Chapter 31 Chromosomes and Chromosomal Abnormalities
This chapter focuses on the approach to chromosomal disorders in pediatric neurology. The various methods of chromosomal analyses are considered first, followed by a description of the various types of chromosomal abnormalities. This discussion is followed by an overview of the clinical approach to chromosomal abnormalities, and then a brief clinical description of chromosomal syndromes relevant to the practice of pediatric neurology. The chapter closes with a look at the future of cytogenetic analysis (see also chapter 30).
Methods of Chromosome Analysis
Chromosome Banding
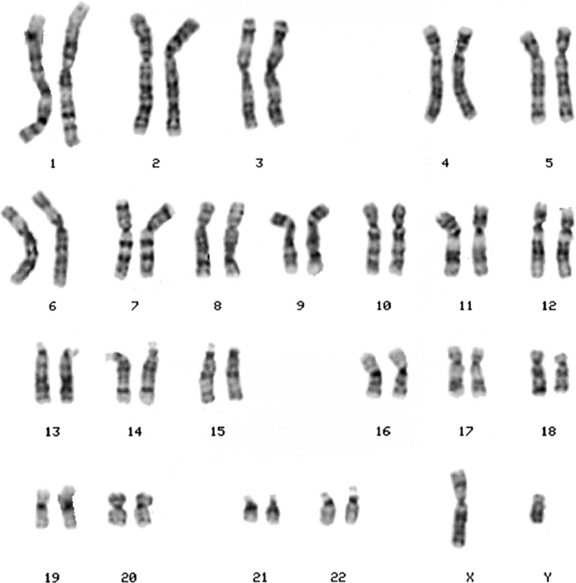
Chromosomes are displayed as a karyotype (Figure 31-1), which is prepared by arranging homologous chromosomes in an orderly fashion, starting from chromosome 1 and ending with chromosome 22, as well as the sex chromosomes. Subsequent developments in laboratory cytogenetics have gradually improved the resolution of chromosomal analysis. As the cell proceeds through mitosis, the chromosome gradually contracts, until anaphase, when the chromatids separate. If cells are collected during early prophase, chromosomes are highly extended, revealing a fine, highly detailed banding pattern. This banding pattern has facilitated recognition of subtle chromosome rearrangements involving small chromosome segments. Even with this approach, however, the resolution is limited to 3–5 million bp (Mb) of DNA, which may include dozens of genes.
Molecular Cytogenetics
The gap between light microscope resolution of chromosome structure and the gene was bridged by the introduction of several molecular cytogenetic techniques. Fluorescence in situ hybridization (FISH) involves hybridizing a fluorescently labeled single-stranded DNA probe to denatured chromosomal DNA on a microscope slide preparation of metaphase chromosomes and/or interphase nuclei prepared from the patient’s sample. After overnight hybridization, the slide is washed and counterstained with a nucleic acid dye (e.g., DAPI, or 4′,6-diamidino-2-phenylindole), allowing the region where hybridization has occurred to be visualized using a fluorescence microscope. FISH is now widely used for clinical diagnostic purposes. There are different types of FISH probes, including locus-specific probes, centromeric probes, and whole-chromosome paint probes. Locus-specific probes are specific for a particular single locus. They are especially useful for identifying subtle submicroscopic deletions and duplications (Figure 31-2). Centromeric probes are specific for unique repetitive DNA sequences (e.g., alpha-satellite sequences) in the centromere of a specific chromosome. They are suitable for making a rapid diagnosis of one of the common aneuploidy syndromes (trisomies 13, 18, and 21, and sex chromosome aneuploidies) using nondividing interphase nuclei. This is particularly useful in a prenatal setting using amniotic fluid or chorionic villi samples (CVS). Whole-chromosome paint probes consist of a cocktail of probes obtained from different regions of a particular chromosome. When this cocktail mixture is used in a single hybridization, the entire relevant chromosome fluoresces (is “painted”). Whole-chromosome paints are useful for characterizing complex chromosomal rearrangements, and for identifying the origin of additional chromosomal material, such as small marker or ring chromosomes.
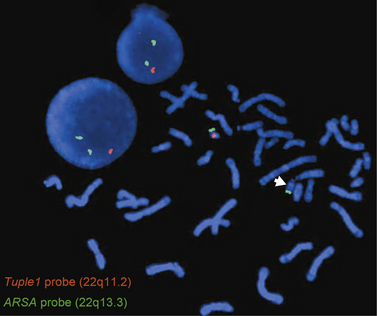
FISH using locus-specific probes has been extremely useful in the detection of “microdeletion syndromes” resulting from deletions of multiple contiguous genes. These are subtle submicroscopic deletions that are below the resolution of the routine G-banded chromosome analysis. Also, two-color and three-color FISH applications are routinely used to diagnose specific deletions, duplications, or other rearrangements, both in metaphase chromosomes and in interphase nuclei. Use of FISH usually requires that the patient either exhibits features consistent with a well-defined syndrome with known chromosomal etiology, or demonstrates an abnormal karyotype. This is because single FISH probes reveal rearrangements only of the segments being interrogated, but do not provide information about the rest of the genome. Another limitation of FISH is the number of probes that can be applied in a simultaneous assay. FISH techniques have been developed utilizing pools of whole-chromosome paint probes for every chromosome to provide a multicolor human karyotype in which each pair of homologous chromosomes can be identified on the basis of its unique color when studied using special computer-based image analysis software (spectral karyotyping and multicolor or M-FISH) [Liehr et al., 2004].
One type of FISH that has the potential to reveal chromosomal imbalances across the genome is comparative genomic hybridization (CGH). In CGH, DNA specimens from the patient and a normal control are differentially labeled with two different fluorescent dyes and hybridized to normal metaphase chromosome spreads. Difference between the fluorescent intensities of the two dyes along the length of any given chromosome will reveal gains and losses of genomic segments [Levy et al., 1998]. The limitations of this technology include many of the same limitations of G-banded chromosome analysis. Thus, like G bands, the resolution of CGH is limited to that of metaphase chromosomes, which is approximately 5 Mb for most clinical applications [Liehr et al., 2004].
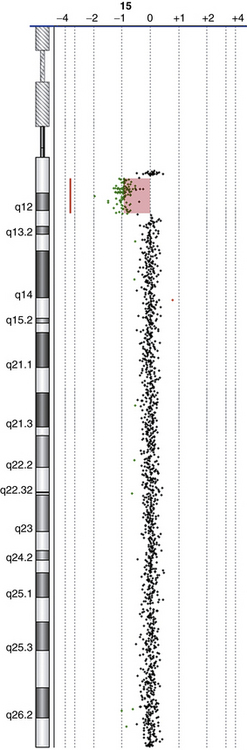
The latest addition to molecular cytogenetic techniques is array CGH, where CGH is applied to an array of DNA targets (probes), each representing a part of the human genome and fixed to a solid support (usually a glass slide). Like CGH, array CGH directly compares DNA content between two differentially labeled DNA specimens (a test or patient, and a reference or normal control), which are labeled and co-hybridized on to the array. Arrays have been constructed with a variety of DNA targets, ranging from bacterial artificial chromosomes (BACs), which are 80–250 thousand bp (kb) long [Shaffer and Bejjani, 2006] to oligonucleotides (oligos), which are 25–80 bp long [Lucito et al., 2003; Ylstra et al., 2006]. Following hybridization and washing to remove unbound DNA, the array is scanned and analyzed using special computer software to measure the relative ratios of fluorescence of the two dyes, and detect gains/losses of genomic regions represented on the array (Figure 31-3). The resolution of array CGH is dependent on the type of probes used (BACs or oligos) and the distance between them. In the past few years, high-resolution whole-genome coverage array CGH platforms have been increasingly used in clinical molecular cytogenetic labs. These provide a relatively quick method of scanning the entire genome for gains and/or losses with significantly higher resolution and greater clinical abnormality yield than was previously possible. This led to the identification of novel genomic disorders in patients with autistic spectrum disorders (ASD), developmental delay (DD), mental retardation (MR), and/or multiple congenital anomalies (MCAs) [Edelmann and Hirschhorn, 2009].
Chromosomal Abnormalities
Deletions and Duplications
A deletion involves loss of part of a chromosome and results in monosomy for that segment of the chromosome, whereas duplication represents the doubling of part of a chromosome, resulting in trisomy for that segment. The result is either decrease (in a deletion) or increase (in a duplication) in gene dosage. In general, duplications appear to be less harmful than deletions. Very large deletions usually are incompatible with survival to term. Deletions or duplications larger than approximately 5 Mb in size can be visualized under the microscope using G-banded chromosome analysis. Clinical syndromes resulting from submicroscopic deletions or duplications (i.e., microdeletions/microduplications) with a size <5 Mb have been identified with the help of molecular cytogenetic techniques, including FISH and array CGH. In these syndromes, groups of contiguous genes are either deleted or duplicated, resulting in a defined set of congenital anomalies. The molecular mechanisms responsible for these microdeletions/microduplications have been extensively studied and are well documented [Gu et al., 2008]. Specific microdeletion/microduplication syndromes of neurologic interest are described later in this chapter.
Translocations
Translocations involve the exchange of genetic material between chromosomes. In a balanced reciprocal translocation the exchange is equal, with no loss or gain of genetic material, though it is possible for a gene to be disrupted at one of the breakpoints. More often, the carrier of a balanced translocation is free of clinical signs or symptoms but is at risk for having offspring with unbalanced chromosomes. The risk for production of unbalanced gametes from a balanced translocation carrier depends on the chromosomes involved, the specific breakpoints of the translocation, and the sex of the carrier. Empirical data are available for some specific translocations [Daniel et al., 1989]. Risks include miscarriage and birth of a liveborn child with congenital anomalies, resulting from chromosome imbalance. The phenotype usually is a complex mixture of the results of loss or gain of at least two chromosome segments and therefore can be difficult to predict.
Marker and Ring Chromosomes
A “marker” chromosome is a rearranged chromosome whose genetic origin is unknown based on its G-banded chromosome morphology. Usually, these chromosomes are present in addition to the normal chromosome complement and are thus called supernumerary marker chromosomes (SMCs). The birth prevalence of SMCs is in the range of 2–7 per 10,000, and 30–50 percent originate from chromosome 15 [Gardner and Sutherland, 2004]. Two-thirds of de novo marker chromosomes can be associated with an abnormal outcome, whereas inherited ones can be passed from generation to generation without apparent clinical effects. Larger markers with more genetically active material are more likely to be of clinical significance. FISH and array CGH have proved very helpful in the precise identification of the genetic origin of SMCs.
Isochromosomes
An “isochromosome” is a chromosome in which one arm is missing and the other duplicated in a mirror-image fashion. The most probable mechanism for the formation of an isochromosome is the misdivision through the centromere in meiosis II, wherein the centromere divides transversely rather than longitudinally. The most commonly encountered isochromosome is that which consists of two long arms of the X chromosome. This accounts for approximately 15 percent of all cases of Turner’s syndrome [Gardner and Sutherland, 2004].
