[level-membership-for-neurology-category]
1 Clinical Neurologic Evaluation
Approach to the Neurologic Evaluation
Overview and Basic Tenets
The subsequent definition of the formal examination may be subdivided into a few major sections. Speech and language are assessed during the history taking. The cognitive part of the examination is often clearly defined with the initial history and often does not require formal mental status testing. However there are a number of clinical neurologic settings where this evaluation is very time consuming and complicated; Chapter 2 is dedicated to this aspect of the patient evaluation. However, when there is no clinical suspicion of either a cognitive or language dysfunction, these more formal testing modalities are not specifically required.
Here the multisystem neurologic examination provides a careful basis for most essential clinical evaluations. Neurologists in training and their colleagues in practice cannot expect to test all possible cognitive elements in each patient that they evaluate. Certain basic elements are required; most of these are readily observable or elicited during initial clinical evaluation. These include documentation of language function, affect, concentration, orientation, and memory. When concerned about the patient’s cognitive abilities, the neurologist must elicit evidence of an apraxia or agnosia and test organizational skills. Once language and cognitive functions are assessed, the neurologist dedicates the remaining portion of the exam to the examination of many functions. These include visual fields, cranial nerves (CNs) (Fig. 1-1), muscle strength, muscle stretch reflexes (MSRs), plantar stimulation, coordination, gait and equilibrium, as well as sensory modalities. These should routinely be examined in an organized rote fashion in order not to overlook an important part of the examination. The patient’s general health, nutritional status, and cardiac function, including the presence or absence of significant arrhythmia, heart murmur, hypertension, or signs of congestive failure, should be noted. If the patient is encephalopathic, it is important to search for subtle signs of infectious, hepatic, renal, or pulmonary disease.
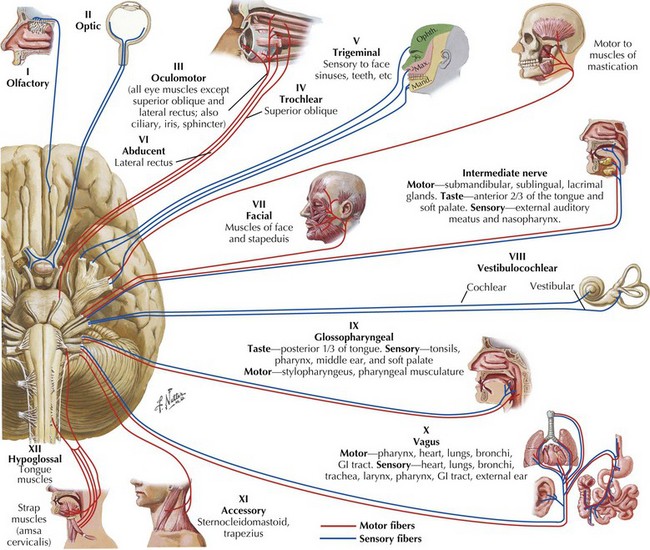
Cranial Nerves: An Introduction
The 12 CNs subserve multiple types of neurologic function (see Fig. 1-1). The cranial nerves are formed by afferent sensory fibers, motor efferent fibers, or mixed fibers traveling to and from brainstem nuclei (Fig. 1-2A and B).
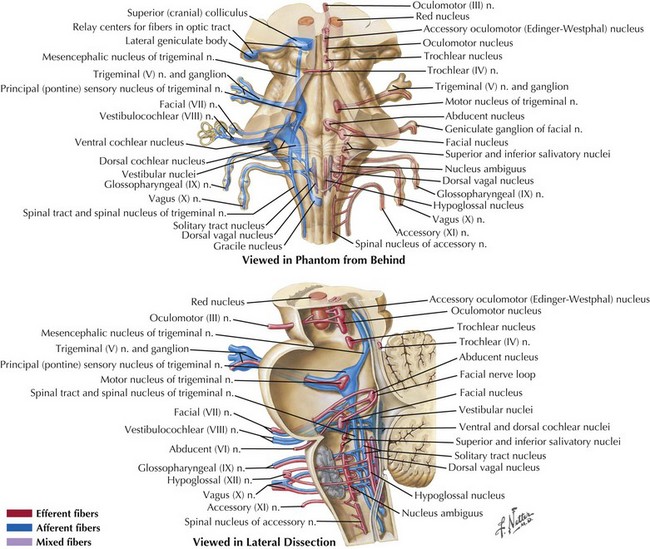
Cranial Nerve Testing
II Optic Nerve
Of all the human sensations, the ability to see one’s family and friends, to read, and appreciate the beauties of nature, it is difficult to imagine life without vision, something that is totally dependent on the second cranial nerve. Obviously many individuals, such as Helen Keller, have vigorously and successfully conquered the challenge of being blind; however, given the choice, vision is one of the most precious of all animal sensations. “Blurred” vision is a common but relatively nonspecific symptom that may relate to dysfunction anywhere along the visual pathway (Fig. 1-3). When examining optic nerve function, it is important to identify any concomitant ocular abnormalities such as proptosis, ptosis, scleral injection (congestion), tenderness, bruits, and pupillary changes.
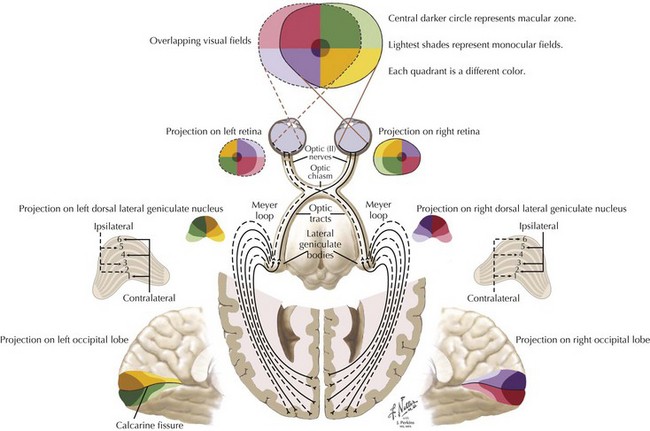
Most visual field changes have localizing value: specific location of the loss, its shape, border sharpness (i.e., how quickly across the field the values change from abnormal to normal). Its concordance with the visual field of the other eye tends to implicate specific areas of the visual system. Localization is possible because details of anatomic organization at different levels predispose to particular types of loss (see Chapter 4).
When one examines the pupils, their shape and size need to be recorded. A side-to-side difference of no more than 1 mm in otherwise round pupils is acceptable as a normal variant. Pupillary responses are tested with a bright flashlight and are primarily mediated by the autonomic innervation of the eye (Fig. 1-4). A normal pupil reacts to light stimulus by constricting with the contralateral constriction of the unstimulated pupil as well. These responses are called the direct and consensual reactions, respectively, and are mediated through parasympathetic innervation to the pupillary sphincter from the Edinger-Westphal nucleus along the oculomotor nerve. The pupils also constrict when shifting focus from a far to a near object (accommodation) and during convergence of the eyes, as when patients are asked to look at their nose.
