CHAPTER 4 Cardiovascular Physiology
Fetal circulation
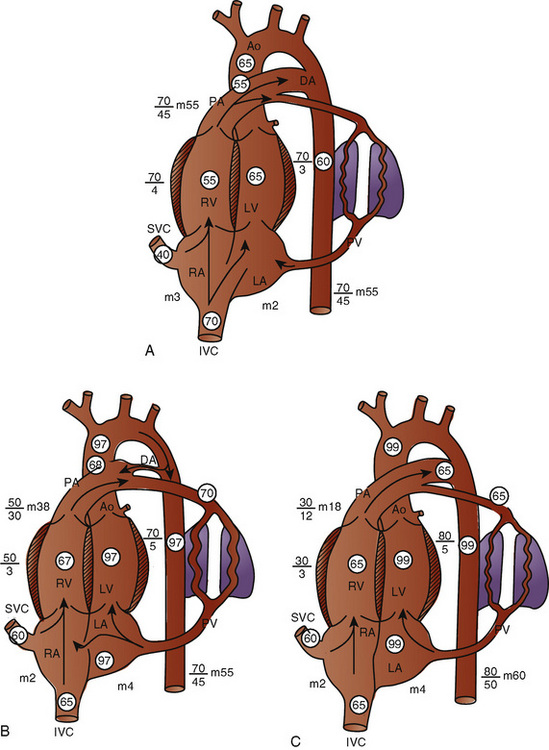
Anatomy
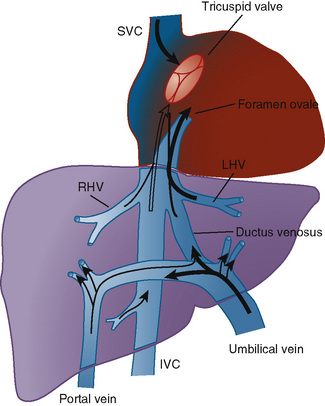
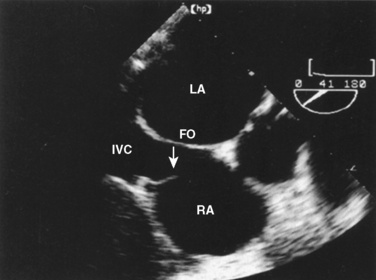
The organ of prenatal respiration is the placenta. It is a large, low-resistance circuit that has an enormous influence on the pattern of fetal blood flow. The lungs are almost completely excluded from fetal circulation, and three special shunts (the ductus venosus, the foramen ovale, and the ductus arteriosus) allow the most oxygenated blood to perfuse the heart and brain (Fig. 4-1). Two umbilical arteries originate from the internal iliac arteries and deliver fetal blood to the placenta. One umbilical vein carries oxygenated blood from the placenta to the fetus. When umbilical venous blood approaches the liver, it can take two pathways. It is estimated that 50% to 60% of umbilical venous blood bypasses the liver via the ductus venosus, while the remainder perfuses the left lobe of the liver. Blood flow to the right lobe of the liver is predominantly from the portal circulation. The right and left hepatic veins, along with the ductus venosus, merge into the suprahepatic inferior vena cava (IVC) (Fig. 4-2). Bypassing the high-resistance hepatic microcirculation, umbilical venous blood in the ductus venosus remains not only more oxygenated, but it also flows at a higher velocity. Within the suprahepatic IVC there are now two streams of blood. The stream of blood with higher velocity is derived from the ductus venosus and drainage from the left hepatic vein. The stream of blood with slower velocity consists of drainage from the right hepatic vein mixed with venous return from the abdominal IVC. Upon entering the right atrium, these two streams of blood diverge. The blood with higher velocity is primarily directed across the foramen ovale to the left atrium. This right-to-left shunt is possible because left atrial pressure is low due to minimal pulmonary venous return. The Eustachian valve is a flap of tissue at the junction of the IVC and the right atrium. It functions to help direct the higher-velocity stream of blood across the foramen ovale and into the left atrium (Fig. 4-3). The lower-velocity stream of blood crosses the tricuspid valve and is ejected by the right ventricle. This anatomic arrangement allows the most oxygenated blood from the umbilical vein to bypass the liver and the right side of heart.
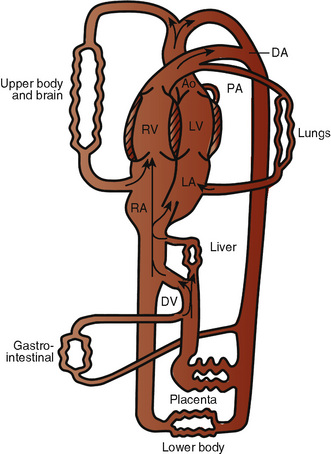
The key features of fetal circulation are shown in Figure 4-4 and listed below:
Fetal Cardiac Output
In the fetus, right and left ventricular outputs are not equal. The ventricles, acting in parallel, pump different amounts of blood, and organs receive blood flow from both ventricles. Thus, it is customary to refer to the combined ventricular output (CVO) of fetal circulation. The fetal CVO is estimated at 400 mL/kg per minute, a value that is similar to that of the neonate but almost threefold higher than it is in adults. The ratio of right ventricular to left ventricular output is approximately 1.3:1 (Kenny et al., 1986). The volume load borne by the right ventricle combined with the high fetal PVR results in significant hypertrophy. After birth, the right ventricle remodels over time under the influence of a series circulation and lowered PVR (Fig. 4-5). The demands of a parallel circulation result in increased myocardial work superimposed on the demands of a fetus having to grow and develop in a cyanotic milieu. Nevertheless, the fetal circulation is an efficient arrangement with adequate physiologic reserve. This is demonstrated by the full spectrum of congenital heart lesions that are well tolerated in utero.
Oxygen Delivery
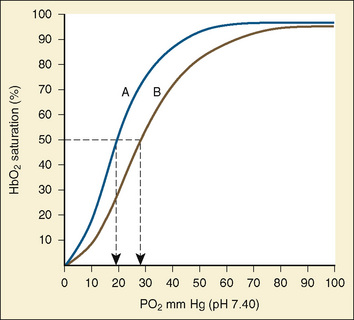
Oxygen transport must be achieved in a relatively hypoxic environment (Lister et al., 1979). How does the fetus ensure adequate oxygen delivery? Fetal hemoglobin (HbF) has unique properties that allow the fetus to transport oxygen despite a low Po2. Approximately 80% of fetal hemoglobin is HbF compared with an adult who has over 90% adult hemoglobin (HbA). The Pao2 at which Hb is 50% saturated is called the P50. Fetal hemoglobin (HbF P50:19 mm Hg) is shifted to the left in comparison with adult hemoglobin (HbA P50:26 mm Hg). A low level of 2,3-diphosphoglycerate (2,3-DPG) and the decreased affinity of HbF for 2,3-DPG cause the leftward shift of HbF. In vitro, HbF has a sigmoidal oxygen dissociation curve similar to that of HbA. The result is that for any given Po2, the oxygen saturation is higher for HbF than for HbA.
Assuming a Pao2 of 30 mm Hg in the blood ejected by the left ventricle, this value falls in the steepest part of the oxygen-hemoglobin dissociation curve. At a Pao2 of 30 mm Hg, fetal hemoglobin would be approximately 70% saturated, and adult hemoglobin would only be 50% saturated (Fig. 4-6). Using the equation for oxygen content of the blood (Cao2) demonstrates how the fetus can achieve levels of oxygen transport that are near those of an adult. Oxygen content of blood is defined by the following equation, Sao2 is the arterial oxygen saturation of hemoglobin, and Hb is the hemoglobin concentration in g/dL:
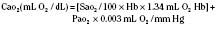
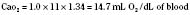
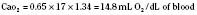
Transitional circulation
At birth, the fetus must make a transition to an adult circulatory system. The fact that the vast majority of newborns make this transition smoothly does not mean that the changes required are inconsequential. On the contrary, the events precipitating the transition from fetal to adult circulation are profound and immediate, requiring the fetus to make dramatic changes to ensure survival. The primary events that occur at birth are the clamping of the umbilical cord and initiation of breathing, with inflation of the lungs with air. These changes markedly alter the resistances in the cardiovascular system, changing the pattern of blood flow through the three vital shunts that characterize the fetal circulation. The fetus moves from a circulation that functions in parallel to one that is in a series. The lungs must now become the organs of oxygen supply and ventilation. Lung inflation and increased oxygen tension lower PVR dramatically, causing increased pulmonary blood flow and increased blood return to the left atrium. Cord clamping removes the low-resistance placenta from the circulation system and raises SVR. Left atrial pressure now exceeds right atrial pressure, closing the flap of tissue covering the foramen ovale. Normal intracardiac pressures keep the tissue flap over the foramen ovale closed, and over weeks it will completely seal. However, permanent occlusion does not occur in up to 25% of adults who retain a small defect or probe patency of the patent foramen ovale (PFO) (Hagen et al., 1984). Left atrial pressure rises for two reasons. First, left atrial volume increases significantly because of the increase in pulmonary blood flow. Second, left-sided pressures in the heart rise because of the increase in SVR. With the increase in systemic blood pressure and fall in PVR, flow through the ductus arteriosus becomes initially bidirectional and then very quickly evolves into a left-to-right shunt. Within the first hours of life the ductus arteriosus begins to close under the influence of the increased oxygen tension, loss of placental prostaglandins, and the more alkalotic environment of the newborn. Permanent closure of the ductus arteriosus takes weeks to occur.
Cardiac output in utero is considered to be the combined output of both ventricles and has been estimated at 400 mL/kg per minute. The right ventricle does more of this work, resulting in its hypertrophied state at birth. After birth, right ventricular work decreases because the volume load is reduced and PVR falls. In a series circulation, the cardiac output is the equal volume of blood ejected by each ventricle. Newborn cardiac output is the same as that in utero but by convention is calculated as 200 mL/kg per minute, which is the output of each ventricle (Lister et al., 1979). In the transition to postnatal life, it is the left ventricle that must cope with increased demands. Left ventricular output increases two- to threefold. This is accomplished by an increase in the left ventricular preload and stroke volume and heart rate. The volume load increase and rise in SVR represent a significant increase from the fetal state (Anderson, 1996). Additionally, as a result of the high beta stimulation associated with labor and delivery, heart rate increases and is near maximum. The newborn must maintain a high cardiac output because its metabolic rate (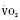 :6-7 mL O2/kg per minute) is double that of an adult (Anderson, 1990). With a large surface-to-mass ratio, the newborn is at a significant disadvantage for maintaining temperature. Its compensation is to use its high metabolic rate to generate heat for temperature homeostasis (Hill and Rahimtulla, 1965). The remainder of the increased
:6-7 mL O2/kg per minute) is double that of an adult (Anderson, 1990). With a large surface-to-mass ratio, the newborn is at a significant disadvantage for maintaining temperature. Its compensation is to use its high metabolic rate to generate heat for temperature homeostasis (Hill and Rahimtulla, 1965). The remainder of the increased 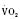 is devoted to growth and the oxygen requirement of the brain, which is proportionally much larger in newborns.
is devoted to growth and the oxygen requirement of the brain, which is proportionally much larger in newborns.
Persistence of fetal circulation most commonly occurs in instances of severe prematurity with respiratory distress syndrome (RDS). Hypoxemia is a potent stimulus, maintaining a patent ductus arteriosus (PDA). Flow through the ductus arteriosus remains possible as long as it is patent. The direction of that flow depends solely on the relative resistances between the systemic and pulmonary circulations. Flow through the PDA is usually left to right, adding an additional volume burden to the lung that is already coping with RDS. Significant RDS can be accompanied by elevations in PVR that are high enough to cause bidirectional or even right-to-left shunting at the PDA. The elevated PVR also raises right heart pressures, causing the same phenomenon to occur at the foramen ovale. A patent foramen ovale (PFO) provides an opportunity for intracardiac right-to-left shunting. Any other type of lung disease (e.g., meconium aspiration or pneumonia) that is severe enough can also cause persistence of the fetal circulation. Nonsteroidal antiinflammatory drugs, via their antiprostaglandin action, can be used to induce closure of a PDA. Indomethacin is the preferred agent (Giroud and Jacobs, 2007).
Pulmonary Blood Flow and Pulmonary Vascular Resistance
At midgestation, PVR is estimated to be tenfold higher than it is 24 hours after an uncomplicated birth. During the last trimester, PVR decreases slightly to levels seven- to eightfold greater than it is 24 hours after delivery (Rudolph, 1979). This reduction results from the physical growth of the pulmonary vasculature that increases the cross-sectional area by more than the corresponding increase in blood flow. Yet, PVR still remains high immediately prior to birth and must fall dramatically in the first postnatal minutes to ensure survival. Pulmonary blood flow increases by an amount corresponding to the decrease in PVR, and pulmonary blood pressure falls by 50% (Fig. 4-7). Thus, the pulmonary vasculature presents a puzzle that flies in the face of the normal principles of cardiovascular embryology. The guiding principle of cardiac and vascular development is that once normal structures form, the correct pathway for blood flow is established. Once flow is established, normal growth ensues. The pulmonary vasculature must grow normally despite greatly reduced flow. The elevated PVR necessary for fetal existence cannot be solely the result of anatomic hypoplasia. If this were the case, the transition to postnatal life would be rocky indeed. Rather, the pulmonary vessels must be of normal size but retain the ability to markedly vasoconstrict. Even more remarkable is that the ductus arteriosus is an outgrowth of the pulmonary arterial system, yet it responds in a completely opposite fashion to the pulmonary vasculature. The very stimuli that raise PVR in utero cause dilation of the ductus arteriosus. The factors that govern pulmonary vasoreactivity have still not been fully elucidated, but our understanding has grown considerably in the last 20 years (Martin et al., 2006).
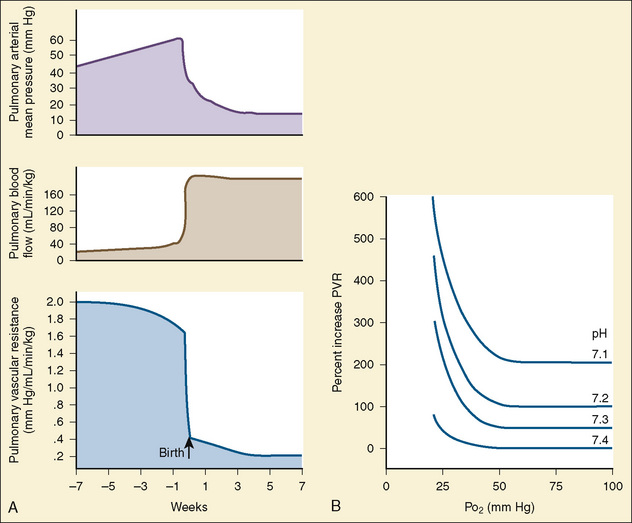
The primary force driving high fetal PVR is hypoxemia. The estimated Po2 in the pulmonary arteries is less than 20 mm Hg. This phenomenon of hypoxemia-induced pulmonary vasoconstriction (HPV) persists into adulthood, where it is vital in maintaining oxygen saturation during one-lung anesthesia. In utero, the mechanism of HPV is not fully understood. Oxygen is a potent stimulator of endothelial- derived vasodilating substances such as nitric oxide and prostacyclin. Nitric oxide activates soluble guanylate cyclase, which increases guanylate 3′-5′-cyclic monophosphate (cGMP). Prostacyclin stimulates adenylate cyclase to increase adenylate or adenosine 3′-5′-cyclic monophosphate (cAMP). Both cGMP and cAMP initiate vasodilation in pulmonary vessels. Under conditions of low oxygen tension, the release of nitric oxide and prostacyclin is presumably attenuated, tipping the balance in favor of vasoconstriction. The specific substances inducing pulmonary vasoconstriction are not well understood. Attention is focused on arachidonic acid and its metabolites and the potent vasoconstrictor endothelin (Tod and Cassin, 1984; Hickey et al., 1985a; Ivy et al., 1996).
At delivery, mechanical and biochemical factors lead to the abrupt fall in PVR (Teitel et al., 1990). Aeration of the previously fluid-filled lungs removes the external compressive force on the pulmonary vasculature. Responding to the sudden rise in oxygen tension, the endothelium secretes potent vasodilators, nitric oxide and prostacyclin. Luminal diameter increases as endothelial and smooth muscle cells become thinner. The increase in blood flow further recruits small lumen vessels, leading to an overall increase in the cross-sectional area of the pulmonary vascular bed. Smooth muscle relaxation occurs in the larger pulmonary vessels. This first, rapid phase of pulmonary vasodilation is followed by a period of remodeling that lasts for months. During this time there is maturation of vascular smooth muscles with continuing modest declines in PVR. PVR approximates adult values by about 2 months of age, and the remodeling process is usually complete by 6 months of age. During the first few years of life, new vessels develop to supply the growing lung parenchyma (Fig. 4-8). It is important to understand that in the early postnatal period, PVR is markedly affected by hypoxia and acidosis (Rudolph and Yuan, 1966). More recently, pain has also been associated with increasing PVR.
Myocardial performance
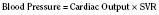
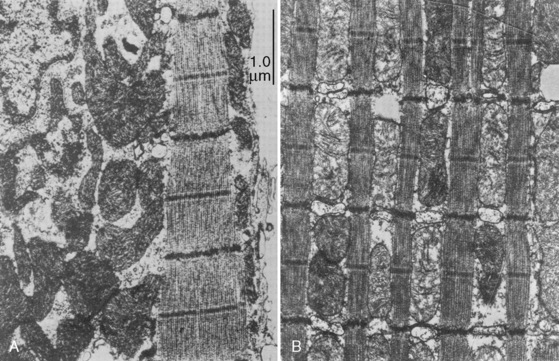
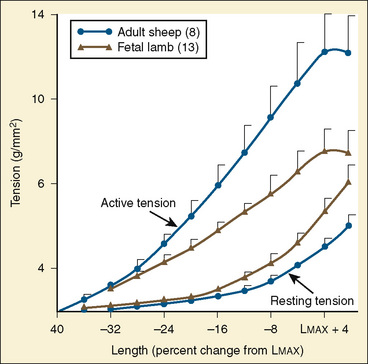
Cardiac cells are known as myocytes. Hyperplasia, which is an increase in cell number, is responsible for growth of the myocardium during fetal life. Hyperplasia continues into the early newborn period, after which increased demands on the heart can only be met by hypertrophy (increase in myocyte size). The functional unit of each myocyte is the myofibril. Myofibrils consist of contractile proteins arranged in repeating units called sarcomeres. The immature myocyte shape is rounded compared with the rodlike appearance of the adult myocyte. Immature myocytes also have a much larger surface-area to volume ratio. Adult myocytes contain multiple repeating rows of longitudinally arranged myofibrils. The newborn myocyte has fewer myofibrils in a more chaotic and scattered intracellular arrangement (Fig. 4-9). Sarcomere volume is only 30% of the newborn myocyte compared with 60% in the adult (Baum and Palmisano, 1997). The T-tubule system is a series of invaginations of the sarcolemma, or cell membrane, bringing it in close contact with the myofibrils. In this way, the action potential can rapidly disperse itself throughout the myocyte. Both the sarcolemma and T-tubule system are relatively well developed in the human newborn. The sarcoplasmic reticulum (SR) is a tubular network regulating the uptake, storage, and release of intracellular calcium. The adult heart relies on the SR to fully regulate calcium transport. Contrastingly, the newborn heart has an underdeveloped sarcoplasmic reticulum. There is more reliance on the sarcolemma and T-tubule system for the appropriate movement of calcium necessary for contraction and relaxation. The newborn has decreased contractile reserve on the basis of reduced sarcomere number and an immature system of calcium transport. The reduction in sarcomeres also reduces the compliance of the immature heart.
Preload
In 1895, the German physiologist Otto Frank published his observations on the relationship of diastolic filling of the heart and the pressure the heart was able to generate during systole (Frank, 1895). Ernest Starling, an English physiologist, conducted the classic experiment in the early 1900s that defined the length-tension relationship for cardiac muscle (Fig. 4-10). Understanding his experiment is important, because in the laboratory he was able to isolate that which is impossible to isolate in vivo. Using a fixed weight to keep afterload constant, he was able to prove that the tension developed was proportional to the length of the muscle strip prior to stimulation. The greater the length of the muscle strip, the greater the tension it was able to develop. The Frank-Starling mechanism, or Starling’s Law of the Heart, is taken from a famous lecture by Dr. Starling himself, in which he stated, “the energy of contraction, however measured, is a function of the length of the muscle fiber” (Starling, 1918).
