CHAPTER 24 Chronic myelogenous leukemia
Introduction
Chronic myelogenous leukemia (CML) is a myeloproliferative neoplasm consistently associated with the BCR-ABL1 fusion gene (reviewed in1). CML is characterized by neutrophilic granulocytosis and pathologic left shift in the peripheral blood (PB) with less than 20% (WHO) or 30% (European LeukemiaNet) blasts in blood and bone marrow (BM).2,3 CML is a disease most frequently occurring in the middle age with a peak in the 5th and 6th decades of life, affecting about 1–2 persons/100 000.3 It may also occur in younger and older patients, but it is rarely seen in childhood.
Increased risk of CML had been observed in persons exposed to ionizing radiation.5 However, history of radiation exposure is unusual in patients with CML so that the exact cause of disease remains unclear in the great majority of patients.5 Inherited predisposition to CML has not been reported, but risk of CML appears to be reduced in persons with HLA-B3 and -B8 phenotype, indicating that some unknown predisposing factors may exist.6
Animal models7 and the success of BCR-ABL tyrosine kinase (TK) inhibitors in inducing a remission of CML8 have impressively demonstrated the relevance of the BCR-ABL1 fusion to the pathogenesis of CML (described in detail in Chapter 21). Monotherapy with BCR-ABL TK inhibitors induces a complete cytogenetic (CCyR) and a major molecular (MMR) remission in the great majority of patients.9,10
Clinical features and course of disease
CML shows a characteristic bi- or triphasic course, starting with a rather indolent chronic phase (CP) and transforming to a usually lethal final acute phase, a dramatic clinical picture called ‘blast crisis’ (Table 24.1). In more than 95% of patients, CML is diagnosed in CP. Rarely, CML is diagnosed in acute phase with a clinical picture of a BCR-ABL1-positive acute myeloid (AML) or lymphatic leukemia (ALL).
During the last 3 decades, the median duration of the CP could be markedly prolonged from less than 511 to more than 10 years.12 Before the use of TK inhibitors, the majority of patients developed blast crisis several years after diagnosis of CML.11,12 After diagnosis of blast crisis, patients die within few months, usually from fatal infection or bleeding or organic failure due to blast infiltration.13
In the majority of patients, blast phase is preceded by an accelerated phase characterized by therapy resistance with increasing leuko- or thrombocytosis, increasing counts of immature myeloid precursors in PB, progressive basophilia, splenomegaly or hepatomegaly, and reduced quality of life. From the time of diagnosis of accelerated phase, the survival time is markedly reduced (median less than 2 years).1
With the application of BCR-ABL specific TK inhibitors for therapy of CML, prognosis and quality of life of patients have dramatically improved since the risk of progression to accelerated or acute phase has been significantly reduced when compared to earlier therapy concepts.10,14
Diagnostic procedures
Genetics
Evidence of the Philadelphia chromosome or the reciprocal translocation t(9;22)(q34;q11.2) confirms the CML diagnosis3 in patients with characteristic morphologic changes in blood and BM (see below). Molecular evidence of the BCR-ABL1 fusion is mandatory in patients without the Philadelphia chromosome. Cytogenetic and molecular studies are also used to control the efficacy of treatment (see Chapter 21 for details). Nowadays, disease is considered therapy-resistant in patients who do not achieve cytogenetic remission2 but patients who achieve a continuing CyCR have an excellent prognosis.15
Whereas additional chromosomal changes besides t(9;22)(q34;q11.2) do not appear to provide independent prognostic information when occurring prior to anti-neoplastic therapy,16 clonal evolution may support diagnosis of therapy resistance or accelerated phase when occurring during the course of CML.17,18
BCR-ABL1 mutations are frequent in patients who become resistant to a therapy with a TK inhibitor.17,19 Therefore, molecular analysis of BCR-ABL1 mutations has become a further standard procedure in patients losing their cytogenetic or molecular response to therapy.2
Morphology
Morphological diagnosis of CML is based on characteristic changes in blood and BM (Table 24.1). Organomegaly (splenomegaly, hepatomegaly) may also be present at diagnosis, indicating extramedullary expansion of disease.16
At diagnosis
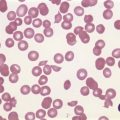
Peripheral blood. The first diagnostic feature usually arousing suspicion of CML is a leukemic PB picture. Granulocytosis with a pathologic left shift without hiatus leukaemicus is the main characteristic (Fig. 24.1). Granulocytosis comprises neutrophils, eosinophils and basophils. Myelocytes usually predominate among immature precursors mobilized into PB, but promyelocytes and myeloblasts may also occur especially in the late chronic or the accelerated phase (Fig. 24.1). The WHO classification defines a blast count of less than 20%, otherwise the disease should be classified as an acute leukemia.3 Some therapy trials use the previous FAB classification definition, where the count of blasts and promyelocytes should be less than 30%.2 Thrombocytosis is a common finding and when excessive (>106/l) may lead to the misdiagnosis of ‘essential thrombocythemia’ (ET).20,21 However, CML with thrombocytosis differ from ET by a pathologic left shift and basophilia in the differential count, characteristic morphologic changes in BM (see below), evidence of the BCR-ABL1 fusion, and in most cases, lack of the JAK2V617F mutation (see Chapters 21 and 23 for details).
