[level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 14
Chronic Kidney Disease Mineral and Bone Disorder
The kidney plays a major role in bone and mineral homeostasis by regulating calcium, phosphorous, parathyroid hormone (PTH), fibroblast growth factor-23 (FGF-23), and calcitriol [1,25-dihydroxyvitamin D3, 1,25(OH)2D3] metabolism. Disordered regulation of mineral metabolism occurs early in the course of chronic kidney disease (CKD) and results in alterations in bone modeling and remodeling. A growing body of evidence demonstrates that cardiovascular calcifications accompany CKD, that cardiovascular disease is the leading cause of mortality in patients with CKD, and that therapies designed to treat the skeletal consequences of CKD affect the progression of vascular pathology; this has led to reclassification of the mineral, skeletal, and vascular complications associated with progressive kidney disease. Together, these alterations are termed CKD mineral and bone disorder (CKD-MBD).1
CKD-MBD is defined as a systemic disorder of mineral and bone metabolism due to CKD that is manifested by one or a combination of the following: (1) abnormalities of calcium, phosphorous, PTH, or vitamin D metabolism; (2) abnormalities in bone histology, linear growth, or strength; or (3) vascular or other soft tissue calcification. Renal osteodystrophy is the specific term used to describe the bone pathology that occurs as a complication of CKD and therefore is one aspect of CKD-MBD. Traditionally, such lesions have been defined according to alterations in bone turnover, ranging from high bone turnover (secondary hyperparathyroidism, osteitis fibrosa) to low bone turnover (adynamic bone disease and osteomalacia). However, alterations in skeletal mineralization and volume are common in patients in CKD1 and may contribute to such outcomes as fractures and skeletal deformities, which may persist despite normalization of bone turnover.2 Bone histomorphometry continues to be the gold standard for the assessment of three essential aspects of bone histology: turnover, mineralization, and volume.1 This chapter summarizes major aspects of the pathogenesis, clinical manifestations, histologic features, and therapeutic interventions currently used in the management of CKD-MBD. Clinical and histologic features of bone disease after successful kidney transplantation are also described.
Pathogenesis of CKD-MBD
Abnormalities of Calcium, Phosphorous, FGF-23, Vitamin D, and PTH Metabolism
Calcium and Phosphorus
1,25(OH)2D3, the most active form of vitamin D, regulates serum calcium levels by increasing intestinal calcium absorption. The kidney generates most circulating 1,25(OH)2D3, converting 25(OH)D to 1,25(OH)2D3 by means of the enzyme 1α-hydroxylase. As renal failure progresses, calcitriol levels and intestinal calcium absorption decline. However, at the same time, rising PTH levels increase 1α-hydroxylase activity, release calcium and phosphorus from bone, and promote renal conservation of calcium, thus maintaining serum calcium levels within the normal range until late in the course of CKD.3,4
Likewise, serum phosphorous levels usually are maintained in the normal range throughout mild to moderate CKD. Elevated serum PTH and FGF-23 levels increase phosphate excretion, thus maintaining overall phosphate balance until the glomerular filtration rate (GFR) declines to 25% to 30% of normal.5,6 In late (stage 4) CKD, hyperphosphatemia ensues and contributes to secondary hyperparathyroidism (Fig. 14-1A).3
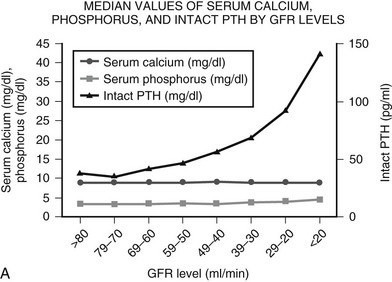
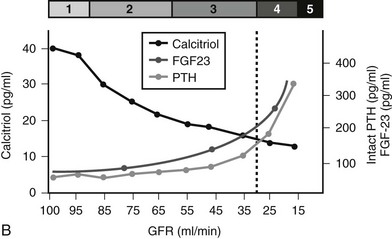
FIGURE 14-1 A, Median levels of calcium, phosphorus, and parathyroid hormone (PTH) per stage of chronic kidney disease (CKD). Median serum levels of calcium and phosphorus stay constant until late in the course of CKD; PTH levels rise before any changes are seen in calcium and phosphorus.
(A, Data from Levin A, et al: Kidney Int 71:31–38, 2007.3)
B, In patients with CKD, serum 1,25(OH)2D3 levels decline early in the course of kidney dysfunction, before any changes in serum calcium or phosphorous concentrations occur and prior to any rise in serum PTH levels.
Fibroblast Growth Factor-23
A recently described phosphaturic hormone, FGF-23, was first identified in patients with tumor-induced osteomalacia7 and autosomal dominant hypophosphatemic rickets.8 In these conditions and in patients with X-linked hypophosphatemia, elevated circulating levels of FGF-23 result in renal phosphate wasting and suppression of 1,25(OH)2D3 production.8,9 FGF-23 is made within bone,10,11 and the presence of a cofactor, Klotho, is essential for its action.12 Serum values increase as CKD progresses, becoming markedly elevated in individuals with end-stage kidney disease (Fig. 14-1B).13,14 In patients with CKD, 1,25(OH)2 D3 levels are inversely related to levels of circulating FGF-23, suggesting that increases in FGF-23 early in the course of CKD may play a role in declining active vitamin D levels.14 FGF-23 levels increase in response to vitamin D sterol therapy,15 and levels may be regulated by phosphorous intake.16,17 FGF-23 levels have been implicated in parathyroid gland regulation (vide infra).18,19
Vitamin D
Serum 1,25(OH)2D3 levels decline early in the course of kidney dysfunction, before any changes in serum calcium or phosphorous concentrations and prior to any rise in serum PTH levels.3,20 In late stages of CKD, phosphate retention and increased serum phosphorous levels directly suppress 1α-hydroxylase activity.16 However, although these factors contribute to declining 1α-hydroxylase activity, current evidence demonstrates that rising FGF-23 levels may be of even greater importance.14
Low circulating levels of 1,25(OH)2D3 have consequences for many tissues. Aside from its effect on intestinal calcium absorption, 1,25(OH)2D3 plays a direct role in the suppression of PTH gene transcription (vide infra). Animal studies also indicate that 1,25(OH)2D3 is essential for normal skeletal physiology—particularly in growing animals—and that this effect may not be mediated by the vitamin D receptor (VDR). Mice who lack the VDR [i.e., mice unable to respond to the actions of 1,25(OH)2D3 through its classical receptor] are phenotypically similar to those lacking the 1α-hydroxylase gene itself [i.e., mice unable to generate 1,25(OH)2D3]; both sets of mice are hypocalcemic with markedly elevated serum PTH levels, parathyroid gland hyperplasia, and rickets.21 However, a diet replete in calcium, phosphorus, and lactate is sufficient to normalize serum calcium, phosphorous, and PTH levels and to prevent the development of rickets in VDR-deficient animals.22 By contrast, this “rescue diet” is unable to completely reverse growth plate abnormalities in 1α-hydroxylase–deficient mice, suggesting that 1,25(OH)2D3, acting through a receptor other than the classical VDR, may be essential for proper growth plate development.23 1,25(OH)2D3 has been shown to regulate the renin-angiotensin system; 1α-hydroxylase–deficient mice demonstrate cardiac hypertrophy and dysfunction, which are reversed with angiotensin-converting enzyme blockade.24,25 Thus, 1,25(OH)2D3 may be essential for cardiac health—a finding that could explain observational data suggesting that active vitamin D sterol therapy improves survival in patients treated with maintenance dialysis.26,27
Native 25-hydroxyvitamin D [25(OH)D] deficiency is prevalent in patients with CKD; low levels of this form of the hormone also contribute to altered mineral metabolism. Vitamin D may be made in the skin or ingested from the diet.28,29 Ultraviolet B (UVB) (290 to 315 nm) photons penetrate the skin and are absorbed by 7-dehydrocholesterol to form previtamin D3, which then spontaneously converts to vitamin D3. Vitamin D3 is extruded from the skin cell into the extracellular space, where it binds vitamin D–binding protein.30 Although vitamin D created in the skin is exclusively of the D3 form, dietary sources of vitamin D and food supplements may contain vitamin D2 (created through UV irradiation of ergosterol in yeast) or D3 (from animal sources, particularly fish). Vitamin D (both D2 and D3) undergoes hydroxylation by the liver, forming 25(OH)D.31 Subsequently, 25(OH)D is taken up by renal tubular cells through a megalin-dependent process and undergoes a second hydroxylation, facilitated by renal 1α-hydroxylase, to 1,25(OH)2D3, a more potent stimulator of gut calcium absorption.32,33 Conversion of 25(OH)D to 1,25(OH)2D3 is independent of 25(OH)D stores in the general population but becomes a substrate-dependent process in patients with CKD.34 Although the actions of 25(OH)D have been underemphasized in CKD, extrarenal 1α-hydroxylase activity may significantly contribute to 1,25(OH)2D3 production, even in anephric patients.35–37 Thus low levels of the precursor, 25(OH)D, exacerbate 1,25(OH)2 D3 deficiency in the context of CKD.
Levels of 25(OH)D below 32 ng/mL are associated with increased PTH levels, reduced bone mineral density (BMD),38 and increased rates of hip fracture39 in the general population and therefore represent insufficient vitamin D storage. It is interesting to note that 25(OH)D levels in the vast majority of the general population meet the definition of D insufficiency, and a large percentage—as many as 57% in one series of medical inpatients40—have serum levels less than 15 ng/mL, thus qualifying as vitamin D deficient. Prevalence is higher in individuals with darker skin pigmentation, with 52% of Hispanic and black adolescents from the same cohort meeting the criteria for vitamin D deficiency.41
Several recent studies have documented a high prevalence of 25(OH)D deficiency in patients with CKD.42,43 Patients with CKD are at increased risk for vitamin D deficiency for several reasons. Many are chronically ill with little outdoor (sunlight) exposure, and CKD dietary restrictions, particularly of dairy products, curtail the intake of vitamin D–rich food.44 When compared with individuals with normal kidney function, patients with CKD display decreased skin synthesis of vitamin D3 in response to sunlight.45 This is exacerbated in individuals with darker skin.46 Proteinuria also contributes to D deficiency in the CKD population; 25(OH)D, in combination with vitamin D–binding protein, is lost in the urine.47,48
Parathyroid Hormone
The human PTH gene is located on chromosome 11 and contains two introns, which separate three exons encoding the 5′ untranslated region (UTR), the prepro region and PTH, and the 3′ untranslated region. The initial translational product of the mRNA is prepro-PTH, a 115 amino acid single-chain polypeptide, which undergoes conversion to pro-PTH (90 amino acids) in the rough endoplasmic reticulum. Six additional residues are removed from the N-terminal of pro-PTH in the Golgi apparatus to form the biologically active 1-84 PTH. PTH then is stored in secretory granules prior to release into the bloodstream.49–51 Sustained increases in PTH secretion occur with progression of CKD. This prolonged stimulation leads to high turnover bone disease and to the development of parathyroid gland hyperplasia.
PTH levels increase in response to alterations in metabolism of calcium, 1,25(OH)2 D3, phosphorus, 25(OH)D, and FGF-23 (Fig. 14-2). With the progression of CKD, additional factors develop that sustain high production of PTH. Among these factors are alterations in the regulation of prepro-PTH gene transcription, post-transcriptional modifications of PTH mRNA, reductions in calcium-sensing receptor (CaSR) and vitamin D receptor expression in the parathyroids, autonomous activity of adenomatous parathyroid glands, and skeletal resistance to the calcemic actions of PTH.
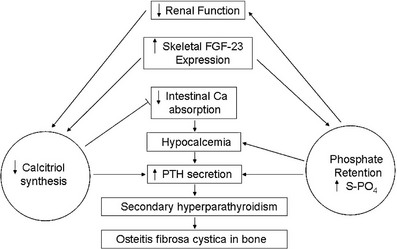
FIGURE 14-2 Pathogenesis of disordered mineral metabolism in chronic kidney disease (CKD). Dark arrows indicate effects of altered parathyroid hormone regulation; open arrows depict changes in fibroblast growth factor (FGF)-23 metabolism.
Calcium is the primary stimulus for PTH release. Calcium directly regulates PTH release via activation of the CaSR (vide infra). Activation of the CaSR decreases PTH release; deactivation increases PTH secretion. Serum calcium levels also regulate pre-pro PTH transcription via two negative calcium-response elements (nCaRE) located far upstream (−2.4 kbp and −3.5 kbp) of the PTH gene,52 and low serum calcium levels increase the stability of PTH mRNA by increasing levels of cytosolic parathyroid proteins, including AUF1,53 which binds to the 3′ UTR of the PTH mRNA. Thus, low levels of serum calcium present in CKD increase the amount of PTH release and the longevity of the PTH transcript. However, serum calcium levels are maintained within the normal range until late in the course of CKD, but serum PTH levels begin to rise much earlier, before any changes in serum calcium are evident.3 This suggests that factors other than calcium are important for the development of secondary hyperparathyroidism. Altered 1,25(OH)2 D3 metabolism may, in fact, be the initiating event early in the course of CKD.54–56
Apart from its effects on serum calcium levels, 1,25(OH)2D directly suppresses PTH levels. In conjunction with the VDR, 1,25(OH)2D3 binds negative vitamin D response elements in the promoter region of the PTH gene, thus inhibiting prepro-PTH gene transcription.57,58 In a positive-feedback loop, 1,25(OH)2D3 itself increases VDR gene expression in the parathyroid gland, further suppressing PTH gene transcription. 1,25(OH)2D3 also increases the expression of the CaSR, the expression of which is reduced in hyperplastic parathyroid tissues obtained from patients with secondary hyperparathyroidism.59 Vitamin D deficiency in animals is associated with decreased expression of CaSR mRNA in parathyroid tissue; 1,25(OH)2D3 therapy increases CaSR mRNA levels in a dose-dependent manner.60 Because 1,25(OH)2D3 is a potent inhibitor of cell proliferation, disturbances in renal calcitriol production and/or changes in VDR expression may be particularly important determinants of the degree of parathyroid hyperplasia and the extent of parathyroid gland enlargement in CKD.61
Phosphorous retention and hyperphosphatemia have been recognized for many years as important factors in the pathogenesis of secondary hyperparathyroidism. Phosphorous retention and hyperphosphatemia indirectly promote the secretion of PTH in several ways. Hyperphosphatemia lowers blood ionized calcium levels as free calcium ions complex with excess inorganic phosphate. The ensuing hypocalcemia stimulates PTH release. Increased phosphorus also impairs renal 1α-hydroxylase activity, which diminishes the conversion of 25(OH)D to 1,25(OH)2D3.16 Finally, phosphorus can directly enhance PTH synthesis. High serum phosphorous levels decrease cytosolic phospholipase A2 (normally increased by CaSR activation), leading to a decrease in arachidonic acid production with a subsequent increase in PTH secretion.62 Hypophosphatemia also decreases PTH mRNA transcript stability in vitro,63 suggesting that phosphorus itself affects serum PTH levels, probably by increasing the stability of the PTH mRNA transcript.
Although the actions of 25(OH)D have been underemphasized in dialyzed patients, evidence suggests that levels of this so-called “storage” form of vitamin D have both indirect and direct effects on PTH secretion. Extrarenal 1α-hydroxylase activity may contribute significantly to calcitriol production, even in anephric patients.35–37 Furthermore, recent evidence suggests the presence of 1α-hydroxylase in the parathyroid glands. 25(OH)D is converted inside the gland to 1,25(OH)2D, thereby suppressing PTH.64 25(OH)D administration suppresses PTH synthesis even when parathyroid gland 1α-hydroxylase is inhibited, indicating that 25(OH)D contributes to PTH suppression independent of its conversion to calcitriol.64 Indeed, recent studies have demonstrated that supplementation with ergocalciferol decreases serum PTH levels in patients with CKD.65,66 Such findings suggest that assessment of vitamin D status should be routinely performed in this patient population.67
Altered FGF-23 synthesis and secretion may contribute to increasing PTH levels through both indirect and direct mechanisms. Levels of FGF-23 rise as CKD progresses13,14 and contribute to declining 1,25(OH)2D levels. Lower 1,25(OH)2D levels, in turn, result in increasing PTH release. FGF-23 levels have been implicated in direct regulation of parathyroid gland function. In vitro and in vivo analysis of parathyroid glands from animals with normal renal function demonstrates that FGF-23 suppresses PTH secretion through a mechanism independent of its actions on vitamin D metabolism (vide infra).18,19
Alterations in parathyroid gland CaSR expression also occur in secondary hyperparathyroidism and may contribute to parathyroid gland hyperplasia. The CaSR is a seven-transmembrane G protein–coupled receptor with a large extracellular N-terminus, which binds acidic amino acids and divalent cations.68 Low extracellular calcium levels result in decreased calcium binding to the receptor, conformational relaxation of the receptor, and a resultant increase in PTH secretion,69 while activation of the receptor by high levels of serum calcium decreases PTH secretion.70,71 Expression of the CaSR is reduced by 30% to 70%, as judged by immunohistochemical methods in hyperplastic parathyroid tissue obtained from human subjects with renal failure.59,72 Decreased expression and activity of CaSR have been linked to decreased responsiveness in PTH secretion due to altered calcium levels.73 This decreased expression of the CaSR results in insensitivity to serum calcium levels with subsequent uncontrolled secretion of PTH. Increased stimulation of the CaSR by calcimimetics has been shown to decrease PTH cell proliferation, implicating the CaSR as a regulator of cell proliferation and PTH secretion.74
The link between the CaSR and vitamin D in cell cycling in the parathyroid gland is incompletely understood. However, some evidence suggests that vitamin D may decrease parathyroid hyperplasia by activating the CaSR. CaSR gene transcription is regulated by vitamin D through two distinct vitamin D response elements in the gene’s promoter region,75 suggesting that alterations in vitamin D metabolism in renal failure could account for changes in calcium sensing by the parathyroid glands, and that vitamin D may act upstream of the CaSR in preventing parathyroid cell hyperplasia.21
Once established, parathyroid enlargement is difficult to reverse because the rate of apoptosis in parathyroid glands is low, and the half-life of parathyroid cells is approximately 30 years.76 Chronic stimulation of parathyroid glands may lead to chromosomal changes that ultimately result in autonomous, unregulated growth and hormone release.77 Hyperplastic parathyroid tissue has been shown to develop inactivation of tumor suppressor genes MEN1, the retinoblastoma protein,78–80 and/or activating mutations of the RET proto-oncogene (MEN2a).81 Chromosomal translocations resulting in the parathyroid promoter driving cell cycle proteins (particularly cyclin D1) have been shown to be present in parathyroid adenomas.82 Even in the absence of somatic mutations, PTH secretion from enlarged parathyroid glands may become uncontrollable owing to the nonsuppressible component of PTH release from a large number of parathyroid cells. This alone may be sufficient to produce hypercalcemia and progressive bone disease in patients with end-stage renal disease.
Pathogenesis of Renal Bone Disease
Abnormalities In Bone Turnover, Mineralization, and Volume
Evaluation of skeletal histology by bone histomorphometry provides a method for understanding the pathophysiology of renal bone disease and a guide to its proper management. As recently recommended by the Kidney Disease Improving Global Outcomes (KDIGO) workgroup, three areas of bone histology are examined: bone turnover, mineralization, and volume, all of which may be altered in patients with CKD.1
Turnover
Traditionally, renal osteodystrophy has been classified primarily by alterations in bone turnover. Because PTH activates the PTH-related protein (PTHrP) receptor on osteocytes and osteoblasts, increasing cellular activity of both osteoblasts and osteoclasts,83,84 excessive levels of circulating PTH result in increased bone turnover (Figs. 14-3A and 14-3B).85 Serum PTH levels are inversely correlated with GFR, and most patients with GFR less than 50 mL/min have increased serum PTH levels and high turnover bone disease.86–88 The presence of CKD, however, markedly attenuates the effects of PTH on bone.89–90 Indeed, serum levels of PTH that are three to five times the normal range are associated with normal bone turnover in patients treated with maintenance dialysis, while similar PTH values in patients with mild to moderate kidney disease are associated with high turnover osteodystrophy.87,91,92 Although the precise mechanisms are poorly understood, uremia has been associated with this “skeletal resistance” to the actions of PTH. Uremic animals and humans display decreased PTH/PTHrP receptor mRNA expression in bone and growth plate.93,94 Hyperphosphatemia and alterations in vitamin D metabolism, among other factors, have been implicated in these changes, and calcitriol administration has been shown to partially restore the calcemic response to PTH in both experimental animals and patients with moderate CKD.95 Despite this “skeletal resistance” to PTH, many patients with end-stage kidney disease display bone biopsy evidence of PTH excess.
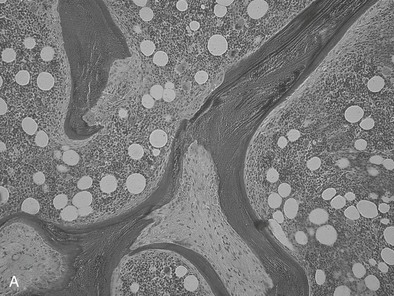
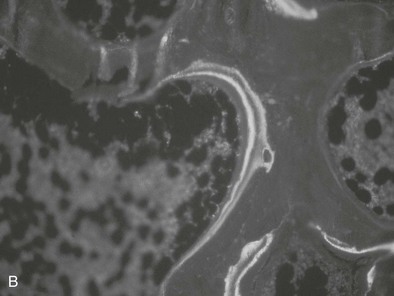
FIGURE 14-3 Bone histology, osteitis fibrosa. A, Under light microscopy, an increase in cellular activity, osteoid accumulation, erosion, and fibrosis are visible. B, An increase in double tetracycline labeling signifies an increase in bone turnover rate.