Cytogenetic Nomenclature
By convention, each chromosome arm is divided into regions, and each region is subdivided into bands and sub-bands, numbered from the centromere outwards. Cytogeneticists describe findings of chromosomal analysis using a standardized system of nomenclature (International System for Human Cytogenetic Nomenclature). Detailed description of this system is beyond the scope of this chapter, but major terms with examples are listed in Table 31-1. The normal male karyotype is designated 46,XY and the normal female karyotype is 46,XX. Any chromosomal abnormality is described after the sex chromosome constitution.
Incidence of Chromosomal Abnormalities
Estimates of the incidence of chromosomal abnormalities vary with the mode of ascertainment and the technology used for chromosome analysis. In general, the incidence falls rapidly from conception to birth. The highest rates have been observed among products of conception from first-trimester spontaneous abortions. Approximately 50 percent of these spontaneous miscarriages have a chromosomal abnormality [Boue et al., 1973]. By birth, the rate of chromosomal abnormalities declines to approximately 0.5–1 percent in liveborn infants, although the rate is much higher (5–10 percent) in stillborn infants [Jacobs et al., 1992].
Clinical Indications for Cytogenetic Analysis
Multiple Congenital Anomalies
Genetic imbalance resulting from a chromosomal abnormality usually leads to aberrant embryonic development. Most commonly, this abnormal development involves multiple tissues, including the brain. Many specific syndromes can be recognized from a constellation of dysmorphic physical features and specific congenital anomalies. The clinician should be familiar with the most common syndromes, especially those resulting from trisomies 13, 18, and 21, as well as the sex chromosome aneuploidies (47,XXX, 47,XXY, and 45,X). Phenotypes resulting from duplication or deletion of smaller amounts of genetic material can be more difficult to identify clinically. Some of the more important syndromes are described in the next section. Some clues to the occurrence of a chromosomal abnormality are provided in Box 31-1. As a rule, chromosomal studies should be performed in a patient who exhibits congenital anomalies involving two or more tissues, in whom a specific alternative diagnosis cannot be established, and if the anomalies are not related to one another as cause and effect (e.g., hydrocephalus resulting from spina bifida).










Developmental Delay and/or Mental Retardation
In some chromosomal abnormalities, the phenotype is primarily that of developmental delay (DD) and/or mental retardation (MR), with few or no congenital anomalies. Sometimes, minor dysmorphic features are present, but these often are not noticed on routine examination. G-banded chromosome analysis and array CGH testing therefore should be considered in the evaluation of a child with unexplained DD/MR. MR is a common condition that affects 1–3 percent of the population, and the cause is established in only 50 percent of the cases [Anderlid et al., 2002; Kriek et al., 2004] (also see chapter 43).
The use of array CGH to analyze the genomes of normal humans led to the discovery of extensive genomic copy number variations (CNVs), both gains and losses, ranging in size from kb to Mb, and not recognized by high-resolution G-banded chromosome analysis [Iafrate et al., 2004; Sebat et al., 2004]. CNVs have been proposed to be a major factor responsible for human diversity [Lupski, 2006]. Through genomic rearrangement of rearrangement-prone regions as a result of the genomic architecture, CNVs can cause genomic disorders due to gains and/or losses of dosage-sensitive gene(s), resulting in a clinical phenotype [Stankiewicz and Beaudet, 2007]. Using array CGH technologies, clinically significant pathogenic CNVs have been reported in up to 17 percent of patients with ASD, DD, MR, and/or MCAs [Stankiewicz and Beaudet, 2007; Edelmann and Hirschhorn, 2009].
Fertility Problems
Chromosomal imbalance most often leads to miscarriage rather than to live birth. Carriers of balanced rearrangements, including translocations or inversions, may therefore come to attention through recurrent miscarriage [Flint and Gibb, 1996; Hook and Cross, 1989]. It is recommended that couples who have experienced two or more unexplained first-trimester miscarriages be offered chromosomal analysis. Finding a balanced rearrangement permits genetic counseling of the couple, including offering prenatal diagnosis for future pregnancies. Other members of the family also may carry the balanced rearrangement and should be offered counseling and testing. Unexplained infertility should prompt a request for chromosome studies, especially for women presenting with primary amenorrhea, and for men presenting with azoospermia.
Prenatal diagnosis
Chromosomal analysis of a developing fetus can be achieved through collection of fetal cells by CVS, amniocentesis, or peripheral umbilical blood sampling (PUBS) [D’Alton and DeCherney, 1993]. CVS involves sampling part of the fetal placenta using a biopsy device either passed through the cervix or inserted by a needle through the mother’s abdomen [Pijpers et al., 1988; Smidt-Jensen and Hahnemann, 1988]. It is performed at 10–12 weeks of gestation. CVS offers the advantage of early testing. Amniocentesis involves sampling amniotic fluid at 16–18 weeks of gestation. Fetal cells in the fluid are cultured and can be used for chromosomal analysis. PUBS is offered after 20 weeks of gestation and involves sampling fetal blood by nicking the umbilical vein under ultrasound guidance [Sermon et al., 2004].
Indications for prenatal testing are listed in Box 31-2. General practice is to offer prenatal testing for pregnancies in which the risk of a chromosomal abnormality exceeds the risk of a complication of the procedure. For couples in which one partner carries a chromosome rearrangement, prenatal testing to detect unbalanced chromosomes can be offered. The actual risk of unbalanced chromosomes in the pregnancy depends on the nature of the rearrangement but generally is greater than 1 percent. The laboratory performing the prenatal testing must be informed of the details of the rearrangement, to ensure that subtle changes are detected. The recurrence risk for future trisomy for a couple who have had one pregnancy affected with trisomy is approximately 1 percent [Lister and Frota-Pessoa, 1980]. This risk is irrespective of the particular chromosome involved in the trisomy. Pregnancies are increasingly being monitored for fetal anomalies by ultrasound or maternal serum screening, with findings indicative of increased risk followed up by prenatal diagnostic testing.







Specific Cytogenetic Syndromes
Polyploidy
Clinical Features
The triploid phenotype is distinct and easily recognized. Polyhydramnios or pre-eclampsia may complicate the pregnancy. The placenta may be large, and hydatidiform changes may be seen. Birth weight usually is low. Syndactyly involving the third and fourth digits is characteristic. Craniofacial features include low-set, malformed ears, hypertelorism, and micrognathia. Cardiac, renal, and central nervous system malformations are common. Males may have dysplastic external genitalia. Studies of the parental origin of the three chromosome sets in triploidy have revealed that a majority of affected persons have two maternal sets, perhaps because of more frequent survival to term of triploid fetuses with two maternal sets of chromosomes (digynic triploids) [McFadden et al., 1993]. Long-term survivors often are mosaics and may have less obvious phenotypic features. Body asymmetry and pigmentary dysplasia may be clues to chromosomal mosaicism in general, including, in some cases, triploidy [Woods et al., 1994].
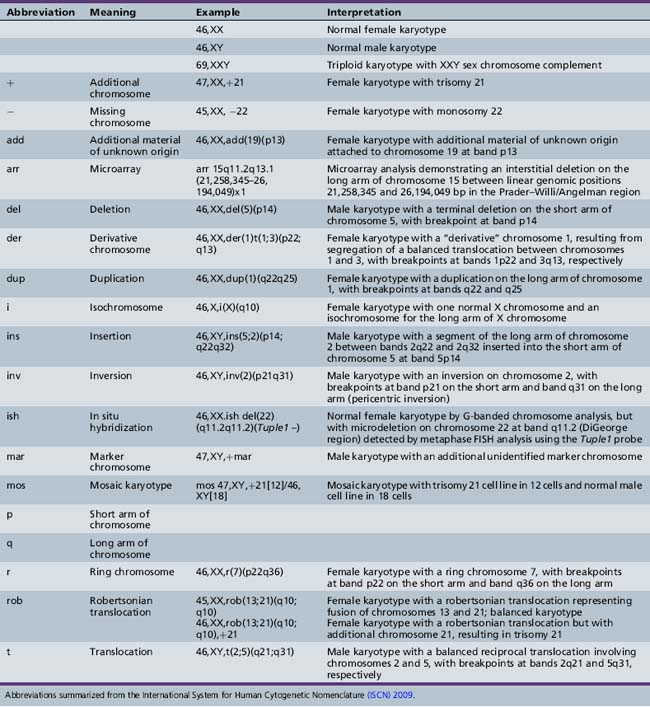
Trisomy 13 (Patau Syndrome)
Cytogenetics
Trisomy 13 occurs in approximately 1 in 7000 live births [Savva et al., 2010]. A majority of affected persons have 47 chromosomes, with an extra copy of chromosome 13. Approximately 5–10 percent have trisomy because of translocation between 13 and another acrocentric chromosome, usually chromosome 14 (robertsonian translocation). Mosaicism occurs in a small proportion of cases and may ameliorate the phenotype. Duplication of part of chromosome 13 resulting from unbalanced translocation can result in abnormal phenotypic features, although not necessarily similar to those seen in full trisomy 13. Advanced maternal age has been a factor in the occurrence of this aneuploidy syndrome.
Clinical Features
Trisomy 13 is associated with congenital anomalies involving most major organ systems (Figure 31-4). Holoprosencephaly is the hallmark central nervous system anomaly [Moerman et al., 1988], occurring in about 80 percent of cases. Infants with trisomy 13 who demonstrate holoprosencephaly usually have accompanying craniofacial anomalies. The eyes may be set closely together (hypotelorism) or even fused in a single orbit (cyclopia). Other ocular anomalies include micro-ophthalmia, iris colobomata, cataracts, and retinal dysplasia. Premaxillary agenesis and cleft lip or palate also may be present. Ulcer-like defects in scalp skin (cutis aplasia) occur commonly. Limb anomalies include postaxial polydactyly in two-thirds of patients and rocker-bottom foot. Congenital heart defects, especially ventricular septal defect (VSD), are common, as are renal anomalies, including cystic dysplasia. The phenotype overlaps to some degree with that of Meckel–Gruber syndrome (encephalocele, polydactyly, polycystic kidney), inherited as an autosomal-recessive trait due to mutation of the MKS1 gene. This overlap underlines the importance of confirming the clinical diagnosis of trisomy 13 by chromosomal analysis.
Management
Few infants with trisomy 13 survive the newborn period, with apnea being the most common cause of death [Rasmussen et al., 2003]. Often the anomalies are too numerous and severe to be corrected. In the absence of life-threatening malformations, however, long-term survival has been well documented, albeit usually with severely impaired cognitive function. Baty and co-workers documented the natural history of this disorder [Baty et al., 1994; Baty et al., 1994a & b].
