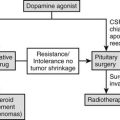
Clinically Nonfunctioning Sellar Masses
Many types of lesions present as a mass within or near the sella turcica (Table 9-1). The majority of sellar masses are pituitary adenomas, even those that are not obviously associated with clinical syndromes. The majority of these clinically nonfunctioning pituitary adenomas are gonadotroph adenomas, but some are relatively silent lactotroph, somatotroph, corticotroph, and thyrotroph adenomas. Determining whether a sellar mass is a pituitary adenoma or other type of sellar mass is important; and if it is a pituitary adenoma, the type of adenoma is significant, because that distinction will determine optimal treatment.
Table 9-1
Cysts (Rathke’s cleft, arachnoid, dermoid, epidermoid)
Infiltrative and granulomatous diseases (sarcoidosis, Langerhans cell histiocytosis, tuberculoma)
Natural History of a Pituitary or Parasellar Mass
Prior to the advent of sensitive pituitary imaging techniques, a wide spectrum of clinical sequelae were evident from the effects of an enlarging mass arising from within the pituitary or its adjacent structures (Table 9-2). Although it is today relatively uncommon for such a mass to be invasive at the time of diagnosis, the relative subtlety of clinical features may delay the anatomic imaging of such a mass.
Table 9-2
Local Neurologic Effects of a Pituitary or Hypothalamic Mass
| Impacted Structure | Clinical Effect |
| Optic tract | Loss of red perception, bitemporal hemianopsia, superior or bitemporal field defect, scotoma, blindness |
| Hypothalamus | Temperature dysregulation, appetite disorders, obesity, thirst disorders, diabetes insipidus, sleep disorders, behavioral dysfunction, autonomic nervous system dysfunction |
| Cavernous sinus | Ptosis, diplopia, ophthalmoplegia, facial numbness |
| Temporal lobe | Uncinate seizures |
| Frontal lobe | Personality disorder, anosmia |
| Central | Headache, hydrocephalus, psychosis, dementia, laughing seizures |
Types of Sellar Masses
Any pituitary adenoma type may first be recognized as a sellar mass, even those that usually cause recognizable clinical syndromes, such as corticotroph, somatotroph, and lactotroph adenomas. In some situations, the clinical syndrome (e.g., Cushing’s syndrome or acromegaly) is present but not recognized; in other situations, the clinical syndrome is relatively subtle or nonexistent. The latter have been referred to as “silent” adenomas.1–3 It is probably more accurate, however, to regard the clinical presentations of these adenomas as a spectrum from clinically obvious (e.g., frank acromegaly), to clinically subtle (e.g., slight increase in ring size but still overall normal appearance), to clinically silent (e.g., no clinical manifestations but a high IGF-1),4,5 to silent (e.g., normal IGF-1 but immunostaining of excised tissue for growth hormone) (Table 9-3).
Table 9-3
Spectrum of Functionality of Pituitary Adenomas
| Term | Description |
| Clinically obvious | Typical physical features of excessive hormonal hypersecretion |
| Clinically subtle | Subtle physical features of excessive hormonal hypersecretion |
| Clinically silent | Elevated serum concentration of pituitary hormone but not even subtle clinical manifestations |
| Silent | Type of adenoma identifiable only by immunostaining; normal serum concentration of hormone normally secreted by that cell type |
Pituitary Hyperplasia
Hyperplasia of anterior pituitary cells can result from several causes and results in generalized enlargement of the pituitary. During pregnancy, lactotroph hyperplasia occurs as the result of estrogen stimulation. When a target gland fails, the resulting lack of feedback inhibition on the corresponding pituitary trophic hormone-secreting cells results in hyperplasia. For example, thyrotroph hyperplasia occurs as a consequence of longstanding and untreated primary hypothyroidism, and gonadotroph hyperplasia occurs as a consequence of longstanding and untreated primary hypogonadism.6–9 When a neuroendocrine tumor secretes growth hormone–releasing hormone excessively, the result is somatotroph hyperplasia.10
Lymphocytic Hypophysitis
Lymphocytic infiltration of the pituitary usually occurs in late pregnancy or postpartum but can also be seen in women who are not pregnant and infrequently in men.11 It is characterized by headaches whose intensity is out of proportion to the size of the lesion and by hypopituitarism, in which adrenal insufficiency is unusually prominent.
Benign Tumors, Nonpituitary
Rathke’s Cleft and Other Cysts
During early embryogenesis, the anterior and intermediate lobes of the pituitary gland arise from Rathke’s pouch. If the pouch fails to obliterate, cystic remnants remain at the interface between the anterior and posterior pituitary lobes. These small cysts (<5 mm) are found in about 20% of pituitary glands at autopsy.12 Occasionally a pituitary adenoma may also contain small cleft cysts.13 The imaging of these cysts on MRI reveals hyperdense or hypodense masses on either TI or T2 images. CT scan reveals the presence of homogenous hypodense areas that may allow differentiation from pituitary adenomas.14 Other sellar cysts include arachnoid, epidermoid, and dermoid cysts. Although these lesions develop mainly in the cerebellopontine angle, they may also occur in the suprasellar region. Clinical features of compression include internal hydrocephalus, visual disturbances, and rarely growth hormone or ACTH deficiency, hyperprolactinemia, and diabetes insipidus.15–17 Rarely, a squamous cell carcinoma may develop in the cyst.18
Craniopharyngiomas
Craniopharyngiomas19 are solid or mixed solid-cystic tumors that arise from remnants of Rathke’s pouch, either intrasellar or suprasellar. About half present clinically during childhood and adolescence, but some do not present until age 70 or 80 years. The major presenting symptoms are growth retardation in children and abnormal vision in adults. Anterior pituitary hormonal deficiencies and diabetes insipidus are also common. MRI often reveals a heterogeneous signal, and CT scan often shows calcifications. When cut, they often ooze a viscous fluid described as looking like “crankcase oil.” Histologically, they show their epithelial origin, either an adamantinomatous or papillary pattern.
The endocrine manifestations of craniopharyngioma usually result from partial or complete pituitary hormonal deficiencies. Growth hormone deficiency, with resultant short stature in childhood, diabetes insipidus, and other anterior pituitary hormonal deficiencies are common. Compression of the pituitary stalk or damage to the dopaminergic neurons in the hypothalamus result in hyperprolactinemia, which sometimes leads to misdiagnosis of a craniopharyngioma as a lactotroph adenoma. Although imaging may not easily distinguish the two lesions,20 a highly asymmetrical mass (especially with preferential posterior or dorsal extension) that does not shrink in response to dopamine agonist treatment should arouse suspicion of craniopharyngioma. A decrease in serum prolactin in response to dopamine agonist treatment, however, does not distinguish between the two. Thus craniopharyngioma may mimic a lactotroph adenoma in imaging, presence of hyperprolactinemia, and response of hyperprolactinemia to dopamine agonist treatment.
The treatment of these lesions is radical surgery, radiotherapy, or a combination of these modalities.21,22 In selected centers, stereotactic irradiation of the mass has been performed with some success. Nevertheless, regardless of which form of therapy is chosen, the ablation of the mass invariably results in anterior and/or posterior pituitary hormone deficits. Postoperative recurrence may occur in about a fifth of patients who undergo radical surgical excision,23 while no appreciable difference is noted in the outcome in those who undergo a subtotal surgical excision followed by radiotherapy. The presence of pure papillary squamous cellular elements may portend a higher surgical recurrence rate.24 The long-term effects of childhood irradiation for these tumors are considered elsewhere.
Meningiomas
These are usually benign and arise from the meninges anywhere within the head. About 20% arise near the sella,25 causing visual impairment and hormonal deficiencies. By MRI, meningiomas typically emit a low signal on T1-weighted images and a high signal on T2-weighted images and exhibit intense enhancement after gadolinium.
Pituicytomas
Pituicytomas are rare, benign tumors that arise from pituicytes,26 which are glial cells of the posterior pituitary. They have no hormonal secretory function and can be diagnosed only histologically by the characteristic histologic pattern of elongated cells in bundles and immunostaining for cell adhesion molecules.