The bone in secondary hyperparathyroidism exhibits a marked increase in turnover with increased numbers of osteoblasts and osteoclasts and variable degrees of peritrabecular fibrosis. Activation of osteoclasts is mediated through PTH96–99; the result is increased resorption of both mineral and matrix along the trabecular surface and within the haversian canals of cortical bone.100 Such increased cellular activity can occur secondary to a nonspecific reaction to local factors, such as insulin-like growth factor-1 (IGF-1), cytokines, or fracture, or as the result of systemic stimuli, such as increased thyroxine or PTH. One characteristic of high bone turnover is increased quantities of woven osteoid, exhibiting haphazard arrangements of collagen fibers in contrast to the usual lamellar pattern of osteoid in normal bone. Woven osteoid can become mineralized in patients with advanced kidney disease in the absence of vitamin D; however, the calcium may be deposited as amorphous calcium phosphate rather than hydroxyapatite.101
At the other end of the spectrum of bone turnover, adynamic bone disease is characterized by normal osteoid volume, an absence of fibrosis, and a reduced bone formation rate, as indicated by a reduced or absent double tetracycline label on bone histomorphometry (Fig. 14-4).102,103 A paucity of osteoblasts and osteoclasts is observed.85 The histologic features of adynamic renal osteodystrophy, in the absence of aluminum deposition in bone, cannot be distinguished from the histologic features of corticosteroid-induced osteoporosis or age-related or postmenopausal osteoporosis. Therefore, it is not possible to determine whether osteoporosis accounts for decreases in osteoblastic activity and bone formation in patients with adynamic renal osteodystrophy unless the amount of trabecular bone is reduced. Decreases in bone mass and histologic evidence of trabecular bone loss are not integral features of the adynamic lesion of renal osteodystrophy when other causes of osteoporosis can be excluded. Adynamic bone is associated with low PTH levels, low alkaline phosphatase levels, high serum calcium levels, and a propensity for increased vascular calcification.104,105
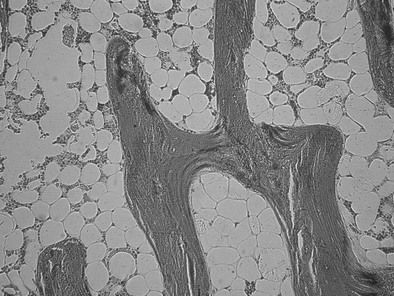
FIGURE 14-4 Bone histology, adynamic bone. Under light microscopy, decreased cellular activity occurs with minimal osteoid accumulation.
In the 1970s and 1980s, aluminum intoxication was largely responsible for the development of adynamic bone and osteomalacia in patients with CKD. Two distinct patterns of aluminum intoxication were identified: (1) absorbed from the aluminum content of water used to prepare dialysate solution,106–108 and (2) intestinal aluminum absorption after ingestion of large doses of aluminum hydroxide.109–115 The neurologic syndrome of “dialysis encephalopathy” and a bone disease manifested by fractures, pain, persistent hypercalcemia, and osteomalacia were the main clinical features. Although the prevalence of aluminum bone disease in developed countries is now very low, the prevalence of adynamic renal osteodystrophy not associated with aluminum intoxication has increased substantially over the past few years in adult patients receiving regular dialysis.116 Currently, adynamic renal osteodystrophy is commonly associated with disorders such as age-related or postmenopausal osteoporosis, steroid-induced osteoporosis, hypoparathyroidism (idiopathic or surgically induced), and diabetes mellitus, and with overtreatment with calcium and vitamin D therapy.117
Because PTH is the major determinant of bone formation and skeletal remodeling in renal failure, oversuppression of PTH secretion can result in adynamic renal osteodystrophy. Approximately 40% of those treated with hemodialysis and more than half of adult patients undergoing peritoneal dialysis have serum PTH levels that are only minimally elevated or that fall within the normal range; such values are typically associated with normal or reduced rates of bone formation and turnover.103 Prolonged treatment with calcium-containing phosphate-binding medications and the use of high-dialysate calcium concentrations also contribute to low bone turnover.118 Calcitriol may directly suppress osteoblastic activity when given intermittently in large doses to patients receiving regular dialysis.119
The long-term consequences of adynamic renal osteodystrophy not attributable to aluminum toxicity remain to be determined, but concerns have been raised about increases in the risk for skeletal fracture and delayed fracture healing due to low rates of bone remodeling.119 The development of soft tissue and vascular calcifications has been associated with adynamic bone disease in cross-sectional studies.105 In prepubertal children, adynamic renal osteodystrophy has been associated with a reduction in linear growth.117
Mineralization
Although renal osteodystrophy has traditionally been defined by lesions in bone turnover, alterations in skeletal mineralization are also prevalent in CKD.92 Increases in unmineralized bone (osteoid) in conjunction with delayed rates of mineral deposition are common.92,100 Defective mineralization associated with low to normal bone turnover is termed osteomalacia.1 Histomorphometric characteristics of osteomalacia include the presence of wide osteoid seams, an increased number of osteoid lamellae, an increase in the trabecular surface covered with osteoid, and a diminished rate of mineralization or bone formation, as assessed by double tetracycline labeling. Fibrosis is often absent.85 In patients on long-term dialysis, osteomalacia that is refractory to vitamin D therapy is most commonly a result of aluminum intoxication.120
Although the mechanisms of skeletal mineralization are incompletely understood, factors such as 25(OH)D deficiency and altered FGF-23 metabolism have been implicated in their pathogenesis. In the general population, nutritional 25(OH)D deficiency results in osteomalacia, and a similar phenotype may occur in patients with CKD. FGF-23 may play a role; both overexpression121–123 and ablation of FGF-23124,125 in mice with normal renal function are associated with abnormal mineralization of osteoid, although by different mechanisms. The phosphaturic effects of increased FGF-23 may cause rickets and osteomalacia through an insufficiency of mineral substrate. The mechanisms leading to impaired mineralization in FGF-23–null animals, which have severe hyperphosphatemia and normal or elevated serum calcium levels, remain uncertain; however, osteomalacia in these animals suggests that FGF-23 may play a direct role in skeletal mineral deposition. Although the ramifications of defective mineralization remain to be established, increased fracture rates and bone deformities are prevalent in patients with CKD despite adequate control of bone turnover. These complications may be due, in part, to alterations in bone mineralization.
Treatment with anticonvulsant therapy may contribute to the development of osteomalacia in patients with kidney disease. Long-term ingestion of phenytoin and/or phenobarbital is associated with a high incidence of osteomalacia in nonuremic patients.126 These findings may be due in part to alterations in vitamin D metabolism.127
Osteitis fibrosa cystica, a finding of secondary hyperparathyroidism, can coexist with defective mineralization in some patients; this pattern is called a “mixed” lesion.128 Patients with mixed lesions often display high serum PTH and alkaline phosphatase levels, along with lower serum calcium levels. Mixed lesions are seen with high turnover bone disease in patients who are developing aluminum toxicity, or in patients with low turnover aluminum-related bone disease during desferoxamine (DFO) therapy (vide infra).129 In these cases, mixed lesions represent a transitional stage between high turnover and low turnover bone disease.
Volume
Because PTH is anabolic at the level of trabecular bone, high levels of serum PTH are typically associated with increases in bone volume, trabecular volume, and trabecular width.92,116,130,131 However, bone volume may also be low (termed osteoporosis), particularly in individuals with underlying age-related bone loss and in those treated with corticosteroids. Osteoporosis in the general population is associated with increased risk for hip fractures and mortality.132 Thus, bone volume is considered a critically important parameter of bone histology. The impact of osteoporosis on morbidity and mortality in the CKD population, however, remains to be defined.
Clinical Manifestations
Bone Pain
Bone pain is a common manifestation of severe bone disease in patients with advanced kidney disease. It usually is insidious in appearance and often is aggravated by weight bearing or a change in posture. Physical findings are often absent. Pain is most common in the lower part of the back, hips, and legs but may occur in the peripheral skeleton. Occasionally, sudden appearance of pain around the knee, ankle, or heel can suggest acute arthritis; such pain is not usually relieved by massage or local heat. Bone pain is more common and often is more marked in patients with aluminum-related bone disease than in those with osteitis fibrosa cystica, but marked variability may be seen from one patient to another.133
In patients on long-term dialysis, carpal tunnel syndrome and chronic arthralgias often occur in association with the deposition of β2-microglobulin amyloid in articular and periarticular structures.134 Arthralgias usually are bilateral and most commonly affect the shoulders, knees, wrists, and small joints of the hand; symptoms typically are worse with inactivity and at night.135,136
Muscle Weakness
The pathogenesis of this myopathy is not clear, and several different mechanisms, including secondary hyperparathyroidism, phosphate depletion,137 abnormal vitamin D metabolism, and aluminum intoxication, have been implicated.138 Improvement in gait posture has been reported in children with moderate renal failure after treatment with 1,25(OH)2D3, and muscle weakness improves rapidly in affected adult patients with end-stage kidney disease.139 Improvement in muscular strength has been observed after treatment with 25(OH)D3, after subtotal parathyroidectomy, after successful renal transplantation, and after chelation therapy with deferoxamine for aluminum intoxication.
Skeletal Deformities
Bone deformities are common in uremic children because their bones undergo growth, modeling, and remodeling. In adult patients, skeletal deformities also arise from abnormal remodeling or recurrent fractures.140 In children, bone deformities of the femora and wrists arise from slipped epiphyses.141 This problem is most common during the preadolescent period and is most frequent in patients with long-standing congenital kidney disease. In adults with kidney disease, particularly those with aluminum-related bone disease, skeletal deformities may be characterized by lumbar scoliosis, kyphosis, and deformities of the thoracic cage.140
Growth Retardation
Growth retardation is the hallmark of CKD in children. Protein and calorie malnutrition, metabolic acidosis, end-organ growth hormone resistance, and renal bone disease are most commonly implicated in growth failure.142 Despite correction of acidosis and anemia, normalization of serum calcium and phosphorous levels, and vitamin D sterol therapy replacement, most children with CKD continue to grow poorly. Growth failure worsens as renal function declines; the average height of children with even mild CKD (GFR 50 to 70 mL/minute/1.73 m2) is 1 standard deviation (SD) below the average for healthy children. Moderate CKD (GFR 25 to 49 mL/minute/1.73 m2) is associated with a height SD of −1.5, and, at the time of initiation of dialysis, the mean height SD is −1.8. Boys, younger patients, and those with prior renal transplants are at greatest risk for growth failure.143
Acidosis has been linked to delayed linear growth in patients with renal tubular acidosis and normal renal function, and correction of metabolic acidosis often leads to acceleration in growth velocity.144 Acidotic rats have been found to have decreased growth hormone (GH) secretion, serum insulin-like growth factor 1 (IGF-1), and hepatic IGF-1 mRNA expression. Moreover, metabolic acidosis has been shown to inhibit the effects of GH in rats with normal and decreased renal function.145–147 Growth plate mRNA levels of GH receptor, IGF-1 receptor, and IGF-1 expression are downregulated, and IGF-binding proteins are upregulated.148 In adults treated with maintenance dialysis, correction of acidosis has been shown to decrease the progression of secondary hyperparathyroidism and to improve skeletal mineralization.149
Calcitriol deficiency has been thought to contribute to growth retardation and bone disease in children with CKD. Secondary hyperparathyroidism remains prevalent in children with advanced renal disease, and osteitis fibrosa continues to be the most common skeletal lesion of renal osteodystrophy in those undergoing regular dialysis despite treatment with daily doses of oral calcitriol.116,150 Secondary hyperparathyroidism contributes to growth retardation, although optimal target values for PTH in children at all stages of CKD remain controversial. In children with moderate CKD, some data indicate that normal growth velocity is achieved when PTH levels are maintained within the normal range151; others have demonstrated a linear correlation between growth and PTH levels in the same patient population, with those with the highest PTH values displaying the highest growth velocity.152 Treatment for secondary hyperparathyroidism with large, intermittent doses of calcitriol and calcium-based phosphate binders has been shown to significantly reduce bone formation and suppress osteoblastic activity in both adults and children.95,153 However, adynamic bone disease may develop and linear bone growth decrease, despite serum PTH levels in the K/DOQI117,154 recommended range during intermittent vitamin D sterol therapy. Maintaining serum PTH levels at between 300 and 500 pg/mL reduces the frequency of these complications.131 The mechanisms by which calcitriol inhibits epiphyseal growth plate cartilage remain poorly understood. However, it is well known that calcitriol exerts dose-dependent inhibitory effects on cell proliferation of chondrocytes and osteoblasts in vitro. In addition, vitamin D sterols increase expression of a number of IGF-binding proteins (IGFBPs). IGFBP-2, -3, -4, and -5 sequester IGF-1 and may exert IGF-1–independent antiproliferative effects through their own receptors.155–159
GH resistance also contributes to impaired linear growth in renal failure. In CKD, poor growth develops despite normal or increased serum GH levels.160,161 Uremia has been associated with diminished hepatic GH receptor and IGF-1 mRNA expression, defects in postreceptor GH-mediated signal transduction,162,163 reductions in serum GH-binding protein levels,164 and increased synthesis and reduced clearance of IGF binding proteins.138,164,165 Improved growth velocity during recombinant human GH (rhGH) therapy has been ascribed to increased bioavailability of IGF-1 to target tissues. Children who are treated with maintenance dialysis respond less well to rhGH therapy than do children with less severe CKD, but the mechanisms for differences in response to GH therapy remain to be determined.
Extraskeletal Calcification
Extraskeletal calcification has been associated with uremia for many years166 and has been included recently in the definition of CKD-MBD.1 Vascular calcifications are associated with increased mortality. These lesions have their origins in CKD prior to dialysis and begin in childhood (Fig. 14-5).167,169 The mortality rate in adults and children with CKD is markedly higher than that in the general population, and cardiovascular disease is the leading cause of death in both children and adults treated with maintenance dialysis.168,170 In contrast to the calcified atherosclerotic plaques that develop in the vascular intima of aging individuals with normal kidney function, uremia facilitates calcification of the tunica media. This form of calcification is associated with decreased distensibility of blood vessels, causing a rigid “lead pipe” pathology that is associated with increased risk for congestive heart failure.1 Electron beam computed tomography (EBCT) is used in assessment of vascular calcifications in the adult population, and measurements in young adults who were treated with maintenance dialysis as children demonstrated that a significant proportion of this population has evidence of vascular calcification.168 Carotid ultrasound measurement of intima medial thickness (IMT) has been validated for the assessment of cardiovascular pathology in children, with increased thickness associated with worsening disease.169,171
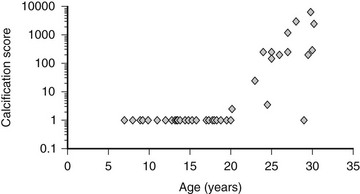
FIGURE 14-5 Coronary artery calcification scores in 39 children and young adults with end-stage renal disease who were treated by dialysis, according to age. Coronary artery calcification was assessed by electron beam computed tomography. The scale on the y-axis is logarithmic. (Data from Goodman WG, et al: Coronary artery calcification in young adults with end-stage renal disease who are undergoing dialysis, N Engl J Med 106:100–105, 2000.)
Hypercalcemia, hyperphosphatemia, elevated levels of the calcium x phosphorous product, and high doses of vitamin D sterols167,168 all have been implicated in the pathogenesis of cardiovascular calcification. However, 40% of adult patients with stage 3 CKD, without these risk factors, show evidence of calcification,172 suggesting a role for uremic factors other than high levels of calcium and phosphorus. Indeed, vascular tissues in the uremic milieu express osteoblast differentiation factors.173 Osteoblasts and vascular smooth muscle cells have a common mesenchymal origin; core binding factor-1 (Cbfa1) is thought to trigger mesenchymal cell-to-osteoblast transformation. Mice that are deficient in Cbfa1 fail to mineralize bone,174 and arteries obtained from patients undergoing renal transplantation show increased levels of this protein.173 Upregulation of the sodium-dependent phosphate transporter PIT-1 likely also contributes to increased calcification,175 and upregulation of pro-mineralization factors such as osteopontin, bone sialoprotein, osteonectin, alkaline phosphatase, type I collagen, and bone morphogenic protein-2 (BMP-2) is potentiated by the uremic milieu.176–179 By contrast, expression of calcification inhibitors such as fetuin A, matrix gla protein, and klotho is suppressed.180–183 Levels of circulating FGF-23 also may contribute, as high levels of the protein are associated with increased mortality in adult dialysis patients.184 Klotho, the cofactor necessary for the actions of FGF-23, has been implicated in vascular calcification. Animals lacking the Klotho gene display elevated levels of calcium, phosphorus, and vitamin D, along with vascular calcification and premature aging. CKD is associated with low circulating levels of Klotho,183 which has been implicated in the regulation of sodium/phosphate co-transport in the aorta.185
Because the pathophysiology of cardiovascular disease in CKD is multifactorial, treatment strategies are also multifaceted and vary according to stage of CKD. Therapies that are effective in early CKD may not be effective in later stages—lipid-lowering agents decrease mortality in adults with CKD186 and in those with stable renal allografts187 but have not been shown to benefit patients treated with dialysis.188 By contrast, normalization of mineral metabolism (attained by avoiding hypercalcemia and hyperphosphatemia, limiting calcium intake, and avoiding adynamic bone) is effective at slowing the progression of cardiovascular calcification in patients treated with maintenance dialysis.154,168,189,190 Thus, at different stages of CKD, the relative importance of individual risk factors and the value of different biomarkers may vary.
Calciphylaxis
This unique syndrome, which is characterized by ischemic necrosis of the skin, subcutaneous fat, and muscles, can develop in patients with advanced renal failure not yet treated by dialysis, in those treated with regular dialysis, and in patients with well-functioning kidney transplants.191 The pathogenesis of this syndrome is uncertain. Extensive medial calcifications of medium-sized arteries are noted; such calcifications commonly exist in patients with renal disease without causing gangrene or ulcerations, and it is not clear that vascular calcifications per se are the cause of the ischemic necrosis. Two distinct types of the syndrome are recognized: proximal calciphylaxis, which affects the thighs, abdomen, and chest wall, and acral calciphylaxis, which involves sites distal to the knees and elbows, such as the toes, fingers, and ankles.192 The former has a terrible prognosis, with death occurring in more than 80% to 90% of affected patients. This syndrome is often accompanied by morbid obesity and hypoalbuminemia. Many patients have severe secondary hyperparathyroidism, and most have a history of severe and uncontrolled hyperphosphatemia.192 Some patients may have defective regulation of coagulation.193 The appearance of this syndrome in renal transplant recipients receiving glucocorticoids suggests that steroids may also play a role. Patients with calciphylaxis frequently die of secondary infection.
A significant number of patients improve after parathyroidectomy, and a few have healed after substantial reductions in levels of serum phosphorus. Parathyroidectomy and aggressive control of serum phosphate levels therefore are indicated in those with evidence of severe secondary hyperparathyroidism. However, although ischemic lesions and medial vascular calcifications are common in uremic patients with diabetes, such lesions rarely improve after parathyroidectomy. Thus, parathyroid surgery should be reserved for those diabetic patients with calciphylaxis who have clear evidence of severe secondary hyperparathyroidism. Calcimimetics,194 as well as sodium-thiosulfate (a calcium chelating agent) and pamidronate,195 have been used effectively in some individuals. Hyperbaric oxygen therapy also has been advocated.195
Dialysis-Related Amyloidosis
Several clinical syndromes that arise as a consequence of dialysis-related amyloidosis can mimic the clinical features of renal osteodystrophy, or amyloidosis can occur concurrently with uremic bone disease. Dialysis-related amyloidosis arises from the deposition in bone and periarticular structures of a specific type of amyloid composed of β2-microglobulin.196 In addition to β2-microglobulin, the amyloid deposits contain advanced glycosylation end products, which may account for their uptake by certain collagen-rich structures.197,198 The frequency of its clinical manifestation rises markedly in patients treated with regular dialysis for longer than 5 to 10 years, and it is much more common in patients who start dialysis when older than 50 years.199 Blood levels of β2-microglobulin are strikingly elevated in all patients with end-stage renal disease because of failure of normal renal clearance and catabolism. Clinical manifestations include (1) carpal tunnel syndrome; (2) destructive or erosive arthropathy involving the large and medium-sized joints, with shoulder, knee, hip, or back pain being common manifestations; (3) spondyloarthropathy, most commonly affecting the cervical spine; and (4) subchondral, thin-walled cysts of bone, most commonly affecting the carpal bones, the humoral and femoral heads, the distal end of the radius, the acetabulum, and the tibial plateau. These subchondral cysts at times are confused with brown tumors of secondary hyperparathyroidism, although their location and occurrence in multiple sites make them quite different from brown tumors. Nonetheless, in a patient who has undergone long-term dialysis, this syndrome may be a potential cause of neuromuscular and periarticular symptoms usually considered to be due to secondary hyperparathyroidism or aluminum accumulation. Parathyroid surgery should be avoided in dialysis patients whose severe musculoskeletal symptoms have not improved with calcitriol therapy; symptoms in these patients actually may be due to dialysis-related amyloidosis. Specific diagnosis of the latter is made from the biopsy demonstration of amyloid composed of β2-microglobulin; however, the diagnosis can be strongly suspected from the clinical features, the presence of multiple thin-walled cysts, or the demonstration in periarticular sites of presumed amyloid tissue on ultrasonography.200 Its management is difficult and largely unsatisfactory, but successful renal transplantation leads to rapid disappearance of symptoms and no further progression of the bone lesions on radiographs.201 In patients who remain on maintenance dialysis, the use of high-flux dialyzer membranes may decrease amyloid accumulation202; the long-term effects of this form of therapy on symptoms remain to be determined.
Diagnostic Evaluations
Phosphorus
Hyperphosphatemia has been found to be an independent risk factor for mortality in patients treated with maintenance dialysis.203 Hemodialysis and continuous ambulatory peritoneal dialysis (CAPD) remove phosphate, but dietary phosphate restriction and the use of phosphate-binding agents are required by 90% to 95% of patients undergoing treatment with dialysis. To prevent the development and progression of secondary hyperparathyroidism and vascular calcification, such measures as dietary phosphate restriction and the use of phosphate-binding medications should be initiated to maintain serum phosphorous levels within age-appropriate levels and calcium phosphorous products lower than 55 mg2/dL.67,204
Calcium
Hypocalcemia often resolves during treatment with calcium-containing phosphate binders, with vitamin D, and with initiation of dialysis. The development of hypercalcemia in patients undergoing regular dialysis warrants prompt and thorough investigation. Conditions associated with hypercalcemia include marked hyperplasia of the parathyroid glands as a result of severe secondary hyperparathyroidism, aluminum-related bone disease, therapy with calcitriol or other vitamin D sterols, administration of large doses of calcium carbonate or other calcium-containing compounds, immobilization, malignancy, and granulomatous disorders, such as sarcoidosis or tuberculosis in which extrarenal production of 1,25(OH)D occurs.102,205,206 Basal serum calcium levels are also higher in patients with adynamic bone than in subjects with other lesions of renal osteodystrophy, and episodes of hypercalcemia are common.92 Because skeletal calcium uptake is limited in adynamic lesions, calcium entering the extracellular fluid from dialysate or after intestinal absorption cannot be buffered adequately in bone, and serum calcium levels rise.207 Maintaining total calcium intake at between 1500 and 2000 mg per day, the use of new-generation vitamin D sterols, and treatment with calcimimetic agents help to limit episodes of hypercalcemia.
