[level-membership-for-neurology-category]
16 Coma, Vegetative State, Brain Death, and Increased Intracranial Pressure
Coma
Clinical Vignette
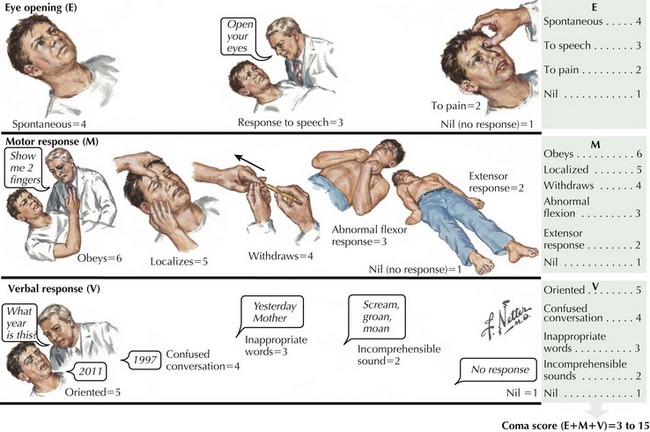
The Glasgow Coma Scale assesses and quantifies the degree of consciousness across three measures: response to verbal commands, response of eye opening, and the nature of motor movements in response to verbal or physical stimuli. Those not responding to verbal commands or opening their eyes with a Glasgow Coma Scale score of 8 or less are defined as being in a coma (Fig. 16-1). The Glasgow Coma Scale is one of the primary predictors of long-term outcomes, especially in cases of head trauma.
Prevalence of the different etiologies of coma varies depending on the population surveyed. For example, head trauma and intoxicants are the major causes in registries based on densely populated high-crime areas. Stroke and cardiac events are the leading etiologies in suburban areas with retirement communities. Overall, trauma, stroke, diffuse anoxic–ischemic brain insult (secondary to cardiorespiratory arrest), and intoxicants are the leading mechanisms for coma. Infections, seizures, and metabolic–endocrine disorders account for the remaining cases (Fig. 16-2).
States that affect cognition and attention without affecting wakefulness such as the various degenerative dementias (characterized by progressive cognitive deterioration) and focal brain lesions (which cause restricted cortical dysfunction) do not fit the definition of coma. Sleep is a normal patterned physiologic disconnection of the cortex from external stimuli and is discussed elsewhere (Chapter 15).
Evaluation and Treatment of the Comatose Patient
The initial evaluation of a patient in coma must occur simultaneously with its management. Any delay in treatment while waiting to determine the exact cause is not acceptable. Clearing the airway and ensuring adequate ventilation and oxygenation with a bag mask or intubation, if needed, must be addressed immediately. Management of hypotension must be prompt, especially in suspected cases of increased intracranial pressure (ICP). Hemodynamic collapse should never be attributed to an intracranial process, and cardiac or circulatory causes need to be sought. These form the “ABCs of coma management”: airway, breathing, and circulation (Fig. 16-3). Immobilizing the neck until a cervical spine injury is excluded is also important in cases of suspected trauma.
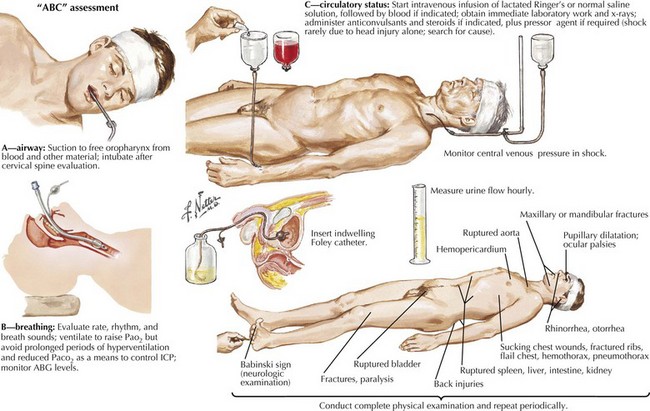
Urgent intravenous antibiotic coverage is indicated for febrile patients because time is crucial in treating meningitis and septicemia (Chapter 48). Lumbar puncture should be performed only after brain imaging has excluded mass lesions that could lead to herniation.