Malignant Tumors
Germ Cell Tumors (Ectopic Pinealomas)
Suprasellar germ cell tumors27 usually occur through the third decade of life and are histologically and biologically similar to germ cell tumors in other anatomic locations, such as germinomas, teratomas, embryonal carcinomas, and choriocarcinomas. They may present with headache, nausea, vomiting, and lethargy (from increased intracranial pressure in patients with pineal lesions), diplopia, hypopituitarism or diabetes insipidus (with suprasellar tumors), and paralysis of upward conjugate gaze. If the tumor is in the pineal, imaging will show a mass in the third ventricle; if the tumor is in the infundibulum, it will be thickened.28 Serum concentrations of human chorionic gonadotropin beta (β-hCG), and/or alpha fetoprotein (AFP) may be increased. Although these lesions are highly malignant and metastasize readily, they are also highly radiosensitive.
Chordomas
Chordomas are slowly growing malignancies that arise from notochord remnants. Chordomas that arise in the clivus, the bone that is the base of the sella turcica, may present with headaches, visual impairment, and anterior pituitary hormonal deficiencies. MRI often shows a heterogeneous sellar mass associated with osteolytic bony erosion and calcification that may or may not be distinct from the normal pituitary. Histologically, they exhibit markers for epithelial cells, including cytokeratin and vimentin. After surgical excision, local invasion and recurrence commonly occur; mean patient survival is about 5 years. Rarely, chordomas become sarcomatous, sometimes after single fraction gamma- or proton-beam radiation,29,30 and then they are more aggressive.
Primary Lymphoma
Primary central nervous system (CNS) lymphoma sometimes involves the pituitary and hypothalamus. A review of 13 patients with pituitary involvement noted neurologic symptoms, including headaches and visual and oculomotor impairment, and/or deficiencies of anterior pituitary hormones and vasopressin.31 MRI shows a sellar mass with variable extrasellar extension.
Metastatic Disease
Metastases to the hypothalamus and pituitary gland occur most commonly with breast cancer in women and lung cancer in men but are encountered with other cancers.32,33 Symptoms include diabetes insipidus, anterior pituitary dysfunction, visual field defects, retro-orbital pain, and ophthalmoplegia.32,34 Reflecting that these are patients with metastatic disease, survival is not long; in 36 patients in one series, average survival was 6 months.33 Up to one quarter of patients with metastatic breast cancer have pituitary metastases. Interestingly, symptomatic pituitary metastases may be the presenting sign of previously undiscovered malignancy and even of malignancy of unknown origin. Although anterior pituitary failure is rare, an isolated metastatic deposit in the pituitary stalk without involvement of the anterior lobe may also present with pituitary failure. Metastases to the posterior pituitary lobe are far more common. About 15% of patients with diabetes insipidus harbor metastases from extrapituitary sources. Unfortunately, imaging of the pituitary mass does not distinguish these deposits from a pituitary adenoma unless extensive bony erosion is present. In fact, metastatic pituitary lesions may masquerade as a pituitary adenoma. In several instances, the diagnosis of pituitary metastasis will be made only by histologic study of the specimen removed at transsphenoidal surgery.
Pituitary Carcinoma
Carcinoma arising from anterior pituitary cells is quite rare. When it does occur, the malignancy can arise from any anterior pituitary cell type. Lactotroph,35 somatotroph, corticotroph,35 thyrotroph,36 and gonadotroph37 carcinomas have been reported. Diagnosis is made by finding a distant extracranial metastasis.
Infiltrative and Granulomatous Diseases
Infiltrative diseases, such as sarcoidosis and Langerhans cell histiocytosis, may cause a sellar mass and often cause widening of the pituitary stalk. Tuberculomas of the sellar region may occur as part of systemic infection or may be isolated to this region. Because these lesions primarily affect the hypothalamus and infundibulum, diabetes insipidus is common, whereas anterior pituitary hormone deficiencies are less frequently encountered. Infiltrative sarcoidosis of the hypothalamic-pituitary unit occurs in most patients with central nervous system sarcoid involvement.38 Typically these patients present with varying degrees of anterior pituitary failure with or without diabetes insipidus.
Abscess
Pituitary abscesses, which are rare, can occur in a normal or diseased pituitary gland. In immunocompromised subjects, they may be caused by fungi (Aspergillus, Nocardia, or Candida albicans) or Pneumocystis carinii. In a series of 24 patients, 16 presented with symptoms and physical findings consistent with a pituitary mass, while only 8 had features suggestive of infection, such as fever, leukocytosis, meningismus.39 MRI is usually unable to distinguish between pituitary abscess and pituitary adenoma, so most patients are diagnosed at the time of surgical exploration.
Etiology of Pituitary Adenomas
All pituitary adenomas appear to be true neoplasms, arising from a somatic mutation of a single progenitor cell that divides repetitively. The evidence for this view comes from studies that show that virtually all pituitary adenomas are monoclonal, that is, arise from a somatic mutation of a single cell. In one study of five women whose pituitary macroadenomas expressed some combination of FSHβ, LHβ, and α subunit and whose peripheral leukocytes were heterozygous for HPRT, the adenomas had predominantly one allele or the other, but not both40 (Fig. 9-1). This study suggests that gonadotroph adenomas arise from a somatic mutation of a single progenitor cell that then proliferates. Other studies present similar evidence that other types of pituitary adenomas are also clonal.41
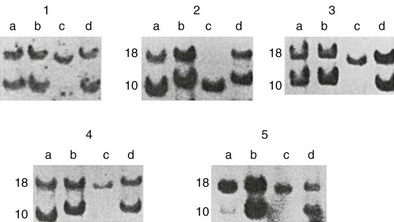
FIGURE 9-1 Demonstration of clonality of pituitary adenomas. Southern blots from extracts of gonadotroph adenomas (c lanes) and peripheral leukocytes (d lanes) of five women who were heterozygous for the HPRT gene. The peripheral leukocytes from all five women expressed both alleles of the HPRT gene, but the adenomas expressed only one allele or the other. (From Alexander JM, Biller BMK, Bikkal H, Zervas NT, Arnold A, Klibanski A: Clinically nonfunctioning pituitary tumors are monoclonal in origin. J Clin Invest 86:336–340, 1990.)
Specific mutations have been identified in association with hereditary pituitary adenomas in multiple endocrine neoplasia type I (MEN 1), Carney complex, and familial isolated acromegaly associated with mutations of aryl hydrocarbon receptor interacting protein (AIP). In MEN 1,42 a mutation of the MEN1 gene results in decreased expression of the tumor suppressor gene menin and development of adenomas of the pituitary, parathyroids, and pancreas. All pituitary adenoma types can occur in MEN 1, most commonly lactotroph and somatotroph adenomas, and rarely including gonadotroph adenomas37,43 and those identified only as clinically nonfunctioning.44–46 In the Carney complex, about half the patients have germ line inactivating mutations in the regulatory subunit type I of the c-AMP-dependent protein kinase A gene (PRKARIA).47 The resulting phenotype consists of somatotroph adenomas, myxomas of the heart, skin, and breast, spotty skin pigmentation (multiple skin lentigines and blue nevi), schwannomas, ovarian cysts, and adrenal, testicular, and thyroid tumors. Mutations of AIP have been found in familial acromegaly in Finland48 but infrequently in familial acromegaly in other countries.
About 40% of somatotroph adenomas are associated with mutations of the gene encoding the alpha subunit of the G stimulatory protein (Gsα), and as a consequence, constitutively activating adenylyl cyclase and increasing cAMP, which is mitogenic to somatotroph cells, thereby resulting in somatotroph adenomas.49 Mutations that cause other pituitary adenomas, including gonadotroph adenomas, are not known. Investigators have searched for other mutations that might be causally related to development of other pituitary adenomas, but none of these has been clearly associated with the pathogenesis of any pituitary adenoma. Three genes have been identified that might be related to the pathogenesis of pituitary adenomas. One is the pituitary tumor transforming gene (PTTG), which was cloned from GH4 cells, a rat pituitary tumor cell line.50 It is overexpressed in the majority of human pituitary adenomas of all cell types compared with nonadenomatous pituitary tissue.51 Another is a truncated form of the fibroblast growth factor receptor 4, which has been identified in all types of human pituitary adenomas. A third is the MEG3 tumor suppressor gene, expression of which is selectively lost in nonfunctioning adenomas by hypermethylation.52
External hormonal stimulation from the hypothalamus is unlikely to be a primary cause of gonadotroph adenomas but might have a secondary effect on adenoma growth and probably has an effect on adenoma secretion, since administration of the GnRH antagonist Nal-Glu GnRH to patients who have gonadotroph adenomas and supranormal serum FSH concentrations lowers FSH levels to normal.53
Clinical Features
Clinically nonfunctioning sellar masses, by definition, do not cause florid syndromes of hormonal excess and as a result often grow unrecognized until they become so large as to cause neurologic symptoms, including abnormalities of vision and oculomotor function (see Table 9-2).
