[level-membership-for-neurosurgery-category]
CHAPTER 364 Cerebral Vasospasm
Vasospasm in Conditions Other Than Aneurysm Rupture
Vasospasm has been observed in head-injured patients both with and without subarachnoid blood clots in the basal subarachnoid cisterns, although the diagnosis has sometimes relied on transcranial Doppler (TCD) blood velocity measurements, which are difficult to interpret in the setting of brain injury.1,2 The clinical significance of posttraumatic vasospasm is debated, and there is not yet any proven value of either monitoring cerebral arterial diameters or routine treatment with calcium channel blockers in the setting of traumatic brain injury associated with SAH.3,4
Epidemiology of Vasospasm
Angiographic vasospasm is common after rupture of an aneurysm, with an overall incidence of 50% to 90%.5 Still useful but rough estimates are that at least moderate vasospasm in at least one cerebral artery will develop in two thirds of patients with ruptured aneurysms, half of these patients will become symptomatic as a result of ischemia, and a cerebral infarct will develop in about half of these patients. An analysis of 2741 patients entered into SAH trials in the 1990s found that cerebral infarction (all causes) had developed in 26% at 6 weeks, which correlated strongly with poor outcome.6 Cerebral infarction was significantly associated with increasing patient age, worse neurological grade on admission, history of hypertension or diabetes mellitus, larger aneurysm, induced hypertension, fever, and a diagnosis of symptomatic vasospasm. With modern SAH management routines, the combined risk for death and permanent disability from vasospasm alone has shrunk to less than 10%, but it still remains one of the leading causes of preventable poor outcome after rupture of an aneurysm (Figs. 364-1 and 364-2).7–9
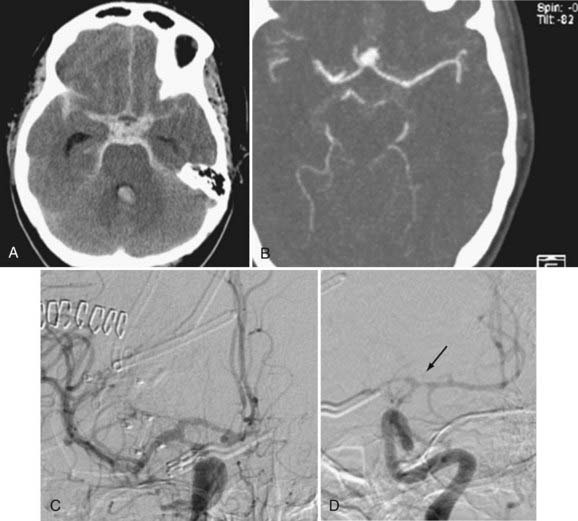
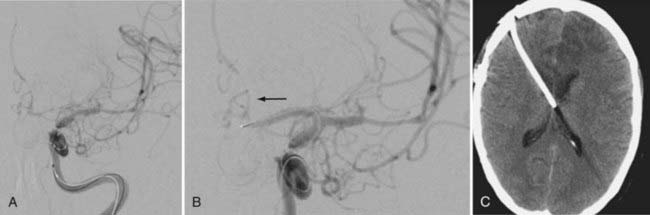
Prediction of Vasospasm
A large volume of persistent subarachnoid clot is the most important risk factor predictive of vasospasm after SAH.10,11 The original Fisher grading scale in which clot volume and distribution on admission computed tomography (CT) are related to risk for vasospasm has been modified and in one single-center study found to have greater predictive value for delayed ischemia and prognosis.12 This modified Fisher scale (Table 364-1) scores hemorrhage noted on CT from 0 to 4: 0, no SAH or intraventricular hemorrhage (IVH) (very low risk for vasospasm); 1, focal or diffuse thin layer of SAH, no IVH (low risk for vasospasm); 2, focal or diffuse thin layer of SAH, IVH present (moderate risk for vasospasm); 3, focal or diffuse thick layer of SAH, no IVH (high risk for vasospasm); and 4, focal or diffuse thick layer of SAH, IVH present (highest risk for vasospasm). A slower rate of subarachnoid clot clearance has also been shown to be an independent predictor of vasospasm, although this is not an easy measurement in clinical practice.11
| 0 | No SAH or IVH: very low risk |
| 1 | Focal or diffuse thin layer of SAH, no IVH: low risk |
| 2 | Focal or diffuse thin layer of SAH, IVH present: moderate risk |
| 3 | Focal or diffuse thick layer of SAH, no IVH: high risk |
| 4 | Focal or diffuse thick layer of SAH, IVH present: very high risk |
IVH, intraventricular hemorrhage; SAH, subarachnoid hemorrhage.
Other risk factors for the development of vasospasm have been identified, including poor neurological grade or loss of consciousness on admission, cigarette smoking, and preexisting hypertension (Table 364-2).13 Factors studied but found to have either an unclear or unlikely relationship with risk for vasospasm include gender, patient age, and aneurysm location. There has been a suggestion that Japanese people may be more susceptible to vasospasm.14 In addition, a small study suggested that cocaine use is an independent risk factor for vasospasm.15
TABLE 364-2 Risk Factors for Vasospasm
Spontaneous perimesencephalic or prepontine SAH (or both) unassociated with aneurysm rupture is typically a low-volume hemorrhage that clears quickly with a low risk for the development of vasospasm.16 Thick and persistent nonaneurysmal SAH, however, is still associated with a risk for vasospasm.
There is some evidence that endovascular coiling, as opposed to microsurgical repair of ruptured aneurysms, is associated with a lower risk for the subsequent development of vasospasm,17 although the difference was not large and a rigorous comparison has not yet been made.
Pathogenesis
Smooth Muscle Contraction
Vasospasm is prolonged cerebral arterial constriction caused by vascular smooth muscle contraction. The hemoglobin released from subarachnoid blood clots triggers the entry and release of calcium and subsequent activation of calcium/calmodulin-dependent myosin light-chain kinase, which in turn leads to phosphorylation of the myosin light chain and induces actin and myosin cross-linkage and mechanical shortening (smooth muscle contraction). Such contraction requires adenosine triphosphate and calcium, and vascular smooth muscle relies more on extracellular than intracellular calcium stores, which enter through voltage-gated and receptor-operated calcium channels. Although myofilament activation depends on calcium and high-energy phosphates, chronic vasospasm, which ensues days later and lasts up to several weeks, does not. The contractile proteins protein kinase C, Rho kinase, and protein tyrosine kinase and their corresponding signal transduction pathways have been implicated in vasospasm models when their activation shifts the contractile mechanism toward increased shortening in the absence of high intracellular calcium levels.18,19 This contiguous and second-phase “chronic” vasospasm is less reversible with pharmacologic vasodilators both in animal models20 and in humans undergoing intra-arterial, pharmacologic vasodilation treatment.
Sustained vasoconstriction is associated not only with functional impairment of the vessel but also with ultrastructural damage to the vascular wall layers, including vacuolization of endothelial cells and loss of tight junctions, breakage of the internal elastic lamina, and patchy myonecrosis in the tunica media.21
Endothelial Injury, Nitric Oxide, and Endothelin-1
Auto-oxidation of the oxyhemoglobin contained in blood clots encasing cerebral arteries produces methemoglobin and superoxide anion radical, which in turn lead to lipid peroxidation.22,23 Harmful hydroxyl radicals and lipid peroxides permeate the vessel wall and injure endothelial and smooth muscle cells.24,25 Damage to the endothelium in particular is thought to play a key role in the establishment of vasospasm, either through the loss of endothelial nitric oxide (NO) synthesis, an important vasodilator and regulator of vascular tone, or through the overproduction of endothelin, a powerful vasoconstrictor peptide.26 These two endothelial-derived substances and the possible imbalance in their production after SAH are at the center of experimental vasospasm research at the present time.
Decreased availability of the simple molecule NO may contribute to the development of vasospasm in the following ways: (1) endothelial NO synthase (eNOS) dysfunction in vasospastic vessels, (2) NO scavenging by oxyhemoglobin; (3) reversal of vasospasm by NO donors, (4) disappearance of neuronal NO synthase (nNOS) activity from the adventitia of vasospastic vessels, and (5) decreased cerebrospinal fluid (CSF) nitrite levels along with increased levels of asymmetric dimethyl-L-arginine, the endogenous inhibitor of NO synthase.27–35
Endothelin-1 (ET-1) is the predominant isoform of endothelin and has the greatest role in vasoconstriction. ET-1 is a 21–amino acid cleavage product of a 212–amino acid peptide precursor, the final step mediated by endothelin-converting enzyme. It is released on the abluminal side of the tunica media, acts on neighboring vascular smooth muscle ETA receptors, and causes profound and sustained vasoconstriction.36 ET-1 levels have been found to be elevated in the CSF of patients in whom vasospasm and brain ischemia develop,37–39 and either inhibition of ET-1 production or antagonism of its effect has prevented vasospasm in animal models.36
Inflammation, Vessel Remodeling, and Vasospasm
Although it has become clear that vasospasm is not a type of vasculitis, there is evidence that inflammatory mechanisms are activated after SAH and may therefore be involved in the development of vasospasm, possibly by contributing to vasocontraction or modifying the vessel wall extracellular matrix and smooth muscle cell phenotype—a process known as vascular “remodeling.”40 Inflammatory cytokines,41,42 intercellular adhesion molecules (ICAMs),43,44 and genetic upregulation of inflammatory, proliferative, and extracellular matrix–regulating genes45–47 have been examined. In humans, elevations of plasma complement C3a and soluble ICAM-1 have been associated with poor outcome and the development of vasospasm, respectively,48,49 and increased intrathecal levels of the cytokine interleukin-6 predicted vasospasm in another small study.50
Although the precise pathogenesis of vasospasm is still the subject of investigation, a prolonged, biphasic vasoconstrictive process in which the second, chronic phase is mechanistically distinct from the first seems most consistent with what has been seen experimentally and in humans. It remains possible that additional processes, including inflammation, may contribute to the pathogenesis of this condition (Table 364-3).
