CHAPTER 9 Cerebral Edema
Overview and Historical Background
Paul Ehrlich was the first to identify a potential barrier between the vasculature and the brain after he observed that intravenous albumin-bound dyes stained all tissues except the brain.1 Goldmann carried Ehrlich’s studies further to demonstrate that dye injected into CSF did not circulate in the systemic circulation.2 These barriers (Table 9-1), which are rate-limiting steps in the movement of water, ions, solutes, and macromolecules between compartments, help the brain regulate its own environment distinct from the rest of the body. The BBB is an essential component of brain homeostasis.
TABLE 9-1 Principal Features of the Blood-Brain Neurovascular Unit
| INTERFACE | LOCATION OF THE BLOOD-BRAIN JUNCTION | FUNCTIONAL OUTCOME |
|---|---|---|
| Blood-brain | Capillary endothelial cell (see Fig. 9-1) | Active transport of most materials. The Na+,K+-ATPase pump on the apical surface of the capillary endothelium transports materials across high-resistance tight junctions. Tight junctions restrict the entry of hydrophilic materials and high-molecular-weight molecules |
| Blood–cerebrospinal fluid | Choroid plexus | Ultrafiltration of plasma with active secretion of cerebrospinal fluid; this high-energy process requires ATP. Essential components are ATPase and carbonic anhydrase |
| Cerebrospinal fluid–venous blood | Arachnoid granulations | Arachnoid granulations transmit cerebrospinal fluid into the cerebral venous sinuses along a pressure gradient |
ATP, adenosine triphosphate; ATPase, adenosine triphosphatase.
The Blood-Brain Barrier
The principal component of the BBB is the endothelial cells that line the cerebral microvasculature (Fig. 9-1).3–10 The tight junctions between adjacent endothelial cells in the brain, which are nonpermissive in comparison to those in the systemic circulation, prevent the paracellular transport of most molecules. Although small substances, such as oxygen and carbon dioxide, and small lipophilic molecules, such as ethanol, may diffuse freely through the lipid membranes that constitute the BBB, larger, bulkier, more complex or hydrophilic molecules require active, transcellular transport mechanisms, potentially on both the luminal (endothelial) and abluminal (brain) membranes, to enter the brain.11 The active transport mechanisms require energy in the form of adenosine triphosphate (ATP).11
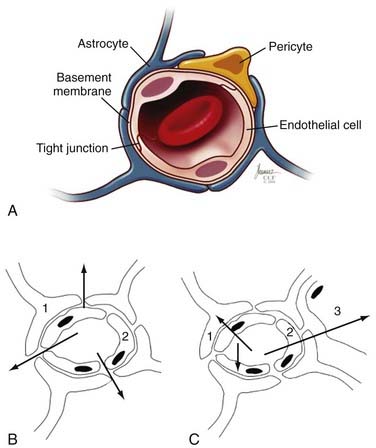
Several studies have identified a variety of molecular mechanisms and metabolic barriers that indicate that the BBB is an exceptionally active system.11–13 For example, endothelial cells contain active peptides, peptidases that inactivate traversing proteins, and numerous intracellular enzymes, such as cytochrome P-450 (1A and 2B isoforms), that inactivate neuroactive and neurotoxic substances.14,15 In general, to traverse the normal BBB, large or hydrophilic molecules require an active transport mechanism—either receptor-mediated or absorptive-mediated transcytosis. In the cerebral endothelium, these mechanisms are less efficient than in systemic (outside the central nervous system [CNS]) endothelial cells, which enhances the potential BBB. Thus, the BBB contains active and passive features that regulate the passage of substances from the systemic circulation into the brain. Similar versions of these tight junctions and permeability restrictions are found between CSF and the brain (except in the circumventricular organs [area postrema, tuber cinereum, and pineal gland], where the endothelium is penetrated more easily to permit secreted neuropeptides to act systemically; see Table 9-1).11–16
The BBB has an extensive surface area—which some researchers have estimated to be approximately 20 m2/1.3 kg of brain. Consequently, no neuron is more than 20 to 25 µm away from a brain capillary, and thus the BBB plays several critical roles in regulation of the brain’s microenvironment.17,18 It manages the entry of nutrients and controls elimination of wastes. The BBB limits distribution and thus contains within the brain generated neuroregulators and neurotransmitters that act centrally while excluding or regulating entry into the brain of similar molecules intended to act peripherally. This reduces or prevents debilitating biochemical crosstalk within the CNS. Finally, the BBB regulates movement of fluid and ions between the circulation and the brain, which permits maintenance of an ideal interstitial fluid that enhances neuronal function. The brain’s interstitial fluid has some similarities with plasma but has a lower protein content and lower concentration of calcium (Ca2+) and potassium (K+) ions and a higher concentration of magnesium (Mg2+), which generates a significant buffering capacity. This limits the effect of fluxes of systemic metabolism, such as occur with exercise, a meal, or starvation.5,11,19 Continuous turnover of interstitial fluid and CSF, which is regulated by the cerebral endothelium and its barriers (see Table 9-1), is critical to homeostasis of the brain’s microenvironment.
