140 |
Disorders of Platelets and Vessel Wall |
Hemostasis is a dynamic process in which the platelet and the blood vessel wall play key roles. Platelets become activated upon adhesion to von Willebrand factor (VWF) and collagen in the exposed subendothelium after injury. Platelet activation is also mediated through shear forces imposed by blood flow itself, particularly in areas where the vessel wall is diseased, and is also affected by the inflammatory state of the endothelium. The activated platelet surface provides the major physiologic site for coagulation factor activation, which results in further platelet activation and fibrin formation. Genetic and acquired influences on the platelet and vessel wall, as well as on the coagulation and fibrinolytic systems, determine whether normal hemostasis or bleeding or clotting symptoms will result.
THE PLATELET
Platelets are released from the megakaryocyte, likely under the influence of flow in the capillary sinuses. The normal blood platelet count is 150,000–450,000/μL. The major regulator of platelet production is the hormone thrombopoietin (TPO), which is synthesized in the liver. Synthesis is increased with inflammation and specifically by interleukin 6. TPO binds to its receptor on platelets and megakaryocytes, by which it is removed from the circulation. Thus a reduction in platelet and megakaryocyte mass increases the level of TPO, which then stimulates platelet production. Platelets circulate with an average life span of 7–10 days. Approximately one-third of the platelets reside in the spleen, and this number increases in proportion to splenic size, although the platelet count rarely decreases to <40,000/μL as the spleen enlarges. Platelets are physiologically very active, but are anucleate, and thus have limited capacity to synthesize new proteins.
Normal vascular endothelium contributes to preventing thrombosis by inhibiting platelet function (Chap. 78). When vascular endothelium is injured, these inhibitory effects are overcome, and platelets adhere to the exposed intimal surface primarily through VWF, a large multimeric protein present in both plasma and in the extracellular matrix of the subendothelial vessel wall. Platelet adhesion results in the generation of intracellular signals that lead to activation of the platelet glycoprotein (Gp) IIb/IIIa (αIIbβ3) receptor and resultant platelet aggregation.
Activated platelets undergo release of their granule contents, which include nucleotides, adhesive proteins, growth factors, and procoagulants that serve to promote platelet aggregation and blood clot formation and influence the environment of the forming clot. During platelet aggregation, additional platelets are recruited to the site of injury, leading to the formation of an occlusive platelet thrombus. The platelet plug is stabilized by the fibrin mesh that develops simultaneously as the product of the coagulation cascade.
THE VESSEL WALL
Endothelial cells line the surface of the entire circulatory tree, totaling 1–6 × 1013 cells, enough to cover a surface area equivalent to about six tennis courts. The endothelium is physiologically active, controlling vascular permeability, flow of biologically active molecules and nutrients, blood cell interactions with the vessel wall, the inflammatory response, and angiogenesis.
The endothelium normally presents an antithrombotic surface (Chap. 78) but rapidly becomes prothrombotic when stimulated, which promotes coagulation, inhibits fibrinolysis, and activates platelets. In many cases, endothelium-derived vasodilators are also platelet inhibitors (e.g., nitric oxide) and, conversely, endothelium-derived vasoconstrictors (e.g., endothelin) can also be platelet activators. The net effect of vasodilation and inhibition of platelet function is to promote blood fluidity, whereas the net effect of vasoconstriction and platelet activation is to promote thrombosis. Thus, blood fluidity and hemostasis are regulated by the balance of antithrombotic/prothrombotic and vasodilatory/vasoconstrictor properties of endothelial cells.
DISORDERS OF PLATELETS
THROMBOCYTOPENIA
Thrombocytopenia results from one or more of three processes: (1) decreased bone marrow production; (2) sequestration, usually in an enlarged spleen; and/or (3) increased platelet destruction. Disorders of production may be either inherited or acquired. In evaluating a patient with thrombocytopenia, a key step is to review the peripheral blood smear and to first rule out “pseudothrombocytopenia,” particularly in a patient without an apparent cause for the thrombocytopenia. Pseudothrombocytopenia (Fig. 140-1B) is an in vitro artifact resulting from platelet agglutination via antibodies (usually IgG, but also IgM and IgA) when the calcium content is decreased by blood collection in ethylenediamine tetraacetic (EDTA) (the anticoagulant present in tubes [purple top] used to collect blood for complete blood counts [CBCs]). If a low platelet count is obtained in EDTA-anticoagulated blood, a blood smear should be evaluated and a platelet count determined in blood collected into sodium citrate (blue top tube) or heparin (green top tube), or a smear of freshly obtained unanticoagulated blood, such as from a finger stick, can be examined.
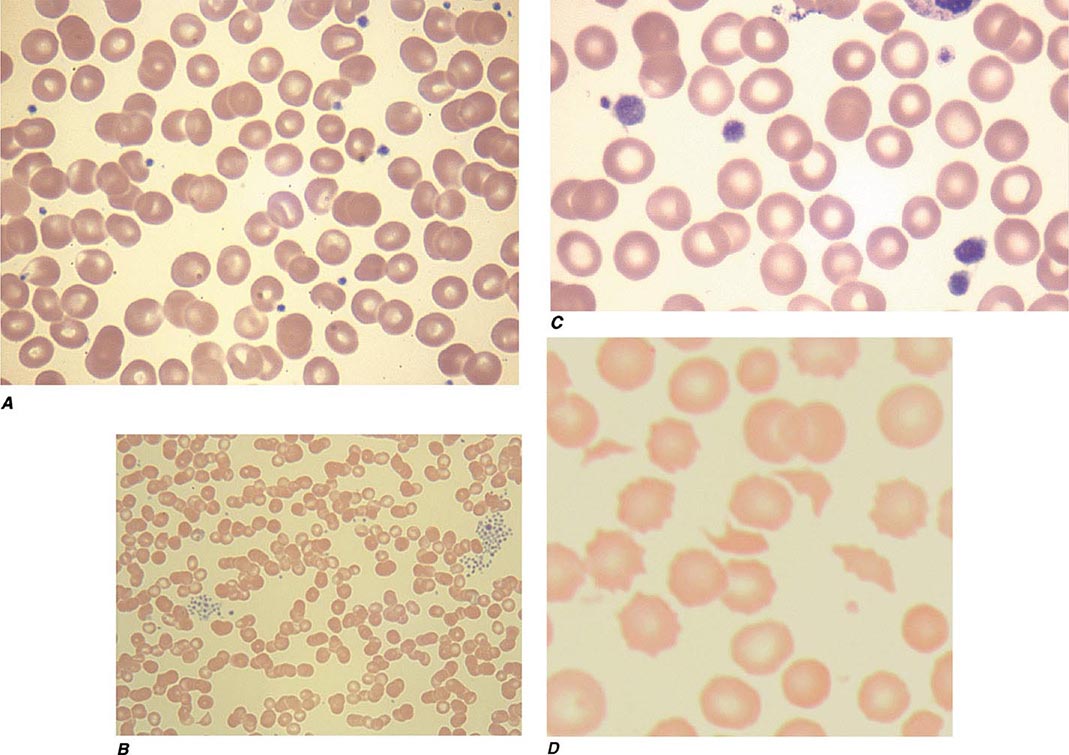
FIGURE 140-1 Photomicrographs of peripheral blood smears. A. Normal peripheral blood. B. Platelet clumping in pseudothrombocytopenia. C. Abnormal large platelet in autosomal dominant macrothrombocytopenia. D. Schistocytes and decreased platelets in microangiopathic hemolytic anemia.
APPROACH TO THE PATIENT:
Thrombocytopenia
The history and physical examination, results of the CBC, and review of the peripheral blood smear are all critical components in the initial evaluation of thrombocytopenic patients (Fig. 140-2). The overall health of the patient and whether he or she is receiving drug treatment will influence the differential diagnosis. A healthy young adult with thrombocytopenia will have a much more limited differential diagnosis than an ill hospitalized patient who is receiving multiple medications. Except in unusual inherited disorders, decreased platelet production usually results from bone marrow disorders that also affect red blood cell (RBC) and/or white blood cell (WBC) production. Because myelodysplasia can present with isolated thrombocytopenia, the bone marrow should be examined in patients presenting with isolated thrombocytopenia who are older than 60 years of age. While inherited thrombocytopenia is rare, any prior platelet counts should be retrieved and a family history regarding thrombocytopenia obtained. A careful history of drug ingestion should be obtained, including nonprescription and herbal remedies, because drugs are the most common cause of thrombocytopenia.
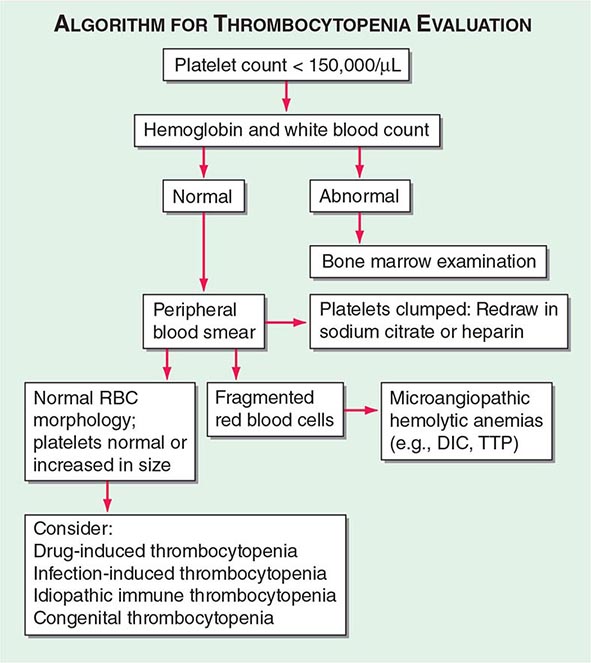
FIGURE 140-2 Algorithm for evaluating the thrombocytopenic patient. DIC, disseminated intravascular coagulation; RBC, red blood cell; TTP, thrombotic thrombocytopenic purpura.
The physical examination can document an enlarged spleen, evidence of chronic liver disease, and other underlying disorders. Mild to moderate splenomegaly may be difficult to appreciate in many individuals due to body habitus and/or obesity but can be easily assessed by abdominal ultrasound. A platelet count of approximately 5000–10,000 is required to maintain vascular integrity in the microcirculation. When the count is markedly decreased, petechiae first appear in areas of increased venous pressure, the ankles and feet in an ambulatory patient. Petechiae are pinpoint, nonblanching hemorrhages and are usually a sign of a decreased platelet number and not platelet dysfunction. Wet purpura, blood blisters that form on the oral mucosa, are thought to denote an increased risk of life-threatening hemorrhage in the thrombocytopenic patient. Excessive bruising is seen in disorders of both platelet number and function.
Infection-Induced Thrombocytopenia Many viral and bacterial infections result in thrombocytopenia and are the most common noniatrogenic cause of thrombocytopenia. This may or may not be associated with laboratory evidence of disseminated intravascular coagulation (DIC), which is most commonly seen in patients with systemic infections with gram-negative bacteria. Infections can affect both platelet production and platelet survival. In addition, immune mechanisms can be at work, as in infectious mononucleosis and early HIV infection. Late in HIV infection, pancytopenia and decreased and dysplastic platelet production are more common. Immune-mediated thrombocytopenia in children usually follows a viral infection and almost always resolves spontaneously. This association of infection with immune thrombocytopenic purpura is less clear in adults.
Bone marrow examination is often requested for evaluation of occult infections. A study evaluating the role of bone marrow examination in fever of unknown origin in HIV-infected patients found that for 86% of patients, the same diagnosis was established by less invasive techniques, notably blood culture. In some instances, however, the diagnosis can be made earlier; thus, a bone marrow examination and culture are recommended when the diagnosis is needed urgently or when other, less invasive methods have been unsuccessful.
Drug-Induced Thrombocytopenia Many drugs have been associated with thrombocytopenia. A predictable decrease in platelet count occurs after treatment with many chemotherapeutic drugs due to bone marrow suppression (Chap. 103e). Drugs that cause isolated thrombocytopenia and have been confirmed with positive laboratory testing are listed in Table 140-1, but all drugs should be suspect in a patient with thrombocytopenia without an apparent cause and should be stopped, or substituted, if possible. A helpful website, Platelets on the Internet (http://www.ouhsc.edu/platelets/ditp.html), lists drugs and supplements reported to have caused thrombocytopenia and the level of evidence supporting the association. Although not as well studied, herbal and over-the-counter preparations may also result in thrombocytopenia and should be discontinued in patients who are thrombocytopenic.
|
DRUGS REPORTED AS DEFINITELY OR PROBABLY CAUSING ISOLATED THROMBOCYTOPENIAa |
aBased on scoring requiring a compatible clinical picture and positive laboratory testing.
Source: Adapted from DM Arnold et al: J Thromb Hemost 11:169, 2013.
Classic drug-dependent antibodies are antibodies that react with specific platelet surface antigens and result in thrombocytopenia only when the drug is present. Many drugs are capable of inducing these antibodies, but for some reason, they are more common with quinine and sulfonamides. Drug-dependent antibody binding can be demonstrated by laboratory assays, showing antibody binding in the presence of, but not without, the drug present in the assay. The thrombocytopenia typically occurs after a period of initial exposure (median length 21 days), or upon reexposure, and usually resolves in 7–10 days after drug withdrawal. The thrombocytopenia caused by the platelet Gp IIb/IIIa inhibitory drugs, such as abciximab, differs in that it may occur within 24 h of initial exposure. This appears to be due to the presence of naturally occurring antibodies that cross-react with the drug bound to the platelet.
Heparin-Induced Thrombocytopenia Drug-induced thrombocytopenia due to heparin differs from that seen with other drugs in two major ways. (1) The thrombocytopenia is not usually severe, with nadir counts rarely <20,000/μL. (2) Heparin-induced thrombocytopenia (HIT) is not associated with bleeding and, in fact, markedly increases the risk of thrombosis. HIT results from antibody formation to a complex of the platelet-specific protein platelet factor 4 (PF4) and heparin. The anti-heparin/PF4 antibody can activate platelets through the FcγRIIa receptor and also activate monocytes and endothelial cells. Many patients exposed to heparin develop antibodies to heparin/PF4, but do not appear to have adverse consequences. A fraction of those who develop antibodies will develop HIT, and a portion of those (up to 50%) will develop thrombosis (HITT).
HIT can occur after exposure to low-molecular-weight heparin (LMWH) as well as unfractionated heparin (UFH), although it is more common with the latter. Most patients develop HIT after exposure to heparin for 5–14 days (Fig. 140-3). It occurs before 5 days in those who were exposed to heparin in the prior few weeks or months (<~100 days) and have circulating anti-heparin/PF4 antibodies. Rarely, thrombocytopenia and thrombosis begin several days after all heparin has been stopped (termed delayed-onset HIT). The “4T’s” have been recommended to be used in a diagnostic algorithm for HIT: thrombocytopenia, timing of platelet count drop, thrombosis and other sequelae such as localized skin reactions, and other causes of thrombocytopenia not evident. Application of the 4T scoring system is very useful in excluding a diagnosis of HIT but will result in overdiagnosis of HIT in situations where thrombocytopenia and thrombosis due to other etiologies are common, such as in the intensive care unit. A scoring model based on broad expert opinion (the HIT Expert Probability [HEP] Score) has improved operating characteristics and may provide better utility as a scoring system.

FIGURE 140-3 Time course of heparin-induced thrombocytopenia (HIT) development after heparin exposure. The timing of development after heparin exposure is a critical factor in determining the likelihood of HIT in a patient. HIT occurs early after heparin exposure in the presence of preexisting heparin/platelet factor 4 (PF4) antibodies, which disappear from circulation by ~100 days following a prior exposure. Rarely, HIT may occur later after heparin exposure (termed delayed-onset HIT). In this setting, heparin/PF4 antibody testing is usually markedly positive. HIT can occur after exposure to either unfractionated (UFH) or low-molecular-weight heparin (LMWH).
LABORATORY TESTING FOR HIT HIT (anti-heparin/PF4) antibodies can be detected using two types of assays. The most widely available is an enzyme-linked immunoassay (ELISA) with PF4/polyanion complex as the antigen. Because many patients develop antibodies but do not develop clinical HIT, the test has a low specificity for the diagnosis of HIT. This is especially true in patients who have undergone cardiopulmonary bypass surgery, where approximately 50% of patients develop these antibodies postoperatively. IgG-specific ELISAs increase specificity but may decrease sensitivity. The other assay is a platelet activation assay, most commonly the serotonin release assay, which measures the ability of the patient’s serum to activate platelets in the presence of heparin in a concentration-dependent manner. This test has lower sensitivity but higher specificity than the ELISA. However, HIT remains a clinical diagnosis.
Immune Thrombocytopenic Purpura Immune thrombocytopenic purpura (ITP; also termed idiopathic thrombocytopenic purpura) is an acquired disorder in which there is immune-mediated destruction of platelets and possibly inhibition of platelet release from the megakaryocyte. In children, it is usually an acute disease, most commonly following an infection, and with a self-limited course. In adults, it is a more chronic disease, although in some adults, spontaneous remission occurs, usually within months of diagnosis. ITP is termed secondary if it is associated with an underlying disorder; autoimmune disorders, particularly systemic lupus erythematosus (SLE), and infections, such as HIV and hepatitis C, are common causes. The association of ITP with Helicobacter pylori infection is unclear.
ITP is characterized by mucocutaneous bleeding and a low, often very low, platelet count, with an otherwise normal peripheral blood cells and smear. Patients usually present either with ecchymoses and petechiae, or with thrombocytopenia incidentally found on a routine CBC. Mucocutaneous bleeding, such as oral mucosa, gastrointestinal, or heavy menstrual bleeding, may be present. Rarely, life-threatening, including central nervous system, bleeding can occur. Wet purpura (blood blisters in the mouth) and retinal hemorrhages may herald life-threatening bleeding.
LABORATORY TESTING IN ITP Laboratory testing for antibodies (serologic testing) is usually not helpful due to the low sensitivity and specificity of the current tests. Bone marrow examination can be reserved for those who have other signs or laboratory abnormalities not explained by ITP or in patients who do not respond to initial therapy. The peripheral blood smear may show large platelets, with otherwise normal morphology. Depending on the bleeding history, iron-deficiency anemia may be present.
Laboratory testing is performed to evaluate for secondary causes of ITP and should include testing for HIV infection and hepatitis C (and other infections if indicated). Serologic testing for SLE, serum protein electrophoresis, immunoglobulin levels to potentially detect hypogammaglobulinemia, selective testing for IgA deficiency or monoclonal gammopathies, and testing for H. pylori infection should be considered, depending on the clinical circumstance. If anemia is present, direct antiglobulin testing (Coombs’ test) should be performed to rule out combined autoimmune hemolytic anemia with ITP (Evans’ syndrome).
Inherited Thrombocytopenia Thrombocytopenia is rarely inherited, either as an isolated finding or as part of a syndrome, and may be inherited in an autosomal dominant, autosomal recessive, or X-linked pattern. Many forms of autosomal dominant thrombocytopenia are now known to be associated with mutations in the nonmuscle myosin heavy chain MYH9 gene. Interestingly, these include the May-Hegglin anomaly, and Sebastian, Epstein’s, and Fechtner syndromes, all of which have distinct distinguishing features. A common feature of these disorders is large platelets (Fig. 140-1C). Autosomal recessive disorders include congenital amegakaryocytic thrombocytopenia, thrombocytopenia with absent radii, and Bernard-Soulier syndrome. The latter is primarily a functional platelet disorder due to absence of Gp Ib-IX-V, the VWF adhesion receptor. X-linked disorders include Wiskott-Aldrich syndrome and a dyshematopoietic syndrome resulting from a mutation in GATA-1, an important transcriptional regulator of hematopoiesis.
THROMBOTIC THROMBOCYTOPENIC PURPURA AND HEMOLYTIC-UREMIC SYNDROME
Thrombotic thrombocytopenic microangiopathies are a group of disorders characterized by thrombocytopenia, a microangiopathic hemolytic anemia evident by fragmented RBCs (Fig. 140-1D) and laboratory evidence of hemolysis, and microvascular thrombosis. They include thrombotic thrombocytopenic purpura (TTP) and hemolytic-uremic syndrome (HUS), as well as syndromes complicating bone marrow transplantation, certain medications and infections, pregnancy, and vasculitis. In DIC, although thrombocytopenia and microangiopathy are seen, a coagulopathy predominates, with consumption of clotting factors and fibrinogen resulting in an elevated prothrombin time (PT) and often activated partial thromboplastin time (aPTT). The PT and aPTT are characteristically normal in TTP or HUS.
Thrombotic Thrombocytopenic Purpura TTP and HUS were previously considered overlap syndromes. However, in the past few years, the pathophysiology of inherited and idiopathic TTP has become better understood and clearly differs from HUS. TTP was first described in 1924 by Eli Moschcowitz and characterized by a pentad of findings that include microangiopathic hemolytic anemia, thrombocytopenia, renal failure, neurologic findings, and fever. The full-blown syndrome is less commonly seen now, probably due to earlier diagnosis. The introduction of treatment with plasma exchange markedly improved the prognosis in patients, with a decrease in mortality from 85–100% to 10–30%.
The pathogenesis of inherited (Upshaw-Schulman syndrome) and idiopathic TTP is related to a deficiency of, or antibodies to, the metalloprotease ADAMTS13, which cleaves VWF. VWF is normally secreted as ultra-large multimers, which are then cleaved by ADAMTS13. The persistence of ultra-large VWF molecules is thought to contribute to pathogenic platelet adhesion and aggregation (Fig. 140-4). This defect alone, however, is not sufficient to result in TTP because individuals with a congenital absence of ADAMTS13 develop TTP only episodically. Additional provocative factors have not been defined. The level of ADAMTS13 activity, as well as antibodies, can now be detected by laboratory assays. Although assays with sufficient sensitivity and specificity to direct clinical management have yet to be clearly defined, ADAMTS13 activity levels of <10% are more clearly associated with idiopathic TTP.
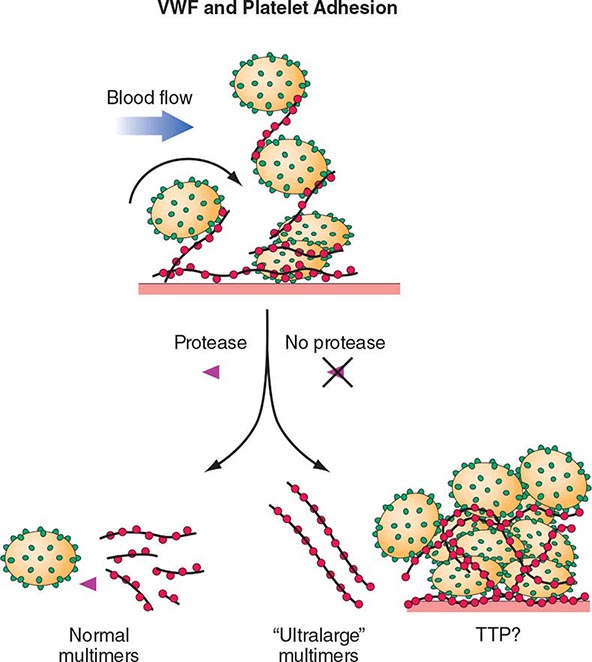
FIGURE 140-4 Pathogenesis of thrombotic thrombocytopenic purpura (TTP). Normally the ultra-high-molecular-weight multimers of von Willebrand factor (VWF) produced by the endothelial cells are processed into smaller multimers by a plasma metalloproteinase called ADAMTS13. In TTP, the activity of the protease is inhibited, and the ultra-high-molecular-weight multimers of VWF initiate platelet aggregation and thrombosis.
Idiopathic TTP appears to be more common in women than in men. No geographic or racial distribution has been defined. TTP is more common in patients with HIV infection and in pregnant women. TTP in pregnancy is not clearly related to ADAMTS13. Medication-related microangiopathic hemolytic anemia may be secondary to antibody formation (ticlopidine and possibly clopidogrel) or direct endothelial toxicity (cyclosporine, mitomycin C, tacrolimus, quinine), although this is not always so clear, and fear of withholding treatment, as well as lack of other treatment alternatives, results in broad application of plasma exchange. However, withdrawal, or reduction in dose, of endothelial toxic agents usually decreases the microangiopathy.
Hemolytic-Uremic Syndrome HUS is a syndrome characterized by acute renal failure, microangiopathic hemolytic anemia, and thrombocytopenia. It is seen predominantly in children and in most cases is preceded by an episode of diarrhea, often hemorrhagic in nature. Escherichia coli O157:H7 is the most frequent, although not only, etiologic serotype. HUS not associated with diarrhea is more heterogeneous in presentation and course. Atypical HUS (aHUS) due to genetic defects that result in chronic complement activation has been defined, and screening for mutations in complement regulatory genes is available.
THROMBOCYTOSIS
Thrombocytosis is almost always due to (1) iron deficiency; (2) inflammation, cancer, or infection (reactive thrombocytosis); or (3) an underlying myeloproliferative process (essential thrombocythemia or polycythemia vera) (Chap. 131) or, rarely, the 5q– myelodysplastic process (Chap. 130). Patients presenting with an elevated platelet count should be evaluated for underlying inflammation or malignancy, and iron deficiency should be ruled out. Thrombocytosis in response to acute or chronic inflammation has not been clearly associated with an increased thrombotic risk. In fact, patients with markedly elevated platelet counts (>1.5 million), usually seen in the setting of a myeloproliferative disorder, have an increased risk of bleeding. This appears to be due, at least in part, to acquired von Willebrand disease (VWD) due to platelet-VWF binding and removal from the circulation.
QUALITATIVE DISORDERS OF PLATELET FUNCTION
Inherited Disorders of Platelet Function Inherited platelet function disorders are thought to be relatively rare, although the prevalence of mild disorders of platelet function is unclear, in part because our testing for such disorders is suboptimal. Rare qualitative disorders include the autosomal recessive disorders Glanzmann’s thrombasthenia (absence of the platelet Gp IIb/IIIa receptor) and Bernard-Soulier syndrome (absence of the platelet Gp Ib-IX-V receptor). Both are inherited in an autosomal recessive fashion and present with bleeding symptoms in childhood.
Platelet storage pool disorder (SPD) is the classic autosomal dominant qualitative platelet disorder. This results from abnormalities of platelet granule formation. It is also seen as a part of inherited disorders of granule formation, such as Hermansky-Pudlak syndrome. Bleeding symptoms in SPD are variable, but often are mild. The most common inherited disorders of platelet function prevent normal secretion of granule content and are termed secretion defects. Few of these abnormalities have been dissected at the molecular level but they likely result from various mutations..
Acquired Disorders of Platelet Function Acquired platelet dysfunction is common, usually due to medications, either intentionally as with antiplatelet therapy or unintentionally as with high-dose penicillins. Acquired platelet dysfunction occurs in uremia. This is likely multifactorial, but the resultant effect is defective adhesion and activation. The platelet defect is improved most by dialysis but may also be improved by increasing the hematocrit to 27–32%, giving DDAVP (0.3 μg/kg), or use of conjugated estrogens. Platelet dysfunction also occurs with cardiopulmonary bypass due to the effect of the artificial circuit on platelets, and bleeding symptoms respond to platelet transfusion. Platelet dysfunction seen with underlying hematologic disorders can result from nonspecific interference by circulating paraproteins or intrinsic platelet defects in myeloproliferative and myelodysplastic syndromes.
VON WILLEBRAND DISEASE
VWD is the most common inherited bleeding disorder. Estimates from laboratory data suggest a prevalence of approximately 1%, but data based on symptomatic individuals suggest that it is closer to 0.1% of the population. VWF serves two roles: (1) as the major adhesion molecule that tethers the platelet to the exposed subendothelium; and (2) as the binding protein for factor VIII (FVIII), resulting in significant prolongation of the FVIII half-life in circulation. The platelet-adhesive function of VWF is critically dependent on the presence of large VWF multimers, whereas FVIII binding is not. Most of the symptoms of VWD are “platelet-like” except in more severe VWD when the FVIII is low enough to produce symptoms similar to those found in FVIII deficiency (hemophilia A).
