CHAPTER 331 Clinical Pathophysiology of Traumatic Brain Injury
The initial mechanical insult of traumatic brain injury (TBI) results in tissue deformation that causes damage to neurons, glia, axons, and blood vessels. This is followed by a more delayed phase of injury, which is mediated by intracellular and extracellular biologic pathways and can be present for minutes, hours, days, and even weeks after the primary insult.1–6 During this phase, many patients experience superimposed secondary insults such as hypoxia, hypotension, cerebral swelling, and the consequences of increased intracranial pressure (ICP). These secondary insults7 further exacerbate TBI and have a profound negative effect on patient outcome. An understanding of the sequelae of TBI and secondary insults is paramount for managing head injury; all current neurosurgical and neurocritical care interventions target the delayed effects of secondary injuries and secondary insults.
In a recent report from the International Mission for Prognosis and Clinical Trial (IMPACT) database, hypoxia and hypotension were present on admission in 20% and 18% of TBI patients.8 These insults can be quantified using a system described by Miller and colleagues.9,10 In this study, 124 patients with mild, moderate, or severe head injury were monitored for the following secondary insults: elevated ICP, reduced cerebral perfusion pressure (CPP), hypotension, hypoxia, jugular venous oxygen desaturation, and presence of pyrexia, tachycardia, and bradycardia. Overall, 90% of patients experienced one or more secondary insults, with 50% sustaining an insult of highest severity grade. Secondary insults were most common in the severely injured group (67 of 68) and occurred less frequently in moderate (7 of 36) and mild (3 of 20) injury groups. The authors found that 50% of patients sustained a secondary insult during transport within the hospital, and repeat secondary insults were common even during intensive care management.7 Five insults consistently correlated with poor outcome in this study: arterial hypotension, reduced CPP, elevated ICP, hypoxemia, and pyrexia.10
Although treating secondary injury mechanisms and preventing secondary insults can be challenging, mortality after severe TBI has significantly improved from 39% in 1984 to 27% in 1996.11 This is due, in large part, to increased recognition of the mechanisms underlying secondary brain injury and optimization of cerebral perfusion and oxygenation to avoid secondary brain insults. Significant advances have been made in prehospital care, resuscitation, and rapid radiologic diagnosis,12 and there is increasing emphasis placed on early and aggressive surgical and medical treatment of intracranial mass lesions, raised ICP, and decreased CPP.13–17
No particular treatment has been shown to improve outcome in a randomized, controlled clinical trial, partly owing to ethical concerns.18–22 However, many authors have shown that protocol-driven therapy for TBI significantly improves patient outcome,23–26 especially when aggressive care27 is provided in specialized neurocritical care settings.28 As a result, various TBI guidelines have been developed and updated to guide patient care. These address topics such as prehospital management,29 penetrating TBI,13 surgical management,30 and critical care management of infants, children, and adolescents31 as well as adults.32 Further improvements in management and outcome of TBI patients will likely depend on improved understanding of the pathophysiology of this extremely complex disease. It is important to note, however, that none of these protocols have proved effective in a randomized clinical trial.
In this chapter, we discuss the pathogenesis of closed head injury and the effect of trauma on cerebral metabolism and circulation. Basic concepts of therapeutic intervention, as they pertain to these processes, are reviewed. Penetrating head injury is described in Chapter 331, and specific medical and surgical management of TBI is discussed in Chapter 326, Chapter 327 to Chapter 328.
Mechanisms of Brain Injury
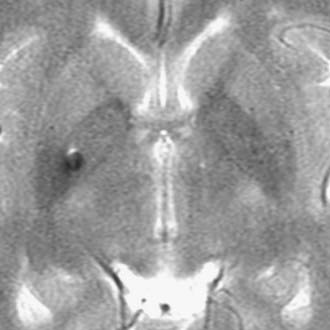
The biomechanics of closed head injury have been extensively described in animals,33–38 human cadavers,39–42 and experimental models of the skull and brain.13,36,43–45 In 1943, Holbourn44 demonstrated the effects of rotational forces on gel housed within a human skull, and 3 years later, Pudenz and Shelden46 visually recorded this phenomenon in a monkey skull replaced with a transparent plastic top. High-speed filming of gel-filled skulls36,47 and high-speed biplanar radiography of cadaveric brains48 have shed additional light on brain deformation after head injury. More recently, computational models49 and magnetic resonance imaging (MRI) techniques have been adapted to study the biomechanical properties of head injury. For example, Bayly and associates have studied the effects of mild linear50 and angular51 head acceleration on brain deformation in healthy volunteers (Fig. 331-1). Their data suggest that mechanical responses are mediated by divisions between brain regions (e.g., central sulcus), dural reflections (e.g., falx cerebri, tentorium cerebelli), and tethering of the brain at the sella and suprasellar regions.
In 1966, Goldsmith defined three physical processes causing head injury52: collision of the head with a solid object at an appreciable velocity, an impulsive load producing sudden motion of the head without significant physical contact, and a static or quasistatic load compressing the head with gradual force. Collision typically results in brain injury through a combination of contact and inertial forces,53 whereas impulsive forces cause inertial loading to the head. Although mild injuries such as concussion may result from this process, impulsive forces typically occur in conjunction with a collision or impact mechanism.54 A static or quasistatic load also involves a contact force, but the speed of impact is minimal or zero. In this scenario, the contribution of inertial forces is negligible, and damage is caused by gradually increasing contact forces trapping the head against a rigid structure.
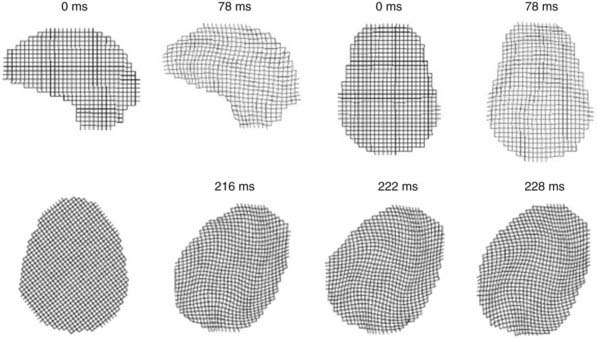
Contact forces typically result in focal injuries such as coup contusions and skull fractures. Inertial loading forces that are primarily translational also result in focal injuries, such as contusions and subdural hematomas, whereas rotational acceleration-deceleration injuries are more likely to result in diffuse injuries ranging from concussion to diffuse axonal injury (DAI). Rotational injuries are particularly concerning because they cause injury to both the cortical surface and deep brain structures. Recently, diffusion tensor imaging (DTI) techniques have been used to better visualize the distribution and severity of white matter fiber injuries after DAI (Fig. 331-2).55,56
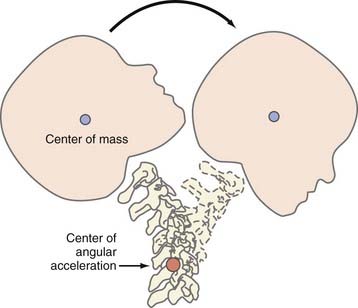
Angular acceleration represents a combination of translational and rotational acceleration and is the most common form of inertial injury. Because of the biomechanical properties of the head and neck, TBI often results in deflection of the head and neck around the middle or lower cervical spine (the center of angular movement; Fig. 331-3). The resultant magnitude of rotation that occurs with this injury depends on the distance between the center of gravity and the center of angulation: the smaller the distance, the larger the rotational component of angulation.57
The extent of injury after TBI is also determined, in large part, by the magnitude and duration of the insult mechanism. In an experimental model of angular acceleration, the influence of duration of the acceleration force, the time to peak acceleration, and the magnitude of acceleration were tested; a threshold value was established below which the impact resulted in concussion rather than a subdural hematoma.33 The study also found that a low magnitude of acceleration with a long duration results in DAI, owing to propagation of the forces deep within the brain. In contrast, a brief, high-velocity impact often results in tearing of superficially located bridging veins and pial vessels, causing subdural hematoma. The former mechanism is typically seen with motor vehicle collisions, whereas the latter occurs in falls or assaults in which the head strikes a broad, hard surface, and inertial loading is the predominant mechanism.33,58
Classification of Head Injury
Clinical condition and level of consciousness after TBI are typically described using the Glasgow Coma Scale (GCS; Table 331-1). This scale has been universally adopted for grading the clinical severity of head injuries and other pathologies that impair consciousness.59 Before the computed tomography (CT) era, GCS score was used to identify patients who should undergo contrast studies to detect intracranial collections of blood. In the modern era, however, GCS plays an important role in categorizing injury severity, allowing for standardized determination of clinical neurological status and detecting episodes of neurological deterioration. It must be noted, however, that the utility of GCS is somewhat limited with modern-day therapies. For example, most patients arrive to the hospital by ambulance unresponsive because of sedation and neuromuscular blockade. In these patients, an accurate postresuscitation score cannot be determined until pharmacologic agents are actively antagonized or metabolized. Similar challenges are faced during intensive care unit (ICU) management because many ICP-lowering therapies render patients heavily sedated, paralyzed, or both. Furthermore, intubation and concomitant injuries resulting in orbital swelling can significantly interfere with accurate eye opening and verbal scoring. In a study by Gale and associates,60 50% of patients could not be assigned an accurate GCS score because of these confounding variables. An alternative scale for scoring clinical condition is the head injury severity scale (Table 331-2), but it faces similar challenges because it is largely based on the GCS score.61
| TEST | SCORE |
|---|---|
| Eye Opening (E) | |
| Spontaneous | 4 |
| Open to voice | 3 |
| Open to pain | 2 |
| None | 1 |
| Best Motor Response (M) | |
| Following commands | 6 |
| Localizing to painful stimulus | 5 |
| Flexion-withdrawal to painful stimulus | 4 |
| Flexor/decorticate posturing to painful stimulus | 3 |
| Extensor/decerebrate posturing to painful stimulus | 2 |
| None | 1 |
| Best Verbal Response (V) | |
| Oriented conversation | 5 |
| Confused/disoriented conversation | 4 |
| Inappropriate words | 3 |
| Incomprehensible sounds | 2 |
| None* | 1 |
| Maximum Score (E + M + V) | 15 |
* Patients who are intubated receive a verbal score of “T,” and the scale is adjusted to 3T-11T.
TABLE 331-2 Head Injury Severity Scale
| INJURY CATEGORY | GLASGOW COMA SCALE SCORE |
|---|---|
| Minimal | 15, no loss of consciousness (LOC) or amnesia |
| Mild | 14-15 with amnesia, brief LOC, or impaired alertness |
| Moderate | 9-13, LOC ≥ 5 minutes or focal neurological deficit |
| Severe | 5-8 |
| Critical | 3-4 |
Adapted from Stein S: Classification of head injury. In Narayan RK, Wilberger JE, Povlishock JT, eds: Neurotrauma. New York: McGraw-Hill; 1996:31-41.
Because of the widespread use of GCS by paramedics, emergency physicians, and surgical and critical care colleagues, it is important for the practicing neurosurgeon to understand the anatomic correlation of the GCS score. One persistent misconception about the GCS is that loss of a flexor response (loss of decorticate rigidity and posturing) represents a lesion that affects descending rubrospinal tracts originating from the upper midbrain, and that the extensor response (decerebrate rigidity and posturing) represents output primarily from the pons and medulla. Although this anatomic relationship was classically described in animal studies from the Sherrington preparations,62,63 more recent studies suggest that these responses in human TBI do not strictly correspond to anatomic lesions. Instead, Greenberg and colleagues found that GCS can be related to the severity of injury at any site in the brain; absent motor response or decerebrate motor posturing can occur with severe, purely cortical, or hemispheric lesions.64
TBI can also be classified anatomically into focal or diffuse injury patterns (Table 331-3). By using CT and MRI to describe injury patterns, anatomic classifications can be used to identify patients at increased risk for secondary insults, delayed neurological worsening, and poor clinical outcome. In 1991, the National Institutes of Health (NIH) Trauma Coma Data Bank (TCDB) introduced a classification system for head injury based on initial CT scan findings (Table 331-4).65 Recognized as the Marshall score, this classification system has been used to design clinical trials, guide patient management, and predict outcome based on radiographic criteria. Recently, Marshall, Maas, and colleagues66 proposed a modified version of the Marshall score (termed the Rotterdam score) to account for additional radiographic criteria that more accurately predict survival from head injury (Table 331-5). These systems describe a strong correlation between CT scan findings (e.g., compression of the basal cisterns, presence of subarachnoid hemorrhage, midline shift) and clinical course, mortality, and functional outcome after TBI.67 Because determining an accurate postresuscitation GCS score is extremely difficult in the current era, it is likely that radiographic scoring systems will play a larger role in predicting outcome and directing care in the acute period following TBI.
TABLE 331-3 Focal Compared with Diffuse Injury
| FOCAL INJURIES | DIFFUSE INJURIES |
|---|---|
| Contusions | Concussion |
| Fracture | Diffuse axonal injury |
| Coup | Moderate |
| Contrecoup | Severe |
| Herniation | |
| Intermediate | |
| Gliding | |
| Hematomas | |
| Epidural | |
| Subdural | |
| Intracerebral |
TABLE 331-4 Classification of Head Injury Based on Initial Computed Tomography Findings: Marshall Score
| CATEGORY | DEFINITION |
|---|---|
| Diffuse injury I | No visible intracranial pathology |
| Diffuse injury II | Cisterns present with midline shift 0-5 mm and/or: |
| Lesion densities present | |
| No high- or mixed-density lesion >25 mL | |
| May include bone fragments and foreign bodies | |
| Diffuse injury III (swelling) | Cisterns compressed or absent with midline shift 0-5 mm; no high- or mixed-density lesion >25mL |
| Diffuse injury IV | Midline shift >5 mm; no high- or mixed-density lesion >25 mL |
| Evacuated mass lesion | Any lesion surgically evacuated |
| Nonevacuated mass lesion | High- or mixed-density lesion >25 mL, not surgically evacuated |
From Marshall L, Bowers S, Klauber M. A new classification of head injury based on computerized tomography. J Neurosurg. 1991;75:514-520.
TABLE 331-5 Classification of Head Injury Based on Initial Computed Tomography Findings: Rotterdam Score
| PREDICTOR VALUE | SCORE |
|---|---|
| Basal Cisterns | |
| Normal | 0 |
| Compressed | 1 |
| Absent | 2 |
| Midline Shift | |
| ≤5 mm | 0 |
| >5 mm | 1 |
| Epidural Mass Lesion | |
| Present | 0 |
| Absent | 1 |
| IVH or Traumatic SAH | |
| Absent | 0 |
| Present | 1 |
| Sum score | +1 |
| Total | 1-6 |
IVH, intraventricular hemorrhage; SAH, subarachnoid hemorrhage.
From Maas A, Hukkelhoven C, Marshall L, Steyerberg E. Prediction of outcome in traumatic brain injury with computed tomographic characteristics: a comparison between the computed tomographic classification and combinations of computed tomographic predictors. Neurosurgery. 57:1173-82, 2005.
Focal Brain Injury
Focal brain injuries typically result in contusions and traumatic intracranial hematomas (see Table 331-3).
Brain Contusion
Brain contusions represent focal regions of subpial hemorrhage and swelling and are present in 31% of patients on initial CT scan.68,69 When the overlying pia mater is compromised, the lesion is termed a laceration, although this distinction is typically not clear. Contusions are most common in regions that contact bony surfaces in the cranial vault during trauma: frontal and temporal poles, orbitofrontal gyri, perisylvian cortices, and inferolateral temporal lobe surfaces.
Contusions can be characterized by mechanism, anatomic location, or adjacent injuries. For example, fracture contusions result from direct contact injuries and occur immediately adjacent to a skull fracture.70 Coup contusions refer to those that occur at the site of impact in the absence of a fracture, whereas contrecoup contusions are those that are diametrically opposite to the point of impact. Gliding contusions are focal hemorrhages involving the cortex and adjacent white matter of the superior margins of the cerebral hemispheres; they are due to rotational mechanisms rather than contact forces.71 Intermediary contusions are lesions that affect deep brain structures, such as the corpus callosum, basal ganglia, hypothalamus, and brainstem.71 Herniation contusions can occur in areas where the medial parts of the temporal lobe contact the tentorial edge (i.e., uncal herniation) or where the cerebellar tonsils contact the foramen magnum (i.e., tonsillar herniation).
Contusions typically result in varying degrees of neurological deficits depending on the area involved. Occasionally, contusions can cause significant mass effect owing to surrounding edema or hemorrhagic progression to an intracerebral hematoma (discussed later). Contusions also represent a significant source of secondary injury to adjacent tissue through release of neurotransmitter and local biochemical changes.5 Adams and colleagues proposed a method for quantifying cerebral contusions (contusion index) caused by nonmissile head injury.72,73 They found that contusions are most severe in the frontal and temporal lobes and do not consistently correlate with injury mechanism. Contusions are more severe when associated with a skull fracture, less severe in patients with DAI, and more severe in patients who do not experience a lucid interval.
Traumatic Intracranial Hematoma
Intracranial hematomas represent mass lesions that are potential therapeutic targets of surgical intervention (as opposed to most contusions). They are more common in patients with skull fractures (Table 331-6). About half of patients with severe TBI and skull fracture have a sizeable intracranial hematoma on initial head CT.10,74 The three major types of traumatic intracranial hematomas are distinguished by their location relative to the meninges: epidural, subdural, and intracerebral.
TABLE 331-6 Relationship between Skull Fracture and Intracranial Hematoma in Patients with Severe, Moderate, and Minor Head Injury

Epidural Hematoma
Epidural hematomas (EDHs) occur in 1% to 2% of TBI patients admitted to the hospital75 and account for 5% to 15% of fatal head injuries.76 They are most common in patients younger than 50 years, although they do occur in all age groups.77–80 In adults, EDH is far less common than subdural or intracerebral hemorrhage. In pediatric patients, however, EDH is relatively more common after TBI. This is likely because of abundant diploic and dural vascularization normally present in infants and young children, notwithstanding the tight adherence of dura to the inner table of the skull.81
EDHs result from vascular injuries to dural vessels or the skull and are often associated with overlying skull fractures. They rarely occur spontaneously in patients with infections,82 sinusitis,83 vascular anomalies,84 or chronic renal failure.85–88 The classic EDH occurs beneath a temporoparietal skull fracture owing to damage to the middle meningeal artery. Separation of dura and bone is thought to occur at the time of injury rather than in a delayed fashion due to stripping of the dura from the inner table as a result of clot enlargement.89 EDHs can be classified by their radiographic progression into three appearances: type I (acute and hyperacute—day 1, associated with “swirl” of unclotted blood), type II (subacute—days 2 to 4, solid), and type III (chronic—days 7 to 20, mixed or lucent with contrast enhancement).90 They occur in 58%, 31%, and 11% of cases, respectively.
The classic clinical course of a patient with EDH was first described by Jacobson in 188691,92: initial loss of consciousness after trauma, transient complete recovery (“lucid interval”), then rapid progression of neurological deterioration. This classic presentation occurs in only 14% to 21% of patients with an EDH.93 Neurological deterioration from an expanding EDH typically results in obtundation, contralateral hemiparesis, ipsilateral oculomotor nerve paresis, decerebrate rigidity, arterial hypertension, cardiac arrhythmias, respiratory disturbances, and finally, apnea and death. Development of these symptoms depends on hematoma size94 and the presence of associated intracranial lesions.95,96 Patients with pure EDHs have an excellent prognosis after surgical evacuation, whereas those with associated intradural lesions experience good outcome in only 44% of cases.97 Although rapid diagnosis and evacuation are critical factors, data suggest that appropriate treatment (by a neurosurgeon, compared with a general surgeon in remote areas) is also important in determining patient outcome.98,99
Morbidity and mortality from a pure EDH is primarily due to delay in diagnosis and appropriate treatment. Lee and associates100 found that patients with epidural hematoma volume greater than 50 cm3 before evacuation experience worse neurological outcome and increased mortality. Delays in diagnosis of EDH are often due to so-called atypical clinical presentations. It is important for the practicing neurosurgeon to be aware, however, that the classic clinical course is far less common and that atypical presentations are far more frequent with EDH. The classic lucid interval is most common in pure EDHs that are very large and demonstrate CT signs of active bleeding (type I).94,101 However, it also occurs in patients harboring subdural hematomas93 or those with mild TBI complicated by meningitis. In fact, a review of 80 consecutive cases of EDH revealed lucid intervals in only 5 patients (6.25%).78 Instead, patients with EDH may be unconscious from the time of initial injury (23% to 44%), may regain consciousness after a brief coma (20% to 28%), or may have no loss of consciousness whatsoever (8% to 24%).77,80,102,103 Typical clinical symptoms of EDH include hemiparesis (contralateral or ipsilateral due to Kernohan’s notch),80,104 decreased level of consciousness, and dilation of the ipsilateral pupil (occurs in less that 50% of cases).101,105,106
EDH in the posterior fossa is a rare finding,107,108 accounting for about 5% of all posttraumatic intracranial mass lesions.109,110 These EDHs are particularly challenging to manage because these patients may remain conscious until late in the evolution of the hematoma, when they may suddenly lose consciousness, become apneic, and die. The neurosurgeon should be aware that these hematomas often extend into the supratentorial compartment by stripping the dura over the transverse sinus, resulting in significant intracranial hemorrhage during surgical evacuation.10 Outcome in patients with posterior fossa EDH is generally better in children and correlates with GCS on admission and CT evidence of hydrocephalus (due to compression of the fourth ventricle).111
Subdural Hematoma
Subdural hematomas (SDHs) are located between the dura and arachnoid layer and may occur as a result of arterial or venous hemorrhage. Classically, SDHs are due to tearing of bridging veins that span the subdural space to drain cortical blood directly into dural sinuses. Many SDHs, however, result from bleeding from other structures adjacent to the subdural space, such as superficial cortical vessels. SDH can be classified as acute, subacute, or chronic, although there is no uniformity of nomenclature.71,112,113 Pathologically, an acute SDH is composed of clot and blood (within 48 hours), a subacute SDH represents a mixture of clotted blood and fluid (2 to 14 days), and a chronic SDH is one that is in fluid phase (>14 days; Table 331-7).71 Clinically, an acute SDH becomes evident within 3 days of injury, subacute between 3 and 21 days, and chronic if more than 21 days pass between injury and clinical presentation. Radiographically, acute SDHs are hyperdense on CT scan, subacute are hyperdense to isodense, and chronic are hypodense relative to adjacent brain.

