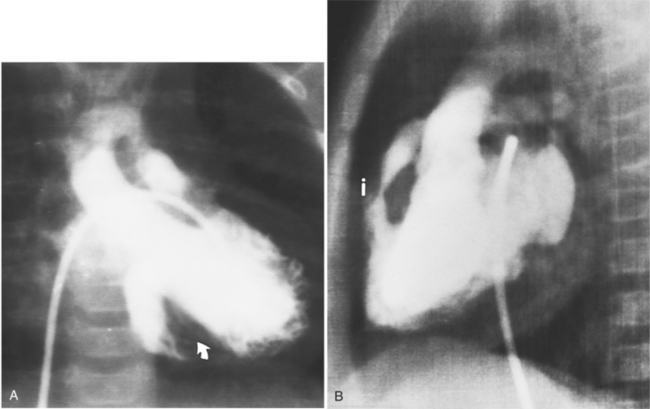
The Fontan Operation
The Fontan procedure was originally performed in patients with tricuspid atresia and the indications for the operation have been extended to palliate many forms of CHD that have a functionally single ventricle (such as hypoplastic left heart syndrome) or cannot undergo a biventricular repair (Table 10-7). A surgically created pathway reroutes the systemic venous return from the inferior vena cava and superior vena cava directly to the pulmonary arteries (Fig. 10-103). The Fontan procedure effectively separates the pulmonary and systemic circulations and creates one circuit of blood flow with the dominant ventricle functioning as the single pumping chamber for the systemic circulation.
TABLE 10-7 Types of congenital heart disease requiring Fontan
| Tricuspid atresia |
| Hypoplastic left heart syndrome |
| Double inlet ventricle |
| Heterotaxy |
| Pulmonary atresia with intact ventricular septum |
| Ebstein anomaly |
| Straddling atrioventricular valve |
| Crossed atrioventricular connections |
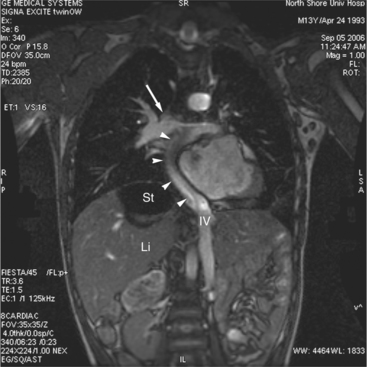
In the early versions of the Fontan operation, the right atrial appendage was directly anastomosed to the main or right pulmonary artery. Over time, progressive right atrial dilatation occurred (Fig. 10-104) within the pathway because of relative stasis of slow flowing blood, leading to complications of right atrial thrombus, pulmonary vein compression, and arrhythmia (Fig. 10-105). The operation has been changed over the years and now the lateral tunnel or extracardiac conduit approaches are used to direct deoxygenated blood from the inferior vena cava and superior vena cava to the pulmonary arteries. Usually the Fontan operation is preceded by the bidirectional Glenn shunt that directs blood from the superior vena cava to the pulmonary arteries (Figures 10-103, 10-106). In some centers, at the time of Fontan completion, a fenestration is created between the inferior vena cava pathway or lateral tunnel and the right atrium to reduce early complications from pulmonary overcirculation and increased pulmonary pressures. The fenestration is later closed with an occlusion device that is placed percutaneously.
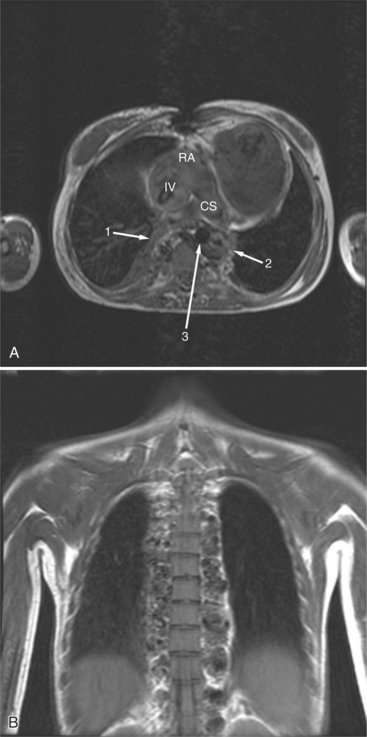
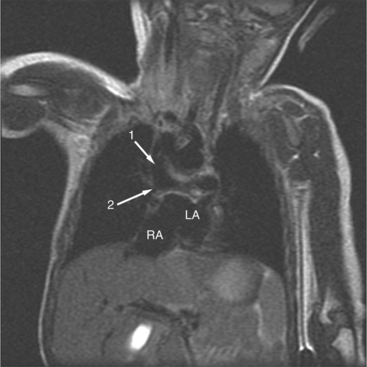
Cardiac MR is now used to assess for all of these possible levels of obstruction as well as quantification of ventricular function, valvular regurgitation, and differential pulmonary blood flow. Both cardiac MR and MDCT are helpful to evaluate for systemic to pulmonary vein collaterals and pulmonary arteriovenous malformations, which can contribute to significant cyanosis and can be treated by coil occlusion. Patients who have older versions of the Fontan surgery and have developed complications are now often referred to cardiac MR for morphologic and functional evaluation in preparation for surgical revision to either the lateral tunnel or extracardiac conduit Fontan pathways (Table 10-8).