Clinical Features and Investigation
Symptoms, Signs, and Differential Diagnosis
Coincident with its delayed onset, symptoms of ischemia resulting from cerebral vasospasm most commonly appear 1 week after aneurysm rupture but often occur later; accordingly, it is important to remain vigilant for this complication for at least 2 weeks after SAH. Regular and careful bedside examination remains the simplest and most effective means of detecting early ischemia in awake, examinable patients; one should concentrate on subtle findings such as diminished attention, changes in verbal output, or a slight but new pronator drift of the upper extremity. Symptomatic vasospasm usually has a gradual onset, sometimes heralded by increased headache and either agitation or somnolence—a change in patient behavior. It then follows a progressive course if untreated. A smaller group of patients will experience precipitous deterioration.51 Signs of symptomatic vasospasm are referable to the territory that has become ischemic and are most easily distinguished when they lateralize to a middle cerebral artery (MCA) territory with monoparesis or hemiparesis and, when the dominant hemisphere is affected, aphasia. Anterior cerebral artery vasospasm can be marked by leg weakness, but because it is often bilateral in distribution, confusion, drowsiness, poverty of speech, and eventually abulia are characteristic signs. Vertebrobasilar vasospasm can also cause a more generalized deterioration, with a reduced level of consciousness being an early sign.
Delayed neurological deterioration after aneurysmal SAH has a number of causes, including increased edema surrounding intracerebral hematomas, contusions, or infarcts; rebleeding of the aneurysm or aneurysmal remnant; hydrocephalus; sepsis, including meningitis and ventriculitis; hyponatremia; hypoxia; and hypotension (Table 364-4). One or more of these conditions can magnify even a focal neurological deficit and therefore easily be mistaken for vasospasm, which has a tendency to be overdiagnosed in the setting of SAH. Conversely, it is recognized that there is a subgroup of patients who suffer a delayed and often global decline in neurological status for which no single underlying cause can be identified. “Cortical spreading depression” has been one proposed mechanism at work in these patients.19
TABLE 364-4 Causes of Delayed Deterioration after Subarachnoid Hemorrhage
Diagnosis
Diagnosis of symptomatic vasospasm requires that the other causes of delayed worsening listed earlier be ruled out with CT and appropriate laboratory investigations. If vasospasm remains the most likely cause of deterioration and treatment by induced hypertension reverses the deficit, the diagnosis can safely be assumed without further testing. Failure to respond in this scenario, as well as evaluation of comatose patients, requires additional testing (Table 364-5).
CBF, cerebral blood flow; CT, computed tomography; MRI, magnetic resonance imaging; SPECT, single-photon emission computed tomography; TCD, transcranial Doppler.
Transcranial Doppler
TCD works on the principle that as an artery narrows, blood flow velocity within it increases. Because examinations are noninvasive, are carried out at the bedside, and can easily be performed on a daily basis, TCD is frequently used to monitor patients with SAH for increases in intracranial blood flow velocity suggestive of incipient vasospasm. There is good general correlation between TCD velocities and vasospasm, with velocities in the MCA greater than 120 cm/sec indicative of some degree of vasospasm and those greater than 200 cm/sec consistent with severe vasospasm. Because other factors can influence velocity, including blood pressure and overall cerebral blood flow (CBF), distinguishing vasospastic from hyperemic increases in blood velocity has been reported to be facilitated by measuring cervical internal carotid artery (ICA) velocity in addition to intracranial blood velocity.52 A “Lindegaard ratio” of VMCA/VICA greater than 3 is consistent with vasospasm (hyperemia is associated with increased velocity in both the MCA and ICA, so the ratio is the same). A similar velocity ratio between the basilar artery and the extracranial vertebral artery has been proposed to improve the sensitivity and specificity of detecting basilar artery vasospasm.53
The clinical utility of TCD is best when MCA values are clearly low (<120 cm/sec) or very high (>200 cm/sec), and the respective negative and positive predictive values for significant angiographic vasospasm in the MCA trunk are close to 90%.54–56 For values in the intermediate range, additional maneuvers may improve its accuracy in detecting vasospasm, such as testing for hyperemic autoregulatory responses to transient, manual carotid compression57,58 or combining the TCD information with CBF measurements.59 TCD does not reliably detect vasospasm in more peripheral branches,60 which may account for its failure to always correlate with perfusion deficits detected on CBF studies.61 Regular surveillance of SAH patients with TCD ultrasonography by skilled technicians is considered helpful in many neurosurgical units, but the results must be considered in the context of the individual patient along with all other information available.
Cerebral Blood Flow and Perfusion
Single-photon emission CT,62 quantitative stable xenon–enhanced CT,59 and positron emission tomography61 can be used to detect cerebral ischemia after SAH, but none are practical investigations that can be easily performed or repeated in critically ill patients. Magnetic resonance imaging to look for either perfusion deficits63 or ischemia on diffusion-weighted images64,65 has the same limitation.
Perfusion CT has been used in patients with SAH66,67 and with greater experience will probably find more widespread use and validation for predicting or diagnosing vasospasm.
Thermal diffusion flowmetry has been used as bedside monitoring in SAH patients to detect vasospasm causing significant reductions in CBF.68 Provided that the white matter location into which the thermal diffusion microprobe is inserted (usually the frontal lobe) is representative of the territory at risk, continuous measurement of CBF values could be particularly useful in patients with high-grade SAH who cannot be assessed neurologically.
Microdialysis Monitoring
Cerebral microdialysis catheters allow continuous bedside measurement of extracellular concentrations of glutamate, lactate, pyruvate, glucose, and glycerol in brain tissue, thereby screening for excitotoxic cell injury characterized by elevations in lactate with respect to glucose and pyruvate levels and an increase in the glycerol concentration. This somewhat demanding monitoring technique provides chemical information about a very small region of the brain, but it has been used, along with cerebral oximetry, to detect ischemia in patients with SAH.69–71
Vascular Imaging
The most practical method of imaging large and medium-sized cerebral arteries is digital subtraction angiography or high-definition CT angiography.72 Catheter-based angiography is best in patients in whom balloon angioplasty is being considered and can accompany the procedure.
Angiographic vasospasm is a concentric narrowing that can be focal, segmental, or diffuse. It is commonly graded as mild (<25%), moderate (25% to 50%), or severe (>50%) in comparison to baseline, prevasospasm imaging. “Early” vasospasm on admission angiography and within 48 hours of aneurysm rupture has been described in a small percentage of SAH patients, although without baseline angiography it is not clear how this diagnosis can be made accurately in all cases.73 Its detection has been associated with the development of cerebral infarction and poor outcome.
Prevention of Vasospasm and Cerebral Protection
General Measures: Fluid Management and Medical Treatment
Patients have a tendency toward volume contraction in the acute stage of SAH,74 and hypovolemia should be carefully avoided. It is not clear that a deliberate attempt to induce hypervolemia with volume expansion therapy is beneficial in terms of prevention of vasospasm or ischemia or even possible in patients with normal renal function.75–78 Patients should be hydrated with at least 3 L of isotonic fluids daily, and there is some evidence that additional colloid infusions (such as albumin) are beneficial.79 Some SAH patients experience excessive natriuresis and are susceptible to the development of hyponatremia during this time (usually related to elevations in brain natriuretic peptide), an electrolyte disturbance that may increase the risk for vasospasm.80 It has been reported that fludrocortisone administration (0.3 mg/day) may help avert this complication.81
Blood transfusion has been associated with a higher likelihood of both vasospasm and poor outcome in SAH patients,12,82 thus suggesting that anemia is a marker for other factors that contribute to SAH-associated morbidity. The optimal hemoglobin concentration in patients with SAH in terms of hematocrit, blood viscosity, and oxygen delivery to the brain is not known with certainty, but is generally accepted to be higher than 9 g/dL.
Systemic blood pressure should be maintained in the normotensive to slightly hypertensive range, provided that the aneurysm has been repaired. In patients with external ventricular drains, cerebral perfusion pressure (CPP) is a more important parameter to monitor and maintain at 70 mm Hg or greater in poorer grade patients.83
Nimodipine, administered orally or via a nasogastric tube, 60 mg every 4 hours and continued for 3 weeks (for patients requiring that length of hospitalization), is standard treatment of patients with aneurysmal SAH because it has a modest but statistically significant beneficial impact on clinical outcome.84 Nimodipine prevents increases in intracellular calcium by blocking dihydropyridine-sensitive (L-type) calcium channels, but its mechanism of effect in patients with SAH is unknown. It can cause temporary depression of blood pressure, in which case the dose can be reduced and, if possible, given more frequently (i.e., 30 mg every 2 hours) (Table 364-6).
| General Measures |
CPP, cerebral perfusion pressure; CSF, cerebrospinal fluid; HMG-CoA, 3-hydroxyl-3-methylglutaryl coenzyme A; ICP, intracranial pressure.
Investigational Preventive Treatments
Prophylactic Balloon Angioplasty
Based on the experimental observation that angioplasty of canine carotid arteries prevented the development of vasospasm when the arteries were subsequently encased in blood clots,85 prophylactic transluminal balloon angioplasty (TBA) was tested in a phase II multicenter, randomized trial.86 One hundred seventy patients with thick subarachnoid clots were enrolled and 85 were randomized to undergo percutaneous TBA within 96 hours of SAH; the major cerebral arteries targeted included the supraclinoid ICA, MCA M1 segments, and the basilar artery. Although there was a trend toward reduced vasospasm in the patients treated prophylactically by percutaneous TBA, outcome was no different from that in the control group, and the safety of this treatment was questionable; 4 patients had a procedure-related vessel perforation, 3 of whom died.
Clot Clearance
Intracisternal thrombolysis with either recombinant tissue plasminogen activator (rt-PA) or urokinase speeds clearance of subarachnoid clots, and there is evidence that this is associated with prevention of vasospasm and less cerebral ischemia.87 However intuitive and hopeful this approach appears, safety concerns about introducing a thrombolytic agent into the subarachnoid space in a recently injured brain has precluded wide acceptance of this approach. “Head shaking” with cisternal thrombolysis88 or with lumboventricular lavage89 has been reported to be beneficial in terms of prevention of vasospasm in single-center studies, as has simple lumbar drainage.90 Finally, one report has suggested that fenestration of the lamina terminalis at the time of aneurysm clipping reduces the incidence of not just subsequent hydrocephalus but also vasospasm from 55% to 30% in patients with thick subarachnoid clots, the proposed mechanism being increased CSF flow and clot clearance in the basal subarachnoid cisterns.91
Intrathecal Vasodilators
Prolonged-release nicardipine implants placed in the subarachnoid space of patients with thick clots and undergoing surgical clipping appeared to significantly prevent vasospasm and ischemia in a single-center, nonrandomized cohort study,92 results similar to those in a previous study in which papaverine pellets were used.93
Magnesium
Magnesium sulfate (MgSO4) has neuroprotective and vasodilatory properties and has therefore been tested for the prevention of vasospasm and ischemia in patients with SAH. One study found no benefit from intravenous MgSO4 infusions,94 another showed various trends toward benefit,95 and a third suggested efficacy equivalent to nimodipine in preventing ischemic damage.96 Larger randomized trials are required.