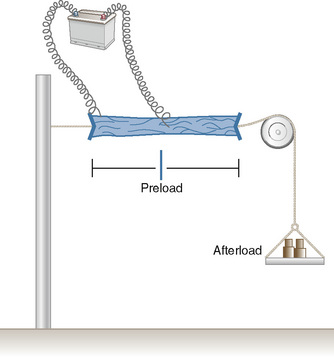
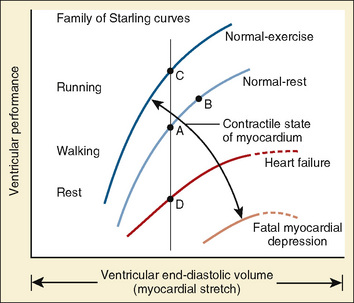
The clinical correlates of length and tension are left ventricular end-diastolic volume (LVEDV) and stroke volume, respectively. In the laboratory, the length of a muscle strip is easily determined because both ends can be fixed. This is very different from the intact heart, where the ventricle is a three-dimensional structure with complex geometry that defies the simple concept of length. Nevertheless, it is clinically appropriate to regard length as LVEDV. The LVEDV represents the loading of the ventricle. It is present before contraction, and therefore has come to be called preload. The force of ventricular contraction increases with increasing preload until a point is reached where the ventricle is over stretched and the force of contraction decreases. This point is the apex of the well-known Starling curve. Force of contraction is not synonymous with contractility, and for all points along a Starling curve, contractility is equal. Changes in the force of contraction, however, occur when preload changes (Fig. 4-11). Laboratory evidence has shown that resting sarcomere distance is 1.6 microns, with optimal conditions occurring at 2.2 microns. Excessive stretch, causing decreased force of contraction, does not occur until sarcomere length reaches 3.5 microns (Sonnenblick, 1974). In the laboratory the force of contraction increases until the sarcomere is stretched to over twice its resting length. If this translated fully to the intact heart, any increase in preload would result in increased force of contraction, because physiologically achieving a doubling of resting sarcomere length is almost impossible. However, well before a doubling of resting sarcomere length is reached, the patient falls on to the descending limb of the Starling curve. The reason is the relationship between volume and pressure, which is known as compliance.
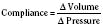
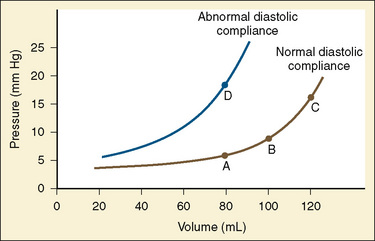
The change in pressure resulting from a change in volume is compliance. Volume is in the numerator of the equation meaning that if a large increase in volume is met by a relatively small rise in pressure, compliance is high. As a completely empty ventricle is filled, the initial rise in LVEDP is small. In this situation the ventricle is said to be compliant. When the ventricle is full, a small increase in volume results in a large increase in pressure. The ventricle is now poorly compliant or “stiff.” As with the Starling curve, it is important to note that moving to different points on the compliance curve does not mean that the intrinsic compliance of the ventricle has changed. A true intrinsic change in compliance will be reflected in a new compliance curve (Fig. 4-12). In adult cardiac medicine it is readily appreciated that ischemia, infarction, or hypertrophy result in a stiffer, or less compliant, ventricle. However, no ventricle, regardless of its intrinsic compliance, has an infinite ability to accept volume. Eventually the inflection point on the curve will be reached and pressure will climb rapidly for any given increase in volume. It is this sharp increase in pressure beyond the inflection point that is responsible for the descending limb of the Staring curve.

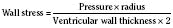
The response of the newborn heart to volume loading (relative insensitivity) has been the source of great confusion (Rudolph, 1974). This confusion stems from experimental work done in the 1970s and is the logical extension of the known structural differences in the newborn heart (Romero and Friedman, 1979; Gilbert, 1980). The response to preload was investigated in fetal sheep for the left and right ventricles. For either ventricle, isolated output rose only slightly with increases in filling pressures. Using a microsphere technique in fetal sheep, alterations in combined cardiac output were measured as blood volume was modulated (Gilbert, 1980). With a decrease of 10% in circulating volume, there was a significant drop in right atrial pressure, as well as in cardiac output. In contrast, cardiac output did not significantly change despite a significant change in right atrial pressure with a 10% increase in circulating blood volume from baseline. These data lead the author to conclude that the fetus operates at the upper end of the Starling curve and possesses limited cardiac reserve.
With only half the amount of sarcomere volume that is in an adult, connective tissue comprises a much greater percentage of the newborn heart. The myofibrils are fewer in number with a disorderly arrangement in the myocyte. There is much more stiff connective tissue. The sum total of these changes makes it plausible that the relatively noncompliant newborn heart does function at the upper end of its Starling curve, where the response to volume loading is blunted. In fact, the original experiments failed to account for an inevitable consequence of increases in preload. An increase in the preload leads to greater stroke volume, and if heart rate is unchanged, cardiac output must increase as well. Blood pressure must now rise unless there is a corresponding drop in SVR. Therefore, an increase in preload leads to an increase in afterload, which then acts to reduce stroke volume. This created the impression that the newborn heart could not increase stroke volume in response to volume loading. Other work, controlling for arterial pressure, shows that the newborn heart is indeed responsive to volume within the limitations of its decreased compliance (Fig. 4-13) (Kirkpatrick et al.; 1976, Hawkins et al., 1989). This has been shown in the laboratory with a sheep model and though echocardiography in the human fetus. The newborn can be likened to the hypertensive adult with diastolic dysfunction. This is not to suggest that the otherwise healthy newborn has diastolic dysfunction, because the newborn heart is not dysfunctional but normal for its stage of development. Rather, the lesson of diastolic dysfunction is that the ventricle is preload dependent. At the low end of the Starling curve, reduced preload is poorly tolerated. The upper end of the Starling curve is flattened, reflecting its reduced compliance. Between these two points is the steepest part of the curve, where increases in LVEDV result in significantly greater stroke volume. Clinical experience observing the response to volume infusion confirms this property of the newborn heart.
Afterload
In addition to preload, there is another loading force that influences myocardial performance. The force that resists the ejection of blood is known as afterload. In isolated muscle strip experiments, the afterload is represented by the weight against which the muscle contracts or shortens. The stretch applied to the isolated muscle strip before contraction is fixed, allowing the experiment to be conducted at a constant length, or preload. Plotting the velocity of myocardial shortening against progressively increasing afterload reveals an inverse relationship. The shape of the curve is exponential with the points of maximal and zero shortening both occurring at physiologically impossible limits. The greatest shortening velocity occurs at zero load. When the load is so great that no muscle fiber shortening can occur, isometric contraction occurs. Between these two extremes, it is intuitive that increasing afterload results in reduced velocity of muscle fiber shortening. Transferring this straightforward laboratory concept to the intact circulation is difficult (Martin et al., 2006). The force generated by the shortening of an isolated muscle strip is directed in only one plane. This is very different from the three-dimensional contraction of the left ventricle with its asymmetric geometry. Additionally, in the laboratory the force that resists the shortening of the muscle is the weight applied to the muscle strip. Physiologically, the force that resists the ejection of blood is a complex interplay between blood pressure, vascular impedance, walls stress, and inertia. Finally, blood pressure and SVR are commonly used measures of afterload, but neither is fully accurate.
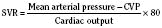
These two hypothetical patients are very different, yet the SVR is similar. The first patient has normal cardiovascular function and the second patient’s cardiovascular function is compromised. A reduction in contractility has caused a fall in cardiac output despite the attempts of the ventricle to compensate with a higher diastolic filling pressure. This example demonstrates how SVR in isolation does not necessarily reflect large changes in loading conditions and cardiac output. The modified Law of Laplace describes a better approximation of afterload (see Preload section p. 91). As discussed in the section on preload, wall stress was demonstrated to be a key component of myocardial oxygen demand. During left ventricular ejection, the value for pressure is the systolic blood pressure. By including the radius and accounting for the compensatory mechanism of left ventricular hypertrophy the modified Law of Laplace provides a more comprehensive view of the forces that resist ventricular ejection. Importantly, it demonstrates the concept that the dilated ventricle with an increased radius must eject blood under increased afterload conditions. Given the inability to easily measure ventricular radius and thickness, the modified Law of Laplace is not a clinical parameter to guide management. It is presented here to help the reader conceptualize afterload. It still cannot describe all the variables that account for afterload in the intact cardiovascular system.
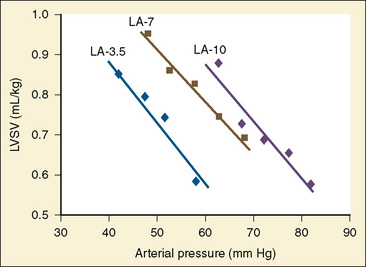
Studies of the intact heart that assess the effect of changes in afterload on cardiac output are difficult to interpret without controlling for the other variables. The most important additional variable is preload. Preload and afterload are linked. For instance, pharmacologically increasing afterload decreases stroke volume. As the heart ejects less blood with each contraction, the end-diastolic volume must increase if venous retum is held constant. With an increase in LVEDV the Starling curve dictates an enhanced ventricular force of contraction, which acts to return stroke volume to its previous level. The general inverse relationship between afterload and stroke volume holds across the spectrum from fetal life to adulthood (Friedman, 1972). The question is whether the immature myocardium is excessively sensitive to increases in afterload. In the laboratory, afterload can be assessed while strictly controlling for preload. These studies have revealed that the fetal myocardium is indeed more sensitive to increases in afterload than the adult myocardium (Friedman, 1972). Animal experiments with an intact circulation also show a decrease in ventricular output under conditions of increasing afterload (Fig. 4-13) (Gilbert, 1982; Thornburg and Morton, 1986). Van Hare and colleagues (1990) used a balloon occluder in the aortas of fetal sheep to modulate afterload while keeping preload constant. Combined ventricular output was measured by transducing aortic flow. They noted that at physiologic mean atrial pressures, there was an inverse linear relationship between mean arterial pressure and stroke volume. This exists for both right and left ventricles. Despite this, a fetus normally makes a smooth transition to postnatal life when the low-resistance placenta is removed. At birth, pulmonary blood flow dramatically increases, raising left-ventricular preload. Stroke volume increases, which leads to an increase in systemic blood pressure. Despite the rise in blood pressure and the loss of the low-resistance placenta, cardiac performance in the newborn is not compromised. The explanation is that much of the rise in afterload is secondary to increased preload. In clinical practice with newborns and young children, it is rare to encounter conditions of purely increased afterload. A more likely situation is significant hypovolemia that results in hypotension. Despite the low afterload state, overall cardiac performance is impaired, because the decrease in afterload is caused by decreased preload. The correct therapy is to restore intravascular volume. Blood pressure improves, and the increased afterload is well tolerated. This example illustrates the interrelationship between these two variables of cardiac output.
It has been shown that when preload is held relatively constant, cardiac output moves inversely within the physiologic range of afterload. The relevance of afterload in the newborn stage might be questioned, because systemic hypertension is so rare. The right ventricle develops in an environment of raised PVR, yet after birth it is more sensitive to increases in afterload. Although there is limited ability of the systemic circulation to become hypertensive, the pulmonary circulation, under appropriately provocative conditions, can revert to its fetal state. Postnatally, the right ventricle begins to remodel in response to decreasing pulmonary artery pressures. Sustained elevations in pulmonary artery pressures decrease right ventricular output and reduce left-ventricular preload (Thornburg and Morton, 1983). Because both ventricles share the septum, the strain on the right ventricle impairs left-ventricular filling and contraction (Rein et al., 1987). Reductions in left-sided preload and contractile force demonstrate how the inability of the right ventricle to cope with increased afterload can lead to decreases in systemic cardiac output. In the newborn stage, the potential for pulmonary hypertension far exceeds that of systemic hypertension. This is the most common scenario for a significant rise in afterload while preload remains relatively unchanged. The strain on the right side of the heart and ventricular interdependence may lead to biventricular failure.
Contractility
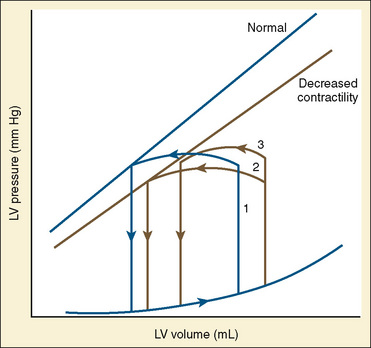
The immature myocardium has a reduced sarcomere concentration combined with a transport system for calcium that is not fully developed. The myofibrillar arrangement is also more disorganized in the infant heart when compared with the adult heart. Mitochondria, the energy powerhouse of the myocyte, are reduced in number in the immature heart (Barth et al., 1992). Based on the above differences, a reduction in contractility is expected. Contractility is measured by the tension developed in the isolated muscle strip. This expected reduction in contractility of the immature myocardium is confirmed in laboratory experiments where preload and afterload are controlled. Across the entire range of loading conditions, fetal cardiac muscle generates less tension than adult myocardium (Fig. 4-14) (Friedman, 1972; Romero et al., 1972). In an intact heart it is not possible to fully separate the influences of different loading conditions. Nevertheless, fractional area shortening of the ventricle measured echocardiographically is used as a measure of contractility. Decreased fractional area shortening has been observed in the fetal heart (St. John Sutton et al., 1984). Effecting a true increase in contractility requires either a greater number of contractile elements or more vigorous action by the contractile elements already present. Increasing the number of contractile elements (hyperplasia and hypertrophy) occurs as a normal part of development. Improving the action of the contractile elements already present requires an understanding of the singular role of calcium. Therapy to improve inotropy exerts its action at the myocyte level by affecting the levels of intracellular calcium.
Calcium and Diastolic Function
A comprehensive understanding of the excitation-contraction coupling mechanism of contraction and relaxation is important for the safe practice of pediatric anesthesia. The relevant points are included here. The formation of actin-myosin cross-bridges occurs under the influence of calcium. Tropomyosin and troponin are inhibitory proteins that prevent the formation of actin-myosin cross-bridges. Calcium induces conformational changes in tropomyosin and troponin that remove their inhibition to actin-myosin cross-bridge formation. This is the basis of contraction and requires major changes to intracellular calcium content. It is estimated that the difference between diastolic and systolic calcium levels is 100-fold (10–7 M to 10–5 M) (Shah et al., 1994). Responsibility for these large changes in intracellular calcium concentration lies with the integrated function of the sarcolemma, the T-tubule system, and the SR. Breaking the actin-myosin cross-bridges and returning the ventricle to its baseline state is an energy-consuming process that requires adenosine triphosphate (ATP) and calcium reuptake. This is primarily accomplished by the removal of calcium into the SR and the sarcolemmal Na+–Ca2+ exchanger (Mahony, 2007). Ryanodine is an inhibitor of SR function. In the presence of ryanodine, fetal and newborn hearts are minimally affected, whereas adult hearts suffer a significant decline in contractility (Penefsky, 1974).
Experimental modulation of calcium handling has been shown to alter ventricular mechanics. In a study with mice that overexpressed the Ca2+ ATPase, the rate of myocardial relaxation was directly correlated with the rate of calcium uptake by the SR (He et al., 1997). In guinea pigs, ryanodine blockade of SR function caused impaired relaxation (Kaufman et al., 1990). Additionally, there were age-dependent changes in the relaxation response. In adult hearts, ryanodine blockade produced a greater impairment of relaxation compared with immature hearts. Concomitantly, there was greater density of Ca2+ pumps, greater calcium-dependent ATPase activity, and greater uptake of calcium in isolated SR vesicles from adult hearts as compared with those isolated from immature hearts. With decreased SR function in the immature heart, the extrusion of calcium across the cell membrane assumes greater importance. The Na+–Ca2+ exchanger provides the primary mechanism for this. The exchanger is sensitive to membrane potential as it exchanges three sodium ions for one calcium ion. Developmental changes in the exchanger function have been demonstrated, and there are differences among species in relative function (Nakanishi and Jarmakani, 1981; Artman, 1992; Boerth et al., 1994). In the rabbit, with its poorly developed SR exchanger, mRNA is significantly elevated in the neonate versus the adult (Artman, 1992). The relative contribution of calcium sequestration between the SR and the exchanger in the human is similar to that of the rabbit (Mahony, 2007). In addition to reduced calcium handling, there may be developmental changes in troponin interactions that effect calcium binding. There is differential expression of cardiac and slow skeletal-muscle isoforms. In fetal hearts, the predominant form is the slow-skeletal form. Shifts to the cardiac isoform are completed in the first year of life. This is significant in that the cardiac isoform has a decreased affinity for calcium when phosphorylated, potentially aiding the removal of calcium from troponin C.
The sum total of these events is to demonstrate that across all age ranges the intracellular handling of calcium is critical for systolic function and even more important for diastolic function. The diastolic characteristics of the immature myocardium have been studied. The immature myocardium has been described as “stiffer” when compared with the adult myocardium. Ventricular compliance increases with maturation (Friedman, 1972; Romero et al., 1972; Kaufman et al., 1990). Measurements of rates of pressure change demonstrate decreased relaxation capabilities in neonatal hearts when compared with adult hearts (Palmisano et al., 1994). There are a number of potential factors involved. Structural and contractile protein changes, extra-cardiac structures, and maturing organelle function have been studied. The diastolic relaxation properties of the ventricle are a key determinant of the ventricle’s compliance. Reductions in calcium reuptake lead to expected decreases in diastolic relaxation (Kaufman et al., 1990). Echocardiographic studies on human fetuses with normal hearts have demonstrated age-related changes in early diastolic flow that are consistent with improved relaxation (Kenny et al., 1986; Harada et al., 1997). Consequently, intracellular calcium homeostasis in the newborn is more dependent on a normal serum ionized calcium level, and the newborn tolerates hypocalcemia poorly. Immaturity of calcium transport leads to a decrease in systolic-force generation and a decrease in diastolic relaxation. The manifestation of impaired diastolic relaxation is a reduction in compliance. Clinically, these issues must be appreciated when interpreting assessments of volume based on pressure readings such as the central venous pressure (CVP).
Heart Rate
The question is whether the newborn who has a heart rate within normal range is fundamentally different than the adult. When corrected for weight, stroke volume is similar across all ages. The high cardiac output of newborns and infants can only be achieved with a heart rate that is significantly higher than in adults. This has created the idea that the newborn is dependent on heart rate. Here it becomes difficult to sort out the isolated effect of heart rate from its effects on loading conditions and contractility. It is not intuitive as to why changes in heart rate alter contractility. The mechanism is known as the force-frequency relationship. Experiments using atrial pacing, while controlling loading conditions, demonstrate an increase in stroke volume with an increase in heart rate (Anderson et al., 1982).With constant loading conditions, the only explanation for increased stroke volume is that contractility has increased. Thus, an increased heart rate improves contractility. The basis for the force-frequency relationship is that an increase in heart rate is accompanied by enhanced release of intracellular calcium (Parilak et al., 2009). There is a suggestion that the force-frequency relationship has minimal effect in newborns but is present during infancy (Wiegerinck et al., 2008). Spontaneous increases in heart rate also improve cardiac output. In this scenario, the increase in heart rate is the result of a neurohumoral stimulus with an effect that cannot be isolated to producing tachycardia. Both preload and contractility can be expected to increase, as long as the increase in heart rate does not lead to deleterious changes in preload or myocardial oxygen supply and demand. The combination of these factors allows the newborn and infant to use heart rate to significantly augment cardiac output.
