[level-membership-for-hematology-oncology-and-palliative-medicine-category]
Chapter 65 Central Nervous System Tumors in Children
This is a pivotal time for RT in the treatment of children with brain tumors. There are fundamental questions that need to be addressed, including the indications for RT in the setting of clinical and molecular risk stratification for children with medulloblastoma and potentially ependymoma and the refinement of total dose and treatment volumes regimens for specific patient groups, including very young children and those with germ cell tumors. Improved knowledge of treatment-related side effects and a wider availability of proton therapy will likely impact how RT is administered and perceptions about the safety of RT in these patients. Despite the increasing availability of advanced treatment technology, children should be treated on clinical trials designed to improve tumor control, objectively study CNS effects, and build a consensus for treatment guidelines. The impact of pediatric CNS tumors on public health is small—there are fewer than 4000 cases diagnosed annually in the United States. CNS tumors represent 20% of all childhood malignancies.1 Most children with brain tumors will require RT during the course of their management. Based on the U.S. standard population data from 2000, which includes 78.8 million individuals aged birth to 19 years, annually there were 2742 cases of neuroepithelial tumors that included 654 cases of juvenile pilocytic astrocytoma, 118 cases of glioblastoma multiforme, 236 cases of ependymoma, and 433 cases of CNS embryonal tumors including medulloblastoma. Nonneuroepithelial tumors of interest to radiation oncologists included 197 cases of CNS germ cell tumors and 118 cases of craniopharyngiomas. These tumors require special care and remain a leading cause of cancer death in children.
Embryonal Central Nervous System Tumors
Embryonal tumors of the CNS are the most common malignant brain tumors in children. They are characterized by their infiltrative nature, aggressive patterns of growth, propensity for dissemination, and common need for intensive combined modality therapy. These tumors are found throughout the CNS and are further classified according to histologic and molecular features into two groups: primitive neuroectodermal tumors (PNET) and atypical teratoid rhabdoid tumors (ATRT). The PNET group includes two distinct entities—medulloblastoma (Fig. 65-1) and pineoblastoma (Fig. 65-2)—named for their site of origin in the posterior fossa and pineal region, respectively. ATRT are rare aggressive embryonal tumors that most commonly arise in young children in the brain and at peripheral soft tissue sites. With some exceptions, the diagnosis of ATRT is made on molecular grounds by demonstrating the deletion or mutation of the SMARCB1 gene found on chromosome 22q11.2.2 Atypical teratoid rhabdoid tumors unknowingly were considered the same as other PNET but are now known to carry a worse prognosis.
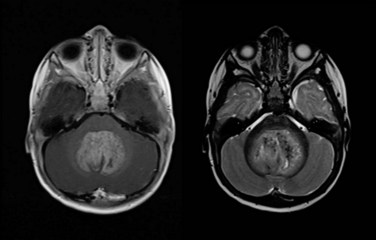
Figure 65-1 Medulloblastoma as shown on axial postgadolinium T1-weighted (left) and T2-weighted (right) MR images.
Medulloblastoma and supratentorial PNET are commonly diagnosed in young children. The relatively large numbers of patients and a cooperative spirit among caregivers have led to considerable understanding and progress in the treatment of these tumors. Patients are risk classified for treatment based on the three most important prognostic factors for tumor control— extent of disease, extent of resection, and histological subtype—and the most important prognostic factor for functional outcome—age at the time of irradiation. The radiation oncologist should be aware of the aggressive histologic variant of medulloblastoma (large cell or anaplastic medulloblastoma) that is more prone to dissemination through the cerebrospinal fluid (CSF).3,4 One also should be aware that there is a less aggressive variant (desmoplastic medulloblastoma) that may not require irradiation under certain conditions.
Medulloblastoma is a heterogeneous tumor and is now recognized as a number of different diseases based on specific molecular markers that have been retrospectively evaluated. Depending on the series, investigators have identified from four to six different subtypes based on the combination of molecular and classic histopathologic evaluation. A report by Northcott and associates5 described four distinct molecular variants of medulloblastoma, including WNT, SHH, and two non-WNT/SHH. The former two have clinical features that are associated with less aggressive phenotypes and the latter two, described as groups C and D in their report, have molecular and clinical features associated with more aggressive phenotypes, including overexpression of MYC and disseminated disease (group C) and the most common cancer isochromosome i17q and children between the ages of 4 and 15 years. One of the more striking findings by this group was the variation in molecular characteristics by age. Many of these findings have been confirmed by others groups,6 which suggests that tumor biologic information will be combined with clinical prognostic data to stratify patients in future trials.
At present, the treatment of embryonal tumors of the CNS is remarkably similar regardless of subtype or tumor location and consists of surgery, postoperative RT, and chemotherapy. The requirement of craniospinal irradiation, regardless of extent of disease, is the element that differentiates the treatment of these tumors. Approximately one third of patients present with neuraxis dissemination at the time of diagnosis as determined by magnetic resonance imaging (MRI) of the brain and spine (Fig. 65-3) or CSF cytology.7 Because the negative predictive value of these staging procedures remains relatively low, and the most common mode of failure remains distant, craniospinal irradiation is a mainstay in the treatment of these tumors with few exceptions.8
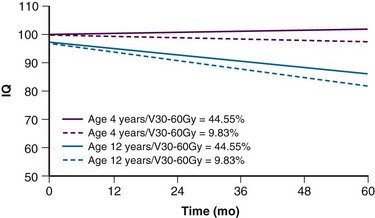
The side effects attributed to RT have been a primary concern for patients with medulloblastoma. The concern has been greatest for patients with average-risk disease, for whom long-term disease control is likely and the side effects of therapy have a lasting impact. The treatment of this tumor is technically demanding and consists of craniospinal irradiation and boost treatment of the anatomic posterior fossa. Until recently, the standard of care for an average-risk patient was postoperative craniospinal irradiation (CSI) to 36 Gy and boost treatment to 54 Gy using conventional fractionation of 1.8 Gy/day. In a cooperative group trial the CSI dose was lowered to 23.4 Gy for average-risk patients in a randomized comparison with 36 Gy CSI. This trial showed an increased risk of relapse in patients receiving the reduced CSI dose.9 Multiple-agent chemotherapy combined with reduced-dose CSI was found to be equivalent to standard-dose CSI and was subsequently adopted as a treatment standard for average-risk patients.10 Cognitive impairment continues to be a significant problem, with intelligence quotient (IQ) loss most commonly seen in children younger than the age of 8 years and those treated for high-risk medulloblastoma in which high-dose craniospinal irradiation (36 Gy) is required.11
The most recently completed Children’s Oncology Group study sought to define the best chemotherapy regimen (lomustine [CCNU], cisplatin, and vincristine vs. cyclophosphamide, cisplatin, and vincristine) to accompany low-dose craniospinal irradiation and the standard posterior fossa boost. No differences were observed in disease control (3-year event-free survival >83% in both arms); however, patients treated with the cyclophosphamide-containing regimen appeared to have more severe toxicity (18% vs. 25%).12 Attention has now turned to combining the two chemotherapy regimens and randomizing patients between 18.0 Gy and 23.4 Gy CSI followed by a second randomization between conventional boost treatment and treatment of less than the entire posterior fossa in an ambitious four-arm randomized trial developed for pediatric patients between the ages of 3 and 8 years. For those older than 8 years, the CSI dose remains at 23.4 Gy. The trial also includes a randomized comparison of conventional posterior fossa boost and boost treatment of the tumor bed using a 1.5-cm clinical target volume (CTV) margin.
Treatment of less than the entire posterior fossa after 23.4 Gy of CSI using focal radiation delivery techniques appears to be as effective as treatment of the entire posterior fossa based on recent trial results from St. Jude Children’s Research Hospital and participating centers involving 84 patients. In this prospective trial the cumulative incidence of posterior fossa failure was only 6.3% at 3 years.13 Treatment of these patients included postoperative CSI to 23.4 Gy, conformal posterior fossa boost to 36 Gy, and focal treatment of the tumor bed with a 2-cm margin. Chemotherapy was administered after RT. Patients on this study experienced a dose reduction to the anatomic posterior fossa of approximately 13%. The risks associated with reducing the CSI dose and limiting the boost treatment to the postoperative tumor bed with a limited margin must be balanced against the observed effects of RT on cognition, neurologic function, and growth and development.
Hyperfractionation has been tested in Europe to reduce toxicity.14 Although these investigations have shown that hyperfractionation may be isoeffective, lacking are the data that indicate side effects are reduced by this strategy. Considering logistics including the need for anesthesia in this often young patient population, investigators demand some certainty that there would be a benefit associated with hyperfractionated treatment before planning future trials.
Current cooperative group trials for very young children (<3 years of age) with medulloblastoma highlight the importance of RT in the treatment of these tumors. The recently completed Children’s Oncology Group protocol for children younger than the age of 3 years with localized medulloblastoma included 20 weeks of combination chemotherapy followed by second surgery when indicated and sequential irradiation of the posterior fossa and primary site before 20 additional weeks of maintenance chemotherapy.15 CSI was been omitted for these patients, and the doses to the posterior fossa (18 and 23.4 Gy) and primary site (45 to 54 Gy) were prescribed based on patient age, response, and risk status. A similar strategy was used in the Pediatric Brain Tumor Consortium (PBTC) protocol for similar patients with one exception: intrathecal chemotherapy (mafosfamide) was given during the first 20 weeks of chemotherapy as a substitute for CSI.16 Both protocols included RT after 20 weeks based on results from prior trials that showed a median time to progression of approximately 24 weeks.17,18 The targeting guidelines used in these studies were largely empirical: investigators attempted to minimize the dose to normal tissues while maintaining an acceptable rate of local tumor control.
