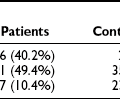Classification and Diagnosis of Diabetes Mellitus
Definition
The term diabetes mellitus does not represent a single disease entity but rather a set of disease states that share certain characteristics. Foremost among these is the presence of elevated plasma glucose levels. As is discussed below, the presence of hyperglycemia in a patient is used both to diagnose diabetes and to guide management decisions, which are directed largely toward avoiding hyperglycemia. The hyperglycemia itself results from a combination of defects in insulin secretion, insulin action, or both.1 An important characteristic of the various disease states that are labeled as diabetes is the development of end-organ damage in vital organs of the body, including the retina, the renal glomerulus, and peripheral nerves. Damage results, at least in part, from the long-term effects of hyperglycemia and are mediated through glycation of tissue proteins, increased activity of the polyol pathway, or other, as yet unrecognized, mechanisms.2 Individual patients vary in their predisposition to develop these so-called microvascular complications. Because of this and because of the length of time they take to develop (frequently decades), the complications of diabetes cannot be used to classify or diagnose the disease. People with diabetes are at considerably greater risk for developing atherosclerotic disease affecting the coronary, cerebrovascular, peripheral arterial, or other parts of the circulation. A cause-and-effect relationship between chronic hyperglycemia and these so-called macrovascular complications of diabetes has not been as clearly established, although evidence linking the two is accumulating.3 Any definition of diabetes that refers only to carbohydrate metabolism is incomplete. Oskar Minkowski is reputed to have first made the association between the insulin-deficient pancreatectomized state of his laboratory dogs and the sweet taste of their urine. It has been suggested that if Minkowski had lacked a sense of taste but possessed a keen sense of smell, he might have smelled the ketones on the breath of his animals and thereby directed diabetes research toward the study of fat metabolism.4 Disordered fat and protein metabolism must be included in a complete definition of the disease, although an emphasis on the pathogenesis of hyperglycemia continues to this day. To define a disease purely in biochemical terms is to diminish the component of the disease that leads to much physical, mental, and psychosocial distress for the many millions of people around the world who live with it every day. Chronic rheumatic diseases such as rheumatoid arthritis are not associated with any biochemical hallmark, and their definition is based largely on patient-derived symptoms and signs. Therefore, it is important to try to include the patient’s perspective in any definition of the chronic disease referred to as diabetes.
Classification
Before 1979, a classification system for diabetes was not well established, and many different terms were used to describe essentially the same clinical entity. Following publication that year of the report of the National Diabetes Data Group (NDDG),5 some order was brought to bear in this area. The recommendations of the NDDG were subsequently endorsed by the World Health Organization (WHO) in a publication in 1980, and minor modifications were later made in a document published in 1985.6 This classification in large part was based on pharmacologic treatment of the disease. Insulin-dependent diabetes mellitus (IDDM) and non–insulin-dependent diabetes mellitus (NIDDM) were the two major forms of diabetes that had been identified. The term insulin-dependent diabetes mellitus was used to describe patients who typically were lean at presentation, were prone to ketosis, and required insulin for survival. The term non–insulin-dependent diabetes mellitus was used to describe patients who typically were overweight or obese at presentation, were not prone to ketosis, and did not require insulin for survival. The NDDG also had categories for gestational diabetes, malnutrition-related diabetes mellitus (MRDM), and a category labeled “other types,” which included certain forms of diabetes for which a cause had been suggested at that time. As the terms IDDM and NIDDM became widely used during the 1980s and 1990s, several problems became apparent. The main problem arose from the fact that many patients with NIDDM ended up at some point in the course of their disease being treated with insulin and being misclassified as IDDM or having the rather confusing term insulin-requiring NIDDM applied to them. In addition, as more information became available on the causes of the various forms of diabetes, it became apparent that a classification based on therapy was not always consistent with new insights into the pathogenesis of the various forms of diabetes. For this reason, the American Diabetes Association (ADA) convened an expert panel in 1995 to address the issue of classification. This panel published its recommendations in 1997,7 and these were subsequently endorsed by a WHO consultation group in a 1998 report.8 The main thrust of this proposal was to move away from a classification based on therapy and toward one based on pathogenesis. Four major categories were proposed: type 1 diabetes, type 2 diabetes, other specific types of diabetes (including categories for which a cause has been established), and gestational diabetes. The details of this system, which are outlined in Table 13-1, are discussed in the following sections.
Table 13-1
Classification of Diabetes Mellitus
A Genetic defects in β cell function
B Genetic defects in insulin action
C Diseases of the exocrine pancreas
G Uncommon forms of immune-mediated diabetes
H Other genetic syndromes sometimes associated with diabetes
From the Report of the Expert Committee on the Diagnosis and Classification of Diabetes Mellitus, Diabetes Care 20(7):1183–1197, 1997.
Type 1 Diabetes
Type 1 diabetes is characterized by the development of a state of complete insulin deficiency. In its fully developed form, patients, if deprived of insulin, will develop ketoacidosis, coma, and death. Biochemical testing reveals the absence of circulating C-peptide (a marker of insulin secretion) despite hyperglycemia. The incidence of type 1 diabetes in the United States is estimated to be approximately 30,000 new cases per year.9 Although the peak incidence occurs in childhood and early adolescence, this form of diabetes can occur at any age. The incidence of the disease shows marked regional variation, with the highest worldwide incidence reported in Scandinavia.10 Epidemiologic and immunologic research has led to the recognition of two major forms of type 1 diabetes based on the presence or absence of certain immunologic markers.