Endothelin Receptor Antagonists
As discussed earlier, ET-1 overproduction from cerebral endothelial cells damaged by SAH is one of the leading theories behind the pathogenesis of cerebral vasospasm. The ETA/B receptor antagonist TAK-044 was found to reduce delayed ischemia with an acceptable safety profile in a phase II randomized controlled trial, but with no apparent effect on outcome.97 The ETA receptor antagonist clazosentan, which showed promising results in a multicenter phase IIa study,98 has now been studied in a randomized, double-blind, placebo-controlled, phase II dose-finding trial (CONSCIOUS-1).99 Treatment with either 1, 5, or 15 mg/hr of intravenous clazosentan was started within 56 hours of SAH and continued up to 14 days. The primary end point, moderate to severe angiographic vasospasm, was significantly reduced in a dose-dependent fashion from 66% in the placebo group to 23% in the 15-mg/hr clazosentan group (risk reduction, 65%; 95% confidence interval, 47% to 78%; P < .0001). There was no clear effect on infarction or outcome, and clazosentan was associated with increased rates of pulmonary edema, hypotension, and anemia. CONSCIOUS-2 is presently under way (fall 2008), a multicenter placebo-controlled phase III study examining the effect of clazosentan, 15 mg/hr given for 14 days after aneurysm clipping, on clinical outcome.
Statins
In a pilot randomized clinical trial, simvastatin (80 mg) given once daily for 14 days appeared to decrease the incidence of radiographic vasospasm and delayed ischemia.100 Reduced vasospasm and cerebral infarction were also found in several cohort studies comparing SAH outcomes in statin users and nonusers at the time of hospital admission.101,102 A phase II placebo-controlled trial of 80 patients randomized to receive either placebo or 40 mg of oral pravastatin for up to 14 days found that statin use reduced vasospasm and severe vasospasm by 32% and 42%, respectively, and reduced vasospastic infarcts and mortality by 83% and 75%, respectively (all significant P values).103 The improvement in early outcome proved durable at 6 months.104 Even though there has been a single negative retrospective statin study, the evidence in total suggests that statins are safe and quite possibly effective in preventing vasospasm, infarction, and poor outcome after aneurysmal SAH. A large, international phase III study investigating the use of simvastatin for aneurysmal subarachnoid hemorrhage (STASH) is currently under way (fall 2008).
Tirilazad Mesylate
Tirilazad, an inhibitor of iron-dependent lipid peroxidation and free radical scavenger, was tested in a series of large-scale randomized controlled trials and became licensed for use in several countries, although not in North America. An analysis of all tirilazad results showed decreased mortality associated with treatment, but nonmortality end points of good outcome did not show benefit except in a post hoc analysis of poor-grade patients.105 Because of gender-specific pharmacokinetics, higher dosages were necessary in women, who appeared to benefit less than men.
Reversal of Vasospasm and Cerebral Ischemia: Rescue Treatments
Triple-H Therapy: Hypervolemia, Hypertension, and Hemodilution
When symptomatic vasospasm is diagnosed or strongly suspected, hemodynamic treatment can commence by additional volume expansion with an isotonic crystalloid infusion, which in euvolemic patients raises CBF in vasospastic territories without a significant change in cardiac indices.106 A replete intravascular volume beyond which additional expansion is probably of no further benefit corresponds to a central venous pressure between 8 and 10 mm Hg or a pulmonary capillary wedge pressure in the range of 14 to 16 mm Hg. Unless patients have a complicated cardiopulmonary status (because of myocardial infarction, heart failure, or pulmonary edema, for example), a pulmonary artery catheter (PAC) is unnecessary.107,108
Induced hypertension is more effective than aggressive hypervolemia in improving cerebral oxygenation in patients with vasospasm and has fewer complications.109 Provided that the ruptured aneurysm has been repaired, symptomatic vasospasm should be treated by the administration of dobutamine or dopamine, which at low to moderate doses have mainly β-agonist, inotropic effects. Another cardiac inotrope, milrinone, has also been used after SAH.110 Blood pressure rises with elevations in cardiac output, which increases CPP. If a prompt blood pressure response does not occur (i.e., to dopamine administered at 10 to 15 µg/kg per minute), a more purely α-agonist vasopressor, such as norepinephrine (titrated to a maximum dose of 20 µg/kg per minute), phenylephrine (titrated to a maximum dose of 180 µg/kg per minute), or a combination of the two, should be added. At high doses, dopamine also causes α-adrenergic agonism; however, it is accompanied by unwanted tachycardia because of β2 receptor activity, which norepinephrine does not possess. In some centers, vasopressors are used before inotropes; the key aspect of treatment is elevating blood pressure, regardless of the agent chosen. Systolic blood pressure of 200 mm Hg or higher or CPP greater than 80 mm Hg is sometimes required, but if ischemic signs persist at systolic pressures greater than 220 mm Hg or CPP higher than 120 mm Hg, hypertensive treatment should be considered to have failed.
It should be noted that hypertension and hypervolemia do not seem to increase the risk for hemorrhage from unsecured, unruptured aneurysms in the acute setting or in their short-term natural history.111
The significant risks of triple-H therapy are cardiac failure and infarction, pulmonary edema, the complications associated with either central venous catheter or PAC placement, and possibly cerebral edema and raised intracranial pressure (ICP).112–114 Risks are greatest in the elderly and patients with intrinsic, preexisting cardiopulmonary disease.
Intra-aortic Balloon Counterpulsation
Originally designed for the management of cardiogenic shock, placement of a transfemoral aortic balloon that inflates on aortic valve closure with each cardiac cycle and augments diastolic flow proximally to the coronary and cranial arteries and distally to the peripheral circulation has been reported to be feasible and effective in patients with combined severe vasospasm and cardiac failure.115,116
Chemical Angioplasty
There has been considerable experience with superselective intra-arterial infusion of the vasodilator papaverine for the management of vasospasm. Although demonstrated to sometimes be effective in dilating spastic arteries, its effect is short-lived and inconsistently associated with clinical improvement. There have been reports of toxicity and other adverse effects, including elevations in ICP.117
Other agents have also been administered intra-arterially for vasospasm, including the calcium channel blocker verapamil (≈3 mg per vessel or ≈20 mg per patient),118 the intracellular calcium antagonist fasudil hydrochloride,119 and the phosphodiesterase inhibitor milrinone.120,121 The calcium antagonists nicardipine and nimodipine have likewise been administered intra-arterially. Although experience with these various agents remains preliminary, most appear to be quite well tolerated in terms of blood pressure and ICP at the doses used. There have been reports of verapamil administration and seizures.122 Although intra-arterial vasodilators are able to reach more distal vessels, the response of vessels exhibiting chronic vasospasm is often blunted, and the duration of their effect is usually temporary.
Balloon Angioplasty
In vitro and in vivo experiments in dogs indicate that TBA results in arterial stretching with an immediate and profound functional impairment in vascular smooth muscle123,124 that lasts several weeks.125
Clinical series suggest that approximately two thirds of patients will show some clinical improvement after TBA, and its risks include vessel perforation, dissection, and occlusion, which complicate between 5% and 10% of procedures.117,126–129 Good results with this technique depend on appropriate timing and expertise (Fig. 364-3).
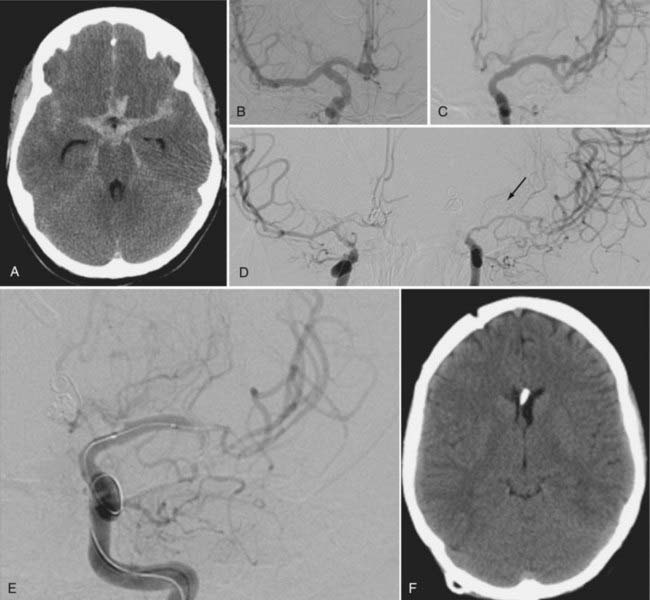
Other Reversal Therapies
In patients with severe and medically refractory vasospasm, sodium nitroprusside, an NO donor, has been administered into the lateral ventricles, either as single injections or as a continuous infusion via a ventriculostomy catheter, but the results have not been consistent or promising.130,131 Cervical sympathetic blockade with regional anesthesia appeared to result in improvement of mild to moderate symptoms of vasospasm, but without reversal of angiographic vasospasm.132
Practical Approach to the Prevention and Treatment of Cerebral Vasospasm
Patients are kept well hydrated with isotonic crystalloid and intermittent albumin infusions (at least 3 L/day, combined), ICP is kept normal with the liberal use of external ventricular drainage, and CPP is optimized to levels higher than 70 mm Hg. No attempt is made to control mild or moderate hypertension (Table 364-7). Daily TCD examination is carried out by a dedicated technician, and values in excess of 200 cm/sec in the MCA are considered indicative of significant angiographic vasospasm in that segment. Regular clinical assessment to search for subtle changes in mentation, verbal output, and arm and hand control is most important. Obtunded or sedated patients being ventilated undergo direct vascular imaging on day 5 as a matter of routine and then again several days later if considered necessary.
TABLE 364-7 Reversal of Vasospasm and Cerebral Ischemia: Stepped-Care Approach
CT, computed tomography; CVP, central venous pressure; TBA, transluminal balloon angioplasty; triple-H therapy, hypervolemia, hypertension, and hemodilution.
Acknowledgment
I would like to thank Ms. Terri Lord for expert assistance in preparation of this chapter.
Amin-Hanjani S, Ogilvy CS, Barker FG. Does intracisternal thrombolysis prevent vasospasm after aneurismal subarachnoid hemorrhage? A meta-analysis. Neurosurgery. 2004;54:326-335.
Chow M, Dumont AS, Kassell NF. Endothelin receptor antagonists and cerebral vasospasm: an update. Neurosurgery. 2002;51:1333-1342.