TABLE 10-8 Complications after the Fontan procedure
| Right atrial enlargement and hepatic dysfunction |
| Fontan pathway narrowing or baffle leaks |
| Ventricular dysfunction and failure |
| Atrioventricular and aortic valvular regurgitation |
| Ventricular outflow tract obstruction |
| Thromboembolic events |
| Pulmonary arteriovenous malformations |
| Pulmonary venous obstruction |
| Systemic to pulmonary vein collateralization |
| Atrial arrhythmia |
In general, patients with CHD are at increased risk for developing thromboembolic complications as a result of sluggish flow through dilated chambers, presence of prosthetic devices, conduits and baffles, and use of central venous catheters and cardiac catheterization. Patients who are status post Fontan are at particularly high risk for development of pulmonary embolism that may be clinically silent, and chronic pulmonary embolus can result in an increase in pulmonary vascular resistance that can lead to failure of the Fontan circulation. These patients require routine surveillance for thrombus throughout the Fontan pathway and pulmonary arteries with either cardiac MR or MDCT angiography.
Many patients with CHD can develop conduction abnormalities or arrhythmias related to the surgical repair (especially following the Mustard or Senning or Fontan operations) or related to abnormal intracardiac connections, such as in congenitally corrected TGA. An indwelling pacemaker and retained pacing wires or leads are contraindications for MR imaging, and MDCT can be used as an alternative method for both morphologic and functional imaging in these patients.
Pulmonary Atresia with Intact Ventricular Septum
Chest Film Abnormalities
The typical features of pulmonary atresia on the chest film are a large cardiac silhouette, a concave pulmonary artery segment, and diminished pulmonary vasculature (Fig. 10-107). The severity of each of these abnormalities depends on the degree of obstruction in the pulmonary circuit and the competence of the tricuspid valve. The pulmonary vessels at birth are visibly small, and in extreme cases, the pulmonary vessels beyond the hila are invisible. The pulmonary artery segment in older children appears concave, reflecting the hypoplasia of the main and left pulmonary arteries. In newborns, the thymus may obscure the entire superior mediastinum so that the true pulmonary artery segment and the hila are not visible. When atresia of the pulmonary valve is associated with a normal-sized main pulmonary artery, the pulmonary artery segment is straight.
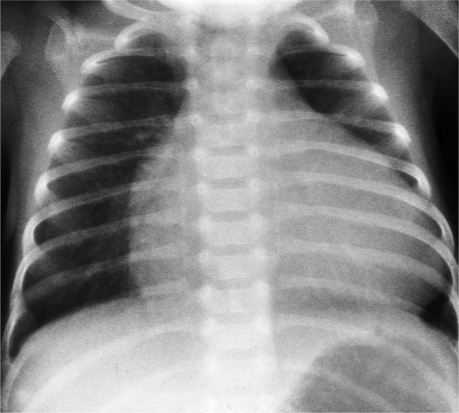
Imaging Evaluation
The primary diagnosis is usually established by echocardiography. The size of the pulmonary arteries may require MRI or angiography before surgery. Depending on the size of the cavity, a hand injection or pressure injection with reduced volume generally shows an irregular and highly trabeculated chamber and documents the pulmonary atresia and intact septum (Fig. 10-108). The amount of contrast material opacifying the right atrium reflects the degree of tricuspid regurgitation. A severely dilated right atrium and reflux into the superior or inferior vena cava are signs of severe tricuspid insufficiency.