Autoimmune Type 1 Diabetes
Autoimmune type 1 diabetes is a prototypic organ-specific autoimmune disorder. Individuals who develop this form of diabetes are born with a genetic predisposition to autoimmune dysfunction, which may manifest in the development of other autoimmune conditions such as Addison’s disease or Hashimoto’s thyroiditis. The genetic predisposition is not well understood but is known to be linked to the major histocompatibility locus on chromosome 6.11 The presence of certain human lymphocyte antigen (HLA) haplotypes appears to predispose the individual to the disease, but other HLA haplotypes appear to be protective. In predisposed individuals, a poorly understood environmental trigger sets off a series of immunologic events that culminate in selective T cell–mediated destruction of the β cells of the pancreatic islet. Many antigens have been investigated as potential triggers for the disease. These include certain viral antigens12 as well as an antigen contained in cow’s milk protein.13 The rate at which β cell destruction occurs varies from individual to individual and may be very brief, as is seen when type 1 diabetes presents in the neonatal period, or may be prolonged, as is seen in what has been called latent autoimmune diabetes in adults.14 Antibodies appear in the circulation early in the process of β cell destruction.15 These autoantibodies are believed to be markers (rather than true instigators) of the immune response. Their presence can help to classify a newly diagnosed patient with diabetes. In several studies, screening for these autoantibodies has led to recognition of autoimmune type 1 diabetes in individuals who otherwise might have been labeled as having type 2 diabetes16,17 (see later). Islet cell antibodies, the first autoantibodies to be discovered, are directed against a range of islet antigens. The best characterized autoantibodies are those directed against glutamic acid decarboxylase (GAD), an enzyme that is involved in γ-aminobutyric acid synthesis.18 Isoforms of GAD are found in the central nervous system and in β cells of the pancreatic islet. Other autoantibodies include antibodies directed against IA-2 and IA-2β, as well as antibodies directed against insulin itself (anti-insulin antibodies). Childhood-onset type 1 diabetes is associated with higher levels of autoantibody in serum. Testing for these autoantibodies is still restricted to a limited number of laboratories. Greater standardization of assays is required before they can be used widely in clinical practice.
Idiopathic Type 1 Diabetes
The term idiopathic type 1 diabetes is used to describe a small subset of individuals with type 1 diabetes who appear not to have an autoimmune basis for their β cell destruction.19 Other features of this subtype include its occurrence predominantly in individuals of African American ethnicity, its lack of HLA association, and its intermittent proneness to ketosis. A number of different terms, including atypical diabetes, Flatbush diabetes, and type 1.5 diabetes, have been used to describe this form of diabetes. The preferred term in the recent literature appears to be ketosis-prone type 2 diabetes.20
Type 2 Diabetes
Type 2 diabetes represents the most common form of diabetes. Current estimates for the U.S. population indicate that almost 20 million people have type 2 diabetes.21 The condition is characterized by hyperglycemia that results from a combination of defects in insulin secretion and insulin action. In any given individual, the degree to which these defects contribute to hyperglycemia may vary. The disease usually has its onset after age 40, although type 2 diabetes is being seen increasingly in young adults and adolescents.22 Although progressive β cell failure is believed by many to be an important part of the natural history of this form of diabetes,23 the β cell destruction is not autoimmune mediated1 and does not progress to the point at which the patient becomes dependent on insulin for survival. Ketoacidosis is unusual in this form of diabetes, and when it occurs, it usually does so in the setting of a major intercurrent illness such as myocardial infarction or stroke, or when treatment with glucocorticoids is provided. Individuals with type 2 diabetes are not at increased risk for autoimmune disease but have a higher prevalence of metabolic abnormalities, including obesity, hypertension, and a typical dyslipidemia that is characterized by hypertriglyceridemia and low levels of high-density lipoprotein cholesterol. This combination of metabolic derangements is associated with a marked increase in risk for atherosclerotic disease. In fact, the prevalence of atherosclerotic disease in people with type 2 diabetes has led to the suggestion that, rather than one leading to the other, the two conditions may share common antecedents.3 Insulin resistance may be an important predisposing factor for both conditions.
The cause of type 2 diabetes remains to be determined. Any pathogenetic model of the disease must include both genetic and environmental factors. Challenges in establishing the cause of type 2 diabetes include the following: (1) The disease lacks an easy-to-define phenotype and instead is characterized by considerable heterogeneity across different ethnic groups; this heterogeneity often is represented by a spectrum ranging from a predominant defect in insulin secretion on the one hand to a predominant defect in insulin action on the other. (2) The relatively late age of onset makes it difficult to establish large kindreds and therefore limits genetic studies. (3) No easy-to-apply methods are available for screening populations for insulin resistance and defective insulin secretion. (4) The pathways that are involved in mediating insulin action are complex and are not fully understood; most authors believe that a single genetic defect will explain only a subset of the disease; it is much more likely that type 2 diabetes represents a set of disorders. Evidence to support a genetic component of the disease comes from the strong concordance for the disease that is seen among monozygotic twins.24 On the other hand, the dramatic increase in incidence and prevalence of type 2 diabetes that accompanies the change to a so-called Westernized lifestyle strongly supports an environmental component as well.25
Other Specific Types of Diabetes
Other specific types of diabetes are included in both the 1979 NDDG and the 1997 American Diabetes Association (ADA) classification systems.5,7 A number of changes occurred in the subtypes of diabetes listed between the two eras. In particular, the form of diabetes that is referred to as maturity-onset diabetes of the young (MODY) has been better defined genetically, and this is reflected in the 1997 ADA classification system.7 Another change resulted from removal of MRDM from the classification and inclusion of fibrocalculous pancreatic diabetes (which was previously a subtype of MRDM) as a disease of the exocrine pancreas. The decision to remove MRDM as a separate entity resulted from an international conference on this subject.26,27 The findings of the conference did not support a direct cause-and-effect relationship between protein-calorie malnutrition and the development of diabetes. Rather, it was thought that the presence of malnutrition could influence the manner in which diabetes might present in an otherwise predisposed individual.
