[level-membership-for-pathology-category]26
Classification and pathogenesis of neurodegenerative diseases
CLASSIFICATION
There are two main groups of neurodegenerative diseases: the movement disorders and the dementia syndromes (Table 26.1).
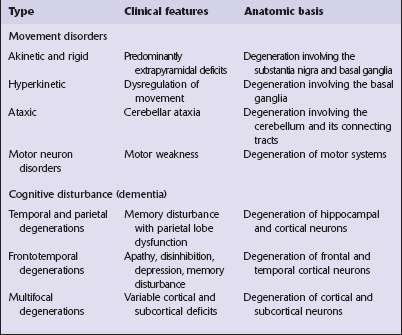
 The clinical features of Parkinson’s disease are most commonly due to Lewy body pathology, but can also be caused by multiple system atrophy or by several disorders characterized by neurofibrillary tangle formation.
The clinical features of Parkinson’s disease are most commonly due to Lewy body pathology, but can also be caused by multiple system atrophy or by several disorders characterized by neurofibrillary tangle formation.




PATHOGENESIS
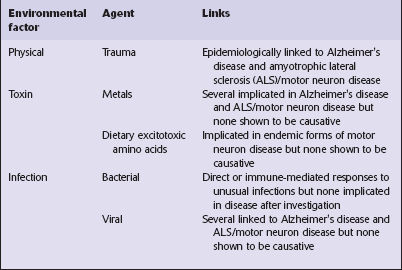
Most neurodegenerative diseases are of unknown cause, but recent work has provided some insights into the mechanisms of neurodegeneration. Many environmental factors, such as toxins and viruses, have in the past been considered as possible causes of neurodegenerative diseases (Table 26.2), but the search for such factors has been largely disappointing and none has been established as causative.
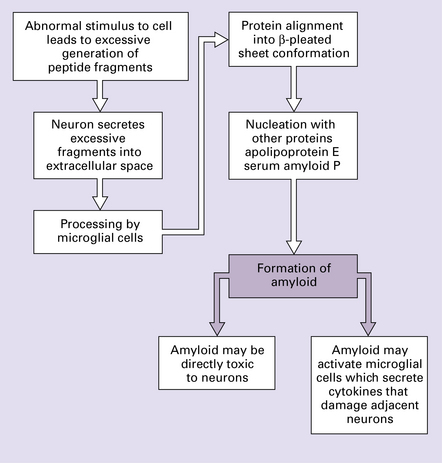
PROTEIN ACCUMULATION AND DEGRADATION


The prion diseases (see Chapter 32) represent a biological phenomenon that is probably unique in which a protein-only transmissible agent is responsible for disease.





EXCITOTOXICITY
Neuronal death may be induced by excessive glutamatergic stimulation of neurons, a process termed excitotoxicity, which has been implicated in the pathogenesis of several degenerative diseases. There are several forms of glutamate receptor, distinguished by their responses to different pharmacologic agonists. In neurodegenerative disease, the vulnerability of certain neuronal systems to damage has been linked to the expression of certain types of glutamate receptor that render neurons vulnerable to excitotoxic damage (Fig. 26.2).
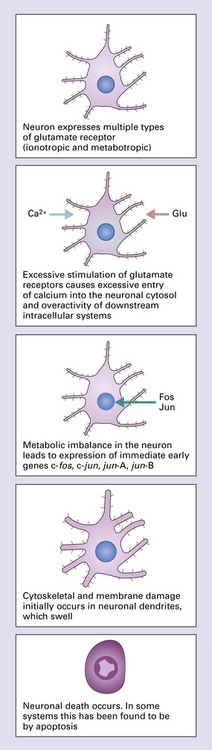
26.2 Excitotoxicity.
INDUCTION OF PROGRAMMED CELL DEATH
The death of neurons in at least some neurodegenerative diseases has been linked to the form of programmed cell death termed apoptosis (Fig. 26.3). The stimuli that trigger this in the different diseases are at present uncertain. Initiating factors under consideration include excitotoxicity, deprivation of neurotrophic growth factors, toxicity of local cytokines, and toxic effects of accumulated proteins. The end result of mitochondrial damage can also result in signaling for apoptosis. However it is important to note that other forms of cell death which are also energy dependent and ‘programmed’ may take place in some situations and are not the same process as conventional apoptotic cell death.