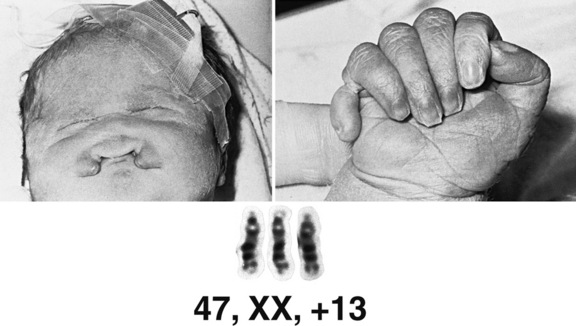
Trisomy 18 (Edwards’ Syndrome)
Clinical Features
Infants with trisomy 18 have low birth weight and microcephaly. Other common features include a prominent occiput, low-set “simple” ears, and a small mouth (Figure 31-5). Hands usually are tightly clenched in a characteristic configuration, with the fourth and fifth fingers overlapping the first and second. Terminal phalanges often are hypoplastic, and rocker-bottom foot may be present. Congenital heart defects and renal anomalies also are common. Brain malformations include heterotopias, agenesis of the corpus callosum, Dandy–Walker malformation, and Arnold–Chiari malformation. Infants commonly are jittery and hypertonic, and have apnea and seizures.
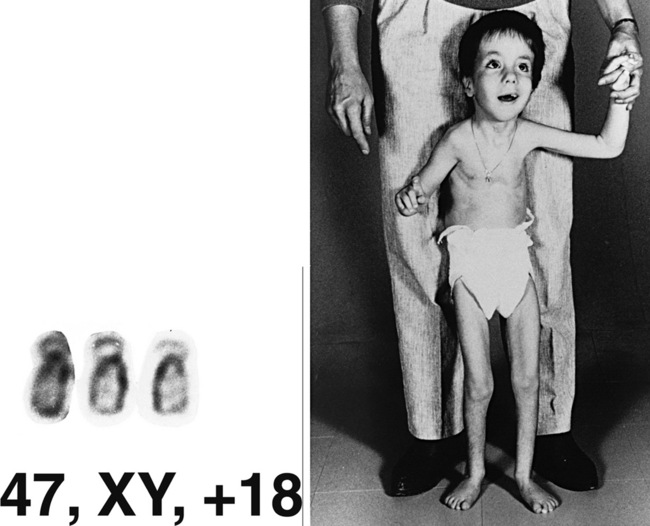
Trisomy 21 (Down Syndrome)
Clinical Features
Down syndrome consists of a set of characteristic physical features and developmental impairment (Figure 31-6). Short stature and brachycephaly are characteristic, and mild microcephaly may be noted. Down syndrome growth charts are available and should be used to monitor growth in affected children [Cronk et al., 1988]. Craniofacial features include upslanted palpebral fissures, epicanthal folds, flat facial profile, and small, low-set ears with narrow ear canals. White speckles (Brushfield spots) may be seen on the iris. A common finding is redundant folds of nuchal skin, which is one of the markers used for prenatal diagnosis by ultrasound examination [Gray and Crane, 1994]. Fingers are short, with incurving of the fifth finger (clinodactyly) and, often, a single transverse palmar crease. A wide space between the first and second toes is a frequent finding. The hallmark neurologic feature of Down syndrome is hypotonia. No gross central nervous system malformation is consistently seen, although lack of normal growth of the brain is typical. Microscopic analysis has revealed impaired myelination, reduced density of neurons, malformed dendritic trees and spines, defective lamination of the cortex, and abnormality of synaptic density [Wisniewski et al., 1990]. Impaired neurologic development is a universal feature [Silverman, 2007], but the degree of impairment varies widely. Children with Down syndrome benefit from early intervention [Fidler and Nadel, 2007; Feeley and Jones, 2008], physical therapy, and being reared in a family setting. An increased frequency of psychiatric problems, such as depression and behavioral problems, including hyperactivity, disruptive behaviors, and repetitive behaviors, have been documented [Visootsak Sherman, 2007; Dykens, 2007], as have sleep problems [Carter et al., 2009]. Linguistic ability may be impaired out of proportion to cognitive impairment. Seizures, including infantile spasms, may be seen with increased frequency [Smigielska-Kuzia et al., 2009]. An increased frequency of dementia, associated with pathologic changes of Alzheimer’s disease, has been described in patients with Down syndrome [Nieuwenhuis-Mark, 2009; Waldman et al., 2008; Deb et al., 2007]. Congenital anomalies commonly associated with Down syndrome include heart and gastrointestinal defects. The most typical heart defect is common atrioventricular (AV) canal, although other anomalies, such as VSD or tetralogy of Fallot, may be seen. Gastrointestinal malformations include duodenal atresia and Hirschsprung’s disease.
Management
The American Academy of Pediatrics has published guidelines for management of children with Down syndrome [American Academy of Pediatrics: Health supervision for children with Down syndrome, 2001]. Children with Down syndrome frequently require surgery for correction of congenital anomalies, such as heart defects or gastrointestinal malformations. They have a markedly increased risk of respiratory infection and sleep-related upper airway obstruction. often requiring antibiotic treatment. The frequency of leukemia is increased in children with Down syndrome. Transient leukemoid reactions also may occur.
Children with Down syndrome are at risk for atlantoaxial dislocation. Whether screening for dislocation should be offered for children with Down syndrome, particularly those who will participate in sports activities, has been the subject of controversy [Atlantoaxial instability in Down syndrome: subject review. American Academy of Pediatrics Committee on Sports Medicine and Fitness, 1995]. All children with Down syndrome should be monitored for neurologic signs of cervical cord compression. Parents of children with Down syndrome should be counseled regarding the natural history of the disorder, opportunities for intervention, and genetic recurrence risks, and should be provided emotional support. Life expectancy for persons with Down syndrome has improved with advances in surgery and medical treatment of complications of the disorder.
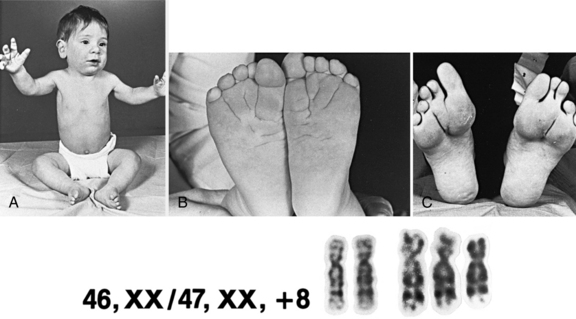
Trisomy 8 (Warkany’s Syndrome)
The only other autosomal trisomy compatible with live birth is trisomy 8. This trisomy is lethal in utero, except as a mosaic karyotype. Phenotypic features include hypertelorism; camptodactyly and other joint contractures; long, slender habitus; absence of patellae; and deep creases of the palms and soles (Figure 31-7). Asymmetric growth, presumably due to chromosomal mosaicism, also may be a feature.
Turner’s Syndrome
Cytogenetics
Turner’s syndrome is associated with a 45,X karyotype, with a single X chromosome. Mosaicism is not uncommon, however, with a separate cell line containing either a normal 46,XX or XY karyotype, or 46 chromosomes including a structurally rearranged X or Y [Crocker, 1992]. Turner’s syndrome occurs in about 1 in 4000 female live births worldwide but it is much more common in stillbirths and miscarriages. Unlike other aneuploidy syndromes, the frequency of Turner’s syndrome does not increase with advancing maternal age.
Clinical Features
Patients with Turner’s syndrome typically have a female phenotype, although those with a cell line including a Y chromosome may have some degree of virilization, often with ambiguous genitalia. At birth, infants may manifest pedal edema or diffuse edema (Figure 31-8). Facial features include small mandible, narrow maxilla, and epicanthal folds. In older children and adults with Turner’s syndrome, short stature and webbing of the neck are seen commonly. The thorax is broad, with increased distance between the nipples. Congenital anomalies include abnormalities of the lymphatic system, cardiac defects, especially coarctation of the aorta and bicuspid aortic valve, and renal anomalies, such as horseshoe kidney.
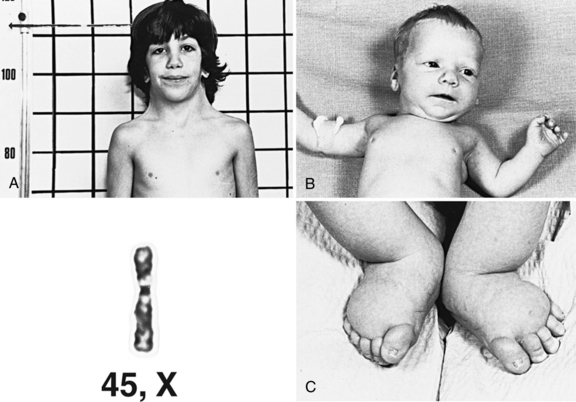
Fig. 31-8 A girl with Turner’s syndrome.
A, At 9 years of age. B, At 1 month of age. C, At birth.
(Courtesy of K Khoury; karyotype courtesy of M Rochon, Sherbrooke, Quebec, Canada.)
Although mental retardation is rare, delays in both gross and fine motor development are common in females with Turner’s syndrome [Nijhuis-van der Sanden et al., 2003]. Some patients display cognitive problems, but difficulties with visuospatial perception are most common [Pennington et al., 1982]. Hearing impairment occurs frequently and children should be monitored for deficits or progression of impairment.
Management
Recommendations for diagnosis and management of Turner’s syndrome have been published [Frias and Davenport, 2003; Health supervision for children with Turner syndrome. American Academy of Pediatrics. Committee on Genetics, 1995]. Newborns should be evaluated for renal and cardiac defects, and monitored if these are found. An increased risk for dissection of the aorta has been reported in affected adults [Elsheikh et al., 2002]. Thyroid autoimmunity may be a feature, so monitoring of thyroid function is recommended. Although not all children with Turner’s syndrome are growth hormone-deficient, significant growth hormone-induced improvement has been demonstrated in affected individuals.
Klinefelter’s Syndrome
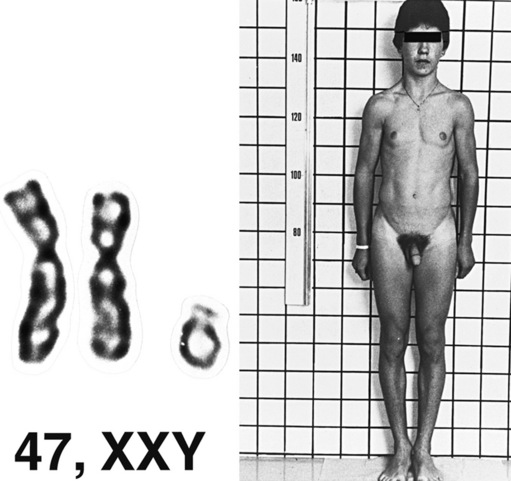
Clinical Features
The diagnosis of Klinefelter’s syndrome usually is not suspected at birth. Affected males tend to be tall, with long limbs (Figure 31-9). They display hypogonadism, and virilization may be incomplete at puberty; gynecomastia develops in some patients. Azoospermia and infertility are characteristic. Breast cancer is 20 times more common in Klinefelter’s syndrome than in the normal male population. As in Turner’s syndrome, mental retardation is not a typical feature of Klinefelter’s syndrome. Learning disabilities, language delay, and behavior problems are reported [Boada et al., 2009; Ross et al., 2009].
