[level-membership-for-neurology-category]
Chapter 15 Chorea, ballism, and athetosis
Introduction
Chorea consists of involuntary, continual, abrupt, rapid, brief, unsustained, irregular movements that flow randomly from one body part to another. Patients can partially and temporarily suppress the chorea and frequently camouflage some of the movements by incorporating them into semipurposeful activities (parakinesia). The inability to maintain voluntary contraction (motor impersistence), such as manual grip (milkmaid grip) or tongue protrusion, is a characteristic feature of chorea and results in dropping of objects and clumsiness. Chorea should be differentiated from “pseudochoreoathetosis,” a movement disorder that is phenomenologically similar to chorea or athetosis (slow chorea) due to loss of proprioception (Sharp et al., 1994). Muscle stretch reflexes are often “hung-up” and “pendular.” Affected patients typically have a peculiar, irregular, and dance-like gait. The pathophysiology of chorea is poorly understood, but in contrast to parkinsonism, dystonia, and other movement disorders, intracortical inhibition of the motor cortex is normal in chorea (Hanajima et al., 1999). In addition, semiquantitative analysis of single photon emission computed tomography in patients with hemichorea due to various causes suggests that there is an increase in activity in the contralateral thalamus, possibly due to disinhibition as a result of loss of normal pallidal inhibitory input (Kim et al., 2002).
Chorea may be a manifestation of a primary neurologic genetic disorder, such as Huntington disease (HD), or it may occur as a neurologic complication of systemic, toxic, or other disorders (Rosenblatt et al., 1998; Cardoso, 2004; Cardoso et al., 2006; Jankovic and Fahn, 2009) (Tables 15.1 and 15.2, Fig. 15.1). Chorea may be seen in normal infants, but these movements usually disappear by age 8 months, and some of these movements may be purposeful (Van der Meer et al., 1995).
| Developmental choreas |
Table 15.2 Differential diagnosis of inherited and sporadic choreas
| Inherited disorders | Sporadic disorders |
|---|---|
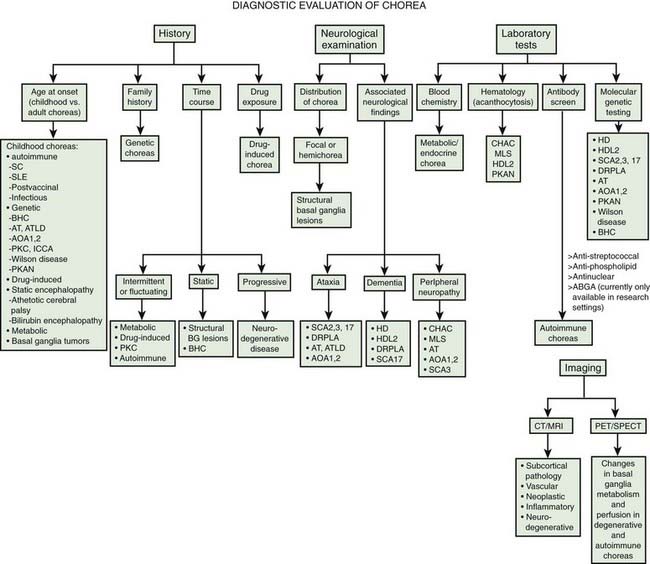
In this chapter we will briefly mention HD, but the focus will be on non-HD causes of chorea, as HD-like phenotypes without the HD genotype are increasingly being described and must be recognized. Several neurodegenerative disorders, some with expanded trinucleotide repeats, have been reported as phenocopies of HD, including spinocerebellar atrophy, particularly SCA2 and SCA3 (Kawaguchi et al., 1994), pure cerebello-olivary degeneration (Fox et al., 2003), and dentatorubral-pallidoluysian atrophy (DRPLA) (see below) (La Spada et al., 1994; Ikeuchi et al., 1995; Komure et al., 1995; Warner et al., 1995; Ross et al., 1997; Rosenblatt et al., 1998; Wild et al., 2008). In a study of 285 patients with clinical features consistent with HD, but who tested negative for HD by a DNA analysis, the following diagnoses were identified: five cases had Huntington disease-like type 4 (HDL4), one had HDL1, one had HDL2, and one patient had Friedreich ataxia (Wild et al., 2008).
Dentatorubral-pallidoluysian atrophy and HD-like disorders
Dentatorubral-pallidoluysian atrophy (DRPLA) is an autosomal dominant neurodegenerative disorder that is particularly prevalent in Japan, but it has been also identified in Europe and in African-American families (“Haw River syndrome”) (Burke et al., 1994; Thomas and Jankovic, 2001; Wardle et al., 2009) (Table 15.3). Usually beginning in the fourth decade, the disorder may occur as an early-onset DRPLA (before 20 years of age), manifested by a variable combination of myoclonus, epilepsy, and mental retardation, or late-onset DRPLA (after 20 years of age), manifested by cerebellar ataxia, choreoathetosis, dystonia, rest and postural tremor, parkinsonism, and dementia (Video 15.1). 
Table 15.3 Clinical features of dentatorubral-pallidoluysion atrophy (DRPLA)
| Early onset | Late onset |
|---|---|
Unstable CAG expansion has been identified as the mutation in the DRPLA (or ATN1) gene on chromosome 12p13.31 (Koide et al., 1994; Komure et al., 1995; Warner et al., 1995; Becher et al., 1997; Ross et al., 1997). The DRPLA gene codes for protein that has been identified as a phosphoprotein, c-Jun NH(2)-terminal kinase, one of the major factors involved in its phosphorylation. In DRPLA, this protein appears to be slowly phosphorylated; thus, it may delay a process that is essential in keeping neurons alive (Okamura-Oho et al., 2003). Similar to HD, there is an inverse correlation between the age at onset and the number of CAG repeats (Ikeuchi et al., 1995). The early onset of DRPLA is associated with greater number of CAG repeats (62–79) as compared to the late-onset type (54–67 repeats) (Ikeuchi et al., 1995). Testing for the various gene mutations will undoubtedly lead to better recognition and appreciation of the spectrum of clinical and pathologic changes associated with these disorders. For example, a family with spastic paraplegia, truncal ataxia, and dysarthria, but without other clinical features of DRPLA, has been found to show homozygosity for an allele that carries intermediate CAG repeats in the DRPLA gene (Kuroharas et al., 1997). The DRPLA gene is expressed predominantly in neurons, but neurons that are vulnerable to degeneration in DRPLA do not selectively express the gene (Nishiyama et al., 1997).
Neuroimaging studies in patients with DRPLA often show evidence of cortical, brainstem, and cerebellar atrophy and widespread white matter changes (Koide et al., 1997; Muñoz et al., 1999, 2004). Neuropathologic findings consist chiefly of degeneration of the dentatorubral system, globus pallidus externa (GPe), subthalamic nucleus, and, to a lesser extent, striatum, substantia nigra, inferior olive, and thalamus (Warner et al., 1994) as well as demyelination and reactive astrogliosis in the cerebral white matter (Muñoz et al., 2004). Involvement of oligodendrocytes in autopsied brains and an increased number of affected glia, as well as larger expansions in CAG in these glia, in transgenic mice might explain the widespread demyelination (Yamada et al., 2002). Several pathologic reports have noted widespread deposition of lipofuscin. Similar to HD and other diseases associated with CAG repeat expansions, DRPLA has also been associated with the formation of perinuclear aggregates that can be prevented by the use of transglutaminase inhibitors such as cystamine and monodansyl cadaverine (Igarashi et al., 1998). These intranuclear inclusions stain intensely with ubiquitin (Becher and Ross, 1998). Subsequent studies have demonstrated accumulation of mutant atrophin-1 in the neuronal nuclei, rather than neuronal intranuclear inclusions, as the predominant pathologic feature in this neurodegenerative disorder (Yamada et al., 2001).
Other Huntington disease-like disorders
An autosomal dominant HD-like neurodegenerative disorder, now classified as HDL1 and mapped to chromosome 20p (Xiang et al., 1998), is a familial prion disease with an expanded PrP. A 192-nucleotide insertion in the region of the prion protein gene (PRNP) encoding an octapeptide repeat in the prion protein, was found in a single family with HD phenotype, suggesting that PRNP mutations can result in HD phenocopies (Moore et al., 2001).
Another disorder, termed HDL2, is characterized by onset in the fourth decade, involuntary movements such as chorea and dystonia as well as other movement disorders (bradykinesia, rigidity, tremor), dysarthria, hyperreflexia, gait abnormality, psychiatric symptoms, weight loss, and dementia with progression from onset to death in about 20 years (Margolis et al., 2001, 2004; Walker et al., 2003a). The disorder appears to be present exclusively or predominantly in individuals of African origin. The neuroimaging and neuropathologic findings are very similar to those in HD, except that there appears to be more severe involvement of the occipital cortex (Greenstein et al., 2007), and the intranuclear inclusions stain with 1C2 but not with anti-huntingtin antibodies. Unlike the family linked to chromosome 20p, seizures are not present in the HDL2 family. All 10 affected family members had a CAG repeat expansion of 50–60 triplets. The gene was later mapped to chromosome 16q24.3 and was found to encode junctophilin-3, a protein of the junctional complex linking the plasma membrane and the endoplasmic reticulum (Holmes et al., 2001). Although acanthocytosis was emphasized by Walker and colleagues (2002, 2003b) in their initial report and in one of three patients reported subsequently (Walker et al., 2003b), we have not been able to confirm the presence of acanthocytes in one member of the original family or in the other members when we carefully examined the peripheral smear.
The mutation associated with HDL2 has been identified as a CTG/CAG trinucleotide repeat expansion within the junctophilin-3 (JPH3) gene. In the normal population, the repeat length ranges from 6 to 27 CTG triplets, whereas affected individuals have 41–58 triplets. One family, previously described as “autosomal dominant chorea–acanthocytosis with polyglutamine-containing neuronal inclusions” (see later) (Walker et al., 2002), was subsequently found to have the triple nucleotide expansion of HDL2 (Stevanin et al., 2003; Walker et al., 2003b). The CTG repeat expansion at the HDL2 locus has been found to be responsible for 2% of patients with typical features of HD but without expanded CAG repeats in the IT15 gene and 0.2% of all HD families, again providing evidence that HD is clinically and genetically heterogeneous (Stevanin et al., 2002). This group later analyzed 252 patients with an HDL phenotype, including 60 with typical HD, who had tested negative for pathologic expansion in the IT15 gene and found two patients that had an abnormal CTG expansion in the JPH3 gene and two other patients with abnormal CAG expansion in the gene coding for TATA-binding protein (TBP/SCA17), important in initiation of transcription (Stevanin et al., 2003; Toyoshima et al., 2004; Schneider et al., 2006). SCA17, categorized as HDL4, has many clinical features that overlap with HD (Schneider et al., 2007; Bech et al., 2010). Thus, the frequency of mutation in either the JPH3 gene or TBP gene among patients with HDL phenotype is about 3%. Initially, the TBP expansion was found in the family with SCA17, characterized clinically by intellectual deterioration, cerebellar ataxia, epilepsy, and chorea. CUG repeat-containing RNA foci, resembling myotonic dystrophy type 1, were detected in neurons of HDL2 brains, suggesting that RNA toxicity may play a role in the pathogenesis of this neurodegenerative disorder (Rudnicki et al., 2007). HLD2 resembles HD clinically and pathologically more than any other disease (Rudnicki et al., 2008).
While the majority of genetic forms of chorea are inherited in an autosomal dominant pattern, a novel autosomal recessive neurodegenerative HDL disorder has been described (Kambouris et al., 2000). Beginning at 3–4 years of age and manifested by chorea, dystonia, ataxia, gait disorder, spasticity, seizures, mutism, intellectual impairment, and bilateral frontal and caudate atrophy, this neurodegenerative disorder has been linked to 4p15.3, different from the 4p16.3 HD locus, but confirmation of this finding is lacking. Although classified as HDL3, because of its AR inheritance and clinical features atypical for HD, it should not be categorized as HDL. In fact the HDL terminology should be replaced with specific disorders in which genetic mutation has been identified.
Some cases of neuronal intranuclear inclusion disease, caused by expanded CAG repeats and characterized by the combination of extrapyramidal signs, lower motor neuron signs, and cognitive and behavioral abnormalities resulting in death by the third decade, also show intranuclear aggregates, similar to the other CAG disorders (Lieberman et al., 1998).
Neuroacanthocytosis
After HD, neuroacanthocytosis is perhaps the most common form of hereditary chorea (Table 15.4). Previously also referred to as chorea–acanthocytosis, it is now recognized that this multisystem, neurodegenerative disorder can be expressed by a wide variety of clinical and laboratory abnormalities, hence the term neuroacanthocytosis (Spitz et al., 1985; Danek et al., 2005; Walker et al., 2006, 2007; Thomas and Jankovic, 2006) (Table 15.5). The term was coined by Jankovic et al. (1985) to replace the old term Levine–Critchley syndrome choreoacanthocytosis to draw attention to the heterogeneous presentation with a variety of hyperkinetic (chorea, dystonia, tics) and hypokinetic (parkinsonism) movement disorders in addition to other neurologic deficits and abnormal laboratory findings. Symptoms usually first begin in the third and fourth decades of life (range: 8–62 years) with lip and tongue biting followed by orolingual (eating) dystonia, motor and phonic tics, generalized chorea, and stereotypies (Video 15.2). Other features include cognitive and personality changes, seizures, dysphagia, dysarthria, vertical ophthalmoparesis, parkinsonism, amyotrophy, areflexia, evidence of axonal neuropathy, and elevated serum creatine kinase without evidence of myopathy. Hardie and colleagues (1991) reviewed the clinical, hematologic, and pathologic findings in 19 patients (10 males and 9 females) with a mean age of 32 years (range: 8–62 years) with more than 3% acanthocytes on peripheral blood smear. Twelve of these patients with neuroacanthocytosis were familial, and seven were sporadic; two had the McLeod phenotype (see later). In their series, Hardie and colleagues (1991) found a variety of movement disorders, including chorea (58%), orofacial dyskinesia (53%), dystonia (47%), vocalizations (47%), tics (42%), and parkinsonism (34%). Although lip and tongue biting was observed in only 16% of the patients, this is a characteristic feature of neuroacanthocytosis and when present, it strongly suggests the diagnosis. The use of a mouth guard has been reported to be effective in the treatment of oral self-mutilation associated with neuroacanthocytosis (Fontenelle and Leite, 2008). Besides movement disorders other associated features included dysarthria (74%); absent or reduced reflexes (68%); dementia (63%); psychiatric problems such as depression, anxiety, and obsessive-compulsive disorder (58%); dysphagia (47%); seizures (42%); muscle weakness and wasting (16%); and elevated creatine phosphokinase (CK) in 58%. Magnetic resonance volumetry and fluorodeoxyglucose positron emission tomography (PET) show striatal atrophy in patients with neuroacanthocytosis (Jung et al., 2001). 
Table 15.4 Clinical and genetic features of neuroacanthocytosis
Table 15.5 Classification of neuroacanthocytosis
| I. Normal lipids |
Although autosomal dominant, X-linked recessive, and sporadic forms of neuroacanthocytosis have been reported, the majority of the reported families indicate autosomal recessive inheritance. Genome-wide scan for linkage in 11 families with autosomal recessive inheritance showed a linkage to a marker on chromosome 9q21, indicating a single locus for the disease (Rubio et al., 1997). Ueno and colleagues (2001) carried out a linkage-free analysis in the region of chromosome 9q21 in the Japanese population and identified a 260 bp deletion in the EST (expressed sequence tags) region K1AA0986 in exon 60, 61 that was homozygous in patients with neuroacanthocytosis and heterozygous in their parents. Further sequencing has identified a polyadenylation site with a protein with 3096 amino acid residues that has been named “chorein” by the authors. This deletion is not found in normal Japanese and European populations (Ueno et al., 2001). In another study by Rampoldi and colleagues (2001) in European patients, a novel gene encoding a 3174-amino-acid protein on chromosome 9q21 with 73 exons was identified. They identified 16 mutations in the chorea acanthocytosis (ChAc) gene, later renamed VPS13A gene. These mutations were identified in various exons. They suggested that chorea acanthocytosis encodes an evolutionarily conserved protein that is involved in protein sorting (Rampoldi et al., 2002). Other single heterozygous mutations have been identified in this gene (Saiki et al., 2003). Molecular analysis by screening all 73 exons of the VPS13A gene showed marked genotype–phenotype heterogeneity (Lossos et al., 2005). The function of the protein product, chorein, is not yet fully understood, but it is probably involved in intracellular protein trafficking. Using antichorein antisera, the expression of chorein in peripheral red blood cells has been found to be absent or markedly reduced in patients with neuroacanthocytosis, but not with McLeod syndrome or with HD (Dobson-Stone et al., 2004). Loss of chorein expression, measured by Western blot analysis has been found to be a reliable diagnostic test for neuroacanthocytosis.
Walker and colleagues (2002) described a family with chorea or parkinsonism as well as cognitive changes, inherited in an autosomal dominant pattern. At autopsy, there was marked degeneration of the striatum and intranuclear inclusion bodies immunoreactive for ubiquitin, expanded polyglutamine CGG repeats, and torsinA. Interestingly, one of the patients had fragile X syndrome, and two had expanded trinucleotide repeats at permutation range, previously associated with postural/kinetic tremor, parkinsonism, ataxia, and cognitive decline (Hagerman et al., 2001). The family reported by Walker and colleagues (2002) turned out to have the trinucleotide repeat expansion associated with HDL2, but subsequent analysis of the family shed doubt on the presence of acanthocytes as a feature of the HDL2 syndrome (Walker et al., 2003a).