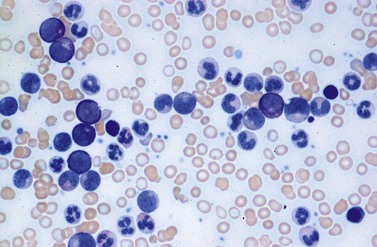
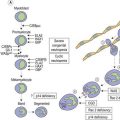
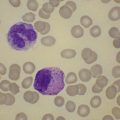
Changes in hematopoiesis. First obvious morphologic feature in microscopic evaluation at low magnification is a marked expansion of neutrophilic granulopoiesis (Fig. 24.2). In the majority of patients, eosinophilic granulopoiesis is also increased. Basophilic granulopoiesis is at least slightly increased in almost all patients (Fig. 24.3). In the majority of patients, granulopoiesis shows a complete maturation (Fig. 24.2), but it is often left shifted with a slight relative increase of myelocytes. In some patients, numbers of promyelocytes and myeloblasts are also increased. The myeloblast count is usually lower than 10%. The neoplastic granulocytes show a reduced leukocyte alkaline phosphatase (LAP) index, which distinguishes them from non-neoplastic granulocytes in leukemoid reactions.22 However, this does not significantly influence their function so that infections due to granulocytic dysfunction are uncommon in CP of CML. In the past, determination of the LAP index of granulocytes was a standard method applied at diagnosis of CML. Molecular investigation for the presence of the BCR-ABL1 fusion has replaced this procedure.
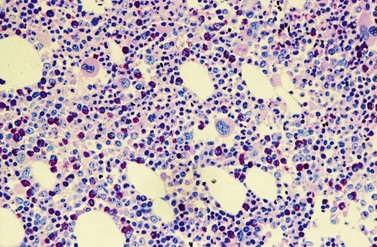
A further characteristic feature is an increase of micromegakaryocytes with a central hypolobulated nucleus, which is due to a reduction of mature hyperploid megakaryocytes23,24 (Figs 24.4 and 24.5). The exact mechanism leading to the lack of hyperploid megakaryocytes is not clear as yet. This feature is pathognomonic for CML and lack of it should question the diagnosis of CML. Some authors have divided CML according to the count of micromegakaryocytes into a megakaryocytic-rich and a granulocytic subtype.23,25 The megakaryocytic-rich subtype differs from the granulocytic by an increased risk of BM fibrosis.
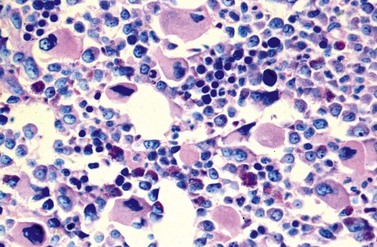
Although erythropoiesis in CML is also BCR-ABL1 positive, it is usually not expanded. In the majority of patients, erythropoiesis is rather reduced, particularly in late chronic, accelerated or blastic phase, correlating with a varying degree of anemia.25
Except for micromegakaryocytes, dysplastic features of hematopoiesis are uncommon in CP of CML. Platelets may show dysfunction with increased risk of bleeding, but thromboembolic events are uncommon.26
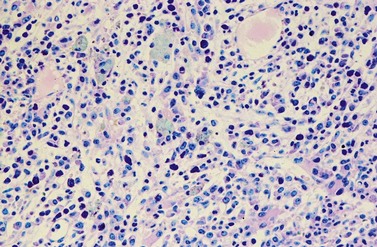
A blast count exceeding 10% of marrow cells is not common at diagnosis, but a typical feature during the course of CML, especially in accelerated or blastic phase (Fig. 24.7). Blasts occur mainly within the proliferation zone of granulopoiesis (myeloblasts; Fig. 24.7), but they may also be observed outside of it, particularly lymphoblasts or megakaryoblasts (Fig. 24.8). By immunohistochemistry, myeloblasts are often but not always CD34 or CD117 positive with co-expression of CD33, myeloperoxidase or lysozyme. Megakaryoblasts are CD42b or CD61, and lymphoblasts have B-precursor related phenotype: TdT, CD34, CD10, CD20 and CD79a positive. Flow cytometry may be useful by demonstrating aberrant CD56 expression in granulopoiesis, which is, however, also observed in other myeloproliferative and myelodysplastic disorders.27 This method can also help in early detection of incipient blast crisis and in differentiating between lymphatic and myeloid blastic transformation.28
Changes of non-hematopoietic tissue. First obvious morphologic feature in microscopic evaluation at low magnification is the replacement of the adipose BM tissue usually to less than 5% of marrow volume (Figs 24.2, 24.5–7). A further typical feature is the occurrence of storage histiocytes resembling Gaucher cells, the so-called pseudo-Gaucher cells29 (Figs 24.2, 24.5). These histiocytes belong to the neoplastic clone,30 and their Gaucher-like appearance is caused by a relative enzyme insufficiency as a result of an excessive phagocytosis of leukemic cells whereas the mesenchymal stem cells, which differentiate into adipocytes, fibroblasts and osteoblasts are not involved in the neoplastic proliferation.31
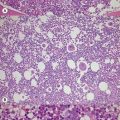
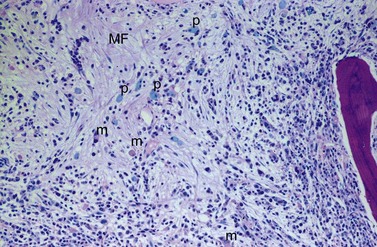
BM fibrosis is another typical feature of CML (Figs 24.5, 24.6).32 It affects only a minority of patients at diagnosis, but a significant proportion of patients develop fibrosis during the course of disease, especially when therapy has lost its efficacy.33,34 In patients not responding to therapy, the cumulative proportion of cases with BM fibrosis amounts to more than 90%.33 Therefore, CML is the second myeloproliferative neoplasm besides primary myelofibrosis (PMF) characterized by a high risk of BM fibrosis.