Other Sex Chromosome Aneuploidies
Two other major sex chromosome aneuploidies are 47,XXX and 47,XYY. The XXX aneuploidy is associated with a female phenotype and tall stature; usually other major physical stigmata are absent. XYY is associated with a male phenotype and tall stature but no other physical features. Learning disabilities and neuromotor impairment occur commonly in 47,XXX females [Bender et al., 2001]. The behavioral phenotype of XYY syndrome has been a source of some controversy because of reports associating the karyotype with criminal behavior. The frequency of learning disabilities and behavioral problems is increased among affected males, although a wide range of cognitive outcomes have been reported [Ross et al., 2009].
Structural Abnormalities
To date, most genomic imbalances have been classified as either benign or pathogenic, and most microdeletion/microduplication syndromes are presumed to be well-defined clinical conditions. Well-known genomic disorders can be phenotypically heterogeneous and variable, however, owing to incomplete penetrance or variable expression. Clinical variability could also be explained in part by other genetic or environmental determinants, modifying factors of other genes, multigenic inheritance, imprinting, and unmasking of recessive genes. Array CGH and genomic copy-number analysis using single-nucleotide polymorphism (SNP) genotyping arrays are proving particularly effective for the investigation of patients with developmental delay, learning disabilities, mental retardation, dysmorphic features, and/or multiple congenital anomalies, and are identifying the probable underlying cause of the disease phenotype in approximately 17 percent of previously undiagnosed cases [Stankiewicz and Beaudet, 2007; Edelmann and Hirschhorn, 2009]. Moreover, a genomic basis to several late-onset disorders, e.g., early-onset Alzheimer’s disease with amyloid angiopathy (EOAD), and adult-onset autosomal-dominant leukodystrophy, has now been defined [Firth et al., 2009]. Table 31-2 lists the microdeletion/microduplication syndromes reported to date, as shown in the DECIPHER database (https://decipher.sanger.ac.uk/application/). In the following section, some of these syndromes that are clinically relevant to the practice of pediatric neurology are discussed.
Table 31-2 Microdeletion/Microduplication Syndromes Listed in the DECIPHER Database
| Syndrome | OMIM Number |
|---|---|
| 12q14 microdeletion syndrome | 166700 |
| 15q13.3 microdeletion syndrome | 612001 |
| 15q24 recurrent microdeletion syndrome | – |
| 15q26 overgrowth syndrome | – |
| 16p11.2 autism susceptibility locus | 611913 |
| 16p11.2–p12.2 microdeletion syndrome | – |
| 16p13.11 recurrent microdeletion (MR/MCA susceptibility locus) | – |
| 16p13.11 recurrent microduplication (uncertain significance) | – |
| 17q21.3 recurrent microdeletion syndrome | 610443 |
| 1p36 microdeletion syndrome | 607872 |
| 1q21.1 recurrent microdeletion (susceptibility locus for neurodevelopmental disorders) | 612474 |
| 1q21.1 recurrent microduplication (possible susceptibility locus for neurodevelopmental disorders) | 612475 |
| 1q21.1 susceptibility locus for thrombocytopenia-absent radius (TAR) syndrome | 274000 |
| 22q11.2 deletion syndrome (velocardiofacial/DiGeorge’s syndrome) | 192430, 188400 |
| 22q11.2 duplication syndrome | 608363 |
| 22q11.2 distal deletion syndrome | 611867 |
| 22q13 deletion syndrome (Phelan–McDermid syndrome) | 606232 |
| 2p15-16.1 microdeletion syndrome | 612513 |
| 2q33.1 deletion syndrome | – |
| 2q37 monosomy | 600430 |
| 3q29 microdeletion syndrome | 609425 |
| 3q29 microduplication syndrome | 611936 |
| 6p deletion syndrome | 612582 |
| 7q11.23 duplication syndrome | 609757 |
| 8p23.1 deletion syndrome | 222400 |
| 9q subtelomeric deletion syndrome | 610253 |
| ATR-16 syndrome | 141750 |
| AZF a, b, and c | 415000 |
| Adult-onset autosomal-dominant leukodystrophy (ADLD) | 169500 |
| Angelman’s syndrome (types 1 and 2) | 105830 |
| Cat-eye syndrome (type I) | 115470 |
| Charcot–Marie–Tooth syndrome type 1A (CMT1A) | 118220 |
| Cri du chat syndrome (5p deletion) | 123450 |
| Early-onset Alzheimer’s disease with cerebral amyloid angiopathy | 605714 |
| Familial adenomatous polyposis | 175100 |
| Hereditary liability to pressure palsies (HNPP) | 162500 |
| Leri–Weill dyschondrostosis (LWD) – SHOX deletion | 127300 |
| Miller–Dieker syndrome (MDS) | 247200 |
| NF1-microdeletion syndrome | 162200 |
| Pelizaeus–Merzbacher disease | 312080 |
| Potocki–Lupski syndrome (17p11.2 duplication syndrome) | 610883 |
| Potocki–Shaffer syndrome | 601224 |
| Prader–Willi syndrome (types 1 and 2) | 176270 |
| RCAD (renal cysts and diabetes) | 137920 |
| Rubinstein–Taybi syndrome | 180849 |
| Smith–Magenis syndrome | 182290 |
| Sotos’ syndrome | 117550 |
| Split hand/foot malformation 1 (SHFM1) | 183600 |
| Steroid sulphatase deficiency (STS) | 300747 |
| WAGR 11p13 deletion syndrome | 194072 |
| Williams–Beuren syndrome (WBS) | 194050 |
| Wolf–Hirschhorn syndrome | 194190 |
| Xq28 (MECP2) duplication | 300260 |
ATR, alpha thalassemia/mental retardation syndrome; AZF, azoospermic factors; MCA, multiple congenital anomalies; MR, mental retardation; WAGR, Wilms tumor, aniridia, genitourinary dysplasia, and mental retardation.
22q11.2 Deletion Syndrome
The 22q11.2 deletion syndrome includes the phenotypes previously called DiGeorge’s syndrome (DGS), velocardiofacial syndrome (VCFS, Shprintzen’s syndrome), conotruncal anomaly face syndrome, many cases of autosomal-dominant Opitz G/BBB syndrome, and Cayler’s cardiofacial syndrome (asymmetric crying facies) [McDonald-McGinn et al., 1997]. The condition is clinically heterogeneous. Congenital heart defects are present in most affected individuals (74 percent), particularly conotruncal malformation. Additional findings include palatal abnormalities and velopharyngeal incompetence (VPI), learning disabilities, immune deficiencies, hypocalcemia, and characteristic facies. Also reported are hearing loss, seizures without hypocalcemia, speech delays, and behavioral difficulties. The 22q11.2 deletion affects an estimated 1:2000 to 1:4000 live births. The deletion occurs in 2 percent of patients with isolated conotruncal heart defects and in 5–8 percent of individuals with isolated cleft palate. Most cases are de novo and approximately 6 percent are familial. The inheritance is autosomal-dominant. Some cases with deletion 10p13p14 have a deletion 22q11.2-like phenotype. The phenotype of the reciprocal microduplication of the 22q11.2 region is mild and highly variable, with familial transmission frequently observed.
Prader–Willi and Angelman’s Syndromes
The recognition of the phenomenon of genomic imprinting has led to the discovery of a new class of genetic disorders associated with aberrations of imprinted genes. The prototype disorders are Prader–Willi and Angelman’s syndromes (Figure 31-10 and Figure 31-11) [Gurrieri and Accadia, 2009]. The features of these syndromes are described in Table 31-3. Prader–Willi syndrome (PWS) is characterized by hypotonia and feeding difficulties in early life, hyperphagia and obesity later, short stature, hypogonadism, and acromicria. Behavior problems are common and psychomotor development is mildy affected. PWS affects 1:5000 to 1:10,000 individuals. Approximately 70–75 percent of the individuals with PWS have a deletion of the paternally contributed 15q11.2q13.1 region, while in Angelman’s syndrome (AS), 70 percent of affected individuals have a deletion of the maternally contributed region. The AS phenotype is completely different and it is characterized by severe mental retardation, absent speech, autistic behavior, unique behavior (inappropriate happy demeanor), gait ataxia, epilepsy, electroencephalogram (EEG) abnormalities (2–3-Hz, large-amplitude slow-wave bursts), microcephaly, and dysmorphic features. The EEG may be abnormal, even in the absence of seizures, however; a normal EEG and/or absence of seizures do not exclude the diagnosis. Approximately 1:40,000 children are affected with AS. Most patients with PWS who do not have the 15q11.2q13.1 deletions have uniparental disomy for chromosome 15, with two maternal copies and no paternal copies. Either mechanism – deletion or uniparental disomy – leads to deficiency of a gene or genes on chromosome 15 that are expressed in the paternal but not the maternal homolog. Paternal uniparental disomy accounts for a low percentage of cases of AS. Mutations in the UBE3A gene (a ubiquitin ligase gene involved in early brain development), located at 15q11.2, have been found in some patients with AS [Kishino et al., 1997]. This gene is imprinted in the brain [Albrecht et al., 1997; Rougeulle et al., 1997], and is the gene responsible for the AS phenotype. Deletion of the paternally expressed HBII-85 C/D box small nucleolar RNA cluster has been implicated in PWS [Sahoo et al., 2008]. A small proportion of patients with PW or AS may have a small deletion or other mutation that leads to aberrant imprinting of the region [Buiting et al., 1995]. These findings have led to major advances in genetic diagnosis of PW and AS. Chromosome 15 deletions usually are submicroscopic but are easily detected by FISH and/or array CGH. Defects in imprinting or uniparental disomy can be identified by studies of patterns of DNA methylation in the region. One particular cloned segment of DNA is methylated in the maternal, but not the paternal, genome. Failure to identify the methylated or nonmethylated copy of the sequence is indicative of deletion, uniparental disomy, or a mutation that alters the imprinting mechanism.
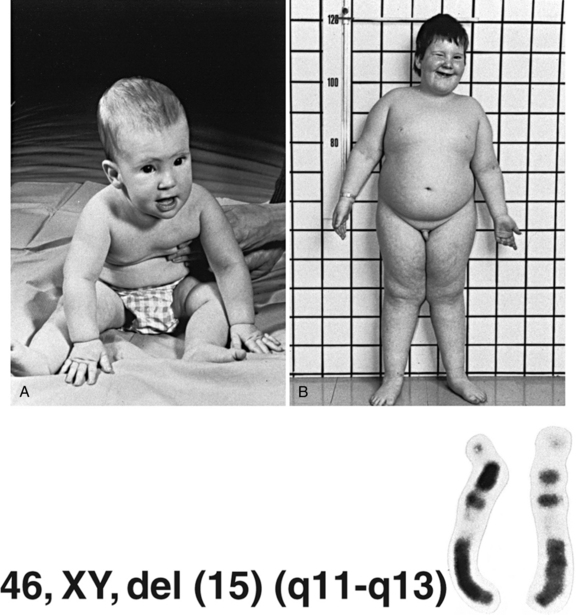
Fig. 31-10 A boy with Prader–Willi syndrome.
A, At 7 months of age. B, At 6 years of age.