Prognosis
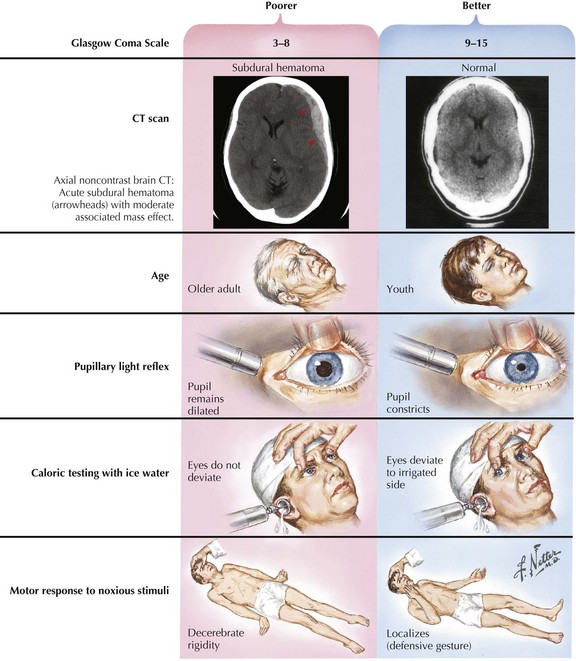
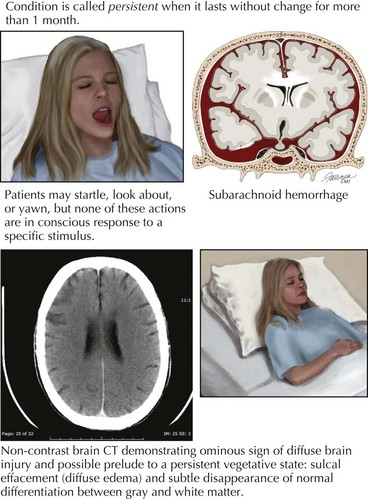
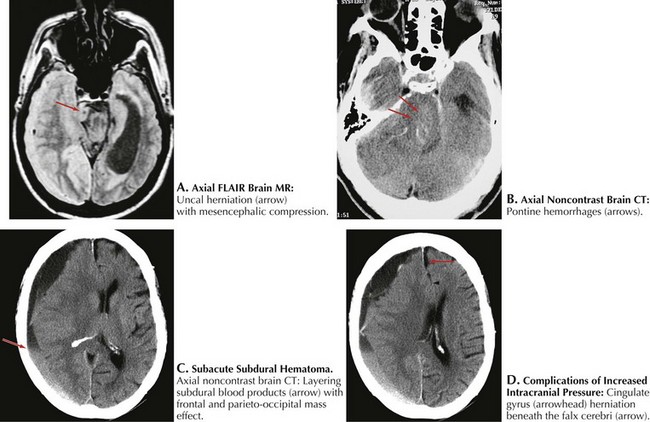
In most instances, coma from head trauma has a better outcome than that from nontraumatic mechanisms or cardiac arrest. Although severe head trauma has a mortality of approximately 50% within the first 48 hours, few surviving patients remain in a permanent vegetative state and most progress toward some degree of functional improvement. Those who remain vegetative usually succumb within 3–5 years. There are rare reports of patients who awaken after a prolonged vegetative period and show some return of functionality. None, however, return to their premorbid status or even an independent state. Signs that correlate with a poor prognosis after head trauma are age older than 60, bilateral pupillary abnormalities or absent oculocephalic reflexes at initial examination in a relatively stable patient. Large volumes of contused brain, large intra- or extra-axial hematomas, and lack of intracranial pressure response to conventional medical treatment (usually associated with compression of basal cisterns on CT) also betoken a poorer prognosis (Fig. 16-4).
Persistent Vegetative State
Clinical Vignette
The vegetative state, minimally conscious states, or post-coma unawareness are terms that describe a state of preserved brainstem and hypothalamic functions with absent or insufficient cortical function to sustain awareness of environment and self. Wakefulness is by definition preserved, and patients may cycle through sleep stages. There is no behavioral evidence of even the simplest reproducible response. Patients may startle, look about, occasionally move a limb, shift position, or yawn, but none of these actions are consistently in response to a specific stimulus (Fig. 16-5). Even the most basic voluntary actions, such as chewing and swallowing, are lost. Once reversible metabolic or exogenous causes have been eliminated, the condition is called persistent when it lasts without change for more than 1 month. It is considered permanent when lasting more than 12 months for traumatic brain injury and more than 3 months for nontraumatic causes. After these time limits, the chance of recovery is exceedingly low and at best progresses to severe disability.