A striking appearance on the right ventriculogram is sinusoids leading to the coronary arteries (see Figure 10-108). These connections between the right ventricle and the coronary arteries alter the coronary flow and produce ischemia. Left coronary artery flow in the normal heart is mainly diastolic. In pulmonary atresia, the coronary flow from the hypertensive right ventricular sinusoids partially reverses the normal antegrade direction in the coronary arteries, leading to inadequate oxygenation of the myocardium and myocardial infarction.
The central pulmonary arteries have a characteristic seagull shape when the pulmonary valve is atretic. The right and left pulmonary arteries are the wings of the bird, the main pulmonary artery is the head, and the atretic valve is the beak (Fig. 10-109). If the pulmonary arteries are confluent, a conduit between the right ventricle and the confluence might be constructed, whereas if the arteries are not confluent, a systemic-pulmonary artery anastomosis might be attempted.
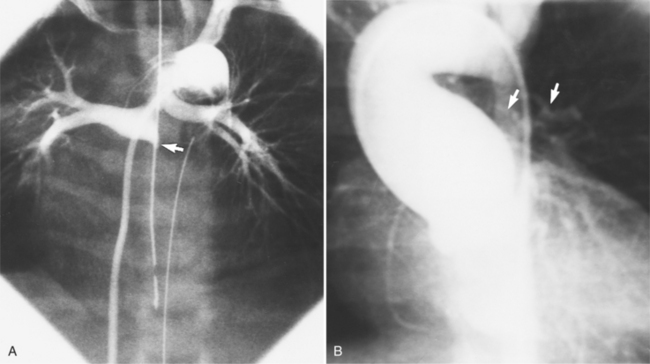
[/level-membership-for-cardiovascular-category][not-level-membership-for-cardiovascular-category]
Chapter 10 Congenital Heart Disease
Over one million adults have congenital heart disease (CHD) in the United States. For the first time, there are now more adults living with CHD than children. The reported incidence of CHD varies widely because of differences in counting minor lesions at birth (such as bicuspid aortic valve, small ventricular septal defects, silent patent ductus arteriosus, and anomalous pulmonary veins), and increasingly better diagnostic methods. The incidence of severe CHD requiring surgery is about 3 per 1000 live births. More accurate diagnosis at presentation and improved medical, surgical, and postoperative care have greatly improved survival so that 90% of children born today with CHD will reach adulthood.
SEGMENTAL ANALYSIS OF CARDIAC MALFORMATIONS
The steps for the segmental diagnostic approach to cardiac malformations are:
Cardiac Axis and Visceral Situs
The situs is determined by chest and abdominal radiographs. Situs solitus and situs inversus are recognized by the asymmetry of the tracheobronchial tree and by the positions of the abdominal organs. Symmetrically lobed lungs (Fig. 10-10), midline liver, gastrointestinal malrotations, asplenia, and polysplenia denote the heterotaxy syndrome (Fig. 10-11), which can also be recognized by isomerism of the atria. Isomerism means that both atria have features of the right atrium or of the left atrium. The visceral-atrial rule is that the right and left atria develop on the same side as the thoracic and abdominal viscera do. In situs solitus, the right atrium is on the right side of the mediastinum and the left atrium is on the left side. In situs inversus, the morphologic right atrium is on the left side and the left atrium lies on the right side. In situs ambiguus, right and left sides cannot be determined because the lungs and abdomen are symmetric. For example, in asplenia there are two right (trilobed) lungs and both atria are morphologically right atria. In polysplenia there are two (bilobed) left lungs and both atria are morphologic left atria.
In most people, the major portion of the heart lies slightly to the left of midline. The cardiac apex denotes the location of the heart within the thorax. Dextrocardia (Fig. 10-12), levocardia (Fig. 10-13), and mesocardia then indicate the possible positions of the heart. Using this terminology, a cardiac malposition is any heart that does not have a leftward cardiac axis in situs solitus. A malposition includes dextrocardia in situs solitus and levocardia in situs inversus (Fig. 10-14), as well as dextrocardia in situs inversus. All these positions represent deviation from normal embryologic development without necessarily implying any hemodynamic or morphologic derangement.
In primary dextrocardia, the main defect is in the heart. There are two types of primary dextrocardia: (1) dextroversion, in which the heart is rotated or pivoted so that its apex lies on the right side with the atria as a fulcrum; and (2) mirror-image dextrocardia. In secondary dextrocardia, the heart is normal but the mediastinum is shifted to the right because of extracardiac abnormalities that involve the lungs, pleura, or skeleton (Box 10-1). Examples of the latter include pneumothorax, congenital herniation of the gastrointestinal tract into the thorax, and thoracolumbar scoliosis (Fig. 10-15).
