CHAPTER 140 Chordomas and Chondrosarcomas
Historical Background
Virchow first described small nodules along the clivus in 1846 and named them “ecchondrosis physaliphora” in 1857 because he hypothesized them to be of cartilaginous origin.1 The following year, Luschka, Henker, and Hasse described similar nodules. Concurrently, Muller postulated that ecchondrosis physaliphora was of notochordal rather than cartilaginous origin. In 1894, Ribbert coined the term “chordoma” and presented a case that supported Muller’s hypothesis. A year later, he experimentally induced chordomas in rabbits by perforating the anterior intervertebral ligament.1,2 In 1909, Harvey Cushing reported successful removal of a chordoma (initially described as a teratoma, case XVII) in a 35-year-old man (although the patient died during reoperation some 6 months later).3,4 Today, chordomas present the same challenges to neurosurgeons that they did 100 years ago; despite the remarkable advances made in operative and imaging techniques, the individual surgeon must often choose between the chance for cure and reduction of morbidity to achieve the greatest quality of life.
Natural History
Chordomas are rare, slow-growing tumors that are believed to arise from rests of vestigial notochordal cells. They represent only 1% to 4% of primary bone tumors,5 with an age-adjusted incidence rate of 0.08 per 100,000 patient-years. There is a twofold greater incidence in males and a fivefold greater incidence in the white population.6 The peak incidence occurs in the eighth decade of life, and development of chordomas is quite rare before the age of 40. One recent, multi-institutional review suggested that anatomic location is evenly divided among the spheno-occipital region (32%), mobile spine (32.8%), and sacrum (29.2%).6 Older, often-cited reports, based on single-institution data, estimated that sacral chordomas constituted 50% of cases whereas 35% and 15% were cranial and spinal chordomas, respectively.5,7 Irrespective of the statistical distribution along the craniospinal axis, the anatomic diversity of skull base chordomas reflects the complex, serpentine course of the notochord within the clivus (Fig. 140-1). The clinical findings and symptomatology depend on the anatomic location of the tumor. Patients with cranial chordomas often have headaches, neck pain, cranial neuropathies, and even endocrine abnormalities, whereas those with spinal chordomas have back pain, pathologic fractures, myelopathy, and radiculopathy. Sacral chordomas tend to be quite large at the time of diagnosis because of the insidious onset of symptoms such as pain and bowel or bladder dysfunction.8
Overall, 5- and 10-year survival rates are 67.6% and 39.9%, respectively.6 Local recurrence is the most important predictor of mortality in patients with sacral chordomas. Distant metastases to the lung, liver, and bone can occur with chordomas, but they generally take place quite late in the disease process and, as a result, probably contribute little to overall mortality.
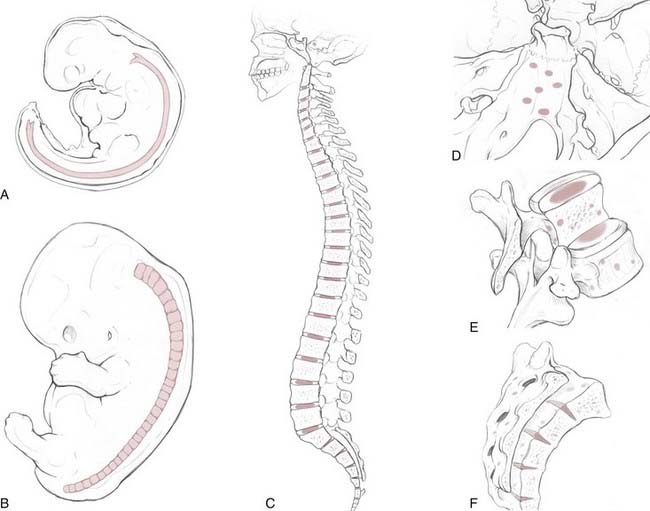