Genetic Defects in β Cell Function
In the past, the term maturity-onset diabetes of the young was used to describe a subset of patients with a form of diabetes characterized by early age of onset of hyperglycemia with an autosomal dominant mode of inheritance. Mutations in certain genes that are involved in regulating insulin secretion have now been shown to be responsible for the hyperglycemia seen in MODY kindreds.28 Six major forms of MODY have been described,29–32 and their clinical and genetic features are outlined in Table 13-2. All forms of MODY are associated with defective insulin secretion without any significant degree of insulin resistance.33,34 In the case of glucokinase, the pathophysiologic mechanism is clear, because the enzyme is involved in phosphorylating glucose, one of the first steps in glucose metabolism. A glucokinase mutation therefore decreases the ability of the β cell to sense glucose. MODY2 is associated with relatively mild hyperglycemia that is usually amenable to treatment with diet and exercise. MODY1 and MODY3, on the other hand, can be associated with more severe hyperglycemia, a greater likelihood that insulin will be required for management, and a higher propensity to develop complications.28 The link between diabetes and mutations in the genes for hepatocyte nuclear factor (HNF)-1α, HNF-1β (hepatocyte transcription factors also expressed in β cells), and HNF-4α (a member of the steroid-thyroid hormone superfamily and an upstream regulator of HNF-1α), appears to involve regulation of expression of the insulin gene.35 It has been suggested that MODY may account for between 2% and 5% of cases of type 2 diabetes.36
Table 13-2
MODY-Related Genes and the Clinical Phenotypes Associated With Mutations in the Genes

From Fajans SS, Bell GI, Polonsky KS: Molecular mechanisms and clinical pathophysiology of maturity-onset diabetes of the young, N Engl J Med 345:971–980, 2001.
Neonatal diabetes is rare and may be transient or permanent.37 Several genetic defects have been identified in these patients, most notably mutations in the adenosine triphosphate (ATP)-sensitive potassium channel (KATP) of the β cell. The KATP channel comprises two subunits (Kir6.2 and SUR1), and mutations in these can result in loss of function (leading to channel closure and neonatal hyperinsulinemic hypoglycemia) or gain of function (leading to channel opening and neonatal diabetes). Although rare, elucidation of these conditions and the molecular mechanisms underlying them has improved our understanding of this area of human biology.38
Other genetic disorders that are associated with impaired β cell function include certain maternally inherited forms of diabetes with a mutation in mitochondrial DNA,39 disorders that lead to impaired conversion of proinsulin to insulin,40 and disorders that lead to synthesis of an aberrant form of the insulin molecule.41 Of the former conditions, diabetes associated with an A-to-G transition at the nucleotide pair 3243 in mitochondrial transfer RNA (tRNA) has been best characterized and appears to have a wide phenotypic expression from type 2 through to type 1 diabetes. The latter two conditions are inherited in an autosomal dominant manner and are associated with relatively mild glucose intolerance.
Genetic Defects in Insulin Action
For insulin to exert its biological effect, it first must bind to its receptor on the cell surface. Following receptor binding, a complex series of postreceptor signaling reactions take place, leading to the hormone’s metabolic and mitogenic effects. Disruption of some of these postreceptor mediators (e.g., insulin receptor substrate-1) has been shown to cause diabetes in animals.42 However, very few human forms of diabetes have been linked clearly to specific genetic defects in the insulin signaling cascade. Leprechaunism and Rabson-Mendenhall syndrome represent rare congenital disorders of the insulin receptor.43 Both syndromes are associated with diabetes and hyperinsulinemia and altered growth in utero. As the insulin signaling cascade is further defined, it is likely that additional forms of diabetes will be found to be caused by genetic defects in insulin action.
The nomenclature associated with clinical syndromes of severe insulin resistance can be confusing.44 The term type A insulin resistance has been used to describe a syndrome in which severe insulin resistance is associated with a skin condition called acanthosis nigricans and with hyperandrogenism in females. Glucose intolerance or overt diabetes may or not be present. The term type B insulin resistance describes a rare syndrome in which autoantibodies to the insulin receptor lead to insulin resistance and hyperinsulinemia (see later). Also associated with insulin resistance are the lipodystrophies; several forms of lipodystrophic diabetes have been characterized,45 and in some cases, their genetic basis has been established and novel therapeutic interventions have been utilized.46
Diseases of the Exocrine Pancreas
Hyperglycemia can occur during an episode of acute pancreatitis and is associated with a poor prognosis. It is unusual for permanent diabetes to develop following a single episode of acute pancreatitis. Furthermore, removal of up to 90% of the pancreas does not always cause diabetes. On the other hand, diabetes has been reported in association with very small pancreatic adenocarcinomas, leading some investigators to speculate that these tumors produce some diabetogenic factor or factors.47 Five percent to 15% of patients with cystic fibrosis develop diabetes.48 Up to half of these patients require insulin chronically or at times of added stress, such as glucocorticoid therapy. Hemochromatosis can cause diabetes.49 Because glucose tolerance may improve with phlebotomy, this condition represents an important, potentially reversible cause of diabetes. Fibrocalculous pancreatic diabetes is seen mainly in the tropics and is associated with abdominal pain and calcification of the pancreas on abdominal imaging.50 The natural history of this form of diabetes has been established recently,51 along with a genetic marker of the disease.52
Drug- or Chemical-Induced Diabetes
Certain compounds are toxic to β cells. These include the rat poison vacor and the anti-Pneumocystis drug pentamidine. The hyperglycemia that results from these agents is usually not reversible. Thiazide diuretics can inhibit insulin secretion by causing hypokalemia. Glucocorticoids and nicotinic acid cause hyperglycemia by impairing insulin action. Protease inhibitors used in the treatment of persons with human immunodeficiency virus infection can cause hyperglycemia via an as yet undetermined mechanism. The so-called atypical or second-generation antipsychotics are associated with metabolic disorders such as overt diabetes mellitus. The ADA recently published a consensus statement highlighting this association and identifying the need for clinical research in this area.53 The pathophysiologic mechanisms underlying the metabolic derangements are beginning to be elucidated.54
Infections
Certain viral infections, including rubella55 and coxsackie B virus,56 have been associated with diabetes. Some studies suggest that a viral infection can trigger autoimmune destruction of β cells in genetically predisposed individuals, leading to autoimmune type 1 diabetes.