(Karyotype courtesy of MG Mattei, Marseilles, France.)
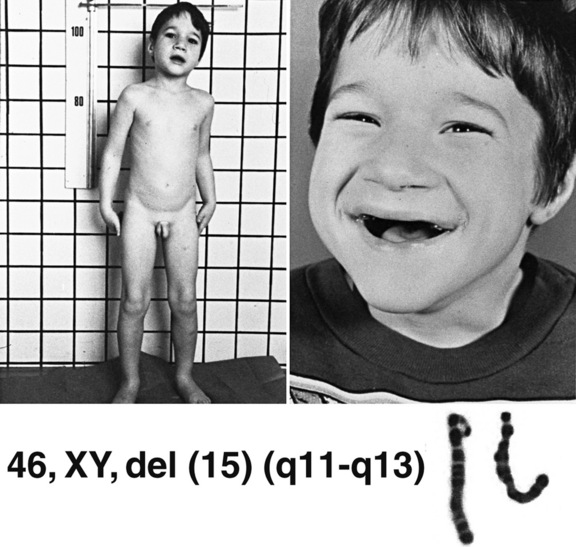
Fig. 31-11 A boy with Angelman’s syndrome at 6 years of age.
(Karyotype courtesy of M Rochon, Sherbrooke, Quebec, Canada.)
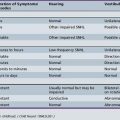
Table 31-3 Comparison of Features of Prader–Willi and Angelman’s Syndromes
| Prader–Willi Syndrome* | Angelman’s Syndrome† | |
|---|---|---|
| Diagnostic criteria |
EEG, electroencephalogram.
* Data from Holm V et al. 1993. Prader–Willi syndrome: consensus diagnostic criteria. Pediatrics 1993;91(8424017):398–402.
† Data from Williams CA et al. Angelman syndrome: consensus for diagnostic criteria. Angelman Syndrome Foundation. Am J Med Genet 1995;56(2):237–238.
(Mutation analysis data from Buiting K et al. Epimutations in Prader-Willi and Angelman syndromes: a molecular study of 136 patients with an imprinting defect. Am J Hum Genet 2003;72[12545427]:571–577; Jiang Y et al. Genetics of Angelman syndrome. Am J Hum Genet 1999;65[10364509]:1–6.)
Imprinted genes are expressed only from one of the two parental alleles (gene pair). In mammals, genomic imprinting is an event in which particular genes are expressed differentially, depending on the parent of origin. Genomic imprints are reversible and lead to differential expression in the course of development. Genomic imprinting is an epigenetic process that involves methylation and histone modifications in order to achieve monoallelic gene expression without altering the genetic sequence. Genome-wide research for imprinted genes in the human genome has identified over 150 candidate imprinted genes [Luedi et al., 2007]. Other examples of human disorders of genomic imprinting include Silver–Russell syndrome, Beckwith–Wiedemann syndrome, Albright hereditary osteodystrophy, and uniparental disomy 14 (maternal and paternal forms).
William–Beuren Syndrome
The William–Beuren syndrome (WBS) is a microdeletion syndrome of chromosome 7 at band q11.23, and occurs in 1:10,000 live births [Tassabehji, 2003]. Cardiovascular disease is present in 80 percent of affected individuals, mostly in the form of supravalvular aortic stenosis (SVAS), peripheral pulmonary stenosis, elastin arteriopathy, and hypertension. The 7q11.23 microdeletion encompasses the elastin gene (ELN), which is also mutated in isolated SVAS. Characteristic facial features include periorbital fullness, long philtrum, wide mouth, full lips, full cheeks, and small, wide-spaced teeth. Affected individuals have mild to moderate MR, specific cognitive profile/learning disabilities, and unique or distinctive behavior/personality characteristics. Growth and endocrine abnormalities (hypercalcemia, hypothyroidism, hypercalciuria), and feeding difficulties in infancy are also common.
1p36 Deletion Syndrome
1p36 deletion syndrome is considered to be the most common subtelomeric microdeletion syndrome with an estimated incidence of 1 in 5000 to 1 in 10,000. It accounts for 0.5–1.2 percent of idiopathic MR [Battaglia et al., 2008]. Clinical findings include a characteristic craniofacial appearance: microbrachycephaly, large and late-closing anterior fontanel, straight eyebrows, deep-set eyes, epicanthic folds, broad nasal bridge, midface hypoplasia, abnormally formed low-set ears, and limb and skeletal defects. DD and MR with absent/poor expressive language are constant features. Affected individuals often face serious physical disabilities that include congenital heart defects (70 percent), cardiomyopathy (25 percent), brain abnormalities (88 percent), seizures (44 percent), and EEG abnormalities (100 percent). Ocular malformations or vision problems and hearing loss are observed approximately 50 percent of affected individuals.
Wolf–Hirschhorn Syndrome
Wolf–Hirschhorn syndrome (WHS) results from a variable-sized deletion on the terminal end of the short arm of chromosome 4 [Bahi-Buisson et al., 2005]. It is characterized by distinctive facial appearance, growth delay, psychomotor retardation, and seizures, and is confirmed by detection of a deletion of the Wolf–Hirschhorn critical region (WHCR) (chromosome 4p16.3). The syndrome has clinical and cytogenetic variability. In some, the deletion is visible by G-banded chromosome analysis; in others, it is cryptic, and molecular cytogenetic analysis is required to make the diagnosis. Characteristic facial features include the “Greek warrior helmet” appearance of the nose (the broad bridge of the nose continuing to the forehead), microcephaly, high forehead with prominent glabella, ocular hypertelorism, epicanthus, highly arched eyebrows, short philtrum, downturned mouth, micrognathia, and poorly formed ears with pits/tags. MR ranges from mild to severe. Other birth defects have been reported in individuals with WHS. One-third of the patients have structural central nervous system defects, and seizures occur in 50–100 percent of affected children. In 75 percent of patients with WHS, the deletion is de novo, and in about 13 percent of patients the deletion results from the unbalanced segregation of a parental balanced translocation. It is now recognized that WHS and Pitt–Rogers–Danks syndrome (PRDS) represent the clinical spectrum associated with a single syndrome [Moretti et al., 2001].
Cri du Chat Syndrome
Cri du chat syndrome is a genetic syndrome resulting from a variable-sized deletion on the terminal end of the short arm of chromosome 5. The first description was made by Lejeune et al., in 1964 [Lejeune et al., 1964]. The incidence ranges from 1:15,000 to 1:50,000. A high-pitched, cat-like cry is among the main clinical features; hence the name of the syndrome. Other frequently described features are microcephaly, broad nasal bridge, epicanthic folds, micrognathia, impaired growth, and severe psychomotor and mental retardation. The syndrome has significant clinical and cytogenetic variability. Clinical analysis of affected individuals and detailed molecular cytogenetic analysis suggest the existence of two critical regions, one on 5p15.2 for facial dysmorphism, microcephaly, and MR, and another on 5p15.3 for the typical cry [Zhang et al., 2005]. Eighty percent of affected individuals are the result of a de novo deletion, and 10 percent are the result of the unbalanced segregation of a parental balanced translocation.
Deletions Involving Distal 6p
Deletions involving distal 6p are relatively rare. These deletions can be divided into two groups: interstitial with breakpoints within the 6p22p24 region, and terminal deletions with breakpoints within the 6p24pter region. The terminal deletion of 6p results in a distinct phenotype, including hypertelorism, downslanting palpebral fissures, flat nasal bridge, Dandy–Walker malformation, congenital heart defect, anterior eye chamber abnormalities, hearing loss, and DD [Lin et al., 2005]. Hydrocephalus has been seen in patients with terminal 6p deletions but the majority has been associated with Dandy–Walker malformation [Murray et al., 1985].
Chromosome 9q Subtelomeric Deletion
The chromosome 9q subtelomeric deletion represents one of the most common subtelomeric deletions (6 percent) [Ravnan et al., 2006]. The syndrome can be caused either by a submicroscopic 9q34.3 deletion, or by mutations in the EHMT1 gene, which is involved in histone methylation. Affected individuals invariably have severe hypotonia, with speech and gross motor delay. Facial features include micro-/brachycephaly, hypertelorism, synophrys, arched eyebrows, midface hypoplasia, short nose with upturned nares, protruding tongue, everted lower lip, and downturned corners of the mouth. Congenital heart defects have been reported in approximately 50 percent of affected individuals. Epilepsy and behavior and sleep disturbances have also been reported in some (10–20 percent) [Kleefstra et al., 2009].
Langer–Giedion Syndrome
Patients with the Langer–Giedion syndrome (LGS), also called tricho-rhino-phalangeal syndrome type II (TRPSII), have a de novo deletion on chromosome 8 at band q24.11, comprising a contiguous deletion of the TPRS and EXT1 genes [Shanske et al., 2008]. The syndrome is characterized by short stature, sparse scalp hair, long nose with a bulbous tip, notched alae nasi, long, flat philtrum, thin lips, cone-shaped epiphyses, and multiple cartilaginous exostoses.
WAGR 11p13 Deletion Syndrome
The cardinal features of the WAGR 11p13 deletion syndrome include Wilms tumor, aniridia, genitourinary abnormalities, and growth and mental retardation. The size of the deletion varies but always includes WT1 and PAX6 genes, which accounts for the oncogenic, ocular, and genitourinary features, respectively. Approximately 30 percent of infants with sporadic aniridia will be positive for the characteristic deletion [Robinson et al., 2008]. The majority of patients have MR, and more than 20 percent of patients also have features of autism [Xu et al., 2008].
Jacobsen’s Syndrome
Jacobsen’s syndrome is a contiguous gene deletion syndrome caused by partial deletion of the long arm of chromosome 11 (11q23.3qter). Typical features include DD, MR, short stature, congenital heart defects, thrombocytopenia, and characteristic dysmorphic facial features. Malformation of heart, kidney, gastrointestinal tract, central nervous system, and skeleton is common. Some of the facial dysmorphism described includes skull deformities, hypertelorism, epicanthic folds, ptosis, broad nasal bridge, and small ears. The deletion is de novo in 85 percent of cases, and in the remaining patients it results from the unbalanced segregation of a parental balanced chromosome rearrangement [Mattina et al., 2009].
Charcot–Marie–Tooth Neuropathy Type 1A and Hereditary Neuropathy with Liability to Pressure Palsies
Charcot–Marie–Tooth neuropathy type 1A (CMT1A) represents 70–80 percent of all CMT1 and results from an approximately 1.5-Mb duplication at 17p11.2, which encompasses the PMP22 gene (peripheral myelin protein 22) [Szigeti et al., 2006]. This protein is predominantly produced by Schwann cells and is a major component of the peripheral nervous system. Reciprocal deletion of the same region results in the milder phenotype of hereditary neuropathy with liability to pressure palsies (HNPP). The duplication is inherited in around two-thirds of individuals and is de novo in the remaining third. Please see Chapter 89 for more details.