Two patients from the original study by Rubio and colleagues (1997) were found to have the McLeod phenotype, an X-linked (Xp21) recessive form of acanthocytosis associated with depression, bipolar and personality disorder, and neurologic manifestations, including chorea, involuntary vocalizations, seizures, motor axonopathy, hemolysis, liver disease, and high creatine kinase levels (Witt et al., 1992; Danek et al., 2001a, 2001b). Neuroimaging usually reveals caudate and occasionally cerebellar atrophy with a rim of increased T2-intensity in the lateral putamen. Functional neuroimaging studies show evidence of downregulation of D2 dopamine receptors. In contrast to the autosomal recessive form of neuroacanthocytosis linked to mutations in VPS13A gene on chromosome 9, patients with McLeod syndrome usually do not exhibit lip-biting or dysphagia. This multisystem disorder is associated with low reactivity of Kell erythrocyte antigens (weak antigenicity of red cells) due to absence of XK, a 37 kDa, 444-amino-acid, membrane protein that forms a complex with the Kell protein. The disorder is caused by different mutations in the XK gene encoding for the XK protein (Ho et al., 1996; Danek et al., 2001a; Jung et al., 2001). Mutations identified by various authors include frame shift mutations in exon 2 at codon 151, deletion at codon 90 in exon 2 and at codon 408 in exon 3, and splicing mutations in intron 2 of the XK gene (Dotti et al., 2000; Ueyama et al., 2000; Danek et al., 2001a; Jung et al., 2001). Rarely, neuroacanthocytosis may be associated with abetaliproteinemia due to mutations in the microsomal triglyceride transfer protein (Sharp et al., 1993). In addition to acanthocytosis, the patients exhibit retinopathy; malabsorption, including that of vitamin E; low serum cholesterol levels; and abnormal serum lipoprotein electrophoresis. Aprebetalipoproteinemia can also cause movement disorders and acanthocytosis (Bohlega et al., 1998).
An examination of wet blood or Wright-stained, fast dry, blood smear usually reveals over 15% of red blood cells as acanthocytes. In mild forms of acanthocytosis, scanning electron microscopy might be required to demonstrate the red blood cell abnormalities (Feinberg et al., 1991). In a recent study of two patients with pathologically proven neuroacanthocytosis, Feinberg and colleagues (1991) noted that the yield in demonstrating acanthocytosis may be increased by using a coverslip because the contact with glass causes the fragile cells to undergo morphologic changes. Diluting the blood with normal saline, incubating the Wright-stained smear with EDTA, using a scanning electron microscope, and other techniques designed to increase “echinocytotic stress” are also helpful (Feinberg et al., 1991; Orrell et al., 1995). The characteristic acanthocytic appearance of red blood cells has been attributed to abnormalities in transmembrane glycoprotein band 3 that can be demonstrated on gel electrophoresis. It is not yet clear how the gene mutation leads to the abnormal morphology of the red cells.
By using high-performance liquid chromatography, fatty acids of erythrocyte membrane proteins were analyzed in six patients with neuroacanthocytosis (Sakai et al., 1991). In comparison with normal controls and patients with HD, erythrocytes of patients with neuroacanthocytosis showed a marked abnormality in the composition of covalently bound fatty acids: an increase in palmitic acid (C16:0) and a decrease in stearic acid (C18:0).
Brain magnetic resonance imaging (MRI) in patients with neuroacanthocytosis usually shows caudate and more generalized brain atrophy, but some cases also show extensive white matter abnormalities (Nicholl et al., 2004). Caudate hypometabolism and atrophy have been demonstrated by PET studies and by neuroimaging. Similar to the findings in Parkinson disease, PET scans in six patients with neuroacanthocytosis showed a reduction to 42% of normal in [18F]dopa uptake in the posterior putamen; in contrast to Parkinson disease, however, there was a marked reduction in the striatal [11C]raclopride (D2) receptor binding (Brooks et al., 1991).
Neuronal loss and gliosis were particularly prominent in the striatum and pallidum but may also affect the thalamus, substantia nigra, and anterior horns of the spinal cord (Rinne et al., 1994b). The neuronal loss in the substantia nigra is most evident in the ventrolateral region, similar to Parkinson disease, but the nigral neuronal loss is more widespread in neuroacanthocytosis (Rinne et al., 1994a). The preservation of the cerebral cortex, cerebellum, subthalamic nucleus, pons, and medulla may serve to differentiate pathologically between neuroacanthocytosis, HD, and DRPLA. Brain biochemical analyses showed low substance P in the substantia nigra and striatum and increased levels of norepinephrine in the putamen and pallidum (DeYebenes et al., 1988).
Neurodegeneration with brain iron accumulation
A group of neurodegenerative disorders, formerly known as Hallervorden–Spatz disease, has been receiving increasing attention as the genetic and pathogenic mechanisms of the various subtypes become elucidated. Because of Hallervorden’s terrible past and his shameless involvement in active euthanasia (Shevell, 2003), this group of disorders has been renamed neurodegeneration with brain iron accumulation (NBIA). The prototype form of NBIA consists of an autosomal recessive disorder characterized by childhood-onset progressive rigidity, dystonia, choreoathetosis, spasticity, optic nerve atrophy, and dementia, and has been associated with acanthocytosis (Malandrini et al., 1996; Racette et al., 2001; Thomas et al., 2004). Although chorea is not a typical feature of NBIA, “senile chorea” has been described in a patient with pathologically proven NBIA type1 (NBIA-1) (Grimes et al., 2000).
The most classic NBIA, NBIA-1, is the pantothenate kinase-associated neurodegeneration (PKAN). Linkage analyses initially localized the NBIA-1 gene on 20p12.3–p13; subsequently, a 7 bp deletion and various missense mutations were identified in the coding sequence of the PANK2 gene, which codes for pantothenate kinase (Zhou et al., 2001; Hayflick et al., 2003). Pantothenate kinase is an essential regulatory enzyme in coenzyme A biosynthesis. It has been postulated that as a result of phosphopantothenate deficiency, cysteine accumulates in the globus pallidus of brains of patients with NBIA-1. It undergoes rapid auto-oxidation in the presence of nonheme iron that normally accumulates in the globus pallidus interna (GPi) and substantia nigra, generating free radicals that are locally neurotoxic. Interestingly, atypical subjects were found to be compound heterozygotes for certain mutations for which classic subjects were homozygous. The disorder with the clinical phenotype of NBIA associated with mutations in the PANK2 gene is now referred to as pantothenate kinase-associated neurodegeneration or PKAN (Thomas et al., 2004). On the basis of an analysis of 123 patients from 98 families with NBIA-1, Hayflick and colleagues (2003) found that “classic Hallervorden–Spatz syndrome” was associated with PANK2 mutation in all cases and that one-third of “atypical” cases had the mutations within the PANK2 gene. Those who had the PANK2 mutation were more likely to have dysarthria and psychiatric symptoms, and all had the typical “eye-of-the-tiger” abnormality on MRI with a specific pattern of hyperintensity within the hypointense GPi (McNeill et al., 2008).
Neuroacanthocytosis and NBIA may overlap in some clinical features. While PKAN may be associated with acanthocytosis, another neuroacanthocytosis syndrome, linked to PKAN, is the hypoprebetalipoproteinemia, acanthocytosis, retinitis pigmentosa, and pallidal degeneration (HARP) syndrome (Orrell et al., 1995). This disorder is associated with dystonia, particularly involving the oromandibular region, rather than chorea and self-mutilation. Indeed, a homozygous nonsense mutation in exon 5 of the PANK2 gene that creates a stop codon at amino acid 371, found in the original HARP patient, establishes that HARP is part of the pantothenate kinase-associated neurodegeneration disease spectrum (Ching et al., 2002; Houlden et al., 2003).
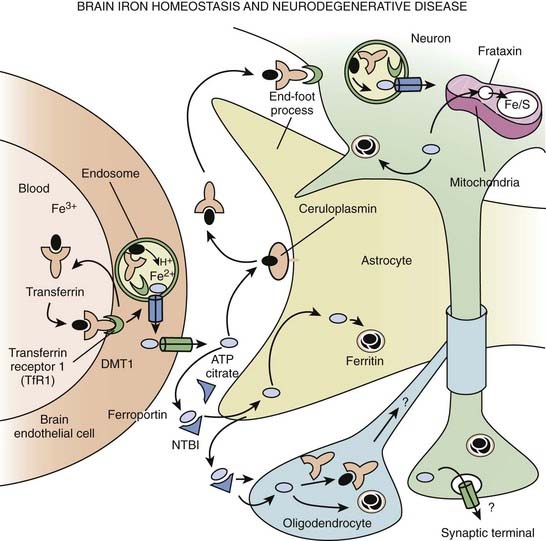
The classification of NBIA is continuously being revised as our understanding of this group of disorders is improving. In addition to PKAN, other forms of NBIA include neuroferritinopathy, infantile neuroaxonal dystrophy, and aceruloplasminemia, and PLA2G6-associated neurodegeneration (PLAN), with mutations in the PLA2G6 gene, on chromosome 22q13.1 (Schneider et al., 2009) (Table 15.6, Fig. 15.2). These disorders are chiefly manifested by childhood-onset axial hypotonia, spasticity, bulbar dysfunction, ataxia, dystonia, and choreoathetosis, as well as MRI changes indicative of iron deposition in the globus pallidus and substantia nigra (Kurian et al., 2008). Previously diagnosed as infantile neuroaxonal dystrophy, now classified as NBIA-2, PLAN may also present as adult-onset, levodopa-responsive dystonia–parkinsonism without iron on brain imaging (McNeill et al., 2008; Paisan-Ruiz et al., 2008; Schneider et al., 2009). Another gene, FA2H, when mutated, has been found to cause not only leukodystrophy and hereditary spastic paraplegia, but also NBIA (Kruer et al., 2010). The FA2H-associated neurodegeneration (FAHN) is characterized by childhood-onset gait impairment, spastic paraparesis, ataxia, and dystonia (Kruer et al., 2010; Schneider and Bhatia, 2010). FA2H is involved in lipid and ceramide metabolism. Another form of NBIA was highlighted by a report of 11 children with a biochemical profile suggestive of dopamine transporter deficiency syndrome (Kurian et al., 2011). Presenting in infancy, this disorder is usually characterized by severe parkinsonism-dystonia syndrome, but chorea, oculomotor deviations, and spasticity may also dominate the clinical phenotype. The CSF ratio of homovanillic acid to 5-hydroxyindoleacetic acid is usually increased. This autosomal recessive disorder has been attributed to homozygous or compound heterozygous SLC6A3 mutations and complete loss of dopamine transporter activity in the basal nuclei (indicated by abnormal DAT Scan SPECT). Regression of symptoms after 6 months of iron chelation with deferiprone has been reported in some patients with NBIA (Forni et al., 2008).
Table 15.6 Differential diagnosis of neurodegeneration with brain iron accumulation (NBIA)
|
• Infantile neuroaxonal dystrophy (NBIA-2)
|
Other familial choreas
Besides HD and neuroacanthocytosis, genetically transmitted choreas include benign hereditary chorea (BHC), a nonprogressive chorea of childhood onset (Wheeler et al., 1993; Kleiner-Fisman and Lang, 2007; Adam and Jankovic, 2010) (Table 15.7). BHC usually starts in early childhood and progresses until the second decade, after which time it remains static or even spontaneously improves. The patients may have a slight motor delay because of chorea, slight gait ataxia, and their handwriting may be impaired, but the disorder is self-limiting after adolescence in most cases, although it may persist as a mild chorea beyond age 60 years. Inherited as an autosomal dominant disorder, BHC has been linked to a marker on chromosome 14q13.1–q21.1 (de Vries et al., 2000; Fernandez et al., 2001; Breedveld et al., 2002) and a novel single nucleotide substitution in the TITF1 gene (also referred to as TTF, Nkx2.1, and T/ebp), coding for a transcription essential for the organogenesis of the lung, thyroid, and basal ganglia, has been identified in one Canadian family (Kleiner-Fisman et al., 2003; Kleiner-Fisman and Lang, 2007; Ferrara et al., 2008) (http://www.ncbi.nlm.nih.gov/sites/GeneTests/). This syndrome has been also described in patients with deletion of not only the TITF1 gene but also the contiguous PAX9 gene (Devos et al., 2006). These mutations should be considered in children and adults with chorea, mental retardation, congenital hypothyroidism, and chronic lung disease, hence the term brain–lung–thyroid syndrome proposed for this disease (Willemsen et al., 2005; Devos et al., 2006). MRI has been generally reported to be unremarkable but some cases showed hypoplastic pallidum, lack of differentiation of medial and lateral components, and bilateral signal hyperintensities on T2-weighted MRI images (Kleiner-Fisman and Lang, 2007). Two autopsied brains from patients with BHC due to TITF1 showed no pathologic abnormalities (Asmus et al., 2005), but specific immunochemical investigations suggested reduced number of striatal and neocortical interneurons, consistent with a defect in migration normally mediated by the TITF1 gene (Kleiner-Fisman et al., 2005).
|
• MRI normal, but may show hypoplastic pallidum, lack of differentiation of medial and lateral components, and bilateral signal hyperintensities on T2-weighted images
|
Adult-onset, autosomal dominant, benign chorea without dementia, initially reported in Japan, in which HD, HDL1, HDL2, DRPLA, SCA17, and mutations in the TITF1 gene were excluded, revealed linkage to a novel locus on chromosome 8q21.3–q23.3 in two Japanese families with “benign hereditary chorea type 2” (BHC2) (Shimohata et al., 2007). Surprisingly, BHC may improve markedly with levodopa (Asmus et al., 2005). The existence of BHC has been questioned because many patients who were initially diagnosed with the disorder were later found to have some other diagnosis, such as myoclonic dystonia, hereditary essential myoclonus, tics, and HD (Schrag et al., 2000).
Essential chorea is a form of adult-onset, nonprogressive chorea without family history of chorea or other symptoms suggestive of HD and without evidence of striatal atrophy on neuroimaging studies. Sometimes referred to as senile chorea, essential chorea usually has its onset after age 60, and in contrast to HD, it is not associated with dementia or positive family history. Some cases of senile chorea, however, have been reported to have pathologic changes identical to HD; others have had predominant degeneration of the putamen rather than the caudate (Friedman and Ambler, 1990). The CAG repeat length should, by definition, be normal, but Ruiz and colleagues (1997) found abnormal CAG expansion in three of six clinically diagnosed cases of senile chorea. Although the authors suggest that some patients with senile chorea have a sporadic form of HD, the term essential (or senile) chorea should be reserved for those patients with late-onset chorea without family history, without dementia, without psychiatric problems, and without CAG expansion. These criteria are necessary in order to separate senile or essential chorea from HD. Hereditary chorea without dementia and with a benign course has been also described (Behan and Bone, 1977).
Choreoathetosis, along with developmental regression, mental retardation, pendular nystagmus, optic atrophy, dysphagia, dystonia, spasticity, and severe bilateral striatal atrophy with response to biotin, is a feature of familial infantile bilateral striatal necrosis (IBSN) (Straussberg et al., 2002; Basel-Vanagaite et al., 2006). IBSN, an autosomal recessive neurodegenerative disorder, was found to be associated with mutation of nup62 on chromosome 19q13.32–13.41 (Basel-Vanagaite et al., 2006).
Ataxia telangiectasia, an autosomal recessive multisystem disease, is another disorder often associated with chorea. In a retrospective analysis of the clinical characteristics in 18 adult patients with ataxia telangiectasia from 9 families and 6 unrelated adults documented with elevated alpha-fetoprotein, chromosomal instability, and mutations in the A-T mutated (ATM) gene and measurements of ATM protein expression and kinase activity, chorea–athetosis was present in 14/18 (78%) cases (Verhagen et al., 2009). Other common abnormalities included dysarthria, oculomotor apraxia, dystonia, rest tremor, neuropathy, immunodeficiency, restricted respiratory function, and malignancies. Not all affected individuals have telangiectasia.
Another disorder, rarely considered in the differential diagnosis of chorea, is familial dyskinesia and facial myokymia (FDFM), characterized by childhood-onset adventitious movements and perioral or periorbital myokymia. These movements may be paroxysmal in early stages, increase in frequency and severity, and become constant in the third decade, without intellectual impairment or shortening of lifespan. Frontotemporal dementia, particularly associated with TAR-DNA binding protein (TDP)-43 abnormalities due to TARDBP mutations, may be associated not only with amyotrophic lateral sclerosis, but also supranuclear gaze palsy and chorea (Kovacs et al., 2009).
Infectious chorea
A variety of infections that affect the central nervous system have been associated with chorea. Acute manifestations of bacterial meningitis, encephalitis, tuberculous meningitis, and aseptic meningitis include movement disorders such as chorea, athetosis, dystonia, or hemiballismus (Alarcon et al., 2000). Human immunodeficiency virus (HIV) has also been reported to cause chorea and other features of HD, either as the result of human immunodeficiency virus encephalitis (Sevigny et al., 2005) or as the result of focal opportunistic infections such as toxoplasmosis (Nath et al., 1987; Gallo et al., 1996; Pardo et al., 1998; Piccolo et al., 1999).
Postinfectious and autoimmune choreas
Sydenham disease
Occasionally still referred to as St Vitus’ chorea or St Vitus’ dance (Krack, 1999), Sydenham chorea was described originally by Thomas Sydenham, an English physician, in 1686. Considered an autoimmune disorder, a consequence of infection with group A β-hemolytic streptococcus (GABHS), the phenomenology has broadened to include not only chorea but also a variety of other neurologic, psychiatric, cardiac, rheumatologic, and other disorders (Swedo, 1994; Cardoso et al., 1997; Cardoso, 2004) (Videos 15.3 and 15.4). Since chorea is only one of many neurologic and non-neurologic manifestations, the term Sydenham disease is preferred, rather than Sydenham chorea, but the latter is used so frequently in the literature that its usage will be difficult to change. Osler, in his seminal paper “On Chorea and Choreiform Affections,” published in 1894, drew attention to the distinction between Sydenham disease and HD (Goetz, 2000). Although the majority of the patients have bilateral involvement, the distribution of chorea is usually asymmetrical, and pure hemichorea can be seen in 20% of all Sydenham disease patients. The individual contractions are slightly longer in Sydenham disease (>100 ms) compared to those in HD (50–100 ms) (Hallett and Kaufman, 1981). Other features of Sydenham disease include dysarthria and decreased verbal output, hypometric saccades, oculogyric crisis, papilledema, central retinal artery occlusion, and seizures. Similar to Tourette syndrome (Kwak et al., 2003b), migraine is more common in children with Sydenham disease than in controls (Teixeira et al., 2005). Unlike arthritis and carditis, which occur soon after streptococcal infection, chorea and various neurobehavioral symptoms may be delayed for 6 months or longer and may be the sole manifestation of rheumatic fever (Stollerman, 1997). In 50 consecutive patients with rheumatic fever, 26% developed chorea, but arthritis was more frequent in patients without chorea (84%) than in those with chorea (31%) (Cardoso et al., 1997). 