Uncommon Forms Of Immune-Mediated Diabetes
The stiff-man syndrome is a rare neurologic syndrome characterized by spasticity of the axial muscles. It is associated with very high titers of anti-GAD antibodies, and up to one third of patients develop diabetes.57 Autoantibodies directed against the insulin receptor represent another rare cause of diabetes (referred to as type B insulin resistance).58 These antibodies have the potential to change from being receptor antagonists (causing insulin resistance) to being receptor agonists (leading to potentially life-threatening hypoglycemia). Spontaneous remission of antibody production can occur. The syndrome is typically seen in African American females in association with other autoimmune diseases (most commonly systemic lupus erythematosus).
Other Genetic Syndromes Sometimes Associated with Diabetes
Many of the genetic syndromes listed under H in Table 13-1 are known to be associated with diabetes. Wolfram’s syndrome, also known as DIDMOAD (reflecting the components of the disorder; diabetes insipidus, diabetes mellitus, optic atrophy, and deafness), is a rare autosomal recessive disorder caused by mutations in the WFS1 gene on the short arm of chromosome 4. Recently, polymorphisms in the WFS1 gene have been linked to an increased risk for type 2 diabetes,59 providing another example of the study of rare disorders improving our understanding of common conditions.
Gestational Diabetes
Gestational diabetes is defined as diabetes with onset or first recognition during pregnancy. The prevalence of gestational diabetes increases in parallel with the prevalence of type 2 diabetes in a population. Recent data from a large U.S. health care organization revealed a prevalence of 7.4 cases per 100 pregnancies.60 Risk factors include age (it is more common among older women), ethnicity (higher rates are seen among women from ethnic groups with a high incidence of type 2 diabetes), prepregnancy body mass index (the risk increases with degree of obesity), parity (the risk increases with the number of previous pregnancies), and family history of diabetes. A previous pregnancy that was complicated by gestational diabetes or a history of delivery of a macrosomic infant also represents a strong risk factor for future gestational diabetes. The diagnosis of gestational diabetes is important because if it is left untreated, adverse fetal or maternal outcomes can occur. The main adverse fetal outcomes are macrosomia and neonatal hypoglycemia.61 Maternal complications include higher rates of dystocia and cesarean section, as well as increased risk for future development of type 2 diabetes62 (among women who revert to normal glucose tolerance after completion of the pregnancy).
No uniformly agreed upon criteria have been identified for the diagnosis of gestational diabetes.63 The criteria that are used most widely in North America were developed on the basis of the ability of plasma glucose levels measured during pregnancy to predict future development of type 2 diabetes and not adverse outcomes of that pregnancy. In addition, the 100 g glucose load that is used in North America is different from the 75 g load that is used in other parts of the world. Finally, no consensus exists as to who should be screened.64 In some countries (e.g., the United States), universal screening is undertaken routinely, whereas in other countries (e.g., those in Europe), only women who are believed to be at high risk are screened.
Diagnosis
In addition to recommending changes in the classification system for diabetes, the Expert Committee of the ADA, in their 1997 report, recommended changes in the diagnostic criteria used for diabetes.7 These recommendations were subsequently endorsed by the WHO8 (Table 13-3). Major changes included a reduction in the fasting plasma glucose cut point used to diagnose diabetes from 140 mg/dL to 126 mg/dL and a recommendation to use fasting plasma glucose rather than an oral glucose tolerance test (OGTT) for diagnosis. The ability to use casual (i.e., random) plasma glucose levels in patients with hyperglycemic symptoms was retained, as was the requirement that in asymptomatic individuals, a diagnosis should be based on testing carried out on more than one occasion. The ADA report based its criteria on use of the fasting plasma glucose level, and the WHO report included equivalent cut points for whole-blood venous and capillary glucose.8 The ADA report introduced the terms normal fasting glucose and impaired fasting glucose. The latter describes patients with fasting plasma glucose levels in the intermediate zone between normal and overt diabetes. Considerable controversy and debate followed publication of the 1997 report, and the ADA re-convened its expert committee in 2003.65 Major areas of controversy, how they were dealt with in the 2003 report, and the current status of the diagnostic criteria are discussed in the remaining sections of this chapter.
Table 13-3
Diagnostic Thresholds for Diabetes and Lesser Degrees of Impaired Glucose Regulation*
| Category | Fasting Plasma Glucose | 2 Hour Plasma Glucose |
| Normal | <100 mg/dL (<5.6 mmol/L) | <140 mg/dL (<7.8 mmol/L) |
| IFG | 100–125 mg/dL (5.6–6.9 mmol/L) | — |
| IGT | — | 140–199 mg/dL (7.8–11.0 mmol/L) |
| Diabetes† | ≥126 mg/dL (≥7.0 mmol/L) | ≥200 mg/dL (≥11.1 mmol/L) |
IFG, Impaired fasting glucose; IGT, impaired glucose tolerance.