Acute Subdural Hematoma
Acute SDHs account for 50% to 60% of all subdural hematomas. They are most common after sudden head movements that occur with assaults or falls.79 In a review of patients with acute SDH, 72% had suffered falls or assault, whereas only 24% experienced a motor vehicle crash.33 Rarely, acute SDH may occur spontaneously (or after minor trauma) in patients receiving chronic anticoagulation therapy114,115 or after rupture of a posterior communicating artery aneurysm (only four of these cases have been seen by the senior author (JPM) in more than 30 years of practice).
Most acute SDHs result from venous vascular injury at the brain surface, resulting in two distinct pathologies.116 The first type of hematoma, produced by contact forces and associated with contusions or lacerations, results from cortical bleeding into the adjacent subdural space and is most common at the temporal pole. This complex of subdural hematoma and damaged and necrotic brain is termed burst lobe. The second type of SDH is located over the cerebral convexity and is produced by inertial forces that tear bridging veins.33,116 The underlying brain damage in this type of injury is usually milder and is primarily due to local ischemia from mass effect or compromised venous outflow. Rapid deterioration, as in the case of classic EDH, may accompany these lesions, especially if cortical arteries are ruptured. Despite the often relatively minor underlying brain damage, prognosis is generally poor in these patients unless the hematoma is rapidly evacuated.117
Cerebral ischemia plays a critical role in the pathology of SDH and has been demonstrated both experimentally118 and in postmortem studies.119 The mechanisms responsible for ischemia after SDH are poorly understood but are likely due to compressive effects of the hematoma and elevated ICP with resultant compromised CPP. Evidence of brain compression resulting in ischemia was reported by Schroder and colleagues120; in two patients with acute SDH, preoperative CBF was in the ischemic range (<18 mL/100 g per minute), and cerebral blood volume (CBV) was about half of normal. These values normalized immediately after surgical evacuation. In a small series of five patients with acute SDHs and low GCS scores (≤5), Verweij and colleagues found ICP between 40 and 80 mm Hg, CPP between 10 and 60 mm Hg, low jugular venous oxygen saturation (40% to 60%), and low cerebral blood flow (CBF) measured with laser Doppler immediately before hematoma evacuation.121 Values began to normalize with elevation of the bone flap, opening of the dura, and subsequent hematoma removal.
Timely clot evacuation (within 4 hours) generally results in significantly improved neurological outcome.122 Patients with initial CT evidence of significant hemispheric or generalized brain swelling have extremely poor outcome with or without early surgery.123 The prognosis of SDH is still poor in many cases. It is thought that the coexisting brain damage (DAI, contusion, laceration) is responsible for poor neurological function after injury. In a subset of patients, compression of the microcirculation and resultant low CBF may explain the poor clinical condition and outcome. Patients who deteriorate after a lucid interval (i.e., patients who “talk and die”) may be the subset of patients that experience this type of insult.120
Chronic Subdural Hematoma
Membrane formation around a chronic SDH is a characteristic process attributed to an inflammatory response from the dura. The inner dural layer is highly vascular, and direct contact with blood products, fibrin, and fibrin degradation products in the subdural cavity is thought to elicit a nonspecific inflammatory reaction.124 Because the arachnoid membrane has a much lower reaction potential, the inner capsule of chronic SDHs has no significant vascularity.125
Secondary enlargement of a chronic SDH is common, but the precise cause is unknown. Gardner proposed that the membrane surrounding a chronic SDH acts as an osmotic barrier, allowing cerebrospinal fluid (CSF) diffusion into the hyperosmotic hematoma.126 Zollinger and Gross also proposed an osmotic mechanism for enlargement of the hematoma; they thought that flow across the membrane was the result of an increase of osmotic pressure from a breakdown of hemoglobin molecules in red cells.127 Weir, however, could not find a significant increase in osmolality of the hematoma with increasing age, nor were there any significant differences between the osmolality of blood and hematoma.128 Sato and Suzuki used light and electron microscopy to examine the capsules of chronic subdural hematomas in 33 cases and found that in patients with neurological deficits, capillary endothelial cells in the capsule had many cytoplasmic protrusions and fenestrations, suggesting high permeability of the capillary wall.129 Observations by Yamashima and Yamamoto130 revealed that gap junctions frequently form between adjacent endothelial cells. Numerous blood components, including red blood cells and plasma, can be seen squeezing or spilling into the interstitial space of the outer membrane. It is no longer thought that an osmotic mechanism plays a significant role in hematoma enlargement. Either repeated microhemorrhages from the neocapillary network in the outer membrane or abnormally high vascular permeability is thought to be responsible for hematoma enlargement.125,131
The presentation and pathophysiology of chronic SDHs are distinct from acute SDH, and neurological signs and symptoms are typically due to extra-axial brain compression rather than neurological injury. In a study by Tanaka and coworkers,132 chronic SDHs were shown to have no significant effect on global ICP, CPP, or CBF, but instead resulted in mechanical distortion of central brain regions (e.g., thalamus) with secondary cortical effects. As a result, neurological recovery from evacuation of chronic SDHs is often excellent. Risk for recurrence of chronic SDH after surgical evacuation has been associated with hematoma width and density, lower GCS at time of presentation, ambulation within the first 72 hours, and pneumocephalus that persists beyond 7 days after surgery.133–135
Intracerebral Hematoma
Intracerebral hematomas (ICHs) account for 20% of all traumatic intracranial hematomas.136 They are associated with extensive lobar contusions, from which they are often difficult to distinguish.137,138 ICHs differ from cerebral contusions in that a large proportion of these lesions are composed of blood, but they often result from growth or coalescence of smaller cerebral contusions. By definition, an ICH is a parenchymal lesion composed of at least two-thirds blood; otherwise, the lesion is described as disrupted tissue with areas of microscopic hemorrhage.139 A hemorrhagic mass should be considered an intracerebral hematoma when there is a homogeneous collection of blood with relatively well-defined margins. Multiple intracerebral hematomas are found in about 20% of TBI cases.140
Because ICHs typically result from rupture of intrinsic cerebral vessels (a small parenchymal artery in most cases), they often arise from cerebral contusions. As a result, most traumatic ICHs occur in the orbitofrontal and temporal lobes, as do most cerebral contusions.109,136,137,141,142 Deeper ICHs, such as those occurring in the basal ganglia and internal capsule, are less common and found in about 2% of TBI patients. ICHs are most common in focal head injuries, such as missile injuries, perforating wounds, and depressed skull fractures.143 In a series of 400 civilian TBI cases with depressed skull fracture, 61% of intracranial hematomas were ICHs.144 Patients on chronic anticoagulation therapy are at increased risk for developing ICH, even after mild head injury.143 Recent studies145,146 suggest that treatment with recombinant factor VIIa may reduce expansion of traumatic intracranial hemorrhages, but randomized trials are needed to determine whether this therapy results in improved neurological outcome.
Diffuse brain injuries are the most common result of TBI and represent a clinical continuum of injury from mild concussion to persistent posttraumatic coma (see Table 331-3).147
Concussion
Concussion is the mildest form of diffuse injury and is thought to be due to rotational acceleration of the head in the absence of significant mechanical contact. In its classic form, patients with concussion experience a transient loss of consciousness followed by a rapid return to a normal state of alertness. However, concussion is not as harmless as previously thought, and repeat concussions often result in some degree of permanent neurological impairment.148–150
The pathophysiology of concussion is poorly understood and may be due to disturbances of consciousness from lesions of the brainstem and diencephalon.151 Recent studies suggest that patients with concussion often have diffuse cerebral hemisphere involvement, and brainstem lesions are far less common.152 DTI reveals signs of cytotoxic edema in the brain despite a normal head CT and GCS of 15.153,154
Diffuse Axonal Injury
DAI results from severe angular and rotational acceleration and deceleration that delivers shear and tensile forces to axons. As a result, DAI is responsible for most TBI patients that are severely impaired despite lack of gross parenchymal contusions, lacerations, or hematomas. DAI was described by Stritch in 1956155 in her report of a series of patients with severe posttraumatic dementia and “diffuse degeneration of the white matter.” Others have described this injury as shearing injury, diffuse damage to the white matter of the immediate impact type, and diffuse white matter shearing injury,1,152,156–158 but DAI is the preferred terminology. Coronal or lateral acceleration injuries produce the most severe DAIs,159 whereas acceleration in the oblique or sagittal plane results in less severe to minimal DAI.
The histologic findings of DAI have been well described and include disruption and swelling of axons, “retraction balls” (swollen proximal ends of severed axons), and punctate hemorrhages in the pons, midbrain, and corpus callosum.160 Many of these abnormalities, including axonal severing, are not present initially but develop over the course of several hours or days after injury.161 In many cases, it is difficult to distinguish axonal damage due to mechanical shearing (primary injury) from damage caused by biochemical and metabolic sequelae of TBI (secondary injury). Animal studies suggest that axonal injury is often secondary or delayed, and there is some confirmation of this in humans.161,162 These findings have critical implications for the treating physician because clinical interventions can occur before or during onset of these biochemical cascades.
DAI is often associated with punctate hemorrhages, termed Strich hemorrhages, that represent bleeding from small cerebral vessels.160 Strich hemorrhages are typically found in areas that experience maximal acceleration forces during trauma: corpus callosum, peri-third ventricular (hypothalamus, columns of the fornix, anterior commissure), internal capsule, basal ganglia, dorsolateral brainstem, and superior cerebellar peduncles. The location and severity of axonal injuries are important determinants of functional recovery. Adams and associates1 developed a grading system for patients with DAI (Table 331-8) that predicts length of coma and persistent neurological deficits.
TABLE 331-8 Neuropathologic Classification of Diffuse Axonal Injury
| GRADE | LOCALIZATION OF LESION |
|---|---|
| I | Axonal injury of parasagittal white matter of cerebral hemispheres |
| II | Grade I plus focal lesion in corpus callosum |
| III | Grade II plus focal lesion in cerebral peduncle |
From Adams JH, Doyle D, Ford I, et al. Diffuse axonal injury in head injury: definition, diagnosis and grading. Histopathology 1989;15:49-59.
DAI lesions are often difficult to visualize on conventional CT and are better imaged using MRI techniques. T2-weighted gradient-echo (GE) imaging is particularly sensitive for hemorrhagic lesions after DAI, whereas diffusion-weighted (DW) sequences are more effective in identifying shear injuries.163,164 DW images detect some lesions that are missed by GE sequences and correlate better with neurological prognosis following TBI.163 Recently, DTI has been used to more effectively characterize white matter lesions after TBI. DTI in the early period after TBI has been shown to correlate with GCS score and eventual outcome at discharge (using modified Rankin score).165 Xu and colleagues56 recently demonstrated that DTI is more accurate than conventional MRI in demonstrating the extent of white matter injury after severe TBI. Benson and colleagues166 found that DTI can be used to more accurately characterize injury severity, and Wang and coworkers55 found that quantitative diffusion tensor tractography in the acute period after head injury is highly predictive of outcome. As imaging techniques (and availability) continue to develop, MRI will likely play a larger role in predicting outcome and directing care for patients with DAI.
Traumatic Subarachnoid Hemorrhage and Posttraumatic Vasospasm
The centripetal theory of Ommaya and Gennarelli suggests that lesion depth is dependent on the force of injury.167 Accordingly, traumatic subarachnoid hemorrhage (SAH) results from relatively severe injury to the brain: high angular acceleration of long duration is necessary to produce a strain that causes rupture of the superficial vessels in subarachnoid cisterns. Traumatic SAH is relatively common after severe TBI, occurring in about 33% to 60% of all cases,67,168,169 and strongly correlates with worse neurological outcome.168,170–173
Posttraumatic vasospasm (PTV) is a significant secondary insult to the injured brain that is an independent predictor of permanent neurological deficit and poor outcome.170,174–181 The incidence of PTV varies by frequency of screening and diagnostic modality but is estimated to be 18.6% to 50% in the anterior circulation170,179,182–190 and 19% to 37% in the posterior circulation.180,191,192 It typically develops between 12 hours and 5 days after injury and lasts anywhere between 12 hours and 30 days.170,180,185–187,189–191,193,194 PTV can also occur in a more delayed fashion and may involve both anterior and posterior circulation arteries.195
In an earlier study, Martin and colleagues used CBF and transcranial Doppler measurements to identify three different circulatory stages after severe head injury: phase I (hypoperfusion), phase II (hyperemia), and phase III (vasospasm) (Table 331-9).186 Phase I occurs on the day of injury (day 0) and is defined by low CBF, normal middle cerebral artery (MCA) velocity, normal hemispheric index (ratio of MCA velocity to internal carotid artery velocity), and normal arteriovenous difference of oxygen (AVDO2). The cerebral metabolic rate of oxygen (CMRO2) is about 50% of normal during this phase and remains depressed during the second and third phases. In phase II (relative hyperemia phase, days 1 to 3), CBF increases, AVDO2 falls, MCA velocity rises, and hemispheric index remains less than 3. In phase III (vasospasm phase, days 4 to 15), there is a fall in CBF, a further increase in MCA velocity, and a pronounced rise in the hemispheric index.
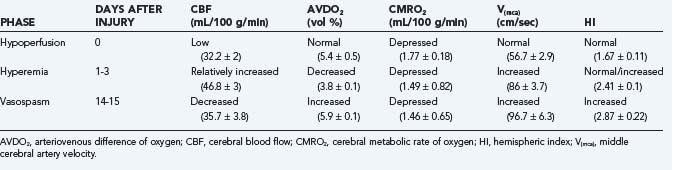
The mechanisms resulting in PTV are poorly understood but may involve extension of intradural blood into CSF spaces (subarachnoid, intraventricular, and subdural).177 Romner and associates187 found anterior circulation PTV incidence increased from 28% to 41% with traumatic SAH, a correlation that has been supported by other authors as well.177,189,196,197 Key differences do exist between traumatic and aneurysmal SAH. For example, traumatic SAH has a different distribution, often involving supratentorial, cortical convexity, sulcal and interhemispheric spaces. PTV also differs in its time course, occurring earlier and resolving more quickly on serial CT than aneurysmal SAH.198 PTV results in decreased CBF with decreased transcranial Doppler velocity. Finally, development of PTV is not always associated with significant SAH and has been reported in patients with extra-axial hematomas177,189,190,199 or no radiographic evidence of SAH whatsoever.190,199
Fukuda and colleagues reported that unlike aneurysmal SAH, in which all low-density areas on CT scans corresponded to vascular territories, low-density areas in patients with traumatic SAH were rarely associated with vascular territories.198 Instead, the low-density areas contained deep-seated or gliding contusions. As discussed in detail later, vasospasm of the large intracranial arteries is accompanied by an increase in CBV, owing to compensatory dilation of the vessels in the microcirculation. Reduced CBF in the presence of increased CBV thus supports the diagnosis of large artery spasm. Schroder and colleagues simultaneously evaluated early CBF and CBV in seven patients with severe head injury.200 These patients were selected from a larger series of 51 patients because they exhibited both nonischemic and ischemic (CBF < 18 mL/100 g per minute) areas on stable xenon CT measurements. Both CBF and CBV were significantly lower in the ischemic zones, indicating that in the early phase after injury, compromise of the microvasculature is the cause of ischemia, rather than vasospasm of the larger conductance vessels. No simultaneous studies have been performed at a later stage (i.e., when the highest incidence of vasospasm is expected).
Intraventricular Hemorrhage
About 25% of patients with severe TBI have CT evidence of intraventricular hemorrhage (IVH). Development of IVH typically requires a large force to the head and is only occasionally present after mild TBI.201 Patients with IVH are also more likely to demonstrate intraparenchymal and basal ganglia hemorrhages.202,203 Postmortem examinations have shown that most patients with primary IVH (no significant parenchymal blood) had a high incidence of damage to the septum pellucidum, choroid plexus, and subependymal vein in the fornix.204 Tearing of these veins is likely the result of negative pressure generated from a sagittal impact to the skull that transiently increases ventricular diameter.202
Although traumatic IVH has the potential of obstructing CSF flow, acute hydrocephalus is an uncommon manifestation. In a review by LeRoux and associates,203 only 4 of 43 patients with traumatic IVH required ventricular drainage for acute hydrocephalus. In a study by Hashimoto and coworkers, 20 of 32 patients with traumatic IVH did not survive their injuries.204
Normal Physiology of Cerebral Metabolism and Circulation
Cerebral Metabolism
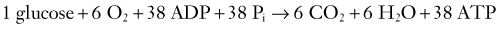
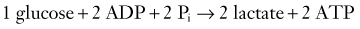
Two terms are common in reference to metabolic turnover of glucose and oxygen: the cerebral metabolic rate of oxygen (CMRO2) and the cerebral metabolic rate of glucose (CMRglc). Under normal circumstances, in awake adults, the CMRO2 is about 3.3 mg/100 g brain tissue per minute,205 and the CMRglc is 5.5 mg/100 g per minute.206 The lactate-oxygen index (LOI) is sometimes used as a measure of the ratio of the amount of glucose metabolized anaerobically to the amount metabolized aerobically (LOI = AVDL/AVDO2), although it does not accurately reflect the stoichiometry of glucose metabolism. AVDL is the arteriovenous difference of lactate, and AVDO2 is the arteriovenous difference of oxygen.206 Both are calculated by subtracting the jugular venous oxygenation (SjvO2) from the systemic arterial oxygenation (SaO2), followed by a correction for hemoglobin oxygen-carrying capacity in the latter:
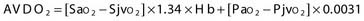
Using 2-fluoro-2-deoxy-D-glucose (FDG)–positron emission tomography (PET), Bergsneider and associates207 described a metabolic ratio (MR = CMRO2/CMRglc) to compare the amount of oxygen versus glucose utilization. Reference values of metabolism are summarized in Table 331-10. The global arteriovenous difference of glucose (AVDglc) has been estimated at 9.6 mL/dL but requires careful interpretation because reliable AVDglc values are difficult to obtain. The absolute AVDglc value is near or within the accuracy limits of plasma glucose levels of most clinical laboratories.
TABLE 331-10 Normal Values of Parameters of Cerebral Metabolism and Circulation in Healthy Adults
| PARAMETER | VALUE |
|---|---|
| CMRO2 | 3.3 ± 0.4 mL/100 g/min |
| CBF (mixed [sub]cortical flow) | 54 ± 12 mL/100 g/min |
| AVDO2 | 6.7 ± 0.8 mL/dL |
| CMRG | 5.5 ± 1.1 mg/100 g/min |
| AVDG | 9.6 ± 1.7 mL/dL |
| LOI | 0.06 ± 0.03 |
| MR | 0.49 ± 0.07 mg O2/mg glucose |
AVDG, arteriovenous difference of glucose; AVDO2, arteriovenous difference of oxygen; CBF, cerebral blood flow; CMRG, cerebral metabolic rate of glucose; CMRO2, cerebral metabolic rate of oxygen; LOI, lactate-oxygen index; MR, metabolic ratio.
Of the total energy generated, 50% is used for neurotransmitter production, release, and uptake (synaptic activity); 25% is used for maintenance and restoration of ion gradients across the cell membrane; and the remaining 25% is used for molecular transport, biosynthesis, and other unidentified processes.208,209 Although glial cells account for 50% of the brain, they have a much lower metabolic rate and account for less than 10% of total cerebral energy expenditure.209 Compared with other organs, the metabolic demand of the brain is high: the brain accounts for only 2% to 3% of total body weight and does not do any mechanical work, yet it receives 20% of cardiac output.