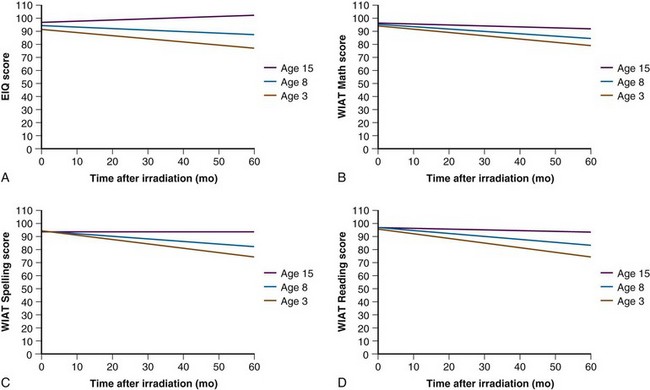
Fear of cognitive deficits has been the driving force in the design of current treatment trials for this young patient population. Age has the greatest impact on cognitive outcome after irradiation (Fig. 65-4). For very young children with medulloblastoma, a persistent decline in cognitive function has been observed by Walter and colleagues.19 The decline is not surprising given the median cranial dose of 35.2 Gy for patients with a median age of 2.6 years and the additive effects of tumor, surgery, and chemotherapy. Because a significant decline has also been observed for older children with medulloblastoma treated with lower craniospinal doses,11 CSI for the youngest children no longer appears to be a reasonable treatment option. Data concerning the effectiveness of yet lower CSI doses are extremely limited.20 These findings form the basis of the most recent trials for very young children. CSI has been omitted with the hope that the side effects of treatment will be reduced enough to provide for a meaningful quality of life among long-term survivors. There is a move to increase the age at which CSI is considered acceptable from age 3 years to 5 years at the time of treatment. Investigators acknowledge that they are trading fewer side effects for an increase in the rate of disseminated failure.
Ependymoma
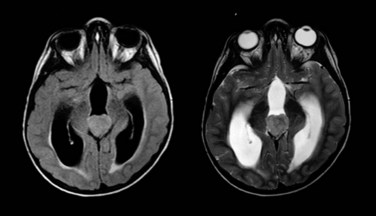
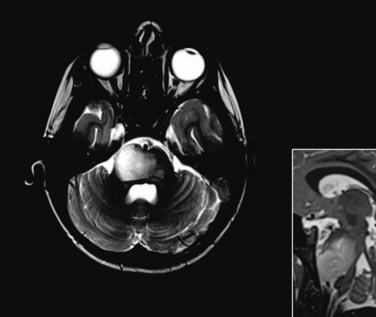
Ependymoma is the third most common CNS tumor in children, affecting approximately 236 individuals younger than the age of 19 years annually in the United States.1 It shares many of the clinical characteristics of more common gliomas, embryonal tumors, and less common germ cell tumors. Ependymoma may occur anywhere within the CNS and has the highest prevalence within the posterior fossa, arising from the floor or roof of the fourth ventricle or cerebellopontine angle, where it is known to invade the brainstem or envelop cranial nerves or vascular structures. Ependymoma also arises within the parenchyma of the cerebral hemispheres in older patients and rarely in the spinal cord. The interval from the onset of symptoms to diagnosis may be influenced by the young age at the time of diagnosis, the perceived slow growth rate of this tumor, and the remitting signs and symptoms of increased intracranial pressure resulting from obstructive hydrocephalus (Fig. 65-5).
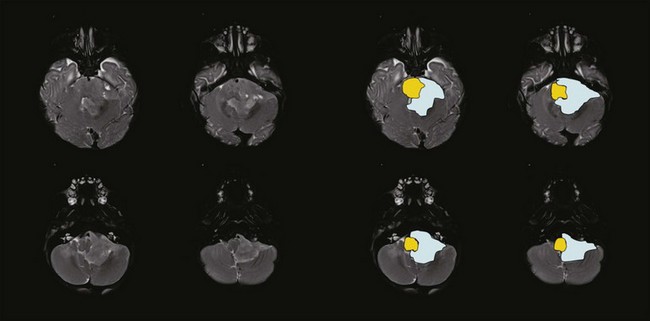
Ependymoma is most commonly diagnosed in children who are younger than 4 years of age. For the past two decades, progress in the treatment of this tumor has been slowed by concerns about late effects. Recent advances in neuroimaging, surgery, and RT have moved the field of neuro-oncology forward in the treatment of this tumor, resulting in more acceptable outcomes and providing new disease control benchmarks. Investigators treating these patients with contemporary surgery and RT have been able to increase event-free survivals measured at 3 years from 55% to 62% to 75% when measured at 7 years with remarkably limited treatment-related effects.21,22
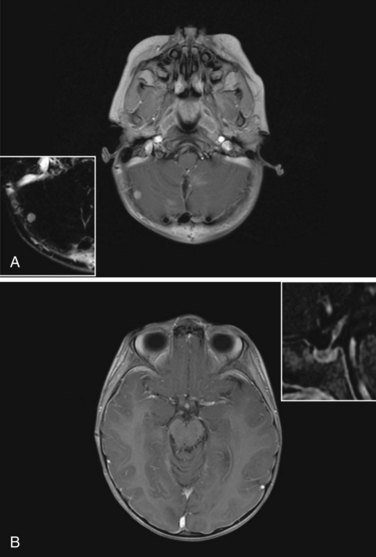
The standard of care for a child with localized ependymoma is to attempt gross-total resection, perform second surgery for potentially resectable residual tumor, and proceed with postoperative RT directed at the primary site using a 10-mm, anatomically confined CTV surrounding the residual tumor and/or tumor bed as defined on postoperative neuroimaging. The recommended total dose is 59.4 Gy using conventional fractionation because these tumors are prone to recur locally. Conventions regarding organs at risk apply; thus, for patients with tumors arising in the lower aspect of the posterior fossa, avoidance of the spinal cord and/or chiasm should be considered after approximately 54 Gy. Special considerations for patients with posterior fossa tumors may apply to very young children (generally those younger than the age of 18 months) with no evidence of residual tumor or patients who are noted to have signs and symptoms of severe brainstem injury after resection (postoperative seizure, hypertension, and ischemic changes on neuroimaging) who are at increased risk for necrosis. The recent trial of the Children’s Oncology Group showed that there was consensus to irradiate children younger than the age of 3 years and as young as 12 months postoperatively. The newest study from the Children’s Oncology Group, known as ACNS0831 and initiated in 2010, deploys a similar treatment algorithm and methods of irradiation with a smaller (5 mm) CTV margin. Children are randomized to receive four cycles of postirradiation chemotherapy (Fig. 65-6).
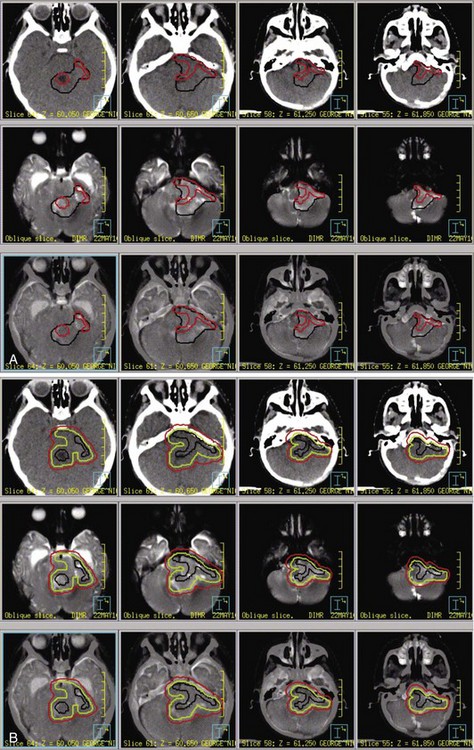
The prognosis for a patient with ependymoma is determined by tumor grade and extent of resection.23,24,25 Patients with overtly anaplastic tumors have inferior disease control when compared with those with differentiated tumors; however, the assignment of tumor grade can be difficult for those cases in which focal anaplasia occurs in the setting of a largely differentiated tumor. All patients should be treated with curative intent regardless of tumor grade. Extent of resection is an unequivocal prognostic factor: patients with substantial residual tumor have the worst prognosis. Because patients with varying amounts of residual tumor have achieved long-term survival after RT and modern surgical resection has altered the definition of near-total and subtotal resection, the volume of residual tumor differentiating between patients with good and poor prognosis remains unknown. It is incumbent on the radiation oncologist to question whether additional resection might be safely achieved in a patient with residual tumor referred for irradiation.
Research performed to evaluate molecular prognostic markers in ependymoma has revealed gain of 1q and homozygous deletion of CDKN2A as valuable predictors of poor survival in this disease26 and other markers associated with more favorable outcomes. Although molecular markers are not likely to supplant standard histopathologic evaluation, they will certainly contribute beneficial information for separating tumors that appear to have features of both differentiated and anaplastic ependymoma and determining which patients might require additional treatment after gross-total resection and irradiation.
High-Grade Astrocytoma
One study has shown a benefit for chemotherapy.27 This study included a randomization of 58 patients with high-grade astrocytoma to local irradiation versus local irradiation plus weekly vincristine followed by 12 month of carmustine, vincristine, and prednisone. The reported 5-year progression-free survival rate was 18% versus 46% favoring the use of chemotherapy, and patients with glioblastoma multiforme (n = 40) had a 5-year progression-free survival of 6% versus 42% favoring the use of chemotherapy. A larger study that included 172 patients randomized between local irradiation versus local irradiation plus preirradiation and postirradiation 8-in-1 chemotherapy (i.e., with vincristine, carmustine, procarbazine, cytarabine, hydroxyurea, cisplatin, dacarbazine, and methylprednisolone) showed no difference between the two study arms with 5-year progression-free survivals of 26% and 33%, respectively.28 This study was illustrative for the lack of concordance between institutional and expert pathology review. Thirty percent of patients in this study were found not to have high-grade glioma, and the survival estimates were in fact lower than originally reported.29
Low-Grade Astrocytoma
Low-grade astrocytomas are found throughout the nervous system in children yet are clustered predominantly in subsites of the cerebellum (cerebellum, cerebellar peduncles, dorsally exophytic brainstem glioma) or diencephalon, including the optic chiasm, optic pathways, hypothalamus, and thalami (Fig. 65-7). Slow-growing tumors located in these critical links of the nervous and endocrine systems result in presenting signs and symptoms that differ markedly from the other types of pediatric tumors that are prone to obstruction of CSF pathways. Loss of vision, dysdiadokinesia, dysmetria, ataxia, tremor, the spectrum of endocrinopathies including precocious puberty, and cognitive dysfunction are a few of the insidious signs and symptoms that are often overlooked in the developing child and lead to well-established deficits.
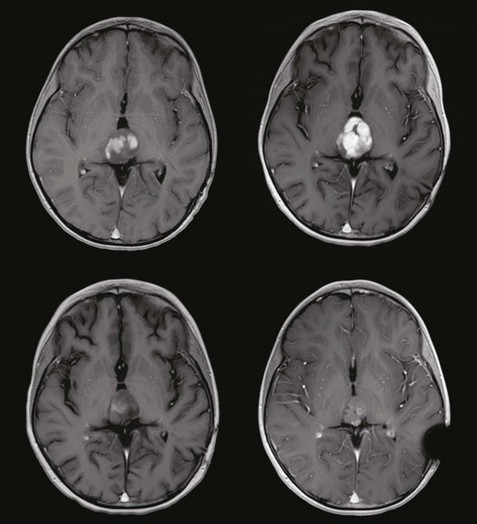
Few inroads have been made in RT for pediatric low-grade astrocytoma in recent years, mainly because most who would be candidates for RT are younger children with optic pathway tumors. Indeed, advances in surgery have resulted in successful primary resection of most cerebellar and peripheral cerebral lesions, with long-term disease control exceeding 90% when measured at 5 years. In the setting of residual tumor after initial resection and in the absence of persistent symptoms, most children should follow a course of observation, with second surgery at the time of progression contingent on the associated risks. In a landmark study conducted from 1991 to 1996 and enrolling 726 patients with the intention of observation after surgery, the progression-free survival at 5 years was 92% for patients after gross-total resection, 53% after near-total resection, and 61% after subtotal resection. More than 75% of patients had juvenile pilocytic astrocytoma, and progression-free survival was lowest in midline (59%) and hypothalamic-chiasmatic tumors (50%) compared with tumors of the cerebellar hemisphere (89%), cerebellar vermis (84%), or cerebral hemisphere (89%).30 Without information regarding the ability of follow-up surgery to render these patients disease free or asymptomatic, a large proportion of patients remain candidates for adjuvant therapy, including external beam RT.
In some centers, chemotherapy is recommended regardless of patient age, and in these settings the patient is seldom seen by the radiation oncologist. Chemotherapy has a track record of success in delaying RT for patients with pediatric low-grade astrocytoma but cannot be considered curative because the majority of patients will eventually require RT.31,32 The results from a landmark study comparing two chemotherapy regimens for pediatric patients with low-grade glioma were surprising when they demonstrated that the progression-free survival rates were less than 50% even when stratified by age.33 The progression-free survival rate estimated at 5 years was 35% for those treated with carboplatin and vincristine versus 48% for those treated with thioguanine, procarbazine, lomustine, and vincristine.
These results pale in comparison to RT series as outlined by Merchant and colleagues,34 in which, in addition to profiling and comparing the results from different chemotherapy and RT series, they reported the results from their prospective series of 78 patients and 10-year event-free (74.3% ± 15.4%) and overall (95.9% ± 5.8%) survival estimates. The cumulative incidence of local failure at 10 years was only 16.4% ± 5.4%.
Indeed, fractionated RT remains the standard of care and CTV margins have been reduced from 10 to 5 mm in the current study of the Children’s Oncology Group. Although single-fraction irradiation (radiosurgery) has been used to treat well-defined areas of residual disease, experience is limited and, if the lesion is resectable, surgery is the preferred approach. Most children with low-grade astrocytoma have WHO grade I tumors, and conventionally fractionated RT to 54 Gy is recommended (see Fig. 65-7). Patients with disseminated low-grade astrocytoma should be approached with curative intent using craniospinal irradiation. Although dissemination is rare at presentation, the natural history of hypothalamic low-grade astrocytoma suggests a pattern of failure that is distant. Spinal imaging might be considered in the evaluation of these patients, especially those with hypothalamic tumors that have a relatively higher propensity to disseminate.
In the future, research will focus on molecular risk factors for disease progression after chemotherapy and it will be important to monitor outcomes after RT based on the same data when corrected for disease progression on chemotherapy and before RT. Research should be directed to help determine which patients are candidates for RT as a frontline approach and improve our understanding of the complications of this modality. A recent study has clarified the incidence and time to onset of clinically significant late effects such as cognitive effects (Fig. 65-8), hearing loss, or endocrinopathy, which appear to be low or predictable.35 In a companion article,34 the risk of vasculopathy was reported to be 4.8% ± 2.7% at 6 years and was higher for those younger than 5 years of age. Despite the number of irradiated children with optic pathway glioma, solid data regarding visual outcomes after irradiation are lacking in the modern era. Awdeh and colleagues36 showed that patients treated with chemotherapy had significantly worse visual acuity at the start of irradiation compared with those who did not receive chemotherapy and that acuity declined significantly in those who received chemotherapy compared with those who did not. Their data suggested a role for decompressive surgery. Those who underwent a single surgical procedure before irradiation had significantly better visual acuity at the start of RT; however, there was no difference in change over time based on surgery.
Brainstem Glioma
Brainstem glioma refers to a diffuse intrinsic tumor most often involving the pons. This tumor has a characteristic appearance on MRI and does not require biopsy. Its outcome is uniformly fatal despite excellent responses to RT and corticosteroids. After years of attempting to escalate dose, alter fractionation, and sensitize this tumor to the effects of radiation, investigators have now turned to more targeted therapies in conjunction with conventionally fractionated RT to 54.0 to 55.8 Gy (Fig. 65-9).
Germ Cell Tumors
CNS germ cell tumors are rare among brain tumors affecting adults and children. Based on data from the Central Brain Tumor Registry of the United States, fewer than 197 are expected to be diagnosed annually in the United States. These tumors affect males more than females.1
The importance of staging cannot be overemphasized in the management of these patients. Poor staging affects classification for outcomes analysis, response evaluation, radiation dose and volume prescriptions, and the cure of these patients. High-quality spinal MRI is required as well as adequate CSF sampling. The negative predictive value of a single lumbar CSF cytologic analysis remains unclear and should be considered an important secondary objective in any major study. Serum and CSF α-fetoprotein and β-human chorionic gonadotropin studies are important for diagnostic and staging purposes, including response evaluation and making decisions regarding the use of surgery and RT; however, each institution and cooperative group has their own criteria for cutoff values and risk classification. Standardization in the use of serum and CSF markers is required as well as in the assays used for their measurement. In addition, markers such as carcinoembryonic antigen, placental alkaline phosphatase, lactate dehydrogenase, and the soluble form of c-KIT, a transmembrane tyrosine kinase receptor, should be investigated.37
Based on the curability of extra-CNS germ cell tumors with multiple-agent chemotherapy it was logical for investigators to consider chemotherapy alone as a treatment option, and this option was pursued for a number of years because of concerns about the effects of RT. There have been two approaches with chemotherapy: chemotherapy alone and chemotherapy as a component of a combined modality approach involving the use of reduced dose and volume irradiation. There have been two successive trials attempting to treat tumors with chemotherapy alone.38,39 These trials differed in the types of agents used as well as their intensity and sequencing. My impression from the first trial is that the event-free survival was short, the progression rate was high, and that there were a noticeable number of toxic deaths. My impression from the second trial was that outcomes were similarly poor and toxicity remained high. The key finding from these trials is that there is a subset of patients who may be effectively treated without irradiation and that salvage with RT, at least for the patients with germinoma, may be feasible. It would be important to find a way to determine which patients might be candidates to avoid RT and to consider this treatment approach only for the youngest patients. Toxic deaths using chemotherapy alone have made a bad impression; however, it has been acknowledged that such studies were carried out at centers that might not have been equipped to handle this type of patient and such treatment intensity.
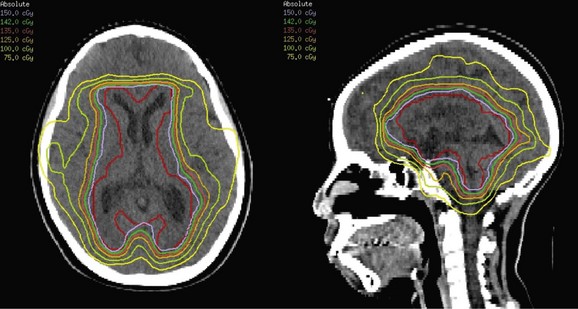
The recent Children’s Oncology Group study for CNS germinoma did not successfully recruit enough patients to meet its objectives and was closed early. It proposed to randomize patients between “standard” RT and combined modality therapy consisting of two cycles of carboplatin and etoposide (induction) followed by RT (complete responders) or further chemotherapy and RT based on response assessment, mainly including those with minimal residual disease, partial responses, or stable disease. There were eight possible treatment groups combining the different possible stages of disease, responses, and randomization arms. For this trial, radiation oncologists developed a consensus for the RT guidelines: standard RT for patients with localized disease was considered ventricular irradiation to 24 Gy, followed by focal irradiation with 21 Gy for a total primary site dose of 45 Gy. Standard RT for patients with disseminated disease was considered craniospinal irradiation to 24 Gy, followed by focal irradiation with 21 Gy, for a total primary site dose of 45 Gy. Patients treated in the combined modality arm would receive response- and risk-adapted RT to include focal irradiation to 30 Gy for patients with localized disease with complete response after induction or focal irradiation to 40.5 Gy for patients with partial response after induction and complete response or minimal residual disease after two additional cycles of cisplatin and cyclophosphamide (Fig. 65-10). Variations to the proposed regimen were included for those patients with neuraxis dissemination at diagnosis. They were to receive combined modality treatment with craniospinal irradiation to 24 Gy, except for those patients who had a complete response to induction, in whom 21 Gy was proposed. In the future study, patients with localized CNS germinoma will undergo evaluation after the second and fourth of four 21-day cycles of carboplatin (600 mg/m2 on day 1) and etoposide (150 mg/m2 on days 1 to 3). Those in complete response will receive whole-ventricle irradiation to 18 Gy and primary site irradiation to 30 Gy, cumulative. Those in less than complete response will receive 24 Gy whole-ventricle irradiation and 36 Gy, cumulative, to the primary site after surgery is performed to determine the histologic response.
Craniopharyngioma
Combined modality therapy (limited surgery and RT) was first proposed by colleagues at the St. Georges and Royal Marsden Hospitals more than 50 years ago. Long-term survivors treated using this approach were found to have good functional outcomes, whereas those treated with primary surgery seldom reached any level of independence and productivity. The Royal Marsden Hospital results for limited surgery and RT remain the standard to which others compare for disease control, reporting 10- and 20-year progression-free survivals of 83% and 79%, respectively.40
Choroid Plexus Tumors
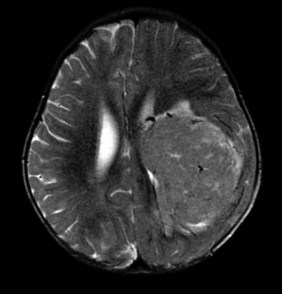
Choroid plexus papilloma is treated with surgery and observation. Choroid plexus carcinomas, because of their vascular nature, are often treated with pre-resection chemotherapy and postoperative focal RT when localized, and residual microscopic or macroscopic tumor is known to remain at the primary site. Craniospinal irradiation has fallen from favor because of the young age at presentation and the effectiveness of chemotherapy but remains an option (Fig. 65-11).
Spinal Cord Tumors
Spinal cord tumors are rare and require special consideration.41,42 The most common presentation is a localized low-grade astrocytoma involving the cervicothoracic spinal cord. Other locations and tumor types include myxopapillary ependymoma, generally involving the cauda equina or conus medullaris, localized ependymoma, and a variety of high-grade neoplasms, including anaplastic astrocytoma, glioblastoma multiforme, and PNET.
Normal Tissue Tolerances and Side Effects
Long-term complications are central to decisions about the use of RT in children with CNS tumors. Despite their significance, until recently, the data have been limited, which has made it difficult to estimate the long-term effects of treatment.43
RT for pediatric CNS tumors may affect neurologic, endocrine, and cognitive function and lead to somatic effects, parenchymal or vascular damage, and secondary neoplasms (Fig. 65-12). The risk of a particular side effect is governed by the age of the patient, time after treatment, location of the primary site, and the prescribed dose and volume. The effects of tumor such as hydrocephalus, irreversible neurologic impairment, and prior treatment, including surgery and chemotherapy, may exceed those expected from RT or RT alone, meaning that combinations of clinical and treatment factors should be considered. Genetic features of the individual may include mutations involving chromosome 17 (NF1), 11q22-q23 (ATM), and chromosome 7 (BRAF).
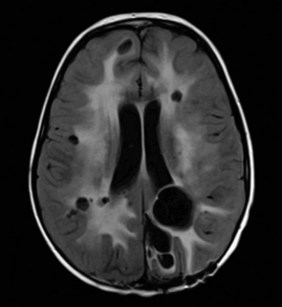
Children with the most common CNS tumors may experience a broad range of side effects. Those effects that appear in the perioperative period from tumor or surgery may contribute significantly to the side effect risk profile of RT when they exceed in scope the expected effects of irradiation, result in side effects normally attributable to irradiation, or reduce the tolerance of normal tissues to irradiation. Poignant examples include vision loss or endocrinopathies resulting from the direct compressive or infiltrative effects of tumor or hydrocephalus, resection of infiltrative tumors in the region of the cerebellum and brainstem to cause cerebellar mutism syndrome,44 and ischemia of normal tissues through the compressive effects of tumor or from tumor resection. In the absence of such factors, predictions regarding side effects are driven by logical factors of treatment site, total dose, and the targeted volume. The weight of age is an order of magnitude greater than the effect of dose in most models that estimate cognitive effects based on dose and volume.
The cognitive effects of RT include effects on global intelligence, learning, memory, attention, and behavior. The adverse cognitive effects of RT in children with brain tumors are generally known only for children who have received whole-brain irradiation as a component of their treatment to dose levels of 18, 24, and 36 Gy. These doses are representative of those customarily used in the treatment of medulloblastoma. Fewer data are available showing the effects of partial brain irradiation in patients with other tumor types,45,46 although data are forthcoming regarding this issue. Most studies have shown a relationship between discrete doses and age at treatment.47 Age- and dose-related decline in IQ was first demonstrated in children who received 36 Gy. These patients had IQ scores approximately 8.2 points lower than those who received 24 Gy, and 12.3 points lower than those who received 18 Gy.48 At least one study found no difference in effect on IQ between doses of 18 and 24 Gy among children older than 6 years but doses over 24 Gy adversely affected IQ.45 Young age at the time of irradiation is the most important prognostic factor regardless of dose. Volume of irradiation is an important prognostic factor for functional outcome.46 Children treated with partial-brain irradiation were more likely to have an IQ greater than 90 points at 5 years (60%) compared with those who received craniospinal irradiation. There is a need for additional data that describe the effects of focal irradiation to more limited volumes. The use of focal radiation techniques in the treatment of children with brain tumors, especially those who do not require craniospinal irradiation, presents a unique opportunity to study the cognitive effects of radiation dose to more limited volumes. The combined advantages of newer treatment methods and more limited volume guidelines have been demonstrated in children with ependymoma. Among 88 patients with localized ependymoma who received RT using a 1-cm CTV margin, there was no decline in IQ, memory, or academic achievement, including 48 children who were younger than the age of 3 years at the time of irradiation.49 These same patients have contributed to separate reports correlating the effects of hydrocephalus and treatment dosimetry on outcome.50,51 Similar encouraging findings have been demonstrated for a variety of patients in the assessment of attention, impulsivity, and reaction time.52
Irradiation of the hypothalamic-pituitary unit leads to endocrine deficiencies. The incidence and time to onset of clinically significant endocrinopathies depends on the total dose to the hypothalamic-pituitary axis, time after irradiation, and other factors, namely, the direct (tumor invasion or mass effect) and indirect (hydrocephalus) effects of tumor. Residents have been taught that the pituitary is the organ at risk for the intracranial portion of the endocrine system and that panhypopituitarism is a foregone conclusion after irradiation of the sellar or suprasellar region. More recent data show the importance of the hypothalamus and the intrinsic and differential sensitivity of the hypothalamic nuclei.53 Among hormone deficiencies observed after RT, growth hormone deficiency is most common, followed by adrenocorticotropic hormone and gonadotropin secretion abnormality, and, finally, less common, hypothyroidism (central). Growth hormone deficiency has been observed after doses as low as 10 Gy and as early as 6 to 12 months after RT at higher doses.54
Hearing loss is a potential complication of RT in children. Radiation may affect the conducting system of the ear. The best example is serous otitis media resulting from mucociliary dysfunction during and after RT. Soft tissue infections of the ear should be treated aggressively, and instrumentation of the external auditory canal should be undertaken cautiously. Sensorineural hearing loss may occur as a result of damage to the cochlea or other parts of the auditory system. Because the doses administered are below the threshold for obvious neurologic impairment of hearing, effects of radiation on the cochlea are more common and usually noted months or years after treatment. The incidence and time course due to radiation is unknown. Most children also receive ototoxic chemotherapy and are therefore at risk for the combined effects of both treatments.55
Dental complications are uncommon but may occur in patients with brain tumors who receive craniospinal irradiation when the salivary glands are subtended by the irradiated volume. Medications used to treat seizure disorders and other neurologic conditions may contribute to tooth decay. Other long-term somatic complications include permanent hair loss, wound healing problems, softening of the bone and ligaments of the spine, and spinal growth impairment and small head size. Patients with optic pathway glioma and, to a lesser extent, craniopharyngioma appear to have a higher incidence of vasculopathy in general, a higher incidence after RT, and an acceleration of preexisting abnormalities. As more children become long-term survivors and neuroimaging improves, subclinical abnormalities on follow-up imaging are seen with increasing frequency, including microangiopathy with calcifications, lacunar infarcts, and transient or permanent enhancing or T2-signal abnormalities, most often but not always in the region that receives the highest dose (see Fig. 65-12). Some changes are more apparent in patients who receive chemotherapy or those known to have suffered ischemia at the time of surgery. Second tumors are an unfortunate complication of RT for CNS tumors. They tend to occur years after treatment, usually when the patient is considered cured. The reported incidence of malignant brain tumors at 10 years is approximately 1% when considering patients with medulloblastoma or ependymoma. Secondary malignancies are a common cause of late morbidity and mortality in medulloblastoma survivors.
1 Statistical Report: Primary Brain Tumors in the United States, 2004-2006. Central Brain Tumor Registry of the United States, 2010.
2 Packer RJ, Biegel JA, Blaney S, et al. Atypical teratoid/rhabdoid tumor of the central nervous system. Report on workshop. J Pediatr Hematol Oncol. 2002;24:337-342.
3 Eberhart CG, Kepner JL, Goldthwaite PT, et al. Histopathologic grading of medulloblastomas. A Pediatric Oncology Group study. Cancer. 2002;94:552-560.
5 Northcott PA, Korshunov A, Witt H, et al. Medulloblastoma comprises four distinct molecular variants. J Clin Oncol. 2011;29:1408-1414.
6 Ellison DW, Kocak M, Dalton J, et al. Definition of disease-risk stratification groups in childhood medulloblastoma using combined clinical, pathologic, and molecular variables. J Clin Oncol. 2011;29:1400-1407.
7 Merchant TE, Wang MH, Haida T, et al. Medulloblastoma. Long-term results for patients treated with definitive radiation therapy during the computed tomography era. Int J Radiat Oncol Biol Phys. 1996;36:29-35.
8 Straathof CS, de Bruin HG, Dippel DW, et al. The diagnostic accuracy of magnetic resonance imaging and cerebrospinal fluid cytology in leptomeningeal metastasis. J Neurol. 1999;246:810-814.
9 Thomas PR, Deutsch M, Kepner JL, et al. Low-stage medulloblastoma. Final analysis of trial comparing standard-dose with reduced-dose neuraxis irradiation. J Clin Oncol. 2000;18:3004-3011.
10 Packer RJ, Goldwein J, Nicholson HS, et al. Treatment of children with medulloblastomas with reduced-dose craniospinal radiation therapy and adjuvant chemotherapy. A Children’s Cancer Group Study. J Clin Oncol. 1999;17:2127-2136.
11 Ris MD, Packer R, Goldwein J, et al. Intellectual outcome after reduced-dose radiation therapy plus adjuvant chemotherapy for medulloblastoma. A Children’s Cancer Group study. J Clin Oncol. 2001;19:3470-3476.
12 Packer RJ, Gajjar A, Vezina G, et al. Phase III study of craniospinal radiation therapy followed by adjuvant chemotherapy for newly diagnosed average-risk medulloblastoma. J Clin Oncol. 2006;24:4202-4208.
13 Merchant TE, Kun LE, Krasin MJ, et al. A multi-institution prospective trial of reduced-dose craniospinal irradiation (23.4 Gy) followed by conformal posterior fossa (36 Gy) and primary site irradiation (55.8 Gy) and dose-intensive chemotherapy for average-risk medulloblastoma. Int J Radiat Oncol Biol Phys. 2003;57:S194-S195.
14 Carrie C, Muracciole X, Gomez F, et al. Conformal radiotherapy, reduced boost volume, hyperfractionnated radiotherapy and on-line quality control in standard risk medulloblastoma without chemotherapy. Results of the French M-SFOP 98 protocol. Int J Radiat Oncol Biol Phys. 2003;57:S195.
15 Ashley D, Merchant T, Strother D, et al. Induction chemotherapy and conformal radiation therapy for very young children with non-metastatic medulloblastoma: P9934. A Children’s Oncology Group Study, submitted for publication, 2011.
16 Blaney SM, Boyett J, Friedman H, et al. Phase I clinical trial of mafosfamide in infants and children aged 3 years or younger with newly diagnosed embryonal tumors. A Pediatric Brain Tumor Consortium study (PBTC-001). J Clin Oncol. 2005;23:525-531.
18 Geyer JR, Zeltzer PM, Boyett JM, et al. Survival of infants with primitive neuroectodermal tumors or malignant ependymomas of the CNS treated with eight drugs in 1 day. A report from the Children’s Cancer Group. J Clin Oncol. 1994;12:1607-1615.
19 Walter AW, Mulhern RK, Gajjar A, et al. Survival and neurodevelopmental outcome of young children with medulloblastoma at St Jude Children’s Research Hospital. J Clin Oncol. 1999;17:3720-3728.
20 Goldwein JW, Radcliffe J, Johnson J, et al. Updated results of a pilot study of low dose craniospinal irradiation plus chemotherapy for children under five with cerebellar primitive neuroectodermal tumors (medulloblastoma). Int J Radiat Biol Phys. 1996;34:899-904.
21 Garvin J, Sposto R, Stanley P, et al. Childhood ependymoma. Improved survival for patients with incompletely resected tumors with the use of pre-irradiation chemotherapy. Presented before the 11th International Symposium on Pediatric Neuro-Oncology, Boston, 2004 [abstract]. Available online at home.comcast.net/~turne038/abstracts.htm Abstract THER 29
22 Merchant TE, Li C, Xiong X, et al. Conformal radiotherapy after surgery for paediatric ependymoma. A prospective study. Lancet Oncol. 2009;10:258-266.
23 Merchant TE, Jenkins JJ, Burger PC, et al. Influence of tumor grade on time to progression after irradiation for localized ependymoma in children. Int J Radiat Oncol Biol Phys. 2002;53:52-57.
24 Pollack IF, Gerszten PC, Martinez AJ, et al. Intracranial ependymomas of childhood. Long-term outcome and prognostic factors. Neurosurgery. 1995;37:655-666. discussion 666-667
26 Korshunov A, Witt H, Hielscher T, et al. Molecular staging of intracranial ependymoma in children and adults. J Clin Oncol. 2010;28:3182-3190.
28 Finlay JL, Boyett JM, Yates AJ, et al. Randomized phase III trial in childhood high-grade astrocytoma comparing vincristine, lomustine, and prednisone with the eight-drugs-in-1-day regimen. Childrens Cancer Group. J Clin Oncol. 1995;13:112-123.
29 Pollack IF, Boyett JM, Yates AJ, et al. The influence of central review on outcome associations in childhood malignant gliomas. Results from the CCG-945 experience. Neuro-oncol. 2003;5:197-207.
30 Wisoff JH, Sanford RA, Holmes E, et al. For the Children’s Oncology Group Impact of surgical resection on low grade gliomas of childhood. A report from the CCG9891/POG9130 low grade astrocytoma study. Proc Am Soc Clin Oncol. 2003;22:100. (abstract 401)
31 Packer RJ, Lange B, Ater J, et al. Carboplatin and vincristine for recurrent and newly diagnosed low-grade gliomas of childhood. J Clin Oncol. 1993;11:850-856.
32 Laithier V, Grill J, Le Deley MC, et al. Progression-free survival in children with optic pathway tumors. Dependence on age and the quality of the response to chemotherapy—results of the first French prospective study for the French Society of Pediatric Oncology. J Clin Oncol. 2003;21:4572-4578.
33 Ater J, Holmes E, Zhou T, et al. Results of COG protocol A9952. A randomized phase 3 study of two chemotherapy regimens for incompletely resected low-grade glioma in young children. Neuro Oncol. 2008;10:451. (abstract LGG18)
34 Merchant TE, Kun LE, Wu S, et al. Phase II trial of conformal radiation therapy for pediatric low-grade glioma. J Clin Oncol. 2009;27:3598-3604.
35 Merchant TE, Conklin HM, Wu S, et al. Late effects of conformal radiation therapy for pediatric patients with low-grade glioma. Prospective evaluation of cognitive, endocrine, and hearing deficits. J Clin Oncol. 2009;27:3691-3697.
36 Awdeh RM, Drewry RD, Kerr NC, et al. Visual outcome after conformal radiation therapy in children with optic pathway glioma. Invest Ophthalmol Vis Sci. 44, 2003. E-Abstract 2232
37 Takeshima H, Kuratsu J. A review of soluble c-kit (s-kit) as a novel tumor marker and possible molecular target for the treatment of CNS germinoma. Surg Neurol. 2003;60:321-324. discussion 324-325
38 Balmaceda C, Heller G, Rosenblum M, et al. Chemotherapy without irradiation—a novel approach for newly diagnosed CNS germ cell tumors. Results of an international cooperative trial. The First International Central Nervous System Germ Cell Tumor Study. J Clin Oncol. 1996;14:2908-2915.
39 Kellie SJ, Boyce H, Dunkel IJ, et al. Primary chemotherapy for intracranial nongerminomatous germ cell tumors. Results of the Second International CNS Germ Cell Study Group protocol. J Clin Oncol. 2004;22:846-853.
40 Rajan B, Ashley S, Gorman C, et al. Craniopharyngioma—long term results following limited surgery and radiotherapy. Radiother Oncol. 1993;26:1-10.
41 Merchant TE, Nguyen D, Thompson SJ, et al. High-grade pediatric spinal cord tumors. Pediatr Neurosurg. 1999;30:1-5.
42 Merchant TE, Kiehna EN, Thompson SJ, et al. Pediatric low-grade and ependymal spinal cord tumors. Pediatr Neurosurg. 2000;32:30-36.
43 Zebrack BJ, Gurney JG, Oeffinger K, et al. Psychological outcomes in long-term survivors of childhood brain cancer. A report from the childhood cancer survivor study. J Clin Oncol. 2004;22:999-1006.
44 Robertson PL, Muraszko KM, Holmes EJ, et al. Incidence and severity of postoperative cerebellar mutism syndrome in children with medulloblastoma. A prospective study by the Children’s Oncology Group. J Neurosurg. 2006;105:444-451.
45 Fuss M, Poljanc K, Hug EB. Full scale IQ (FSIQ) changes in children treated with whole brain and partial brain irradiation. A review and analysis. Strahlenther Onkol. 2000;176:573-581.
46 Hoppe-Hirsch E, Brunet L, Laroussinie F, et al. Intellectual outcome in children with malignant tumors of the posterior fossa. Influence of the field of irradiation and quality of surgery. Childs Nerv Syst. 1995;11:340-345.
47 Mulhern RK, Hancock J, Fairclough D, et al. Neuropsychological status of children treated for brain tumors. Critical review and integrative analysis. Med Pediatr Oncol. 1992;20:181-191.
49 Merchant TE, Mulhern RK, Krasin MJ, et al. Preliminary results from a phase II trial of conformal radiation therapy and the evaluation of radiation-related CNS effects for pediatric patients with localized ependymoma. J Clin Oncol. 2004;22:3156-3162.
50 Merchant TE, Lee H, Zhu J, et al. The effects of hydrocephalus on IQ before and after focal irradiation of children with localized infratentorial ependymoma. J Neurosurg Pediatr. 2004;101:159-168.
51 Merchant TE, Kiehna EN, Li C, et al. Radiation dosimetry predicts IQ after conformal radiation therapy in pediatric patients with localized ependymoma. Int J Radiat Oncol Biol Phys. 2005;63:1546-1554.
52 Kiehna EN, Mulhern RK, Li C, et al. Changes in attentional performance of children and young adults with localized primary brain tumors after conformal radiation therapy. J Clin Oncol. 2006;24:5283-5290.
53 Robinson IC, Fairhall KM, Hendry JH, et al. Differential radiosensitivity of hypothalamo-pituitary function in the young adult rat. J Endocrinol. 2001;169:519-526.
54 Merchant TE, Goloubeva O, Pritchard DL, et al. Radiation dose-volume effects on growth hormone secretion. Int J Radiat Oncol Biol Phys. 2002;52:1264-1270.
55 Hua C, Bass JK, Khan R, et al. Hearing loss after radiotherapy for pediatric brain tumors. Effect of cochlear dose. Int J Radiat Oncol Biol Phys. 2008;72:892-899.
1 Statistical Report: Primary Brain Tumors in the United States, 2004-2006. Central Brain Tumor Registry of the United States, 2010.
2 Packer RJ, Biegel JA, Blaney S, et al. Atypical teratoid/rhabdoid tumor of the central nervous system. Report on workshop. J Pediatr Hematol Oncol. 2002;24:337-342.
3 Eberhart CG, Kepner JL, Goldthwaite PT, et al. Histopathologic grading of medulloblastomas. A Pediatric Oncology Group study. Cancer. 2002;94:552-560.
4 Giangaspero F, Rigobello L, Badiali M, et al. Large-cell medulloblastomas. A distinct variant with highly aggressive behavior. Am J Surg Pathol. 1992;16:687-693.
5 Northcott PA, Korshunov A, Witt H, et al. Medulloblastoma comprises four distinct molecular variants. J Clin Oncol. 2011;29:1408-1414.
6 Ellison DW, Kocak M, Dalton J, et al. Definition of disease-risk stratification groups in childhood medulloblastoma using combined clinical, pathologic, and molecular variables. J Clin Oncol. 2011;29:1400-1407.
7 Merchant TE, Wang MH, Haida T, et al. Medulloblastoma. Long-term results for patients treated with definitive radiation therapy during the computed tomography era. Int J Radiat Oncol Biol Phys. 1996;36:29-35.
8 Straathof CS, de Bruin HG, Dippel DW, et al. The diagnostic accuracy of magnetic resonance imaging and cerebrospinal fluid cytology in leptomeningeal metastasis. J Neurol. 1999;246:810-814.
9 Thomas PR, Deutsch M, Kepner JL, et al. Low-stage medulloblastoma. Final analysis of trial comparing standard-dose with reduced-dose neuraxis irradiation. J Clin Oncol. 2000;18:3004-3011.
10 Packer RJ, Goldwein J, Nicholson HS, et al. Treatment of children with medulloblastomas with reduced-dose craniospinal radiation therapy and adjuvant chemotherapy. A Children’s Cancer Group Study. J Clin Oncol. 1999;17:2127-2136.
11 Ris MD, Packer R, Goldwein J, et al. Intellectual outcome after reduced-dose radiation therapy plus adjuvant chemotherapy for medulloblastoma. A Children’s Cancer Group study. J Clin Oncol. 2001;19:3470-3476.
12 Packer RJ, Gajjar A, Vezina G, et al. Phase III study of craniospinal radiation therapy followed by adjuvant chemotherapy for newly diagnosed average-risk medulloblastoma. J Clin Oncol. 2006;24:4202-4208.
13 Merchant TE, Kun LE, Krasin MJ, et al. A multi-institution prospective trial of reduced-dose craniospinal irradiation (23.4 Gy) followed by conformal posterior fossa (36 Gy) and primary site irradiation (55.8 Gy) and dose-intensive chemotherapy for average-risk medulloblastoma. Int J Radiat Oncol Biol Phys. 2003;57:S194-S195.
14 Carrie C, Muracciole X, Gomez F, et al. Conformal radiotherapy, reduced boost volume, hyperfractionnated radiotherapy and on-line quality control in standard risk medulloblastoma without chemotherapy. Results of the French M-SFOP 98 protocol. Int J Radiat Oncol Biol Phys. 2003;57:S195.
15 Merchant TE, Pritchard DL, Vargo JA, et al. Radiation therapy for the treatment of childhood medulloblastoma. The rationale for current techniques, strategies, and dose-volume considerations. Electromedica. 2001;69:69-71.
16 Blaney SM, Boyett J, Friedman H, et al. Phase I clinical trial of mafosfamide in infants and children aged 3 years or younger with newly diagnosed embryonal tumors. A Pediatric Brain Tumor Consortium study (PBTC-001). J Clin Oncol. 2005;23:525-531.
17 Duffner PK, Horowitz ME, Krischer JP, et al. Postoperative chemotherapy and delayed radiation in children less than three years of age with malignant brain tumors. N Engl J Med. 1993;328:1725-1731.
18 Geyer JR, Zeltzer PM, Boyett JM, et al. Survival of infants with primitive neuroectodermal tumors or malignant ependymomas of the CNS treated with eight drugs in 1 day. A report from the Children’s Cancer Group. J Clin Oncol. 1994;12:1607-1615.
19 Walter AW, Mulhern RK, Gajjar A, et al. Survival and neurodevelopmental outcome of young children with medulloblastoma at St Jude Children’s Research Hospital. J Clin Oncol. 1999;17:3720-3728.
20 Goldwein JW, Radcliffe J, Johnson J, et al. Updated results of a pilot study of low dose craniospinal irradiation plus chemotherapy for children under five with cerebellar primitive neuroectodermal tumors (medulloblastoma). Int J Radiat Biol Phys. 1996;34:899-904.
21 Garvin J, Sposto R, Stanley P, et al. Childhood ependymoma. Improved survival for patients with incompletely resected tumors with the use of pre-irradiation chemotherapy. Presented before the 11th International Symposium on Pediatric Neuro-Oncology, Boston, 2004 [abstract]. Available online at home.comcast.net/~turne038/abstracts.htm Abstract THER 29
22 Merchant TE, Li C, Xiong X, et al. Conformal radiotherapy after surgery for paediatric ependymoma. A prospective study. Lancet Oncol. 2009;10:258-266.
23 Merchant TE, Jenkins JJ, Burger PC, et al. Influence of tumor grade on time to progression after irradiation for localized ependymoma in children. Int J Radiat Oncol Biol Phys. 2002;53:52-57.
24 Pollack IF, Gerszten PC, Martinez AJ, et al. Intracranial ependymomas of childhood. Long-term outcome and prognostic factors. Neurosurgery. 1995;37:655-666. discussion 666-667
25 Sutton LN, Goldwein J, Perilongo G, et al. Prognostic factors in childhood ependymomas. Pediatr Neurosurg. 1990;16:57-65.
26 Korshunov A, Witt H, Hielscher T, et al. Molecular staging of intracranial ependymoma in children and adults. J Clin Oncol. 2010;28:3182-3190.
27 Sposto R, Ertel IJ, Jenkin RD, et al. The effectiveness of chemotherapy for treatment of high grade astrocytoma in children. Results of a randomized trial. A report from the Childrens Cancer Study Group. J Neurooncol. 1989;7:165-177.
28 Finlay JL, Boyett JM, Yates AJ, et al. Randomized phase III trial in childhood high-grade astrocytoma comparing vincristine, lomustine, and prednisone with the eight-drugs-in-1-day regimen. Childrens Cancer Group. J Clin Oncol. 1995;13:112-123.
29 Pollack IF, Boyett JM, Yates AJ, et al. The influence of central review on outcome associations in childhood malignant gliomas. Results from the CCG-945 experience. Neuro-oncol. 2003;5:197-207.
30 Wisoff JH, Sanford RA, Holmes E, et al. For the Children’s Oncology Group Impact of surgical resection on low grade gliomas of childhood. A report from the CCG9891/POG9130 low grade astrocytoma study. Proc Am Soc Clin Oncol. 2003;22:100. (abstract 401)
31 Packer RJ, Lange B, Ater J, et al. Carboplatin and vincristine for recurrent and newly diagnosed low-grade gliomas of childhood. J Clin Oncol. 1993;11:850-856.
32 Laithier V, Grill J, Le Deley MC, et al. Progression-free survival in children with optic pathway tumors. Dependence on age and the quality of the response to chemotherapy—results of the first French prospective study for the French Society of Pediatric Oncology. J Clin Oncol. 2003;21:4572-4578.
33 Ater J, Holmes E, Zhou T, et al. Results of COG protocol A9952. A randomized phase 3 study of two chemotherapy regimens for incompletely resected low-grade glioma in young children. Neuro Oncol. 2008;10:451. (abstract LGG18)
34 Merchant TE, Kun LE, Wu S, et al. Phase II trial of conformal radiation therapy for pediatric low-grade glioma. J Clin Oncol. 2009;27:3598-3604.
35 Merchant TE, Conklin HM, Wu S, et al. Late effects of conformal radiation therapy for pediatric patients with low-grade glioma. Prospective evaluation of cognitive, endocrine, and hearing deficits. J Clin Oncol. 2009;27:3691-3697.
36 Awdeh RM, Drewry RD, Kerr NC, et al. Visual outcome after conformal radiation therapy in children with optic pathway glioma. Invest Ophthalmol Vis Sci. 44, 2003. E-Abstract 2232
37 Takeshima H, Kuratsu J. A review of soluble c-kit (s-kit) as a novel tumor marker and possible molecular target for the treatment of CNS germinoma. Surg Neurol. 2003;60:321-324. discussion 324-325
38 Balmaceda C, Heller G, Rosenblum M, et al. Chemotherapy without irradiation—a novel approach for newly diagnosed CNS germ cell tumors. Results of an international cooperative trial. The First International Central Nervous System Germ Cell Tumor Study. J Clin Oncol. 1996;14:2908-2915.
39 Kellie SJ, Boyce H, Dunkel IJ, et al. Primary chemotherapy for intracranial nongerminomatous germ cell tumors. Results of the Second International CNS Germ Cell Study Group protocol. J Clin Oncol. 2004;22:846-853.
40 Rajan B, Ashley S, Gorman C, et al. Craniopharyngioma—long term results following limited surgery and radiotherapy. Radiother Oncol. 1993;26:1-10.
41 Merchant TE, Nguyen D, Thompson SJ, et al. High-grade pediatric spinal cord tumors. Pediatr Neurosurg. 1999;30:1-5.
42 Merchant TE, Kiehna EN, Thompson SJ, et al. Pediatric low-grade and ependymal spinal cord tumors. Pediatr Neurosurg. 2000;32:30-36.
43 Zebrack BJ, Gurney JG, Oeffinger K, et al. Psychological outcomes in long-term survivors of childhood brain cancer. A report from the childhood cancer survivor study. J Clin Oncol. 2004;22:999-1006.
44 Robertson PL, Muraszko KM, Holmes EJ, et al. Incidence and severity of postoperative cerebellar mutism syndrome in children with medulloblastoma. A prospective study by the Children’s Oncology Group. J Neurosurg. 2006;105:444-451.
45 Fuss M, Poljanc K, Hug EB. Full scale IQ (FSIQ) changes in children treated with whole brain and partial brain irradiation. A review and analysis. Strahlenther Onkol. 2000;176:573-581.
46 Hoppe-Hirsch E, Brunet L, Laroussinie F, et al. Intellectual outcome in children with malignant tumors of the posterior fossa. Influence of the field of irradiation and quality of surgery. Childs Nerv Syst. 1995;11:340-345.
47 Mulhern RK, Hancock J, Fairclough D, et al. Neuropsychological status of children treated for brain tumors. Critical review and integrative analysis. Med Pediatr Oncol. 1992;20:181-191.
48 Silber JH, Radcliffe J, Peckham V, et al. Whole-brain irradiation and decline in intelligence. The influence of dose and age on IQ score. J Clin Oncol. 1992;10:1390-1396.
49 Merchant TE, Mulhern RK, Krasin MJ, et al. Preliminary results from a phase II trial of conformal radiation therapy and the evaluation of radiation-related CNS effects for pediatric patients with localized ependymoma. J Clin Oncol. 2004;22:3156-3162.
50 Merchant TE, Lee H, Zhu J, et al. The effects of hydrocephalus on IQ before and after focal irradiation of children with localized infratentorial ependymoma. J Neurosurg Pediatr. 2004;101:159-168.
51 Merchant TE, Kiehna EN, Li C, et al. Radiation dosimetry predicts IQ after conformal radiation therapy in pediatric patients with localized ependymoma. Int J Radiat Oncol Biol Phys. 2005;63:1546-1554.
52 Kiehna EN, Mulhern RK, Li C, et al. Changes in attentional performance of children and young adults with localized primary brain tumors after conformal radiation therapy. J Clin Oncol. 2006;24:5283-5290.
53 Robinson IC, Fairhall KM, Hendry JH, et al. Differential radiosensitivity of hypothalamo-pituitary function in the young adult rat. J Endocrinol. 2001;169:519-526.
54 Merchant TE, Goloubeva O, Pritchard DL, et al. Radiation dose-volume effects on growth hormone secretion. Int J Radiat Oncol Biol Phys. 2002;52:1264-1270.
55 Hua C, Bass JK, Khan R, et al. Hearing loss after radiotherapy for pediatric brain tumors. Effect of cochlear dose. Int J Radiat Oncol Biol Phys. 2008;72:892-899.
56 Merchant TE, Palmer SL, Lukose R, et al. Critical combinations of radiation dose and volume predict IQ and academic achievement after craniospinal irradiation in children. In J Radiat Oncol Biol Phys. 2010;78:S17.
[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
Chapter 65 Central Nervous System Tumors in Children
This is a pivotal time for RT in the treatment of children with brain tumors. There are fundamental questions that need to be addressed, including the indications for RT in the setting of clinical and molecular risk stratification for children with medulloblastoma and potentially ependymoma and the refinement of total dose and treatment volumes regimens for specific patient groups, including very young children and those with germ cell tumors. Improved knowledge of treatment-related side effects and a wider availability of proton therapy will likely impact how RT is administered and perceptions about the safety of RT in these patients. Despite the increasing availability of advanced treatment technology, children should be treated on clinical trials designed to improve tumor control, objectively study CNS effects, and build a consensus for treatment guidelines. The impact of pediatric CNS tumors on public health is small—there are fewer than 4000 cases diagnosed annually in the United States. CNS tumors represent 20% of all childhood malignancies.1 Most children with brain tumors will require RT during the course of their management. Based on the U.S. standard population data from 2000, which includes 78.8 million individuals aged birth to 19 years, annually there were 2742 cases of neuroepithelial tumors that included 654 cases of juvenile pilocytic astrocytoma, 118 cases of glioblastoma multiforme, 236 cases of ependymoma, and 433 cases of CNS embryonal tumors including medulloblastoma. Nonneuroepithelial tumors of interest to radiation oncologists included 197 cases of CNS germ cell tumors and 118 cases of craniopharyngiomas. These tumors require special care and remain a leading cause of cancer death in children.
Embryonal Central Nervous System Tumors
Embryonal tumors of the CNS are the most common malignant brain tumors in children. They are characterized by their infiltrative nature, aggressive patterns of growth, propensity for dissemination, and common need for intensive combined modality therapy. These tumors are found throughout the CNS and are further classified according to histologic and molecular features into two groups: primitive neuroectodermal tumors (PNET) and atypical teratoid rhabdoid tumors (ATRT). The PNET group includes two distinct entities—medulloblastoma (Fig. 65-1) and pineoblastoma (Fig. 65-2)—named for their site of origin in the posterior fossa and pineal region, respectively. ATRT are rare aggressive embryonal tumors that most commonly arise in young children in the brain and at peripheral soft tissue sites. With some exceptions, the diagnosis of ATRT is made on molecular grounds by demonstrating the deletion or mutation of the SMARCB1 gene found on chromosome 22q11.2.2 Atypical teratoid rhabdoid tumors unknowingly were considered the same as other PNET but are now known to carry a worse prognosis.
Figure 65-1 Medulloblastoma as shown on axial postgadolinium T1-weighted (left) and T2-weighted (right) MR images.
Medulloblastoma and supratentorial PNET are commonly diagnosed in young children. The relatively large numbers of patients and a cooperative spirit among caregivers have led to considerable understanding and progress in the treatment of these tumors. Patients are risk classified for treatment based on the three most important prognostic factors for tumor control— extent of disease, extent of resection, and histological subtype—and the most important prognostic factor for functional outcome—age at the time of irradiation. The radiation oncologist should be aware of the aggressive histologic variant of medulloblastoma (large cell or anaplastic medulloblastoma) that is more prone to dissemination through the cerebrospinal fluid (CSF).3,4 One also should be aware that there is a less aggressive variant (desmoplastic medulloblastoma) that may not require irradiation under certain conditions.
Medulloblastoma is a heterogeneous tumor and is now recognized as a number of different diseases based on specific molecular markers that have been retrospectively evaluated. Depending on the series, investigators have identified from four to six different subtypes based on the combination of molecular and classic histopathologic evaluation. A report by Northcott and associates5 described four distinct molecular variants of medulloblastoma, including WNT, SHH, and two non-WNT/SHH. The former two have clinical features that are associated with less aggressive phenotypes and the latter two, described as groups C and D in their report, have molecular and clinical features associated with more aggressive phenotypes, including overexpression of MYC and disseminated disease (group C) and the most common cancer isochromosome i17q and children between the ages of 4 and 15 years. One of the more striking findings by this group was the variation in molecular characteristics by age. Many of these findings have been confirmed by others groups,6 which suggests that tumor biologic information will be combined with clinical prognostic data to stratify patients in future trials.