Magnesium
The net intestinal absorption of magnesium generally is normal or only very slightly reduced in patients with renal failure,204 yet serum magnesium levels often increase with advanced kidney disease as the result of reduced renal excretion. During hemodialysis, serum magnesium levels generally are increased if the dialysate magnesium concentration is maintained at 1.75 mEq/L; however, magnesium levels remain within the upper range of normal with dialysate magnesium concentrations of 0.5 mEq/L. The use of magnesium-containing laxatives or antacids should be avoided as these may cause an abrupt increase in serum magnesium levels in patients with kidney disease.208 Serum magnesium levels should be measured frequently and regularly if magnesium-containing medications are used. Rarely, hypomagnesemia can develop in uremic patients with severe malabsorption or diarrhea.204
Alkaline Phosphatase
Serum alkaline phosphatase values are fair markers of the severity of secondary hyperparathyroidism in patients with renal failure. Osteoblasts normally express large amounts of the bone isoenzyme of alkaline phosphatase, and serum levels usually are elevated when osteoblastic activity and bone formation rates are increased. High levels generally reflect the extent of histologic change in patients with high turnover lesions of renal osteodystrophy, and values frequently correlate with serum PTH levels.209 Serum total alkaline phosphatase measurements are also useful for monitoring the skeletal response to treatment with vitamin D sterols in patients with osteitis fibrosa; values that decrease over several months usually indicate histologic improvement.117 Bone-specific alkaline phosphatase activity may be useful in predicting the histologic lesions of renal osteodystrophy, but whether these values are superior to total alkaline phosphatase levels remains to be demonstrated. Serum alkaline phosphatase levels may increase early in the course of treatment for aluminum-related bone disease with the chelating agent deferoxamine. Levels also may increase during therapy with recombinant human growth hormone in pediatric patients with renal failure.210
Assays for bone-specific alkaline phosphatase and measurements of serum osteocalcin levels provide additional information about the level of osteoblastic activity in patients with CKD.211 Osteocalcin levels generally are elevated in renal failure, but values may help distinguish between patients with high turnover and those with low turnover skeletal lesions.211,212 If these assays are not available, measurement of the heat-stabile and heat-labile fractions of alkaline phosphatase helps to separate skeletal from hepatic causes of elevated total alkaline phosphatase levels.
Parathyroid Hormone
Over the past few years, a series of observations have highlighted important shortcomings of the first-generation immunometric assays (IMAs) for measuring PTH (1st PTH-IMAs). Studies by D’Amour et al.213–215 demonstrated that 1st PTH-IMAs detect not only the intact hormone, but also PTH fragments truncated at the aminoterminus. Indeed, most of the detection antibodies, which usually are directed against epitopes within the aminoterminus of the hormone, detect not only PTH(1-84), but also one or several amino-truncated fragments of the PTH molecule, some of which co-elute from reverse-phase high-performance liquid chromatography (HPLC) columns with synthetic PTH(7-84).214 By contrast, a more recently developed second-generation immunometric PTH assay (2nd PTH-IMA), using detection antibodies, which interact with epitopes comprising the first four aminoterminal amino acids of human PTH, recognizes only PTH(1-84) and possibly PTH fragments that are truncated at the carboxyterminus, but not PTH(7-84).69,216,217 PTH levels determined by 1st PTH-IMAs overestimate the concentration of PTH(1-84) by 40% to 50% in healthy individuals with normal renal function and in those with varying degrees of CKD (Fig. 14-6). Unlike 1st PTH-IMAs, more recently developed 2nd PTH-IMAs do not detect these large PTH fragments.69,216–219 Consistent with this assessment, human PTH(1-34), but not human PTH(2-34) or other aminoterminally truncated fragments of human PTH(1-34), cross-react with the detection antibody.69,216,217 Faugere et al.218 suggested that 2nd PTH-IMAs and, mainly, the ratio between PTH(1-84) and amino-truncated PTH fragments (calculated from the differences in PTH levels determined between 1st and 2nd PTH-IMAs) could be better predictors of bone turnover than 1st PTH-IMA218; however, these findings were not confirmed by subsequent investigations.130,219 When the crucial role of serum PTH concentrations in the diagnosis and treatment of renal osteodystrophy is considered, 2nd PTH-IMA may provide important new insights into the physiology of parathyroid gland function. At present, however, measurements of PTH using 1st or 2nd PTH-IMAs have shown similar accuracy for predicting bone turnover in patients undergoing maintenance dialysis. Current data do not yet support the claim that 2nd PTH-IMAs provide an advantage over 1st PTH-IMAs in the diagnosis of different subtypes of renal bone disease.218
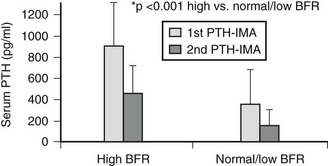
FIGURE 14-6 Parathyroid hormone (PTH) levels by different assays according to bone histology in patients treated with maintenance dialysis.
Although these molecules detected by PTH-IMAs are still uncharacterized, in vitro and in vivo experimental data indicate that one or more aminoterminally truncated PTH(1-84) fragments antagonize the calcemic actions of PTH(1-84) and diminish bone cell activity, and therefore may modulate bone metabolism. Indeed, synthetic PTH(7-84), which appears to be similar to naturally occurring circulating aminoterminally truncated PTH fragments,220 has hypocalcemic properties in vivo.216 Furthermore, PTH(7-84) inhibits the formation of TRAP-positive bone resorbing cells in vitro,221 and it inhibits bone formation in vivo.222 In addition, Nguyen-Yamamoto et al.223 demonstrated that PTH(7-84), with or without a mixture of other carboxyterminal PTH-fragments, inhibited the calcemic effects of PTH(1-34) in vivo, indicating that these actions are not mediated through the PTH/PTHrP receptor, but instead via a receptor that interacts only with carboxyterminal portions of PTH. Thus, a growing body of evidence suggests that at least some of the different carboxyterminal PTH fragments have biological activity.
Aluminum
Aluminum toxicity occurs in dialysis patients or patients with CKD with GFR less than 30 mL/min/1.73 m2 because aluminum that is absorbed from the gut, from the dialysate, or from parenteral infusions is inadequately excreted by the diseased kidney. Accumulation occurs in various tissues, including bone, liver, brain, and parathyroid glands, and can produce toxicity such as dialysis encephalopathy, osteomalacia, and microcytic anemia. The gold standard for the diagnosis of aluminum bone disease is a bone biopsy demonstrating increased aluminum staining of the bone surface (>15% to 25%) with histologic evidence of adynamic bone or osteomalacia. The presence of aluminum deposits in the bone and liver does not correlate with plasma levels224; however, plasma aluminum levels are useful for monitoring patients who are undergoing long-term dialysis therapy and receiving aluminum-containing phosphate-binding agents for prolonged periods. DFO should be administered to symptomatic patients with aluminum levels between 60 and 200 µg/L or a positive DFO test. The DFO infusion test is performed by infusing 5 mg/kg of DFO during the last hour of the dialysis session. Serum aluminum is measured before DFO infusion and 2 days later, before the next dialysis session. To prevent DFO-induced neurotoxicity, DFO should not be administered if serum aluminum concentrations are greater than 200 µg/L.130,225
Radiography
Radiographic Features of Osteitis Fibrosa Cystica
The most consistent radiographic feature of secondary hyperparathyroidism is the presence of subperiosteal erosions.226,227 The degree of subperiosteal erosion can correlate with serum PTH and alkaline phosphatase levels, but radiographs can be normal in patients with moderate to severe histologic features of osteitis fibrosa cystica on bone biopsy.228
In pediatric patients, metaphyseal changes (i.e., growth zone lesions that are termed “rickets-like lesions”) are common.226 Radiographic changes arising from secondary hyperparathyroidism may be difficult to differentiate from true rachitic abnormalities. However, the radiographic features of slipped epiphyses resulting from osteitis fibrosa are present in uremic children but absent in rickets resulting from vitamin D deficiency.141 These findings are best detected by hand radiographs, and several techniques, including the use of fine-grain films and magnification with a hand lens, are used to enhance sensitivity.229 Subperiosteal erosions also occur in the distal ends of clavicles, on the surface of the ischium and pubis, at the sacroiliac joints, and at the junction of the metaphysis and diaphysis of long bones.227,228 Subperiosteal erosions can be found in patients with aluminum-related bone disease.230 This finding represents the residuals of earlier hyperparathyroidism with osteitis fibrosa cystica, which, owing to aluminum toxicity, are unable to remineralize during treatment with vitamin D sterols or with parathyroidectomy.230
Radiographic Features of Osteomalacia
Radiographic features of osteomalacia are less specific and less common than those of secondary hyperparathyroidism. Typical rachitic lesions, with widening of the epiphyseal growth plate and other deformities, can occur in children with open epiphyses.226 Looser’s zones or pseudofractures, the only pathognomonic radiographic features of osteomalacia in adult patients, are rare in renal patients with osteomalacia; they occur as straight, wide bands of radiolucency that are perpendicular to the long axis of the bone. Fractures, particularly of the ribs, vertebral bodies, and hips, are more common in patients with osteomalacia than in patients with osteitis fibrosa cystica or mixed osteodystrophy.231 Decreased bone density is another common radiographic feature present in patients with advanced kidney disease and may arise from secondary hyperparathyroidism, osteomalacia, or osteoporosis.232 Paradoxically, localized osteosclerosis is common in patients with kidney disease and is more frequent in patients with osteitis fibrosa cystica.
Bone Biopsy
Although not routinely performed in the clinical setting, a bone biopsy should be considered in all patients with CKD who have fractures with minimal trauma (pathologic fractures), suspected aluminum bone disease, or persistent hypercalcemia despite serum PTH levels between 400 and 600 pg/mL.154 For bone labeling, a 2 day course of tetracycline is administered at 15 mg/kg/day (divided into twice- or thrice-daily doses). Phosphate binders should be held during labeling because they may interfere with gut absorption of tetracycline. Fourteen days later, the 2 day course is repeated. For children younger than 8 years, tetracycline dosage usually is kept below 10 mg/kg/day to avoid toxicity. Histochemical staining procedures demonstrate the deposition of abnormal components such as iron, aluminum, and oxalate within bone.116,233
Treatment of CKD-MBD
Dietary Manipulation of Calcium and Phosphorus
The development of hyperphosphatemia occurs in the vast majority of patients with advanced renal insufficiency. Hyperphosphatemia and an elevated calcium-phosphorous ion product have been reported as independent risk factors for vascular calcification and mortality in adult dialysis patients.189,190,203 Thus, treatment goals include maintaining serum phosphorous levels within normal limits for age and avoiding a calcium-phosphorous ion product above 55 mg2/dL2. The average phosphorous intake of both adults and children in the U.S. population is approximately 1500 to 2000 mg/day, and 60% to 70% of the dietary intake is absorbed. In the early stages of renal failure, dietary phosphate restriction is sufficient in preventing hyperphosphatemia. However, strict adherence to dietary phosphate restriction is often difficult because low-phosphate diets are unpalatable, especially to older children and adults, and because phosphorous intake is directly linked to protein intake, with 10 to 12 mg of phosphorus accompanying each gram of protein. Adequate protein intake is necessary for growth in children and for maintenance of lean body mass in adults. Current dietary recommendations suggest that adults with CKD ingest between 0.8 to 1 g/kg/day of protein, and that children, depending on age, ingest anywhere from 1 to 2.5 g/kg/day.67,154 This translates to a minimum phosphate ingestion of 800 mg/day in an 80 kg person.
Patients treated with dialysis require dietary phosphorous restriction, in addition to phosphate-binder therapy (vide infra), because standard prescription peritoneal dialysis and hemodialysis remove insufficient amounts of phosphate (300 to 400 mg/day for peritoneal dialysis and 800 mg/treatment for hemodialysis) to maintain normal serum phosphorous levels. The use of daily, slow, continuous hemodialysis in some centers has been associated with excellent control of serum phosphorous levels, often allowing phosphate-binding agents to be discontinued.234,235 Indeed, some patients have developed hypophosphatemia and have required the addition of phosphorus to the dialysate solution to prevent the long-term consequences of hypophosphatemia.234,235
Phosphate-Binding Agents
Phosphate-binding agents reduce intestinal phosphate absorption by forming poorly soluble complexes with phosphorus in the intestinal tract. Aluminum-containing phosphate binders were used frequently in the past, but long-term treatment led to bone disease, encephalopathy, and anemia.236 The use of aluminum-containing phosphate binders, therefore, should be restricted to the treatment of patients with severe hyperphosphatemia (>7 mg/dL) associated with hypercalcemia or an elevated calcium-phosphorous ion product, because both conditions will be aggravated by calcium-containing compounds. In such cases, the dose of aluminum hydroxide should not exceed 30 mg/kg/day, and the lowest possible dose should be given only for a limited period of approximately 4 to 6 weeks.237 Plasma aluminum levels should be monitored regularly. Concomitant intake of citrate-containing compounds should be avoided, because citrate increases intestinal aluminum absorption238 and increases the risk for acute aluminum intoxication.239,240 Constipation is a common adverse effect and can be relieved by stool softeners.
To avoid aluminum-related bone disease and encephalopathy, the use of aluminum-free phosphate binders has been advocated. Among these, calcium-containing salts are used worldwide for control of hyperphosphatemia and serve as a source of supplemental calcium. Several calcium salts, including calcium carbonate, calcium acetate, and calcium citrate, are commercially available. Calcium carbonate is the most commonly used compound, and studies in adults and children have shown its efficacy in controlling serum phosphorous levels.102,241,242 The recommended dose is proportional to the phosphorous content of the meal and is adjusted to achieve acceptable serum levels of calcium and phosphorus. Large doses of calcium carbonate may lead to hypercalcemia, particularly in patients treated with vitamin D or those with adynamic bone.243,244 Hypercalcemia usually is reversible with reductions in the dose of oral calcium salts, dose of vitamin D sterol, and dialysate calcium concentrations. To avoid the development and progression of cardiovascular calcifications, it is currently recommended that elemental calcium intake should not exceed 2 g/day, with less than 1500 mg of calcium given as calcium-containing phosphorous binders.67
Comparison studies between calcium carbonate and calcium acetate have demonstrated that an equivalent dose of calcium acetate binds twice as much phosphorus, but the relative incidence of hypercalcemia varies among studies.245–247 Calcium citrate is an effective phosphate-binding agent but should be used with caution in patients with renal failure because of enhanced intestinal aluminum absorption when given in combination with aluminum-containing phosphate binders.248 Calcium ketoglutarate is less calcemic than calcium carbonate and has anabolic benefits, but gastrointestinal side effects and high cost of therapy often limit its use.249
To limit the vascular calcification risks associated with the use of calcium salts and the bone and neurologic toxicity associated with aluminum hydroxide, alternative phosphate binders have been developed. Sevelamer hydrochloride (RenaGel), a calcium- and aluminum-free hydrogel of cross-linked poly-allylamine, has been shown to lower serum phosphorus, the calcium-phosphorous ion product, and PTH without inducing hypercalcemia in adult patients treated with hemodialysis.250–252 Sevelamer also halts the progression of vascular calcification, although such lesions increase during calcium-containing binder therapy in adult hemodialysis patients.253 In addition to its effects on serum phosphorous levels, sevelamer has been shown to decrease concentrations of total serum cholesterol and low-density lipoprotein cholesterol, but to increase high-density lipoprotein levels.252 These effects may offer additional benefits in reducing cardiovascular complications in patients with end-stage renal disease. Acidosis may occur in patients treated with sevelamer; thus, a new form of sevelamer, sevelamer carbonate, has been introduced recently. This new compound is as effective a phosphate binder as sevelamer hydrochloride, with less potential to induce acidosis.254
Other alternative phosphate-binding agents include magnesium, iron, and lanthanum compounds. Magnesium carbonate lowers serum phosphorous levels, but magnesium-free dialysate solutions should be used in those treated with dialysis.139,255 Large doses, however, result in diarrhea, limiting the use of this compound as a single agent. Iron compounds, such as stabilized polynuclear iron hydroxide and ferric polymaltose complex, have proved to be effective phosphate binders in short-term studies in adults with CKD.256,257 Clinical trials have demonstrated that lanthanum carbonate also effectively controls serum phosphorous and PTH levels without increasing serum calcium values. Lanthanum carbonate lowers serum phosphorous and PTH levels without causing hypercalcemia, adynamic bone disease, or osteomalacia.258–260 However, lanthanum is a heavy metal that accumulates in animal livers.261 Lanthanum also accumulates in the bone of dialysis patients, where its presence persists despite discontinuation for as long as 2 years.262 Additional long-term studies therefore needed to confirm the absence of toxicity before this agent is recommended for widespread use.
Vitamin D Therapy
Assessment and Treatment of 25(OH)D Deficiency: Measurement of 25(OH)D levels and treatment of vitamin D deficiency are an important part of the management of hyperparathyroidism in patients with CKD. The current classification system stratifies vitamin D deficiency into three categories67: (1) severe deficiency, defined as a serum level less than 5 ng/mL; (2) mild deficiency, equivalent to serum concentrations of 5 to 15 ng/mL; and (3) vitamin D insufficiency, with levels between 16 and 30 ng/mL. Thus, ergocalciferol treatment should be initiated in patients with CKD when 25(OH)D levels fall below 30 ng/mL. Severe deficiency (<5 ng/mL) should be treated with 50,000 IU orally, once a week for 12 weeks, then 50,000 IU orally once a month for a total of 6 months. Alternatively, 500,000 IU may be given as a single intramuscular dose. Serum 25(OH)D levels in the range of 5 to 15 ng/mL (so-called mild deficiency) should be treated with 50,000 IU of ergocalciferol orally once a week for 4 weeks, followed by 50,000 IU orally once a month for a total of 6 months. Vitamin D insufficiency (serum levels between 16 and 30 ng/mL) should be treated with 50,000 IU of ergocalciferol orally once a month for 6 months. In D-deficient patients, serum 25(OH)D levels should be rechecked after completion of the 6 month course of therapy.67
Treatment With Active Vitamin D Sterols: As mentioned previously, active vitamin D sterols act through a variety of pathways to decrease PTH production—by increasing calcium absorption in the gut and kidney, by binding to the CaSR, by increasing skeletal sensitivity to PTH, and by altering prepro-PTH transcription. Calcitriol (Rocaltrol) has been widely used for many years to control secondary hyperparathyroidism in both adults and children. The efficacy of daily oral doses of calcitriol for the treatment of patients with symptomatic renal osteodystrophy has been documented in several clinical trials.263,264 Bone pain diminishes, muscle strength and gait-posture improve, and osteitis fibrosa frequently resolves partially or completely.120 When measured by reliable assays, serum PTH levels decrease in patients who respond favorably to treatment. Doses of oral calcitriol in most clinical trials have ranged from 0.25 to 1.5 µg/day. In patients with CKD, initial doses are determined by target PTH levels and specific stage of kidney disease.206 In dialysis patients, 1,25(OH)2D3 given thrice weekly by IV injection or by oral pulse therapy is effective in reducing serum PTH levels.153,265 Dosage regimens range from 0.5 to 1.0 µg to 3.5 to 4.0 µg three times weekly or 2.0 to 5.0 µg twice weekly. Low doses should be used initially, and dosage adjustments should be based on frequent measurements of serum calcium, phosphorous, and PTH levels.
Oral 1α-(OH)D3 (alfacalcidol) undergoes 25-hydroxylation in the liver to form calcitriol,266,267 and this agent is used widely in Europe, Japan, and Canada. Calcitriol and 1α-(OH)D3 are similarly effective for the treatment of secondary hyperparathyroidism in patients with CKD.
19-Nor-1α,25(OH)2D2 (paricalcitol) is effective in controlling serum PTH levels in adult patients with stages 3 and 4 CKD,268 as well as in dialysis patients. The long-term consequences of therapy with paricalcitol in conjunction with the use of calcium-containing binders for vascular calcification and cardiovascular complications remain to be determined. However, in a large cohort of patients undergoing hemodialysis, higher survival rates were observed in dialyzed patients treated with paricalcitol when compared with those receiving calcitriol.269
Another new vitamin analogue, 1α-(OH)D2 (1α-D2, doxercalciferol), is equipotent to 1α-(OH)D3 in intestinal calcium absorption and bone calcium mobilization in rats.270 A comparative trial of calcitriol and doxercalciferol in the control of secondary hyperparathyroidism in pediatric patients revealed no differences between the two vitamin D sterols in the control of secondary hyperparathyroidism or the development of hypercalcemia.131 Doxercalciferol also effectively controls secondary hyperparathyroidism in adult patients with stable CKD.271 Similar to paricalcitol, a survival advantage has been observed in adult hemodialysis patients receiving paricalcitol over those treated with calcitriol.272
Active vitamin D therapy has been associated with protective effects on both the heart and the kidney. Active vitamin D sterols ameliorate cardiac hypertrophy in animals,24 and calcitriol therapy improves cardiac systolic function in hemodialysis patients.273 Administration of active vitamin D sterols reduces proteinuria, fibrosis, and podocyte hypertrophy in subtotally nephrectomized rats,274 and paricalcitol treatment decreases proteinuria in CKD patients.275 These effects may be mediated by suppression of the renin-angiotensin system (RAS); indeed in vitro studies have demonstrated that calcitriol, paricalcitol, and doxercalciferol all suppress the RAS to a similar degree.276 However, all activated vitamin D analogues may also increase FGF-23 secretion. Although the consequences of these increased levels in dialyzed patients remain to be completely determined, current evidence suggests that excessive circulating FGF-23 is associated with increased mortality rates.184
A growing body of evidence suggests additional health benefits of 25(OH)D therapy, primarily due to its immune regulatory role. Current observations also suggest a role for 25(OH)D in improving survival in patients treated with maintenance dialysis.277
Calcimimetics
Cinacalcet, an allosteric activator of the calcium-sensing receptor, is now available for the treatment of secondary hyperparathyroidism. This small organic molecule reduces serum PTH levels and has been shown to decrease the calcium-phosphorous ion product in adult patients treated with maintenance dialysis, regardless of the specific phosphate-binding agent.74,278 Experiments in uremic rats have demonstrated that calcimimetics are able to halt the progression of parathyroid cell hyperplasia74; the antiproliferative effect of this agent shows promise for use of the molecule as a “medical parathyroidectomy.” Studies in animals suggest that the use of calcimimetic agents may play a role in reversing the process of vascular calcification279; however, such effects need to be further evaluated in humans. Owing to the presence of the calcium-sensing receptor on the growth plate, these agents are not approved and should be used with caution in growing children.