The notochord is an evolutionarily conserved structure (hence, the phylum Chordata) that arises early in embryonic development from the dorsal organizer region. It plays a critical role in many aspects of development, such as determining the dorsal-ventral axis of the neural tube (through secretion of the sonic hedgehog homologue [SHH] gene product), left-right asymmetries, and arterial-versus-venous fates of early blood vessels. The notochord is made up of highly vacuolated cells (the physaliphorous cells) that contain a hydrated protein matrix and are surrounded by a thick basement membrane. The protein matrix absorbs water, which allows the cells to swell and exert pressure on the basement membrane. In this fashion, the notochord provides structural support to the developing embryo. In animals with a bony spine, the notochord involutes as the adjacent somites form the vertebral bodies. Some notochordal cells persist and form the nucleus pulposus of the intervertebral disks, whereas others survive as “rests” within the clivus, sacrum, and vertebrae.9,10 The relationship between these cellular rests and the development of later chordomas is complex. Along the dorsal clivus, for example, approximately 2% of the population, based on autopsies (Ribbert’s theory, see Bailey and Bagdasar1), have benign growths termed ecchordosis physaliphora (i.e., the ecchondrosis of Virchow). Rarely, they can grow and become symptomatic giant ecchordoses or intradural benign chordomas.11,12 Similarly, benign notochordal tumor is a recently recognized entity found within the clivus (11.5% of autopsies) and vertebral bodies (14% of autopsies) that can expand and become a classic chordoma.13,14 These tumors are immunohistochemically distinct from fetal notochord rests.13 However, whether all classic chordomas arise from benign tumor precursors is unknown.
Radiography
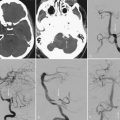
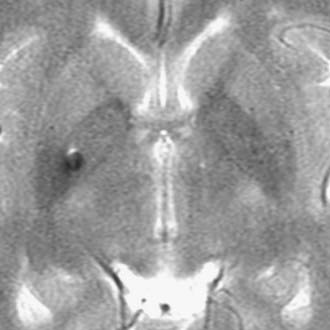
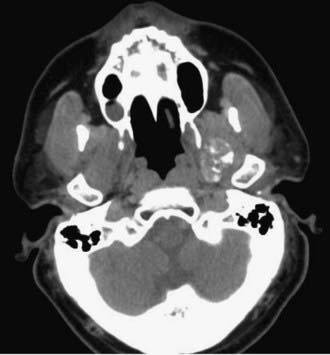
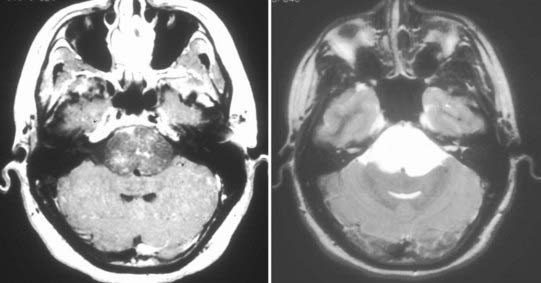
Chordomas and chondrosarcomas are indistinguishable from one another on radiographic examination, except for the fact that chondrosarcomas tend to arise from a paramedian location and chordomas are nearly always found along or near the midline.15–18 One caveat in this regard is that chordomas can distort their midline origin by becoming quite large before they are detected and growing asymmetrically in one direction or another. This asymmetric growth may extend into the nasopharynx, orbits, or sella or along the cranial nerves, with dorsal deflection of the brainstem. Although chondrosarcomas typically arise from the skull base, they can also arise from the meninges. On non–contrast-enhanced computed tomography (CT), both chordomas and chondrosarcomas are slightly hyperdense masses with variable amounts of calcification (Fig. 140-2). Bone windows often demonstrate significant bony erosion. For chordomas, the erosion is predominantly along the clivus and petrous apex. Tumors of both types show variable enhancement on contrast-enhanced CT. Chordomas and chondrosarcomas are both isointense to hypointense on T1-weighted magnetic resonance imaging (MRI), again with variable enhancement. On T2-weighted MRI these tumors are hyperintense, but they may have some heterogeneity in signal intensity because of calcifications and bony sequestrations (Fig. 140-3). Benign ecchordosis physaliphora is radiographically similar but does not show enhancement.19,20
Pathology
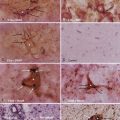
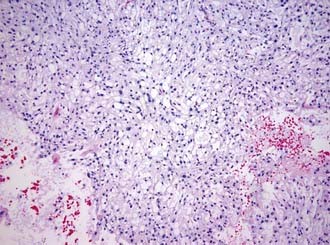