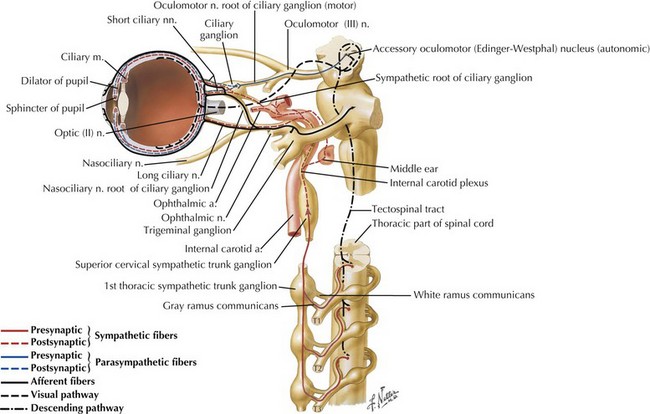
A number of pathophysiologic mechanisms lead to mydriasis (pupillary dilatation) (Table 1-1). Atropine-like eye drops, often used for their ability to produce pupillary dilation, inadvertent ocular application of certain nebulized bronchodilators, and placement of a scopolamine anti-motion patch with inadvertent leak into the conjunctiva are occasionally overlooked as potential causes for an otherwise asymptomatic, dilated, poorly reactive pupil. Other medications may also lead to certain atypical light reactions. The presence of bilateral dilated pupils, in an otherwise neurologically intact patient, is unlikely to reflect significant neuropathology. In contrast, the presence of prominent pupillary constriction most likely reflects the use of narcotic analogs or parasympathomimetic drugs, such as those typically used to treat glaucoma.
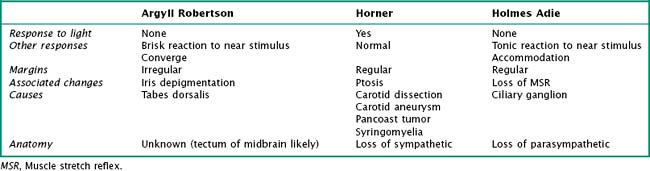
Horner Syndrome
Sympathetic efferent fibers originate within the hypothalamus and traverse the brainstem and cervical spinal cord, then exit the upper thoracic levels and course rostrally to reach the superior cervical ganglia (see Fig. 1-4). Subsequently, these sympathetic fibers track with the carotid artery within the neck to reenter the cranium and subsequently reach their destination innervating the eye’s pupillodilator musculature. Typically, patients with Horner syndrome have an ipsilateral loss of sweating in the face (anhidrosis), a constricted pupil (miosis), and an upper lid droop from loss of innervation to Muller’s muscle, a small smooth muscle lid elevator (ptosis). The levator palpebra superioris, a striated muscle innervated by the oculomotor nerve CN-III, is not affected (Fig. 1-5).
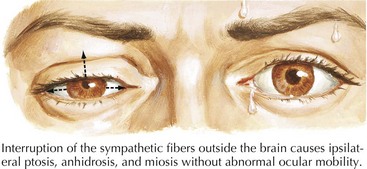
Optic Fundus
Papilledema is characterized by elevation and blurring of the optic disk, absence of venous pulsations, and hemorrhages adjacent to and on the disk (Fig. 1-6). The finding of papilledema indicates increased intracranial pressure of any cause, including brain tumors, subarachnoid hemorrhage, metabolic processes, pseudotumor cerebri, and venous sinus thrombosis.
III, IV, VI Oculomotor, Trochlear, and Abducens Nerves
In order to identify isolated EOM dysfunction, it is most accurate to test each eye individually describing the observed specific loss of EOM function. For example, when the eye cannot be turned laterally, the condition is labeled as an abduction paresis, as opposed to CN-VI palsy. This is because the responsible lesion can be at any one of three sites, namely, cranial nerve, neuromuscular junction, or muscle per se. A more detailed assessment of these cranial nerves is available in Section II, Chapter 5.
The medial longitudinal fasciculus (MLF) is responsible for controlling EOM function because it provides a means to modify central horizontal conjugate gaze circuits. The medial longitudinal fasciculus connects CN-III on one side and CN-VI on the opposite side. Understanding the circuit of horizontal conjugate gaze helps clinicians appreciate the relation between the frontal eye fields and the influence it exerts on horizontal conjugate gaze (see Fig. 1-6) as well the reflex relation between the ocular and vestibular systems (Fig. 1-7).
Ice-water caloric stimulation provides another option to study vestibular ocular MLF pathways. This is primarily used for the examination of comatose patients; on very rare occasions, it is extremely helpful for rousing a patient presenting with a suspected nonorganic, that is, feigned coma. Patients are placed at an elevation of approximately 45°. Next, the tympanic membranes are checked for intactness, and then 25–50 mL of ice water is gradually infused into each ear. A normal response in the awake patient, after left ear stimulation, is to observe slow deviation of the eyes to the left followed by rapid movement (nystagmus) to the right (see Fig. 1-10). In contrast, the comatose patient with an intact brainstem has a persistent ipsilateral deviation of the eyes to the site of stimulation with loss of the rapid eye movement component to the opposite side.
V Trigeminal Nerve
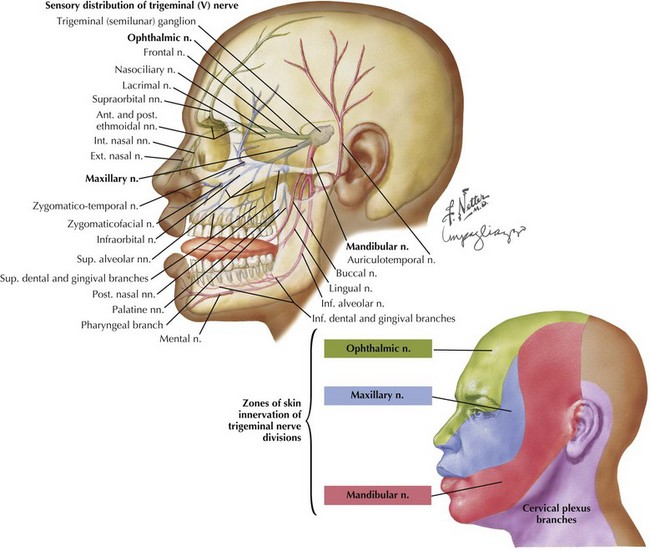
Our ability to perceive various stimuli applied to the face depends almost entirely on this nerve; whether as a warning to protect oneself from subzero cold, something potentially threatening to our eyesight, or the pleasurable sensation from the kiss of a beloved one, all forms of sensations applied to the face are tracked to our brain through the trigeminal nerve (Fig. 1-8). The primary sensory portion of this nerve has three divisions, ophthalmic, maxillary, and mandibular; they respectively supply approximately one third of the face from top to bottom, as well as the anterior aspects of the scalp. The angle of the jaw is spared within the trigeminal mandibular division territory. This provides an important landmark to differentiate patients with conversion disorders or obvious secondary gain as they are not anatomically sophisticated and will report they have lost sensation in this.