Regulation of Cerebral Blood Flow
Normally, CBF is regulated to provide an adequate supply of substrates to the brain. This is primarily accomplished through changes in caliber of resistance vessels, arterioles with a diameter of 30 to 300 µm.210 This process, termed autoregulation,211 occurs in response to metabolic signals (metabolic autoregulation), blood pressure (pressure autoregulation), and viscosity (viscosity autoregulation) and reacts to changes in the arterial partial pressure of CO2 (carbon dioxide reactivity).
Pressure Autoregulation
In a healthy person, pressure autoregulation is extremely effective in maintaining relatively constant CBF in the CPP range of 50 to 150 mm Hg (Fig. 331-4). When CPP drops below these limits, vessels are maximally dilated and can no longer maintain adequate CBF in the context of dropping CPP. When CPP exceeds these limits, vessels can no longer remain constricted due to high intravascular pressure and experienced forced dilation; this condition is called pressure breakthrough.
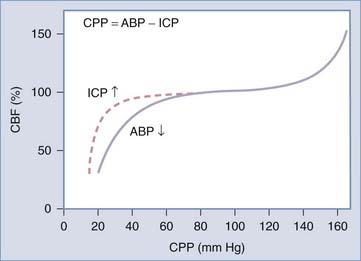
Defining autoregulation as constancy of CBF despite changes in CPP is not completely accurate however, because small changes in CBF normally occur during CPP changes within the limits of autoregulation. Therefore, some authors instead define pressure autoregulation in terms of cerebrovascular resistance (CVR), that is, the change in CVR (calculated as CPP/CBF) that occurs in response to a given change in CPP.212 Pressure autoregulation is considered intact when 0 < Δ% CPP/ Δ% CVR < 2.213–215
Viscosity Autoregulation
Poiseuille’s equation can also be used to describe the effects of viscosity on CBF. Blood viscosity changes with variations in hematocrit, gamma globulin, and plasma fibrinogen. Increased viscosity results in increased cerebral vascular resistance [CVR = 8 × 1 × viscosity/π·r4], which triggers an autoregulatory response. Under these circumstances, viscosity autoregulation responds by vasodilating and vasoconstricting in response to changes in serum viscosity to maintain relatively constant CBF.216–218
Theories of Autoregulation
Metabolic coupling is thought to be due to vasoactive metabolites released from active neurons. This concept was first introduced in 1890 by Roy and Sherrington: “The chemical products of cerebral metabolism contained in the lymph, which bathes the walls of the arterioles of the brain, can cause variations in the caliber of the cerebral vessels. In the reaction the brain possesses an intrinsic mechanism by which its vascular supply can be varied locally in correspondence with local variations of functional activity.”219 The identity of these vasoactive agents, however, remains unclear. Agents that are directly influenced by local energy metabolism, such as CO2, H+, O2, adenosine, and the ions K+ and Ca2+, have been proposed.210,220,221 However, there are problems with each of these compounds as a sole factor.209
The vascular endothelium plays an important role in maintaining the normal physiologic function of the blood vessel wall by releasing relaxing and contracting factors. Endothelial-derived relaxing factor–nitric oxide (EDRF-NO) is the primary mediator of endothelin-dependent relaxation.222 The role of EDRF-NO in autoregulation is controversial, and many authors think its main role is maintenance of basal CBF.223,224 Garthwaite and colleagues, however, found that EDRF-NO may mediate a functional coupling of metabolism and CBF in certain types of neural activation.225 During glutamate activation of the N-methyl-D-aspartate (NMDA) receptors, they noted calcium-dependent release of a substance with properties similar to that of EDRF-NO. The observed vasodilation after glutamate activation was inhibited by administration of both an NMDA antagonist and an NO synthase inhibitor, suggesting coupling of neuronal activation and CBF through EDRF-NO release.
Goadsby and colleagues simultaneously studied cerebral neuronal activity and local blood flow in cats by the induction of spreading depression (a wave of depolarization).226 Neuronal activity and CBF in the parietal cortex were measured simultaneously. They found that intravenous administration of NG-nitro-L-arginine methyl ester (1-NAME), a potent NO synthase inhibitor, resulted in a complete blockade of the hyperemia associated with spreading depression, but did not cause a change in either resting cell firing or spreading depression-evoked increases in firing rate.
Rapid elevation of transmural pressure triggers vasoconstriction (the Bayliss effect), a response that is prevented by removal of the endothelium.227,228 Pressure autoregulation is thought to be mediated by the endothelium. Two major endothelium-derived contracting factors can be identified: thromboxane A2 and endothelin.223 Martinez-Orgado and coworkers studied the influence of endothelial factors on pressure autoregulation in a porcine model.229 Middle cerebral arteries from 3- to 4-day-old piglets were cannulated, and diameter changes after transmural pressure variation were measured. Segments with endothelium showed vasodilation during pressure decrease and vasoconstriction during pressure increase, whereas segments without endothelium responded passively to pressure change. Their results suggested an endothelium-dependent autoregulatory mechanism, with involvement of NO and K-Ca channels in vasodilation during transmural pressure decrease and mediation of vasoconstriction by endothelin-1 (through endothelin A receptors) and prostanoids during pressure increase.
Winn and colleagues have extensively studied the role of adenosine in regulating pial vessel diameter and vascular resistance and have implicated its role as a metabolic trigger for autoregulation.230–234 The mechanisms of its actions are not completely understood but may include nitric oxide release from astrocytes,235 recruitment of cyclic guanosine monophosphate–dependent pathways,236 changes in blood-brain barrier permeability to excitatory amino acids,237 and direct activity on arteriolar adenosine receptors.232
Carbon Dioxide Reactivity
The effects of hypocarbia and hypercarbia are mediated by pH changes in the perivascular space. The effects of CO2 changes on blood vessel diameter persist for less than 20 to 24 hours; at a constant level of PacO2, the pH in the perivascular space normalizes, and cerebral blood vessel diameter returns to baseline.238 To maintain a constant supply of substrates to support metabolism (CMRO2), changes in CBF due to hypocarbia or hypercarbia are compensated for by changes in AVDO2. Again, this is fundamentally different from metabolic, pressure, and viscosity autoregulation, whereby AVDO2 remains constant and CBF is “tuned” to metabolism.
Cerebral Blood Flow and Cerebral Blood Volume
Alterations in vascular caliber not only affect brain perfusion but also change CBV. Changes in CBV can have dramatic effects on ICP, which may be critical when managing patients with severe TBI. CBV is determined by CBF, and the mean transit time of blood through the cerebral vasculature (MTT): CBV = CBF × MTT. Most of the intracranial blood volume is present in vessels with diameters between 30 and 300 µm. Changes in vessel caliber primarily occur in arterioles (200 µm), whereas the diameter of venules remains more or less constant.210
Cerebral Circulation and Metabolism after Severe Head Injury: Mechanisms of Secondary Injury
Disturbances of Cerebral Metabolism
In patients with severe TBI that are comatose, CMRO2 is typically reduced from a normal value of 3.2 mL/100 g per minute to between 1.2 and 2.3 mL/100 g per minute. Soustiel and associates recently correlated CMRO2 after head injury with GCS and neurological outcome.239 Inadequate CBF, cerebral hypoxia, and mitochondrial failure following TBI compromise normal oxidative metabolism and shift cells to ATP generation through anaerobic glycolytic pathways. As a result of this shift, lactate production increases, as does the metabolic ratio (MR): MR = CMRglc/CMRO2, or LOI. This shift toward hyperglycolysis has been demonstrated in both animal and clinical studies after severe head injury.153,207,240–243
One consequence of hyperglycolysis following TBI is cerebral acidosis, which occurs frequently after severe injuries. Although lactic acid is a byproduct of anaerobic metabolism, it may have neuroprotective effects in the injured brain. For example, acidosis shifts the hemoglobin-oxygen dissociation curve to the right, improving oxygen delivery to damaged tissue. Also, acidosis optimizes tissue pH for glycolysis and causes vasodilation, which maximizes available collateral flow to the damaged region. Despite these effects, however, the recovery potential of tissue in the presence of high lactate and resultant acidosis is poor. The mechanisms by which acidosis results in cell damage likely involve electrolyte disturbances, delayed reversal of decreased CBF, and denaturation of vital proteins.244–246
In the past, increased anaerobic glycolytic turnover was thought to simply result from compromised oxygen supply. This was largely based on the work of Vink and coworkers,247 who demonstrated normal mitochondrial function after severe head injury. However, calcium-free media was used in these studies, and more recent experiments have shown that TBI results in significant mitochondrial dysfunction. This mitochondrial damage appears to occur through calcium-mediated interference of respiratory chain–linked functions. This may help explain decreased AVDO2 and increased anaerobic glycolytic turnover in patients with severe TBI. However, the fact that initial elevations of CSF lactate normalize within 12 hours of injury, before the onset of relative hyperemia and low AVDO2, suggests that CSF lactic acidosis is still primarily due to cerebral ischemia.248
Recent evidence suggests that lactic acid may represent a fuel source for the injured brain.13,239,249,250 Following head injury, a condition of “relative hyperglycolysis” occurs during which glucose uptake is in excess of what would be predicted from oxygen consumption and lactate production.13,207,249,251,252 In other words, glucose is used in the brain through a pathway that is not accounted for by oxidative metabolism or glycolysis. Recent clinical studies after severe TBI have demonstrated that this is likely due to increased flux through the pentose phosphate cycle (PPC). In a recent glucose-labeling study by Dusick and colleagues,253 patients with severe TBI demonstrated an increase in PPC flux from 6.9% (control data) to 19.6%. It has been hypothesized that this increased PPC flux provides additional NADPH, which allows for recycling of the free radical scavenger glutathione and promotes fatty acid synthesis.254–256 The PPC also produces substrates necessary for DNA repair and replication and messenger RNA synthesis, which may promote amino acid and neurotransmitter production.
Disturbances of Cerebral Blood Flow
Cerebral Ischemia
Cerebral ischemia occurs when CBF is inadequate to meet the metabolic demands of the brain. This represents a significant mechanism of secondary injury that can dramatically worsen patient outcome. Eighty percent of patients who die after severe TBI have histologic evidence of cerebral ischemia.257 In one study, Bouma and associates found that cerebral ischemia (CBF < 18 mL/dL with high AVDO2 values) in the first 4 to 12 hours after injury occurs in 20% to 33% of patients with severe TBI and is predictive of worse outcome.91
Cerebral ischemia is most common in patients with acute SDH and diffuse cerebral swelling. TBI significantly lowers the brain’s threshold for ischemia—ischemic insults that are well-tolerated under normal conditions can have devastating effects after head injury.4 This increased vulnerability is likely due to the combined effects of abnormal excitatory neurotransmitter release, metabolic derangements, and biochemical imbalances.220
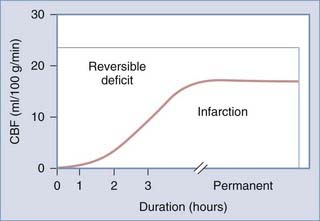
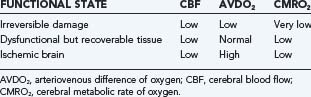
When CBF begins to decline, due to failure of pressure autoregulation or severe hypotension, protective mechanisms are recruited to increase extraction of oxygen. This results in increased AVDO2 from baseline values of about 6.7 mL/100 mL to a maximum of about 13 mL/100 mL.220 Once maximized, further reduction of CBF results in neuronal abnormalities such as synaptic dysfunction (50% of energy expenditure). This is followed by membrane failure, sodium-potassium pump arrest, cell swelling and dysfunction, and eventual cell death. Using an arterial occlusion model of ischemia, the CBF threshold at which these changes occur is at 18 mL/100 g per minute.258 Following severe TBI, however, this threshold is likely elevated to about 20 mL/100 g per minute.259 Neuronal dysfunction and death depend on both the duration and magnitude of CBF depression after injury (Fig. 331-5). Langfitt and Obrist suggested three hypothetical states of brain dysfunction after head injury: brain regions that are irreversibly damaged, brain regions that are dysfunctional, and ischemic brain (Table 331-11).260
Although microdialysis (MD) probes can be used to monitor markers of cerebral metabolism, these do not always correlate with cerebral ischemia. This was recently demonstrated by Vespa and associates,261 who performed both MD probe and PET monitoring in patients with severe TBI. MD data demonstrated a 25% incidence of elevated lactate-to-pyruvate ratio (to above 40) and a 2.4% incidence of ischemia; however, only 1% of patients had ischemia on PET. The authors concluded that TBI results in a “metabolic crisis” that may affect intracellular metabolic pathways, but this does not necessarily represent cerebral ischemia.
Impaired Cerebrovascular Reactivity
Although cerebrovascular reactivity and autoregulation are often abnormal after severe head injury, there is significant variability with respect to the incidence, magnitude, and duration of this disturbance. Derangements are often confined to local brain regions262–265 and may affect different mechanisms of cerebrovascular regulation in varying ways.220 For example, CO2 reactivity is often preserved in the presence of impaired pressure autoregulation.262,266,267
Metabolic Autoregulation
Although comatose patients typically experience reduction of CMRO2 from a normal value of 3.3 mL/100 g per minute to about 2.1 mL/100 g per minute,268 this is not always accompanied by a proportional decrease in CBF. This disturbance in metabolic coupling has been demonstrated in both pediatric and adult TBI patients and is most common in the first 24 hours after injury.215,269,270 In fact, only 45% of patients with severe TBI demonstrate normal metabolic coupling after injury.270
Metabolic uncoupling occurs when CBF exceeds CMRO2, a phenomenon termed luxury perfusion or hyperemia. Obrist and colleagues defined luxury perfusion in comatose head-injured patients as CBF above 33 mL/100 g per minute; CBF of 33 to 55 mL/100 g per minute represents relative hyperemia, and CBF above 55 mL/100 g per minute is absolute hyperemia.270 These limits were based on reports that patients with severe TBI normally demonstrate CMRO2 values that are half of normal (i.e., 1.6 mL/100 g per minute). Therefore, CBF values above half of normal suggest uncoupling of metabolism and flow, and values that are more than 2 standard deviations above the mean CBF found in head-injured patients reflect absolute hyperemia. Similarly, an AVDO2 value of less than 4 has been considered a threshold value for uncoupling, with CBF exceeding metabolic demand. Hyperemia is most prevalent between 1 and 5 days after TBI.271, 272
Hyperemia is a critical concept in TBI because it is strongly associated with diffuse cerebral swelling and elevated ICP.270,273 It should be noted that these observations were based on measurements of CBF, and because of the complex physiology after TBI, it is likely that CBF does not accurately reflect CBV. Subsequent studies by Bouma and associates274 found that although CBV is significantly increased in many patients with elevated ICP, increased CBV does not consistently correlate with radiographic evidence of cerebral swelling. In these patients, therefore, factors other than vascular engorgement (e.g., cytotoxic edema) may play a critical role. In fact, in patients with acute SDH, elevated ICP, and cerebral ischemia, CBV is half of normal. In the acute setting after TBI, therefore, severely increased ICP is likely not due to vascular engorgement.
Hyperemia or vascular engorgement has long been implicated in diffuse cerebral swelling and raised ICP in children after severe head injury. This was based, in large part, on observations by Bruce and colleagues273 of diffuse bilateral swelling on CT in 63 of 214 children with severe head injury. Density of deep frontal white matter (expressed in Hounsfield units) during and after resolution of swelling was measured in 12 children in this study. Although there was a significant difference between these two time points, no density differences were found between children with edema and controls.273 Nonetheless, the concept of hyperemia has influenced therapy for the treatment of high ICP in young children. Vigorous hyperventilation to lower ICP has been advocated because it was thought that children have adequate reserve to protect from induced ischemia. However, prolonged hyperventilation may have detrimental effects in children after TBI. In a study of 23 children with severe TBI, Skippen and coworkers275 demonstrated regions of ischemia (defined as CBF < 18 mL/100 g per minute) in 73.1% of the patients when PacO2 was decreased below 25 mm Hg.
Defining hyperemia after severe head injury requires an understanding of normal CBF levels in children. In a previous publication, we discussed the findings of Suzuki and colleagues,276 who obtained CBF data in a large series of normal unanesthetized children using the xenon-133 method.277 As illustrated in Figure 331-6, CBF is at its lowest value shortly after birth (mean, 40 mL/100 g per minute), then increases to a peak at 3 to 4 years of age (108 mL/100 g per minute, double the normal adult value), then decreases to 71 mL/100 g per minute at about 9 years of age, after which it slowly reaches adult levels. Similar findings were reported by Chiron and associates,278 although peak CBF values were not as pronounced in this study. Therefore, when interpreting CBF measurements, values should be referenced to age-appropriate normal values. Fortunately, vigorous hyperventilation has been largely abandoned by most practitioners, and current guidelines only recommend short-term use in the setting of acute neurological deterioration or ICP elevation.
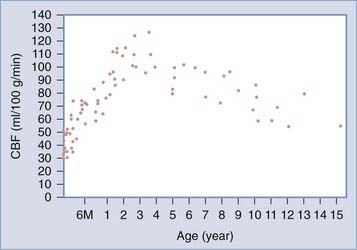
FIGURE 331-6 Changes in mean regional cerebral blood flow (CBF) with age.
(From Suzuki T, Ishii I, Inoue S, et al: [Reproducibility of a non-invasive quantitative assessment of cerebral blood flow using 123I-IMP SPECT]. Kaku Igaku. 1990;27:1331.)
It is important to note that luxury perfusion is defined in terms of CMRO2, and many assume that cerebral metabolism must be depressed after severe TBI because it is primarily oxidative in nature. However, CBF is functionally coupled to CMRglc,279,280 and what is termed “luxury” may actually be an appropriate response to increased glucose turnover (hyperglycolysis) after TBI.207,281 As suggested by Bergsneider and colleagues,207 the metabolic state of the brain should be differentially defined in terms of glucose and oxygen metabolism. This is particularly important in the management of severe TBI because certain therapeutic interventions (e.g., hyperventilation) may affect so-called luxury CBF that is, in fact, essential for ATP generation.
Pressure Autoregulation
Because many neurocritical care interventions can affect vascular hemodynamics, pressure autoregulation after severe TBI is a topic of significant interest.215,266,267,282,283 Enevoldsen and Jensen265 found that pressure autoregulation is usually intact during the first few days after head injury and is only temporarily dysfunctional, with no apparent effect on outcome. Others have found that pressure autoregulation is always intact during the first 36 hours after injury, but then absent in 50% of patients between 36 and 96 hours after injury.267 Overgaard and Tweed283 related autoregulatory capacity to clinical outcome and found that patients had a better outcome when autoregulation was intact in the first 7 days after injury; however, no such correlation could be found after the first week.
Bouma and associates282 performed autoregulation tests in 117 severely head-injured patients. CBF measurements were used to monitor the effects of increases (through administration of phenylephrine, an α-adrenergic agonist that does not affect cerebral vessels)284–286 and decreases (through administration of trimethaphan camsylate, a ganglion blocker without effect on cerebral vessels)287 in mean arterial pressure (MAP). Autoregulation was intact if the percentage change in CBF per percentage change in CVR was between 0% and 2%.213–215 After performing 158 tests in 117 adult patients, they found that autoregulation was intact in 51% of patients. However, no specific temporal pattern or relation to clinical status could be determined.
The mechanisms responsible for abnormal pressure autoregulation after TBI are poorly understood. One clinical study suggests that brainstem lesions from severe TBI may affect an “autoregulatory center” that is necessary for normal pressure autoregulation.266 In a study of multimodality evoked potentials in severely head-injured children, Muizelaar and associates215 did not find a relationship between autoregulatory status and lesion site. In this study, pressure autoregulation was defective in 15 of 37 children.
Endothelial damage may be responsible for the perturbation of pressure autoregulation. In experimental brain injury, endothelial lesions are often present with severe ischemia or trauma.288 Oxygen radicals generated after injury may also cause endothelial damage.289,290 There are data suggesting that impairment of vascular reactivity can be prevented with administration of oxygen radical scavengers.291,292
Viscosity Autoregulation
Few studies have specifically evaluated the status of viscosity autoregulation after severe TBI.216,217 Viscosity autoregulation was first suggested by Muizelaar and associates218 following observations in cats, and subsequent clinical studies have evaluated the response of CBF to bolus administration of mannitol.17 In patients with intact pressure autoregulation, 18% demonstrate a 10% increase in CBF following mannitol administration. On the other hand, 79% of patients with defective pressure autoregulation experience greater than 10% increase in CBF after mannitol administration. In total, 38% of patients demonstrated impaired viscosity autoregulation after head injury. Mannitol resulted in at least 10% decrease in ICP in 86% of patients with intact pressure autoregulation, but in only 35% of those with impaired pressure autoregulation (Table 331-12). These findings suggest a close correlation between the response of CBF and ICP to mannitol and the status of viscosity autoregulation.