Macroscopically, chordomas and chondrosarcomas are nodular, exophytic masses with a smooth whitish or occasionally reddish appearance.1,21 They often display a fibrous outer layer that is contiguous with the fibrous septations found within the tumor. This pattern of growth gives the tumor a lobulated appearance in cross section. These tumors lack a thick capsule. Internally, the tumors are soft, gray, and translucent but generally lack significant areas of necrosis. In addition to their local bony destruction, these tumors will extend out into adjacent cancellous bone, thus making oncologic excision difficult if not impossible. Microscopically, chordomas can be divided into three main histochemical types: conventional, chondroid, and dedifferentiated. Conventional chordomas are composed of sheets or cords of cells with eosinophilic cytoplasm embedded in a mucinous matrix. The appearance of these cells can range from having a homogeneous cytoplasm completely lacking in vacuoles to a cytoplasm markedly engorged with vacuoles, the so-called physaliphorous or bubble-bearing cells (Fig. 140-4). Differentiation into cartilaginous or, occasionally, osseous-appearing cells may occur focally or throughout the tumor and, if extensive, distinguishes the chondroid chordoma subtype from conventional chordomas. Rarely, chordomas undergo malignant transformation, which gives rise to dedifferentiated chordomas. In this subtype there is a predominant sarcomatous appearance with malignant spindle cells, increased atypia, and increased mitotic index. Immunohistochemically, chordomas are reactive for epithelial membrane antigen, cytokeratins, α-fetoprotein, and rarely, S-100.
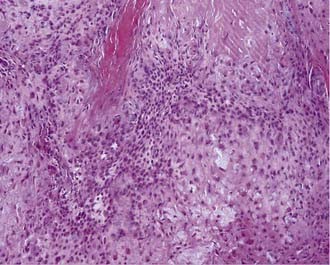
Chondrosarcomas can also be divided into three subtypes: classic or conventional, mesenchymal, and dedifferentiated. Conventional chondrosarcomas are composed of large, often multinucleated cells within a cartilaginous matrix (Fig. 140-5). Their pathology has three histologic grades. The degree of atypia, number of mitoses, nuclear-to-cytoplasmic ratio, and amount of cartilaginous matrix correlate with the aggressiveness of the tumor, with grade III being the most aggressive. Mesenchymal chondrosarcomas have islands of cartilage mixed with primitive mesenchymal cells, and undifferentiated tumors have a marked sarcomatous appearance. Both mesenchymal and undifferentiated chondrosarcomas have a more aggressive course. Immunohistochemically, chondrosarcomas lack the epithelial antigens expressed by chordomas despite a histologic appearance similar to that of chondroid chondromas.
Cytogenetics
Numerous studies of the cytogenetics of chordomas have been published.22,23 The most common chromosomal abnormalities identified are chromosome 1 monosomy and gain of chromosome 7. Additional chromosomal abnormalities (either gain or loss) described in the literature involve 1q, 2p, 3p, 5p, 9p, 10, 12q, 13q, 17, and 20q. One recent study suggested that an abnormality in the 6p32 region may have a specific association with the genesis of chordoma (rather than progression or recurrence). Although most chordomas are of sporadic origin, case reports of potentially familial chordoma lines have been published. These studies show no consistent cytogenetic change consistent with the heterogeneity seen in sporadic cases. To date, no consistent cytogenetic profile predicts tumor biology or response to therapy.