*When both tests are performed, IFG or IGT should be diagnosed only if diabetes is not diagnosed by the other test.
†A diagnosis of diabetes needs to be confirmed on a separate day.
What Level of Fasting Plasma Glucose Constitutes Diabetes?
The development of microvascular changes, particularly in the retina, is one of the hallmarks of diabetes mellitus. Because these complications can take many years to develop, it is not possible to use their presence to diagnose the condition. Instead, the level of plasma glucose that confers a risk for occurrence of these changes is used for diagnostic purposes. At the time the 1997 report was written, the best estimate of this level was fasting plasma glucose of 126 mg/dL. This value was based on data from three epidemiological studies involving Pima Indians, Egyptians, and a U.S. national sample.7 In all three studies, there appeared to be a threshold of risk (i.e., a level below which the risk for retinopathy was negligible, and above which the risk rose steeply). In the 2003 report of the expert committee, reference was made to a separate analysis of Pima Indian data, suggesting that the threshold at which retinopathy risk increased might be lower than that previously reported.66 However, the presence of a threshold was not disputed, and the committee did not recommend a change in the 126 mg/dL cut point for diagnosis. A recent analysis of data from two cross-sectional Australian cohort studies and one longitudinal U.S. cohort study has raised serious doubt about the presence of a retinopathy threshold (Fig. 13-1).67 All three studies used multiple-field digital retinal photography to identify retinopathy, a more sophisticated method than that used in earlier studies that informed the ADA expert committee decision. The risk for retinopathy in all three studies appeared to be continuous across the entire range of plasma glucose, with up to 10% of individuals demonstrating some degree of retinopathy at levels of fasting plasma glucose well within the range considered normal. These data are consistent with an earlier report from the Diabetes Prevention Program, which also identified retinopathy in individuals with impaired glucose tolerance.68 Taken together, these data suggest that current diagnostic criteria may need further revision.
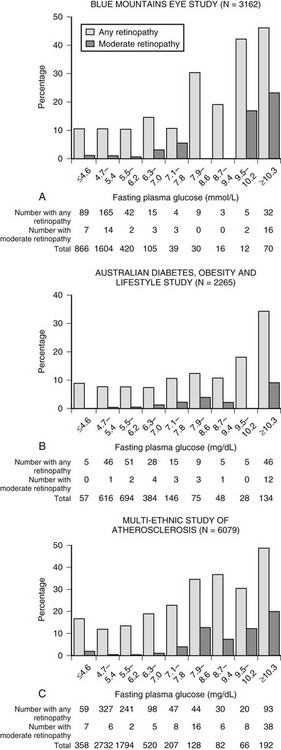
FIGURE 13-1 Prevalence of retinopathy across a wide range of fasting plasma glucose levels in three epidemiologic studies using digital retinal photography to screen the retina. (Data from Wong TY, Liew G, Tapp RJ, et al: Relation between fasting glucose and retinopathy for diagnosis of diabetes: three population-based cross-sectional studies, Lancet 371:736–743, 2008.)
What Level of Fasting Plasma Glucose Constitutes Normality?
Choosing a cut point to define the upper limit of normal fasting glucose (and the lower limit of impaired fasting glucose) is an arbitrary decision. The expert committee in its 1997 report chose a value of 110 mg/dL despite the fact that many laboratories at the time were using 100 mg/dL as their definition of normality.7 In its 2003 report, the expert committee recommended that the cut point should be revised to 100 mg/dL.65 This recommendation was based on receiver operator characteristic (ROC) curve analyses of the ability of various baseline levels of fasting plasma glucose to predict future diabetes. ROC curves were derived from cohort studies available to the committee at the time but were not published in the report. A factor that undoubtedly influenced the committee was a desire to achieve “equivalence” between impaired fasting glucose and impaired glucose tolerance. The term equivalence can be considered as cross-sectional or prospective. In cross-sectional terms, complete equivalence would amount to those patients with impaired fasting glucose also having impaired glucose tolerance. This was not the case when the cut point was at 110 mg/dL and still is not the case with the cut point at 100 mg/dL. In fact, it has been suggested that these two states of pre-diabetes may be different biological entities, with altered insulin action and altered β cell function contributing in different ways to their pathogenesis.69 A lesser degree of cross-sectional equivalence would be a situation in which population prevalences of impaired fasting glucose and impaired glucose tolerance are the same. Lowering the cut point to 100 mg/dL did help move things toward equivalence with this definition. In prospective terms, equivalence amounts to an equal ability of these two intermediate states of hyperglycemia to predict future events, such as overt diabetes and cardiovascular disease or mortality. This is the point about which most of the controversy has arisen. A large European epidemiologic consortium published data clearly demonstrating that 2 hour plasma glucose following an OGTT was superior to fasting plasma glucose in predicting future cardiovascular events.70 Members argued that the OGTT should not be discarded from clinical practice or from epidemiologic research.
A large number of studies have looked at the ability of fasting plasma glucose to predict future diabetes. One of these, the Baltimore Longitudinal Study of Aging (BLSA), followed a cohort of more than 800 subjects with serial OGTTs for up to 20 years.71 Investigators documented the natural history of progression from normal glucose tolerance to intermediate states of hyperglycemia to overt diabetes. Many subjects reverted to normal from intermediate states of hyperglycemia, but in general, a slow, steady progression to diabetes was seen, with male gender and obesity increasing risk. By altering the threshold that was used to define the lower limit of impaired fasting glucose, BLSA investigators showed that much of the “discrepancy” between rates of progression from impaired fasting glucose versus impaired glucose tolerance to overt diabetes was a function of the threshold used rather than of any inherent biological difference between the two states of hyperglycemia. Using the definition of a threshold that we discussed previously, one would expect that the cut point between normal and impaired fasting glucose would be associated with a significant increase in risk. This is not the case. Several investigators have demonstrated that risk for future diabetes and risk for cardiovascular events increase across the continuum of glucose levels from normal through impaired fasting glucose or impaired glucose tolerance.72–75 There does not appear to be a threshold of risk.