Atrial Morphology
With rare exceptions, the morphology of the atria corresponds closely with the situs of the tracheobronchial tree and the abdominal viscera. Of the various criteria for distinguishing between right and left atria, the most reliable are the shape of the atrial appendage (see Figure 10-1) and the connection to the inferior vena cava (Box 10-2). The right atrial appendage is broad and pyramidal, whereas the left atrial appendage is thin with a narrow neck. The inferior vena cava almost always connects with the right atrium. This is true even in the “absence” of the inferior vena cava and azygos continuation. In this entity, there is no intrahepatic portion of the cava but the hepatic veins connect to the subdiaphragmatic portion of the inferior vena cava, which joins the right atrium.
Ventricular Morphology
When the right and left ventricles are normal, identification of the two ventricles is relatively simple (Box 10-3). The normal right ventricle has coarse, trabeculated walls when compared with the smooth-walled left ventricle. The right ventricle has a contractile muscle called the conus or infundibulum between the tricuspid and pulmonary valves (see Figure 10-2), whereas the left ventricle has mitral-aortic continuity with no intervening muscle (Fig. 10-16). The right ventricle has trabeculations and papillary muscles on its septum, whereas in the left ventricle these structures are not present on the septum. A bicuspid (mitral) atrioventricular valve is a part of the left ventricle, whereas a tricuspid atrioventricular valve is part of the right ventricle, although either of these valves may have a cleft or be absent.
Box 10-3 Normal ventricles
| Right Ventricle | Left Ventricle |
|---|---|
| Coarse, trabeculated walls | Smooth walls |
| Contractile muscle (conus, infundibulum between tricuspid and pulmonary valves) | Mitral aortic continuity with no intervening muscle |
| Trabeculation and papillary muscles on septum | Septum free from trabeculations and papillary muscles |
| Tricuspid atrioventricular valve | Bicuspid (mitral) atrioventricular valve |
| Complex triangular shape | Spheroidal shape |
There is general agreement that an inflow tract must be present for a chamber to be considered a ventricle. The trabecular portion determines whether the chamber is of the right or left ventricular type. In these instances, the single ventricle consists of one large chamber that receives both atrioventricular valves. (Note that this definition excludes mitral or tricuspid atresia.) If only the trabecular and outflow segments are present, this structure is called an outlet chamber. Examples of such hearts are the univentricular heart of the left ventricular type, with or without a rudimentary outflow chamber.
Atrioventricular Connections
The atrioventricular connections are called concordant when the right atrium connects to the right ventricle (see Figure 10-4) and the left atrium connects to the left ventricle. When the right atrium connects to the left ventricle and the left atrium connects to the right ventricle, the ventricles are discordant in relation to the atria (see Figure 10-5). In the heterotaxy syndrome, in which either two right atria or two left atria may exist, the atrioventricular connection is ambiguous. This schema is less clear when either atresia of one of the atrioventricular valves exists or when one of the atrioventricular valves straddles the interventricular septum. When there is a double-inlet or a straddling atrioventricular valve, the tensor apparatus (the attachments of the chordae tendineae) may connect to either side of the interventricular septum.
Relations of the Great Arteries
The position of the aorta is described relative to the pulmonary artery in both the anteroposterior and lateral planes (Fig. 10-17). In the normal heart, the aorta is to the right of and posterior to the pulmonary artery (see Figure 10-6). An anterior aorta to the right of the pulmonary artery is common in transposition of the great arteries (TGA; see Figures 10-7, 10-9). An aorta to the left of and anterior to the pulmonary artery is typical in, but not diagnostic of, corrected transposition (Fig. 10-18). Then there is a characteristic leftward convexity of the aorta (Fig. 10-19).