Some features of Sydenham disease overlap with those of Tourette syndrome. Although tics are often differentiated from chorea by the presence of premonitory sensation, reported by more than 90% of patients with Tourette syndrome (Kwak et al., 2003a), some patients with chorea also report sensory symptoms (Rodopman-Arman et al., 2004). There is controversy as to whether pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections (PANDAS), discussed in more detail in Chapter 16, which may be associated with motor and phonic tics, is a variant of Sydenham disease or a separate entity (Kurlan, 2004; Kurlan et al., 2008; Singer et al., 2008).
Behavioral problems associated with Sydenham disease include irritability, emotional lability, obsessive-compulsive disorder, hyperactivity, learning disorders, and other behavioral problems that are typically observed in patients with Tourette syndrome. One study compared 56 patients with Sydenham disease with 50 rheumatic fever patients and 50 healthy subjects and found that obsessive-compulsive behavior was present in 19%, obsessive-compulsive disorder in 23.2%, and attention deficit-hyperactivity disorder in 30.4%, all significantly more frequently than in the rheumatic fever or healthy controls (Maia et al., 2005). The neurobehavioral symptoms usually begin within 2–4 weeks after the onset of the choreic movements. The disorder tends to resolve spontaneously in 3–4 months but may persist in half of the patients during a 3-year follow-up (Cardoso et al., 1999). Female gender and the presence of carditis might be risk factors for persistent disease. In some cases of Sydenham disease, the chorea recurs during pregnancy (chorea gravidarum) or when the patient is exposed to estrogen. Sydenham disease, once relatively common, is now encountered relatively rarely in developed countries (Eshel et al., 1993). Recurrences of Sydenham disease are not associated with anti-basal ganglia antibodies (Harrison et al., 2004).
In addition to occasionally elevated titers of antistreptolysin O (ASO), which is not specific for GABHS infection as it can also reflect group G, the majority of patients with Sydenham disease have been found to have IgG antibodies reacting with neurons in the caudate and subthalamic nuclei (Kiessling et al., 1993; Swedo et al., 1993). These antineuronal antibodies are found in nearly all patients with Sydenham disease (Swedo, 1994; Abraham et al., 1997; Mittleman, 1997). The rheumatic B-cell alloantigen D8/17 is also frequently found in patients with rheumatic chorea, but it is not clear whether this could be used as a diagnostic test (Feldman et al., 1993). Anti-basal ganglia antibodies have been identified in patients with acute and persistent Sydenham disease, providing further evidence that the disease is an antibody-mediated disorder (Church et al., 2002).
MRI is usually normal in patients with Sydenham disease except for selective enlargement of the caudate, putamen, and globus pallidus (Giedd et al., 1995). In contrast to other choreic disorders, PET scans in patients with Sydenham disease indicate increased rather than decreased striatal glucose consumption (Weindl et al., 1993).
The standard of care for all children diagnosed with Sydenham disease, even in cases of isolated chorea, is secondary prevention with penicillin to reduce the risk of recurrences of chorea but especially to reduce the likelihood that future GABHS infections could cause carditis and permanent valvular damage (Panamonta et al., 2007; Cardoso, 2008). Current recommendations in the United States are for treatment until age 21 years with either monthly intramuscular or daily oral penicillin. Although cephalosporins are equally effective, penicillin, 500–1000 mg of penicillin G four times per day or one intramuscular injection of 600 000 to 1.2 million units of benzathine penicillin, is considered the drug of choice for pharyngitis caused by GABHS infection (Garvey and Swedo, 1997). Despite an adequate (10-day) course, the bacteriologic failure rate is as high as 15%, and some patients develop rheumatic fever. Therefore, oral rifampin 20 mg/kg every 24 hours for four doses is recommended during the last 4 days of the 10-day course of penicillin therapy. Alternatively, oral rifampin 10 mg/kg can be given every 12 hours for eight doses with one dose of intramuscular benzathine penicillin G. Another alternative is oral clindamycin 20 mg/kg/day in three doses for 10 days. The best prevention of rheumatic fever is accurate diagnosis and adequate treatment of the initial acute pharyngitis. Penicillin prophylaxis is advisable in all patients for at least 10 years after rheumatic fever. Symptomatic treatment usually consists of antidopaminergic drugs such as tetrabenazine, valproic acid, and carbamazepine until the condition resolves spontaneously. A double-blind, placebo-controlled study of prednisone showed beneficial effects on Sydenham disease (Paz et al., 2006).
Other autoimmune choreas
Besides Sydenham disease, another poststreptococcal disorder which may include chorea, but more commonly is characterized by parkinsonism and dystonia is poststreptococcal acute disseminated encephalomyelitis (PSADEM), associated with the anti-basal ganglia antibodies (Dale et al., 2001). Although both Sydenham disease and PSADEM are associated with MRI T2 hyperintensities, the lesions described in PSADEM had normal T1 sequences and the white matter and brainstem are additionally involved. Chorea has been also recognized in a variety of other autoimmune processes, such as systemic lupus erythematosus (SLE). Chorea occurs in 2% of patients with SLE, and choreic movements precede the diagnosis of SLE in 22% of these cases. Choreic movements associated with pregnancy (chorea gravidarum) and with birth control pills probably result from a common pathogenesis, and chorea gravidarum may be the first manifestation of SLE or may represent as a variant of Sydenham disease. Chorea in SLE has been associated with the presence of antiphospholipid antibodies, a heterogeneous group of antibodies that produce platelet endothelial dysfunction and promote thrombogenesis. The primary antiphospholipid syndrome is characterized by the presence of antiphospholipid antibodies in patients who have autoimmune phenomena but insufficient clinical or serologic features to be classified as having SLE. Antiphospholipid antibody syndrome is defined by a clinical event of thrombosis or miscarriage in the setting of persistently present and sufficiently high titers of antibodies, either anticardiolipin IgG or IgM, lupus anticoagulant, or anti-B2-glycoprotein 1 (“Sapporo” criteria ) (Orzechowski et al., 2008). In a cohort of 1000 patients who met the Sapporo criteria for definite antiphospholipid syndrome, chorea occurred in 13 (1.3%) patients. In addition to a hypercoagulable state, a variety of neurologic and movement disorders, such as chorea, ballism, dystonia, parkinsonism and paroxysmal dyskinesias have been associated with the antiphospholipid syndrome (Gutrecht et al., 1991; Lang et al., 1991; Cervera et al., 1997; Martino et al., 2006; Wu et al., 2007; Huang et al., 2008; Orzechowski et al., 2008). Other clinical features include spontaneous abortions, arthralgias, Raynaud phenomenon, digital infarctions, transient ischemic attacks, and stroke (Sanna et al., 2006). Antiphospholipid antibodies present in patients with this syndrome include lupus anticoagulant, anticardiolipin antibody, and anti-β2-glycoprotein-I antibodies. Anticardiolipin antibodies, frequently found in SLE, are absent in patients with Sydenham disease. Contralateral striatal hypermetabolism was documented by an 18F-fluorodeoxyglucose PET scan in a 23-year-old woman with alternating hemichorea and antiphospholipid syndrome (Furie et al., 1994). The striatal D1 and D2 receptor binding, measured by PET scans, was normal in one patient with SLE (Turjanski et al., 1995).
Other choreas
One of the most common forms of chorea encountered in a movement disorder clinic is drug-induced chorea associated with the use of dopaminergic or antidopaminergic drugs, anticonvulsants, and other drugs (Hyde et al., 1991; Jankovic, 1995). Levodopa-induced dyskinesia is often manifested by stereotypies, dystonia, and myclonus as well as chorea (Jankovic, 2005) (Video 15.5). Although the dominant hyperkinetic movement disorder seen in patients with tardive dyskinesia is stereotypy, some patients have associated chorea, and chorea is the chief manifestation in children with withdrawal emergent syndrome. Chorea, often accompanied by ataxia and deafness, has also been associated with mitochondrial DNA mutations (Nelson et al., 1995) and other evidence of mitochondrial cytopathies, including mutations in the POLG gene (Caer et al., 2005; Wong et al., 2008). The disorder of early-onset ataxia with oculomotor apraxia and hypoalbuminemia due to mutations in the aprataxin gene on chromosome 19p13 has also been associated with chorea (Shimazaki et al., 2002). 
Besides vasculitis, chorea has been associated with a variety of other vascular etiologies, but few have been documented pathologically. The topic of vascular chorea has been reviewed in a report of an 85-year-old woman who developed progressive dementia and chorea at age 70 (Bhatia et al., 1994). At autopsy, her brain showed neostriatal neuronal loss and gliosis associated with congophilic angiopathy and atherosclerosis. Hemichorea has been also reported following endarterectomy for carotid stenosis (Galea et al., 2008).
During the 1980s, there was an increase in the number of children with chorea as sequelae to cardiac surgery, but with modification of treatment strategies during the perioperative period, the incidence of this complication has subsequently decreased (Robinson et al., 1988). This post-pump chorea appears to be associated with prolonged time on pump, deep hypothermia, and circulatory arrest (Medlock et al., 1993; Newburger et al., 1993; du Plessis et al., 2002) (Videos 15.6 and 15.7). Others have suggested that hypoxia rather than hypothermia is critical in the development of CHAP syndrome (choreathetosis and orofacial dyskinesia, hypotonia, and pseudobulbar signs) after surgery for congenital heart disease. In most patients, the chorea persists, and fewer than 25% improve with antidopaminergic therapy such as haloperidol. Steroid-responsive chorea was described in a patient after heart transplant (Blunt et al., 1994). Although the chorea may improve, long-term studies have shown that these children have persistent deficits in memory, attention, and language (du Plessis et al., 2002). 
A variety of systemic (Janavs and Aminoff, 1998), metabolic, and neurodegenerative disorders can be associated with chorea, such as hypocalcemia or hypercalcemia, hyperglycemia (Ahlskog et al., 2001; Chu et al., 2002; Oh et al., 2002; Pisani et al., 2005; Sitburana and Ondo, 2006), hyperthyroidism (Pozzan et al., 1992), B12 deficiency (Pacchetti et al., 2002), Lesch–Nyhan syndrome (Jankovic et al., 1988; Jinnah et al., 1994; Ernst et al., 1996), propionic acidemia (Nyhan et al., 1999), and other metabolic disorders such as glutaric aciduria, gluscose transporter type 1 deficiency (Pérez-Dueñas et al., 2009), and GM1 gangliosidosis (Shulman et al., 1995; Stacy and Jankovic, 1995).
Chorea (with or without associated ballism) has been reported as the presenting feature of renal failure (Kujawa et al., 2001) and paraneoplastic striatal encephalitis (Tani et al., 2000; Vernino et al., 2002; Kinirons et al., 2003; Grant and Graus, 2009; Kleinig et al., 2009). In a series of 16 patients with paraneoplastic chorea, 11 had small-cell carcinoma, all had CRMP-5 IgG, and 6 had ANNA-1 (anti-Hu) antibodies (Vernino et al., 2002). Another metabolic cause of chorea is liver disease, particularly chronic acquired hepatocerebral degeneration (Thobois et al., 2002) (Video 15.8). Many of the metabolic choreas are associated with abnormalities on MRI scans. For example, hepatocerebral degeneration and hyperglycemic chorea are often associated with high signal intensity on T1-weighted MRI, involving the striatum and pallidum (Ahlskog et al., 2001; Thobois et al., 2002; Sitburana and Ondo, 2006). In a report of two patients with hyperglycemic hemichorea–hemiballism, Chu and colleagues (2002) found high signal intensities on T1- and T2-weighted images as well as on diffusion-weighted MRI accompanied by a reduction in diffusion coefficient, suggestive of hyperviscosity, rather than petechial hemorrhages, as the mechanism of edema in the striatum. This is also supported by another study of seven patients with hyperglycemic choreoathetosis using MRI and MR spectroscopy (Kandiah et al., 2009). Interestingly, the presence of high acanthocyte count may predispose patients with diabetes to develop hyperglycemic chorea (Pisani et al., 2005). Other disorders frequently associated with this MRI abnormality include manganese toxicity, Wilson disease, abnormal calcium metabolism, neurofibromatosis, hypoxia, and hemorrhage. Chorea has been associated with a variety of other causes, including cerebrospinal fluid leak (Mokri et al., 2006). 
Another disorder associated with chorea and frequently misdiagnosed as Huntington disease is neuroferritinopathy, a progressive but potentially treatable disorder caused by mutations in the ferritin light chain gene (FTL1), located on 19q13.3–q13.4. Patients may be initially diagnosed as “idiopathic dystonia,” “Parkinson disease,” “Huntington disease,” and a variety of other disorders. In a study of 41 genetically homogeneous subjects with the 460InsA mutation in FTL1, the mean age at onset was 39.4 ± 13.3 years (range: 13–63), beginning with chorea in 50%, focal lower limb dystonia in 42.5%, and parkinsonism in 7.5% (Chinnery et al., 2007). The disease progressed over a 5–10-year period, eventually leading to aphonia, dysphagia and severe, often asymmetrical, motor disability, and finally dementia in the advanced stages. A characteristic action-specific facial dystonia was present in 65%. Serum ferritin levels were low in the majority of males and postmenopausal females, but may be normal, particularly in premenopausal females. MRI typically shows gradient echo T2* hypointensity in red nuclei, substantia nigra, and globus pallidus; brain imaging was abnormal in all affected individuals and one presymptomatic carrier, with T1 hyperintensity in the globus pallidus and posterior putamen (Chinnery et al., 2007).
Treatment of chorea
The first step in the treatment of chorea is the identification of a specific etiology. Chorea has been treated successfully with drugs that interfere with central dopaminergic function, such as the dopamine receptor-blocking drugs (neuroleptics), reserpine, and tetrabenazine (Jankovic and Beach, 1997; Chatterjee and Frucht, 2003; Kenney and Jankovic, 2006; Sitburana and Ondo, 2006; Fasano and Bentivoglio, 2009; Jankovic, 2009). Indeed, tetrabenazine provides the most effective relief of chorea with only minimal, dose-related side effects, such as drowsiness, insomnia, depression, and parkinsonism. Tetrabenazine is effective not only for the treatment of chorea associated with HD (Ondo et al., 2002; Huntington Study Group, 2006), but also for choreatic disorders associated with tardive dyskinesia (Ondo et al., 1999), cerebral palsy (CP), and post-pump encephalopathy (Chatterjee and Frucht, 2003) (Video 15.6). While some studies have suggested that sodium valproate may be effective in the treatment of chorea (Daoud et al., 1990; Hoffman and Feinberg, 1990), other reports have been less conclusive (Sethi and Patel, 1990). Levetiracetam has been reported to markedly improve a patient with CP and postinfectious chorea (Recio et al., 2005). 
There is no consensus as to the optimal management of autoimmune chorea. Patients with Sydenham disease should be treated with penicillin prophylactically to prevent rheumatic fever. Symptomatic suppression of chorea with dopamine antagonists may lessen disability. Anticoagulation, immunosuppressants, and plasmapheresis have been utilized with variable success, and the frequent occurrence of spontaneous remissions makes the results of treatment difficult to interpret. Until prospective therapeutic trials can be designed, careful selection of treatment that best seems to suit the severity of the patient’s illness might be indicated. The presence of true vasculitis might require more aggressive management. Steroids are sometimes recommended for the treatment of autoimmune chorea, and this treatment was found effective also in chorea associated with heart transplant (Blunt et al., 1994). Stereotactic surgery is occasionally needed in very severe and disabling cases of hemichorea/hemiballism (Krauss and Mundinger, 2000).
Ballism
Chorea, athetosis, and ballism represent a continuum of involuntary, hyperkinetic movement disorders. Ballism is a form of forceful, flinging, high-amplitude, coarse, chorea (Videos 15.9 and 15.10). Ballism and chorea are often interrelated and may occur in the same patient (Harbord and Kobayashi, 1991). The involuntary movement usually affects only one side of the body; the term hemiballism is used to describe unilateral ballism. Although various structural lesions have been associated with ballism (Rossetti et al., 2003), damage to the subthalamic nucleus and the pallidosubthalamic pathways appears to play a critical role in the expression of this hyperkinetic movement disorder (Guridi and Obeso, 2001). Subthalamotomy has been found to ameliorate the motor disturbances in human and experimental parkinsonism, but the procedure can produce transient or permanent hemiballism (Bergman et al., 1990; Aziz et al., 1991; Guridi and Obeso, 2001; Chen et al., 2002). 
When caused by a hemorrhagic or ischemic stroke, the movement disorder is often preceded by hemiparesis. Anterior parietal artery stroke, without any evidence of involvement of the basal ganglia, thalamus, or subthalamic nucleus, has been associated with contralateral hemiballism and neurogenic pain (Rossetti et al., 2003). This and other similar cases suggest that the lesion associated with hemiballism may extend beyond the contralateral subthalamic nucleus and connecting structures into the adjacent internal capsule. Less common causes of hemiballism include abscess, arteriovenous malformation, cerebral trauma, hyperosmotic hyperglycemia (Ahlskog et al., 2001), multiple sclerosis, and tumor (Glass et al., 1984). Rarely, ballism occurs bilaterally (paraballism), usually due to bilateral basal ganglia strokes or calcification (Inbody and Jankovic, 1986; Vidakovic et al., 1994). In a series of 21 patients with hemiballism, hemichorea, or both, an identifiable cause was found in all (Dewey and Jankovic, 1989). Stroke was the most common cause, followed by tumors, abscesses, encephalitis, vasculitis, and other causes. In a series of 23 patients with hemiballism and 2 with biballism, Vidakovic and colleagues (1994) found ischemic and hemorrhagic strokes to be the most common causes of hemiballism. Only two patients had “pure” hemiballism, and only six showed a lesion in the subthalamic nucleus on neuroimaging studies. The prognosis for spontaneous remission was good; nine patients completely recovered, and in seven additional patients, there was complete recovery of the ballism, but mild chorea had persisted. While hemichorea–hemiballism is usually contralateral to a basal ganglia lesion, ipsilateral hemichorea–hemiballism has been described in patients with contralateral hemiparesis (Krauss et al., 1999). The mechanism of this peculiar phenomenon is unknown. Survival rates following vascular hemiballismus are similar to those of vascular disease, with only 32% survival and 27% stroke-free survival 150 months following the onset of the movement disorder (Ristic et al., 2002).