Clatterbuck RE, Gailloud P, Ogata L, et al. Prevention of cerebral vasospasm by a humanized anti-CD11/CD18 monoclonal antibody administered after experimental subarachnoid hemorrhage in nonhuman primates. J Neurosurg. 2003;99:376-382.
Clatterbuck RE, Gailloud P, Tierney T, et al. Controlled release of a nitric oxide donor for the prevention of delayed cerebral vasospasm following experimental subarachnoid hemorrhage in nonhuman primates. J Neurosurg. 2005;103:745-751.
Dorhout Mees SM, Rinkel GJE, Feigin VL, et al. Calcium antagonists for aneurysmal subarachnoid hemorrhage. Stroke. 2009;39:514-515.
Fergusen S, MacDonald RL. Predictors of cerebral infarction in patients with aneurysmal subarachnoid hemorrhage. Neurosurgery. 2007;60:658-667.
Friedman JA, Goerss SJ, Meyer FB, et al. Volumetric quantification of Fisher grade 3 aneurysmal subarachnoid hemorrhage: a novel method to predict symptomatic vasospasm on admission computerized tomography scans. J Neurosurg. 2002;97:401-407.
Gonzalez NR, Boscardin WJ, Glenn T, et al. Vasospasm probability index: a combination of transcranial Doppler velocities, cerebral blood flow, and clinical risk factors to predict cerebral vasospasm after aneurysmal subarachnoid hemorrhage. J Neurosurg. 2007;107:1101-1112.
Hansen-Schwartz J, Vajkoczy P, Macdonald RL, et al. Cerebral vasospasm: looking beyond vasoconstriction. Trends Pharmacol Sci. 2007;28:252-256.
Harrod CG, Bendok BR, Batjer HH. Prediction of cerebral vasospasm in patients presenting with aneurysmal subarachnoid hemorrhage: a review. Neurosurgery. 2005;56:633-654.
Hoh BL, Carter BS, Ogilvy CS. Risk of hemorrhage from unsecured, unruptured aneurysms during and after hypertensive hypervolemic therapy. Neurosurgery. 2002;50:1207-1212.
Iuliano BA, Pluta RM, Jung C, et al. Endothelial dysfunction in a primate model of cerebral vasospasm. J Neurosurg. 2004;100:287-294.
Jestaedt L, Pham M, Bartsch AJ, et al. The impact of balloon angioplasty on the evolution of vasospasm-related infarction after aneurysm subarachnoid hemorrhage. Neurosurgery. 2008;62:610-617.
Jung CS, Oldfield EH, Harvey-White J, et al. Association of an endogenous inhibitor of nitric oxide synthase with cerebral vasospasm in patients with aneurysmal subarachnoid hemorrhage. J Neurosurg. 2007;107:945-950.
Kasuya H, Onda H, Sasahara A, et al. Application of nicardipine prolonged-release implants: analysis of 97 consecutive patients with acute subarachnoid hemorrhage. Neurosurgery. 2005;56:895-902.
Kramer AH, Hehir M, Nathan B, et al. A comparison of 3 radiographic scales for the prediction of delayed ischemia and prognosis following subarachnoid hemorrhage. J Neurosurg. 2008;109:199-207.
Lennihan L, Mayer SA, Fink ME, et al. Effect of hypervolemic therapy on cerebral blood flow after subarachnoid hemorrhage. A randomized controlled trial. Stroke. 2000;31:383-391.
Lynch JR, Wang H, McGirt MJ, et al. Simvastatin reduces vasospasm after aneurysmal subarachnoid hemorrhage. Results of a pilot randomized clinical trial. Stroke. 2005;36:2024-2026.
Lysakowski C, Walder B, Costanza MC, et al. Transcranial Doppler versus angiography in patients with vasospasm due to a ruptured cerebral aneurysm. A systematic review. Stroke. 2001;32:2292-2298.
Macdonald RL, Kassell NF, Mayer S, et al. Clazosentan to overcome neurological ischemia and infarction occurring after subarachnoid hemorrhage (CONSCIOUS-1). Stroke. 2008;39:3015-3021.
Oertel M, Boscardin WJ, Obrist WD, et al. Posttraumatic vasospasm: the epidemiology, severity, and time course of an underestimated phenomenon: a prospective study performed in 299 patients. J Neurosurg. 2005;103:812-824.
Raabe A, Beck J, Keller M, et al. Relative importance of hypertension compared with hypervolemia for increasing cerebral oxygenation in patients with cerebral vasospasm after subarachnoid hemorrhage. J Neurosurg. 2005;103:974-981.
Rabinstein AA, Pichelmann MA, Friedman JA, et al. Symptomatic vasospasm and outcomes following aneurysmal subarachnoid hemorrhage: a comparison between surgical repair and endovascular coil occlusion. J Neurosurg. 2003;98:319-325.
Ricardo J, Komotar BE, Zacharia ML, et al. Controversies in the endovascular management of cerebral vasospasm after intracranial aneurysm rupture and future directions for therapeutic approaches. Neurosurgery. 2008;62:897-905.
Rinkel GJE, Feigin VL, Algra A, et al. Hypervolemia in aneurysmal subarachnoid hemorrhage. Stroke. 2005;36:1104-1105.
Schmid-Elsaesser R, Kunz M, Zausinger S, et al. Intravenous magnesium versus nimodipine in the treatment of patients with aneurysmal subarachnoid hemorrhage: a randomized study. Neurosurgery. 2006;58:1054-1065.
Tseng MY, Czosnyka M, Richards H, et al. Effects of acute treatment with pravastatin on cerebral vasospasm, autoregulation, and delayed ischemic deficits after aneurysmal subarachnoid hemorrhage. A phase II randomized placebo-controlled trial. Stroke. 2005;36:1627-1632.
Vajkoczy P, Horn P, Thome C, et al. Regional cerebral blood flow monitoring in the diagnosis of delayed ischemia following aneurysmal subarachnoid hemorrhage. J Neurosurg. 2003;98:1227-1234.
Vikman P, Ansar S, Edvinsson L. Transcriptional regulation of inflammatory and extracellular matrix-regulating genes in cerebral arteries following experimental subarachnoid hemorrhage in rats. J Neurosurg. 2007;107:1015-1022.
Zwienenberg-Lee M, Hartman J, Rudisill N, et al. Effect of prophylactic transluminal balloon angioplasty on cerebral vasospasm and outcome in patients with Fisher grade III subarachnoid hemorrhage. Results of a phase II multicenter, randomized, clinical trial. Stroke. 2008;39:1759-1765.
1 Fukuda T, Hasue M, Ito H. Does traumatic subarachnoid hemorrhage caused by diffuse brain injury cause delayed ischemic brain damage? Comparison with subarachnoid hemorrhage caused by ruptured intracranial aneurysms. Neurosurgery. 1998;43:1040-1049.
2 Oertel M, Boscardin WJ, Obrist WD, et al. Posttraumatic vasospasm: the epidemiology, severity, and time course of an underestimated phenomenon: a prospective study performed in 299 patients. J Neurosurg. 2005;103:812-824.
3 Langham J, Goldfrad C, Teasdale G, et al. Calcium channel blockers for acute traumatic brain injury. Cochrane Database Syst Rev. 2003;4:CD000565.
4 Armin SS, Colohan AR, Zhang JH. Traumatic subarachnoid haemorrhage: our current understanding and its revolution over the past half century. Neurol Res. 2006;28:445-452.
5 Dorsch NWC, King MT. A review of cerebral vasospasm in aneurysmal subarachnoid haemorrhage: I. Incidence and effects. J Clin Neurosci. 1994;1:19-26.
6 Fergusen S, MacDonald RL. Predictors of cerebral infarction in patients with aneurysmal subarachnoid hemorrhage. Neurosurgery. 2007;60:658-667.
7 Kassell NF, Tomer JC, Haley ECJr, et al. The International Cooperative Study on the Timing of Aneurysm Surgery: I. Overall management results. J Neurosurg. 1990;73:18-36.
8 Broderick JP, Brott TG, Duldner JE, et al. Initial and recurrent bleeding are the major causes of death following subarachnoid hemorrhage. Stroke. 1994;25:1342-1347.
9 Proust F, Hannequin D, Langlois O, et al. Causes of morbidity and mortality after ruptured aneurysm surgery in a series of 230 patients: the importance of control angiography. Stroke. 1995;26:1553-1557.
10 Friedman JA, Goerss SJ, Meyer FB, et al. Volumetric quantification of Fisher grade 3 aneurysmal subarachnoid hemorrhage: a novel method to predict symptomatic vasospasm on admission computerized tomography scans. J Neurosurg. 2002;97:401-407.
11 Reilly C, Amidei C, Tolentino J, et al. Clot volume and clearance rate as independent predictors of vasospasm after aneurysmal subarachnoid hemorrhage. J Neurosurg. 2004;101:255-261.
12 Kramer AH, Hehir M, Nathan B, et al. A comparison of 3 radiographic scales for the prediction of delayed ischemia and prognosis following subarachnoid hemorrhage. J Neurosurg. 2008;109:199-207.
13 Harrod CG, Bendok BR, Batjer HH. Prediction of cerebral vasospasm in patients presenting with aneurysmal subarachnoid hemorrhage: a review. Neurosurgery. 2005;56:633-654.
14 Mocco J, Ransom ER, Komotar RJ, et al. Racial differences in cerebral vasospasm: a systematic review of the literature. Neurosurgery. 2006;58:305-314.
15 Conway JE, Tamargo RJ. Cocaine use is an independent risk factor for cerebral vasospasm after aneurysmal subarachnoid hemorrhage. Stroke. 2001;32:2338-2343.
16 Schwartz TH, Solomon RA. Perimesencephalic nonaneurysmal subarachnoid hemorrhage: review of the literature. Neurosurgery. 1996;39:433-440.
17 Rabinstein AA, Pichelmann MA, Friedman JA, et al. Symptomatic vasospasm and outcomes following aneurysmal subarachnoid hemorrhage: a comparison between surgical repair and endovascular coil occlusion. J Neurosurg. 2003;98:319-325.
18 Koide M, Nishizawa S, Ohta S, et al. Chronological changes of the contractile mechanism in prolonged vasospasm after subarachnoid hemorrhage: from protein kinase C to protein tyrosine kinase. Neurosurgery. 2002;51:1468-1476.
19 Hansen-Schwartz J, Vajkoczy P, Macdonald RL, et al. Cerebral vasospasm: looking beyond vasoconstriction. Trends Pharmacol Sci. 2007;28:252-256.