Increased Intracranial Pressure and Cerebral Herniation
Rostrocaudal Signs of Brain Compromise
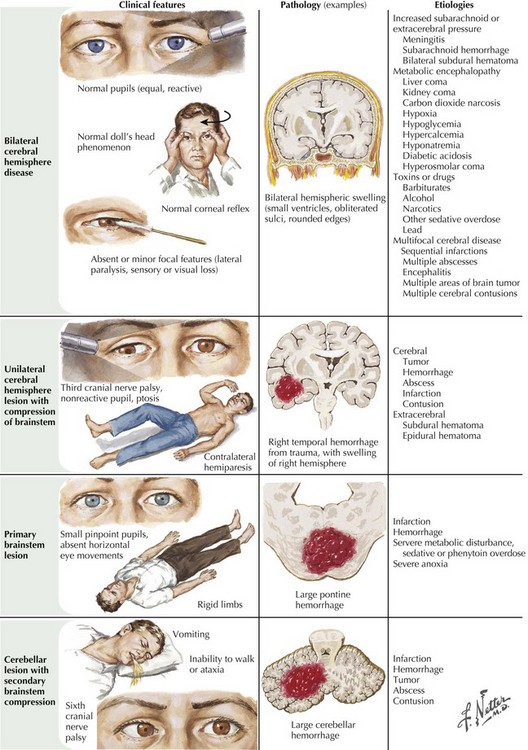
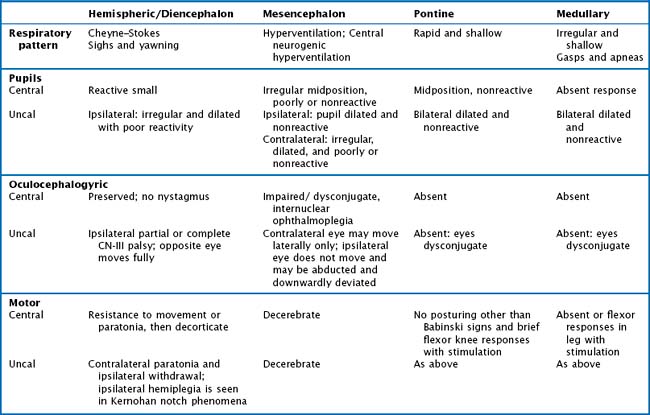
The ascending reticular formation, excited by sensory input, mediates arousal and consciousness to the cortex via the thalamic nuclei. Lesions that cause coma are at one of three levels along the neuraxis: bilateral cerebral cortex, the thalami, or the upper brainstem. The classic concept of herniation and coma produced by brain mass lesions pertains to a hemispheric process that ultimately causes “rostrocaudal” deterioration of function, gradually coursing down the hemispheres into the medulla. Although these “stages” rarely manifest symmetrically in a strict and clearly delineated sequential pattern, this paradigm remains useful for evaluating deteriorating patients with evolving neurologic signs. In addition to the level of consciousness, important physical examination elements include pupillary size and reactivity, reflexive eye movements, limb posturing, and breathing pattern (Fig. 16-4, Table 16-1).