TABLE 331-12 Changes in Intracranial Pressure (ICP) and Cerebral Blood Flow (CBF) Larger than 10% of Baseline after Mannitol Administration
| AUTOREGULATION INTACT | AUTOREGULATION DEFECTIVE | |
|---|---|---|
| Decreased ICP | 86% | 36% |
| Decreased CBF | 18% | 79% |
Data from Muizelaar JP, Lutz HA, Becker DP. Effect of mannitol on ICP and CBF and correlation with pressure autoregulation in severely head-injured patients. J Neurosurg. 1984;61:700-706.
Hypertonic saline also has beneficial effects on ICP and CPP, likely due to similar mechanisms involving serum osmolarity and blood rheology. Although some authors advocate its use as resuscitation fluid or an intermittent therapy for elevated ICP,293–297 there are insufficient data to support its general use in current TBI guidelines.298
Carbon Dioxide Reactivity
CO2 reactivity is typically preserved after severe head injury. Although it may be mildly decreased in the first 24 hours, in most cases it returns to a normal 3% change per mm Hg CO2 after day 1.266,267,270,283,299–301 In experimental studies, CO2 reactivity is transiently decreased in the first few hours after injury but then normalizes.13,302 Patients with persistently impaired CO2 reactivity after TBI typically die or survive with severe neurological deficits.270,283 The pathologic mechanisms leading to disturbed CO2 regulation are poorly understood but may involve free radical pathways. In a feline model of TBI, free radical scavengers have been shown to prevent the loss of CO2 reactivity following injury.302
Marion and colleagues271 used stable xenon-enhanced CT to determine CO2 vasoresponsivity in 17 patients with severe TBI. Hemispheric, lobar, basal ganglia, and midbrain CBF values before and after hyperventilation were used to determine CO2 reactivity as the percentage change in CBF per mm Hg PcO2. This study found that 16 of 17 patients had significant differences between global reactivity and regional CO2 reactivity in at least one brain region. These differences were sometimes as large as 50%, particularly in patients with SDH or diffuse cerebral contusions. The clinical implications of this finding are discussed later in this chapter.
Cerebral Blood Flow, Cerebral Blood Volume, Arteriovenous Difference in Oxygen, and Autoregulation
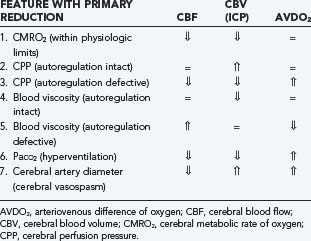
The autoregulatory status of patients with TBI determines the effects of many therapeutic interventions, particularly those that manipulate ICP or CPP. As discussed previously, CBF is influenced by vascular diameter, blood viscosity, and CPP, whereas CBV is determined by vascular diameter alone. Therefore, alterations in CBF, CBV (ICP), and AVDO2 in response to a variety of physiologic and pathologic conditions can be understood (Table 331-13):
Elevated Intracranial Pressure
Elevated ICP (>20 mm Hg) is a common complication of severe TBI that is persistent in 50% of patients with intracranial mass lesions and 33% of those with diffuse injuries.303 ICP higher than 20 is a significant independent determinant of outcome,304 and a recent study of 233 patients found ICP greater than 15 to be one of five independent risk factors for death.305 Patients whose ICP is maintained below 20 mm Hg have significantly better outcome.306 A recent systematic review found that ICP of 20 to 40 increased mortality by 3.5-fold, and ICP above 40 increases mortality by 6.9-fold; raised but reducible ICP increases mortality by 3- to 4-fold, and refractory ICP is associated with an odds ratio for death of 114.3.307 The number and duration of elevated ICP episodes significantly correlates with worse functional outcome and death after TBI.303,308,309
Intracranial pressure is often measured from CSF pressure, which is defined as the pressure one must exert against a needle introduced into the CSF space to prevent escape of fluid.310 Four parameters describe the static and dynamic CSF pressure: (1) the rate of CSF production, (2) the variable compliance given by the exponential relationship of CSF pressure to volume, (3) the outflow resistance, and (4) the intradural sinus pressure.311
The Monro-Kellie doctrine states that the total volume of intracranial contents (CBV, CSF, and brain parenchyma) is constant. An increase in one of the three compartments must be accompanied by an equal decrease in one of the other compartments to maintain constant ICP.13,312–314 Much of this compensation occurs by translocation of CSF and venous blood from the intracranial vault, but at a certain point (decompensation), this volume-buffering capacity is exhausted, and an exponential pressure rise occurs with further volume addition (Fig. 331-7). Marmarou and associates plotted this curve on a semilogarithmic scale to create a straight line (Fig. 331-8).311 The slope of this line is the pressure-volume index (PVI), the amount of volume that must be added or withdrawn from the craniospinal axis to increase or decrease ICP 10-fold:
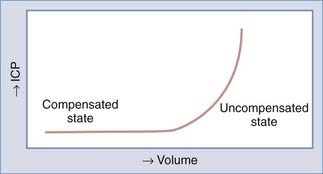
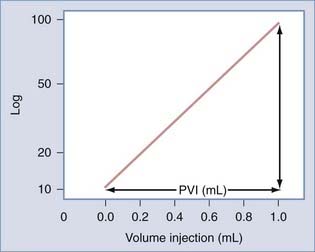
FIGURE 331-8 Relation between pressure and volume plotted on a semilogarithmic scale. PVI, pressure-volume index.
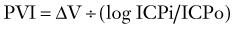
Change in volume is represented by ΔV, ICPo is the pressure before volume change, and ICPi is the pressure after volume change. Thus, PVI is a measure of compliance (ΔV/ΔP) or tightness of the brain. Brain compliance can be estimated by injecting or withdrawing small quantities of fluid into or from the CSF space with simultaneous recording of ICP. This involves repeated injections of 2 to 3 mL into the CSF space, typically through lumbar infusion. Compliance can also be estimated or derived by analyzing the slope and amplitude of the ICP pressure wave.315–317
Normal PVI is 26 ± 4 mL318: 26 mL of volume raises ICP from 1 to 10 mm Hg, but the same volume also raises ICP from 10 to 100 mm Hg. Conversely, the addition of only 6.4 mL increases ICP from 10 mm Hg to the treatment threshold of 20 mm Hg, demonstrating the sensitivity of ICP to volume changes.319,320
Another measure of brain compliance is the volume-pressure ratio (VPR). VPR represents the slope of the pressure-volume curve at a given level of ICP and is defined as the change in ICP achieved by a 1-mL bolus addition of CSF volume.321,322 Under steady-state circumstances, VPR is related to PVI by the following equation: VPR = Po/(0.434 PVI).319
The precise sequence of events leading to ICP elevation and the exact contribution of each component remain elusive. The following five pathways of intracranial volume increase were described by Marmarou319: (1) CSF system, (2) CBV, (3) blood-brain barrier damage–associated edema (vasogenic edema), (4) neurotoxic edema, and (5) ischemic edema. CSF components (CSF resistance to outflow and absorption) account for about one third of ICP elevation. This component can increase substantially in patients with SAH who experience outflow resistance owing to blockage of CSF flow through arachnoid villi. The remaining two thirds of ICP elevation is attributed to a vascular component: increased blood volume (pathway 2) and increased tissue water (vasogenic, neurotoxic, and ischemic edema—pathways 3, 4, and 5).323 Neurotoxic and ischemic edema are thought to be of cellular origin, whereas edema due to blood-brain barrier damage is extracellular in nature.
Klatzo categorized cellular edema into neurotoxic and ischemic.324 Ischemic edema is primarily due to disturbance of cellular osmoregulation from ionic pump failure. Neurotoxic edema is caused by excessive release of excitatory amino acids, loss of calcium and potassium homeostasis, and generation of free radicals. In a series of experiments at the Medical College of Virginia, Barzo and colleagues325,326 investigated the role of vasogenic and cellular edema in a rodent model of diffuse brain injury. Using diffusion-weighted MRI to determine the apparent diffusion coefficient and T1 sequencing to map total brain water, they found a biphasic development of vasogenic and cellular edema. The apparent diffusion coefficient increased with vasogenic edema and decreased with cytotoxic edema. Initially (i.e., in the first 40 minutes), an increase in the apparent diffusion coefficient was observed, consistent with an increase in extracellular fluid. This increase in extracellular volume is thought to be the result of blood-brain barrier compromise. Total water content was increased slightly 1 hour after injury. Forty to 60 minutes after injury, however, the apparent diffusion coefficient started to decrease, reaching a minimum in the second week after TBI. Total brain water remained increased, suggesting delayed intracellular fluid accumulation or cellular edema. No distinction between neurotoxic and ischemic edema could be made in this study.
Secondary Displacement of the Brain
Any discrete, expanding intracranial mass lesion can lead to brain herniation or shift of brain tissue through existing rigid openings in the dura and skull, thereby causing brain compression and raised ICP. Displacement of the brain by expanding lesions and raised ICP are important mechanisms of secondary injury. Brain herniation occurs in five major patterns, and each is associated with a characteristic clinical presentation (Table 331-14).143
TABLE 331-14 Sites of Brain Herniation, Structures Involved, and Resulting Clinical Symptoms
| SITE OF HERNIATION | STRUCTURES INVOLVED | CLINICAL SYMPTOMS |
|---|---|---|
| Subfalcine (cingulate) | Cingulate gyrus Pericallosal arteries |
Leg weakness |
| Lateral (uncal) | Oculomotor nerve | Ptosis |
| Cerebral peduncle | Ipsilateral mydriasis | |
| Posterior cerebral artery | Contralateral hemiparesis | |
| Decreased consciousness | ||
| Posterior (tectal) | Superior colliculi | Bilateral ptosis |
| Upward gaze paralysis | ||
| Central (axial) | Perforating branches from basilar artery | Depressed consciousness |
| Impaired eye movement | ||
| Midbrain, pons, medulla | Respiratory irregularity, apnea | |
| Reticular formation | Hypertension | |
| Bradycardia | ||
| Tonsillar | Medulla | Apnea |
Lateral (Uncal) Tentorial Herniation
Ritter and collegues327 have suggested that decreased brainstem perfusion may result in mydriasis after TBI. In this study, brainstem CBF measurements from 162 patients with severe TBI revealed significantly reduced brainstem blood flow in patients with bilateral nonreactive pupils. The rapid response of mydriasis to mannitol infusion after TBI supports the hypothesis that pupillary dilation is due to brainstem ischemia and not mechanical compression (oculomotor nerve manipulation during surgery can result in pupillary abnormalities lasting many months). Although the role of compression cannot be ruled out in traumatic mydriasis, pupillary asymmetry after TBI is often an indicator of neurological deterioration that should prompt immediate therapeutic intervention.
Central (Axial) Herniation
Central or axial herniation is defined as a downward shift of the brainstem toward the foramen magnum. Because of this downward progression, the brainstem is elongated in its anterior-posterior diameter, and central perforating branches of the basilar artery become stretched. This can result in brainstem ischemia and hemorrhage, the latter occasionally resulting from reversal of the displacement by operative decompression. Clinically, central herniation results in impaired consciousness and a Cushing response to brainstem ischemia (arterial hypertension, bradycardia, and respiratory irregularity). It should be noted that brainstem ischemia is not always present in patients with a Cushing response,328 and a Cushing variant response (tachycardia or systolic hypotension with absence of the classic Cushing triad) should not give a false sense of security in the presence of posterior fossa lesions.
Significance of Extracranial Injury
Hypotension is one of the five most powerful indicators of outcome after TBI2,304 and strongly correlates with poor neurological recovery.2,304,329–331 A single episode of hypotension after TBI increases morbidity and doubles mortality,2 whereas two or more episodes increase the relative risk for mortality to 8.1.330 During the initial resuscitation phase, hypotension, and not hypoxia, significantly affects patient outcome.330 The total duration of hypotensive episodes is a significant predictor of morbidity and mortality.332 In a review of the TCDB, hypotension occurred in 35% of 717 patients with severe TBI and resulted in a 150% increase in mortality.2 In patients with intact pressure autoregulation, hypotension may trigger cerebral vasodilation with subsequent ICP elevation. Therefore, a management protocol that aggressively prevents hypotension has been shown to significantly improve outcome after TBI.333,334Currently published guidelines recommend maintaining systemic blood pressure (SBP) above 90 mm Hg after TBI.32 However, more recent reports from the IMPACT study suggest that this threshold should be raised to 135 mm Hg. In this report, “best outcomes” were found in patients with SBP of about 135 mm Hg and MAP of about 90 mm Hg, with lower or higher levels corresponding to worse neurological outcome.335
Hypoxemia is also one of five powerful indictors of patient outcome after TBI.2 Severe oxygen desaturation (<60%) during transport to the hospital is associated with a 3.5-fold increase in mortality,336 and duration of in-hospital oxygen desaturation (<90%) is an independent predictor of mortality.332 A more recent study of TBI patients transported by helicopter from the scene of an accident found that a secondary hypoxic insult during transportation occurred in 24.7% and corresponded to a mortality rate of 37%.337 In this study, in patients with severe TBI, hypoxia occurred in 35% and doubled patient mortality.338 Data from the TCDB demonstrated an increase in mortality after severe TBI from 27% to 50% when hypoxia occurred. Data from the IMPACT study have also demonstrated the strong correlation between postinjury hypoxia and poor neurological outcome,331 and current published guidelines recommend maintaining oxygen saturation above 90% and PaO2 above 60 mm Hg at all times.32
TBI followed by hypoxia results in widespread edema, ischemic changes, and poor functional outcome.339,340 The effects of hypoxia are not limited to disturbance in substrate supply but may also involve changes in ICP. During a hypoxic episode, CBF is increased by compensatory vasodilation to support CMRO2. This results in increased CBV and resultant elevation of ICP, particularly in patients with poor brain compliance (PVI). In normal adults, a PaO2 of 40 mm Hg increases CBF by 35%, and a PaO2 of 35 mm Hg increases CBF by 70%.341 An even more detrimental CBF response to hypoxia has been described in a feline model of fluid percussion injury: higher levels of injury (>2 atmospheres) severely impaired cerebrovascular reactivity such that hypoxia resulted in an actual decrease in CBF in some animals.342
Other Causes of Secondary Cerebral Damage
Pyrexia
Hyperthermia after TBI is a significant source of secondary injury that is strongly associated with worse neurological outcome.332,343–347 For each degree Celsius elevation in body temperature, cerebral metabolism increases by 10% to 13%. In patients with impaired metabolic autoregulation, this can result in significant brain injury because CBF does not increase proportionally. On the other hand, when autoregulation is intact and CBF does increase proportionally, fever leads to increased CBV with resultant elevation of ICP and reduction of CPP.348–351 In animal models, temperatures above 39°C are associated with increased histologic evidence of brain injury.346
In a study of 846 patients with severe TBI, pyrexia was one of six factors strongly associated with outcome at 1 year.352 It has been estimated that 68% of TBI patients experience temperature greater than 38.0°C,353 and 50% experience temperatures greater than 38.5°C.354 The incidence and duration of fever episodes are significantly associated with injury severity.354 TBI patients with fever are 4.7 times more likely to have an ICU stay longer than 12 days.355 An analysis of 4259 patients managed in a neurology or neurosurgery ICU found that elevated body temperature is an independent predictor of ICU stay, hospital stay, functional outcome, and mortality.356
Anemia
Because hemoglobin-bound oxygen is necessary to meet tissue metabolic demand, anemia can have devastating consequences to the injured brain. In a recent report from the IMPACT group, anemia on admission strongly correlates with worse outcome.357 The ideal hemoglobin after TBI remains unknown, however, and likely differs among patients during their hospital course. Lower hemoglobin levels improve blood viscosity but may compromise oxygen delivery, whereas elevated levels provide better oxygen-carrying capacity but cause the blood to become more viscous. Although one would expect a U-shaped relationship between hemoglobin and neurological outcome, IMPACT investigators found a linear relationship and were unable to determine a particular threshold for therapy. Of note, some authors have reported improved tissue oxygenation with transfusion,358–360 whereas others have demonstrated better outcome with lower levels.361 Age of transfused blood may also play an important factor; in a study of 66 patients with severe head injury, brain tissue oxygenation improved with blood transfusion only if erythrocytes had been stored for less than 19 days before administration.358 Currently, there is insufficient evidence to support an ideal or target hemoglobin level after TBI (discussed later).
Hyperglycemia
Hyperglycemia after TBI also correlates with worse neurological outcome and is more common in patients with higher injury severity.362,363 The source of hyperglycemia in more severe injuries is likely hepatic owing to a sympathoadrenal response to injury.364
Worse neurological outcome in patients with hyperglycemia has been attributed to increased anaerobic metabolism of glucose through glycolysis, resulting in secondary cellular injury from lactic acidosis.248,365 A randomized study of glucose versus saline alimentation after severe head injury revealed no difference in plasma or CSF levels of glucose but showed an increase in CSF lactate in patients receiving glucose support.366 Of note, the buffer tromethamine counteracts acidosis and is effective in experimental367 brain injury but failed in clinical studies.368
Hypomagnesemia
Severe TBI is associated with decreased serum and increased CSF levels of magnesium.369,370 Although these derangements strongly correlate with worse neurological outcome, aggressive correction of hypomagnesemia does not have beneficial effects.370 In fact, a recent randomized clinical trial of 499 patients with severe TBI found that magnesium infusion after severe TBI did not have neuroprotective benefit and was instead associated with increased mortality.371 Therefore, it is likely that hypomagnesemia following TBI is an indicator of injury severity rather than a target for therapy.
Injuries to the Scalp and Skull
Injuries to the skull can result in fractures over the cortical surface (convexity fractures) or along the skull base (basilar fractures). Convexity fractures are often categorized as linear, depressed, or penetrating-perforating and are related to the nature of impact. For example, depressed skull fractures typically result from impact of small objects with surface area of less than 2 square inches; the resultant strain is concentrated immediately beneath the impact site.52 In contrast, energy deposited over a wider surface area results in linear skull fractures.372 Penetrating or perforating injures are often caused by high-energy impact of a small object on the skull. The resultant patterns of linear skull fractures are determined by bone density, local thickness, and presence of sutures. Cortical damage from convexity skull fractures is highly variable and depends largely on the degree of acceleration or deceleration of the head on impact. For example, although severe skull fractures may occur with crush injuries, there is typically minor brain injury in these patients owing to minimal brain motion within the cranial vault.
Basilar skull fractures are often caused by direct impact to the occiput, mastoid prominence, supraorbital area, or facial bones. Because of the thinness of the basilar skull, fractures may also occur as a result of stress waves propagating from a more remote site of impact.53
Basic Concepts of Therapy For Severe Head Injury
Intracranial Pressure Management
Elevated ICP is an independent predictor of poor outcome and mortality after TBI, and aggressive management has long been a cornerstone of head injury management. Although published protocols and popular practice indicate aggressively treating ICP when it exceeds 20 mm Hg, it is important to interpret this value in the appropriate clinical context. For example, it is possible for patients to experience transtentorial herniation with an ICP of 15 mm Hg in the context of a middle fossa lesion. Conversely, adequate CPP can be maintained in patients with ICP between 20 and 30 mm Hg, and elevated ICP may not result in brain tissue ischemia (i.e., brain tissue oxygenation). Despite these observations, however, there is sufficient evidence to suggest that ICP above 20 mm Hg should trigger therapeutic intervention.180,303,304,306,308,309,373,374 ICP-lowering strategies primarily include reduction of CBV and CSF volume.
Reducing Cerebral Blood Volume
Hyperventilation is an effective means of lowering ICP by inducing vasoconstriction and causing a reduction in CBV. However, this is achieved by dramatically reducing CBF (recall that vessel diameter will affect CBF by a power of 4) in the context of unchanged CMRO2 and CMRglc. Because many brain regions experience ischemia after TBI,275 hyperventilation can result in superimposed cellular injury and loss. In a randomized clinical trial, Muizelaar and associates375 found that prophylactic hyperventilation can have detrimental effects on patient outcome. Moreover, it was shown that cerebral vessels become unresponsive to sustained hyperventilation 24 hours after institution of hypocapnia. In the absence of a continuous cerebral monitor for ischemia (e.g., brain tissue oxygen probe, jugular bulb monitor376), hyperventilation for reduction of ICP should primarily be used as a temporizing measure during acute neurological deterioration until urgent definitive therapy (e.g., surgical evacuation of an expanding hematoma) can be delivered.
Three methods exist for lowering ICP through vasoconstriction without reducing CBF. First, decreasing blood viscosity in patients with intact viscosity autoregulation results in decreased ICP without significant changes in CBF. The sustained ICP-lowering effects of hypertonic saline and mannitol are likely due, in part, to its impact on blood viscosity.214,377,378 Second, in patients with intact pressure autoregulation, increasing cerebral perfusion pressure will trigger vasoconstriction with resultant decreases in CBV and ICP; CBF remains constant under these circumstances.379 Third, sedation (e.g., propofol, pentobarbiturate infusion) reduces CMRO2, which not only protects cells from secondary injuries and insults but also reduces ICP through metabolic autoregulatory pathways.
Reducing Cerebrospinal Fluid Volume
As predicted by the Monro-Kellie doctrine, drainage of intracranial CSF reduces ICP.313,314 This effect is particularly dramatic in patients with poor intracranial compliance (low PVI, exponential portion of the pressure-volume curve), in whom a small decrease in intracranial volume can significantly decrease ICP. It should be noted, however, that the therapeutic effects of ventricular drainage can be limited by ventricular collapse in patients with diffuse cerebral swelling.