Parathyroidectomy
In many cases when parathyroid surgery is needed and undertaken, the tumor has become monoclonal and growth autonomous.77,280 Patients with severe hyperparathyroidism often are unresponsive to vitamin D therapy, developing hypercalcemia and hyperphosphatemia without reduction in PTH values or parathyroid gland size.280 Clinical features that indicate the need for parathyroidectomy are as follows: the presence of hyperplasia and/or hypertrophy of the parathyroid glands (as documented by the presence of biochemical and radiographic features and, if necessary, the findings of osteitis fibrosa cystica on bone biopsy), elevated serum PTH levels unresponsive to vitamin D sterol therapy, persistent hypercalcemia, pruritus unresponsive to dialysis or other medical treatment, progressive extraskeletal calcification, severe skeletal pain or fractures, and calciphylaxis. Aluminum-related bone disease must be ruled out first in patients receiving low-dose calcitriol with persistent hypercalcemia.281 Other causes of hypercalcemia, such as sarcoidosis, malignancy-related hypercalcemia, intake of calcium supplements, and the presence of adynamic/aplastic bone lesions not related to aluminum, should also be considered.100,282
When the decision has been made to perform parathyroid surgery, it is essential to avoid a marked postoperative fall in serum calcium levels caused by the “hungry bone” syndrome. Because of the severity of the bone disease, this fall can be much more marked and prolonged than after parathyroidectomy for primary hyperparathyroidism. Renal patients should receive daily oral calcitriol (0.5 to 1.0 µg) or some sort of intravenous active vitamin D sterol for 2 to 6 days before parathyroid surgery and during the postoperative period to stimulate intestinal calcium absorption and to maximize the effectiveness of oral calcium salts. Within 24 to 36 hours after surgery, marked hypocalcemia with serum calcium levels below 7 to 8 mg/dL may develop. This condition may be associated with serious symptoms, including seizures resulting in fractures and tendon avulsion. For reasons that are still unclear, these seizures most often occur during the last 1 to 2 hours of a hemodialysis procedure or immediately thereafter. To reduce the risk for convulsions, an infusion containing calcium gluconate should be started in the operating room, upon removal of the parathyroid glands. Calcium gluconate should be initiated at a rate of 100 mg of calcium ion per hour. Serum calcium should be measured every 4 to 6 hours and the calcium gluconate infusion rate increased if the serum calcium level continues to fall. The infusion rate may exceed 200 mg/hr. Enteral calcium carbonate is initiated once the patient is able to tolerate oral intake, and doses as high as 1.0 g (elemental calcium) given four to six times daily, along with vitamin D sterol in excess of 1.0 to 2.0 µg/day (for calcitriol, doses of other agents vary according to their potency), are often needed for patients with marked hypocalcemia. The intravenous calcium drip is weaned as soon as the oral intake of calcium salts is able to maintain normal serum calcium levels. The duration of intravenous calcium requirements varies greatly between patients—most patients require IV therapy for 2 to 3 days, but severe hypocalcemia may persist for several weeks or months, necessitating permanent central catheter access for daily home infusions of 800 to 1000 mg of calcium ion. Serum phosphorous levels may fall to subnormal levels postoperatively; phosphate treatment will markedly aggravate the hypocalcemia, and patients should not be treated with phosphate unless serum phosphorus falls to below 2.0 mg/dL.283–286
Hyperplastic parathyroid glands may be infiltrated with ethanol or calcitriol to cause sclerosis of the parathyroid tissue. This technique has been used at some centers287,288 with variable efficacy in reducing hyperplastic tissue. This technique is used currently by only a few centers worldwide.
Growth Hormone Therapy
Recombinant human growth hormone (rhGH) should be considered in children with growth failure that does not respond to optimization of nutrition, correction of acidosis, and control of renal osteodystrophy. Serum phosphorous and PTH levels should be well controlled prior to the initiation of rhGH in children with CKD. Serum phosphorous levels should be less than 1.5 times the upper limit for age and PTH-IMA-1 levels less than 1.5 times the upper target values for the CKD stage before rhGH therapy is begun.154 GH therapy will increase serum PTH levels during the first months of therapy; therefore, serum PTH levels should be monitored monthly. GH therapy should be discontinued temporarily if PTH levels exceed three times the upper target value for the CKD stage.154
Bone Disease After Successful Kidney Transplantation
Hypercalcemia is not uncommon after renal transplantation. During the first several months, it can be quite severe, and patients with severe secondary hyperparathyroidism before renal transplantation are at greatest risk. More often, hypercalcemia may be less severe, with serum calcium levels between 10.5 and 12.0 mg/dL, and usually resolves within the first 12 months.289 Parathyroidectomy should be considered when serum calcium levels are persistently above 12.5 mg/dL for longer than 1 year after transplantation.290 Calcimimetic agents may be useful in preventing the need for parathyroidectomy in these patients.291
Hypophosphatemia may occur early in the post-transplant period, mainly in patients with severe secondary hyperparathyroidism, although persistent post-transplant elevation of serum FGF-23 levels also may contribute.292,293 The clinical manifestations are variable; some patients complain of malaise, fatigue, and muscle weakness.294,295 Phosphorous supplementation is required when values are persistently below 2.0 mg/dL, primarily in pediatric patients.
Significant bone loss has been shown to occur as early as 3 to 6 months after kidney transplantation.296,297 Several factors, including persistent alterations in mineral metabolism, prolonged immobilization, and the use of immunosuppressive agents required to maintain graft function, have been implicated in the development of bone disease after transplantation. Osteonecrosis, or avascular necrosis, is by far the most debilitating skeletal complication associated with organ transplantation. In approximately 15% of patients, osteonecrosis will develop within 3 years of renal transplantation.298,299 The occurrence of osteonecrosis in inpatients after cardiac, hepatic, and bone marrow transplantation suggests that glucocorticoids play a critical role in the pathogenesis of this disorder.300,301
In both adult and pediatric kidney recipients, bone histologic changes associated with secondary hyperparathyroidism have been shown to resolve within 6 months after kidney transplantation.297 However, some patients have persistently elevated rates of bone turnover, and others develop adynamic lesions, despite moderately elevated serum PTH levels.302 Bone biopsy data from pediatric kidney recipients indicate that 67% of patients with stable graft function have features of normal bone formation; 10% have adynamic bone lesion, and 23% have bone lesions characteristic of secondary hyperparathyroidism.303 Bone resorption typically is increased,304 leading to a net loss of bone mass over time. Serum PTH levels are unable to discriminate between adynamic, normal, and increased bone turnover in the pediatric transplant population.303 The use of maintenance corticosteroids has been implicated in these alterations; steroids decrease intestinal calcium absorption, enhance urinary calcium excretion, inhibit osteoblastic activity, decrease bone formation, and increase osteoclastic activity and bone resorption.305–308 Likewise, cyclosporine has been reported to increase both bone formation and bone resorption and to reduce cancellous bone volume in the rat.309,310 By contrast, azathioprine has been shown to have minimal impact on skeletal remodeling.311 The role of other immunosuppressive agents, such as mycophenolate mofetil, as potential modifiers of bone formation and bone resorption has not been evaluated.
Although bone turnover may return to normal, defective skeletal mineralization is present in most pediatric transplant recipients.303 Osteoid volume is increased, and mineral apposition rate is markedly reduced.303 Although the factors responsible for the persistent increase in osteoid formation remain to be fully explained, corticosteroid use may contribute, as may persistent imbalances in PTH, vitamin D, and FGF-23 metabolism.293
After successful kidney transplantation with standard immunosuppressive regimens (daily corticosteroids, calcineurin inhibitor, and anti-metabolite), growth may be accelerated by an improvement in kidney function, but catch-up growth may not be observed even in children who undergo transplantation very early in life.143 Moreover, height deceleration occurs in approximately 75% of patients who undergo transplantation before the age of 15 years.312 The cause of persistent growth retardation is not completely understood, but immunosuppressive agents, persistent secondary hyperparathyroidism, altered vitamin D and FGF-23 metabolism, and the persistence of defective skeletal mineralization all may contribute. Children receiving steroid-free immunosuppressive regimens, those treated with alternate-day steroids, and those with better height SD at the time of transplant attain the greatest final adult height.143,312–314 Used in children with significant height deficit after kidney transplantation, rhGH has produced a substantial increase in linear growth within the first year of therapy, but the magnitude of growth response may decline thereafter.315
Cardiovascular disease continues to be the leading cause of death after renal transplantation. In the post-transplant period, the presence of hypertension is strongly linked to increased IMT and poor vessel distensibility in children.316 Alterations in bone and mineral metabolism also may contribute, as impaired kidney function persists in most patients during the post-transplant period.67,143 Indeed, EBCT data indicate that vascular calcifications do not regress posttransplantation and do contribute to the burden of cardiovascular disease in this population.317
References
1. Moe, S, Drueke, T, Cunningham, J, et al. Definition, evaluation, and classification of renal osteodystrophy: a position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int. 2006;69:1945–1953.
2. Groothoff, JW, Offringa, M, Van Eck-Smit, BL, et al. Severe bone disease and low bone mineral density after juvenile renal failure. Kidney Int. 2003;63:266–275.
3. Levin, A, Bakris, GL, Molitch, M, et al. Prevalence of abnormal serum vitamin D, PTH, calcium, and phosphorus in patients with chronic kidney disease: results of the study to evaluate early kidney disease. Kidney Int. 2007;71:31–38.
4. Avasthi, G, Singh, HP, Katyal, JC, et al. Copper, zinc, calcium and magnesium in chronic renal failure. J Assoc Physicians India. 1991;39:531–534.
5. Goldman, R, Bassett, SH. Phosphorus excretion in renal failure. J Clin Invest. 1954;33:1623–1628.
6. Coburn, JW, Popovtzer, MM, Massry, SG, et al. The physicochemical state and renal handling of divalent ions in chronic renal failure. Arch Intern Med. 1969;124:302–311.
7. Shimada, T, Mizutani, S, Muto, T, et al. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci U S A. 2001;98:6500–6505.
8. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet. 2000;26:345–348.
9. Larsson, T, Zahradnik, R, Lavigne, J, et al. Immunohistochemical detection of FGF-23 protein in tumors that cause oncogenic osteomalacia. Eur J Endocrinol. 2003;148:269–276.
10. Mirams, M, Robinson, BG, Mason, RS, et al. Bone as a source of FGF23: regulation by phosphate? Bone. 2004;35:1192–1199.
11. Yoshiko, Y, Wang, H, Minamizaki, T, et al. Mineralized tissue cells are a principal source of FGF23. Bone. 2007;40:1565–1573.
12. Urakawa, I, Yamazaki, Y, Shimada, T, et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. 2006;444:770–774.
13. Larsson, T, Nisbeth, U, Ljunggren, O, et al. Circulating concentration of FGF-23 increases as renal function declines in patients with chronic kidney disease, but does not change in response to variation in phosphate intake in healthy volunteers. Kidney Int. 2003;64:2272–2279.
14. Gutierrez, O, Isakova, T, Rhee, E, et al. Fibroblast growth factor-23 mitigates hyperphosphatemia but accentuates calcitriol deficiency in chronic kidney disease. J Am Soc Nephrol. 2005;16:2205–2215.
15. Kolek, OI, Hines, ER, Jones, MD, et al. 1alpha,25-Dihydroxyvitamin D3 upregulates FGF23 gene expression in bone: the final link in a renal-gastrointestinal-skeletal axis that controls phosphate transport. Am J Physiol Gastrointest Liver Physiol. 2005;289:G1036–G1042.
16. Portale, AA, Booth, BE, Halloran, BP, et al. Effect of dietary phosphorus on circulating concentrations of 1,25-dihydroxyvitamin D and immunoreactive parathyroid hormone in children with moderate renal insufficiency. J Clin Invest. 1984;73:1580–1589.
17. Antoniucci, DM, Yamashita, T, Portale, AA. Dietary phosphorus regulates serum fibroblast growth factor-23 concentrations in healthy men. J Clin Endocrinol Metab. 2006;91:3144–3149.
18. Krajisnik, T, Bjorklund, P, Marsell, R, et al. Fibroblast growth factor-23 regulates parathyroid hormone and 1alpha-hydroxylase expression in cultured bovine parathyroid cells. J Endocrinol. 2007;195:125–131.
19. Ben-Dov, IZ, Galitzer, H, Lavi-Moshayoff, V, et al. The parathyroid is a target organ for FGF23 in rats. J Clin Invest. 2007;117:4003–4008.
20. Martinez, I, Saracho, R, Montenegro, J, et al. A deficit of calcitriol synthesis may not be the initial factor in the pathogenesis of secondary hyperparathyroidism. Nephrol Dial Transplant. 1996;11(suppl 3):22–28.
21. Li, YC, Pirro, AE, Amling, M, et al. Targeted ablation of the vitamin D receptor: an animal model of vitamin D-dependent rickets type II with alopecia. Proc Natl Acad Sci U S A. 1997;94:9831–9835.
22. Amling, M, Priemel, M, Holzmann, T, et al. Rescue of the skeletal phenotype of vitamin D receptor-ablated mice in the setting of normal mineral ion homeostasis: formal histomorphometric and biomechanical analyses. Endocrinology. 1999;140:4982–4987.
23. Panda, DK, Miao, D, Bolivar, I, et al. Inactivation of the 25-hydroxyvitamin D 1alpha-hydroxylase and vitamin D receptor demonstrates independent and interdependent effects of calcium and vitamin D on skeletal and mineral homeostasis. J Biol Chem. 2004;279:16754–16766.
24. Bodyak, N, Ayus, JC, Achinger, S, et al. Activated vitamin D attenuates left ventricular abnormalities induced by dietary sodium in Dahl salt-sensitive animals. Proc Natl Acad Sci U S A. 2007;104:16810–16815.
25. Zhou, C, Lu, F, Cao, K, et al. Calcium-independent and 1,25(OH)2D3-dependent regulation of the renin-angiotensin system in 1alpha-hydroxylase knockout mice. Kidney Int. 2008;74:170–179.
26. Teng, M, Wolf, M, Ofsthun, MN, et al. Activated injectable vitamin D and hemodialysis survival: a historical cohort study. J Am Soc Nephrol. 2005;16:1115–1125.
27. Teng, M, Wolf, M, Lowrie, E, et al. Survival of patients undergoing hemodialysis with paricalcitol or calcitriol therapy. N Engl J Med. 2003;349:446–456.
28. Holick, MF, Garabedian, M. Vitamin D: photobiology, metabolism, mechanism of action, and clinical applications. In: Favus MJ, ed. Primer on the metabolic bone diseases and disorders of mineral metabolism. Washington, DC: American Society for Bone and Mineral Research; 2006:129–137.
29. DeLuca, HF. Overview of general physiologic features and functions of vitamin D. Am J Clin Nutr. 2004;80S:1689S–1696S.
30. Holick, MF. Resurrection of vitamin D deficiency and rickets. J Clin Invest. 2006;116:2062–2072.
31. Holick, MF. Vitamin D deficiency. N Engl J Med. 2007;357:266–281.
32. Nykjaer, A, Dragun, D, Walther, D, et al. An endocytic pathway essential for renal uptake and activation of the steroid 25-(OH) vitamin D3. Cell. 1999;96:507–515.
33. Leheste, JR, Melsen, F, Wellner, M, et al. Hypocalcemia and osteopathy in mice with kidney-specific megalin gene defect. FASEB J. 2003;17:247–249.
34. Zehnder, D, Landray, MJ, Wheeler, DC, et al. Cross-sectional analysis of abnormalities of mineral homeostasis, vitamin D and parathyroid hormone in a cohort of pre-dialysis patients. The Chronic Renal Impairment in Birmingham (CRIB) study. Nephron Clin Pract. 2007;107:c109–c116.
35. Lambert, PW, Stern, PH, Avioli, RC, et al. Evidence for extrarenal production of 1 alpha,25-dihydroxyvitamin D in man. J Clin Invest. 1982;69:722–725.
36. Dusso, A, Lopez-Hilker, S, Rapp, N, et al. Extra-renal production of calcitriol in chronic renal failure. Kidney Int. 1988;34:368–375.
37. Dusso, AS, Finch, J, Brown, A, et al. Extrarenal production of calcitriol in normal and uremic humans. J Clin Endocrinol Metab. 1991;72:157–164.
38. Khaw, KT, Sneyd, MJ, Compston, J. Bone density parathyroid hormone and 25-hydroxyvitamin D concentrations in middle aged women. BMJ. 1992;305:273–277.
39. LeBoff, MS, Kohlmeier, L, Hurwitz, S, et al. Occult vitamin D deficiency in postmenopausal US women with acute hip fracture. JAMA. 1999;281:1505–1511.
40. Thomas, MK, Lloyd-Jones, DM, Thadhani, RI, et al. Hypovitaminosis D in medical inpatients. N Engl J Med. 1998;338:777–783.
41. Gordon, CM, DePeter, KC, Feldman, HA, et al. Prevalence of vitamin D deficiency among healthy adolescents. Arch Pediatr Adolesc Med. 2004;158:531–537.
42. Stavroulopoulos, A, Porter, CJ, Roe, SD, et al. Relationship between vitamin D status, parathyroid hormone levels and bone mineral density in patients with chronic kidney disease stages 3 and 4. Nephrology (Carlton). 2008;13:63–67.
43. Reichel, H, Deibert, B, Schmidt-Gayk, H, et al. Calcium metabolism in early chronic renal failure: implications for the pathogenesis of hyperparathyroidism. Nephrol Dial Transplant. 1991;6:162–169.
44. Coburn, JW, Koppel, MH, Brickman, AS, et al. Study of intestinal absorption of calcium in patients with renal failure. Kidney Int. 1973;3:264–272.
45. Holick, MF. Vitamin D and the kidney. Kidney Int. 1987;32:912–929.
46. Clemens, TL, Adams, JS, Henderson, SL, et al. Increased skin pigment reduces the capacity of skin to synthesise vitamin D3. Lancet. 1982;1:74–76.
47. Koenig, KG, Lindberg, JS, Zerwekh, JE, et al. Free and total 1,25-dihydroxyvitamin D levels in subjects with renal disease. Kidney Int. 1992;41:161–165.
48. Saha, H. Calcium and vitamin D homeostasis in patients with heavy proteinuria. Clin Nephrol. 1994;41:290–296.
49. Vasicek, TJ, McDevitt, BE, Freeman, MW, et al. Nucleotide sequence of the human parathyroid hormone gene. Proc Natl Acad Sci U S A. 1983;80:2127–2131.
50. Reis, A, Hecht, W, Groger, R, et al. Cloning and sequence analysis of the human parathyroid hormone gene region. Hum Genet. 1990;84:119–124.
51. Fraser, RA, Kronenberg, HM, Pang, PK, et al. Parathyroid hormone messenger ribonucleic acid in the rat hypothalamus. Endocrinology. 1990;127:2517–2522.
52. Okazaki, T, Ando, K, Igarashi, T, et al. Conserved mechanism of negative gene regulation by extracellular calcium. Parathyroid hormone gene versus atrial natriuretic polypeptide gene. J Clin Invest. 1992;89:1268–1273.
53. Moallem, E, Kilav, R, Silver, J, et al. RNA-Protein binding and post-transcriptional regulation of parathyroid hormone gene expression by calcium and phosphate. J Biol Chem. 1998;273:5253–5259.
54. Saggese, G, Federico, G, Cinquanta, L. In vitro effects of growth hormone and other hormones on chondrocytes and osteoblast-like cells. Acta Paediatr Suppl. 1993;82(suppl 391):54–59.
55. Scharla, SH, Strong, DD, Mohan, S, et al. 1,25-Dihydroxyvitamin D3 differentially regulates the production of insulin-like growth factor I (IGF-I) and IGF-binding protein-4 in mouse osteoblasts. Endocrinology. 1991;129:3139–3146.
56. Akiyama, H, Hiraki, Y, Shigeno, C, et al. 1 alpha,25-dihydroxyvitamin D3 inhibits cell growth and chondrogenesis of a clonal mouse EC cell line, ATDC5. J Bone Miner Res. 1996;11:22–28.
57. Silver, J, Russell, J, Sherwood, LM. Regulation by vitamin D metabolites of messenger ribonucleic acid for preproparathyroid hormone in isolated bovine parathyroid cells. Proc Natl Acad Sci U S A. 1985;82:4270–4273.
58. Silver, J, Naveh-Many, T, Mayer, H, et al. Regulation by vitamin D metabolites of parathyroid hormone gene transcription in vivo in the rat. J Clin Invest. 1986;78:1296–1301.
59. Kifor, O, Moore, FD, Jr., Wang, P, et al. Reduced immunostaining for the extracellular Ca2+-sensing receptor in primary and uremic secondary hyperparathyroidism. J Clin Endocrinol Metab. 1996;81:1598–1606.
60. Brown, AJ, Zhong, M, Finch, J, et al. Rat calcium-sensing receptor is regulated by vitamin D but not by calcium. Am J Physiol. 1996;270(3 Pt 2):F454–F460.
61. Szabo, A, Merke, J, Beier, E, et al. 1,25(OH)2 vitamin D3 inhibits parathyroid cell proliferation in experimental uremia. Kidney Int. 1989;35:1049–1056.
62. Almaden, Y, Canalejo, A, Ballesteros, E, et al. Regulation of arachidonic acid production by intracellular calcium in parathyroid cells: effect of extracellular phosphate. J Am Soc Nephrol. 2002;13:693–698.
63. Silver, J, Kilav, R, Sela-Brown, A, et al. Molecular mechanisms of secondary hyperparathyroidism. Pediatr Nephrol. 2000;14:626–628.
64. Ritter, CS, Armbrecht, HJ, Slatopolsky, E, et al. 25-Hydroxyvitamin D(3) suppresses PTH synthesis and secretion by bovine parathyroid cells. Kidney Int. 2006;70:654–659.
65. Zisman, AL, Hristova, M, Ho, LT, et al. Impact of ergocalciferol treatment of vitamin D deficiency on serum parathyroid hormone concentrations in chronic kidney disease. Am J Nephrol. 2007;27:36–43.
66. Chandra, P, Binongo, JN, Ziegler, TR, et al. Cholecalciferol (vitamin D3) therapy and vitamin D insufficiency in patients with chronic kidney disease: a randomized controlled pilot study. Endocr Pract. 2008;14:10–17.
67. K/DOQI clinical practice guidelines for bone metabolism and disease in chronic kidney disease. Am J Kidney Dis. 2003;42(4 suppl 3):S1–S201.
68. Brown, EM, Gamba, G, Riccardi, D, et al. Cloning and characterization of an extracellular Ca(2+)-sensing receptor from bovine parathyroid. Nature. 1993;366:575–580.
69. John, MR, Goodman, WG, Gao, P, et al. A novel immunoradiometric assay detects full-length human PTH but not amino-terminally truncated fragments: implications for PTH measurements in renal failure. J Clin Endocrinol Metab. 1999;84:4287–4290.
70. Freichel, M, Zink-Lorenz, A, Holloschi, A, et al. Expression of a calcium-sensing receptor in a human medullary thyroid carcinoma cell line and its contribution to calcitonin secretion. Endocrinology. 1996;137:3842–3848.
71. Kirkwood, JR, Ozonoff, MB, Steinbach, HL. Epiphyseal displacement after metaphyseal fracture in renal osteodystrophy. Am J Roentgenol Radium Ther Nucl Med. 1972;115:547–554.
72. Martin-Salvago, M, Villar-Rodriguez, JL, Palma-Alvarez, A, et al. Decreased expression of calcium receptor in parathyroid tissue in patients with hyperparathyroidism secondary to chronic renal failure. Endocr Pathol. 2003;14:61–70.
73. Brown, AJ, Zhong, M, Ritter, C, et al. Loss of calcium responsiveness in cultured bovine parathyroid cells is associated with decreased calcium receptor expression. Biochem Biophys Res Commun. 1995;212:861–867.