Integrating Preload, Afterload, and Contractility
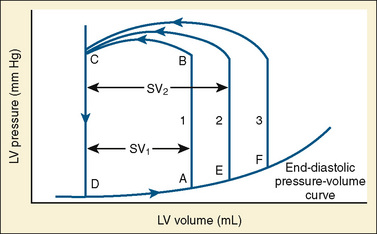
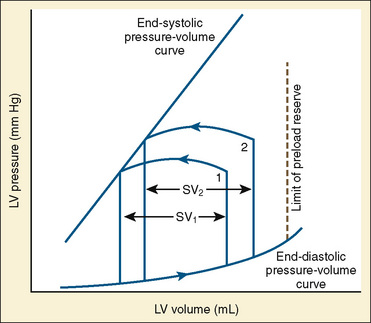
The determinants of myocardial performance, while discussed in isolation, are actually intricately linked. Diagrammatically, the determinants of myocardial performance can be represented by ventricular pressure-volume loops (Suga et al., 1973). The loop shows the pressure and ventricular volume changes that occur during one cardiac cycle. Increases in preload while afterload is held constant result in greater stroke volume (Fig. 4-15). This simple curve does not provide any information about contractility. The end systolic pressure-volume relationship (ESPVR) is a family of curves that is generated by rapidly altering preload or afterload. The curves create a series of points that are connected to become the ESPVR line. The slope of this line represents contractility (Fig. 4-16). The pressure-volume loop illustrates the concept that movement along the ESPVR line reflects changes in loading conditions while contractility remains constant. In Figure 4-16, the increases in afterload result in a series of points that are connected to become the ESPVR line. The slope of the ESPVR line represents contractility. The two figures of ventricular pressure-volume loops represent an idealized situation where preload and afterload can be manipulated independent of each other. In the intact organism, preload and afterload are linked. The interdependence of preload and afterload is shown in a new ventricular pressure-volume loop (Fig. 4-17).
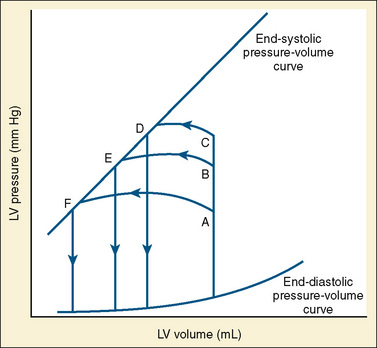
In states of decreased contractility, the ventricular pressure-volume loop displays the limitations of the failing heart. Analysis of the ventricular pressure-volume loop shows that the failing ventricle is sensitive to changes in loading conditions (Fig. 4-18). The ESPVR line has shifted down and to the right. With preload held constant, stroke volume is reduced. The body’s response to this situation is to increase LVEDV in an attempt to preserve stroke volume. Stroke volume has improved at the expense of a higher end-diastolic pressure. The ventricular pressure-volume loop demonstrates why afterload reduction is the cornerstone of medical therapy for the failing heart. Any increases in afterload come at the expense of stroke volume, with a limited ability for compensatory changes in preload.
Developmental aspects of cardiomyocyte structure and function
Subcellular Structures
Sarcolemmal Ion Channels
Numerous voltage-dependent and ligand-gated ion channels reside in the sarcolemma, and a full discussion far exceeds the space available in this chapter. Although developmental changes in the inward sodium current have been noted in several species, there appear to be few developmental changes in the human atrium (Sakakibara et al., 1992). A variety of inward and outward potassium channels exists. Although developmental changes have been documented, methodologic differences and differences among species do not allow application to human development (Sanchez-Chapula et al., 1994; Xie et al., 1997; Morrissey et al., 2005). Na+,K+ ATPase maintains the sodium gradient across the cell and is inhibited by the cardiac glycosides, such as digitalis. There are developmental changes in the isoform distribution of the enzyme subunits, and this enzyme has less activity in immature myocardium. Calcium handling is crucial to myocardial contraction. Calcium channel (Ica) density increases two to threefold in the developing rabbit, although the voltage sensitive activation is similar to that of humans (Osaka, 1991; Huynh et al., 1992; Wetzel et al., 1993). In one study of human atrial myocytes (presumably from ill children), younger hearts had decreased calcium channel density, and in another study, more rapid inactivation of calcium current was evident (Hatem et al., 1995; Roca et al., 1996). However, in the realm of human myocardial development, many of these hearts were fairly mature when studied. The Na+–Ca+ exchanger, which can serve to bring calcium either into or out of the cell, has higher activity in immature myocardium in a variety of species (Artman et al., 1995). This is a purported source of additional calcium entry into the contractile apparatus in immature myocardial cells that have relatively deficient sarcoplasmic reticulum. There is increased activity of the Na+–H+ exchanger in immature myocardium, and this has been implicated as a factor in the greater resistance of immature myocardium to acidosis (Downing et al., 1966; Haworth et al., 1997).
Transverse Tubules
Transverse tubules, or T-tubules, invaginations of the sarcolemma in ventricular, but not atrial, myocardial cells have been mentioned in the discussion of myocardial performance. They allow transmission of the action potential, with its attendant ion shifts, to all parts of the cell, which allows rapid activation of the entire cell. T-tubules are thus a required component of larger cells (Gotoh, 1983). Maturation of T-tubules is associated with the increased size of mature myocardial cells, with the sarcolemmal calcium channels further from the contractile apparatus. Species that have a relatively mature myocardium at birth have well-developed T-tubules at birth, whereas animals with immature myocardium at birth do not. T-tubules first appear at about 30 weeks gestation in humans (Kim et al., 1992).
Mitochondria
Mitochondria increase in size, relative volume, and internal compactness and complexity during myocardial development, and their growth may continue into the postnatal period (Barth et al., 1992). In some species, maturation is postnatal. Maturation of mitochondria mirrors the shift from a primary carbohydrate energy source of immature myocardium to the primary long-chain free fatty-acid energy source of mature myocardium (Fisher et al., 1980, 1981).
Cytoskeleton
An intracellular construction of microtubules and microfilaments links the contractile elements, T-tubules, sarcolemma, mitochondria, and nucleus. This scaffolding organizes the subcellular components that participate in cell signaling and allows transmission of the force of contraction to be applied to the myocyte. Mutations in several of these can be responsible for several familial cardiomyopathic conditions. The cytoskeleton not only undergoes modification with myocardial development, but microfilaments also play a role in the adaptive response to mechanical loading of the heart (Small et al., 1992; van der Loop et al., 1995; Schroder et al., 2002). One of the most important roles of the cytoskeleton is to link the thick filaments. Titin, the largest protein in the human, extends from the Z-disc to the M-line of the sarcomere. It both aligns the thick filament and has a spring-like function that determines passive tension. Titin isoforms are under developmental regulation, with fetal myocardium having the more compliant N2BA isoform, which is then replaced with the stiffer isoforms. The conversion of isoforms is species dependent, but it correlates with the shift from the more compliant fetal myocardial cells (when studied removed from the surrounding matrix) to the less compliant cells of the adult.
Sarcoplasmic Reticulum
The tubular network of SR regulates intracytosolic calcium concentration. The ryanodine receptor is located in the SR, as are a variety of proteins and channels that regulate calcium. Calcium regulation by the SR is critically important for the release of calcium (contraction) and its re-uptake (diastole). In the mature cardiac myocyte, the SR is the primary source of calcium to the cell. The amount of SR is significantly reduced in immature myocardium, as are indices of SR function, in a variety of species (Maylie, 1982; Nakanishi et al., 1987; Nassar et al., 1987).
Contractile Proteins
These proteins compose the thin filaments of the I-band (actin, the troponin complex and tropomyosin) and the thick filaments (myosin and titin) are composed of contractile proteins. All of these proteins have developmentally regulated changes in isoform expression. Additionally, their expression is specific to cell type (e.g., cardiac vs. noncardiac or atrial vs. ventricular) and is regulated by physiologic signaling, such as thyroid hormone or diabetes (Baum et al., 1989).
Myosin
Myosin is the most abundant contractile protein and is responsible for transducing chemical energy (ATP) into mechanical energy. It is composed of two heavy and two light chains. Different heavy-chain myosin isoforms have different ATPase activity, conferring different calcium sensitivity. Developmental changes in myosin are species specific. In humans, the V3 isoform is most common in the fetus (90% at 30 weeks, gestation), decreases in the neonate, and reaccumulates beginning in the second month of life to become predominant in the adult. The V3 isoform, consisting of two β heavy chains, has the lowest ATPase activity of the various myosin types and consumes less ATP for the same amount of force generation (Cummins et al., 1980). Atrial myosin, with its briefer contraction, is primarily the V1 isoform (consisting of two α heavy chains). Regulation of the myosin heavy chain is at the transcriptional level and appears to account, at least in part, for the rapid conditioning of a ventricle after the placement of a pulmonary artery band (Lompre et al., 1984). The myosin light chain, located near the head of the heavy chain, has a variety of subunits that regulate myosin ATPase activity and that are also under developmental regulation.
Actin
The thin filament consists of two intertwined bands of actin monomers. Both skeletal and cardiac forms of actin are present during human cardiac development. In the early embryonic heart the skeletal form predominates (more than 80%). The cardiac form is present in the early stages of heart tube development and gradually increases. The onset of rhythmic contraction coincides with the disappearance of the skeletal isoform. In humans, cardiac actin increases to approximately 50% in the first decade, but the physiologic implication of this shift is not known (Boheler et al., 1991). Skeletal actin rapidly increases after the imposition of an acute pressure load to the ventricle before declining slowly.
Tropomyosin
Tropomyosin lies in the groove between the two actin bands and, depending on its deformation by troponin, either permits or prevents the interaction of actin and myosin. There are two isomers, and the isomer distribution depends on the intrinsic heart rate (and therefore the species). In humans, the amount of β isoform increases from 5% in the fetal ventricle to 10% in the adult, but the physiologic implication of this shift is not known (Humphreys and Cummins, 1984).
Troponins
The three distinct but functionally coupled proteins of the troponin complex (troponins T, C, and I) confer calcium sensitivity to actin-myosin cross-bridge formation, and each has multiple developmental and species specificity (Anderson et al., 1991). Troponin T binds the complex to tropomyosin; troponin C binds calcium; and troponin I regulates the interaction of the complex with tropomyosin, binding to troponin C during systole and to actin during diastole. Four forms of cardiac troponin T are present in the human as the result of alternative splicing of a single gene, and their expression is developmentally regulated (Anderson et al., 1991). All isoforms are expressed in the fetal heart, but only one (cTnT3) is expressed in the adult. In failing human hearts, including those of children, cTnT1 and cTnT4 are upregulated (Saba et al., 1996). These isoform shifts likely affect contractility, as they modify the calcium sensitivity of the contractile apparatus. Troponin I has both cardiac and slow skeletal forms. Both are found in fetal hearts. Approximately 70% of troponin I is the skeletal form, but the isoform shift to the adult form of troponin I (all cardiac) is complete in the human by 9 months of age (Sasse et al., 1993). Phosphorylation of the cardiac isoform, but not the skeletal isoform, decreases the sensitivity of troponin C and the myofilament for calcium, as well as the affinity of troponin I for troponin C, with both decreases altering contractile performance. The slow skeletal form of troponin I in neonates may contribute to the resistance of the neonatal myofilament to deactivation at acid pH, perhaps contributing to the greater recovery of neonatal myocardium from an acidotic insult (Solaro et al., 1986). Although there are several isoforms of troponin C, only the cardiac isoform is expressed in cardiac muscle.
Myocardial Energy
Newborn sheep have a higher myocardial oxygen consumption than adult sheep. These metabolic demands of the developing heart are met by a capillary network of higher density than that found in adults (Fisher et al., 1982).
The immature myocardium uses lactate as a primary energy source. This is in contrast to the mature myocardium, where fatty acids are the primary energy source. To be metabolized, fatty acids must first be transported into the mitochondria. This is accomplished by carnitine palmitoyl-CoA transferase. Activity of this enzyme is reduced in the immature myocardium, suggesting the necessity of carbohydrate as the primary energy source (Fisher et al., 1980, 1981).
Electrophysiology
The sinus node can initially be identified as a horseshoe-shaped structure at the lateral junction of the superior vena cava and the right atrium. As the heart develops, it assumes the elongated spindle shape that is seen in the mature heart. Using voltage-sensitive dyes, spontaneous cellular depolarization has been observed prior to the development of contractile function. Although spontaneous depolarization is also seen in the developing atrium and bulboventricular portion, cells that are destined to form the sinus node have a higher intrinsic rate of depolarization (Pickoff, 2007).
Discrete connections between the sinus node and the atrioventricular (AV) node are a matter for debate. The lack of an identified cell type insulated from the surrounding atrial myocardium has been considered evidence of a lack of specialized internodal conducting pathways (Anderson et al., 1981). Preferential conduction along bands of atrial myocytes in the anterior septum may be to the result of a more organized cellular alignment (Racker, 1989). More recent evidence from studies of developing human hearts has demonstrated three internodal pathways identified by antibody staining (Blom et al., 1999).
Functional changes in the electrophysiologic properties of the atrium have been described. Action potential duration in human neonates is shorter than in adults. This is relevant in that it may explain the highest incidence in childhood of atrial arrhythmias in the fetus and neonate (Pickoff, 2007). Once these dysrhythmias are terminated in the newborn, there is a low incidence of recurrence.
Delay in contraction between the atrium and ventricle is seen before the development of the AV node. Action-potential recordings from the AV canal area of the developing chicken heart demonstrate characteristics that are to be expected of slowing areas of conduction—namely, a slow rate of rise and a prolonged duration. These characteristics are further linked to differential expression of acetylcholinesterase. Expression is higher in the AV canal region compared with expression in the free wall of the atrium or ventricle (Mikawa and Hurtado, 2007; Pickoff, 2007).
AV nodal cells are distinct from the other embryonic myocardial cells. They have differential expression of calcium-regulatory proteins and membrane channels (Mikawa and Hurtado, 2007). Formation of the AV node is an incompletely understood complex series of events that likely involve modulation by the gene NK×2-5 (Mikawa and Hurtado, 2007; Briggs et al., 2008).
Formation of the Purkinje system is an area of active investigation. Purkinje-like function is seen after cardiac looping (Pickoff, 2007). However, the network is still developing in the fetal heart. Studies suggest a paracrine interaction between the fetal myocardium and the cardiac endothelial cells. Purkinje fiber development is seen primarily in two sites, periarterially and subendocardially. Phenotypical recruitment of a beating myocyte to a conduction fiber is thought to be modulated by endothelin-1 (ET-1). The ET-1–induced expression of conduction-tissue markers in myocytes is dependent on dosage and inhibited by ET-1 antagonists (Mikawa and Hurtado, 2007). Exposure of myocytes to ET-1 is also associated with downward regulation of markers found primarily on muscle cells.
In chicken embryos, conduction velocities increase with age and are related to the emergence of fast-conducting sodium channels (Shigenobu and Sperelakis, 1971; Pickoff, 2007). Findings have been similar in mammalian systems (Rosen et al., 1981; Pickoff, 2007). Of note is the maintenance of conduction slowing from atrium to ventricle seen in the primitive heart. In a canine model, measurement of AV-nodal conduction demonstrated that conduction was faster in immature hearts, but the AV node, not the ventricular myocardium, remained the site of conduction slowing with differential atrial pacing (McCormack et al., 1988).
Through fetal magnetocardiography, conduction times have been noted to increase with gestational age (Kahler et al., 2002; Stinstra et al., 2002; van Leeuwen et al., 2004). This is thought to be related to myocardial growth (van Leeuwen et al., 2004). Intracardiac conduction times have been measured in children and compared with those in adults. There were no age-related differences in atrial, AV-nodal, or His Purkinje-conduction times (Gillette et al., 1975).
Central Nervous System Regulation of Cardiovascular Function
The autonomic nervous system is developed in the human by about 27 weeks’ gestation. Parasympathetic innervation precedes sympathetic innervation (Fig. 4-19). In sheep, at least, the postnatal increase in contractile function with adrenergic stimulation is a consequence of higher resting adrenergic tone rather than a maturation of the myocardium (Teitel et al., 1985). The β-adrenergic/adenylate cyclase system develops during fetal life. The density of the β-adrenergic receptor density peaks at term and declines postnatally, mirroring the maturation of cardiac sympathetic innervation. Calcium channels in neonatal myocardium do, however, respond less to β-adrenergic stimulation than they do in the adult, and isoproterenol stimulation of adenyl cyclase is blunted in the late fetus and neonate (Osaka and Joyner, 1992). In the late term fetus and the neonate, coupling of the β-receptor and adenylate cyclase may be decreased, because differential β-adrenergic response to isoproterenol and forskolin, a direct activator of adenylate cyclase, has been observed (Schumacher et al., 1982; Tanaka and Shigenobu, 1990). This finding may help explain the decreased response of calcium channels to β-adrenergic stimulation in the neonatal myocardium (Baum and Palmisano, 1997).
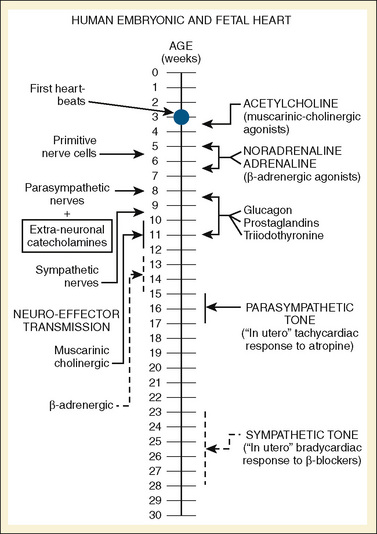
FIGURE 4-19 A timeline of human fetal autonomic development.
(From Papp JG: Autonomic responses and neurohumeral control in the early antenatal heart, Basic Res Cardiol 83:2, 1988.)
Vagal myelination in humans continues through the fetal period and reaches adult levels by about 50 weeks postconceptual age (Sachis et al., 1982). In humans, parasympathetic input to the heart is from the superior, inferior, and thoracic branches of the vagus nerve (Hildreth et al., 2009). The relative balance of sympathetic and parasympathetic stimulation is reflected in the decreasing mean heart rate as a child reaches adolescence.
The aortic arch and carotid bodies have baroreceptors that provide the afferent limb of the feedback circuit. Stimulation of these receptors sends impulses to the cardioinhibitory and vasomotor centers of the medulla. In turn, the efferent signals result in decreased blood pressure, vasodilation, and slowing of the heart rate. Arterial baroreflexes are present and operational in healthy and critically ill human neonates; human neonates, even preterm neonates, have well-developed vagally mediated cardiac responses to hypoxemia and other stimuli, although the course of maturation is unclear (Thoresen et al., 1991; Buckner et al., 1993). Decreasing sensitivity in studies of awake animals suggests a decreasing role for heart rate in the control of blood pressure in aging animals (Palmisano et al., 1990).
Stretch receptors in the myocardium also provide afferent signals to the central nervous system. Within the atrium there are two types of stretch receptors. Type A receptors are sensitive to pressure and are activated by atrial systole. Type B receptors are sensitive to volume and fire during ventricular systole. They have opposing effects on the sympathetic nervous system, with Type A receptors stimulating sympathetic activity. Secretion of vasopressin is inhibited by stimulation of stretch receptors. Stretch receptors are also found in the ventricular myocardium and when activated cause hypotension and bradycardia (Teitel et al., 2007).
Pulmonary Vascular Development
Pulmonary vessels are described by their relation to the bronchioles. Terminal bronchioles are the smallest purely conductive airways. Beyond them lie respiratory bronchioles, with dual conductive and gas-exchange functions, and then more distally, the alveolar units. Resistance in the pulmonary vasculature is at the level of the small pulmonary arteries (approximately fifth to sixth generation), which are defined as preacinar if they are proximal to the terminal bronchioles and intraacinar if they course parallel to the respiratory bronchioles and alveolar units. Conductive airways and preacinar arteries are fully present by 16 weeks’ gestation (Hislop and Reid, 1973). There is a thickened muscular layer in the medial wall of preacinar vessels, most prominent at the fifth and sixth generation arteries (Hlastala et al., 1998). Beyond the preacinar arteries there is a transition zone of vessels at the level of the respiratory bronchioles with incomplete muscularization in the medial layer. The medial layer of these incompletely muscularized vessels contains pericytes (mesenchymal cells capable of differentiation) and precursor smooth muscle cells. Compared with a complete muscular layer, partial muscularization is believed necessary to aid the gas exchange function of respiratory bronchioles because alveolar development at birth is very immature. Arteries associated with immature alveoli are completely free of a muscular medial layer at birth.