Cerebral Perfusion Pressure Management
Maintaining adequate CPP after TBI has been shown to improve outcome in multiple studies.380,381 A retrospective analysis of 392 patients found MAP of less than 70 mm Hg and CPP of less than 60 mm Hg to be independent predictors of poor outcome.382 Another review of 427 patients found CPP of less than 60 mm Hg to be strongly associated with worse outcome.383 Cerebral microdialysis studies suggest that ischemic insults to the brain occur when CPP trends below 50 mm Hg.384 A study of almost 400 patients confirmed previous observations that CPP below 60 mm Hg is potentially hazardous in adults but failed to demonstrate that higher CPP was beneficial.385 Some studies suggest that CPP of 70 to 80 mm Hg is the threshold below which there may be significant risk for poor outcome in patients, particularly if CPP falls further.19,22
The effects of CPP on ICP depend heavily on the status of pressure autoregulation. When autoregulation is intact, elevated CPP results in vasoconstriction, decreased CBV, and decreased ICP in the context of normal CBF. On the other hand, decreased CPP can result in vasodilation and resultant ICP elevation.386,387 When autoregulation is defective, the cerebrovascular system becomes “pressure passive”: CBF decreases linearly with CPP and may reach ischemic levels, whereas increasing CPP can result in concomitant elevations of ICP.387
Increased CPP appears somewhat beneficial because of its effect on PVI in patients with intact autoregulation. The PVI tends to be higher when CPP is increased. Reduction of the diameter of the cerebrovascular compartment is thought to contribute to the increased brain compliance with increased CPP. The increased transmural pressure (leading to stiffening of the brain) is offset by this vasoconstriction. In patients with defective autoregulation, however, higher transmural pressure is not offset by arteriolar vasoconstriction and is consequently transmitted to the distal parts of the brain, leading to forced vascular distention in the microcirculation and stiffening of the brain.387
A randomized trial of CPP-targeted therapy (CPP > 70 mm Hg) compared with ICP-targeted therapy (CPP > 50 mm Hg, ICP < 20 mm Hg) found no significant difference in neurological outcome between the two strategies, but a higher morbidity with CPP therapy due to a five-fold increased risk for acute lung injury.388,389 As a result, current adult guidelines recommend maintaining CPP between 50 and 70 mm Hg.16
Hemoglobin, Hematocrit, and Blood Viscosity
Maximal cerebral oxygenation occurs when CPP, hemoglobin oxygen saturation, vessel diameter, and blood viscosity are optimized. Management of hematocrit can be challenging, however, because increasing hematocrit has the benefit of improved oxygen-carrying capacity but the drawback of increased blood viscosity. Historically, most practicing neurosurgeons have targeted a hematocrit of 30% to strike a balance between these competing factors,390 but recent studies have brought this practice into question. A retrospective review of 169 patients with severe TBI found that blood transfusion is associated with worse outcome and that the number of days with hematocrit below 30% is associated with improved neurological outcome (although extremely low levels were detrimental.361
The Transfusion Requirements in Critical Care (TRICC) group391 randomized 838 critically ill patients to a transfusion threshold of either 21% (to maintain hematocrit between 21% and 27%) or 30% (to maintain hematocrit between 30% and 36%). They found no overall differences in mortality between the two groups, although patients with clinically significant cardiac disease benefitted from higher treatment goals. A subgroup analysis of patients with moderate or severe TBI echoed the results of the larger patient population,392 but these subgroups were small and not adequately powered for analysis of transfusion thresholds in TBI. Therefore, an ideal hematocrit for patients with TBI is currently not known. It is possible that this target value is dynamic rather than static (and universal) and may differ among patients across time. Recently, multimodal brain monitoring has shed light on the effects of blood transfusion on brain tissue oxygenation.358–360
Brain Protection: Barbiturate Coma and Hypothermia
If therapeutic interventions to improve oxygen delivery are insufficient, brain protection strategies can be implemented to decrease CMRO2. One highly effective technique is infusion of barbiturates to “induce coma.” The dramatic ICP-lowering effects of barbiturate coma are due to alterations in vascular tone, suppression of metabolism, and inhibition of free radical lipid peroxidation. Most important, this therapy couples CBF to regional metabolic demands such that lower metabolic requirements result in decreased CBF, CBV, and ICP. In a series of 25 patients with ICP above 40 mm Hg, Marshall and colleagues309,393 reported better ICP control and improved clinical outcome with barbiturate therapy. Patients in whom ICP control was achieved with barbiturate therapy had a 92% survival rate and a 50% good recovery rate, whereas those that did not respond to barbiturate therapy experienced a mortality rate of 83%. Although barbiturate therapy is 2 to 4 times more likely to control ICP,306 its prophylactic use has been associated with hypotension and worse outcome.394,395 Therefore, barbiturate infusion should primarily be used in hemodynamically stable patients with elevated ICP that is “refractory to maximum standard medical and surgical treatment.”15
Hypothermia produces a balanced reduction in energy production and consumption, resulting in proportional decrease of CMRO2 and CBF. Tokutomi and coworkers studied the effects of cooling to 33° C on ICP/CPP, brain oxygenation, resting energy expenditure, and cardiac output in 31 patients with severe TBI.396 They found that ICP decreases sharply between 36° C and 35° C, but did not significantly change between 35° C and 33° C. CPP was highest between 36° C and 35° C, but decreased with cooling below 35° C. Resting energy expenditure and cardiac output decreased progressively between 37° C and 33° C, whereas mixed and jugular venous oxygen saturation remained normal. Oxygen delivery and consumption decreased to “abnormally low” levels below 35° C. Maintaining temperature between 35° C and 35.5° C maximally reduced ICP while maintaining CPP, cardiac dynamics, and oxygen delivery.
Prophylactic hypothermia for management of TBI was recently reviewed in a meta-analysis397 that included six studies.398–403 Although all studies were classified as level II data, concerns surrounding “confounding and effect modifying factors” resulted in the following level III recommendation: prophylactic hypothermia maintained over 48 hours may decrease mortality and is associated with better long-term outcome. However, pooled data did not reveal an overall difference in mortality between normothermia and prophylactic hypothermia.397 Despite promising data from a phase II study,404 a recent trial of prophylactic hypothermia in pediatric TBI has suggested detrimental effects.405 Currently, a phase III study is being conducted in children younger than 16 years to further investigate this therapy using a modified protocol that includes earlier and more prolonged cooling followed by slower rewarming. Prophylactic hypothermia has also been studied in adult TBI populations,400,406,407 which have demonstrated trends toward improved outcome. A revised trial is currently underway to further study the role of prophylactic hypothermia in more select adult TBI populations. At present, the literature only supports the use of hypothermia for refractory ICP when medical and surgical interventions have been maximized.
Adams JH, Doyle D, Ford I, et al. Diffuse axonal injury in head injury: definition, diagnosis and grading. Histopathology. 1989;15:49.
Barzo P, Marmarou A, Fatouros P, et al. Contribution of vasogenic and cellular edema to traumatic brain swelling measured by diffusion-weighted imaging. J Neurosurg. 1997;87:900.
Bayly PV, Cohen TS, Leister EP, et al. Deformation of the human brain induced by mild acceleration. J Neurotrauma. 2005;22:845.
Bouma GJ, Muizelaar JP, Fatouros P. Pathogenesis of traumatic brain swelling: role of cerebral blood volume. Acta Neurochir Suppl. 1998;71:272.
Bouma GJ, Muizelaar JP. Cerebral blood flow, cerebral blood volume, and cerebrovascular reactivity after severe head injury. J Neurotrauma. 1992;9(suppl 1):S333.
Cloots RJ, Gervaise HM, van Dommelen JA, et al. Biomechanics of traumatic brain injury: influences of the morphologic heterogeneities of the cerebral cortex. Ann Biomed Eng. 2008;36:1203.
Czosnyka M, Guazzo E, Whitehouse M, et al. Significance of intracranial pressure waveform analysis after head injury. Acta Neurochir (Wien). 1996;138:531.
Diringer MN, Reaven NL, Funk SE, et al. Elevated body temperature independently contributes to increased length of stay in neurologic intensive care unit patients. Crit Care Med. 2004;32:1489.
Eisenberg HM, Gary HEJr, Aldrich EF, et al. Initial CT findings in 753 patients with severe head injury. A report from the NIH Traumatic Coma Data Bank. J Neurosurg. 1990;73:688.
Enevoldsen EM, Cold G, Jensen FT, et al. Dynamic changes in regional CBF, intraventricular pressure, CSF pH and lactate levels during the acute phase of head injury. J Neurosurg. 1976;44:191.
Hebert PC, Wells G, Blajchman MA, et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. Transfusion Requirements in Critical Care Investigators, Canadian Critical Care Trials Group. N Engl J Med. 1999;340:409.
Huisman TA, Schwamm LH, Schaefer PW, et al. Diffusion tensor imaging as potential biomarker of white matter injury in diffuse axonal injury. AJNR Am J Neuroradiol. 2004;25:370.
Jaggi JL, Obrist WD, Gennarelli TA, et al. Relationship of early cerebral blood flow and metabolism to outcome in acute head injury. J Neurosurg. 1990;72:176.
Jones PA, Andrews PJ, Midgley S, et al. Measuring the burden of secondary insults in head-injured patients during intensive care. J Neurosurg Anesthesiol. 1994;6:4.
Kontos HA, Wei EP, Navari RM, et al. Responses of cerebral arteries and arterioles to acute hypotension and hypertension. Am J Physiol. 1978;234:H371.
Maas AI, Hukkelhoven CW, Marshall LF, et al. Prediction of outcome in traumatic brain injury with computed tomographic characteristics: a comparison between the computed tomographic classification and combinations of computed tomographic predictors. Neurosurgery. 2005;57:1173.
Marion DW, Darby J, Yonas H. Acute regional cerebral blood flow changes caused by severe head injuries. J Neurosurg. 1991;74:407.
Marshall LF, Marshall SB, Klauber MR, et al. The diagnosis of head injury requires a classification based on computed axial tomography. J Neurotrauma. 1992;9(suppl 1):S287.
Martin NA, Patwardhan RV, Alexander MJ, et al. Characterization of cerebral hemodynamic phases following severe head trauma: hypoperfusion, hyperemia, and vasospasm. J Neurosurg. 1997;87:9.
Muizelaar JP, Wei EP, Kontos HA, et al. Cerebral blood flow is regulated by changes in blood pressure and in blood viscosity alike. Stroke. 1986;17:44.
Obrist WD, Langfitt TW, Jaggi JL, et al. Cerebral blood flow and metabolism in comatose patients with acute head injury. Relationship to intracranial hypertension. J Neurosurg. 1984;61:241.
Oertel M, Boscardin WJ, Obrist WD, et al. Posttraumatic vasospasm: the epidemiology, severity, and time course of an underestimated phenomenon: a prospective study performed in 299 patients. J Neurosurg. 2005;103:812.
Povlishock JT, Becker DP, Cheng CL, et al. Axonal change in minor head injury. J Neuropathol Exp Neurol. 1983;42:225.
Teasdale G, Jennett B. Assessment of coma and impaired consciousness. A practical scale. Lancet. 1974;2:81.
Vespa P, Bergsneider M, Hattori N, et al. Metabolic crisis without brain ischemia is common after traumatic brain injury: a combined microdialysis and positron emission tomography study. J Cereb Blood Flow Metab. 2005;25:763.
1 Adams JH, Doyle D, Ford I, et al. Diffuse axonal injury in head injury: definition, diagnosis and grading. Histopathology. 1989;15:49.
2 Chesnut RM, Marshall LF, Klauber MR, et al. The role of secondary brain injury in determining outcome from severe head injury. J Trauma. 1993;34:216.
3 Hovda DA, Becker DP, Katayama Y. Secondary injury and acidosis. J Neurotrauma. 1992;9(Suppl 1):S47.
4 Jenkins LW, Moszynski K, Lyeth BG, et al. Increased vulnerability of the mildly traumatized rat brain to cerebral ischemia: the use of controlled secondary ischemia as a research tool to identify common or different mechanisms contributing to mechanical and ischemic brain injury. Brain Res. 1989;477:211.
5 Katayama Y, Becker DP, Tamura T, et al. Massive increases in extracellular potassium and the indiscriminate release of glutamate following concussive brain injury. J Neurosurg. 1990;73:889.
6 Povlishock JT, Becker DP, Cheng CL, et al. Axonal change in minor head injury. J Neuropathol Exp Neurol. 1983;42:225.
7 Andrews PJ, Piper IR, Dearden NM, et al. Secondary insults during intrahospital transport of head-injured patients. Lancet. 1990;335:327.
8 McHugh GS, Engel DC, Butcher I, et al. Prognostic value of secondary insults in traumatic brain injury: results from the IMPACT study. J Neurotrauma. 2007;24:287.
9 Jones PA, Andrews PJ, Midgley S, et al. Measuring the burden of secondary insults in head-injured patients during intensive care. J Neurosurg Anesthesiol. 1994;6:4.
10 Miller J, Piper I, Jones P. Pathophysiology of head injury. In: Narayan RK, Wilberger JE, Povlishock JT, editors. Neurotrauma. New York: McGraw-Hill; 1996:61.
11 Lu J, Marmarou A, Choi S, et al. Mortality from traumatic brain injury. Acta Neurochir Suppl. 2005;95:281.
12 Klauber MR, Marshall LF, Toole BM, et al. Cause of decline in head-injury mortality rate in San Diego County, California. J Neurosurg. 1985;62:528.
13 Part 1. Guidelines for the management of penetrating brain injury. Introduction and methodology. J Trauma. 2001;51:S3.
14 Practice Parameter. Antiepileptic drug treatment of posttraumatic seizures. Brian Injury Special Interest Group of the American Academy of Physical Medicine and Rehabilitation. Arch Phys Med Rehabil. 1998;79:594.
15 Brain Trauma Foundation, American Association of Neurological Surgeons, Congress of Neurological Surgeons. Guidelines for the management of severe traumatic brain injury: anesthetics, analgesics, and sedatives. J Neurotrauma. 2007;24:S71.
16 Brain Trauma Foundation, American Association of Neurological Surgeons, Congress of Neurological Surgeons. Guidelines for the management of severe traumatic brain injury: cerebral perfusion thresholds. J Neurotrauma. 2007;24:S59.
17 Brain Trauma Foundation, American Association of Neurological Surgeons, Congress of Neurological Surgeons. Guidelines for the management of severe traumatic brain injury: intracranial pressure thresholds. J Neurotrauma. 2007;24:S55.
18 Cortbus F, Jones PA, Miller JD, et al. Cause, distribution and significance of episodes of reduced cerebral perfusion pressure following head injury. Acta Neurochir (Wien). 1994;130:117.
19 McGraw C. A cerebral perfusion pressure greater than 80 mmHg is more beneficial. In: Hoff JT, Betz A, editors. Intracranial Pressure, Vol VII. Berlin: Springer-Verlag; 1989:839.
20 Rosner MJ. Introduction to cerebral perfusion pressure management. Neurosurg Clin N Am. 1995;6:761.
21 Rosner MJ. Role of cerebral perfusion pressure in acute brain trauma. Crit Care Med. 1996;24:1274.
22 Rosner MJ, Rosner SD, Johnson AH. Cerebral perfusion pressure: management protocol and clinical results. J Neurosurg. 1995;83:949.
23 Elf K, Nilsson P, Enblad P. Outcome after traumatic brain injury improved by an organized secondary insult program and standardized neurointensive care. Crit Care Med. 2002;30:2129.
24 Fakhry SM, Trask AL, Waller MA, et al. Management of brain-injured patients by an evidence-based medicine protocol improves outcomes and decreases hospital charges. J Trauma. 2004;56:492.
25 Palmer S, Bader MK, Qureshi A, et al. The impact on outcomes in a community hospital setting of using the AANS traumatic brain injury guidelines. American Associations for Neurologic Surgeons. J Trauma. 2001;50:657.
26 Patel HC, Menon DK, Tebbs S, et al. Specialist neurocritical care and outcome from head injury. Intensive Care Med. 28, 2002.
27 Bulger EM, Nathens AB, Rivara FP, et al. Management of severe head injury: institutional variations in care and effect on outcome. Crit Care Med. 2002;30:1870.
28 Patel HC, Bouamra O, Woodford M, et al. Trends in head injury outcome from 1989 to 2003 and the effect of neurosurgical care: an observational study. Lancet. 2005;366:1538.
29 Badjatia N, Carney N, Crocco TJ, et al. Guidelines for the prehospital management of traumatic brain injury. Prehosp Emerg Care. 2007;12:S1.
30 Bullock MR, Chesnut R, Ghajar J, et al. Guidelines for the surgical management of traumatic brain injury. Neurosurgery. 2006;58:S2.
31 Adelson PD, Bratton SL, Carney NA, et al. Guidelines for the acute medical management of severe traumatic brain injury in infants, children, and adolescents. Pediatr Crit Care Med. 2003;4:S1.
32 Brain Trauma Foundation, American Association of Neurological Surgeons, Congress of Neurological Surgeons. Guidelines for the management of severe traumatic brain injury: blood pressure and oxygenation. J Neurotrauma. 2007;24:S7.
33 Gennarelli TA, Thibault LE. Biomechanics of acute subdural hematoma. J Trauma. 1982;22:680.
34 Gennarelli TA, Thibault LE, Tipperman R, et al. Axonal injury in the optic nerve: a model simulating diffuse axonal injury in the brain. J Neurosurg. 1989;71:244.
35 Margulies SS, Thibault LE. A proposed tolerance criterion for diffuse axonal injury in man. J Biomech. 1992;25:917.
36 Margulies SS, Thibault LE, Gennarelli TA. Physical model simulations of brain injury in the primate. J Biomech. 1990;23:832.
37 Ommaya AK, Grubb RLJr, Naumann RA. Coup and contre-coup injury: observations on the mechanics of visible brain injuries in the rhesus monkey. J Neurosurg. 1971;35:503.
38 Ommaya AK, Hirsch AE. Tolerances for cerebral concussion from head impact and whiplash in primates. J Biomech. 1971;4:13.
39 Gurdjian ES, Lissner HR. Photoelastic confirmation of the presence of shear strains at the craniospinal junction in closed head injury. J Neurosurg. 1961;18:58.
40 Gurdjian ES, Lissner HR, Evans FG, et al. Intracranial pressure and acceleration accompanying head impacts in human cadavers. Surg Gynecol Obstet. 1961;113:185.
41 Nahum AM. The prediction of maxillofacial trauma. Trans Sect Otolaryngol Am Acad Ophthalmol Otolaryngol. 1977;84:ORL932.
42 Stalnaker RL, Fogle JL. Driving point impedance characteristics of the head. J Biomech. 1971;4:127.
43 Bradshaw DR, Ivarsson J, Morfey CL, et al. Simulation of acute subdural hematoma and diffuse axonal injury in coronal head impact. J Biomech. 2001;34:85.
44 Holbourn AHS. The mechanics of head injuries. Lancet. 1943;2:438.
45 Ivarsson J, Viano DC, Lovsund P, et al. Strain relief from the cerebral ventricles during head impact: experimental studies on natural protection of the brain. J Biomech. 2000;33:181.
46 Pudenz RH, Shelden CH. The Lucite calvarium—a method for direct observation of the brain. J Neurosurg. 1946;3:487.
47 Meaney DF, Smith DH, Ross DT, et al. Biomechanical analysis of experimental diffuse axonal injury in the miniature pig. J Neurotrauma. 1995;12:689.
48 Hardy WN, Foster CD, Mason MJ, et al. Investigation of head injury mechanisms using neutral density technology and high-speed biplanar x-ray. Strapp Car Crash J. 2001;45:337.
49 Cloots RJ, Gervaise HM, van Dommelen JA, et al. Biomechanics of traumatic brain injury: influences of the morphologic heterogeneities of the cerebral cortex. Ann Biomed Eng. 2008;36:1203.
50 Bayly PV, Cohen TS, Leister EP, et al. Deformation of the human brain induced by mild acceleration. J Neurotrauma. 2005;22:845.
51 Sabet AA, Christoforou E, Zatlin B, et al. Deformation of the human brain induced by mild angular head acceleration. J Biomech. 2008;41:307.
52 Goldsmith W. The physical processes producing head injury. In: Head Injury Conference. Philadelphia.
53 Gennarelli T, Meaney D. Mechanisms of primary head injury. In: Wilkins R, Rengachary S, editors. Neurosurgery. 2nd ed. New York: McGraw-Hill; 1996:2611.
54 McLean AJ. Brain injury without head impact? J Neurotrauma. 1995;12:621.
55 Wang JY, Bakhadirov K, Devous MDSr, et al. Diffusion tensor tractography of traumatic diffuse axonal injury. Arch Neurol. 2008;65:619.