Other causes of ballism include encephalitis, Sydenham disease, SLE, basal ganglia calcifications, tuberous sclerosis, and overlap between Fisher syndrome and Guillain–Barré syndrome (Odaka et al., 1999; Postuma and Lang, 2003). In addition to structural lesions, metabolic disorders, such as hyperglycemia (Sitburana and Ondo, 2006), nutritional vitamin D deficiency (Fernandez et al., 2007), and certain drugs, such as phenytoin and lamotrigine (Yetimalar et al., 2007), have been associated with ballism.
Treatment of ballism
The frequent occurrence of spontaneous remission makes the assessment of therapy difficult. Dopamine receptor-blocking drugs such as haloperidol, chlorpromazine, pimozide, and atypical neuroleptics have been used most frequently (Dewey and Jankovic, 1989; Bashir and Manyam, 1994; Shannon, 2005). Dopamine-depleting drugs, such as reserpine and tetrabenazine, have also been used successfully (Jankovic and Beach, 1997). Tetrabenazine is our preferred drug for its rapid onset of action and its effectiveness, without the danger of inducing tardive dyskinesia if chronic antidopaminergic treatment is needed (Sitburana and Ondo, 2006). Other drugs that are sometimes beneficial in the treatment of ballism include sodium valproate and clonazepam. Finally, ventrolateral thalamotomy and other stereotactic surgeries may be needed to control violent and disabling contralateral hemiballism (Cardoso et al., 1995; Krauss and Mundinger, 2000). Although effective and relatively safe, this procedure should be used only as a last resort in patients with disabling and medically intractable movement disorder.
Athetosis
Athetosis is a slow form of chorea that consists of writhing movements resembling dystonia, but in contrast to dystonia, these movements are not sustained, patterned, repetitive, or painful. Originally described by Hammond in acquired hemidystonia and by Shaw in CP, athetosis should be viewed as a movement disorder separate from dystonia (Morris et al., 2002a). The relationship of athetosis to chorea is highlighted not only by the continuously changing direction of movement but also by the observation that chorea often evolves into athetosis or vice versa. In some patients, particularly children, chorea and athetosis often coexist, hence the term choreoathetosis. Dystonia, particularly involving the trunk causing opisthotonic posturing, also frequently accompanies athetosis, particularly in children with CP (Video 15.11). In contrast to idiopathic dystonia, athetosis associated with perinatal brain injury often causes facial grimacing and spasms, particularly during speaking and eating, and the bulbar function is usually impaired. 
Athetosis is typically present in children with CP, but may be caused by many various etiologies (Kyllerman, 1982; Kyllerman et al., 1982; Foley, 1983; Murphy et al., 1995; Goddard-Finegold, 1998; Morris et al., 2002b; Cowan et al., 2003; Ashwal et al., 2004; Koman et al., 2004; Bax et al., 2006; Keogh and Badawi, 2006). In a study of 431 children diagnosed with CP, 26.2% had hemiplegia, 34.4% had diplegia, 18.6% had quadriplegia, 14.4% had dyskinesia, and 3.9% had ataxia (Bax et al., 2006). In a study of 1817 of the 2357 (77%) children born preterm (22–32 weeks of gestation) who survived at least 5 years, CP was diagnosed in 9%, and 5% had severe, 9% moderate, and 25% minor disability; cognitive and motor disability was most common in children born at 24–28 weeks of gestation (49%) (Larroque et al., 2008). In addition to motor disorders manifested chiefly by weakness and hypertonia (i.e., spasticity, rigidity, athetosis, dystonia), patients with CP may also have cognitive impairment, epilepsy, visual and hearing problems, and other neurologic deficits. Although many patients with dyskinetic CP have well-preserved intellectual function, most of them have a variety of comorbidities. In one study the following comorbidities were documented: nonverbal 22.2% (54/243), active afebrile seizure disorder 16.9% (41/243), severe auditory impairment 11.5% (28/243), cortical blindness 9.5% (23/243), and gavage feeding requirement 7.8% (19/243) (Shevell et al., 2009). Children with ataxic-hypotonic, spastic quadriplegic, and dyskinetic CP subtypes experienced about five times the numerical burden (i.e., frequency) of comorbidities compared to children with the spastic diplegic or hemiplegic variants; dyskinetic children had a particularly high frequency of nonverbal skills (8/16, 50%) and auditory impairment (6/16, 38%).
Although there has been a steady decline in infant mortality, the incidence of CP has remained unchanged. Because of the higher frequency of premature births, the frequency of certain types of CP, such as spastic diplegia, has been increasing. In one study of children born at 25 or fewer completed weeks of gestation, half of the patients at 30 months of age were considered disabled, 18% were diagnosed with CP, and 24% had gait difficulties (Wood et al., 2000).
Kernicterus, once a common cause of CP due to bilirubin encephalopathy associated with neonatal jaundice, is now quite rare, although still an important cause of childhood disabilities in developing countries (Maisels, 2009). Besides delayed developmental milestones and athetotic or dystonic movements, patients with kernicterus often exhibit vertical ophthalmoparesis, deafness, and dysplasia of the dental enamel. Hearing abnormalities, very common in chronic bilirubin encephalopathy, can be present as the only finding of kernicterus. The high-frequency sensory neural hearing loss has been attributed to damage to the cochlear nuclei and auditory nerves, possibly as a result of bilirubin’s interference with intracellular calcium homeostasis (Shapiro and Nakamura, 2001). Unconjugated bilirubin can also cause damage to endoplasmic reticulum membranes leading to neuronal excitotoxicity and mitochondrial energy failure.
Although improved perinatal care has reduced the frequency of birth-related injuries, birth asphyxia with anoxia is still a relatively common cause of CP (Kuban and Leviton, 1994; Cowan et al., 2003). Expression of tyrosine hydroxylase was reduced in the putamen in cases of acute kernicterus and in the globus pallidus of acute and chronic post-kernicterus (Hachiya and Hayashi, 2008).
The cause of CP is multifactorial. Intrauterine insults, particularly chorioamnionitis and prolonged rupture of membranes (Murphy et al., 1995), might be responsible for many of the cases of CP. In one study of 351 full-term infants with neonatal encephalopathy, early seizures or both, excluding infants with congenital malformations and obvious chromosomal disorders, MRI showed evidence of an acute insult in 69–80% (Cowan et al., 2003). The higher figure correlated with evidence of perinatal asphyxia. The vast majority of children with neurologic deficits associated with perinatal asphyxia show abnormalities within the basal ganglia with shrinkage of the striatum. In addition, defects in myelination are often associated with a marbled gross appearance (hypermyelination-status marmoratus) or dysmyelination.
The border zones between the major cerebral arteries (“watershed”) is most vulnerable to asphyxia (Folkerth, 2005). In the striatum, the excitatory glutamate receptors and GABAergic neurons are particularly sensitive to asphyxia. Striatal neurons also die by glutamate-mediated excitotoxicity in which apoptosis may be delayed over days to weeks. Leukomalacia was the most common MRI abnormality, found in 42.5%, followed by basal ganglia lesions in 12.8%, cortical/subcortical lesions in 9.4%, malformations in 9.1%, and infarcts in 7.4%. Although athetosis is usually associated with perinatal brain injury, the neuroimaging studies often fail to show basal ganglia pathology.
Both above-normal and below-normal weight at birth are also significant risk factors for CP (Jarvis et al., 2003). These data strongly suggest that events in the immediate perinatal period are most important in the neonatal brain injury. An analysis of 58 brains of patients with clinical diagnosis of CP showed wide morphologic variation, but the authors were able to classify the brains into three major categories: thinned cerebral mantle (n = 10), hydrocephalus (n = 3), and microgyria–pachygyria (n = 45) (Tsusi et al., 1999). Of the 19 brains that were examined microscopically, four showed heterotopic gray matter, three showed cortical folding (cortical dysplasia), and three showed neuronal cytomegaly. The majority of the examined brains showed a variable degree of laminal disorganization in the cortex and disorientation of neurons, suggesting impaired neuronal migration during cortical development. Because 5–10% of patients with athetoid CP have a family history, genetic factors are considered important in the pathogenesis of this disorder (Fletcher and Foley, 1993). In a study based on a Swedish registry, 40% of cases of CP were thought to have a genetic basis (Costeff, 2004).
A growing number of studies also draw attention to inflammation and coagulation abnormalities in children with CP. The increased concentrations of interleukins, tumor necrosis factor, reactive antibodies to lupus anticoagulant, anticardiolipin, antiphospholipid, antithrombin III, epidermal growth factor, and other abnormal cytokine patterns may play an important role in the etiology of CP (Nelson et al., 1998; Kaukola et al., 2004). Kadhim and colleagues (2001) suggest that an early macrophage reaction and associated cytokine production and coagulation necrosis, coupled with intrinsic vulnerability of the immature oligodendrocyte, lead to periventricular leukomalacia, the most common neuropathologic changes found in premature infants who develop CP. Although infection and inflammation, along with free radicals, can activate the process that leads to periventricular leukomalacia and even to delayed progression (Scott and Jankovic, 1996), the cause or pathogenesis of CP is still not well understood.
Often referred to as static encephalopathy, the neurologic deficit associated with CP may progress with time. Motor development curves derived by assessing patients with the Gross Motor Function Measure, used to prognosticate gross motor function in patients with CP, indicate that, depending on their level of impairment (levels I to V) 3–10 years after birth, the natural course becomes static (Rosenbaum et al., 2002). We and others, however, found that some patients continue to progress, and others may progress after a period of static course. In about half of the patients with CP, the abnormal movements become apparent within the first year of life, but in some cases, they might not appear until the fifth decade or even later. The mechanism by which such “delayed-onset” movement disorder becomes progressive after decades of a static course is unknown, but aberrant regeneration and sprouting of nerve fibers has been considered (Scott and Jankovic, 1996). In contrast to the other forms of CP (e.g., diplegic or spastic and hemiplegic), the athetoid variety, which constitutes only about a quarter of all cases, is usually not associated with significant cognitive impairment or epilepsy. Although here we emphasize athetosis, the most common movement disorder in patients with CP is spasticity (Albright, 1995).
Many other disorders associated with developmental delay and intellectual disability can cause athetosis. Chromosomal microarray has been recommended for genetic testing of individuals with unexplained developmental delay/intellectual disability, autism spectrum disorders, or multiple congenital anomalies (Miller et al., 2010). Some are due to errors in metabolism and include acidurias, lipidoses, and Lesch–Nyhan syndrome (Jankovic et al., 1988; Stacy and Jankovic, 1995) (Table 15.1). Finally, athetotic movements, or “pseudoathetosis,” can be seen in patients with severe proprioceptive deficit (Sharp et al., 1994).
Treatment of athetosis
Athetosis usually does not respond well to pharmacologic therapy. Because dopa-responsive dystonia is sometimes confused with athetoid CP, it is prudent to treat all these patients with levodopa. If levodopa fails to provide any meaningful benefit, then anticholinergic drugs should be tried in the same manner as when treating dystonia. Although generally recommended, physical therapy might or might not prevent contractures, and its role in altering the eventual outcome is uncertain (Palmer et al., 1988). Other complications of CP, such as carpal tunnel syndrome, cervical spondylosis with radiculopathy, and myelopathy, require independent assessment and treatment (Hirose and Kadoya, 1984; Treves and Korczyn, 1986). Bilateral GPi deep brain stimulation has been reported to produce about 24.4% improvement in the Burke–Fahn–Marsden scale in adult patients with dystonia–choreoathetosis associated with CP (Vidailhet et al., 2009).
Abraham S., O’Gorman M., Shulman S.T. Anti-nuclear antibodies in Sydenham’s chorea. Adv Exp Med Biol. 1997;418:153-156.
Adam O., Jankovic J. Benign hereditary chorea. In: Walker R.H., editor. The Differential Diagnosis of Chorea. New York: Oxford University Press, 2010. in press
Ahlskog J.E., Nishimo H., Evidente V.G.H., et al. Persistent chorea triggered by hyperglycemic crisis in diabetics. Mov Disord. 2001;16:890-898.
Alarcon F., Duenas G., Cevallos N., et al. Movement disorders in 30 patients with tuberculous meningitis. Mov Disord. 2000;15:561-569.
Albright A.L. Spastic cerebral palsy. Approaches to drug treatment. CNS Drugs. 1995;4:17-27.
Allen F.H.Jr., Krabbe S.M., Corcoran P.A. A new phenotype (McLeod) in the Kell blood-group system. Vox Sang. 1961;6:555-560.
Ashwal S., Russman B.S., Blasco P.A., et al. Practice parameter: Diagnostic assessment of the child with cerebral palsy: Report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2004;62:851-863.
Asmus F., Horber V., Pohlenz J., et al. A novel TITF-1 mutation causes benign hereditary chorea with response to levodopa. Neurology. 2005;64:1952-1954.
Aziz T.Z., Peggs D., Sambrook M.A., Crossman A.R. Lesion of the subthalamic nucleus for the alleviation of 1-methyl-4-phenyl- 1,2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism in the primate. Mov Disord. 1991;6:288-292.
Basel-Vanagaite L., Muncher L., Straussberg R., et al. Mutated nup62 causes autosomal recessive infantile bilateral striatal necrosis. Ann Neurol. 2006;60:214-222.
Bashir K., Manyam B.V. Clozapine for the control of hemiballism. Clin Neuropharmacol. 1994;17:477-480.
Bassen F.A., Kornzweig A.L. Malformation of the erythrocytes in a case of atypical retinitis pigmentosa. Blood. 1950;5:381-387.
Bax M., Tydeman C., Flodmark O. Clinical and MRI correlates of cerebral palsy: the European Cerebral Palsy Study. JAMA. 2006;296:1602-1608.
Bech S., Petersen T., Nørremølle A., et al. Huntington’s disease-like and ataxia syndromes: identification of a family with a de novo SCA17/TBP mutation. Parkinsonism Relat Disord. 2010;16:12-15.
Becher M.W., Ross C.A. Intranuclear neuronal inclusions in DRPLA. Mov Disord. 1998;13:852-953.
Becher M.W., Rubinsztein D.C., Leggo J., et al. Dentatorubral and pallidoluysian atrophy (DRPLA): Clinical and neuropathological findings in genetically confirmed North American and European pedigrees. Mov Disord. 1997;12:519-530.
Behan P.O., Bone A. Hereditary chorea without dementia. J Neurol Neurosurg Psychiatry. 1977;40:687-691.
Bergman H., Wichmann T., Delong M.R. Reversal of experimental parkinsonism by lesions of the subthalamic nucleus. Science. 1990;249:1436-1438.
Bhatia K.P., Lera G., Luthert P.J., Marsden C.D. Vascular chorea: Case report with pathology. Mov Disord. 1994;9:447-450.
Blunt S.B., Brooks D.J., Kennard C. Steroid-responsive chorea in childhood following cardiac transplantation. Mov Disord. 1994;9:112-113.
Bohlega S., Riley W., Powe J., et al. Neuroacanthocytosis and aprebetalipoproteinemia. Neurology. 1998;50:1912-1914.
Breedveld G.J., Percy A.K., MacDonald M.E., et al. Clinical and genetic heterogeneity in benign hereditary chorea. Neurology. 2002;59:579-584.
Brooks D.J., Ibanez V., Playford E.D., et al. Presynaptic and postsynaptic striatal dopaminergic function in neuroacanthocytosis: A positron emission tomographic study. Ann Neurol. 1991;30:166-171.
Burke J.R., Wingfield M.S., Lewis K.E., et al. The Haw River syndrome: Entatorubropallidoluysian atrophy in an African-American family. Nat Genet. 1994;7:521-524.
Caer M., Viala K., Levy R., et al. Adult-onset chorea and mitochondrial cytopathy. Mov Disord. 2005;20:490-492.
Cardoso F. Chorea: Non-genetic causes. Curr Opin Neurol. 2004;17:433-436.
Cardoso F. Sydenham’s chorea. Curr Treat Options Neurol. 2008;10:230-235.
Cardoso F., Eduardo C., Silva A.P., Mota C.C. Chorea in 50 consecutive patients with rheumatic fever. Mov Disord. 1997;12:701-703.
Cardoso F., Jankovic J., Grossman R.G., Hamilton W.J. Outcome following stereotactic thalamotomy for dystonia and hemiballism. Neurosurgery. 1995;36:501-508.
Cardoso F., Seppi K., Mair K.J., et al. Seminar on choreas. Lancet Neurol. 2006;5:589-602.
Cardoso F., Vargas A.P., Oliveira L.D., et al. Persistent Sydenham’s chorea. Mov Disord. 1999;14:805-807.
Cervera R., Asherson R.A., Font J., et al. Chorea in the antiphospholipid syndrome: Clinical, radiologic, and immunologic characteristics of 50 patients from our clinics and the recent literature. Medicine. 1997;76:203-212.
Chatterjee A., Frucht S.J. Tetrabenazine in the treatment of severe pediatric chorea. Mov Disord. 2003;18:703-706.
Chen C.C., Lee S.T., Wu T., et al. Hemiballism after subthalamotomy in patients with Parkinson’s disease: Report of 2 cases. Mov Disord. 2002;17:1367-1371.
Ching K.H.L., Westway S.K., Gitschier J., et al. HARP syndrome is allelic with pantothenate-kinase-associated neurodegeneration. Neurology. 2002;58:1673-1674.
Chinnery P.F., Crompton D.E., Birchall D., et al. Clinical features and natural history of neuroferritinopathy caused by the FTL1 460InsA mutation. Brain. 2007;130:110-119.
Chu K., Kang D.W., Kim D.E., et al. Diffusion-weighted and gradient echo magnetic resonance findings of hemichorea-hemiballismus associated with diabetic hyperglycemia: A hyperviscosity syndrome? Arch Neurol. 2002;59:448-452.