20 Megyesi JF, Vollrath B, Cook DA, et al. Long-term effects of in vivo angioplasty in normal and vasospastic canine carotid arteries: pharmacological and morphological analyses. J Neurosurg. 1999;91:100-108.
21 Findlay JM, Weir BKA, Kanamaru K, et al. Arterial wall changes in cerebral vasospasm. Neurosurgery. 1989;25:736-746.
22 Sasaki T, Wakai S, Asano T, et al. The effect of a lipid hydroperoxide of arachidonic acid on the canine basilar artery: an experimental study on cerebral vasospasm. J Neurosurg. 1981;54:357-365.
23 Kamezaki T, Yanaka K, Nagase S, et al. Increased levels of lipid peroxides as predictive of symptomatic vasospasm and poor outcome after aneurysmal subarachnoid hemorrhage. J Neurosurg. 2002;97:1302-1305.
24 Ohta T, Satoh G, Kuroiwa T. The permeability change of major cerebral arteries in experimental vasospasm. Neurosurgery. 1992;30:331-336.
25 Foley PL, Takenaka K, Kassell NF, et al. Cytotoxic effects of bloody cerebrospinal fluid on cerebral endothelial cells in culture. J Neurosurg. 1994;81:87-92.
26 Iuliano BA, Pluta RM, Jung C, et al. Endothelial dysfunction in a primate model of cerebral vasospasm. J Neurosurg. 2004;100:287-294.
27 Moon CT, Gajdusek C, London S, et al. Expression of endothelial nitric oxide synthase after exposure to perivascular blood. Neurosurgery. 2001;48:1328-1334.
28 Pluta RM, Jung CS, Harvey-White J, et al. In vitro and in vivo effects of probucol on hydrolysis of asymmetric dimethyl L-arginine and vasospasm in primates. J Neurosurg. 2005;103:731-738.
29 Tierney TS, Clatterbuck RE, Lawson C, et al. Prevention and reversal of experimental posthemorrhagic vasospasm by the periadventitial administration of nitric oxide from a controlled-release polymer. Neurosurgery. 2001;49:945-953.
30 Rabikian P, Clatterbuck RE, Eberhart CG, et al. Prevention of experimental cerebral vasospasm by intracranial delivery of a nitric oxide donor from a controlled-release polymer. Toxicity and efficacy studies in rabbits and rats. Stroke. 2002;33:2681-2686.
31 Pradilla G, Thai QA, Legnani FG, et al. Delayed intracranial delivery of a nitric oxide donor from a controlled-release polymer prevents experimental cerebral vasospasm in rabbits. Neurosurgery. 2004;55:1393-1400.
32 Clatterbuck RE, Gailloud P, Tierney T, et al. Controlled release of a nitric oxide donor for the prevention of delayed cerebral vasospasm following experimental subarachnoid hemorrhage in nonhuman primates. J Neurosurg. 2005;103:745-751.
33 Santhanam AVR, Smith LA, Akiyama M, et al. Role of endothelial NO synthase phosphorylation in cerebrovascular protective effect of recombinant erythropoietin during subarachnoid hemorrhage–induced cerebral vasospasm. Stroke. 2005;36:2731-2737.
34 Vatter H, Weidauer S, Dias S, et al. Persistence of the nitric oxide–dependent vasodilator pathway of cerebral vessels after experimental subarachnoid hemorrhage. Neurosurgery. 2007;60:179-188.
35 Jung CS, Oldfield EH, Harvey-White J, et al. Association of an endogenous inhibitor of nitric oxide synthase with cerebral vasospasm in patients with aneurysmal subarachnoid hemorrhage. J Neurosurg. 2007;107:945-950.
36 Chow M, Dumont AS, Kassell NF. Endothelin receptor antagonists and cerebral vasospasm: an update. Neurosurgery. 2002;51:1333-1342.
37 Seifert V, Löffler BM, Zimmermann M, et al. Endothelin concentrations in patients with aneurysmal subarachnoid hemorrhage: correlation with cerebral vasospasm, delayed ischemic neurological deficits, and volume of hematoma. J Neurosurg. 1995;82:55-62.
38 Juvela S. Plasma endothelin concentrations after aneurysmal subarachnoid hemorrhage. J Neurosurg. 2000;92:390-400.
39 Mascia L, Fedorko L, Stewart DJ, et al. Temporal relationship between endothelin-1 concentrations and cerebral vasospasm in patients with aneurysmal subarachnoid hemorrhage. Stroke. 2001;32:1185-1190.
40 Dumont AS, Dumont RJ, Chow MM, et al. Cerebral vasospasm after subarachnoid hemorrhage: putative role of inflammation. Neurosurgery. 2003;53:123-135.
41 Sasaki T, Kasuya H, Onda H, et al. Role of p-38 mitogen–activated protein kinase on cerebral vasospasm after subarachnoid hemorrhage. Stroke. 2004;35:1466-1470.
42 Bowman G, Dixit S, Bonneau RH, et al. Neutralizing antibody against interleukin-6 attenuates posthemorrhagic vasospasm in the rat femoral artery model. Neurosurgery. 2004;54:719-726.
43 Clatterbuck RE, Gailloud P, Ogata L, et al. Prevention of cerebral vasospasm by a humanized anti-CD11/CD18 monoclonal antibody administered after experimental subarachnoid hemorrhage in nonhuman primates. J Neurosurg. 2003;99:376-382.
44 Pradilla G, Wang PP, Legnani RG, et al. Prevention of vasospasm by anti-CD11/CD18 monoclonal antibody therapy following subarachnoid hemorrhage in rabbits. J Neurosurg. 2004;101:88-92.
45 Macdonald RL, Zhang ZD, Ono S, et al. Up-regulation of parathyroid hormone receptor in cerebral arteries after subarachnoid hemorrhage in monkeys. Neurosurgery. 2002;50:1083-1093.
46 Vikman P, Beg S, Khurana T, et al. Gene expression and molecular changes in cerebral arteries following subarachnoid hemorrhage in the rat. J Neurosurg. 2006;105:438-444.
47 Vikman P, Ansar S, Edvinsson L. Transcriptional regulation of inflammatory and extracellular matrix–regulating genes in cerebral arteries following experimental subarachnoid hemorrhage in rats. J Neurosurg. 2007;107:1015-1022.
48 Mack WJ, Ducruet AF, Hickman ZL, et al. Early plasma complement C3a levels correlate with functional outcome after aneurysmal subarachnoid hemorrhage. Neurosurgery. 2007;61:255-261.
49 Mocco J, Mack WJ, Kim GH, et al. Rise in serum soluble intercellular adhesion molecule-1 levels with vasospasm following aneurysmal subarachnoid hemorrhage. J Neurosurg. 2002;97:537-541.
50 Schoch B, Regel JP, Wichert M, et al. Analysis of intrathecal interleukin-6 as a potential predictive factor for vasospasm in subarachnoid hemorrhage. Neurosurgery. 2007;60:828-836.
51 Fisher CM, Roberson GH, Ojemann RG. Cerebral vasospasm with ruptured saccular aneurysm—the clinical manifestations. Neurosurgery. 1977;1:245-258.
52 Lindegaard KF, Bakke SJ, Sorteberg W, et al. A non-invasive Doppler ultrasound method for the evaluation of patients with subarachnoid hemorrhage. Acta Radiol. 1986;369:96-98.
53 Sviri GE, Ghodke B, Britz GW, et al. Transcranial Doppler grading criteria for basilar artery vasospasm. Neurosurgery. 2006;59:360-366.
54 Vora YY, Suarez-Almazor M, Steinke DE, et al. Role of transcranial Doppler monitoring in the diagnosis of cerebral vasospasm after subarachnoid hemorrhage. Neurosurgery. 1999;44:1237-1248.
55 Lysakowski C, Walder B, Costanza MC, et al. Transcranial Doppler versus angiography in patients with vasospasm due to a ruptured cerebral aneurysm. A systematic review. Stroke. 2001;32:2292-2298.
56 Mariak Z, Krejza J, Swiercz M, et al. Accuracy of transcranial color Doppler ultrasonography in the diagnosis of middle cerebral artery spasm determined by receiver operating characteristic analysis. J Neurosurg. 2002;96:323-330.
57 Lam JM, Smielweski P, Czosnyka M, et al. Predicting delayed ischemic deficits after aneurysmal subarachnoid hemorrhage using a transient hyperemic response test of cerebral autoregulation. Neurosurgery. 2000;47:819-826.
58 Ratsep T, Asser T. Cerebral hemodynamic impairment after aneurysmal subarachnoid hemorrhage as evaluated using transcranial Doppler ultrasonography: relationship to delayed cerebral ischemia and clinical outcome. J Neurosurg. 2001;95:393-401.
59 Gonzalez NR, Boscardin WJ, Glenn T, et al. Vasospasm probability index: a combination of transcranial Doppler velocities, cerebral blood flow, and clinical risk factors to predict cerebral vasospasm after aneurysmal subarachnoid hemorrhage. J Neurosurg. 2007;107:1101-1112.
60 Okada Y, Shima T, Nishida M, et al. Comparison of transcranial Doppler investigation of aneurysmal vasospasm with digital subtraction angiographic and clinical findings. Neurosurgery. 1999;45:443-450.
61 Minhas PW, Menon DK, Smielewski P, et al. Positron emission tomographic cerebral perfusion disturbances and transcranial Doppler findings among patients with neurological deterioration and subarachnoid hemorrhage. Neurosurgery. 2003;52:1017-1024.
62 Egge A, Sjoholm H, Waterloo K, et al. Serial single-photon emission computed tomographic and transcranial Doppler measurements for evaluation of vasospasm after aneurysmal subarachnoid hemorrhage. Neurosurgery. 2005;57:237-242.
63 Hertel F, Walter C, Bettag M, et al. Perfusion-weighted magnetic resonance imaging in patients with vasospasm: a useful new tool in the management of patients with subarachnoid hemorrhage. Neurosurgery. 2005;56:28-35.
64 Condette-Auliac S, Bracard S, Anxionnat R, et al. Vasospasm after subarachnoid hemorrhage. Interest in diffusion-weighted MR imaging. Stroke. 2001;32:1818-1824.
65 Weidauer S, Lanfermann H, Raabe A, et al. Impairment of cerebral perfusion and infarct patterns attributable to vasospasm after aneurysmal subarachnoid hemorrhage. A prospective MRI and DSA study. Stroke. 2007;38:1831-1836.