Smith–Magenis Syndrome and Potocki–Lupski Syndrome
Approximately 90 percent of individuals with the Smith–Magenis syndrome (SMS) have a deletion on chromosome 17 at band p11.2 that encompasses the RAI1 gene. The remaining 5–10 percent of cases carry a mutation in the RAI1 gene. Physical features include short stature, obesity, craniofacial dysmorphism, and small hands and feet. Behavior disturbances, especially sleep problems and self-injurious behavior, are frequently reported. The self-injurious behavior includes self-hitting, self-biting and/or skin picking, inserting foreign objects in body orifices (polyembolokoilamania), and pulling nails (onchotillomania). Self-hug or spasmodic upper-body squeeze, hand licking, and page flipping (“lick and flip”) constitute stereotypic behavior that appears to be specific for SMS. All the affected individuals have mild to severe learning disabilities [Elsea and Girirajan, 2008]. The phenotypic features may be subtle in infancy and early childhood. The facial appearance is characterized by a broad, square-shaped face, brachycephaly, prominent forehead, synophrys, mildly upslanting palpebral fissures, deep-set eyes, broad nasal bridge, midfacial hypoplasia, short, full-tipped nose, flat nasal bridge, micrognathia in infancy changing to relative prognathia with age, and fleshy, everted upper lip. With progressing age, the facial appearance becomes more distinctive and coarse. The reciprocal duplication of this 17p11.2 region has been reported (Potocki–Lupski syndrome). The most frequent features of this syndrome are hypotonia in infancy, DD, language and cognitive impairment, autistic features, poor feeding and failure to thrive in infancy, oral-pharyngeal dysphagia, obstructive and central sleep apnea, structural cardiovascular abnormalities, EEG abnormalities, and hypermetropia. Most have short stature and mild to normal facies [Potocki et al., 2007]. Variability in the phenotype is observed. It is expected that persons with large duplications that encompass the more distal CMT1A region will have a more severe phenotype, including peripheral neuropathy.
Miller–Dieker Syndrome
Miller–Dieker syndrome represents a microdeletion syndrome spanning the LIS1 gene at 17p13.3, which results in severe lissencephaly with characteristic facial changes, other more variable malformations, and severe neurologic and developmental abnormalities. The facial features consist of high and prominent forehead, bitemporal hollowing, short nose with upturned nares, protuberant upper lip with downturned vermillion border, and small jaw [Cardoso et al., 2002]. The reciprocal duplication results in DD, hypotonia, and facial dysmorphism. In contrast to the patients with the deletion, those with the duplication have neither gross brain malformations nor lissencephaly [Roos et al., 2009].
Neurofibromatosis Type 1
Approximately 5 percent of patients with neurofibromatosis type 1 (NF1) have deletions of the entire NF1 gene and contiguous genes at 17q11.2, resulting in the NF1 microdeletion syndrome. NF1 with large deletions is more likely to have dysmorphic features, cardiac anomalies, connective tissue dysplasia, and MR [Mensink et al., 2006]. Patients with reciprocal microduplications have been reported.
Alagille’s Syndrome
Alagille’s syndrome (AGS) is a complex multisystem disorder involving primarily the liver, heart, eyes, face, and skeleton. The clinical features are highly variable, even within families. The major clinical manifestations of AGS are cholestasis, congenital cardiac defects, posterior embryotoxon in the eye, typical facial features, and butterfly vertebrae. Renal and central nervous abnormalities also occur. The two genes associated with AGS are JAG1 and NOTCH2 [Krantz et al., 1999; McDaniell et al., 2006]. Sequence analysis of JAG1 detects mutations in over 88 percent of individuals who meet clinical diagnostic criteria, whereas FISH and/or array CGH analyses detect a microdeletion encompassing JAG1 at 20p12.2 in approximately 7 percent of affected individuals.
Potocki–Shaffer Syndrome
Potocki–Shaffer syndrome (PSS) is a contiguous gene deletion syndrome that results from a deletion of the 11p11.2p12 region [Wu et al., 2000]. The clinical features can include DD, MR, multiple exostoses, parietal foramina, enlarged anterior fontanel, minor craniofacial anomalies, ophthalmologic anomalies, and genital anomalies in males. Parietal foramina and multiple exostoses are the primary characteristic of this syndrome. Larger deletions of proximal 11p may result in features of PSS and WAGR.
X-Linked Ichthyosis Due to Steroid Sulphatase Enzyme Deficiency
Males with X-linked ichthyosis due to steroid sulphatase enzyme deficiency have generalized scaling that usually starts shortly after birth. In 90 percent of cases, it is caused by a microdeletion encompassing the STS gene (Xp22.31). In 5 percent of cases, the deletion is extensive enough to involve adjacent genes, resulting in learning disabilities, autism, and epilepsy in some of the affected boys [Gohlke et al., 2000].
Loss of Function of the MECP2 Gene/Duplication of the MECP2 Region (Xq28)
Loss of function of the MECP2 gene results in Rett’s syndrome in females, and both syndromic and nonsyndromic forms of MR in males. Duplication of the MECP2 region (Xq28) in males is associated with severe MR and progressive spasticity. This duplication spans the L1CAM and MECP2 genes [Van Esch et al., 2005].
Autistic Spectrum Disorders
About 5–10 percent of patients with ASDs are associated with chromosomal abnormalities or monogenic disorders [Folstein and Rosen-Sheidley, 2001; Jacquemont et al., 2006]. They are also the most frequently identified cause of DD and/or MR, and are often seen in conjunction with growth retardation, dysmorphic features, and various congenital anomalies [Aradhya et al., 2007]. According to Fernandez et al., ASDs are etiologically heterogeneous [Fernandez et al., 2009]. About 10 percent are associated with a mendelian syndrome. Another 5–7 percent are associated with the maternal 15q11.2q13.1 duplication. Teratogens have also been implicated. The remainder of affected individuals are presumed to have multifactorial forms of ASD. More recently, de novo CNVs have been observed in 7–10 percent of sporadic ASD, and in 2–3 percent of affected individuals from multiplex families. The most frequent are 15q11.2q13.1 duplication, and 2q37 and 22q13.3 deletions [Folstein and Rosen-Sheidley, 2001]. The following are some of the recently reported genomic disorders described as clinically significant CNVs sometimes associated with ASDs.
Maternal duplication of the 15q11.2q13.1 region
Chromosome 15q11.2q13.1 contains a cluster of imprinted genes essential for normal development. Deficiencies in paternal or maternal 15q11.2q13.1 result in PWS or AS, respectively, as previously discussed. Maternal duplication of the 15q11.2q13.1 region leads to a distinct condition that often includes autism. Despite incomplete penetrance of autism in this syndrome, this duplication is the leading cytogenetic cause of autism [Hogart et al., 2009]. Recently, the 15q11.2q13.1 duplication has been associated with a distinct pattern of mitochondrial abnormalities that include a deficiency of complex III [Frye, 2009].
15q13.3 microdeletion
Manifestations of the 15q13.3 microdeletion include idiopathic generalized epilepsy, autism, intellectual disability, and schizophrenia. The condition has highly variable intra- and interfamilial phenotypic heterogeneity, with some carriers having no clinical manifestations [Mulley and Dibbens, 2009]. Cardiac defects have also been reported. Patients with the reciprocal duplication do not share a recognizable phenotype [van Bon et al., 2009].
15q24 microdeletion syndrome
The 15q24 microdeletion syndrome has a distinct clinical phenotype with specific facial features, DD, microcephaly, and digital and genital anomalies. Facial features include hypertelorism, strabismus, downslanted palpebral fissures, epicanthic folds, full lower lip, high frontal hairline, broad medial eyebrows, long/smooth philtrum, and palate and ear anomalies. Umbilical and inguinal hernias, genital anomalies in males, and musculoskeletal anomalies have also been reported [Klopocki et al., 2008].
Recurrent 16p11.2 microdeletions and microduplications
Recurrent 16p11.2 microdeletions and microduplications appear to be associated with approximately 1 percent of unexplained, idiopathic, and nonsyndromic autism [Weiss et al., 2008]. The phenotypic spectrum probably also includes MR, DD, variable nonspecific dysmorphism, and/or possibly other primary psychiatric disorders.
16p11.2p12.2 microdeletion
The 16p11.2p12.2 microdeletion should be distinguished from the 16p11.2 microdeletion. All reported patients with the 16p11.2p12.2 microdeletion have shared a common distal breakpoint at 16p12.2, but the proximal breakpoint varies [Ballif et al., 2007; Hempel et al., 2009]. Common features of this microdeletion syndrome include facial dysmorphism, orofacial clefting, heart defects, frequent ear infections, short stature, feeding difficulties, hypotonia, and DD. The facial features include flat face, deep and low-set eyes, and posteriorly rotated ears.
Deletions and reciprocal duplications of the chromosome 16p13.11 region
Deletions and reciprocal duplications of the chromosome 16p13.11 region have recently been reported in several cases of autism and MR. Current clinical data indicate that deletions that are either de novo or inherited from clinically normal parents are likely to be pathogenic, whereas the clinical significance of the duplications is still uncertain [Hannes et al., 2009].
22q13 microdeletion syndrome (phelan–mcdermid syndrome)
The 22q13 microdeletion syndrome (Phelan–McDermid syndrome) is a chromosome microdeletion characterized by neonatal hypotonia, global DD, normal to accelerated growth, absent to severely delayed speech, and dysmorphic features [Phelan, 2008]. Reported features include dolicocephaly, long eyelashes, large or unusual ears, large hands, dysplastic toe nails, full cheeks, bulbous nose, and pointed chin. Behavior tends to be autistic-like.
17q21.31 microdeletion syndrome
17q21.31 is a new microdeletion syndrome with recognizable clinical phenotype of MR, hypotonia, and characteristic facies, including long hypotonic face, tubular nose with bulbous nasal tip, large low-set ears, and blepharophimosis. The prevalence of this new syndrome has been estimated to be between 1 in 13,000 and 1 in 20,000. The reciprocal microduplication appears to have unspecific features, such as hypotonia and feeding difficulties in the newborn period [Sharp et al., 2006; Kirchhoff et al., 2007].
Interstitial microdeletion at 2p15p16.1
The clinical phenotype of the interstitial microdeletion at 2p15p16.1 includes moderate to severe intellectual disability, autistic features, poor motor and speech development, microcephaly, structural brain anomalies, renal anomalies, digital anomalies, vision impairment, and a distinctive pattern of craniofacial features. The reported structural brain anomalies include cortical dysplasia/pachygyria. Dysmorphic craniofacial features include microcephaly, bitemporal narrowing, wide inner canthal distance, short downslanted palpebral fissures, epicanthic folds, prominent nasal tip, long straight eyelashes, smooth philtrum, and everted lower lip [Rajcan-Separovic et al., 2007].
Terminal deletions of the long arm of chromosome 2 (2q37)
Terminal deletions of the long arm of chromosome 2 (2q37) have a recognizable clinical phenotype, including MR (mild to severe), seizure disorder, autistic features, a recognizable pattern of dysmorphism, major malformations, and a pattern of findings described as Albright hereditary osteodystrophy-like (AHO) metacarpal/metatarsal shortening phenotype. Other phenotypes associated with 2q37 deletions include Wilms tumor and urogenital anomalies, epilepsy, and eczema [Falk and Casas, 2007].