Diseases that affect normal pulmonary blood flow patterns have far-reaching consequences. In fetuses with abnormal restriction of pulmonary blood flow, there is often hypoplasia of the pulmonary arterial system. The degree of hypoplasia can be mild to fulminant, with atresia of all or parts of the pulmonary arterial system. For example, with tetralogy of Fallot, the level of pulmonary outflow obstruction is typically in the infundibulum of the right ventricular outflow tract. The resulting decrease in pulmonary blood flow affects the main and branch pulmonary arteries, which can be variably hypoplastic. Despite being hypoplastic, the vasculature has the ability to remodel and grow with improved blood flow. In patients who receive an early modified Blalock-Taussig shunt that provides augmentation of pulmonary blood flow, growth of the pulmonary arteries is seen. Also, in patients with hypoplastic pulmonary arteries, patch augmentation can result in reversal of pulmonary artery hypoplasia (Agnoletti et al., 2004).
Disruption of normal pulmonary vasculature has also been observed when the perturbation to pulmonary flow is on the venous side. In hypoplastic left heart syndrome, blood returning to the left atrium must cross over to the right atrium via the foramen ovale. If the foramen ovale is widely patent, blood returning via the pulmonary veins to the left atrium is shunted across to the right atrium. Right-sided structures are often enlarged to accommodate the flow. However, if the patent foramen ovale is restrictive, pulmonary venous pressure rises and development of the pulmonary arterial vasculature is compromised. There is pressure rather than volume stress on the pulmonary vasculature. At birth, the increase in pulmonary blood flow with lung expansion leads quickly to left-atrial hypertension, compromising pulmonary blood flow and resulting in severe hypoxemia. Even if the emergent situation is optimally treated, surgical mortality remains high. At autopsy, the pulmonary veins of these infants are thickened and “arterialized” (Rychik et al., 1999). In juxtaposition to the pulmonary hypoplasia seen in patients with reduced pulmonary blood flow, patients with increased pulmonary blood flow may develop enlarged pulmonary arteries. In an uncommon variant of tetralogy of Fallot, the pulmonary valve is absent, resulting in free pulmonary insufficiency. This increased volume of blood flowing back and forth through the main and branch pulmonary arteries leads to massive dilation.
The most common scenario of altered postnatal pulmonary blood flow occurs in patients with left-to-right shunts. Despite the increased pulmonary blood flow in the first few months of life, PVR continues to fall, leading to even greater pulmonary blood flow. The consequence of increased flow through the same cross-sectional vascular area is pulmonary hypertension. Because resistance is defined as pressure divided by flow, the diagnosis of pulmonary hypertension is not synonymous with increased PVR. The pulmonary vasculature accepts the increased blood flow at the expense of a rise in pressure. Resistance only increases when pressure remains high and pulmonary blood flow falls. Untreated, the natural result of this situation is a condition of pulmonary hypertension, elevated PVR, and fixed, irreversible pulmonary vascular disease. This is known as Eisenmenger’s syndrome. In 1897, Dr. Eisenmenger described a 32-year-old male with cyanosis and exercise intolerance who died from massive hemoptysis (Eisenmenger, 1897). The autopsy revealed a large ventricular septal defect (VSD) and overriding aorta. It was the first demonstration of congenital heart disease causing pulmonary vascular changes.
Heath and Edwards (1958) first described the following histologic changes in the pulmonary vasculature that are created by excessive blood flow and pressure:
Abnormal pulmonary vascular remodeling begins at birth if there is excessive pulmonary blood flow (Hall and Haworth, 1992). The Heath and Edwards scale is prognostic but limited, because pathologic stages are not correlated with clinical parameters. Rabinovitch overcame this problem by correlating pathologic severity with the hemodynamic state and assigning a simple three-stage grading system (Rabinovitch, 1999):
Grade A or mild grade B changes are histologically equivalent to Heath-Edwards Stage I. Grade C changes are associated with Heath-Edwards stage II or III and the possibility of irreversible changes in the pulmonary vasculature. Severe grade B or grade C disease is predictive of postoperative pulmonary hypertensive problems after surgery to repair the defect (Rabinovitch et al., 1984).
Any discussion of the pulmonary vasculature would be incomplete without exploring the role of the endothelium. Far from being a passive structure, modern understanding of the endothelium has evolved to recognize its singular place in the regulation of vascular tone (Rabinovitch, 1999). Exposure to high flow has multiple negative effects. Endothelial barrier function is compromised, which leads to the release of growth factors that increase vascular smooth muscle and deposition of subendothelial matrix proteins. Luminal diameter eventually decreases with superimposed increased vascular reactivity. Normally antithrombogenic, damaged endothelium reacts abnormally with marginating platelets, resulting in activation of the coagulation system. The endothelium is a key source of the endogenous vasodilators prostacyclin and nitric oxide. When subjected to high flow and shear stress, the release of the endogenous vasodilators is increased, but it may not be enough of an increase to counteract the increased vascular reactivity when pulmonary blood flow is high.
Assessment of the cardiovascular system
The evaluation of a pediatric patient for anesthesia requires knowledge of age-appropriate history, physical findings, and laboratory data. Age correction is implicit in all facets of the cardiovascular examination. A heart rate of 110 beats per minute in a 6-month-old infant, for example, is normal, not “sinus tachycardia, normal for age,” as one often sees when cardiovascular data are reported by adult cardiologists. This section relates to the examination of the child with a nominally healthy and normal cardiovascular system (see Chapter 36, Systemic Disorders; issues of the child with congenital heart disease are discussed in Chapter 20, Anesthesia for Congenital Heart Surgery).
Physical Examination
“Failure to thrive” is determined by plotting patient data on weight, height, and head circumference growth charts. Specific growth charts are available, for example, for children with Down’s syndrome, where normal growth may not mirror that of the population at large (Cronk et al., 1988). More important than the current position on growth charts is the child’s trajectory. The tenth percentile may be adequate for an individual patient. However, it becomes worrisome if the child’s growth had previously been recorded at the fiftieth percentile. In general, most conditions causing failure to thrive will result first in loss of weight, followed by loss of height, preserving head circumference if at all possible. Certain metabolic disorders, however, show a preferential loss in height first.
Vital signs need to be corrected for age, as indicated above. Normal values for heart rate, respiratory rate, and blood pressure are shown in Tables 4-1 and 4-2 and Figures 4-20 and 4-21. Blood pressure cuffs need to be appropriately sized to avoid artifact. Although it was originally taught that the cuff width should be approximately two-thirds the length of the humerus, the current recommendation is now similar to that for adults. The width of the cuff should be approximately 40% to 50% of the circumference (approximately 125% to 155% of the diameter) of the limb where the pressure is being measured and long enough to approximately encircle it. The cardiovascular physical examination begins with inspection, and proceeds to palpation and then auscultation.
| Age | Respiratory Rate (min–1) |
| Birth-6 weeks | 45-60 |
| 6 weeks-2 years | 40 |
| 2-6 years | 30 |
| 6-10 years | 25 |
| > 10 years | 20 |
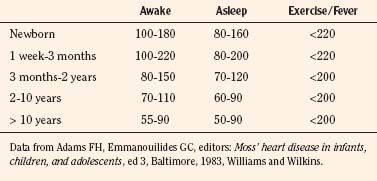
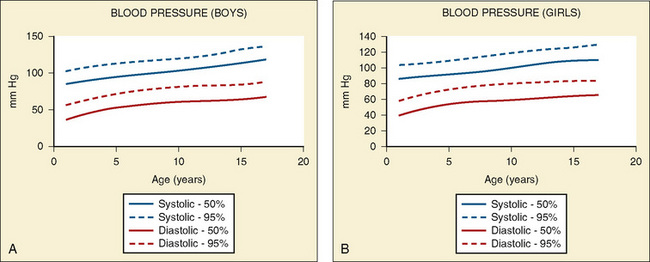
(Data from National High Blood Pressure Education Program Working Group on High Blood Pressure in Children and Adolescents: The fourth report on the diagnosis, evaluation, and treatment of high blood pressure in children and adolescents. Available at www.nhlbi.nih.gov/guidelines/hypertension/child_tbl.htm.)
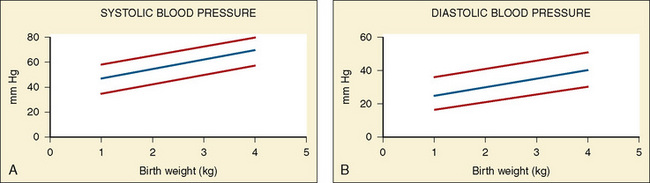
Auscultation
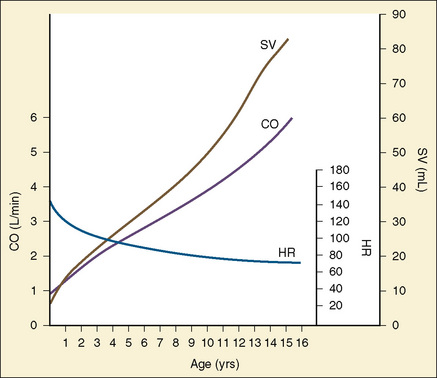
Are the lungs clear without wheezing or rales, both of which could result from cardiac dysfunction? Are there any murmurs? Murmurs are almost universally heard in all children at some point, as heart sounds readily transmit through the thinner pediatric thorax. The differentiation between functional (innocent) murmurs and pathologic murmurs is often easy, but it can at times be challenging. A further discussion of murmurs is included in Chapter 36, Systemic Disorders. Figure 4-22 summarizes the changes in heart rate, cardiac output (CO), and stroke volume (SV) that occur in childhood.
Chest Radiograph
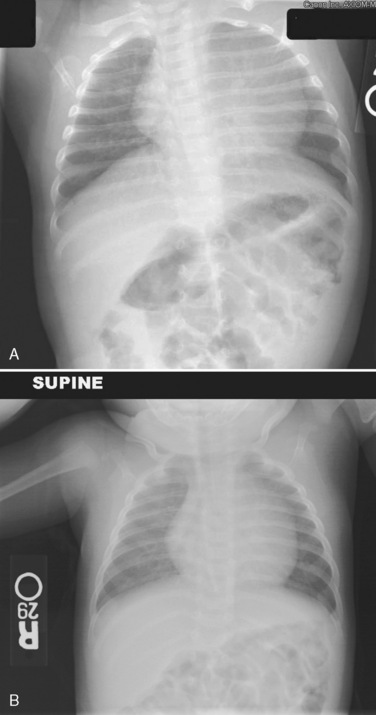
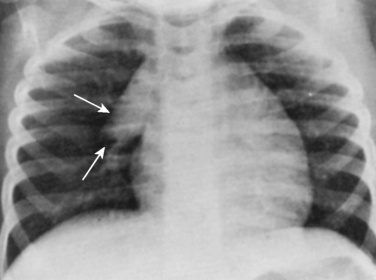
Evidence of heart disease on the routine chest radiograph includes cardiomegaly (heart failure or large left-to-right shunts), decreased pulmonary blood flow (right-to-left shunts), abnormal cardiac silhouette (small pulmonary artery segment with tetralogy of Fallot), widened angle at the carina (large left atrium), pruning (decreased peripheral pulmonary arteries, as in pulmonary arterial hypertension), and rib notching (in children over about age 5 years with coarctation). Artifacts are more commonly seen in children. Given the inability of young children to cooperate, films taken during exhalation are more common in children, and the cardiac silhouette may appear artifactually large (Fig. 4-23). Although the lungs may appear abnormally small in an expiratory film, a more sensitive finding on a film taken in exhalation is buckling of the trachea. The shadow of a normal large thymus in infants can overlay the heart shadow, tempting a diagnosis of cardiomegaly. Sometimes a “sail sign” can be seen at the inferolateral border of the thymus, where it separates from the cardiac shadow, clearly identifying it as the thymus and not the heart (Fig. 4-24).
Electrocardiogram
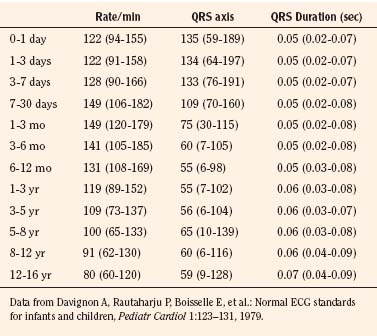
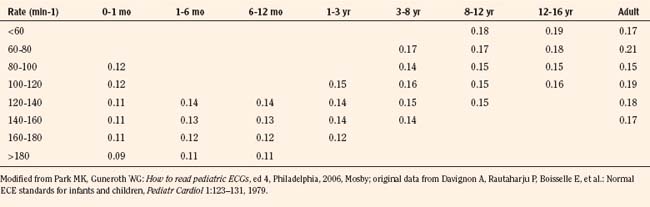
Normal values of the electrocardiogram are dependent on age and heart rate. With development and growth, neonatal right-ventricular predominance is replaced with left ventricular predominance, the heart rate slows, and all durations and intervals lengthen. Between the ages of 3 and 8 years, the electrocardiogram of a child looks similar to that of the adult, with the exception of right precordial T waves, which are normally inverted until about age 10 years. Upright T waves over the right precordium in children younger than 10 years are an indication of right ventricular hypertrophy. Normal values for heart rate, QRS width, frontal-plane QRS axis, and PR interval are shown in Tables 4-3 and 4-4. It can be seen that the ranges of normal are fairly wide. In addition, these values are for children who are awake or resting. Heart rates in anesthetized children are likely to be somewhat lower. The QT interval (QTc), corrected by Bazett’s formula (QTc = QT/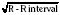 ), has an upper limit of normal of 0.44 seconds in infants and children 6 months of age and older. The upper limit of normal is 0.47 seconds in the first week of life and 0.45 in the first 6 months.
), has an upper limit of normal of 0.44 seconds in infants and children 6 months of age and older. The upper limit of normal is 0.47 seconds in the first week of life and 0.45 in the first 6 months.
Cardiac Catheterization
A cardiac catheterization includes one or more of the following:
Although major advances have been made in deriving physiologic measures of cardiac function by means of echocardiography and magnetic resonance imaging (MRI), cardiac catheterization remains the gold standard for many of these, in addition to providing images of radiologic anatomy and function (Odegard et al., 2004). In addition, the number of interventional procedures performed or proposed is expanding, while the patient population is increasingly complex. New procedures, including stenting of a variety of vessels, closure of atrial and ventricular septal defects, percutaneous placement of prosthetic valves, and hybrid repairs of defects jointly in the catheterization laboratory by both cardiac surgeon and an interventional cardiologist have expanded therapeutic options for patients (Kapoor et al., 2006; Galantowicz et al., 2008; Lurz et al., 2008). Transvascular replacement of pulmonary and aortic valves is also on the near horizon. These increasingly complex procedures require the attendance of an anesthesiologist, and the care rendered transcends beyond keeping the child from moving. The physiologic state of children undergoing such procedures can be tenuous, and the anesthetic care is provided in an environment that is foreign to many anesthesiologists (Cua et al., 2007) (See Chapter 21, Anesthesia for Children with Congenital Heart Disease Undergoing Non-Cardiac Surgery, Closed Cardiac Procedures, and Cardiac Catheterization). The environment may become increasingly foreign, because there are proposals to develop MRI suites for cardiac catheterization procedures in order to lower the radiation exposure to patients. More and more complex electrophysiologic procedures are also being done, some of which can take many hours to complete. All of these factors have contributed to a general shift from sedation toward general anesthesia in the pediatric cardiac catheterization laboratory. Additionally, transesophageal echocardiography guidance is required for many of the procedures, which requires endotracheal intubation.
Vitiello and colleagues (1998) studied pediatric cardiac catheterization laboratory complications in a consecutive series of almost 5000 patients. One or more complications occurred in 8.8% of children. Vascular complications were most common (in 3.8%) and death occurred in 0.14%, most commonly in infants. With the large introducer sheaths needed for many newer interventional procedures, one might expect the incidence of vascular complications to increase.
Pressure
Normal intracardiac pressures are shown in Tables 4-5 and 4-6. The difference between pressures obtained on both sides of a valve, referred to as the gradient, is obtained with two separate catheters, or more commonly by withdrawing a single catheter across the valve. Unlike valve area that is fixed (and can be measured by echocardiography or derived from hemodynamic data), the gradient varies depending on the cardiac output—the higher the output, the higher the gradient for a specific valve area. Similarly, the gradient for a fixed lesion can decrease in the face of falling cardiac output.
TABLE 4-5 Normal Values of Intracardiac Cardiac and Vascular Pressures (in mm Hg)
| Location | Term Newborns | Infants and Children |
| Right atrium | m = 0-4 | a = 5-10 v = 4-8 m = 2-6 |
| Right ventricle | 35-50/1-5 | 15-25/2-5 |
| Pulmonary artery | 35-80/20-50 m = 25-60 |
15-25/8-12 10-6 |
| Pulmonary wedge | m = 3-6 | a = 6-12 v = 8-15 m = 5-10 |
| Left atrium | m = 3-6 | a = 6-12 v = 8-15 m = 5-10 |
| Left ventricle | 80-130/5-10 | |
| Systemic artery | 65-80/45-60 m = 60-65 |
90-130/60-80 m = 70-95 |
a, A wave; m, mean; v, v wave.
Data from Rudolph AM: Congenital Disease of the Heart, Chicago, 1974, Mosby.
TABLE 4-6 Normal Hemodynamic Variables Beyond Infancy
| Location | Average | Range |
| Mean right atrial pressure (CVP) (mm Hg) | 3 | 1-5 |
| Right ventricle pressure (mm Hg) | ||
| Systolic | 25 | 17-32 |
| Diastolic | 5 | 1-7 |
| Pulmonary artery pressure (mm Hg) | ||
| Systolic | 25 | 9-19 |
| Diastolic | 10 | 17-32 |
| Mean | 15 | 4-13 |
| Mean pulmonary wedge pressure (mm Hg) | 9 | 6-12 |
| Mean left atrial pressure (mm Hg) | 8 | 2-12 |
| Cardiac index (L/min/m2) | 3.5 | 2.5-4.2 |
| Stroke volume index (mL/m2) | 45 | |
| Oxygen consumption (mL/min/m2) | 150 | 110-150 |
| Vascular resistance index | ||
| Pulmonary (Wood units/m2) | 1-3 | |
| Pulmonary (dynes•sec•cm–5•m2) | 80-240 | |
| Systemic (Wood units/m2) | 10-20 | |
| Systemic (dynes•sec•cm−5•m2) | 800-1600 | |
Pressures are in mm Hg.
Data from Rudolph AM: Congenital disease of the heart, Chicago, 1974, Mosby.
Oxygen Content and Saturation
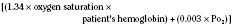
where (0.003 × Po2) reflects the amount of oxygen dissolved in plasma. This is the oxygen content in milliliters of oxygen per 100 mL of blood. Sometimes the content is referenced in milliliters per liter, in which case everything is multiplied by 10. Because the contribution of oxygen in plasma is small, this component is typically neglected in calculations done at physiologic Po2. Oxygen saturation, the fraction of hemoglobin that is bound to oxygen, is readily measured in samples of blood from each cardiac chamber or vessel by oximetry.
Oxygen Consumption
Oxygen consumption is required to derive cardiac output by the Fick equation. Historically, oxygen consumption was measured by comparing the volume of oxygen in a timed sample of expiratory gas collected in a large (Douglas) bag with that inspired over the same time (Rudolph, 2001). This is cumbersome and was replaced by a simpler method that uses a mouthpiece or a head hood, making it usable even in infants (Lister et al., 1974). In most centers, however, oxygen consumption (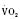 ) is simply derived from a nomogram that uses age, gender, and heart rate (LaFarge and Miettinen, 1970). In any event, after the immediate newborn period, oxygen consumption in the resting child is approximately 150 mL/min/m2; during the first 3 weeks of life, it is approximately 120-130 mL/min/m2 (Rudolph, 2001). When a child is under general anesthesia, oxygen consumption reliably decreases (Table 4-7).
) is simply derived from a nomogram that uses age, gender, and heart rate (LaFarge and Miettinen, 1970). In any event, after the immediate newborn period, oxygen consumption in the resting child is approximately 150 mL/min/m2; during the first 3 weeks of life, it is approximately 120-130 mL/min/m2 (Rudolph, 2001). When a child is under general anesthesia, oxygen consumption reliably decreases (Table 4-7).