Pupillary Reactivity and Eye Movements
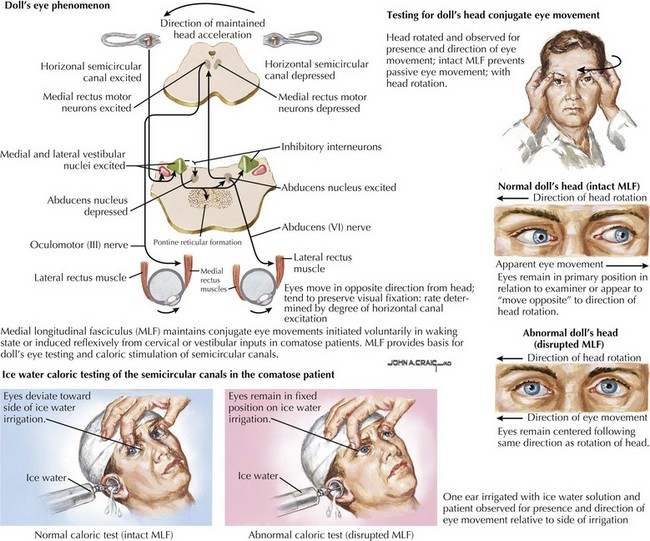
When pressure onto or across the diencephalon exists, loss of wakefulness results, but patients may transiently continue to withdraw appropriately from uncomfortable stimuli and to resist passive limb movements. Pupils are small and retain reactivity, although at times blunted and subtle to detect. Although there is no visual fixation, eye movements are conjugate and full. As pressure mounts across the thalami onto the mesencephalon, pupillary and eye movement abnormalities appear. Involvement of CN-III or its nucleus initially causes irregular and poorly reactive pupils (corectopia). Eventually, eye movements are disrupted by CN-III or CN-VI lesions or from involvement of the medial longitudinal fasciculus (MLF). The MLF, a paracentral dorsally located tract coursing up the vestibular nuclei to the CN-III nucleus, maintains conjugate eye movements either initiated voluntarily in the waking state or induced reflexively from cervical or vestibular inputs in comatose patients. This pathway provides the basis of doll’s-eyes testing or caloric stimulation testing of the semicircular canals. An intact MLF system keeps the eyes from moving passively when the examiner rolls the head to one side. The eyes remain in their primary position in relation to the examiner or seem to move to the opposite side in relation to the head rolling. With unilateral caloric stimulation of the ears, the eyes deviate conjugately to one side or another, depending on the water temperature used for irrigation. The direction of the convection current induced in the semicircular canals by different temperatures determines the direction of eye movement. With the head maintained in the neutral position, cold water causes the eye to deviate to the side of the stimulated ear while warm water causes deviation away from the stimulated ear. Disruption of the MLF system causes an abnormal or absent responses of these reflexive eye movements (Fig. 16-6). Therefore oculocephalic testing checks the integrity of a large portion of the brainstem from the vestibular nuclei to the mesencephalic third-nerve nucleus.
Breathing
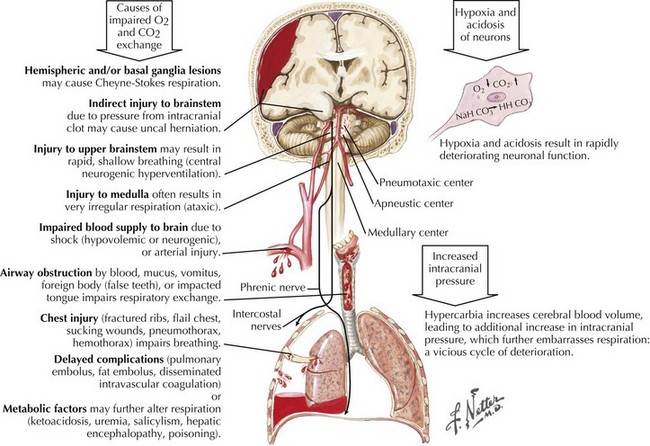
Respiratory patterns also change with worsening levels of consciousness in coma (Fig. 16-7). The earliest breathing alterations are Cheyne–Stokes respirations. Hemispheric forebrain structures serve to regulate breathing by mechanisms independent of CO2 accumulation. With bilateral cerebral cortex damage, this breathing control is lost, and CO2-driven breathing is accentuated with only modest CO2 accumulations, thus inducing an increased rate and depth of respiration. This reactive hyperpnea leads to an eventual decrease in arterial CO2 and, again without forebrain control, loss of respiratory drive. The ensuing apnea then allows CO2 to reaccumulate and the cycle to repeat itself, resulting in hyperpnea of a crescendo–decrescendo pattern, alternating with intervening episodes of brief apnea.