VWD has been classified into three major types, with four subtypes of type 2 (Table 140-2; Fig. 140-5). By far the most common type of VWD is type 1 disease, with a parallel decrease in VWF protein, VWF function, and FVIII levels, accounting for at least 80% of cases. Patients have predominantly mucosal bleeding symptoms, although postoperative bleeding can also be seen. Bleeding symptoms are very uncommon in infancy and usually manifest later in childhood with excessive bruising and epistaxis. Because these symptoms occur commonly in childhood, the clinician should particularly note bruising at sites unlikely to be traumatized and/or prolonged epistaxis requiring medical attention. Menorrhagia is a common manifestation of VWD. Menstrual bleeding resulting in anemia should warrant an evaluation for VWD and, if negative, functional platelet disorders. Frequently, mild type 1 VWD first manifests with dental extractions, particularly wisdom tooth extraction, or tonsillectomy.
|
LABORATORY DIAGNOSIS OF VON WILLEBRAND DISEASE (VWD) |
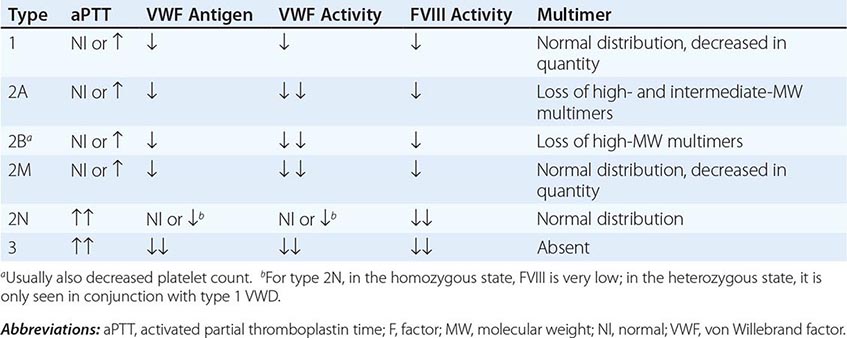
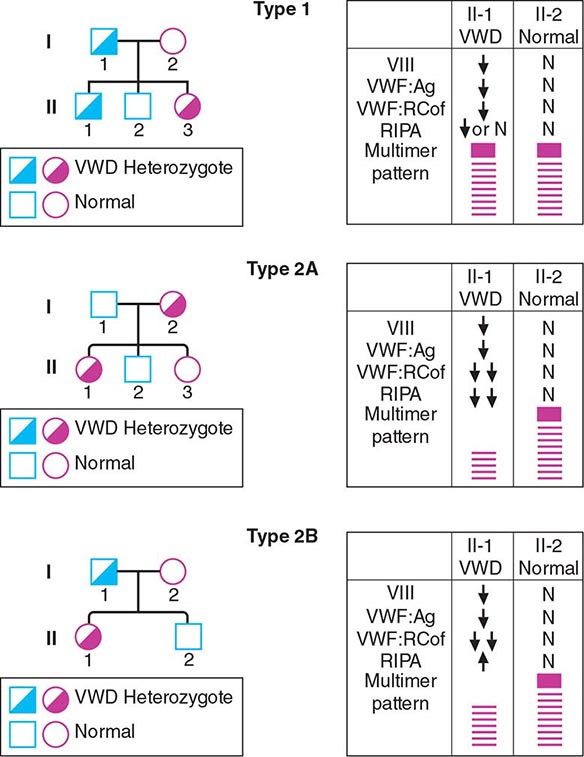
FIGURE 140-5 Pattern of inheritance and laboratory findings in von Willebrand disease (VWD). The assays of platelet function include a coagulation assay of factor VIII bound and carried by von Willebrand factor (VWF), abbreviated as VIII; immunoassay of total VWF protein (VWF:Ag); bioassay of the ability of patient plasma to support ristocetin-induced agglutination of normal platelets (VWF:RCoF); and ristocetin-induced aggregation of patient platelets, abbreviated RIPA. The multimer pattern illustrates the protein bands present when plasma is electrophoresed in a polyacrylamide gel. The II-1 and II-2 columns refer to the phenotypes of the second-generation offspring.
Not all patients with low VWF levels have bleeding symptoms. Whether patients bleed or not will depend on the overall hemostatic balance they have inherited, along with environmental influences and the type of hemostatic challenges they experience. Although the inheritance of VWD is autosomal, many factors modulate both VWF levels and bleeding symptoms. These have not all been defined, but include blood type, thyroid hormone status, race, stress, exercise, and hormonal (both endogenous and exogenous) influences. Patients with type O blood have VWF protein levels of approximately one-half that of patients with AB blood type; and, in fact, the normal range for patients with type O blood overlaps that which has been considered diagnostic for VWD. A mildly decreased VWF level should be viewed more as a risk factor for bleeding than as an actual disease.
Patients with type 2 VWD have functional defects; thus, the VWF antigen measurement is significantly higher than the test of function. For types 2A, 2B, and 2M VWD, platelet-binding and/or collagen binding VWF activity is decreased. In type 2A VWD, the impaired function is due either to increased susceptibility to cleavage by ADAMTS13, resulting in loss of intermediate- and high-molecular-weight multimers, or to decreased secretion of these multimers by the cell. Type 2B VWD results from gain-of-function mutations that result in increased spontaneous binding of VWF to platelets in circulation, with subsequent clearance of this complex by the reticuloendothelial system. The resulting VWF in the patients’ plasma lacks the highest molecular-weight multimers, and the platelet count is usually modestly reduced. Type 2M occurs as a consequence of a group of mutations that cause dysfunction but do not affect multimer structure.
Type 2N VWD is due to mutations in VWF that affect binding of FVIII. As FVIII is stabilized by binding to VWF, the FVIII in patients with type 2N VWD has a very short half-life, and the FVIII level is markedly decreased. This is sometimes termed autosomal hemophilia. Type 3 VWD, or severe VWD, describes patients with virtually no VWF protein and FVIII levels <10%. Patients experience mucosal and joint bleeding, surgery-related bleeding, and other bleeding symptoms. Some patients with type 3 VWD, particularly those with large VWF gene deletions, are at risk of developing antibodies to infused VWF.
Acquired VWD is a rare disorder, most commonly seen in patients with underlying lymphoproliferative disorders, including monoclonal gammopathies of underdetermined significance (MGUS), multiple myeloma, and Waldenström’s macroglobulinemia. It is seen most commonly in the setting of MGUS and should be suspected in patients, particularly elderly patients, with a new onset of severe mucosal bleeding symptoms. Laboratory evidence of acquired VWD is found in some patients with aortic valvular disease. Heyde’s syndrome (aortic stenosis with gastrointestinal bleeding) is attributed to the presence of angiodysplasia of the gastrointestinal tract in patients with aortic stenosis. The shear stress on blood passing through the stenotic aortic valve appears to produce a change in VWF, making it susceptible to serum proteases. Consequently, large multimer forms are lost, leading to an acquired type 2 VWD, but return when the stenotic valve is replaced.
DISORDERS OF THE VESSEL WALL
The vessel wall is an integral part of hemostasis, and separation of a fluid phase is artificial, particularly in disorders such as TTP or HIT that clearly involve the endothelium as well. Inflammation localized to the vessel wall, such as vasculitis, and inherited connective tissue disorders are abnormalities inherent to the vessel wall.
METABOLIC AND INFLAMMATORY DISORDERS Acute febrile illnesses may result in vascular damage. This can result from immune complexes containing viral antigens or the viruses themselves. Certain pathogens, such as the rickettsiae causing Rocky Mountain spotted fever, replicate in endothelial cells and damage them. Vascular purpura may occur in patients with polyclonal gammopathies but more commonly in those with monoclonal gammopathies, including Waldenström’s macroglobulinemia, multiple myeloma, and cryoglobulinemia. Patients with mixed cryoglobulinemia develop a more extensive maculopapular rash due to immune complex–mediated damage to the vessel wall.
Patients with scurvy (vitamin C deficiency) develop painful episodes of perifollicular skin bleeding as well as more systemic bleeding symptoms. Vitamin C is needed to synthesize hydroxyproline, an essential constituent of collagen. Patients with Cushing’s syndrome or on chronic glucocorticoid therapy develop skin bleeding and easy bruising due to atrophy of supporting connective tissue. A similar phenomenon is seen with aging, where following minor trauma, blood spreads superficially under the epidermis. This has been termed senile purpura. It is most common on skin that has been previously damaged by sun exposure.
Henoch-Schönlein, or anaphylactoid, purpura is a distinct, self-limited type of vasculitis that occurs in children and young adults. Patients have an acute inflammatory reaction with IgA and complement components in capillaries, mesangial tissues, and small arterioles leading to increased vascular permeability and localized hemorrhage. The syndrome is often preceded by an upper respiratory infection, commonly with streptococcal pharyngitis, or is triggered by drug or food allergies. Patients develop a purpuric rash on the extensor surfaces of the arms and legs, usually accompanied by polyarthralgias or arthritis, abdominal pain, and hematuria from focal glomerulonephritis. All coagulation tests are normal, but renal impairment may occur. Glucocorticoids can provide symptomatic relief but do not alter the course of the illness.
INHERITED DISORDERS OF THE VESSEL WALL Patients with inherited disorders of the connective tissue matrix, such as Marfan’s syndrome, Ehlers-Danlos syndrome, and pseudoxanthoma elasticum, frequently report easy bruising. Inherited vascular abnormalities can result in increased bleeding. This is notably seen in hereditary hemorrhagic telangiectasia (HHT, or Osler-Weber-Rendu disease), a disorder where abnormal telangiectatic capillaries result in frequent bleeding episodes, primarily from the nose and gastrointestinal tract. Arteriovenous malformation (AVM) in the lung, brain, and liver may also occur in HHT. The telangiectasia can often be visualized on the oral and nasal mucosa. Signs and symptoms develop over time. Epistaxis begins, on average, at the age of 12 and occurs in >95% of affected individuals by middle age. Two genes involved in the pathogenesis are eng (endoglin) on chromosome 9q33-34 (so-called HHT type 1), associated with pulmonary AVM in 40% of cases, and alk1 (activin-receptor-like kinase 1) on chromosome 12q13, associated with a much lower risk of pulmonary AVM.
ACKNOWLEDGMENT
Robert Handin, MD, contributed this chapter in the 16th edition and some materials from his chapter are included here.
141 |
Coagulation Disorders |
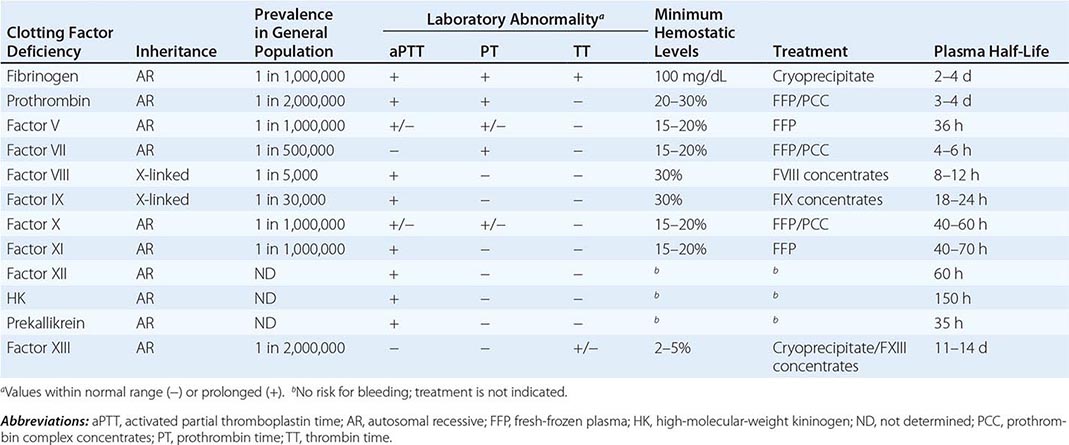
Deficiencies of coagulation factors have been recognized for centuries. Patients with genetic deficiencies of plasma coagulation factors exhibit life-long recurrent bleeding episodes into joints, muscles, and closed spaces, either spontaneously or following an injury. The most common inherited factor deficiencies are the hemophilias, X-linked diseases caused by deficiency of factor (F) VIII (hemophilia A) or FIX (hemophilia B). Rare congenital bleeding disorders due to deficiencies of other factors, including FII (prothrombin), FV, FVII, FX, FXI, and FXIII, and fibrinogen are commonly inherited in an autosomal recessive manner (Table 141-1). Advances in characterization of the molecular bases of clotting factor deficiencies have contributed to better understanding of the disease phenotypes and may eventually allow more targeted therapeutic approaches through the development of small molecules, recombinant proteins, or cell and gene-based therapies.
|
GENETIC AND LABORATORY CHARACTERISTICS OF INHERITED COAGULATION DISORDERS |
Commonly used tests of hemostasis provide the initial screening for clotting factor activity (Fig. 141-1), and disease phenotype often correlates with the level of clotting activity. An isolated abnormal prothrombin time (PT) suggests FVII deficiency, whereas a prolonged activated partial thromboplastin time (aPTT) indicates most commonly hemophilia or FXI deficiency (Fig. 141-1). The prolongation of both PT and aPTT suggests deficiency of FV, FX, FII, or fibrinogen abnormalities. The addition of the missing factor at a range of doses to the subject’s plasma will correct the abnormal clotting times; the result is expressed as a percentage of the activity observed in normal subjects.
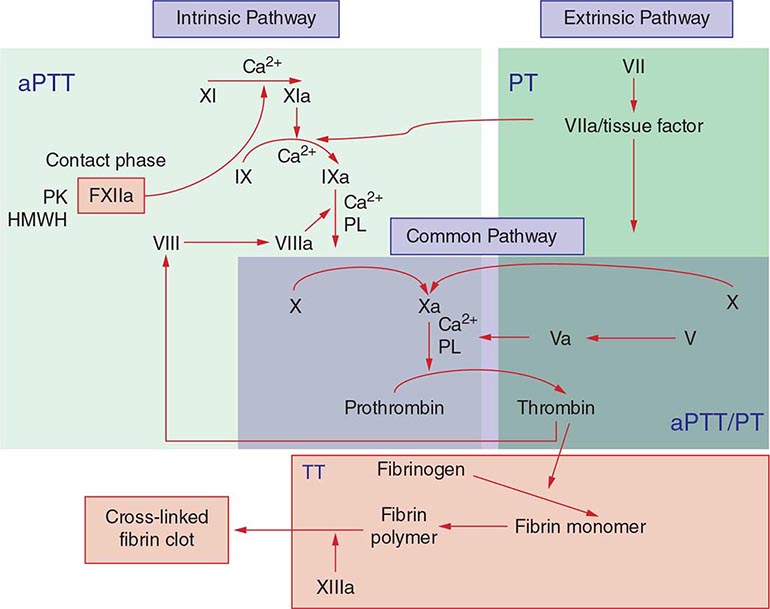
FIGURE 141-1 Coagulation cascade and laboratory assessment of clotting factor deficiency by activated partial prothrombin time (aPTT), prothrombin time (PT), thrombin time (TT), and phospholipid (PL).
Acquired deficiencies of plasma coagulation factors are more frequent than congenital disorders; the most common disorders include hemorrhagic diathesis of liver disease, disseminated intravascular coagulation (DIC), and vitamin K deficiency. In these disorders, blood coagulation is hampered by the deficiency of more than one clotting factor, and the bleeding episodes are the result of perturbation of both primary (coagulation) and secondary (e.g., platelet and vessel wall interactions) hemostasis.
The development of antibodies to coagulation plasma proteins, clinically termed inhibitors, is a relatively rare disease that often affects hemophilia A or B and FXI-deficient patients on repetitive exposure to the missing protein to control bleeding episodes. Inhibitors also occur among subjects without genetic deficiency of clotting factors (e.g., in the postpartum setting as a manifestation of underlying autoimmune or neoplastic disease or idiopathically). Rare cases of inhibitors to thrombin or FV have been reported in patients receiving topical bovine thrombin preparation as a local hemostatic agent in complex surgeries. The diagnosis of inhibitors is based on the same tests as those used to diagnose inherited plasma coagulation factor deficiencies. However, the addition of the missing protein to the plasma of a subject with an inhibitor does not correct the abnormal aPTT and/or PT tests (known as mixing tests). This is the major laboratory difference between deficiencies and inhibitors. Additional tests are required to measure the specificity of the inhibitor and its titer.
The treatment of these bleeding disorders often requires replacement of the deficient protein using recombinant or purified plasma-derived products or fresh-frozen plasma (FFP). Therefore, it is imperative to arrive at a proper diagnosis to optimize patient care without unnecessary exposure to suboptimal treatment and the risks of bloodborne disease.
HEMOPHILIA
PATHOGENESIS AND CLINICAL MANIFESTATIONS
Hemophilia is an X-linked recessive hemorrhagic disease due to mutations in the F8 gene (hemophilia A or classic hemophilia) or F9 gene (hemophilia B). The disease affects 1 in 10,000 males worldwide, in all ethnic groups; hemophilia A represents 80% of all cases. Male subjects are clinically affected; women, who carry a single mutated gene, are generally asymptomatic. Family history of the disease is absent in ~30% of cases, and in these cases, 80% of the mothers are carriers of the de novo mutated allele. More than 500 different mutations have been identified in the F8 or F9 genes of patients with hemophilia A or B, respectively. One of the most common hemophilia A mutations results from an inversion of the intron 22 sequence, and it is present in 40% of cases of severe hemophilia A. Advances in molecular diagnosis now permit precise identification of mutations, allowing accurate diagnosis of women carriers of the hemophilia gene in affected families.
Clinically, hemophilia A and hemophilia B are indistinguishable. The disease phenotype correlates with the residual activity of FVIII or FIX and can be classified as severe (<1%), moderate (1–5%), or mild (6–30%). In the severe and moderate forms, the disease is characterized by bleeding into the joints (hemarthrosis), soft tissues, and muscles after minor trauma or even spontaneously. Patients with mild disease experience infrequent bleeding that is usually secondary to trauma. Among those with residual FVIII or FIX activity >25% of normal, the disease is discovered only by bleeding after major trauma or during routine presurgery laboratory tests. Typically, the global tests of coagulation show only an isolated prolongation of the aPTT assay. Patients with hemophilia have normal bleeding times and platelet counts. The diagnosis is made after specific determination of FVIII or FIX clotting activity.
Early in life, bleeding may present after circumcision or rarely as intracranial hemorrhages. The disease is more evident when children begin to walk or crawl. In the severe form, the most common bleeding manifestations are the recurrent hemarthroses, which can affect every joint but mainly affect knees, elbows, ankles, shoulders, and hips. Acute hemarthroses are painful, and clinical signs are local swelling and erythema. To avoid pain, the patient may adopt a fixed position, which leads eventually to muscle contractures. Very young children unable to communicate verbally show irritability and a lack of movement of the affected joint. Chronic hemarthroses are debilitating, with synovial thickening and synovitis in response to the intraarticular blood. After a joint has been damaged, recurrent bleeding episodes result in the clinically recognized “target joint,” which then establishes a vicious cycle of bleeding, resulting in progressive joint deformity that in critical cases requires surgery as the only therapeutic option. Hematomas into the muscle of distal parts of the limbs may lead to external compression of arteries, veins, or nerves that can evolve to a compartment syndrome.
Bleeding into the oropharyngeal spaces, central nervous system (CNS), or retroperitoneum is life threatening and requires immediate therapy. Retroperitoneal hemorrhages can accumulate large quantities of blood with formation of masses with calcification and inflammatory tissue reaction (pseudotumor syndrome) and also result in damage to the femoral nerve. Pseudotumors can also form in bones, especially long bones of the lower limbs. Hematuria is frequent among hemophilia patients, even in the absence of genitourinary pathology. It is often self-limited and may not require specific therapy.
|
HEMOPHILIA |
Without treatment, severe hemophilia has a limited life expectancy. Advances in the blood fractionation industry during World War II resulted in the realization that plasma could be used to treat hemophilia, but the volumes required to achieve even modest elevation of circulating factor levels limit the utility of plasma infusion as an approach to disease management. The discovery in the 1960s that the cryoprecipitate fraction of plasma was enriched for FVIII, and the eventual purification of FVIII and FIX from plasma, led to the introduction of home infusion therapy with factor concentrates in the 1970s. The availability of factor concentrates resulted in a dramatic improvement in life expectancy and in quality of life for people with severe hemophilia. However, the contamination of the blood supply with hepatitis viruses and, subsequently, HIV resulted in widespread transmission of these bloodborne infections within the hemophilia population; complications of HIV and of hepatitis C are now the leading causes of death among U.S. adults with severe hemophilia. The introduction of viral inactivation steps in the preparation of plasma-derived products in the mid-1980s greatly reduced the risk of HIV and hepatitis, and the risks were further reduced by the successful production of recombinant FVIII and FIX proteins, both licensed in the 1990s. It is uncommon for hemophilic patients born after 1985 to have contracted either hepatitis or HIV, and for these individuals, life expectancy is approximately 65 years. In fact, since 1998, no evidence of new infections with viral hepatitis or HIV has been reported in patients using blood products. Factor replacement therapy for hemophilia can be provided either in response to a bleeding episode or as a prophylactic treatment. Primary prophylaxis is defined as a strategy for maintaining the missing clotting factor at levels ~1% or higher on a regular basis in order to prevent bleeds, especially the onset of hemarthroses. Hemophilic boys receiving regular infusions of FVIII (3 days/week) or FIX (2 days/week) can reach puberty without detectable joint abnormalities. Prophylaxis has become gradually more common in young patients. The Centers for Disease Control and Prevention reported that 51% of children with severe hemophilia who are younger than age 6 years receive prophylaxis, increasing considerably from 33% in 1995. Although highly recommended, the high cost and difficulties in accessing peripheral veins in young patients and the potential infectious and thrombotic risks of long-term central vein catheters are important limiting factors for many young patients. Emerging data show that prophylaxis is also increasing among adults with severe hemophilia.
General considerations regarding the treatment of bleeds in hemophilia include the following: (1) Treatment should begin as soon as possible because symptoms often precede objective evidence of bleeding; because of the superior efficacy of early therapeutic intervention, classic symptoms of bleeding into the joint in a reliable patient, headaches, or automobile or other accidents require prompt replacement and further laboratory investigation. (2) Drugs that hamper platelet function, such as aspirin or aspirin- containing drugs, should be avoided; to control pain, drugs such as ibuprofen or propoxyphene are preferred. FVIII and FIX are dosed in units. One unit is defined as amount of FVIII (100 ng/mL) or FIX (5 μg/mL) in 1 mL of normal plasma. One unit of FVIII per kilogram of body weight increases the plasma FVIII level by 2%. One can calculate the dose needed to increase FVIII levels to 100% in a 70-kg severe hemophilia patient (<1%) using the simple formula below. Thus, 3500 units of FVIII will raise the circulating level to 100%.
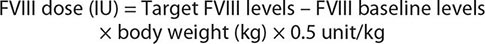
The doses for FIX replacement are different from those for FVIII, because FIX recovery after infusion is usually only 50% of the predicted value. Therefore, the formula for FIX replacement is as follows:
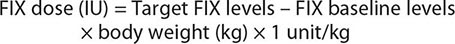
The FVIII half-life of 8–12 h requires injections twice a day to maintain therapeutic levels, whereas the FIX half-life is longer, ~24 h, so that once-a-day injection is sufficient. In specific situations such as after surgery, continuous infusion of factor may be desirable because of its safety in achieving sustained factor levels at a lower total cost.
Cryoprecipitate is enriched with FVIII protein (each bag contains ~80 IU of FVIII) and was commonly used for the treatment of hemophilia A decades ago; it is still in use in some developing countries, but because of the risk of bloodborne diseases, this product should be avoided in hemophilia patients when factor concentrates are available.
Mild bleeds such as uncomplicated hemarthroses or superficial hematomas require initial therapy with factor levels of 30–50%. Additional doses to maintain levels of 15–25% for 2 or 3 days are indicated for severe hemarthroses, especially when these episodes affect the “target joint.” Large hematomas, or bleeds into deep muscles, require factor levels of 50% or even higher if the clinical symptoms do not improve, and factor replacement may be required for a period of 1 week or longer. The control of serious bleeds including those that affect the oropharyngeal spaces, CNS, and the retroperitoneum require sustained protein levels of 50–100% for 7–10 days. Prophylactic replacement for surgery is aimed at achieving normal factor levels (100%) for a period of 7–10 days; replacement can then be tapered depending on the extent of the surgical wounds. Oral surgery is associated with extensive tissue damage that usually requires factor replacement for 1–3 days coupled with oral antifibrinolytic drugs.
NONTRANSFUSION THERAPY IN HEMOPHILIA
DDAVP (1-Amino-8-D-Arginine Vasopressin) DDAVP is a synthetic vasopressin analog that causes a transient rise in FVIII and von Willebrand factor (VWF), but not FIX, through a mechanism involving release from endothelial cells. Patients with moderate or mild hemophilia A should be tested to determine if they respond to DDAVP before a therapeutic application. DDAVP at doses of 0.3 μg/kg body weight, over a 20-min period, is expected to raise FVIII levels by two- to threefold over baseline, peaking between 30 and 60 min after infusion. DDAVP does not improve FVIII levels in severe hemophilia A patients, because there are no stores to release. Repeated dosing of DDAVP results in tachyphylaxis because the mechanism is an increase in release rather than de novo synthesis of FVIII and VWF. More than three consecutive doses become ineffective, and if further therapy is indicated, FVIII replacement is required to achieve hemostasis.
Antifibrinolytic Drugs Bleeding in the gums, gastrointestinal tract, and during oral surgery requires the use of oral antifibrinolytic drugs such as ε-amino caproic acid (EACA) or tranexamic acid to control local hemostasis. The duration of the treatment depending on the clinical indication is 1 week or longer. Tranexamic acid is given at doses of 25 mg/kg three to four times a day. EACA treatment requires a loading dose of 200 mg/kg (maximum of 10 g) followed by 100 mg/kg per dose (maximum 30 g/d) every 6 h. These drugs are not indicated to control hematuria because of the risk of formation of an occlusive clot in the lumen of genitourinary tract structures.
COMPLICATIONS
Inhibitor Formation The formation of alloantibodies to FVIII or FIX is currently the major complication of hemophilia treatment. The prevalence of inhibitors to FVIII is estimated to be between 5 and 10% of all cases and ~20% of severe hemophilia A patients. Inhibitors to FIX are detected in only 3–5% of all hemophilia B patients. The high-risk group for inhibitor formation includes severe deficiency (>80% of all cases of inhibitors), familial history of inhibitor, African descent, mutations in the FVIII or FIX gene resulting in deletion of large coding regions, or gross gene rearrangements. Inhibitors usually appear early in life, at a median of 2 years of age, and after 10 cumulative days of exposure. However, intensive replacement therapy such as for major surgery, intracranial bleeding, or trauma increases the risk of inhibitor formation for patients of all ages and degree of clinical severity, which requires close laboratory monitoring in the following weeks.