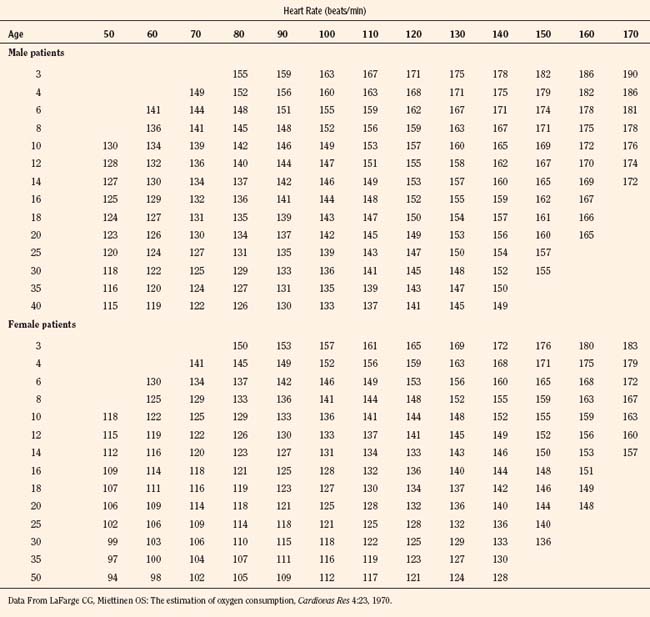
Cardiac Output
Cardiac output is derived from the Fick principle as follows:
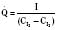
where 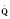 is the flow rate, I is the amount of indicator added and CI1 and CI2 are the concentrations of indicator before and after the addition (or subtraction) of indicator I. Using oxygen extraction in the systemic circulation as the indicator:
is the flow rate, I is the amount of indicator added and CI1 and CI2 are the concentrations of indicator before and after the addition (or subtraction) of indicator I. Using oxygen extraction in the systemic circulation as the indicator:
where 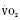 is oxygen consumption, CaO2 is the content of oxygen in the systemic arteries and Cmvo2 is the mixed venous oxygen content.
is oxygen consumption, CaO2 is the content of oxygen in the systemic arteries and Cmvo2 is the mixed venous oxygen content.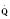 now represents cardiac output. The units of
now represents cardiac output. The units of 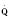 are L/min, and the units of CaO2 and Cmvo2 are mL oxygen/L blood. However, in children,
are L/min, and the units of CaO2 and Cmvo2 are mL oxygen/L blood. However, in children, 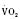 is typically expressed as mL/min/m2, so that the resulting number represents cardiac index rather than cardiac output. Because pulmonary flow and systemic flow are almost identical in normal individuals, oxygen uptake across the pulmonary circulation provides identical results as oxygen consumption across the systemic circulation. When the components of oxygen content are substituted, the equation becomes:
is typically expressed as mL/min/m2, so that the resulting number represents cardiac index rather than cardiac output. Because pulmonary flow and systemic flow are almost identical in normal individuals, oxygen uptake across the pulmonary circulation provides identical results as oxygen consumption across the systemic circulation. When the components of oxygen content are substituted, the equation becomes:
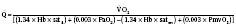
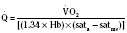
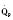 ) and systemic flows (
) and systemic flows (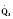 ) need to be calculated independently. In these cases:
) need to be calculated independently. In these cases: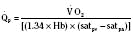
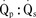 ). This becomes somewhat more complex in the case of multiple levels of shunt or bidirectional shunting. Dividing equation 4.5 by equation 4.4 results in the pulmonary:systemic flow ratio:
). This becomes somewhat more complex in the case of multiple levels of shunt or bidirectional shunting. Dividing equation 4.5 by equation 4.4 results in the pulmonary:systemic flow ratio: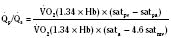
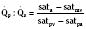
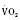 cancel. Thus, if only an estimation of
cancel. Thus, if only an estimation of 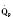 :
: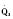 is of interest,
is of interest, 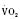 has to be neither measured nor assumed. As an example, presume a ventricular septal defect with a left-to-right shunt. The mixed venous (right atrial) saturation is 70%, the pulmonary arterial saturation is 85%, and the pulmonary venous and systemic arterial saturations are 100%. Substituting into equation 4.7 shows that the
has to be neither measured nor assumed. As an example, presume a ventricular septal defect with a left-to-right shunt. The mixed venous (right atrial) saturation is 70%, the pulmonary arterial saturation is 85%, and the pulmonary venous and systemic arterial saturations are 100%. Substituting into equation 4.7 shows that the 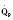 :
: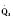 is: (100-70)/(100-85), or 30/15, or 2:1. There is twice as much pulmonary blood flow as systemic.
is: (100-70)/(100-85), or 30/15, or 2:1. There is twice as much pulmonary blood flow as systemic.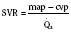
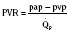
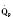 and
and 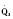 . In general, pulmonary capillary wedge, left atrial, or left ventricular end-diastolic pressures can be substituted for pulmonary venous pressure, assuming the absence of obstructions to pulmonary venous or left atrial drainage. Pediatric cardiologists have historically expressed vascular resistance in terms of Wood units. Wood units are derived when the units are pressure in mm Hg and blood flow in L/min (or, more typically, L/min/m2) to give a resistance index. Multiplying Wood units by approximately 80 converts to SI units (dyne•cm–5/sec). Normal values (Wood units) in children are systemic vascular resistance index of 20, with a range of 15 to 30 (10 to 15 in neonates, with adult levels by 12 to 18 months of age), and pulmonary vascular resistance of 1 to 3 in infants older than 6 to 8 weeks of age, after the normal postnatal fall.
. In general, pulmonary capillary wedge, left atrial, or left ventricular end-diastolic pressures can be substituted for pulmonary venous pressure, assuming the absence of obstructions to pulmonary venous or left atrial drainage. Pediatric cardiologists have historically expressed vascular resistance in terms of Wood units. Wood units are derived when the units are pressure in mm Hg and blood flow in L/min (or, more typically, L/min/m2) to give a resistance index. Multiplying Wood units by approximately 80 converts to SI units (dyne•cm–5/sec). Normal values (Wood units) in children are systemic vascular resistance index of 20, with a range of 15 to 30 (10 to 15 in neonates, with adult levels by 12 to 18 months of age), and pulmonary vascular resistance of 1 to 3 in infants older than 6 to 8 weeks of age, after the normal postnatal fall.Echocardiography
For many infants and children, echocardiography has supplanted the need for cardiac catheterization, and many children now have cardiac surgery based on the results of echocardiography rather than catheterization. For some problems, such as AV valve anatomy, echocardiography is distinctly superior to catheterization (see www.expertconsult.com for further discussion). The thin chests and lack of hyperinflated lungs in children mean that echocardiographic views in children are often superior to those in adults. In common parlance, echocardiography also includes the use of Doppler to assess blood flow and to infer intracardiac pressures, shunts, and pressure gradients (see Doppler Echocardiography Basics, p. 110).
Modern echocardiography is derived from sonar used on ships. A brief electrical impulse is sent to a transducer, which converts (transduces) it and emits it as high frequency sound. As the sound wave encounters a change in density, such as a cardiac structure, some of the sound energy continues on and some is reflected. The echo transducer acts as a receiver and converts the transmitted sound to electrical energy, which is sent to an image processor and displayed on a screen. Because the speed of sound through the body is both known and constant, the distance of the structure from the transducer is easily calculated. This process occurs many times each second. The original format was A-mode echocardiography, where A stood for amplitude. The intensity of the reflected wave was shown as the height or amplitude, like a series of mountain peaks. The first clinical use was echoencephalography, where a beam was directed across the skull looking for shifts of midline structures. The next modification was B-mode echocardiography, where the intensity of the reflected wave was shown as the brightness of a dot. If the reflected waves were recorded on a rolling piece of paper, the series of dots would meld into a series of moving lines, with each line representing a separate cardiac structure with its own distinctive movement pattern. This was the M-mode echocardiograph. M-mode echocardiography was particularly useful for measuring vessel and chamber size and thickness. Although it showed only a one-dimensional representation of the heart (an “ice pick view”), ventricular function could be estimated by ventricular end-systolic and end-diastolic dimensions or by measuring systolic time intervals. The next major advance was two-dimensional echocardiography. Several (32, then 64) separate B-mode ultrasound emitting transducers were mounted together such that a fan-shaped series of lines of light and dark was created. These were repeated rapidly and formed a real-time image of the moving heart. This was exactly analogous to a black-and-white television set, where there is not really a picture but rather a series of rapidly refreshed lines of bright dots that the eyes and brain integrate into a picture. Quality improved with the development of phased array transducers, which generated multiple B-mode lines electronically. These transducers were miniaturized so that they could be mounted onto a gastroscope, resulting in transesophageal echocardiography. Current pediatric transesophageal probes are useful down to infants who weigh approximately 3 kg. Finally, electronic manipulation has allowed the representation of three-dimensional views of the heart. This is currently available on transesophageal probes, although as of this writing, it is not available on pediatric transesophageal probes. Intraoperative transesophageal echocardiography has been shown in several studies to improve intraoperative surgical repairs in a cost-effective manner and is routine in centers with the capability (Sutherland et al., 1989; Muhiudeen et al., 1992; Bettex et al., 2005). This technique is not totally without risk, however. In addition to the risks of local pharyngeal and esophageal injury, which attend its use in both children and adults, introduction or manipulation of the probe can cause acute and massive obstruction of the easily compressible aorta or large bronchi in infants.
Evaluation of Cardiac Function by Echocardiography
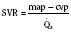
Contractility
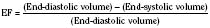
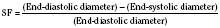
The normal value for SF is >0.28, with a range of 0.28 to 0.44 (Gutgesell et al., 1977). The concept of SF was developed when the only echocardiographic modality available was M-mode, which utilizes an echo representation in only one dimension. A variety of mathematical manipulations were derived to convert a one-dimensional measurement into three-dimensions, which would allow for expression of contractility as the already recognized concept of ejection fraction. This was particularly useful to adult cardiologists, who routinely used EF derived from other methods. However, this conversion also resulted in a cubing of any inherent error in the measurement and made these formulae somewhat less than adequate. Advances in two-dimensional echocardiography have improved the ability to generate a realistic measure of ejection fraction (three-dimensional) from echocardiography (two-dimensional); however, SF remains in common use, particularly among pediatric cardiologists. Velocity of circumferential fiber shortening (Vcf), is a somewhat outdated measure of contractility, and also suffers from being dependent on afterload. In circumferences/sec, it is:
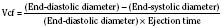
Additional measures of left-ventricular contractility can be derived from echocardiographic measures in conjunction with hemodynamic measures. These include the end-systolic pressure-volume relationship popularized by Suga and colleagues (1973). However, this is not done routinely because it requires measures of hemodynamic data and ventricular volume data at multiple levels of ventricular volume to derive the slope of the line connecting the end-systolic pressure volume points. Unlike the other measures of ventricular function, it is load independent.
Doppler Echocardiography Basics
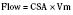
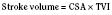
and
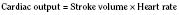
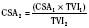
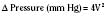
Readers are referred to textbooks of echocardiography for a complete derivation of the modified Bernoulli equation. Assuming good alignment of the Doppler beam and the direction of blood flow, peak velocity is now easily converted into the pressure gradient across the lesion (Fig 4-25).
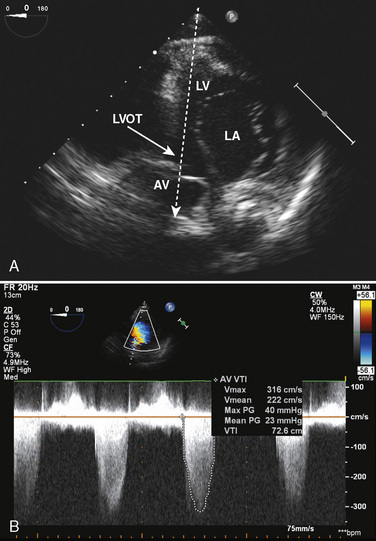
Cardiac magnetic resonance imaging
MRI has proven safe in pediatric patients. There are no known harmful effects from exposure to the magnetic field. Implanted devices containing metal can make MRI unsafe. The dangers of implanted metal exposed to a magnetic field are threefold. First, the implanted metallic devices may heat up. Second, under the influence of a magnetic field, implanted metal devices may move, causing tissue damage in the area of implantation. This is relevant to the pediatric cardiac population because patients often have various stents, coils, sternal wires, pacemakers, and other foreign material. Manufacturers attempt to make their devices “MRI compatible” but there are no hard and fast rules. The presence of a pacemaker generally contraindicates MRI imaging, although most other foreign material is safe. Foreign material implanted in pediatric cardiac patients is usually weakly ferromagnetic, which results in little heat being generated. The caveat is that the implanted device may move in the presence of a strong magnetic field. Manufacturers and clinicians often wait 4 to 6 weeks after implantation to use MRI. During this time it is believed that the device becomes fixed by surrounding fibrosis and cannot move in response to a magnetic field. The 4-to-6–week recommendation is arbitrary and not based on good data. Finally, implanted devices may pose negligible risk to the patient, but they can cause image artifacts. The discovery of possible dangerous foreign material on preoperative questioning should delay the scan while advice is sought from the manufacturer and MRI personnel. MRI demands a still patient who is confined in a small tube. Children who have not reached adolescence require some form of sedation that covers the spectrum from “light” sedation to general anesthesia, with or without the airway being secured. The reader is referred to Chapter 33, Anesthesia and Sedation for Pediatric Procedures Outside the Operating Room, for a more complete discussion on the challenges of anesthesia and sedation in remote locations such as the MRI suite.
The lesions most commonly sought by MRI are coarctation, vascular rings, aortic arch abnormalities, and branch pulmonary artery stenosis. The common feature of these lesions is that they usually all lie beyond the reach of the echocardiography probe. The original cardiovascular imaging technique was a technique called spin echo, which has been improved and may now be called fast or turbo-spin echo. Flowing blood appeared black, resulting in the term “black blood imaging.” The technique was good for imaging structural abnormalities of the heart and vascular system, but it only provided still images that could not display cardiac function. After the diagnosis of a lesion on two-dimensional imaging, a three-dimensional reconstruction can display the lesion in astounding detail and clarity (Fig. 4-26). Intravenous contrast (gadolinium) can be injected rapidly to create magnetic resonance angiography and better demonstrate the pathology (Fig. 4-27). The obvious drawback to MRI for stenotic vascular lesions is that no information about the pressure gradient can be obtained. Sometimes, on visualization, the stenosis appears so severe that it demands repair. However, in cases in which even more information is needed, cardiac catheterization remains the benchmark for visualization and measurement of pressure gradients.The ability of MRI to accurately diagnose lesions of the thoracic vasculature is obvious, but a more exciting application of cardiac MRI testing is in the quantitative and qualitative assessments of cardiac function. In contrast to “black blood imaging,” gradient echo MRI produces bright blood images and is done at high speed, which allows the creation of a cine loop that displays the entire cardiac cycle in real time (Fig. 4-28). Freezing the image at end-diastole and end-systole allows the calculation of ventricular volumes. The ventricle is divided into “slices,” and the blood volume of each slice is calculated by multiplying its cross-sectional area and thickness. Knowing precisely end-diastolic and end-systolic volumes allows the calculation of stroke volume, ejection fraction, and cardiac output. Very good correlation has been made with other methods of calculating cardiac output (Bellenger et al., 2000; Ioannidis et al., 2002). In fact, the ability of MRI to detect small changes in ventricular volume (10 mL) exceeds that of echocardiography.
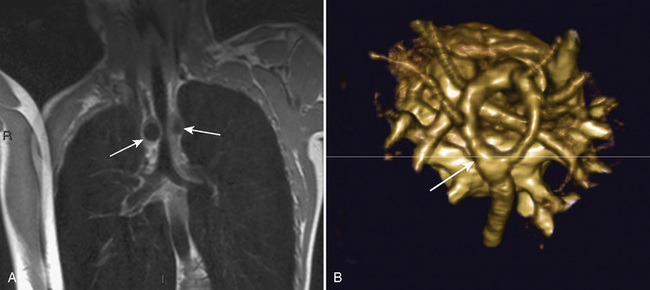
In addition to static measurements of ventricular volume, MRI can also measure blood-flow velocity, which allows the calculation of blood flow. Blood flowing through a magnetic field produces a phase shift proportional to its velocity. Plotting blood-flow velocity versus time (one cardiac cycle) creates a curve. The integration of the area under the curve is stroke volume. Calculation of cardiac output by MRI compares favorably with the Fick and thermodilution methods (Hundley et al., 1995). Measuring stroke volume in the aorta and pulmonary artery simultaneously allows the measurement of pulmonary to systemic (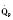 :
: ) ratio. For pediatric patients with septal defects or intracardiac mixing and single ventricle physiology, accurate noninvasive measurements of
) ratio. For pediatric patients with septal defects or intracardiac mixing and single ventricle physiology, accurate noninvasive measurements of  :
: ratios can now be made. The agreement with oximetry-based calculations obtained in the cardiac catheterization laboratory is very good (Beerbaum et al., 2001).
ratios can now be made. The agreement with oximetry-based calculations obtained in the cardiac catheterization laboratory is very good (Beerbaum et al., 2001).
Although less relevant to pediatric patients, MRI can also be used to assess wall motion, diagnose ischemic heart disease, and evaluate myocardial viability. Wall motion can be assessed qualitatively by analyzing a real-time image of ventricular contraction. For these purposes, the left ventricle is divided into the same anatomic regions recommended for echocardiographic assessment (Shanewise et al., 1999). Function is graded as normal, variably hypokinetic, akinetic, or dyskinetic. Quantification of regional wall motion can be done by measuring myocardial thickening throughout the cardiac cycle, but this is time consuming. Dobutamine-stress MRI operates under the same principles as other noninvasive stress tests. Regional wall-motion abnormalities that result from coronary stenoses are unmasked by increasing the inotropic and chronotropic state of the heart. The regional wall-motion defects are assessed qualitatively. Lastly, akinetic areas of myocardium may consist of fixed scar (nonviable myocardium) or hibernating (viable myocardium) tissue, which take up and release intravenous contrast differently. Myocardial delayed enhancement is a technique that allows the differentiation of viable, chronically ischemic myocardium from that which is permanently damaged (Fig. 4-29).
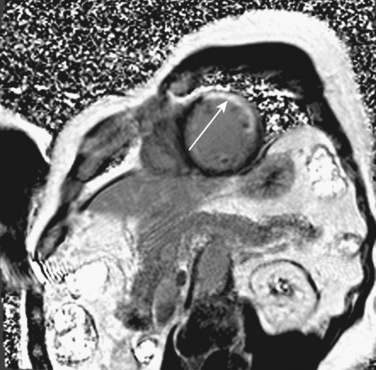
Effects of anesthesia on the cardiovascular system
Anesthetic Effects on Ion Currents
Both volatile anesthetics and several intravenous anesthetics can affect many of the voltage-dependent myocardial ion currents, although studies in immature myocardium are limited. Halothane, for example, can inhibit ICa,L the L-type calcium current in fetal as well as adult myocardium (Fig. 4-30). Baum and Klitzner (1991) have shown that both halothane and isoflurane can decrease the height of the action potential in neonatal right-ventricular papillary muscle, consistent with an effect on trans-sarcolemmal calcium entry. A variety of anesthetics are known to shift the activation and inactivation kinetics of ICa,L. These include halothane and ketamine (Baum et al., 1994). These might decrease calcium entry via ICa,L in cells with more negative resting potential, such as immature myocardial cells.
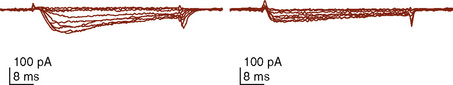
BAY K8644, a calcium-channel agonist, can only partially prevent or reverse halothane- or isoflurane-induced depression in right-ventricular papillary muscles of neonatal rabbits (Baum and Klitzner, 1993). This is consistent with the view that mechanisms other than decreased trans-sarcolemmal calcium entry contribute to anesthetic-induced myocardial depression. Halothane, even in clinically appropriate doses, reversibly inhibits Na+ -Ca2+ exchange in neonatal ventricular myocytes. This provides for an additional mechanism for the more pronounced volatile anesthetic-induced depression of immature myocardium, with its increased reliance on Na+-Ca2+ exchange relative to adult myocardium (Baum et al., 1994).
A variety of volatile and intravenous anesthetics can affect the various K+ channels, with implications for arrhythmia generation and anesthetic-induced myocardial preconditioning (Baum, 1993; Buljubasic et al., 1996; Stadnicka et al., 1997; Stadnicka et al., 2000; Fujimoto et al., 2002; Suzuki et al., 2003). However, there is no information on the effects in the young or immature heart, and the phenomenon of anesthesia-induced preconditioning, mediated at least in part by potassium current, has not been fully evaluated in immature myocardium.