VII Facial Nerve
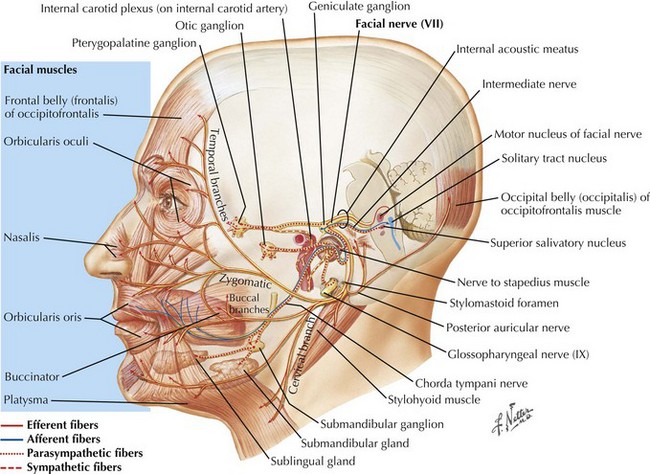
Facial expression is one of our very important innate human attributes allowing one to demonstrate a very broad spectrum of human emotions, especially happiness and sorrow; these are primarily dependent on the facial nerve (Fig. 1-9). The motor functions of CN-VII are tested by asking patients to wrinkle their forehead, close their eyes, and smile. Whistling and puffing up the cheeks are other techniques to test for subtle weakness. When unilateral peripheral weakness affects the facial nerve after it leaves the brainstem, the face may look “ironed out,” and when the patient smiles, the contralateral healthy facial muscle pulls up the opposite half of the mouth while the affected side remains motionless. Patients often cannot keep water in their mouths, and saliva may constantly drip from the paralyzed side. With peripheral CN-VII palsies, patients are also unable to close their ipsilateral eye or wrinkle their foreheads on the affected side. However, although the lid cannot close, the eyeball rolls up into the head, removing the pupil from observation. This is known as the Bell phenomena.
VIII Cochlear and Vestibular Nerves (Auditory [Cochlear] Nerve)
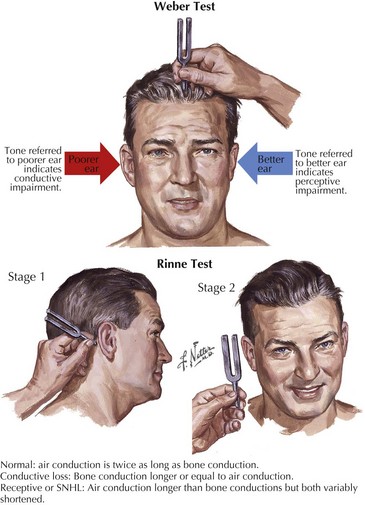
Initially a vibrating tuning fork is placed on the vertex of the skull, Weber test, allowing bone conduction to be assessed. Here the patient is asked to decide whether one ear perceives the sound created by the vibration better than the other (Fig. 1-11). If the patient has nerve deafness, the vibrations are still appreciated more in the normal ear. In contrast, with conduction deafness, the vibrations are better appreciated in the abnormal ear.
Vestibular Nerve
The vestibular system can be tested indirectly by evaluating for nystagmus during testing of ocular movements or by positional techniques, such as the Barany maneuver, that induce nystagmus in cases of benign positional vertigo (BPV) where inner ear dysfunction is caused by otolith displacement into the semicircular canals (Fig. 1-12). Here the patient is seated on an examining table and the eyes are observed for the presence of spontaneous nystagmus. If none is present, the examiner rapidly lays the patient back down, with the head slightly extended and concomitantly turning the head laterally. If after a few seconds’ delay, the patient develops the typical symptoms of vertigo with a characteristic delayed rotary, eventually fatiguing nystagmus, the study is positive.
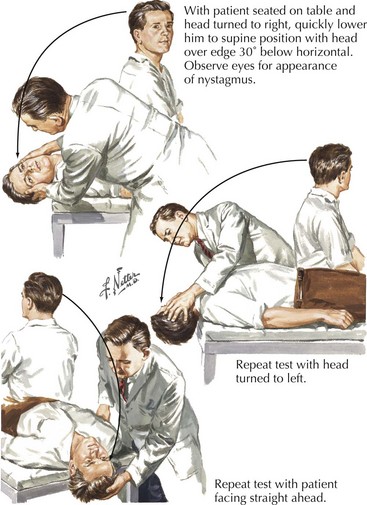
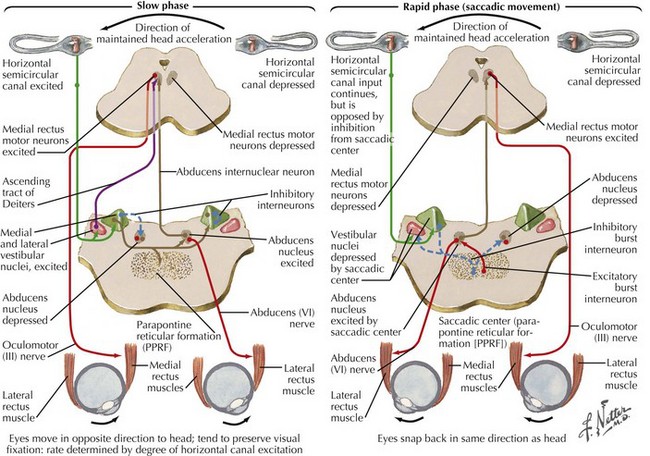
Eye movements depend on two primary components, the induced voluntary frontal eye fields and the primary reflex-driven vestibular–ocular movement controlled by multiple connections (Fig. 1-10; see also Fig. 1-7). The ability to maintain conjugate eye movements and a visual perspective on the surrounding world is an important brainstem function. It requires inputs from receptors in muscles, joints, and the cupulae of the inner ear. Therefore, when the patient has dysfunction involving any portion of the vestibular-ocular or cerebellar axis, the maintenance of basic visual orientation is challenged. Nystagmus is a compensatory process that attempts to help maintain visual fixation.
Traditionally, when one describes nystagmus, the fast phase direction becomes the designated title (see Fig. 1-10). For example, left semicircular canal stimulation produces a slow nystagmus to the left, with a fast component to the right. As a result, the nystagmus is referred to as right beating nystagmus. Direct stimulation of the semicircular canals or its direct connections, that is, the vestibular nuclei, often induces a torsional nystagmus. This is described as clockwise or counterclockwise, according to the fast phase.
IX, X, XI Glossopharyngeal, Vagus, and Accessory Nerves
The most common complaints related to glossopharyngeal-vagal system dysfunction include swallowing difficulties (dysphagia) and changes in voice (dysphonia). A patient with a glossopharyngeal nerve paresis presents with flattening of the palate on the affected side. When the patient is asked to produce a sound, the uvula is drawn to the unaffected side (Fig. 1-13). Indirect mirror examination of the vocal cords may demonstrate paralysis of the ipsilateral cord. The traditional test for gag reflex, placing a tongue depressor on the posterior pharynx, is of equivocal significance at best, because the gag response varies significantly and patients evidence wide varieties of tolerance to this stimulus. Preservation of swallowing reflexes is best tested by giving the patient 30 mL of fluid to drink through a straw while seated at 90°. Patients with compromised swallowing reflexes develop a “wet cough” and regurgitate fluids through their nose. Intracranial or proximal spinal accessory nerve damage limits the ability to turn the head to the opposite side (weakness of the ipsilateral sternocleidomastoid muscles and trapezius muscle). More distal accessory nerve damage is most commonly seen following surgical misadventures during biopsying a lymph node from the posterior triangle of the neck, sparing the sternocleidomastoid but affecting the trapezius, causing dysfunction and winging of the scapula.