Ventriculoarterial Connections
The pulmonary valve is part of the pulmonary artery (not part of the right ventricle) and the aortic valve is part of the aorta. When the pulmonary artery or the aorta is related to, or overrides more than 50% of, a particular ventricle, it is defined as being connected to that ventricle. This association is particularly strong when there is a continuity between an atrioventricular valve and the semilunar valve. Concordant connections exist when the left ventricle is connected to the aorta and the right ventricle to the pulmonary artery (see Figures 10-2, 10-8). Discordant connections result when the left ventricle is connected to the pulmonary artery and the right ventricle to the aorta (see Figure 10-9). This latter connection is also called transposition. When both great arteries arise predominantly from one ventricle, there is a double-outlet right ventricle or double-outlet left ventricle. The final type of arterial connection is a single-outlet heart, of which there are three varieties:
The ventricular outflow tracts may be one of four distinct types:
Certain types of conus are associated with particular malformations:
Associated Malformations
Cardiotyping
At this point in the segmental analysis of the heart, the atrial and ventricular situs, the atrioventricular connections, the ventriculoarterial connections, and the position of the aorta are known. This data can be expressed in a notation developed by Van Praagh to categorize all possible types of hearts (Box 10-4). The situs of the atria, as indicated by the position of the abdominal viscera and the trachea and lungs, is designated as solitus (S), inversus (I), or ambiguus (A). The ventricular situs is characterized as a D-loop (D), L-loop (L), or undiagnosed loop (X). The position of the aorta in relation to the pulmonary valve is to the right (D), to the left (L), or directly anterior (A). These three letters are written in sequence (atrial situs, ventricular situs, and aortic situs). Examples are a normal heart (S,D,D), a dextrotransposition of the great arteries (S,D,D), and a congenitally corrected TGA or levotransposition (S,L,L) in which the D- and L- indicate the aortic position.
Box 10-4 Cardiotypes
DETERMINE ATRIAL SITUS BY ANALYZING ABDOMEN AND TRACHEOBRONCHIAL TREE
| S | solitus |
| I | inversus |
| A | ambiguus |
DETERMINE VENTRICULAR SITUS
| D | D-loop or solitus |
| L | L-loop or inverted |
| X | X-loop or undiagnosed |
DETERMINE AORTIC SITUS IN RELATIONS TO PULMONARY VALVE
| D | Normally related great arteries with aorta to the right of the pulmonary artery |
| L | Inverted great arteries with aorta to left of pulmonary artery |
| A | Aortic valve is directly anterior to pulmonary valve |
NOTATION (ATRIA, VENTRICLES, GREAT ARTERIES)
| Normal | (S,D,D) |
| D-transportation of great arteries | (S,D,D) |
| L-transportation of great arteries | (S,L,L) |
| Situs inversus | (I,L,L) |
| Asplenia, dextrocardia, transposition of great arteries | (A,L,L) |
MALPOSITIONS AND ABNORMAL CONNECTIONS
Cardiac Connections and Positions
Dextrocardia
Dextrocardia signifies that the apex of the heart is directed toward the right. Primary dextrocardia exists because of an embryologic abnormality. This type of dextrocardia can exist with any type of situs position (Fig. 10-21). When dextrocardia exists with situs inversus, the atrial and ventricular relations are a mirror image of their positions in the usual situs solitus. When the dextrocardia exists in situs solitus, the term isolated dextrocardia is frequently applied. It is clear then that dextrocardia can occur in situs solitus, inversus, and ambiguus. Many associated cardiac anomalies exist in primary dextrocardia. Frequent conditions include ventricular septal defect, TGA, corrected TGA, double-outlet right ventricle, and juxtaposition of the atrial appendages.
The goal of echocardiography, magnetic resonance imaging (MRI), and angiography is to define the position and location of each chamber of the heart and their connections and relations with one another and with the great arteries. In those malpositioned hearts in which the location of the interventricular septum is not known before angiography, posteroanterior and lateral projections serve as initial guidelines. Frequently, the projections can be reversed for a malposition; that is, those structures that are normally best seen in the left anterior oblique projection in the normal heart would be studied in the right anterior oblique projection in dextrocardia. As a rule, the dextrocardia itself does not cause clinical problems but rather the associated malformations mandate medical or surgical alleviation.
Levocardia
Strictly speaking, levocardia means that the cardiac apex is left sided. Isolated levocardias are those hearts that are left sided when situs inversus is present. This anomaly occurs in less than 1% of all patients with congenital cardiac malformation compared with a 2% incidence of dextrocardia in patients with CHD. With levocardia, the position of the thoracic and abdominal organs ranges from partial to complete situs inversus and also to heterotaxy (Fig. 10-22). Severe malformations are always associated with levocardia and frequently include ventricular septal defect, complete atrioventricular canal defects, and pulmonary stenosis or atresia. Isolated levocardia may be suspected on the chest film with a right-sided stomach bubble and left-sided liver shadow and a left cardiac apex. In contrast, in extrinsic levocardia the heart is intrinsically normal but the mediastinum is shifted from skeletal or pulmonary abnormalities (Fig. 10-23).