Treatment
Surgical Approach and Technique
The appropriate surgical approach to clival chordomas and chondrosarcomas is based primarily on the location and size of the tumor (Fig. 140-6). If sufficiently large, two approaches may be used to remove the bulk of the tumor safely. Under all circumstances, careful preoperative planning is essential. It is important to identify which anatomic compartment or compartments the tumor occupies,25 the presence or absence of natural planes, and the relationship of the tumor to vital surrounding structures such as the cranial nerves, cavernous sinus, and brainstem. Based on these characteristics, the surgeon can then choose the appropriate approach or approaches that will safely provide the largest view with the least morbidity.
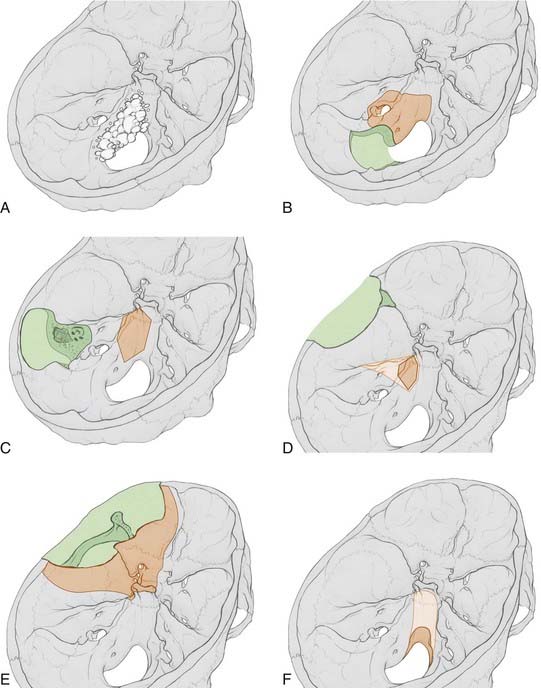
FIGURE 140-6 Skull base approaches to clival tumors. The amount of bone removed (green) and the working area obtained (orange) for a variety of standard skull base approaches are illustrated. A, An example of a large clival chordoma growing asymmetrically along the skull base is provided for comparison with each illustrated approach. Of note, large tumors can involve multiple compartments and require a combination of standard skull base approaches for adequate resection. B, Far lateral transcondylar transtubercular approach to the lower petroclival region. The working area illustrated represents the quantity of exposure obtained after C1 hemilaminectomy, partial resection of the condyle (to the hypoglossal canal), and removal of the jugular tuberculum. An additional mastoidectomy, resection of the lateral mass of C1, and complete resection of the condyle will increase exposure by just 30%. C, Transcrusal transpetrosal approach to the mid petroclival region. This approach provides up to four times the exposure of a retrolabyrinthine transpetrosal approach, along with hearing preservation. In contrast, more extensive transpetrosal approaches, such as the transotic or transcochlear approaches, provide little additional exposure but have much higher morbidity.24 D, Middle fossa approach to the mid and upper petroclival region. The working area illustrated depicts the area obtained after drilling out Kawase’s triangle. Removal of the bone of Glasscock’s triangle or the zygoma adds no additional exposure, whereas removing the cochlea will double it along the petrous bone. E, Frontotemporal approach to the parasellar region. This standard approach can be tailored to reach a variety of anterior or anterolateral regions of the skull base. It provides only limited access to the upper clivus. F, Transoral with (light orange) or without (dark orange) a Le Fort I osteotomy approach to the clivus. A standard transoral approach allows only limited access to the lower clivus. The addition of a palatal split will increase exposure to the lower clivus somewhat, whereas the addition of a Le Fort I osteotomy will increase clival exposure from 8% to 85%.
Variations on and combinations of these approaches may be necessary, depending on individual variation in the location and size of the tumor, as well as associated comorbid conditions. For example, in a patient with intact hearing and a tumor extending posteriorly from the midclivus, a transcrusal transpetrosal approach may be chosen over the transcochlear version. The transcrusal approach has a lower risk of injury to the facial and auditory nerves and provides an almost identical operative field.24,26 The exact approach chosen should provide the largest surgical corridor with the least amount of morbidity to minimize brain and cranial nerve retraction.