74. Wada, M, Furuya, Y, Sakiyama, J, et al. The calcimimetic compound NPS R-568 suppresses parathyroid cell proliferation in rats with renal insufficiency. Control of parathyroid cell growth via a calcium receptor. J Clin Invest. 1997;100:2977–2983.
75. Canaff, L, Hendy, GN. Human calcium-sensing receptor gene. Vitamin D response elements in promoters P1 and P2 confer transcriptional responsiveness to 1,25-dihydroxyvitamin D. J Biol Chem. 2002;277:30337–30350.
76. Lloyd, HM, Parfitt, AM, Jacobi, JM, et al. The parathyroid glands in chronic renal failure: a study of their growth and other properties made on the basis of findings in patients with hypercalcemia. J Lab Clin Med. 1989;114:358–367.
77. Arnold, A, Brown, MF, Urena, P, et al. Monoclonality of parathyroid tumors in chronic renal failure and in primary parathyroid hyperplasia. J Clin Invest. 1995;95:2047–2053.
78. Agarwal, SK, Guru, SC, Heppner, C, et al. Menin interacts with the AP1 transcription factor JunD and represses JunD-activated transcription. Cell. 1999;96:143–152.
79. Chandrasekharappa, SC, Guru, SC, Manickam, P, et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science. 1997;276:404–407.
80. Cryns, VL, Thor, A, Xu, HJ, et al. Loss of the retinoblastoma tumor-suppressor gene in parathyroid carcinoma. N Engl J Med. 1994;330:757–761.
81. Mulligan, LM, Marsh, DJ, Robinson, BG, et al. Genotype-phenotype correlation in multiple endocrine neoplasia type 2: report of the International RET Mutation Consortium. J Intern Med. 1995;238:343–346.
82. Motokura, T, Bloom, T, Kim, HG, et al. A novel cyclin encoded by a bcl1-linked candidate oncogene. Nature. 1991;350:512–515.
83. Atkins, D, Peacock, M. A comparison of the effects of the calcitonins, steroid hormones and thyroid hormones on the response of bone to parathyroid hormone in tissue culture. J Endocrinol. 1975;64:573–583.
84. Lee, K, Deeds, JD, Bond, AT, et al. In situ localization of PTH/PTHrP receptor mRNA in the bone of fetal and young rats. Bone. 1993;14:341–345.
85. Sherrard, DJ. Renal osteodystrophy. Semin Nephrol. 1986;6:56–67.
86. Malluche, HH, Ritz, E, Lange, HP, et al. Changes of bone histology during maintenance hemodialysis at various levels of dialysate Ca concentration. Clin Nephrol. 1976;6:440–447.
87. Hamdy, NA, Kanis, JA, Beneton, MN, et al. Effect of alfacalcidol on natural course of renal bone disease in mild to moderate renal failure. BMJ. 1995;310:358–363.
88. Norman, ME, Mazur, AT, Borden, S, et al. Early diagnosis of juvenile renal osteodystrophy. J Pediatr. 1980;97:226–232.
89. Massry, SG, Friedler, RM, Coburn, JW. Excretion of phosphate and calcium. Physiology of their renal handling and relation to clinical medicine. Arch Intern Med. 1973;131:828–859.
90. Galceran, T, Martin, KJ, Morrissey, JJ, et al. Role of 1,25-dihydroxyvitamin D on the skeletal resistance to parathyroid hormone. Kidney Int. 1987;32:801–807.
91. Mathias, R, Salusky, I, Harman, W, et al. Renal bone disease in pediatric and young adult patients on hemodialysis in a children’s hospital. J Am Soc Nephrol. 1993;3:1938–1946.
92. Salusky, IB, Ramirez, JA, Oppenheim, W, et al. Biochemical markers of renal osteodystrophy in pediatric patients undergoing CAPD/CCPD. Kidney Int. 1994;45:253–258.
93. Urena, P, Ferreira, A, Morieux, C, et al. PTH/PTHrP receptor mRNA is down-regulated in epiphyseal cartilage growth plate of uraemic rats. Nephrol Dial Transplant. 1996;11:2008–2016.
94. Sanchez, CP, Salusky, IB, Kuizon, BD, et al. Growth of long bones in renal failure: roles of hyperparathyroidism, growth hormone and calcitriol. Kidney Int. 1998;54:1879–1887.
95. Massry, SG, Stein, R, Garty, J, et al. Skeletal resistance to the calcemic action of parathyroid hormone in uremia: role of 1,25 (OH)2 D3. Kidney Int. 1976;9:467–474.
96. Parfitt, AM. The actions of parathyroid hormone on bone: relation to bone remodeling and turnover, calcium homeostasis, and metabolic bone disease. Part IV of IV parts: The state of the bones in uremic hyperaparathyroidism—the mechanisms of skeletal resistance to PTH in renal failure and pseudohypoparathyroidism and the role of PTH in osteoporosis, osteopetrosis, and osteofluorosis. Metabolism. 1976;25:1157–1188.
97. Parfitt, AM. The actions of parathyroid hormone on bone: relation to bone remodeling and turnover, calcium homeostasis, and metabolic bone disease. Part I of IV parts: Mechanisms of calcium transfer between blood and bone and their cellular basis: morphological and kinetic approaches to bone turnover. Metabolism. 1976;25:809–844.
98. Parfitt, AM. The actions of parathyroid hormone on bone: relation to bone remodeling and turnover, calcium homeostasis, and metabolic bone disease. Part III of IV parts: PTH and osteoblasts, the relationship between bone turnover and bone loss, and the state of the bones in primary hyperparathyroidism. Metabolism. 1976;25:1033–1069.
99. Parfitt, AM. The actions of parathyroid hormone on bone: relation to bone remodeling and turnover, calcium homeostasis, and metabolic bone diseases. Part II of IV parts: PTH and bone cells: bone turnover and plasma calcium regulation. Metabolism. 1976;25:909–955.
100. Malluche, H, Faugere, MC. Renal bone disease 1990: an unmet challenge for the nephrologist. Kidney Int. 1990;38:193–211.
101. El-Husseini, AA, El-Agroudy, AE, El-Sayed, M, et al. A prospective randomized study for the treatment of bone loss with vitamin D during kidney transplantation in children and adolescents. Am J Transplant. 2004;4:2052–2057.
102. Salusky, IB, Coburn, JW, Foley, J, et al. Effects of oral calcium carbonate on control of serum phosphorus and changes in plasma aluminum levels after discontinuation of aluminum-containing gels in children receiving dialysis. J Pediatr. 1986;108(5 Pt 1):767–770.
103. Goodman, WG, Ramirez, JA, Belin, TR, et al. Development of adynamic bone in patients with secondary hyperparathyroidism after intermittent calcitriol therapy. Kidney Int. 1994;46:1160–1166.
104. Ott, SM, Maloney, NA, Klein, GL, et al. Aluminum is associated with low bone formation in patients receiving chronic parenteral nutrition. Ann Intern Med. 1983;98:910–914.
105. London, GM, Marty, C, Marchais, SJ, et al. Arterial calcifications and bone histomorphometry in end-stage renal disease. J Am Soc Nephrol. 2004;15:1943–1951.
106. Ward, MK, Feest, TG, Ellis, HA, et al. Osteomalacic dialysis osteodystrophy: evidence for a water-borne aetiological agent, probably aluminium. Lancet. 1978;1:841–845.
107. Parkinson, IS, Ward, MK, Feest, TG, et al. Fracturing dialysis osteodystrophy and dialysis encephalopathy. An epidemiological survey. Lancet. 1979;1:406–409.
108. Pierides, AM, Edwards, WG, Jr., Cullum, UX, Jr., et al. Hemodialysis encephalopathy with osteomalacic fractures and muscle weakness. Kidney Int. 1980;18:115–124.
109. Nathan, E, Pedersen, SE. Dialysis encephalopathy in a non-dialysed uraemic boy treated with aluminium hydroxide orally. Acta Paediatr Scand. 1980;69:793–796.
110. Felsenfeld, AJ, Gutman, RA, Llach, F, et al. Osteomalacia in chronic renal failure: a syndrome previously reported only with maintenance dialysis. Am J Nephrol. 1982;2:147–154.
111. Griswold, WR, Reznik, V, Mendoza, SA, et al. Accumulation of aluminum in a nondialyzed uremic child receiving aluminum hydroxide. Pediatrics. 1983;71:56–58.
112. Kaye, M. Oral aluminum toxicity in a non-dialyzed patient with renal failure. Clin Nephrol. 1983;20:208–211.
113. Andreoli, SP, Bergstein, JM, Sherrard, DJ. Aluminum intoxication from aluminum-containing phosphate binders in children with azotemia not undergoing dialysis. N Engl J Med. 1984;310:1079–1084.
114. Sedman, AB, Miller, NL, Warady, BA, et al. Aluminum loading in children with chronic renal failure. Kidney Int. 1984;26:201–204.
115. Kaehny, WD, Hegg, AP, Alfrey, AC. Gastrointestinal absorption of aluminum from aluminum-containing antacids. N Engl J Med. 1977;296:1389–1390.
116. Salusky, IB, Coburn, JW, Brill, J, et al. Bone disease in pediatric patients undergoing dialysis with CAPD or CCPD. Kidney Int. 1988;33:975–982.
117. Kuizon, BD, Goodman, WG, Jüppner, H, et al. Diminished linear growth during intermittent calcitriol therapy in children undergoing CCPD. Kidney Int. 1998;53:205–211.
118. Cohen-Solal, ME, Sebert, JL, Boudailliez, B, et al. Non-aluminic adynamic bone disease in non-dialyzed uremic patients: a new type of osteopathy due to overtreatment? Bone. 1992;13:1–5.
119. Atsumi, K, Kushida, K, Yamazaki, K, et al. Risk factors for vertebral fractures in renal osteodystrophy. Am J Kidney Dis. 1999;33:287–293.
120. Ott, SM, Maloney, NA, Coburn, JW, et al. The prevalence of bone aluminum deposition in renal osteodystrophy and its relation to the response to calcitriol therapy. N Engl J Med. 1982;307:709–713.
121. De Beur, SM, Finnegan, RB, Vassiliadis, J, et al. Tumors associated with oncogenic osteomalacia express genes important in bone and mineral metabolism. J Bone Miner Res. 2002;17:1102–1110.
122. Jonsson, KB, Zahradnik, R, Larsson, T, et al. Fibroblast growth factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia. N Engl J Med. 2003;348:1656–1663.
123. Nelson, AE, Bligh, RC, Mirams, M, et al. Clinical case seminar: Fibroblast growth factor 23: a new clinical marker for oncogenic osteomalacia. J Clin Endocrinol Metab. 2003;88:4088–4094.
124. Shimada, T, Kakitani, M, Yamazaki, Y, et al. Targeted ablation of FGF23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest. 2004;113:561–568.
125. Sitara, D, Razzaque, MS, Hesse, M, et al. Homozygous ablation of fibroblast growth factor-23 results in hyperphosphatemia and impaired skeletogenesis, and reverses hypophosphatemia in Phex-deficient mice. Matrix Biol. 2004;23:421–432.
126. Genuth, SM, Klein, L, Rabinovich, S, et al. Osteomalacia accompanying chronic anticonvulsant therapy. J Clin Endocrinol Metab. 1972;35:378–386.
127. Pierides, AM, Ellis, HA, Ward, M, et al. Barbiturate and anticonvulsant treatment in relation to osteomalacia with haemodialysis and renal transplantation. Br Med J. 1976;1:190–193.
128. Wang, M, Hercz, G, Sherrard, DJ, et al. Relationship between intact 1-84 parathyroid hormone and bone histomorphometric parameters in dialysis patients without aluminum toxicity. Am J Kidney Dis. 1995;26:836–844.
129. NIDDK. USRDS 1994 Annual Report. 2008.
130. Salusky, IB, Goodman, WG, Kuizon, BD, et al. Similar predictive value of bone turnover using first- and second-generation immunometric PTH assays in pediatric patients treated with peritoneal dialysis. Kidney Int. 2003;63:1801–1808.
131. Salusky, IB, Goodman, WG, Sahney, S, et al. Sevelamer controls parathyroid hormone-induced bone disease as efficiently as calcium carbonate without increasing serum calcium levels during therapy with active vitamin D sterols. J Am Soc Nephrol. 2005;16:2501–2508.
132. Ho, AY, Kung, AW. Determinants of peak bone mineral density and bone area in young women. J Bone Miner Metab. 2005;23:470–475.
133. Bouillon, R, Van, CS, Carmeliet, G. Intestinal calcium absorption: Molecular vitamin D mediated mechanisms. J Cell Biochem. 2003;88:332–339.
134. Noel, LH, Zingraff, J, Bardin, T, et al. Tissue distribution of dialysis amyloidosis. Clin Nephrol. 1987;27:175–178.
135. Kleinman, KS, Coburn, JW. Amyloid syndromes associated with hemodialysis. Kidney Int. 1989;35:567–575.
136. Hampl, H, Lobeck, H, Bartel-Schwarze, S, et al. Clinical, morphologic, biochemical, and immunohistochemical aspects of dialysis-associated amyloidosis. ASAIO Trans. 1987;33:250–259.
137. Baker, LR, Ackrill, P, Cattell, WR, et al. Iatrogenic osteomalacia and myopathy due to phosphate depletion. Br Med J. 1974;3:150–152.
138. Powell, DR. Effects of renal failure on the growth hormone-insulin-like growth factor axis. J Pediatr. 1997;131(1 Pt 2):S13–S16.
139. Zanello, SB, Collins, ED, Marinissen, MJ, et al. Vitamin D receptor expression in chicken muscle tissue and cultured myoblasts. Horm Metab Res. 1997;29:231–236.
140. Wright, RS, Hunt, SM. A radioimmunoassay for 17-alpha 20 beta-dihydroxy-4-pregnen-3-one: its use in measuring changes in serum levels at ovulation in Atlantic salmon (Salmo salar), coho salmon (Oncorhynchus kisutch), and rainbow trout (Salmo gairdneri). Gen Comp Endocrinol. 1982;47:475–482.
141. Mehls, O, Ritz, E, Krempien, B, et al. Slipped epiphyses in renal osteodystrophy. Arch Dis Child. 1975;50:545–554.
142. Tonshoff, B, Schaefer, F, Mehls, O. Disturbance of growth hormone–insulin-like growth factor axis in uraemia. Implications for recombinant human growth hormone treatment. Pediatr Nephrol. 1990;4:654–662.
143. North American Pediatric Renal Transplant Cooperative Study (NAPRTCS) 2006 Annual Report. 2006.
144. Nash, MA, Torrado, AD, Greifer, I, et al. Renal tubular acidosis in infants and children. Clinical course, response to treatment, and prognosis. J Pediatr. 1972;80:738–748.
145. Challa, A, Chan, W, Krieg, RJ, Jr., et al. Effect of metabolic acidosis on the expression of insulin-like growth factor and growth hormone receptor. Kidney Int. 1993;44:1224–1227.
146. Maniar, S, Kleinknecht, C, Zhou, X, et al. Growth hormone action is blunted by acidosis in experimental uremia or acid load. Clin Nephrol. 1996;46:72–76.
147. Challa, A, Krieg, RJ, Jr., Thabet, MA, et al. Metabolic acidosis inhibits growth hormone secretion in rats: mechanism of growth retardation. Am J Physiol. 1993;265(4 Pt 1):E547–E553.
148. Green, J, Maor, G. Effect of metabolic acidosis on the growth hormone/IGF-I endocrine axis in skeletal growth centers. Kidney Int. 2000;57:2258–2267.
149. Lefebvre, A, de Vernejoul, MC, Gueris, J, et al. Optimal correction of acidosis changes progression of dialysis osteodystrophy. Kidney Int. 1989;36:1112–1118.
150. Goodman, WG, Salusky, IB. Evolution of secondary hyperparathyroidism during oral calcitriol therapy in pediatric renal osteodystrophy. Contrib Nephrol. 1991;90:189–195.
151. Waller, SC, Ridout, D, Cantor, T, et al. Parathyroid hormone and growth in children with chronic renal failure. Kidney Int. 2005;67:2338–2345.
152. Schmitt, CP, Ardissino, G, Testa, S, et al. Growth in children with chronic renal failure on intermittent versus daily calcitriol. Pediatr Nephrol. 2003;18:440–444.
153. Martin, KJ, Ballal, HS, Domoto, DT, et al. Pulse oral calcitriol for the treatment of hyperparathyroidism in patients on continuous ambulatory peritoneal dialysis: preliminary observations. Am J Kidney Dis. 1992;19:540–545.
154. K/DOQI clinical practice guidelines for bone metabolism and disease in children with chronic kidney disease. Am J Kidney Dis. 2005;46(4 suppl 1):S1–S121.
155. Nickerson, T, Huynh, H. Vitamin D analogue EB1089-induced prostate regression is associated with increased gene expression of insulin-like growth factor binding proteins. J Endocrinol. 1999;160:223–229.
156. Miyakoshi, N, Richman, C, Kasukawa, Y, et al. Evidence that IGF-binding protein-5 functions as a growth factor. J Clin Invest. 2001;107:73–81.
157. Richman, C, Baylink, DJ, Lang, K, et al. Recombinant human insulin-like growth factor-binding protein-5 stimulates bone formation parameters in vitro and in vivo. Endocrinology. 1999;140:4699–4705.
158. Longobardi, L, Torello, M, Buckway, C, et al. A novel insulin-like growth factor (IGF)-independent role for IGF binding protein-3 in mesenchymal chondroprogenitor cell apoptosis. Endocrinology. 2003;144:1695–1702.
159. Collard, TJ, Guy, M, Butt, AJ, et al. Transcriptional upregulation of the insulin-like growth factor binding protein IGFBP-3 by sodium butyrate increases IGF-independent apoptosis in human colonic adenoma-derived epithelial cells. Carcinogenesis. 2003;24:393–401.
160. Samaan, NA, Freeman, RM. Growth hormone levels in severe renal failure. Metabolism. 1970;19:102–113.
161. Tonshoff, B, Veldhuis, JD, Heinrich, U, et al. Deconvolution analysis of spontaneous nocturnal growth hormone secretion in prepubertal children with preterminal chronic renal failure and with end-stage renal disease. Pediatr Res. 1995;37:86–93.
162. Tonshoff, B, Sammet, A, Sanden, I, et al. Outcome and prognostic determinants in the hemolytic uremic syndrome of children. Nephron. 1994;68:63–70.
163. Schaefer, F, Chen, Y, Tsao, T, et al. Impaired JAK-STAT signal transduction contributes to growth hormone resistance in chronic uremia. J Clin Invest. 2001;108:467–475.
164. Tonshoff, B, Cronin, MJ, Reichert, M, et al. Reduced concentration of serum growth hormone (GH)-binding protein in children with chronic renal failure: correlation with GH insensitivity. The European Study Group for Nutritional Treatment of Chronic Renal Failure in Childhood. The German Study Group for Growth Hormone Treatment in Chronic Renal Failure. J Clin Endocrinol Metab. 1997;82:1007–1013.
165. Powell, DR, Liu, F, Baker, BK, et al. Modulation of growth factors by growth hormone in children with chronic renal failure. The Southwest Pediatric Nephrology Study Group. Kidney Int. 1997;51:1970–1979.
166. Conger, JD, Hammond, WS, Alfrey, AC, et al. Pulmonary calcification in chronic dialysis patients. Clinical and pathologic studies. Ann Intern Med. 1975;83:330–336.
167. Milliner, DS, Zinsmeister, AR, Lieberman, E, et al. Soft tissue calcification in pediatric patients with end-stage renal disease. Kidney Int. 1990;38:931–936.
168. Goodman, WG, Goldin, J, Kuizon, BD, et al. Coronary-artery calcification in young adults with end-stage renal disease who are undergoing dialysis. N Engl J Med. 2000;342:1478–1483.
169. Oh, J, Wunsch, R, Turzer, M, et al. Advanced coronary and carotid arteriopathy in young adults with childhood-onset chronic renal failure. Circulation. 2002;106:100–105.
170. Chavers, BM, Li, S, Collins, AJ, et al. Cardiovascular disease in pediatric chronic dialysis patients. Kidney Int. 2002;62:648–653.
171. Mitsnefes, MM, Kimball, TR, Kartal, J, et al. Cardiac and vascular adaptation in pediatric patients with chronic kidney disease: role of calcium-phosphorus metabolism. J Am Soc Nephrol. 2005;16:2796–2803.
172. Russo, D, Palmiero, G, De Blasio, AP, et al. Coronary artery calcification in patients with CRF not undergoing dialysis. Am J Kidney Dis. 2004;44:1024–1030.
173. Moe, SM, Duan, D, Doehle, BP, et al. Uremia induces the osteoblast differentiation factor Cbfa1 in human blood vessels. Kidney Int. 2003;63:1003–1011.
174. Ducy, P, Zhang, R, Geoffroy, V, et al. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell. 1997;89:747–754.
175. Jono, S, McKee, MD, Murry, CE, et al. Phosphate regulation of vascular smooth muscle cell calcification. Circ Res. 2000;87:E10–E17.
176. Ahmed, S, O’Neill, KD, Hood, AF, et al. Calciphylaxis is associated with hyperphosphatemia and increased osteopontin expression by vascular smooth muscle cells. Am J Kidney Dis. 2001;37:1267–1276.
177. Bostrom, K. Insights into the mechanism of vascular calcification. Am J Cardiol. 2001;88(2A):20E–22E.
178. Moe, SM, O’Neill, KD, Duan, D, et al. Medial artery calcification in ESRD patients is associated with deposition of bone matrix proteins. Kidney Int. 2002;61:638–647.
179. Chen, NX, O’Neill, KD, Duan, D, et al. Phosphorus and uremic serum up-regulate osteopontin expression in vascular smooth muscle cells. Kidney Int. 2002;62:1724–1731.
180. Schafer, C, Heiss, A, Schwarz, A, et al. The serum protein alpha 2-Heremans-Schmid glycoprotein/fetuin-A is a systemically acting inhibitor of ectopic calcification. J Clin Invest. 2003;112:357–366.
181. Schinke, T, Amendt, C, Trindl, A, et al. The serum protein alpha2-HS glycoprotein/fetuin inhibits apatite formation in vitro and in mineralizing calvaria cells. A possible role in mineralization and calcium homeostasis. J Biol Chem. 1996;271:20789–20796.
182. Sweatt, A, Sane, DC, Hutson, SM, et al. Matrix Gla protein (MGP) and bone morphogenetic protein-2 in aortic calcified lesions of aging rats. J Thromb Haemost. 2003;1:178–185.
183. Koh, N, Fujimori, T, Nishiguchi, S, et al. Severely reduced production of klotho in human chronic renal failure kidney. Biochem Biophys Res Commun. 2001;280:1015–1020.
184. Gutierrez, O, Mannstadt, M, Isakova, T, et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med. 2008;359:584–592.
185. Miyamoto, K, Ito, M, Segawa, H, Kuwahata, M. Molecular targets of hyperphosphataemia in chronic renal failure. Nephrol Dial Transplant. 2003;18(suppl 3):iii79–iii80.