Anesthetic Effects on the Conduction System
In vitro, infant rabbit hearts are more resistant than adult rabbit hearts to the direct sinus-node pacemaker depression of halothane and isoflurane (Palmisano et al., 1994). Maximal depression of the sinus node is about 10%, suggesting that indirect effects rather than direct effects are primarily responsible for the bradycardia seen during clinical anesthesia. It is likely that cholinergic effects play a major role, as baseline cholinergic tone is present in neonates and infants, and under halothane and nitrous-oxide anesthesia there is a dose-related increase in heart rate with atropine (Palmisano et al., 1991b).
For both infant and adult hearts, the sinus rate is more resistant to the effects of halothane and isoflurane than are other measurements of cardiac function (Palmisano et al., 1994). These anesthetics decrease spontaneous pacemaker discharge by decreasing the rate of diastolic depolarization and increasing the action potential duration (Bosnjak and Kampine, 1983). In the adult heart, where it has been studied, halothane decreases the rate of diastolic depolarization and moves the maximal diastolic depolarization (Vm) closer to threshold potential. These two effects counterbalance with little effect on sinus rate (Hauswirth and Schaer, 1967).
Halothane prolongs AV conduction time more than isoflurane, and the effect in infants is greater than it is in adults (Palmisano et al., 1994). The age difference for isoflurane is much less marked than for halothane. There has been little information evaluating the effects of the newer inhalational agents on sinus-node function in healthy children. Sevoflurane and isoflurane have little effects on sinus node function or AV conduction in children with pre-excitation conditions such as Wolff-Parkinson-White syndrome, suggesting they have little if any effects on normal tissue (Chang et al., 1996; Sharpe et al., 1999). In a study of in vitro rabbit hearts, propofol had no effects on atrial or AV conduction, although it did prolong AV conduction in adult hearts of rabbits (Wu et al., 1997).
Anesthetic Effects on Myocardial Metabolism
Reactivity of the coronary vasculature to a variety of physiologic and pharmacologic stimuli has been shown to be present in newborn animals of a variety of species (Toma et al., 1985; Downing and Chen, 1986; Buss et al., 1987; Hickey et al., 1988; Ascuitto et al., 1992). In infant rabbit and in vitro fetal lamb hearts, both halothane and isoflurane vasodilate coronary arteries and result in increased coronary flow, and these effects are similar to those in adult hearts of those animals (Palmisano et al., 1994; Davis et al., 1995). Isoflurane decreases oxygen consumption, which coupled with increased coronary flow, results in relative overperfusion (Hickey et al., 1988; Stowe et al., 1991; Palmisano et al., 1994). Because isoflurane causes a greater decrease in heart rate in adults, the decrement in oxygen consumption is more pronounced in adult hearts than in neonatal hearts (Palmisano et al., 1994). However, when heart rates are kept similar, there are no age differences. In the hypoxic, stressed, neonatal lamb, neither halothane nor isoflurane alter redistribution of blood flow to vital organs, including the heart (Cameron et al., 1985; Brett et al., 1989).
Myocardial flow in the neonatal lamb decreases at 1 MAC isoflurane (from 250 to 88 mL/100g per minute), but this fall is in exact proportion to the decrease in myocardial oxygen consumption, resulting in unchanged myocardial oxygen extraction and endocardial-to-epicardial flow ratios (Brett et al., 1987). Consistent with this, 1.5% halothane was not found to affect steady state levels of myocardial high energy phosphates or intracellular pH in neonatal myocardium, despite a decrement in myocardial performance (McAuliffe and Hickey, 1987). This indicates that uncoupling of oxidative phosphorylation does not account for volatile anesthetics’ depressant effect on myocardial function.
Anesthetic Effects on Systolic Function
Young hearts show an increased susceptibility to myocardial depression from the volatile anesthetics (Cook et al., 1981). Although it has been suggested that the apparent increased hemodynamic depression in the young human heart may be the result of differences in anesthetic uptake and distribution, several studies have indicated that the increased hemodynamic effects of the volatile anesthetics are a result of increased direct action on the myocardium in the immature heart (Barash et al., 1978; Boudreaux et al., 1984; Schieber et al., 1986; Murray et al., 1992).
A major effect on myocardial contractility is via limitation of calcium availability to the contractile apparatus. Trans-sarcolemmal and sarcoplasmic reticular calcium flux are altered with the net effect of depleting intracellular stores (Nakao et al., 1989; Wilde et al., 1991; Frazer and Lynch, 1992; Schmidt et al., 1993; Wilde et al., 1993). Halothane depresses contractility more than isoflurane does (Lynch, 1986; Krane and Su, 1987; Baum and Klitzner, 1991; Palmisano et al., 1994). Halothane decreases peak intracellular calcium concentration more than isoflurane does and is a more potent depressant of contractile function in vitro in both neonatal and adult hearts (Komai and Rusy, 1987; Krane and Su, 1989; Lynch, 1990; Bosnjak et al., 1992; Pan and Potter, 1992; Palmisano et al., 1994). Although there were no age effects of isoflurane, halothane was more depressant to neonatal hearts than adult hearts. However, there may be some dependence on species, because a study in isolated rat atrium did not find that the depression was dependent on age (Rao et al., 1986).
Studies in neonatal lambs have shown that both halothane and isoflurane decrease cardiac output to the same degree that they decrease myocardial oxygen consumption (Cameron et al., 1985; Brett et al., 1987; Brett et al., 1989). Isoflurane at 1 MAC decreased blood pressure primarily by decreasing cardiac output rather than by affecting vascular resistance (Brett et al., 1987). Results in human neonates, infants, and children are more variable, probably because of differing techniques and the confounders of changes in heart rate and afterload. Nicodemus, in an early study, showed increased hypotension in neonates and less hypotension in older children (Nicodemus et al., 1969).
Murray et al. (1992), using echocardiographic indexes of cardiac function, could show no difference between halothane and isoflurane at equipotent concentrations. Sevoflurane has been shown to cause less myocardial depression in young children than halothane, although very young infants were not studied (Holzman et al., 1996).
Anesthetic Effects on Diastolic Function
Anesthetic effects of calcium flux that might impair systolic function could also affect diastolic function, which requires temporary reuptake of released calcium into stores. Indexes of diastolic relaxation show a more depressant effect of halothane than isoflurane, and the effects are greater in infant rabbit hearts than in adult rabbit hearts (Palmisano et al., 1994). There was no age effect seen with isoflurane. This prominent effect of halothane may be a reflection of immature myocardium’s limited capacity to remove calcium from the contractile proteins. The principle mechanism for relaxation in adult myocardium is sequestration of calcium in the sarcoplasmic reticulum, and this is relatively undeveloped in immature myocardium; thus, these hearts may depend more on removal via Na+-Ca2+ exchange (Hoerter et al., 1981; Bers and Bridge, 1989; Fisher and Tate, 1992). It is possible that developmental changes in the actin-regulatory proteins could also affect anesthetic mediation of relaxation.
Anesthetic Effects on Autonomic Control
Halothane, isoflurane, fentanyl, sevoflurane, and nitrous oxide all depress baroreceptor control of heart rate through the central nervous system, autonomic ganglia, and the heart (Biscoe and Millar, 1966; Duke et al., 1977; Duncan et al., 1981; Seagard et al., 1982; Seagard et al., 1983; Kotrly et al., 1984; Murat et al., 1988; Murat et al., 1989). The effect of halothane is more pronounced in younger animals when animals are made pharmacologically hypertensive, but there does not seem to be an age effect when animals are made pharmacologically hypotensive (Wear et al., 1982; Dise et al., 1991; Palmisano et al., 1991a). In (anesthetized) infants undergoing ligation of a patent ductus arteriosus, a heart rate change is typically not seen with acute alterations in blood pressure (Gregory, 1982). Constant showed that while halothane preserves cardiac vagal activity in children, it does not preserve baroreceptor activity any better than sevoflurane (Constant et al., 1999; Constant et al., 2004). Murat showed that isoflurane-mediated tachycardia may be less pronounced in neonates (Murat et al., 1989). Although acute increases in the inspiratory concentration of desflurane or isoflurane can cause an abrupt increase in heart rate and systemic blood pressure from stimulation of the tracheobronchial tree, this effect is not seen with acute increases in sevoflurane concentration (Ebert et al., 1995).
Hemodynamic Effects of Specific Agents
Preanesthetic Medications
Appropriate preanesthetic management of pediatric patients has been an area of much interest for quite a few years. Preoperative education, parental presence, and pharmacologic agents have all been used and all have a place in providing a smoother and safer induction. These are considered in Chapter 9 (Preoperative Preparation) and only cardiac effects of pharmacologic agents will be considered here. Although numerous oral, intramuscular, and intranasal drugs have been proposed as pediatric premedicants, the current most popular agent is oral midazolam. Intravenous midazolam can decrease cardiac output when it is combined with intravenous morphine (Shekerdemian et al., 1997). Midazolam in routine oral doses of 0.5 to 1.0 mg/kg is well tolerated hemodynamically, even in children with cardiac disease. In a study by Masue and colleagues (2003), larger doses of 1.5 mg/kg did not cause any overall decrease in blood pressure, heart rate, or oxygen saturation, although a small number of patients did have a decrease in blood pressure or saturation (6% and 4%, respectively). This was likely related to baseline agitation or underlying cyanotic heart disease confounding the measurements (Masue et al., 2003). If used for sedation in the intensive care unit, abrupt cessation after several days of use can result in cardiovascular withdrawal phenomena. Audenaert et al. (1995) used Doppler echocardiography to compare three premedication regimens in children. They compared oral premedication (meperidine, 3 mg/kg + pentobarbital 4 mg/kg), nasal premedication (ketamine, 5 mg/kg + midazolam, 0.2 mg/kg), and rectal premedication (methohexital, 30 mg/kg). All had relatively modest effects if any. Meperidine + pentobarbital decreased heart rate, mean arterial pressure, and cardiac index. Ketamine + midazolam had no significant cardiovascular effects, and methohexital increased heart rate with a consequent decrease in stroke volume, but without additional effects.
Inhalational Anesthetics
To some extent, all the currently used volatile anesthetics are myocardial depressants. Many studies in humans suffer from studying relatively older children, where differences in myocardial function from adults would be expected to be limited if at all. In general, halothane decreases blood pressure by decreasing myocardial contractility without a compensatory rise in heart rate. Thus, cardiac output decreases. Isoflurane, desflurane, and sevoflurane decrease blood pressure by decreasing left-ventricular afterload. Cardiac output is also maintained by these three agents because they preserve autonomic function and baroreceptor-mediated tachycardia. Children older than age 3 years will have an increase in heart rate with sevoflurane but no change in cardiac output, whereas halothane results in a lower blood pressure and no change in heart rate (Piat et al., 1994; Sarner et al., 1995; Kern et al., 1997). The greatest decrease in blood pressure and the least increase in heart rate with sevoflurane occur in infants younger than 6 months of age (Lerman et al., 1994). The myocardial effects of volatile anesthetics may be more pronounced in the myopathic heart, which mirrors clinical experience (Hettrick et al., 1997).
Ejection fraction and cardiac index are decreased at 1.25 MAC by both isoflurane and halothane (Murray et al., 1987). Halothane has a more pronounced effect on contractility than isoflurane or sevoflurane. In a group of children (not neonates) with congenital heart disease, sevoflurane and isoflurane maintained cardiac output with minimal effect on contractility. Sevoflurane decreased contractility less than halothane. Isoflurane, as it did in other studies, increased heart rate and lowered systemic vascular resistance. Halothane depressed contractility, cardiac output, mean arterial pressure, and systemic vascular resistance (Rivenes et al., 2001). The addition of nitrous oxide to halothane or isoflurane at 1.0 MAC does not seem to change contractility, although it may decrease heart rate, blood pressure, and cardiac index (Murray et al., 1988). Cardiac output has been shown to improve, particularly with halothane, with the administration of atropine in several studies (Miller and Friesen, 1988; Murray et al., 1989). The effect of atropine on cardiac output was because of its effect on heart rate.
Sevoflurane has gained widespread acceptance in the practice of pediatric anesthesia. It produces less tachycardia than isoflurane, as well as less myocardial depression and fewer arrhythmias than halothane (Lerman et al., 1990; Frink et al., 1992; Holzman et al., 1996; Paris et al., 1997; Wodey et al., 1997). Sevoflurane does not cause heart rate or cardiac output to change appreciably; however; it does lower systemic vascular resistance and blood pressure compared with those values of awake patients.
Desflurane has been shown to either decrease or increase heart rate before an incision is made (Taylor and Lerman, 1991; Zwass et al., 1992).
Nitrous oxide is a direct myocardial depressant; however, this is likely offset by an increase in sympathetic tone (Ebert and Kampine, 1989). In the intact animal, it is a very mild cardiac depressant, and its effects are similar to its effects in infants and adults. Its use in infants does not result in an increase in pulmonary vascular resistance (Hickey et al., 1986). The effects of sympathetic stimulation that can be seen with nitrous oxide in adults are absent in young children (Murray et al., 1988).
Xenon has significant potential as a general anesthetic, and it has been shown to not have significant cardiac affects in vitro, however both clinical and pediatric uses are very limited (Stowe et al., 2000).
Opioids
Opioids have long been used in the field of pediatric cardiac anesthesia for their cardiovascular stability, and even a high dose of an opioid has minimal or no effect on heart rate, cardiac output, PVR, mean arterial pressure, and SVR (Robinson and Gregory, 1981; Hickey et al., 1985c; Hansen and Hickey, 1986). As when they are used in adults, opioids used in pediatric patients can blunt the increases in PVR that are associated with tracheal suctioning. The effects of high-dose sufentanil are qualitatively similar to those of high-dose fentanyl (Hickey and Hansen, 1984; Davis et al., 1987). Other opioids have not been studied as intensively in infants and young children. However, given their similar effects in adults, similar findings can be expected in children.
Propofol
Propofol can cause decrease in blood pressure and heart rate, even in healthy children (Short and Aun, 1991). Hannallah and coworkers, however, noted no significant hemodynamic differences when induction/maintenance was done with propofol/propofol infusion, propofol/halothane, thiopentone/halothane, or halothane/halothane (1994). Intracardiac hemodynamic values, including shunts, measured in the catheterization laboratory tend to remain unchanged with propofol (Gozal et al., 2001).
Ketamine
Ketamine has long history of use in the arena of pediatric anesthesia. It can be given orally or intramuscularly as a premedication, or in can be given intravenously to induce or maintain anesthesia. Ketamine’s benefits particularly concern its hemodynamic stability. Although it is a direct myocardial depressant, possibly related to its effects on ICa,L (Baum et al., 1991b; Baum et al., 1994), its actions as a sympathomimetic preserve myocardial function. However, in hearts that are depleted of catecholamine or beta-blocked, one would expect to see a more prominent depressant effect. In the pediatric cardiac catheterization laboratory, ketamine has been shown to have little hemodynamic effects if given as a 2 mg/kg bolus or as an infusion of 50 to 75 mcg/kg per minute (Morray et al., 1984; Oklu et al., 2003). Compared with an infusion of propofol, ketamine causes an increase in systemic arterial pressure and has fewer effects on shunting, because PVR and SVR were unchanged. After cardiac surgery, ketamine at a dose of 2 mg/kg has also been shown to cause no change in heart rate, cardiac output, PVR, or SVR (Hickey et al., 1985b). This statement, however, presumes adequate ventilation. That said, a study in children with pulmonary hypertension showed that even with sevoflurane and spontaneous ventilation, ketamine did not change pulmonary arterial pressure or PVR (Williams et al., 2007). Two studies in children have evaluated the effects of ketamine at altitude (Denver and Albuquerque) and found significant increases in pulmonary vascular resistance with ketamine (Berman et al., 1990; Wolfe et al., 1991).
Regional Anesthetics
Regional anesthesia is discussed more fully in Chapter 16 (Regional Anesthesia). Both spinal and epidural anesthesia with local anesthetics in children have minimal hemodynamic effects compared with the vasodilation noted in adults. Routine prophylactic fluid loading is not required in pediatric practice. A caudal and thoracic epidural block with local anesthetic and fentanyl had no effect unless epinephrine (5 mcg/mL) was added, when it was associated with increased cardiac output accompanied by decreased arterial blood pressure and SVR (Raux et al., 2004). There are no hemodynamic differences noted between spinal or epidural anesthesia (combined with general anesthesia) when used for pediatric cardiac anesthesia (Hammer et al., 2000). Regional anesthesia has such a negligible hemodynamic effect in children that high or even total spinal anesthesia has in fact been suggested by some groups for use in pediatric cardiac anesthesia (Finkel et al., 2003).
Summary
For questions and answers on topics in this chapter, go to “Chapter Questions” at www.expertconsult.com.
Agnoletti G., Boudjemline Y., Bonnet D., et al. Surgical reconstruction of occluded pulmonary arteries in patients with congenital heart disease: effects on pulmonary artery growth. Circulation. 2004;109:2314-2318.
Anderson P.A.. Myocardial Development. Long W.A., editor. Fetal and neonatal cardiology. Philadelphia: WB Saunders; 1990:17-38.
Anderson P.A. The heart and development. Semin Perinatol. 1996;20:482-509.
Anderson P.A., Manring A., Glick K.L., et al. Biophysics of the developing heart. III. A comparison of the left ventricular dynamics of the fetal and neonatal lamb heart. Am J Obstet Gyn. 1982;143:195-203.
Anderson P.A., Malouf N.N., Oakeley A.E., et al. Troponin T isoform expression in humans: a comparison among normal and failing adult heart, fetal heart, and adult and fetal skeletal muscle. Circ Res. 1991;69:1226-1233.
Anderson R.H., Ho S.Y., Smith A., et al. The internodal atrial myocardium. Anatom Record. 1981;201:75-82.
Artman M. Sarcolemmal Na +-Ca2+ exchange activity and exchanger immunoreactivity in developing rabbit hearts. Am J Physiol. 1992;263:H1506-H1513.
Artman M., Ichikawa H., Avkiran M., et al. Na + /Ca2 + exchange current density in cardiac myocytes from rabbits and guinea pigs during postnatal development. Am J Physiol. 1995;268:H1714-H1722.
Ascuitto R.J., Ross-Ascuitto N.T., Ramage D., et al. Acetylcholine-induced coronary vasoconstriction and negative inotropy in the neonatal pig heart. Pediatr Res. 1992;32:236-242.
Audenaert S.M., Wagner Y., Montgomery C.L., et al. Cardiorespiratory effects of premedication in children. Anesth Analg. 1995;80:506-510.
Barash P.G., Glanz S., Katz J.D., et al. Ventricular function in children during halothane anesthesia. Anesthesiology. 1978;49:79-85.
Barth E., Stammler G., Speiser B., et al. Ultrastructural quantitation of mitochondria and myofilaments in cardiac muscle from 10 different animal species including man. J Mol Cell Cardiol. 1992;24:669-681.
Baum V.C. Distinctive effects of three intravenous anesthetics on the inward rectifier (IK1) and the delayed rectifier (IK) potassium currents in myocardium: implications for the mechanism of action. Anesth Analg. 1993;76:18-23.
Baum V.C., Clark W.A., Pelligrino D.A. Cardiac myosin isoenzyme shifts in non-insulin treated spontaneously diabetic rats. Diabetes Res. 1989;10:187-190.
Baum V.C., Klitzner T.S. Excitation-contraction coupling in neonatal myocardium: effects of halothane and isoflurane. Dev Pharmacol Ther. 1991;16:99-107.
Baum V.C., Klitzner T.S. Effect of calcium channel agonist (BAY K8644) on volatile anesthetic-mediated depression in neonatal rabbit papillary muscle. Dev Pharmacol Ther. 1993;20:1-8.
Baum V.C., Palmisano B.W. The immature heart and anesthesia. Anesthesiology. 1997;87:1529-1548.
Baum V.C., Tecson M.E. Ketamine inhibits transsarcolemmal calcium entry in guinea pig myocardium: direct evidence by single cell voltage clamp. Anesth Analg. 1991;73:804-807.
Baum V.C., Wetzel G.T. Sodium-calcium exchange in neonatal myocardium: reversible inhibition by halothane. Anesth Analg. 1994;78:1105-1109.