The clinical diagnosis of an inhibitor is suspected when patients do not respond to factor replacement at therapeutic doses. Inhibitors increase both morbidity and mortality in hemophilia. Because early detection of an inhibitor is critical to a successful correction of the bleeding or to eradication of the antibody, most hemophilia centers perform annual screening for inhibitors. The laboratory test required to confirm the presence of an inhibitor is an aPTT with a mix (with normal plasma). In most hemophilia patients, a 1:1 mix with normal plasma completely corrects the aPTT. In inhibitor patients, the aPTT on a 1:1 mix is abnormally prolonged, because the inhibitor neutralizes the FVIII clotting activity of the normal plasma. The Bethesda assay uses a similar principle and defines the specificity of the inhibitor and its titer. The results are expressed in Bethesda units (BU), in which 1 BU is the amount of antibody that neutralizes 50% of the FVIII or FIX present in normal plasma after 2 h of incubation at 37°C. Clinically, inhibitor patients are classified as low responders or high responders, which provides guidelines for optimal therapy. Therapy for inhibitor patients has two goals: the control of acute bleeding episodes and the eradication of the inhibitor. For the control of bleeding episodes, low responders, those with titer <5 BU, respond well to high doses of human or porcine FVIII (50–100 U/kg), with minimal or no increase in the inhibitor titers. However, high-responder patients, those with initial inhibitor titer >10 BU or an anamnestic response in the antibody titer to >10 BU even if low titer initially, do not respond to FVIII or FIX concentrates. The control of bleeding episodes in high-responder patients can be achieved by using concentrates enriched for prothrombin, FVII, FIX, FX (prothrombin complex concentrates [PCCs] or activated PCCs [aPCCs]), and more recently recombinant activated factor VII (FVIIa) known as “bypass agents” (Fig. 141-1). The rates of therapeutic success have been higher for FVIIa than for PCC or aPCC. For eradication of the inhibitory antibody, immunosuppression alone is not effective. The most effective strategy is the immune tolerance induction (ITI) based on daily infusion of missing protein until the inhibitor disappears, typically requiring periods longer than 1 year, with success rates of approximately 60%. The management of patients with severe hemophilia and inhibitors resistant to ITI is challenging. The use of anti-CD20 monoclonal antibody (rituximab) combined with ITI was thought to be effective. Although this therapy may reduce the inhibitor titers in some cases, sustained eradication is uncommon and may require two to three infusions weekly of clotting factor concentrates.
Novel Therapeutic Approaches in Development for Hemophilia Clinical studies using long-acting clotting factors with prolonged half-lives are in the late phase of clinical testing, and these new-generation products (for FVIII and FIX) may facilitate prophylaxis by requiring fewer injections to maintain circulating levels above 1%.
The use of recombinant interleukin 11 in patients with moderate or mild hemophilia A unresponsive to DDAVP has been tested in early-phase clinical trials and may be an alternate therapeutic strategy for clinical situations that require transient increases in FVIII levels.
Gene therapy trials for hemophilia B using adeno-associated viral vectors are ongoing, and initial data are promising (Chap. 91e).
INFECTIOUS DISEASES
Hepatitis C virus (HCV) infection is the major cause of morbidity and the second leading cause of death in hemophilia patients exposed to older clotting factor concentrates. The vast majority of young patients treated with plasma-derived products from 1970 to 1985 became infected with HCV. It has been estimated that >80% of patients older than 20 years of age are HCV antibody positive as of 2006. The comorbidity of the underlying liver disease in hemophilia patients is clear when these individuals require invasive procedures; correction of both genetic and acquired (secondary to liver disease) deficiencies may be needed. Infection with HIV also swept the population of patients using plasma-derived concentrates two decades ago. Co-infection of HCV and HIV, present in almost 50% of hemophilia patients, is an aggravating factor for the evolution of liver disease. The response to HCV antiviral therapy in hemophilia is restricted to <30% of patients and even poorer among those with both HCV and HIV infection. End-stage liver disease requiring organ transplantation may be curative for both the liver disease and for hemophilia.
EMERGING CLINICAL PROBLEMS IN AGING HEMOPHILIA PATIENTS
There has been continuous improvement of the management of hemophilia since the increase in the population of adults living beyond middle age in the developing world. The life expectancy of a patient with severe hemophilia is only ~10 years shorter than the general male population. In patients with mild or moderate hemophilia, life expectancy is approaching that of the male population without coagulopathy. Elderly hemophilia patients have different problems compared to the younger generation; they have more severe arthropathy and chronic pain, due to suboptimal treatment, and high rates of HCV and/or HIV infections.
Early data indicate that mortality from coronary artery disease is lower in hemophilia patients than the general male population. The underlying hypocoagulability probably provides a protective effect against thrombus formation, but it does not prevent atherogenesis. Similar to the general population, these patients are exposed to cardiovascular risk factors such as age, obesity, and smoking. Moreover, physical inactivity, hypertension, and chronic renal disease are commonly observed in hemophilia patients. In HIV patients on combined antiretroviral therapy, there may be a further increase in the risk of cardiovascular disease. Therefore, these patients should be carefully considered for preventive and therapeutic approaches to minimize the risk of cardiovascular disease.
Excessive replacement therapy should be avoided, and it is prudent to slowly infuse factor concentrates. Continuous infusion of clotting factor is preferable to bolus dosing in patients with cardiovascular risk factors undergoing invasive procedures. The management of an acute ischemic event and coronary revascularization should include the collaboration of hematologists and internists. The early assumption that hemophilia would protect against occlusive vascular disease may change in this aging population. Cancer is a common cause of mortality in aging hemophilia patients because they are at risk for HIV- and HCV-related malignancies. Hepatocellular carcinoma (HCC) is the most prevalent primary liver cancer and a common cause of death in HIV-negative patients. The recommendations for cancer screening for the general population should be the same for age-matched hemophilia patients. Among those with high-risk HCV, a semiannual or annual ultrasound and α fetoprotein are recommended for HCC. Screening for urogenital neoplasm in the presence of hematuria or hematochezia may be delayed due to the underlying bleeding disease, thus preventing early intervention. Multidisciplinary interaction should facilitate the attempts to ensure optimal cancer prevention and treatment recommendations for those with hemophilia.
MANAGEMENT OF CARRIERS OF HEMOPHILIA
Usually hemophilia carriers, with factor levels of ~50% of normal, have not been considered to be at risk for bleeding. However, a wide range of values (22–116%) have been reported due to random inactivation of the × chromosomes (lyonization). Therefore, it is important to measure the factor level of carriers to recognize those at risk of bleeding and to optimize preoperative and postoperative management. During pregnancy, both FVIII and FIX levels increase gradually until delivery. FVIII levels increase approximately two- to threefold compared to nonpregnant women, whereas an FIX increase is less pronounced. After delivery, there is a rapid fall in the pregnancy-induced rise of maternal clotting factor levels. This represents an imminent risk of bleeding that can be prevented by infusion of factor concentrate to levels of 50–70% for 3 days in the setting of vaginal delivery and up to 5 days for cesarean section. In mild cases, the use of DDAVP and/or antifibrinolytic drugs is recommended.
FACTOR XI DEFICIENCY
Factor XI is a zymogen of an active serine protease (FIXa) in the intrinsic pathway of blood coagulation that activates FIX (Fig. 141-1). There are two pathways for the formation of FXIa. In an aPTT-based assay, the protease is the result of activation by FXIIa in conjunction with high-molecular-weight kininogen and kallikrein. In vivo data suggest that thrombin is the physiologic activator of FXI. The generation of thrombin by the tissue factor/factor VIIa pathway activates FXI on the platelet surface that contributes to additional thrombin generation after the clot has formed and thus augments resistance to fibrinolysis through a thrombin-activated fibrinolytic inhibitor (TAFI).
Factor XI deficiency is a rare bleeding disorder that occurs in the general population at a frequency of one in a million. However, the disease is highly prevalent among Ashkenazi and Iraqi Jewish populations, reaching a frequency of 6% as heterozygotes and 0.1–0.3% as homozygotes. More than 65 mutations in the FXI gene have been reported, whereas fewer mutations (two to three) are found among affected Jewish populations.
Normal FXI clotting activity levels range from 70 to 150 U/dL. In heterozygous patients with moderate deficiency, FXI ranges from 20 to 70 U/dL, whereas in homozygous or double heterozygote patients, FXI levels are <1–20 U/dL. Patients with FXI levels <10% of normal have a high risk of bleeding, but the disease phenotype does not always correlate with residual FXI clotting activity. A family history is indicative of the risk of bleeding in the propositus. Clinically, the presence of mucocutaneous hemorrhages such as bruises, gum bleeding, epistaxis, hematuria, and menorrhagia are common, especially following trauma. This hemorrhagic phenotype suggests that tissues rich in fibrinolytic activity are more susceptible to FXI deficiency. Postoperative bleeding is common but not always present, even among patients with very low FXI levels.
FXI replacement is indicated in patients with severe disease required to undergo a surgical procedure. A negative history of bleeding complications following invasive procedures does not exclude the possibility of an increased risk for hemorrhage.
RARE BLEEDING DISORDERS
Collectively, the inherited disorders resulting from deficiencies of clotting factors other than FVIII, FIX, and FXI (Table 141-1) represent a group of rare bleeding diseases. The bleeding symptoms in these patients vary from asymptomatic (dysfibrinogenemia or FVII deficiency) to life-threatening (FX or FXIII deficiency). There is no pathognomonic clinical manifestation that suggests one specific disease, but overall, in contrast to hemophilia, hemarthrosis is a rare event and bleeding in the mucosal tract or after umbilical cord clamping is common. Individuals heterozygous for plasma coagulation deficiencies are often asymptomatic. The laboratory assessment for the specific deficient factor following screening with general coagulation tests (Table 141-1) will define the diagnosis.
Replacement therapy using FFP or prothrombin complex concentrates (containing prothrombin, FVII, FIX, and FX) provides adequate hemostasis in response to bleeds or as prophylactic treatment. The use of PCC should be carefully monitored and avoided in patients with underlying liver disease, or those at high risk for thrombosis because of the risk of DIC.
FAMILIAL MULTIPLE COAGULATION DEFICIENCIES
There are several bleeding disorders characterized by the inherited deficiency of more than one plasma coagulation factor. To date, the genetic defects in two of these diseases have been characterized, and they provide new insights into the regulation of hemostasis by gene-encoding proteins outside blood coagulation.
Combined Deficiency of FV and FVIII Patients with combined FV and FVIII deficiency exhibit ~5% of residual clotting activity of each factor. Interestingly, the disease phenotype is a mild bleeding tendency, often following trauma. An underlying mutation has been identified in the endoplasmic reticulum/Golgi intermediate compartment (ERGIC-53) gene, a mannose-binding protein localized in the Golgi apparatus that functions as a chaperone for both FV and FVIII. In other families, mutations in the multiple coagulation factor deficiency 2 (MCFD2) gene have been defined; this gene encodes a protein that forms a Ca2+ -dependent complex with ERGIC-53 and provides cofactor activity in the intracellular mobilization of both FV and FVIII.
Multiple Deficiencies of Vitamin K–Dependent Coagulation Factors Two enzymes involved in vitamin K metabolism have been associated with combined deficiency of all vitamin K–dependent proteins, including the procoagulant proteins prothrombin, VII, IX, and × and the anticoagulant proteins C and S. Vitamin K is a fat-soluble vitamin that is a cofactor for carboxylation of the gamma carbon of the glutamic acid residues in the vitamin K–dependent factors, a critical step for calcium and phospholipid binding of these proteins (Fig. 141-2). The enzymes γ-glutamylcarboxylase and epoxide reductase are critical for the metabolism and regeneration of vitamin K. Mutations in the genes encoding the γ-carboxylase (GGCX) or vitamin K epoxide reductase complex 1 (VKORC1) result in defective enzymes and thus in vitamin K–dependent factors with reduced activity, varying from 1 to 30% of normal. The disease phenotype is characterized by mild to severe bleeding episodes present from birth. Some patients respond to high doses of vitamin K. For severe bleeding, replacement therapy with FFP or PCC may be necessary to achieve full hemostatic control.
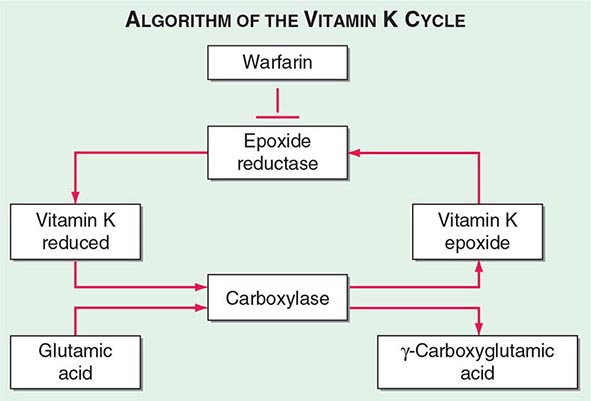
FIGURE 141-2 The vitamin K cycle. Vitamin K is a cofactor for the formation of γ-carboxyglutamic acid residues on coagulation proteins. Vitamin K–dependent γ-glutamylcarboxylase, the enzyme that catalyzes the vitamin K epoxide reductase, regenerates reduced vitamin K. Warfarin blocks the action of the reductase and competitively inhibits the effects of vitamin K.
DISSEMINATED INTRAVASCULAR COAGULATION
DIC is a clinicopathologic syndrome characterized by widespread intravascular fibrin formation in response to excessive blood protease activity that overcomes the natural anticoagulant mechanisms. There are several underlying pathologies associated with DIC (Table 141-2).
|
COMMON CLINICAL CAUSES OF DISSEMINATED INTRAVASCULAR COAGULATION |
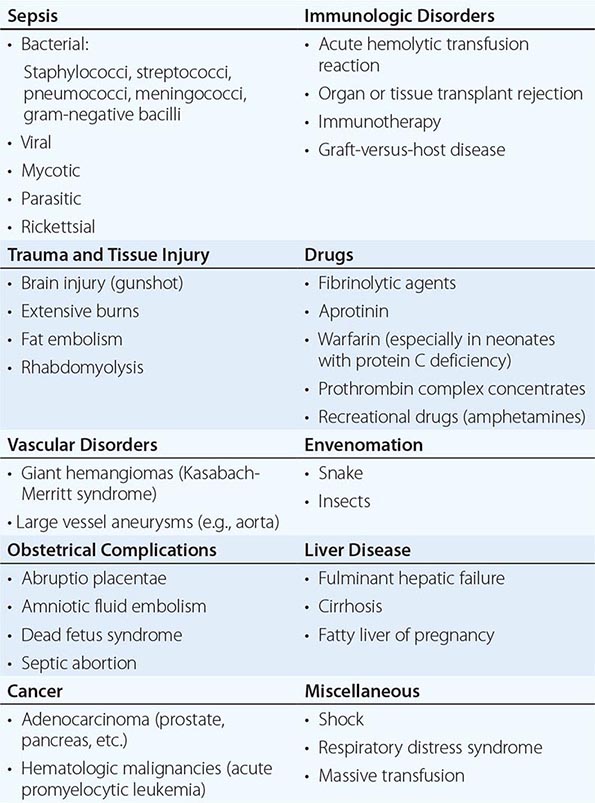
The most common causes are bacterial sepsis, malignant disorders such as solid tumors or acute promyelocytic leukemia, and obstetric causes. DIC is diagnosed in almost one-half of pregnant women with abruptio placentae or with amniotic fluid embolism. Trauma, particularly to the brain, can also result in DIC. The exposure of blood to phospholipids from damaged tissue, hemolysis, and endothelial damage are all contributing factors to the development of DIC in this setting. Purpura fulminans is a severe form of DIC resulting from thrombosis of extensive areas of the skin; it affects predominantly young children following viral or bacterial infection, particularly those with inherited or acquired hypercoagulability due to deficiencies of the components of the protein C pathway. Neonates homozygous for protein C deficiency also present high risk for purpura fulminans with or without thrombosis of large vessels.
The central mechanism of DIC is the uncontrolled generation of thrombin by exposure of the blood to pathologic levels of tissue factor (Fig. 141-3). Simultaneous suppression of physiologic anticoagulant mechanisms and abnormal fibrinolysis further accelerate the process. Together, these abnormalities contribute to systemic fibrin deposition in small and midsize vessels. The duration and intensity of the fibrin deposition can compromise the blood supply of many organs, especially the lung, kidney, liver, and brain, with consequent organ failure. The sustained activation of coagulation results in consumption of clotting factors and platelets, which in turn leads to systemic bleeding. This is further aggravated by secondary hyperfibrinolysis. Studies in animals demonstrate that the fibrinolytic system is indeed suppressed at the time of maximal activation of coagulation. Interestingly, in patients with acute promyelocytic leukemia, a severe hyperfibrinolytic state often occurs in addition to the coagulation activation. The release of several proinflammatory cytokines such as interleukin 6 and tumor necrosis factor α plays a central role in mediating the coagulation defects in DIC and symptoms associated with systemic inflammatory response syndrome (SIRS).
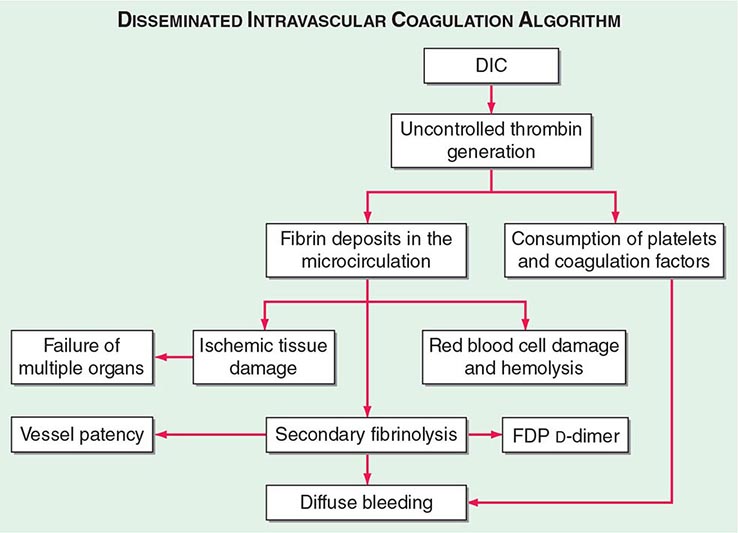
FIGURE 141-3 The pathophysiology of disseminated intravascular coagulation (DIC). Interactions between coagulation and fibrinolytic pathways result in bleeding and thrombosis in the microcirculation in patients with DIC. FDP, fibrin degradation product.
Clinical manifestations of DIC are related to the magnitude of the imbalance of hemostasis, to the underlying disease, or to both. The most common findings are bleeding ranging from oozing from venipuncture sites, petechiae, and ecchymoses to severe hemorrhage from the gastrointestinal tract, lung, or into the CNS. In chronic DIC, the bleeding symptoms are discrete and restricted to skin or mucosal surfaces. The hypercoagulability of DIC manifests as the occlusion of vessels in the microcirculation and resulting organ failure. Thrombosis of large vessels and cerebral embolism can also occur. Hemodynamic complications and shock are common among patients with acute DIC. The mortality ranges from 30 to >80% depending on the underlying disease, the severity of the DIC, and the age of the patient.
The diagnosis of clinically significant DIC is based on the presence of clinical and/or laboratory abnormalities of coagulation or thrombocytopenia. The laboratory diagnosis of DIC should prompt a search for the underlying disease if it is not already apparent. There is no single test that establishes the diagnosis of DIC. The laboratory investigation should include coagulation tests (aPTT, PT, thrombin time [TT]) and markers of fibrin degradation products (FDPs), in addition to platelet and red cell count and analysis of the blood smear. These tests should be repeated over a period of 6–8 h because an initially mild abnormality can change dramatically in patients with severe DIC.
Common findings include the prolongation of PT and/or aPTT; platelet counts μ100,000/μL, or a rapid decline in platelet numbers; the presence of schistocytes (fragmented red cells) in the blood smear; and elevated levels of FDP. The most sensitive test for DIC is the FDP level. DIC is an unlikely diagnosis in the presence of normal levels of FDP. The D-dimer test is more specific for detection of fibrin—but not fibrinogen—degradation products and indicates that the cross-linked fibrin has been digested by plasmin. Because fibrinogen has a prolonged half-life, plasma levels diminish acutely only in severe cases of DIC. High-grade DIC is also associated with levels of antithrombin III or plasminogen activity <60% of normal.
Chronic DIC Low-grade, compensated DIC can occur in clinical situations including giant hemangioma, metastatic carcinoma, or the dead fetus syndrome. Plasma levels of FDP or D-dimers are elevated. aPTT, PT, and fibrinogen values are within the normal range or high. Mild thrombocytopenia or normal platelet counts are also common findings. Red cell fragmentation is often detected but at a lower degree than in acute DIC.
Differential Diagnosis The differential diagnosis between DIC and severe liver disease is challenging and requires serial measurements of the laboratory parameters of DIC. Patients with severe liver disease are at risk for bleeding and manifest laboratory features including thrombocytopenia (due to platelet sequestration, portal hypertension, or hypersplenism), decreased synthesis of coagulation factors and natural anticoagulants, and elevated levels of FDP due to reduced hepatic clearance. However, in contrast to DIC, these laboratory parameters in liver disease do not change rapidly. Other important differential findings include the presence of portal hypertension or other clinical or laboratory evidence of an underlying liver disease.
Microangiopathic disorders such as thrombotic thrombocytopenic purpura present an acute clinical onset of illness accompanied by thrombocytopenia, red cell fragmentation, and multiorgan failure. However, there is no consumption of clotting factors or hyperfibrinolysis.
Over the last few years, several clinical trials on immune therapies for neoplasias using monoclonal antibodies or gene-modified T cells targeting tumor-specific antigens showed unwanted inflammatory responses with increased cytokine release. These complications are sometimes associated with increased D-dimers and decreased fibrinogen levels, cytopenias, and liver dysfunction; thus, careful screening tests for DIC are indicated.
VITAMIN K DEFICIENCY Vitamin K–dependent proteins are a heterogenous group, including clotting factor proteins and also proteins found in bone, lung, kidney, and placenta. Vitamin K mediates posttranslational modification of glutamate residues to γ-carboxylglutamate, a critical step for the activity of vitamin K–dependent proteins for calcium binding and proper assembly to phospholipid membranes (Fig. 141-2). Inherited deficiency of the functional activity of the enzymes involved in vitamin K metabolism, notably the GGCX or VKORC1 (see above), results in bleeding disorders. The amount of vitamin K in the diet is often limiting for the carboxylation reaction; thus recycling of the vitamin K is essential to maintain normal levels of vitamin K–dependent proteins. In adults, low dietary intake alone is seldom reason for severe vitamin K deficiency but may become common in association with the use of broad-spectrum antibiotics. Disease or surgical interventions that affect the ability of the intestinal tract to absorb vitamin K, either through anatomic alterations or by changing the fat content of bile salts and pancreatic juices in the proximal small bowel, can result in significant reduction of vitamin K levels. Chronic liver diseases such as primary biliary cirrhosis also deplete vitamin K stores. Neonatal vitamin K deficiency and the resulting hemorrhagic disease of the newborn have been almost entirely eliminated by routine administration of vitamin K to all neonates. Prolongation of PT values is the most common and earliest finding in vitamin K–deficient patients due to reduction in prothrombin, FVII, FIX, and FX levels. FVII has the shortest half-life among these factors that can prolong the PT before changes in the aPTT. Parenteral administration of vitamin K at a total dose of 10 mg is sufficient to restore normal levels of clotting factor within 8–10 h. In the presence of ongoing bleeding or a need for immediate correction before an invasive procedure, replacement with FFP or PCC is required. The latter should be avoided in patients with severe underlying liver disorders due to high risk of thrombosis. The reversal of excessive anticoagulant therapy with warfarin or warfarin-like drugs can be achieved by minimal doses of vitamin K (1 mg orally or by intravenous injection) for asymptomatic patients. This strategy can diminish the risk of bleeding while maintaining therapeutic anticoagulation for an underlying prothrombotic state.
In patients with life-threatening bleeds, the use of recombinant factor VIIa in nonhemophilia patients on anticoagulant therapy has been shown to be effective at restoring hemostasis rapidly, allowing emergency surgical intervention. However, patients with underlying vascular disease, vascular trauma and other comorbidities are at risk for thromboembolic complications that affect both arterial and venous systems. Thus, the use of factor VIIa in this setting is limited to administration of low doses given for only a limited number of injections. Close monitoring for vascular complications is highly indicated.
COAGULATION DISORDERS ASSOCIATED WITH LIVER FAILURE The liver is central to hemostasis because it is the site of synthesis and clearance of most procoagulant and natural anticoagulant proteins and of essential components of the fibrinolytic system. Liver failure is associated with a high risk of bleeding due to deficient synthesis of procoagulant factors and enhanced fibrinolysis. Thrombocytopenia is common in patients with liver disease, and may be due to congestive splenomegaly (hypersplenism) or immune-mediated shortened platelet lifespan (primary biliary cirrhosis). In addition, several anatomic abnormalities secondary to underlying liver disease further promote the occurrence of hemorrhage (Table 141-3). Dysfibrinogenemia is a relatively common finding in patients with liver disease due to impaired fibrin polymerization. The development of DIC concomitant to chronic liver disease is not uncommon and may enhance the risk for bleeding. Laboratory evaluation is mandatory for an optimal therapeutic strategy, either to control ongoing bleeding or to prepare patients with liver disease for invasive procedures. Typically, these patients present with prolonged PT, aPTT, and TT depending on the degree of liver damage, thrombocytopenia, and normal or slight increase of FDP. Fibrinogen levels are diminished only in fulminant hepatitis, decompensated cirrhosis, or advanced liver disease, or in the presence of DIC. The presence of prolonged TT and normal fibrinogen and FDP levels suggest dysfibrinogenemia. FVIII levels are often normal or elevated in patients with liver failure, and decreased levels suggest superimposing DIC. Because FV is only synthesized in the hepatocyte and is not a vitamin K–dependent protein, reduced levels of FV may be an indicator of hepatocyte failure. Normal levels of FV and low levels of FVII suggest vitamin K deficiency. Vitamin K levels may be reduced in patients with liver failure due to compromised storage in hepatocellular disease, changes in bile acids, or cholestasis that can diminish the absorption of vitamin K. Replacement of vitamin K may be desirable (10 mg given by slow intravenous injection) to improve hemostasis.
|
COAGULATION DISORDERS AND HEMOSTASIS IN LIVER DISEASE |
Abbreviations: DIC, disseminated intravascular coagulation; EACA, ε-aminocaproic acid.
Treatment with FFP is the most effective to correct hemostasis in patients with liver failure. Infusion of FFP (5–10 mL/kg; each bag contains ~200 mL) is sufficient to ensure 10–20% of normal levels of clotting factors but not correction of PT or aPTT. Even high doses of FFP (20 mL/kg) do not correct the clotting times in all patients. Monitoring for clinical symptoms and clotting times will determine if repeated doses are required 8–12 h after the first infusion. Platelet concentrates are indicated when platelet counts are <10,000–20,000/μL to control an ongoing bleed or immediately before an invasive procedure if counts are <50,000/μL. Cryoprecipitate is indicated only when fibrinogen levels are less than 100 mg/mL; dosing is six bags for a 70-kg patient daily. Prothrombin complex concentrate infusion in patients with liver failure should be avoided due to the high risk of thrombotic complications. The safety of the use of antifibrinolytic drugs to control bleeding in patients with liver failure is not yet well defined and should be avoided.
LIVER DISEASE AND THROMBOEMBOLISM The clinical bleeding phenotype of hemostasis in patients with stable liver disease is often mild or even asymptomatic. However, as the disease progresses, the hemostatic balance is less stable and more easily disturbed than in healthy individuals. Furthermore, the hemostatic balance is compromised by comorbid complications such as infections and renal failure (Fig. 141-4). Based on the clinical bleeding complications in patients with cirrhosis and laboratory evidence of hypocoagulation such as a prolonged PT/aPTT, it has long been assumed that these patients are protected against thrombotic disease. Cumulative clinical experience, however, has demonstrated that these patients are at risk for thrombosis, especially those with advanced liver disease. Although hypercoagulability could explain the occurrence of venous thrombosis, according to Virchow’s triad, hemodynamic changes and damaged vasculature may also be a contributing factor, and both processes may potentially also occur in patients with liver disease. Liver-related thrombosis, in particular, thrombosis of the portal and mesenteric veins, is common in patients with advanced cirrhosis. Hemodynamic changes, such as decreased portal flow, and evidence that inherited thrombophilia may enhance the risk for portal vein thrombosis in patients with cirrhosis suggest that hypercoagulability may play a role as well. Patients with liver disease develop deep vein thrombosis and pulmonary embolism at appreciable rates (ranging from 0.5 to 1.9%). The implication of these findings is relevant to the erroneous exclusion of thrombosis in patients with advanced liver disease, even in the presence of prolongation of routine clotting times, and caution should be advised on overcorrection of these laboratory abnormalities.