Marrow fibrosis usually starts focally (patchily), followed by transformation to diffuse extensive fibrosis developing during course of disease.34 As in PMF, the fiber-producing cells (fibroblasts) are not part of the neoplastic clone.31 However, their fiber production is up-regulated by growth factors produced by the neoplastic hematopoiesis, particularly platelet-derived growth factor and transforming growth factor-β, resulting in increased collagen type I and III (reticulin) fiber deposits.35
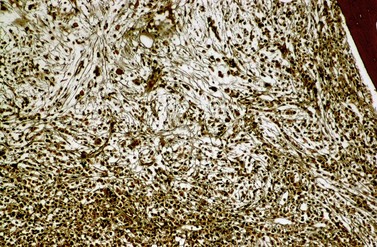
Fibrosis becomes visible after silver impregnation (Fig. 24.6). Various methods were presented with respect to grading of marrow fibrosis (see Chapter 3), but a worldwide uniform approach to diagnosis and quantification of marrow fibrosis does not exist so far. In general, more than 5% of marrow volume per µm thickness of tissue section with fiber deposits >104 mm/mm3 are uncommon in normal healthy subjects so that this limit can be applied to diagnose BM fibrosis.36 In fibrotic BM regions in CML, the density of the fiber deposits usually exceeds 104 mm/mm3.36
BM fibrosis correlates with an increased megakaryocyte count, splenomegaly and mobilization of normoblasts in PB.25 It may also correlate with an excess of blasts. Extensive diffuse fibrosis may be accompanied with osteosclerosis.
Another characteristic feature that CML shares with other neoplastic disorders is an increased BM vascularity37 (Fig. 24.7). It results from an increased production of pro-angiogenic growth factors by the neoplastic clone, especially the vascular endothelial growth factor A (VEGF). It has been found that BCR-ABL1 oncoprotein could in vitro induce VEGF promoter activity and increased VEGF protein levels in Ba/F3 cells as well as promote the expression of functionally active hypoxia-inducible factor-1 (HIF-1), a major transcriptional regulator of VEGF gene expression.38 Furthermore, the pluripotent neoplastic stem cell appears to be able to differentiate into endothelial cells.39 Thus, CML seems to be able to induce BM hypervascularity as a part of the neoplastic proliferation.
Extramedullary manifestations. Up to 70% of CML patients at diagnosis show a varying degree of hepato- and splenomegaly.16 Spleen enlargement results from an infiltration of red pulp cords by neoplastic hematopoiesis, mainly granulopoiesis. Hepatomegaly is a result of intrasinusoidal and periportal infiltration by hematopoiesis.
Phases of disease
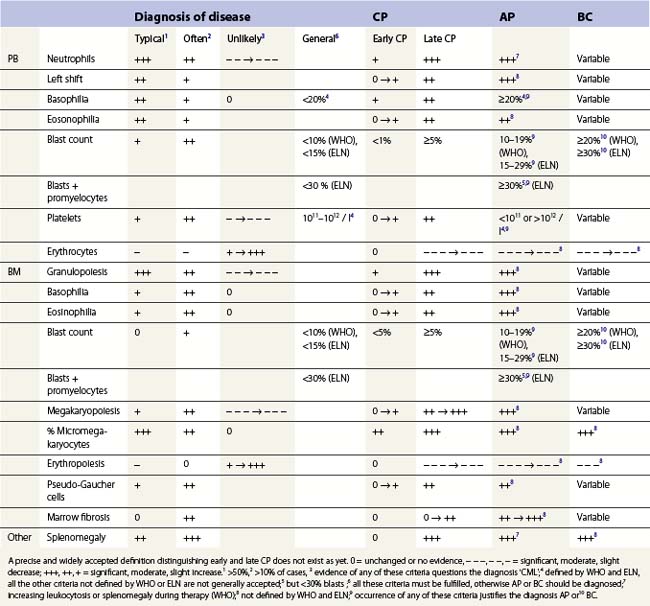
The phase of disease as defined by blood and BM morphology is still the most important prognostic factor of CML and its prognostic significance has not been abolished by current therapy, including allogenic stem cell transplantation and TK inhibitors. However, worldwide uniform approach to the definitions of these phases has not been achieved as yet. Two definitions are widely applied (Table 24.1), the criteria proposed by the WHO, and the definitions recommended by the European LeukemiaNet.2,3
Chronic phase (CP)
CML is usually diagnosed in chronic phase (CP). It is defined by less than 10% (WHO) or 15% (European LeukemiaNet) blasts within blood and BM, and less than 20% basophils in the PB. If these criteria are not fulfilled, the diagnosis of accelerated or acute phase is made. During therapy, persistent thrombocytopenia <100 × 109/l unrelated to treatment should not occur. Furthermore, the WHO classification mentions additional following criteria for CP CML: absence of therapy-resistant progressive disease (increasing splenomegaly, increasing white blood cell count (WBC), thrombocytosis >1000 × 109/l) and absence of cytogenetic evidence of clonal evolution.3
At diagnosis, BM fibrosis may occur in up to 30% of patients otherwise fulfilling criteria of CP (Figs 24.5, 24.6). Currently, BM fibrosis is not considered as a significant factor in the definition of the phase of disease. However, the prognosis of patients presenting with fibrosis appears to be rather poor, resembling accelerated phase.34 Therefore, as long as a final consensus of opinions does not exist and as long as these patients do not show other signs of acceleration, we recommend omitting differentiation between chronic and accelerated phase in these patients and report the findings as ‘CML with fibrosis’.