Church A.J., Cardoso F., Dale R.C., et al. Anti-basal ganglia antibodies in acute and persistent Sydenham’s chorea. Neurology. 2002;59:227-231.
Costeff H. Estimated frequency of genetic and nongenetic causes of congenital idiopathic cerebral palsy in west Sweden. Ann Hum Genet. 2004;68:515-520.
Cowan F., Rutherford M., Groenendaal F., et al. Origin and timing of brain lesions in term infants with neonatal encephalopathy. Lancet. 2003;361:736-742.
Dale R.C., Church A.J., Cardoso F., et al. Poststreptococcal acute disseminated encephalomyelitis with basal ganglia involvement and auto-reactive antibasal ganglia antibodies. Ann Neurol. 2001;50:588-595.
Danek A., Jung H.H., Melone M.A., et al. Neuroacanthocytosis: New developments in a neglected group of dementing disorders. J Neurol Sci. 2005;229–230:171-186.
Danek A., Rubio J.P., Rampoldi L., et al. McLeod neuroacanthocytosis: Genotype and phenotype. Ann Neurol. 2001;50:755-764.
Danek A., Tison F., Rubio J., et al. The chorea of McLeod syndrome. Mov Disord. 2001;16:882-889.
Daoud A.S., Zaki M., Shakir R., Al-Saleh Q. Effectiveness of sodium valproate in the treatment of Sydenham’s chorea. Neurology. 1990;40:1140-1141.
Devos D., Vuillaume I., de Becdelievre A., et al. New syndromic form of benign hereditary chorea is associated with a deletion of TITF-1 and PAX-9 contiguous genes. Mov Disord. 2006;21:2237-2240.
de Vries B.B., Arts W.F., Breedveld G.J., et al. Benign hereditary chorea of early onset maps to chromosome 14q. Am J Hum Genet. 2000;66:136-142.
Dewey R.B., Jankovic J. Hemiballism-hemichorea: Clinical and pharmacologic findings in 21 patients. Arch Neurol. 1989;46:862-867.
DeYebenes J.G., Brin M.F., Mena M.A., et al. Neurochemical findings in neuroacanthocytosis. Mov Disord. 1988;3:300-312.
Dobson-Stone C., Velayos-Baeza A., Filippone L.A., et al. Chorein detection for the diagnosis of chorea-acanthocytosis. Ann Neurol. 2004;56:299-302.
Dotti M.T., Battisti C., Malandrini A., et al. McLeod syndrome and neuroacanthocytosis with a novel mutation in the XK gene. Mov Disord. 2000;15(6):1282-1284.
du Plessis A.J., Bellinger D.C., Gauvreau K., et al. Neurologic outcome of choreoathetoid encephalopathy after cardiac surgery. Pediatr Neurol. 2002;27:9-17.
Ernst M., Zametkin A.J., Matochik J.A., et al. Presynaptic dopaminergic deficits in Lesch-Nyhan disease. N Engl J Med. 1996;334:1568-1572.
Eshel G., Lahat E., Azizi E., et al. Chorea as a manifestation of rheumatic fever: A 30-year survey (1960–1990). Eur J Pediatr. 1993;152:645-646.
Fasano A., Bentivoglio A.R. Tetrabenazine. Expert Opin Pharmacother. 2009;10:2883-2896.
Feinberg T.E., Cianci C.D., Morrow J.S., et al. Diagnostic tests for choreoacanthocytosis. Neurology. 1991;41:1000-1006.
Feldman B.M., Zabriskie J.B., Silverman E.D., Laxer R.M. Diagnostic use of B-cell alloantigen D8/17 in rheumatic chorea. J Pediatr. 1993;123:84-86.
Fernandez M., Raskind W., Matsushita M., et al. Hereditary benign chorea: Clinical and genetic features of a distinct disease. Neurology. 2001;57:106-110.
Fernandez R., Ambika A., Dure L.S. Nutritional vitamin D deficiency presenting as hemichorea. J Child Neurol. 2007;22:74-76.
Ferrara A.M., De Michele G., Salvatore E., et al. A novel NKX2.1 mutation in a family with hypothyroidism and benign hereditary chorea. Thyroid. 2008;18:1005-1009.
Fletcher N.A., Foley J. Parental age, genetic mutation, and cerebral palsy. J Med Genet. 1993;30:44-46.
Foley J. The athetoid syndrome: A review of a personal series. J Neurol Neurosurg Psychiatry. 1983;46:289-298.
Folkerth R.D. Neuropathologic substrate of cerebral palsy. J Child Neurol. 2005;20:940-949.
Fontenelle L.F., Leite M.A. Treatment-resistant self-mutilation, tics, and obsessive-compulsive disorder in neuroacanthocytosis: a mouth guard as a therapeutic approach. J Clin Psychiatry. 2008;69:1186-1187.
Forni G.L., Balocco M., Cremonesi L., et al. Regression of symptoms after selective iron chelation therapy in a case of neurodegeneration with brain iron accumulation. Mov Disord. 2008;23:904-907.
Fox S.H., Nieves A., Bergeron C., Lang A.E. Pure cerebello-olivary degeneration of Marie, Foix, and Alajouanine presenting with progressive cerebellar ataxia, cognitive decline, and chorea. Mov Disord. 2003;18:1550-1554.
Friedman J.H., Ambler M. A case of senile chorea. Mov Disord. 1990;5:251-253.
Furie R., Ishikawa T., Dhawan V., Eidelberg D. Alternating hemichorea in primary antiphospholipid syndrome: Evidence for contralateral striatal hypermetabolism. Neurology. 1994;44:2197-2199.
Galea I., Norwood F., Phillips M.J., et al. Pearls & Oy-sters: resolution of hemichorea following endarterectomy for severe carotid stenosis. Neurology. 2008;71:e80-e82.
Gallo B.V., Shulman L.M., Weiner W.J., et al. HIV encephalitis presenting with severe generalized chorea. Neurology. 1996;46:1163-1165.
Garvey M.A., Swedo S.E. Sydenham’s chorea. Clinical and therapeutic update. Adv Exp Med Biol. 1997;418:115-120.
Giedd J.N., Rapoport J.L., Kruesi M.J.P., et al. Sydenham’s chorea: Magnetic resonance imaging of the basal ganglia. Neurology. 1995;45:2199-2202.
Glass J.P., Jankovic J., Borit A. Hemiballismus and metastatic brain tumor. Neurology. 1984;34:204-207.
Goddard-Finegold J. Perinatal aspects of cerebral palsy. In: Miller G., Clark G.D., editors. The Cerebral Palsies. Boston: Butterworth-Heinemann; 1998:151-173.
Goetz C.G. William Osler: On chorea: On Charcot. Ann Neurol. 2000;47:404-407.
Grant R., Graus F. Paraneoplastic movement disorders. Mov Disord. 2009;24:1715-1724.
Greenstein P.E., Vonsattel J.P., Margolis R.L., Joseph J.T. Huntington’s disease like-2 neuropathology. Mov Disord. 2007;22:1416-1423.
Grimes D.A., Lang A.E., Bergeron C. Late adult onset chorea with typical pathology of Hallervorden-Spatz syndrome. J Neurol Neurosurg Psychiatry. 2000;69:392-395.
Guridi J., Obeso J.A. The subthalamic nucleus, hemiballismus and Parkinson’s disease: Reappraisal of a neurosurgical dogma. Brain. 2001;124:5-19.
Gutrecht J.A., Kattwinkel N., Stillman M.J. Retinal migraine, chorea, and retinal artery thrombosis in a patient with primary antiphospholipid antibody syndrome. J Neurol. 1991;238:55-56.
Hachiya Y., Hayashi M. Bilirubin encephalopathy: A study of neuronal subpopulations and neurodegenerative mechanisms in 12 autopsy cases. Brain Dev. 2008;30:269-278.
Hagerman R.J., Leehey M., Heinrichs W., et al. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology. 2001;57:127-130.
Hallett M., Kaufman C. Physiological observations in Sydenham’s chorea. J Neurol Neurosurg Psychiatry. 1981;44:829-832.
Hanajima R., Ugawa Y., Terao Y., et al. Intracortical inhibition of the motor cortex is normal in chorea. J Neurol Neurosurg Psychiatry. 1999;66:783-786.
Harbord M.G., Kobayashi J.S. Fever producing ballismus in patients with choreoathetosis. J Child Neurol. 1991;6:49-52.
Hardie R.J., Pullon H.W.H., Harding A.E., et al. Neuroacanthocytosis: A clinical, haematological and pathological study of 19 cases. Brain. 1991;114:13-49.
Harrison N.A., Church A., Nisbet A., et al. Late recurrences of Sydenham’s chorea are not associated with anti-basal ganglia antibodies. J Neurol Neurosurg Psychiatry. 2004;75:1478-1479.
Hayflick S.J., Westaway S.K., Levinson B., et al. Genetic, clinical, and radiographic delineation of Hallervorden-Spatz syndrome. N Engl J Med. 2003;348:33-40.
Hirose G., Kadoya S. Cervical spondylotic radiculo-myelopathy in patients with athetoid-dystonic cerebral palsy: Clinical evaluation and surgical treatment. J Neurol Neurosurg Psychiatry. 1984;47:775-780.
Ho M.F., Chalmers R.M., Davis M.B., et al. A novel point mutation in the McLeod syndrome gene in neuroacanthocytosis. Ann Neurol. 1996;39:672-675.
Hoffman A.S., Feinberg T.E. Successful treatment of age-related chorea with sodium valproate. J Am Geriatr Soc. 1990;38:56-58.
Holmes S.E., O’Hearn E., Rosenblatt A., et al. A repeat expansion in the gene encoding junctophilin-3 is associated with Huntington disease-like 2. Nat Genet. 2001;29:377-378.
Houlden H., Lincoln S., Farrer M., et al. Compound heterozygous PANK2 mutations confirm HARP and Hallervorden-Spatz syndromes are allelic. Neurology. 2003;61:1423-1426.
Huang Y.C., Lyu R.K., Chen S.T., et al. Parkinsonism in a patient with antiphospholipid syndrome –case report and literature review. J Neurol Sci. 2008;267:166-169.
Huntington Study Group. Tetrabenazine as antichorea therapy in Huntington disease: a randomized controlled trial. Neurology. 2006;66:366-372.
Hyde T.M., Hotson J.R., Kleinman J.E. Differential diagnosis of choreiform tardive dyskinesia. J Neuropsychiatry. 1991;3:255-268.
Igarashi S., Koide R., Shimohata T., et al. Suppression of aggregate formation and apoptosis by transglutaminase inhibitors in cells expressing truncated DRPLA proteins with an expanded polyglutamine stretch. Nat Genet. 1998;18:111-117.
Ikeuchi T., Koide R., Tanaka H., et al. Dentatorubral-pallidoluysian atrophy: Clinical features are closely related to unstable expansions of trinucleotide (CAG) repeat. Ann Neurol. 1995;37:769-775.
Inbody S., Jankovic J. Hyperkinetic mutism: Bilateral ballism and basal ganglia calcification. Neurology. 1986;36:825-827.
Janavs J., Aminoff M.J. Dystonia and chorea in acquired systemic disorders. J Neurol Neurosurg Psychiatry. 1998;65:436-445.
Jankovic J. Tardive syndromes and other drug-induced movement disorders. Clin Neuropharmacol. 1995;18:197-214.
Jankovic J. Motor fluctuations and dyskinesias in Parkinson’s disease: Clinical manifestations. Mov Disord. 2005;20(Suppl 11):S11-S16.
Jankovic J. Treatment of hyperkinetic movement disorders. Lancet Neurol. 2009;8:844-856.
Jankovic J., Beach J. Long-term effects of tetrabenazine in hyperkinetic movement disorders. Neurology. 1997;48:358-362.
Jankovic J., Caskey T.C., Stout J.T., Butler I. Lesch-Nyhan syndrome: A study of motor behavior and CSF monoamine turnover. Ann Neurol. 1988;23:466-469.
Jankovic J., Fahn S. Choreas. In: Rowland L.P., Pedley T., editors. Merritt’s Textbook of Neurology. Philadelphia, PA: Lippincott Williams & Wilkins, 2009.
Jankovic J., Killian J.M., Spitz M.C. Neuroacanthocytosis syndrome and choreo-acanthocytosis (Levine-Critchley syndrome). Neurology. 1985;35:1679.
Jarvis S., Glinianaia S.V., Torrioli M.G., et al. Surveillance of Cerebral Palsy in Europe (SCPE) collaboration of European Cerebral Palsy Registers. Cerebral palsy and intrauterine growth in single births: European collaborative study. Lancet. 2003;362:1106-1111.
Jinnah H.A., Wojcik B.E., Hunt M., et al. Dopamine deficiency in a genetic mouse model of Lesch-Nyhan disease. J Neurosci. 1994;14:1164-1175.
Jung H.H., Hergerberg M., Kneifel S., et al. McLeod syndrome: A novel mutation, predominant psychiatric manifestations, and distinct striatal imaging findings. Ann Neurol. 2001;49:384-392.
Kadhim H., Tabarki B., Verellen G., et al. Inflammatory cytokines in the pathogenesis of periventricular leukomalacia. Neurology. 2001;56:1278-1284.
Kambouris M., Bohlega S., Al-Tahan A., Meyer B.F. Localization of the gene for a novel autosomal recessive neurodegenerative Huntington-like disorder to 4p15.3. Am J Hum Genet. 2000;66:445-452.
Kandiah N., Tan K., Lim C.C., Venketasubramanian N. Hyperglycemic choreoathetosis: role of the putamen in pathogenesis. Mov Disord. 2009;24:915-919.
Kaukola T., Satyaraj E., Patel D.D., et al. Cerebral palsy is characterized by protein mediators in cord serum. Ann Neurol. 2004;55:186-194.
Kawaguchi Y., Okamoto T., Taniwaki M., et al. CAG expansions in a novel gene from Machado-Joseph disease at chromosome 14q32.1. Nat Genet. 1994;8:221-228.
Kenney C., Jankovic J. Tetrabenazine in the treatment of hyperkinetic movement disorders. Expert Rev Neurother. 2006;6:7-17.
Keogh J.M., Badawi N. The origins of cerebral palsy. Curr Opin Neurol. 2006;19:129-134.
Kiessling L.S., Marcotte A.C., Culpepper L. Antineuronal antibodies in movement disorders. Pediatrics. 1993;92:39-43.
Kim J.-S., Lee K.-S., Le K.-H., et al. Evidence of thalamic disinhibition in patients with hemichorea: Semiquantitative analysis using SPECT. J Neurol Neurosurg Psychiatry. 2002;72:329-333.
Kinirons P., Fulton A., Keoghan M., et al. Paraneoplastic limbic encephalitis (PLE) and chorea associated with CRMP-5 neuronal antibody. Neurology. 2003;61:1623-1624.
Kleiner-Fisman G., Rogaeva E., Halliday W., et al. Benign hereditary chorea: Clinical, genetic, and pathological findings. Ann Neurol. 2003;54:244-247.
Kleiner-Fisman G., Calingasan N.Y., Putt M., et al. Alterations of striatal neurons in benign hereditary chorea. Mov Disord. 2005;20:1353-1357.
Kleiner-Fisman G., Lang A.E. Benign hereditary chorea revisited: A journey to understanding. Mov Disord. 2007;22:2297-2305.
Kleinig T.J., Thompson P.D., Kneebone C.S. Chorea, transverse myelitis, neuropathy, and a distinctive MRI: Paraneoplastic manifestations of probable small cell lung cancer. J Clin Neurosci. 2009;16:964-966.
Koide R., Ikeuchi T., Onodera O., et al. Unstable expansion of CAG repeat in hereditary dentatorubral-pallidoluysian atrophy (DRPLA). Nat Genet. 1994;6:9-13.
Koide R., Onodera O., Ikeuchi T., et al. Atrophy of the cerebellum and brainstem in dentatorubral pallidoluysian atrophy: Influence of CAG repeat size on MRI findings. Neurology. 1997;49:1605-1612.
Koman L.A., Smith B.P., Shilt J.S. Cerebral palsy. Lancet. 2004;363:1619-1631.
Komure O., Sano A., Nishino N., et al. DNA analysis in hereditary dentatorubral-pallidoluysian atrophy: Correlation between CAG repeat length and phenotypic variation and the molecular basis anticipation. Neurology. 1995;45:143-149.
Kovacs G.G., Murrell J.R., Horvath S., et al. TARDBP variation associated with frontotemporal dementia, supranuclear gaze palsy, and chorea. Mov Disord. 2009;24:1843-1847.
Krack P. Relicts of dancing mania: The dancing procession of Echternach. Neurology. 1999;55:2169-2172.
Krauss J.K., Mundinger F. Surgical treatment of hemiballism and hemichorea. In: Krauss J.K., Jankovic J., Grossman R.G., editors. Surgery for Movement Disorders. Philadelphia: Lippincott Williams & Wilkins; 2000:397-403.
Krauss J.K., Pohle T., Borremans J.J. Hemichorea and hemiballism associated with contralateral hemiparesis and ipsilateral basal ganglia lesions. Mov Disord. 1999;14:497-501.
Kruer M.C., Paisán-Ruiz C., Boddaert N., et al. Defective FA2H leads to a novel form of neurodegeneration with brain iron accumulation (NBIA). Ann Neurol. 2010;68:611-618.
Kuban K.C.K., Leviton A. Cerebral palsy. N Engl J Med. 1994;330:188-195.
Kujawa K.A., Niemi V., Tomasi M.A., et al. Ballistic-choreic movement as the presenting feature of renal cancer. Arch Neurol. 2001;58:1133-1135.
Kurian M.A., Morgan N.V., MacPherson L., et al. Phenotypic spectrum of neurodegeneration associated with mutations in the PLA2G6 gene (PLAN). Neurology. 2008;70:1623-1629.
Kurian M.A., Li Y., Zhen J., et al. Clinical and molecular characterisation of hereditary dopamine transporter deficiency syndrome: an observational cohort and experimental study. Lancet Neurol. 2011;10(1):54-62.
Kurlan R. The PANDAS hypothesis: Losing its bite? Mov Disord. 2004;19:371-374.