186. Tonelli, M, Keech, A, Shepherd, J, et al. Effect of pravastatin in people with diabetes and chronic kidney disease. J Am Soc Nephrol. 2005;16:3748–3754.
187. Holdaas, H, Fellstrom, B, Cole, E, et al. Long-term cardiac outcomes in renal transplant recipients receiving fluvastatin: the ALERT extension study. Am J Transplant. 2005;5:2929–2936.
188. Wanner, C, Krane, V, Marz, W, et al. Atorvastatin in patients with type 2 diabetes mellitus undergoing hemodialysis. N Engl J Med. 2005;353:238–248.
189. Block, GA, Spiegel, DM, Ehrlich, J, et al. Effects of sevelamer and calcium on coronary artery calcification in patients new to hemodialysis. Kidney Int. 2005;68:1815–1824.
190. Block, GA, Raggi, P, Bellasi, A, et al. Mortality effect of coronary calcification and phosphate binder choice in incident hemodialysis patients. Kidney Int. 2007;71:438–441.
191. Gipstein, RM, Coburn, JW, Adams, DA, et al. Calciphylaxis in man. A syndrome of tissue necrosis and vascular calcification in 11 patients with chronic renal failure. Arch Intern Med. 1976;136:1273–1280.
192. Bleyer, AJ, Choi, M, Igwemezie, B, et al. A case control study of proximal calciphylaxis. Am J Kidney Dis. 1998;32:376–383.
193. Goldsmith, DJ. Calciphylaxis, thrombotic diathesis and defects in coagulation regulation. Nephrol Dial Transplant. 1997;12:1082–1083.
194. Mohammed, IA, Sekar, V, Bubtana, AJ, et al. Proximal calciphylaxis treated with calcimimetic “Cinacalcet.”. Nephrol Dial Transplant. 2008;23:387–389.
195. Rogers, NM, Teubner, DJ, Coates, PT. Calcific uremic arteriolopathy: advances in pathogenesis and treatment. Semin Dial. 2007;20:150–157.
196. Bardin, T, Kuntz, D, Zingraff, J, et al. Synovial amyloidosis in patients undergoing long-term hemodialysis. Arthritis Rheum. 1985;28:1052–1058.
197. Hou, FF, Chertow, GM, Kay, J, et al. Interaction between beta 2-microglobulin and advanced glycation end products in the development of dialysis related-amyloidosis. Kidney Int. 1997;51:1514–1519.
198. Miyata, T, Inagi, R, Iida, Y, et al. Involvement of beta 2-microglobulin modified with advanced glycation end products in the pathogenesis of hemodialysis-associated amyloidosis. Induction of human monocyte chemotaxis and macrophage secretion of tumor necrosis factor-alpha and interleukin-1. J Clin Invest. 1994;93:521–528.
199. Van Ypersele de Strihou, C, Jadoul, M, Malghem, J, et al. Effect of dialysis membrane and patient’s age on signs of dialysis-related amyloidosis. The Working Party on Dialysis Amyloidosis. Kidney Int. 1991;39:1012–1019.
200. McMahon, LP, Radford, J, Dawborn, JK. Shoulder ultrasound in dialysis related amyloidosis. Clin Nephrol. 1991;35:227–232.
201. Bindi, P, Chanard, J. Destructive spondyloarthropathy in dialysis patients: an overview. Nephron. 1990;55:104–109.
202. Ayli, M, Ayli, D, Azak, A, et al. The effect of high-flux hemodialysis on dialysis-associated amyloidosis. Ren Fail. 2005;27:31–34.
203. Block, GA, Hulbert-Shearon, TE, Levin, NW, et al. Association of serum phosphorus and calcium x phosphate product with mortality risk in chronic hemodialysis patients: a national study. Am J Kidney Dis. 1998;31:607–617.
204. Alfrey, AC, Miller, NL, Butkus, D. Evaluation of body magnesium stores. J Lab Clin Med. 1974;84:153–162.
205. Brown, EM, Wilson, RE, Eastman, RC, et al. Abnormal regulation of parathyroid hormone release by calcium in secondary hyperparathyroidism due to chronic renal failure. J Clin Endocrinol Metab. 1982;54:172–179.
206. Salusky, IB, Fine, RN, Kangarloo, H, et al. “High-dose” calcitriol for control of renal osteodystrophy in children on CAPD. Kidney Int. 1987;32:89–95.
207. Kurz, P, Monier-Faugere, MC, Bognar, B, et al. Evidence for abnormal calcium homeostasis in patients with adynamic bone disease. Kidney Int. 1994;46:855–861.
208. Guillot, AP, Hood, VL, Runge, CF, et al. The use of magnesium-containing phosphate binders in patients with end-stage renal disease on maintenance hemodialysis. Nephron. 1982;30:114–117.
209. Sherrard, DJ, Hercz, G, Pei, Y, et al. The spectrum of bone disease in end-stage renal failure—an evolving disorder. Kidney Int. 1993;43:436–442.
210. Van Renen, MJ, Hogg, RJ, Sweeney, AL, et al. Accelerated growth in short children with chronic renal failure treated with both strict dietary therapy and recombinant growth hormone. Pediatr Nephrol. 1992;6:451–458.
211. Charhon, SA, Delmas, PD, Malaval, L, et al. Serum bone Gla-protein in renal osteodystrophy: comparison with bone histomorphometry. J Clin Endocrinol Metab. 1986;63:892–897.
212. Epstein, S, Traberg, H, Raja, R, et al. Serum and dialysate osteocalcin levels in hemodialysis and peritoneal dialysis patients and after renal transplantation. J Clin Endocrinol Metab. 1985;60:1253–1256.
213. Brossard, JH, Cloutier, M, Roy, L, et al. Accumulation of a non-(1–84) molecular form of parathyroid hormone (PTH) detected by intact PTH assay in renal failure: importance in the interpretation of PTH values. J Clin Endocrinol Metab. 1996;81:3923–3929.
214. Lepage, R, Roy, L, Brossard, JH, et al. A non-(1–84) circulating parathyroid hormone (PTH) fragment interferes significantly with intact PTH commercial assay measurements in uremic samples. Clin Chem. 1998;44:805–809.
215. Brossard, JH, Yamamoto, LN, D’Amour, P. Parathyroid hormone metabolites in renal failure: bioactivity and clinical implications. Semin Dial. 2002;15:196–201.
216. Slatopolsky, E, Finch, J, Clay, P, et al. A novel mechanism for skeletal resistance in uremia. Kidney Int. 2000;58:753–761.
217. Gao, P, Scheibel, S, D’Amour, P, et al. Development of a novel immunoradiometric assay exclusively for biologically active whole parathyroid hormone 1-84: implications for improvement of accurate assessment of parathyroid function. J Bone Miner Res. 2001;16:605–614.
218. Monier-Faugere, MC, Geng, Z, Mawad, H, et al. Improved assessment of bone turnover by the PTH-(1–84)/large C-PTH fragments ratio in ESRD patients. Kidney Int. 2001;60:1460–1468.
219. Coen, G, Bonucci, E, Ballanti, P, et al. PTH 1-84 and PTH “7-84” in the noninvasive diagnosis of renal bone disease. Am J Kidney Dis. 2002;40:348–354.
220. D’Amour, P, Brossard, JH, Rousseau, L, et al. Structure of non-(1-84) PTH fragments secreted by parathyroid glands in primary and secondary hyperparathyroidism. Kidney Int. 2005;68:998–1007.
221. Divieti, P, John, MR, Jüppner, H, et al. Human PTH-(7-84) inhibits bone resorption in vitro via actions independent of the type 1 PTH/PTHrP receptor. Endocrinology. 2002;143:171–176.
222. Langub, MC, Monier-Faugere, MC, Wang, G, et al. Administration of PTH-(7-84) antagonizes the effects of PTH-(1-84) on bone in rats with moderate renal failure. Endocrinology. 2003;144:1135–1138.
223. Nguyen-Yamamoto, L, Rousseau, L, Brossard, JH, et al. Synthetic carboxyl-terminal fragments of parathyroid hormone (PTH) decrease ionized calcium concentration in rats by acting on a receptor different from the PTH/PTH-related peptide receptor. Endocrinology. 2001;142:1386–1392.
224. Alfrey, AC. Aluminum metabolism. Kidney Int Suppl. 1986;18:S8–S11.
225. Andress, DL, Endres, DB, Maloney, NA, et al. Comparison of parathyroid hormone assays with bone histomorphometry in renal osteodystrophy. J Clin Endocrinol Metab. 1986;63:1163–1169.
226. Wright, RS, Mehls, O, Ritz, E, et al. Musculoskeletal manifestation of chronic renal failure, dialysis and transplantation. In: Bacon P, Hadler N, eds. Renal Manifestations in Rheumatic Disease. London: Butterworth Publishers; 1982:352.
227. Dent, CE, Hodson, CJ. Radiological changes associated with certain metabolic bone diseases. Br J Radiol. 1954;27:605–618.
228. Parfitt, AM, Kleerekoper, M, Cruz, C. Reduced phosphate reabsorption unrelated to parathyroid hormone after renal transplantation: implications for the pathogenesis of hyperparathyroidism in chronic renal failure. Miner Electrolyte Metab. 1986;12:356–362.
229. Meema, HE, Rabinovich, S, Meema, S, et al. Improved radiological diagnosis of azotemic osteodystrophy. Radiology. 1972;102:1–10.
230. Shimada, H, Nakamura, M, Marumo, F. Influence of aluminium on the effect of 1 alpha (OH)D3 on renal osteodystrophy. Nephron. 1983;35:163–170.
231. Simpson, W, Ellis, HA, Kerr, DN, et al. Bone disease in long-term haemodialysis: the association of radiological with histological abnormalities. Br J Radiol. 1976;49:105–110.
232. Parfitt, AM, Massry, SG, Winfield, AC. Osteopenia and fractures occurring during maintenance hemodialysis. A new form of renal osteodystrophy. Clin Orthop Relat Res. 1972;87:287–302.
233. Mallette, LE, Patten, BM, Engel, WK. Neuromuscular disease in secondary hyperparathyroidism. Ann Intern Med. 1975;82:474–483.
234. Mucsi, I, Hercz, G, Uldall, R, et al. Control of serum phosphate without any phosphate binders in patients treated with nocturnal hemodialysis. Kidney Int. 1998;53:1399–1404.
235. Fischbach, M, Terzic, J, Menouer, S, et al. Intensified and daily hemodialysis in children might improve statural growth. Pediatr Nephrol. 2006;21:1746–1752.
236. Goodman, WG. Aluminum and renal osteodystrophy. In: Bushinsky DA, ed. Renal Osteodystrophy. Philadelphia: Lippincott-Raven; 1998:317.
237. Salusky, IB, Foley, J, Nelson, P, et al. Aluminum accumulation during treatment with aluminum hydroxide and dialysis in children and young adults with chronic renal disease. N Engl J Med. 1991;324:527–531.
238. Coburn, JW, Mischel, MG, Goodman, WG, et al. Calcium citrate markedly enhances aluminum absorption from aluminum hydroxide. Am J Kidney Dis. 1991;17:708–711.
239. Bakir, AA, Hryhorczuk, DO, Berman, E, et al. Acute fatal hyperaluminemic encephalopathy in undialyzed and recently dialyzed uremic patients. ASAIO Trans. 1986;32:171–176.
240. Portale, AA, Booth, BE, Tsai, HC, et al. Reduced plasma concentration of 1,25-dihydroxyvitamin D in children with moderate renal insufficiency. Kidney Int. 1982;21:627–632.
241. Alon, U, Davidai, G, Bentur, L, et al. Oral calcium carbonate as phosphate-binder in infants and children with chronic renal failure. Miner Electrolyte Metab. 1986;12:320–325.
242. Andreoli, SP, Dunson, JW, Bergstein, JM. Calcium carbonate is an effective phosphorus binder in children with chronic renal failure. Am J Kidney Dis. 1987;9:206–210.
243. Salusky, IB, Kuizon, BD, Belin, TR, et al. Intermittent calcitriol therapy in secondary hyperparathyroidism: a comparison between oral and intraperitoneal administration. Kidney Int. 1998;54:907–914.
244. Salusky, IB, Goodman, WG. Adynamic renal osteodystrophy: is there a problem? J Am Soc Nephrol. 2001;12:1978–1985.
245. Pflanz, S, Henderson, IS, McElduff, N, et al. Calcium acetate versus calcium carbonate as phosphate-binding agents in chronic haemodialysis. Nephrol Dial Transplant. 1994;9:1121–1124.
246. Caravaca, F, Santos, I, Cubero, JJ, et al. Calcium acetate versus calcium carbonate as phosphate binders in hemodialysis patients. Nephron. 1992;60:423–427.
247. Wallot, M, Bonzel, KE, Winter, A, et al. Calcium acetate versus calcium carbonate as oral phosphate binder in pediatric and adolescent hemodialysis patients. Pediatr Nephrol. 1996;10:625–630.
248. Cushner, HM, Copley, JB, Lindberg, JS, et al. Calcium citrate, a nonaluminum-containing phosphate-binding agent for treatment of CRF. Kidney Int. 1988;33:95–99.
249. Birck, R, Zimmermann, E, Wassmer, S, et al. Calcium ketoglutarate versus calcium acetate for treatment of hyperphosphataemia in patients on maintenance haemodialysis: a cross-over study. Nephrol Dial Transplant. 1999;14:1475–1479.
250. Slatopolsky, EA, Burke, SK, Dillon, MA. RenaGel, a nonabsorbed calcium- and aluminum-free phosphate binder, lowers serum phosphorus and parathyroid hormone. The RenaGel Study Group. Kidney Int. 1999;55:299–307.
251. Bleyer, AJ, Burke, SK, Dillon, M, et al. A comparison of the calcium-free phosphate binder sevelamer hydrochloride with calcium acetate in the treatment of hyperphosphatemia in hemodialysis patients. Am J Kidney Dis. 1999;33:694–701.
252. Chertow, GM, Burke, SK, Dillon, MA, et al. Long-term effects of sevelamer hydrochloride on the calcium x phosphate product and lipid profile of haemodialysis patients. Nephrol Dial Transplant. 1999;14:2907–2914.
253. Chertow, GM, Burke, SK, Raggi, P. Sevelamer attenuates the progression of coronary and aortic calcification in hemodialysis patients. Kidney Int. 2002;62:245–252.
254. Delmez, J, Block, G, Robertson, J, et al. A randomized, double-blind, crossover design study of sevelamer hydrochloride and sevelamer carbonate in patients on hemodialysis. Clin Nephrol. 2007;68:386–391.
255. O’Donovan, R, Baldwin, D, Hammer, M, et al. Substitution of aluminium salts by magnesium salts in control of dialysis hyperphosphataemia. Lancet. 1986;1:880–882.
256. Hergesell, O, Ritz, E. Phosphate binders on iron basis: a new perspective? Kidney Int Suppl. 1999;73:S42–S45.
257. Hergesell, O, Ritz, E. Stabilized polynuclear iron hydroxide is an efficient oral phosphate binder in uraemic patients. Nephrol Dial Transplant. 1999;14:863–867.
258. Hutchison, AJ, Speake, M, Al-Baaj, F. Reducing high phosphate levels in patients with chronic renal failure undergoing dialysis: a 4-week, dose-finding, open-label study with lanthanum carbonate. Nephrol Dial Transplant. 2004;19:1902–1906.
259. Finn, WF, Joy, MS, Hladik, G. Efficacy and safety of lanthanum carbonate for reduction of serum phosphorus in patients with chronic renal failure receiving hemodialysis. Clin Nephrol. 2004;62:193–201.
260. D’Haese, PC, Spasovski, GB, Sikole, A, et al, A multicenter study on the effects of lanthanum carbonate (Fosrenol) and calcium carbonate on renal bone disease in dialysis patients. Kidney Int Suppl, 2003;85:S73–S78.
261. Slatopolsky, E, Liapis, H, Finch, J. Progressive accumulation of lanthanum in the liver of normal and uremic rats. Kidney Int. 2005;68:2809–2813.
262. Spasovski, GB, Sikole, A, Gelev, S, et al. Evolution of bone and plasma concentration of lanthanum in dialysis patients before, during 1 year of treatment with lanthanum carbonate and after 2 years of follow-up. Nephrol Dial Transplant. 2006;21:2217–2224.
263. Baker, LR, Muir, JW, Sharman, VL, et al. Controlled trial of calcitriol in hemodialysis patients. Clin Nephrol. 1986;26:185–191.
264. Berl, T, Berns, AS, Hufer, WE, et al. 1,25 dihydroxycholecalciferol effects in chronic dialysis. A double-blind controlled study. Ann Intern Med. 1978;88:774–780.
265. Fukagawa, M, Okazaki, R, Takano, K, et al. Regression of parathyroid hyperplasia by calcitriol-pulse therapy in patients on long-term dialysis. N Engl J Med. 1990;323:421–422.
266. Pierides, AM, Ellis, HA, Simpson, W, et al. Variable response to long-term 1alpha-hydroxycholecalciferol in haemodialysis osteodystrophy. Lancet. 1976;1:1092–1095.
267. Kanis, JA, Henderson, RG, Heynen, G, et al. Renal osteodystrophy in nondialysed adolescents. Long-term treatment with 1alpha-hydroxycholecalciferol. Arch Dis Child. 1977;52:473–481.
268. Coyne, D, Acharya, M, Qiu, P, et al. Paricalcitol capsule for the treatment of secondary hyperparathyroidism in stages 3 and 4 CKD. Am J Kidney Dis. 2006;47:263–276.
269. Dobrez, DG, Mathes, A, Amdahl, M, et al. Paricalcitol-treated patients experience improved hospitalization outcomes compared with calcitriol-treated patients in real-world clinical settings. Nephrol Dial Transplant. 2004;19:1174–1181.
270. Sjoden, G, Smith, C, Lindgren, U, et al. 1 alpha-hydroxyvitamin D2 is less toxic than 1 alpha-hydroxyvitamin D3 in the rat. Proc Soc Exp Biol Med. 1985;178:432–436.
271. Andress, DL, Norris, KC, Coburn, JW, et al. Intravenous calcitriol in the treatment of refractory osteitis fibrosa of chronic renal failure. N Engl J Med. 1989;321:274–279.
272. Tentori, F, Hunt, WC, Stidley, CA, et al. Mortality risk among hemodialysis patients receiving different vitamin D analogs. Kidney Int. 2006;70:1858–1865.
273. Singh, NP, Sahni, V, Garg, D, et al. Effect of pharmacological suppression of secondary hyperparathyroidism on cardiovascular hemodynamics in predialysis CKD patients: A preliminary observation. Hemodial Int. 2007;11:417–423.
274. Schwarz, U, Amann, K, Orth, SR, et al. Effect of 1,25(OH)2 vitamin D3 on glomerulosclerosis in subtotally nephrectomized rats. Kidney Int. 1998;53:1696–1705.
275. Agarwal, R, Acharya, M, Tian, J, et al. Antiproteinuric effect of oral paricalcitol in chronic kidney disease. Kidney Int. 2005;68:2823–2828.
276. Nakane, M, Ma, J, Ruan, X, et al. Mechanistic analysis of VDR-mediated renin suppression. Nephron Physiol. 2007;107:35–44.
277. Wolf, M, Shah, A, Gutierrez, O, et al. Vitamin D levels and early mortality among incident hemodialysis patients. Kidney Int. 2007;72:1004–1013.
278. Wada, M, Nagano, N. Control of parathyroid cell growth by calcimimetics. Nephrol Dial Transplant. 2003;18(suppl 3):iii13–iii17.
279. Lopez, I, Guilera-Tejero, E, Mendoza, FJ, et al. Calcimimetic R-568 decreases extraosseous calcifications in uremic rats treated with calcitriol. J Am Soc Nephrol. 2006;17:795–804.
280. Quarles, LD, Yohay, DA, Carroll, BA, et al. Prospective trial of pulse oral versus intravenous calcitriol treatment of hyperparathyroidism in ESRD. Kidney Int. 1994;45:1710–1721.
281. Froment, DP, Molitoris, BA, Buddington, B, et al. Site and mechanism of enhanced gastrointestinal absorption of aluminum by citrate. Kidney Int. 1989;36:978–984.
282. Jorna, FH, Tobe, TJ, Huisman, RM, et al. Early identification of risk factors for refractory secondary hyperparathyroidism in patients with long-term renal replacement therapy. Nephrol Dial Transplant. 2004;19:1168–1173.
283. Andress, DL, Ott, SM, Maloney, NA, et al. Effect of parathyroidectomy on bone aluminum accumulation in chronic renal failure. N Engl J Med. 1985;312:468–473.
284. De Vernejoul, MC, Marchais, S, London, G, et al. Increased bone aluminum deposition after subtotal parathyroidectomy in dialyzed patients. Kidney Int. 1985;27:785–791.
285. Kurokawa, K, Akizawa, T, Suzuki, M, et al. Effect of 22-oxacalcitriol on hyperparathyroidism of dialysis patients: results of a preliminary study. Nephrol Dial Transplant. 1996;11(suppl 3):121–124.
286. Charytan, C, Coburn, JW, Chonchol, M, et al. Cinacalcet hydrochloride is an effective treatment for secondary hyperparathyroidism in patients with CKD not receiving dialysis. Am J Kidney Dis. 2005;46:58–67.
287. De Barros Gueiros, JE, Chammas, MC, Gerhard, R, et al. Percutaneous ethanol (PEIT) and calcitrol (PCIT) injection therapy are ineffective in treating severe secondary hyperparathyroidism. Nephrol Dial Transplant. 2004;19:657–663.
288. Fukagawa, M, Kitaoka, M, Tominaga, Y, et al. Guidelines for percutaneous ethanol injection therapy of the parathyroid glands in chronic dialysis patients. Nephrol Dial Transplant. 2003;18(suppl 3):iii31–iii33.
289. Diethelm, AG, Edwards, RP, Whelchel, JD. The natural history and surgical treatment of hypercalcemia before and after renal transplantation. Surg Gynecol Obstet. 1982;154:481–490.
290. D’Alessandro, AM, Melzer, JS, Pirsch, JD, et al. Tertiary hyperparathyroidism after renal transplantation: operative indications. Surgery. 1989;106:1049–1055.
291. Bergua, C, Torregrosa, JV, Fuster, D, et al. Effect of cinacalcet on hypercalcemia and bone mineral density in renal transplanted patients with secondary hyperparathyroidism. Transplantation. 2008;86:413–417.
292. Bhan, I, Shah, A, Holmes, J, et al. Post-transplant hypophosphatemia: Tertiary “Hyper-Phosphatoninism”? Kidney Int. 2006;70:1486–1494.
293. Evenepoel, P, Naesens, M, Claes, K, et al. Tertiary “hyperphosphatoninism” accentuates hypophosphatemia and suppresses calcitriol levels in renal transplant recipients. Am J Transplant. 2007;7:1193–1200.
