[level-membership-for-anesthesiology-category]
Cerebral protection
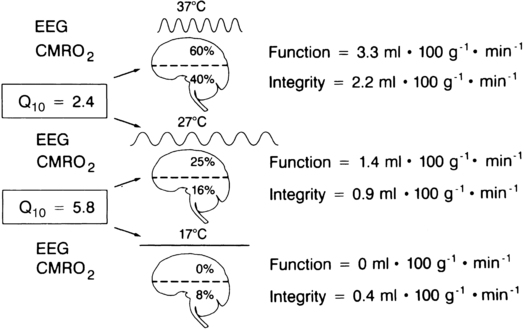
The traditional concept of cerebral metabolism is illustrated in Figure 131-1. Cerebral metabolism may be divided into a functional component and a cellular integrity component. The functional component comprises 60% of neuronal O2 use. This component is responsible for generating action potentials and may be assessed by evaluating the electroencephalogram. The cellular integrity component consists of the remaining 40% of O2 utilized for protein synthesis and other activities geared toward maintaining cellular integrity.
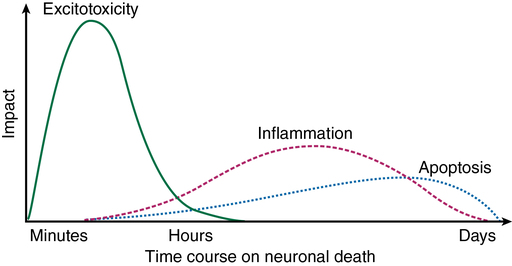
New evidence paints a much more complex picture of cerebral ischemia, in which an initial ischemic event may trigger a process of neuronal demise that continues long after the inciting event has resolved (Figure 131-2). Excitotoxicity is a cascade of glutamate-mediated neuronal demise that occurs shortly after the onset of neuronal ischemia. Apoptosis (programmed cell death via proteases) and inflammation are initiated by the ischemic event and continue to contribute to neuronal death for days. In this newer model of cerebral ischemia, it may be possible to limit ischemic damage by invoking cerebral protective therapies before, during, or after an ischemic event (Table 131-1). The currently available evidence in support of the use of cerebral protection is derived from a mixture of human experiments and animal data extrapolated to human subjects.
Table 131-1
Evidence-Based Status of Plausible Interventions to Reduce Perioperative Ischemic Brain Injury
| Efficacy in Experimental Animals | Efficacy in Humans | Sustained Protection in | ||||
| Intervention | Preischemic | Postischemic | Preischemic | Postischemic | Animals | Humans |
| Hypothermia | ||||||
| Mild | ++ | ++ | ± | ++* | ++ | ++ |
| Moderate | −−− | −−− | −− | −− | −−− | |
| Hyperventilation | −− | −− | −− | −− | −− | −− |
| Normoglycemia | ++ | −− | + | + | ++ | −− |
| Hyperbaric O2 | ++ | −− | −− | ± | −− | −− |
| Barbiturates | ++ | − | + | + | ++ | −− |
| Propofol | ++ | + | − | −− | −− | − |
| Etomidate | −−− | −− | −− | −− | −− | −− |
| N2O | − | −− | −− | −− | −− | −− |
| Isoflurane | ++ | −− | −− | −− | ++ | −− |
| Sevoflurane | −− | −− | −− | ++ | −− | |
| Desflurane | ++ | −− | −− | −− | −− | −− |
| Lidocaine | ++ | −− | + | −− | −− | −− |
| Ketamine | ++ | −− | −− | −− | −− | −− |
| Glucocorticoids | −−− | −− | −− | −− | −− | −− |
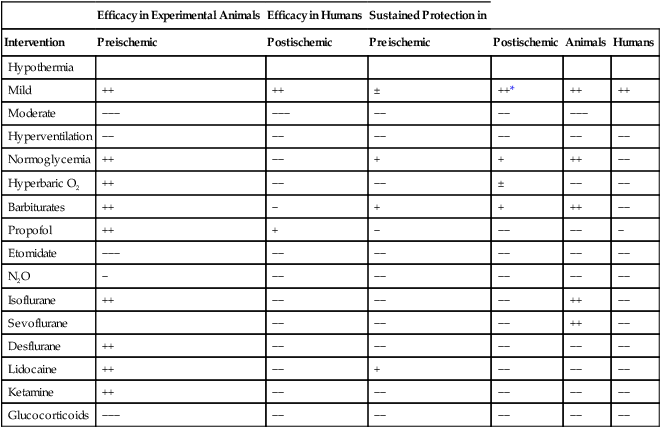
*Out-of-hospital ventricular fibrillation cardiac arrest.
(Adapted, with permission, from Fukuda S, Warner DS. Cerebral protection. Br J Anaesth. 2007;99:10-17.)
Conclusion
Protection of the nervous system from ischemic insult via pharmacologic and physiologic means has been a long-sought-after goal of anesthesiology. The current cerebral protective armamentarium has few proven interventions and many speculative ones. Box 131-1 provides a reasonable framework for addressing an ischemic insult based on the current level of knowledge.
[/level-membership-for-anesthesiology-category][not-level-membership-for-anesthesiology-category]
Cerebral protection
The traditional concept of cerebral metabolism is illustrated in Figure 131-1. Cerebral metabolism may be divided into a functional component and a cellular integrity component. The functional component comprises 60% of neuronal O2 use. This component is responsible for generating action potentials and may be assessed by evaluating the electroencephalogram. The cellular integrity component consists of the remaining 40% of O2 utilized for protein synthesis and other activities geared toward maintaining cellular integrity.
New evidence paints a much more complex picture of cerebral ischemia, in which an initial ischemic event may trigger a process of neuronal demise that continues long after the inciting event has resolved (Figure 131-2). Excitotoxicity is a cascade of glutamate-mediated neuronal demise that occurs shortly after the onset of neuronal ischemia. Apoptosis (programmed cell death via proteases) and inflammation are initiated by the ischemic event and continue to contribute to neuronal death for days. In this newer model of cerebral ischemia, it may be possible to limit ischemic damage by invoking cerebral protective therapies before, during, or after an ischemic event (Table 131-1). The currently available evidence in support of the use of cerebral protection is derived from a mixture of human experiments and animal data extrapolated to human subjects.
Table 131-1
Evidence-Based Status of Plausible Interventions to Reduce Perioperative Ischemic Brain Injury
| Efficacy in Experimental Animals | Efficacy in Humans | Sustained Protection in | ||||
| Intervention | Preischemic | Postischemic | Preischemic | Postischemic | Animals | Humans |
| Hypothermia | ||||||
| Mild | ++ | ++ | ± | ++* | ++ | ++ |
| Moderate | −−− | −−− | −− | −− | −−− | |
| Hyperventilation | −− | −− | −− | −− | −− | −− |
| Normoglycemia | ++ | −− | + | + | ++ | −− |
| Hyperbaric O2 | ++ | −− | −− | ± | −− | −− |
| Barbiturates | ++ |