Endocrinologic Features
Hormonal Excess—Gonadotroph Adenomas
Serum Concentrations of Gonadotropins and Their Subunits: Although gonadotroph adenomas are typically clinically nonfunctioning, because they secrete inefficiently and because their secretory products—intact gonadotropins and their subunits—usually do not cause a clinical syndrome, they often can be identified by their basal and stimulated secretory products (Table 9-4). Gonadotroph adenomas often basally secrete sufficient intact FSH to result in a supranormal serum FSH concentration. In a series of 38 men who had clinically nonfunctioning pituitary adenomas, 10 had supranormal serum FSH concentrations.54 The degree of FSH elevation may range from minimal to 10 times the upper limit of normal. Intact FSH secreted by gonadotroph adenomas appears to be normal or nearly normal in size,55 charge,56 and biological activity in vitro.57 In contrast, gonadotroph adenomas uncommonly produce supranormal serum concentrations of intact LH, but when they do, the serum testosterone concentration is elevated.58–60 About 15% of men who have gonadotroph adenomas have supranormal basal serum concentrations of gonadotropin subunits α, FSHβ, or LHβ.54 Administration of synthetic TRH to patients who have gonadotroph adenomas often produces an increase in the serum concentrations of intact gonadotropins and their subunits, especially of the LHβ subunit.54,61

Clinical Syndromes: Gonadotroph adenomas sometimes result in recognizable clinical syndromes (Table 9-5). One syndrome that is being recognized with increasing frequency is ovarian hyperstimulation when a gonadotroph adenoma secretes intact FSH.62–66 Continuous secretion of FSH by the adenoma, in contrast to cyclical secretion by normal gonadotroph cells, results in very large ovaries, oligomenorrhea, and multiple large cysts and widened endometrial stripe, all detected by pelvic ultrasound (Fig. 9-2). This clinical picture can be mistaken for polycystic ovarian syndrome, but administration of a superactive GnRH analog to a patient with the gonadotroph adenoma results in increased, rather than decreased, FSH secretion and ovarian size and function.67 Typically in these patients, the serum FSH concentration is elevated and the LH concentration is suppressed, and the concentrations of α subunit and estradiol are elevated. The estradiol concentration is often higher than 500 pg/mL and sometimes as high as 2000 pg/mL. Excision of the gonadotroph adenoma can lead to restoration of normal gonadotropin secretion and ovarian function, and pregnancy can occur.67,68
Table 9-5
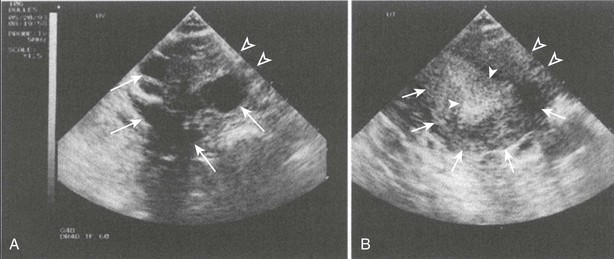
FIGURE 9-2 Ultrasound of ovaries (A) and uterus (B) in a 39-year-old woman who had a gonadotroph adenoma secreting FSH and causing ovarian hyperstimulation. In A, the closed arrows indicate large ovarian cysts. In B, the thin arrows indicate the uterus, and the wide arrows indicate the thickened endometrium. The distance between the open arrows is 1 cm. (From Djerassi A, Coutifaris C, West VA, Asa SL, Kapoor SC, Pavlou SN, Snyder PJ: Gonadotroph adenoma in a premenopausal woman secreting follicle-stimulating hormone and causing ovarian hyperstimulation. J Clin Endocrinol Metab 80:591–594, 1995.)
Other clinical presentations of gonadotroph adenomas are less common. One is pituitary apoplexy following GnRH or GnRH analog administration to patients with a gonadotroph adenoma. Recent reports describe discovery of previously unrecognized gonadotroph adenomas when GnRH superactive analogs were administered to treat prostate cancer.69–71 Enlargement of a gonadotroph adenoma, without apoplexy, was also reported when a superactive GnRH analog was administered for prostate cancer.72 Another presentation is large testicular size in a hypogonadal man. Yet another presentation is premature puberty in a boy whose gonadotroph adenoma secretes intact LH.73,74
Hormonal Excess—Other Pituitary Adenomas
Although corticotroph, somatotroph, and lactotroph adenomas often secrete efficiently and therefore usually result in classic clinical syndromes, a minority of these adenomas secrete inefficiently and do not cause a classic clinical syndrome and therefore present as a clinically silent sellar mass. In some of these, subtle features of the syndrome can be detected, and in others no clinical features can be detected, but the serum concentration of the pituitary or target gland hormone is elevated. The latter are termed clinically silent adenomas.4,5
Diagnosis
Utility of Diagnosing A Clinically Nonfunctioning Sellar Mass
Identifying the specific type of sellar mass is desirable in distinguishing a pituitary from a nonpituitary lesion and in providing a marker by which to monitor the treatment response. Distinguishing the lesion as of pituitary rather than nonpituitary origin is of value because it influences treatment. If surgery is indicated, for example, a pituitary lesion is almost always approached transsphenoidally, no matter how large, because it is situated below the diaphragm sella, but a meningioma should be approached transcranially if it arises above the diaphragm sella. If the lesion is a somatotroph adenoma, identified by an elevated serum concentration of IGF-1, medical treatment may be an option. Finding a tumor marker such as an elevated IGF-1, characteristic of a somatotroph adenoma, or an elevated serum FSH, characteristic of a gonadotroph adenoma, not only identifies the lesion as of somatotroph or gonadotroph origin but also provides a means by which to follow the response to treatment. For example, when the serum FSH concentration is elevated prior to surgery, the decrease after surgery correlates with reduction in adenoma mass seen by imaging.75
Imaging of The Sellar Region
Magnetic resonance imaging is currently the best imaging technique for the sellar region. If MRI shows a lesion that is clearly above the sella and distant from the pituitary, it is safe to say it is not a pituitary lesion, but for a lesion within the sella, with or without extrasellar extension, MRI does not distinguish a pituitary adenoma from other sellar lesions or one kind of pituitary adenoma from another (Fig. 9-3). Some MRI features suggest a greater likelihood of one type of lesion than another, but none is pathognomonic. For example, a sellar mass with a thin rim of tissue that emits a strong signal after gadolinium administration and a core that emits little signal suggests a cystic lesion, such as a Rathke’s cleft cyst, but a largely cystic pituitary adenoma cannot be excluded. Another example is a sellar mass that is somewhat irregular in shape and emits a heterogeneous signal, which is typical of a craniopharyngioma, but a pituitary adenoma can also present similarly. A converse example is an invasive lesion of the clivus, which is typical of a chordoma, but some pituitary adenomas extend primarily into the clivus and therefore can be mistaken for a chordoma. Because of the limitations of imaging in distinguishing types of sellar lesions, endocrinologic testing is invariably necessary.
Endocrinologic Tests
Sellar mass lesions should be evaluated by measurement of serum concentrations of pituitary hormones and related target gland hormones to determine if the lesion is of pituitary or nonpituitary origin, and if pituitary, the cell of origin. A prolactin concentration above 100 ng/mL, and especially above 200 ng/mL, suggests a lactotroph adenoma76; an elevated IGF-1 concentration suggests a somatotroph adenoma even if the patient does not exhibit features of acromegaly; an elevated 24-hour urine cortisol suggests a corticotroph adenoma even if the patient does not exhibit features of hypercortisolemia; and an elevated serum T4 associated with a TSH value that is not suppressed suggests a thyrotroph adenoma.