294. Bonomini, V, Feletti, C, Di Felica, A, et al. Bone remodelling after renal transplantation (RT). Adv Exp Med Biol. 1984;178:207–216.
295. Nielsen, HE, Melsen, F, Christensen, MS. Aseptic necrosis of bone following renal transplantation. Clinical and biochemical aspects and bone morphometry. Acta Med Scand. 1977;202:27–32.
296. Grotz, WH, Mundinger, FA, Gugel, B, et al. Bone mineral density after kidney transplantation. A cross-sectional study in 190 graft recipients up to 20 years after transplantation. Transplantation. 1995;59:982–986.
297. Julian, BA, Laskow, DA, Dubovsky, J, et al. Rapid loss of vertebral mineral density after renal transplantation. N Engl J Med. 1991;325:544–550.
298. Slatopolsky, E, Martin, K. Glucocorticoids and renal transplant osteonecrosis. Adv Exp Med Biol. 1984;171:353–359.
299. Parfrey, PS, Farge, D, Parfrey, NA, et al. The decreased incidence of aseptic necrosis in renal transplant recipients—a case control study. Transplantation. 1986;41:182–187.
300. Isono, SS, Woolson, ST, Schurman, DJ. Total joint arthroplasty for steroid-induced osteonecrosis in cardiac transplant patients. Clin Orthop Relat Res. 1987;217:201–208.
301. Enright, H, Haake, R, Weisdorf, D. Avascular necrosis of bone: a common serious complication of allogeneic bone marrow transplantation. Am J Med. 1990;89:733–738.
302. Velasquez-Forero, F, Mondragon, A, Herrero, B, et al. Adynamic bone lesion in renal transplant recipients with normal renal function. Nephrol Dial Transplant. 1996;11(suppl 3):58–64.
303. Sanchez, CP, Salusky, IB, Kuizon, BD, et al. Bone disease in children and adolescents undergoing successful renal transplantation. Kidney Int. 1998;53:1358–1364.
304. Lindberg, JS, Culleton, B, Wong, G, et al. Cinacalcet HCl, an oral calcimimetic agent for the treatment of secondary hyperparathyroidism in hemodialysis and peritoneal dialysis: a randomized, double-blind, multicenter study. J Am Soc Nephrol. 2005;16:800–807.
305. Allen, DB, Goldberg, BD. Stimulation of collagen synthesis and linear growth by growth hormone in glucocorticoid-treated children. Pediatrics. 1992;89:416–421.
306. Root, AW, Bongiovanni, AM, Eberlein, WR. Studies of the secretion and metabolic effects of human growth hormone in children with glucocorticoid-induced growth retardation. J Pediatr. 1969;75:826–832.
307. Ortoft, G, Oxlund, H. Qualitative alterations of cortical bone in female rats after long-term administration of growth hormone and glucocorticoid. Bone. 1996;18:581–590.
308. Wehrenberg, WB, Bergman, PJ, Stagg, L, et al. Glucocorticoid inhibition of growth in rats: partial reversal with somatostatin antibodies. Endocrinology. 1990;127:2705–2708.
309. Aubia, J, Serrano, S, Marinoso, L, et al. Osteodystrophy of diabetics in chronic dialysis: a histomorphometric study. Calcif Tissue Int. 1988;42:297–301.
310. Movsowitz, C, Epstein, S, Fallon, M, et al. Cyclosporin-A in vivo produces severe osteopenia in the rat: effect of dose and duration of administration. Endocrinology. 1988;123:2571–2577.
311. Bryer, HP, Isserow, JA, Armstrong, EC, et al. Azathioprine alone is bone sparing and does not alter cyclosporin A-induced osteopenia in the rat. J Bone Miner Res. 1995;10:132–138.
312. Hokken-Koelega, AC, van Zaal, MA, van Bergen, W, et al. Final height and its predictive factors after renal transplantation in childhood. Pediatr Res. 1994;36:323–328.
313. Sarwal, MM, Yorgin, PD, Alexander, S, et al. Promising early outcomes with a novel, complete steroid avoidance immunosuppression protocol in pediatric renal transplantation. Transplantation. 2001;72:13–21.
314. Sarwal, MM, Vidhun, JR, Alexander, SR, et al. Continued superior outcomes with modification and lengthened follow-up of a steroid-avoidance pilot with extended daclizumab induction in pediatric renal transplantation. Transplantation. 2003;76:1331–1339.
315. Fine, RN, Yadin, O, Nelson, PA, et al. Recombinant human growth hormone treatment of children following renal transplantation. Pediatr Nephrol. 1991;5:147–151.
316. Mitsnefes, MM, Kimball, TR, Witt, SA, et al. Abnormal carotid artery structure and function in children and adolescents with successful renal transplantation. Circulation. 2004;110:97–101.
317. Ishitani, MB, Milliner, DS, Kim, DY, et al. Early subclinical coronary artery calcification in young adults who were pediatric kidney transplant recipients. Am J Transplant. 2005;5:1689–1693.
[/level-membership-for-endocrinology-diabetes-and-metabolism-category][not-level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 14
Chronic Kidney Disease Mineral and Bone Disorder
The kidney plays a major role in bone and mineral homeostasis by regulating calcium, phosphorous, parathyroid hormone (PTH), fibroblast growth factor-23 (FGF-23), and calcitriol [1,25-dihydroxyvitamin D3, 1,25(OH)2D3] metabolism. Disordered regulation of mineral metabolism occurs early in the course of chronic kidney disease (CKD) and results in alterations in bone modeling and remodeling. A growing body of evidence demonstrates that cardiovascular calcifications accompany CKD, that cardiovascular disease is the leading cause of mortality in patients with CKD, and that therapies designed to treat the skeletal consequences of CKD affect the progression of vascular pathology; this has led to reclassification of the mineral, skeletal, and vascular complications associated with progressive kidney disease. Together, these alterations are termed CKD mineral and bone disorder (CKD-MBD).1
CKD-MBD is defined as a systemic disorder of mineral and bone metabolism due to CKD that is manifested by one or a combination of the following: (1) abnormalities of calcium, phosphorous, PTH, or vitamin D metabolism; (2) abnormalities in bone histology, linear growth, or strength; or (3) vascular or other soft tissue calcification. Renal osteodystrophy is the specific term used to describe the bone pathology that occurs as a complication of CKD and therefore is one aspect of CKD-MBD. Traditionally, such lesions have been defined according to alterations in bone turnover, ranging from high bone turnover (secondary hyperparathyroidism, osteitis fibrosa) to low bone turnover (adynamic bone disease and osteomalacia). However, alterations in skeletal mineralization and volume are common in patients in CKD1 and may contribute to such outcomes as fractures and skeletal deformities, which may persist despite normalization of bone turnover.2 Bone histomorphometry continues to be the gold standard for the assessment of three essential aspects of bone histology: turnover, mineralization, and volume.1 This chapter summarizes major aspects of the pathogenesis, clinical manifestations, histologic features, and therapeutic interventions currently used in the management of CKD-MBD. Clinical and histologic features of bone disease after successful kidney transplantation are also described.
Pathogenesis of CKD-MBD
Abnormalities of Calcium, Phosphorous, FGF-23, Vitamin D, and PTH Metabolism
Calcium and Phosphorus
1,25(OH)2D3, the most active form of vitamin D, regulates serum calcium levels by increasing intestinal calcium absorption. The kidney generates most circulating 1,25(OH)2D3, converting 25(OH)D to 1,25(OH)2D3 by means of the enzyme 1α-hydroxylase. As renal failure progresses, calcitriol levels and intestinal calcium absorption decline. However, at the same time, rising PTH levels increase 1α-hydroxylase activity, release calcium and phosphorus from bone, and promote renal conservation of calcium, thus maintaining serum calcium levels within the normal range until late in the course of CKD.3,4
Likewise, serum phosphorous levels usually are maintained in the normal range throughout mild to moderate CKD. Elevated serum PTH and FGF-23 levels increase phosphate excretion, thus maintaining overall phosphate balance until the glomerular filtration rate (GFR) declines to 25% to 30% of normal.5,6 In late (stage 4) CKD, hyperphosphatemia ensues and contributes to secondary hyperparathyroidism (Fig. 14-1A).3
FIGURE 14-1 A, Median levels of calcium, phosphorus, and parathyroid hormone (PTH) per stage of chronic kidney disease (CKD). Median serum levels of calcium and phosphorus stay constant until late in the course of CKD; PTH levels rise before any changes are seen in calcium and phosphorus.
(A, Data from Levin A, et al: Kidney Int 71:31–38, 2007.3)
B, In patients with CKD, serum 1,25(OH)2D3 levels decline early in the course of kidney dysfunction, before any changes in serum calcium or phosphorous concentrations occur and prior to any rise in serum PTH levels.
Fibroblast Growth Factor-23
A recently described phosphaturic hormone, FGF-23, was first identified in patients with tumor-induced osteomalacia7 and autosomal dominant hypophosphatemic rickets.8 In these conditions and in patients with X-linked hypophosphatemia, elevated circulating levels of FGF-23 result in renal phosphate wasting and suppression of 1,25(OH)2D3 production.8,9 FGF-23 is made within bone,10,11 and the presence of a cofactor, Klotho, is essential for its action.12 Serum values increase as CKD progresses, becoming markedly elevated in individuals with end-stage kidney disease (Fig. 14-1B).13,14 In patients with CKD, 1,25(OH)2 D3 levels are inversely related to levels of circulating FGF-23, suggesting that increases in FGF-23 early in the course of CKD may play a role in declining active vitamin D levels.14 FGF-23 levels increase in response to vitamin D sterol therapy,15 and levels may be regulated by phosphorous intake.16,17 FGF-23 levels have been implicated in parathyroid gland regulation (vide infra).18,19
Vitamin D
Serum 1,25(OH)2D3 levels decline early in the course of kidney dysfunction, before any changes in serum calcium or phosphorous concentrations and prior to any rise in serum PTH levels.3,20 In late stages of CKD, phosphate retention and increased serum phosphorous levels directly suppress 1α-hydroxylase activity.16 However, although these factors contribute to declining 1α-hydroxylase activity, current evidence demonstrates that rising FGF-23 levels may be of even greater importance.14
Low circulating levels of 1,25(OH)2D3 have consequences for many tissues. Aside from its effect on intestinal calcium absorption, 1,25(OH)2D3 plays a direct role in the suppression of PTH gene transcription (vide infra). Animal studies also indicate that 1,25(OH)2D3 is essential for normal skeletal physiology—particularly in growing animals—and that this effect may not be mediated by the vitamin D receptor (VDR). Mice who lack the VDR [i.e., mice unable to respond to the actions of 1,25(OH)2D3 through its classical receptor] are phenotypically similar to those lacking the 1α-hydroxylase gene itself [i.e., mice unable to generate 1,25(OH)2D3]; both sets of mice are hypocalcemic with markedly elevated serum PTH levels, parathyroid gland hyperplasia, and rickets.21 However, a diet replete in calcium, phosphorus, and lactate is sufficient to normalize serum calcium, phosphorous, and PTH levels and to prevent the development of rickets in VDR-deficient animals.22 By contrast, this “rescue diet” is unable to completely reverse growth plate abnormalities in 1α-hydroxylase–deficient mice, suggesting that 1,25(OH)2D3, acting through a receptor other than the classical VDR, may be essential for proper growth plate development.23 1,25(OH)2D3 has been shown to regulate the renin-angiotensin system; 1α-hydroxylase–deficient mice demonstrate cardiac hypertrophy and dysfunction, which are reversed with angiotensin-converting enzyme blockade.24,25 Thus, 1,25(OH)2D3 may be essential for cardiac health—a finding that could explain observational data suggesting that active vitamin D sterol therapy improves survival in patients treated with maintenance dialysis.26,27
Native 25-hydroxyvitamin D [25(OH)D] deficiency is prevalent in patients with CKD; low levels of this form of the hormone also contribute to altered mineral metabolism. Vitamin D may be made in the skin or ingested from the diet.28,29 Ultraviolet B (UVB) (290 to 315 nm) photons penetrate the skin and are absorbed by 7-dehydrocholesterol to form previtamin D3, which then spontaneously converts to vitamin D3. Vitamin D3 is extruded from the skin cell into the extracellular space, where it binds vitamin D–binding protein.30 Although vitamin D created in the skin is exclusively of the D3 form, dietary sources of vitamin D and food supplements may contain vitamin D2 (created through UV irradiation of ergosterol in yeast) or D3 (from animal sources, particularly fish). Vitamin D (both D2 and D3) undergoes hydroxylation by the liver, forming 25(OH)D.31 Subsequently, 25(OH)D is taken up by renal tubular cells through a megalin-dependent process and undergoes a second hydroxylation, facilitated by renal 1α-hydroxylase, to 1,25(OH)2D3, a more potent stimulator of gut calcium absorption.32,33 Conversion of 25(OH)D to 1,25(OH)2D3 is independent of 25(OH)D stores in the general population but becomes a substrate-dependent process in patients with CKD.34 Although the actions of 25(OH)D have been underemphasized in CKD, extrarenal 1α-hydroxylase activity may significantly contribute to 1,25(OH)2D3 production, even in anephric patients.35–37 Thus low levels of the precursor, 25(OH)D, exacerbate 1,25(OH)2 D3 deficiency in the context of CKD.
Levels of 25(OH)D below 32 ng/mL are associated with increased PTH levels, reduced bone mineral density (BMD),38 and increased rates of hip fracture39 in the general population and therefore represent insufficient vitamin D storage. It is interesting to note that 25(OH)D levels in the vast majority of the general population meet the definition of D insufficiency, and a large percentage—as many as 57% in one series of medical inpatients40—have serum levels less than 15 ng/mL, thus qualifying as vitamin D deficient. Prevalence is higher in individuals with darker skin pigmentation, with 52% of Hispanic and black adolescents from the same cohort meeting the criteria for vitamin D deficiency.41
Several recent studies have documented a high prevalence of 25(OH)D deficiency in patients with CKD.42,43 Patients with CKD are at increased risk for vitamin D deficiency for several reasons. Many are chronically ill with little outdoor (sunlight) exposure, and CKD dietary restrictions, particularly of dairy products, curtail the intake of vitamin D–rich food.44 When compared with individuals with normal kidney function, patients with CKD display decreased skin synthesis of vitamin D3 in response to sunlight.45 This is exacerbated in individuals with darker skin.46 Proteinuria also contributes to D deficiency in the CKD population; 25(OH)D, in combination with vitamin D–binding protein, is lost in the urine.47,48
Parathyroid Hormone
The human PTH gene is located on chromosome 11 and contains two introns, which separate three exons encoding the 5′ untranslated region (UTR), the prepro region and PTH, and the 3′ untranslated region. The initial translational product of the mRNA is prepro-PTH, a 115 amino acid single-chain polypeptide, which undergoes conversion to pro-PTH (90 amino acids) in the rough endoplasmic reticulum. Six additional residues are removed from the N-terminal of pro-PTH in the Golgi apparatus to form the biologically active 1-84 PTH. PTH then is stored in secretory granules prior to release into the bloodstream.49–51 Sustained increases in PTH secretion occur with progression of CKD. This prolonged stimulation leads to high turnover bone disease and to the development of parathyroid gland hyperplasia.
PTH levels increase in response to alterations in metabolism of calcium, 1,25(OH)2 D3, phosphorus, 25(OH)D, and FGF-23 (Fig. 14-2). With the progression of CKD, additional factors develop that sustain high production of PTH. Among these factors are alterations in the regulation of prepro-PTH gene transcription, post-transcriptional modifications of PTH mRNA, reductions in calcium-sensing receptor (CaSR) and vitamin D receptor expression in the parathyroids, autonomous activity of adenomatous parathyroid glands, and skeletal resistance to the calcemic actions of PTH.
FIGURE 14-2 Pathogenesis of disordered mineral metabolism in chronic kidney disease (CKD). Dark arrows indicate effects of altered parathyroid hormone regulation; open arrows depict changes in fibroblast growth factor (FGF)-23 metabolism.
Calcium is the primary stimulus for PTH release. Calcium directly regulates PTH release via activation of the CaSR (vide infra). Activation of the CaSR decreases PTH release; deactivation increases PTH secretion. Serum calcium levels also regulate pre-pro PTH transcription via two negative calcium-response elements (nCaRE) located far upstream (−2.4 kbp and −3.5 kbp) of the PTH gene,52 and low serum calcium levels increase the stability of PTH mRNA by increasing levels of cytosolic parathyroid proteins, including AUF1,53 which binds to the 3′ UTR of the PTH mRNA. Thus, low levels of serum calcium present in CKD increase the amount of PTH release and the longevity of the PTH transcript. However, serum calcium levels are maintained within the normal range until late in the course of CKD, but serum PTH levels begin to rise much earlier, before any changes in serum calcium are evident.3 This suggests that factors other than calcium are important for the development of secondary hyperparathyroidism. Altered 1,25(OH)2 D3 metabolism may, in fact, be the initiating event early in the course of CKD.54–56
Apart from its effects on serum calcium levels, 1,25(OH)2D directly suppresses PTH levels. In conjunction with the VDR, 1,25(OH)2D3 binds negative vitamin D response elements in the promoter region of the PTH gene, thus inhibiting prepro-PTH gene transcription.57,58 In a positive-feedback loop, 1,25(OH)2D3 itself increases VDR gene expression in the parathyroid gland, further suppressing PTH gene transcription. 1,25(OH)2D3 also increases the expression of the CaSR, the expression of which is reduced in hyperplastic parathyroid tissues obtained from patients with secondary hyperparathyroidism.59 Vitamin D deficiency in animals is associated with decreased expression of CaSR mRNA in parathyroid tissue; 1,25(OH)2D3 therapy increases CaSR mRNA levels in a dose-dependent manner.60 Because 1,25(OH)2D3 is a potent inhibitor of cell proliferation, disturbances in renal calcitriol production and/or changes in VDR expression may be particularly important determinants of the degree of parathyroid hyperplasia and the extent of parathyroid gland enlargement in CKD.61
Phosphorous retention and hyperphosphatemia have been recognized for many years as important factors in the pathogenesis of secondary hyperparathyroidism. Phosphorous retention and hyperphosphatemia indirectly promote the secretion of PTH in several ways. Hyperphosphatemia lowers blood ionized calcium levels as free calcium ions complex with excess inorganic phosphate. The ensuing hypocalcemia stimulates PTH release. Increased phosphorus also impairs renal 1α-hydroxylase activity, which diminishes the conversion of 25(OH)D to 1,25(OH)2D3.16 Finally, phosphorus can directly enhance PTH synthesis. High serum phosphorous levels decrease cytosolic phospholipase A2 (normally increased by CaSR activation), leading to a decrease in arachidonic acid production with a subsequent increase in PTH secretion.62 Hypophosphatemia also decreases PTH mRNA transcript stability in vitro,63 suggesting that phosphorus itself affects serum PTH levels, probably by increasing the stability of the PTH mRNA transcript.
Although the actions of 25(OH)D have been underemphasized in dialyzed patients, evidence suggests that levels of this so-called “storage” form of vitamin D have both indirect and direct effects on PTH secretion. Extrarenal 1α-hydroxylase activity may contribute significantly to calcitriol production, even in anephric patients.35–37 Furthermore, recent evidence suggests the presence of 1α-hydroxylase in the parathyroid glands. 25(OH)D is converted inside the gland to 1,25(OH)2D, thereby suppressing PTH.64 25(OH)D administration suppresses PTH synthesis even when parathyroid gland 1α-hydroxylase is inhibited, indicating that 25(OH)D contributes to PTH suppression independent of its conversion to calcitriol.64 Indeed, recent studies have demonstrated that supplementation with ergocalciferol decreases serum PTH levels in patients with CKD.65,66 Such findings suggest that assessment of vitamin D status should be routinely performed in this patient population.67
Altered FGF-23 synthesis and secretion may contribute to increasing PTH levels through both indirect and direct mechanisms. Levels of FGF-23 rise as CKD progresses13,14 and contribute to declining 1,25(OH)2D levels. Lower 1,25(OH)2D levels, in turn, result in increasing PTH release. FGF-23 levels have been implicated in direct regulation of parathyroid gland function. In vitro and in vivo analysis of parathyroid glands from animals with normal renal function demonstrates that FGF-23 suppresses PTH secretion through a mechanism independent of its actions on vitamin D metabolism (vide infra).18,19
Alterations in parathyroid gland CaSR expression also occur in secondary hyperparathyroidism and may contribute to parathyroid gland hyperplasia. The CaSR is a seven-transmembrane G protein–coupled receptor with a large extracellular N-terminus, which binds acidic amino acids and divalent cations.68 Low extracellular calcium levels result in decreased calcium binding to the receptor, conformational relaxation of the receptor, and a resultant increase in PTH secretion,69 while activation of the receptor by high levels of serum calcium decreases PTH secretion.70,71 Expression of the CaSR is reduced by 30% to 70%, as judged by immunohistochemical methods in hyperplastic parathyroid tissue obtained from human subjects with renal failure.59,72 Decreased expression and activity of CaSR have been linked to decreased responsiveness in PTH secretion due to altered calcium levels.73 This decreased expression of the CaSR results in insensitivity to serum calcium levels with subsequent uncontrolled secretion of PTH. Increased stimulation of the CaSR by calcimimetics has been shown to decrease PTH cell proliferation, implicating the CaSR as a regulator of cell proliferation and PTH secretion.74
The link between the CaSR and vitamin D in cell cycling in the parathyroid gland is incompletely understood. However, some evidence suggests that vitamin D may decrease parathyroid hyperplasia by activating the CaSR. CaSR gene transcription is regulated by vitamin D through two distinct vitamin D response elements in the gene’s promoter region,75 suggesting that alterations in vitamin D metabolism in renal failure could account for changes in calcium sensing by the parathyroid glands, and that vitamin D may act upstream of the CaSR in preventing parathyroid cell hyperplasia.21
Once established, parathyroid enlargement is difficult to reverse because the rate of apoptosis in parathyroid glands is low, and the half-life of parathyroid cells is approximately 30 years.76 Chronic stimulation of parathyroid glands may lead to chromosomal changes that ultimately result in autonomous, unregulated growth and hormone release.77 Hyperplastic parathyroid tissue has been shown to develop inactivation of tumor suppressor genes MEN1, the retinoblastoma protein,78–80 and/or activating mutations of the RET proto-oncogene (MEN2a).81 Chromosomal translocations resulting in the parathyroid promoter driving cell cycle proteins (particularly cyclin D1) have been shown to be present in parathyroid adenomas.82 Even in the absence of somatic mutations, PTH secretion from enlarged parathyroid glands may become uncontrollable owing to the nonsuppressible component of PTH release from a large number of parathyroid cells. This alone may be sufficient to produce hypercalcemia and progressive bone disease in patients with end-stage renal disease.