Kurlan R., Johnson D., Kaplan E.L., and the Tourette Syndrome Study Group. Streptococcal infection and exacerbations of childhood tics and obsessive-compulsive symptoms: a prospective blinded cohort study. Pediatrics. 2008;121:1188-1197.
Kuroharas K., Kuroda Y., Maruyama H., et al. Homozygosity for an allele carrying intermediate CAG repeats in the dentatorubral-pallidoluysian atrophy (DRPLA) gene results in spastic paraplegia. Neurology. 1997;48:1087-1090.
Kwak C., Dat Vuong K., Jankovic J. Premonitory sensory phenomenon in Tourette’s syndrome. Mov Disord. 2003;18:1530-1533.
Kwak C., Vuong K.D., Jankovic J. Migraine headache in patients with Tourette syndrome. Arch Neurol. 2003;60:1595-1598.
Kyllerman M. Dyskinetic cerebral palsy: II. Pathogenetic risk factors and intrauterine growth. Acta Pediatr Scand. 1982;71:551-558.
Kyllerman M., Bager B., Bensch J., et al. Dyskinetic cerebral palsy: I. Clinical categories, associated neurological abnormalities and incidences. Acta Pediatr Scand. 1982;71:543-550.
La Spada A.R., Paulson H.L., Fischbeck K.H. Trinucleotide repeat expansion in neurological disease. Ann Neurol. 1994;36:814-822.
Lang A.E., Sethi K.D., Provias J.P., Deck J.H. What is it? Case 1,1991: A severe and fatal systemic illness first presenting with a movement disorder. Mov Disord. 1991;6:362-370.
Larroque B., Ancel P.Y., Marret S., et alEPIPAGE Study group. Neurodevelopmental disabilities and special care of 5-year-old children born before 33 weeks of gestation (the EPIPAGE study): a longitudinal cohort study. Lancet. 2008;371:813-820.
Lieberman A.P., Robitaille Y., Trojanowski J.Q., et al. Polyglutamine-containing aggregates in neuronal intranuclear inclusion disease. Lancet. 1998;351:884.
Lossos A., Dobson-Stone C., Monaco A.P., et al. Early clinical heterogeneity in choreoacanthocytosis. Arch Neurol. 2005;62:611-614.
Maia D.P., Teixeira A.L.Jr., Quintao Cunningham M.C., Cardoso F. Obsessive compulsive behavior, hyperactivity, and attention deficit disorder in Sydenham chorea. Neurology. 2005;64:1799-1801.
Maisels M.J. Neonatal hyperbilirubinemia and kernicterus – not gone but sometimes forgotten. Early Hum Dev. 2009;85(11):727-732.
Malandrini A., Fabrizi G.M., Bartalucci P., et al. Clinicopathological study of familial late infantile Hallervorden-Spatz disease: A particular form of neuroacanthocytosis. Childs Nerv Syst. 1996;12:155-160.
Margolis R.L., Holmes S.E., Rosenblatt A., et al. Huntington’s disease-like 2 (HDL2) in North America and Japan. Ann Neurol. 2004;56:670-674.
Margolis R.L., O’Hearn E., Rosenblatt A., et al. A disorder similar to Huntington’s disease is associated with a novel CAG repeat expansion. Ann Neurol. 2001;50:373-380.
Mars H., Lewis L.A., Robertson A.L.Jr., et al. Familial hypo-beta-lipoproteinemia: a genetic disorder of lipid metabolism with nervous system involvement. Am J Med. 1969;46(6):886-900.
Martino D., Chew N.K., Mir P., et al. Atypical movement disorders in antiphospholipid syndrome. Mov Disord. 2006;21:944-949.
McNeill A., Birchall D., Hayflick S.J., et al. T2* and FSE MRI distinguishes four subtypes of neurodegeneration with brain iron accumulation. Neurology. 2008;70:1614-1619.
Medlock M.D., Cruse R.S., Winek S.J., et al. A 10-year experience with postpump chorea. Ann Neurol. 1993;34:820-826.
Miller D.T., Adam M.P., Aradhya S., et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86:749-764.
Mittleman B.B. Cytokine networks in Sydenham’s chorea and PANDAS. Adv Exp Med Biol. 1997;418:933-935.
Mokri B., Ahlskog J.E., Luetmer P.H. Chorea as a manifestation of spontaneous CSF leak. Neurology. 2006;67:1490-1491.
Moore R.C., Xiang F., Monaghan J., et al. Huntington disease phenocopy is a familial prion disease. Am J Hum Genet. 2001;69:1385-1388.
Morris J.G.L., Jankelowitz S.K., Fung V.S.C., et al. Athetosis: I. Historical considerations. Mov Disord. 2002;17:1278-1280.
Morris J.G.L., Grattan-Smith P., Jankelowitz S.K., et al. Athetosis: II. The syndrome of mild athetoid cerebral palsy. Mov Disord. 2002;17:1281-1287.
Muñoz E., Campdelacreu J., Ferrer I., et al. Severe cerebral white matter involvement in a case of dentatorubropallidoluysian atrophy studied at autopsy. Arch Neurol. 2004;61:946-949.
Muñoz E., Mila M., Sanchez A., et al. Dentatorubropallidoluysian atrophy in a Spanish family: A clinical, radiological, pathological, and genetic study. J Neurol Neurosurg Psychiatry. 1999;67:811-814.
Murphy D.J., Sellers S., MacKenzie I.Z., et al. Case-control study of antenatal and intrapartum risk factors for cerebral palsy in very preterm singleton babies. Lancet. 1995;346:1449-1454.
Nath A., Jankovic J., Pettigrew L.C. Movement disorders and AIDS. Neurology. 1987;37:36-41.
Nelson I., Hanna M.G., Alsanjari N., et al. A new mitochondrial DNA mutation associated with progressive dementia and chorea: A clinical, pathological, and molecular genetic study. Ann Neurol. 1995;37:400-403.
Nelson K.B., Dambrosia J.M., Grether J.K., Phillips T.M. Neonatal cytokines and coagulation factors in children with cerebral palsy. Ann Neurol. 1998;44:665-675.
Newburger J.W., Jonas R.A., Wernovsky G., et al. A comparison of the perioperative neurologic effects of hypothermic circulatory arrest versus low-flow cardiopulmonary bypass in infant heart surgery. N Engl J Med. 1993;329:1057-1064.
Nicholl D.J., Sutton I., Dotti M.T., et al. White matter abnormalities on MRI in neuroacanthocytosis. J Neurol Neurosurg Psychiatry. 2004;75:1200-1201.
Nishiyama K., Nakamura K., Murayama S., et al. Regional and cellular expression of dentatorubral-pallidoluysian atrophy gene in brains of normal and affected individuals. Ann Neurol. 1997;41:599-605.
Nyhan W.L., Bay C., Beyer E.W., Mazi M. Neurologic nonmetabolic presentation of propionic acidemia. Arch Neurol. 1999;56:1143-1147.
Odaka M., Yuki N., Hirata K. Bilateral ballism in a patient with overlapping Fisher’s and Guillain-Barré syndromes. J Neurol Neurosurg Psychiatry. 1999;67:206-208.
Oh S.-H., Lee K.-Y., Im J.-H., Lee M.-S. Chorea associated with non-ketotic hyperglycemia and hyperintensity basal ganglia lesion on T1-weighted brain MRI study: A meta-analysis of 53 cases including four present cases. J Neurol Sci. 2002;200:57-62.
Okamura-Oho Y., Miyashita T., Nagao K., et al. Dentatorubral-pallidoluysian atrophy protein is phosphorylated by c-Jun NH(2)-terminal kinase. Hum Mol Genet. 2003;12:1535-1542.
Ondo W.G., Hanna P.A., Jankovic J. Tetrabenazine treatment for tardive dyskinesia: Assessment by randomized videotape protocol. Am J Psychiatry. 1999;156:1279-1281.
Ondo W.G., Tintner R., Thomas M., Jankovic J. Tetrabenazine treatment for Huntington’s disease-associated chorea. Clin Neuropharmacol. 2002;25:300-302.
Orrell R.W., Amrolia P.J., Heald A., et al. Acanthocytosis, retinitis pigmentosa, and pallidal degeneration: A report of three patients, including the second reported case with hypoprebetalipoproteinemia (HARP) syndrome. Neurology. 1995;45:487-492.
Orzechowski N.M., Wolanskyj A.P., Ahlskog J.E., et al. Antiphospholipid antibody-associated chorea. J Rheumatol. 2008;35:2165-2170.
Pacchetti C., Cristina S., Nappi G., et al. Reversible chorea and focal dystonia in vitamin B12 deficiency. N Engl J Med. 2002;347:295.
Paisan-Ruiz C., Bhatia K.P., Li A., et al. Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann Neurol. 2008;65(1):19-23.
Palmer F.B., Shapiro B.K., Wachtel R.C., et al. The effects of physical therapy on cerebral palsy: A controlled trial in infants with spastic diplegia. N Engl J Med. 1988;318:803-808.
Panamonta M., Chaikitpinyo A., Auvichayapat N., et al. Evolution of valve damage in Sydenham’s chorea during recurrence of rheumatic fever. Int J Cardiol. 2007;119:73-79.
Pardo J., Marcos A., Bhathal H., et al. Chorea as a form of presentation of human immunodeficiency virus-associated dementia complex. Neurology. 1998;50:568-569.
Paz J.A., Silva C.A., Marques-Dias M.J. Randomized double-blind study with prednisone in Sydenham’s chorea. Pediatr Neurol. 2006;34:264-269.
Pérez-Dueñas B., Prior C., Ma O., et al. Childhood chorea with cerebral hypotrophy. A treatable GLUT1 energy failure syndrome. Arch Neurol. 2009;66:1410-1414.
Piccolo I., Causarano R., Sterzi R., et al. Chorea in patients with AIDS. Acta Neurol Scand. 1999;100:332-336.
Pisani A., Diomedi M., Rum A., et al. Acanthocytosis as a predisposing factor for non-ketotic hyperglycaemia induced chorea-ballism. J Neurol Neurosurg Psychiatry. 2005;76:1717-1719.
Postuma R.B., Lang A.E. Hemiballism: revisiting a classic disorder. Lancet Neurol. 2003;2:661-668.
Pozzan G.B., Battistella P.A., Rigon F., et al. Hyperthyroid-induced chorea in an adolescent girl. Brain Dev. 1992;14:126-127.
Racette B.A., Perry A., D’Avossa G., Perlmutter J.S. Late-onset neurodegeneration with brain iron accumulation type 1: Expanding the clinical spectrum. Mov Disord. 2001;16:1148-1152.
Rampoldi L., Danek A., Monaco A.P. Clinical features and molecular basis of neuroacanthocytosis. J Mol Med. 2002;80:475-491.
Rampoldi L., Dobson-Stone C., Rubio J.P., et al. A conserved sorting-associated protein is mutant in chorea-acanthocytosis. Nat Genet. 2001;28:119-120.
Recio M.V., Hauser R.A., Louis E.D., et al. Chorea in a patient with cerebral palsy: Treatment with levetiracetam. Mov Disord. 2005;20:762-764.
Rinne J.O., Daniel S.E., Scaravilli F., et al. Nigral degeneration in neuroacanthocytosis. Neurology. 1994;44:1629-1632.
Rinne J.O., Daniel S.E., Scaravilli F., et al. The neuropathological features of neuroacanthocytosis. Mov Disord. 1994;9:297-304.
Ristic A., Marinkovic J., Dragasevic N., et al. Long-term prognosis of vascular hemiballismus. Stroke. 2002;33:2109-2111.
Robinson R.O., Samuels M., Pohls K.R.E. Choreic syndrome after cardiac surgery. Arch Dis Child. 1988;63:1466-1469.
Rodopman-Arman A., Yazgan Y., Berkem M., Eraksoy M. Are sensory phenomena present in Sydenham’s chorea? Evaluation of 13 cases. Neuropediatrics. 2004;35:242-245.
Rosenbaum P.L., Walter S.D., Hanna S.E., et al. Prognosis for gross motor function in cerebral palsy: Creation of motor development curves. JAMA. 2002;288:1357-1363.
Rosenblatt A., Ranen N.G., Rubinsztein D.C., et al. Patients with features similar to Huntington’s disease, without CAG expansion in huntingtin. Neurology. 1998;51:215-220.
Ross C.A., Margolis R.L., Rosenblatt A., et al. Huntington disease and the related disorder, dentatorubral-pallidoluysian atrophy (DRPLA). Medicine. 1997;76:305-308.
Rossetti A.O., Ghika J.A., Vingerhoets F., et al. Neurogenic pain and abnormal movements contralateral to an anterior parietal artery stroke. Arch Neurol. 2003;60:1004-1006.
Rubio J.P., Danek A., Stone C., et al. Chorea-acanthocytosis: Genetic linkage to chromosome 9q21. Am J Hum Genet. 1997;61:899-908.
Rudnicki D.D., Holmes S.E., Lin M.W., et al. Huntington’s disease-like 2 is associated with CUG repeat-containing RNA foci. Ann Neurol. 2007;61:272-282.
Rudnicki D.D., Pletnikova O., Vonsattel J.P., et al. A comparison of huntington disease and huntington disease-like 2 neuropathology. J Neuropathol Exp Neurol. 2008;67:366-374.
Ruiz G., Gomez-Tortosa E., del Barrio A., et al. Senile chorea: A multicenter prospective study. Acta Neurol Scand. 1997;95:180-183.
Saiki S., Sakai K., Kitagawa Y., et al. Mutation in the CHAC gene in a family of autosomal dominant chorea-acanthocytosis. Neurology. 2003;61:1614-1616.
Sakai T., Antoku Y., Iwashita H., et al. Chorea-acanthocytosis: Abnormal composition of covalently bound fatty acids of erythrocyte membrane proteins. Ann Neurol. 1991;29:664-669.
Sanna G., D’Cruz D., Cuadrado M.J. Cerebral manifestations in the antiphospholipid (Hughes) syndrome. Rheum Dis Clin North Am. 2006;32:465-490.
Schneider S.A., Bhatia K.P. Three faces of the same gene: FA2H links neurodegeneration with brain iron accumulation, leukodystrophies, and hereditary spastic paraplegias. Ann Neurol. 2010;68:575-577.
Schneider S.A., Bhatia K.P., Hardy J. Complicated recessive dystonia parkinsonism syndromes. Mov Disord. 2009;24:490-499.
Schneider S.A., van de Warrenburg B.P., Hughes T.D., et al. Phenotypic homogeneity of the Huntington disease-like presentation in a SCA17 family. Neurology. 2006;67:1701-1703.
Schneider S.A., Walker R.H., Bhatia K.P. The Huntington’s disease-like syndromes: what to consider in patients with a negative Huntington’s disease gene test. Nat Clin Pract Neurol. 2007;3:517-525.
Schrag A., Quinn N.P., Bhatia K.P., et al. Benign hereditary chorea: Entity or syndrome? Mov Disord. 2000;15:280-288.
Scott B., Jankovic J. Delayed-onset progressive movement disorders. Neurology. 1996;46:68-74.
Sethi K.D., Patel B.P. Inconsistent response to divalproex sodium in hemichorea/hemiballism. Neurology. 1990;40:1630-1631.
Sevigny J.J., Chin S.S., Milewski Y., et al. HIV encephalitis simulating Huntington’s disease. Mov Disord. 2005;20:610-613.
Shannon K.M. Hemiballismus. Curr Treat Options Neurol. 2005;7:203-210.
Shapiro S.M., Nakamura H. Bilirubin and the auditory system. J Perinatol. 2001;21(Suppl 1):S52-S55.
Sharp D., Blinderman L., Combs K.A., et al. Cloning and gene defects in microsomal triglyceride transfer protein associated with abetalipoproteinemia. Nature. 1993;365(6441):65-69.
Sharp F.R., Rando T.A., Greenberg S.A., et al. Pseudochoreoathetosis: Movements associated with loss of proprioception. Arch Neurol. 1994;51:1103-1109.
Shevell M. Hallervorden and history. N Engl J Med. 2003;348:3-4.
Shevell M.I., Dagenais L., Hall N., REPACQ Consortium. Comorbidities in cerebral palsy and their relationship to neurologic subtype and GMFCS level. Neurology. 2009;72:2090-2096.
Shimazaki H., Takiyama Y., Sakoe K., et al. Early-onset ataxia with ocular motor apraxia and hypoalbuminemia: The aprataxin gene mutations. Neurology. 2002;59:590-595.
Shimohata T., Hara K., Sanpei K., et al. Novel locus for benign hereditary chorea with adult onset maps to chromosome 8q21.3 q23.3. Brain. 2007;130:2302-2309.
Shulman L.M., Lang A.E., Jankovic J., et al. What is it? Case 1, 1995: Psychosis, dementia, chorea, ataxia, and supranuclear gaze dysfunction. Mov Disord. 1995;10:257-262.
Singer H.S., Gause C., Morris C., Lopez P., Tourette Syndrome Study Group. Serial immune markers do not correlate with clinical exacerbations in pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections. Pediatrics. 2008;121:1198-1205.
Sitburana O., Ondo W.G. Tetrabenazine for hyperglycemic-induced hemichorea-hemiballismus. Mov Disord. 2006;21:2023-2025.
Spitz M.C., Jankovic J., Killian J.M. Familial tic disorder, parkinsonism, motor neuron disease, and acanthocytosis: A new syndrome. Neurology. 1985;35:366-377.
Stacy M., Jankovic J. Rare childhood movement disorders associated with metabolic and neurodegenerative disorders. In: Roberts M.M., Eapen V., editors. Movement and Allied Disorders in Childhood. Chichester, England: John Wiley; 1995:177-197.
Stevanin G., Camuzat A., Holmes S.E., et al. CAG/CTG repeat expansions at the Huntington’s disease-like 2 locus are rare in Huntington’s disease patients. Neurology. 2002;58:965-967.
Stevanin G., Fujigasaki H., Lebre A.S., et al. Huntington’s disease-like phenotype due to trinucleotide repeat expansions in the TBP and JPH3 genes. Brain. 2003;126:1599-1603.
Stollerman G.H. Rheumatic fever. Lancet. 1997;349:935-942.
Straussberg R., Shorer Z., Weitz R., et al. Familial infantile bilateral striatal necrosis: Clinical features and response to biotin treatment. Neurology. 2002;59:983-989.