Heterotaxy and the Syndromes of Asplenia and Polysplenia
Heterotaxy is the failure of the developing embryo to establish normal left-right asymmetry. Asplenia and polysplenia are part of this spectrum. In these patients the thoracic and abdominal contents have a degree of symmetry, unlike those in situs solitus or inversus in which right- and left-sided organs exist together. In the thorax, both lungs may be trilobed with bilateral epiarterial bronchi, or both lungs may be bilobed with bilateral hypoarterial bronchi. In the abdomen, the asymmetry is also frequently lost. The liver may be midline. The attachment of the mesentery, which usually runs from the left upper quadrant to the right lower quadrant, may have a midline attachment. The spleen may be absent (asplenia), bilobed with multiple accessory spleens, or multiple small spleens (Fig. 10-24) may be found throughout the mesentery (polysplenia). Situs ambiguus exists either when the right and left sides of the lungs, heart, and abdomen are similar or where a right-left relationship is difficult to identify. Boxes 10-5 and 10-6 summarize the characteristics of asplenia and polysplenia.)
Splenic anomalies with malpositions and malformations in multiple organ systems have been recognized since 1826 when Martin and later Ivemark described the absence of the spleen in cyanotic CHD. Complex cardiac malformations are typical when the type of thoracic and abdominal situs abnormality is uncertain or has features of both situs solitus and situs inversus (Table 10-1).
TABLE 10-1 Cardiovascular abnormalities in asplenia and polysplenia
| Abnormality | Asplenia (%) | Polysplenia (%) |
|---|---|---|
| SUPERIOR VENA CAVA | ||
| Bilateral | 53 | 33 |
| Right | 34 | 33 |
| Left | 10 | 33 |
| Uncertain | 3 | — |
| INFERIOR VENA CAVA | ||
| Right sided | 60 | — |
| Left sided | 28 | — |
| Uncertain | 12 | — |
| Azygos continuation | — | 84 |
| Anomalous pulmonary veins | 84 | 50 |
| Total anomalous connection | 72 | — |
| Partial anomalous connection | 12 | — |
| CARDIAC APEX | ||
| Left | 56 | 58 |
| Right | 41 | 42 |
| Uncertain | 3 | — |
| AORTIC ARCH | ||
| Left | 56 | 33 |
| Right | 38 | 67 |
| Unknown | 6 | — |
| Great vessels | ||
| Normally related | 19 | 84 |
| Transposition of great arteries | 72 | 8 |
| Double-outlet right ventricle | 9 | 8 |
| PULMONARY VALVE | ||
| Normal | 22 | 58 |
| Stenosis | 34 | 33 |
| Atresia | 44 | 9 |
| Patent ductus arteriosus | 56 | 50 |
| Absent coronary sinus | 85 | 42 |
| Single ventricle | 44 | 8 |
| Ventricular septal defects | 90* | 67 |
* Of the ventricular septal defects in asplenia, 84% were of the atrioventricular canal type.
Modified from Rose V, Izukawa T, Moes CAF: Syndromes of asplenia and polysplenia; a review of cardiac and non-cardiac malformations in 60 cases with special reference to diagnosis and prognosis. Br Heart J 37:840-852, 1975.
Segmental analysis of the defects in hearts associated with asplenia begins with the atria and the atrial septum. On the chest film, the external contours of the heart frequently do not conform to the expected heart chambers (Fig. 10-25). Almost all these hearts show a common atrioventricular valve, frequently associated with separate, large atrial septal defects in the primum and secundum location. The size and location of these atrial defects are such that the malformation is called a common atrium. The ventricles also almost invariably have major malformations. About one fourth of the ventricles are inverted (as seen in corrected transposition), and half of the hearts have a univentricular chamber with a rudimentary outflow tract.
Anomalies of the great vessels, including TGA and double-outlet right ventricle, have an incidence of 3% to 30%. Angiographically, the posteroanterior and lateral projections are best to allow identification of their right-left relationships. Anomalies in the ventricular septum and semilunar valve stenosis are common, so that filming is also done with the x-ray beam parallel to the interventricular septum with cranial angulation. Because two thirds of persons with asplenia have anomalous systemic venous or pulmonary venous connections, or both, these malformations frequently complicate catheterization.