Early CP
A clear definition of ‘early’ and ‘late’ CP does not exist so far, although it is well accepted that less and more advanced stages of CP do exist.40 The success of BCR-ABL TK inhibitors in inducing remission in CML has led to an increased awareness of CML and its symptoms, so that earlier stages of this disease are diagnosed more frequently. Patients with early chronic phase of CML (eCP) show relatively low WBC (e.g. <20 × 109/l), less obvious left shift in the differential count, often with only few myelocytes, no or minimal thrombocytosis (platelets <600 × 109/l), no organomegaly, no anemia and usually no general symptoms. In the PB, basophil count is often slightly increased, the eosinophil count is variable. Sometimes, thrombocytosis is the only conspicuous change in the PB. BM shows a varying increase of micromegakaryocytes with hypolobulated nucleus (Figs 24.3 and 24.4). There is often no or only a slight increase of granulopoiesis with slight left shift due to a relative increase of myelocytes and promyelocytes, as well as a slight relative increase of eosinophilic and/or basophilic granulopoiesis (Figs 24.3 and 24.4). Morphology of erythropoiesis and adipose marrow tissue do not appear altered (no significant reduction). BM fibrosis, blast excess and splenomegaly are uncommon. Among the morphologic changes, relative increase of basophils within the PB and the increase of micromegakaryocytes with hypolobulated nucleus in BM are most characteristic and should be followed by test for the BCR-ABL1 fusion. CML in eCP is often detected by chance, due to the alterations of the blood cell counts.
Late chronic phase (lCP)
In BM biopsy, replacement of erythropoiesis and adipose marrow due to excessive expansion of neutro-, eosino- and basophilic granulopoiesis is characteristic; adipose BM tissue is usually lower than 2% of BM volume (Fig. 24.2). The BM blast count often exceeds 5%. Micromegakaryocytes with hypolobulated nuclei may be excessively increased and they may show a larger variation of size than those seen in eCP.23 Focal BM fibrosis may occur, and it is usually associated with a marked increase of megakaryopoiesis.
Accelerated phase (AP)
Accelerated phase (AP) is seen only in a minority of patients at CML diagnosis, but it is a common finding when therapy has lost its efficacy.2,33 By definition, AP differs from lCP in four variables: (1) the percentages of blasts in PB and (2) the percentages of blasts in BM, (3) the thrombocyte counts, and (4) the basophil counts in PB. A basophil count ≥20% of leukocytes or a persistent thrombocytopenia <100 × 109/l unrelated to therapy are generally accepted criteria. Blast counts of 10–19% (WHO) or 15–29% (European LeukemiaNet) within blood or BM are further requirements.2,3 The European LeukemiaNet has defined a BM or blood blast + promyelocyte count >30% with less than 30% blasts in blood and BM as a further fifth criterion for AP2 whereas the WHO has proposed therapy-resistant progressive disease (increasing splenomegaly, increasing WBC, thrombocytosis >1000 × 109/l) or cytogenetic evidence of clonal evolution as additional criteria.3 Occurrence of any of these criteria justifies the diagnosis of AP. In most recent therapeutic trials, the criteria proposed by the European LeukemiaNet are used whereas the criteria proposed by the WHO classification are applied less commonly.
The BM alterations in AP resemble those observed in lCP. The BM blast or basophil counts are usually higher, and focal clusters of blasts may occur (Fig. 24.7). Focal accumulations of more than 30 blast cells should give a suspicion of evolving blast crisis and should therefore be mentioned in the pathology report. Megakaryopoiesis is usually significantly increased (Fig. 24.8), but megakaryocytes rarely exceed 50% of BM cells.23 In a minority of patients, megakaryopoiesis may be reduced. BM fibrosis is a typical finding affecting up to 90% of patients (Figs 24.5 and 24.6).33 In the majority of patients, fibrosis correlates with a markedly increased count of megakaryocytes. Osteosclerosis may also occur, especially in advanced stage of BM fibrosis.23 AP may also show dysplastic features of hematopoiesis resembling those observed in myelodysplastic syndromes (MDS). After detection of AP, the overall survival time of patients is usually reduced to less than 2 years.41 In the majority of patients, there is a continuous transition from lCP to AP of disease. The criteria for diagnosis of AP are based on the experience collected from patients treated with interferon-α or chemotherapy. Optimization of AP criteria for patients treated with TK inhibitors are pending so far.