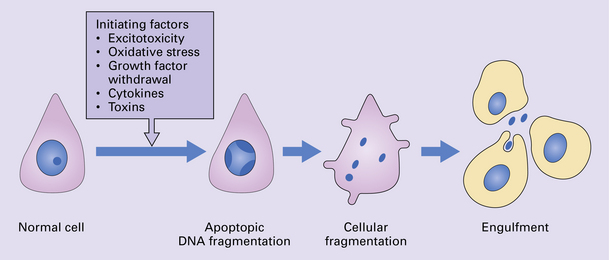
26.3 Apoptosis.
This is believed to be a common mode of cell death in many degenerative diseases. A variety of initiating stimuli can induce programmed cell death. In experimental systems, initiation is followed by reduced neuronal metabolism and expression of immediate early genes. Endonucleases are synthesized and cause nucleosomal fragmentation. The neuron undergoes fragmentation, and these fragments are removed by engulfment by adjacent cells.
GENETIC FACTORS
Many genes responsible for neurodegenerative diseases have been characterized and the identification of such genes has led to major advances in the understanding of disease pathogenesis. A large number neurodegenerative diseases can be caused by single gene mutations but these account for a very small proportion of cases. A genetic abnormality that underlies several neurodegenerative diseases is microsatellite repeat instability leading to expansion of the number of tandem triplet repeats that occur in specific regions of certain genes (Fig. 26.4). This leads to interference with RNA processing and is common to several degenerative diseases. Some neurodegenerative diseases result from interactions between multiple susceptibility factors (e.g. at least 10 genes in addition to environmental factors such as head injury are involved in the development of Alzheimer’s disease) (Fig. 26.5).
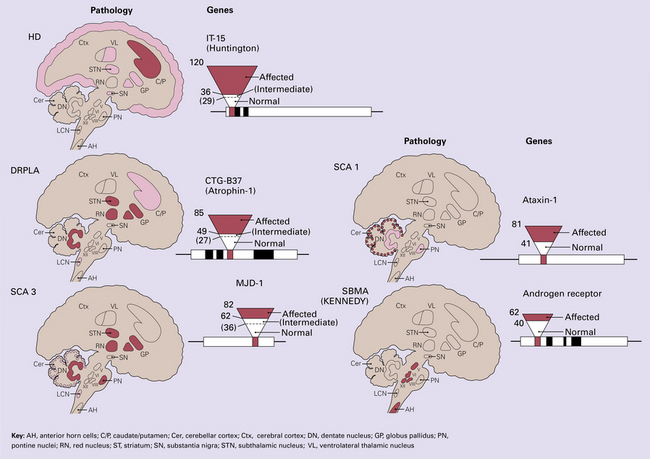
26.4 Neurodegenerative diseases caused by triplet repeat expansions.
Huntington disease (HD), dentatorubropallidoluysial atrophy (DRPLA), spinocerebellar atrophy type 1 (SCA 1), type 3 (SCA 3) and spinobulbar muscular atrophy (SBMA; Kennedy syndrome) are all examples of diseases caused by expansion of triplet repeat regions in key genes. Normal patients have a relatively small number of repeats within these regions. Expansion beyond a particular threshold (which varies for different genes) leads to clinical expression of disease. In some diseases, the deleterious effects of the expansion are probably mediated by inactivation of the protein encoded by the gene, and in other diseases by a toxic action of the abnormal gene product. AH, anterior horn cells; C/P, caudate/putamen; Cer, cerebellar cortex; Ctx, cerebral cortex; DN, dentate nucleus; GP, globus pallidus; PN, pontine nuclei; RN, red nucleus; ST, striatum; SN, substantia nigra; STN, subthalamic nucleus; VL, ventrolateral thalamic nucleus.