7q11.23 microduplication
The 7q11.23 microduplication is the exact reciprocal of the common WBS deletion with an estimated incidence of 1:13,000 to 1:20,000. The main clinical feature is variable speech/language delay with cognitive disabilities ranging from normal to moderate MR. Autism has been reported in some patients. Specific dysmorphic features noted include high and broad nose, short philtrum, thin lips and straight eyebrows. Triplications of this region have a similar phenotype but are more severe than observed in patients with the duplication of the same region [Depienne et al., 2007].
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Albrecht U., Sutcliffe J.S., Cattanach B.M., et al. Imprinted expression of the murine Angelman syndrome gene, Ube3a, in hippocampal and Purkinje neurons. Nat Genet. 1997;17(9288101):75-78.
American Academy of Pediatrics. Health supervision for children with Down syndrome. Pediatrics. 2001;107(2):442-449.
American Academy of Pediatrics. Committee on Genetics. Health supervision for children with Turner syndrome. Pediatrics. 1995;96(6):1166-1173.
American Academy of Pediatrics. Committee on Sports Medicine and Fitness. Atlantoaxial instability in Down syndrome: subject review. Pediatrics. 1995;96(1 Pt 1):151-154.
Anderlid B.-M., Schoumans J., Anneren G., et al. Subtelomeric rearrangements detected in patients with idiopathic mental retardation. Am J Med Genet. 2002;107(11840483):275-284.
Aradhya S., Manning M.A., Splendore A., et al. Whole-genome array-CGH identifies novel contiguous gene deletions and duplications associated with developmental delay, mental retardation, and dysmorphic features. Am J Med Genet A. 2007;143A(17568414):1431-1441.
Bahi-Buisson N., Ville D., Eisermann M., et al. Epilepsy in chromosome aberrations. Arch Pediatr. 2005;12(4):449-458.
Ballif B.C., Hornor S.A., Jenkins E., et al. Discovery of a previously unrecognized microdeletion syndrome of 16p11.2-p12.2. Nat Genet. 2007;39(17704777):1071-1073.
Battaglia A., Eugene Hoyme H., Dallapiccola B., et al. Further delineation of deletion 1p36 syndrome in 60 patients: a recognizable phenotype and common cause of developmental delay and mental retardation. Pediatrics. 2008;121(18245432):404-410.
Baty B.J., Blackburn B.L., Carey J.C. Natural history of trisomy 18 and trisomy 13: I. Growth, physical assessment, medical histories, survival, and recurrence risk. Am J Med Genet. 1994;49(2):175-188.
Baty B.J., Jorde L.B., Blackburn B.L., et al. Natural history of trisomy 18 and trisomy 13: II. Psychomotor development. Am J Med Genet. 1994;49(2):189-194.
Bender B.G., Linden M.G., Harmon R.J. Neuropsychological and functional cognitive skills of 35 unselected adults with sex chromosome abnormalities. Am J Med Genet. 2001;102(4):309-313.
Boada R., Janusz J., Hutaff-Lee C., et al. The cognitive phenotype in Klinefelter syndrome: a review of the literature including genetic and hormonal factors. Dev Disabil Res Rev. 2009;15(4):284-294.
Boue J.G., Boue A., Lazar P., et al. Outcome of pregnancies following a spontaneous abortion with chromosomal anomalies. Am J Obstet Gynecol. 1973;116(4715938):806-812.
Buiting K., Gross S., Lich C., et al. Epimutations in Prader-Willi and Angelman syndromes: a molecular study of 136 patients with an imprinting defect. Am J Hum Genet. 2003;72(12545427):571-577.
Buiting K., Saitoh S., Gross S., et al. Inherited microdeletions in the Angelman and Prader-Willi syndromes define an imprinting centre on human chromosome 15. Nat Genet. 1995;9(7795645):395-400.
Cardoso C., Leventer R.J., Dowling J.J., et al. Clinical and molecular basis of classical lissencephaly: Mutations in the LIS1 gene (PAFAH1B1). Hum Mutat. 2002;19(1):4-15.
Carter M., McCaughey E., Annaz D., et al. Sleep problems in a Down syndrome population. Arch Dis Child. 2009;94(4):308-310.
Crocker M. Rearrangements of the X chromosome and Turner syndrome. Hum Genet. 1992;90(1427778):185-186.
Cronk C., Crocker A.C., Pueschel S.M., et al. Growth charts for children with Down syndrome: 1 month to 18 years of age. Pediatrics. 1988;81(2962062):102-110.
D’Alton M.E., DeCherney A.H. Prenatal diagnosis. N Engl J Med. 1993;328(2):114-120.
Daniel A., Hook E.B., Wulf G. Risks of unbalanced progeny at amniocentesis to carriers of chromosome rearrangements: data from United States and Canadian laboratories. Am J Med Genet. 1989;33(2750783):14-53.
Deb S., Hare M., Prior L. Symptoms of dementia among adults with Down’s syndrome: a qualitative study. J Intellect Disabil Res. 2007;51(Pt 9):726-739.
Depienne C., Heron D., Betancur C., et al. Autism, language delay and mental retardation in a patient with 7q11 duplication. J Med Genet. 2007;44(17400790):452-458.
Dykens E.M. Psychiatric and behavioral disorders in persons with Down syndrome. Ment Retard Dev Disabil Res Rev. 2007;13(3):272-278.
Edelmann L., Hirschhorn K. Clinical utility of array CGH for the detection of chromosomal imbalances associated with mental retardation and multiple congenital anomalies. Ann N Y Acad Sci. 2009;1151(19154522):157-166.
Elsea S.H., Girirajan S. Smith-Magenis syndrome. Eur J Hum Genet. 2008;16(4):412-421.
Elsheikh M., Dunger D.B., Conway G.S., et al. Turner’s syndrome in adulthood. Endocr Rev. 2002;23(11844747):120-140.
Falk R.E., Casas K.A. Chromosome 2q37 deletion: clinical and molecular aspects. Am J Med Genet C Semin Med Genet. 2007;145C(4):357-371.
Feeley K., Jones E. Strategies to address challenging behaviour in young children with Down syndrome. Downs Syndr Res Pract. 2008;12(2):153-163.
Fernandez B.A., Roberts W., Chung B., et al. Phenotypic Spectrum Associated with De Novo and Inherited Deletions and Duplications at 16p11.2 in Individuals Ascertained for Diagnosis of Autism Spectrum Disorder. J Med Genet. 2009.
Fidler D.J., Nadel L. Education and children with Down syndrome: neuroscience, development, and intervention. Ment Retard Dev Disabil Res Rev. 2007;13(3):262-271.
Firth H.V., Richards S.M., Bevan A.P., et al. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am J Hum Genet. 2009;84(4):524-533.
Flint S., Gibb D.M. Recurrent second trimester miscarriage. Curr Opin Obstet Gynecol. 1996;8(8979017):449-453.
Folstein S.E., Rosen-Sheidley B. Genetics of autism: complex aetiology for a heterogeneous disorder. Nat Rev Genet. 2001;2(11733747):943-955.
Frias J.L., Davenport M.L. Health supervision for children with Turner syndrome. Pediatrics. 2003;111(3):692-702.
Frye R.E. 15q11.2–13 duplication, mitochondrial dysfunction, and developmental disorders. J Child Neurol. 2009;24(19535813):1316-1320.
Gardner R.J.M., Sutherland G.R. Chromosome abnormalities and genetic counseling. Oxford: Oxford University Press; 2004.
Gohlke B.C., Haug K., Fukami M., et al. Interstitial deletion in Xp22.3 is associated with X linked ichthyosis, mental retardation, and epilepsy. J Med Genet. 2000;37(8):600-602.
Gray D.L., Crane J.P. Optimal nuchal skin-fold thresholds based on gestational age for prenatal detection of Down syndrome. Am J Obstet Gynecol. 1994;171(7977535):1282-1286.
Gurrieri F., Accadia M. Genetic imprinting: the paradigm of Prader-Willi and Angelman syndromes. Endocr Dev. 2009;14:20-28.
Gu W., Zhang F., Lupski J.R. Mechanisms for human genomic rearrangements. Pathogenetics. 2008;1(19014668):4.
Hannes F.D., Sharp A.J., Mefford H.C., et al. Recurrent reciprocal deletions and duplications of 16p13.11: the deletion is a risk factor for MR/MCA while the duplication may be a rare benign variant. J Med Genet. 2009;46(18550696):223-232.
Hempel M., Rivera Brugues N., Wagenstaller J., et al. Microdeletion syndrome 16p11.2-p12.2: clinical and molecular characterization. Am J Med Genet A. 2009;149A(10):2106-2112.
Hogart A., Leung K.N., Wang N.J., et al. Chromosome 15q11–13 duplication syndrome brain reveals epigenetic alterations in gene expression not predicted from copy number. J Med Genet. 2009;46(18835857):86-93.
Holm V.A., Cassidy S.B., Butler M.G., et al. Prader-Willi syndrome: consensus diagnostic criteria. Pediatrics. 1993;91(8424017):398-402.
Hook E.B., Cross P.K. Maternal age-specific rates of chromosome abnormalities at chorionic villus study: a revision. Am J Hum Genet. 1989;45(3):474-477.
Hsiao C.C., Tsao L.Y., Chen H.N., et al. Changing clinical presentations and survival pattern in trisomy 18. Pediatr Neonatol. 2009;50(4):147-151.
Iafrate A.J., Feuk L., Rivera M.N., et al. Detection of large-scale variation in the human genome. Nat Genet. 2004;36(9):949-951.
ISCN 2009: An international system for human cytogenetic nomenclature (2009). L.G. Shaffer, M.L. Slovak, L.J. Campbell. Karger; 2009.
Jacobs P.A., Browne C., Gregson N., et al. Estimates of the frequency of chromosome abnormalities detectable in unselected newborns using moderate levels of banding. J Med Genet. 1992;29(2):103-108.
Jacquemont M.L., Sanlaville D., Redon R., et al. Array-based comparative genomic hybridisation identifies high frequency of cryptic chromosomal rearrangements in patients with syndromic autism spectrum disorders. J Med Genet. 2006;43(16840569):843-849.
Jiang Y., Lev-Lehman E., Bressler J., et al. Genetics of Angelman syndrome. Am J Hum Genet. 1999;65(10364509):1-6.
Kirchhoff M., Bisgaard A.-M., Duno M., et al. A 17q21.31 microduplication, reciprocal to the newly described 17q21.31 microdeletion, in a girl with severe psychomotor developmental delay and dysmorphic craniofacial features. Eur J Med Genet. 2007;50(17576104):256-263.
Kishino T., Lalande M., Wagstaff J. UBE3A/E6-AP mutations cause Angelman syndrome. Nat Genet. 1997;15(8988171):70-73.
Kleefstra T., van Zelst-Stams W.A., Nillesen W.M., et al. Further clinical and molecular delineation of the 9q subtelomeric deletion syndrome supports a major contribution of EHMT1 haploinsufficiency to the core phenotype. J Med Genet. 2009;46(9):598-606.