56 Xu J, Rasmussen IA, Lagopoulos J, et al. Diffuse axonal injury in severe traumatic brain injury visualized using high-resolution diffusion tensor imaging. J Neurotrauma. 2007;24:753.
57 Halliday A. Pathophysiology. In: Marion DW, editor. Traumatic Brain Injury. New York: Thieme Medical; 1999:29.
58 Adams JH, Graham DI, Murray LS, et al. Diffuse axonal injury due to nonmissile head injury in humans: an analysis of 45 cases. Ann Neurol. 1982;12:557.
59 Teasdale G, Jennett B. Assessment of coma and impaired consciousness. A practical scale. Lancet. 1974;2:81.
60 Gale JL, Dikmen S, Wyler A, et al. Head injury in the Pacific Northwest. Neurosurgery. 1983;12:487.
61 Stein S. Classification of head injury. In: Narayan RK, Wilberger JE, Povlishock JT, editors. Neurotrauma. New York: McGraw-Hill; 1996:31.
62 Denny-Brown D. Selected Writings of Sir Charles Sherrington. Oxford, UK: Oxford University Press; 1979.
63 Eccles J, Gibson W. Sherrington, His Life and Thought. Berlin and Heidelberg: Springer International; 1979.
64 Greenberg RP, Stablein DM, Becker DP. Noninvasive localization of brain-stem lesions in the cat with multimodality evoked potentials: correlation with human head-injury data. J Neurosurg. 1981;54:740.
65 Marshall LF, Marshall SB, Klauber MR, et al. The diagnosis of head injury requires a classification based on computed axial tomography. J Neurotrauma. 1992;9(suppl 1):S287.
66 Maas AI, Hukkelhoven CW, Marshall LF, et al. Prediction of outcome in traumatic brain injury with computed tomographic characteristics: a comparison between the computed tomographic classification and combinations of computed tomographic predictors. Neurosurgery. 2005;57:1173.
67 Eisenberg HM, Gary HEJr, Aldrich EF, et al. Initial CT findings in 753 patients with severe head injury. A report from the NIH Traumatic Coma Data Bank. J Neurosurg. 1990;73:688.
68 Bullock R, Golek J, Blake G. Traumatic intracerebral hematoma—which patients should undergo surgical evacuation? CT scan features and ICP monitoring as a basis for decision making. Surg Neurol. 1989;32:181.
69 Lobato RD, Cordobes F, Rivas JJ, et al. Outcome from severe head injury related to the type of intracranial lesion. A computerized tomography study. J Neurosurg. 1983;59:762.
70 Teasdale G, Mathew P. Mechanism of cerebral concussion, contusion, and other effects of head injury. In Youmans J, editor: Neurological Surgery, 4th ed, Philadelphia: WB Saunders, 1996.
71 Graham DI. Neuropathology of head injury. In: Narayan RK, Wilberger JE, Povlishock JT, editors. Neurotrauma. New York: McGraw-Hill; 1996:43.
72 Adams JH, Doyle D, Graham DI, et al. The contusion index: a reappraisal in human and experimental non-missile head injury. Neuropathol Appl Neurobiol. 1985;11:299.
73 Adams JH, Scott G, Parker LS, et al. The contusion index: a quantitative approach to cerebral contusions in head injury. Neuropathol Appl Neurobiol. 1980;6:319.
74 Miller JD. Minor, moderate and severe head injury. Neurosurg Rev. 1986;9:135.
75 Lindenberg R. Trauma of meninges and brain. Pathol Nerv Syst. 1971;2:1705.
76 Maloney A. Clinical and pathological observations in fatal head injuries—a five-year study of 172 cases. Br J Surg. 1969;56:23.
77 Cordobes F, Lobato RD, Rivas JJ, et al. Observations on 82 patients with extradural hematoma. Comparison of results before and after the advent of computerized tomography. J Neurosurg. 1981;54:179.
78 Heiskanen O. Epidural hematoma. Surg Neurol. 1975;4:23.
79 Marshall LF, Gautille R, Klauber MR, et al. The outcome of severe closed head injury. J Neurosurg. 1991;75:S28.
80 Reale F, Delfini R, Mencattini G. Epidural hematomas. J Neurosurg Sci. 1984;28:9.
81 Gallagher JP, Browder EJ. Extradural hematoma. Experience with 167 patients. J Neurosurg. 1968;29:1.
82 Griffiths S, Jatavallabhula NS, Mitchell RD. Spontaneous extradural haematoma associated with craniofacial infections: case report and review of the literature. Br J Neurosurg. 2002;16:188.
83 Papadopoulos M, Dyer A, Hardwidge C. Spontaneous extradural haematoma with sinusitis. J R Soc Med. 2001;94:588.
84 Sanchis J, Orozco M, Cabanes J. Spontaneous extradural haematomas. J Neurol Neurosurg Psychiatry. 1975;38:577.
85 Shahlaie K, Fox A, Butani L, et al. Spontaneous epidural hemorrhage in chronic renal failure. A case report and review. Pediatr Nephrol. 2004;19:1168.
86 Shimokawa S, Hayashi T, Anegawa S, et al. [Spontaneous epidural hematoma in a patient undergoing hemodialysis: a case report]. No To Shinkei. 2003;55:163.
87 Sulowicz W, Krasniak A, Goscinski I, et al. [Long-term good results of surgical treatment for spontaneous epi- and subdural hematoma in a female patient on maintenance hemodialysis]. Przegl Lek. 2000;57:764.
88 Talalla A, Halbrook H, Barbour BH, Kurze T. Subdural hematoma associated with long-term hemodialysis for chronic renal disease. JAMA. 1970;212:1847.
89 Teasdale G. Traumatic acute intracranial hematoma: Comment. In: Palmer J, editor. Manual of Neurosurgery. New York: Churchill Livingstone; 1996:544.
90 Zimmerman RA, Bilaniuk LT. Computed tomographic staging of traumatic epidural bleeding. Radiology. 1982;144:809.
91 Bouma GJ, Muizelaar JP, Choi SC, et al. Cerebral circulation and metabolism after severe traumatic brain injury: the elusive role of ischemia. J Neurosurg. 1991;75:685.
92 Jacobson W. On middle meningeal hemorrhage. Guy’s Hosp Rep. 1886;43:147.
93 van den Brink WA, Zwienenberg M, Zandee SM, et al. The prognostic importance of the volume of traumatic epidural and subdural haematomas revisited. Acta Neurochir (Wien). 1999;141:509.
94 Lobato RD, Rivas JJ, Cordobes F, et al. Acute epidural hematoma: an analysis of factors influencing the outcome of patients undergoing surgery in coma. J Neurosurg. 1988;68:48.
95 Sahuquillo-Barris J, Lamarca-Ciuro J, Vilalta-Castan J, et al. Epidural hematoma and diffuse axonal injury. Neurosurgery. 1985;17:378.
96 Seelig JM, Marshall LF, Toutant SM, et al. Traumatic acute epidural hematoma: unrecognized high lethality in comatose patients. Neurosurgery. 1984;15:617.
97 Haselsberger K, Pucher R, Auer LM. Prognosis after acute subdural or epidural haemorrhage. Acta Neurochir (Wien). 1988;90:111.
98 Wester K. Decompressive surgery for “pure” epidural hematomas: does neurosurgical expertise improve the outcome? Neurosurgery. 1999;44:495.
99 Wester T, Fevang LT, Wester K. Decompressive surgery in acute head injuries: where should it be performed? J Trauma. 1999;46:914.
100 Lee E, Hung Y, Wang L, et al. Factors influencing the functional outcome of patients with acute epidural hematomas: analysis of 200 patients undergoing surgery. J Trauma. 1998;45:946.
101 Rivas JJ, Lobato RD, Sarabia R, et al. Extradural hematoma: analysis of factors influencing the courses of 161 patients. Neurosurgery. 1988;23:44.
102 Cooper P. Acute traumatic intracranial hematomas. In: Wilkins R, Rengachary S, editors. Neurosurgery. New York: McGraw-Hill; 1985:1657.
103 Jamieson KG, Yelland JD. Extradural hematoma. Report of 167 cases. J Neurosurg. 1968;29:13.
104 Hooper R. Observations on extradural haemorrhage. Br J Surg. 1959;47:71.
105 Bricolo AP, Pasut LM. Extradural hematoma: toward zero mortality. A prospective study. Neurosurgery. 1984;14:8.
106 Kuday C, Uzan M, Hanci M. Statistical analysis of the factors affecting the outcome of extradural haematomas: 115 cases. Acta Neurochir (Wien). 1994;131:203.
107 Ciembroniewicz JE. Subdural hematoma of the posterior fossa. Review of the literature with addition of three cases. J Neurosurg. 1965;22:465.
108 Zuccarello M, Pardatscher K, Andrioli GC, et al. Epidural hematomas of the posterior cranial fossa. Neurosurgery. 1981;8:434.
109 Rivano C, Borzone M, Carta F, et al. Traumatic intracerebral hematomas: 72 cases surgically treated. J Neurosurg Sci. 1980;24:77.
110 Wright RL. Traumatic hematomas of the posterior cranial fossa. J Neurosurg. 1966;25:402.
111 Malik NK, Makhdoomi R, Indira B, et al. Posterior fossa extradural hematoma: our experience and review of the literature. Surg Neurol. 2007;68:155.
112 Bullock R, Teasdale G. Surgical management of traumatic intracranial hematomas. In: Braakman R, editor. Handbook of Clinical Neurology, Vol 15. Amsterdam: Elsevier; 1990:249.
113 Jennett B, Teasdale G. Management of Head Injuries. Philadelphia: Davis; 1981.
114 Kawamata T, Takeshita M, Kubo O, et al. Management of intracranial hemorrhage associated with anticoagulant therapy. Surg Neurol. 1995;44:438.
115 Wintzen AR, de Jonge H, Loeliger EA, et al. The risk of intracerebral hemorrhage during oral anticoagulant treatment: a population study. Ann Neurol. 1984;16:553.
116 Jamieson KG, Yelland JD. Surgically treated traumatic subdural hematomas. J Neurosurg. 1972;37:137.
117 Seelig JM, Becker DP, Miller JD, et al. Traumatic acute subdural hematoma: major mortality reduction in comatose patients treated within four hours. N Engl J Med. 1981;304:1511.
118 Miller JD, Bullock R, Graham DI, et al. Ischemic brain damage in a model of acute subdural hematoma. Neurosurgery. 1990;27:433.
119 Macpherson P, Graham DI. Correlation between angiographic findings and the ischaemia of head injury. J Neurol Neurosurg Psychiatry. 1978;41:122.
120 Schroder ML, Muizelaar JP, Kuta AJ. Documented reversal of global ischemia immediately after removal of an acute subdural hematoma. Report of two cases. J Neurosurg. 1994;80:324.
121 Verweij BH, Muizelaar JP, Vinas FC. Hyperacute measurement of intracranial pressure, cerebral perfusion pressure, jugular venous oxygen saturation, and laser Doppler flowmetry, before and during removal of traumatic acute subdural hematoma. J Neurosurg. 2001;95:569.
122 Seelig JM, Greenberg RP, Becker DP, et al. Reversible brain-stem dysfunction following acute traumatic subdural hematoma: a clinical and electrophysiological study. J Neurosurg. 1981;55:516.
123 Sawauchi S, Abe T. The effect of haematoma, brain injury, and secondary insult on brain swelling in traumatic acute subdural haemorrhage. Acta Neurochir (Wien). 2008;150:531.
124 Apfelbaum RI, Guthkelch AN, Shulman K. Experimental production of subdural hematomas. J Neurosurg. 1974;40:336.
125 Markwalder TM. Chronic subdural hematomas: a review. J Neurosurg. 1981;54:637.
126 Gardner W. Traumatic subdural hematoma with particular reference to the latent interval. Arch Neurol Psychiatry. 1932;27:847.
127 Zollinger R, Gross R. Traumatic subdural hematoma, an explanation of the late onset of pressure symptoms. JAMA. 103, 1934.
128 Weir B. The osmolality of subdural hematoma fluid. J Neurosurg. 1971;34:528.
129 Sato S, Suzuki J. Ultrastructural observations of the capsule of chronic subdural hematoma in various clinical stages. J Neurosurg. 1975;43:569.
130 Yamashima T, Yamamoto S. Clinicopathological classification of chronic subdural hematoma. Zentralbl Neurochir. 1985;46:304.
131 Goodman JC. Pathophysiology. In: Marion DW, editor. Traumatic Brain Injury. New York: Thieme Medical; 1999:143.
132 Tanaka A, Nakayama Y, Yoshinaga S. Cerebral blood flow and intracranial pressure in chronic subdural hematomas. Surg Neurol. 1997;47:346.
133 Abouzari M, Rashidi A, Rezaii J, et al. The role of postoperative patient posture in the recurrence of traumatic chronic subdural hematoma after burr-hole surgery. Neurosurgery. 2007;61:794.
134 Amirjamshidid A, Abouzari M, Eftekhar B, et al. Outcome and recurrence rates in chronic subdural haematoma. Br J Neurosurg. 2007;21:272.
135 Yamamoto HHY, Hamada H, Hayashi N, et al. Independent predictors of recurrence of chronic subdural hematoma: results of multivariate analysis performed using a logistic regression model. J Neurosurg. 2003;98:1217.
136 Zimmerman RA, Bilaniuk LT, Gennarelli T, et al. Cranial computed tomography in diagnosis and management of acute head trauma. AJR Am J Roentgenol. 1978;131:27.
137 Gudeman SK, Kishore PR, Miller JD, et al. The genesis and significance of delayed traumatic intracerebral hematoma. Neurosurgery. 1979;5:309.
138 Ribas G, Jane JA. Traumatic contusions and intracerebral hematomas. New York. Jane JA, Anderson D, Torner J, et al, editors. Central Nervous System Trauma Status Report 1991. 1992:S265.
139 Becker DP, Doberstein C, Hovda D. Craniocerebral trauma: mechanisms, management, and the cellular response to injury. In: Current Concepts. Kalamazoo, Michigan: UpJohn; 1994.
140 Chesnut R, Servadei F. Surgical treatment of post-traumatic mass lesions. In: Marion DW, editor. Traumatic Brain Injury. New York: Thieme Medical; 1999:81.
141 Baratham G, Dennyson WG. Delayed traumatic intracerebral haemorrhage. J Neurol Neurosurg Psychiatry. 1972;35:698.
142 McLaurin RL. Epilepsy and contact sports. Factors contraindicating participation. JAMA. 1973;225:285.
143 Miller J, Becker DP. Pathophysiology of head injury. In: Youmans J, editor. Neurological Surgery. Philadelphia: WB Saunders; 1982:1896.
144 Miller JD, Jennett WB. Complications of depressed skull fracture. Lancet. 1968;2:991.
145 White CE, Schrank AE, Baskin TW, et al. Effects of recombinant activated factor VII in traumatic nonsurgical intracranial hemorrhage. Curr Surg. 2006;63:310.
146 Zaaroor M, Soustiel JF, Brenner B, et al. Administration off label of recombinant factor-VIIa (rFVIIa) to patients with blunt or penetrating brain injury without coagulopathy. Acta Neurochir (Wien). 2008;150:663.
147 Gennarelli T. Cerebral concussion and diffuse brain injuries. In: Cooper P, editor. Head Injury. Philadelphia: Williams & Wilkins; 1983:137.
148 Guskiewicz KM, McCrea M, Marshall SW, et al. Cumulative effects associated with recurrent concussion in collegiate football players: the NCAA Concussion Study. JAMA. 2003;290:2549.
149 Kelly JP, Nichols JS, Filley CM, et al. Concussion in sports. Guidelines for the prevention of catastrophic outcome. JAMA. 1991;266:2867.
150 Wall SE, Williams WH, Cartwright-Hatton S, et al. Neuropsychological dysfunction following repeat concussions in jockeys. J Neurol Neurosurg Psychiatry. 2006;77:518.
151 Cairns H. Disturbances of consciousness with lesions of the brain-stem and diencephalon. Brain. 1952;75:109.
152 Adams H, Mitchell DE, Graham DI, et al. Diffuse brain damage of immediate impact type. Its relationship to ‘primary brain-stem damage’ in head injury. Brain. 1977;100:489.
153 Inglese M, Makani S, Johnson G, et al. Diffuse axonal injury in mild traumatic brain injury: a diffusion tensor imaging study. J Neurosurg. 2005;103:298.
154 Wilde EA, McCauley SR, Hunter JV, et al. Diffusion tensor imaging of acute mild traumatic brain injury in adolescents. Neurology. 2008;70:948.
155 Strich SJ. Diffuse degeneration of the cerebral white matter in severe dementia following head injury. J Neurol Neurosurg Psychiatry. 1956;19:163.
156 Adams JH. Pathology of nonmissile head injury. Neuroimaging Clin N Am. 1991;1:397.
157 Peerless SJ, Rewcastle NB. Shear injuries of the brain. Can Med Assoc J. 1967;96:577.
158 Zimmerman RA, Bilaniuk LT, Genneralli T. Computed tomography of shearing injuries of the cerebral white matter. Radiology. 1978;127:393.
159 Gennarelli TA, Thibault LE, Adams JH, et al. Diffuse axonal injury and traumatic coma in the primate. Ann Neurol. 1982;12:564.
160 Strich S. Shearing of nerve fibers as a cause of brain damage due to head injury: a pathological study of twenty cases. Lancet. 1961;2:443.
161 Povlishock JT, Jenkins LW. Are the pathobiological changes evoked by traumatic brain injury immediate and irreversible? Brain Pathol. 1995;5:415.
162 Povlishock JT, Christman CW. The pathobiology of traumatically induced axonal injury in animals and humans: a review of current thoughts. J Neurotrauma. 1995;12:555.
163 Ezaki Y, Tsutsumi K, Morikawa M, et al. Role of diffusion-weighted magnetic resonance imaging in diffuse axonal injury. Acta Radiol. 2006;47:733.
164 Huisman TA, Sorensen AG, Hergan K, et al. Diffusion-weighted imaging for the evaluation of diffuse axonal injury in closed head injury. J Comput Assist Tomogr. 2003;27:5.
165 Huisman TA, Schwamm LH, Schaefer PW, et al. Diffusion tensor imaging as potential biomarker of white matter injury in diffuse axonal injury. AJNR Am J Neuroradiol. 2004;25:370.
166 Benson RR, Meda SA, Vasudevan S, et al. Global white matter analysis of diffusion tensor images is predictive of injury severity in traumatic brain injury. J Neurotrauma. 2007;24:446.
167 Ommaya AK, Gennarelli TA. Cerebral concussion and traumatic unconsciousness: correlation of experimental and clinical observations of blunt head injuries. Brain. 1974;97:633.
168 Mattioli C, Beretta L, Gerevini S, et al. Traumatic subarachnoid hemorrhage on the computerized tomography scan obtained at admission: a multicenter assessment of the accuracy of diagnosis and the potential impact on patient outcome. J Neurosurg. 2003;98:37.
169 Morris GF, Bullock R, Marshall SB, et al. Failure of the competitive N-methyl-D-aspartate antagonist Selfotel (CGS 19755) in the treatment of severe head injury: results of two phase III clinical trials. The Selfotel Investigators. J Neurosurg. 1999;91:737.
170 Compton JS, Teddy PJ. Cerebral arterial vasospasm following severe head injury: a transcranial Doppler study. Br J Neurosurg. 1987;1:435.
171 Kakarieka A, Braakman R, Schakel EH. Clinical significance of the finding of subarachnoid blood on CT scan after head injury. Acta Neurochir (Wien). 1994;129:1.
172 Kistler JP, Crowell RM, Davis KR, et al. The relation of cerebral vasospasm to the extent and location of subarachnoid blood visualized by CT scan: a prospective study. Neurology. 1983;33:424.
173 Taneda M, Kataoka K, Akai F, et al. Traumatic subarachnoid hemorrhage as a predictable indicator of delayed ischemic symptoms. J Neurosurg. 1996;84:762.
174 Kordestani RK, Martin NA, McBride DQ. Cerebral hemodynamic disturbances following penetrating craniocerebral injury and their influence on outcome. Neurosurg Clin N Am. 1995;6:657.
175 Lee JH, Martin NA, Alsina G, et al. Hemodynamically significant cerebral vasospasm and outcome after head injury: a prospective study. J Neurosurg. 1997;87:221.
176 Marshall LF, Bruce DA, Bruno L, et al. Vertebrobasilar spasm: a significant cause of neurological deficit in head injury. J Neurosurg. 1978;48:560.
177 Martin NA, Doberstein C, Alexander M, et al. Posttraumatic cerebral arterial spasm. J Neurotrauma. 1995;12:897.
178 Pasqualin A, Vivenza C, Rosta L, et al. Cerebral vasospasm after head injury. Neurosurgery. 1984;15:855.
179 Suwanwela C, Suwanwela N. Intracranial arterial narrowing and spasm in acute head injury. J Neurosurg. 1972;36:314.
180 Vajramani GV, Chandramouli BA, Jayakumar PN, et al. Evaluation of posttraumatic vasospasm, hyperaemia, and autoregulation by transcranial colour-coded duplex sonography. Br J Neurosurg. 1999;13:468.