Acute phase, blast crisis (BC)
Blast crisis (BC) is defined by the percentage of blasts in blood and BM. Whereas the WHO defines BC as ≥20% blasts in blood or BM, the European Leukemia Net applies a blast count ≥30%, which is used in current European studies.2 Furthermore, the WHO classification mentions extramedullary blast proliferation or large foci or clusters of blasts in the BM biopsy as indicators of BC.3
In the majority of patients BC evolves from AP, but occasionally it emerges suddenly from CP without obvious transient AP.42 BC is rarely detected at diagnosis of CML. It usually occurs during the course of disease, especially after therapy failure. BC was more common during earlier applied treatment modalities, for example, treatment with chemotherapy.11 Acquired loss of p53 appears to play a central role in a significant proportion of patients.43
In about 70% of patients BC is myeloid and in about 30% it is of lymphoid origin, fulfilling, respectively, the criteria of acute myeloid or B-lymphoblastic leukemia. Megakaryoblastic crisis may occur, particularly in patients with prior excess of megakaryopoiesis. Erythroblastic crisis is rare. In about 20–30% of patients, BC shows mixed phenotype involving both myeloblasts/megakaryoblasts and lymphoblasts.44
Prognostic factors in CML
TK inhibitors (imatinib mesylate and other drugs) have significantly improved the survival time of patients due to a reduction of the risk of transformation into AP or BC.14 However, this has not substantially improved the survival time in AP or BC.41 Thus, AP and BC are still the main disease-related causes of death in CML patients. To predict the outcome in CP, prognostic scores were presented in the literature, comprising clinical, morphological and genetic variables (Table 24.2). Age, spleen size, platelet count and percentage of blasts in blood have been established as prognostic factors at CML diagnosis.16,45 Dynamics and degree of hematologic, cytogenetic and molecular remission are important prognostic factors during therapy. Other variables, such as clonal evolution or BM fibrosis, may also be significant. However, these variables are not considered in the majority of recent therapy studies. The established scoring systems are based mainly on observation of patients treated with interferon-α or chemotherapy.16,45 Due to excellent survival time of patients treated with BCR-ABL specific TK inhibitors, it appears to be too early to make definite recommendations concerning prognostic scoring and overall survival time during this new type of treatment.
| Sokal index45 | Hasford score16 | |
|---|---|---|
| Age (years) | + 0.116 × (age − 43.3) | + 0.666 if age ≥50 years |
| Spleen size (below costal margin) | + 0.0345 × (spleen size [cm] − 7.51) | + 0.042 × spleen size [cm] |
| Thrombocyte count | + 0.188 × ((platelets × 109/l: 700))2 − 0.563 | + 1.0956 if platelets ≥1500 × 109/l |
| % Blasts (differential blood count) | + 0.0887 × (blast count [%] − 2.1) | + 0.0584 × blast count [%] |
| % Basophils (differential count) | − not applied − | + 0.20399 if >3% basophils |
| % Eosinophils (differential count) | − not applied − | + 0.0413 × eosinophils [%] |
| Score value W | = esum of score values | = sum of score values × 1000 |
| Risk groups: low | if W <0.8 | if W ≤780 |
| intermediate | if 0.8 ≤W ≤1.2 | if 780 < W ≤1480 |
| high | if W >1.2 | if W >1480 |
* Based on observations of patients treated with interferon-α or chemotherapy. A risk score based on results of treatment with BCR-ABL specific TK inhibitors is not available so far.
Differential diagnoses
CML can be mimicked by numerous other neoplastic or non-neoplastic disorders with leuko-/thrombocytosis or left shift in the PB (Box 24.1). Molecular evaluation of hematopoiesis in such cases is mandatory, since evidence or lack of the BCR-ABL1 fusion allows a clear distinction. Nevertheless, careful morphologic evaluation usually allows correct diagnosis in the most cases. Furthermore, the course of CML may be complicated by the evolution of BCR-ABL1 negative myeloid neoplasms leading to a misdiagnosis or a misclassification of the phase of CML.
Box 24.1 Differential diagnosis of CML
Neoplastic disorders
Atypical CML
In the past, it was assumed that a minority of cases morphologically and clinically corresponding to CML may lack the BCR-ABL1 fusion. Therefore, it is has been accepted that a BCR-ABL1 negative leukemic disease similar to CML exists, but should be distinguished from it by applying the term ‘atypical CML’.46 It has been shown that BCR-ABL1 negative patients poorly respond to treatment and show an unfavorable prognosis.47
Although atypical CML shares many morphologic features with BCR-ABL1 positive CML, it differs in several characteristic features allowing discrimination. Firstly, increase of basophils in blood or BM and thrombocytosis are uncommon in atypical CML.46 Although atypical CML may also show an increase of micromegakaryocytes, megakaryocytes show a wider variation in size and morphology, may contain hyperlobulated nuclei or may be multinucleated. Erythropoiesis may be slightly increased and it may show macrocytic changes or dysplastic features. Granulopoiesis in atypical CML frequently shows dysplastic features, such as an abnormal chromatin clumping, which is very uncommon in CP of CML.48 Nevertheless, cases by morphology almost indistinguishable from BCR-ABL1 positive CML exist, thus requiring molecular exclusion of the BCR-ABL1 fusion.