66 van der Schaaf I, Wermer MJ, van der Graaf Y, et al. Prognostic value of cerebral perfusion–computed tomography in the acute stage after subarachnoid hemorrhage for the development of delayed cerebral ischemia. Stroke. 2006;37:409-413.
67 Sviri GE, Britz GW, Lewis DH, et al. Dynamic perfusion computed tomography in the diagnosis of cerebral vasospasm. Neurosurgery. 2006;59:319-325.
68 Vajkoczy P, Horn P, Thome C, et al. Regional cerebral blood flow monitoring in the diagnosis of delayed ischemia following aneurysmal subarachnoid hemorrhage. J Neurosurg. 2003;98:1227-1234.
69 Unterberg AW, Sakowitz OW, Sarrafzadeh AS, et al. Role of bedside microdialysis in the diagnosis of cerebral vasospasm following aneurysmal subarachnoid hemorrhage. J Neurosurg. 2001;94:740-749.
70 Kett-White R, Hutchinson PJ, Al-Rawi PG, et al. Adverse cerebral events detected after subarachnoid hemorrhage using brain oxygen and microdialysis probes. Neurosurgery. 2002;50:1213-1222.
71 Skjoth-Rasmussen J, Schulz M, Kristensen SR, et al. Delayed neurological deficits detected by an ischemic pattern in the extracellular cerebral metabolites in patients with aneurysmal subarachnoid hemorrhage. J Neurosurg. 2004;100:8-15.
72 Otawara Y, Ogasawara K, Ogawa A, et al. Evaluation of vasospasm after subarachnoid hemorrhage by use of multislice computed tomographic angiography. Neurosurgery. 2002;51:939-943.
73 Baldwin ME, Macdonald RL, Huo D, et al. Early vasospasm on admission angiography in patients with aneurysmal subarachnoid hemorrhage is a predictor for in-hospital complications and poor outcome. Stroke. 2004;35:2506-2511.
74 Nakagawa A, Su CC, Sato K, et al. Evaluation of changes in circulating blood volume during acute and very acute stages of subarachnoid hemorrhage: implications for the management of hypovolemia. J Neurosurg. 2002;97:268-271.
75 Lennihan L, Mayer SA, Fink ME, et al. Effect of hypervolemic therapy on cerebral blood flow after subarachnoid hemorrhage. A randomized controlled trial. Stroke. 2000;31:383-391.
76 Egge A, Waterloo K, Sjoholm H, et al. Prophylactic hyperdynamic postoperative fluid therapy after aneurysmal subarachnoid hemorrhage: a clinical, prospective, randomized, controlled study. Neurosurgery. 2001;49:593-606.
77 Treggiari MM, Walder B, Suter PM, et al. Systematic review of the prevention of delayed ischemic neurological deficits with hypertension, hypervolemia, and hemodilution therapy following subarachnoid hemorrhage. J Neurosurg. 2003;98:978-984.
78 Rinkel GJE, Feigin VL, Algra A, et al. Hypervolemia in aneurysmal subarachnoid hemorrhage. Stroke. 2005;36:1104-1105.
79 Suarez JI, Shannon L, Zaidat OO, et al. Effect of human albumin administration on clinical outcome and hospital cost in patients with subarachnoid hemorrhage. J Neurosurg. 2004;100:585-590.
80 McGirt MJ, Blessing R, Nimjee SM, et al. Correlation of serum brain natriuretic peptide with hyponatremia and delayed ischemic neurological deficits after subarachnoid hemorrhage. Neurosurgery. 2004;54:1369-1374.
81 Mori T, Katayama Y, Kawamata T, et al. Improved efficiency of hypervolemic therapy with inhibition of natriuresis by fludrocortisones in patients with aneurysmal subarachnoid hemorrhage. J Neurosurg. 1999;91:947-952.
82 Smith MJ, Le Roux PD, Elliott JP, et al. Blood transfusion and increased risk for vasospasm and poor outcome after subarachnoid hemorrhage. J Neurosurg. 2004;101:1-7.
83 Bailes JE, Spetzler RF, Hadley MN, et al. Management and morbidity of poor-grade aneurysm patients. J Neurosurg. 1990;72:559-566.
84 Dorhout Mees SM, Rinkel GJE, Feigin VL, et al. Calcium antagonists for aneurysmal subarachnoid hemorrhage. Stroke. 2009;39:514-515.
85 Megyesi JF, Findlay JM, Vollrath B, et al. In vivo angioplasty prevents the development of vasospasm in canine carotid arteries: pharmacological and morphological analyses. Stroke. 1997;28:1216-1224.
86 Zwienenberg-Lee M, Hartman J, Rudisill N, et al. Effect of prophylactic transluminal balloon angioplasty on cerebral vasospasm and outcome in patients with Fisher grade III subarachnoid hemorrhage. Results of a phase II multicenter, randomized, clinical trial. Stroke. 2008;39:1759-1765.
87 Amin-Hanjani S, Ogilvy CS, Barker FG. Does intracisternal thrombolysis prevent vasospasm after aneurismal subarachnoid hemorrhage? A meta-analysis. Neurosurgery. 2004;54:326-335.
88 Kawamoto S, Tsutsumi K, Yoshikawa G, et al. Effectiveness of the head-shaking method combined with cisternal irrigation with urokinase in preventing cerebral vasospasm after subarachnoid hemorrhage. J Neurosurg. 2004;100:236-243.
89 Hanggi D, Liersch J, Turowski B, et al. The effect of lumboventricular lavage and simultaneous low-frequency head-motion therapy after severe subarachnoid hemorrhage: results of a single center prospective phase II trial. J Neurosurg. 2008;108:1192-1199.
90 Klimo PJr, Kestle JRW, MacDonald JD, et al. Marked reduction of cerebral vasospasm with lumbar drainage of cerebrospinal fluid after subarachnoid hemorrhage. J Neurosurg. 2004;100:215-224.
91 Andaluz N, Zuccarello M. Fenestration of the lamina terminalis as a valuable adjunct in aneurysm surgery. Neurosurgery. 2004;55:1050-1059.
92 Kasuya H, Onda H, Sasahara A, et al. Application of nicardipine prolonged-release implants: analysis of 97 consecutive patients with acute subarachnoid hemorrhage. Neurosurgery. 2005;56:895-902.
93 Dalbasti T, Karabiyikoglu M, Ozdamar N, et al. Efficacy of controlled-release papaverine pellets in preventing symptomatic cerebral vasospasm. J Neurosurg. 2001;95:44-50.
94 Veyna RS, Seyfried D, Burke DG, et al. Magnesium sulfate therapy after aneurysmal subarachnoid hemorrhage. J Neurosurg. 2002;96:510-514.
95 Stippler S, Crago E, Levy EI, et al. Magnesium infusion for vasospasm prophylaxis after subarachnoid hemorrhage. J Neurosurg. 2006;105:723-729.
96 Schmid-Elsaesser R, Kunz M, Zausinger S, et al. Intravenous magnesium versus nimodipine in the treatment of patients with aneurysmal subarachnoid hemorrhage: a randomized study. Neurosurgery. 2006;58:1054-1065.
97 Shaw MDM, Vermeulen V, Murray GD, et al. Efficacy and safety of the endothelinA/B receptor antagonist TAK-044 in treating subarachnoid hemorrhage: a report by the Steering Committee on behalf of the IK/Netherlands/Eire TAK-044 Subarachnoid Haemorrhage Study Group. J Neurosurg. 2000;93:992-997.
98 Vajkoczy P, Meyer B, Weidauer S, et al. Clazosentan (AXV-034343), a selective endothelin A receptor antagonist, in the prevention of cerebral vasospasm following severe aneurysmal subarachnoid hemorrhage; results of a randomized, double-blind, placebo-controlled, multicenter phase IIa study. J Neurosurg. 2005;103:9-17.
99 Macdonald RL, Kassell NF, Mayer S, et al. Clazosentan to overcome neurological ischemia and infarction occurring after subarachnoid hemorrhage (CONSCIOUS-1). Stroke. 2008;39:3015-3021.
100 Lynch JR, Wang H, McGirt MJ, et al. Simvastatin reduces vasospasm after aneurysmal subarachnoid hemorrhage. Results of a pilot randomized clinical trial. Stroke. 2005;36:2024-2026.
101 Parra A, Kreiter KT, Williams S, et al. Effect of prior statin use on functional outcome and delayed vasospasm after acute aneurysmal subarachnoid hemorrhage: a matched controlled cohort study. Neurosurgery. 2005;56:476-484.
102 McGirt MJ, Blessing R, Alexander MJ, et al. Risk of cerebral vasospasm after subarachnoid hemorrhage reduced by statin therapy: a multivariate analysis of an institutional experience. J Neurosurg. 2006;10:671-674.
103 Tseng MY, Czosnyka M, Richards H, et al. Effects of acute treatment with pravastatin on cerebral vasospasm, autoregulation, and delayed ischemic deficits after aneurysmal subarachnoid hemorrhage. A phase II randomized placebo-controlled trial. Stroke. 2005;36:1627-1632.
104 Tseng MY, Hutchinson PJ, Czosnyka M, et al. Effects of acute pravastatin treatment on intensity of rescue therapy, length of inpatient stay, and 6-month outcome in patients after aneurysmal subarachnoid hemorrhage. Stroke. 2007;38:1545-1550.
105 Dorsch NW, Kassell NF, Sinkula MS, et al. Metaanalysis of trials of tirilazad mesylate in aneurysmal SAH. Acta Neurochir Suppl. 2001;77:233-235.
106 Jost SC, Diringer MN, Zazulia AR, et al. Effect of normal saline bolus on cerebral blood flow in regions with low baseline flow in patients with vasospasm following subarachnoid hemorrhage. J Neurosurg. 2005;103:25-30.
107 Shure D. Pulmonary artery catheters—Peace at last? N Engl J Med. 2006;354:2273-2274.
108 Mutoh T, Kazumata K, Ajiki M, et al. Goal-directed fluid management by bedside transpulmonary hemodynamic monitoring after subarachnoid hemorrhage. Stroke. 2007;38:3218-3224.
109 Raabe A, Beck J, Keller M, et al. Relative importance of hypertension compared with hypervolemia for increasing cerebral oxygenation in patients with cerebral vasospasm after subarachnoid hemorrhage. J Neurosurg. 2005;103:974-981.
110 Naidech A, Du Y, Kreiter KT, et al. Dobutamine versus milrinone after subarachnoid hemorrhage. Neurosurgery. 2005;56:21-27.