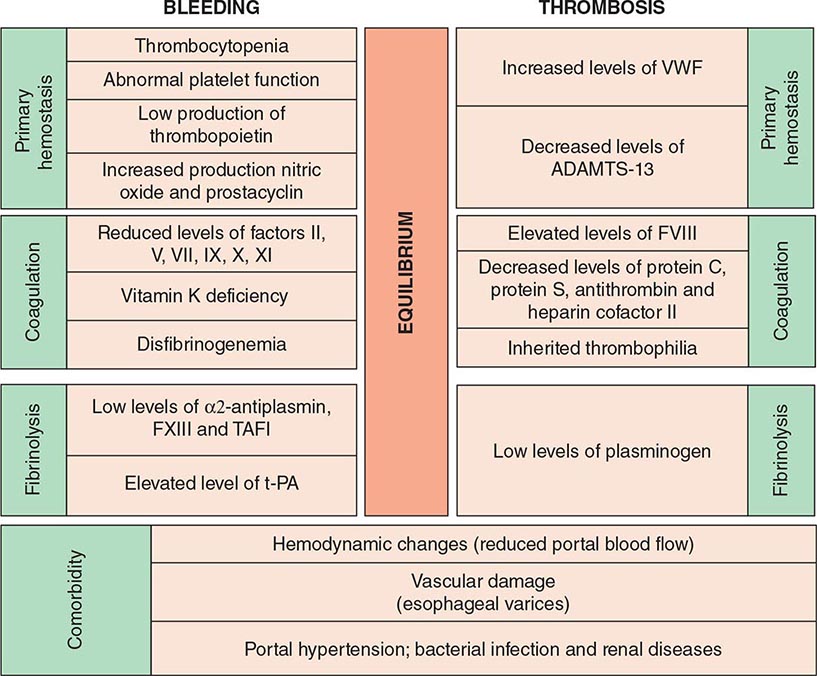
FIGURE 141-4 Balance of hemostasis in liver disease. TAFI, thrombin-activated fibrinolytic inhibitor; t-PA, tissue plasminogen activator; VWF, von Willebrand factor.
ACQUIRED INHIBITORS OF COAGULATION FACTORS An acquired inhibitor is an immune-mediated disease characterized by the presence of an autoantibody against a specific clotting factor. FVIII is the most common target of antibody formation, and is sometimes referred to as acquired hemophilia A, but inhibitors to prothrombin, FV, FIX, FX, and FXI are also reported. Acquired inhibitor to FVIII occurs predominantly in older adults (median age of 60 years), but occasionally in pregnant or postpartum women with no previous history of bleeding. In 50% of patients with inhibitors, no underlying disease is identified at the time of diagnosis. In the remaining patients, the causes are autoimmune diseases, malignancies (lymphomas, prostate cancer), dermatologic diseases, and pregnancy. Bleeding episodes occur commonly in soft tissues, the gastrointestinal or urinary tracts, and skin. In contrast to hemophilia, hemarthrosis is rare in these patients. Retroperitoneal hemorrhages and other life-threatening bleeding may appear suddenly. The overall mortality in untreated patients ranges from 8 to 22%, and most deaths occur within the first few weeks after presentation. The diagnosis is based on the prolonged aPTT with normal PT and TT. The aPTT remains prolonged after mixture of the test plasma with equal amounts of pooled normal plasma for 2 h at 37°C. The Bethesda assay using FVIII-deficient plasma as performed for inhibitor detection in hemophilia will confirm the diagnosis. Major bleeding is treated with bypass products such as PCC/aPCC or recombinant FVIIa. In contrast to hemophilia, inhibitors in nonhemophilic patients are typically responsive to immune suppression, and therapy should be initiated early for most cases. The first choice includes steroid or a combination of steroid with cytotoxic therapy (e.g., cyclophosphamide), with complete eradication of the inhibitors in more than 70% of patients. High-dose intravenous γ-globulin and anti-CD20 monoclonal antibody have been reported to be effective in patients with autoantibodies to FVIII; however, there is no firm evidence that these alternatives are superior to the first line of immunosuppressive drugs. Notably, relapse of the inhibitor to FVIII is relatively common (up to 20%) within the first 6 months following withdrawal of immunosuppression. Thus, after eradication, patients should be followed up regularly for early therapeutic intervention when indicated or prior to invasive procedure.
Topical plasma-derived bovine and human thrombin are commonly used in the United States and worldwide. These effective hemostatic sealants are used during major surgery such as for cardiovascular, thoracic, neurologic, pelvic, and trauma indications, as well as in the setting of extensive burns. The development of antibody formation to the xenoantigen or its contaminant (bovine clotting protein) has the potential to show cross-reactivity with human clotting factors that may hamper their function and induce bleeding.
Clinical features of these antibodies include bleeding from a primary hemostatic defect or coagulopathy that sometimes can be life threatening. The clinical diagnosis of these acquired coagulopathies is often complicated by the fact that the bleeding episodes may be detectable during or immediately following major surgery and could be assumed to be due to the procedure itself.
Notably, the risk of this complication is further increased by repeated exposure to topical thrombin preparations. Thus, a careful medical history of previous surgical interventions that may have occurred even decades earlier is critical to assessing risk.
The laboratory abnormalities are reflected by combined prolongation of the aPTT and PT that often fails to improve by transfusion of FFP and vitamin K. The abnormal laboratory tests cannot be corrected by mixing a test with equal parts of normal plasma that denotes the presence of inhibitory antibodies. The diagnosis of a specific antibody is obtained by the determination of the residual activity of human FV or other suspected human clotting factor. There are no commercially available assays specific for bovine thrombin coagulopathy.
There are no established treatment guidelines. Platelet transfusions have been used as a source of FV replacement for patients with FV inhibitors. Frequent injections of FFP and vitamin K supplementation may function as co-adjuvant rather than an effective treatment of the coagulopathy itself. Experience with recombinant FVIIa as a bypass agent is limited, and outcomes have been generally poor. Specific treatments to eradicate the antibodies based on immunosuppression with steroids, intravenous immunoglobulin, or serial plasmapheresis have been sporadically reported. Patients should be advised to avoid any topical thrombin sealant in the future.
Novel plasma-derived and recombinant human thrombin preparations for topical hemostasis have been approved by the U.S. Food and Drug Administration. These preparations have demonstrated hemostatic efficacy with reduced immunogenicity compared to the first generation of bovine thrombin products.
The presence of lupus anticoagulant can be associated with venous or arterial thrombotic disease. However, bleeding has also been reported in lupus anticoagulant; it is due to the presence of antibodies to prothrombin, which results in hypoprothrombinemia. Both disorders show a prolonged PTT that does not correct on mixing. To distinguish acquired inhibitors from lupus anticoagulant, note that the dilute Russell’s viper venom test and the hexagonal-phase phospholipids test will be negative in patients with an acquired inhibitor and positive in patients with lupus anticoagulants. Moreover, lupus anticoagulant interferes with the clotting activity of many factors (FVIII, FIX, FXII, FXI), whereas acquired inhibitors are specific to a single factor.
142 |
Arterial and Venous Thrombosis |
OVERVIEW OF THROMBOSIS
GENERAL OVERVIEW
Thrombosis, the obstruction of blood flow due to the formation of clot, may result in tissue anoxia and damage, and it is a major cause of morbidity and mortality in a wide range of arterial and venous diseases and patient populations. In 2009 in the United States, an estimated 785,000 people had a new coronary thrombotic event, and about 470,000 had a recurrent ischemic episode. Each year, approximately 795,000 people have a new or recurrent stroke. It is estimated that 300,000–600,000 people each year have a pulmonary embolism or deep venous thrombotic event. In the nondiseased state, physiologic hemostasis reflects a delicate interplay between factors that promote and inhibit blood clotting, favoring the former. This response is crucial as it prevents uncontrolled hemorrhage and exsanguination following injury. In specific settings, the same processes that regulate normal hemostasis can cause pathologic thrombosis, leading to arterial or venous occlusion. Importantly, many commonly used therapeutic interventions may also alter the thrombotic–hemostatic balance adversely.
Hemostasis and thrombosis primarily involve the interplay among three factors: the vessel wall, coagulation proteins, and platelets. Many prevalent acute vascular diseases are due to thrombus formation within a vessel, including myocardial infarction, thrombotic cerebrovascular events, and venous thrombosis. Although the end result is vessel occlusion and tissue ischemia, the pathophysiologic processes governing these pathologies have similarities as well as distinct differences. While many of the pathways regulating thrombus formation are similar to those that regulate hemostasis, the processes triggering thrombosis and, often, perpetuating the thrombus may be distinct and can vary in different clinical and genetic settings. In venous thrombosis, primary hypercoagulable states reflecting defects in the proteins governing coagulation and/or fibrinolysis or secondary hypercoagulable states involving abnormalities of blood vessels and blood flow or stasis lead to thrombosis. By contrast, arterial thrombosis is highly dependent on the state of the vessel wall, the platelet, and factors related to blood flow.
ARTERIAL THROMBOSIS
OVERVIEW OF ARTERIAL THROMBOSIS
In arterial thrombosis, the platelets and abnormalities of the vessel wall typically play a key role in vessel occlusion. Arterial thrombus forms via a series of sequential steps in which platelets adhere to the vessel wall, additional platelets are recruited, and thrombin is activated (Fig. 142-1). The regulation of platelet adhesion, activation, aggregation, and recruitment will be described in detail below. In addition, while the primary function of platelets is regulation of hemostasis, our understanding of their role in other processes, such as immunity, wound healing, and inflammation, continues to grow.
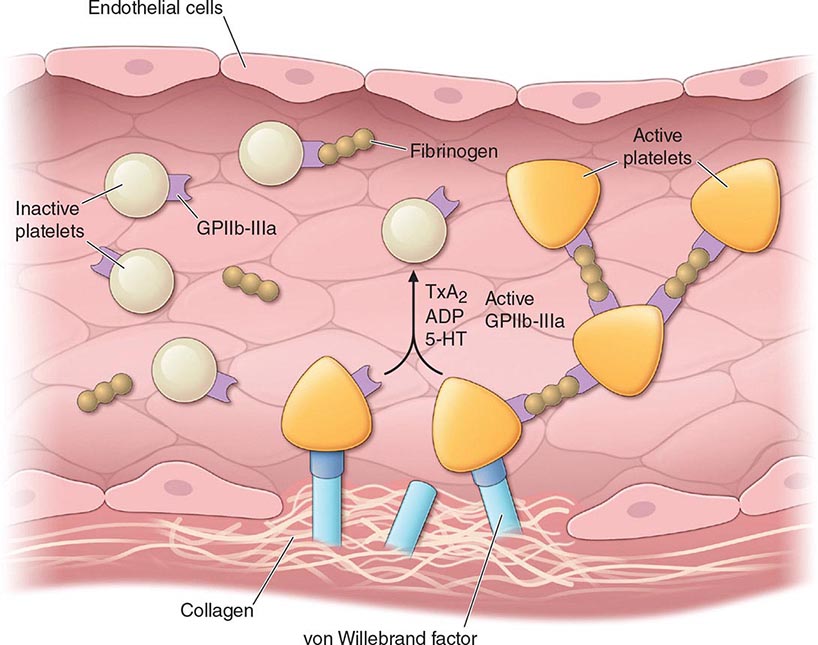
FIGURE 142-1 Platelet activation and thrombosis. Platelets circulate in an inactive form in the vasculature. Damage to the endothelium and/or external stimuli activates platelets that adhere to the exposed subendothelial von Willebrand factor and collagen. This adhesion leads to activation of the platelet, shape change, and the synthesis and release of thromboxane (TxA2), serotonin (5-HT), and adenosine diphosphate (ADP). Platelet stimuli cause conformational change in the platelet integrin glycoprotein (GP) IIb/IIIa receptor, leading to the high-affinity binding of fibrinogen and the formation of a stable platelet thrombus.
ARTERIAL THROMBOSIS AND VASCULAR DISEASE
Arterial thrombosis is a major cause of morbidity and mortality both in the United States and, increasingly, worldwide. Although the rates have declined in the United States, the overall burden remains high and accounts for approximately 33% of deaths. Overall, coronary heart disease is estimated to cause about 1 of every 5 deaths in the United States. In addition to the 785,000 Americans who will have a new coronary event, an additional 195,000 silent first myocardial infarctions are projected to occur annually. Although the rate of strokes has fallen by a third, each year, about 795,000 people experience a new or recurrent stroke, although not all are caused by thrombotic occlusion of the vessel. Approximately 610,000 strokes are first events and 185,000 are recurrent events; it is estimated that 1 of every 18 deaths in the United States is due to stroke.
THE PLATELET
Many processes in platelets have parallels with other cell types, such as the presence of specific receptors and signaling pathways; however, unlike most cells, platelets lack a nucleus and are unable to adapt to changing biologic settings by altered gene transcription. Platelets sustain limited protein synthetic capacity from megakaryocyte-derived and intracellularly transported microRNA (miRNA) and messenger RNA (mRNA). Most of the molecules needed to respond to various stimuli, however, are maintained in storage granules and membrane compartments.
Platelets are disc-shaped, very small, anucleate cells (1–5 μm in diameter) that circulate in the blood at concentrations of 200–400,000/μL, with an average lifespan of 7–10 days. Platelets are derived from megakaryocytes, polyploidal hematopoietic cells found in the bone marrow. The primary regulator of platelet formation is thrombopoietin (TPO). The precise mechanism by which megakaryocytes produce and release fully formed platelets is unclear, but the process likely involves formation of proplatelets, pseudopod-like structures generated by the evagination of the cytoplasm from which platelets bud. Platelet granules are synthesized in megakaryocytes before thrombopoiesis and contain an array of prothrombotic, proinflammatory, and antimicrobial mediators. The two major types of platelet granules, alpha and dense, are distinguished by their size, abundance, and content. Alpha-granules contain soluble coagulation proteins, adhesion molecules, growth factors, integrins, cytokines, and inflammatory modulators. Platelet dense-granules are smaller than alpha-granules and less abundant. Whereas alpha-granules contain proteins that may be more important in the inflammatory response, dense-granules contain high concentrations of small molecules, including adenosine diphosphate (ADP) and serotonin, that influence platelet aggregation.
Platelet Adhesion (See Fig. 142-1) The formation of a thrombus is initiated by the adherence of platelets to the damaged vessel wall. Damage exposes subendothelial components responsible for triggering platelet reactivity, including collagen, von Willebrand factor, fibronectin, and other adhesive proteins, such as vitronectin and thrombospondin. The hemostatic response may vary, depending on the extent of damage, the specific proteins exposed, and flow conditions. Certain proteins are expressed on the platelet surface that subsequently regulate collagen-induced platelet adhesion, particularly under flow conditions, and include glycoprotein (GP) IV, GPVI, and the integrin α2β1. The platelet GPIb-IX-V complex adhesive receptor is central both to platelet adhesion and to the initiation of platelet activation. Damage to the blood vessel wall exposes subendothelial von Willebrand factor and collagen to the circulating blood. The GPIb-IX-V complex binds to the exposed von Willebrand factor, causing platelets to adhere (Fig. 142-1). In addition, the engagement of the GPIb-IX-V complex with ligand induces signaling pathways that lead to platelet activation. von Willebrand factor–bound GPIb-IX-V promotes a calcium-dependent conformational change in the GPIIb/IIIa receptor, transforming it from an inactive low-affinity state to an active high-affinity receptor for fibrinogen.
Platelet Activation The activation of platelets is controlled by a variety of surface receptors that regulate various functions in the activation process. Platelet receptors control many distinct processes and are stimulated by a wide variety of agonists and adhesive proteins that result in variable degrees of activation. In general terms, the stimulation of platelet receptors triggers two specific processes: (1) activation of internal signaling pathways that lead to further platelet activation and granule release and (2) the capacity of the platelet to bind to other adhesive proteins/platelets. Both of these processes contribute to the formation of a thrombus. Stimulation of nonthrombotic receptors results in platelet adhesion or interaction with other vascular cells including endothelial cells, neutrophils, and mononuclear cells.
Many families and subfamilies of receptors are found on platelets that regulate a variety of platelet functions. These include the seven transmembrane receptor family, which is the main agonist- stimulated receptor family. Several seven transmembrane receptors are found on platelets, including the ADP receptors, prostaglandin receptors, lipid receptors, and chemokine receptors. Receptors for thrombin comprise the major seven transmembrane receptors found on platelets. Among this last group, the first identified was the protease activation receptor 1 (PAR1). The PAR class of receptors has a distinct mechanism of activation that involves specific cleavage of the N-terminus of thrombin, which, in turn, acts as a ligand for the receptor. Other PAR receptors are present on platelets, including PAR2 (not activated by thrombin) and PAR4. Adenosine receptors are responsible for transduction of ADP-induced signaling events, which are initiated by the binding of ADP to purinergic receptors on the platelet surface. There are several distinct ADP receptors, classified as P2X1, P2Y1, and P2Y12. The activation of both the P2Y12 and P2Y1 receptors is essential for ADP-induced platelet aggregation. The thienopyridine derivatives, clopidogrel and prasugrel, are clinically used inhibitors of ADP-induced platelet aggregation.
Platelet Aggregation Activation of platelets results in a rapid series of signal transduction events, including tyrosine kinase, serine/threonine kinase, and lipid kinase activation. In unstimulated platelets, the major platelet integrin GPIIb/IIIa is maintained in an inactive conformation and functions as a low-affinity adhesion receptor for fibrinogen. This integrin is unique as it is only expressed on platelets. After stimulation, the interaction between fibrinogen and GPIIb/IIIa forms intercellular connections between platelets, leading to the formation of a platelet aggregate (Fig. 142-1). A calcium-sensitive conformational change in the extracellular domain of GPIIb/IIIa enables the high-affinity binding of soluble plasma fibrinogen as a result of a complex network of inside-out signaling events. The GPIIb/IIIa receptor serves as a bidirectional conduit with GPIIb/IIIa-mediated signaling (outside-in) occurring immediately after the binding of fibrinogen. This leads to additional intracellular signaling that further stabilizes the platelet aggregate and transforms platelet aggregation from a reversible to an irreversible process (inside-out).
THE ROLE OF PLATELETS AND THROMBOSIS IN INFLAMMATION
Inflammation plays an important role during the acute thrombotic phase of acute coronary syndromes. In the setting of acute upper respiratory infections, people are at higher risk of myocardial infarction and thrombotic stroke. Patients with acute coronary syndromes have not only increased interactions between platelets (homotypic aggregates), but also increased interactions between platelets and leukocytes (heterotypic aggregates) detectable in circulating blood. These latter aggregates form when platelets are activated and adhere to circulating leukocytes. Platelets bind via P-selectin (CD62P) expressed on the surface of activated platelets to the leukocyte receptor, P-selectin glycoprotein ligand 1 (PSGL-1). This association leads to increased expression of CD11b/CD18 (Mac-1) on leukocytes, which itself supports interactions with platelets partially via bivalent fibrinogen linking this integrin with its platelet surface counterpart, GPIIb/IIIa. Platelet surface P-selectin also induces the expression of tissue factor on monocytes, which promotes fibrin formation.
In addition to platelet–monocyte aggregates, the immunomodulator, soluble CD40 ligand (CD40L or CD154), also reflects a link between thrombosis and inflammation. The CD40 ligand is a trimeric transmembrane protein of the tumor necrosis factor family and, with its receptor CD40, is an important contributor to the inflammatory process leading both to thrombosis and atherosclerosis. While many immunologic and vascular cells have been found to express CD40 and/or CD40 ligand, in platelets, CD40 ligand is rapidly translocated to the surface after stimulation and is upregulated in the newly formed thrombus. The surface-expressed CD40 ligand is cleaved from the platelet to generate a soluble fragment (soluble CD40 ligand).
Links have also been established among platelets, infection, immunity, and inflammation. Bacterial and viral infections are associated with a transient increase in the risk of acute thrombotic events, such as acute myocardial infarction and stroke. In addition, platelets contribute significantly to the pathophysiology and high mortality rates of sepsis. The expression, functionality, and signaling pathways of toll-like receptors (TLRs) have been established in platelets. Stimulation of platelet TLR2, TLR3, and TLR4 directly and indirectly activates the platelet’s thrombotic and inflammatory responses, and live bacteria induce a proinflammatory response in platelets in a TLR2-dependent manner, suggesting a mechanism by which specific bacteria and bacterial components can directly activate platelet-dependent thrombosis.
GENETICS OF ARTERIAL THROMBOSIS
![]() Some studies have associated arterial thrombosis with genetic variants (Table 142-1A); however, the associations have been weak and not confirmed in larger series. Platelet count and mean platelet volume have been studied by genome-wide association studies (GWAS), and this approach identified signals located to noncoding regions. Of 15 quantitative trait loci associated with mean platelet volume and platelet count, one located at 12q24 is also a risk locus for coronary artery disease.
Some studies have associated arterial thrombosis with genetic variants (Table 142-1A); however, the associations have been weak and not confirmed in larger series. Platelet count and mean platelet volume have been studied by genome-wide association studies (GWAS), and this approach identified signals located to noncoding regions. Of 15 quantitative trait loci associated with mean platelet volume and platelet count, one located at 12q24 is also a risk locus for coronary artery disease.
|
HERITABLE CAUSES OF ARTERIAL AND VENOUS THROMBOSIS |
In the area of genetic variability and platelet function, studies have primarily dealt with pharmacogenetics, the field of pharmacology dealing with the interindividual variability in drug response based on genetic determinants (Table 142-2). This focus has been driven by the wide variability among individuals in terms of response to antithrombotic drugs and the lack of a common explanation for this variance. The best described is the issue of “aspirin resistance,” although heterogeneity for other antithrombotics (e.g., clopidogrel) has also been extensively examined. Primarily, platelet-dependent genetic determinants have been defined at the level of (1) drug effect, (2) drug compliance, and (3) drug metabolism. Many candidate platelet genes have been studied for their interaction with antiplatelet and antithrombotic agents.
|
GENETIC VARIATION AND PHARMACOGENETIC RESPONSES TO PLATELET INHIBITORS |
Many patients have an inadequate response to the inhibitory effects of aspirin. Heritable factors contribute to the variability; however, ex vivo tests of residual platelet responsiveness after aspirin administration have not provided firm evidence for a pharmacogenetic interaction between aspirin and COX1 or other relevant platelet receptors. As such, currently, there is no clinical indication for genotyping to optimize aspirin’s antiplatelet efficiency. For the platelet P2Y12 receptor inhibitor clopidogrel, additional data suggest that genetics may affect the drug’s responsiveness and utility. The responsible genetic variant appears not to be the expected P2Y12 receptor but an enzyme responsible for drug metabolism. Clopidogrel is a prodrug, and liver metabolism by specific cytochrome P450 enzymes is required for activation. The genes encoding the CYP-dependent oxidative steps are polymorphic, and carriers of specific alleles of the CYP2C19 and CYP3A4 loci have increased platelet aggregability. Increased platelet activity has also been specifically associated with the CYP2C19*2 allele, which causes loss of platelet function in select patients. Because these are common genetic variants, this observation has been shown to be clinically relevant in large studies. In summary, although the loss-of-function polymorphisms in CYP2C19 is the strongest individual variable affecting pharmacokinetics and antiplatelet response to clopidogrel, it only accounts for 5–12% of the variability in ADP-induced platelet aggregation on clopidogrel. In addition, genetic variables do not appear to significantly contribute to the clinical outcomes of patients treated with the P2Y12 receptor antagonists prasugrel or ticagrelor.
VENOUS THROMBOSIS
OVERVIEW OF VENOUS THROMBOSIS
Coagulation is the process by which thrombin is activated and soluble plasma fibrinogen is converted into insoluble fibrin. These steps account for both normal hemostasis and the pathophysiologic processes influencing the development of venous thrombosis. The primary forms of venous thrombosis are deep vein thrombosis (DVT) in the extremities and the subsequent embolization to the lungs (pulmonary embolism), referred to together as venous thromboembolic disease. Venous thrombosis occurs due to heritable causes (Table 142-1B) and acquired causes (Table 142-3).
|
ACQUIRED CAUSES OF VENOUS THROMBOSIS |
DEEP VENOUS THROMBOSIS AND PULMONARY EMBOLISM
More than 200,000 new cases of venous thromboembolism occur each year. Of these cases, up to 30% of patients die within 30 days and one-fifth suffer sudden death due to pulmonary embolism; 30% go on to develop recurrent venous thromboembolism within 10 years. Data from the Atherosclerosis Risk in Communities (ARIC) study reported a 9% 28-day fatality rate from DVT and a 15% fatality rate from pulmonary embolism. Pulmonary embolism in the setting of cancer has a 25% fatality rate. The mean incidence of first DVT in the general population is 5 per 10,000 person-years; the incidence is similar in males and females when adjusting for factors related to reproduction and birth control and increases dramatically with age from 2 to 3 per 10,000 person-years at 30–49 years of age to 20 at 70–79 years of age.
OVERVIEW OF THE COAGULATION CASCADE AND ITS ROLE IN VENOUS THROMBOSIS
Coagulation is defined as the formation of fibrin by a series of linked enzymatic reactions in which each reaction product converts the subsequent inactive zymogen into an active serine protease (Fig. 142-2). This coordinated sequence is called the coagulation cascade and is a key mechanism for regulating hemostasis. Central to the function of the coagulation cascade is the principle of amplification: due to a series of linked enzymatic reactions, a small stimulus can lead to much greater quantities of fibrin, the end product that prevents hemorrhage at the site of vascular injury. In addition to the known risk factors relevant to hypercoagulopathy, stasis, and vascular dysfunction, newer areas of research have identified contributions from procoagulant microparticles, inflammatory cells, microvesicles, and fibrin structure.
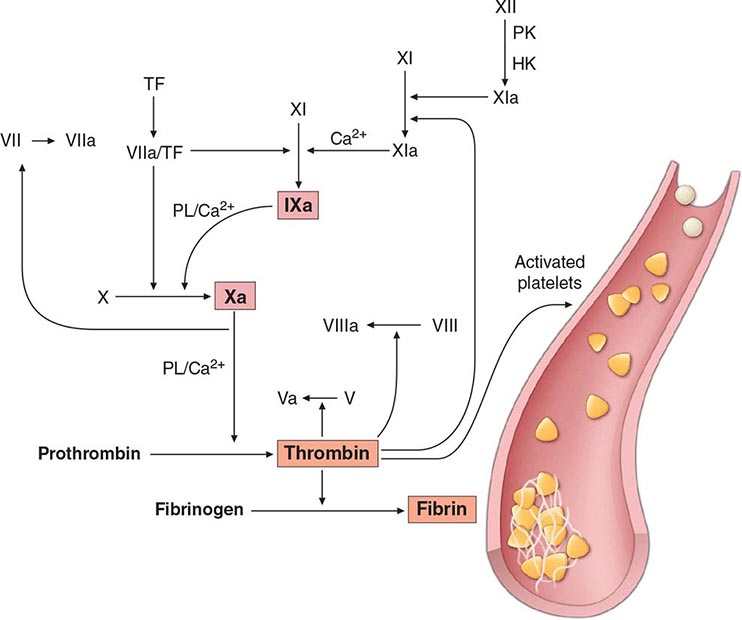
FIGURE 142-2 Summary of the coagulation pathways. Specific coagulation factors (“a” indicates activated form) are responsible for the conversion of soluble plasma fibrinogen into insoluble fibrin. This process occurs via a series of linked reactions in which the enzymatically active product subsequently converts the downstream inactive protein into an active serine protease. In addition, the activation of thrombin leads to stimulation of platelets. HK, high-molecular-weight kininogen; PK, prekallikrein; TF, tissue factor.
