[level-membership-for-pulmolory-and-respiratory-category]9
CHEMICAL CONTROL OF BREATHING
Introduction
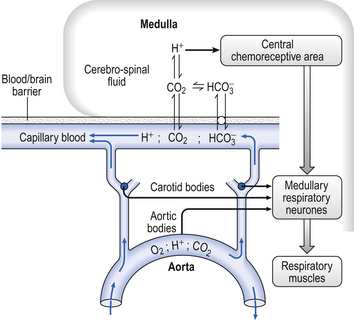
It is rare for excess CO2 or lack of O2 to occur alone: they usually occur together, and the whole chemoreceptor system is shown schematically in Figure 9.1.
Oxygen lack
The term for a lack of oxygen in any gas mixture or solution is hypoxia. Lack of O2 in arterial blood is termed hypoxaemia. Total absence of O2 is anoxia. It is very easy to change the amount of a gas in the arterial blood by utilizing the powerful gas-transporting properties of the lungs. Simply giving a subject a gas mixture to breathe will result in his or her arterial blood taking on the composition of that gas mixture within remarkably few breaths. The rate at which equilibrium is reached depends on the solubility of the gas in body fluids, and this has important consequences in anaesthesia. However, for the gases we are concerned with here equilibrium is approached within a few dozen breaths. The chemoreceptors that sense lack of arterial O2 are the carotid bodies and the aortic bodies. In humans it is the carotid bodies that are mainly responsible for the respiratory response. They are small (5.0 mm diameter) nodules of glomus tissue (Latin glomerus, a skein or ball of thread, i.e. a knot of capillaries) situated near the bifurcation of each common carotid artery. Unlike the carotid bodies, which mainly respond to Pao2, the aortic bodies are stimulated by reductions in arterial O2 content, e.g. carbon monoxide poisoning and anaemia affect them more. So it seems that the aortic bodies are sensitive to the total amount of O2 delivered to them, and the carotid bodies are sensitive to Pao2. The carotid bodies are situated close to the baroreceptor region of the carotid arteries, which help to regulate blood pressure, and are frequently confused with them. The carotid bodies are not baroreceptors.
Histology, embryology and anatomy of the carotid bodies
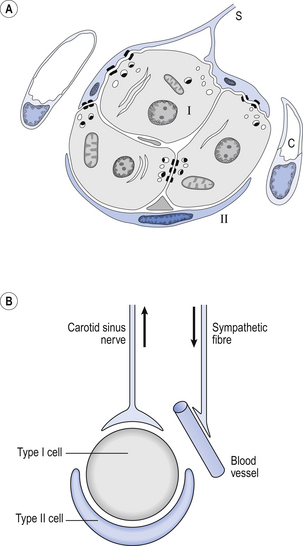
The function of the carotid bodies is related to their unusual structure. They have an extremely high metabolic rate (about three times that of the brain) but their rate of perfusion by blood from the carotid arteries is even higher: 10 times that which would be expected. This blood flows through capillaries (Fig. 9.2A and B) which surround the sensory elements (the glomus or type I cells) that monitor blood Po2. The type I cells seem to be supported by type II (sustentacular) cells, whose function is still not clear. The type I cells send their information to the brain via the carotid sinus nerve, a branch of the glossopharyngeal nerve (Fig. 9.2A and B), which also provides them with sympathetic and parasympathetic innervation. A separate supply of sympathetic fibres from the nearby superior cervical ganglion innervates the carotid bodies’ blood vessels.
Hypoxic stimulation
Activity in the carotid bodies is measured experimentally as the frequency of discharge of action potentials in the carotid sinus nerve. Increased activity expresses itself in the whole animal as an increase in ventilation. Hypoxia stimulates peripheral chemoreceptors, which is unusual, as the activity of almost all other organs is depressed by it.