At present, the treatment of embryonal tumors of the CNS is remarkably similar regardless of subtype or tumor location and consists of surgery, postoperative RT, and chemotherapy. The requirement of craniospinal irradiation, regardless of extent of disease, is the element that differentiates the treatment of these tumors. Approximately one third of patients present with neuraxis dissemination at the time of diagnosis as determined by magnetic resonance imaging (MRI) of the brain and spine (Fig. 65-3) or CSF cytology.7 Because the negative predictive value of these staging procedures remains relatively low, and the most common mode of failure remains distant, craniospinal irradiation is a mainstay in the treatment of these tumors with few exceptions.8
The side effects attributed to RT have been a primary concern for patients with medulloblastoma. The concern has been greatest for patients with average-risk disease, for whom long-term disease control is likely and the side effects of therapy have a lasting impact. The treatment of this tumor is technically demanding and consists of craniospinal irradiation and boost treatment of the anatomic posterior fossa. Until recently, the standard of care for an average-risk patient was postoperative craniospinal irradiation (CSI) to 36 Gy and boost treatment to 54 Gy using conventional fractionation of 1.8 Gy/day. In a cooperative group trial the CSI dose was lowered to 23.4 Gy for average-risk patients in a randomized comparison with 36 Gy CSI. This trial showed an increased risk of relapse in patients receiving the reduced CSI dose.9 Multiple-agent chemotherapy combined with reduced-dose CSI was found to be equivalent to standard-dose CSI and was subsequently adopted as a treatment standard for average-risk patients.10 Cognitive impairment continues to be a significant problem, with intelligence quotient (IQ) loss most commonly seen in children younger than the age of 8 years and those treated for high-risk medulloblastoma in which high-dose craniospinal irradiation (36 Gy) is required.11
The most recently completed Children’s Oncology Group study sought to define the best chemotherapy regimen (lomustine [CCNU], cisplatin, and vincristine vs. cyclophosphamide, cisplatin, and vincristine) to accompany low-dose craniospinal irradiation and the standard posterior fossa boost. No differences were observed in disease control (3-year event-free survival >83% in both arms); however, patients treated with the cyclophosphamide-containing regimen appeared to have more severe toxicity (18% vs. 25%).12 Attention has now turned to combining the two chemotherapy regimens and randomizing patients between 18.0 Gy and 23.4 Gy CSI followed by a second randomization between conventional boost treatment and treatment of less than the entire posterior fossa in an ambitious four-arm randomized trial developed for pediatric patients between the ages of 3 and 8 years. For those older than 8 years, the CSI dose remains at 23.4 Gy. The trial also includes a randomized comparison of conventional posterior fossa boost and boost treatment of the tumor bed using a 1.5-cm clinical target volume (CTV) margin.
Treatment of less than the entire posterior fossa after 23.4 Gy of CSI using focal radiation delivery techniques appears to be as effective as treatment of the entire posterior fossa based on recent trial results from St. Jude Children’s Research Hospital and participating centers involving 84 patients. In this prospective trial the cumulative incidence of posterior fossa failure was only 6.3% at 3 years.13 Treatment of these patients included postoperative CSI to 23.4 Gy, conformal posterior fossa boost to 36 Gy, and focal treatment of the tumor bed with a 2-cm margin. Chemotherapy was administered after RT. Patients on this study experienced a dose reduction to the anatomic posterior fossa of approximately 13%. The risks associated with reducing the CSI dose and limiting the boost treatment to the postoperative tumor bed with a limited margin must be balanced against the observed effects of RT on cognition, neurologic function, and growth and development.
Hyperfractionation has been tested in Europe to reduce toxicity.14 Although these investigations have shown that hyperfractionation may be isoeffective, lacking are the data that indicate side effects are reduced by this strategy. Considering logistics including the need for anesthesia in this often young patient population, investigators demand some certainty that there would be a benefit associated with hyperfractionated treatment before planning future trials.
Current cooperative group trials for very young children (<3 years of age) with medulloblastoma highlight the importance of RT in the treatment of these tumors. The recently completed Children’s Oncology Group protocol for children younger than the age of 3 years with localized medulloblastoma included 20 weeks of combination chemotherapy followed by second surgery when indicated and sequential irradiation of the posterior fossa and primary site before 20 additional weeks of maintenance chemotherapy.15 CSI was been omitted for these patients, and the doses to the posterior fossa (18 and 23.4 Gy) and primary site (45 to 54 Gy) were prescribed based on patient age, response, and risk status. A similar strategy was used in the Pediatric Brain Tumor Consortium (PBTC) protocol for similar patients with one exception: intrathecal chemotherapy (mafosfamide) was given during the first 20 weeks of chemotherapy as a substitute for CSI.16 Both protocols included RT after 20 weeks based on results from prior trials that showed a median time to progression of approximately 24 weeks.17,18 The targeting guidelines used in these studies were largely empirical: investigators attempted to minimize the dose to normal tissues while maintaining an acceptable rate of local tumor control.
Fear of cognitive deficits has been the driving force in the design of current treatment trials for this young patient population. Age has the greatest impact on cognitive outcome after irradiation (Fig. 65-4). For very young children with medulloblastoma, a persistent decline in cognitive function has been observed by Walter and colleagues.19 The decline is not surprising given the median cranial dose of 35.2 Gy for patients with a median age of 2.6 years and the additive effects of tumor, surgery, and chemotherapy. Because a significant decline has also been observed for older children with medulloblastoma treated with lower craniospinal doses,11 CSI for the youngest children no longer appears to be a reasonable treatment option. Data concerning the effectiveness of yet lower CSI doses are extremely limited.20 These findings form the basis of the most recent trials for very young children. CSI has been omitted with the hope that the side effects of treatment will be reduced enough to provide for a meaningful quality of life among long-term survivors. There is a move to increase the age at which CSI is considered acceptable from age 3 years to 5 years at the time of treatment. Investigators acknowledge that they are trading fewer side effects for an increase in the rate of disseminated failure.
Ependymoma
Ependymoma is the third most common CNS tumor in children, affecting approximately 236 individuals younger than the age of 19 years annually in the United States.1 It shares many of the clinical characteristics of more common gliomas, embryonal tumors, and less common germ cell tumors. Ependymoma may occur anywhere within the CNS and has the highest prevalence within the posterior fossa, arising from the floor or roof of the fourth ventricle or cerebellopontine angle, where it is known to invade the brainstem or envelop cranial nerves or vascular structures. Ependymoma also arises within the parenchyma of the cerebral hemispheres in older patients and rarely in the spinal cord. The interval from the onset of symptoms to diagnosis may be influenced by the young age at the time of diagnosis, the perceived slow growth rate of this tumor, and the remitting signs and symptoms of increased intracranial pressure resulting from obstructive hydrocephalus (Fig. 65-5).
Ependymoma is most commonly diagnosed in children who are younger than 4 years of age. For the past two decades, progress in the treatment of this tumor has been slowed by concerns about late effects. Recent advances in neuroimaging, surgery, and RT have moved the field of neuro-oncology forward in the treatment of this tumor, resulting in more acceptable outcomes and providing new disease control benchmarks. Investigators treating these patients with contemporary surgery and RT have been able to increase event-free survivals measured at 3 years from 55% to 62% to 75% when measured at 7 years with remarkably limited treatment-related effects.21,22
The standard of care for a child with localized ependymoma is to attempt gross-total resection, perform second surgery for potentially resectable residual tumor, and proceed with postoperative RT directed at the primary site using a 10-mm, anatomically confined CTV surrounding the residual tumor and/or tumor bed as defined on postoperative neuroimaging. The recommended total dose is 59.4 Gy using conventional fractionation because these tumors are prone to recur locally. Conventions regarding organs at risk apply; thus, for patients with tumors arising in the lower aspect of the posterior fossa, avoidance of the spinal cord and/or chiasm should be considered after approximately 54 Gy. Special considerations for patients with posterior fossa tumors may apply to very young children (generally those younger than the age of 18 months) with no evidence of residual tumor or patients who are noted to have signs and symptoms of severe brainstem injury after resection (postoperative seizure, hypertension, and ischemic changes on neuroimaging) who are at increased risk for necrosis. The recent trial of the Children’s Oncology Group showed that there was consensus to irradiate children younger than the age of 3 years and as young as 12 months postoperatively. The newest study from the Children’s Oncology Group, known as ACNS0831 and initiated in 2010, deploys a similar treatment algorithm and methods of irradiation with a smaller (5 mm) CTV margin. Children are randomized to receive four cycles of postirradiation chemotherapy (Fig. 65-6).
The prognosis for a patient with ependymoma is determined by tumor grade and extent of resection.23,24,25 Patients with overtly anaplastic tumors have inferior disease control when compared with those with differentiated tumors; however, the assignment of tumor grade can be difficult for those cases in which focal anaplasia occurs in the setting of a largely differentiated tumor. All patients should be treated with curative intent regardless of tumor grade. Extent of resection is an unequivocal prognostic factor: patients with substantial residual tumor have the worst prognosis. Because patients with varying amounts of residual tumor have achieved long-term survival after RT and modern surgical resection has altered the definition of near-total and subtotal resection, the volume of residual tumor differentiating between patients with good and poor prognosis remains unknown. It is incumbent on the radiation oncologist to question whether additional resection might be safely achieved in a patient with residual tumor referred for irradiation.
Research performed to evaluate molecular prognostic markers in ependymoma has revealed gain of 1q and homozygous deletion of CDKN2A as valuable predictors of poor survival in this disease26 and other markers associated with more favorable outcomes. Although molecular markers are not likely to supplant standard histopathologic evaluation, they will certainly contribute beneficial information for separating tumors that appear to have features of both differentiated and anaplastic ependymoma and determining which patients might require additional treatment after gross-total resection and irradiation.
High-Grade Astrocytoma
One study has shown a benefit for chemotherapy.27 This study included a randomization of 58 patients with high-grade astrocytoma to local irradiation versus local irradiation plus weekly vincristine followed by 12 month of carmustine, vincristine, and prednisone. The reported 5-year progression-free survival rate was 18% versus 46% favoring the use of chemotherapy, and patients with glioblastoma multiforme (n = 40) had a 5-year progression-free survival of 6% versus 42% favoring the use of chemotherapy. A larger study that included 172 patients randomized between local irradiation versus local irradiation plus preirradiation and postirradiation 8-in-1 chemotherapy (i.e., with vincristine, carmustine, procarbazine, cytarabine, hydroxyurea, cisplatin, dacarbazine, and methylprednisolone) showed no difference between the two study arms with 5-year progression-free survivals of 26% and 33%, respectively.28 This study was illustrative for the lack of concordance between institutional and expert pathology review. Thirty percent of patients in this study were found not to have high-grade glioma, and the survival estimates were in fact lower than originally reported.29
[/not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