111 Hoh BL, Carter BS, Ogilvy CS. Risk of hemorrhage from unsecured, unruptured aneurysms during and after hypertensive hypervolemic therapy. Neurosurgery. 2002;50:1207-1212.
112 Rosenwasser RH, Jallo JI, Getch CC, et al. Complications of Swan-Ganz catheterization for hemodynamic monitoring in patients with subarachnoid hemorrhage. Neurosurgery. 1995;37:872-876.
113 Miller JA, Dacey RGJr, Diringer MN. Safety of hypertensive hypervolemic therapy with phenylephrine in the treatment of delayed ischemic deficits after subarachnoid hemorrhage. Stroke. 1995;26:2260-2266.
114 Amin-Hanjani S, Schwartz RB, Sathi S, et al. Hypertensive encephalopathy as a complication of hyperdynamic therapy for vasospasm: report of two cases. Neurosurgery. 1999;44:1113-1116.
115 Apostolides PJ, Greene KA, Zambramski JM, et al. Intra-aortic balloon pump counterpulsation in the management of concomitant cerebral vasospasm and cardiac failure after subarachnoid hemorrhage: technical case report. Neurosurgery. 1996;38:1056-1060.
116 Nussbaum ES, Sebring LA, Ganz WF, et al. Intra-aortic balloon counterpulsation augments cerebral blood flow in the patient with cerebral vasospasm: a xenon-enhanced computed tomography study. Neurosurgery. 1998;42:206-214.
117 Ricardo J, Komotar BE, Zacharia ML, et al. Controversies in the endovascular management of cerebral vasospasm after intracranial aneurysm rupture and future directions for therapeutic approaches. Neurosurgery. 2008;62:897-905.
118 Keuskamp J, Murali R, Chao KH. High-dose intraarterial verapamil in the treatment of cerebral vasospasm after aneurysmal subarachnoid hemorrhage. J Neurosurg. 2008;108:458-463.
119 Tanaka K, Minami H, Kota M, et al. Treatment of cerebral vasospasm with intra-arterial fasudil hydrochloride. Neurosurgery. 2005;56:214-223.
120 Arakawa Y, Kikuta KI, Hojo M, et al. Milrinone for the treatment of cerebral vasospasm after subarachnoid hemorrhage: report of seven cases. Neurosurgery. 2001;48:723-730.
121 Fraticelli AT, Cholley BP, Losser MR, et al. Milrinone for the treatment of cerebral vasospasm after aneurysmal subarachnoid hemorrhage. Stroke. 2008;39:893-898.
122 Westhout F, Nwagwu C. Intra-arterial verapamil-induced seizures: case report and review of the literature. Surg Neurol. 2007;67:483-486.
123 Chan PDS, Findlay JM, Vollrath B, et al. Pharmacological and morphological effects of in vitro transluminal balloon angio plasty on normal and vasospastic canine basilar arteries. J Neurosurg. 1995;83:522-530.
124 Megyesi JF, Findlay JM, Vollrath B, et al. In vivo angioplasty prevents the development of vasospasm in canine carotid arteries: pharmacological and morphological analyses. Stroke. 1997;28:1216-1224.
125 Megyesi JF, Vollrath B, Cook DA, et al. Long-term effects of in vivo angioplasty in normal and vasospastic canine carotid arteries: pharmacological and morphological analyses. J Neurosurg. 1999;91:100-108.
126 Eskridge JM, McAuliffe W, Song JK, et al. Balloon angioplasty for the treatment of vasospasm: rsults of first 50 cases. Neurosurgery. 1998;42:510-517.
127 Bejjani GK, Bank WO, Olan WJ, et al. The efficacy and safety of angioplasty for cerebral vasospasm after subarachnoid hemorrhage. Neurosurgery. 1998;42:979-987.
128 Polin RS, Coenen VA, Hansen CA, et al. Efficacy of transluminal angioplasty for the management of symptomatic cerebral vasospasm following aneurysmal subarachnoid hemorrhage. J Neurosurg. 2000;92:284-290.
129 Jestaedt L, Pham M, Bartsch AJ, et al. The impact of balloon angioplasty on the evolution of vasospasm-related infarction after aneurysm subarachnoid hemorrhage. Neurosurgery. 2008;62:610-617.
130 Thomas JE, McGinnis G. Safety of intraventricular sodium nitroprusside and thiosulfate for the treatment of cerebral vasospasm in the intensive care unit setting. Stroke. 2002;33:486-492.
131 Raabe A, Zimmerman M, Setzer M, et al. Effect of intraventricular sodium nitroprusside on cerebral hemodynamics and oxygenation in poor-grade aneurysm patients with severe, medically refractor vasospasm. Neurosurgery. 2002;50:1006-1014.
132 Treggiari MM, Romand JA, Martin JP, et al. Cervical sympathetic block to reverse delayed ischemic neurological deficits after aneurysmal subarachnoid hemorrhage. Stroke. 2003;34:961-967.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
CHAPTER 364 Cerebral Vasospasm
Vasospasm in Conditions Other Than Aneurysm Rupture
Vasospasm has been observed in head-injured patients both with and without subarachnoid blood clots in the basal subarachnoid cisterns, although the diagnosis has sometimes relied on transcranial Doppler (TCD) blood velocity measurements, which are difficult to interpret in the setting of brain injury.1,2 The clinical significance of posttraumatic vasospasm is debated, and there is not yet any proven value of either monitoring cerebral arterial diameters or routine treatment with calcium channel blockers in the setting of traumatic brain injury associated with SAH.3,4
Epidemiology of Vasospasm
Angiographic vasospasm is common after rupture of an aneurysm, with an overall incidence of 50% to 90%.5 Still useful but rough estimates are that at least moderate vasospasm in at least one cerebral artery will develop in two thirds of patients with ruptured aneurysms, half of these patients will become symptomatic as a result of ischemia, and a cerebral infarct will develop in about half of these patients. An analysis of 2741 patients entered into SAH trials in the 1990s found that cerebral infarction (all causes) had developed in 26% at 6 weeks, which correlated strongly with poor outcome.6 Cerebral infarction was significantly associated with increasing patient age, worse neurological grade on admission, history of hypertension or diabetes mellitus, larger aneurysm, induced hypertension, fever, and a diagnosis of symptomatic vasospasm. With modern SAH management routines, the combined risk for death and permanent disability from vasospasm alone has shrunk to less than 10%, but it still remains one of the leading causes of preventable poor outcome after rupture of an aneurysm (Figs. 364-1 and 364-2).7–9
Prediction of Vasospasm
A large volume of persistent subarachnoid clot is the most important risk factor predictive of vasospasm after SAH.10,11 The original Fisher grading scale in which clot volume and distribution on admission computed tomography (CT) are related to risk for vasospasm has been modified and in one single-center study found to have greater predictive value for delayed ischemia and prognosis.12 This modified Fisher scale (Table 364-1) scores hemorrhage noted on CT from 0 to 4: 0, no SAH or intraventricular hemorrhage (IVH) (very low risk for vasospasm); 1, focal or diffuse thin layer of SAH, no IVH (low risk for vasospasm); 2, focal or diffuse thin layer of SAH, IVH present (moderate risk for vasospasm); 3, focal or diffuse thick layer of SAH, no IVH (high risk for vasospasm); and 4, focal or diffuse thick layer of SAH, IVH present (highest risk for vasospasm). A slower rate of subarachnoid clot clearance has also been shown to be an independent predictor of vasospasm, although this is not an easy measurement in clinical practice.11
| 0 | No SAH or IVH: very low risk |
| 1 | Focal or diffuse thin layer of SAH, no IVH: low risk |
| 2 | Focal or diffuse thin layer of SAH, IVH present: moderate risk |
| 3 | Focal or diffuse thick layer of SAH, no IVH: high risk |
| 4 | Focal or diffuse thick layer of SAH, IVH present: very high risk |
IVH, intraventricular hemorrhage; SAH, subarachnoid hemorrhage.
Other risk factors for the development of vasospasm have been identified, including poor neurological grade or loss of consciousness on admission, cigarette smoking, and preexisting hypertension (Table 364-2).13 Factors studied but found to have either an unclear or unlikely relationship with risk for vasospasm include gender, patient age, and aneurysm location. There has been a suggestion that Japanese people may be more susceptible to vasospasm.14 In addition, a small study suggested that cocaine use is an independent risk factor for vasospasm.15
TABLE 364-2 Risk Factors for Vasospasm
Spontaneous perimesencephalic or prepontine SAH (or both) unassociated with aneurysm rupture is typically a low-volume hemorrhage that clears quickly with a low risk for the development of vasospasm.16 Thick and persistent nonaneurysmal SAH, however, is still associated with a risk for vasospasm.
There is some evidence that endovascular coiling, as opposed to microsurgical repair of ruptured aneurysms, is associated with a lower risk for the subsequent development of vasospasm,17 although the difference was not large and a rigorous comparison has not yet been made.
Pathogenesis
Smooth Muscle Contraction
Vasospasm is prolonged cerebral arterial constriction caused by vascular smooth muscle contraction. The hemoglobin released from subarachnoid blood clots triggers the entry and release of calcium and subsequent activation of calcium/calmodulin-dependent myosin light-chain kinase, which in turn leads to phosphorylation of the myosin light chain and induces actin and myosin cross-linkage and mechanical shortening (smooth muscle contraction). Such contraction requires adenosine triphosphate and calcium, and vascular smooth muscle relies more on extracellular than intracellular calcium stores, which enter through voltage-gated and receptor-operated calcium channels. Although myofilament activation depends on calcium and high-energy phosphates, chronic vasospasm, which ensues days later and lasts up to several weeks, does not. The contractile proteins protein kinase C, Rho kinase, and protein tyrosine kinase and their corresponding signal transduction pathways have been implicated in vasospasm models when their activation shifts the contractile mechanism toward increased shortening in the absence of high intracellular calcium levels.18,19 This contiguous and second-phase “chronic” vasospasm is less reversible with pharmacologic vasodilators both in animal models20 and in humans undergoing intra-arterial, pharmacologic vasodilation treatment.
Sustained vasoconstriction is associated not only with functional impairment of the vessel but also with ultrastructural damage to the vascular wall layers, including vacuolization of endothelial cells and loss of tight junctions, breakage of the internal elastic lamina, and patchy myonecrosis in the tunica media.21
Endothelial Injury, Nitric Oxide, and Endothelin-1
Auto-oxidation of the oxyhemoglobin contained in blood clots encasing cerebral arteries produces methemoglobin and superoxide anion radical, which in turn lead to lipid peroxidation.22,23 Harmful hydroxyl radicals and lipid peroxides permeate the vessel wall and injure endothelial and smooth muscle cells.24,25 Damage to the endothelium in particular is thought to play a key role in the establishment of vasospasm, either through the loss of endothelial nitric oxide (NO) synthesis, an important vasodilator and regulator of vascular tone, or through the overproduction of endothelin, a powerful vasoconstrictor peptide.26 These two endothelial-derived substances and the possible imbalance in their production after SAH are at the center of experimental vasospasm research at the present time.