Molecular Events in Cerebral Edema
Cerebral edema is a common end result of a variety of neurological and systemic disorders. Most classifications of cerebral edema describe four categories (summarized in Table 9-2): cytotoxic, or cellular swelling secondary to cell injury; vasogenic, which results from vascular leakage through a disrupted BBB and consequently increased fluid and altered concentrations of ions, peptides, and macromolecules in the extracellular space; interstitial, which occurs with transependymal flow of CSF in patients with hydrocephalus; and osmotic, when the brain is hyperosmolar relative to plasma and thus induces water to flow passively across an intact BBB along its concentration gradient. It may be difficult to separate edema into these distinct classes in every patient because more than one type may be present simultaneously as a result of the nature and timing of the underlying disorder (see Table 9-2). Because interstitial edema and osmotic edema have fewer causes or are uncommon in neurosurgical patients, our principal focus in this chapter is on vasogenic and cytotoxic edema.
Tissue swelling—edema—may be intracellular or extracellular. It has the potential to result in profound shifts in the relative volumes occupied by the cellular and interstitial elements. Continued redistribution of water, ions, peptides, and other neuroactive substances within and between the cells of the CNS (neurons, glia, microglia, and endothelial cells) may exacerbate the primary cause of the edema. These failures lead to a variety of molecular events and cascades that potentiate cerebral and BBB dysfunction, some of which are summarized later and are discussed in greater detail in several recent, specialized monographs.4,5,20
Vasogenic Edema
Vasogenic edema may share some mechanisms with cytotoxic and other forms of brain edema. However, the principal source of edema formation is abnormal permeability of the BBB.21 The most common source is a primary or secondary brain tumor, in which case the nascent microvessels are deficient in tight junctions. This “brain-tumor barrier” is an incompetent obstacle that permits leakage of plasma ultrafiltrate into the brain’s extracellular space.18,22,23
The edema associated with brain tumors results from this passive deficiency and from cellular invasion and migration.10,24,25 In addition, many tumors have active mechanisms to promote vascular permeability and neovascularity. The most widely studied permeability and angiogenic agent secreted by tumor cells is vascular endothelial growth factor (VEGF), which induces capillary permeability, endothelial proliferation, and migration and organization of new capillaries that lack tight junctions.26 Additional chemokines, cytokines, growth factors, and inflammatory mediators that play similar or complementary roles in blood-tumor permeability and angiogenesis have been identified. For example, angiopoietin-1, angiopoietin-2, fibroblast growth factor, hepatocyte growth factor/scatter factor, platelet-derived growth factor, interleukin-3 (IL-3), IL-4, IL-8, transforming growth factor-α (TGF-α), TGF-β, a variety of adhesion molecules and proteases such as urokinase plasminogen activator, multiple matrix metalloproteinases, integrins αvβ3 and αvβ5, and even oncogenes such as mutated Ras and tumor suppressor gene products such as Tp53 and vhl protein can affect BBB function. Many of these are discussed elsewhere in this volume.