Baum V.C., Wetzel G.T., Klitzner T.S. Effects of halothane and ketamine on activation and inactivation of myocardial calcium current. J Cardiovasc Pharmacol. 1994;23:799-805.
Beerbaum P., Korperich H., Barth P., et al. Non-invasive quantification of left-to-right shunt in pediatric patients: phase contrast cine magnetic resonance imaging compared with invasive oximetry. Circulation. 2001;103:2476-2482.
Bellenger N.G., Davies L.C., Francis J.M., et al. Reduction in sample size for studies of remodeling in heart failure by the use of cardiovascular magnetic resonance. J Cardiovasc Magn Reson. 2000;2:271-278.
Berman W.Jr, Fripp R.R., Rubler M., et al. Hemodynamic effects of ketamine in children undergoing cardiac catheterization. Pediatr Cardiol. 1990;11:72-76.
Bers D.M., Bridge J.H.B. Relaxation of rabbit ventricular muscle by Na-Ca exchange and sarcoplasmic reticulum calcium pump. Cardiovasc Res. 1989;65:334-342.
Bettex D.A., Pretre R., Jenni R., et al. Cost-effectiveness of routine intraoperative transesophageal echocardiography in pediatric cardiac surgery. Anesth Analg. 2005;100:1271-1275.
Biscoe T.J., Millar R.A. The effects of cyclopropane, halothane and ether on central baroreceptor pathways. J Physiol (London). 1966;184:535-559.
Blom N.A., Gittenberger-de Groot A.C., DeRuiter M.C., et al. Development of the cardiac conduction tissue in human embryos using HNK-1 antigen expression: possible relevance for understanding of abnormal atrial automaticity. Circulation. 1999;99:800-806. [erratum appears in Circulation 112 (4): e71, 2005 Jul 26]
Boerth S.R., Zimmer D.B., Artman M. Steady-state mRNA levels of the sarcolemmal Na(+)-Ca2 + exchanger peak near birth in developing rabbit and rat hearts. Circ Res. 1994;74:354-359.
Boheler K.R., Carrier L., de la Bastie D., et al. Skeletal actin mRNA increases in the human heart during ontogenic development and is the major isoform of control and failing adult hearts. J Clin Invest. 1991;88:323-330.
Bosnjak Z.J., Aggrawal A., Turner L.A., et al. Differential effects of halothane, enflurane, and isoflurane on Ca2+ transients and papillary muscle tension in guinea pigs. Anesthesiology. 1992;76:123-131.
Bosnjak Z.J., Kampine J.P. Effects of halothane, enflurane, and isoflurane on the SA node. Anesthesiology. 1983;58:314-321.
Boudreaux J.P., Schieber R.A., Cook D.R. Hemodynamic effects of halothane in the newborn piglet. Anesth Analg. 1984;63:731-737.
Brett C.M., Teitel D.F., Heymann M.A., et al. The cardiovascular effects of isoflurane in lambs. Anesthesiology. 1987;67:60-65.
Brett C.M., Teitel D.F., Heymann M.A., et al. The young lamb can increase cardiovascular performance during isoflurane anesthesia. Anesthesiology. 1989;71:751-756.
Briggs L.E., Takeda M., Cuadra A.E., et al. Perinatal loss of Nkx2–5 results in rapid conduction and contraction defects. Circ Res. 2008;103:580-590.
Buckner P., Maidens J.M., Finer N. Characterization of the neonatal heart rate baroreflex during and after ECMO. Early Hum Dev. 1993;32:49-61.
Buljubasic N., Marijic J., Berczi V., et al. Differential effects of etomidate, propofol, and midazolam on calcium and potassium channel currents in canine myocardial cells. Anesthesiology. 1996;85:1092-1099.
Buss D.D., Hennemann W.W.III, Posner P. Maturation of coronary responsiveness to exogeneous adenosine in the rabbit. Basic Res Cardiol. 1996;82:290-296.
Cameron C.B., Gregory G.A., Rudolph A.M., et al. The cardiovascular and metabolic effects of halothane in normoxic and hypoxic newborn lambs. Anesthesiology. 1985;62:732-737.
Chang R.K., Stevenson W.G., Wetzel G.T., et al. Effects of isoflurane on electrophysiological measurements in children with the Wolff-Parkinson-White syndrome. Pacing Clin Electrophysiol. 1996;19:1082-1088.
Constant I., Dubois M.C., Piat V., et al. Changes in EEG and autonomic cardiovascular activity during induction in anesthesia with sevoflurane compared to halothane in children. Anesthesiology. 1999;91:1604-1615.
Constant I., Laude D., Hentzgen E., et al. Does halothane really preserve cardiac baroreflex better than sevoflurane? A noninvasive study of spontaneous baroreflex in children anesthetized with sevoflurane versus halothane. Anesth Analg. 2004;99:360-369.
Cook D.R., Brandom B.W., Shiu G., et al. The inspired median effective dose, brain concentration at anesthesia, and cardiovascular index for halothane in young rats. Anesth Analg. 1981;60:182-185.
Cronk C., Crocker A.C., Pueschel S.M., et al. Growth charts for children with Down syndrome: 1 month to 18 years of age. Pediatrics. 1988;81:102-110.
Cua C.L., Galantowicz M., Turner D.R., et al. Palliation via hybrid procedure of a 1.4-kg patient with a hypoplastic left heart. Congen Heart Dis. 2007;2:191-193.
Cummins P., Price K.M., Littler W.A. Foetal myosin light chain in human ventricle. J Muscle Res Cell Motil. 1980:357-366.
Davis D.A., Speziali G., Wagerle C., et al. Effects of halothane on the immature lamb heart. Ann Thorac Surg. 1995;59:695-698.
Davis P.J., Cook D.R., Stiller R.L., et al. Pharmacodynamics and pharmacokinetics of high-dose sufentanil in infants and children undergoing cardiac surgery. Anesth Analg. 1987;66:203-208.
Dise T.L., Stolfi A., Clarkson C.W., et al. Rate-dependent effects of lidocaine on His-Purkinje conduction in the intact neonatal heart: characterization and amplification by N-acetylprocainamide. J Pharmacol Exp Ther. 1991;259:535-542.
Downing S.E., Chen V. Dissociation of adenosine from metabolic regulation of coronary flow in the lamb. Am J Physiol. 1986;251:H40-H46.
Downing S.E., Talner N.S., Gardner T.H. Influences of arterial oxygen tension and pH on cardiac function in the newborn lamb. Am J Physiol. 1966;211:1203-1208.
Duke P.C., Fownes D., Wage J.G. Halothane depresses baroreflex control of heart rate in man. Anesthesiology. 1977;46:184-187.
Duncan P.G., Gregory G.A., Wade J.G. The effect of nitrous oxide on baroreceptor function in newborn and adult rabbits. Can Anaesth Soc J. 1981;28:339-341.
Ebert T.J., Kampine J.P. Nitrous oxide augments sympathetic outflow: direct evidence from human peroneal nerve recordings. Anesth Analg. 1989;69:444-449.
Ebert T.J., Muzi M., Lopatka C.W. Neurocirculatory responses to sevoflurane in humans: a comparison to desflurane. Anesthesiology. 1995;83:88-95.
Eisenmenger V. Die angeborenen defecte der kammerscheidewand des herzens. Z Klin Med. 1897;32:1-28.
Finkel J., Boltz M., Conran A. Haemodynamic changes during high spinal anaesthesia in children having open heart surgery. Paediatr Anaesth. 2003;13:48-52.
Fisher D.J., Heymann M.A., Rudolph A.M. Myocardial oxygen and carbohydrate consumption in fetal lambs in utero and in adult sheep. Am J Physiol. 1980;238:H399-H405.
Fisher D.J., Heymann M.A., Rudolph A.M. Myocardial consumption of oxygen and carbohydrates in newborn sheep. Pediatr Res. 1981;15:843-846.
Fisher D.J., Heymann M.A., Rudolph A.M. Regional myocardial blood flow and oxygen delivery in fetal, newborn, and adult sheep. Am J Physiol. 1982;243:H729-H731.
Fisher D.J., Tate C.A., Phillips S. Developmental regulation of the sarcoplasmic reticulum calcium pump in the rabbit heart. Pediatr Res. 1992;31:474-479.
Frank O. Dynamik des herzmuskels. A Biol. 1895;32:370.
Frazer M.J., Lynch C. Halothane and isoflurane effects on Ca2 + fluxes of isolated myocardial sarcoplasmic reticulum. Anesthesiology. 1992;77:316-323.
Friedman W.F. The intrinsic physiologic properties of the developing heart. Prog Cardiovasc Dis. 1972;15:87-111.
Frink E.J.Jr, Malan T.P., Atlas M., et al. Clinical comparison of sevoflurane and isoflurane in healthy patients. Anesth Analg. 1992;74:241-245.
Fujimoto K., Bosnjak Z.J., Kwok W.M. Isoflurane-induced facilitation of the cardiac sarcolemmal K(ATP) channel. Anesthesiology. 2002;97:57-65.
Galantowicz M., Cheatham J.P., Phillips A., et al. Hybrid approach for hypoplastic left heart syndrome: intermediate results after the learning curve. Ann Thorac Surg. 2008;85:2063-2070.
Gilbert R.D. Control of fetal cardiac output during changes in blood volume. Am J Physiol. 1980;238:H80-H86.
Gilbert R.D. Effects of afterload and baroreceptors on cardiac function in fetal sheep. J Dev Physiol. 1982;4:299-309.
Gillette P.C., Reitman M.J., Gutgesell H.P., et al. Intracardiac electrography in children and young adults. Am Heart J. 1975;89:36-44.
Giroud J.M., Jacobs J.P. Evolution of strategies for management of the patent arterial duct. Cardiol Young. 2007;17(Suppl 2):68-74. [see comment],
Gotoh T. Quantitative studies on the ultrastructural differentiation and growth of mammalian cardiac muscle cells: the atria and ventricles of the cat. Acta Anat (Basel). 1983;115:168-177.
Gozal D., Rein A.J., Nir A., et al. Propofol does not modify the hemodynamic status of children with intracardiac shunts undergoing cardiac catheterization. Pediatr Cardiol. 2001;22:488-490.
Gregory G.A. The baroresponses of preterm infants during halothane anaesthesia. Can Anaesth Soc J. 1982;29:105-107.
Gutgesell H.P., Paquet M., Duff D.F., et al. Evaluation of left ventricular size and function by echocardiography: results in normal children. Circulation. 1977;56:457-462.
Hagen P.T., Scholz D.G., Edwards W.D. Incidence and size of patent foramen ovale during the first 10 decades of life: an autopsy study of 965 normal hearts. Mayo Clin Proc. 1984;59:17-20.
Hall S.M., Haworth S.G. Onset and evolution of pulmonary vascular disease in young children: abnormal postnatal remodeling studied in lung biopsies. J Pathol. 1992;166:183-193.
Hammer G.B., Ngo K., Macario A. A retrospective examination of regional plus general anesthesia in children undergoing open heart surgery. Anesth Analg. 2000;90:1020-1024.
Hannallah R.S., Britton J.T., Schafer P.G., et al. Propofol anaesthesia in paediatric ambulatory patients: a comparison with thiopentone and halothane. Can J Anaesth. 1994;41:12-18.
Hansen D.D., Hickey P.R. Anesthesia for hypoplastic left heart syndrome: use of high-dose fentanyl in 30 neonates. Anesth Analg. 1986;65:127-132.
Harada K., Rice M.J., Shiota T., et al. Gestational age- and growth-related alterations in fetal right and left ventricular diastolic filling patterns. Am J Cardiol. 1997;79:173-177.
Hatem S.N., Sweeten T., Vetter V., et al. Evidence for presence of Ca2+ channel-gated Ca2 + stores in neonatal human atrial myocytes. Am J Physiol. 1995;268:H1195-H1201.
Hauswirth O., Schaer H. Effects of halothane on the sino-atrial node. J Pharmacol Exp Ther. 1967;158:36-39.
Hawkins J., Van Hare G.F., Schmidt K.G., et al. Effects of increasing afterload on left ventricular output in fetal lambs. Circ Res. 1989;65:127-134.
Haworth R.S., Yasutake M., Brooks G., et al. Cardiac Na+-H + exchanger during postnatal development in the rat: changes in mRNA expression and sarcolemmal activity. J Mol Cell Cardiol. 1997;29:321-332.
He H., Giordano F.J., Hilal-Dandan R., et al. Overexpression of the rat sarcoplasmic reticulum Ca2+ ATPase gene in the heart of transgenic mice accelerates calcium trans-ients and cardiac relaxation. J Clin Invest. 1997;100:380-389.
Heath D., Edwards J.E. The pathology of hypertensive pulmonary vascular disease: a description of six grades of structural changes in the pulmonary arteries with special reference to congenital cardiac septal defects. Circulation. 1958;18(4 part 1):533-547.
Hettrick D.A., Pagel P.S., Kersten J.R., et al. The effects of isoflurane and halothane on left ventricular afterload in dogs with dilated cardiomyopathy. Anesth Analg. 1997;85:979-986.
Hickey K.A., Rubanyi G., Paul R.J., et al. Characterization of a coronary vasoconstrictor produced by cultured endothelial cells. Am J Physiol. 1985;248:C550-C556.
Hickey P.R., Hansen D.D., Cramolini G.M., et al. Pulmonary and systemic hemodynamic responses to ketamine in infants with normal and elevated pulmonary vascular resistance. Anesthesiology. 1985;62:287-293.
Hickey P.R., Hansen D.D., Wessel D.L., et al. Pulmonary and systemic hemodynamic responses to fentanyl in infants. Anesth Analg. 1985;64:483-486.
Hickey P.R., Hansen D.D. Fentanyl- and sufentanil-oxygen-pancuronium anesthesia for cardiac surgery in infants. Anesth Analg. 1984;63:117-124.
Hickey P.R., Hansen D.D., Strafford M., et al. Pulmonary and systemic hemodynamic effects of nitrous oxide in infants with normal and elevated pulmonary vascular resistance. Anesthesiology. 1986;65:374-378.
Hickey R.F., Sybert P.E., Verrier E.D., et al. Effects of halothane, enflurane, and isoflurane on coronary blood flow autoregulation and coronary vascular reserve in the canine heart. Anesthesiology. 1988;68:21-30.
Hildreth V., Anderson R.H., Henderson D.J. Autonomic innervation of the developing heart: origins and function. Clin Anat. 2009;22:36-46.
Hill J.R., Rahimtulla K.A. Heat balance and the metabolic rate of new-born babies in relation to environmental temperature; and the effect of age and of weight on basal metabolic rate. J Physiol. 1965;180:239-265.
Hislop A., Reid L. Pulmonary arterial development during childhood: branching pattern and structure. Thorax. 1973;28:129-135.
Hlastala M.P., Chornuk M.A., Self D.A., et al. Pulmonary blood flow redistribution by increased gravitational force. J Appl Physiol. 1998;84:1278-1288.
Hoerter J., Mazet F., Vassort G. Perinatal growth of the rabbit cardiac cell: possible implications for the mechanism of relaxation. J Mol Cell Cardiol. 1981;13:725-740.
Holzman R.S., van der Velde M.E., Kaus S.J., et al. Sevoflurane depresses myocardial contractility less than halothane during induction of anesthesia in children. Anesthesiology. 1996;85:1260-1267.
Humphreys J.E., Cummins P. Regulatory proteins of the myocardium: atrial and ventricular tropomyosin and troponin-I in the developing and adult bovine and human heart. J Mol Cel Cardiol. 1984;16:643-657.
Hundley W.G., Li H.F., Hillis L.D., et al. Quantitation of cardiac output with velocity-encoded, phase-difference magnetic resonance imaging. Am J Cardiol. 1995;75:1250-1255.
Huynh T.V., Chen F., Wetzel G.T., et al. Developmental changes in membrane Ca2 + and K+ currents in fetal, neonatal, and adult rabbit ventricular myocytes. Circ Res. 1992;70:508-515.
Ioannidis J.P., Trikalinos T.A., Danias P.G. Electrocardiogram-gated single photon emission computed tomography versus cardiac magnetic resonance imaging for the assessment of left ventricular volumes and ejection fraction: a meta-analysis. J Am Coll Cardiol. 2002;39:2059-2068.
Ivy D.D., Kinsella J.P., Abman S.H. Endothelin blockade augments pulmonary vasodilation in the ovine fetus. J Appl Physiol. 1996;81:2481-2487.
Kahler C., Schleussner E., Grimm B., et al. Fetal magnetocardiography: development of the fetal cardiac time intervals. Prenat Diagn. 2002;22:408-414.
Kapoor M.C., Sharma S., Sharma V.P., et al. Anesthesia for percutaneous transcatheter closure or perimembranous ventricular septal defect. J Cardiothorac Vasc Anesth. 2006;20:202-208.
Kaufman T.M., Horton J.W., White D.J., et al. Age-related changes in myocardial relaxation and sarcoplasmic reticulum function. Am J Physiol. 1990;259:H309-H316.
Kenny J.F., Plappert T., Doubilet P., et al. Changes in intracardiac blood flow velocities and right and left ventricular stroke volumes with gestational age in the normal human fetus: a prospective Doppler echocardiographic study. Circulation. 1986;74:1208-1216.
Kern C., Erb T., Frei F.J. Haemodynamic responses to sevoflurane compared with halothane during inhalational induction in children. Paediatr Anaesth. 1997;7:439-444.
Kim H.D., Kim D.J., Lee I.J., et al. Human fetal heart development after mid-term: morphometry and ultrastructural study. J Mol Cell Cardiol. 1992;24:949-965.
Kirkpatrick S.E., Pitlick P.T., Naliboff J., et al. Frank-Starling relationship as an important determinant of fetal cardiac output. Am J Physiol. 1976;231:495-500.
Komai H., Rusy B.F. Negative inotropic effects of isoflurane and halothane in rabbit papillary muscles. Anesthesiology. 1987;66:29-33.
Kotrly K.J., Ebert T.J., Vucins E., et al. Baroreceptor reflex control of heart rate during isoflurane anesthesia in humans. Anesthesiology. 1984;60:173-179.
Krane E.J., Su J.Y. Comparison of the effects of halothane on newborn and adult rabbit myocardium. Anesth Analg. 1987;66:1240-1244.
Krane E.J., Su J.Y. Comparison of the effects of halothane on skinned myocardial fibers from newborn and adult rabbit. I. Effects on contractile proteins. Anesthesiology. 1989;70:76-81.
LaFarge C.G., Miettinen O.S. The estimation of oxygen consumption. Cardiovasc Res. 1970;4:23-30.
Lerman J., Oyston J.P., Gallagher T.M., et al. The minimum alveolar concentration (MAC) and hemodynamic effects of halothane, isoflurane, and sevoflurane in newborn swine. Anesthesiology. 1990;73:717-721.
Lerman J., Sikich N., Kleinman S., et al. The pharmacology of sevoflurane in infants and children. Anesthesiology. 1994;80:814-824.
Lister G., Hoffman J.I., Rudolph A.M. Oxygen uptake in infants and children: a simple method for measurement. Pediatrics. 1974;53:656-662.
Lister G., Walter T.K., Versmold H.T., et al. Oxygen delivery in lambs: cardiovascular and hematologic development. Am J Physiol. 1979;237:H668-H675.
Lompre A.M., Nadal-Ginard B., Mahdavi V. Expression of the cardiac ventricular alpha- and beta-myosin heavy chain genes is developmentally and hormonally regulated. J Biol Chem. 1984;259:6437-6446.
Lurz P., Coats L., Khambaskone S., et al. Percutaneous pulmonary valve implantation: impact of evolving technology and learning curve on clinical outcome. Circulation. 2008;117:1964-1972.
Lynch C. Differential depression of myocardial contractility by halothane and isoflurane in vitro. Anesthesiology. 1986;64:620-631.
Lynch C. Differential depression of myocardial contractility by volatile anesthetics in vitro: comparison with uncouplers of excitation-contraction coupling. J Cardiovasc Pharmacol. 1990;15:655-665.
Mahony L.. Development of myocardial structure and function. Allen H.D., Driscoll D.J., Shaddy R.E., Feltes T.F., editors. Heart disease in infants, children, and adolescents. Philadelphia,: Wolter-Kluwer / Lippincott Williams and Wilkins; 2007:573-591.