Pathogenesis of Renal Bone Disease
Abnormalities In Bone Turnover, Mineralization, and Volume
Evaluation of skeletal histology by bone histomorphometry provides a method for understanding the pathophysiology of renal bone disease and a guide to its proper management. As recently recommended by the Kidney Disease Improving Global Outcomes (KDIGO) workgroup, three areas of bone histology are examined: bone turnover, mineralization, and volume, all of which may be altered in patients with CKD.1
Turnover
Traditionally, renal osteodystrophy has been classified primarily by alterations in bone turnover. Because PTH activates the PTH-related protein (PTHrP) receptor on osteocytes and osteoblasts, increasing cellular activity of both osteoblasts and osteoclasts,83,84 excessive levels of circulating PTH result in increased bone turnover (Figs. 14-3A and 14-3B).85 Serum PTH levels are inversely correlated with GFR, and most patients with GFR less than 50 mL/min have increased serum PTH levels and high turnover bone disease.86–88 The presence of CKD, however, markedly attenuates the effects of PTH on bone.89–90 Indeed, serum levels of PTH that are three to five times the normal range are associated with normal bone turnover in patients treated with maintenance dialysis, while similar PTH values in patients with mild to moderate kidney disease are associated with high turnover osteodystrophy.87,91,92 Although the precise mechanisms are poorly understood, uremia has been associated with this “skeletal resistance” to the actions of PTH. Uremic animals and humans display decreased PTH/PTHrP receptor mRNA expression in bone and growth plate.93,94 Hyperphosphatemia and alterations in vitamin D metabolism, among other factors, have been implicated in these changes, and calcitriol administration has been shown to partially restore the calcemic response to PTH in both experimental animals and patients with moderate CKD.95 Despite this “skeletal resistance” to PTH, many patients with end-stage kidney disease display bone biopsy evidence of PTH excess.
FIGURE 14-3 Bone histology, osteitis fibrosa. A, Under light microscopy, an increase in cellular activity, osteoid accumulation, erosion, and fibrosis are visible. B, An increase in double tetracycline labeling signifies an increase in bone turnover rate.
The bone in secondary hyperparathyroidism exhibits a marked increase in turnover with increased numbers of osteoblasts and osteoclasts and variable degrees of peritrabecular fibrosis. Activation of osteoclasts is mediated through PTH96–99; the result is increased resorption of both mineral and matrix along the trabecular surface and within the haversian canals of cortical bone.100 Such increased cellular activity can occur secondary to a nonspecific reaction to local factors, such as insulin-like growth factor-1 (IGF-1), cytokines, or fracture, or as the result of systemic stimuli, such as increased thyroxine or PTH. One characteristic of high bone turnover is increased quantities of woven osteoid, exhibiting haphazard arrangements of collagen fibers in contrast to the usual lamellar pattern of osteoid in normal bone. Woven osteoid can become mineralized in patients with advanced kidney disease in the absence of vitamin D; however, the calcium may be deposited as amorphous calcium phosphate rather than hydroxyapatite.101
At the other end of the spectrum of bone turnover, adynamic bone disease is characterized by normal osteoid volume, an absence of fibrosis, and a reduced bone formation rate, as indicated by a reduced or absent double tetracycline label on bone histomorphometry (Fig. 14-4).102,103 A paucity of osteoblasts and osteoclasts is observed.85 The histologic features of adynamic renal osteodystrophy, in the absence of aluminum deposition in bone, cannot be distinguished from the histologic features of corticosteroid-induced osteoporosis or age-related or postmenopausal osteoporosis. Therefore, it is not possible to determine whether osteoporosis accounts for decreases in osteoblastic activity and bone formation in patients with adynamic renal osteodystrophy unless the amount of trabecular bone is reduced. Decreases in bone mass and histologic evidence of trabecular bone loss are not integral features of the adynamic lesion of renal osteodystrophy when other causes of osteoporosis can be excluded. Adynamic bone is associated with low PTH levels, low alkaline phosphatase levels, high serum calcium levels, and a propensity for increased vascular calcification.104,105
FIGURE 14-4 Bone histology, adynamic bone. Under light microscopy, decreased cellular activity occurs with minimal osteoid accumulation.
In the 1970s and 1980s, aluminum intoxication was largely responsible for the development of adynamic bone and osteomalacia in patients with CKD. Two distinct patterns of aluminum intoxication were identified: (1) absorbed from the aluminum content of water used to prepare dialysate solution,106–108 and (2) intestinal aluminum absorption after ingestion of large doses of aluminum hydroxide.109–115 The neurologic syndrome of “dialysis encephalopathy” and a bone disease manifested by fractures, pain, persistent hypercalcemia, and osteomalacia were the main clinical features. Although the prevalence of aluminum bone disease in developed countries is now very low, the prevalence of adynamic renal osteodystrophy not associated with aluminum intoxication has increased substantially over the past few years in adult patients receiving regular dialysis.116 Currently, adynamic renal osteodystrophy is commonly associated with disorders such as age-related or postmenopausal osteoporosis, steroid-induced osteoporosis, hypoparathyroidism (idiopathic or surgically induced), and diabetes mellitus, and with overtreatment with calcium and vitamin D therapy.117
Because PTH is the major determinant of bone formation and skeletal remodeling in renal failure, oversuppression of PTH secretion can result in adynamic renal osteodystrophy. Approximately 40% of those treated with hemodialysis and more than half of adult patients undergoing peritoneal dialysis have serum PTH levels that are only minimally elevated or that fall within the normal range; such values are typically associated with normal or reduced rates of bone formation and turnover.103 Prolonged treatment with calcium-containing phosphate-binding medications and the use of high-dialysate calcium concentrations also contribute to low bone turnover.118 Calcitriol may directly suppress osteoblastic activity when given intermittently in large doses to patients receiving regular dialysis.119
The long-term consequences of adynamic renal osteodystrophy not attributable to aluminum toxicity remain to be determined, but concerns have been raised about increases in the risk for skeletal fracture and delayed fracture healing due to low rates of bone remodeling.119 The development of soft tissue and vascular calcifications has been associated with adynamic bone disease in cross-sectional studies.105 In prepubertal children, adynamic renal osteodystrophy has been associated with a reduction in linear growth.117
Mineralization
Although renal osteodystrophy has traditionally been defined by lesions in bone turnover, alterations in skeletal mineralization are also prevalent in CKD.92 Increases in unmineralized bone (osteoid) in conjunction with delayed rates of mineral deposition are common.92,100 Defective mineralization associated with low to normal bone turnover is termed osteomalacia.1 Histomorphometric characteristics of osteomalacia include the presence of wide osteoid seams, an increased number of osteoid lamellae, an increase in the trabecular surface covered with osteoid, and a diminished rate of mineralization or bone formation, as assessed by double tetracycline labeling. Fibrosis is often absent.85 In patients on long-term dialysis, osteomalacia that is refractory to vitamin D therapy is most commonly a result of aluminum intoxication.120
Although the mechanisms of skeletal mineralization are incompletely understood, factors such as 25(OH)D deficiency and altered FGF-23 metabolism have been implicated in their pathogenesis. In the general population, nutritional 25(OH)D deficiency results in osteomalacia, and a similar phenotype may occur in patients with CKD. FGF-23 may play a role; both overexpression121–123 and ablation of FGF-23124,125 in mice with normal renal function are associated with abnormal mineralization of osteoid, although by different mechanisms. The phosphaturic effects of increased FGF-23 may cause rickets and osteomalacia through an insufficiency of mineral substrate. The mechanisms leading to impaired mineralization in FGF-23–null animals, which have severe hyperphosphatemia and normal or elevated serum calcium levels, remain uncertain; however, osteomalacia in these animals suggests that FGF-23 may play a direct role in skeletal mineral deposition. Although the ramifications of defective mineralization remain to be established, increased fracture rates and bone deformities are prevalent in patients with CKD despite adequate control of bone turnover. These complications may be due, in part, to alterations in bone mineralization.
Treatment with anticonvulsant therapy may contribute to the development of osteomalacia in patients with kidney disease. Long-term ingestion of phenytoin and/or phenobarbital is associated with a high incidence of osteomalacia in nonuremic patients.126 These findings may be due in part to alterations in vitamin D metabolism.127
Osteitis fibrosa cystica, a finding of secondary hyperparathyroidism, can coexist with defective mineralization in some patients; this pattern is called a “mixed” lesion.128 Patients with mixed lesions often display high serum PTH and alkaline phosphatase levels, along with lower serum calcium levels. Mixed lesions are seen with high turnover bone disease in patients who are developing aluminum toxicity, or in patients with low turnover aluminum-related bone disease during desferoxamine (DFO) therapy (vide infra).129 In these cases, mixed lesions represent a transitional stage between high turnover and low turnover bone disease.
Volume
Because PTH is anabolic at the level of trabecular bone, high levels of serum PTH are typically associated with increases in bone volume, trabecular volume, and trabecular width.92,116,130,131 However, bone volume may also be low (termed osteoporosis), particularly in individuals with underlying age-related bone loss and in those treated with corticosteroids. Osteoporosis in the general population is associated with increased risk for hip fractures and mortality.132 Thus, bone volume is considered a critically important parameter of bone histology. The impact of osteoporosis on morbidity and mortality in the CKD population, however, remains to be defined.
Clinical Manifestations
Bone Pain
Bone pain is a common manifestation of severe bone disease in patients with advanced kidney disease. It usually is insidious in appearance and often is aggravated by weight bearing or a change in posture. Physical findings are often absent. Pain is most common in the lower part of the back, hips, and legs but may occur in the peripheral skeleton. Occasionally, sudden appearance of pain around the knee, ankle, or heel can suggest acute arthritis; such pain is not usually relieved by massage or local heat. Bone pain is more common and often is more marked in patients with aluminum-related bone disease than in those with osteitis fibrosa cystica, but marked variability may be seen from one patient to another.133
In patients on long-term dialysis, carpal tunnel syndrome and chronic arthralgias often occur in association with the deposition of β2-microglobulin amyloid in articular and periarticular structures.134 Arthralgias usually are bilateral and most commonly affect the shoulders, knees, wrists, and small joints of the hand; symptoms typically are worse with inactivity and at night.135,136
Muscle Weakness
The pathogenesis of this myopathy is not clear, and several different mechanisms, including secondary hyperparathyroidism, phosphate depletion,137 abnormal vitamin D metabolism, and aluminum intoxication, have been implicated.138 Improvement in gait posture has been reported in children with moderate renal failure after treatment with 1,25(OH)2D3, and muscle weakness improves rapidly in affected adult patients with end-stage kidney disease.139 Improvement in muscular strength has been observed after treatment with 25(OH)D3, after subtotal parathyroidectomy, after successful renal transplantation, and after chelation therapy with deferoxamine for aluminum intoxication.
Skeletal Deformities
Bone deformities are common in uremic children because their bones undergo growth, modeling, and remodeling. In adult patients, skeletal deformities also arise from abnormal remodeling or recurrent fractures.140 In children, bone deformities of the femora and wrists arise from slipped epiphyses.141 This problem is most common during the preadolescent period and is most frequent in patients with long-standing congenital kidney disease. In adults with kidney disease, particularly those with aluminum-related bone disease, skeletal deformities may be characterized by lumbar scoliosis, kyphosis, and deformities of the thoracic cage.140
Growth Retardation
Growth retardation is the hallmark of CKD in children. Protein and calorie malnutrition, metabolic acidosis, end-organ growth hormone resistance, and renal bone disease are most commonly implicated in growth failure.142 Despite correction of acidosis and anemia, normalization of serum calcium and phosphorous levels, and vitamin D sterol therapy replacement, most children with CKD continue to grow poorly. Growth failure worsens as renal function declines; the average height of children with even mild CKD (GFR 50 to 70 mL/minute/1.73 m2) is 1 standard deviation (SD) below the average for healthy children. Moderate CKD (GFR 25 to 49 mL/minute/1.73 m2) is associated with a height SD of −1.5, and, at the time of initiation of dialysis, the mean height SD is −1.8. Boys, younger patients, and those with prior renal transplants are at greatest risk for growth failure.143
Acidosis has been linked to delayed linear growth in patients with renal tubular acidosis and normal renal function, and correction of metabolic acidosis often leads to acceleration in growth velocity.144 Acidotic rats have been found to have decreased growth hormone (GH) secretion, serum insulin-like growth factor 1 (IGF-1), and hepatic IGF-1 mRNA expression. Moreover, metabolic acidosis has been shown to inhibit the effects of GH in rats with normal and decreased renal function.145–147 Growth plate mRNA levels of GH receptor, IGF-1 receptor, and IGF-1 expression are downregulated, and IGF-binding proteins are upregulated.148 In adults treated with maintenance dialysis, correction of acidosis has been shown to decrease the progression of secondary hyperparathyroidism and to improve skeletal mineralization.149
Calcitriol deficiency has been thought to contribute to growth retardation and bone disease in children with CKD. Secondary hyperparathyroidism remains prevalent in children with advanced renal disease, and osteitis fibrosa continues to be the most common skeletal lesion of renal osteodystrophy in those undergoing regular dialysis despite treatment with daily doses of oral calcitriol.116,150 Secondary hyperparathyroidism contributes to growth retardation, although optimal target values for PTH in children at all stages of CKD remain controversial. In children with moderate CKD, some data indicate that normal growth velocity is achieved when PTH levels are maintained within the normal range151; others have demonstrated a linear correlation between growth and PTH levels in the same patient population, with those with the highest PTH values displaying the highest growth velocity.152 Treatment for secondary hyperparathyroidism with large, intermittent doses of calcitriol and calcium-based phosphate binders has been shown to significantly reduce bone formation and suppress osteoblastic activity in both adults and children.95,153 However, adynamic bone disease may develop and linear bone growth decrease, despite serum PTH levels in the K/DOQI117,154 recommended range during intermittent vitamin D sterol therapy. Maintaining serum PTH levels at between 300 and 500 pg/mL reduces the frequency of these complications.131 The mechanisms by which calcitriol inhibits epiphyseal growth plate cartilage remain poorly understood. However, it is well known that calcitriol exerts dose-dependent inhibitory effects on cell proliferation of chondrocytes and osteoblasts in vitro. In addition, vitamin D sterols increase expression of a number of IGF-binding proteins (IGFBPs). IGFBP-2, -3, -4, and -5 sequester IGF-1 and may exert IGF-1–independent antiproliferative effects through their own receptors.155–159
GH resistance also contributes to impaired linear growth in renal failure. In CKD, poor growth develops despite normal or increased serum GH levels.160,161 Uremia has been associated with diminished hepatic GH receptor and IGF-1 mRNA expression, defects in postreceptor GH-mediated signal transduction,162,163 reductions in serum GH-binding protein levels,164 and increased synthesis and reduced clearance of IGF binding proteins.138,164,165 Improved growth velocity during recombinant human GH (rhGH) therapy has been ascribed to increased bioavailability of IGF-1 to target tissues. Children who are treated with maintenance dialysis respond less well to rhGH therapy than do children with less severe CKD, but the mechanisms for differences in response to GH therapy remain to be determined.
Extraskeletal Calcification
Extraskeletal calcification has been associated with uremia for many years166 and has been included recently in the definition of CKD-MBD.1 Vascular calcifications are associated with increased mortality. These lesions have their origins in CKD prior to dialysis and begin in childhood (Fig. 14-5).167,169 The mortality rate in adults and children with CKD is markedly higher than that in the general population, and cardiovascular disease is the leading cause of death in both children and adults treated with maintenance dialysis.168,170 In contrast to the calcified atherosclerotic plaques that develop in the vascular intima of aging individuals with normal kidney function, uremia facilitates calcification of the tunica media. This form of calcification is associated with decreased distensibility of blood vessels, causing a rigid “lead pipe” pathology that is associated with increased risk for congestive heart failure.1 Electron beam computed tomography (EBCT) is used in assessment of vascular calcifications in the adult population, and measurements in young adults who were treated with maintenance dialysis as children demonstrated that a significant proportion of this population has evidence of vascular calcification.168 Carotid ultrasound measurement of intima medial thickness (IMT) has been validated for the assessment of cardiovascular pathology in children, with increased thickness associated with worsening disease.169,171
FIGURE 14-5 Coronary artery calcification scores in 39 children and young adults with end-stage renal disease who were treated by dialysis, according to age. Coronary artery calcification was assessed by electron beam computed tomography. The scale on the y-axis is logarithmic. (Data from Goodman WG, et al: Coronary artery calcification in young adults with end-stage renal disease who are undergoing dialysis, N Engl J Med 106:100–105, 2000.)
Hypercalcemia, hyperphosphatemia, elevated levels of the calcium x phosphorous product, and high doses of vitamin D sterols167,168 all have been implicated in the pathogenesis of cardiovascular calcification. However, 40% of adult patients with stage 3 CKD, without these risk factors, show evidence of calcification,172 suggesting a role for uremic factors other than high levels of calcium and phosphorus. Indeed, vascular tissues in the uremic milieu express osteoblast differentiation factors.173 Osteoblasts and vascular smooth muscle cells have a common mesenchymal origin; core binding factor-1 (Cbfa1) is thought to trigger mesenchymal cell-to-osteoblast transformation. Mice that are deficient in Cbfa1 fail to mineralize bone,174 and arteries obtained from patients undergoing renal transplantation show increased levels of this protein.173 Upregulation of the sodium-dependent phosphate transporter PIT-1 likely also contributes to increased calcification,175 and upregulation of pro-mineralization factors such as osteopontin, bone sialoprotein, osteonectin, alkaline phosphatase, type I collagen, and bone morphogenic protein-2 (BMP-2) is potentiated by the uremic milieu.176–179 By contrast, expression of calcification inhibitors such as fetuin A, matrix gla protein, and klotho is suppressed.180–183 Levels of circulating FGF-23 also may contribute, as high levels of the protein are associated with increased mortality in adult dialysis patients.184 Klotho, the cofactor necessary for the actions of FGF-23, has been implicated in vascular calcification. Animals lacking the Klotho gene display elevated levels of calcium, phosphorus, and vitamin D, along with vascular calcification and premature aging. CKD is associated with low circulating levels of Klotho,183 which has been implicated in the regulation of sodium/phosphate co-transport in the aorta.185
Because the pathophysiology of cardiovascular disease in CKD is multifactorial, treatment strategies are also multifaceted and vary according to stage of CKD. Therapies that are effective in early CKD may not be effective in later stages—lipid-lowering agents decrease mortality in adults with CKD186 and in those with stable renal allografts187 but have not been shown to benefit patients treated with dialysis.188 By contrast, normalization of mineral metabolism (attained by avoiding hypercalcemia and hyperphosphatemia, limiting calcium intake, and avoiding adynamic bone) is effective at slowing the progression of cardiovascular calcification in patients treated with maintenance dialysis.154,168,189,190 Thus, at different stages of CKD, the relative importance of individual risk factors and the value of different biomarkers may vary.
Calciphylaxis
This unique syndrome, which is characterized by ischemic necrosis of the skin, subcutaneous fat, and muscles, can develop in patients with advanced renal failure not yet treated by dialysis, in those treated with regular dialysis, and in patients with well-functioning kidney transplants.191 The pathogenesis of this syndrome is uncertain. Extensive medial calcifications of medium-sized arteries are noted; such calcifications commonly exist in patients with renal disease without causing gangrene or ulcerations, and it is not clear that vascular calcifications per se are the cause of the ischemic necrosis. Two distinct types of the syndrome are recognized: proximal calciphylaxis, which affects the thighs, abdomen, and chest wall, and acral calciphylaxis, which involves sites distal to the knees and elbows, such as the toes, fingers, and ankles.192 The former has a terrible prognosis, with death occurring in more than 80% to 90% of affected patients. This syndrome is often accompanied by morbid obesity and hypoalbuminemia. Many patients have severe secondary hyperparathyroidism, and most have a history of severe and uncontrolled hyperphosphatemia.192 Some patients may have defective regulation of coagulation.193 The appearance of this syndrome in renal transplant recipients receiving glucocorticoids suggests that steroids may also play a role. Patients with calciphylaxis frequently die of secondary infection.
A significant number of patients improve after parathyroidectomy, and a few have healed after substantial reductions in levels of serum phosphorus. Parathyroidectomy and aggressive control of serum phosphate levels therefore are indicated in those with evidence of severe secondary hyperparathyroidism. However, although ischemic lesions and medial vascular calcifications are common in uremic patients with diabetes, such lesions rarely improve after parathyroidectomy. Thus, parathyroid surgery should be reserved for those diabetic patients with calciphylaxis who have clear evidence of severe secondary hyperparathyroidism. Calcimimetics,194 as well as sodium-thiosulfate (a calcium chelating agent) and pamidronate,195 have been used effectively in some individuals. Hyperbaric oxygen therapy also has been advocated.195
Dialysis-Related Amyloidosis
Several clinical syndromes that arise as a consequence of dialysis-related amyloidosis can mimic the clinical features of renal osteodystrophy, or amyloidosis can occur concurrently with uremic bone disease. Dialysis-related amyloidosis arises from the deposition in bone and periarticular structures of a specific type of amyloid composed of β2-microglobulin.196 In addition to β2-microglobulin, the amyloid deposits contain advanced glycosylation end products, which may account for their uptake by certain collagen-rich structures.197,198 The frequency of its clinical manifestation rises markedly in patients treated with regular dialysis for longer than 5 to 10 years, and it is much more common in patients who start dialysis when older than 50 years.199 Blood levels of β2-microglobulin are strikingly elevated in all patients with end-stage renal disease because of failure of normal renal clearance and catabolism. Clinical manifestations include (1) carpal tunnel syndrome; (2) destructive or erosive arthropathy involving the large and medium-sized joints, with shoulder, knee, hip, or back pain being common manifestations; (3) spondyloarthropathy, most commonly affecting the cervical spine; and (4) subchondral, thin-walled cysts of bone, most commonly affecting the carpal bones, the humoral and femoral heads, the distal end of the radius, the acetabulum, and the tibial plateau. These subchondral cysts at times are confused with brown tumors of secondary hyperparathyroidism, although their location and occurrence in multiple sites make them quite different from brown tumors. Nonetheless, in a patient who has undergone long-term dialysis, this syndrome may be a potential cause of neuromuscular and periarticular symptoms usually considered to be due to secondary hyperparathyroidism or aluminum accumulation. Parathyroid surgery should be avoided in dialysis patients whose severe musculoskeletal symptoms have not improved with calcitriol therapy; symptoms in these patients actually may be due to dialysis-related amyloidosis. Specific diagnosis of the latter is made from the biopsy demonstration of amyloid composed of β2-microglobulin; however, the diagnosis can be strongly suspected from the clinical features, the presence of multiple thin-walled cysts, or the demonstration in periarticular sites of presumed amyloid tissue on ultrasonography.200 Its management is difficult and largely unsatisfactory, but successful renal transplantation leads to rapid disappearance of symptoms and no further progression of the bone lesions on radiographs.201 In patients who remain on maintenance dialysis, the use of high-flux dialyzer membranes may decrease amyloid accumulation202; the long-term effects of this form of therapy on symptoms remain to be determined.