Suspicion that the lesion is a gonadotroph adenoma depends on the absence of findings suggestive of another adenoma type, as well as the presence of specific combinations of intact gonadotropins and their subunits (see Table 9-4). The combinations differ somewhat in men and women. In a man who has a pituitary macroadenoma, elevated basal serum concentrations of intact gonadotropins and/or their subunits alone or in combination with responses of any of these to TRH is strong evidence that the adenoma is of gonadotroph origin. An elevated basal FSH concentration is common. In a woman of postmenopausal age, elevated basal serum concentrations of intact FSH or gonadotropin subunits are usually of little diagnostic value, because either the adenoma or the nonadenomatous postmenopausal gonadotroph cells could be the source. However, a gonadotroph adenoma is likely if intact FSH is markedly elevated but LH is not at all elevated, or if one of the gonadotropin subunits is distinctly elevated but intact FSH and LH are not elevated.61 More commonly, however, the diagnosis depends on finding an LHβ subunit response to TRH. In a woman of premenopausal age, ovarian hyperstimulation, including elevated serum estradiol concentration, as discussed earlier, elevated FSH out of proportion to LH levels, or elevated basal α subunit concentration all point to the gonadotroph nature of the sellar mass.
Gonadotroph adenomas can usually be readily distinguished from pituitary enlargement due to gonadotroph hyperplasia that results from longstanding primary hypogonadism. The pituitary enlargement seen with primary hypogonadism is not as prominent as that observed with gonadotroph adenomas at the time of presentation. In primary hypogonadism, LH as well as FSH is elevated, and neither intact gonadotropins nor their subunits respond to TRH.77
Histologic Evaluation
Even when the identity of a sellar mass cannot be determined in vivo, pathologic examination of excised tissue can usually make this identification. Pituitary adenomas have a characteristic appearance that differs from that of the normal pituitary and of other sellar masses. By light microscopy, pituitary adenoma cells do not exhibit the normal pituitary glandular pattern but are arranged in cords or sheets,78,79 sometimes interspersed with varying amounts of fibrous tissue. In an adenoma, cells are usually homogenously similar in size, but cell size varies considerably among adenomas. Immunostaining for pituitary hormones (GH, ACTH, prolactin, FSHβ, LHβ and TSHβ, and α) usually identifies the adenoma type.
Approach to The Patient With A Sellar Mass
The clinical approach to the patient harboring a pituitary mass is compounded by the observation that the incidence of incidental silent pituitary microadenomas discovered at autopsy is between 10% and 20%. Pituitary cysts, hemorrhages, and infarctions are also not uncommonly discovered at autopsy. With the widespread sensitive imaging techniques, asymptomatic pituitary lesions are being identified with increasing frequency.80 Pituitary abnormalities compatible with the diagnosis of pituitary microadenomas are detectable in about 10% of the normal adult population. Considering the differential diagnosis of the intrasellar mass discussed previously, and recognizing that most observed lesions represent pituitary adenomas, several issues should be considered in the management of these masses. Of particular concern is whether the mass is hormonally functional, and whether local mass effects are apparent at the time of diagnosis or develop in the future.80
Evaluation of pituitary mass function is important, as the onset of symptoms and signs related to disordered hormone secretion are often insidious and may remain unnoticed for years. Clinical evaluation for changes compatible with ACTH, GH, or PRL hypersecretion or hyposecretion may reveal long-term serious systematic complications, and each may require distinct therapeutic approaches.81 In the absence of clinical features of a humoral hypersecretory syndrome, recommendations for cost-effective laboratory screening are debatable. The incidence of hormone-secreting tumors in asymptomatic subjects with incidental pituitary masses is low, and low-grade asymptomatic hormone hypersecretion (e.g., for PRL or α subunits) carries questionable long-term risk.
In the absence of evidence for hormone oversecretion, the presence of (or potential for) local compressive effects must be considered. The risk for macroadenoma enlargement towards a compressive macroadenoma is low, so that a decision to operate may be confidently postponed. For hypothalamic or parasellar masses of uncertain origin, a histologic tissue examination may be the only direct approach to yielding an accurate diagnosis. Although distinguishing MRI or CT features may be helpful in the differential diagnosis of the nonpituitary sellar mass, the final diagnosis usually remains elusive until pathologic confirmation is obtained. The benefits of pituitary surgery must also be weighed carefully against the potential side effects,82 although endoscopic approaches may now facilitate safer access to sellar tissue for histologic diagnosis or resection.83 If surgery is not indicated, subsequent imaging studies can determine the lesion’s slow growth rate, if any. In the absence of tumor growth, the interval between scans may be prolonged, and surgery should not be recommended in these asymptomatic cases. When an incidentally asymptomatic macroadenoma is diagnosed, visual field and pituitary function should be comprehensively evaluated. If these are found to be normal, imaging should be repeated. Progressive enlargement or impingement of vital structures will indicate the need for surgical intervention.
Treatment
Surgery
Transsphenoidal surgery using the operating microscope replaced transcranial surgery in the 1970s as the preferred treatment for sellar masses thought to be below the diaphragm sella, because that approach allowed excision of more tissue with fewer serious complications. In the past decade, an increasing number of neurosurgeons have been using endoscopic surgery84–86 instead of or in addition to the operating microscope, and now several series report experience with more than 100 patients who have undergone a procedure involving endoscopy. Some surgeons employ the endoscope exclusively.87–89 Others use transsphenoidal surgery with the operative microscope for primary resection and then use an endoscope for assistance in extending surgery within and beyond the sella.90 Other techniques for guiding surgery are intraoperative MRI, to determine the extent of sellar mass remaining after initial debulking,91 and intraoperative Doppler to allow operating near and within the cavernous sinuses without damaging the internal carotid arteries.92 Neurosurgeons are using these and other techniques to extend the reach of surgery in and around the sella to include the cavernous sinuses and suprasellar regions. Some neurosurgeons are now operating on suprasellar lesions, such as craniopharyngiomas, via a transsphenoidal approach.93–95
Efficacy of Surgery
Efficacy of surgery for a sellar mass can be judged by the amount of the mass excised, reduction in serum concentrations of secretory products that were elevated before surgery, improvement in vision, and restoration of normal pituitary function. In one series of 230 patients whose visual fields were abnormal before transsphenoidal surgery, the fields improved in 73%, remained the same in 23%, and worsened in 4%.96 In another series of 113 pituitary adenomas that extended beyond the sella, 81% of those with visual field defects before transsphenoidal surgery experienced improvement in fields after surgery, 19% remained the same, and none worsened.97 In patients with gonadotroph adenomas who had elevated serum FSH concentrations before transsphenoidal surgery, improvement in vision was paralleled by a decrease in FSH hypersecretion.75 Few reports of endoscopic surgery describe the degree of mass removal or change in postoperative vision.
Complications of Surgery
Serious complications of transsphenoidal and endoscopic surgery are uncommon when the procedures are performed by surgeons who have great experience with these procedures but are greater when the adenoma is very large and the surgeon has performed fewer procedures. In a survey in which neurosurgeons were asked to report their own experience with transsphenoidal surgery (Table 9-6), serious complications reported by the 958 respondents included some that were serious, including carotid artery injury (1.1%), central nervous system injury (1.3%), loss of vision (1.8%), ophthalmoplegia (1.4%), hemorrhage or swelling of the residual tumor (2.9%), cerebrospinal fluid leak (3.9%), meningitis (1.5%), and death (0.9%).82 The chances of anterior pituitary insufficiencies (19.4%) and diabetes insipidus (17.8%) were higher. The incidence of each complication was higher among neurosurgeons who were less experienced. Among neurosurgeons who reported performing fewer than 200 transsphenoidal procedures, 1.2% of procedures resulted in death, but among neurosurgeons who reported performing more than 500 procedures, only 0.2% resulted in death. Although these results are based on retrospective self-reporting via questionnaire, they provide a broader assessment of complications of transsphenoidal surgery than that provided by individual pituitary surgeons,97,98 whose complication rates are closer to those of the most experienced group above.82
Table 9-6
Relationship of a Neurosurgeon’s Experience in Performing Transsphenoidal Surgery to the Rate of Complications
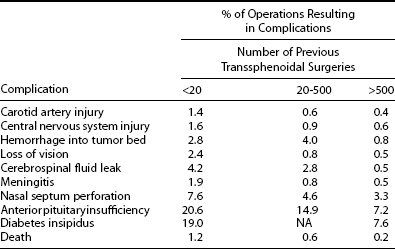
Data were collected from participating surgeons by questionnaire.
Adapted from Ciric I, Ragin A, Baumgartner C, Pierce D: Complications of transsphenoidal surgery: results of a national survey, review of the literature, and personal experience. Neurosurg 40:225–237, 1997.