The coagulation cascade is primarily initiated by vascular injury exposing tissue factor to blood components (Fig. 142-2). Tissue factor may also be found in bloodborne cell-derived microparticles and, under pathophysiologic conditions, in leukocytes or platelets. Plasma factor VII (FVII) is the ligand for and is activated (FVIIa) by binding to tissue factor exposed at the site of vessel damage. The binding of FVII/VIIa to tissue factor activates the downstream conversion of factor × (FX) to active FX (FXa). In an alternative reaction, the FVII/FVIIa–tissue factor complex initially converts FIX to FIXa, which then activates FX in conjunction with its cofactor factor VIII (FVIIIa). Factor Xa with its cofactor FVa converts prothrombin to thrombin, which then converts soluble plasma fibrinogen to insoluble fibrin, leading to clot or thrombus formation. Thrombin also activates FXIII to FXIIIa, a transglutaminase that covalently cross-links and stabilizes the fibrin clot. Formation of thrombi is affected by mechanisms governing fibrin structure and stability including specific fibrinogen variants and how they alter fibrin formation, strength and structure.
Several antithrombotic factors also regulate coagulation; these include antithrombin, tissue factor pathway inhibitor (TFPI), heparin cofactor II, and protein C/protein S. Under normal conditions, these factors limit the production of thrombin to prevent the perpetuation of coagulation and thrombus formation. Typically, after the clot has caused occlusion at the damaged site and begins to expand toward adjacent uninjured vessel segments, the anticoagulant reactions governed by the normal endothelium become pivotal in limiting the extent of this hemostatically protective clot.
RISK FACTORS FOR VENOUS THROMBOSIS
The risk factors for venous thrombosis are primarily related to hypercoagulability, which can be genetic (Table 142-1) or acquired, or due to immobilization and venous stasis. Independent predictors for recurrence include increasing age, obesity, malignant neoplasm, and acute extremity paresis. It is estimated that 5–8% of the U.S. population has a genetic risk factor known to predispose to venous thrombosis. Often, multiple risk factors are present in a single individual. Significant risk is incurred by major orthopedic, abdominal, or neurologic surgeries. Moderate risk is promoted by prolonged bedrest; certain types of cancer, pregnancy, hormone replacement therapy, or oral contraceptive use; and other sedentary conditions such as long-distance plane travel. It has been reported that the risk of developing a venous thromboembolic event doubles after air travel lasting 4 h, although the absolute risk remains low (1 in 6000). The relative risk of venous thromboembolism among pregnant or postpartum women is 4.3, and the overall incidence (absolute risk) is 199.7 per 100,000 woman-years.
GENETICS OF VENOUS THROMBOSIS
![]() (See Table 142-2) Less common causes of venous thrombosis are those due to genetic variants. These abnormalities include loss-of-function mutations of endogenous anticoagulants as well as gain-of-function mutations of procoagulant proteins. Heterozygous antithrombin deficiency and homozygosity of the factor V Leiden mutation significantly increase the risk of venous thrombosis. While homozygous protein C or protein S deficiencies are rare and may lead to fatal purpura fulminans, heterozygous deficiencies are associated with a moderate risk of thrombosis. Activated protein C impairs coagulation by proteolytic degradation of FVa. Patients resistant to the activity of activated protein C may have a point mutation in the FV gene located on chromosome 1, a mutant denoted factor V Leiden. Mildly increased risk has been attributed to elevated levels of procoagulant factors, as well as low levels of tissue factor pathway inhibitor. Polymorphisms of methylene tetrahydrofolate reductase as well as hyperhomocysteinemia have been shown to be independent risk factors for venous thrombosis, as well as arterial vascular disease; however, many of the initial descriptions of genetic variants and their associations with thromboembolism are being questioned in larger, more current studies.
(See Table 142-2) Less common causes of venous thrombosis are those due to genetic variants. These abnormalities include loss-of-function mutations of endogenous anticoagulants as well as gain-of-function mutations of procoagulant proteins. Heterozygous antithrombin deficiency and homozygosity of the factor V Leiden mutation significantly increase the risk of venous thrombosis. While homozygous protein C or protein S deficiencies are rare and may lead to fatal purpura fulminans, heterozygous deficiencies are associated with a moderate risk of thrombosis. Activated protein C impairs coagulation by proteolytic degradation of FVa. Patients resistant to the activity of activated protein C may have a point mutation in the FV gene located on chromosome 1, a mutant denoted factor V Leiden. Mildly increased risk has been attributed to elevated levels of procoagulant factors, as well as low levels of tissue factor pathway inhibitor. Polymorphisms of methylene tetrahydrofolate reductase as well as hyperhomocysteinemia have been shown to be independent risk factors for venous thrombosis, as well as arterial vascular disease; however, many of the initial descriptions of genetic variants and their associations with thromboembolism are being questioned in larger, more current studies.
FIBRINOLYSIS AND THROMBOSIS
Specific abnormalities in the fibrinolytic system have been associated with enhanced thrombosis. Factors such as elevated levels of tissue plasminogen activator (tPA) and plasminogen activator inhibitor type 1 (PAI-1) have been associated with decreased fibrinolytic activity and an increased risk of arterial thrombotic disease. Specific genetic variants have been associated with decreased fibrinolytic activity, including the 4G/5G insertion/deletion polymorphism in the (plasminogen activator type 1) PAI-1 gene. Additionally, the 311-bp Alu insertion/deletion in tPA’s intron 8 has been associated with enhanced thrombosis; however, genetic abnormalities have not been associated consistently with altered function or tPA levels, raising questions about the relevant pathophysiologic mechanism. Thrombin-activatable fibrinolysis inhibitor (TAFI) is a carboxypeptidase that regulates fibrinolysis; elevated plasma TAFI levels have been associated with an increased risk of both DVT and cardiovascular disease.
The metabolic syndrome also is accompanied by altered fibrinolytic activity. This syndrome, which comprises abdominal fat (central obesity), altered glucose and insulin metabolism, dyslipidemia, and hypertension, has been associated with atherothrombosis. The mechanism for enhanced thrombosis appears to be due both to altered platelet function and to a procoagulant and hypofibrinolytic state. One of the most frequently documented prothrombotic abnormalities reported in this syndrome is an increase in plasma levels of PAI-1.
In addition to contributing to platelet function, inflammation plays a role in both coagulation-dependent thrombus formation and thrombus resolution. Both polymorphonuclear neutrophils and monocytes/macrophages contribute to multiple overlapping thrombotic functions, including fibrinolysis, chemokine and cytokine production, and phagocytosis.
THE DISTINCTION BETWEEN ARTERIAL AND VENOUS THROMBOSIS
Although there is overlap, venous thrombosis and arterial thrombosis are initiated differently, and clot formation progresses by somewhat distinct pathways. In the setting of stasis or states of hypercoagulability, venous thrombosis is activated with the initiation of the coagulation cascade primarily due to exposure of tissue factor; this leads to the formation of thrombin and the subsequent conversion of fibrinogen to fibrin. In the artery, thrombin formation also occurs, but thrombosis is primarily promoted by the adhesion of platelets to an injured vessel and stimulated by exposed extracellular matrix (Figs. 142-1 and 142-2). There is wide variation in individual responses to vascular injury, an important determinant of which is the predisposition an individual has to arterial or venous thrombosis. This concept has been supported indirectly in prothrombotic animal models in which there is poor correlation between the propensity to develop venous versus arterial thrombosis.
Despite considerable progress in understanding the role of hypercoagulable states in venous thromboembolic disease, the contribution of hypercoagulability to arterial vascular disease is much less well understood. Although specific thrombophilic conditions, such as factor V Leiden and the prothrombin G20210A mutation, are risk factors for DVT, pulmonary embolism, and other venous thromboembolic events, their contribution to arterial thrombosis is less well defined. In fact, to the contrary, many of these thrombophilic factors have not been found to be clinically important risk factors for arterial thrombotic events, such as acute coronary syndromes.
Clinically, although the pathophysiology is distinct, arterial and venous thrombosis do share common risk factors, including age, obesity, cigarette smoking, diabetes mellitus, arterial hypertension, hyperlipidemia, and metabolic syndrome. Select genetic variants, including those of the glutathione peroxidase gene, have also been associated with arterial and venous thrombo-occlusive disease. Importantly, arterial and venous thrombosis may both be triggered by pathophysiologic stimuli responsible for activating inflammatory and oxidative pathways.
The diagnosis and treatment of ischemic heart disease are discussed in Chap. 293. Stroke diagnosis and management are discussed in Chap. 330. The diagnosis and management of DVT and pulmonary embolus are discussed in Chap. 300.
143 |
Antiplatelet, Anticoagulant, and Fibrinolytic Drugs |
Thromboembolic disorders are major causes of morbidity and mortality. Thrombosis can occur in arteries or veins. Arterial thrombosis is the most common cause of acute myocardial infarction (MI), ischemic stroke, and limb gangrene. Venous thromboembolism encompasses deep vein thrombosis (DVT), which can lead to postthrombotic syndrome, and pulmonary embolism (PE), which can be fatal or can result in chronic thromboembolic pulmonary hypertension.
Most arterial thrombi are superimposed on disrupted atherosclerotic plaque because plaque rupture exposes thrombogenic material in the plaque core to the blood. This material then triggers platelet aggregation and fibrin formation, which results in the generation of a platelet-rich thrombus that can temporarily or permanently occlude blood flow. In contrast, venous thrombi rarely form at sites of obvious vascular disruption. Although they can develop after surgical trauma to veins or secondary to indwelling venous catheters, venous thrombi usually originate in the valve cusps of the deep veins of the calf or in the muscular sinuses. Sluggish blood flow reduces the oxygen supply to the avascular valve cusps. Endothelial cells lining these valve cusps become activated and express adhesion molecules on their surface. Tissue factor–bearing leukocytes and microparticles adhere to these activated cells and induce coagulation. DNA extruded from neutrophils forms neutrophil extracelluar traps (NETs) that provide a scaffold that traps red blood cells, promotes platelet adhesion and activation, and augments coagulation. Local thrombus formation is exacerbated by reduced clearance of activated clotting factors as a result of impaired blood flow. If the thrombi extend from the calf veins into the popliteal and more proximal veins of the leg, thrombus fragments can dislodge, travel to the lungs, and produce a PE.
Arterial and venous thrombi are composed of platelets, fibrin, and trapped red blood cells, but the proportions differ. Arterial thrombi are rich in platelets because of the high shear in the injured arteries. In contrast, venous thrombi, which form under low shear conditions, contain relatively few platelets and are predominantly composed of fibrin and trapped red cells. Because of the predominance of platelets, arterial thrombi appear white, whereas venous thrombi are red in color, reflecting the trapped red cells.
Antithrombotic drugs are used for prevention and treatment of thrombosis. Targeting the components of thrombi, these agents include (1) antiplatelet drugs, (2) anticoagulants, and (3) fibrinolytic agents (Fig. 143-1). With the predominance of platelets in arterial thrombi, strategies to attenuate arterial thrombosis focus mainly on antiplatelet agents, although, in the acute setting, often include anticoagulants and fibrinolytic agents. Anticoagulants are the mainstay of prevention and treatment of venous thromboembolism because fibrin is the predominant component of venous thrombi. Antiplatelet drugs are less effective than anticoagulants in this setting because of the limited platelet content of venous thrombi. Fibrinolytic therapy is used in selected patients with venous thromboembolism. For example, patients with massive or submassive PE can benefit from systemic or catheter-directed fibrinolytic therapy. Pharmaco-mechanical therapy also is used to restore blood flow in patients with extensive DVT involving the iliac and/or femoral veins.
FIGURE 143-1 Classification of antithrombotic drugs.
ANTIPLATELET DRUGS
ROLE OF PLATELETS IN ARTERIAL THROMBOSIS
In healthy vasculature, circulating platelets are maintained in an inactive state by nitric oxide (NO) and prostacyclin released by endothelial cells lining the blood vessels. In addition, endothelial cells also express CD39 on their surface, a membrane-associated ecto-adenosine diphosphatase (ADPase) that degrades ADP released from activated platelets. When the vessel wall is damaged, release of these substances is impaired and subendothelial matrix is exposed. Platelets adhere to exposed collagen via α2β1 and glycoprotein (Gp) V1 and to von Willebrand factor (VWF) via Gp Ibα and Gp IIb/IIIa (αIIbβ3)—receptors that are constitutively expressed on the platelet surface. Adherent platelets undergo a change in shape, secrete ADP from their dense granules, and synthesize and release thromboxane A2. Released ADP and thromboxane A2, which are platelet agonists, activate ambient platelets and recruit them to the site of vascular injury (Fig. 143-2).
FIGURE 143-2 Coordinated role of platelets and the coagulation system in thrombogenesis. Vascular injury simultaneously triggers platelet activation and aggregation and activation of the coagulation system. Platelet activation is initiated by exposure of subendothelial collagen and von Willebrand factor (VWF), onto which platelets adhere. Adherent platelets become activated and release ADP and thromboxane A2, platelet agonists that activate ambient platelets and recruit them to the site of injury. When platelets are activated, glycoprotein IIb/IIIa on their surface undergoes a conformational change that enables it to ligate fibrinogen and/or VWF and mediate platelet aggregation. Coagulation is triggered by tissue factor exposed at the site of injury. Tissue factor triggers thrombin generation. As a potent platelet agonist, thrombin amplifies platelet recruitment to the site of injury. Thrombin also converts fibrinogen to fibrin, and the fibrin strands then weave the platelet aggregates together to form a platelet/fibrin thrombus.
Disruption of the vessel wall also exposes tissue factor–expressing cells to the blood. Tissue factor binds factor VIIa and initiates coagulation. Activated platelets potentiate coagulation by providing a surface that binds clotting factors and supports the assembly of activation complexes that enhance thrombin generation. In addition to converting fibrinogen to fibrin, thrombin serves as a potent platelet agonist and recruits more platelets to the site of vascular injury. Thrombin also amplifies its own generation by feedback activation of factors V, VIII, and XI and solidifies the fibrin network by activating factor XIII, which then cross-links the fibrin strands.
When platelets are activated, Gp IIb/IIIa, the most abundant receptor on the platelet surface, undergoes a conformational change that enables it to bind fibrinogen and, under high shear conditions, VWF. Divalent fibrinogen or multivalent VWF molecules bridge adjacent platelets together to form platelet aggregates. Fibrin strands, generated through the action of thrombin, then weave these aggregates together to form a platelet/fibrin mesh.
Antiplatelet drugs target various steps in this process. The commonly used drugs include aspirin, ADP receptor inhibitors, which include the thienopyridines (clopidogrel and prasugrel) and ticagrelor, dipyridamole, and Gp IIb/IIIa antagonists.
ASPIRIN
The most widely used antiplatelet agent worldwide is aspirin. As a cheap and effective antiplatelet drug, aspirin serves as the foundation of most antiplatelet strategies.
Mechanism of Action Aspirin produces its antithrombotic effect by irreversibly acetylating and inhibiting platelet cyclooxygenase (COX)-1 (Fig. 143-3), a critical enzyme in the biosynthesis of thromboxane A2. At high doses (~1 g/d), aspirin also inhibits COX-2, an inducible COX isoform found in endothelial cells and inflammatory cells. In endothelial cells, COX-2 initiates the synthesis of prostacyclin, a potent vasodilator and inhibitor of platelet aggregation.
FIGURE 143-3 Site of action of antiplatelet drugs. Aspirin inhibits thromboxane A2 (TXA2) synthesis by irreversibly acetylating cyclooxygenase-1 (COX-1). Reduced TXA2 release attenuates platelet activation and recruitment to the site of vascular injury. Clopidogrel and prasugrel irreversibly block P2Y12, a key ADP receptor on the platelet surface; cangrelor and ticagrelor are reversible inhibitors of P2Y12. Abciximab, eptifibatide, and tirofiban inhibit the final common pathway of platelet aggregation by blocking fibrinogen and von Willebrand factor binding to activated glycoprotein (Gp) IIb/IIIa. Vorapaxar inhibits thrombin-mediated platelet activation by targeting protease-activated receptor-1 (PAR-1), the major thrombin receptor on human platelets.
Indications Aspirin is widely used for secondary prevention of cardiovascular events in patients with coronary artery, cerebrovascular, or peripheral vascular disease. Compared with placebo, aspirin produces a 25% reduction in the risk of cardiovascular death, MI, or stroke. Aspirin is also used for primary prevention in patients whose estimated annual risk of MI is >1%, a point where its benefits are likely to outweigh harms. This includes patients older than age 40 years with two or more major risk factors for cardiovascular disease or men older than age 45 years and women over the age of 55 years with one or more such risk factors. Aspirin is equally effective in men and women. In men, aspirin mainly reduces the risk of MI, whereas in women, aspirin lowers the risk of stroke.
Dosages Aspirin is usually administered at doses of 75–325 mg once daily. Higher doses of aspirin are not more effective than lower aspirin doses, and some analyses suggest reduced efficacy with higher doses. Because the side effects of aspirin are dose-related, daily aspirin doses of 75–100 mg are recommended for most indications. When rapid platelet inhibition is required, an initial aspirin dose of at least 160 mg should be given.
Side Effects The most common side effects are gastrointestinal and range from dyspepsia to erosive gastritis or peptic ulcers with bleeding and perforation. These side effects are dose-related. Use of enteric-coated or buffered aspirin in place of plain aspirin does not eliminate gastrointestinal side effects. The overall risk of major bleeding with aspirin is 1–3% per year. The risk of bleeding is increased two- to threefold when aspirin is given in conjunction with other antiplatelet drugs, such as clopidogrel, or with anticoagulants, such as warfarin. When dual or triple therapy is prescribed, low-dose aspirin should be given (75–100 mg daily). Eradication of Helicobacter pylori infection and administration of proton pump inhibitors may reduce the risk of aspirin-induced upper gastrointestinal bleeding in patients with peptic ulcer disease.
Aspirin should not be administered to patients with a history of aspirin allergy characterized by bronchospasm. This problem occurs in ~0.3% of the general population but is more common in those with chronic urticaria or asthma, particularly in individuals with nasal polyps or chronic rhinitis. Hepatic and renal toxicity are observed with aspirin overdose.
Aspirin Resistance Clinical aspirin resistance is defined as the failure of aspirin to protect patients from ischemic vascular events. This is not a helpful definition because it is made after the event occurs. Furthermore, it is not realistic to expect aspirin, which only blocks thromboxane A2–induced platelet activation, to prevent all vascular events.
Aspirin resistance has also been described biochemically as failure of the drug to produce its expected inhibitory effects on tests of platelet function, such as thromboxane A2 synthesis or arachidonic acid-induced platelet aggregation. Potential causes of aspirin resistance include poor compliance, reduced absorption, drug-drug interaction with ibuprofen, and overexpression of COX-2. Unfortunately, the tests for aspirin resistance have not been well standardized, and there is little evidence that they identify patients at increased risk of recurrent vascular events, or that resistance can be reversed by giving higher doses of aspirin or by adding other antiplatelet drugs. Until such information is available, testing for aspirin resistance remains a research tool.
ADP RECEPTOR ANTAGONISTS
The ADP receptor antagonists include the thienopyridines (clopidogrel and prasugrel) and ticagrelor. All of these drugs target P2Y12, the key ADP receptor on platelets.
Thienopyridines • MECHANISM OF ACTION The thienopyridines are structurally related drugs that selectively inhibit ADP-induced platelet aggregation by irreversibly blocking P2Y12 (Fig. 143-3). Clopidogrel and prasugrel are prodrugs that require metabolic activation by the hepatic cytochrome P450 (CYP) enzyme system. Prasugrel is about 10-fold more potent than clopidogrel and has a more rapid onset of action because of better absorption and more streamlined metabolic activation.
INDICATIONS When compared with aspirin in patients with recent ischemic stroke, recent MI, or a history of peripheral arterial disease, clopidogrel reduced the risk of cardiovascular death, MI, and stroke by 8.7%. Therefore, clopidogrel is more effective than aspirin but is also more expensive. In some patients, clopidogrel and aspirin are combined to capitalize on their capacity to block complementary pathways of platelet activation. For example, the combination of aspirin plus clopidogrel is recommended for at least 4 weeks after implantation of a bare metal stent in a coronary artery and for at least a year in those with a drug-eluting stent. Concerns about late in-stent thrombosis with drug-eluting stents have led some experts to recommend long-term use of clopidogrel plus aspirin for the latter indication. However, these recommendations are likely to change because the risk of late stent thrombosis is decreasing with the newer generation of drug-eluting coronary stents.
The combination of clopidogrel and aspirin is also effective in patients with unstable angina. Thus, in 12,562 such patients, the risk of cardiovascular death, MI, or stroke was 9.3% in those randomized to the combination of clopidogrel and aspirin and 11.4% in those given aspirin alone. This 20% relative risk reduction with combination therapy was highly statistically significant. However, combining clopidogrel with aspirin increases the risk of major bleeding to about 2% per year. This bleeding risk persists even if the daily dose of aspirin is ≤100 mg. Therefore, the combination of clopidogrel and aspirin should only be used when there is a clear benefit. For example, this combination has not proven to be superior to clopidogrel alone in patients with acute ischemic stroke or to aspirin alone for primary prevention in those at risk for cardiovascular events.
Prasugrel was compared with clopidogrel in 13,608 patients with acute coronary syndromes who were scheduled to undergo percutaneous coronary intervention. The incidence of the primary efficacy endpoint, a composite of cardiovascular death, MI, or stroke, was significantly lower with prasugrel than with clopidogrel (9.9% and 12.1%, respectively), mainly reflecting a reduction in the incidence of nonfatal MI. The incidence of stent thrombosis also was significantly lower with prasugrel (1.1% and 2.4%, respectively). However, these advantages were at the expense of significantly higher rates of fatal bleeding (0.4% and 0.1%, respectively) and life-threatening bleeding (1.4% and 0.9%, respectively) with prasugrel. Because patients older than age 75 years and those with a history of prior stroke or transient ischemic attack have a particularly high risk of bleeding, prasugrel should generally be avoided in older patients, and the drug is contraindicated in those with a history of cerebrovascular disease. Caution is required if prasugrel is used in patients weighing less than 60 kg or in those with renal impairment.
When prasugrel was compared with clopidogrel in 7243 patients with unstable angina or MI without ST-segment elevation, prasugrel failed to reduce the rate of the primary efficacy endpoint, which was a composite of cardiovascular death, MI, and stroke. Because of the negative results of this study, prasugrel is reserved for patients undergoing percutaneous coronary intervention. In this setting, prasugrel is usually given in conjunction with aspirin. To reduce the risk of bleeding, the daily aspirin dose should be ≤100 mg.
DOSING Clopidogrel is given once daily at a dose of 75 mg. Loading doses of clopidogrel are given when rapid ADP receptor blockade is desired. For example, patients undergoing coronary stenting are often given a loading dose of 300 mg, which produces inhibition of ADP-induced platelet aggregation in about 6 h; loading doses of 600 or 900 mg produce an even more rapid effect. After a loading dose of 60 mg, prasugrel is given once daily at a dose of 10 mg. Patients older than age 75 years or weighing less than 60 kg should receive a lower daily prasugrel dose of 5 mg.
SIDE EFFECTS The most common side effect of clopidogrel and prasugrel is bleeding. Because of its greater potency, bleeding is more common with prasugrel than clopidogrel. To reduce the risk of bleeding, clopidogrel and prasugrel should be stopped 5–7 days before major surgery. In patients taking clopidogrel or prasugrel who present with serious bleeding, platelet transfusion may be helpful.
Hematologic side effects, including neutropenia, thrombocytopenia, and thrombotic thrombocytopenic purpura, are rare.
THIENOPYRIDINE RESISTANCE The capacity of clopidogrel to inhibit ADP-induced platelet aggregation varies among subjects. This variability reflects, at least in part, genetic polymorphisms in the CYP isoenzymes involved in the metabolic activation of clopidogrel. Most important of these is CYP2C19. Clopidogrel-treated patients with the loss-of-function CYP2C19*2 allele exhibit reduced platelet inhibition compared with those with the wild-type CYP2C19*1 allele and experience a higher rate of cardiovascular events. This is important because estimates suggest that up to 25% of whites, 30% of African Americans, and 50% of Asians carry the loss-of-function allele, which would render them resistant to clopidogrel. Even patients with the reduced function CYP2C19*3, *4, or *5 alleles may derive less benefit from clopidogrel than those with the full-function CYP2C19*1 allele. Concomitant administration of clopidogrel with proton pump inhibitors, which are inhibitors of CYP2C19, produces a small reduction in the inhibitory effects of clopidogrel on ADP-induced platelet aggregation. The extent to which this interaction increases the risk of cardiovascular events remains controversial.
In contrast to their effect on the metabolic activation of clopidogrel, CYP2C19 polymorphisms appear to be less important determinants of the activation of prasugrel. Thus, no association was detected between the loss-of-function allele and decreased platelet inhibition or increased rate of cardiovascular events with prasugrel. The observation that genetic polymorphisms affecting clopidogrel absorption or metabolism influence clinical outcomes raises the possibilities that pharmacogenetic profiling may be useful to identify clopidogrel-resistant patients and that point-of-care assessment of the extent of clopidogrel-induced platelet inhibition may help detect patients at higher risk for subsequent cardiovascular events. Clinical trials designed to evaluate these possibilities have thus far been negative. Although administration of higher doses of clopidogrel can overcome a reduced response to clopidogrel, the clinical benefit of this approach is uncertain. Instead, prasugrel or ticagrelor may be better choices for these patients.
Ticagrelor As an orally active inhibitor of P2Y12, ticagrelor differs from the thienopyridines in that ticagrelor does not require metabolic activation and it produces reversible inhibition of the ADP receptor.
MECHANISM OF ACTION Like the thienopyridines, ticagrelor inhibits P2Y12. Because it does not require metabolic activation, ticagrelor has a more rapid onset and offset of action than clopidogrel, and it produces greater and more predictable inhibition of ADP-induced platelet aggregation than clopidogrel.
INDICATIONS When compared with clopidogrel in patients with acute coronary syndromes, ticagrelor produced a greater reduction in the primary efficacy endpoint—a composite of cardiovascular death, MI, and stroke at 1 year—than clopidogrel (9.8% and 11.7%, respectively; p = .001). This difference reflected a significant reduction in both cardiovascular death (4.0% and 5.1%, respectively; p = .001) and MI (5.8% and 6.9%, respectively; p = .005) with ticagrelor compared with clopidogrel. Rates of stroke were similar with ticagrelor and clopidogrel (1.5% and 1.3%, respectively), and no difference in rates of major bleeding was noted. When minor bleeding was added to the major bleeding results, however, ticagrelor showed an increase relative to clopidogrel (16.1% and 14.6%, respectively; p = .008). Ticagrelor also was superior to clopidogrel in patients with acute coronary syndrome who underwent percutaneous coronary intervention or cardiac surgery. Based on these observations, some guidelines give ticagrelor preference over clopidogrel, particularly in higher risk patients.
DOSING Ticagrelor is initiated with an oral loading dose of 180 mg followed by 90 mg twice daily. The dose does not require adjustment in patients with renal impairment, but the drug should be used with caution in patients with hepatic disease and in those receiving potent inhibitors or inducers of CYP3A4 because ticagrelor is metabolized in the liver via CYP3A4. Ticagrelor is usually administered in conjunction with aspirin; the daily aspirin dose should not exceed 100 mg.