Chronic neutrophilic leukemia
Chronic neutrophilic leukemia is a very rare disease. It shares with CML features of neutrophilic granulocytosis and splenomegaly, and (few) immature precursors of granulopoiesis may also occur in PB. Usually, it can be distinguished from CML by lack of basophilia and lack of micromegakaryocytes with hypolobulated nuclei. However, in patients with a BCR-ABL1 fusion at e19a2, which results in a p230 oncoprotein, neutrophilia is a characteristic feature49 so that molecular exclusion of the BCR-ABL1 fusion is mandatory for discrimination from CML.
Chronic eosinophilic leukemia
CML may show a marked eosinophilia requiring distinction from chronic eosinophilic leukemia (CEL), another rare myeloproliferative neoplasm (see Chapter 25 for details). CML usually differs from CEL by a higher basophil count in blood and BM as well as by the presence of micromegakaryocytes with hypolobulated nucleus. However, some cases with CEL may also show a slight basophilia or micromegakaryocytes and differential diagnosis will rely on molecular studies.
Chronic myelomonocytic leukemia
Although the absolute monocyte count within the PB is usually slightly increased, relative monocytosis exceeding 10% in the differential count and significant increase of monocytes in BM are uncommon in CML. However, in cases with a BCR-ABL1 fusion at e1a2 with resulting P190 oncoprotein, monocytosis is frequent.49
Primary myelofibrosis
In early cellular phase of PMF, marked leukocytosis, pathologic left shift and thrombocytosis in blood, and marked increase of granulopoiesis may mimic CML. In its advanced stage, excessive BM fibrosis with marked increase of megakaryopoiesis or excess of blasts may cause problems in distinguishing from AP of CML. Most important morphologic differences to BCR-ABL1 positive CML are megakaryocytes and basophils. In PMF, megakaryocytes are usually larger than normal, containing an enlarged hypolobulated nucleus with low density of chromatin (see Chapter 23). Basophilia in blood or BM is uncommon even in advanced stage of PMF, but it is a typical feature of AP of CML. PMF frequently shows a JAK2V617F mutation (see Chapter 21), which is uncommon in BCR-ABL1 positive CML and in atypical CML. However, in patients with a BCR-ABL1 fusion at e19a2 which results in a p230 oncoprotein, basophilia may be missing or insignificant49 so that molecular exclusion of the BCR-ABL1 fusion is also recommended for PMF cases.
Polycythemia vera (PV)
Long-standing PV may transform into a disease resembling CML with marked leukocytosis and anemia. Eosinophilia, basophilia, left shift in the PB, BM fibrosis, and splenomegaly may also occur. CML-like PV differs from CML by giant megakaryocytes which do not occur in CML, and by the JAK2V617F mutation (see Chapters 21 and 22). BCR-ABL1 positive CML occurring during the course of PV is extremely rare.50 Nevertheless, if doubts remain, PB should be examined for the BCR-ABL1 fusion.
Essential thrombocythemia
In a minority of cases, BCR-ABL1 positive CML may show a marked thrombocytosis that may exceed 2000 × 109/l, whereas leukocytosis is mild or lacking. However, megakaryopoiesis significantly differs from that usually seen in ET; the megakaryocytes are not enlarged as it is characteristic in ET and their morphology resembles that of ‘classic’ CML. Therefore, it is nowadays accepted that BCR-ABL1 positive ET does not exist; BCR-ABL1 positive MPN presenting with thrombocythemia only is regarded as a variant of CML.51
Myelodysplastic syndromes
Nevertheless, patients with MDS with a del(5q) (see Chapter 20 for details) often show a thrombocytosis; they may also show a slight leukocytosis. Atypical megakaryocytes with hypolobulated nucleus similar to those observed in CML occur in the BM, the granulopoiesis may be increased, and erythropoiesis is often reduced.52 Most important differences between MDS with del(5q) and CML are a less prominent proliferation of granulopoiesis, lower counts of eosinophils and basophils, no continuous left shift in the blood, and macrocytosis of erythrocytes. A cytogenetic evaluation provides final diagnosis.
Acute leukemia
By definition, AML is distinguished from CML by the blast count within blood and bone marrow (see Chapter 18). BCR-ABL1 positive primary AML is rare.53 If residual hematopoiesis shows morphologic features characteristic for CML and the BCR-ABL1 fusion is detected in a case presenting as AML, it is more likely a BC of CML rather than a primary AML. BCR-ABL1 positive acute lymphoblastic leukemia (ALL) occurs more frequently (see Chapter 19). Nevertheless, it cannot be excluded that some BCR-ABL1 positive B-precursor ALL in adults may be lymphatic BC of CML, particularly if p210 BCR-ABL1 is detected.
1 Vardiman JW. Chronic myelogenous leukmia, BCR-ABL1+. Am J Clin Pathol. 2009;132:250-260.
2 Baccarani M, Saglio G, Goldman J, et al. Evolving concepts in the management of chronic myeloid leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood. 2006;108:1809-1820.
3 Swerdlow SH, Campo E, Harris NL, et al. WHO Classification of Tumours of Haematopoietic and Lymphatic Tissues. Lyon: IARC; 2008.
4 Rohrbacher M, Hasford J. Epidemiology of chronic myeloid leukaemia (CML). Best Pract Res Clin Haematol. 2009;22:295-302.