Swedo S.E. Sydenham’s chorea. A model for childhood autoimmune neuropsychiatric disorders. JAMA. 1994;272:1788-1791.
Swedo S.E., Leonard H.L., Schapiro M.B., et al. Sydenham’s chorea: Physical and psychological symptoms of St Vitus dance. Pediatrics. 1993;91:706-713.
Tani T., Piao Y.-S., Mori S., et al. Chorea resulting from paraneoplastic striatal encephalitis. J Neurol Neurosurg Psychiatry. 2000;69:512-515.
Teixeira A.L.Jr., Meira F.C.A., Maia D.P., et al. Migraine headache in patients with Sydenham’s chorea. Cephalalgia. 2005;25:542-544.
Thobois S., Giraud P., Debat P., et al. Orofacial dyskinesias in a patient with primary biliary cirrhosis: A clinicopathological case report and review. Mov Disord. 2002;17:415-419.
Thomas M., Hayflick S.J., Jankovic J. Clinical heterogeneity of neurodegeneration with iron accumulation-1 (Hallervorden-Spatz syndrome) and pantothenate kinase associated neurodegeneration (PKAN). Mov Disord. 2004;19:36-42.
Thomas M., Jankovic J. Dentatorubropallidoluysian atrophy in North American families. Ann Neurol. 2001;50(suppl 1):S65.
Thomas M., Jankovic J. Neuroacanthocytosis. Ed.-in-chief. Noseworthy J., editor. Neurological Therapeutics: Principles and Practice, second ed. Abingdon, Oxon, UK: Informa Healthcare. 2006:2882-2889.
Toyoshima Y., Yamada M., Onodera O., et al. SCA17 homozygote showing Huntington’s disease-like phenotype. Ann Neurol. 2004;55:281-286.
Treves T., Korczyn A.D. Progressive dystonia and paraparesis in cerebral palsy. Eur Neurol. 1986;25:148-153.
Tsusi Y., Nagahama M., Mizutani A. Neuronal migration disorders in cerebral palsy. Neuropathology. 1999;19:14-27.
Turjanski N., Weeks R., Dolan R., et al. Striatal D1 and D2 receptor binding in patients with Huntington’s disease and other choreas. A PET study. Brain. 1995;118:689-696.
Ueno S., Maruki Y., Nakamura M., et al. The gene encoding a newly discovered protein, chorein, is mutated in chorea-acanthocytosis. Nat Genet. 2001;28:121-122.
Ueyama H., Kumamoto T., Nagao S., et al. A novel mutation of the McLeod syndrome gene in a Japanese family. J Neurol Sci. 2000;176(2):151-154.
Van der Meer, van der Weel, Lee D.N. The functional significance of arm movements in neonates. Science. 1995;267:693-695.
Verhagen M.M., Abdo W.F., Willemsen M.A., et al. Clinical spectrum of ataxia-telangiectasia in adulthood. Neurology. 2009;73:430-437.
Vernino S., Tuite P., Adler C.H., et al. Paraneoplastic chorea associated with CRMP-5 neuronal antibody and lung carcinoma. Ann Neurol. 2002;51:625-630.
Vidailhet M., Yelnik J., Lagrange C., et al. Bilateral pallidal deep brain stimulation for the treatment of patients with dystonia-choreoathetosis cerebral palsy: a prospective pilot study. for the French SPIDY-2 Study Group. Lancet Neurol, 8. 2009:709-717.
Vidakovic A., Dragasevic N., Kostic V.S. Hemiballism: Report of 25 cases. J Neurol Neurosurg Psychiatry. 1994;57:945-949.
Walker R.H., Danek A., Dobson-Stone C., et al. Developments in neuroacanthocytosis: Expanding the spectrum of choreatic syndromes. Mov Disord. 2006;21:1794-1805.
Walker R.H., Jankovic J., O’Hearn E., Margolis R.L. Phenotypic features of Huntington disease-like 2. Mov Disord. 2003;18:1527-1530.
Walker R.H., Jung H.H., Dobson-Stone C., et al. Neurologic phenotypes associated with acanthocytosis. Neurology. 2007;68:92-98.
Walker R.H., Morgello S., Davidoff-Feldman B., et al. Autosomal dominant chorea-acanthocytosis with polyglutamine-containing neuronal inclusions. Neurology. 2002;58:1031-1037.
Walker R.H., Rasmussen A., Rudnicki D., et al. Huntington’s disease-like 2 can present as chorea-acanthocytosis. Neurology. 2003;61:1002-1004.
Wardle M., Morris H.R., Robertson N.P. Clinical and genetic characteristics of non-Asian dentatorubral-pallidoluysian atrophy: A systematic review. Mov Disord. 2009;24:1636-1640.
Warner T.T., Lennox G.G., Janota I., Harding A.E. Autosomal-dominant dentatorubropallido-luysian atrophy in the United Kingdom. Mov Disord. 1994;9:289-296.
Warner T.T., Williams L.D., Walker R.W.H., et al. A clinical and molecular genetic study of dentatorubropallidoluysian atrophy in four European families. Ann Neurol. 1995;37:452-459.
Weindl A., Kuwert T., Leenders K.L., et al. Increased striatal glucose consumption in Sydenham’s chorea. Mov Disord. 1993;8:437-444.
Wheeler P.G., Weaver D.D., Dobyns W.B. Benign hereditary chorea. Pediatr Neurol. 1993;9:337-340.
Willemsen M.A., Breedveld G.J., Wouda S., et al. Brain-thyroid-lung syndrome: a patient with a severe multi-system disorder due to a de novo mutation in the thyroid transcription factor 1 gene. Eur J Pediatr. 2005;164:28-30.
Wild E.J., Mudanohwo E.E., Sweeney M.G., et al. Huntington’s disease phenocopies are clinically and genetically heterogeneous. Mov Disord. 2008;23:716-720.
Witt T.N., Danek A., Hein M.U., et al. McLeod syndrome: A distinct form of neuroacanthocytosis. J Neurol. 1992;239:302-306.
Wong L.J., Naviaux R.K., Brunetti-Pierri N., et al. Molecular and clinical genetics of mitochondrial diseases due to POLG mutations. Hum Mutat. 2008;29:E150-E172.
Wood N.S., Marlow N., Costeloe K., et al. Neurologic and developmental disability after extremely preterm birth. N Engl J Med. 2000;343:378-384.
Wu S.W., Graham B., Gelfand M.J., et al. Clinical and positron emission tomography findings of chorea associated with primary antiphospholipid antibody syndrome. Mov Disord. 2007;22:1813-1815.
Xiang F., Almqvist E.W., Huq M., et al. A Huntington disease-like neurodegenerative disorder maps to chromosome 20p. Am J Hum Genet. 1998;63:1431-1438.
Yamada M., Sato T., Tsuji S., Takahashi H. Oligodendrocytic polyglutamine pathology in dentatorubral-pallidoluysian atrophy. Ann Neurol. 2002;52:670-674.
Yamada M., Wood J.D., Shimohata T., et al. Widespread occurrence of intranuclear atrophin-1 accumulation in the central nervous system neurons of patients with dentatorubral-pallidoluysian atrophy. Ann Neurol. 2001;49:14-23.
Yetimalar Y., Seckin M., Secil Y., Basoglu M. Lamotrigine-induced bilateral ballism. Mov Disord. 2007;22:1832-1833.
Zhou B., Westaway S.K., Levinson B., et al. A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndrome. Nat Genet. 2001;28:345-349.
Appendix
Testing for various causes of chorea is available at the following:
Neuroacanthocytosis
Loss of chorein expression has been found to be a reliable diagnostic test and the analysis by Western blot test is available without charge by sending 20–30 mL of EDTA blood to Dr B. Baker, Munich, Germany (Benedikt.Bader@med.uni-muenchen.de) through the support of the Advocacy for Neuroacanthocytosis patients (http://www.naadvocacy.org).
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 15 Chorea, ballism, and athetosis
Introduction
Chorea consists of involuntary, continual, abrupt, rapid, brief, unsustained, irregular movements that flow randomly from one body part to another. Patients can partially and temporarily suppress the chorea and frequently camouflage some of the movements by incorporating them into semipurposeful activities (parakinesia). The inability to maintain voluntary contraction (motor impersistence), such as manual grip (milkmaid grip) or tongue protrusion, is a characteristic feature of chorea and results in dropping of objects and clumsiness. Chorea should be differentiated from “pseudochoreoathetosis,” a movement disorder that is phenomenologically similar to chorea or athetosis (slow chorea) due to loss of proprioception (Sharp et al., 1994). Muscle stretch reflexes are often “hung-up” and “pendular.” Affected patients typically have a peculiar, irregular, and dance-like gait. The pathophysiology of chorea is poorly understood, but in contrast to parkinsonism, dystonia, and other movement disorders, intracortical inhibition of the motor cortex is normal in chorea (Hanajima et al., 1999). In addition, semiquantitative analysis of single photon emission computed tomography in patients with hemichorea due to various causes suggests that there is an increase in activity in the contralateral thalamus, possibly due to disinhibition as a result of loss of normal pallidal inhibitory input (Kim et al., 2002).
Chorea may be a manifestation of a primary neurologic genetic disorder, such as Huntington disease (HD), or it may occur as a neurologic complication of systemic, toxic, or other disorders (Rosenblatt et al., 1998; Cardoso, 2004; Cardoso et al., 2006; Jankovic and Fahn, 2009) (Tables 15.1 and 15.2, Fig. 15.1). Chorea may be seen in normal infants, but these movements usually disappear by age 8 months, and some of these movements may be purposeful (Van der Meer et al., 1995).
| Developmental choreas |
Table 15.2 Differential diagnosis of inherited and sporadic choreas
| Inherited disorders | Sporadic disorders |
|---|---|
In this chapter we will briefly mention HD, but the focus will be on non-HD causes of chorea, as HD-like phenotypes without the HD genotype are increasingly being described and must be recognized. Several neurodegenerative disorders, some with expanded trinucleotide repeats, have been reported as phenocopies of HD, including spinocerebellar atrophy, particularly SCA2 and SCA3 (Kawaguchi et al., 1994), pure cerebello-olivary degeneration (Fox et al., 2003), and dentatorubral-pallidoluysian atrophy (DRPLA) (see below) (La Spada et al., 1994; Ikeuchi et al., 1995; Komure et al., 1995; Warner et al., 1995; Ross et al., 1997; Rosenblatt et al., 1998; Wild et al., 2008). In a study of 285 patients with clinical features consistent with HD, but who tested negative for HD by a DNA analysis, the following diagnoses were identified: five cases had Huntington disease-like type 4 (HDL4), one had HDL1, one had HDL2, and one patient had Friedreich ataxia (Wild et al., 2008).
Dentatorubral-pallidoluysian atrophy and HD-like disorders
Dentatorubral-pallidoluysian atrophy (DRPLA) is an autosomal dominant neurodegenerative disorder that is particularly prevalent in Japan, but it has been also identified in Europe and in African-American families (“Haw River syndrome”) (Burke et al., 1994; Thomas and Jankovic, 2001; Wardle et al., 2009) (Table 15.3). Usually beginning in the fourth decade, the disorder may occur as an early-onset DRPLA (before 20 years of age), manifested by a variable combination of myoclonus, epilepsy, and mental retardation, or late-onset DRPLA (after 20 years of age), manifested by cerebellar ataxia, choreoathetosis, dystonia, rest and postural tremor, parkinsonism, and dementia (Video 15.1).
Table 15.3 Clinical features of dentatorubral-pallidoluysion atrophy (DRPLA)
| Early onset | Late onset |
|---|---|
Unstable CAG expansion has been identified as the mutation in the DRPLA (or ATN1) gene on chromosome 12p13.31 (Koide et al., 1994; Komure et al., 1995; Warner et al., 1995; Becher et al., 1997; Ross et al., 1997). The DRPLA gene codes for protein that has been identified as a phosphoprotein, c-Jun NH(2)-terminal kinase, one of the major factors involved in its phosphorylation. In DRPLA, this protein appears to be slowly phosphorylated; thus, it may delay a process that is essential in keeping neurons alive (Okamura-Oho et al., 2003). Similar to HD, there is an inverse correlation between the age at onset and the number of CAG repeats (Ikeuchi et al., 1995). The early onset of DRPLA is associated with greater number of CAG repeats (62–79) as compared to the late-onset type (54–67 repeats) (Ikeuchi et al., 1995). Testing for the various gene mutations will undoubtedly lead to better recognition and appreciation of the spectrum of clinical and pathologic changes associated with these disorders. For example, a family with spastic paraplegia, truncal ataxia, and dysarthria, but without other clinical features of DRPLA, has been found to show homozygosity for an allele that carries intermediate CAG repeats in the DRPLA gene (Kuroharas et al., 1997). The DRPLA gene is expressed predominantly in neurons, but neurons that are vulnerable to degeneration in DRPLA do not selectively express the gene (Nishiyama et al., 1997).
Neuroimaging studies in patients with DRPLA often show evidence of cortical, brainstem, and cerebellar atrophy and widespread white matter changes (Koide et al., 1997; Muñoz et al., 1999, 2004). Neuropathologic findings consist chiefly of degeneration of the dentatorubral system, globus pallidus externa (GPe), subthalamic nucleus, and, to a lesser extent, striatum, substantia nigra, inferior olive, and thalamus (Warner et al., 1994) as well as demyelination and reactive astrogliosis in the cerebral white matter (Muñoz et al., 2004). Involvement of oligodendrocytes in autopsied brains and an increased number of affected glia, as well as larger expansions in CAG in these glia, in transgenic mice might explain the widespread demyelination (Yamada et al., 2002). Several pathologic reports have noted widespread deposition of lipofuscin. Similar to HD and other diseases associated with CAG repeat expansions, DRPLA has also been associated with the formation of perinuclear aggregates that can be prevented by the use of transglutaminase inhibitors such as cystamine and monodansyl cadaverine (Igarashi et al., 1998). These intranuclear inclusions stain intensely with ubiquitin (Becher and Ross, 1998). Subsequent studies have demonstrated accumulation of mutant atrophin-1 in the neuronal nuclei, rather than neuronal intranuclear inclusions, as the predominant pathologic feature in this neurodegenerative disorder (Yamada et al., 2001).
Other Huntington disease-like disorders
An autosomal dominant HD-like neurodegenerative disorder, now classified as HDL1 and mapped to chromosome 20p (Xiang et al., 1998), is a familial prion disease with an expanded PrP. A 192-nucleotide insertion in the region of the prion protein gene (PRNP) encoding an octapeptide repeat in the prion protein, was found in a single family with HD phenotype, suggesting that PRNP mutations can result in HD phenocopies (Moore et al., 2001).
Another disorder, termed HDL2, is characterized by onset in the fourth decade, involuntary movements such as chorea and dystonia as well as other movement disorders (bradykinesia, rigidity, tremor), dysarthria, hyperreflexia, gait abnormality, psychiatric symptoms, weight loss, and dementia with progression from onset to death in about 20 years (Margolis et al., 2001, 2004; Walker et al., 2003a). The disorder appears to be present exclusively or predominantly in individuals of African origin. The neuroimaging and neuropathologic findings are very similar to those in HD, except that there appears to be more severe involvement of the occipital cortex (Greenstein et al., 2007), and the intranuclear inclusions stain with 1C2 but not with anti-huntingtin antibodies. Unlike the family linked to chromosome 20p, seizures are not present in the HDL2 family. All 10 affected family members had a CAG repeat expansion of 50–60 triplets. The gene was later mapped to chromosome 16q24.3 and was found to encode junctophilin-3, a protein of the junctional complex linking the plasma membrane and the endoplasmic reticulum (Holmes et al., 2001). Although acanthocytosis was emphasized by Walker and colleagues (2002, 2003b) in their initial report and in one of three patients reported subsequently (Walker et al., 2003b), we have not been able to confirm the presence of acanthocytes in one member of the original family or in the other members when we carefully examined the peripheral smear.
The mutation associated with HDL2 has been identified as a CTG/CAG trinucleotide repeat expansion within the junctophilin-3 (JPH3) gene. In the normal population, the repeat length ranges from 6 to 27 CTG triplets, whereas affected individuals have 41–58 triplets. One family, previously described as “autosomal dominant chorea–acanthocytosis with polyglutamine-containing neuronal inclusions” (see later) (Walker et al., 2002), was subsequently found to have the triple nucleotide expansion of HDL2 (Stevanin et al., 2003; Walker et al., 2003b). The CTG repeat expansion at the HDL2 locus has been found to be responsible for 2% of patients with typical features of HD but without expanded CAG repeats in the IT15 gene and 0.2% of all HD families, again providing evidence that HD is clinically and genetically heterogeneous (Stevanin et al., 2002). This group later analyzed 252 patients with an HDL phenotype, including 60 with typical HD, who had tested negative for pathologic expansion in the IT15 gene and found two patients that had an abnormal CTG expansion in the JPH3 gene and two other patients with abnormal CAG expansion in the gene coding for TATA-binding protein (TBP/SCA17), important in initiation of transcription (Stevanin et al., 2003; Toyoshima et al., 2004; Schneider et al., 2006). SCA17, categorized as HDL4, has many clinical features that overlap with HD (Schneider et al., 2007; Bech et al., 2010). Thus, the frequency of mutation in either the JPH3 gene or TBP gene among patients with HDL phenotype is about 3%. Initially, the TBP expansion was found in the family with SCA17, characterized clinically by intellectual deterioration, cerebellar ataxia, epilepsy, and chorea. CUG repeat-containing RNA foci, resembling myotonic dystrophy type 1, were detected in neurons of HDL2 brains, suggesting that RNA toxicity may play a role in the pathogenesis of this neurodegenerative disorder (Rudnicki et al., 2007). HLD2 resembles HD clinically and pathologically more than any other disease (Rudnicki et al., 2008).