An even broader assessment is provided by a study using the Nationwide Inpatient Sample, 1996-2000, of 5497 operations at 538 hospitals by 825 surgeons, which also demonstrated that surgeons who performed more transsphenoidal operations had fewer complications than those who performed fewer.99 Odds ratio for one or more complications of surgery or perioperative care was 0.76 for a fivefold larger case load per surgeon (95% confidence interval 0.65 to 0.89; P = 0.005). The lowest quartile of surgeons in this survey performed only one transsphenoidal procedure a year and the highest only eight or more per year, illustrating dramatically that this survey represented a much broader sampling than that of the primarily pituitary neurosurgeons above.
Complication rates are also greater in patients who have had prior pituitary surgery than in those who never had, and even greater in those whose prior surgery was via craniotomy than in those whose prior surgery was transsphenoidal.100 The types of complications that occur after endoscopic surgery are similar to those that occur after transsphenoidal surgery,87–89 but no reports directly compare the two approaches.
Radiation
Radiation therapy has been used to treat pituitary adenomas, including clinically nonfunctioning adenomas, for decades. The technique that had been used for much of this period employed a supervoltage source to deliver a total of 45 to 50 Gy in daily 2-Gy doses via three external portals. Much of our information about the long-term effects of radiation on the adenomas and surrounding tissues is based on patients treated by this technique. This technique has now been supplanted by techniques in which the radiation is delivered stereotactically to attempt to minimize the amount of radiation to which the brain is exposed. Current techniques employ radiation from one of several sources: protons from a cyclotron, high-energy x-rays from a linear accelerator, or gamma radiation from a 60Co source (Table 9-7). Radiation from a linear accelerator and cyclotron can be administered either as a single dose or multiple fractions over the course of several weeks. Single-dose techniques are often referred to, inappropriately, as “radiosurgery.” All of the techniques use computer-generated models based on magnetic resonance imaging, so that the radiation conforms to boundaries of the lesion.

Efficacy of Radiation
Studies of pre-stereotactic radiation administered following surgery for a pituitary macroadenoma generally showed efficacy in preventing regrowth of the adenoma.101–103 In one study of men who had radiation therapy following surgery for clinically nonfunctioning pituitary macroadenomas, only 7% of the 63 patients who received radiation following surgery developed new visual impairment requiring additional treatment during the subsequent 15 years, but 66% of the 63 who did not receive radiation developed new visual impairment.103
Several series have been reported in recent years describing the efficacy of stereotactic methods of radiation on preventing recurrence of pituitary adenomas and other sellar tumors. In series of patients treated with fractionated radiotherapy using a linear accelerator104,105 or proton beam106 and observed for a median of 40 months or more, adenoma size was reduced or stable in 90% to 100%. In a review of 25 studies involving 1621 patients, of whom 452 had clinically nonfunctioning pituitary adenomas and who were treated with single-dose radiation from a linear accelerator, gamma source, or proton beam, adenoma size was controlled in about 90% during a follow-up that was more than 40 months in half.107 In a report from a single center at which 100 clinically nonfunctioning adenomas were treated with single-dose radiation from a gamma source and followed for a median of 45 months, adenoma volume decreased or remained stable in 92% of patients.108
Complications of Radiation
The long-term side effects after pre-stereotactic radiation include hypopituitarism and neurologic deficits. Hypopituitarism, in several studies, began about a year or more after radiation, and by 10 years afterwards, about 50% of patients had a deficiency of ACTH, TSH, or LH.109–111 Neurologic side effects occurred less commonly. Blindness due to optic neuritis,112 brain tumors, and cerebrovascular accidents attributed to accelerated local atherosclerosis were reported as case reports and in some series,113,114 but other series have reported no neurologic sequelae.115 Although decreased cognitive function has been reported anecdotally after radiation, one systematic study did not confirm this effect.116
Although current radiotherapy techniques offer the theoretical advantage of targeting the tumor by stereotactic means, pituitary deficiencies and neurologic complications also occur with these techniques. In the series of 100 patients with clinically nonfunctioning adenomas treated with single-dose gamma radiation and followed for a median of 45 months, 20% developed new hypopituitarism.108 In the review of 35 studies involving 1621 patients, optic neuropathy occurred in about 1%, other cranial neuropathies in 1.3%, and parenchymal brain damage in about 0.8%.107
Pharmacologic Treatment
Several drugs have been administered in attempts to treat gonadotroph adenomas, but none has been found that reduces their size consistently and substantially. Although dopamine does not decrease gonadotropin secretion to an appreciable degree in normal subjects, bromocriptine has been reported to reduce the secretion of intact gonadotropins and α subunit in a few patients, and even to improve vision in one, but not to reduce adenoma size.117 CV 205-504 has been reported to reduce secretion and adenoma size in occasional patients.118 Cabergoline has been reported to reduce α subunit concentration in a single patient with a gonadotroph adenoma119 and to decrease adenoma volume by 10% to 18% in 7 of 13 other patients with gonadotroph adenomas.120
The somatostatin analog, octreotide, has been used to treat gonadotroph adenomas, because gonadotroph adenomas express somatostatin receptors and because of the demonstration that somatostatin itself may decrease secretion by gonadotroph adenomas in vitro. Although there have been occasional reports of dramatic decreases in size of gonadotroph adenomas associated with octreotide administration121,122 and some improvement in vision, the majority of patients have little if any improvement in adenoma size or vision.121–123 In a report of ten patients with clinically nonfunctioning adenomas, a combination of cabergoline and octreotide decreased adenoma size by 30% only in the six with evidence of secretory activity of FSH, LH, or their subunits.124
Several agonist analogs of GnRH have been administered to patients with gonadotroph adenomas, based on the rationale that chronic administration of these agonists causes down-regulation of GnRH receptors on, and decreased secretion of FSH and LH from, normal gonadotroph cells. Administration of GnRH agonist analogs to patients with gonadotroph adenomas, however, generally produces either an agonist effect or no effect on secretion and no effect on adenoma size.125,126 Administration for 1 week of the GnRH antagonist, Nal-Glu GnRH, to men with gonadotroph adenomas reduced their elevated FSH concentrations to normal.53 However, when Nal-Glu administration was continued for 6 months, although FSH remained suppressed, adenoma size did not decrease.127
Observation
Observation alone is a reasonable course even for patients who have sellar masses extending outside of the sella and elevating the optic chiasm, as long as the mass is not associated with neurologic symptoms, especially for patients whose surgical risk is high or who prefer not to have surgery until necessary. In a series of 40 patients with clinically nonfunctioning sellar masses, 24 “macro” and 16 “micro,” who were followed for a mean of 42 months, the 48-month probability of enlargement was 19% for the micro lesions and 44% for the macros.128 New or worse visual field defects were observed in 67% of the macro lesions that increased in size.
References
1. Horvath, E, Kovacs, K, Killinger, DW, et al. Silent corticotropic adenomas of the human pituitary gland. A histologic study, immunocytologic, and ultrastructural study. Am J Pathol. 1980;98:617–638.
2. Jouanneau, E, Ducluzeau, PH, Tilikete, C, et al. Should silent corticotroph-cell adenoma be classified as a non-functional pituitary adenoma? Neurochirurgie. 2001;47:128–132.
3. Klibanski, A, Zervas, NT, Kovacs, K, et al. Clinically silent hypersecretion of growth hormone in patients with pituitary tumors. J Neurosurg. 1987;66:806–811.
4. Sakharova, AA, Dimaraki, EV, Chandler, WF, et al. Clinically silent somatotropinomas may be biochemically active. J Clin Endocrinol Metab. 2005;90:2117–2121.
5. Yamada, S, Sano, T, Stefaneanu, L, et al. Endocrine and morphological study of a clinically silent somatotroph adenoma of the human pituitary. J Clin Endocrinol Metab. 1993;76:352–356.
6. Groff, TR, Shulkin, BL, Utiger, RD, et al. Amenorrhea-galactorrhea, hyperprolactinemia, and suprasellar pituitary enlargement as presenting features of primary hypothyroidism. Obstet Gynecol. 1984;63:86S.
7. Samaan, NA, Stephans, AV, Danziger, J, et al. Reactive pituitary abnormalities in patients with Klinefelter’s and Turner’s syndromes. Arch Intern Med. 1979;139:198–201.
8. Scheithauer, BW, Kovacs, K, Horvath, E, et al. The pituitary in Turner syndrome. Endocr Pathol. 2005;16:195–200.
9. Scheithauer, BW, Moschopulos, M, Kovacs, K, et al. The pituitary in Klinefelter syndrome. Endocr Pathol. 2005;16:133–138.