5 Corso A, Lazzarino M, Morra E, et al. Chronic myelogenous leukemia and exposure to ionizing radiation – a retrospective study of 443 patients. Ann Hematol. 1995;70:79-82.
6 Posthuma EF, Falkenburg JH, Apperley JF, et al. HLA-B8 and HLA-A3 coexpressed with HLA-B8 are associated with a reduced risk of the development of chronic myeloid leukemia. Chronic Leukemia Working Party of the EBMT. Blood. 1999;93:3863-3865.
7 Zhang X, Ren R. Bcr-Abl efficiently induces a myeloproliferative disease and production of excess interleukin-3 and granulocyte-macrophage colony-stimulating factor in mice: a novel model for chronic myelogenous leukemia. Blood. 1998;92:3829-3840.
8 Jabbour E, Fava C, Kantarjian H. Advances in the biology and therapy of patients with chronic myeloid leukaemia. Best Pract Res Clin Haematol. 2009;22:395-407.
9 Hughes TP, Kaeda J, Branford S, et al. Frequency of major molecular responses to imatinib or interferon alfa plus cytarabine in newly diagnosed chronic myeloid leukemia. N Engl J Med. 2003;349:1423-1432.
10 O’Brien SG, Guilhot F, Larson RA, et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2003;348:994-1004.
11 Interferon alfa versus chemotherapy for chronic myeloid leukemia: a meta-analysis of seven randomized trials: Chronic Myeloid Leukemia Trialists’ Collaborative Group. J Natl Cancer Inst. 1997;89:1616-1620.
12 Hehlmann R, Berger U, Pfirrmann M, et al. Drug treatment is superior to allografting as first-line therapy in chronic myeloid leukemia. Blood. 2007;109:4686-4692.
13 Silver RT. The blast phase of chronic myeloid leukaemia. Best Pract Res Clin Haematol. 2009;22:387-394.
14 Druker BJ, Guilhot F, O’Brien SG, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355:2408-2417.
15 Piazza RG, Magistroni V, Franceschino A, et al. The achievement of durable complete cytogenetic remission in late chronic and accelerated phase patients with CML treated with imatinib mesylate predicts for prolonged response at 6 years. Blood Cells Mol Dis. 2006;37:111-115.
16 Hasford J, Pfirrmann M, Hehlmann R, et al. A new prognostic score for survival of patients with chronic myeloid leukemia treated with interferon alfa. Writing Committee for the Collaborative CML Prognostic Factors Project Group. J Natl Cancer Inst. 1998;90:850-858.
17 Hochhaus A, Kreil S, Corbin AS, et al. Molecular and chromosomal mechanisms of resistance to imatinib (STI571) therapy. Leukemia. 2002;16:2190-2196.
18 Cortes JE, Talpaz M, Giles F, et al. Prognostic significance of cytogenetic clonal evolution in patients with chronic myelogenous leukemia on imatinib mesylate therapy. Blood. 2003;101:3794-3800.
19 Jabbour E, Kantarjian H, Jones D, et al. Frequency and clinical significance of BCR-ABL mutations in patients with chronic myeloid leukemia treated with imatinib mesylate. Leukemia. 2006;20:1767-1773.
20 Michiels JJ, Berneman Z, Schroyens W, et al. Philadelphia (Ph) chromosome-positive thrombocythemia without features of chronic myeloid leukemia in peripheral blood: natural history and diagnostic differentiation from Ph-negative essential thrombocythemia. Ann Hematol. 2004;83:504-512.
21 Rice L, Popat U. Every case of essential thrombocythemia should be tested for the Philadelphia chromosome. Am J Hematol. 2005;78:71-73.
22 Okun DB, Tanaka KR. Leukocyte alkaline phosphatase. Am J Hematol. 1978;4:293-299.
23 Georgii A, Buesche G, Kreft A. The histopathology of chronic myeloproliferative diseases. Baillière’s Clin Haematol. 1998;11:721-749.
24 Jacobsson S, Wadenvik H, Kutti J, Swolin B. Low megakaryocyte ploidy in Ph-positive chronic myelogenous leukemia measured by flow cytometry. Am J Clin Pathol. 1999;111:185-190.
25 Thiele J, Kvasnicka HM, Fischer R. Bone marrow histopathology in chronic myelogenous leukemia (CML) – evaluation of distinctive features with clinical impact. Histol Histopathol. 1999;14:1241-1256.
26 Savage DG, Szydlo RM, Goldman JM. Clinical features at diagnosis in 430 patients with chronic myeloid leukaemia seen at a referral centre over a 16-year period. Br J Haematol. 1997;96:111-116.
27 Lanza F, Bi S, Castoldi G, Goldman JM. Abnormal expression of N-CAM (CD56) adhesion molecule on myeloid and progenitor cells from chronic myeloid leukemia. Leukemia. 1993;7:1570-1575.
28 Urbano-Ispizua A, Cervantes F, Matutes E, et al. Immunophenotypic characteristics of blast crisis of chronic myeloid leukaemia: correlations with clinico-biological features and survival. Leukemia. 1993;7:1349-1354.
29 Busche G, Majewski H, Schlue J, et al. Frequency of pseudo-Gaucher cells in diagnostic bone marrow biopsies from patients with Ph-positive chronic myeloid leukaemia. Virchows Arch. 1997;430:139-148.