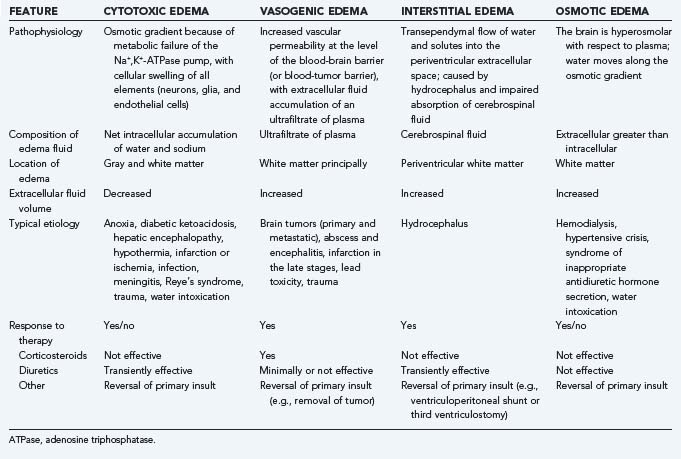
The most thoroughly studied mechanism that produces cerebral edema is the vasogenic edema mediated by tumor production of a macromolecular protein initially identified as vascular permeability factor (VPF) and later, after its angiogenic activity had been identified, as VEGF. VPF/VEGF was initially identified by Senger and Dvorak in 1983.27 Their landmark study demonstrated that the ascites caused by the intraperitoneal injection of hepatocarcinoma cells into guinea pigs was a product of excessive permeability of the small vessels that line the peritoneal cavity and, furthermore, that a protein secreted by the tumor and acting on the vessels was responsible for the enhanced vascular permeability. Hepatocarcinoma lines that did not produce the protein did not cause ascites. In addition, in an in vivo biologic assay of cutaneous vascular permeability, the enhanced vascular permeability produced by the protein was blocked by antibodies to the partially isolated protein. Secretion of the same protein was subsequently shown to occur in several systemic and CNS tumors.28–30 Bruce and colleagues and Heiss and associates demonstrated that vascular permeability is increased by conditioned media from cultures of high-grade gliomas and meningiomas, the types of human brain tumors that most commonly produce clinically significant cerebral edema. In addition, they demonstrated that antibodies to VPF/VEGF block the permeability-enhancing effects of conditioned media from tumor types that produce cerebral edema and showed that glucocorticoids block the permeability-enhancing effects of VPF/VEGF on the vessel wall and inhibit tumor cell production of VPF/VEGF.29,31 CNS tumors that are frequently associated with marked edema, such as glioblastomas, meningiomas, and metastases, are found to contain high levels of VPF/VEGF gene expression, whereas the types of CNS tumors that are not commonly associated with significant cerebral edema do not usually produce levels of VPF/VEGF mRNA higher than found in normal brain.30 In clinical studies, the concept that VPF/VEGF is a principal mediator of peritumoral edema is confirmed by the reduction in contrast enhancement (vascular permeability) and surrounding cerebral edema on imaging studies by treatment of glioblastoma patients with bevacizumab, an anti-VEGF antibody (Fig. 9-2).32,33
Cytotoxic Edema
Cytotoxic edema occurs after cerebral infarction or ischemia, meningitis, Reye’s syndrome, trauma, seizures, and water intoxication. There are several distinct or overlapping mechanisms that underlie cytotoxic cerebral edema. One common mechanism of cytotoxic edema associated with cerebral ischemia implicates a direct role for excess glutamate.34–36 After hypoxia, neuronal swelling results from sodium influx through α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptors and kainite receptor activity and from influx of chloride ion and water, which enter passively.34–36 The glial swelling associated with glutamate toxicity is due to enhanced sodium entry into astrocytes by glutamate transporter hyperactivity.11,12,17,37 Combined neuronal and astrocytic swelling also involves a variety of additional and complementary mechanisms, including dysfunction of the sodium-potassium adenosine triphosphatase (Na+,K+-ATPase) pump, which permits sodium influx, and activation of the Na+/H+ and Cl−/HCO3− exchangers and the Na+/K+/2Cl− cotransporter.11,12,17,37 The altered extracellular potassium ion concentration that occurs, for example, with primary epilepsy or with seizures after ischemia causes swelling through the Na+/K+/2Cl− cotransporter.38–40 The same transmitter, because it passively allows passage of ammonium ions, has been implicated in the cerebral edema associated with hepatic encephalopathy. Aquaporin-4, a member of the family of aquaporin water channels, is enriched in astroglial end-feet and appears to play a central role in the entry of water into astrocytes.41–43 Furthermore, the Kir4.1 potassium channel that colocalizes with aquaporin-4 regulates the extracellular potassium concentration. Elevated levels of potassium ions passing through Kir4.1 channels depolarize astrocytes (as they do neurons), which enhances astroglial uptake of sodium and bicarbonate through the Na+/HCO3− cotransporter.19 This increases intracellular osmolarity and allows water to move passively into the cell through aquaporin-4 channels. Aquaporin-1 and aquaporin-4 are overexpressed in primary and secondary brain tumors and enhance water uptake.41–43