Klopocki E., Graul-Neumann L.M., Grieben U., et al. A further case of the recurrent 15q24 microdeletion syndrome, detected by array CGH. Eur J Pediatr. 2008;167(8):903-908.
Krantz I.D., Piccoli D.A., Spinner N.B. Clinical and molecular genetics of Alagille syndrome. Curr Opin Pediatr. 1999;11(6):558-564.
Kriek M., White S.J., Bouma M.C., et al. Genomic imbalances in mental retardation. J Med Genet. 2004;41(15060096):249-255.
Lejeune J., Lafourcade J., De Grouchy J., et al. Partial Deletion of the Short Arm of Chromosome 5. Individualization of a New Morbid State. Sem Hop. 1964;40:1069-1079.
Levy B., Dunn T.M., Kaffe S., et al. Clinical applications of comparative genomic hybridization. Genet Med. 1998;1(1):4-12.
Liehr T., Starke H., Weise A., et al. Multicolor FISH probe sets and their applications. Histol Histopathol. 2004;19(1):229-237.
Lin R.J., Cherry A.M., Chen K.C., et al. Terminal deletion of 6p results in a recognizable phenotype. Am J Med Genet A. 2005;136(15940702):162-168.
Lister T.J., Frota-Pessoa O. Recurrence risks for Down syndrome. Hum Genet. 1980;55(6450157):203-208.
Lucito R., Healy J., Alexander J., et al. Representational oligonucleotide microarray analysis: a high-resolution method to detect genome copy number variation. Genome Res. 2003;13(10):2291-2305.
Luedi P.P., Dietrich F.S., Weidman J.R., et al. Computational and experimental identification of novel human imprinted genes. Genome Res. 2007;17(18055845):1723-1730.
Lupski J.R. Genome structural variation and sporadic disease traits. Nat Genet. 2006;38(9):974-976.
Mattina T., Perrotta C.S., Grossfeld P. Jacobsen syndrome. Orphanet J Rare Dis. 2009;4:9.
McDaniell R., Warthen D.M., Sanchez-Lara P.A., et al. NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the notch signaling pathway. Am J Hum Genet. 2006;79(1):169-173.
McDonald-McGinn D.M., LaRossa D., Goldmuntz E., et al. The 22q11.2 deletion: screening, diagnostic workup, and outcome of results; report on 181 patients. Genet Test. 1997;1(2):99-108.
McFadden D.E., Kwong L.C., Yam I.Y., et al. Parental origin of triploidy in human fetuses: evidence for genomic imprinting. Hum Genet. 1993;92(5):465-469.
Mensink K.A., Ketterling R.P., Flynn H.C., et al. Connective tissue dysplasia in five new patients with NF1 microdeletions: further expansion of phenotype and review of the literature. J Med Genet. 2006;43(2):e8.
Moerman P., Fryns J.P., van der Steen K., et al. The pathology of trisomy 13 syndrome. A study of 12 cases. Hum Genet. 1988;80(3198112):349-356.
Moretti P., Ferrari M., Di Battista S., et al. The 4P-syndrome. Case description and literature review. Minerva Pediatr. 2001;53(1):23-28.
Mulley J.C., Dibbens L.M. Chipping away at the common epilepsies with complex genetics: the 15q13.3 microdeletion shows the way. Genome Med. 2009;1(19341504):33.
Murray J.C., Johnson J.A., Bird T.D. Dandy-Walker malformation: etiologic heterogeneity and empiric recurrence risks. Clin Genet. 1985;28(4064366):272-283.
Nieuwenhuis-Mark R.E. Diagnosing Alzheimer’s dementia in Down syndrome: problems and possible solutions. Res Dev Disabil. 2009;30(5):827-838.
Nijhuis-van der Sanden M.W.G., Eling P.A.T.M., Otten B.J. A review of neuropsychological and motor studies in Turner Syndrome. Neurosci Biobehav Rev. 2003;27(12946685):329-338.
Pennington B.F., Bender B., Puck M., et al. Learning disabilities in children with sex chromosome anomalies. Child Dev. 1982;53(7140426):1182-1192.
Phelan M.C. Deletion 22q13.3 syndrome. Orphanet J Rare Dis. 2008;3:14.
Pijpers L., Jahoda M.G., Reuss A., et al. Transabdominal chorionic villus biopsy in second and third trimesters of pregnancy to determine fetal karyotype. BMJ. 1988;297(3140935):822-823.
Potocki L., Bi W., Treadwell-Deering D., et al. Characterization of Potocki-Lupski syndrome (dup(17)(p11.2p11.2)) and delineation of a dosage-sensitive critical interval that can convey an autism phenotype. Am J Hum Genet. 2007;80(17357070):633-649.
Rajcan-Separovic E., Harvard C., Liu X., et al. Clinical and molecular cytogenetic characterisation of a newly recognised microdeletion syndrome involving 2p15–16.1. J Med Genet. 2007;44(16963482):269-276.
Rasmussen S.A., Wong L-YC, Yang Q., et al. Population-based analyses of mortality in trisomy 13 and trisomy 18. Pediatrics. 2003;111(12671111):777-784.
Ravnan J.B., Tepperberg J.H., Papenhausen P., et al. Subtelomere FISH analysis of 11 688 cases: an evaluation of the frequency and pattern of subtelomere rearrangements in individuals with developmental disabilities. J Med Genet. 2006;43(16199540):478-489.
Robinson D.O., Howarth R.J., Williamson K.A., et al. Genetic analysis of chromosome 11p13 and the PAX6 gene in a series of 125 cases referred with aniridia. Am J Med Genet A. 2008;146A(18241071):558-569.
Roos L., Jonch A.E., Kjaergaard S., et al. A new microduplication syndrome encompassing the region of the Miller-Dieker (17p13 deletion) syndrome. J Med Genet. 2009;46(10):703-710.
Ross J.L., Zeger M.P., Kushner H., et al. An extra X or Y chromosome: contrasting the cognitive and motor phenotypes in childhood in boys with 47,XYY syndrome or 47,XXY Klinefelter syndrome. Dev Disabil Res Rev. 2009;15(4):309-317.
Rougeulle C., Glatt H., Lalande M. The Angelman syndrome candidate gene, UBE3A/E6-AP, is imprinted in brain. Nat Genet. 1997;17(9288088):14-15.
Sahoo T., del Gaudio D., German J.R., et al. Prader-Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nat Genet. 2008;40(6):719-721.
Savva G.M., Walker K., Morris J.K. The maternal age-specific live birth prevalence of trisomies 13 and 18 compared to trisomy 21 (Down syndrome). Prenat Diagn. 2010;30(1):57-64.
Sebat J., Lakshmi B., Troge J., et al. Large-scale copy number polymorphism in the human genome. Science. 2004;305(15273396):525-528.
Sermon K., Van Steirteghem A., Liebaers I. Preimplantation genetic diagnosis. Lancet. 2004;363(15145639):1633-1641.
Shaffer L.G., Bejjani B.A. Medical applications of array CGH and the transformation of clinical cytogenetics. Cytogenet Genome Res. 2006;115(3–4):303-309.
Shaffer L.G., Slovak M.L., Campbell L.J. An International System for Human Cytogenic Nomenclature. Basel: Karger; 2009.
Shanske A.L., Patel A., Saukam S., et al. Clinical and molecular characterization of a patient with Langer-Giedion syndrome and mosaic del(8)(q22.3q24.13). Am J Med Genet A. 2008;146A(24):3211-3216.
Sharp A.J., Hansen S., Selzer R.R., et al. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat Genet. 2006;38(16906162):1038-1042.
Silverman W. Down syndrome: cognitive phenotype. Ment Retard Dev Disabil Res Rev. 2007;13(3):228-236.
Smidt-Jensen S., Hahnemann N. Transabdominal chorionic villus sampling for fetal genetic diagnosis. Technical and obstetrical evaluation of 100 cases. Prenat Diagn. 1988;8(1):7-17.
Smigielska-Kuzia J., Sobaniec W., Kulak W., et al. Clinical and EEG features of epilepsy in children and adolescents in Down syndrome. J Child Neurol. 2009;24(4):416-420.
Stankiewicz P., Beaudet A.L. Use of array CGH in the evaluation of dysmorphology, malformations, developmental delay, and idiopathic mental retardation. Curr Opin Genet Dev. 2007;17(17467974):182-192.
Szigeti K., Garcia C.A., Lupski J.R. Charcot-Marie-Tooth disease and related hereditary polyneuropathies: molecular diagnostics determine aspects of medical management. Genet Med. 2006;8(2):86-92.
Tassabehji M. Williams-Beuren syndrome: a challenge for genotype-phenotype correlations. Hum Mol Genet. 2003;12(Spec No 2):R229-R237.
van Bon B.W.M., Mefford H.C., Menten B., et al. Further delineation of the 15q13 microdeletion and duplication syndromes: a clinical spectrum varying from non-pathogenic to a severe outcome. J Med Genet. 2009;46(19372089):511-523.
Van Esch H., Bauters M., Ignatius J., et al. Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am J Hum Genet. 2005;77(3):442-453.
Visootsak J., Sherman S. Neuropsychiatric and behavioral aspects of trisomy 21. Curr Psychiatry Rep. 2007;9(2):135-140.
Waldman H.B., Perlman S.P., Roesch R.E. Down syndrome and Alzheimer’s disease: children grow older. Gen Dent. 2008;56(5):420-422.
Weiss L.A., Shen Y., Korn J.M., et al. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med. 2008;358(18184952):667-675.
Williams C.A., Angelman H., Clayton-Smith J., et al. Angelman syndrome: consensus for diagnostic criteria. Angelman Syndrome Foundation. Am J Med Genet. 1995;56(2):237-238.
Wisniewski K.E., Kida E., Gordon-Majszak W., et al. Altered amyloid beta-protein precursor processing in brains of patients with neuronal ceroid lipofuscinosis. Neurosci Lett. 1990;120(2127306):94-96.
Woods C.G., Bankier A., Curry J., et al. Asymmetry and skin pigmentary anomalies in chromosome mosaicism. J Med Genet. 1994;31(7815438):694-701.
Wu Y.Q., Badano J.L., McCaskill C., et al. Haploinsufficiency of ALX4 as a potential cause of parietal foramina in the 11p11.2 contiguous gene-deletion syndrome. Am J Hum Genet. 2000;67(5):1327-1332.
Xu S., Han J.C., Morales A., et al. Characterization of 11p14-p12 deletion in WAGR syndrome by array CGH for identifying genes contributing to mental retardation and autism. Cytogenet Genome Res. 2008;122(19096215):181-187.
Ylstra B., van den Ijssel P., Carvalho B., et al. BAC to the future! or oligonucleotides: a perspective for micro array comparative genomic hybridization (array CGH). Nucleic Acids Res. 2006;34(16439806):445-450.
Zhang X., Snijders A., Segraves R., et al. High-resolution mapping of genotype-phenotype relationships in cri du chat syndrome using array comparative genomic hybridization. Am J Hum Genet. 2005;76(2):312-326.