10. Thorner, MO, Perryman, RL, Cronin, MJ, et al. Somatotroph hyperplasia, successful treatment of acromegaly by removal of a pancreatic islet tumor secreting a growth hormone-releasing factor. J Clin Invest. 1982;70(5):965–977.
11. Thodou, E, Asa, SL, Kontogeorgos, G, et al. Lymphocytic hypophysitis: clinicopathologic findings. J Clin Endocrinol Metab. 1995;80:2302–2311.
12. el-Mahdy, W, Powell, M. Transsphenoidal management of 28 symptomatic Rathke’s cleft cysts, with special reference to visual and hormonal recovery. Neurosurgery. 1998;42:7–16.
13. Nishio, S, Mizuno, J, Barrow, DL, et al. Pituitary tumors composed of adenohypophysial adenoma and Rathke’s cleft cyst elements: a clinicopathological study. Neurosurgery. 1987;21:371–377.
14. Mukherjee, JJ, Islam, N, Kaltsas, G, et al. Clinical, radiological and pathological features of patients with Rathke’s cleft cysts: tumors that may recur. J Clin Endocrinol Metab. 1997;82:2357–2362.
15. Baskin, DS, Wilson, CB. Transsphenoidal treatment of non-neoplastic intrasellar cysts. A report of 38 cases. J Neurosurg. 1984;60:8–13.
16. Yamakawa, K, Shitara, N, Genka, S, et al. Clinical course and surgical prognosis of 33 cases of intracranial epidermoid tumors. Neurosurgery. 1989;24:568–573.
17. Lewis, AJ, Cooper, PW, Kassel, EE, et al. Squamous cell carcinoma arising in a suprasellar epidermoid cyst. Case report. J Neurosurg. 1983;59:538–541.
18. Schlachter, LB, Tindall, GT, Pearl, GS. Granular cell tumor of the pituitary gland associated with diabetes insipidus. Neurosurgery. 1980;6:418–421.
19. Karavitaki, N, Cudlip, S, Adams, CB, et al. Craniopharyngiomas. Endocr Rev. 2006;27:371–397.
20. Pigeau, I, Sigal, R, Halimi, P, et al. MRI features of craniopharyngiomas at 1.5 Tesla. A series of 13 cases. J Neuroradiol. 1988;15:276–287.
21. Yasargil, MG, Curcic, M, Kis, M, et al. Total removal of craniopharyngiomas. Approaches and long-term results in 144 patients. J Neurosurg. 1990;73:3–11.
22. Wen, BC, Hussey, DH, Staples, J, et al. A comparison of the roles of surgery and radiation therapy in the management of craniopharyngiomas. Int J Radiat Oncol Biol Phys. 1989;16:17–24.
23. Adamson, TE, Wiestler, OD, Kleihues, P, et al. Correlation of clinical and pathological features in surgically treated craniopharyngiomas. J Neurosurg. 1990;73:12–17.
24. Honegger, J, Buchfelder, M, Fahlbusch, R. Surgical treatment of craniopharyngiomas: endocrinological results. J Neurosurg. 1999;90:251–257.
25. Ciric, I, Rosenblatt, S. Suprasellar meningiomas. Neurosurgery. 2001;49:1372–1377.
26. Figarella-Branger, D, Dufour, H, Fernandez, C, et al. Pituicytomas, a mis-diagnosed benign tumor of the neurohypophysis: report of three cases. Acta Neuropathol. 2002;104:313–319.
27. Marsden, HB, Birch, JM, Swindell, R. Germ cell tumours of childhood: a review of 137 cases. J Clin Pathol. 1981;34:879–883.
28. Leger, J, Velasquez, A, Garel, C, et al. Thickened pituitary stalk on magnetic resonance imaging in children with central diabetes insipidus. J Clin Endocrinol Metab. 1999;84:1954–1960.
29. Hara, T, Kawahara, N, Tsuboi, K, et al. Sarcomatous transformation of clival chordoma after charged-particle radiotherapy. Report of two cases. J Neurosurg. 2006;105:136–141.
30. Tsuboi, Y, Hayashi, N, Kurimoto, M, et al. Malignant transformation of clival chordoma after gamma knife surgery—case report. Neurol Med Chir (Tokyo). 2007;47:479–482.
31. Giustina, A, Gola, M, Doga, M, et al. Clinical review 136: primary lymphoma of the pituitary: an emerging clinical entity. J Clin Endocrinol Metab. 2001;86:4567–4575.
32. Morita, A, Meyer, FB, Laws, ER, Jr. Symptomatic pituitary metastases. J Neurosurg. 1998;89:69–73.
33. Schubiger, O, Haller, D. Metastases to the pituitary-hypothalamic axis. Neuroradiology. 1992;34:131–134.
34. Fassett, DR, Couldwell, WT. Metastases to the pituitary gland. Neurosurg Focus. 2004;16:E8.
35. Pernicone, PJ, Scheithauer, BW, Sebo, TJ, et al. Pituitary carcinoma. A clinicopathologic study of 15 cases. Cancer. 1996;79:804–812.
36. Mixson, AJ, Friedman, TC, Katz, DA, et al. Thyrotropin-secreting pituitary carcinoma. J Clin Endocrinol Metab. 1993;76:529–533.
37. Benito, M, Asa, SL, Livolsi, VA, et al. Gonadotroph tumor associated with multiple endocrine neoplasia type 1. J Clin Endocrinol Metab. 2005;90:570–574.
38. Cannavo, S, Romano, C, Buffa, R, et al. Granulomatous sarcoidotic lesion of hypothalamic-pituitary region associated with Rathke’s cleft cyst. J Endocrinol Invest. 1997;20:77–81.
39. Vates, GE, Berger, MS, Wilson, CB. Diagnosis and management of pituitary abscess: a review of twenty-four cases. J Neurosurg. 2001;95:233–241.
40. Alexander, JM, Biller, BMK, Bikkal, H, et al. Clinically nonfunctioning pituitary tumors are monoclonal in origin. J Clin Invest. 1990;86:336–340.
41. Melmed, S. Mechanisms for pituitary tumorigenesis: the plastic pituitary. J Clin Invest. 2003;112(11):1603–1618.
42. Chandrasekhapappa, SC, Guru, SC, Manickam, P, et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science. 1997;276:404–407.
43. Sztal-Mazer, S, Topliss, DJ, Simpson, RW, et al. Gonadotroph adenoma in multiple endocrine neoplasia type 1. Endocr Pract. 2008;14:592–594.
44. Bassett, JH, Forbes, SA, Pannett, AA, et al. Characterization of mutations in patients with multiple endocrine neoplasia type 1. Am J Hum Genet. 1998;62:232–244.
45. Cebrian, A, Ruiz-Llorente, S, Cascon, A, et al. Mutational and gross deletion study of the MEN1 gene and correlation with clinical features in Spanish patients. J Med Genet. 2003;40:e72.
46. Verges, B, Boureille, F, Goudet, P, et al. Pituitary disease in MEN type 1 (MEN1): data from the France-Belgium MEN1 multicenter study. J Clin Endocrinol Metab. 2002;87:457–465.
47. Carney, JA, Gordon, H, Carpenter, PC, et al. The complex of myxomas, spotty pigmentation, and endocrine overactivity. Medicine. 1985;64:270–283.
48. Vierimaa, O, Georgitsi, M, Lehtonen, R, et al. Pituitary adenoma predisposition caused by germline mutations in the AIP gene. Science. 2006;312:1228–1230.
49. Spada, A, Arosio, M, Bochicchio, D, et al. Clinical, biochemical, and morphological correlates in patients bearing growth hormone–secreting pituitary tumors with or without constitutively active adenylyl cyclase. J Clin Endocrinol Metab. 1990;71:1421–1426.
50. Pei, L, Melmed, S. Isolation and characterization of a pituitary tumor transforming gene (PTTG). Mol Endocrinol. 1997;11:433–441.
51. Zhang, X, Horwitz, GA, Heaney, AP, et al. Pituitary tumor transforming gene (PTTG) expression in pituitary adenomas. J Clin Endocrinol Metab. 1999;84:761–767.
52. Gejman, R, Batista, DL, Zhong, Y, et al. Selective loss of MEG3 expression and intergenic differentially methylated region hypermethylation in the MEG3/DLK1 locus in human clinically nonfunctioning pituitary adenomas. J Clin Endocrinol Metab. 2008;93:4119–4125.