Other mechanisms that lead to or enhance cytotoxic edema occur after hypoxic, ischemic, or traumatic brain injury. Loss of intracellular ATP and glutamate release enhance the influx of calcium ions. Efflux of one calcium ion is associated with the uptake of three sodium ions, which potentiates the osmotic gradient and draws more water into the cell. Excess intracellular calcium ions can initiate apoptosis, activate inflammatory cascades through activation of immediate early genes such as c-fos and c-jun, and generate the release of a variety of cytokines, free radicals, and proteases that act on surrounding neuronal and glial cells, the extracellular matrix, and the cerebral endothelium.17,36,37,44–46 Furthermore, nitric oxide (NO), which serves as a vasodilator but may also exert toxic effects through interactions with superoxide anions to generate peroxynitrite anions (ONOO−), may exacerbate cerebral edema in certain circumstances.47 NO is synthesized by nitric oxide synthase (NOS), which has three distinct forms. Neuronal NOS produces toxic free radicals early after cytotoxic injury; endothelial NOS produces NO that enhances blood flow through vasodilation; and inducible NOS, produced by macrophages and microglia, can exacerbate injury through generation of NO—and free radicals—24 to 48 hours after the initiating insult has passed.3,47
Inflammation may also figure prominently in disruption of the BBB. For example, inflammatory cascades enhance the expression of bradykinin, substance P, leukotrienes, serotonin, and histamine, all of which nonselectively open the BBB to varying degrees.3,5,48 Lipopolysaccharides, released during infection, induce the production of tumor necrosis factor and reactive oxygen species from microglia with potent effects on BBB permeability and can worsen cytotoxic injury.3,5,11 Finally, Simard and coworkers recently identified an NCCA, nonselective cation channel or NCCa-ATP channel, that when opened by depletion of ATP, causes cytotoxic edema associated with ischemia. This channel is regulated by sulfonylurea receptor 1, which can be blocked by low doses of glibenclamide and thus provides a potential new approach to treat cerebral edema associated with cerebral infarction and brain trauma.49–51
Neuroimaging Studies and Classification of Cerebral Edema
The principal types of cerebral edema—cytotoxic, interstitial, and vasogenic—can be characterized in patients, at least to some degree, through neuroimaging studies, especially magnetic resonance imaging (MRI) (Figs. 9-3 to 9-5; see Fig. 9-2).52–54
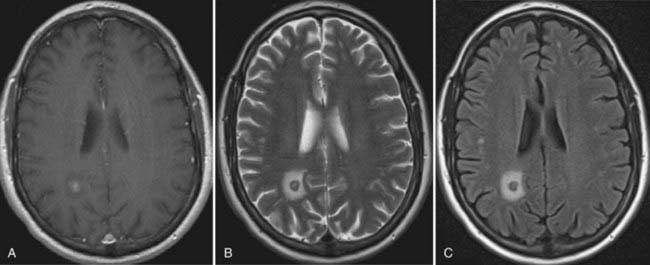
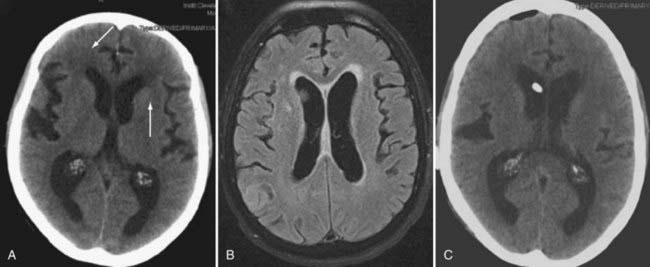
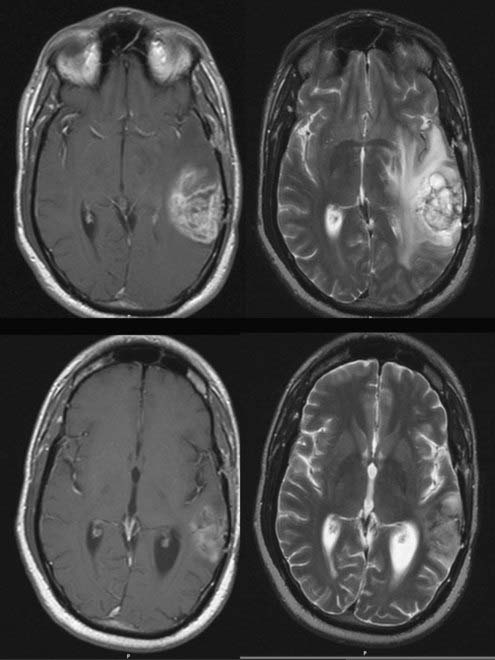
In vasogenic edema (see Figs. 9-2 and 9-3), the edematous tissue is hypodense with respect to normal brain on both non–contrast-enhanced computed tomography and T1-weighted MRI. This feature is highlighted after the administration of contrast material.53 On imaging, the edema primarily affects the white matter and tends to spare the gray matter, unlike cytotoxic edema. T2-weighted and fluid-attenuated inversion recovery (FLAIR) sequences accentuate the appearance of vasogenic edema and can, in FLAIR sequences, distinguish edema from normal brain water content, such as CSF. FLAIR images, in particular, are also useful to demonstrate the extent of tumor cell migration beyond the enhancing bulk of tumor because these MRI sequences are especially sensitive to the changes in brain water content induced by migrating or invading cells. Because vasogenic edema is primarily extracellular, the interstitial spaces also appear enlarged, a feature that increases the apparent diffusion coefficient (ADC) on diffusion studies.52,53 MRI perfusion sequences can also demonstrate the enhanced vascularity and permeability associated with tumor angiogenesis, alone or with provocative testing, such as with the administration of dexamethasone.23 Finally, dynamic, contrast-enhanced MRI can be used to quantify the permeability of the BBB and assess the effects of treatments that target the BBB.53,54
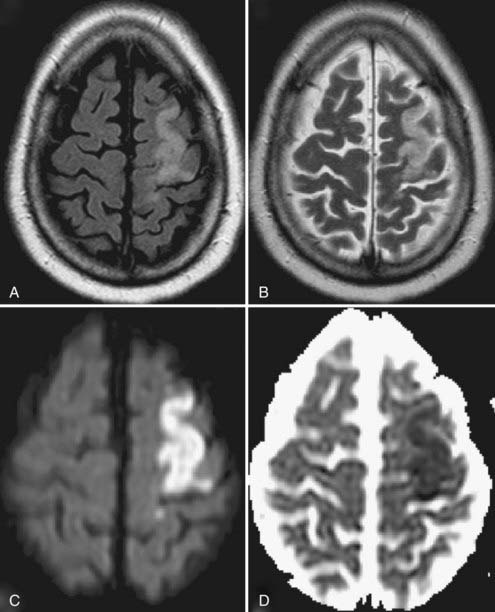
Cytotoxic edema, found in patients with ischemic (see Fig. 9-4) or hemorrhagic stroke and traumatic brain injury,55,56 induces a rapid intracellular influx of water because of energy failure that evolves clinically and neuroradiographically.52 Within 12 hours, there is loss of the visible gray-white junction and gyral edema, and at 12 to 24 hours, T2-weighted images show hyperintensity. The changes in diffusion-weighted MRI signal intensity may occur rapidly (within minutes after the insult); acute infarcts have a lower ADC than normal brain does, and these ADC maps sensitively indicate early cytotoxic edema.42