While the majority of genetic forms of chorea are inherited in an autosomal dominant pattern, a novel autosomal recessive neurodegenerative HDL disorder has been described (Kambouris et al., 2000). Beginning at 3–4 years of age and manifested by chorea, dystonia, ataxia, gait disorder, spasticity, seizures, mutism, intellectual impairment, and bilateral frontal and caudate atrophy, this neurodegenerative disorder has been linked to 4p15.3, different from the 4p16.3 HD locus, but confirmation of this finding is lacking. Although classified as HDL3, because of its AR inheritance and clinical features atypical for HD, it should not be categorized as HDL. In fact the HDL terminology should be replaced with specific disorders in which genetic mutation has been identified.
Some cases of neuronal intranuclear inclusion disease, caused by expanded CAG repeats and characterized by the combination of extrapyramidal signs, lower motor neuron signs, and cognitive and behavioral abnormalities resulting in death by the third decade, also show intranuclear aggregates, similar to the other CAG disorders (Lieberman et al., 1998).
Neuroacanthocytosis
After HD, neuroacanthocytosis is perhaps the most common form of hereditary chorea (Table 15.4). Previously also referred to as chorea–acanthocytosis, it is now recognized that this multisystem, neurodegenerative disorder can be expressed by a wide variety of clinical and laboratory abnormalities, hence the term neuroacanthocytosis (Spitz et al., 1985; Danek et al., 2005; Walker et al., 2006, 2007; Thomas and Jankovic, 2006) (Table 15.5). The term was coined by Jankovic et al. (1985) to replace the old term Levine–Critchley syndrome choreoacanthocytosis to draw attention to the heterogeneous presentation with a variety of hyperkinetic (chorea, dystonia, tics) and hypokinetic (parkinsonism) movement disorders in addition to other neurologic deficits and abnormal laboratory findings. Symptoms usually first begin in the third and fourth decades of life (range: 8–62 years) with lip and tongue biting followed by orolingual (eating) dystonia, motor and phonic tics, generalized chorea, and stereotypies (Video 15.2). Other features include cognitive and personality changes, seizures, dysphagia, dysarthria, vertical ophthalmoparesis, parkinsonism, amyotrophy, areflexia, evidence of axonal neuropathy, and elevated serum creatine kinase without evidence of myopathy. Hardie and colleagues (1991) reviewed the clinical, hematologic, and pathologic findings in 19 patients (10 males and 9 females) with a mean age of 32 years (range: 8–62 years) with more than 3% acanthocytes on peripheral blood smear. Twelve of these patients with neuroacanthocytosis were familial, and seven were sporadic; two had the McLeod phenotype (see later). In their series, Hardie and colleagues (1991) found a variety of movement disorders, including chorea (58%), orofacial dyskinesia (53%), dystonia (47%), vocalizations (47%), tics (42%), and parkinsonism (34%). Although lip and tongue biting was observed in only 16% of the patients, this is a characteristic feature of neuroacanthocytosis and when present, it strongly suggests the diagnosis. The use of a mouth guard has been reported to be effective in the treatment of oral self-mutilation associated with neuroacanthocytosis (Fontenelle and Leite, 2008). Besides movement disorders other associated features included dysarthria (74%); absent or reduced reflexes (68%); dementia (63%); psychiatric problems such as depression, anxiety, and obsessive-compulsive disorder (58%); dysphagia (47%); seizures (42%); muscle weakness and wasting (16%); and elevated creatine phosphokinase (CK) in 58%. Magnetic resonance volumetry and fluorodeoxyglucose positron emission tomography (PET) show striatal atrophy in patients with neuroacanthocytosis (Jung et al., 2001).
Table 15.4 Clinical and genetic features of neuroacanthocytosis
Table 15.5 Classification of neuroacanthocytosis
| I. Normal lipids |
Although autosomal dominant, X-linked recessive, and sporadic forms of neuroacanthocytosis have been reported, the majority of the reported families indicate autosomal recessive inheritance. Genome-wide scan for linkage in 11 families with autosomal recessive inheritance showed a linkage to a marker on chromosome 9q21, indicating a single locus for the disease (Rubio et al., 1997). Ueno and colleagues (2001) carried out a linkage-free analysis in the region of chromosome 9q21 in the Japanese population and identified a 260 bp deletion in the EST (expressed sequence tags) region K1AA0986 in exon 60, 61 that was homozygous in patients with neuroacanthocytosis and heterozygous in their parents. Further sequencing has identified a polyadenylation site with a protein with 3096 amino acid residues that has been named “chorein” by the authors. This deletion is not found in normal Japanese and European populations (Ueno et al., 2001). In another study by Rampoldi and colleagues (2001) in European patients, a novel gene encoding a 3174-amino-acid protein on chromosome 9q21 with 73 exons was identified. They identified 16 mutations in the chorea acanthocytosis (ChAc) gene, later renamed VPS13A gene. These mutations were identified in various exons. They suggested that chorea acanthocytosis encodes an evolutionarily conserved protein that is involved in protein sorting (Rampoldi et al., 2002). Other single heterozygous mutations have been identified in this gene (Saiki et al., 2003). Molecular analysis by screening all 73 exons of the VPS13A gene showed marked genotype–phenotype heterogeneity (Lossos et al., 2005). The function of the protein product, chorein, is not yet fully understood, but it is probably involved in intracellular protein trafficking. Using antichorein antisera, the expression of chorein in peripheral red blood cells has been found to be absent or markedly reduced in patients with neuroacanthocytosis, but not with McLeod syndrome or with HD (Dobson-Stone et al., 2004). Loss of chorein expression, measured by Western blot analysis has been found to be a reliable diagnostic test for neuroacanthocytosis.
Walker and colleagues (2002) described a family with chorea or parkinsonism as well as cognitive changes, inherited in an autosomal dominant pattern. At autopsy, there was marked degeneration of the striatum and intranuclear inclusion bodies immunoreactive for ubiquitin, expanded polyglutamine CGG repeats, and torsinA. Interestingly, one of the patients had fragile X syndrome, and two had expanded trinucleotide repeats at permutation range, previously associated with postural/kinetic tremor, parkinsonism, ataxia, and cognitive decline (Hagerman et al., 2001). The family reported by Walker and colleagues (2002) turned out to have the trinucleotide repeat expansion associated with HDL2, but subsequent analysis of the family shed doubt on the presence of acanthocytes as a feature of the HDL2 syndrome (Walker et al., 2003a).
Two patients from the original study by Rubio and colleagues (1997) were found to have the McLeod phenotype, an X-linked (Xp21) recessive form of acanthocytosis associated with depression, bipolar and personality disorder, and neurologic manifestations, including chorea, involuntary vocalizations, seizures, motor axonopathy, hemolysis, liver disease, and high creatine kinase levels (Witt et al., 1992; Danek et al., 2001a, 2001b). Neuroimaging usually reveals caudate and occasionally cerebellar atrophy with a rim of increased T2-intensity in the lateral putamen. Functional neuroimaging studies show evidence of downregulation of D2 dopamine receptors. In contrast to the autosomal recessive form of neuroacanthocytosis linked to mutations in VPS13A gene on chromosome 9, patients with McLeod syndrome usually do not exhibit lip-biting or dysphagia. This multisystem disorder is associated with low reactivity of Kell erythrocyte antigens (weak antigenicity of red cells) due to absence of XK, a 37 kDa, 444-amino-acid, membrane protein that forms a complex with the Kell protein. The disorder is caused by different mutations in the XK gene encoding for the XK protein (Ho et al., 1996; Danek et al., 2001a; Jung et al., 2001). Mutations identified by various authors include frame shift mutations in exon 2 at codon 151, deletion at codon 90 in exon 2 and at codon 408 in exon 3, and splicing mutations in intron 2 of the XK gene (Dotti et al., 2000; Ueyama et al., 2000; Danek et al., 2001a; Jung et al., 2001). Rarely, neuroacanthocytosis may be associated with abetaliproteinemia due to mutations in the microsomal triglyceride transfer protein (Sharp et al., 1993). In addition to acanthocytosis, the patients exhibit retinopathy; malabsorption, including that of vitamin E; low serum cholesterol levels; and abnormal serum lipoprotein electrophoresis. Aprebetalipoproteinemia can also cause movement disorders and acanthocytosis (Bohlega et al., 1998).
An examination of wet blood or Wright-stained, fast dry, blood smear usually reveals over 15% of red blood cells as acanthocytes. In mild forms of acanthocytosis, scanning electron microscopy might be required to demonstrate the red blood cell abnormalities (Feinberg et al., 1991). In a recent study of two patients with pathologically proven neuroacanthocytosis, Feinberg and colleagues (1991) noted that the yield in demonstrating acanthocytosis may be increased by using a coverslip because the contact with glass causes the fragile cells to undergo morphologic changes. Diluting the blood with normal saline, incubating the Wright-stained smear with EDTA, using a scanning electron microscope, and other techniques designed to increase “echinocytotic stress” are also helpful (Feinberg et al., 1991; Orrell et al., 1995). The characteristic acanthocytic appearance of red blood cells has been attributed to abnormalities in transmembrane glycoprotein band 3 that can be demonstrated on gel electrophoresis. It is not yet clear how the gene mutation leads to the abnormal morphology of the red cells.
By using high-performance liquid chromatography, fatty acids of erythrocyte membrane proteins were analyzed in six patients with neuroacanthocytosis (Sakai et al., 1991). In comparison with normal controls and patients with HD, erythrocytes of patients with neuroacanthocytosis showed a marked abnormality in the composition of covalently bound fatty acids: an increase in palmitic acid (C16:0) and a decrease in stearic acid (C18:0).
Brain magnetic resonance imaging (MRI) in patients with neuroacanthocytosis usually shows caudate and more generalized brain atrophy, but some cases also show extensive white matter abnormalities (Nicholl et al., 2004). Caudate hypometabolism and atrophy have been demonstrated by PET studies and by neuroimaging. Similar to the findings in Parkinson disease, PET scans in six patients with neuroacanthocytosis showed a reduction to 42% of normal in [18F]dopa uptake in the posterior putamen; in contrast to Parkinson disease, however, there was a marked reduction in the striatal [11C]raclopride (D2) receptor binding (Brooks et al., 1991).
Neuronal loss and gliosis were particularly prominent in the striatum and pallidum but may also affect the thalamus, substantia nigra, and anterior horns of the spinal cord (Rinne et al., 1994b). The neuronal loss in the substantia nigra is most evident in the ventrolateral region, similar to Parkinson disease, but the nigral neuronal loss is more widespread in neuroacanthocytosis (Rinne et al., 1994a). The preservation of the cerebral cortex, cerebellum, subthalamic nucleus, pons, and medulla may serve to differentiate pathologically between neuroacanthocytosis, HD, and DRPLA. Brain biochemical analyses showed low substance P in the substantia nigra and striatum and increased levels of norepinephrine in the putamen and pallidum (DeYebenes et al., 1988).
Neurodegeneration with brain iron accumulation
A group of neurodegenerative disorders, formerly known as Hallervorden–Spatz disease, has been receiving increasing attention as the genetic and pathogenic mechanisms of the various subtypes become elucidated. Because of Hallervorden’s terrible past and his shameless involvement in active euthanasia (Shevell, 2003), this group of disorders has been renamed neurodegeneration with brain iron accumulation (NBIA). The prototype form of NBIA consists of an autosomal recessive disorder characterized by childhood-onset progressive rigidity, dystonia, choreoathetosis, spasticity, optic nerve atrophy, and dementia, and has been associated with acanthocytosis (Malandrini et al., 1996; Racette et al., 2001; Thomas et al., 2004). Although chorea is not a typical feature of NBIA, “senile chorea” has been described in a patient with pathologically proven NBIA type1 (NBIA-1) (Grimes et al., 2000).
The most classic NBIA, NBIA-1, is the pantothenate kinase-associated neurodegeneration (PKAN). Linkage analyses initially localized the NBIA-1 gene on 20p12.3–p13; subsequently, a 7 bp deletion and various missense mutations were identified in the coding sequence of the PANK2 gene, which codes for pantothenate kinase (Zhou et al., 2001; Hayflick et al., 2003). Pantothenate kinase is an essential regulatory enzyme in coenzyme A biosynthesis. It has been postulated that as a result of phosphopantothenate deficiency, cysteine accumulates in the globus pallidus of brains of patients with NBIA-1. It undergoes rapid auto-oxidation in the presence of nonheme iron that normally accumulates in the globus pallidus interna (GPi) and substantia nigra, generating free radicals that are locally neurotoxic. Interestingly, atypical subjects were found to be compound heterozygotes for certain mutations for which classic subjects were homozygous. The disorder with the clinical phenotype of NBIA associated with mutations in the PANK2 gene is now referred to as pantothenate kinase-associated neurodegeneration or PKAN (Thomas et al., 2004). On the basis of an analysis of 123 patients from 98 families with NBIA-1, Hayflick and colleagues (2003) found that “classic Hallervorden–Spatz syndrome” was associated with PANK2 mutation in all cases and that one-third of “atypical” cases had the mutations within the PANK2 gene. Those who had the PANK2 mutation were more likely to have dysarthria and psychiatric symptoms, and all had the typical “eye-of-the-tiger” abnormality on MRI with a specific pattern of hyperintensity within the hypointense GPi (McNeill et al., 2008).
Neuroacanthocytosis and NBIA may overlap in some clinical features. While PKAN may be associated with acanthocytosis, another neuroacanthocytosis syndrome, linked to PKAN, is the hypoprebetalipoproteinemia, acanthocytosis, retinitis pigmentosa, and pallidal degeneration (HARP) syndrome (Orrell et al., 1995). This disorder is associated with dystonia, particularly involving the oromandibular region, rather than chorea and self-mutilation. Indeed, a homozygous nonsense mutation in exon 5 of the PANK2 gene that creates a stop codon at amino acid 371, found in the original HARP patient, establishes that HARP is part of the pantothenate kinase-associated neurodegeneration disease spectrum (Ching et al., 2002; Houlden et al., 2003).
The classification of NBIA is continuously being revised as our understanding of this group of disorders is improving. In addition to PKAN, other forms of NBIA include neuroferritinopathy, infantile neuroaxonal dystrophy, and aceruloplasminemia, and PLA2G6-associated neurodegeneration (PLAN), with mutations in the PLA2G6 gene, on chromosome 22q13.1 (Schneider et al., 2009) (Table 15.6, Fig. 15.2). These disorders are chiefly manifested by childhood-onset axial hypotonia, spasticity, bulbar dysfunction, ataxia, dystonia, and choreoathetosis, as well as MRI changes indicative of iron deposition in the globus pallidus and substantia nigra (Kurian et al., 2008). Previously diagnosed as infantile neuroaxonal dystrophy, now classified as NBIA-2, PLAN may also present as adult-onset, levodopa-responsive dystonia–parkinsonism without iron on brain imaging (McNeill et al., 2008; Paisan-Ruiz et al., 2008; Schneider et al., 2009). Another gene, FA2H, when mutated, has been found to cause not only leukodystrophy and hereditary spastic paraplegia, but also NBIA (Kruer et al., 2010). The FA2H-associated neurodegeneration (FAHN) is characterized by childhood-onset gait impairment, spastic paraparesis, ataxia, and dystonia (Kruer et al., 2010; Schneider and Bhatia, 2010). FA2H is involved in lipid and ceramide metabolism. Another form of NBIA was highlighted by a report of 11 children with a biochemical profile suggestive of dopamine transporter deficiency syndrome (Kurian et al., 2011). Presenting in infancy, this disorder is usually characterized by severe parkinsonism-dystonia syndrome, but chorea, oculomotor deviations, and spasticity may also dominate the clinical phenotype. The CSF ratio of homovanillic acid to 5-hydroxyindoleacetic acid is usually increased. This autosomal recessive disorder has been attributed to homozygous or compound heterozygous SLC6A3 mutations and complete loss of dopamine transporter activity in the basal nuclei (indicated by abnormal DAT Scan SPECT). Regression of symptoms after 6 months of iron chelation with deferiprone has been reported in some patients with NBIA (Forni et al., 2008).
Table 15.6 Differential diagnosis of neurodegeneration with brain iron accumulation (NBIA)
|
• Infantile neuroaxonal dystrophy (NBIA-2)
|
Other familial choreas
Besides HD and neuroacanthocytosis, genetically transmitted choreas include benign hereditary chorea (BHC), a nonprogressive chorea of childhood onset (Wheeler et al., 1993; Kleiner-Fisman and Lang, 2007; Adam and Jankovic, 2010) (Table 15.7). BHC usually starts in early childhood and progresses until the second decade, after which time it remains static or even spontaneously improves. The patients may have a slight motor delay because of chorea, slight gait ataxia, and their handwriting may be impaired, but the disorder is self-limiting after adolescence in most cases, although it may persist as a mild chorea beyond age 60 years. Inherited as an autosomal dominant disorder, BHC has been linked to a marker on chromosome 14q13.1–q21.1 (de Vries et al., 2000; Fernandez et al., 2001; Breedveld et al., 2002) and a novel single nucleotide substitution in the TITF1 gene (also referred to as TTF, Nkx2.1, and T/ebp), coding for a transcription essential for the organogenesis of the lung, thyroid, and basal ganglia, has been identified in one Canadian family (Kleiner-Fisman et al., 2003; Kleiner-Fisman and Lang, 2007; Ferrara et al., 2008) (http://www.ncbi.nlm.nih.gov/sites/GeneTests/). This syndrome has been also described in patients with deletion of not only the TITF1 gene but also the contiguous PAX9 gene (Devos et al., 2006). These mutations should be considered in children and adults with chorea, mental retardation, congenital hypothyroidism, and chronic lung disease, hence the term brain–lung–thyroid syndrome proposed for this disease (Willemsen et al., 2005; Devos et al., 2006). MRI has been generally reported to be unremarkable but some cases showed hypoplastic pallidum, lack of differentiation of medial and lateral components, and bilateral signal hyperintensities on T2-weighted MRI images (Kleiner-Fisman and Lang, 2007). Two autopsied brains from patients with BHC due to TITF1 showed no pathologic abnormalities (Asmus et al., 2005), but specific immunochemical investigations suggested reduced number of striatal and neocortical interneurons, consistent with a defect in migration normally mediated by the TITF1 gene (Kleiner-Fisman et al., 2005).
|
• MRI normal, but may show hypoplastic pallidum, lack of differentiation of medial and lateral components, and bilateral signal hyperintensities on T2-weighted images
|
[/not-level-membership-for-neurology-category]