Decreased availability of the simple molecule NO may contribute to the development of vasospasm in the following ways: (1) endothelial NO synthase (eNOS) dysfunction in vasospastic vessels, (2) NO scavenging by oxyhemoglobin; (3) reversal of vasospasm by NO donors, (4) disappearance of neuronal NO synthase (nNOS) activity from the adventitia of vasospastic vessels, and (5) decreased cerebrospinal fluid (CSF) nitrite levels along with increased levels of asymmetric dimethyl-L-arginine, the endogenous inhibitor of NO synthase.27–35
Endothelin-1 (ET-1) is the predominant isoform of endothelin and has the greatest role in vasoconstriction. ET-1 is a 21–amino acid cleavage product of a 212–amino acid peptide precursor, the final step mediated by endothelin-converting enzyme. It is released on the abluminal side of the tunica media, acts on neighboring vascular smooth muscle ETA receptors, and causes profound and sustained vasoconstriction.36 ET-1 levels have been found to be elevated in the CSF of patients in whom vasospasm and brain ischemia develop,37–39 and either inhibition of ET-1 production or antagonism of its effect has prevented vasospasm in animal models.36
Inflammation, Vessel Remodeling, and Vasospasm
Although it has become clear that vasospasm is not a type of vasculitis, there is evidence that inflammatory mechanisms are activated after SAH and may therefore be involved in the development of vasospasm, possibly by contributing to vasocontraction or modifying the vessel wall extracellular matrix and smooth muscle cell phenotype—a process known as vascular “remodeling.”40 Inflammatory cytokines,41,42 intercellular adhesion molecules (ICAMs),43,44 and genetic upregulation of inflammatory, proliferative, and extracellular matrix–regulating genes45–47 have been examined. In humans, elevations of plasma complement C3a and soluble ICAM-1 have been associated with poor outcome and the development of vasospasm, respectively,48,49 and increased intrathecal levels of the cytokine interleukin-6 predicted vasospasm in another small study.50
Although the precise pathogenesis of vasospasm is still the subject of investigation, a prolonged, biphasic vasoconstrictive process in which the second, chronic phase is mechanistically distinct from the first seems most consistent with what has been seen experimentally and in humans. It remains possible that additional processes, including inflammation, may contribute to the pathogenesis of this condition (Table 364-3).
Clinical Features and Investigation
Symptoms, Signs, and Differential Diagnosis
Coincident with its delayed onset, symptoms of ischemia resulting from cerebral vasospasm most commonly appear 1 week after aneurysm rupture but often occur later; accordingly, it is important to remain vigilant for this complication for at least 2 weeks after SAH. Regular and careful bedside examination remains the simplest and most effective means of detecting early ischemia in awake, examinable patients; one should concentrate on subtle findings such as diminished attention, changes in verbal output, or a slight but new pronator drift of the upper extremity. Symptomatic vasospasm usually has a gradual onset, sometimes heralded by increased headache and either agitation or somnolence—a change in patient behavior. It then follows a progressive course if untreated. A smaller group of patients will experience precipitous deterioration.51 Signs of symptomatic vasospasm are referable to the territory that has become ischemic and are most easily distinguished when they lateralize to a middle cerebral artery (MCA) territory with monoparesis or hemiparesis and, when the dominant hemisphere is affected, aphasia. Anterior cerebral artery vasospasm can be marked by leg weakness, but because it is often bilateral in distribution, confusion, drowsiness, poverty of speech, and eventually abulia are characteristic signs. Vertebrobasilar vasospasm can also cause a more generalized deterioration, with a reduced level of consciousness being an early sign.
Delayed neurological deterioration after aneurysmal SAH has a number of causes, including increased edema surrounding intracerebral hematomas, contusions, or infarcts; rebleeding of the aneurysm or aneurysmal remnant; hydrocephalus; sepsis, including meningitis and ventriculitis; hyponatremia; hypoxia; and hypotension (Table 364-4). One or more of these conditions can magnify even a focal neurological deficit and therefore easily be mistaken for vasospasm, which has a tendency to be overdiagnosed in the setting of SAH. Conversely, it is recognized that there is a subgroup of patients who suffer a delayed and often global decline in neurological status for which no single underlying cause can be identified. “Cortical spreading depression” has been one proposed mechanism at work in these patients.19
TABLE 364-4 Causes of Delayed Deterioration after Subarachnoid Hemorrhage
Diagnosis
Diagnosis of symptomatic vasospasm requires that the other causes of delayed worsening listed earlier be ruled out with CT and appropriate laboratory investigations. If vasospasm remains the most likely cause of deterioration and treatment by induced hypertension reverses the deficit, the diagnosis can safely be assumed without further testing. Failure to respond in this scenario, as well as evaluation of comatose patients, requires additional testing (Table 364-5).
CBF, cerebral blood flow; CT, computed tomography; MRI, magnetic resonance imaging; SPECT, single-photon emission computed tomography; TCD, transcranial Doppler.
Transcranial Doppler
TCD works on the principle that as an artery narrows, blood flow velocity within it increases. Because examinations are noninvasive, are carried out at the bedside, and can easily be performed on a daily basis, TCD is frequently used to monitor patients with SAH for increases in intracranial blood flow velocity suggestive of incipient vasospasm. There is good general correlation between TCD velocities and vasospasm, with velocities in the MCA greater than 120 cm/sec indicative of some degree of vasospasm and those greater than 200 cm/sec consistent with severe vasospasm. Because other factors can influence velocity, including blood pressure and overall cerebral blood flow (CBF), distinguishing vasospastic from hyperemic increases in blood velocity has been reported to be facilitated by measuring cervical internal carotid artery (ICA) velocity in addition to intracranial blood velocity.52 A “Lindegaard ratio” of VMCA/VICA greater than 3 is consistent with vasospasm (hyperemia is associated with increased velocity in both the MCA and ICA, so the ratio is the same). A similar velocity ratio between the basilar artery and the extracranial vertebral artery has been proposed to improve the sensitivity and specificity of detecting basilar artery vasospasm.53
The clinical utility of TCD is best when MCA values are clearly low (<120 cm/sec) or very high (>200 cm/sec), and the respective negative and positive predictive values for significant angiographic vasospasm in the MCA trunk are close to 90%.54–56 For values in the intermediate range, additional maneuvers may improve its accuracy in detecting vasospasm, such as testing for hyperemic autoregulatory responses to transient, manual carotid compression57,58 or combining the TCD information with CBF measurements.59 TCD does not reliably detect vasospasm in more peripheral branches,60 which may account for its failure to always correlate with perfusion deficits detected on CBF studies.61 Regular surveillance of SAH patients with TCD ultrasonography by skilled technicians is considered helpful in many neurosurgical units, but the results must be considered in the context of the individual patient along with all other information available.
Cerebral Blood Flow and Perfusion
Single-photon emission CT,62 quantitative stable xenon–enhanced CT,59 and positron emission tomography61 can be used to detect cerebral ischemia after SAH, but none are practical investigations that can be easily performed or repeated in critically ill patients. Magnetic resonance imaging to look for either perfusion deficits63 or ischemia on diffusion-weighted images64,65 has the same limitation.
Perfusion CT has been used in patients with SAH66,67 and with greater experience will probably find more widespread use and validation for predicting or diagnosing vasospasm.
Thermal diffusion flowmetry has been used as bedside monitoring in SAH patients to detect vasospasm causing significant reductions in CBF.68 Provided that the white matter location into which the thermal diffusion microprobe is inserted (usually the frontal lobe) is representative of the territory at risk, continuous measurement of CBF values could be particularly useful in patients with high-grade SAH who cannot be assessed neurologically.
Microdialysis Monitoring
Cerebral microdialysis catheters allow continuous bedside measurement of extracellular concentrations of glutamate, lactate, pyruvate, glucose, and glycerol in brain tissue, thereby screening for excitotoxic cell injury characterized by elevations in lactate with respect to glucose and pyruvate levels and an increase in the glycerol concentration. This somewhat demanding monitoring technique provides chemical information about a very small region of the brain, but it has been used, along with cerebral oximetry, to detect ischemia in patients with SAH.69–71
Vascular Imaging
The most practical method of imaging large and medium-sized cerebral arteries is digital subtraction angiography or high-definition CT angiography.72 Catheter-based angiography is best in patients in whom balloon angioplasty is being considered and can accompany the procedure.
Angiographic vasospasm is a concentric narrowing that can be focal, segmental, or diffuse. It is commonly graded as mild (<25%), moderate (25% to 50%), or severe (>50%) in comparison to baseline, prevasospasm imaging. “Early” vasospasm on admission angiography and within 48 hours of aneurysm rupture has been described in a small percentage of SAH patients, although without baseline angiography it is not clear how this diagnosis can be made accurately in all cases.73 Its detection has been associated with the development of cerebral infarction and poor outcome.
Prevention of Vasospasm and Cerebral Protection
General Measures: Fluid Management and Medical Treatment
Patients have a tendency toward volume contraction in the acute stage of SAH,74 and hypovolemia should be carefully avoided. It is not clear that a deliberate attempt to induce hypervolemia with volume expansion therapy is beneficial in terms of prevention of vasospasm or ischemia or even possible in patients with normal renal function.75–78 Patients should be hydrated with at least 3 L of isotonic fluids daily, and there is some evidence that additional colloid infusions (such as albumin) are beneficial.79 Some SAH patients experience excessive natriuresis and are susceptible to the development of hyponatremia during this time (usually related to elevations in brain natriuretic peptide), an electrolyte disturbance that may increase the risk for vasospasm.80 It has been reported that fludrocortisone administration (0.3 mg/day) may help avert this complication.81
Blood transfusion has been associated with a higher likelihood of both vasospasm and poor outcome in SAH patients,12,82 thus suggesting that anemia is a marker for other factors that contribute to SAH-associated morbidity. The optimal hemoglobin concentration in patients with SAH in terms of hematocrit, blood viscosity, and oxygen delivery to the brain is not known with certainty, but is generally accepted to be higher than 9 g/dL.
[/not-level-membership-for-neurosurgery-category]