181 Wilkins RH, Odom GL. Intracranial arterial spasm associated with craniocerebral trauma. J Neurosurg. 1970;32:626.
182 Chan KH, Dearden NM, Miller JD. The significance of posttraumatic increase in cerebral blood flow velocity: a transcranial Doppler ultrasound study. Neurosurgery. 1992;30:697.
183 Grolimund P, Weber M, Seiler RW, et al. Time course of cerebral vasospasm after severe head injury. Lancet. 1988;1:1173.
184 Hadani M, Bruk B, Ram Z, et al. Transiently increased basilar artery flow velocity following severe head injury: a time course transcranial Doppler study. J Neurotrauma. 1997;14:629.
185 Martin NA, Doberstein C, Zane C, et al. Posttraumatic cerebral arterial spasm: transcranial Doppler ultrasound, cerebral blood flow, and angiographic findings. J Neurosurg. 1992;77:575.
186 Martin NA, Patwardhan RV, Alexander MJ, et al. Characterization of cerebral hemodynamic phases following severe head trauma: hypoperfusion, hyperemia, and vasospasm. J Neurosurg. 1997;87:9.
187 Romner B, Bellner J, Kongstad P, et al. Elevated transcranial Doppler flow velocities after severe head injury: cerebral vasospasm or hyperemia? J Neurosurg. 1996;85:90.
188 Rozsa L, Gombi R, Szabo S, et al. Vasospasm after head injury studied by transcranial Doppler sonography. Radiol Diagn (Berl). 1989;30:151.
189 Weber M, Grolimund P, Seiler RW. Evaluation of posttraumatic cerebral blood flow velocities by transcranial Doppler ultrasonography. Neurosurgery. 1990;27:106.
190 Zubkov AY, Pilkington AS, Bernanke DH, et al. Posttraumatic cerebral vasospasm: clinical and morphological presentations. J Neurotrauma. 1999;16:763.
191 Oertel M, Boscardin WJ, Obrist WD, et al. Posttraumatic vasospasm: the epidemiology, severity, and time course of an underestimated phenomenon. A prospective study performed in 299 patients. J Neurosurg. 2005;103:812.
192 Soustiel JF, Shik V. Posttraumatic basilar artery vasospasm. Surg Neurol. 2004;62:201.
193 Armonda RA, Bell RS, Vo AH, et al. Wartime traumatic cerebral vasospasm: recent review of combat casualties. Neurosurgery. 2006;59:1215.
194 Soustiel JF, Shik V, Feinsod M. Basilar vasospasm following spontaneous and traumatic subarachnoid haemorrhage: clinical implications. Acta Neurochir (Wien). 2002;144:137.
195 Shahlaie K, Boggan JE, Latchaw RE, et al. Post-traumatic vasospasm detected by continuous brain tissue oxygen monitoring: treatment with intra-arterial verapamil and balloon angioplasty. Neurocrit Care. 2009;10:61-69.
196 Sander D, Klingelhofer J. Cerebral vasospasm following post-traumatic subarachnoid hemorrhage evaluated by transcranial Doppler ultrasonography. J Neurol Sci. 1993;119:1.
197 Steiger HJ, Aaslid R, Stooss R, et al. Transcranial Doppler monitoring in head injury: relations between type of injury, flow velocities, vasoreactivity, and outcome. Neurosurgery. 1994;34:79.
198 Fukuda T, Hasue M, Ito H. Does traumatic subarachnoid hemorrhage caused by diffuse brain injury cause delayed ischemic brain damage? Comparison with subarachnoid hemorrhage caused by ruptured intracranial aneurysms. Neurosurgery. 1998;43:1040.
199 Zubkov AY, Lewis AI, Raila FA, et al. Risk factors for the development of post-traumatic cerebral vasospasm. Surg Neurol. 2000;53:126.
200 Schroder ML, Muizelaar JP, Fatouros PP, et al. Regional cerebral blood volume after severe head injury in patients with regional cerebral ischemia. Neurosurgery. 1998;42:1276.
201 Karavelis A, Sirmos C. Primary post-traumatic intraventricular hemorrhage. J Neurosurg Sci. 1995;39:253.
202 Fujitsu K, Kuwabara T, Muramoto M, et al. Traumatic intraventricular hemorrhage: report of twenty-six cases and consideration of the pathogenic mechanism. Neurosurgery. 1988;23:423.
203 LeRoux PD, Haglund MM, Newell DW, et al. Intraventricular hemorrhage in blunt head trauma: an analysis of 43 cases. Neurosurgery. 1992;31:678.
204 Hashimoto T, Nakamura N, Ke R, et al. [Traumatic intraventricular hemorrhage in severe head injury]. No Shinkei Geka. 1992;20:209.
205 Kety SS, Schmidt CF. The nitrous oxide method for the quantitative determination of cerebral blood flow in man: theory, procedure and normal values. J Clin Invest. 1948;27:476.
206 Gibbs E, Lennox W, Nims L, et al. Arterial and cerebral venous blood: arterial-venous differences in man. J Biol Chem. 1942;144:325.
207 Bergsneider M, Hovda DA, Shalmon E, et al. Cerebral hyperglycolysis following severe traumatic brain injury in humans: a positron emission tomography study. J Neurosurg. 1997;86:241.
208 Astrup J. Energy-requiring cell functions in the ischemic brain. Their critical supply and possible inhibition in protective therapy. J Neurosurg. 1982;56:482.
209 Siesjo BK. Cerebral circulation and metabolism. J Neurosurg. 1984;60:883.
210 Kontos HA, Wei EP, Navari RM, et al. Responses of cerebral arteries and arterioles to acute hypotension and hypertension. Am J Physiol. 1978;234:H371.
211 Lassen NA. Cerebral blood flow and oxygen consumption in man. Physiol Rev. 1959;39:183.
212 Heistad D, Kontos H. Cerebral circulation. In: Sheperd J, Abboud F, editors. Handbook of Physiology, Vol 3. Bethesda, MD: American Physiologic Society; 1983:137.
213 Bouma GJ, Muizelaar JP. Relationship between cardiac output and cerebral blood flow in patients with intact and with impaired autoregulation. J Neurosurg. 1990;73:368.
214 Muizelaar JP, Lutz HA3rd, Becker DP. Effect of mannitol on ICP and CBF and correlation with pressure autoregulation in severely head-injured patients. J Neurosurg. 1984;61:700.
215 Muizelaar JP, Marmarou A, DeSalles AA, et al. Cerebral blood flow and metabolism in severely head-injured children. Part 1: Relationship with GCS score, outcome, ICP, and PVI. J Neurosurg. 1989;71:63.
216 Hudak ML, Jones MDJr, Popel AS, et al. Hemodilution causes size-dependent constriction of pial arterioles in the cat. Am J Physiol. 1989;257:H912.
217 Hudak ML, Koehler RC, Rosenberg AA, et al. Effect of hematocrit on cerebral blood flow. Am J Physiol. 1986;251:H63.
218 Muizelaar JP, Wei EP, Kontos HA, et al. Cerebral blood flow is regulated by changes in blood pressure and in blood viscosity alike. Stroke. 1986;17:44.
219 Roy CS, Sherrington CS. On the regulation of the blood supply of the liver. J Physiol. 1890.
220 Bouma G. Cerebral Circulation After Severe Head Injury: A Clinical Study, in. Amsterdam: University of Amsterdam; 1993.
221 Kuschinsky W, Wahl M. Local chemical and neurogenic regulation of cerebral vascular resistance. Physiol Rev. 1978;58:656.
222 Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373.
223 Davies MG, Hagen PO. The vascular endothelium. A new horizon. Ann Surg. 1993;218:593.
224 Tanaka K. Is nitric oxide really important for regulation of the cerebral circulation? Yes or no? Keio J Med. 1996;45:14.
225 Garthwaite J, Charles SL, Chess-Williams R. Endothelium-derived relaxing factor release on activation of NMDA receptors suggests role as intercellular messenger in the brain. Nature. 1988;336:385.
226 Goadsby PJ, Kaube H, Hoskin KL. Nitric oxide synthesis couples cerebral blood flow and metabolism. Brain Res. 1992;595:167.
227 Rubanyi GM. Endothelium-dependent pressure-induced contraction of isolated canine carotid arteries. Am J Physiol. 1988;255:H783.
228 Rubanyi GM, Freay AD, Kauser K, et al. Mechanoreception by the endothelium: mediators and mechanisms of pressure- and flow-induced vascular responses. Blood Vessels. 1990;27:246.
229 Martinez-Orgado J, Gonzalez R, Alonso MJ, et al. Endothelial factors and autoregulation during pressure changes in isolated newborn piglet cerebral arteries. Pediatr Res. 1998;44:161.
230 Berne RM, Winn HR, Rubio R. The local regulation of cerebral blood flow. Prog Cardiovasc Dis. 1981;24:243.
231 Ibayashi S, Ngai AC, Meno JR, et al. Effects of topical adenosine analogs and forskolin on rat pial arterioles in vivo. J Cereb Blood Flow Metab. 1991;11:72.
232 Ngai AC, Winn HR. Effects of adenosine and its analogues on isolated intracerebral arterioles. Extraluminal and intraluminal application. Circ Res. 1993;73:448.
233 Winn HR, Morii S, Berne RM. The role of adenosine in autoregulation of cerebral blood flow. Ann Biomed Eng. 1985;13:321.
234 Winn HR, Rubio GR, Berne RM. The role of adenosine in the regulation of cerebral blood flow. J Cereb Blood Flow Metab. 1981;1:239.
235 Janigro D, Wender R, Ransom G, et al. Adenosine-induced release of nitric oxide from cortical astrocytes. Neuroreport. 1996;7:1640.
236 West GA, Meno JR, Nguyen TS, et al. cGMP-dependent and not cAMP-dependent kinase is required for adenosine-induced dilation of intracerebral arterioles. J Cardiovasc Pharmacol. 2003;41:444.
237 Grant GA, Meno JR, Nguyen TS, et al. Adenosine-induced modulation of excitatory amino acid transport across isolated brain arterioles. J Neurosurg. 2003;98:554.
238 Muizelaar JP, van der Poel HG, Li ZC, et al. Pial arteriolar vessel diameter and CO2 reactivity during prolonged hyperventilation in the rabbit. J Neurosurg. 1988;69:923.
239 Soustiel JF, Glenn TC, Shik V, et al. Monitoring of cerebral blood flow and metabolism in traumatic brain injury. J Neurotrauma. 2005;22:955.
240 Andersen B, Marmarou A. Isolated stimulation of glycolysis following traumatic brain injury. In: Hoff J, Betz A, editors. Intracranial Pressure, Vol VII. Berlin: Springer-Verlag; 1989:575.
241 Andersen BJ, Marmarou A. Post-traumatic selective stimulation of glycolysis. Brain Res. 1992;585:184.
242 Sunami K, Nakamura T, Ozawa Y, et al. Hypermetabolic state following experimental head injury. Neurosurg Rev. 1989;12(suppl 1):400.
243 Yoshino A, Hovda DA, Kawamata T, et al. Dynamic changes in local cerebral glucose utilization following cerebral concussion in rats: evidence of a hyper- and subsequent hypometabolic state. Brain Res. 1991;561:106.
244 Brain injury: neurochemical aspects. In: Central Nervous System Trauma Status Report. Bethesda, MD: National Institutes of Health; 1985.
245 Kalimo H, Rehncrona S, Soderfeldt B, et al. Brain lactic acidosis and ischemic cell damage: 2. Histopathology. J Cereb Blood Flow Metab. 1981;1:313.
246 Paljarvi L. Brain lactic acidosis and ischemic cell damage: a topographic study with high-resolution light microscopy of early recovery in a rat model of severe incomplete ischemia. Acta Neuropathol. 1984;64:89.
247 Vink R, Head VA, Rogers PJ, et al. Mitochondrial metabolism following traumatic brain injury in rats. J Neurotrauma. 1990;7:21.
248 De Salles AA, Muizelaar JP, Young HF. Hyperglycemia, cerebrospinal fluid lactic acidosis, and cerebral blood flow in severely head-injured patients. Neurosurgery. 1987;21:45.
249 Glenn TC, Kelly DF, Boscardin WJ, et al. Energy dysfunction as a predictor of outcome after moderate or severe head injury: indices of oxygen, glucose, and lactate metabolism. J Cereb Blood Flow Metab. 2003;23:1239.
250 Vespa PM, McArthur D, O’Phelan K, et al. Persistently low extracellular glucose correlates with poor outcome 6 months after human traumatic brain injury despite a lack of increased lactate: a microdialysis study. J Cereb Blood Flow Metab. 2003;23:865.
251 Bergsneider M, Hovda DA, Lee SM, et al. Dissociation of cerebral glucose metabolism and level of consciousness during the period of metabolic depression following human traumatic brain injury. J Neurotrauma. 2000;17:389.
252 Jaggi JL, Obrist WD, Gennarelli TA, et al. Relationship of early cerebral blood flow and metabolism to outcome in acute head injury. J Neurosurg. 1990;72:176.
253 Dusick JR, Glenn TC, Lee WN, et al. Increased pentose phosphate pathway flux after clinical traumatic brain injury: a [1,2-(13)C(2)]glucose labeling study in humans. J Cereb Blood Flow Metab. 2007;27:1593-1602.
254 Arundine M, Tymianski M. Molecular mechanisms of glutamate-dependent neurodegeneration in ischemia and traumatic brain injury. Cell Mol Life Sci. 2004;61:657.
255 Hall ED, Detloff MR, Johnson K, et al. Peroxynitrite-mediated protein nitration and lipid peroxidation in a mouse model of traumatic brain injury. J Neurotrauma. 2004;21:9.
256 Lewen A, Matz P, Chan PH. Free radical pathways in CNS injury. J Neurotrauma. 2000;17:871.
257 Adams J, Graham D. The pathology of blunt head injury. In: Critchley M, O’Leary J, Jennet B, editors. Scientific Foundation of Neurology. London: W Heineman; 1972:488.
258 Jones TH, Morawetz RB, Crowell RM, et al. Thresholds of focal cerebral ischemia in awake monkeys. J Neurosurg. 1981;54:773.
259 Schroder ML, Muizelaar JP, Kuta AJ, et al. Thresholds for cerebral ischemia after severe head injury: relationship with late CT findings and outcome. J Neurotrauma. 1996;13:17.
260 Langfitt T, Obrist W. Cerebral blood flow and metabolism after intracranial trauma. In: Krayenbuhl H, editor. Progress in Neurological Surgery. Basel: S Karger; 1981:10.
261 Vespa P, Bergsneider M, Hattori N, et al. Metabolic crisis without brain ischemia is common after traumatic brain injury: a combined microdialysis and positron emission tomography study. J Cereb Blood Flow Metab. 2005;25:763.
262 Bruce DA, Langfitt TW, Miller JD, et al. Regional cerebral blood flow, intracranial pressure, and brain metabolism in comatose patients. J Neurosurg. 1973;38:131.
263 Cold GE, Jensen FT, Malmros R. The cerebrovascular CO2 reactivity during the acute phase of brain injury. Acta Anaesthesiol Scand. 1977;21:222.
264 Cold GE, Jensen FT, Malmros R. The effects of PaCO2 reduction on regional cerebral blood flow in the acute phase of brain injury. Acta Anaesthesiol Scand. 1977;21:359.
265 Enevoldsen EM, Jensen FT. Reproducibility of regional cerebral blood flow measurements in acute severe head injury. J Neurosurg. 1978;49:366.
266 Enevoldsen EM, Jensen FT. Autoregulation and CO2 responses of cerebral blood flow in patients with acute severe head injury. J Neurosurg. 1978;48:689.
267 Fieschi C, Battistini N, Beduschi A, et al. Regional cerebral blood flow and intraventricular pressure in acute head injuries. J Neurol Neurosurg Psychiatry. 1974;37:1378.
268 Roberston C. Nitrous oxide saturation technique for CBF measurement. In: Narayan RK, Wilberger JE, Povlishock JT, editors. Neurotrauma. New York: McGraw-Hill; 1996:487.
269 Lassen NA. The luxury-perfusion syndrome and its possible relation to acute metabolic acidosis localised within the brain. Lancet. 1966;2:1113.
270 Obrist WD, Langfitt TW, Jaggi JL, et al. Cerebral blood flow and metabolism in comatose patients with acute head injury. Relationship to intracranial hypertension. J Neurosurg. 1984;61:241.
271 Marion DW, Darby J, Yonas H. Acute regional cerebral blood flow changes caused by severe head injuries. J Neurosurg. 1991;74:407.
272 Zane C, Khanna R, Martin NA. Patterns of cerebral blood flow and transcranial Doppler ultrasound velocities following head injury [abstract]. J Neurosurg. 1992;76:399A.
273 Bruce DA, Alavi A, Bilaniuk L, et al. Diffuse cerebral swelling following head injuries in children: the syndrome of “malignant brain edema.”. J Neurosurg. 1981;54:170.
274 Bouma GJ, Muizelaar JP, Fatouros P. Pathogenesis of traumatic brain swelling: role of cerebral blood volume. Acta Neurochir Suppl. 1998;71:272.
275 Skippen P, Seear M, Poskitt K, et al. Effect of hyperventilation on regional cerebral blood flow in head-injured children. Crit Care Med. 1997;25:1402.
276 Suzuki T, Ishii I, Inoue S, et al. [Reproducibility of a non-invasive quantitative assessment of cerebral blood flow using 123I-IMP SPECT]. Kaku Igaku. 1990;27:1331.
277 Zwienenberg M, Muizelaar JP. Severe pediatric head injury: the role of hyperemia revisited. J Neurotrauma. 1999;16:937.
278 Chiron C, Raynaud C, Maziere B, et al. Changes in regional cerebral blood flow during brain maturation in children and adolescents. J Nucl Med. 1992;33:696.
279 Cox SB, Woolsey TA, Rovainen CM. Localized dynamic changes in cortical blood flow with whisker stimulation corresponds to matched vascular and neuronal architecture of rat barrels. J Cereb Blood Flow Metab. 1993;13:899.
280 Gonzalez MF, Sharp FR. Vibrissae tactile stimulation: (14C) 2-deoxyglucose uptake in rat brainstem, thalamus, and cortex. J Comp Neurol. 1985;231:457.
281 Hovda DA, Lee SM, Smith ML, et al. The neurochemical and metabolic cascade following brain injury: moving from animal models to man. J Neurotrauma. 1995;12:903.
282 Bouma GJ, Muizelaar JP. Cerebral blood flow, cerebral blood volume, and cerebrovascular reactivity after severe head injury. J Neurotrauma. 1992;9(suppl 1):S333.
283 Overgaard J, Tweed WA. Cerebral circulation after head injury. 1. Cerebral blood flow and its regulation after closed head injury with emphasis on clinical correlations. J Neurosurg. 1974;41:531.
284 Allen GS. Cerebral arterial spasm. Part 8: The treatment of delayed cerebral arterial spasm in human beings. Surg Neurol. 1976;6:71.
285 Allen GS, Gross CJ. Cerebral arterial spasm. Part 7: In vitro effects of alpha adrenergic agents on canine arteries from six anatomical sites and six blocking agents on serotonin-induced contractions of the canine basilar artery. Surg Neurol. 1976;6:63.
286 Hoff JT, Harper M, Sengupta D, et al. Effect of alpha-adrenergic blockade on response of cerebral circulation to hypocapnia in the baboon. Lancet. 1972;2:1337.
287 Taylor P. Ganglionic stimulating and blocking agents. In: Gilman AG, Goodman L, Rall T, editors. The Pharmacological Basis of Therapeutics. New York: Macmillan; 1985:215.
288 Wei EP, Dietrich WD, Povlishock JT, et al. Functional, morphological, and metabolic abnormalities of the cerebral microcirculation after concussive brain injury in cats. Circ Res. 1980;46:37.
289 Beckman JS, Beckman TW, Chen J, et al. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci U S A. 1990;87:1620.
290 Ignarro L, Byrns R, Wood K. Pharmacological and biochemical properties of endothelium-derived relaxing factor (EDRF): evidence that EDRF is closely related to nitric oxide (NO) radical. Circulation. 1986;74:287.
291 Ellis EF, Dodson LY, Police RJ. Restoration of cerebrovascular responsiveness to hyperventilation by the oxygen radical scavenger n-acetylcysteine following experimental traumatic brain injury. J Neurosurg. 1991;75:774.
292 Kontos HA, Wei EP. Oxygen-dependent mechanisms in cerebral autoregulation. Ann Biomed Eng. 1985;13:329.
293 Forsyth LL, Liu-DeRyke X, Parker DJr, et al. Role of hypertonic saline for the management of intracranial hypertension after stroke and traumatic brain injury. Pharmacotherapy. 2008;28:469.
294 Himmelseher S. Hypertonic saline solutions for treatment of intracranial hypertension. Curr Opin Anaesthesiol. 2007;20:414.