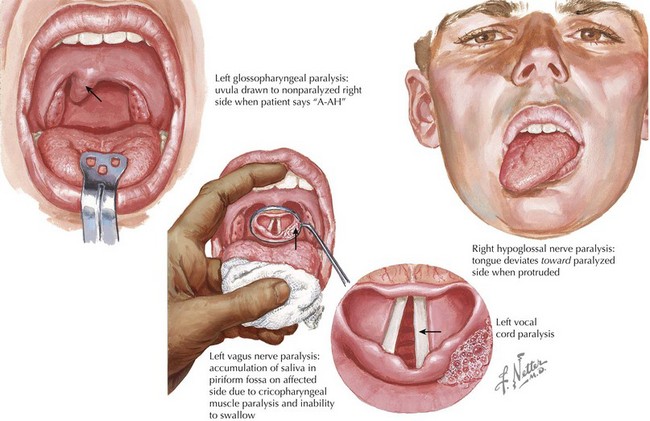
XII Hypoglossal Nerve
Damage to the hypoglossal nucleus or its nerve produces tongue atrophy and fasciculations The fasciculations usually are seen best on the lateral aspects of the tongue. If the nerve damage is unilateral, the tongue often deviates to that side (see Fig. 1-13). Two means to test for subtle weakness include asking the patient to push against a tongue depressor held by the examiner and having the patient push the tongue into the cheek.
Gait Evaluation
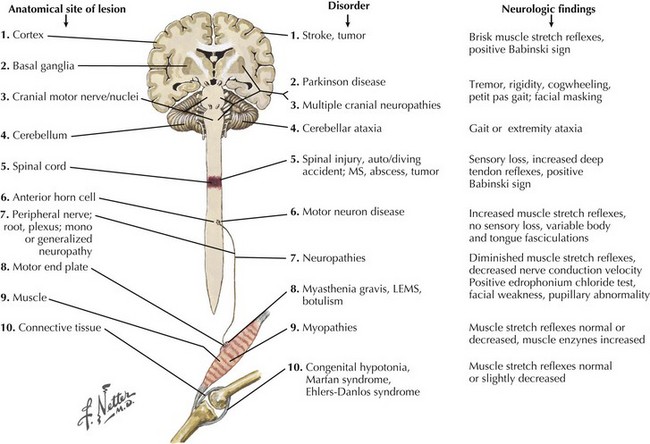
Whenever possible, the neurologic clinician is encouraged to personally greet the patient, watching them arise from their chair and initiate their gate. Next, before moving to the examination room the patient needs to be observed walking in the hallway. On occasion it is important to observe the patient on stairs particularly if there is a query about proximal weakness. A smooth gait requires multiple inputs from the cerebellum and primary motor and sensory systems. Gait disorders provide a very broad differential diagnostic challenge that results from lesions in any part of the neuraxis (Fig. 1-14).

Frontal lobe (Fig. 1-14,D) processes including tumors and normal-pressure hydrocephalus lead to apraxia, spasticity, and leg weakness. Spasticity per se is a nonspecific marker of corticospinal tract disorders that may arise with various neurologic lesions between the frontal lobe and the distal spinal cord (Fig. 1-15). Various neurodegenerative conditions, particularly those affecting the basal ganglia, such as Parkinson disease (Fig. 1-15,A1–3), are some of the most common causes of gait difficulties. These are typically manifested by slowness initiating gait, small steps, and eventually gait festination, wherein once patients begin to accelerate their walking, they take increasingly more rapid but paradoxically smaller steps. There is an innate, almost wax-like rigidity to their stooped body carriage, including the frozen posture of one or both arms that usually lack the normal arm swing. Very occasionally, a change in posture from the seated position to attempted gait will be manifested by a dystonic posturing, which may be indicative of another genetic disorder, dystonia musculorum deformans or paroxysmal choreoathetosis.
Cerebellar disorders related to midline anterior cerebellar vermis lesions or various heredofamilial spinocerebellar entities lead to a broad-base gait ataxia (Fig. 1-14,C1–2). The patient is asked to walk in tandem, with one foot in front of the other. It is an effective means to elicit a subtle disequilibrium often related to midline cerebellar dysfunction such as with simple entities, including alcohol intoxication.
Myelopathies with posterior column dysfunction, such as vitamin B12 deficiency, present with loss of proprioception function. These particularly affect the patient’s gait in dark environments, as do some of the peripheral neuropathies, especially those with a primary sensory ganglionopathy (Fig. 1-14,F1). Testing for the presence of a Romberg sign is an excellent clinical marker for these disorders. Here patients are asked to stand in place with their eyes open, gain their equilibrium, and then close their eyes. Individuals with various proprioceptive disorders are unable to maintain their balance when visual clues are withdrawn; such a condition is referred to as a positive Romberg sign. One of the earliest signs, and at times a prominent sign of a myopathy, is needing to push off the arms of a chair when arising to walk. When these individuals do walk their gait may be a broad-based gait mimicking an anterior cerebellar lesion. When viewed from the side the curve of their low back is accentuated, i.e. hyperlordotic. Both the wide base and the hyperlordosis are representative of weakness of the most proximal muscle groups—the iliopsoas, quadriceps, and glutei—as well as the paraspinal axial musculature.
Very often, in this setting there is not one specific mechanism either operative or identifiable. A number of patients have a multifaceted source related to the gradual aging (graying) of multiple neurologic systems. One source that always requires consideration is the possibility of orthostatic hypotension. Most commonly, this relates to medications; however, one of the neurodegenerative disorders, multiple system atrophy (see Chapter 34), may present in this fashion. Thus, it is important to carefully check blood pressures in the supine posture, when seated, then immediately on standing, and then every 30 seconds thereafter until the pressure is stabilized. A persistent drop in blood pressure of 20–30/15 mm Hg is usually regarded as significant in this setting.
It is important to ask about the circumstances accompanying the gait decline. Does the individual scuff a foot because of a spastic leg that interferes with a smooth alteration of individual legs? What settings lead to a fall? Does one catch one’s toe on a rug as with subtle spasticity (Fig. 1-14,B1–2) or feel a leg give out going downstairs secondary to weakness of the quadriceps femoris muscle (Fig. 1-14,F2)? Having such information, the examiner can then easily try to reproduce the circumstances that lead to the falls.
Typically, gait function is tested under several conditions, including walking straight, walking at least 10 yards in open space, making turns, maneuvering through a tight corridor, attempting tandem gait, or in low light settings as well as on the stairs. The normal degree of foot separation (the base) is widened when proprioception or midline cerebellar vermis function is compromised. Occasionally, having the patient climb stairs reveals a subtle degree of iliopsoas weakness as found in various peripheral motor unit disorders (particularly myopathies) and, less commonly, neuromuscular junction or proximal peripheral neuropathies (Fig. 1-14,G). Finally, the appearance of spasticity may be enhanced by having the patient walk longer distances and even asking him or her to walk several blocks and return to the clinic. Rarely, this uncovers an unsuspected corticospinal tract lesion. Chapter 32 expands on the clinical evaluation of gait disorders.