53. Daneshdoost, L, Pavlou, S, Molitch, ME. Inhibition of follicle-stimulating hormone secretion from gonadotroph adenomas by repetitive administration of a gonadotropin-releasing hormone antagonist. J Clin Endocrinol Metab. 1990;71:92–97.
54. Daneshdoost, L, Gennarelli, TA, Bashey, HM, et al. Identification of gonadotroph adenomas in men with clinically nonfunctioning adenomas by the LHβ subunit response to TRH. J Clin Endocrinol Metab. 1993;77:1352–1355.
55. Snyder, PJ, Bashey, HM, Kim, SU, et al. Secretion of uncombined subunits of luteinizing hormone by gonadotroph cell adenomas. J Clin Endocrinol Metab. 1984;59:1169–1175.
56. Chappel, SC, Bashey, HM, Snyder, PJ. Similar isoelectric profiles of FSH from gonadotroph cell adenomas and non-adenomatous pituitaries. Acta Endocrinol. 1986;113:311–316.
57. Galway, AB, Hsueh, JW, Daneshdoost, L, et al. Gonadotroph adenomas in men produce biologically active follicle-stimulating hormone. J Clin Endocrinol Metab. 1990;71:907–912.
58. Snyder, PJ, Sterling, FH. Hypersecretion of LH and FSH by a pituitary adenoma. J Clin Endocrinol Metab. 1976;42:544–550.
59. Peterson, RD, Kourides, IA, Horwith, M, et al. Luteinizing hormone and α-subunit-secreting pituitary tumor: positive feedback of estrogen. J Clin Endocrinol Metab. 1981;51:692–698.
60. Klibanski, A, Deutsch, PJ, Jameson, JL, et al. Luteinizing hormone-secreting pituitary tumor: biosynthetic characterization and clinical studies. J Clin Endocrinol Metab. 1987;64:536–542.
61. Daneshdoost, L, Gennarelli, TA, Bashey, HM, et al. Recognition of gonadotroph adenomas in women. N Engl J Med. 1991;324:589–594.
62. Djerassi, A, Coutifaris, C, West, VA, et al. Gonadotroph adenoma in a premenopausal woman secreting follicle-stimulating hormone and causing ovarian hyperstimulation. J Clin Endocrinol Metab. 1995;80:591–594.
63. Christin-Maitre, S, Rongieres-Bertrand, C, Kottler, ML, et al. A spontaneous and severe hyperstimulation of the ovaries revealing a gonadotroph adenoma. J Clin Endocrinol Metab. 1998;83:3450–3453.
64. Mor, E, Rodi, IA, Bayrak, A, et al. Diagnosis of pituitary gonadotroph adenomas in reproductive-aged women. Fertil Steril. 2005;84:757.
65. Valimaki, MJ, Tiitinen, A, Alfthan, H, et al. Ovarian hyperstimulation caused by gonadotroph adenoma secreting follicle-stimulating hormone in 28-year-old woman. J Clin Endocrinol Metab. 1999;84:4204–4208.
66. Cooper, O, Geller, JL, Melmed, S. Ovarian hyperstimulation syndrome caused by an FSH-secreting pituitary adenoma. Nat Clin Pract Endocrinol Metab. 2008;4:234–238.
67. Castelbaum, AJ, Bigdeli, H, Post, KD, et al. Exacerbation of ovarian hyperstimulation by leuprolide reveals a gonadotroph adenoma. Fertil Steril. 2002;78:1311–1313.
68. Sugita, T, Seki, K, Nagai, Y, et al. Successful pregnancy and delivery after removal of gonadotrope adenoma secreting follicle-stimulating hormone in a 29-year-old amenorrheic woman. Gynecol Obstet Invest. 2005;59:138–143.
69. Chanson, P, Schaison, G. Pituitary apoplexy caused by GnRH-agonist treatment revealing gonadotroph adenoma. J Clin Endocrinol Metab. 1995;80:2267–2268.
70. Reznik, Y, Chapon, F, Lahlou, N, et al. Pituitary apoplexy of a gonadotroph adenoma following gonadotrophin releasing hormone agonist therapy for prostatic cancer. J Endocrinol Invest. 1997;20:566–568.
71. Davis, A, Goel, S, Picolos, M, et al. Pituitary apoplexy after leuprolide. Pituitary. 2006;9:263–265.
72. Massoud, W, Paparel, P, Lopez, JG, et al. Discovery of a pituitary adenoma following treatment with a gonadotropin-releasing hormone agonist in a patient with prostate cancer. Int J Urol. 2006;13:87–88.
73. Ambrosi, B, Basstti, M, Ferrario, R, et al. Precocious puberty in a boy with a PRL, LH- and FSH-secreting pituitary tumour: hormonal and immunocytochemical studies. Acta Endocrinol. 1990;122:569–576.
74. Faggiano, M, Criscuolo, T, Perrone, I, et al. Sexual precocity in a boy due to hypersecretion of LH and prolactin by a pituitary adenoma. Acta Endocrinol. 1983;102:167–172.
75. Harris, RI, Schatz, NJ, Gennarelli, T, et al. Follicle-stimulating hormone-secreting pituitary adenomas: correlation of reduction of adenoma size with reduction of adenoma size with reduction of hormone hypersecretion after transsphenoidal surgery. J Clin Endocrinol Metab. 1983;56:1288–1293.
76. Karavitaki, N, Thanabalasingham, G, Shore, HC, et al. Do the limits of serum prolactin in disconnection hyperprolactinaemia need re-definition? A study of 226 patients with histologically verified non-functioning pituitary macroadenoma. Clin Endocrinol (Oxf). 2006;65:524–529.
77. Snyder, PJ, Muzyka, R, Johnson, J, et al. Thyrotropin-releasing hormone provokes abnormal follicle-stimulating hormone (FSH) and luteinizing hormone responses in men who have pituitary adenomas and FSH hypersecretion. J Clin Endocrinol Metab. 1980;51:744–748.
78. Trouillas, J, Girod, C, Sassolas, G, et al. The human gonadotropic adenoma pathologic diagnosis and hormonal correlations in 26 tumors. Semin Diagn Pathol. 1986;3:42–57.
79. Horvath, E, Kovacs, K. Gonadotroph adenomas of the human pituitary sex-related fine-structural dichotomy. Am J Pathol. 1984;117:429–440.
80. Greenman, Y, Melmed, S. Diagnosis and management of nonfunctioning pituitary tumors. Annu Rev Med. 1996;47:95–106.
81. Shimon, I, Melmed, S. Management of pituitary tumors. Ann Intern Med. 1998;129:472–483.
82. Ciric, I, Ragin, A, Baumgartner, C, et al. Complications of transsphenoidal surgery: results of a national survey, review of the literature, and personal experience. Neurosurg. 1997;40:225–237.
83. Jho, HD, Carrau, RL. Endoscopic endonasal transsphenoidal surgery: experience with 50 patients. J Neurosurg. 1997;87:44–51.
84. Cappabianca, P, Cavallo, LM, Colao, A, et al. Endoscopic endonasal transsphenoidal approach: outcome analysis of 100 consecutive procedures. Minim Invasive Neurosurg. 2002;45:193–200.
85. Jho, HD. Endoscopic transsphenoidal surgery. J Neurooncol. 2001;54:187–195.
86. Kawamata, T, Iseki, H, Ishizaki, R, et al. Minimally invasive endoscope-assisted endonasal trans-sphenoidal microsurgery for pituitary tumors: experience with 215 cases comparing with sublabial trans-sphenoidal approach. Neurol Res. 2002;24:259–265.
87. Dehdashti, AR, Ganna, A, Karabatsou, K, et al. Pure endoscopic endonasal approach for pituitary adenomas: early surgical results in 200 patients and comparison with previous microsurgical series. Neurosurgery. 2008;62:1006–1015.
88. Senior, BA, Ebert, CS, Bednarski, KK, et al. Minimally invasive pituitary surgery. Laryngoscope. 2008;118:1842–1855.
89. Frank, G, Pasquini, E, Farneti, G, et al. The endoscopic versus the traditional approach in pituitary surgery. Neuroendocrinology. 2006;83:240–248.
90. Fatemi, N, Dusick, JR, de Paiva Neto, MA, et al. The endonasal microscopic approach for pituitary adenomas and other parasellar tumors: a 10-year experience. Neurosurgery. 2008;63:244–256.