SIDE EFFECTS In addition to bleeding, the most common side effects of ticagrelor are dyspnea, which can occur in up to 15% of patients, and asymptomatic ventricular pauses. The dyspnea, which tends to occur soon after initiating ticagrelor, is usually self-limiting and mild in intensity. The mechanism responsible for this side effect is unknown.
To reduce the risk of bleeding, ticagrelor should be stopped 5–7 days prior to major surgery. Platelet transfusions are unlikely to be of benefit in patients with ticagrelor-related bleeding because the drug will bind to P2Y12 on the transfused platelets.
DIPYRIDAMOLE
Dipyridamole is a relatively weak antiplatelet agent on its own, but an extended-release formulation of dipyridamole combined with low-dose aspirin, a preparation known as Aggrenox, is used for prevention of stroke in patients with transient ischemic attacks.
Mechanism of Action By inhibiting phosphodiesterase, dipyridamole blocks the breakdown of cyclic adenosine monophosphate (AMP). Increased levels of cyclic AMP reduce intracellular calcium and inhibit platelet activation. Dipyridamole also blocks the uptake of adenosine by platelets and other cells. This produces a further increase in local cyclic AMP levels because the platelet adenosine A2 receptor is coupled to adenylate cyclase (Fig. 143-4).
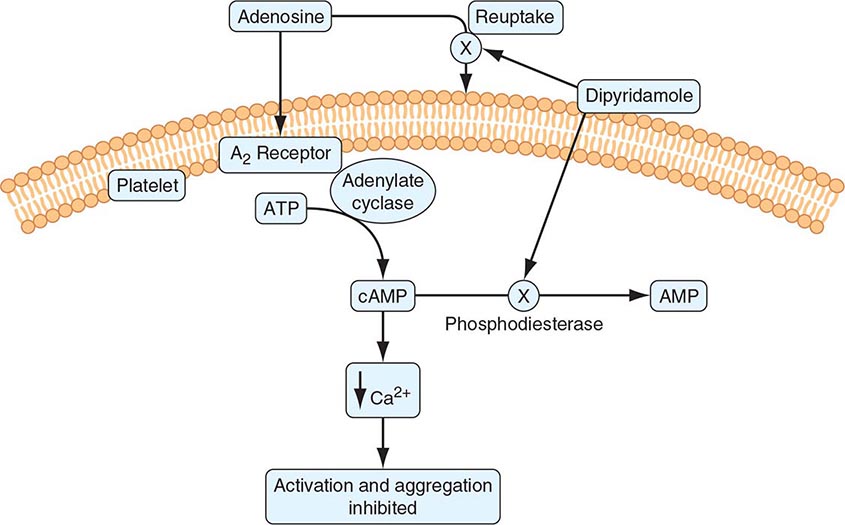
FIGURE 143-4 Mechanism of action of dipyridamole. Dipyridamole increases levels of cyclic AMP (cAMP) in platelets by (1) blocking the reuptake of adenosine and (2) inhibiting phosphodiesterase-mediated cyclic AMP degradation. By promoting calcium uptake, cyclic AMP reduces intracellular levels of calcium. This, in turn, inhibits platelet activation and aggregation.
Indications Dipyridamole plus aspirin was compared with aspirin or dipyridamole alone, or with placebo, in patients with an ischemic stroke or transient ischemic attack. The combination reduced the risk of stroke by 22.1% compared with aspirin and by 24.4% compared with dipyridamole. A second trial compared dipyridamole plus aspirin with aspirin alone for secondary prevention in patients with ischemic stroke. Vascular death, stroke, or MI occurred in 13% of patients given combination therapy and in 16% of those treated with aspirin alone. Another trial randomized 20,332 patients with noncardioembolic ischemic stroke to either Aggrenox or clopidogrel. The primary efficacy endpoint of recurrent stroke occurred in 9.0% of those given Aggrenox and in 8.8% of patients treated with clopidogrel. Although this difference was not statistically significant, the study failed to meet the prespecified margin to claim noninferiority of Aggrenox relative to clopidogrel. These results have dampened enthusiasm for the use of Aggrenox.
Because of its vasodilatory effects and the paucity of data supporting the use of dipyridamole in patients with symptomatic coronary artery disease, Aggrenox should not be used for stroke prevention in such patients. Clopidogrel is a better choice in this setting.
Dosing Aggrenox is given twice daily. Each capsule contains 200 mg of extended-release dipyridamole and 25 mg of aspirin.
Side Effects Because dipyridamole has vasodilatory effects, it must be used with caution in patients with coronary artery disease. Gastrointestinal complaints, headache, facial flushing, dizziness, and hypotension can also occur. These symptoms often subside with continued use of the drug.
GP IIB/IIIA RECEPTOR ANTAGONISTS
As a class, parenteral Gp IIb/IIIa receptor antagonists have an established niche in patients with acute coronary syndromes. The three agents in this class are abciximab, eptifibatide, and tirofiban.
Mechanism of Action A member of the integrin family of adhesion receptors, Gp IIb/IIIa is found on the surface of platelets and megakaryocytes. With about 80,000 copies per platelet, Gp IIb/IIIa is the most abundant receptor. Consisting of a noncovalently linked heterodimer, Gp IIb/IIIa is inactive on resting platelets. When platelets are activated, inside-outside signal transduction pathways trigger a conformational activation of the receptor. Once activated, Gp IIb/IIIa binds adhesive molecules, such as fibrinogen and, under high shear conditions, VWF. Binding is mediated by the Arg-Gly-Asp (RGD) sequence found on the α chains of fibrinogen and on VWF, and by the Lys-Gly-Asp (KGD) sequence located within a unique dodecapeptide domain on the γ chains of fibrinogen. Once bound, fibrinogen and/or VWF bridge adjacent platelets together to induce platelet aggregation.
Although abciximab, eptifibatide, and tirofiban all target the Gp IIb/IIIa receptor, they are structurally and pharmacologically distinct (Table 143-1). Abciximab is a Fab fragment of a humanized murine monoclonal antibody directed against the activated form of Gp IIb/IIIa. Abciximab binds to the activated receptor with high affinity and blocks the binding of adhesive molecules. In contrast, eptifibatide and tirofiban are synthetic small molecules. Eptifibatide is a cyclic heptapeptide that binds Gp IIb/IIIa because it incorporates the KGD motif, whereas tirofiban is a nonpeptidic tyrosine derivative that acts as an RGD mimetic. Abciximab has a long half-life and can be detected on the surface of platelets for up to 2 weeks; eptifibatide and tirofiban have short half-lives.
|
FEATURES OF GPIIB/IIIA ANTAGONISTS |
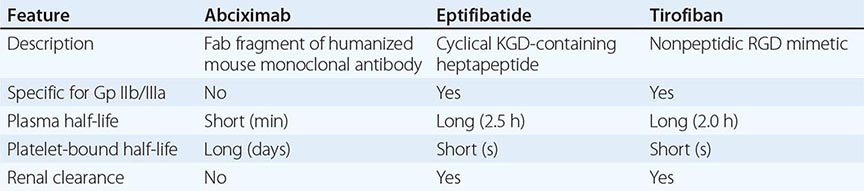
Whereas eptifibatide and tirofiban are specific for Gp IIb/IIIa, abciximab also inhibits the closely related αvβ3 receptor, which binds vitronectin, and αMβ2, a leukocyte integrin. Inhibition of αvβ3 and αMβ2 may endow abciximab with anti-inflammatory and/or antiproliferative properties that extend beyond platelet inhibition.
Indications Abciximab and eptifibatide are used in patients undergoing percutaneous coronary interventions, particularly those who have not been pretreated with an ADP receptor antagonist. Tirofiban is used in high-risk patients with unstable angina. Eptifibatide also can be used for this indication.
Dosing All of the Gp IIb/IIIa antagonists are given as an IV bolus followed by an infusion. The recommended dose of abciximab is a bolus of 0.25 mg/kg followed by an infusion of 0.125 μg/kg per minute to a maximum of 10 μg/kg for 12 h. Eptifibatide is given as two 180 μg/kg boluses given 10 min apart, followed by an infusion of 2.0 μg/kg per minute for 18–24 h. Tirofiban is started at a rate of 0.4 μg/kg per minute for 30 min; the drug is then continued at a rate of 0.1 μg/kg per minute for up to 18 h. Because these agents are cleared by the kidneys, the doses of eptifibatide and tirofiban must be reduced in patients with renal insufficiency. Thus, the eptifibatide infusion is reduced to 1 μg/kg per minute in patients with a creatinine clearance below 50 mL/min, whereas the dose of tirofiban is cut in half for patients with a creatinine clearance below 30 mL/min.
Side Effects In addition to bleeding, thrombocytopenia is the most serious complication. Thrombocytopenia is immune-mediated and is caused by antibodies directed against neoantigens on Gp IIb/IIIa that are exposed upon antagonist binding. With abciximab, thrombocytopenia occurs in up to 5% of patients. Thrombocytopenia is severe in ~1% of these individuals. Thrombocytopenia is less common with the other two agents, occurring in ~1% of patients.
NEW ANTIPLATELET AGENTS
New agents in advanced stages of development include cangrelor, a parenteral, rapidly acting, reversible inhibitor of P2Y12, and vorapaxar, an orally active inhibitor of protease-activated receptor 1 (PAR-1), the major thrombin receptor on platelets (Fig. 143-3).
Cangrelor An adenosine analogue, cangrelor binds reversibly to P2Y12 and inhibits its activity. The drug has a half-life of 3–6 min and is given IV as a bolus followed by an infusion. When stopped, platelet function recovers within 60 min. A trial comparing cangrelor with placebo during percutaneous coronary interventions and a study comparing cangrelor with clopidogrel after such procedures revealed little or no advantage of cangrelor. A third trial compared cangrelor (given as an IV bolus of 30 μg/kg followed by an infusion of 4 μg/kg per minute for at least 2 h, or for the duration of the procedure, whichever was longer) with a loading dose of clopidogrel (300 or 600 mg) in 11,145 patients undergoing urgent or elective percutaneous coronary intervention. The rate of the primary efficacy endpoint, a composite of death, MI, ischemia-driven revascularization, and stent thrombosis, was 4.7% in the cangrelor group and 5.9% in the clopidogrel group (p = .005). The rates of severe bleeding, the primary safety endpoint, were 0.16% and 0.11% in the cangrelor and clopidogrel groups, respectively. Using the same efficacy endpoint, a prespecified meta-analysis of the three trials revealed a relative risk reduction of 19% with cangrelor compared with clopidogrel (3.8% and 4.7%, respectively) and a 40% reduction in stent thrombosis (0.5% and 0.8%, respectively) with no significant increase in serious bleeding. Based on these data, cangrelor is currently under regulatory review.
Vorapaxar An orally active PAR-1 antagonist, vorapaxar is slowly eliminated with a half-life of about 200 h. When compared with placebo in 12,944 patients with acute coronary syndrome without ST-segment elevation, vorapaxar failed to significantly reduce the primary efficacy endpoint, a composite of cardiovascular death, MI, stroke, recurrent ischemia requiring rehospitalization, and urgent coronary revascularization. Moreover, vorapaxar was associated with increased rates of bleeding, including intracranial bleeding.
In a second trial, vorapaxar was compared with placebo for secondary prevention in 26,449 patients with prior MI, ischemic stroke, or peripheral arterial disease. Overall, vorapaxar reduced the risk for cardiovascular death, MI, or stroke by 13%, but doubled the risk of intracranial bleeding. In the prespecified subgroup of 17,779 patients with prior MI, however, vorapaxar reduced the risk for cardiovascular death, MI, or stroke by 20% compared with placebo (from 9.7% to 8.1%, respectively). The rate of intracranial hemorrhage was higher with vorapaxar than with placebo (0.6% and 0.4%, respectively; p = .076) as was the rate of moderate or severe bleeding (3.4% and 2.1%, respectively; P <0.0001). Based on these data, the drug is under consideration for regulatory approval in MI patients under the age of 75 years who have no history of stroke or transient ischemic attack and have a weight over 60 kg.
ANTICOAGULANTS
There are both parenteral and oral anticoagulants. The parenteral anticoagulants include heparin, low-molecular-weight heparin (LMWH), fondaparinux (a synthetic pentasaccharide), lepirudin, desirudin, bivalirudin, and argatroban. Currently available oral anticoagulants include warfarin; dabigatran etexilate, an oral thrombin inhibitor; and rivaroxaban and apixaban, oral factor Xa inhibitors. Edoxaban, a third oral factor Xa inhibitor, is undergoing regulatory review.
PARENTERAL ANTICOAGULANTS
Heparin A sulfated polysaccharide, heparin is isolated from mammalian tissues rich in mast cells. Most commercial heparin is derived from porcine intestinal mucosa and is a polymer of alternating D-glucuronic acid and N-acetyl-D-glucosamine residues.
MECHANISM OF ACTION Heparin acts as an anticoagulant by activating antithrombin (previously known as antithrombin III) and accelerating the rate at which antithrombin inhibits clotting enzymes, particularly thrombin and factor Xa. Antithrombin, the obligatory plasma cofactor for heparin, is a member of the serine protease inhibitor (serpin) superfamily. Synthesized in the liver and circulating in plasma at a concentration of 2.6 ± 0.4 μM, antithrombin acts as a suicide substrate for its target enzymes.
To activate antithrombin, heparin binds to the serpin via a unique pentasaccharide sequence that is found on one-third of the chains of commercial heparin (Fig. 143-5). Heparin chains without this pentasaccharide sequence have little or no anticoagulant activity. Once bound to antithrombin, heparin induces a conformational change in the reactive center loop of antithrombin that renders it more readily accessible to its target proteases. This conformational change enhances the rate at which antithrombin inhibits factor Xa by at least two orders of magnitude but has little effect on the rate of thrombin inhibition. To catalyze thrombin inhibition, heparin serves as a template that binds antithrombin and thrombin simultaneously. Formation of this ternary complex brings the enzyme in close apposition to the inhibitor, thereby promoting the formation of a stable covalent thrombin-antithrombin complex.
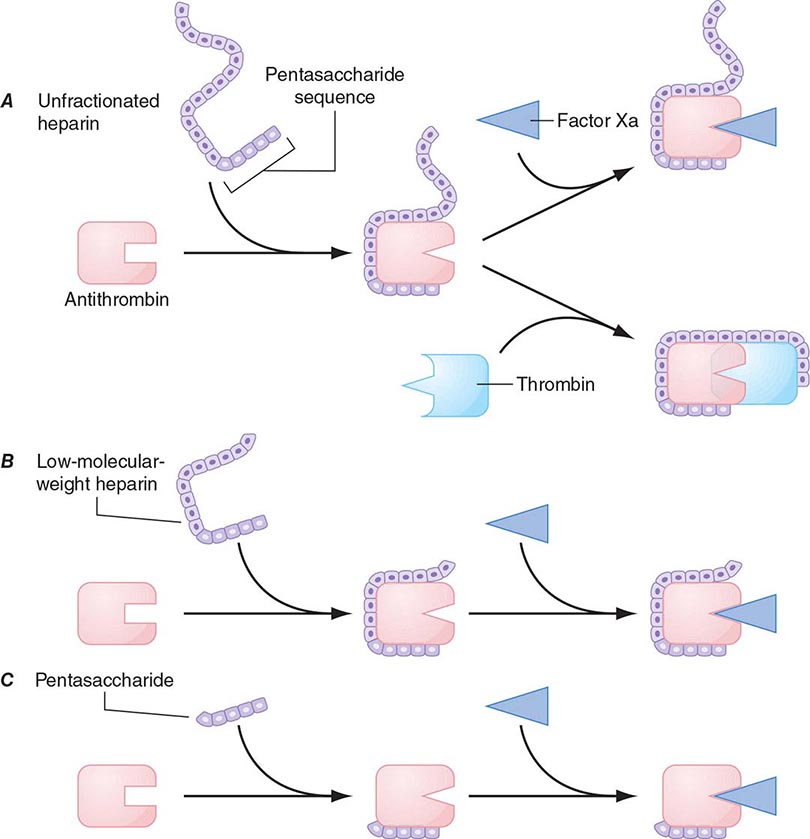
FIGURE 143-5 Mechanism of action of heparin, low-molecular-weight heparin (LMWH), and fondaparinux, a synthetic pentasaccharide. A. Heparin binds to antithrombin via its pentasaccharide sequence. This induces a conformational change in the reactive center loop of antithrombin that accelerates its interaction with factor Xa. To potentiate thrombin inhibition, heparin must simultaneously bind to antithrombin and thrombin. Only heparin chains composed of at least 18 saccharide units, which corresponds to a molecular weight of 5400, are of sufficient length to perform this bridging function. With a mean molecular weight of 15,000, all of the heparin chains are long enough to do this. B. LMWH has greater capacity to potentiate factor Xa inhibition by antithrombin than thrombin because, with a mean molecular weight of 4500–5000, at least half of the LMWH chains are too short to bridge antithrombin to thrombin. C. The pentasaccharide only accelerates factor Xa inhibition by antithrombin because the pentasaccharide is too short to bridge antithrombin to thrombin.
Only pentasaccharide-containing heparin chains composed of at least 18 saccharide units (which correspond to a molecular weight of 5400) are of sufficient length to bridge thrombin and antithrombin together. With a mean molecular weight of 15,000, and a range of 5000–30,000, almost all of the chains of unfractionated heparin are long enough to do so. Consequently, by definition, heparin has equal capacity to promote the inhibition of thrombin and factor Xa by antithrombin and is assigned an anti-factor Xa to anti-factor IIa (thrombin) ratio of 1:1.
Heparin causes the release of tissue factor pathway inhibitor (TFPI) from the endothelium. A factor Xa–dependent inhibitor of tissue factor–bound factor VIIa, TFPI may contribute to the antithrombotic activity of heparin. Longer heparin chains induce the release of more TFPI than shorter ones.
PHARMACOLOGY Heparin must be given parenterally. It is usually administered SC or by continuous IV infusion. When used for therapeutic purposes, the IV route is most often employed. If heparin is given SC for treatment of thrombosis, the dose of heparin must be high enough to overcome the limited bioavailability associated with this method of delivery.
In the circulation, heparin binds to the endothelium and to plasma proteins other than antithrombin. Heparin binding to endothelial cells explains its dose-dependent clearance. At low doses, the half-life of heparin is short because it binds rapidly to the endothelium. With higher doses of heparin, the half-life is longer because heparin is cleared more slowly once the endothelium is saturated. Clearance is mainly extrarenal; heparin binds to macrophages, which internalize and depolymerize the long heparin chains and secrete shorter chains back into the circulation. Because of its dose-dependent clearance mechanism, the plasma half-life of heparin ranges from 30 to 60 min with bolus IV doses of 25 and 100 units/kg, respectively.
Once heparin enters the circulation, it binds to plasma proteins other than antithrombin, a phenomenon that reduces its anticoagulant activity. Some of the heparin-binding proteins found in plasma are acute-phase reactants whose levels are elevated in ill patients. Others, such as high-molecular-weight multimers of VWF, are released from activated platelets or endothelial cells. Activated platelets also release platelet factor 4 (PF4), a highly cationic protein that binds heparin with high affinity. The large amounts of PF4 found in the vicinity of platelet-rich arterial thrombi can neutralize the anticoagulant activity of heparin. This phenomenon may attenuate heparin’s capacity to suppress thrombus growth.
Because the levels of heparin-binding proteins in plasma vary from person to person, the anticoagulant response to fixed or weight-adjusted doses of heparin is unpredictable. Consequently, coagulation monitoring is essential to ensure that a therapeutic response is obtained. This is particularly important when heparin is administered for treatment of established thrombosis because a subtherapeutic anticoagulant response may render patients at risk for recurrent thrombosis, whereas excessive anticoagulation increases the risk of bleeding.
MONITORING THE ANTICOAGULANT EFFECT Heparin therapy can be monitored using the activated partial thromboplastin time (aPTT) or anti-factor Xa level. Although the aPTT is the test most often used for this purpose, there are problems with this assay. aPTT reagents vary in their sensitivity to heparin, and the type of coagulometer used for testing can influence the results. Consequently, laboratories must establish a therapeutic aPTT range with each reagent-coagulometer combination by measuring the aPTT and anti-factor Xa level in plasma samples collected from heparin-treated patients. For most of the aPTT reagents and coagulometers in current use, therapeutic heparin levels are achieved with a two- to threefold prolongation of the aPTT.
Anti-factor Xa levels also can be used to monitor heparin therapy. With this test, therapeutic heparin levels range from 0.3 to 0.7 units/mL. Although this test is gaining in popularity, anti-factor Xa assays have yet to be standardized, and results can vary widely between laboratories.
Up to 25% of heparin-treated patients with venous thromboembolism require >35,000 units/d to achieve a therapeutic aPTT. These patients are considered heparin resistant. It is useful to measure anti-factor Xa levels in heparin-resistant patients because many will have a therapeutic anti-factor Xa level despite a subtherapeutic aPTT. This dissociation in test results occurs because elevated plasma levels of fibrinogen and factor VIII, both of which are acute-phase proteins, shorten the aPTT but have no effect on anti-factor Xa levels. Heparin therapy in patients who exhibit this phenomenon is best monitored using anti-factor Xa levels instead of the aPTT. Patients with congenital or acquired antithrombin deficiency and those with elevated levels of heparin-binding proteins may also need high doses of heparin to achieve a therapeutic aPTT or anti-factor Xa level. If there is good correlation between the aPTT and the anti-factor Xa levels, either test can be used to monitor heparin therapy.
DOSING For prophylaxis, heparin is usually given in fixed doses of 5000 units SC two or three times daily. With these low doses, coagulation monitoring is unnecessary. In contrast, monitoring is essential when the drug is given in therapeutic doses. Fixed-dose or weight-based heparin nomograms are used to standardize heparin dosing and to shorten the time required to achieve a therapeutic anticoagulant response. At least two heparin nomograms have been validated in patients with venous thromboembolism and reduce the time required to achieve a therapeutic aPTT. Weight-adjusted heparin nomograms have also been evaluated in patients with acute coronary syndromes. After an IV heparin bolus of 5000 units or 70 units/kg, a heparin infusion rate of 12–15 units/kg per hour is usually administered. In contrast, weight-adjusted heparin nomograms for patients with venous thromboembolism use an initial bolus of 5000 units or 80 units/kg, followed by an infusion of 18 units/kg per h. Thus, patients with venous thromboembolism appear to require higher doses of heparin to achieve a therapeutic aPTT than do patients with acute coronary syndromes. This may reflect differences in the thrombus burden. Heparin binds to fibrin, and the amount of fibrin in patients with extensive DVT is greater than that in those with coronary thrombosis.
Heparin manufacturers in North America have traditionally measured heparin potency in USP units, with a unit defined as the concentration of heparin that prevents 1 mL of citrated sheep plasma from clotting for 1 h after calcium addition. In contrast, manufacturers in Europe measure heparin potency with anti-Xa assays using an international heparin standard for comparison. Because of problems with heparin contamination with oversulfated chondroitin sulfate, which the USP assay system does not detect, North American heparin manufacturers now use the anti-Xa assay to assess heparin potency. The use of international units in place of USP units results in a 10% reduction in heparin doses, which is a difference unlikely to affect patient care because monitoring will help to ensure that a therapeutic anticoagulant response has been achieved.
LIMITATIONS Heparin has pharmacokinetic and biophysical limitations (Table 143-2). The pharmacokinetic limitations reflect heparin’s propensity to bind in a pentasaccharide-independent fashion to cells and plasma proteins. Heparin binding to endothelial cells explains its dose-dependent clearance, whereas binding to plasma proteins results in a variable anticoagulant response and can lead to heparin resistance.
|
PHARMACOKINETIC AND BIOPHYSICAL LIMITATIONS OF HEPARIN |
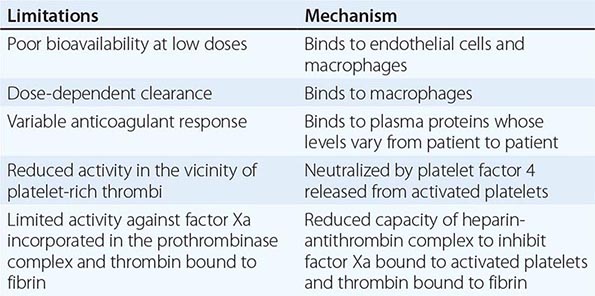
The biophysical limitations of heparin reflect the inability of the heparin-antithrombin complex to inhibit factor Xa when it is incorporated into the prothrombinase complex, the complex that converts prothrombin to thrombin, and to inhibit thrombin bound to fibrin. Consequently, factor Xa bound to activated platelets within platelet-rich thrombi has the potential to generate thrombin, even in the face of heparin. Once this thrombin binds to fibrin, it too is protected from inhibition by the heparin-antithrombin complex. Clot-associated thrombin can then trigger thrombus growth by locally activating platelets and amplifying its own generation through feedback activation of factors V, VIII, and XI. Further compounding the problem is the potential for heparin neutralization by the high concentrations of PF4 released from activated platelets within the platelet-rich thrombus.
SIDE EFFECTS The most common side effect of heparin is bleeding. Other complications include thrombocytopenia, osteoporosis, and elevated levels of transaminases.
Bleeding The risk of bleeding rises as the dose of heparin is increased. Concomitant administration of drugs that affect hemostasis, such as antiplatelet or fibrinolytic agents, increases the risk of bleeding, as does recent surgery or trauma. Heparin-treated patients with serious bleeding can be given protamine sulfate to neutralize the heparin. Protamine sulfate, a mixture of basic polypeptides isolated from salmon sperm, binds heparin with high affinity, and the resultant protamine-heparin complexes are then cleared. Typically, 1 mg of protamine sulfate neutralizes 100 units of heparin. Protamine sulfate is given IV. Anaphylactoid reactions to protamine sulfate can occur, and drug administration by slow IV infusion is recommended to reduce the risk.
THROMBOCYTOPENIA Heparin can cause thrombocytopenia. Heparin-induced thrombocytopenia (HIT) is an antibody-mediated process that is triggered by antibodies directed against neoantigens on PF4 that are exposed when heparin binds to this protein. These antibodies, which are usually of the IgG isotype, bind simultaneously to the heparin-PF4 complex and to platelet Fc receptors. Such binding activates the platelets and generates platelet microparticles. Circulating microparticles are prothrombotic because they express anionic phospholipids on their surface and can bind clotting factors and promote thrombin generation.
The clinical features of HIT are illustrated in Table 143-3. Typically, HIT occurs 5–14 days after initiation of heparin therapy, but it can manifest earlier if the patient has received heparin within the past 3 months. A platelet count below 100,000/μL or a 50% decrease in the platelet count from the pretreatment value should raise the suspicion of HIT in those receiving heparin. HIT is more common in surgical patients than in medical patients and, like many autoimmune disorders, occurs more frequently in females than in males.
|
FEATURES OF HEPARIN-INDUCED THROMBOCYTOPENIA |
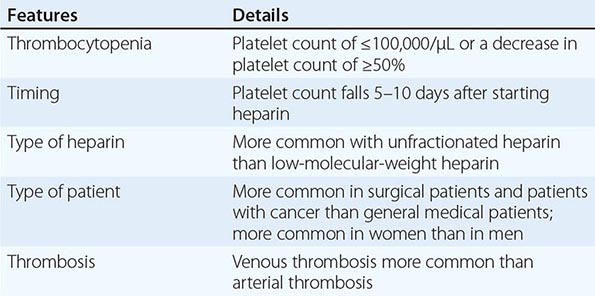
HIT can be associated with thrombosis, either arterial or venous. Venous thrombosis, which manifests as DVT and/or PE, is more common than arterial thrombosis. Arterial thrombosis can manifest as ischemic stroke or acute MI. Rarely, platelet-rich thrombi in the distal aorta or iliac arteries can cause critical limb ischemia.