Variation from the Classic Rostrocaudal Paradigm
Unilateral cerebral lesions can cause asymmetric pressures leading to medial temporal lobe (uncal) herniation through the tentorial incisure, with direct compression of the midbrain (Fig. 16-8). In this case, the diencephalic features described above are not seen; instead, rapid loss of consciousness is immediately followed by a decerebrate posture. This is usually heralded by a compressive palsy of CN-III as it exits the ventral aspect of the midbrain and runs between the superior cerebellar and posterior cerebral arteries across the top of the tentorium. The initial sign is pupillary dilation, followed by ophthalmoplegia and ptosis with downward displacement of the abducted globe. Hemiplegia ipsilateral to the lesion may result from compression of the contralateral anteriorly located pyramidal tract against the anterior edge of the tentorium (Kernohan notch phenomenon). Further increase in pressure causes stretching of the pontine penetrators off the basilar artery or venous congestion with paramedian hemorrhages and usually irreversible worsening (Fig. 16-8). A sudden worsening and increase in ICP may also result from posterior cerebral artery compression with occipital lobe infarction. Finally, the cingulate gyrus may herniate beneath the falx cerebri, compressing the ipsilateral or contralateral anterior cerebral artery and causing infarction in its distribution (Fig. 16-8).
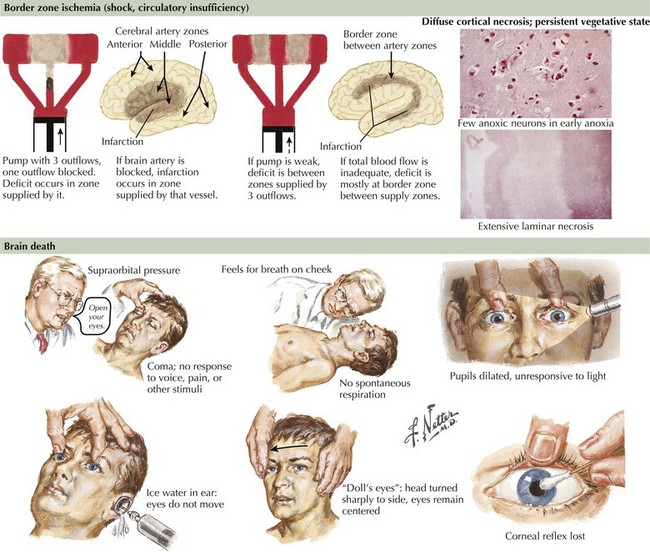
Brain Death
Brain Death Criteria
Bullock R, Chestnut F, Clifton G, et al. Guidelines for the management of severe head injury. J Neurotrauma. 2000;17:471-553.
Conrad GR, Sinha P. Scintigraphy as a confirmatory test of brain death. Semin Nucl Med. 2003;33:312-323.
American Electroencephalographic Society. Guideline three: minimum technical standards for EEG recording in suspected cerebral death. J Clin Neurophysiol. 1994;11:10-13.
Wijdicks EFM, Hijdra A, Young GB, et al. Practice Parameter: Prediction of outcome in comatose survivors after cardiopulmonary resuscitation (an evidence-based review): Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2006;67:203-210. A systematic review of the outcomes in coma after cardiopulmonary arrest identifying the factors that most reliably predict a poor prognosis
Hypothermia After Cardiac Arrest Study Group. Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. New England Journal of Medicine. 2002;346(8):549-556.
Levy DE, Bates D, Corona JJ, et al. Prognosis in non-traumatic coma. Ann Intern Med. 1981;94:293-301. Landmark paper that details the examination of postanoxic coma and the various findings that predict outcomes
Muizelaar JP, Marmarou A, Ward JD, et al. Adverse effects of prolonged hyperventilation in patients with severe head injury: a randomized clinical trial. J Neurosurg. 1991;75:731-739.
Piatt J, Schiff SJ. High dose barbiturate therapy in neurosurgery and intensive care. Neurosurgery. 1984;15:427-444.
Presidents Commission on Guidelines for the Determination of Death. Neurology. 1982;32:395.
Posner JB, Saper CB, Schiff ND, et al. The Diagnosis of Stupor and Coma. (Contemporary Neurology Series. 71). 4th ed. New York: Oxford University Press; 2007. An expanded and exhaustive edition of the classic monogram on the pathophysiology of coma and its various etiologies. Detailed description of the associated vascular and anatomic pathology. It ends with a small section on the approach to the unconscious patient and treatment
Qureshi AI, Kirmani JF, Xavier AR, et al. Computed tomographic angiography for diagnosis of brain death. Neurology. 2004;24(62):652-653.
Report of the Quality Standards Subcommittee of the American Academy of Neurology 1995;1-7.