Martin L.D., Nyhan D., Wetzel R.C.. Regulation of pulmonary vascular resistance and blood flow. Nichols D.G., Ungerleider R.M., Spevak P.J., et al, editors. Critical heart disease in infants and children. Philadelphia: Mosby Elsevier; 2006:73-112.
Masue T., Shimonaka H., Fukao I., et al. Oral high-dose midazolam premedication for infants and children undergoing cardiovascular surgery. Paediatr Anaesth. 2003;13:662-667.
Maylie J.G. Excitation-contraction coupling in neonatal and adult myocardium of cat. Am J Physiol. 1982;242:H834-H843.
McAuliffe J.J., Hickey P.R. The effect of halothane on the steady-state levels of high energy phosphates in the neonatal heart. Anesthesiology. 1987;67:231-235.
McCormack J., Gelband H., Xu H., et al. Atrioventricular nodal function in the immature canine heart. Pediatr Res. 1988;23:99-103. [corrected and republished article orginally printed in Pediatr Res 22 (5): 616—620, 1987 Nov]
Mikawa T., Hurtado R. Development of the cardiac conduction system. Semin Cell Dev Biol. 2007;18:90-100.
Miller B.R., Friesen R.H. Oral atropine premedication in infants attenuates cardiovascular depression during halothane anesthesia. Anesth Analg. 1988;67:180-185.
Morray J.P., Lynn A.M., Stamm S.J., et al. Hemodynamic effects of ketamine in children with congenital heart disease. Anesth Analg. 1984;63:895-899.
Morrissey A., Parachuru L., Leung M., et al. Expression of ATP-sensitive K+ channel subunits during perinatal maturation in the mouse heart. Pediatr Res. 2005;58:185-192.
Muhiudeen I.A., Roberson D.A., Silverman N.H., et al. Intraoperative echocardiography for evaluation of congenital heart defects in infants and children. Anesthesiology. 1992;76:165-172.
Murat I., Lapeyre G., Saint-Maurice C. Isoflurane attenuates baroreflex control of heart rate in human neonates. Anesthesiology. 1989;70:395-400.
Murat I., Levron J., Berg A., et al. Effects of fentanyl on baroreceptor reflex control of heart rate in newborn infants. Anesthesiology. 1988;68:717-722.
Murray D., Forbes R., Murphy K., et al. Nitrous oxide: cardiovascular effects in infants and small children during halothane and isoflurane anesthesia. Anesth Analg. 1988;67:1059-1064.
Murray D.J., Forbes R.B., Dillman J.B., et al. Haemodynamic effects oof atropine during halothane or isoflurane anaesthesia in infants and small children. Can J Anaesth. 1989;36:295-300.
Murray D.J., Forbes R.B., Mahoney L.T. Comparative hemodynamic depression of halothane versus isoflurane in neonates and infants: an echocardiographic study. Anesth Analg. 1992;74:329-337.
Murray D.J., Vandewalker G., Matherne G.P., et al. Pulsed Doppler and two-dimensional echocardiography: comparison of halothane and isoflurane on cardiac function in infants and small children. Anesthesiology. 1987;67:211-217.
Nakanishi T., Jarmakani J.M. Effect of extracellular sodium on mechanical function in the newborn rabbit. Dev Pharmacol Ther. 1981;2:188-200.
Nakanishi T., Okuda H., Kamata K., et al. Development of myocardial contractile system in the fetal rabbit. Pediatr Res. 1987;22:201-207.
Nakao S., Hirata H., Kagawa Y. Effects of volatile anesthetics on cardiac calcium channels. Acta Anaesth Scand. 1989;33:326-330.
Nassar R., Reedy M.C., Anderson P.A. Developmental changes in the ultrastructure and sarcomere shortening of the isolated rabbit ventricular myocyte. Circ Res. 1987;61:465-483.
Nicodemus H.F., Nassiri-Rahimi C., Bachman L., et al. Median effective dose (ED50) of halothane in adults and children. Anesthesiology. 1969;31:344-348.
Odegard K.C., DiNardo J.A., Tasai-Goodman B., et al. Anaesthesia considerations for cardiac MRI in infants and small children. Paediatr Anaesth. 2004;14:471-476.
Oklu E., Bulutcu F.S., Yalcin Y., et al. Which anesthetic agent alters the hemodynamic status during pediatric catheterization? Comparison of propofol versus ketamine. J Cardiothorac Vasc Anesth. 2003;17:686-690.
Osaka T., Joyner R.W. Developmental changes in the beta-adrenergic modulation of calcium currents in rabbit ventricular cells. Circ Res. 1992;70:104-115.
Palmisano B.W., Clifford P.S., Coon R.L., et al. Development of baroreflex control of heart rate in swine. Pediatr Res. 1990;27:148-152.
Palmisano B.W., Clifford P.S., Hoffman R.G., et al. Depression of baroreflex control of heart rate by halothane in growing piglets. Anesthesiology. 1991;75:512-519.
Palmisano B.W., Setlock M.A., Brown M.P., et al. Dose-response for atropine and heart rate in infants and children anesthetized with halothane and nitrous oxide. Anesthesiology. 1991;75:238-242.
Palmisano B.W., Mehner R.W., Stowe D.F., et al. Direct myocardial effects of halothane and isoflurane: comparison between adult and infant rabbits. Anesthesiology. 1994;81:718-729.
Pan B.S., Potter J.D. Two genetically expressed troponin T fragments representing alpha and beta isoforms exhibit functional differences. J Biol Chem. 1992;267:23052-23056.
Parilak L.D., Taylor D.G., Song Y., et al. Contribution of frequency-augmented inward Ca2 + current to myocardial contractility. Can J Pharmacol. 2009;87:69-75.
Paris S.T., Cafferkey M., Tarling M., et al. Comparison of sevoflurane and halothane for outpatient dental anaesthesia in children. Br J Anaesth. 1997;79:280-284.
Penefsky Z.J. Studies on mechanism of inhibition of cardiac muscle contractile tension by ryanodine: mechanical response. Pflugers Arch. 1974;347:173-184.
Piat V., Dubois M.C., Johanet S., et al. Induction and recovery characteristics and hemodynamic responses to sevoflurane and halothane in children. Anesth Analg. 1994;79:840-844.
Pickoff A.S.. Development and function of the cardiac conduction system. Allen H.D., Driscoll D.J., Shaddy R.E., Feltes T.F., editors. Heart disease in infants, children, and adolescents. Philadelphia: Wolter-Kluwer/Lippincott Williams and Wilkins; 2007:240-252.
Rabinovitch M. Pulmonary hypertension: pathophysiology as a basis for clinical decision making. J Heart Lung Transplant. 1999;18:1041-1053.
Rabinovitch M., Keane J.F., Norwood W.I., et al. Vascular structure in lung tissue obtained at biopsy correlated with pulmonary hemodynamic findings after repair of congenital heart defects. Circulation. 1984;69:655-667.
Racker D.K. Atrioventricular node and input pathways: a correlated gross anatomical and histological study of the canine atrioventricular junctional region. Anatom Record. 1989;224:336-354.
Rao C.C., Boyer M.S., Krishna G., et al. Increased sensitivity of the isometric contraction of the neonatal isolated rat atria to halothane, isoflurane, and enflurane. Anesthesiology. 1986;64:13-18.
Raux O., Rochette A., Morau E., et al. The effects of spread of block and adrenaline on cardiac output after epidural anesthesia in young children: a randomized, double-blind, prospective study. Anesth Analg. 2004;98:948-955.
Rein A.J., Sanders S.P., Colan S.D., et al. Left ventricular mechanics in the normal newborn. Circulation. 1987;76:1029-1036.
Rivenes S.M., Lewin M.B., Stayer S.A., et al. Cardiovascular effects of sevoflurane, isoflurane, halothane, and fentanyl-midazolam in children with congenital heart disease. Anesthesiology. 2001;94:223-229.
Robinson S., Gregory G.A. Fentanyl-air-oxygen anesthesia for ligation of patent ductus arteriosus in preterm infants. Anesth Analg. 1981;60:331-334.
Roca T.P., Pigott J.D., Clarkson C.W., et al. L-type calcium current in pediatric and adult human atrial myocytes: evidence for developmental changes in channel inactivation. Pediatr Res. 1996;40:462-468.
Romero T., Covell J., Friedman W.F. A comparison of pressure-volume relations of the fetal, newborn, and adult heart. Am J Physiol. 1972;222:1285-1290.
Romero T.E., Friedman W.F. Limited left ventricular response to volume overload in the neonatal period: a comparative study with the adult animal. Pediatr Res. 1979;13:910-915.
Rosen M.R., Legato M.J., Weiss R.M. Developmental changes in impulse conduction in the canine heart. Am J Physiol. 1981;240:H546-H554.
Rudolph A.M. Congenital Diseases of the Heart. Chicago: Year Book, 1974.
Rudolph A.M. Fetal and neonatal pulmonary circulation. Annu Rev Physiol. 1979;41:383-395.
Rudolph A.M. Congenital diseases of the heart: clinical-physiological considerations. Armonk, NY: Futura, 2001.
Rudolph A.M., Yuan S. Response of the pulmonary to hypoxia and H + ion concentration changes. J Clin Invest. 1966;45:399-411.
Rychik J., Rome J.J., Collins M.H., et al. The hypoplastic left heart syndrome with intact atrial septum: atrial morphology, pulmonary vascular histopathology and outcome. J Am Coll Cardiol. 1999;34:554-560.
Saba Z., Nassar R., Ungerleider R.M., et al. Cardiac troponin T isoform expression correlates with pathophysiological descriptors in patients who underwent corrective surgery for congenital heart disease. Circulation. 1996;94:472-476.
Sachis P.N., Armstrong D.L., Becker L.E., et al. Myelination of the human vagus nerve from 24 weeks postconceptional age to adolescence. J Neuropath Exp Neurol. 1982;41:466-472.
Sakakibara Y., Wasserstrom J.A., Furukawa T., et al. Characterization of the sodium current in single human atrial myocytes. Circ Res. 1992;71:535-546.
Sanchez-Chapula J., Elizalde A., Navarro-Polanco R., et al. Differences in outward currents between neonatal and adult rabbit ventricular cells. Am J Physiol. 1994;266:H1184-H1194.
Sarner J.B., Levine M., Davis P.J., et al. Clinical characteristics of sevoflurane in children: a comparison with halothane. Anesthesiology. 1995;82:38-46.
Sasse S., Brand N.J., Kyprianou P., et al. Troponin I gene expression during human cardiac development and in end-stage heart failure. Circ Res. 1993;72:932-938.
Schieber R.A., Nannoum A., Sugden A., et al. Hemodynamic effects of isoflurane in the newborn piglet: comparison with halothane. Anesth Analg. 1986;65:633-638.
Schmidt U., Schwinger R.H.G., Bohm S., et al. Evidence for an interaction of halothane with the L-type Ca2+ channel in human myocardium. Anesthesiology. 1993;79:332-339.
Schroder E.A., Tobita K., Tinney J.P., et al. Microtubule involvement in the adaptation to altered mechanical load in developing chick myocardium. Circ Res. 2002;91:353-359.
Schumacher W.A., Sheppard J.R., Mirkin B.L. Biological maturation and beta-adrenergic effectors: pre- and postnatal development of the adenylate cyclase system in the rabbit heart. J Pharmacol Exp Ther. 1982;223:587-593.
Seagard J.L., Elegbe E.O., Hoop F.A., et al. Effects of isoflurane on the baroreceptor reflex. Anesthesiology. 1983;59:511-520.
Seagard J.L., Hopp F.A., Donegan J.H., et al. Halothane and the carotid sinus reflex: evidence for multiple sites of action. Anesthesiology. 1982;57:191-202.
Shah A.M., Mebazaa A., Wetzel R.C., et al. Novel cardiac myofilament desensitizing factor released by endocardial and vascular endothelial cells. Circulation. 1994;89:2492-2497.
Shanewise J.S., Cheung A.T., Aronson S., et al. ASE / SCA guidelines for performing a comprehensive intraoperative multiplane transesophageal echocardiography examination: recommendations of the American Society of Echocardiography Council for Intraoperative Echocardiography and the Society of Cardiovascular Anesthesiologists Task Force for Certification in Perioperative Transesophageal Echocardiography. Anesth Analg. 1999;89:870-884.
Sharpe M.D., Cuillerier D.J., Lee J.K., et al. Sevoflurane has no effect on sinoatrial node function or on normal atrioventricular and accessory pathway conduction in Wolff-Parkinson-White syndrome during alfentanil/midazolam anesthesia. Anesthesiology. 1999;90:60-65.
Shekerdemian L., Bush A., Redington A. Cardiovascular effects of intravenous midazolam after open heart surgery. Arch Dis Child. 1997;76:57-561.
Shigenobu K., Sperelakis N. Development of sensitivity to tetrodotoxin of chick embryonic hearts with age. J Mol Cell Cardiol. 1971;3:271-286.
Short S.M., Aun C.S. Haemodynamic effects of propofol in children. Anaesthesia. 1991;46:783-785.
Small J.V., Furst D.O., Thornell L.E. The cytoskeletal lattice of muscle cells. Euro J Biochem. 1992;208:559-572.
Solaro R.J., Kumar P., Blanchard E.M., et al. Differential effects of pH on calcium activation of myofilaments of adult and perinatal dog hearts: evidence for developmental differences in thin filament regulation. Circ Res. 1986;58:721-729.
Sonnenblick E.H. Myocardial ultrastructure in the normal and failing heart. In: Braunwald E., editor. The myocardium: failure and infarction. New York: HP Publishing; 1974:3-13.
John Sutton M.G.St., Gewitz M.H., Shah B., et al. Quantitative assessment of growth and function of the cardiac chambers in the normal human fetus: a prospective longitudinal echocardiographic study. Circulation. 1984;69:645-654.
Stadnicka A., Bosnjak Z.J., Kampine J.P., et al. Effects of sevoflurane on inward rectifier K + current in guinea pig ventricular cardiomyocytes. Am J Physiol. 1997;273(1 Pt 2):H324-H332.
Stadnicka A., Bosnjak Z.J., Kampine J.P., et al. Modulation of cardiac inward rectifier K(+)current by halothane and isoflurane. Anesth Analg. 2000;90:824-833.
Starling E.H. The Linacre lecture on the law of the heart. London: Longmans, Green, and Co, 1918.
Stinstra J., Golbach E., van Leeuwen P., et al. Multicentre study of fetal cardiac time intervals using magnetocardiography. BJOG. 2002;109:1235-1243.
Stowe D.F., Marijic J., Bosnjak Z.J., et al. Direct comparative effects of halothane, enflurane, and isoflurane on oxygen supply and demand in isolated hearts. Anesthesiology. 1991;74:1087-1095.
Stowe D.F., Rehmert G.C., Kwok W.M., et al. Xenon does not alter cardiac function or major cation currents in isolated guinea pig hearts or myocytes. Anesthesiology. 2000;92:516-522.
Suga H., Sagawa K., Shoukas A.A. Load independence of the instantaneous pressure-volume ratio of the canine left ventricle and effects of epinephrine and heart rate on the ratio. Circ Res. 1973;32:314-322.
Sutherland G.R., Balaji S., Monro J.L. Potential value of intraoperative Doppler colour flow mapping in operations for complex intracardiac shunting. Br Heart J. 1989;62:467-469.
Suzuki A., Bosnjak Z.J., Kwok W.M. The effects of isoflurane on the cardiac slowly activating delayed-rectifier potassium channel in guinea pig ventricular myocytes. Anesth Analg. 2003;96:1308-1315.
Tanaka H., Shigenobu K. Role of beta-adrenoceptor-adenylate cyclase system in the developmental decrease in sensitivity to isoprenaline in foetal and neonatal rat heart. Br J Pharmacol. 1990;100:138-142.
Taylor R.H., Lerman J. Minimum alveolar concentration of desflurane and hemodynamic responses in neonates, infants, and children. Anesthesiology. 1991;75:975-979.
Teitel D.F., Cassidy S.C., Fineman J.R. Circulation physiology. In: Allen H.D., Driscoll D.J., Shaddy R.E., Feltes T.F., editors. Heart disease in infants, children, and adolescents. Philadelphia: Wolter-Kluwer / Lippincott Williams and Wilkins,; 2007:617-629.
Teitel D.F., Iwamoto H.S., Rudolph A.M. Changes in the pulmonary circulation during birth-related events. Pediatr Res. 1990;27:372-378.
Teitel D.F., Sidi D., Chin T., et al. Developmental changes in myocardial contractile reserve in the lamb. Pediatr Res. 1985;19:948-955.
Thoresen M., Cowan F., Walloe L. Cardiovascular responses to tilting in healthy newborn babies. Early Human Dev. 1991;26:213-222.
Thornburg K.L., Morton M.J. Filling and arterial pressures as determinants of RV stroke volume in the sheep fetus. Am J Physiol. 1983;244:H656-H663.
Thornburg K.L., Morton M.J. Filling and arterial pressures as determinants of left ventricular stroke volume in fetal lambs. Am J Physiol. 1986;251:H961-H968.
Tod M.L., Cassin S. Perinatal pulmonary responses to arachidonic acid during normoxia and hypoxemia. J Appl Physiol. 1984;57:977-983.
Toma B.S., Wangler R.D., DeWitt D.F., et al. Effect of development on coronary vasodilator reserve in the isolated guinea pig heart. Circ Res. 1985;57:538-544.
Van Hare G.F., Hawkins J.A., Schmidt K.G., et al. The effects of increasing mean arterial pressure on left ventricular output in newborn lambs. Circ Res. 1990;67:78-83.
van der Loop F.T., Schaart G., Langmann H., et al. Rearrangement of intercellular junctions and cytoskeletal proteins during rabbit myocardium development. Eur J Cell Biol. 1995;68:62-69.
Van Leeuwen P., Lange S., Klein A., et al. Dependency of magnetocardiographically determined fetal cardiac time intervals on gestational age, gender and postnatal biometrics in healthy pregnancies. BMC Pregnancy Childbirth. 2004;4:6.
Vitiello R., McCrindle B.W., Nykanen D., et al. Complications associated with pediatric cardiac catheterization. J Am Coll Cardiol. 1998;32:1433-1440.
Wear R., Robinson S., Gregory G. The effect of halothane on the baroresponse of adult and baby rabbits. Anesthesiology. 1982;56:188-191.
Wetzel G.T., Chen F., Klitzner T.S. Ca2+ channel kinetics in acutely isolated fetal, neonatal, and adult rabbit cardiac myocytes. Circ Res. 1993;72:1065-1074.
Wiegerinck R.F., Cojoc A., Zeidenweber C.M., et al. Force frequency relationship of the human ventricle increases during early postnatal development. Pediatr Res. 2008. (in press)
Wilde D.W., Davidson B.A., Smith M.D., et al. Effects of isoflurane and enflurane on intracellular Ca2 + mobilization in isolated cardiac myocytes. Anesthesiology. 1993;79:73-82.
Wilde D.W., Knight P.R., Sheth N., et al. Halothane alters control of intracellular Ca2+ mobilization in single rat ventricular myocytes. Anesthesiology. 1991;75:1075-1086.
Williams G.D., Philip B.M., Chu L.F., et al. Ketamine does not increase pulmonary vascular resistance in children with pulmonary hypertension undergoing sevoflurane anesthesia and spontaneous ventilation. Anesth Analg. 2007;105:1578-1584.
Wodey E., Pladys P., Copin C., et al. Comparative hemodynamic depression of sevoflurane versus halothane in infants: an echocardiographic study. Anesthesiology. 1997;87:795-800.
Wolfe R.R., Loehr J.P., Schaffer M.S., et al. Hemodynamic effects of ketamine, hypoxemia and hyperoxia in children with surgically treated congenital heart disease residing greater than or equal to 1,200 meters above sea level. Am J Cardiol. 1991;67:84-87.
Wu M-H, Su M-J, Sun S.S-M. Age-related propofol effects on electrophysiologic properties of isolated hearts. Anesth Analg. 1997;84:964-971.
Xie L.H., Takano M., Noma A. Development of inwardly rectifying K + channel family in rat ventricular myocytes. Am J Physiol. 1997;272:H1741-H1750.
Zwass M.S., Fisher D.M., Welborn L.G., et al. Induction and maintenance characteristics of anesthesia with desflurane and nitrous oxide in infants and children. Anesthesiology. 1992;76:373-378.