Treatment of Cerebral Edema
Most current treatments of cerebral edema are indirect and focused on amelioration of the effects of edema, whereas more specific treatments are directed at the causative disease or condition (see Table 9-2).21 For cytotoxic, osmotic, and interstitial edema, the main therapeutic intervention is to reverse the cause. In interstitial edema, resolution of transependymal CSF flow requires temporary or permanent diversion of spinal fluid (see Fig. 9-5). In vasogenic edema, the volume of extracellular fluid is a function of the relative rates of production and resorption of extracellular fluid. Higher pressure within the tumor and permeable tumor vessels initiates hydrostatic flow away from the tumor margin and into the extracellular space surrounding it until it reaches (convects to) the ventricles and subarachnoid spaces. There are dynamic limits to resorption: local capillaries absorb extracellular fluid slowly (estimated at 0.0086 mL/hr/cm3), and normal astrocytic cells have a defined capacity to absorb extravasated protein, ions, and water.5,19,21,57–59 These resorptive mechanisms may often be overwhelmed when the BBB is defective and are not generally capable, without adjunctive methods, of handling the amount of extracellular fluid commonly produced with most tumor types.58–61
Several agents, including glucocorticoids, diuretics, and mannitol and other osmotic agents (reviewed in detail elsewhere21), have a moderate effect when used to control peritumoral edema. Although ineffective for cytotoxic edema and only modestly but transiently efficacious in the short-term treatment of interstitial edema, glucocorticoids can improve the neurological symptoms and signs caused by vasogenic edema, especially in patients with brain tumors, in whom the clinical features often result from the mass contributed by the peritumoral edema. Experimental evidence indicates that the effects of glucocorticoids are primarily reduced vascular permeability of vessels rather than reduced VEGF production.26,31 This inhibition of the effects of VPF/VEGF on the vasculature is associated with interference of VEGF’s action on vessels and requires the glucocorticoid receptor.26,31 Thus, the effects of steroids in tumor-induced vasogenic edema is to restrict permeability of the BBB to macromolecules. By contrast, steroids are not effective when the BBB is not functional.3,26,31,58
Diuretics, such as the loop diuretic furosemide, act by inducing systemic dehydration, which may reverse osmotic flow. This therapeutic effect is limited in time and efficacy.21 The carbonic anhydrase inhibitor acetazolamide decreases CSF production by reducing hydrogen and bicarbonate production. Like diuretics, acetazolamide’s efficacy is modest and short in duration, and systemic metabolic derangements may occur.21 Osmotic agents such as glycerol, mannitol, hypertonic saline, and urea create an osmotic gradient between the brain and blood, down which extracellular water flows.21 Because this activity is limited temporally, osmotherapy is generally reserved for settings of acute edema with mass effect and is used as a temporary measure to provide time for the initiation of definitive treatment to eliminate the cause of the edema. Furthermore, prolonged use of osmotic agents can lead to the accumulation of solute (mannitol, glycerol) within the tissues, which can then serve as a “reverse sink” and produce a circumstance in which the edema is refractory to further osmotic therapy. Agents such as mannitol or hypertonic saline may also have other actions, such as vasoconstriction, enhanced cerebral blood flow, and altered rheology, among others, that can alter the content or effect of edema. Many free radical scavengers and neuroprotective agents that have shown promise in the laboratory or in animal models have generally failed in human trials. Whether new agents that modulate the activity of aquaporins, corticotropin-releasing hormone, sulfonylurea receptor 1, or other pathways will prove effective awaits new human trials.62–64 New agents that can mitigate the secondary neural damage caused by cerebral edema and can do so without the side effects of the current therapies are clearly needed.
Abbott NJ. Astrocyte-endothelial interactions and blood-brain barrier permeability. J Anat. 2002;200:629-638.
Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7:41-53.
Al-Okaili RN, Krejza J, Wang S, et al. Advanced MR imaging techniques in the diagnosis of intraaxial brain tumors in adults. Radiographics. 2006;26(suppl 1):S173-S189.
Amiry-Moghaddam M, Ottersen OP. The molecular basis of water transport in the brain. Nat Rev Neurosci. 2003;4:991-1001.
Ballabh P, Braun A, Nedergaard M. The blood-brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol Dis. 2004;16:1-13.
Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57:173-185.
Heiss JD, Papavassiliou E, Merrill MJ, et al. Mechanism of dexamethasone suppression of brain tumor-associated vascular permeability in rats. Involvement of the glucocorticoid receptor and vascular permeability factor. J Clin Invest. 1996;98:1400-1408.
Kaal EC, Vecht CJ. The management of brain edema in brain tumors. Curr Opin Oncol. 2004;16:593-600.
King LS, Kozono D, Agre P. From structure to disease: the evolving tale of aquaporin biology. Nat Rev Mol Cell Biol. 2004;5:687-698.
Manley GT, Fujimura M, Ma T, et al. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med. 2000;6:159-163.
Marmarou A, Signoretti S, Fatouros PP, et al. Predominance of cellular edema in traumatic brain swelling in patients with severe head injuries. J Neurosurg. 2006;104:720-730.
Merrill MJ, Oldfield EH. A reassessment of vascular endothelial growth factor in central nervous system pathology. J Neurosurg. 2005;103:853-868.
Schlageter KE, Molnar P, Lapin GD, et al. Microvessel organization and structure in experimental brain tumors: microvessel populations with distinctive structural and functional properties. Microvas Res. 1999;58:312-328.
Seifert G, Shilling K, Steinhauser C. Astrocyte dysfunction in neurological disorders: a molecular perspective. Nat Rev Neurosci. 2006;7:194-206.
Senger DR, Galli SJ, Dvorak AM, et al. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science. 1983;219:983-985.
Simard JM, Chen M, Tarasov KV, et al. Newly expressed SUR1-regulated NC(Ca-ATP) channel mediates cerebral edema after ischemic stroke. Nat Med. 2006;12:433-440.
Strange K. Cellular volume homeostasis. Adv Physiol Educ. 2004;28:155-159.
Wolburg H, Lippolt A. Tight junctions of the blood-brain barrier: development, composition, and regulation. Vasc Pharmacol. 2002;38:323-337.
Yong VW. Metalloproteinases: mediators of pathology and regeneration in the CNS. Nat Rev Neurosci. 2005;6:931-944.
1 Ehrlich P. Uber die Beziehungen von chemische Constitution, Vertheilung, und pharmakologisher Wirkung. In: Collected Studies in Immunity. New York: John Wiley; 1906:567-595.
2 Goldmann EE. Vitalfarburg am Zentralnervensystem. Abh Preuss Wiss Phys-Math. 1913;1:1-60.
3 Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57:173-185.
4 Amiry-Moghaddam M, Ottersen OP. The molecular basis of water transport in the brain. Nat Rev Neurosci. 2003;4:991-1001.
5 Go KG. The normal and pathological physiology of brain water. Adv Tech Stand Neurosurg. 1997;23:47-142.
6 Lee SW, Kim WJ, Choi YK, et al. SSeCKS regulates angiogenesis and tight junction formation in blood-brain barrier. Nat Med. 2003;9:900-906.
7 Ballabh P, Braun A, Nedergaard M. The blood-brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol Dis. 2004;16:1-13.
8 Abbott NJ. Dynamics of CNS barriers: evolution, differentiation, and modulation. Cell Mol Neurobiol. 2005;25:5-23.
9 Cserr HF, Bundgaard M. Blood-brain interfaces in vertebrates: a comparative approach. Am J Physiol. 1984;246:R277-R288.
10 Carmeliet P, Tessier-Lavigne M. Common cues in vascular and axon guidance. Nature. 2005;436:193-200.
11 Volterra A, Meldolesi J. Astrocytes, from brain glue to communication elements: the revolution continues. Nat Rev Neurosci. 2005;6:626-640.
12 Zonta M, Angulo MC, Gobbo S, et al. Neuron-to astrocyte signaling is central to the dynamic control of brain microcirculation. Nat Neurosci. 2003;6:43-50.
13 Takano T, Tian GF, Peng W, et al. Astrocyte-mediated control of cerebral blood flow. Nat Neurosci. 2006;9:260-267.
14 Mann GE, Yudlilevich DL, Sobrevia L. Regulation of amino acid and glucose transport in endothelial and smooth muscle cells. Physiol Rev. 2003;83:183-252.
15 Leybaert L. Neurobarrier coupling in the brain: a partner of neurovascular and neurometabolic coupling. J Cereb Blood Flow Metab. 2005;25:2-16.
16 O’Kane R, Hawkins RA. Na+-dependent transport of large neutral amino acids occurs at the abluminal membrane of the blood-brain barrier. Am J Physiol Endocrinol Metab. 2003;285:E1167-E1173.
17 Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7:41-53.
18 Schlageter KE, Molnar P, Lapin GD, et al. Microvessel organization and structure in experimental brain tumors: microvessel populations with distinctive structural and functional properties. Microvas Res. 1999;58:312-328.
19 Saadoun S, Papadopoulos MC, Krishna S. Water transport becomes uncoupled from K+ siphoning in brain contusion, bacterial meningitis, and brain tumours: immunohistochemical case review. J Clin Pathol. 2003;56:972-975.
20 Strange K. Cellular volume homeostasis. Adv Physiol Educ. 2004;28:155-159.
21 Kaal EC, Vecht CJ. The management of brain edema in brain tumors. Curr Opin Oncol. 2004;16:593-600.
22 Wilkinson ID, Jellineck DA, Levy D, et al. Dexamethasone and enhancing solitary cerebral mass lesions: alterations in perfusion and blood-tumor barrier kinetics shown by magnetic resonance imaging. Neurosurgery. 2006;58:640-648.
23 Bastin ME, Carpenter TK, Armitage PA, et al. Effects of dexamethasone on cerebral perfusion and water diffusion in patients with high-grade glioma. AJNR Am J Neuroradiol. 2006;27:402-408.
24 Kim H, Lee JM, Park JS, et al. Dexamethasone coordinately regulates angiopoietin-1 and VEGF: a mechanism of glucocorticoid-induced stabilization of the blood-brain barrier. Biochem Biophys Res Commun. 2008;372:243-248.
25 Ramsauer M, Krause D, Dermietzel R. Angiogenesis of the blood-brain barrier in vitro and the function of the cerebral pericytes. FASEB J. 2002;16:1274-1278.
26 Merrill MJ, Oldfield EH. A reassessment of vascular endothelial growth factor in central nervous system pathology. J Neurosurg. 2005;103:853-868.
27 Senger DR, Galli SJ, Dvorak AM, et al. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science. 1983;219:983-985.
28 Senger DR, Perruzzi CA, Feder J, et al. A highly conserved vascular permeability factor secreted by a variety of human and rodent tumor cell lines. Cancer Res. 1986;46:5629-5632.
29 Bruce JN, Criscuolo GR, Merrill MJ, et al. Vascular permeability induced by protein product of malignant brain tumors: inhibition by dexamethasone. J Neurosurg. 1987;67:880-884.
30 Berkman RA, Merrill MJ, Reinhold WC, et al. Expression of the vascular permeability factor/vascular endothelial growth factor gene in central nervous system neoplasms. J Clin Invest. 1993;91:153-159.
31 Heiss JD, Papavassiliou E, Merrill MJ, et al. Mechanism of dexamethasone suppression of brain tumor-associated vascular permeability in rats. Involvement of the glucocorticoid receptor and vascular permeability factor. J Clin Invest. 1996;98:1400-1408.
32 Ananthnarayan S, Bahng J, Roring J, et al. Time course of imaging changes of GBM during extended bevacizumab treatment. J Neurooncol. 2008;88:339-347.
33 Norden AD, Drappatz J, Wen PY. Antiangiogenic therapies for high-grade glioma. Nat Rev Neurol. 2009;5:610-620.
34 Hansson E, Ronnback L. Astrocyte receptors and second messenger systems. Adv Mol Cell Biol. 2004;51:475-501.
35 Maragakis NJ, Rothstein JD. Glutamate transporters in neurologic disease. Arch Neurol. 2001;58:365-370.
36 Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges, and opportunities in stroke. Nat Rev Neurosci. 2003;4:399-415.
37 Seifert G, Shilling K, Steinhauser C. Astrocyte dysfunction in neurological disorders: a molecular perspective. Nat Rev Neurosci. 2006;7:194-206.
38 Tian GF, Azmi H, Takano T, et al. An astrocytic basis of epilepsy. Nat Med. 2005;11:973-981.
39 Kimelberg HK. Astrocytic swelling in cerebral ischemia as a possible cause of injury and target for therapy. Glia. 2005;50:389-397.
40 Davies DC. Blood-brain barrier breakdown in septic encephalopathy and brain tumors. J Anat. 2002;200:639-646.
41 Manley GT, Fujimura M, Ma T, et al. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med. 2000;6:159-163.
42 Saadoun S, Papadopoulos MC, Davies DC, et al. Aquaporin-4 expression is increased in oedematous brain tumors. J Neurol Neurosurg Psychiatry. 2002;72:262-265.
43 Hiyashi Y, Edwards NA, Proescholdt MA, et al. Regulation and function of aquaporin-1 in glioma cells. Neoplasia. 2007;9:777-787.
44 Jian LK, Rosenberg GA. Matrix metalloproteinases and free oxygen radicals in cerebral ischemia. Free Radic Biol Med. 2005;39:71-80.
45 Yong VW. Metalloproteinases: mediators of pathology and regeneration in the CNS. Nat Rev Neurosci. 2005;6:931-944.
46 Banks WA. Blood-brain barrier transport of cytokines: a mechanism for neuropathology. Curr Pharm Des. 2005;11:973-984.
47 Endres M, Laufs U, Liao JK, et al. Targeting eNOS for stroke protection. Trends Neurosci. 2004;27:283-289.
48 Dietrich JB. Endothelial cells of the blood-brain barrier: a target for glucocorticoids and estrogens? Front Biosci. 2004;9:684-693.
49 Simard JM, Tsymbalyuk N, Tsymbalyuk O, et al. Glibenclamide is superior to decompressive craniectomy in a rat model of malignant stroke. Stroke. 2010;41:531-537.
50 Simard JM, Geng Z, Woo SK, et al. Glibenclamide reduces inflammation, vasogenic edema, and caspase-3 activation after subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2009;29:317-330.
51 Simard JM, Chen M, Tarasov KV, et al. Newly expressed SUR1-regulated NC(Ca-ATP) channel mediates cerebral edema after ischemic stroke. Nat Med. 2006;12:433-440.
52 Srinivasan A, Goyal M, Al Azri F, et al. State-of-the-art imaging of acute stroke. Radiographics. 2006;26 (suppl 1):S75-S95.
53 Al-Okaili RN, Krejza J, Wang S, et al. Advanced MR imaging techniques in the diagnosis of intraaxial brain tumors in adults. Radiographics. 2006;26(suppl 1):S173-S189.
54 Nielsen T, Mouridsen K, Maxwell RJ, et al. Segmentation of dynamic contrast enhanced magnetic resonance imaging data. Acta Oncol. 2008;25:1-6.
55 Marmarou A, Signoretti S, Fatouros PP, et al. Predominance of cellular edema in traumatic brain swelling in patients with severe head injuries. J Neurosurg. 2006;104:720-730.
56 Marmarou A. A review of progress in understanding the pathophysiology and treatment of brain edema. Neurosurg Focus. 2007;22(5):E1.
57 Zunkeler B, Carson RE, Olson J, et al. Quantification and pharmacokinetics of blood-brain barrier disruption in humans. J Neurosurg. 1996;85:1056-1065.
58 Abbott NJ. Evidence for bulk flow of brain interstitial fluid: significance for physiology and pathology. Neurochem Int. 2004;45:545-552.
59 Abbott NJ. Astrocyte-endothelial interactions and blood-brain barrier permeability. J Anat. 2002;200:629-638.
60 Bundgaard M, Abbott NJ. All vertebrates started out with a glial blood-brain barrier 4-500 million years ago. Glia. 2008;56:699-708.
61 Wolburg H, Lippolt A. Tight junctions of the blood-brain barrier: development, composition, and regulation. Vasc Pharmacol. 2002;38:323-337.
62 Hauger RL, Risbrough V, Brauns O, et al. Corticotropin releasing factor (CRF) receptor signaling in the central nervous system: new molecular targets. CNS Neurol Disord Drug Targets. 2006;5:453-479.
63 King LS, Kozono D, Agre P. From structure to disease: the evolving tale of aquaporin biology. Nat Rev Mol Cell Biol. 2004;5:687-698.
64 Rothstein JD, Patel S, Regan MR, et al. β-Lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433:73-77.