CHAPTER 202 Cerebellar Astrocytomas
Historical Perspective
Harvey Cushing in 1931 described his experience with 76 cerebellar astrocytomas and was the first to note that they represented a group separate from other astrocytomas. He remarked on their cerebellar location, frequent cystic appearance, occurrence at a young age, and excellent prognosis after surgical resection.1 In this series, the mean age at initial evaluation was 13 years, but most of his patients had been symptomatic for 2 or more years and exhibited blindness secondary to chronic papilledema. Cushing himself suggested that improved diagnostic techniques would allow diagnosis at a younger age, and the development of computed tomography (CT) and magnetic resonance imaging (MRI) has fulfilled this prediction.
The first histologic description of these tumors was provided by Bergstrand in 1932.2 He found that many of the cells in these masses were unipolar or bipolar spongioblasts, reminiscent of cells found during the late embryonic stage of development. He suggested that these were not true neoplasms but congenital malformations. Bucy and Gustafson recognized cerebellar astrocytoma as a true neoplastic entity and first described Rosenthal’s fibers as a histologic feature.3 In the 1940s, the designation “spongioblastoma” was used to describe many of these tumors, but in 1977, the term juvenile pilocytic astrocytoma was introduced by Russell and Rubinstein.4 This term remains in the current World Health Organization classification of brain tumors.
Etiology
The cause of these tumors remains obscure. The predominance of the lesion during childhood prompted Cushing to suggest a congenital origin, and in fact, infants younger than 1 year have been reported with cerebellar astrocytoma.5 Although many patients give a history of some antecedent trauma, this is hardly surprising in a population composed of children. It is likely that the injury merely calls attention to a preexisting problem; no link between trauma or any other external etiologic factor and cerebellar astrocytoma has ever been established. There does not appear to be an increased familial incidence, and they are mainly sporadic. The incidence is equal by gender.
A germline mutation or deletion of the neurofibromatosis type 1 (NF1) gene predisposes individuals to the development of a variety of tumors, including cerebellar astrocytoma.6 A role has been postulated for deletion of the long arm of chromosome 17q or mutation in the NF1 locus in the pathogenesis of sporadic pilocytic astrocytoma, but this hypothesis has yet to be confirmed.7 Most recently, microarray technology has provided a rapid method to screen for multiple gene abnormalities in tumors. A single novel duplication in chromosome band 7q34 was identified in 20 of 28 low-grade astrocytomas.8 One known oncogene (BRAF) maps to this region.
Patient Characteristics
Incidence
Cerebellar astrocytoma accounts for between 12% and 28% of all pediatric brain tumors. Approximately half of all tumors in children arise within the infratentorial compartment, and of these, astrocytoma accounts for about a third and is equal in incidence to medulloblastoma/primitive neuroectodermal tumor.8
Age
Roughly 70% of cerebellar astrocytomas are diagnosed in children.9 Cerebellar astrocytomas may occur in adults, but the outlook is less favorable, and most likely adult tumors are a different biologic entity, similar to astrocytomas in other locations. In children, the age at diagnosis has been steadily declining as imaging techniques improve and become more readily available. In 1971, the average age at admission to the hospital was 8.9 years.10 More recent series published in the era of modern imaging report the average age to be 7 years.11 Cerebellar astrocytoma is rare in infants. A midline cerebellar tumor in a young child or infant is more likely to be a medulloblastoma or ependymoma.
Clinical Findings
The typical history is characteristic and indistinguishable from that of other extrinsic posterior fossa masses apart from the duration of symptoms. Usually, the child has nonspecific symptoms of raised intracranial pressure arising from obstructive hydrocephalus. The symptoms are frequently insidious and, in retrospect, may have been present intermittently for years. The slow growth of these tumors allows gradual displacement of the adjacent cerebellum and brainstem, and alarming symptoms usually await the development of massive tumors and hydrocephalus. In small children, the cranium can expand to accommodate the increased intracranial volume with little increase in pressure. The early symptoms that do occur are often nonspecific and frequently attributed to viral illness, migraine, gastrointestinal disease, or psychiatric problems. The duration of symptoms before definitive diagnosis has fallen from 18.7 months reported by Ilgren and Stiller9 to 5.8 months in a more recent report.12 A long history of headache or vomiting in the context of a posterior fossa mass suggests the diagnosis of astrocytoma, whereas a history of just a few days or weeks makes a more rapidly growing tumor such as ependymoma or medulloblastoma more probable. The abundance of CT and MRI scanners in the developed world has resulted in an increased incidence of asymptomatic cerebellar astrocytomas found as incidental lesions on imaging performed for unrelated indications such as trauma.
Headache is the most common symptom in children with posterior fossa tumors and occurs in virtually 100% in some series.10 Nonspecific headache is common in children, but the headache of a posterior fossa tumor is characteristic. It is more often frontal than suboccipital at first. When the headache becomes localized to the suboccipital region, it is often described along with neck pain or stiffness and suggests chronic tonsillar herniation. Headaches occurring only in the morning and subsiding with activity are particularly suggestive. The combination of hypoventilation associated with sleep and increased intracranial pressure with recumbence provokes the headache on awakening, and the pain may awaken the child from sleep. Coughing, sneezing, or straining at stool may cause headache, and some children may become constipated to avoid discomfort.