Muscle Strength Evaluation
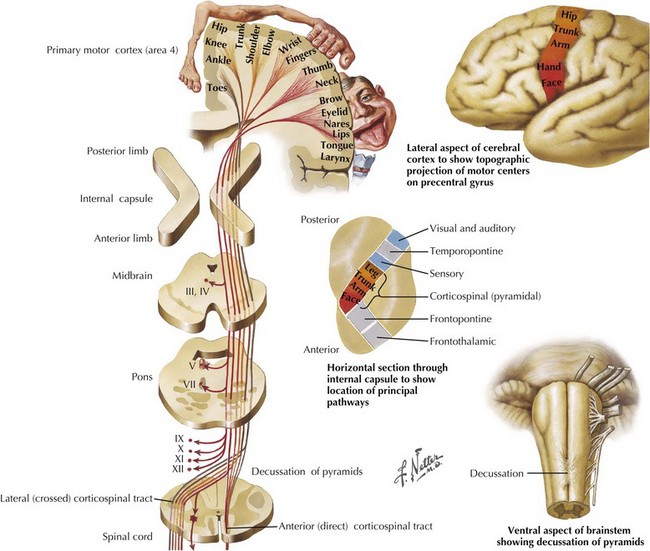
Weakness is one of the most common complaints of patients seeking neurologic care. The motor pathways encompass multiple anatomic areas within the CNS, including the cerebral cortex and important subcortical structures such as the basal ganglia, the brainstem, the cerebellum, the spinal cord, and the peripheral motor unit (Fig. 1-16). Although complaints of generalized weakness, fatigue, or both often are not caused by a specific neurologic condition, the possibility of multiple sclerosis in younger individuals and Parkinson disease in older patients always needs to be considered. When the patient is significantly overweight or has a neuromuscular disorder, sleep apnea needs consideration as a cause of fatigue or a feeling of “weakness.” Peripheral motor unit disorders are important considerations for the differential diagnosis of a patient with generalized weakness. These include processes affecting the anterior horn cell (i.e., amyotrophic lateral sclerosis), peripheral nerve (i.e., Guillain–Barré syndrome or chronic inflammatory demyelinating disorders), neuromuscular junction (including Lambert–Eaton myasthenic syndrome [LEMS]), or muscle cells (various myopathies).
Focal weakness often has a subtle character that frequently is not recognized by the patient as loss of motor strength. Dropping objects or clumsy handwriting may represent a single peripheral nerve lesion such as a radial neuropathy leading to a wrist drop. Tripping on rugs or steps may be the expression of a peroneal nerve lesion causing a foot drop (Fig. 1-14,F3). In contrast, dramatic whole limb weakness is obvious and of greater patient concern, often leading to immediate medical attention as occurs with a stroke. Bilateral motor loss without cognitive or visual difficulties is most commonly due to lesions affecting the spinal cord or the peripheral nervous system and muscle.
When analyzing the complaint of weakness, the physician must consider the presence or absence of associated neurologic complaints or difficulties, such as language, speech, and visual changes; gait dysfunction; difficulty with rising from chairs and associated movements; and alteration in sensation. The neurologist testing for strength must search for evidence of atrophy and fasciculations, or spasticity. Equally important is the need to note the degree of patient effort and cooperation, as well as to consider associated problems that may compromise the testing, such as pain and skin or orthopedic lesions. Formal strength testing must be conducted in a systematic manner evaluating successive areas of the motor unit beginning at the brain and proceeding distally to the individual muscles per se (see Fig. 1-16). Here one places an initial focus on the major muscle groups, such as the flexors and extensors, to seek out any areas of weakness. More specific muscle testing is particularly useful when distinguishing between lesions of the nerve root, plexus, or mononeuropathies (Table 1-2).
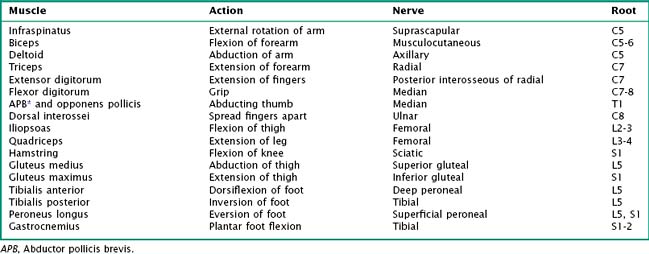
Grading Weakness
The traditional, most widely used British system for quantifying degrees of weakness is based on a scoring range of 0 to 5, with 5 being normal. The extremes of grading are easy to understand, although the subtle grading between 4 and 5 (i.e., >4, 4, >4, or <5) may be slightly different depending on the examiner’s own strength (Table 1-3). Other systems judges the patient to have mild (<1), moderate (<2), severe (<3), or total paralysis (<4) strength, and this grading is viewed by some of us to be a simpler and more reproducible methodology. When testing individual muscles of the patient, the examiner must recognize that this is not an athletic match but rather a determination of whether the patient has normal strength. There is a significant range of normal, and a sense of that latitude can be gained only by examining multiple individuals.
Table 1-3 Grading System for Clinical Documentation of Degree of Weakness
| Grade | Clinical Findings |
|---|---|
| 0 | No movement (complete paralysis) |
| 1 | Able to move a muscle but no movement of limb |
| 2 | Minor movement of limb but inability to overcome gravity |
| 3 | Moderate weakness; movement of limb against gravity |
| 4 | Mild weakness; some resistance against mild pressure |
| 5 | Normal; resistance against moderate pressure |
Adapted from Brain. Aids to the Examination of the Peripheral Nervous System. 4th ed. Philadelphia: WB Saunders; 2000.
Motor Lesions
Myelopathies
A very careful examination is crucial in order to define the presence of a sensory level; this is often best documented by using pin and temperature modalities. One must either sit the patient up or turn them on their side, carefully moving the sensory stimulus from the buttocks to the neck to see if there is a sudden change in degree of perception characteristic of a “sensory level.” Failure to perform this evaluation may lead to missing a treatable spinal cord lesion. Detailed knowledge of the specific sensory territories of the nerve root dermatomes (Fig. 1-17) is very helpful when assessing potential spinal cord lesions. Looking for a sweat level is also sometimes helpful because the skin below the level of a significant spinal cord lesion will be noticeably drier from loss of autonomic sympathetic innervation. Acute lower extremity weakness is also seen with the Guillain–Barré syndrome or other acute generalized polyneuropathies. These disorders may mimic a primary spinal cord lesion.