Where Next?
So where does this leave us, and how do we define normality in plasma glucose terms? The answer would appear to be that when one is dealing with a continuous variable associated with a continuous increase in risk, any attempt to “draw a line in the sand” is fraught with problems. Changing definitions of diabetes and impaired fasting glucose over the past 10 years have generated a huge number of research papers but may not have served clinicians or patients all that well.76 In this regard, the lower limit of impaired fasting plasma glucose still is considered 6.1 mmol/L (110 mg/dL) in many parts of Europe (where clinicians look to the WHO rather than the ADA for direction). Terms such as pre-diabetes or dysglycemia77 are additional examples of the confusion that occurs in this area at present. In the wake of several diabetes prevention studies demonstrating positive results with either lifestyle78,79 or with pharmacologic interventions,80,81 the real question should be this: How do we present future risk for events to our patients in a way that will motivate them to put on their trainers and embark on useful lifestyle modification? It may be that a presentation of global risk (incorporating several risk factors for vascular disease) might work better than documentation of individual states of altered glucose and blood pressure and lipid abnormalities in an obese individual. A recent commentary from two leading participants in the ADA expert committee saga presents a “back to the future” scenario, suggesting that we may need to abandon all definitions of altered states of glucose tolerance and revert to a (pre-1979) world, where people either have diabetes or do not.82 Watch for another re-convening of the expert committee in the not too distant future!
References
1. Kahn, SE. The relative contributions of insulin resistancce and beta-cell dysfunction to the pathophysiology of type 2 diabetes. Diabetologia. 2003;46:3–19.
2. Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820.
3. Stern, M. Do non-insulin-dependent diabetes mellitus and cardiovascular disease share common antecedents? Ann Intern Med. 1996;124:110–116.
4. McGarry, J. What if Minkowski had been ageusic? An alternative angle on diabetes. Science. 1992;258:766–770.
5. National Diabetes Data Group. Classification and diagnosis of diabetes mellitus and other categories of glucose intolerance. Diabetes. 1979;28:1039–1057.
6. World Health Organization. Diabetes Mellitus: Report of a WHO study group. Geneva: World Health Organization; 1985.
7. Report of the Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Diabetes Care. 1997;20:1183–1197.
8. Alberti, KGMM, Zimmet, PZ. Definition, diagnosis, and classification of diabetes mellitus and its complications. Part 1: Diagnosis and classification of diabetes mellitus. Provisional report of a WHO consultation. Diabet Med. 1998;15:539–553.
9. LaPorte, R, Matsushima, M, Chang, Y. Prevalence and incidence of insulin-dependent diabetes. In: Diabetes in America. Bethesda, MD: National Institutes of Health; 1995:37–46.
10. Harjutsalo, V, Sjoberg, L, Tuomilehto, J. Time trends in the incidence of type 1 diabetes in Finnish children: a cohort study. Lancet. 2008;371:1777–1782.
11. Buzzetti, R, Quattrocchi, C, Nistico, L. Dissecting the genetics of type 1 diabetes: Relevance for familial clustering and differences in incidence. Diabetes Metab Rev. 1998;14:111–128.
12. Dahlquist, G. The aetiology of type 1 diabetes: An epidemiological perspective. Acta Paediatr. 1998;425:5–10.
13. Scott, F, Norris, J, Hubert, K. Milk and type 1 diabetes: Examining the evidence and broadening the focus. Diabetes Care. 1996;19:379–383.
14. Zimmet, P, Tuomi, T, Mackay, R, et al. Latent autoimmune diabetes mellitus in adults (LADA): The role of antibodies to glutamic acid decarboxylase in diagnosis and prediction of insulin dependency. Diabet Med. 1994;11:299–303.
15. Bingley, PJ, Williams, AJ, Gale, EA. Optimized autoantibody-based risk assessment in family members: implications for future intervention trials. Diabetes Care. 1999;22:1796–1801.
16. Molbak, A, Christau, B, Marner, B, et al. Incidence of insulin-dependent diabetes mellitus in age groups over 30 years in Denmark. Diabet Med. 1994;11:650–655.
17. Humphrey, A, McCarty, D, Mackay, I, et al. Autoantibodies to glutamic acid decarboxylase and phenotypic features associated with early insulin treatment in individuals with adult-onset diabetes mellitus. Diabet Med. 1998;15:113–119.
18. Willis, J, Scott, R, Brown, L, et al. Islet cell antibodies and antibodies against glutamic acid decarboxylase in newly diagnosed adult-onset diabetes mellitus. Diabetes Res Clin Prac. 1996;33:89–97.
19. Maldonado, M, Hampe, CS, Gaur, LK, et al. Ketosis-prone diabetes: dissection of a heterogeneous syndrome using an immunogenetic and beta-cell functional classification, prospective analysis and clinical outcomes. J Clin Endocrinol Metab. 2003;88:5090–5098.
20. Umpierrez, GE, Kitabchi, AE. Ketosis-prone type 2 diabetes mellitus. Ann Intern Med. 2006;144:350–357.
21. Cowie, C, Rust, KF, Byrd-Holt, DD, et al. Prevalence of diabetes and impaired fasting glucose in adults in the US population. Diabetes Care. 2006;29:1263–1268.
22. Mokdad, AH, Bowman, BA, Ford, ES, et al. The continuing epidemics of obesity and diabetes in the United States. JAMA. 2001;286:1195–1200.