The diagnosis of HIT is established using enzyme-linked assays to detect antibodies against heparin-PF4 complexes or with platelet activation assays. Enzyme-linked assays are sensitive but can be positive in the absence of any clinical evidence of HIT. The most specific diagnostic test is the serotonin release assay. This test is performed by quantifying serotonin release when washed platelets loaded with labeled serotonin are exposed to patient serum in the absence or presence of varying concentrations of heparin. If the patient serum contains the HIT antibody, heparin addition induces platelet activation and serotonin release.
Management of HIT is outlined in Table 143-4. Heparin should be stopped in patients with suspected or documented HIT, and an alternative anticoagulant should be administered to prevent or treat thrombosis. The agents most often used for this indication are parenteral direct thrombin inhibitors, such as lepirudin, argatroban, or bivalirudin, or factor Xa inhibitors, such as fondaparinux.
|
MANAGEMENT OF HEPARIN-INDUCED THROMBOCYTOPENIA |
Abbreviation: INR, international normalized ratio.
Patients with HIT, particularly those with associated thrombosis, often have evidence of increased thrombin generation that can lead to consumption of protein C. If these patients are given warfarin without a concomitant parenteral anticoagulant to inhibit thrombin or thrombin generation, the further decrease in protein C levels induced by the vitamin K antagonist can trigger skin necrosis. To avoid this problem, patients with HIT should be treated with a direct thrombin inhibitor or fondaparinux until the platelet count returns to normal levels. At this point, low-dose warfarin therapy can be introduced, and the thrombin inhibitor can be discontinued when the anticoagulant response to warfarin has been therapeutic for at least 2 days.
Osteoporosis Treatment with therapeutic doses of heparin for >1 month can cause a reduction in bone density. This complication has been reported in up to 30% of patients given long-term heparin therapy, and symptomatic vertebral fractures occur in 2–3% of these individuals.
Heparin causes bone loss both by decreasing bone formation and by enhancing bone resorption. Thus, heparin affects the activity of both osteoblasts and osteoclasts.
Elevated levels of transaminases Therapeutic doses of heparin are frequently associated with modest elevations in the serum levels of hepatic transaminases without a concomitant increase in the level of bilirubin. The levels of transaminases rapidly return to normal when the drug is stopped. The mechanism responsible for this phenomenon is unknown.
Low-Molecular-Weight Heparin Consisting of smaller fragments of heparin, LMWH is prepared from unfractionated heparin by controlled enzymatic or chemical depolymerization. The mean molecular weight of LMWH is about 5000, one-third the mean molecular weight of unfractionated heparin. LMWH has advantages over heparin (Table 143-5) and has replaced heparin for most indications.
|
ADVANTAGES OF LMWH OVER HEPARIN |
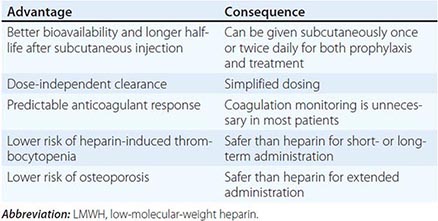
MECHANISM OF ACTION Like heparin, LMWH exerts its anticoagulant activity by activating antithrombin. With a mean molecular weight of 5000, which corresponds to about 17 saccharide units, at least half of the pentasaccharide-containing chains of LMWH are too short to bridge thrombin to antithrombin (Fig. 143-5). However, these chains retain the capacity to accelerate factor Xa inhibition by antithrombin because this activity is largely the result of the conformational changes in antithrombin evoked by pentasaccharide binding. Consequently, LMWH catalyzes factor Xa inhibition by antithrombin more than thrombin inhibition. Depending on their unique molecular weight distributions, LMWH preparations have anti-factor Xa to anti-factor IIa ratios ranging from 2:1 to 4:1.
PHARMACOLOGY Although usually given SC, LMWH also can be administered IV if a rapid anticoagulant response is needed. LMWH has pharmacokinetic advantages over heparin. These advantages reflect the fact that shorter heparin chains bind less avidly to endothelial cells, macrophages, and heparin-binding plasma proteins. Reduced binding to endothelial cells and macrophages eliminates the rapid, dose-dependent, and saturable mechanism of clearance that is a characteristic of unfractionated heparin. Instead, the clearance of LMWH is dose-independent and its plasma half-life is longer. Based on measurement of anti-factor Xa levels, LMWH has a plasma half-life of ~4 h. LMWH is cleared almost exclusively by the kidneys, and the drug can accumulate in patients with renal insufficiency.
LMWH exhibits about 90% bioavailability after SC injection. Because LMWH binds less avidly to heparin-binding proteins in plasma than heparin, LMWH produces a more predictable dose response, and resistance to LMWH is rare. With a longer half-life and more predictable anticoagulant response, LMWH can be given SC once or twice daily without coagulation monitoring, even when the drug is given in treatment doses. These properties render LMWH more convenient than unfractionated heparin. Capitalizing on this feature, studies in patients with venous thromboembolism have shown that home treatment with LMWH is as effective and safe as in-hospital treatment with continuous IV infusions of heparin. Outpatient treatment with LMWH streamlines care, reduces health care costs, and increases patient satisfaction.
MONITORING In the majority of patients, LMWH does not require coagulation monitoring. If monitoring is necessary, anti-factor Xa levels must be measured because most LMWH preparations have little effect on the aPTT. Therapeutic anti-factor Xa levels with LMWH range from 0.5 to 1.2 units/mL when measured 3–4 h after drug administration. When LMWH is given in prophylactic doses, peak anti-factor Xa levels of 0.2–0.5 units/mL are desirable.
Indications for LMWH monitoring include renal insufficiency and obesity. LMWH monitoring in patients with a creatinine clearance of ≤50 mL/min is advisable to ensure that there is no drug accumulation. Although weight-adjusted LMWH dosing appears to produce therapeutic anti-factor Xa levels in patients who are overweight, this approach has not been extensively evaluated in those with morbid obesity. It may also be advisable to monitor the anticoagulant activity of LMWH during pregnancy because dose requirements can change, particularly in the third trimester. Monitoring should also be considered in high-risk settings, such as in patients with mechanical heart valves who are given LMWH for prevention of valve thrombosis, and when LMWH is used in treatment doses in infants or children.
DOSING The doses of LMWH recommended for prophylaxis or treatment vary depending on the LMWH preparation. For prophylaxis, once-daily SC doses of 4000–5000 units are often used, whereas doses of 2500–3000 units are given when the drug is administered twice daily. For treatment of venous thromboembolism, a dose of 150–200 units/kg is given if the drug is administered once daily. If a twice-daily regimen is used, a dose of 100 units/kg is given. In patients with unstable angina, LMWH is given SC on a twice-daily basis at a dose of 100–120 units/kg.
SIDE EFFECTS The major complication of LMWH is bleeding. Meta-analyses suggest that the risk of major bleeding is lower with LMWH than with unfractionated heparin. HIT and osteoporosis are less common with LMWH than with unfractionated heparin.
Bleeding Like the situation with heparin, bleeding with LMWH is more common in patients receiving concomitant therapy with antiplatelet or fibrinolytic drugs. Recent surgery, trauma, or underlying hemostatic defects also increase the risk of bleeding with LMWH.
Although protamine sulfate can be used as an antidote for LMWH, protamine sulfate incompletely neutralizes the anticoagulant activity of LMWH because it only binds the longer chains of LMWH. Because longer chains are responsible for catalysis of thrombin inhibition by antithrombin, protamine sulfate completely reverses the anti-factor IIa activity of LMWH. In contrast, protamine sulfate only partially reverses the anti-factor Xa activity of LMWH because the shorter pentasaccharide-containing chains of LMWH do not bind to protamine sulfate. Consequently, patients at high risk for bleeding may be more safely treated with continuous IV unfractionated heparin than with SC LMWH.
Thrombocytopenia The risk of HIT is about fivefold lower with LMWH than with heparin. LMWH binds less avidly to platelets and causes less PF4 release. Furthermore, with lower affinity for PF4 than heparin, LMWH is less likely to induce the conformational changes in PF4 that trigger the formation of HIT antibodies.
LMWH should not be used to treat HIT patients because most HIT antibodies exhibit cross-reactivity with LMWH. This in vitro cross-reactivity is not simply a laboratory phenomenon because there are case reports of thrombosis when HIT patients were switched from heparin to LMWH.
Osteoporosis Because the risk of osteoporosis is lower with LMWH than with heparin, LMWH is the better choice for extended treatment.
Fondaparinux A synthetic analogue of the antithrombin-binding pentasaccharide sequence, fondaparinux differs from LMWH in several ways (Table 143-6). Fondaparinux is licensed for thromboprophylaxis in general medical or surgical patients and in high-risk orthopedic patients and as an alternative to heparin or LMWH for initial treatment of patients with established venous thromboembolism. Although widely used in Europe, as an alternative to heparin or LMWH in patients with acute coronary syndromes, fondaparinux is not licensed for this indication in the United States.
|
COMPARISON OF LMWH AND FONDAPARINUX |
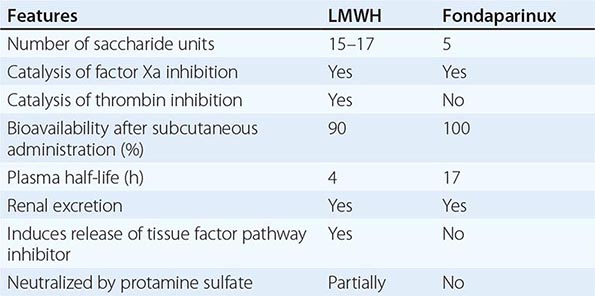
MECHANISM OF ACTION As a synthetic analogue of the antithrombin-binding pentasaccharide sequence found in heparin and LMWH, fondaparinux has a molecular weight of 1728. Fondaparinux binds only to antithrombin (Fig. 143-5) and is too short to bridge thrombin to antithrombin. Consequently, fondaparinux catalyzes factor Xa inhibition by antithrombin and does not enhance the rate of thrombin inhibition.
PHARMACOLOGY Fondaparinux exhibits complete bioavailability after SC injection. With no binding to endothelial cells or plasma proteins, the clearance of fondaparinux is dose independent and its plasma half-life is 17 h. The drug is given SC once daily. Because fondaparinux is cleared unchanged via the kidneys, it is contraindicated in patients with a creatinine clearance <30 mL/min and should be used with caution in those with a creatinine clearance <50 mL/min.
Fondaparinux produces a predictable anticoagulant response after administration in fixed doses because it does not bind to plasma proteins. The drug is given at a dose of 2.5 mg once daily for prevention of venous thromboembolism. For initial treatment of established venous thromboembolism, fondaparinux is given at a dose of 7.5 mg once daily. The dose can be reduced to 5 mg once daily for those weighing <50 kg and increased to 10 mg for those >100 kg. When given in these doses, fondaparinux is as effective as heparin or LMWH for initial treatment of patients with DVT or PE and produces similar rates of bleeding.
Fondaparinux is used at a dose of 2.5 mg once daily in patients with acute coronary syndromes. When this prophylactic dose of fondaparinux was compared with treatment doses of enoxaparin in patients with non-ST-segment elevation acute coronary syndrome, there was no difference in the rate of cardiovascular death, MI, or stroke at 9 days. However, the rate of major bleeding was 50% lower with fondaparinux than with enoxaparin, a difference that likely reflects the fact that the dose of fondaparinux was lower than that of enoxaparin. In acute coronary syndrome patients who require percutaneous coronary intervention, there is a risk of catheter thrombosis with fondaparinux unless adjunctive heparin is given.
SIDE EFFECTS Fondaparinux does not cause HIT because it does not bind to PF4. In contrast to LMWH, there is no cross-reactivity of fondaparinux with HIT antibodies. Consequently, fondaparinux appears to be effective for treatment of HIT patients, although large clinical trials supporting its use are lacking.
The major side effect of fondaparinux is bleeding. There is no antidote for fondaparinux. Protamine sulfate has no effect on the anticoagulant activity of fondaparinux because it fails to bind to the drug. Recombinant activated factor VII reverses the anticoagulant effects of fondaparinux in volunteers, but it is unknown whether this agent controls fondaparinux-induced bleeding.
Parenteral Direct Thrombin Inhibitors Direct thrombin inhibitors bind directly to thrombin and block its interaction with its substrates. Approved parenteral direct thrombin inhibitors include recombinant hirudins (lepirudin and desirudin), argatroban, and bivalirudin (Table 143-7). Lepirudin and argatroban are licensed for treatment of patients with HIT, desirudin is licensed for thromboprophylaxis after elective hip arthroplasty, and bivalirudin is approved as an alternative to heparin in patients undergoing percutaneous coronary intervention, including those with HIT.
|
COMPARISON OF THE PROPERTIES OF LEPIRUDIN, BIVALIRUDIN, AND ARGATROBAN |
LEPIRUDIN AND DESIRUDIN Recombinant forms of hirudin, lepirudin, and desirudin are bivalent direct thrombin inhibitors that interact with the active site and exosite 1, the substrate-binding site on thrombin. For rapid anticoagulation, lepirudin is given by continuous IV infusion, but the drug can be given SC. Lepirudin has a plasma half-life of 60 min after IV infusion and is cleared by the kidneys. Consequently, lepirudin accumulates in patients with renal insufficiency. For thromboprophylaxis, desirudin is given SC twice daily in fixed doses; the half-life of desirudin is 2–3 h after SC injection.
A high proportion of lepirudin-treated patients develop antibodies against the drug; antibody formation is rare with SC desirudin. Although lepirudin-directed antibodies rarely cause problems, in a small subset of patients, they can delay lepirudin clearance and enhance its anticoagulant activity. Serious bleeding has been reported in some of these patients.
Lepirudin is usually monitored using the aPTT, and the dose is adjusted to maintain an aPTT that is 1.5–2.5 times the control. The aPTT is not an ideal test for monitoring lepirudin therapy because the clotting time plateaus with higher drug concentrations. Although the clotting time with ecarin, a snake venom that converts prothrombin to meizothrombin, provides a better index of lepirudin dose than the aPTT, the ecarin clotting time has yet to be standardized. When used for thromboprophylaxis, desirudin does not require monitoring.
ARGATROBAN A univalent inhibitor that targets the active site of thrombin, argatroban is metabolized in the liver. Consequently, this drug must be used with caution in patients with hepatic insufficiency. Argatroban is not cleared via the kidneys, so this drug is safer than lepirudin for HIT patients with renal insufficiency.
Argatroban is administered by continuous IV infusion and has a plasma half-life of ~45 min. The aPTT is used to monitor its anticoagulant effect, and the dose is adjusted to achieve an aPTT 1.5–3 times the baseline value, but not to exceed 100 s. Argatroban also prolongs the international normalized ratio (INR), a feature that can complicate the transitioning of patients to warfarin. This problem can be circumvented by using the levels of factor × to monitor warfarin in place of the INR. Alternatively, argatroban can be stopped for 2–3 h before INR determination.
BIVALIRUDIN A synthetic 20-amino-acid analogue of hirudin, bivalirudin is a divalent thrombin inhibitor. Thus, the N-terminus of bivalirudin interacts with the active site of thrombin, whereas its C-terminus binds to exosite 1. Bivalirudin has a plasma half-life of 25 min, the shortest half-life of all the parenteral direct thrombin inhibitors. Bivalirudin is degraded by peptidases and is partially excreted via the kidneys. When given in high doses in the cardiac catheterization laboratory, the anticoagulant activity of bivalirudin is monitored using the activated clotting time. With lower doses, its activity can be assessed using the aPTT.
Bivalirudin is licensed as an alternative to heparin in patients undergoing percutaneous coronary intervention. Bivalirudin also has been used successfully in HIT patients who require percutaneous coronary intervention or cardiac bypass surgery.
ORAL ANTICOAGULANTS
Current oral anticoagulant practice dates back almost 60 years to when the vitamin K antagonists were discovered as a result of investigations into the cause of hemorrhagic disease in cattle. Characterized by a decrease in prothrombin levels, this disorder is caused by ingestion of hay containing spoiled sweet clover. Hydroxycoumarin, which was isolated from bacterial contaminants in the hay, interferes with vitamin K metabolism, thereby causing a syndrome similar to vitamin K deficiency. Discovery of this compound provided the impetus for development of other vitamin K antagonists, including warfarin.
For many years, the vitamin K antagonists were the only available oral anticoagulants. This situation changed with the introduction of new oral anticoagulants, including dabigatran, which targets thrombin, and rivaroxaban, apixaban, and edoxaban, which target factor Xa.
Warfarin A water-soluble vitamin K antagonist initially developed as a rodenticide, warfarin is the coumarin derivative most often prescribed in North America. Like other vitamin K antagonists, warfarin interferes with the synthesis of the vitamin K–dependent clotting proteins, which include prothrombin (factor II) and factors VII, IX, and X. The synthesis of the vitamin K–dependent anticoagulant proteins, proteins C and S, is also reduced by vitamin K antagonists.
MECHANISM OF ACTION All of the vitamin K–dependent clotting factors possess glutamic acid residues at their N termini. A posttranslational modification adds a carboxyl group to the γ-carbon of these residues to generate γ-carboxyglutamic acid. This modification is essential for expression of the activity of these clotting factors because it permits their calcium-dependent binding to negatively charged phospholipid surfaces. The γ-carboxylation process is catalyzed by a vitamin K–dependent carboxylase. Thus, vitamin K from the diet is reduced to vitamin K hydroquinone by vitamin K reductase (Fig. 143-6). Vitamin K hydroquinone serves as a cofactor for the carboxylase enzyme, which in the presence of carbon dioxide replaces the hydrogen on the γ-carbon of glutamic acid residues with a carboxyl group. During this process, vitamin K hydroquinone is oxidized to vitamin K epoxide, which is then reduced to vitamin K by vitamin K epoxide reductase.
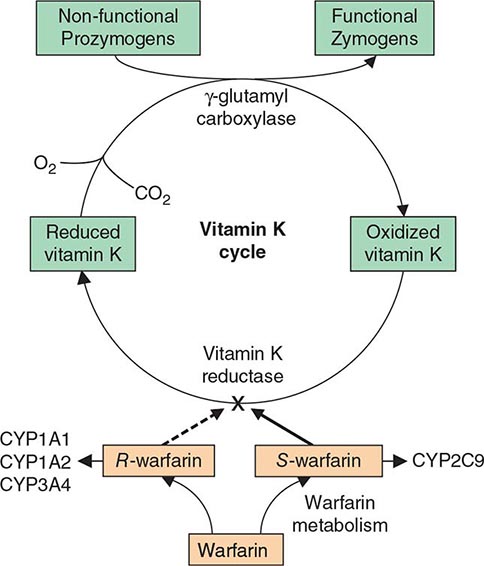
FIGURE 143-6 Mechanism of action of warfarin. A racemic mixture of S– and R-enantiomers, S-warfarin is most active. By blocking vitamin K epoxide reductase, warfarin inhibits the conversion of oxidized vitamin K into its reduced form. This inhibits vitamin K–dependent γ-carboxylation of factors II, VII, IX, and × because reduced vitamin K serves as a cofactor for a γ-glutamyl carboxylase that catalyzes the γ-carboxylation process, thereby converting prozymogens to zymogens capable of binding calcium and interacting with anionic phospholipid surfaces. S-warfarin is metabolized by CYP2C9. Common genetic polymorphisms in this enzyme can influence warfarin metabolism. Polymorphisms in the C1 subunit of vitamin K reductase (VKORC1) also can affect the susceptibility of the enzyme to warfarin-induced inhibition, thereby influencing warfarin dosage requirements.
Warfarin inhibits vitamin K epoxide reductase (VKOR), thereby blocking the γ-carboxylation process. This results in the synthesis of vitamin K–dependent clotting proteins that are only partially γ-carboxylated. Warfarin acts as an anticoagulant because these partially γ-carboxylated proteins have reduced or absent biologic activity. The onset of action of warfarin is delayed until the newly synthesized clotting factors with reduced activity gradually replace their fully active counterparts.
The antithrombotic effect of warfarin depends on a reduction in the functional levels of factor × and prothrombin, clotting factors that have half-lives of 24 and 72 h, respectively. Because the antithrombotic effect of warfarin is delayed, patients with established thrombosis or at high risk for thrombosis require concomitant treatment with a rapidly acting parenteral anticoagulant, such as heparin, LMWH, or fondaparinux, for at least 5 days.
PHARMACOLOGY Warfarin is a racemic mixture of R and S isomers. Warfarin is rapidly and almost completely absorbed from the gastrointestinal tract. Levels of warfarin in the blood peak about 90 min after drug administration. Racemic warfarin has a plasma half-life of 36–42 h, and more than 97% of circulating warfarin is bound to albumin. Only the small fraction of unbound warfarin is biologically active.
Warfarin accumulates in the liver where the two isomers are metabolized via distinct pathways. CYP2C9 mediates oxidative metabolism of the more active S isomer (Fig. 143-6). Two relatively common variants, CYP2C9*2 and CYP2C9*3, encode an enzyme with reduced activity. Patients with these variants require lower maintenance doses of warfarin. Approximately 25% of Caucasians have at least one variant allele of CYP2C9*2 or CYP2C9*3, whereas those variant alleles are less common in African Americans and Asians (Table 143-8). Heterozygosity for CYP2C9*2 or CYP2C9*3 decreases the warfarin dose requirement by 20–30% relative to that required in subjects with the wild-type CYP2C9*1/*1 alleles, whereas homozygosity for the CYP2C9*2 or CYP2C9*3 alleles reduces the warfarin dose requirement by 50–70%.
|
FREQUENCIES OF CYP2C9 GENOTYPES AND VKORC1 HAPLOTYPES IN DIFFERENT POPULATIONS AND THEIR EFFECT ON WARFARIN DOSE REQUIREMENTS |
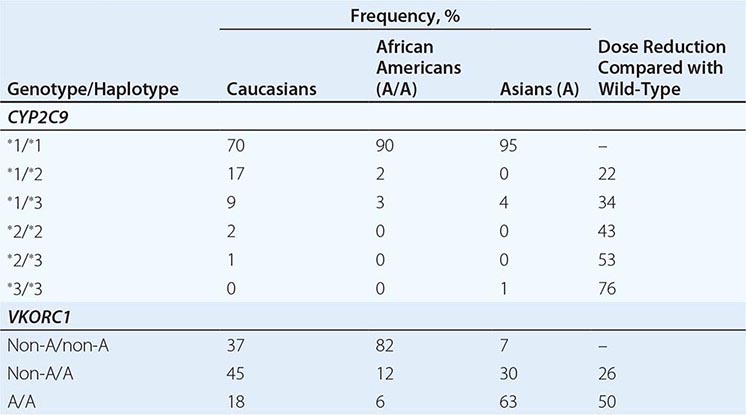
Consistent with their decreased warfarin dose requirement, subjects with at least one CYP2C9 variant allele are at increased risk for bleeding. Compared with individuals with no variant alleles, the relative risks for warfarin-associated bleeding in CYP2C9*2 or CYP2C9*3 carriers are 1.9 and 1.8, respectively.
Polymorphisms in VKORC1 also can influence the anticoagulant response to warfarin. Several genetic variations of VKORC1 are in strong linkage disequilibrium and have been designated as non-A haplotypes. VKORC1 variants are more prevalent than variants of CYP2C9. Asians have the highest prevalence of VKORC1 variants, followed by Caucasians and African Americans (Table 143-8). Polymorphisms in VKORC1 likely explain 30% of the variability in warfarin dose requirements. Compared with VKORC1 non-A/non-A homozygotes, the warfarin dose requirement decreases by 25 and 50% in A haplotype heterozygotes and homozygotes, respectively. These findings prompted the Food and Drug Administration to amend the prescribing information for warfarin to indicate that lower initiation doses should be considered for patients with CYP2C9 and VKORC1 genetic variants. In addition to genotype data, other pertinent patient information has been incorporated into warfarin dosing algorithms. Although such algorithms help predict suitable warfarin doses, it remains unclear whether better dose identification improves patient outcome in terms of reducing hemorrhagic complications or recurrent thrombotic events.
In addition to genetic factors, the anticoagulant effect of warfarin is influenced by diet, drugs, and various disease states. Fluctuations in dietary vitamin K intake affect the activity of warfarin. A wide variety of drugs can alter absorption, clearance, or metabolism of warfarin. Because of the variability in the anticoagulant response to warfarin, coagulation monitoring is essential to ensure that a therapeutic response is obtained.
MONITORING Warfarin therapy is most often monitored using the prothrombin time, a test that is sensitive to reductions in the levels of prothrombin, factor VII, and factor X. The test is performed by adding thromboplastin, a reagent that contains tissue factor, phospholipid, and calcium, to citrated plasma and determining the time to clot formation. Thromboplastins vary in their sensitivity to reductions in the levels of the vitamin K–dependent clotting factors. Thus, less sensitive thromboplastins will trigger the administration of higher doses of warfarin to achieve a target prothrombin time. This is problematic because higher doses of warfarin increase the risk of bleeding.
The INR was developed to circumvent many of the problems associated with the prothrombin time. To calculate the INR, the patient’s prothrombin time is divided by the mean normal prothrombin time, and this ratio is then multiplied by the international sensitivity index (ISI), which is an index of the sensitivity of the thromboplastin used for prothrombin time determination to reductions in the levels of the vitamin K–dependent clotting factors. Highly sensitive thromboplastins have an ISI of 1.0. Most current thromboplastins have ISI values that range from 1.0 to 1.4.
Although the INR has helped to standardize anticoagulant practice, problems persist. The precision of INR determination varies depending on reagent-coagulometer combinations. This leads to variability in the INR results. Also complicating INR determination is unreliable reporting of the ISI by thromboplastin manufacturers. Furthermore, every laboratory must establish the mean normal prothrombin time with each new batch of thromboplastin reagent. To accomplish this, the prothrombin time must be measured in fresh plasma samples from at least 20 healthy volunteers using the same coagulometer that is used for patient samples.
For most indications, warfarin is administered in doses that produce a target INR of 2.0–3.0. An exception is patients with mechanical heart valves, particularly those in the mitral position or older ball and cage valves in the aortic position, where a target INR of 2.5–3.5 is recommended. Studies in atrial fibrillation demonstrate an increased risk of cardioembolic stroke when the INR falls to <1.7 and an increase in bleeding with INR values >4.5. These findings highlight the fact that vitamin K antagonists have a narrow therapeutic window. In support of this concept, a study in patients receiving long-term warfarin therapy for unprovoked venous thromboembolism demonstrated a higher rate of recurrent venous thromboembolism with a target INR of 1.5–1.9 compared with a target INR of 2.0–3.0.
DOSING Warfarin is usually started at a dose of 5–10 mg. Lower doses are used for patients with CYP2C9 or VKORC1 polymorphisms, which affect the pharmacodynamics or pharmacokinetics of warfarin and render patients more sensitive to the drug. The dose is then titrated to achieve the desired target INR. Because of its delayed onset of action, patients with established thrombosis or those at high risk for thrombosis are given concomitant initial treatment with a rapidly acting parenteral anticoagulant, such as heparin, LMWH, or fondaparinux. Early prolongation of the INR reflects reduction in the functional levels of factor VII. Consequently, concomitant treatment with the parenteral anticoagulant should be continued until the INR has been therapeutic for at least 2 consecutive days. A minimum 5-day course of parenteral anticoagulation is recommended to ensure that the levels of factor Xa and prothrombin have been reduced into the therapeutic range with warfarin.
Because warfarin has a narrow therapeutic window, frequent coagulation monitoring is essential to ensure that a therapeutic anticoagulant response is maintained. Even patients with stable warfarin dose requirements should have their INR determined every 3–4 weeks. More frequent monitoring is necessary when new medications are introduced because so many drugs enhance or reduce the anticoagulant effects of warfarin.