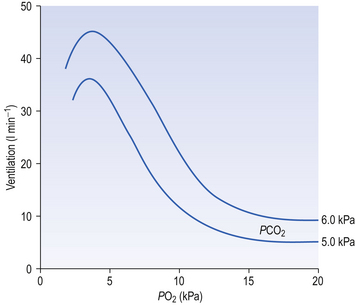
The effect of decreasing a subject’s arterial Po2 by giving them increasingly hypoxic gas to breathe is shown in Figure 9.3.
Hypercapnic stimulation
1. Chemoreceptors have a very high metabolic rate and so rapidly use up O2 supplied to them.
2. They have a very high blood flow, gram for gram 40 times that of the brain.
3. Pao2 must be reduced considerably before there is stimulation of breathing, but then the increase is large.
4. Increasing Pao2 above normal (13 kPa) by inhaling O2-rich mixtures only produces a small reduction in breathing by depressing chemoreceptor activity.
5. Increasing arterial [H+] does not have a great effect on central chemoreceptors but stimulates peripheral chemoreceptors.
6. Peripheral chemoreceptors are much less sensitive to increases in Paco2 than are the central chemoreceptors. (The central chemoreceptors are ‘protected’ from changes in arterial [H+] by the blood–brain barrier.)
7. Sympathetic activity has only a small effect on chemoreceptor blood flow and sensitivity during hypoxic stimulation.
Hypoxia and breathing
The answer to the above question is that it would be a waste of time having a more sensitive detector of O2 lack because the shape of the oxyhaemoglobin dissociation curve would defeat its sensitivity. You can see from the oxyhaemoglobin dissociation curve shown in Figure 8.2 (p. 103) that even if Po2 is reduced to 8 kPa, haemoglobin is still 90% saturated. Also, Po2 can rise to infinity and haemoglobin can only be 100% saturated. This useful situation means that ventilation of the lungs can halve or double without the amount of O2 being carried changing very much. But by the same token, a mechanism that relied on O2 saturation to control breathing under normal circumstances would lack sensitivity, because saturation does not change much over a large range of partial pressure.
Hypoxic stimulation of breathing is also opposed by changes in CO2 and [H+], because as breathing begins to be stimulated CO2 is washed out of the blood, arterial H+ falls and the drive to breathe from these two sources is reduced, producing what is sometimes called the hypocapnic brake (Fig. 9.4). Just how powerful a drive to breathe hypoxia can be is demonstrated if this braking effect is prevented by adding CO2 to the inspired air to keep its levels constant in the blood. Under these circumstances hypoxia produces 10 times the effect produced if CO2 is allowed to be washed out.
Long-term hypoxic stimulation and anaesthesia
For example, in adult human subjects hypoxia lasting for about an hour produces an immediate increase in ventilation (in 3–5 minutes) followed by a decrease to a steady state level higher than the control, normoxic level. This occurs even when Paco2 is kept constant and this phenomenon is called hypoxic ventilatory decline. This may be due to hypoxic CNS depression. On a longer time scale, in animals at least, there is a ventilatory acclimatization characterized by a time dependent increase in ventilation which stabilizes at a value greater than the response to acute hypoxia (Fig. 9.5). This response is somewhat confusingly called acclimatization to short-term hypoxia (ASTH) if only to distinguish it from acclimatization to long-term hypoxia (ALTH) which involves the situation found in natives or very long-term residents at altitude. The major mechanism of ASTH appears to be an increased sensitivity of the carotid body to hypoxia (Fig. 9.5) and not, as was once thought, changes in the CSF surrounding the central chemoreceptors.
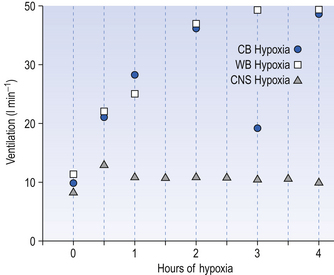
However, in the longer lasting clinical condition, or at altitude, arterial Po2 is reduced, causing stimulation of the peripheral chemoreceptors, which in turn increases ventilation. In the high-altitude situation this hyperventilation washes out CO2 from the blood and cerebrospinal fluid and they become more alkaline, reducing the drive to breathe (mainly at the central chemoreceptors) below the increased level that is appropriate for the reduced atmospheric Po2. After a day or two the active transport system of the blood–brain barrier returns the [H+] of the CSF to normal. This restored drive from CO2 and the extra drive from O2 lack goes part-way to achieving the required ventilation. Within a few weeks at altitude the kidneys excrete extra 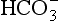 and restore blood [H+] which, together with the hypoxic drive to the peripheral chemoreceptors, stimulates breathing to an appropriate level.
and restore blood [H+] which, together with the hypoxic drive to the peripheral chemoreceptors, stimulates breathing to an appropriate level.