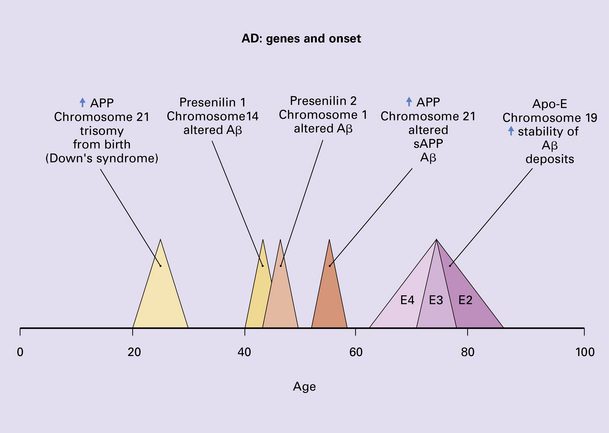
26.5 Genes involved in Alzheimer’s disease.
Genetic factors are important in modulating degenerative diseases, as exemplified by the understanding of the contribution of genetic factors to the etiology of familial and sporadic Alzheimer’s disease. Trisomy 21 is believed to cause an accumulation of abnormal amyloid from about 20 years of age, predisposing to early-onset Alzheimer’s disease in Down syndrome. Mutations in three key genes, presenilin-1, presenilin-2, and APP, predispose to early-onset Alzheimer’s disease. The possession of an apolipoprotein ε4 allele is a risk factor for late-onset Alzheimer’s disease.





REFERENCES
Abeliovich, A. Parkinson’s disease: Mitochondrial damage control. Nature.. 2010;463:744–745.
Ali, Y.O., Kitay, B.M., Zhai, R.G. Dealing with misfolded proteins: examining the neuroprotective role of molecular chaperones in neurodegeneration. Molecules.. 2010;15:6859–6887.
Barnum, C.J., Tansey, M.G. Modeling neuroinflammatory pathogenesis of Parkinson’s disease. Prog Brain Res.. 2010;184:113–132.
Bekris, L.M., Mata, I.F., Zabetian, C.P. The genetics of Parkinson disease. J Geriatr Psychiatry Neurol.. 2010;23:228–242.
Bekris, L.M., Yu, C.E., Bird, T.D., et al. Genetics of Alzheimer disease. J Geriatr Psychiatry Neurol.. 2010;23:213–227.
Bertram, L., Lill, C.M., Tanzi, R.E. The genetics of Alzheimer disease: back to the future. Neuron.. 2010;68:270–281.
Brouwer, J.R., Willemsen, R., Oostra, B.A. Microsatellite repeat instability and neurological disease. Bioessays.. 2009;31:71–83.
Brown, G.C. Mechanisms of inflammatory neurodegeneration: iNOS and NADPH oxidase. Biochem Soc Trans.. 2007;35:1119–1121.
Chen-Plotkin, A.S., Lee, V.M., Trojanowski, J.Q. TAR DNA-binding protein 43 in neurodegenerative disease. Nat Rev Neurol.. 2010;6:211–220.
Cuervo, A.M., Wong, E.S., Martinez-Vicente, M. Protein degradation, aggregation, and misfolding. Mov Disord.. 2010;25:S49–S54.
Cummings, J.L. Toward a molecular neuropsychiatry of neurodegenerative diseases. Ann Neurol.. 2003;54:147–154.
Double, K.L., Reyes, S., Werry, E.L., et al. Selective cell death in neurodegeneration: why are some neurons spared in vulnerable regions? Prog Neurobiol.. 2010;92:316–329.
Gendron, T.F., Josephs, K.A., Petrucelli, L. Transactive response DNA-binding protein 43 (TDP-43): mechanisms of neurodegeneration. Neuropathol Appl Neurobiol.. 2010;36:97–112.
Glass, C.K., Saijo, K., Winner, B., et al. Mechanisms underlying inflammation in neurodegeneration. Cell.. 2010;140:918–934.
Glick, D., Barth, S., Macleod, K.F. Autophagy: cellular and molecular mechanisms. J Pathol.. 2010;221:3–12.
Graeber, M.B., Moran, L.B. Mechanisms of cell death in neurodegenerative diseases: fashion, fiction, and facts. Brain Pathol.. 2002;12(3):385–390.
Hardy, J., Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science.. 2002;297:353–356.
Higgins, G.C., Beart, P.M., Shin, Y.S., et al. Oxidative stress: emerging mitochondrial and cellular themes and variations in neuronal injury. J Alzheimers Dis.. 2010;20:S453–S473.
Huang, Q., Figueiredo-Pereira, M.E. Ubiquitin/proteasome pathway impairment in neurodegeneration: therapeutic implications. Apoptosis.. 2010;15:1292–1311.