23. Rudenski, A, Hadden, D, Atkinson, A, et al. Natural history of pancreatic islet β-cell function in type 2 diabetes mellitus studied over six years by homeostasis model assessment. Diabet Med. 1988;5:36–41.
24. Barnett, A, Eff, C, Leslie, R, et al. Diabetes in identical twins: A study of 200 pairs. Diabetologia. 1981;20:87–93.
25. Knowler, W, Saad, M, Pettitt, D, et al. Determinants of diabetes mellitus in the Pima Indians. Diabetes Care. 1993;16:216–227.
26. Hoet, J, Tripathy, B, Rao, R, et al. Malnutrition and diabetes in the tropics. Diabetes Care. 1996;19:1014–1017.
27. Tripathy, B, Samal, K. Overview and consensus statement on diabetes in tropical areas. Diabetes Metab Rev. 1997;13:63–76.
28. Fajans, SS, Bell, GI, Polonsky, KS. Molecular mechanisms and clinical pathophysiology of maturity-onset diabetes of the young. N Engl J Med. 2001;345:971–980.
29. Yamagata, K, Furuta, H, Oda, N, et al. Mutations in the hepatocyte nuclear factor-4-alpha gene in maturity-onset diabetes of the young (MODY 1). Nature. 1996;384:458–460.
30. Froguel, P, Vaxillaire, M, Sun, F, et al. Close linkage of glucokinase locus on chromosome 7p to early-onset non-insulin-dependent diabetes. Nature. 1992;356:162–164.
31. Yamagata, K, Oda, N, Kaisaki, P, et al. Mutations in the hepatocyte nuclear factor-1-alpha gene in maturity-onset diabetes of the young (MODY 3). Nature. 1996;384:455–458.
32. Stoffers, D, Ferrer, J, Clarke, W, et al. Early-onset type-II diabetes mellitus (MODY 4) linked to IPF1. Nat Genet. 1997;117:138–139.
33. Byrne, M, Sturis, J, Menzel, S, et al. Altered insulin secretory response to glucose in diabetic and nondiabetic subjects with mutations in the diabetes susceptibility gene MODY 3 on chromosome 20. Diabetes. 1996;45:1503–1510.
34. Clement, K, Pueyo, M, Vaxillaire, M, et al. Assessment of insulin sensitivity in glucokinase-deficient subjects. Diabetologia. 1996;39:82–90.
35. Habener, J, Stoffers, D. A newly discovered role of transcription factors involved in pancreas development and the pathogenesis of diabetes mellitus. Proc Assoc Am Physicians. 1998;110:12–21.
36. Velho, G, Froguel, P. Maturity-onset diabetes of the young (MODY), MODY genes and non-insulin-dependent diabetes mellitus. Diabetes Metab. 1997;23:34–37.
37. Sperling, MA. Neonatal diabetes mellitus: from understudy to center stage. Curr Opin Pediatr. 2005;17:512–518.
38. Sperling, MA. ATP-sensitive potassium channels—neonatal diabetes and beyond. N Engl J Med. 2006;355:507–510.
39. Walker, M, Turnbull, D. Mitochondrial related diabetes: a clinical perspective. Diabet Med. 1997;14:1007–1009.
40. O’Rahilly, S, Gray, H, Humphreys, P, et al. Impaired processing of prohormones associated with abnormalities of glucose homeostasis and adrenal function. N Engl J Med. 1995;333:1386–1390.
41. Haneda, M, Polonsky, K, Bergenstal, R, et al. Familial hyperinsulinemia due to a structurally abnormal insulin: definition of an emerging new clinical syndrome. N Engl J Med. 1984;310:1288–1294.
42. Bruning, J, Winnay, J, Bonner-Weir, S, et al. Development of a novel polygenic model of NIDDM in mice heterozygous for IR and IRS-1 null alleles. Cell. 1997;88:561–572.
43. Taylor, S. Lilly lecture: Molecular mechanisms of insulin resistance: lessons from patients with mutations in the insulin-receptor gene. Diabetes. 1992;41:1473–1490.
44. Tritos, NA, Mantzoros, CS. Syndromes of severe insulin resistance. J Clin Endocrinol Metab. 1998;83:3025–3030.
45. Joffe, BI, Panz, VR, Raal, FJ. From lipodystrophy syndromes to diabetes mellitus. Lancet. 2001;357:1379–1381.
46. Arioglu, E, Duncan-Morin, J, Sebring, N, et al. Efficacy and safety of troglitazone in the treatment of lipodystrophy syndromes. Ann Intern Med. 2000;133:263–274.
47. Gullo, L, Pezzilli, R, Morselli-Labate, A, Group, IPCS. Diabetes and the risk of pancreatic cancer. N Engl J Med. 1994;331:81–84.
48. Moran, A, Doherty, L, Wang, X, Thomas, W. Abnormal glucose metabolism in cystic fibrosis. Pediatrics. 1998;133:10–17.
49. Phelps, G, Chapman, I, Hall, P, et al. Prevalence of genetic haemochromatosis among diabetic patients. Lancet. 1989;2:233–234.
50. Yajnik, C, Shelgikar, K, Naik, S, et al. The ketoacidosis-resistance in fibro-calculous-pancreatic-diabetes. Diabetes Res Clin Pract. 1993;15:149–156.
51. Mohan, V, Barman, KK, Rajan, VS, et al. Tropical calcific pancreatitis is a prediabetic stage of fibrocalculous pancreatic diabetes (FCPD) longitudinal follow-up study. Diabetologia. 2003;46(suppl 2):A18.
52. SPINK1 is a susceptibility gene for fibrocalculous pancreatic diabetes in subjects from the Indian subcontinent. Am J Hum Genet. 2002;71:964–968.