Sazbon L, Zagreba F, Ronen J, et al. Course and outcome of patients in vegetative state of non-traumatic aetiology. J Neurol Neurosurg Psychiatry. 1993;56:407-409.
Teasdale G, Jennet B. Assessment of coma and impaired consciousness: a practical scale. Lancet. 1974;ii:81-84. The first description of the Glasgow Coma Scale that has acquired widespread use and has subsequently been shown to be a reliable tool in predicting outcome in head trauma
The Multi-Society Task Force on PVS. Medical aspects of the persistent vegetative state: parts I and II. N Engl J Med. 1994;330:1499-1508. 1572-1579
Vahedi K, Hofmeijer J, Juettler E, et alfor the DECIMAL, DESTINY, and HAMLET investigators. Early decompressive surgery in malignant infarction of the middle cerebral artery: a pooled analysis of three randomized controlled trials. Lancet Neurol. 2007;6:315-322. Data pooled from three different studies showing that decompressive hemicraniectomy more than doubles the chance of survival from “malignant” middle cerebral artery stroke, and likely improves outcomes regardless of the side affected. However, most survivors are left with significant disability
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
16 Coma, Vegetative State, Brain Death, and Increased Intracranial Pressure
Coma
Clinical Vignette
The Glasgow Coma Scale assesses and quantifies the degree of consciousness across three measures: response to verbal commands, response of eye opening, and the nature of motor movements in response to verbal or physical stimuli. Those not responding to verbal commands or opening their eyes with a Glasgow Coma Scale score of 8 or less are defined as being in a coma (Fig. 16-1). The Glasgow Coma Scale is one of the primary predictors of long-term outcomes, especially in cases of head trauma.
Prevalence of the different etiologies of coma varies depending on the population surveyed. For example, head trauma and intoxicants are the major causes in registries based on densely populated high-crime areas. Stroke and cardiac events are the leading etiologies in suburban areas with retirement communities. Overall, trauma, stroke, diffuse anoxic–ischemic brain insult (secondary to cardiorespiratory arrest), and intoxicants are the leading mechanisms for coma. Infections, seizures, and metabolic–endocrine disorders account for the remaining cases (Fig. 16-2).
States that affect cognition and attention without affecting wakefulness such as the various degenerative dementias (characterized by progressive cognitive deterioration) and focal brain lesions (which cause restricted cortical dysfunction) do not fit the definition of coma. Sleep is a normal patterned physiologic disconnection of the cortex from external stimuli and is discussed elsewhere (Chapter 15).
Evaluation and Treatment of the Comatose Patient
The initial evaluation of a patient in coma must occur simultaneously with its management. Any delay in treatment while waiting to determine the exact cause is not acceptable. Clearing the airway and ensuring adequate ventilation and oxygenation with a bag mask or intubation, if needed, must be addressed immediately. Management of hypotension must be prompt, especially in suspected cases of increased intracranial pressure (ICP). Hemodynamic collapse should never be attributed to an intracranial process, and cardiac or circulatory causes need to be sought. These form the “ABCs of coma management”: airway, breathing, and circulation (Fig. 16-3). Immobilizing the neck until a cervical spine injury is excluded is also important in cases of suspected trauma.
Urgent intravenous antibiotic coverage is indicated for febrile patients because time is crucial in treating meningitis and septicemia (Chapter 48). Lumbar puncture should be performed only after brain imaging has excluded mass lesions that could lead to herniation.
Prognosis
In most instances, coma from head trauma has a better outcome than that from nontraumatic mechanisms or cardiac arrest. Although severe head trauma has a mortality of approximately 50% within the first 48 hours, few surviving patients remain in a permanent vegetative state and most progress toward some degree of functional improvement. Those who remain vegetative usually succumb within 3–5 years. There are rare reports of patients who awaken after a prolonged vegetative period and show some return of functionality. None, however, return to their premorbid status or even an independent state. Signs that correlate with a poor prognosis after head trauma are age older than 60, bilateral pupillary abnormalities or absent oculocephalic reflexes at initial examination in a relatively stable patient. Large volumes of contused brain, large intra- or extra-axial hematomas, and lack of intracranial pressure response to conventional medical treatment (usually associated with compression of basal cisterns on CT) also betoken a poorer prognosis (Fig. 16-4).