Jellinger, K.A. Recent advances in our understanding of neurodegeneration. J Neural Transm.. 2009;116:1111–1162.
Jones, N. PINK1 targets dysfunctional mitochondria for autophagy in Parkinson disease. Nat Rev Neurol.. 2010;6:181.
Kovacs, G.G., Botond, G., Budka, H. Protein coding of neurodegenerative dementias: the neuropathological basis of biomarker diagnostics. Acta Neuropathol (Berl).. 2010;119:389–408.
Kwong, L.K., Neumann, M., Sampathu, D.M., et al. TDP-43 proteinopathy: the neuropathology underlying major forms of sporadic and familial frontotemporal lobar degeneration and motor neuron disease. Acta Neuropathol (Berl).. 2007;114:63–70.
Lazarov, O., Mattson, M.P., Peterson, D.A., et al. When neurogenesis encounters aging and disease. Trends Neurosci.. 2010;33:569–579.
Layfield, R., Alban, A., Mayer, R.J., et al. The ubiquitin protein catabolic disorders. Neuropathol Appl Neurobiol.. 2001;27(3):171–179.
Li, L.B., Bonini, N.M. Roles of trinucleotide-repeat RNA in neurological disease and degeneration. Trends Neurosci.. 2010;33:292–298.
Mackenzie, I.R., Rademakers, R., Neumann, M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol.. 2010;9:995–1007.
Matsuda, N., Tanaka, K. Uncovering the roles of PINK1 and parkin in mitophagy. Autophagy.. 2010;6:952–954.
Rogers, N., Paine, S., Bedford, L., et al. The ubiquitin-proteasome system: contributions to cell death or survival in neurodegeneration. Neuropathol Appl Neurobiol.. 2010;36:113–124.
Schapira, A.H. Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol.. 2008;7:97–109.
Shin, J., Charizanis, K., Swanson, M.S. Pathogenic RNAs in microsatellite expansion disease. Neurosci Lett.. 2009;466:99–102.
Spillantini, M.G., Goedert, M. The alpha-synucleinopathies: Parkinson’s disease, dementia with Lewy bodies, and multiple system atrophy. Ann N Y Acad Sci.. 2000;920:16–27.
Swerdlow, R.H. The neurodegenerative mitochondriopathies. J Alzheimers Dis.. 2009;17:737–751.
Tansey, M.G., Goldberg, M.S. Neuroinflammation in Parkinson’s disease: its role in neuronal death and implications for therapeutic intervention. Neurobiol Dis.. 2010;37:510–518.
Todd, P.K., Paulson, H.L. RNA-mediated neurodegeneration in repeat expansion disorders. Ann Neurol.. 2010;67:291–300.
Vives-Bauza, C., Przedborski, S. Mitophagy: the latest problem for Parkinson’s disease. Trends Mol Med.. 2011;17:158–165.
Wadsworth, J.D., Collinge, J. Molecular pathology of human prion disease. Acta Neuropathol (Berl).. 2011;121:69–77.
Witte, M.E., Geurts, J.J., de Vries, H.E., et al. Mitochondrial dysfunction: a potential link between neuroinflammation and neurodegeneration? Mitochondrion.. 2010;10:411–418.
Wong, E., Cuervo, A.M. Autophagy gone awry in neurodegenerative diseases. Nat Neurosci.. 2010;13:805–811.
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]26
Classification and pathogenesis of neurodegenerative diseases
CLASSIFICATION
There are two main groups of neurodegenerative diseases: the movement disorders and the dementia syndromes (Table 26.1).
The clinical features of Parkinson’s disease are most commonly due to Lewy body pathology, but can also be caused by multiple system atrophy or by several disorders characterized by neurofibrillary tangle formation.
PATHOGENESIS
Most neurodegenerative diseases are of unknown cause, but recent work has provided some insights into the mechanisms of neurodegeneration. Many environmental factors, such as toxins and viruses, have in the past been considered as possible causes of neurodegenerative diseases (Table 26.2), but the search for such factors has been largely disappointing and none has been established as causative.
PROTEIN ACCUMULATION AND DEGRADATION
The prion diseases (see Chapter 32) represent a biological phenomenon that is probably unique in which a protein-only transmissible agent is responsible for disease.
[/not-level-membership-for-pathology-category]