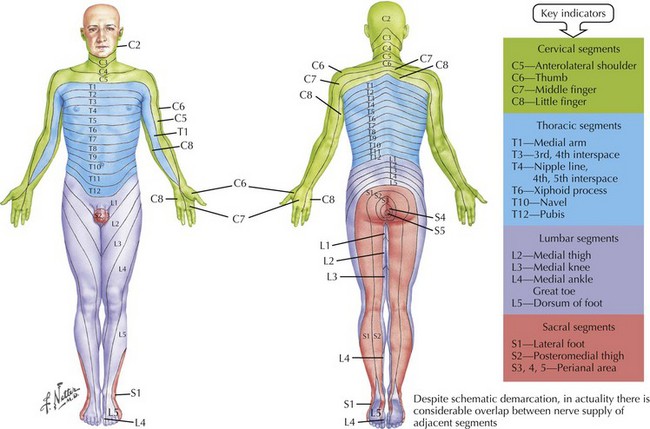
Nerve Root, Plexus, or Peripheral Nerve
The presence of cervical or lumbosacral pain with concomitant focal extremity numbness or weakness is characteristic of a radiculopathy. Interspinal disc herniation and spinal stenosis are the most common processes affecting individual nerve roots. Because sensory examination is the most subjective part of the neurologic examination, occasionally it is difficult to clearly define. Sometimes the patient, per se, can provide the most accurate assessment by using his or her finger to outline the area of diminished sensation. It often then becomes clear that the pattern of sensory loss specifically fits the distribution of a particular peripheral nerve or nerve root dermatome. Knowledge of the cutaneous sensory supply of peripheral nerves is essential to perform an accurate and useful clinical sensory examination (Fig. 1-18).
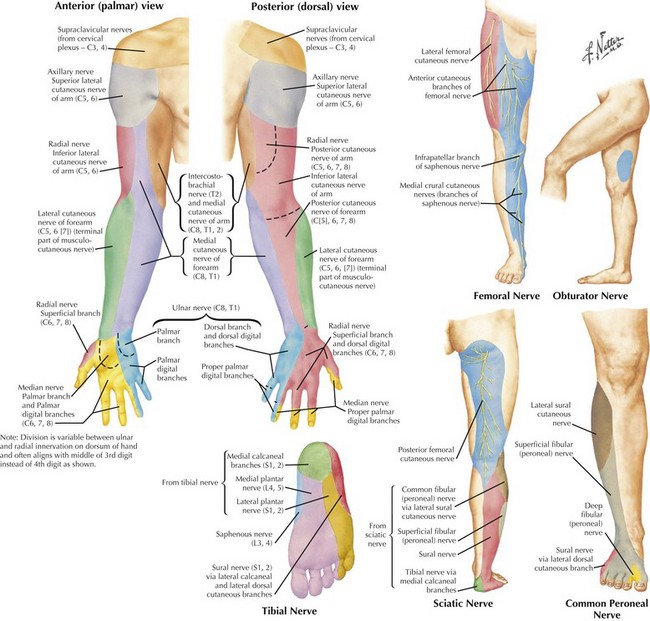
Motor Tone
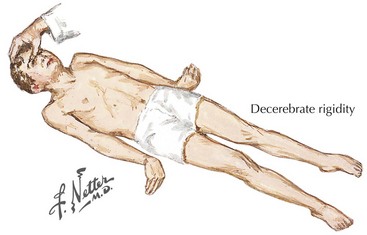
Decerebrate Rigidity
When there is total loss of a motor neuron inhibition, as may occur with an upper brainstem injury, the syndrome of decerebrate rigidity develops. Here, a simple noxious stimulus leads to bilateral extension in unison of all four extremities, with the arms pronated and the legs adducted (Fig. 1-19) rotated inward. Most commonly, one sees this in the setting of cardiac arrest or from shear injuries to the brainstem resulting from severe head injuries, most typically from automobile accidents or battlefield injuries. When these patients survive 1 to 3 months, and are otherwise totally unresponsive, they are said to be in a persistent vegetative state.
Muscle Stretch Reflexes, Clonus, and the Babinski Sign
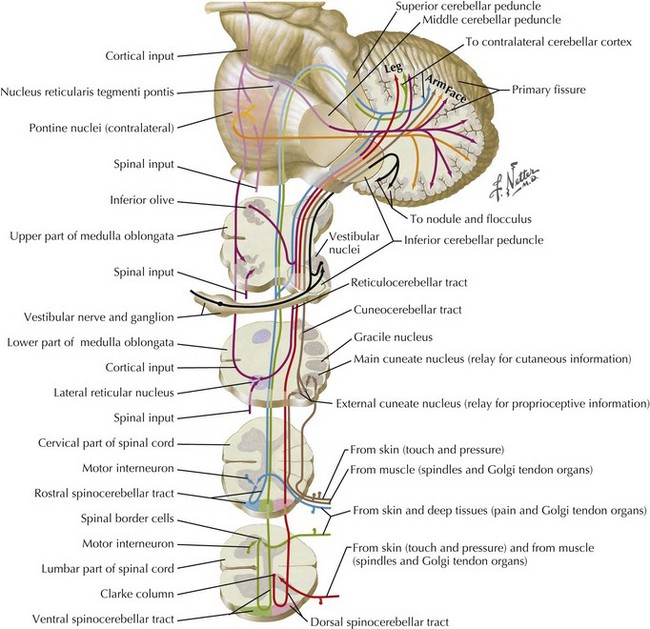
Both Ia and Ib peripheral sensory nerve afferents join the posterior columns of the spinal cord, entering through the dorsal root ganglia. Their primary function is to convey information from touch and pressure receptors. Therefore, although the muscle spindles and Golgi tendon organs cannot be specifically tested, some of their spinal cord connections can be clinically evaluated by testing position and vibration sensory modalities. Additionally, the Ia and Ib afferents convey similar information to the cerebellum via the posterior spinocerebellar tract that travels into the cerebellum through the inferior cerebellar peduncle (Fig. 1-20). In isolation, it is difficult to assess the contribution of each tract specifically to motor control.
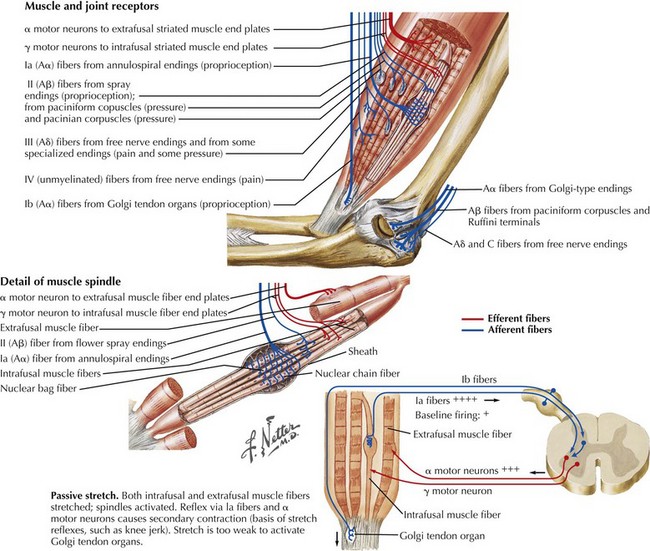
With simple passive stretching, such as occurs with tapping the patellar tendon at the knee, the intrafusal muscle spindle is activated, leading to a direct stimulus to the large alpha motor neurons. These in turn stimulate the extrafusal skeletal muscle fibers, leading to the clinically observed muscle contraction (Fig. 1-21). If the afferent sensory or efferent motor limb of this nerve supply is damaged, the muscle stretch reflex (MSR) is affected and may be diminished or lost, as occurs with many peripheral neuropathies. These reflexes are sometimes inappropriately referred to as deep tendon reflexes (DTRs) when in fact their physiologic basis primarily depends on the intrafusal muscle spindle fibers, not the Golgi tendon organs. MSR is a more accurate term.