30 Anastasi J, Musvee T, Roulston D, et al. Pseudo-Gaucher histiocytes identified up to 1 year after transplantation for CML are BCR/ABL-positive. Leukemia. 1998;12:233-237.
31 Carrara RC, Orellana MD, Fontes AM, et al. Mesenchymal stem cells from patients with chronic myeloid leukemia do not express BCR-ABL and have absence of chimerism after allogeneic bone marrow transplant. Braz J Med Biol Res. 2007;40:57-67.
32 Nowell PC, Kant JA, Finan JB, et al. Marrow fibrosis associated with a Philadelphia chromosome. Cancer Genet Cytogenet. 1992;59:89-92.
33 Buesche G, Hehlmann R, Hecker H, et al. Marrow fibrosis, indicator of therapy failure in chronic myeloid leukemia – prospective long-term results from a randomized-controlled trial. Leukemia. 2003;17:2444-2453.
34 Buesche G, Ganser A, Schlegelberger B, et al. Marrow fibrosis and its relevance during imatinib treatment of chronic myeloid leukemia. Leukemia. 2007;21:2420-2427.
35 Kimura A, Katoh O, Hyodo H, et al. Platelet derived growth factor expression, myelofibrosis and chronic myelogenous leukemia. Leuk Lymphoma. 1995;18:237-242.
36 Buesche G, Georgii A, Duensing A, et al. Evaluating the volume ratio of bone marrow affected by fibrosis: a parameter crucial for the prognostic significance of marrow fibrosis in chronic myeloid leukemia. Hum Pathol. 2003;34:391-401.
37 Korkolopoulou P, Viniou N, Kavantzas N, et al. Clinicopathologic correlations of bone marrow angiogenesis in chronic myeloid leukemia: a morphometric study. Leukemia. 2003;17:89-97.
38 Mayerhofer M, Valent P, Sperr WR, et al. BCR/ABL induces expression of vascular endothelial growth factor and its transcriptional activator, hypoxia inducible factor-1alpha, through a pathway involving phosphoinositide 3-kinase and the mammalian target of rapamycin. Blood. 2002;100:3767-3775.
39 Kvasnicka HM, Wickenhauser C, Thiele J, et al. Mixed chimerism of bone marrow vessels (endothelial cells, myofibroblasts) following allogeneic transplantation for chronic myelogenous leukemia. Leuk Lymphoma. 2003;44:321-328.
40 Palandri F, Iacobucci I, Quarantelli F, et al. Long-term molecular responses to imatinib in patients with chronic myeloid leukemia: comparison between complete cytogenetic responders treated in early and in late chronic phase. Haematologica. 2007;92:1579-1580.
41 Kantarjian HM, O’Brien S, Cortes JE, et al. Treatment of Philadelphia chromosome-positive, accelerated-phase chronic myelogenous leukemia with imatinib mesylate. Clin Cancer Res. 2002;8:2167-2176.
42 Alimena G, Breccia M, Latagliata R, et al. Sudden blast crisis in patients with Philadelphia chromosome-positive chronic myeloid leukemia who achieved complete cytogenetic remission after imatinib therapy. Cancer. 2006;107:1008-1013.
43 Calabretta B, Perrotti D. The biology of CML blast crisis. Blood. 2004;103:4010-4022.
44 Reid AG, De Melo VA, Elderfield K, et al. Phenotype of blasts in chronic myeloid leukemia in blastic phase – analysis of bone marrow trephine biopsies and correlation with cytogenetics. Leuk Res. 2009;33:418-425.
45 Sokal JE, Cox EB, Baccarani M, et al. Prognostic discrimination in ‘good-risk’ chronic granulocytic leukemia. Blood. 1984;63:789-799.
46 Bennett JM, Catovsky D, Daniel MT, et al. The chronic myeloid leukaemias: guidelines for distinguishing chronic granulocytic, atypical chronic myeloid, and chronic myelomonocytic leukaemia. Proposals by the French-American-British Cooperative Leukaemia Group. Br J Haematol. 1994;87:746-754.
47 Onida F, Ball G, Kantarjian HM, et al. Characteristics and outcome of patients with Philadelphia chromosome negative, bcr/abl negative chronic myelogenous leukemia. Cancer. 2002;95:1673-1684.
48 Invernizzi R, Custodi P, de Fazio P, et al. The syndrome of abnormal chromatin clumping in leucocytes: clinical and biological study of a case. Haematologica. 1990;75:532-536.
49 Pane F, Intrieri M, Quintarelli C, et al. BCR/ABL genes and leukemic phenotype: from molecular mechanisms to clinical correlations. Oncogene. 2002;21:8652-8667.
50 Mirza I, Frantz C, Clarke G, et al. Transformation of polycythemia vera to chronic myelogenous leukemia. Arch Pathol Lab Med. 2007;131:1719-1724.
51 Damaj G, Delabesse E, Le Bihan C, et al. Typical essential thrombocythaemia does not express bcr-abelson fusion transcript. Br J Haematol. 2002;116:812-816.
52 Washington LT, Doherty D, Glassman A, et al. Myeloid disorders with deletion of 5q as the sole karyotypic abnormality: the clinical and pathologic spectrum. Leuk Lymphoma. 2002;43:761-765.
53 Mrozek K, Heerema NA, Bloomfield CD. Cytogenetics in acute leukemia. Blood Rev. 2004;18:115-136.