295 Tseng MY, Al-Rawi PG, Pickard JD, et al. Effect of hypertonic saline on cerebral blood flow in poor-grade patients with subarachnoid hemorrhage. Stroke. 2003;34:1389.
296 White H, Cook D, Venkatesh B. The role of hypertonic saline in neurotrauma. Eur J Anaesthesiol Suppl. 2008;42:104.
297 White H, Cook D, Venkatesh B. The use of hypertonic saline for treating intracranial hypertension after traumatic brain injury. Anesth Analg. 2006;102:1836.
298 Brain Trauma Foundation, American Association of Neurological Surgeons, Congress of Neurological Surgeons. Guidelines for the management of severe traumatic brain injury: hyperosmolar therapy. J Neurotrauma. 2007;24:S14.
299 Cold GE, Jensen FT. Cerebral autoregulation in unconscious patients with brain injury. Acta Anaesthesiol Scand. 1978;22:270.
300 Enevoldsen EM, Cold G, Jensen FT, et al. Dynamic changes in regional CBF, intraventricular pressure, CSF pH and lactate levels during the acute phase of head injury. J Neurosurg. 1976;44:191.
301 Messeter K, Nordstrom CH, Sundbarg G, et al. Cerebral hemodynamics in patients with acute severe head trauma. J Neurosurg. 1986;64:231.
302 Zimmerman RA, Muizelaar JP, Wei EP, et al. Acute cerebral arteriolar responses following cold injury. In: Cervos-Navarro J, Ferszt R, editors. Stroke and Microcirculation. New York: Raven Press; 1987:303.
303 Becker DP, Miller JD, Ward JD, et al. The outcome from severe head injury with early diagnosis and intensive management. J Neurosurg. 1977;47:491.
304 Marmarou A, Anderson RL, Ward JD, et al. Impact of ICP instability and hypotension on outcome in patients with severe head trauma. J Neurosurg. 1991;75:S159.
305 Schreber M, Aoki N, Scott BG, et al. Determinants of mortality in patients with severe blunt injury. Arch Surg. 2002;137:285.
306 Eisenberg HM, Frankowski RF, Contant CF, et al. High-dose barbiturate control of elevated intracranial pressure in patients with severe head injury. J Neurosurg. 1988;69:15.
307 Treggiari MM, Schutz N, Yanez ND, et al. Role of intracranial pressure values and patterns in predicting outcome in traumatic brain injury: a systematic review. Neurocrit Care. 2007;6:104.
308 Marmarou A, Anderson R, Ward J. The traumatic coma databank: monitoring of ICP. In: Hoff JT, Betz A, editors. Intracranial Pressure, Vol VII. New York: Springer-Verlag; 1989:549.
309 Marshall LF, Smith RW, Shapiro HM. The outcome with aggressive treatment in severe head injuries. Part II: acute and chronic barbiturate administration in the management of head injury. J Neurosurg. 1979;50:26.
310 Cohadon F, Castel J, Nouillant A. Volume pressure relationship in clinical and experimental condition of raised ICP. In: Lundgerg N, Ponten U, Brock M, editors. Intracranial Pressure, Vol II. Berlin: Springer-Verlag; 1975:107.
311 Marmarou A, Shulman K, Rosende RM. A nonlinear analysis of the cerebrospinal fluid system and intracranial pressure dynamics. J Neurosurg. 1978;48:332.
312 Ekstedt J. CSF hydrodynamic studies in man. 2. Normal hydrodynamic variables related to CSF pressure and flow. J Neurol Neurosurg Psychiatry. 1978;41:345.
313 Kellie G. On death from cold, and on congestions of the brain: an account of the appearances observed in the dissection of two of three individuals presumed to have perished in the storm of 3rd November. 1821; with some reflections on the pathology of the brain. Trans Med Chir Soc Edinb. 84, 1824.
314 Monro A. Observations on the Structure and Function of the Nervous System. Edinburgh: Creech and Johnson; 1823.
315 Czosnyka M, Guazzo E, Whitehouse M, et al. Significance of intracranial pressure waveform analysis after head injury. Acta Neurochir (Wien). 1996;138:531.
316 Czosnyka M, Price DJ, Williamson M. Monitoring of cerebrospinal dynamics using continuous analysis of intracranial pressure and cerebral perfusion pressure in head injury. Acta Neurochir (Wien). 1994;126:113.
317 Wilkinson HA, Schuman N, Ruggiero J. Nonvolumetric methods of detecting impaired intracranial compliance or reactivity: pulse width and wave form analysis. J Neurosurg. 1979;50:758.
318 Shapiro K, Marmarou A, Shulman K. Characterization of clinical CSF dynamics and neural axis compliance using the pressure-volume index: I. The normal pressure-volume index. Ann Neurol. 1980;7:508.
319 Marmarou A. Pathophysiology of intracranial pressure. In: Narayan RK, Wilberger JE, Povlishock JT, editors. Neurotrauma. New York: McGraw-Hill; 1996:413.
320 Yoshihara M, Bandoh K, Marmarou A. Cerebrovascular carbon dioxide reactivity assessed by intracranial pressure dynamics in severely head injured patients. J Neurosurg. 1995;82:386.
321 Miller JD, Garibi J, Pickard JD. Induced changes of cerebrospinal fluid volume. Effects during continuous monitoring of ventricular fluid pressure. Arch Neurol. 1973;28:265.
322 Miller JD, Pickard JD. Intracranial volume pressure studies in patients with head injury. Injury. 1974;5:265.
323 Marmarou A, Maset AL, Ward JD, et al. Contribution of CSF and vascular factors to elevation of ICP in severely head-injured patients. J Neurosurg. 1987;66:883.
324 Klatzo I. Presidential address. Neuropathological aspects of brain edema. J Neuropathol Exp Neurol. 1967;26:1.
325 Barzo P, Marmarou A, Fatouros P, et al. Biphasic pathophysiological response of vasogenic and cellular edema in traumatic brain swelling. Acta Neurochir Suppl. 1997;70:119.
326 Barzo P, Marmarou A, Fatouros P, et al. Contribution of vasogenic and cellular edema to traumatic brain swelling measured by diffusion-weighted imaging. J Neurosurg. 1997;87:900.
327 Ritter AM, Muizelaar JP, Barnes T, et al. Brain stem blood flow, pupillary response, and outcome in patients with severe head injuries. Neurosurgery. 1999;44:941.
328 Rowan JO, Teasdale G. Brain stem blood flow during raised intracranial pressure. Acta Neurol Scand Suppl. 1977;64:520.
329 Fearnside MR, Cook RJ, McDougall P, et al. The Westmead Head Injury Project outcome in severe head injury. A comparative analysis of pre-hospital, clinical and CT variables. Br J Neurosurg. 1993;7:267.
330 Manley G, Knudson MM, Morabito D, et al. Hypotension, hypoxia, and head injury: frequency, duration, and consequences. Arch Surg. 2001;136:1118.
331 Murray GD, Butcher I, McHugh GS, et al. Multivariable prognostic analysis in traumatic brain injury: results from the IMPACT study. J Neurotrauma. 2007;24:329.
332 Jones PA, Andrews PJ, Midgley S, et al. Measuring the burden of secondary insults in head-injured patients during intensive care. J Neurosurg Anesthesiol. 1994;6:4.
333 Vassar MJ, Fischer RP, O’Brien PE, et al. A multicenter trial for resuscitation of injured patients with 7.5% sodium chloride. The effect of added dextran 70. The Multicenter Group for the Study of Hypertonic Saline in Trauma Patients. Arch Surg. 1993;128:1003.
334 Vassar MJ, Perry CA, Holcroft JW. Analysis of potential risk associated with 7.5% sodium chloride resuscitation of traumatic shock. Arch Surg. 1990;125:1309.
335 Butcher I, Maas AI, Lu J, et al. Prognostic value of admission blood pressure in traumatic brain injury: results from the IMPACT study. J Neurotrauma. 2007;24:294.
336 Stochetti N, Furlan A, Volta F. Hypoxemia and arterial hypotension at the accident scene in head injury. J Trauma. 1996;40:764.
337 Chi JH, Knudson MM, Vassar MJ, et al. Prehospital hypoxia affects outcome in patients with traumatic brain injury: a prospective multicenter study. J Trauma. 2006;61:1134.
338 Miller JD, Becker DP. Secondary insults to the injured brain. J R Coll Surg Edinb. 1982;27:292.
339 Ishige N, Pitts LH, Hashimoto T, et al. Effect of hypoxia on traumatic brain injury in rats: Part 1. Changes in neurological function, electroencephalograms, and histopathology. Neurosurgery. 1987;20:848.
340 Ishige N, Pitts LH, Pogliani L, et al. Effect of hypoxia on traumatic brain injury in rats: Part 2. Changes in high energy phosphate metabolism. Neurosurgery. 1987;20:854.
341 Kety SS, Schmidt CF. The effects of altered arterial tensions of carbon dioxide and oxygen on cerebral blood flow and cerebral oxygen consumption of normal young men. J Clin Invest. 1948;27:484.
342 Lewelt W, Jenkins LW, Miller JD. Effects of experimental fluid-percussion injury of the brain on cerebrovascular reactivity of hypoxia and to hypercapnia. J Neurosurg. 1982;56:332.
343 Behr R, Erlingspiel D, Becker A. Early and longtime modifications of temperature regulation after severe head injury: prognostic implications. Ann N Y Acad Sci. 1997;813:722.
344 Chatzipanteli K, Alonso OF, Kraydieh S, et al. Importance of posttraumatic hypothermia and hyperthermia on the inflammatory response after fluid percussion brain injury: biochemical and immunocytochemical studies. J Cereb Blood Flow Metab. 2000;20:531.
345 Dietrich WD. The importance of brain temperature in cerebral injury. J Neurotrauma. 1992;9:S475.
346 Dietrich WD, Alonso O, Halley M, et al. Delayed post-traumatic brain hyperthermia worsens outcome after fluid percussion brain injury: a light and electron microscopic study in rats. Neurosurgery. 1996;38:533.
347 Thompson HJ, Tkacs NC, Saatman KE, et al. Hyperthermia following traumatic brain injury: a critical evaluation. Neurobiol Dis. 2003;12:163.
348 Malkinson TJ, Veale WL, Cooper KE. Fever and intracranial pressures. Brain Res Bull. 1985;15:315.
349 Rossi S, Zanier ER, Mauri I, et al. Brain temperature, body core temperature, and intracranial pressure in acute cerebral damage. J Neurol Neurosurg Psychiatry. 2001;71:448.
350 Soukup J, Zauner A, Doppenberg EM, et al. The importance of brain temperature in patients after severe head injury: relationship to intracranial pressure, cerebral perfusion pressure, cerebral blood flow, and outcome. J Neurotrauma. 2002;19:559.
351 Stochetti N, Protti A, Lattuada M, et al. Impact of pyrexia on neurochemistry and cerebral oxygenation after acute brain injury. J Neurol Neurosurg Psychiatry. 2005;76:1135.
352 Jiang JY, Gao GY, Li WP, et al. Early indicators of prognosis in 846 cases of severe traumatic brain injury. J Neurotrauma. 2002;19:869.
353 Albrecht RF, Wass C, Lanier WL. Occurrence of potentially detrimental temperature alterations in hospitalized patients at risk for brain injury. Mayo Clin Proc. 1998;73:629.
354 Kilpatrick MM, Lowry DW, Firlik AD, et al. Hyperthermia in the neurosurgical intensive care unit. Neurosurgery. 2000;47:850.
355 Stochetti N, Rossi S, Zanier ER, et al. Pyrexia in head-injured patients admitted to intensive care. Intensive Care Med. 2002;28:1555.
356 Diringer MN, Reaven NL, Funk SE, et al. Elevated body temperature independently contributes to increased length of stay in neurologic intensive care unit patients. Crit Care Med. 2004;32:1489.
357 Van Beek JG, Mushkudiani NA, Steyerberg EW, et al. Prognostic value of admission laboratory parameters in traumatic brain injury: results from the IMPACT study. J Neurotrauma. 2007;24:315.
358 Leal-Noval SR, Munoz-Gomez M, Arellano-Orden V, et al. Impact of age of transfused blood on cerebral oxygenation in male patients with severe traumatic brain injury. Crit Care Med. 2008;36:1290.
359 Leal-Noval SR, Rincon-Ferrari MD, Marin-Niebla A, et al. Transfusion of erythrocyte concentrates produces a variable increment on cerebral oxygenation in patients with severe traumatic brain injury: a preliminary study. Intensive Care Med. 2006;32:1733.
360 Smith MJ, Stiefel MF, Magge S, et al. Packed red blood cell transfusion increases local cerebral oxygenation. Crit Care Med. 2005;33:1104.
361 Carlson AP, Schermer CR, Lu SW. Retrospective evaluation of anemia and transfusion in traumatic brain injury. J Trauma. 2006;61:567.
362 Lam AM, Winn HR, Cullen BF, et al. Hyperglycemia and neurological outcome in patients with head injury. J Neurosurg. 1991;75:545.
363 Young B, Ott L, Dempsey R, et al. Relationship between admission hyperglycemia and neurological outcome of severely brain-injured patients. Ann Surg. 1989;210:466.
364 Rosner MJ, Newsome HH, Becker DP. Mechanical brain injury: the sympathoadrenal response. J Neurosurg. 1984;61:76.
365 Sieber FE, Traystman RJ. Special issues: glucose and the brain. Crit Care Med. 1992;20:104.
366 Robertson CS, Goodman JC, Narayan RK, et al. The effect of glucose administration on carbohydrate metabolism after head injury. J Neurosurg. 1991;74:43.
367 Rosner MJ, Becker DP. Experimental brain injury: successful therapy with the weak base, tromethamine. With an overview of CNS acidosis. J Neurosurg. 1984;60:961.
368 Wolf AL, Levi L, Marmarou A, et al. Effect of THAM upon outcome in severe head injury: a randomized prospective clinical trial. J Neurosurg. 1993;78:54.
369 Kafadar AM, Sanus GZ, Is M, et al. Prolonged elevation of magnesium in the cerebrospinal fluid of patients with severe head injury. Neurol Res. 2007;29:824.
370 Stippler M, Fischer MR, Puccio AM, et al. Serum and cerebrospinal fluid magnesium in severe traumatic brain injury outcome. J Neurotrauma. 2007;24:1347.
371 Temkin NR, Anderson GD, Winn HR, et al. Magnesium sulfate for neuroprotection after traumatic brain injury: a randomised controlled trial. Lancet Neurol. 2007;6:29.
372 Yoganandan N, Pintar FA, Sances AJr, et al. Biomechanics of skull fracture. J Neurotrauma. 1995;12:659.
373 Chesnut R. Treating raised intracranial pressure in head injury. In: Narayan RK, Wilberger JE, Povlishock JT, editors. Neurotrauma. New York: McGraw-Hill; 1996:445.
374 Schreiber M, Aoki N, Scott BG, et al. Determinants of mortality in patients with severe blunt injury. Arch Surg. 2002;137:285.
375 Muizelaar JP, Marmarou A, Ward JD, et al. Adverse effects of prolonged hyperventilation in patients with severe head injury: a randomized clinical trial. J Neurosurg. 1991;75:731.
376 Gopinath SP, Robertson CS, Contant CF, et al. Jugular venous desaturation and outcome after head injury. J Neurol Neurosurg Psychiatry. 1994;57:717.
377 Burke AM, Quest DO, Chien S, et al. The effects of mannitol on blood viscosity. J Neurosurg. 1981;55:550.
378 Muizelaar JP, Wei EP, Kontos HA, et al. Mannitol causes compensatory cerebral vasoconstriction and vasodilation in response to blood viscosity changes. J Neurosurg. 1983;59:822.
379 Muizelaar JP, Ward JD, Marmarou A, et al. Cerebral blood flow and metabolism in severely head-injured children. Part 2: Autoregulation. J Neurosurg. 1989;71:72.
380 Chesnut RM. Avoidance of hypotension: condition sine qua non of successful severe head-injury management. J Trauma. 1997;42:S4.
381 Rosner MJ, Rosner SD, Johnson AH. Cerebral perfusion pressure: management protocol and clinical results. J Neurosurg. 1995;83:949.
382 Clifton GL, Miller ER, Choi SC, et al. Fluid thresholds and outcome from severe brain injury. Crit Care Med. 2002;30:739.
383 Juul N, Morris GF, Marshall SB, et al. Intracranial hypertension and cerebral perfusion pressure: influence on neurological deterioration and outcome in severe head injury. The Executive Committee of the International Selfotel Trial. J Neurosurg. 2000;92:1.
384 Nordstrom CH, Reinstrup P, Xu W, et al. Assessment of the lower limit for cerebral perfusion pressure in severe head injuries by bedside monitoring of regional energy metabolism. Anesthesiology. 2003;98:809.
385 Juul N, Morris GF, Marshall SB, et al. Intracranial hypertension and cerebral perfusion pressure: influence on neurological deterioration and outcome in severe head injury. The Executive Committee of the International Selfotel Trial. J Neurosurg. 2000;92:1.
386 Bouma GJ, Muizelaar JP. Relationship between cardiac output and cerebral blood flow in patients with intact and with impaired autoregulation. J Neurosurg. 1990;73:368.
387 Bouma GJ, Muizelaar JP, Bandoh K, et al. Blood pressure and intracranial pressure-volume dynamics in severe head injury: relationship with cerebral blood flow. J Neurosurg. 1992;77:15.
388 Contant CF, Valadka AB, Gopinath SP, et al. Adult respiratory distress syndrome: a complication of induced hypertension after severe head injury. J Neurosurg. 2001;95:560.
389 Robertson CS, Valadka AB, Hannay HJ, et al. Prevention of secondary ischemic insults after severe head injury. Crit Care Med. 1999;27:2086.
390 Kee DB, Wood JH. Rheology of the cerebral circulation. Neurosurgery. 1984;15:125.
391 Hebert PC, Wells G, Blajchman MA, et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. Transfusion Requirements in Critical Care Investigators, Canadian Critical Care Trials Group. N Engl J Med. 1999;340:409.
392 McIntyre LA, Fergusson DA, Hutchison JS, et al. Effect of a liberal versus restrictive transfusion strategy on mortality in patients with moderate to severe head injury. Neurocrit Care. 2006;5:4.
393 Marshall LF, Smith RW, Shapiro HM. The outcome with aggressive treatment in severe head injuries. Part I: the significance of intracranial pressure monitoring. J Neurosurg. 1979;50:20.
394 Schwartz ML, Tator CH, Rowed DW, et al. The University of Toronto head injury treatment study: a prospective, randomized comparison of pentobarbital and mannitol. Can J Neurol Sci. 1984;11:434.
395 Ward JD, Becker DP, Miller JD, et al. Failure of prophylactic barbiturate coma in the treatment of severe head injury. J Neurosurg. 1985;62:383.
396 Tokutomi T, Morimoto K, Miyagi T, et al. Optimal temperature for the management of severe traumatic brain injury: effect of hypothermia on intracranial pressure, systemic and intracranial hemodynamics, and metabolism. Neurosurgery. 2003;52:102.
397 Brain Trauma Foundation, American Association of Neurological Surgeons, Congress of Neurological Surgeons. Guidelines for the management of severe traumatic brain injury: Prophylactic hypothermia. J Neurotrauma. 2007;24:S21.
398 Aibiki M, Maekawa S, Yokono S. Moderate hypothermia improves imbalances of thromboxane A2 and prostaglandin I2 production after traumatic brain injury in humans. Crit Care Med. 2000;28:3902.
399 Clifton GL, Allen S, Barrodale P, et al. A phase II study of moderate hypothermia in severe brain injury. J Neurotrauma. 1993;10:263.
400 Clifton GL, Miller ER, Choi SC, et al. Lack of effect of induction of hypothermia after acute brain injury. N Engl J Med. 2001;344:556.
401 Jiang J, Yu M, Zhu C. Effect of long-term mild hypothermia therapy in patients with severe traumatic brain injury: 1-year follow-up review of 87 cases. J Neurosurg. 2000;93:546.
402 Marion DW, Penrod LE, Kelsey SF, et al. Treatment of traumatic brain injury with moderate hypothermia. N Engl J Med. 1997;336:540.
403 Qiu WS, Liu WG, Shen H, et al. Therapeutic effect of mild hypothermia on severe traumatic brain injury. Chin J Traumatol. 2005;8:27.
404 Adelson PD, Ragheb J, Kanev P, et al. Phase II clinical trial of moderate hypothermia after severe traumatic brain injury in children. Neurosurgery. 2005;56:740.
405 Hutchison JS, Ward RE, Lacroix J, et al. Hypothermia therapy after traumatic brain injury in children. N Engl J Med. 2008;358:2447.
406 Clifton GL, Allen S, Barrodale P, et al. A phase II study of moderate hypothermia in severe brain injury. J Neurotrauma. 1993;10:263.
407 McIntyre LA, Fergusson DA, Hebert PC, et al. Prolonged therapeutic hypothermia after traumatic brain injury in adults: a systematic review. JAMA. 2003;289:2992.