Once the appropriate approach has been performed, it is imperative to first debulk the tumor to avoid retraction injury to adjacent structures and provide adequate exposure of the tumor margins. This is generally relatively easy because chordomas and chondrosarcomas have a soft gelatinous core that can readily be aspirated. However, calcifications and sequestrations may be present and necessitate the use of rongeurs. Once the bulk of the tumor has been removed, an attempt can be made to remove the residual fibrous outer layer, again with care taken not to injure adjacent neural structures. Cancellous bone, whenever possible, should be resected until the bone begins to bleed in “normal” fashion because chordomas tend to extend long fingers into cancellous bone. It should be noted that violation of the tumor bed can cause seeding of the tumor.27 However, total resection determined at the time of surgery does not predict the future presence or absence of local recurrence; in one recent case series,28 intraoperative gross total resection was associated with the presence of residual tumor on postoperative MRI in approximately 50% of cases (Fig. 140-7).
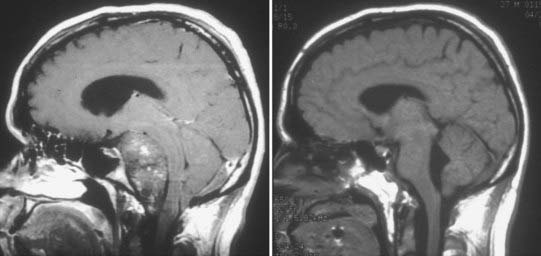
FIGURE 140-7 Large clival chordoma. Sagittal T1-weighted magnetic resonance images of the large clival chordoma illustrated in Figure 140-3 before (left) and after (right) resection. The preoperative scan is a contrast-enhanced image and demonstrates a modest heterogeneous pattern of enhancement. This large tumor involved several different anatomic compartments and required a combination of skull base approaches. The postoperative image demonstrates nearly total resection with decompression of the brainstem.
Surgical Outcomes
Determining accurate morbidity and mortality statistics is difficult; chordomas and chondrosarcomas are rare tumors, which limits the number of patients available for study. Furthermore, there is considerable variability in specific treatment paradigms, length of follow-up, and the method of determining gross total versus partial resection among reported case series. Another potential complicating factor is that some authors lump together patients with chordomas and chondrosarcomas. Some studies suggest that chondrosarcomas are much less aggressive and have a higher rate of recurrence-free survival.28 Nevertheless, one consistent observation across most studies (reviewed by Muro and colleagues8) is that primary tumors, as a group, fare better after resection than do recurrent or previously treated tumors, regardless of whether intraoperative gross total resection was achieved. This suggests that the individual biology of the tumor heavily influences outcome. Reported mortality rates at 5 years range from 33% to 75%.29–33 The lowest survival rates30 included patients who underwent biopsy only before RT. Surgical morbidity occurred in 13% to 90% of patients and included new or persistent neurological deficits, cerebrospinal fluid leaks, meningitis, and death.
Radiation Therapy
Before the advent of three-dimensional planning and dose delivery, it was not possible with conventional RT to administer a therapeutic dose without exceeding the tolerance dose, defined as the radiation dose with a greater than 5% chance of serious complications in critical surrounding structures such as the brainstem and optic nerves. These tumors were therefore considered radioresistant because of poor long-term outcomes.34,35 With the widespread introduction of three-dimensional conformal photon RT and intensity-modulated RT, therapeutic doses of 70 to 80 Gy can now be given safely without a high risk of injury to surrounding structures. One recent case series that used Gamma Knife radiosurgery to treat chordomas and chondrosarcomas reported 5- and 10-year survival rates of 80% and 53%, with progression-free survival rates at 5 and 10 years of 43% and 38%, respectively.36 The low number of patients and combination of tumor types limits the interpretation.
Protons and charged ions have also been used for the treatment of chordomas. These particles have a low entry dose, lack an exit dose, and have the maximal dose deposited at the end of the beam’s range. This type of Bragg peak allows a higher energy dose to be given to the tumor while minimizing injury to surrounding tissues. A recent review looked at two reports of proton beam radiation for the treatment of chordomas and chondrosarcomas.37 Progression-free survival rates at 5 years were similar to those of surgery for both chordomas (50% to 64%) and chondrosarcomas (95% to 100%). Complication rates could not be directly compared with those of standard photon therapy, but brainstem injury has been reported at doses of greater than 60 Gy to the brainstem.38–40
Almefty K, Pravdenkova S, Colli BO, et al. Chordoma and chondrosarcoma: similar, but quite different, skull base tumors. Cancer. 2007;110:2457-2467.
Al-Mefty O, Borba LA. Skull base chordomas: a management challenge. J Neurosurg. 1997;86:182-189.
Arnautovic KI, Al-Mefty O. Surgical seeding of chordomas. Neurosurg Focus. 2001;10(3):E7.
Bayrakli F, Guney I, Kilic T, et al. New candidate chromosomal regions for chordoma development. Surg Neurol. 2007;68:425-430.
Brada M, Pijls-Johannesma M, De Ruysscher D. Proton therapy in clinical practice: current clinical evidence. J Clin Oncol. 2007;25:965-970.
Catton C, O’Sullivan B, Bell R, et al. Chordoma: long-term follow-up after radical photon irradiation. Radiother Oncol. 1996;41:67-72.
Debus J, Schulz-Ertner D, Schad L, et al. Stereotactic fractionated radiotherapy for chordomas and chondrosarcomas of the skull base. Int J Radiat Oncol Biol Phys. 2000;47:591-596.
Erdem E, Angtuaco EC, Van Hemert R, et al. Comprehensive review of intracranial chordoma. Radiographics. 2003;23:995-1009.
Forsyth PA, Cascino TL, Shaw EG, et al. Intracranial chordomas: a clinicopathological and prognostic study of 51 cases. J Neurosurg. 1993;78:741-747.
Fuller DB, Bloom JG. Radiotherapy for chordoma. Int J Radiat Oncol Biol Phys. 1988;15:331-339.
Hasegawa T, Ishii D, Kida Y, et al. Gamma Knife surgery for skull base chordomas and chondrosarcomas. J Neurosurg. 2007;107:752-757.
Heffelfinger MJ, Dahlin DC, MacCarty CS, et al. Chordomas and cartilaginous tumors at the skull base. Cancer. 1973;32:410-420.
Horgan MA, Anderson GJ, Kellogg JX, et al. Classification and quantification of the petrosal approach to the petroclival region. J Neurosurg. 2000;93:108-112.
Katayama Y, Tsubokawa T, Hirasawa T, et al. Intradural extraosseous chordoma in the foramen magnum region. Case report. J Neurosurg. 1991;75:976-979.
Kaylie DM, Horgan MA, Delashaw JB, et al. Hearing preservation with the transcrusal approach to the petroclival region. Otol Neurotol. 2004;25:594-598.
Lanzino G, Dumont AS, Lopes MB, et al. Skull base chordomas: overview of disease, management options, and outcome. Neurosurg Focus. 2001;10(3):E12.
Letson GD, Muro-Cacho CA. Genetic and molecular abnormalities in tumors of the bone and soft tissues. Cancer Control. 2001;8:239-251.
Martin JJ, Niranjan A, Kondziolka D, et al. Radiosurgery for chordomas and chondrosarcomas of the skull base. J Neurosurg. 2007;107:758-764.
McMaster ML, Goldstein AM, Bromley CM, et al. Chordoma: incidence and survival patterns in the United States, 1973-1995. Cancer Causes Control. 2001;12:1-11.
Meyers SP, Hirsch WLJr, Curtin HD, et al. Chordomas of the skull base: MR features. AJNR Am J Neuroradiol. 1992;13:1627-1636.
Salisbury JR, Deverell MH, Cookson MJ, et al. Three-dimensional reconstruction of human embryonic notochords: clue to the pathogenesis of chordoma. J Pathol. 1993;171:59-62.
Samii A, Gerganov VM, Herold C, et al. Chordomas of the skull base: surgical management and outcome. J Neurosurg. 2007;107:319-324.
Tzortzidis F, Elahi F, Wright D, et al. Patient outcome at long-term follow-up after aggressive microsurgical resection of cranial base chordomas. Neurosurgery. 2006;59:230-237.
Wolfe JT3rd, Scheithauer BW. “Intradural chordoma” or “giant ecchordosis physaliphora”? Report of two cases. Clin Neuropathol. 1987;6:98-103.
Yamaguchi T, Suzuki S, Ishiiwa H, et al. Benign notochordal cell tumors: a comparative histological study of benign notochordal cell tumors, classic chordomas, and notochordal vestiges of fetal intervertebral discs. Am J Surg Pathol. 2004;28:756-761.
1 Bailey P, Bagdasar D. Intracranial chordoblastoma. Am J Pathol. 1929;5:439-449.
2 Spjut HJ, Luse SA. Chordoma: an electron microscopic study. Cancer. 1964;17:643-656.
3 Bailey P. Intracranial Tumors, 2nd ed. Springfield, IL: Charles C Thomas; 1948.
4 Cushing H. The Pituitary Body and Its Disorders, Clinical States Produced by Disorder. Philadelphia: J.B.: Lippincott; 1912.
5 Chugh R, Tawbi H, Lucas DR, et al. Chordoma: the nonsarcoma primary bone tumor. Oncologist. 2007;12:1344-1350.
6 McMaster ML, Goldstein AM, Bromley CM, et al. Chordoma: incidence and survival patterns in the United States, 1973-1995. Cancer Causes Control. 2001;12:1-11.
7 Heffelfinger MJ, Dahlin DC, MacCarty CS, et al. Chordomas and cartilaginous tumors at the skull base. Cancer. 1973;32:410-420.
8 Muro K, Das S, Raizer JJ. Chordomas of the craniospinal axis: multimodality surgical, radiation and medical management strategies. Expert Rev Neurother. 2007;7:1295-1312.
9 Dias MS. Normal and abnormal development of the spine. Neurosurg Clin N Am. 2007;18:415-429.
10 Salisbury JR, Deverell MH, Cookson MJ, et al. Three-dimensional reconstruction of human embryonic notochords: clue to the pathogenesis of chordoma. J Pathol. 1993;171:59-62.
11 Macdonald RL, Cusimano MD, Deck JH, et al. Cerebrospinal fluid fistula secondary to ecchordosis physaliphora. Neurosurgery. 1990;26:515-518.
12 Macdonald RL, Deck JH. Immunohistochemistry of ecchordosis physaliphora and chordoma. Can J Neurol Sci. 1990;17:420-423.
13 Yamaguchi T, Suzuki S, Ishiiwa H, et al. Benign notochordal cell tumors: a comparative histological study of benign notochordal cell tumors, classic chordomas, and notochordal vestiges of fetal intervertebral discs. Am J Surg Pathol. 2004;28:756-761.
14 Yamaguchi T, Yamato M, Saotome K. First histologically confirmed case of a classic chordoma arising in a precursor benign notochordal lesion: differential diagnosis of benign and malignant notochordal lesions. Skeletal Radiol. 2002;31:413-418.
15 Erdem E, Angtuaco EC, Van Hemert R, et al. Comprehensive review of intracranial chordoma. Radiographics. 2003;23:995-1009.
16 Lanzino G, Dumont AS, Lopes MB, et al. Skull base chordomas: overview of disease, management options, and outcome. Neurosurg Focus. 2001;10(3):E12.
17 Machida T, Aoki S, Sasaki Y, et al. Magnetic resonance imaging of clival chordomas. Acta Radiol Suppl. 1986;369:167-169.
18 Meyers SP, Hirsch WLJr, Curtin HD, et al. Chordomas of the skull base: MR features. AJNR Am J Neuroradiol. 1992;13:1627-1636.
19 Katayama Y, Tsubokawa T, Hirasawa T, et al. Intradural extraosseous chordoma in the foramen magnum region. Case report. J Neurosurg. 1991;75:976-979.
20 Wolfe JT3rd, Scheithauer BW. “Intradural chordoma” or “giant ecchordosis physaliphora”? Report of two cases. Clin Neuropathol. 1987;6:98-103.
21 Burger PC, Scheithauer BW, Vogel FS. Surgical Pathology of the Nervous System, 4th ed. New York: Churchill Livingstone; 2002.
22 Bayrakli F, Guney I, Kilic T, et al. New candidate chromosomal regions for chordoma development. Surg Neurol. 2007;68:425-430.
23 Letson GD, Muro-Cacho CA. Genetic and molecular abnormalities in tumors of the bone and soft tissues. Cancer Control. 2001;8:239-251.
24 Horgan MA, Anderson GJ, Kellogg JX, et al. Classification and quantification of the petrosal approach to the petroclival region. J Neurosurg. 2000;93:108-112.
25 Al-Mefty O, Borba LA. Skull base chordomas: a management challenge. J Neurosurg. 1997;86:182-189.
26 Kaylie DM, Horgan MA, Delashaw JB, et al. Hearing preservation with the transcrusal approach to the petroclival region. Otol Neurotol. 2004;25:594-598.
27 Arnautovic KI, Al-Mefty O. Surgical seeding of chordomas. Neurosurg Focus. 2001;10(3):E7.
28 Almefty K, Pravdenkova S, Colli BO, et al. Chordoma and chondrosarcoma: similar, but quite different, skull base tumors. Cancer. 2007;110:2457-2467.
29 Colli BO, Al-Mefty O. Chordomas of the skull base: follow-up review and prognostic factors. Neurosurg Focus. 2001;10(3):E1.
30 Forsyth PA, Cascino TL, Shaw EG, et al. Intracranial chordomas: a clinicopathological and prognostic study of 51 cases. J Neurosurg. 1993;78:741-747.
31 Samii A, Gerganov VM, Herold C, et al. Chordomas of the skull base: surgical management and outcome. J Neurosurg. 2007;107:319-324.
32 Sekhar LN, Pranatartiharan R, Chanda A, et al. Chordomas and chondrosarcomas of the skull base: results and complications of surgical management. Neurosurg Focus. 2001;10(3):E2.
33 Tzortzidis F, Elahi F, Wright D, et al. Patient outcome at long-term follow-up after aggressive microsurgical resection of cranial base chordomas. Neurosurgery. 2006;59:230-237.
34 Catton C, O’Sullivan B, Bell R, et al. Chordoma: long-term follow-up after radical photon irradiation. Radiother Oncol. 1996;41:67-72.
35 Fuller DB, Bloom JG. Radiotherapy for chordoma. Int J Radiat Oncol Biol Phys. 1988;15:331-339.
36 Hasegawa T, Ishii D, Kida Y, et al. Gamma Knife surgery for skull base chordomas and chondrosarcomas. J Neurosurg. 2007;107:752-757.
37 Brada M, Pijls-Johannesma M, De Ruysscher D. Proton therapy in clinical practice: current clinical evidence. J Clin Oncol. 2007;25:965-970.
38 Debus J, Hug EB, Liebsch NJ, et al. Brainstem tolerance to conformal radiotherapy of skull base tumors. Int J Radiat Oncol Biol Phys. 1997;39:967-975.
39 Debus J, Schulz-Ertner D, Schad L, et al. Stereotactic fractionated radiotherapy for chordomas and chondrosarcomas of the skull base. Int J Radiat Oncol Biol Phys. 2000;47:591-596.
40 Martin JJ, Niranjan A, Kondziolka D, et al. Radiosurgery for chordomas and chondrosarcomas of the skull base. J Neurosurg. 2007;107:758-764.