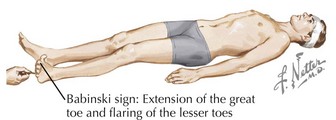
The Babinski sign is an important pathologic reflex that is elicited at the lateral, plantar surface of the foot using subtle, very careful stroking with a tongue depressor or the base of a key. The great toe extends, and the remaining toes fan out (Fig. 1-22). A more exaggerated response, known as triple flexion, includes flexion of the hip, knee, and foot, often with a Babinski response. Because this reflex primarily depends on sensory stimulation of the foot, a kind, gentle, nonirritating stimulus is best to obtain an accurate response. It absolutely does not require excessive or painful pressure. With sensitive or ticklish patients, appropriate responses can usually be obtained from a careful stimulation of the lateral outside, not plantar, surface of the foot. However, some patients have a withdrawal response wherein the foot and entire set of toes dorsiflex. This is often overcome by separately pulling down on the middle toe while carefully stimulating the sole in traditional fashion.
Sensory Examination
Classic Syndromes of Peripheral Sensory Dysfunction
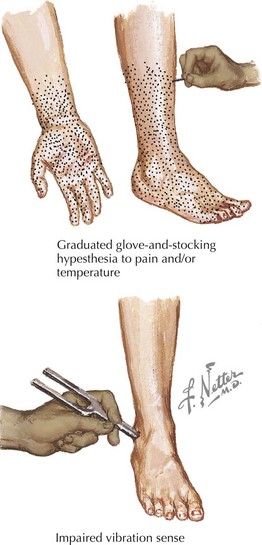
Generalized polyneuropathies typically present with symptoms of numbness and tingling at the tips of the toes and, later, fingers, that is, a stocking-glove distribution (Fig. 1-23). Eventually, this loss will gradually spread proximally past the ankles and wrists into the legs and forearms but usually not above the knees and elbows. On examination with a cold object, a pin (for small fiber function), a tuning fork, and position sense (if large fibers are also involved), the examiner notes a distal loss that is maximum in the periphery and gradually reaches normal at a more proximal site.
Individual mononeuropathies are typified by symptoms and findings specific to a single peripheral nerve (see Fig. 1-18). For example, the patient notes numbness in the thumb, index, middle fingers, and adjacent lateral aspect of the fourth finger if the median nerve is involved. In carpal tunnel syndrome with entrapment of the median nerve at the wrist, the examination results are often subtly abnormal, only with loss of fine discriminatory function with subtle diminution of two-point discrimination. Sometimes, one can employ a reflex hammer to percuss directly over the entrapment site. If there is a focal area of peripheral nerve injury, this action commonly elicits brief paresthesiae distal to the percussion site and within the specific distribution of the sensory fibers of that nerve, in this case the median. This maneuver is known as the Tinel sign; the name applies to instances wherein this simple provocative test defines the lesion site for any mononeuropathy.
Radiculopathies frequently are characterized by more subjective, often intermittent but sometimes persistent, symptoms confined to the dermatomal patterns of one specific nerve root (see Fig. 1-17). Pain is the most common symptom, starting in the neck, shoulder, and low back, often radiating down along the limb in a specific dermatomal distribution. The most common and classic example in the cervical region is at the C7 nerve root where there are paresthesiae primarily involving the index and middle fingers. Often there is a concomitant diminution in triceps muscle strength as well as loss of the triceps reflex. In the low back, the L5 nerve root is the classic example, with numbness in the first and second toes and the lateral calf and accompanying weakness of both the tibialis anterior and tibialis posterior muscles. However as the knee jerk relates to the L4 nerve root, and the ankle jerk to the S1 root, the examiner has to test a less commonly utilized reflex, namely the internal hamstring that has an L5 root innervation. When there is sensory involvement along the lateral aspect of the foot and the small toe with absence of the Achilles reflex, an S1 root lesion is most likely.
Spinal Cord Syndromes
Transverse Complete
The site of a spinal cord lesion is defined by identifying the exact distribution of specific motor and sensory deficits of the various sensory modalities (Fig. 1-24). A complete lesion of the spinal cord leads to total loss of function distal to the site of the abnormality. A distinct level of sensory loss can be discerned with tests for loss of pain and/or temperature sensations, associated with loss of sweating below the lesion level. Concomitantly, all muscles subserved by anterior horn cells distal to the site of the lesion experience paralysis. Distinct partial cord syndromes are briefly described below and discussed further in Section VII (see also Fig. 44-13).
Thalamic Involvement
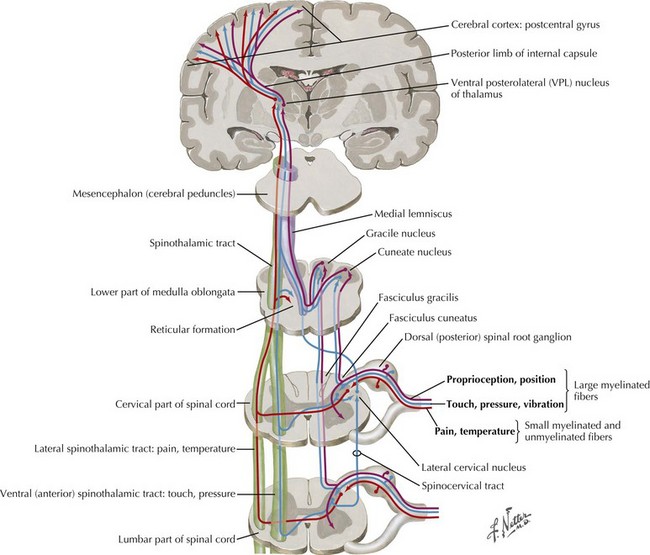
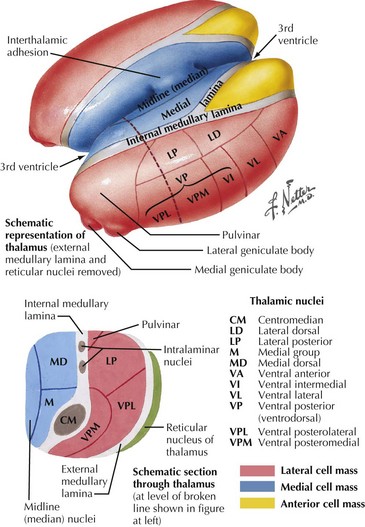
The ventral posterior lateral and ventral posterior medial thalamic nuclei are the two major sensory relay nuclei (Fig. 1-25). Lesions in these areas can cause loss of sensation to all modalities involving the entire contralateral half of the body. This most commonly occurs in patients with lacunar or hemorrhagic infarcts. Initially presenting with a relatively tolerable numbness, eventually the damage incurred from the stroke may produce an unpleasant, sometimes disabling, hyperpathic sensory alteration known as the thalamic pain syndrome. Rarely, this loss of sensation can lead to a limb deafferentiation sensory choreoathetosis. Lesions within the corona radiata, undercutting the parietal cortex, can cause similar, although often less extensive, findings.