The features of the heart in polysplenia are quite variable, and in fact, there is occasionally no cardiac malformation. With a femoral vein approach, you can recognize azygos continuation by the course of the catheter around the azygos arch (Figures 10-10A, 10-26). You should not make the diagnosis of tricuspid atresia if the catheter tip fails to pass leftward through the heart above the diaphragm but instead should continue advancing the catheter superiorly until it goes around the azygos arch.
Atrioventricular Discordance
In the schema of segmental cardiac analysis, after recognition of the atria and ventricles, the next step is to determine whether the connections between them are concordant or discordant. Atrioventricular discordance means that the right atrium is connected to the left ventricle and the left atrium is connected to the right ventricle (see Figures 10-4, 10-5). Implicit in this definition is the presence of two atria, two atrioventricular valves, and two ventricles. This diagnosis is not appropriate when atrial identification is indeterminate in situs ambiguus or when there is a single common atrioventricular valve. Similarly, distinct right and left ventricles are necessary for this definition, although a ventricular septal defect may exist.
Congenitally Corrected Transposition of the Great Vessels (Levotransposition of the Great Arteries)
In 1875 Rokitansky reported a form of transposition in which blood passed in normal serial fashion through the pulmonary and systemic circuits. The right atrium was connected to the left ventricle, which was connected to the pulmonary artery. On the oxygenated side of the lungs, the left atrium was connected to the right ventricle, which was connected to the aorta. The atrioventricular valves always correspond with their ventricles, even when there is atrioventricular discordance. That is, the mitral valve is a left ventricular structure, and the tricuspid valve is a right ventricular structure. In congenitally corrected transposition of the great vessels, the aorta lies to the left of and anterior to the pulmonary artery, whereas the pulmonary valve lies to the right and posterior. The aortic valve is usually somewhat anterior to the pulmonary valve, although the two great vessels may be exactly lateral to each other. The ascending aorta frequently has an unusual course, passing in a direction toward the left shoulder so that occasionally a distinctive contour in the left side of the mediastinum is visible on the chest film.
If there are no other defects, this malformation causes no hemodynamic problems and may go undetected during a normal life span. Unfortunately, associated malformations are the rule, and their site and severity determine the clinical course. Ventricular septal defects are frequent (Figures 10-27, 10-28) and may be large enough to cause pulmonary arterial hypertension. These defects are usually in the membranous septum adjacent to the pulmonary valve; muscular defects and supracristal defects are less common. Generally, the left-sided atrioventricular valve (i.e., the valve between the left atrium and the right ventricle) is displaced slightly into the ventricle in a manner resembling Ebstein anomaly. If the displacement is more than a few millimeters (because the tricuspid valve is usually displaced to the apex by that amount), the diagnosis of Ebstein anomaly is quite likely. Pulmonary stenosis is frequently associated with ventricular septal defect and may be caused by a malformed valve, a subpulmonary membrane, aneurysms of the membranous ventricular septum, or rarely, accessory tissue in the atrioventricular valve or a muscular bar in the subpulmonary region.
Imaging establishes the atrioventricular connections, the morphology of the ventricles, and the position of the aorta and pulmonary artery. The position of the venous and arterial catheters frequently give the first clue to a corrected transposition (Fig. 10-29). In situs solitus and levocardia, the venous catheter passes through the heart in the midline to reach the pulmonary arteries. The catheter in the pulmonary artery is posterior to its usual location, which is where the aorta should be in normal hearts. The retrograde arterial catheter has a distinctive curve in the ascending aorta as its course becomes convex medially and to the left before entering the heart (see Figure 10-18). On the lateral view, the aortic catheter is anterior and superior to the venous catheter. The venous and arterial catheters indicate the fundamental relationship between the aorta and the pulmonary artery in corrected transposition with situs solitus and levocardia; the pulmonary artery lies to the right and posterior, whereas the aorta is anterior and to the left.
In the sagittal view, the left ventricle appears to “stand on its apex” with a conical shape whose apex is in the diaphragmatic-sternal angle. The anterior wall of the left ventricle extends superiorly into a distinctive pouch that is characteristic of inverted ventricles, namely the anteriorly placed left ventricle (Fig. 10-30). This recess is separate from both the mitral and pulmonary valves and is the most anterior and superior structure of either ventricle. The outflow portion of the left ventricle in the lateral projection is posterior and connects to a pulmonary artery, which is beside or posterior to the aorta. The posterior wall of the left ventricle beneath the pulmonary valve is the membranous septum, and the anterior wall forms a neck above the blind recess and below the pulmonary valve.