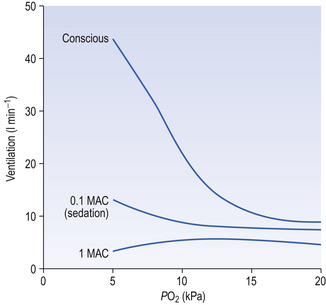
Anaesthetists measure anaesthetic effect in terms of MAC (minimal alveolar concentration for anaesthesia). The peripheral chemoreceptors are extremely sensitive to inhalation anaesthetics (Fig. 9.6). The consequences of this for patients with lung disease receiving anaesthesia are important. They cannot respond when challenged by hypoxia and, if of the ‘blue bloater’ type, who has already lost his drive to breathe from CO2, will stop breathing when anaesthesia abolishes his drive from hypoxia. You can see from Figure 9.6 that quite low levels of anaesthesia, of the order of those found in the postoperative period, when the patient appears able to look after himself, can seriously blunt the response to hypoxia.
Carbon dioxide excess
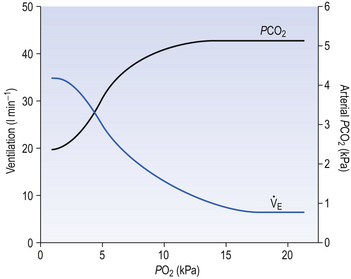
Ambient air normally contains very little CO2 (0.03%) and, unlike reductions in Pao2, any increase in inhaled CO2 stimulates breathing in a linear manner (Fig. 9.7) until levels are reached which act as an effective anaesthetic. (CO2 has been used in this way in clinical practice.)
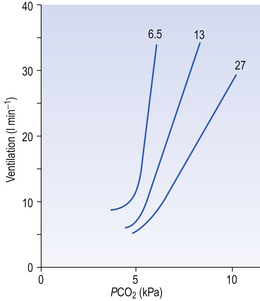
Fig. 9.7 The ventilatory response to asphyxia. The response to varying levels of CO2 at fixed levels of O2 is shown. This relationship might equally well be demonstrated with fixed levels of CO2 and varying levels of O2 (Fig. 9.3).
The site of central chemoreceptors
Although no discrete structures such as the carotid bodies have been identified as central chemoreceptors, perfusing the ventrolateral surfaces of the medulla with acidic solutions or solutions with a high Pco2 stimulates breathing. Intracellular recordings in the neurons 500 μm or so below the surface of the brain in the regions shown in Figure 9.8 reveal that the frequency of discharge of these cells increases as the acidity or Pco2 of the interstitial fluid surrounding them increases. This leads to the question, is it CO2 or H+ produced by the acidifying effects of CO2 that is the stimulus? A great deal of careful research indicates that the specific stimulus to the central chemoreceptor neurons is intracellular [H+], which is determined primarily by the Pco2 of the cerebrospinal fluid.
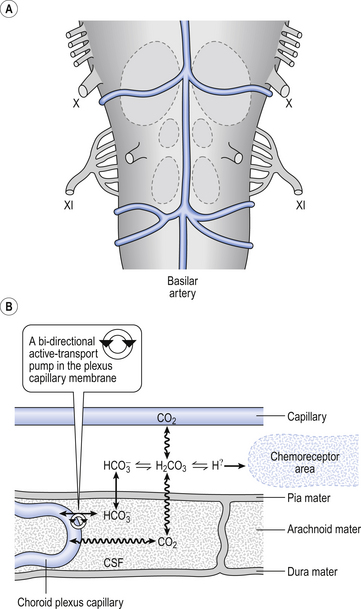
Fig. 9.8 Central chemoreceptive areas of the brain. (A) These are not the traditional ‘respiratory centres’ dealt with in Chapter 11. (B) Their environment is closely controlled by the blood–brain barrier which is permeable to passive diffusion of CO2 and actively transports 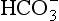 .
.
Blood/CSF relationships
As well as being acidic, CO2 is a highly diffusible gas which is important in the relationship between arterial blood and cerebrospinal fluid (CSF) which is established across the blood–brain barrier. The activity of the blood–brain barrier makes the CSF bathing the brain and spinal cord the most closely controlled environment in the body. Lipid-soluble molecules such as O2 and CO2 diffuse freely between blood plasma and brain. Ions such as H+ and 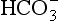 move under strict control, and are often pumped against their concentration gradients by active transport when it is necessary to control the environment of the brain. The capillaries whose walls form the blood–brain barrier are specialized to produce the CSF from plasma in a region known as the choroid plexus (Fig. 9.8B).
move under strict control, and are often pumped against their concentration gradients by active transport when it is necessary to control the environment of the brain. The capillaries whose walls form the blood–brain barrier are specialized to produce the CSF from plasma in a region known as the choroid plexus (Fig. 9.8B).
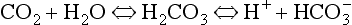
to the right, producing H+, which is probably the specific stimulus of the central chemoreceptors. It is difficult to see at first glance why the above reaction, which produces both acid H+ and base (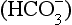 ), acidifies a solution (but remember, it is the ratio of H+/
), acidifies a solution (but remember, it is the ratio of H+/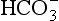 that determines acidity, and there is a lower concentration of H+ than
that determines acidity, and there is a lower concentration of H+ than 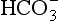 in plasma; therefore, the addition of one of each of the ions has a bigger effect on H+ (see p. 117). Because H+ passes through the blood-brain barrier with difficulty, increases in arterial H+ do not affect the central chemoreceptors if arterial Pco2 is kept constant.
in plasma; therefore, the addition of one of each of the ions has a bigger effect on H+ (see p. 117). Because H+ passes through the blood-brain barrier with difficulty, increases in arterial H+ do not affect the central chemoreceptors if arterial Pco2 is kept constant.
The ion-pumping activity of the blood–brain barrier is particularly important in compensating for chronic disturbances of the composition of the CSF, such as occur during long stays at altitude or in chronic lung disease. Small and acute decreases in blood CO2, caused by singing, for example, do not depress breathing because of the horizontal part of the Pco2/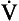 E curve (Fig. 9.7). On average, increasing people’s Pco2 by 0.3 kPa doubles their minute ventilation.
E curve (Fig. 9.7). On average, increasing people’s Pco2 by 0.3 kPa doubles their minute ventilation.
Asphyxia
The overall effect of changes in all three was described by a formula devised by Gray in 1945:
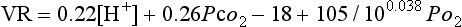
where VR is the ratio of ventilation during asphyxia to unstimulated ventilation. This formula is more important as an illustration that no single factor controls ventilation than as a quantitative estimate.
The way in which hypoxia and hypercapnia combine to stimulate breathing is shown in Figure 9.7, where each curve represents a Pco2/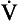 E relationship at a different Po2. With progressive hypoxia the curves are seen to steepen, producing a greater ventilatory response than would be produced by the simple sum of the two stimuli. On the other hand, it is not unusual for the arterial Po2, Pco2 or [H+] of patients to be changed independently by their disease. Most usually this change consists of a fall in Po2 while Pco2 is maintained close to normal.
E relationship at a different Po2. With progressive hypoxia the curves are seen to steepen, producing a greater ventilatory response than would be produced by the simple sum of the two stimuli. On the other hand, it is not unusual for the arterial Po2, Pco2 or [H+] of patients to be changed independently by their disease. Most usually this change consists of a fall in Po2 while Pco2 is maintained close to normal.
Chemical control of breathing determines minute ventilation, with changes taking place over a matter of one or more minutes. The pattern of breathing that makes up this minute ventilation is determined by the neural control of ventilation, which can bring about changes in pattern in fractions of a second. This process is dealt with in Chapter 10.
Further reading
[/level-membership-for-pulmolory-and-respiratory-category][not-level-membership-for-pulmolory-and-respiratory-category]9
CHEMICAL CONTROL OF BREATHING
Introduction
It is rare for excess CO2 or lack of O2 to occur alone: they usually occur together, and the whole chemoreceptor system is shown schematically in Figure 9.1.
Oxygen lack
The term for a lack of oxygen in any gas mixture or solution is hypoxia. Lack of O2 in arterial blood is termed hypoxaemia. Total absence of O2 is anoxia. It is very easy to change the amount of a gas in the arterial blood by utilizing the powerful gas-transporting properties of the lungs. Simply giving a subject a gas mixture to breathe will result in his or her arterial blood taking on the composition of that gas mixture within remarkably few breaths. The rate at which equilibrium is reached depends on the solubility of the gas in body fluids, and this has important consequences in anaesthesia. However, for the gases we are concerned with here equilibrium is approached within a few dozen breaths. The chemoreceptors that sense lack of arterial O2 are the carotid bodies and the aortic bodies. In humans it is the carotid bodies that are mainly responsible for the respiratory response. They are small (5.0 mm diameter) nodules of glomus tissue (Latin glomerus, a skein or ball of thread, i.e. a knot of capillaries) situated near the bifurcation of each common carotid artery. Unlike the carotid bodies, which mainly respond to Pao2, the aortic bodies are stimulated by reductions in arterial O2 content, e.g. carbon monoxide poisoning and anaemia affect them more. So it seems that the aortic bodies are sensitive to the total amount of O2 delivered to them, and the carotid bodies are sensitive to Pao2. The carotid bodies are situated close to the baroreceptor region of the carotid arteries, which help to regulate blood pressure, and are frequently confused with them. The carotid bodies are not baroreceptors.
Histology, embryology and anatomy of the carotid bodies
The function of the carotid bodies is related to their unusual structure. They have an extremely high metabolic rate (about three times that of the brain) but their rate of perfusion by blood from the carotid arteries is even higher: 10 times that which would be expected. This blood flows through capillaries (Fig. 9.2A and B) which surround the sensory elements (the glomus or type I cells) that monitor blood Po2. The type I cells seem to be supported by type II (sustentacular) cells, whose function is still not clear. The type I cells send their information to the brain via the carotid sinus nerve, a branch of the glossopharyngeal nerve (Fig. 9.2A and B), which also provides them with sympathetic and parasympathetic innervation. A separate supply of sympathetic fibres from the nearby superior cervical ganglion innervates the carotid bodies’ blood vessels.
Hypoxic stimulation
Activity in the carotid bodies is measured experimentally as the frequency of discharge of action potentials in the carotid sinus nerve. Increased activity expresses itself in the whole animal as an increase in ventilation. Hypoxia stimulates peripheral chemoreceptors, which is unusual, as the activity of almost all other organs is depressed by it.
The effect of decreasing a subject’s arterial Po2 by giving them increasingly hypoxic gas to breathe is shown in Figure 9.3.
Hypercapnic stimulation
1. Chemoreceptors have a very high metabolic rate and so rapidly use up O2 supplied to them.
2. They have a very high blood flow, gram for gram 40 times that of the brain.
3. Pao2 must be reduced considerably before there is stimulation of breathing, but then the increase is large.
4. Increasing Pao2 above normal (13 kPa) by inhaling O2-rich mixtures only produces a small reduction in breathing by depressing chemoreceptor activity.
5. Increasing arterial [H+] does not have a great effect on central chemoreceptors but stimulates peripheral chemoreceptors.
6. Peripheral chemoreceptors are much less sensitive to increases in Paco2
[/not-level-membership-for-pulmolory-and-respiratory-category]