Other signs and symptoms may include macrocephaly, personality change, torticollis, and dizziness. Diplopia from sixth nerve palsy may accompany hydrocephalus. Other cranial neuropathies suggest brainstem involvement. Tumors in the vermis tend to come to medical attention at a younger age, and patients complain mainly of headache and vomiting. Laterally placed tumors in the cerebellar hemisphere are more likely to occur in older patients and are more likely to be associated with dysmetria and tremor.5
Imaging Features
Cushing described the classic appearance of a cyst associated with a discrete mural nodule found at surgery. With modern imaging this picture has changed, and this classic appearance is present in less than 50% of cases.1,13 Cerebellar astrocytomas appear on CT or MRI in one of several predominant forms:
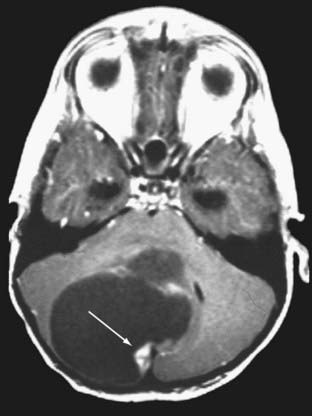
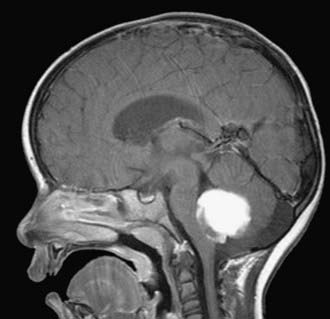
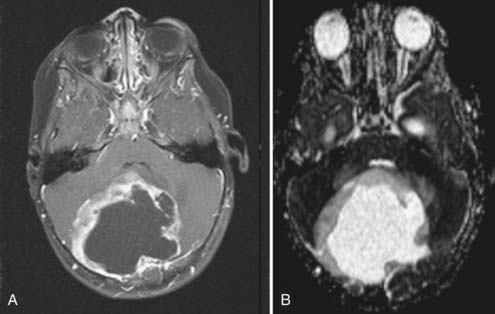
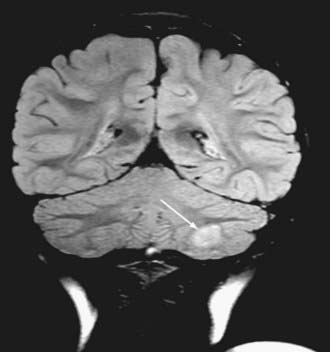
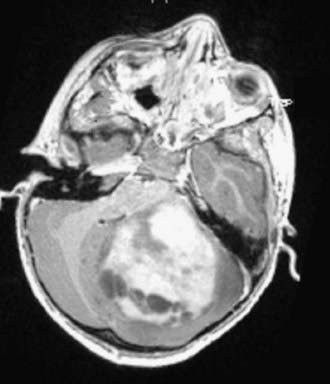
It is estimated that tumors are strictly confined to the cerebellum in 76% and involve the brainstem or cerebellar peduncle (“transitional forms”) in 24%.11
Unusual radiographic findings have been reported. Leptomeningeal dissemination at diagnosis is extremely rare but has been reported at the time of recurrence.19 Some degree of radiographic tonsillar herniation is common, particularly with large tumors, and cervical hydrosyringomyelia has been reported as well, presumably from interference with CSF flow at the foramen magnum.19 A case of a densely calcified cerebellar astrocytoma in an infant has been published as well.20
In summary, the appearance of a cerebellar astrocytoma in a child is sufficiently characteristic that the diagnosis is usually made preoperatively. A combination of radiographic and clinical findings makes the diagnosis even more accurate, and neural network programs have exhibited accuracy as high as 95% in predicting tumor type with pediatric posterior fossa tumors.21
Surgical and Perioperative Management
The primary goal of surgery for cerebellar astrocytoma is total resection of the mass. If contrast-enhanced MRI performed 6 months after surgery, when the postoperative changes have resolved, shows no residual tumor, the patient is very likely cured.22 This is possible in about 88% of cases.11 Total resection is usually straightforward in patients with cystic tumors and a laterally placed mural nodule, but it may be limited by involvement of the brainstem or cerebellar peduncle.
Severe symptoms are generally the result of obstructive hydrocephalus rather than the mass itself, and considerable debate has centered on the management of hydrocephalus. Options include steroids followed by tumor removal,23 external ventricular drainage,24 placement of a shunt before removal of the tumor,25 and preoperative endoscopic third ventriculostomy.26,27 Advocates of preoperative CSF diversion point out that with this strategy, time is afforded to prepare the patient and family for the definitive procedure, perform diagnostic tests, and provide a safer surgical procedure once the increased intracranial pressure has been relieved. Others point out the potential disadvantages: (1) only about 20% of patients with cerebellar astrocytomas ultimately require shunts postoperatively,28 and routine placement of shunts condemns the entire group to the possibility of shunt-related complications; (2) a preoperative shunt delays definitive treatment; (3) shunts serve as a potential route for dissemination should the tumor prove to be malignant; and (4) upward transtentorial herniation has been reported after acute decompression of the supratentorial compartment when the posterior fossa mass is still present.25,29 Endoscopic third ventriculostomy carries a rate of severe complications as high as 9% and is unlikely to relieve any component of hydrocephalus caused by absorptive failure after tumor removal.26
McLaurin concluded that these risks and benefits probably balance each other out and that either approach is acceptable.30 Most neurosurgeons prefer to remove the tumor, place a ventriculostomy during the surgery, and insert a shunt in the postoperative period if needed. Performance of a ventriculostomy at the bedside may be required as an emergency but should probably be followed by prompt tumor removal.
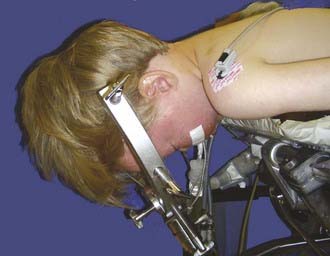
For most posterior fossa tumors in children, the prone position is recommended. The risk for air emboli with the sitting position is largely eliminated, and the surgeon’s arms become less fatigued. The method of head support depends on the age of the patient. Infants and very young children are probably best supported with a padded horseshoe headrest, and care must be taken to avoid pressure on the eyes, forehead, and malar eminences. Older children (≥2 years) and those with high vermian lesions in whom head flexion is desirable are best managed with pin fixation. The Sugita headrest is useful for younger children because multiple fixation points are used and thus require less force. The standard Mayfield device may be used in older children (Fig. 202-6). Patients with long-standing hydrocephalus may have very thin skulls, so care should be taken when adjusting the force of the pins. Severe flexion may result in kinking of the endotracheal tube, high inflating airway pressure, and venous bleeding. Nasotracheal intubation and communication with the anesthesiologist are helpful.
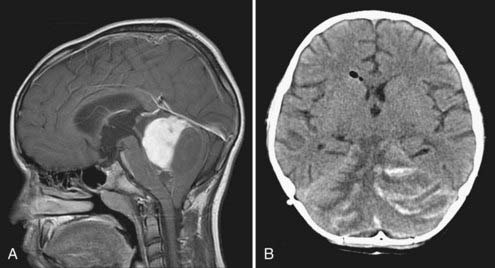
Laterally placed tumors with a cyst and a mural nodule may be approached by a transverse corticectomy through the cerebellar folia. Midline tumors usually require at least some splitting of the cerebellar vermis. A cerebellomedullary fissure approach has been described and allows relatively small inferior tumors to be removed without splitting the vermis, but it is unclear whether this avoids mutism or other potential problems.31 Tumors with a significant solid portion in the superior vermis may pose particular problems. They may be approached by splitting the superior vermis or by going through a supracerebellar infratentorial corridor as done for pineal region tumors. This approach involves sacrificing at least some of the bridging veins to the sinuses of the tentorium. If these veins are stretched, there is risk of tearing them from the straight sinus, which can result in profuse bleeding that is difficult to control. Sacrifice of these veins is usually well tolerated, but in some individuals unpredictable venous infarction, swelling, and hemorrhage may occur (Fig. 202-7).32
Complications
Operative and perioperative mortality has been reported to range from 4.7% to 8%%.11,33 Older series report higher mortality rates. Operative deaths have been due to acute hydrocephalus, respiratory compromise resulting from brainstem manipulation,10 and sudden cardiac arrest.
Clinical or subclinical air embolism can occur even in the prone position but is quite rare.34 It usually occurs with the craniectomy but may become apparent at any point during the procedure, including insertion of a pin fixation device. The most sensitive modality is Doppler monitoring, followed by a drop in end-tidal CO2. The low incidence of clinically significant air embolism in the prone position does not justify the routine use of atrial catheters. Treatment includes lowering the head, flooding the operative field with saline, and waxing all bone edges while the anesthesiologist ventilates with 100% O2.
Surgery on the posterior fossa is painful, and management of such pain in the immediate postoperative period may be difficult.35 Older children may be treated with nonsedating doses of an intravenous narcotic. In a younger child and infant, the mainstay remains acetaminophen supplemented with codeine. Ketorolac and oral opioids may be used after the immediate postoperative period (about 48 hours).
Delayed development of altered consciousness or focal neurological signs in the postoperative period should prompt imaging studies. In addition to the problems of hydrocephalus and hematoma, a peculiar syndrome of “pseudobulbar palsy” has been described in which stupor, irritability, emotional lability, dysarthria, mutism, nystagmus, and facial diplegia may occur about 48 hours after surgery for midline posterior fossa tumors.36 This syndrome tends to resolve over a period of several weeks to months and has been attributed to retraction-induced edema of the cerebellar peduncles, upper pons, and midbrain. It may be difficult to distinguish this syndrome from decompensated hydrocephalus, particularly in a younger child. If repeat scanning shows ventricular enlargement, it may be appropriate to place a shunt.
Mutism occurs in 5% to 30% of patients undergoing surgery for large vermian tumors. It is most commonly seen after surgery for medulloblastoma but may occur after surgery for cerebellar astrocytoma as well.37 Typically, the patient speaks for a day or so after the procedure and then becomes abruptly mute but retains receptive language function. The deficit recovers with time, usually over a few weeks to 6 months. Recovery is characterized by immediate return of full words and sentences rather than just sounds, thus suggesting “inhibition of speech” rather than apraxia, dysphasia, or dysarthria. Patients are often left with a scanning, cerebellar-type speech. The substrate appears to be damage to the dentate nuclei bilaterally.38
Aseptic meningitis is another well-recognized postoperative syndrome. This complication was known to Cushing, who described headache, fever, photophobia, nuchal rigidity, and CSF pleocytosis occurring in association with blood-stained spinal fluid.1 He recommended serial spinal taps to clear the CSF. The syndrome typically appears 5 to 7 days after surgery, often as steroids are being weaned. CSF should be obtained by spinal tap or from the shunt or ventriculostomy to rule out bacterial infection, and usually antibiotics are begun pending culture results. If these prove to be negative, an increase in the corticosteroid dose for several days followed by a 2-week taper generally relieves the symptoms.
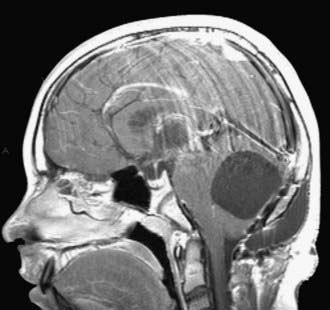
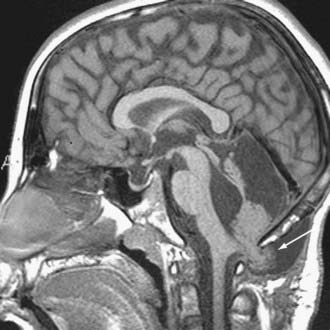
Other rare complications include stress ulcer and late spinal instability. Late development of a cervical syrinx may occur, presumably from scarring of the outlet of the fourth ventricle. The cystic cavity from the tumor resection may also wall off and exert a mass effect, which requires fenestration (Fig. 202-8). Rarely, the residual cerebellum may herniate through the dural opening and necessitate repair (Fig. 202-9).
Pathology
Several classification schemes have been proposed for the histology of these tumors, but their value is uncertain.39 They generally divide these tumors into pilocytic and fibrillary (or diffuse) cell types.
The term pilocytic was originally introduced to describe elongated, bipolar astrocytes that literally look hair-like as a result of their growth within fiber tracts. The majority of the astrocytes that make up cerebellar tumors in children, however, do not display these features. The characteristic histologic pattern of pilocytic astrocytoma consists of a biphasic growth pattern in which islands of densely packed astrocytes with coarse processes alternate with fields in which a microcystic honeycombed pattern is formed by the delicate anastomosing branches of the stellate forms. This pattern is readily seen on routine hematoxylin and eosin staining and is accentuated with glial fibrillary acidic protein (GFAP) stains (Figs. 202-10 and 202-11). A vascular “wickerwork” consisting of a collection of thin-walled vessels encased in adventitia may be seen.40 This is more characteristic of cerebellar astrocytomas than of astrocytomas in other locations. Rosenthal’s fibers are often abundant in regions of dense growth and appear to be GFAP-negative glial filaments that have undergone degradation or biochemical change (Fig. 202-12).41 Rosenthal’s fibers may be seen in either pilocytic or fibrillary astrocytomas and are not specific for astrocytomas because they may occur in the capsule of craniopharyngiomas and in Alexander’s disease. Pilocytic astrocytomas account for 65% to 80% of cerebellar astrocytomas in children.42
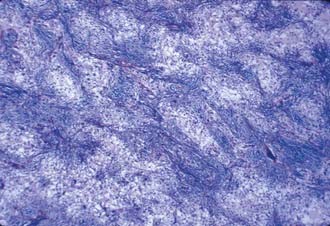
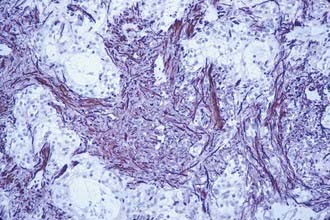
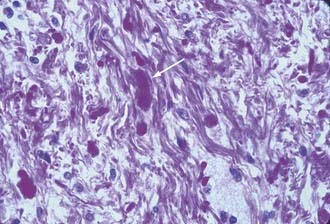
FIGURE 202-12 Cerebellar fibrillary astrocytoma containing a Rosenthal fiber in the center of the field (hematoxylin and eosin, ×400).
Fibrillary or diffuse astrocytomas are characterized by a more tightly compacted array of cells with mild to moderate degrees of morphologic atypia. They are composed of a monophasic population of astrocytes with coarser processes. They may also have an abundance of Rosenthal’s fibers (Fig. 202-12). Flow cytometry may show more aneuploid/tetraploid DNA content than in the pilocytic variant.42 This type accounts for roughly 30% of cerebellar astrocytomas.
Despite multiple attempts to correlate the prognostic significance of these histologic variants, no clear conclusion has emerged. Some reports have suggested a favorable prognosis with the pilocytic variant (79% 20-year survival rate) and a poor prognosis with the diffuse type (7% 20-year survival rate).42 Others have found no difference in outcome.39 This may reflect the fact that many tumors display features of both types, thus making classification unreliable. Series that include adult patients have also led to confusion.
Mixed glial tumors occur. They are most frequently composed of a mixture of neoplastic astrocytes and oligodendroglia, ependymal cells, and even ganglion cells. Malignant astrocytomas are relatively rare and account for only 2% of cerebellar astrocytomas. It must be noted, however, that unlike astrocytic gliomas in other locations, the presence of pleomorphism, mitoses, endothelial proliferation, and necrosis in small areas of a benign cerebellar astrocytoma is not unusual.29 This must be considered by the surgeon and neuropathologist and not be misinterpreted as malignancy on a frozen section.
Radiation Therapy
Given the excellent outcome expected after surgical excision, it is generally agreed that radiation therapy is not indicated after gross surgical resection.23 Furthermore, numerous reports have described late malignant transformation of recurrent astrocytomas that had been irradiated 10 to 28 years previously.43,44 Recurrences generally arise from partially resected lesions, and in many cases the recurrent tumor can be totally resected at reoperation.25 Radiation may improve the outcome in patients with subtotally resected lesions,45 but some of these lesions may remain static or even regress spontaneously without any therapy at all.46
In general, radiation therapy should be reserved for multiply recurrent or progressive low-grade tumors that cannot be safely resected. This usually means those with infiltration of the brainstem or cerebellar peduncle. Malignant cerebellar astrocytomas may disseminate, and craniospinal irradiation should be considered.47
Outcome
The overall risk for recurrence appears to increase steadily with time. A 20-year survival rate of 80% may be expected for the group as a whole,48 and 12 patients from Bailey’s original series were monitored for 28 to 50 years without a recurrence.49 The most robust predictor of outcome with this disease is the extent of surgical resection. Earlier series with long follow-up are difficult to interpret because of the lack of modern imaging techniques, and more recent series using MRI as a measure of resection lack long follow-up. Older studies often relied on the surgeon’s impression of the extent of resection, which is notoriously inaccurate. Even state-of-the art imaging can be confusing immediately after surgery, and postoperative changes may be indistinguishable from residual tumor.50 Total radiographic resection is achieved in approximately 57% to 80% of modern cases but is limited by involvement of the brainstem or cerebellar peduncle.22,51 With total resection, the 5-year survival rate approaches 100% if the patient survives the initial operation. Delayed death may occur as a result of shunt malfunction.33 Cerebellar astrocytomas have been reported to recur as long as 39 years after initial diagnosis and do not follow Collins’ law for congenital brain tumors.52 It is unclear how often such late recurrences occur after MRI-documented total resection.
The long-term prognosis for incompletely excised benign cerebellar astrocytomas in children remains largely unpredictable.46 In part, this may be due to different classification schemes, which may consider some cerebellar astrocytomas with brainstem involvement to be exophytic brainstem tumors rather than cerebellar tumors. Tumors may remain static for years, progress, or spontaneously regress.53 Progression of subtotally resected cerebellar astrocytomas occurs in 27% to 50% of patients.22,33,51 The fact that many of these tumors may remain static or regress makes a “wait and see” strategy justifiable in patients with typical benign histology and nonprogressive or residual tumors after primary tumor resection.
Other prognostic factors are less predictive. Histologic subtype (pilocytic or diffuse fibrillary) is probably not an independent predictor once the extent of resection is removed as a variable. Similarly, reports that tumor location in the hemisphere versus the cerebellar vermis carries a more favorable prognosis and reports that cystic tumors fare better than solid tumors do not hold up in multivariate analysis.22,33,51 Typical cerebellar astrocytomas are unusual in the adult population but do occur. They are more likely than childhood tumors to show malignant features, but if they have benign histology, they have the same favorable prognosis seen in childhood.54,55
Malignant cerebellar astrocytomas behave much like their counterparts in other brain locations and do poorly. The average survival is 14 months, and 50% will disseminate.47
Surveillance
No universally accepted guidelines exist on the need for or frequency of surveillance imaging in children with cerebellar astrocytoma. The rationale for performing surveillance MRI is that early detection of recurrence may result in timely treatment and a better outcome, but this has been difficult to prove. Children with totally resected cerebellar astrocytomas demonstrated by MRI at 3 months after surgery have such a low risk for recurrence that they do not appear to benefit from surveillance.22,56 Nonetheless, it is the practice of most neuro-oncology groups to recommend periodic imaging for 5 years in most cases. Subtotally resected lesions and those in which postoperative imaging is equivocal should probably be imaged on a periodic basis, even though there is no evidence that early detection before symptomatic recurrence results in improved outcome. A schedule of 12, 18, 42, and 66 months, followed by one later image, has been proposed.22
Treatment of Recurrence
A documented recurrence is treated by repeat surgery if possible.57 Small suspected recurrences may be carefully monitored because they may be difficult to find at surgery and may not progress. Tumors with anaplastic features, multiply recurrent tumors, and tumors that are unresectable without significant risk may be considered for radiation therapy. Although rare, leptomeningeal dissemination may occur even with low-grade lesions, usually at the time of a recurrence,19 but it may take place at any time. Diagnosis may be difficult unless it is suspected. Craniospinal irradiation and chemotherapy may be indicated (Fig. 202-13). Treatment, at times, results in clinical improvement and considerable disease control.58
Quality of Life
Anecdotally, children who undergo treatment of cerebellar astrocytomas may expect full and productive lives.49 Children who have undergone surgery for cerebellar astrocytoma often have residual clumsiness, ataxia, and dysmetria, particularly if there has been involvement of the cerebellar peduncle and these signs and symptoms predated the surgery. These complications are usually well tolerated, but the child may switch handedness if dysmetria affects writing with the dominant hand. Patients who have suffered transient cerebellar mutism appear to have significantly more ataxic dysarthric speech and slower speech than do patients who did not have this postoperative complication.59,60
It is generally accepted that the cerebellum has little role in cognitive function, but this has been called into question. Recent work has implicated cerebellar dysfunction in disorders of memory,61 verbal encoding,62 and affective behavior.63 Children who have undergone surgery for cerebellar astrocytomas generally do well, but recent work has demonstrated some mild alteration in cognitive function when compared with controls, particularly in measurements of motor speed, attention, and executive functions.64 These problems are not entirely explained by medical complications and are not correlated with location of the tumor within the cerebellum.65 Overall, the patients generally function well. The average full-scale IQ was 98.2 in a small group of my patients, and the Karnofsky Performance Index score was greater than 90 in 82% of patients in a large series.33 When formal quality-of-life assessments have been carried out, significant differences between patients and controls have been demonstrated in areas such as socializing, thus suggesting that psychosocial factors have been underappreciated in this group of children.66
Abdollahzadeh M, Hoffman H, Blazer S, et al. Benign cerebellar astrocytoma in childhood: experience at the Hospital for Sick Children, 1980-1992. Childs Nerv Syst. 1994;10:380.
Alpers C, Davis R, Wilson C. Persistence and late malignant transformation of childhood cerebellar astrocytoma. J Neurosurg. 1982;57:548.
Austin E, Alvord EJ. Recurrences of cerebellar astrocytomas: a violation of Collins’ law. J Neurosurg. 1988;68:41.
Beebe D, Ris M, Armstrong F, et al. Cognitive and adaptive outcome in low-grade pediatric cerebellar astrocytomas: evidence of diminished cognitive and adaptive functioning in National Collaborative Research Studies (CCG 9891/POG 9130). J Clin Oncol. 2005;23:5198.
Benesch M, Eder H, Sovinz P, et al. Residual or recurrent cerebellar low-grade glioma in children after tumor resection: is re-treatment needed? A single center experience from 1983-2003. Pediatr Neurosurg. 2006;42:159.
Bernhardtsen T, Laursen H, Bojsen-Moller M, et al. Sub-classification of low-grade cerebellar astrocytoma: is it clinically meaningful? Childs Nerv Syst. 2003;19:729.
Civitello L, Packer R, Rorke L, et al. Leptomeningeal dissemination of low-grade gliomas in childhood. Neurology. 1988;38:562.
de Ribaupierre S, Ryser C, Villemure J, et al. Cerebellar lesions: is there a lateralisation effect on memory deficits? Acta Neurochir (Wien). 2008;150:545.
Desai K, Nadkarni T, Muzumdar D, et al. Prognostic factors for cerebellar astrocytomas in children: a study of 102 cases. Pediatr Neurosurg. 2001;35:311.
Favre J, Deruaz J, de Tribolet N. Pilocytic cerebellar astrocytoma in adults: case report. Surg Neurol. 1993;39:360.
Geissinger J. Astrocytomas of the cerebellum in children. Arch Neurol. 1971;24:125.
Gjerris F, Agerlin N, Borgesen S, et al. Epidemiology and prognosis in children treated for intracranial tumours in Denmark 1960-1984. Childs Nerv Syst. 1998;14:302.
Hassounah M, Siqueira E, Haider A, et al. Cerebellar astrocytoma: report of 13 cases aged over 20 years and review of the literature. Br J Neurosurg. 1996;10:365.
Kusano Y, Tanaka Y, Takasuna H, et al. Transient cerebellar mutism caused by bilateral damage to the dentate nuclei after the second posterior fossa surgery. Case report. J Neurosurg. 2006;104:329.
Morelli D, Pirotte B, Lubansu A, et al. Persistent hydrocephalus after early surgical management of posterior fossa tumors in children: is routine preoperative endoscopic third ventriculostomy justified? J Neurosurg. 2005;103:247.
Palma L, Celli P, Mariottini A. Long-term follow-up of childhood cerebellar astrocytomas after incomplete resection with particular reference to arrested growth or spontaneous tumor regression. Acta Neurochir (Wien). 2004;146:581.
Pencalet P, Maixner W, Sainte-Rose C, et al. Benign cerebellar astrocytomas in children. J Neurosurg. 1999;90:265.
Piat JJ, Kellogg J. A hazard of combining the infratentorial supracerebellar and the cerebellomedullary fissure approaches: cerebellar venous insufficiency. Pediatr Neurosurg. 2000;33:243.
Stein B, Tenner M, Fraser R. Hydrocephalus following removal of cerebellar astrocytomas in children. J Neurosurg. 1972;36:763.
Steinbok P, Poskitt K, Hendson G. Spontaneous regression of cerebellar astrocytoma after subtotal resection. Childs Nerv Syst. 2006;22:572.
Wang Z, Sutton L, Cnaan A, et al. Proton MR spectroscopy of pediatric cerebellar tumors. AJNR Am J Neuroradiol. 1995;16:1821.
1 Cushing H. Experiences with the cerebellar astrocytoma. A critical review of seventy-six cases. Surg Gynecol Obstet. 1931;52:129.
2 Bergstrand H. Ueber das Sog. Astrocytom im Kleinhirn. Virchows Arch Pathol Anat. 1932;287:2.
3 Bucy P, Gustafson W. Structure, nature and classification of the cerebellar astrocytomas. Am J Cancer. 1939;35:227.
4 Russell D, Rubinstein L. Pathology of Tumors of the Nervous System, 4th ed. London: Edward Arnold; 1977.
5 Gol A, McKissock W. The cerebellar astrocytomas. A report on 98 verified cases. J Neurosurg. 1959;16:287.
6 Dunn I, Agarwalla P, Papanastassiou A, et al. Multiple pilocytic astrocytomas of the cerebellum in a 17-year-old patient with neurofibromatosis type I. Childs Nerv Syst. 2007;23:1191.
7 von Deimling A, Louis D, Menon A, et al. Deletions on the long arm of chromosome 17 in pilocytic astrocytoma. Acta Neuropathol. 1993;86:81.
8 Sievert A, EM J, Gai X, et al. Duplication of 7q34 in pediatric low-grade astrocytomas detected by high-density single-nucleotide polymorphism–based genotype arrays results in a novel BRAF fusion gene. Brain Pathol. 2009;19:449.
9 Ilgren E, Stiller C. Cerebellar astrocytomas. J Neurooncol. 1987;4:293.
10 Geissinger J. Astrocytomas of the cerebellum in children. Arch Neurol. 1971;24:125.
11 Pencalet P, Maixner W, Sainte-Rose C, et al. Benign cerebellar astrocytomas in children. J Neurosurg. 1999;90:265.
12 Desai K, Nadkarni T, Muzumdar D, et al. Prognostic factors for cerebellar astrocytomas in children:a study of 102 cases. Pediatr Neurosurg. 2001;35:311.
13 Abdollahzadeh M, Hoffman H, Blazer S, et al. Benign cerebellar astrocytoma in childhood: experience at the Hospital for Sick Children, 1980-1992. Childs Nerv Syst. 1994;10:380.
14 Beni-Adani L, Gomori M, Spektor S, et al. Cyst wall enhancement in pilocytic astrocytoma: neoplastic or reactive phenomena. Pediatr Neurosurg. 2000;32:234.
15 Rumboldt A, Camacho D, Lake D, et al. Apparent diffusion coefficients for differentiation of cerebellar tumors in children. AJNR Am J Neuroradiol. 2006;6:1362.
16 Arai K, Sato N, Aoki J, et al. MR signal of the solid portion of pilocytic astrocytoma on T2-weighted images: is it useful for differentiation from medulloblastoma? Neuroradiology. 2006;48:233.
17 Wang Z, Sutton L, Cnaan A, et al. Proton MR spectroscopy of pediatric cerebellar tumors. AJNR Am J Neuroradiol. 1995;16:1821.
18 Thomas B, Krishnamoorthy T, Radhakrishnan V, et al. Advanced MR imaging in Lhermitte-Duclos disease: moving closer to pathology and pathophysiology. Neuroradiology. 2007;49:733.
19 Zorlu F, Selek U, Akyuz C, et al. Spinal seeding of a pilocytic astrocytoma following multiple subtotal resections. Pediatr Neurosurg. 2005;41:248.
20 Shuto T, Ohtsubo Y, Sekido K, et al. Rapidly growing calcified cerebellar astrocytoma in infants. Childs Nerv Syst. 1996;12:107.
21 Arle J, Morriss C, Wang Z, et al. Prediction of posterior fossa tumor type in children by means of magnetic resonance image properties, spectroscopy, and neural networks. J Neurosurg. 1997;86:755.
22 Sutton L, Cnaan A, Klatt L, et al. Post-operative surveillance imaging in children with cerebellar astrocytomas. J Neurosurg. 1996;84:721.
23 Lapras C, Palet J, Lapras CJ, et al. Cerebellar astrocytomas in childhood. Childs Nerv Syst. 1986;2:55.
24 Kessler l, Dugan P, Concanon J. Systemic metastases of medulloblastoma promoted by shunting. Surg Neurol. 1975;3:147.
25 Albright L, Reigel D. Management of hydrocephalus secondary to posterior fossa tumors. J Neurosurg. 1977;46:52.
26 Morelli D, Pirotte B, Lubansu A, et al. Persistent hydrocephalus after early surgical management of posterior fossa tumors in children: is routine preoperative endoscopic third ventriculostomy justified? J Neurosurg. 2005;103:247.
27 Sainte-Rose C, Cinalli G, Roux F, et al. Management of hydrocephalus in pediatric patients with posterior fossa tumors: the role of endoscopic third ventriculostomy. J Neurosurg. 2001;95:791.
28 Stein B, Tenner M, Fraser R. Hydrocephalus following removal of cerebellar astrocytomas in children. J Neurosurg. 1972;36:763.
29 Ilgren E, Stiller C. Cerebellar astrocytomas. Part II. Pathologic features indicative of malignancy. Clin Neuropathol. 1987;6:201.
30 McLaurin R. On the use of precraniotomy shunting in the management of posterior fossa tumors in children. A cooperative study. In: Chapman P, editor. Concepts in Pediatric Neurosurgery, Vol 6. Basel: S Karger; 1985:1.
31 Kellogg J, Piat JJ. Resection of fourth ventricle tumors without splitting the vermis: the cerebellomedullary fissure approach. Pediatr Neurosurg. 1997;27:28.
32 Piat JJ, Kellogg J. A hazard of combining the infratentorial supracerebellar and the cerebellomedullary fissure approaches: cerebellar venous insufficiency. Pediatr Neurosurg. 2000;33:243.
33 Due-Tonnessen B, Helseth E, Scheie D, et al. Long-term outcome after resection of benign cerebellar astrocytomas in children and young adults (0-19 years): report of 110 consecutive cases. Pediatr Neurosurg. 2002;37:71.
34 Shenkin H, Goldfedder P. Air embolism from exposure of the posterior cranial fossa in prone position. JAMA. 1969;210:726.
35 Gottschalk A, Yaster M. Pain management after craniotomy: Part I. Contemp Neurosurg. 2008;30:1.
36 Wisoff J, Epstein F. Pseudobulbar palsy after posterior fossa operation in children. Neurosurgery. 1984;15:707.
37 Humphreys R. Mutism after posterior fossa tumor surgery. Concepts Pediatr Neurosurg. 1988;9:57.
38 Kusano Y, Tanaka Y, Takasuna H, et al. Transient cerebellar mutism caused by bilateral damage to the dentate nuclei after the second posterior fossa surgery. Case report. J Neurosurg. 2006;104:329.
39 Bernhardtsen T, Laursen H, Bojsen-Moller M, et al. Sub-classification of low-grade cerebellar astrocytoma: is it clinically meaningful? Childs Nerv Syst. 2003;19:729.
40 Sato K, Rorke L. Vascular bundles and wickerworks in childhood brain tumors. Pediatr Neurosci. 1989;15:105.
41 Janzer R, Friede R. Do Rosenthal fibers contain glial fibrillary acid protein? Acta Neuropathol. 1981;55:75.
42 Hayostek C, Shaw E, Scheithauer B, et al. Astrocytomas of the cerebellum. A comparative clinicopathologic study of pilocytic and diffuse astrocytomas. Cancer. 1993;72:856.
43 Alpers C, Davis R, Wilson C. Persistence and late malignant transformation of childhood cerebellar astrocytoma. J Neurosurg. 1982;57:548.
44 Budka H. Partially resected and irradiated cerebellar astrocytoma of childhood: Malignant evolution after 28 years. Acta Neurochir (Wien). 1975;32:139.
45 Akyol F, Atahan I, Zorlu F, et al. Results of post-operative or exclusive radiotherapy in grade I and grade II cerebellar astrocytoma patients. Radiother Oncol. 1992;23:245.
46 Palma L, Celli P, Mariottini A. Long-term follow-up of childhood cerebellar astrocytomas after incomplete resection with particular reference to arrested growth or spontaneous tumor regression. Acta Neurochir (Wein). 2004;146:581.
47 Bristot R, Santoro A, Raco A, et al. Malignant cerebellar astrocytomas: clinico-pathological remarks on 10 cases. J Neurosurg Sci. 1999;43:271.
48 Gjerris F, Agerlin N, Borgesen S, et al. Epidemiology and prognosis in children treated for intracranial tumours in Denmark 1960-1984. Childs Nerv Syst. 1998;14:302.
49 Bucy P, Thieman PW. Astrocytomas of the cerebellum. A study of a series of patients operated upon over 28 years ago. Arch Neurol. 1968;18:14.
50 Rollins N, Nisen P, Shapiro K. The use of early postoperative MR in detecting residual juvenile cerebellar pilocytic astrocytoma. AJNR Am J Neuroradiol. 1998;19:151.
51 Benesch M, Eder H, Sovinz P, et al. Residual or recurrent cerebellar low-grade glioma in children after tumor resection: is re-treatment needed? A single center experience from 1983-2003. Pediatr Neurosurg. 2006;42:159.
52 Austin E, Alvord EJ. Recurrences of cerebellar astrocytomas: a violation of Collins’ law. J Neurosurg. 1988;68:41.
53 Steinbok P, Poskitt K, Hendson G. Spontaneous regression of cerebellar astrocytoma after subtotal resection. Childs Nerv Syst. 2006;22:572.
54 Favre J, Deruaz J, de Tribolet N. Pilocytic cerebellar astrocytoma in adults: case report. Surg Neurol. 1993;39:360.
55 Hassounah M, Siqueira E, Haider A, et al. Cerebellar astrocytoma: report of 13 cases aged over 20 years and review of the literature. Br J Neurosurg. 1996;10:365.
56 Steinbok P, Hentschel S, Cochrane D, et al. Value of postoperative surveillance imaging in the management of children with some common brain tumors. J Neurosurg. 1996;84:726.
57 Lapras C, Patet J, Mottolese C, et al. Cerebellar astrocytomas in children. Prog Exp Tumor Res. 1987;30:128.
58 Civitello L, Packer R, Rorke L, et al. Leptomeningeal dissemination of low-grade gliomas in childhood. Neurology. 1988;38:562.
59 Huber J, Bradley K, Spiegler B, et al. Long-term effects of transient cerebellar mutism after cerebellar astrocytoma or medulloblastoma tumor resection in childhood. Childs Nerv Syst. 2006;22:132.
60 Huber J, Bradley K, Spiegler B, et al. Long-term neuromotor speech deficits in survivors of childhood posterior fossa tumors: effects of tumor type, radiation, age at diagnosis and survival years. J Child Neurol. 2007;22:848.
61 de Ribaupierre S, Ryser C, Villemure J, et al. Cerebellar lesions: is there a lateralisation effect on memory deficits? Acta Neurochir (Wien). 2008;150:545.
62 Ackermann H. Cerebellar contributions to speech production and speech perception: psycholinguistic and neurobiological perspectives. Trends Neurosci. 2008;31:265.
63 Mariën P, Baillieux H, De Smet H, et al. Cognitive, linguistic and affective disturbances following a right superior cerebellar artery infarction: a case study. Cortex. 2009;45:527.
64 Ronning C, Sundet K, Due-Tonnessen B, et al. Persistent cognitive dysfunction secondary to cerebellar injury in patients treated for posterior fossa tumors in childhood. Pediatr Neurosurg. 2005;41:15.
65 Beebe D, Ris M, Armstrong F, et al. Cognitive and adaptive outcome in low-grade pediatric cerebellar astrocytomas: evidence of diminished cognitive and adaptive functioning in National Collaborative Research Studies (CCG 9891/POG 9130). J Clin Oncol. 2005;23:5198.
66 Pompili A, Caperle M, Pace A, et al. Quality-of-life assessment in patients who had been surgically treated for cerebellar pilocytic astrocytoma in childhood. J Neurosurg. 2002;96:229.