SIDE EFFECTS Like all anticoagulants, the major side effect of warfarin is bleeding. A rare complication is skin necrosis. Warfarin crosses the placenta and can cause fetal abnormalities. Consequently, warfarin should not be used during pregnancy.
Bleeding At least half of the bleeding complications with warfarin occur when the INR exceeds the therapeutic range. Bleeding complications may be mild, such as epistaxis or hematuria, or more severe, such as retroperitoneal or gastrointestinal bleeding. Life-threatening intracranial bleeding can also occur.
To minimize the risk of bleeding, the INR should be maintained in the therapeutic range. In asymptomatic patients whose INR is between 3.5 and 10, warfarin should be withheld until the INR returns to the therapeutic range. If the INR is over 10, oral vitamin K should be administered, at a dose of 2.5–5 mg, although there is no evidence that doing so reduces the bleeding risk. Higher doses of oral vitamin K (5–10 mg) produce more rapid reversal of the INR but may render patients temporarily resistant to warfarin when the drug is restarted. Patients with serious bleeding need more aggressive treatment. These patients should be given 5–10 mg of vitamin K by slow IV infusion. Additional vitamin K should be given until the INR is in the normal range. Treatment with vitamin K should be supplemented with fresh-frozen plasma as a source of the vitamin K–dependent clotting proteins. Four factor prothrombin complex concentrates, which contain all four vitamin K–dependent clotting proteins, are the treatment of choice for (1) life-threatening bleeds, (2) rapid restoration of the INR into the normal range in patients requiring urgent surgery or intervention, and (3) patients who cannot tolerate the volume load of fresh-frozen plasma.
Warfarin-treated patients who experience bleeding when their INR is in the therapeutic range require investigation into the cause of the bleeding. Those with gastrointestinal or genitourinary bleeding often have an underlying lesion.
Skin necrosis A rare complication of warfarin, skin necrosis usually is seen 2–5 days after initiation of therapy. Well-demarcated erythematous lesions form on the thighs, buttocks, breasts, or toes. Typically, the center of the lesion becomes progressively necrotic. Examination of skin biopsies taken from the border of these lesions reveals thrombi in the microvasculature.
Warfarin-induced skin necrosis is seen in patients with congenital or acquired deficiencies of protein C or protein S. Initiation of warfarin therapy in these patients produces a precipitous fall in plasma levels of proteins C or S, thereby eliminating this important anticoagulant pathway before warfarin exerts an antithrombotic effect through lowering of the functional levels of factor × and prothrombin. The resultant procoagulant state triggers thrombosis. Why the thrombosis is localized to the microvasculature of fatty tissues is unclear.
Treatment involves discontinuation of warfarin and reversal with vitamin K, if needed. An alternative anticoagulant, such as heparin or LMWH, should be given in patients with thrombosis. Protein C concentrate can be given to protein C–deficient patients to accelerate healing of the skin lesions; fresh-frozen plasma may be of value if protein C concentrate is unavailable and for those with protein S deficiency. Occasionally, skin grafting is necessary when there is extensive skin loss.
Because of the potential for skin necrosis, patients with known protein C or protein S deficiency require overlapping treatment with a parenteral anticoagulant when initiating warfarin therapy. Warfarin should be started in low doses in these patients, and the parenteral anticoagulant should be continued until the INR is therapeutic for at least 2–3 consecutive days.
Pregnancy Warfarin crosses the placenta and can cause fetal abnormalities or bleeding. The fetal abnormalities include a characteristic embryopathy, which consists of nasal hypoplasia and stippled epiphyses. The risk of embryopathy is highest if warfarin is given in the first trimester of pregnancy. Central nervous system abnormalities can also occur with exposure to warfarin at any time during pregnancy. Finally, maternal administration of warfarin produces an anticoagulant effect in the fetus that can cause bleeding. This is of particular concern at delivery when trauma to the head during passage through the birth canal can lead to intracranial bleeding. Because of these potential problems, warfarin is contraindicated in pregnancy, particularly in the first and third trimesters. Instead, heparin, LMWH, or fondaparinux can be given during pregnancy for prevention or treatment of thrombosis.
Warfarin does not pass into the breast milk. Consequently, warfarin can safely be given to nursing mothers.
Special problems Patients with a lupus anticoagulant and those who need urgent or elective surgery present special challenges. Although observational studies suggested that patients with thrombosis complicating the antiphospholipid antibody syndrome required higher intensity warfarin regimens to prevent recurrent thromboembolic events, two randomized trials showed that targeting an INR of 2.0–3.0 is as effective as higher intensity treatment and produces less bleeding. Monitoring warfarin therapy can be problematic in patients with antiphospholipid antibody syndrome if the lupus anticoagulant prolongs the baseline INR; factor × levels can be used instead of the INR in such patients.
There is no need to stop warfarin before procedures associated with a low risk of bleeding; these include dental cleaning, simple dental extraction, cataract surgery, or skin biopsy. For procedures associated with a moderate or high risk of bleeding, warfarin should be stopped 5 days before the procedure to allow the INR to return to normal levels. Patients at high risk for thrombosis, such as those with mechanical heart valves, can be bridged with once- or twice-daily SC injections of LMWH when the INR falls to <2.0. The last dose of LMWH should be given 12–24 h before the procedure, depending on whether LMWH is administered twice or once daily. After the procedure, treatment with warfarin can be restarted.
New Oral Anticoagulants New oral anticoagulants are now available as alternatives to warfarin. These include dabigatran, which targets thrombin, and rivaroxaban, apixaban, and edoxaban, which target factor Xa. All of these drugs have a rapid onset and offset of action and have half-lives that permit once- or twice-daily administration. Designed to produce a predictable level of anticoagulation, the new oral agents are more convenient to administer than warfarin because they are given in fixed doses without routine coagulation monitoring.
MECHANISM OF ACTION The new oral anticoagulants are small molecules that bind reversibly to the active site of their target enzyme. Table 143-9 summarizes the distinct pharmacologic properties of these agents.
|
COMPARISON OF THE PHARMACOLOGIC PROPERTIES OF THE NEW ORAL ANTICOAGULANTS |
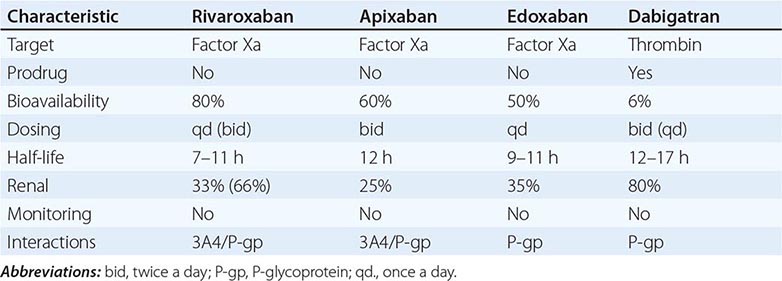
INDICATIONS The new oral anticoagulants have been compared with warfarin for stroke prevention in patients with nonvalvular atrial fibrillation in four randomized trials that enrolled 71,683 patients. A meta-analysis of these data demonstrates that compared with warfarin, the new agents significantly reduce stroke or systemic embolism by 19% (p = .001), primarily driven by a 51% reduction in hemorrhagic stroke (p <.0001), and are associated with a 10% reduction in mortality (p <.0001). New oral anticoagulants reduce intracranial hemorrhage by 52% compared with warfarin (p <.0001), but increase gastrointestinal bleeding by about 24% (p = .04). Overall, the new agents demonstrate a favorable benefit-to-risk profile compared with warfarin, and their relative efficacy and safety are maintained across a wide spectrum of atrial fibrillation patients, including those over the age of 75 years and those with a prior history of stroke. Based on these findings, dabigatran, rivaroxaban, and apixaban are licensed as alternatives to warfarin for stroke prevention in nonvalvular atrial fibrillation, and edoxaban is under regulatory consideration for this indication. Nonvalvular atrial fibrillation is defined as that occurring in patients without mechanical heart valves or severe rheumatic valvular disease, particularly mitral stenosis and/or regurgitation.
Dabigatran, rivaroxaban, and apixaban have been compared with enoxaparin for thromboprophylaxis after elective hip or knee arthroplasty. Currently, only rivaroxaban and apixaban are licensed for this indication in the United States. Rivaroxaban and dabigatran are also licensed for treatment of DVT or PE. Apixaban and edoxaban have also been investigated for treatment of patients with venous thromboembolism, but have not yet been approved for this indication. Rivaroxaban is licensed in Europe for prevention of recurrent ischemic events in patients who have been stabilized after an acute coronary syndrome. In this setting, rivaroxaban is usually administered in conjunction with dual antiplatelet therapy with aspirin and clopidogrel.
DOSING For stroke prevention in patients with nonvalvular atrial fibrillation, rivaroxaban is given at a dose of 20 mg once daily with a dose reduction to 15 mg once daily in patients with a creatinine clearance of 15–49 mL/min; dabigatran is given at a dose of 150 mg twice daily with a dose reduction to 75 mg twice daily in those with a creatinine clearance of 15–30 mL/min; and apixaban is given at a dose of 5 mg twice daily with a dose reduction to 2.5 mg twice daily for patients with a creatinine >1.5 g/dL, for those 80 years of age or older, or for patients who weigh <60 kg.
For thromboprophylaxis after elective hip or knee replacement surgery, rivaroxaban is given at a dose of 10 mg once daily, whereas apixaban is given at a dose of 2.5 mg twice daily. For treatment of patients with DVT or PE, rivaroxaban is started at a dose of 15 mg twice daily for 3 weeks; the dose is then reduced to 20 mg once daily thereafter. After a minimum of a 5 day course of treatment with heparin or LMWH, dabigatran is given at a dose of 150 mg twice daily.
MONITORING Although designed to be administered without routine monitoring, there are situations where determination of the anticoagulant activity of the new oral anticoagulants can be helpful. These include assessment of adherence, detection of accumulation or overdose, identification of bleeding mechanisms, and determination of activity prior to surgery or intervention. For qualitative assessment of anticoagulant activity, the prothrombin time can be used for factor Xa inhibitors and the aPTT for dabigatran. Rivaroxaban and edoxaban prolong the prothrombin time more than apixaban. In fact, because apixaban has such a limited effect on the prothrombin time, anti-factor Xa assays are needed to assess its activity. The effect of the drugs on tests of coagulation varies depending on the time that the blood is drawn relative to the timing of the last dose of the drug and the reagents used to perform the tests. Chromogenic anti-factor Xa assays and a dilute thrombin clotting time with appropriate calibrators provide quantitative assays to measure the plasma levels of the factor Xa inhibitors and dabigatran, respectively.
SIDE EFFECTS Like all anticoagulants, bleeding is the most common side effect of the new oral anticoagulants. The new agents are associated with less intracranial bleeding than warfarin. The increased risk of intracranial bleeding with warfarin likely reflects the reduction in functional levels of factor VII, which precludes efficient thrombin generation at sites of microvascular bleeding in the brain. Because the new oral anticoagulants target downstream coagulation enzymes, they produce less impairment of hemostatic plug formation at sites of vascular injury.
A downside of the new oral anticoagulants is the increased risk of gastrointestinal bleeding. This likely occurs because unabsorbed active drug in the gut exacerbates bleeding from lesions. Although dabigatran etexilate is a prodrug, only 7% is absorbed. Although the remainder passes through the gut, at least two-thirds is metabolically activated to dabigatran by gut esterases.
Dyspepsia occurs in up to 10% of patients treated with dabigatran; this problem improves with time and can be minimized by administering the drug with food. Dyspepsia is rare with rivaroxaban, apixaban, and edoxaban.
PERIPROCEDURAL MANAGEMENT Like warfarin, the new oral anticoagulants must be stopped before procedures associated with a moderate or high risk of bleeding. The drugs should be held for 1–2 days, or longer if renal function is impaired. Assessment of residual anticoagulant activity before procedures associated with a high bleeding risk is prudent.
MANAGEMENT OF BLEEDING There are no specific antidotes for the new oral anticoagulants. With minor bleeding, holding one or two doses of drug is usually sufficient. The approach to serious bleeding is similar to that with warfarin except that vitamin K administration is of no benefit. Thus, the anticoagulant and antiplatelet drugs should be held, the patient should be resuscitated with fluids and blood products as necessary, and, if possible, the bleeding site should be identified and managed. Coagulation testing will determine the extent of anticoagulation. and renal function should be assessed so that the half-life of the drug can be calculated. Timing of the last dose of anticoagulant is important; administration of oral activated charcoal may help to prevent absorption of drug administered in the past 2–4 h. If bleeding continues or is life-threatening, procoagulants, such as prothrombin complex concentrate (either unactivated or activated) or factor VIIa, can be administered, although the evidence of their effectiveness is limited. Dialysis removes dabigatran from the circulation in patients with renal impairment; dialysis does not remove rivaroxaban, apixaban, or edoxaban because unlike dabigatran, these drugs are highly protein-bound.
PREGNANCY As small molecules, the new oral anticoagulants can all pass through the placenta. Consequently, these agents are contraindicated in pregnancy, and when used by women of childbearing potential, appropriate contraception is important.
ONGOING INVESTIGATIONS Although the lack of antidotes has created concern about the risk of bleeding events in patients taking the new oral anticoagulants, emerging postmarketing data suggest that the rates of bleeding in the real-world setting are similar to those reported in the trials. Nonetheless, specific antidotes are under development. These include a humanized mouse monoclonal antibody fragment against dabigatran and a recombinant variant of factor Xa that serves as a decoy for the oral factor Xa inhibitors. Neither agent is currently available for clinical use.
FIBRINOLYTIC DRUGS
ROLE OF FIBRINOLYTIC THERAPY
Fibrinolytic drugs can be used to degrade thrombi and are administered systemically or can be delivered via catheters directly into the substance of the thrombus. Systemic delivery is used for treatment of acute MI, acute ischemic stroke, and most cases of massive PE. The goal of therapy is to produce rapid thrombus dissolution, thereby restoring antegrade blood flow. In the coronary circulation, restoration of blood flow reduces morbidity and mortality rates by limiting myocardial damage, whereas in the cerebral circulation, rapid thrombus dissolution decreases the neuronal death and brain infarction that produce irreversible brain injury. For patients with massive PE, the goal of thrombolytic therapy is to restore pulmonary artery perfusion.
Peripheral arterial thrombi and thrombi in the proximal deep veins of the leg are most often treated using catheter-directed thrombolytic therapy. Catheters with multiple side holes can be used to enhance drug delivery. In some cases, intravascular devices that fragment and extract the thrombus are used to hasten treatment. These devices can be used alone or in conjunction with fibrinolytic drugs.
MECHANISM OF ACTION
Currently approved fibrinolytic agents include streptokinase; acylated plasminogen streptokinase activator complex (anistreplase); urokinase; recombinant tissue-type plasminogen activator (rtPA), which is also known as alteplase or activase; and two recombinant derivatives of rtPA, tenecteplase and reteplase. All of these agents act by converting plasminogen, the zymogen, to plasmin, the active enzyme (Fig. 143-7). Plasmin then degrades the fibrin matrix of thrombi and produces soluble fibrin degradation products.
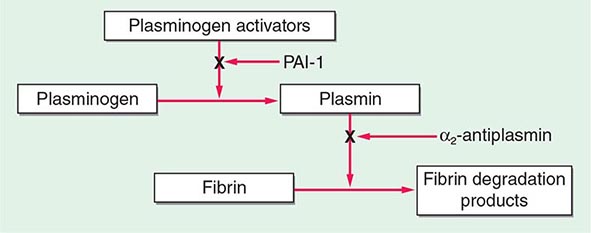
FIGURE 143-7 The fibrinolytic system and its regulation. Plasminogen activators convert plasminogen to plasmin. Plasmin then degrades fibrin into soluble fibrin degradation products. The system is regulated at two levels. Type 1 plasminogen activator inhibitor (PAI-1) regulates the plasminogen activators, whereas α2-antiplasmin serves as the major inhibitor of plasmin.
Endogenous fibrinolysis is regulated at two levels. Plasminogen activator inhibitors, particularly the type 1 form (PAI-1), prevent excessive plasminogen activation by regulating the activity of tPA and urokinase-type plasminogen activator (uPA). Once plasmin is generated, it is regulated by plasmin inhibitors, the most important of which is α2-antiplasmin. The plasma concentration of plasminogen is twofold higher than that of α2-antiplasmin. Consequently, with pharmacologic doses of plasminogen activators, the concentration of plasmin that is generated can exceed that of α2-antiplasmin. In addition to degrading fibrin, unregulated plasmin can also degrade fibrinogen and other clotting factors. This process, which is known as the systemic lytic state, reduces the hemostatic potential of the blood and increases the risk of bleeding.
The endogenous fibrinolytic system is geared to localize plasmin generation to the fibrin surface. Both plasminogen and tPA bind to fibrin to form a ternary complex that promotes efficient plasminogen activation. In contrast to free plasmin, plasmin generated on the fibrin surface is relatively protected from inactivation by α2-antiplasmin, a feature that promotes fibrin dissolution. Furthermore, C-terminus lysine residues, exposed as plasmin degrades fibrin, serve as binding sites for additional plasminogen and tPA molecules. This creates a positive feedback that enhances plasmin generation. When used pharmacologically, the various plasminogen activators capitalize on these mechanisms to a lesser or greater extent.
Plasminogen activators that preferentially activate fibrin-bound plasminogen are considered fibrin-specific. In contrast, nonspecific plasminogen activators do not discriminate between fibrin-bound and circulating plasminogen. Activation of circulating plasminogen results in the generation of unopposed plasmin that can trigger the systemic lytic state. Alteplase and its derivatives are fibrin-specific plasminogen activators, whereas streptokinase, anistreplase, and urokinase are nonspecific agents.
STREPTOKINASE
Unlike other plasminogen activators, streptokinase is not an enzyme and does not directly convert plasminogen to plasmin. Instead, streptokinase forms a 1:1 stoichiometric complex with plasminogen. Formation of this complex induces a conformational change in plasminogen that exposes its active site (Fig. 143-8). The streptokinase-plasminogen complex then converts additional plasminogen to plasmin.
FIGURE 143-8 Mechanism of action of streptokinase. Streptokinase binds to plasminogen and induces a conformational change in plasminogen that exposes its active site. The streptokinase/plasmin(ogen) complex then serves as the activator of additional plasminogen.
Streptokinase has no affinity for fibrin, and the streptokinase- plasminogen complex activates both free and fibrin-bound plasminogen. Activation of circulating plasminogen generates sufficient amounts of plasmin to overwhelm α2-antiplasmin. Unopposed plasmin not only degrades fibrin in the occlusive thrombus but also induces a systemic lytic state.
When given systemically to patients with acute MI, streptokinase reduces mortality. For this indication, the drug is usually given as an IV infusion of 1.5 million units over 30–60 min. Patients who receive streptokinase can develop antibodies against the drug, as can patients with prior streptococcal infection. These antibodies can reduce the effectiveness of streptokinase.
Allergic reactions occur in ~5% of patients treated with streptokinase. These may manifest as a rash, fever, chills, and rigors. Although anaphylactic reactions can occur, these are rare. Transient hypotension is common with streptokinase and has been attributed to plasmin-mediated release of bradykinin from kininogen. The hypotension usually responds to leg elevation and administration of IV fluids and low doses of vasopressors, such as dopamine or norepinephrine.
ANISTREPLASE
To generate this drug, streptokinase is combined with equimolar amounts of Lys-plasminogen, a plasmin-cleaved form of plasminogen with a Lys residue at its N terminus. The active site of Lys-plasminogen that is exposed upon combination with streptokinase is then masked with an anisoyl group. After IV infusion, the anisoyl group is slowly removed by deacylation, giving the complex a half-life of ~100 min. This allows drug administration via a single bolus infusion.
Although it is more convenient to administer, anistreplase offers few mechanistic advantages over streptokinase. Like streptokinase, anistreplase does not distinguish between fibrin-bound and circulating plasminogen. Consequently, it too produces a systemic lytic state. Likewise, allergic reactions and hypotension are just as frequent with anistreplase as they are with streptokinase.
When anistreplase was compared with alteplase in patients with acute MI, reperfusion was obtained more rapidly with alteplase than with anistreplase. Improved reperfusion was associated with a trend toward better clinical outcomes and reduced mortality rate with alteplase. These results and the high cost of anistreplase have dampened the enthusiasm for its use.
UROKINASE
Urokinase is a two-chain serine protease derived from cultured fetal kidney cells with a molecular weight of 34,000. Urokinase converts plasminogen to plasmin directly by cleaving the Arg560-Val561 bond. Unlike streptokinase, urokinase is not immunogenic and allergic reactions are rare. Urokinase produces a systemic lytic state because it does not discriminate between fibrin-bound and circulating plasminogen.
Despite many years of use, urokinase has never been systemically evaluated for coronary thrombolysis. Instead, urokinase is often employed for catheter-directed lysis of thrombi in the deep veins or the peripheral arteries. Because of production problems, the availability of urokinase is limited.
ALTEPLASE
A recombinant form of single-chain tPA, alteplase has a molecular weight of 68,000. Alteplase is rapidly converted into its two-chain form by plasmin. Although single- and two-chain forms of tPA have equivalent activity in the presence of fibrin, in its absence, single-chain tPA has tenfold lower activity.
Alteplase consists of five discrete domains (Fig. 143-9); the N-terminus A chain of two-chain alteplase contains four of these domains. Residues 4 through 50 make up the finger domain, a region that resembles the finger domain of fibronectin; residues 50 through 87 are homologous with epidermal growth factor, whereas residues 92 through 173 and 180 through 261, which have homology to the kringle domains of plasminogen, are designated as the first and second kringle, respectively. The fifth alteplase domain is the protease domain; it is located on the C-terminus B chain of two-chain alteplase.
FIGURE 143-9 Domain structures of alteplase (tPA), tenecteplase (TNK-tPA), desmoteplase (b-PA), and reteplase (r-PA). The finger (F), epidermal growth factor (EGF), first and second kringles (K1 and K2, respectively), and protease (P) domains are illustrated. The glycosylation site (Y) on K1 has been repositioned in tenecteplase to endow it with a longer half-life. In addition, a tetra-alanine substitution in the protease domain renders tenecteplase resistant to type 1 plasminogen activator inhibitor (PAI-1) inhibition. Desmoteplase differs from alteplase and tenecteplase in that it lacks a K2 domain. Reteplase is a truncated variant that lacks the F, EGF, and K1 domains.
The interaction of alteplase with fibrin is mediated by the finger domain and, to a lesser extent, by the second kringle domain. The affinity of alteplase for fibrin is considerably higher than that for fibrinogen. Consequently, the catalytic efficiency of plasminogen activation by alteplase is two to three orders of magnitude higher in the presence of fibrin than in the presence of fibrinogen. This phenomenon helps to localize plasmin generation to the fibrin surface.
Although alteplase preferentially activates plasminogen in the presence of fibrin, alteplase is not as fibrin-selective as was first predicted. Its fibrin specificity is limited because like fibrin, (DD)E, the major soluble degradation product of cross-linked fibrin, binds alteplase and plasminogen with high affinity. Consequently, (DD)E is as potent as fibrin as a stimulator of plasminogen activation by alteplase. Whereas plasmin generated on the fibrin surface results in thrombolysis, plasmin generated on the surface of circulating (DD)E degrades fibrinogen. Fibrinogen degradation results in the accumulation of fragment X, a high-molecular-weight clottable fibrinogen degradation product. Incorporation of fragment × into hemostatic plugs formed at sites of vascular injury renders them susceptible to lysis. This phenomenon may contribute to alteplase-induced bleeding.
A trial comparing alteplase with streptokinase for treatment of patients with acute MI demonstrated significantly lower mortality with alteplase than with streptokinase, although the absolute difference was small. The greatest benefit was seen in patients age <75 years with anterior MI who presented <6 h after symptom onset.
For treatment of acute MI or acute ischemic stroke, alteplase is given as an IV infusion over 60–90 min. The total dose of alteplase usually ranges from 90 to 100 mg. Allergic reactions and hypotension are rare, and alteplase is not immunogenic.
TENECTEPLASE
Tenecteplase is a genetically engineered variant of tPA and was designed to have a longer half-life than tPA and to be resistant to inactivation by PAI-1. To prolong its half-life, a new glycosylation site was added to the first kringle domain (Fig. 143-9). Because addition of this extra carbohydrate side chain reduced fibrin affinity, the existing glycosylation site on the first kringle domain was removed. To render the molecule resistant to inhibition by PAI-1, a tetra-alanine substitution was introduced at residues 296–299 in the protease domain, the region responsible for the interaction of tPA with PAI-1.
Tenecteplase is more fibrin-specific than tPA. Although both agents bind to fibrin with similar affinity, the affinity of tenecteplase for (DD)E is significantly lower than that of tPA. Consequently, (DD)E does not stimulate systemic plasminogen activation by tenecteplase to the same extent as tPA. As a result, tenecteplase produces less fibrinogen degradation than tPA.
For coronary thrombolysis, tenecteplase is given as a single IV bolus. In a large phase III trial that enrolled >16,000 patients, the 30-day mortality rate with single-bolus tenecteplase was similar to that with accelerated-dose tPA. Although rates of intracranial hemorrhage were also similar with both treatments, patients given tenecteplase had fewer noncerebral bleeds and a reduced need for blood transfusions than those treated with tPA. The improved safety profile of tenecteplase likely reflects its enhanced fibrin specificity.
RETEPLASE
Reteplase is a is a single-chain, recombinant tPA derivative that lacks the finger, epidermal growth factor, and first kringle domains (Fig. 143-9). This truncated derivative has a molecular weight of 39,000. Reteplase binds fibrin more weakly than tPA because it lacks the finger domain. Because it is produced in Escherichia coli, reteplase is not glycosylated. This endows it with a plasma half-life longer than that of tPA. Consequently, reteplase is given as two IV boluses, which are separated by 30 min. Clinical trials have demonstrated that reteplase is at least as effective as streptokinase for treatment of acute MI, but the agent is not superior to tPA.
NEWER FIBRINOLYTIC AGENTS
Two new drugs are under investigation. These include desmoteplase (Fig. 143-9), a recombinant form of the full-length plasminogen activator isolated from the saliva of the vampire bat, and alfimeprase, a truncated form of fibrolase, an enzyme isolated from the venom of the southern copperhead snake. Clinical studies with these agents have been disappointing. Desmoteplase, which is more fibrin-specific than tPA, was investigated for treatment of acute ischemic stroke. Patients presenting 3–9 h after symptom onset were randomized to one of two doses of desmoteplase or to placebo. Overall response rates were low and no different with desmoteplase than with placebo. The mortality rate was higher in the desmoteplase arms.
Alfimeprase is a metalloproteinase that degrades fibrin and fibrinogen in a plasmin-independent fashion. In the circulation, alfimeprase is rapidly inhibited by α2-macroglobulin. Consequently, the drug must be delivered via a catheter directly into the thrombus. Studies of alfimeprase for treatment of peripheral arterial occlusion or for restoration of flow in blocked central venous catheters were stopped due to lack of efficacy. The disappointing results with desmoteplase and alfimeprase highlight the challenges of introducing new fibrinolytic drugs.
CONCLUSIONS AND FUTURE DIRECTIONS
Thrombosis involves a complex interplay among the vessel wall, platelets, the coagulation system, and the fibrinolytic pathways. Activation of coagulation also triggers inflammatory pathways that may exacerbate thrombosis. A better understanding of the biochemistry of blood coagulation and advances in structure-based drug design have identified new targets and resulted in the development of novel antithrombotic drugs. Well-designed clinical trials have provided detailed information on which drugs to use and when to use them. Despite these advances, however, thromboembolic disorders remain a major cause of morbidity and mortality. Therefore, the search for better targets and more potent antiplatelet, anticoagulant, and fibrinolytic drugs continues.