Cortical Sensory Involvement
The parietal lobe receives topographically organized sensory inputs from the thalamic nuclei, brainstem, spinal cord, and peripheral nerves (see Fig. 1-24). An important function of the parietal lobe is the integration of this information with other sensory and motor information to formulate body awareness. In the purest form of cortical sensory dysfunctions, patients are unable to differentiate the location of their toes or fingers in space, make a distinction between one and two points, or employ stereognostic discrimination to allow differentiation of various objects placed in their hands, such as differing coin sizes. In addition, these individuals are unable to recognize numbers traced on the palm (graphesthesia).
Bates B. A Guide to Physical Examination and History Taking, 4th ed. Baltimore, MD: JB Lippincott Co; 1987.
Bear MF, Connors BW, Paradiso MA. Neuroscience, Exploring the Brain. Baltimore, MD: Lippincott Williams & Wilkins; 2007.
2000 Brain. Aids to the Examination of the Peripheral Nervous System. 4th ed. Philadelphia, PA: WB Saunders; 2000.
Brazis P, Masdeau J, Biller J. Localization in Clinical Neurology, 5th ed. Baltimore, MD: Lippincott Williams & Wilkins; 2006.
Kandel ER, Schwartz JH, Jessell TM. Principles of Neural Science, 4th ed. New York, NY: McGraw-Hill, Health Professions Division; 2000.
Luria AR. Higher Cortical Functions in Man, 2nd ed. New York, NY: Basic Books, Inc; 1980.
Mayo Clinic Department of Neurology. Mayo Clinic Examinations in Neurology, 7th ed. St. Louis, MO: Mosby; 1998.
Benarroch EE, Daube JR, Flemming KD, Westmoreland BF. Mayo Clinic Medical Neurosciences: Organized by Neurologic Systems and Levels, 5th ed. St. Helier, NJ: Informa; 2008.
Miller NR, Newman NJ, Biousse V, Kerrison JB. Walsh & Hoyt’s Clinical Neuro-Ophthalmology: The Essentials. Baltimore, MD: Lippincott Williams & Wilkins; 2007.
Parent A. Carpenter’s Human Neuroanatomy, 9th ed. Baltimore, MD: Williams & Wilkins; 1996.
Peters A, Jones EG, editors. Cerebral Cortex: Vol 4. Association and Auditory Cortices. New York, NY: Plenum Press, 1985.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
1 Clinical Neurologic Evaluation
Approach to the Neurologic Evaluation
Overview and Basic Tenets
The subsequent definition of the formal examination may be subdivided into a few major sections. Speech and language are assessed during the history taking. The cognitive part of the examination is often clearly defined with the initial history and often does not require formal mental status testing. However there are a number of clinical neurologic settings where this evaluation is very time consuming and complicated; Chapter 2 is dedicated to this aspect of the patient evaluation. However, when there is no clinical suspicion of either a cognitive or language dysfunction, these more formal testing modalities are not specifically required.
Here the multisystem neurologic examination provides a careful basis for most essential clinical evaluations. Neurologists in training and their colleagues in practice cannot expect to test all possible cognitive elements in each patient that they evaluate. Certain basic elements are required; most of these are readily observable or elicited during initial clinical evaluation. These include documentation of language function, affect, concentration, orientation, and memory. When concerned about the patient’s cognitive abilities, the neurologist must elicit evidence of an apraxia or agnosia and test organizational skills. Once language and cognitive functions are assessed, the neurologist dedicates the remaining portion of the exam to the examination of many functions. These include visual fields, cranial nerves (CNs) (Fig. 1-1), muscle strength, muscle stretch reflexes (MSRs), plantar stimulation, coordination, gait and equilibrium, as well as sensory modalities. These should routinely be examined in an organized rote fashion in order not to overlook an important part of the examination. The patient’s general health, nutritional status, and cardiac function, including the presence or absence of significant arrhythmia, heart murmur, hypertension, or signs of congestive failure, should be noted. If the patient is encephalopathic, it is important to search for subtle signs of infectious, hepatic, renal, or pulmonary disease.
Cranial Nerves: An Introduction
The 12 CNs subserve multiple types of neurologic function (see Fig. 1-1). The cranial nerves are formed by afferent sensory fibers, motor efferent fibers, or mixed fibers traveling to and from brainstem nuclei (Fig. 1-2A and B).
Cranial Nerve Testing
II Optic Nerve
Of all the human sensations, the ability to see one’s family and friends, to read, and appreciate the beauties of nature, it is difficult to imagine life without vision, something that is totally dependent on the second cranial nerve. Obviously many individuals, such as Helen Keller, have vigorously and successfully conquered the challenge of being blind; however, given the choice, vision is one of the most precious of all animal sensations. “Blurred” vision is a common but relatively nonspecific symptom that may relate to dysfunction anywhere along the visual pathway (Fig. 1-3). When examining optic nerve function, it is important to identify any concomitant ocular abnormalities such as proptosis, ptosis, scleral injection (congestion), tenderness, bruits, and pupillary changes.
Most visual field changes have localizing value: specific location of the loss, its shape, border sharpness (i.e., how quickly across the field the values change from abnormal to normal). Its concordance with the visual field of the other eye tends to implicate specific areas of the visual system. Localization is possible because details of anatomic organization at different levels predispose to particular types of loss (see Chapter 4).
When one examines the pupils, their shape and size need to be recorded. A side-to-side difference of no more than 1 mm in otherwise round pupils is acceptable as a normal variant. Pupillary responses are tested with a bright flashlight and are primarily mediated by the autonomic innervation of the eye (Fig. 1-4). A normal pupil reacts to light stimulus by constricting with the contralateral constriction of the unstimulated pupil as well. These responses are called the direct and consensual reactions, respectively, and are mediated through parasympathetic innervation to the pupillary sphincter from the Edinger-Westphal nucleus along the oculomotor nerve. The pupils also constrict when shifting focus from a far to a near object (accommodation) and during convergence of the eyes, as when patients are asked to look at their nose.
A number of pathophysiologic mechanisms lead to mydriasis (pupillary dilatation) (Table 1-1). Atropine-like eye drops, often used for their ability to produce pupillary dilation, inadvertent ocular application of certain nebulized bronchodilators, and placement of a scopolamine anti-motion patch with inadvertent leak into the conjunctiva are occasionally overlooked as potential causes for an otherwise asymptomatic, dilated, poorly reactive pupil. Other medications may also lead to certain atypical light reactions. The presence of bilateral dilated pupils, in an otherwise neurologically intact patient, is unlikely to reflect significant neuropathology. In contrast, the presence of prominent pupillary constriction most likely reflects the use of narcotic analogs or parasympathomimetic drugs, such as those typically used to treat glaucoma.
Horner Syndrome
Sympathetic efferent fibers originate within the hypothalamus and traverse the brainstem and cervical spinal cord, then exit the upper thoracic levels and course rostrally to reach the superior cervical ganglia (see Fig. 1-4). Subsequently, these sympathetic fibers track with the carotid artery within the neck to reenter the cranium and subsequently reach their destination innervating the eye’s pupillodilator musculature. Typically, patients with Horner syndrome have an ipsilateral loss of sweating in the face (anhidrosis), a constricted pupil (miosis), and an upper lid droop from loss of innervation to Muller’s muscle, a small smooth muscle lid elevator (ptosis). The levator palpebra superioris, a striated muscle innervated by the oculomotor nerve CN-III, is not affected (Fig. 1-5).