91. Nimsky, C, von Keller, B, Ganslandt, O, et al. Intraoperative high-field magnetic resonance imaging in transsphenoidal surgery of hormonally inactive pituitary macroadenomas. Neurosurgery. 2006;59:105–114.
92. Dusick, JR, Esposito, F, Malkasian, D, et al. Avoidance of carotid artery injuries in transsphenoidal surgery with the Doppler probe and micro-hook blades. Neurosurgery. 2007;60:322–328.
93. Gardner, PA, Prevedello, DM, Kassam, AB, et al. The evolution of the endonasal approach for craniopharyngiomas. J Neurosurg. 2008;108:1043–1047.
94. Laufer, I, Anand, VK, Schwartz, TH. Endoscopic, endonasal extended transsphenoidal, transplanum transtuberculum approach for resection of suprasellar lesions. J Neurosurg. 2007;106:400–406.
95. de Divitiis, E, Cappabianca, P, Cavallo, LM, et al. Extended endoscopic transsphenoidal approach for extrasellar craniopharyngiomas. Neurosurgery. 2007;61:219–227.
96. Trautmann, JC, Laws, ER. Visual status after transsphenoidal surgery at the Mayo Clinic, 1971–1982. Am J Ophthalmol. 1983;96:200–208.
97. Black, PM, Zervas, NT, Candia, GL. Incidence and management of complications of transsphenoidal operations for pituitary adenomas. Neurosurg. 1987;20:920–924.
98. Wilson, CB. A decade of pituitary microsurgery. JNeurosurg. 1984;61:814–833.
99. Barker, FG, 2nd., Klibanski, A, Swearingen, B. Transsphenoidal surgery for pituitary tumors in the United States, 1996–2000: mortality, morbidity, and the effects of hospital and surgeon volume. J Clin Endocrinol Metab. 2003;88:4709–4719.
100. Laws, ER, Jr., Fode, NC, Redmond, MJ. Transsphenoidal surgery following unsuccessful prior therapy. J Neurosurg. 1985;63:823–829.
101. Zaugg, M, Adamman, O, Pescia, R, et al. External irradiation of macroinvasive pituitary adenomas with telecobalt: a retrospective study with long-term follow-up in patients irradiated with doses mostly of between 40–45 Gy. Int J Radiat Oncol Biol Phys. 1995;32:671–680.
102. McCord, MW, Buatti, JM, Fennel, EM, et al. Radiotherapy for pituitary adenoma: long-term outcome and sequelae. Int J Radiat Oncol Biol Phys. 1997;39:437–444.
103. Gittoes, NJL, Bates, AS, Tse, W, et al. Radiotherapy for non-functioning pituitary adenomas. Clin Endocrinol. 1998;48:331–337.
104. Colin, P, Jovenin, N, Delemer, B, et al. Treatment of pituitary adenomas by fractionated stereotactic radiotherapy: a prospective study of 110 patients. Int J Radiat Oncol Biol Phys. 2005;62:333–341.
105. Mackley, HB, Reddy, CA, Lee, SY, et al. Intensity-modulated radiotherapy for pituitary adenomas: the preliminary report of the Cleveland Clinic experience. Int J Radiat Oncol Biol Phys. 2007;67:232–239.
106. Ronson, BB, Schulte, RW, Han, KP, et al. Fractionated proton beam irradiation of pituitary adenomas. Int J Radiat Oncol Biol Phys. 2006;64:425–434.
107. Sheehan, JP, Niranjan, A, Sheehan, JM, et al. Stereotactic radiosurgery for pituitary adenomas: an intermediate review of its safety, efficacy, and role in the neurosurgical treatment armamentarium. J Neurosurg. 2005;102:678–691.
108. Mingione, V, Yen, CP, Vance, ML, et al. Gamma surgery in the treatment of nonsecretory pituitary macroadenoma. J Neurosurg. 2006;104:876–883.
109. Snyder, PJ, Fowble, B, Schatz, NJ, et al. Hypopituitarism following radiation therapy of pituitary adenomas. Am J Med. 1986;81:457–462.
110. Littley, MD, Shalet, SM, Beardwell, CG, et al. Hypopituitarism following external radiotherapy for pituitary tumours in adults. Q J Med. 1970;145:160.
111. Nelson, P, Goodman, M, Flickenger, J, et al. Endocrine function in patients with large pituitary tumors treated with operative decompression and radiation therapy. Neurosurg. 1989;24:398–400.
112. Millar, JL, Spry, NA, Lamb, DS, et al. Blindness in patients after external beam irradiation for pituitary adenoma: two cases occurring after small daily fractional doses. Clin Oncol. 1991;3:291–294.
113. Brada, M, Ford, D, Ashley, S, et al. Risk of second brain tumour after conservative surgery and radiotherapy for pituitary adenoma. Brit Med J. 1993;304:1343–1346.
114. Fisher, BJ, Gaspar, LE, Noone, B. Radiation therapy of pituitary adenoma: delayed sequelae. Radiology. 1993;187:843–846.
115. Dowsett, RJ, Fowble, B, Sergott, RC, et al. Results of radiotherapy in the treatment of acromegaly: lack of ophthalmologic complications. Int J Radiat Oncol Biol Phys. 1990;19:453–459.
116. van Beek, AP, van den Bergh, AC, van den Berg, LM, et al. Radiotherapy is not associated with reduced quality of life and cognitive function in patients treated for nonfunctioning pituitary adenoma. Int J Radiat Oncol Biol Phys. 2007;68:986–991.
117. Stewart, PM, Kane, KF, Stewart, SE, et al. Depot long-acting somatostatin analog (Sandostatin-LAR) is an effective treatment for acromegaly. J Clin Endocrinol Metab. 1995;80:3267–3272.
118. Connor, SE, Penney, CC. MRI in the differential diagnosis of a sellar mass. Clin Radiol. 2003;58:20–31.
119. Giusti, M, Bocca, L, Florio, T, et al. Cabergoline modulation of alpha-subunits and FSH secretion in a gonadotroph adenoma. J Endocrinol Invest. 2000;23:463–466.
120. Lohmann, T, Trantakis, C, Biesold, M, et al. Minor tumour shrinkage in nonfunctioning pituitary adenomas by long-term treatment with the dopamine agonist cabergoline. Pituitary. 2001;4:173–178.
121. Warnet, A, Harris, AG, Renard, E, et al. A prospective multicenter trial of octreotide in 24 patients with visual defects caused by nonfunctioning and gonadotropin-secreting pituitary adenomas. French Multicenter Octreotide Study Group. Neurosurgery. 1997;41:786–795.
122. Sy, RAG, Bernstein, R, Chynn, KY, et al. Reduction in size of a thyrotropin- and gonadotropin-secreting pituitary adenoma treated with octreotide acetate (somatostatin analog). J Clin Endocrinol Metab. 1992;74:690–694.
123. Katznelson, L, Oppenheim, DS, Coughlin, F, et al. Chronic somatostatin analog administration in patients with alpha subunit-secreting pituitary adenomas. J Clin Endocrinol Metab. 1992;75:1318–1325.
124. Andersen, M, Bjerre, P, Schroder, HD, et al. In vivo secretory potential and the effect of combination therapy with octreotide and cabergoline in patients with clinically non-functioning pituitary adenomas. Clin Endocrinol (Oxf). 2001;54:23–30.
125. Roman, SH, Goldstein, M, Kourides, IA, et al. The luteinizing hormone-releasing hormone (LHRH) agonist d-TRT6-PRO9-NEt LHRH increased rather than lowered LH and alpha subunit levels in a patient with an LH-secreting pituitary tumor. J Clin Endocrinol Metab. 1984;58:313–319.
126. Klibanski, A, Jameson, JL, Biller, BMK, et al. Gonadotropin and alpha subunit responses to chronic gonadotropin-releasing hormone analog administration in patients with glycoprotein hormone-secreting pituitary tumors. J Clin Endocrinol Metab. 1989;68:81–86.
127. McGrath, GA, Goncalves, RJ, Udupa, JK, et al. New technique for quantitation of pituitary adenoma size: use in evaluating treatment of gonadotroph adenomas with gonadotropin-releasing hormone antagonist. J Clin Endocrinol Metab. 1993;76:1363–1368.
128. Karavitaki, N, Collison, K, Halliday, J, et al. What is the natural history of nonoperated nonfunctioning pituitary adenomas? Clin Endocrinol (Oxf). 2007;67:938–943.