53. American Diabetes Association. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27:596–601.
54. Ader, M, Kim, SP, Catalano, KJ, et al. Metabolic dysregulation with atypical antipsychotics occurs in the absence of underlying disease: a placebo-controlled study of olanzapine and risperidone in dogs. Diabetes. 2005;54:862–871.
55. Forrest, J, Menser, M, Burgess, JA. High frequency of diabetes mellitus in young patients with congenital rubella. Lancet. 1971;2:332–334.
56. King, M, Bidwell, D, Shikh, A, et al. Coxsackie-B-virus-specific IgM responses in children with insulin-dependent (juvenile-onset; type 1) diabetes mellitus. Lancet. 1983;1:1397–1399.
57. Solimena, M, De Camilli, P. Autoimmunity to glutamic acid decarboxylase (GAD) in stiff-man syndrome and insulin-dependent diabetes mellitus. Trends Neurosci. 1991;14:452–457.
58. Arioglu, E, Andewelt, A, Diabo, C, et al. Clinical course of the syndrome of autoantibodies to the insulin receptor (Type B insulin resistance): a 28-year perspective. Medicine. 2002;8:87–100.
59. Sandhu, MS, Weedon, MN, Fawcett, KA, et al. Common variants in WFS1 confer risk of type 2 diabetes. Nat Genet. 2007;39:951–953.
60. Lawrence, JM, Contreras, R, Chen, W, Sacks, DA. Trends in the prevalence of preexisting diabetes and gestational diabetes mellitus among a racially/ethically diverse population of pregnant women, 1999–2005. Diabetes Care. 2008;31:899–904.
61. Persson, B, Hanson, U. Neonatal morbidities in gestational diabetes mellitus. Diabetes Care. 1998;21:B79–B84.
62. Dornhorst, A, Rossi, M. Risk and prevention of type 2 diabetes in women with gestational diabetes. Diabetes Care. 1998;21:B43–B49.
63. Coustan, D, Carpenter, M. The diagnosis of gestational diabetes. Diabetes Care. 1998;21:B5–B8.
64. Carr, S. Screening for gestational diabetes mellitus. Diabetes Care. 1998;21:B14–B18.
65. The Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Follow-up report on the diagnosis of diabetes mellitus. Diabetes Care. 2003;26:3160–3167.
66. Gabir, MM, Hanson, RL, Dabelea, D, et al. Plasma glucose and prediction of microvascular disease and mortality. Diabetes Care. 2000;23:1113–1118.
67. Wong, TY, Liew, G, Tapp, RJ, et al. Relation between fasting glucose and retinopathy for diagnosis of diabetes: three population-based cross-sectional studies. Lancet. 2008;371:736–743.
68. Diabetes Prevention Program Research Group. The prevalence of retinopathy in impaired glucose tolerance and recent-onset diabetes in the Diabetes Prevention Program. Diabet Med. 2007;24:137–144.
69. Abdul-Ghani, MA, Triphany, D, DeFronzo, RA. Contributions of β-cell dysfunction and insulin resistance to the pathogenesis of impaired glucose tolerance and impaired fasting glucose. Diabetes Care. 2006;29:1130–1139.
70. The DECODE Study Groupthe European Epidemiology Study Group. Glucose tolerance and cardiovascular mortality: comparison of fasting and 2-hour diagnostic criteria. Arch Intern Med. 2001;161:397–404.
71. Meigs, JB, Muller, DC, Nathan, DM, et al. The natural history of progression from normal glucose tolerance to type 2 diabetes in the Baltimore Longitudinal Study of Aging. Diabetes. 2003;52:1475–1484.
72. Dinneen, S, Maldonado, D, Leibson, C, et al. Effects of changing diagnostic criteria on the risk of developing diabetes. Diabetes Care. 1998;21:1408–1413.
73. Nichols, GA, Hillier, TA, Brown, JB. Progression from newly acquired impaired fasting glucose to type 2 diabetes. Diabetes Care. 2007;30:228–233.
74. Tirosh, A, Shai, I, Tekes-Manova, D, et al. Normal fasting plasma glucose levels and type 2 diabetes in young men. N Engl J Med. 2005;353:1454–1462.
75. Khaw, KT, Wareham, N, Bingham, S, et al. Association of hemoglobin A1c with cardiovascular disease and mortality in adults: the European prospective investigation into cancer in Norfolk. Ann Intern Med. 2004;141:413–420.
76. Davidson, MB, Landsman, PB, Alexander, CM. Lowering the criterion for impaired fasting glucose will not provide clinical benefit. Diabetes Care. 2003;26:3329–3330.
77. Gerstein, H, Yusuf, S. Dysglycaemia and risk of cardiovascular disease. Lancet. 1996;347:949–950.
78. Tuomilehto, J, Lindstrom, J, Eriksson, JG, et al. Prevention of Type 2 diabetes by changes in lifestyle among subjects with impaired glucose tolerance. N Engl J Med. 2001;344:1343–1350.
79. Diabetes Prevention Program Research Group. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346:393–403.
80. Chiasson, JL, Josse, RG, Gomis, R, et al. Acarbose for prevention of type 2 diabetes mellitus: the STOP-NIDDM randomised trial. Lancet. 2002;359:2072–2077.
81. Gerstein, HC, Yusuf, S, Bosch, J, et al. The DREAM (Diabetes Reduction Assessment with ramipril and rosiglitazone Medication) Trial Investigators, Effect of rosiglitazone on the frequency of diabetes in patients with impaired glucose tolerance or impaired fasting glucose: a randomised controlled trial. Lancet. 2006;368:1096–1105.
82. Genuth, S, Kahn, R. A step backward—or is it forward? Diabetes Care. 2008;31:1093–1096.