In the frontal view, the right ventricle is to the left of and slightly superior to the left ventricle (Fig. 10-31). In this position, the right ventricle has an oval to triangular shape and the usual coarse trabeculations. The tricuspid annulus is in the posteroanterior plane separated from the aortic valve by the muscular infundibulum. This morphologic right ventricle connects with the left atrium. The crista supraventricularis in this projection is the medial wall of the infundibulum above the tricuspid valve. In the lateral projection, the crista is the posterior wall of the infundibulum and separates the tricuspid from the aortic valve.
About one third of patients with corrected transposition have tricuspid regurgitation (i.e., from the right ventricle connected to the left atrium). The apically displaced tricuspid leaflets of Ebstein anomaly are usually the cause of the regurgitation, but there are occasionally other leaflet abnormalities. When there is severe regurgitation in infants, the details of the leaflets and the origin of their insertion are frequently difficult to identify. If technical factors such as arrhythmia and catheter position can be excluded, it should be presumed that severe regurgitation into the left atrium is associated with a “left-sided” Ebstein anomaly (Fig. 10-32). The tricuspid annulus is adjacent to the right coronary artery, which may be opacified during the ventriculogram.
Coronary Artery Patterns
The coronary anatomy in congenitally corrected transposition of the great vessels is unique to inverted ventricles. The right coronary artery supplies the morphologic right ventricle and the left coronary artery provides an anterior descending branch in the interventricular sulcus and a variable circumflex branch over the morphologic left ventricle (Fig. 10-33). In congenitally corrected TGA, the right coronary artery passes to the left and inferior in the atrioventricular groove between the left atrium and right ventricle. Distally, this artery branches into the atrioventricular nodal branch, the posterior descending artery, and a variable set of branches to the inferior portion of the left ventricle (Fig. 10-34). The marginal branches over the right ventricular epicardial surface tend to be large with numerous branches. In contrast, the left coronary artery lies anterior and to the right of the right coronary artery. The left main coronary artery continues mainly as the anterior descending branch, which has numerous septal and diagonal branches. The circumflex artery in the atrioventricular groove between the right atrium and left ventricle tends to be vestigial. The position of the coronary arteries within the thorax may be different because of dextrocardia or other relative rotations, but the coronary distribution corresponds uniquely to the respective ventricle. When confusing ventricular morphology does not allow identification of the right or left ventricle, visualization of the coronary arteries permits accurate identification of the ventricles.
Isolated Ventricular Inversion
Atrioventricular discordance with ventriculoarterial concordance is termed isolated ventricular inversion. The segmental connections for the venous side of the heart are right atrium to left ventricle to aorta, and for the systemic side left atrium to right ventricle to pulmonary artery. As originally reported by the Van Praaghs (1966), the pulmonary artery arises anteriorly and to the left of the aorta. A large subaortic ventricular septal defect is adjacent to the septal leaf of the tricuspid valve. The aorta may also originate anterior to the pulmonary artery.
Ventriculoarterial Discordance
Complete Dextrotransposition of the Great Arteries
In 1797 Baillie described the heart of an infant in which the aorta connected to the right ventricle and the pulmonary artery to the left ventricle. The term transposition of the aorta and pulmonary artery is ascribed to Farre in 1814. Since that time, there has been controversy about whether it should be defined by the abnormal anteroposterior position of the great arteries or by the abnormal connections to the ventricles. TGA is a ventriculoarterial abnormality in which the aorta originates above the right ventricle and the pulmonary artery originates over the left ventricle.
Ventricular septal defects occur in about one third of babies with transposition (see Figure 10-7A), and when present, may result in congestive heart failure from the large blood flow. Extracardiac shunts may occur, as in patent ductus arteriosus or with bronchopulmonary connections to the pulmonary vascular bed. The ductus arteriosus remains patent in one fourth to one half of infants who do not receive prostaglandin E1 and allows blood to flow from the pulmonary artery to the aorta if the pulmonary vascular resistance is high and from the aorta to the pulmonary artery when the high fetal pulmonary artery pressures fall below the systemic blood pressure.
Besides the ventricular septal defect, the other major associated malformation is obstruction to blood entering the pulmonary arteries. About one fourth of those with TGA have some form of pulmonary stenosis (Fig. 10-35). The site of obstruction is usually in the subpulmonary region and it has a variety of causes:
