[level-membership-for-neonatal-and-perinatal-medicine-category]
Patricia M. Kenney, Deandra Hoover, Luther C. Williams and Victor Iskersky
Congenital heart disease (CHD) is the most common life-threatening birth defect encountered in the neonatal intensive care unit (NICU). Although the incidence of these conditions has remained constant at approximately 1% of all infants born in the United States, the methods of diagnosis and treatment have undergone tremendous change over the past several decades. 37 It is the responsibility of the practitioner to recognize the presence of CHD and to provide an accurate diagnose and treatment. This chapter reviews the physiology of neonatal circulation, the pathophysiology of congenital heart disease, and the most current evidence-based treatments.
CONGENITAL HEART DISEASE: OVERVIEW
History
In 1892, Dr. William Osler wrote that congenital heart disease was of “limited clinical interest as in a large proportion of cases the anomaly is not compatible with life, and in others, nothing can be done to remedy the defect or even relieve the symptoms.”8 Osler encouraged Dr. Maude Abbott in her CHD finding, and she, along with Dr. Helen Taussig and Dr. Alfred Blalock, suggested surgery to help these “blue babies.”35 This opened up the field of surgical treatment of cyanotic malformations of the heart. In 1938, Dr. Robert Gross was the first to successfully ligate a patent ductus arteriosus (PDA) in a 7-year-old girl at Boston’s Children’s Hospital. The first successful United States heart transplant was done at Stanford University by Dr. Norman Shumway in 1968. 7 What remarkable progress has been made in the area of congenital heart disease. This is the direct result of advances in pediatric and fetal cardiology, cardiac surgery, neonatology, and neonatal intensive care nursing.
Incidence and Survival
Each year, approximately 40,000 babies, or just under 1% of all babies born in the United States, are diagnosed with congenital heart disease. 3 Highly sensitive echocardiography has led to the detection of more trivial forms of congenital heart disease such as tiny ventricular septal defects. This inclusion has led to the higher incidence figures in recent years. The incidence of moderate to severe structural congenital heart defects (in liveborn infants) is 6 to 8 per 1000 live births. 8 Of these infants, approximately 3 per 1000 live births will have CHD that results in death or requires cardiac surgery during the first year of life. 25 Advances in diagnostic imaging, cardiac surgery, and neonatal intensive care have led to significant decreases in mortality rates. The death rate from all congenital heart defects declined nearly 32% between 1994 and 2004. 37
Embryology
The heart is one of the earliest differentiating and functioning organs. In human embryos, the heart begins to beat at about 22 to 23 days of life. Blood begins flowing through the heart in the fourth week of life. The heart develops from the cardiogenic mesoderm that originally lies above the cranial end of the neural tube. The heart forms initially as a simple paired tube inside the pericardial cavity. When the embryonic disk folds, the heart is carried into the correct anatomic position in the chest cavity. A key aspect of heart development is the septation of the heart into separate chambers. This complex process converts this simple tube into a four-chambered heart. Cardiogenesis is such as intricate, complex process that it is of little wonder that congenital heart defects occur.
Physiology
The physiologic changes that occur during the transition from intrauterine to extrauterine life have been well documented. To develop a clear understanding of the various congenital heart defects, knowledge of the basic principles of fetal circulation must be established.
FETAL CIRCULATION
Fetal circulation is intended to use the placenta for gas exchange, whereas postnatal circulation uses the lungs for gas exchange (Figure 24-1). Highly oxygenated blood from the mother enters the fetal circulation through the vein in the umbilical cord. This blood enters the inferior vena cava via the ductus venosus. Nearly one half of the umbilical venous blood goes through the liver and reaches the inferior vena cava via the hepatic veins. Inside the fetal heart, blood enters the right atrium, the chamber on the upper right side of the heart. Most of the blood flows to the left side through the foramen ovale, a special fetal opening between the left and right atria. Blood then passes into the left ventricle and then to the aorta, the large artery coming from the heart. From the aorta, blood is sent to the head and upper extremities. Therefore the brain (via the brachiocephalic vessels) and the heart (via the coronary arteries) are perfused with relatively highly oxygenated blood. After circulation, the blood returns to the right atrium through the superior vena cava. Nearly a third of the blood entering the right atrium stays in the right side of the heart, eventually flowing into the pulmonary artery.
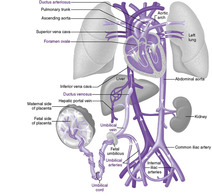 |
| FIGURE 24-1
(Modified from Patton KT, Thibodeau GA: Anatomy and physiology, ed 7, St Louis, 2010, Mosby.)
|
Fetal lungs are not used for breathing. The work of exchanging oxygen and carbon dioxide is done by the placenta. Because of high pulmonary vascular resistance (PVR), fetal circulation shunts most of the blood away from the lungs. Blood is shunted from the pulmonary artery to the aorta through a connecting blood vessel called the ductus arteriosus.29 Blood then enters the placental circulation and is resaturated.
CHANGES THAT OCCUR IN THE FETAL CIRCULATION WITH BIRTH
In utero, systemic vascular resistance (SVR) is low, primarily because of low resistance in the placenta. Conversely, the PVR is high, with the constricted and hypertrophied pulmonary arterioles being relatively resistant to blood flow. At birth, the placenta is removed from the circulation, thereby greatly increasing the SVR.
Both the labor process and the first few breaths of life begin the termination of fetal circulation and the transition to newborn circulation. Initiation of respirations produces increased oxygen tension, which decreases PVR and increases pulmonary blood flow. The ductus arteriosus is extremely sensitive to the oxygen content of the blood. As the neonatal Pa o2 rises, the connection between the aorta and the pulmonary artery (ductus arteriosus) is no longer needed and begins to close. Functional closure occurs at approximately 72 hours of life, and anatomic closure usually occurs between 1 and 2 weeks after birth. The circulation in the lungs then increases, and more blood flows into the left atrium of the heart. This increased pressure in the left atrium causes the foramen ovale to close. Anatomic closure of the foramen ovale can take several months. Finally, with the clamping of the umbilical cord, umbilical venous flow ceases and the ductus venosus begins to close, with anatomic closure taking approximately 1 to 2 weeks. 29
Once these changes occur, the newborn’s circulation resembles that of an adult (Figure 24-2). Desaturated blood returns to the heart by the inferior and superior venae cavae and enters the right atrium, right ventricle, pulmonary artery, and pulmonary circulation in which oxygen and carbon dioxide are exchanged. The saturated blood then returns to the heart through the pulmonary venous system and enters the left atrium, left ventricle, and ultimately the aorta and systemic arterial system. However, PVR and pressures in the right ventricle and pulmonary system remain elevated in the neonate because of the hypertrophy of the pulmonary vessels. This hypertrophy slowly resolves so that pulmonary vascular resistance and right heart pressures decrease to lower levels between 1 and 2 months of age.
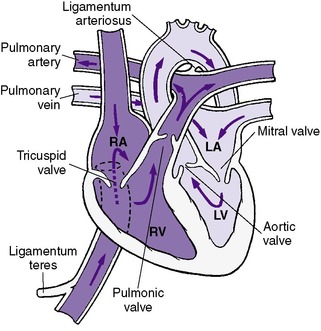 |
| FIGURE 24-2
(Modified from Hockenberry MJ, Wilson D: Wong’s essentials of pediatric nursing, ed 8, St Louis, 2009, Mosby.)
|
Etiology
The development of the cardiovascular system represents a complicated interaction between form and function. In most cases, the cause(s) of abnormal cardiac development is unknown. Traditionally, the etiology of congenital heart defects has been viewed as multifactorial, involving a complex interaction between genetic and environmental factors. Studies are being done on maternal zinc deficiency and its association with an increased risk for fetal heart malformations. These studies postulate that the mechanism of action is the suppression of HNK-1 expression. 19Table 24-1 lists the most common environmental risk factors and their associated cardiac malformations. Box 24-1 lists less common risk factors associated with CHD. Certain chronic illnesses in the mother contribute to the risk for CHD. For example, women with diabetes are at increased risk for having an infant with a heart defect. However, this risk can be greatly reduced by strict control of maternal blood sugar levels. More recent studies have found that the single greatest risk factor for congenital heart defects is genetic. Table 24-2 shows chromosomal syndromes and their associated congenital heart defects.
| Potential Teratogen | Frequency of Cardiovascular Disease (%) | Most Common Malformations |
|---|---|---|
| DRUGS | ||
| Alcohol | 25-30 | Ventricular septal defect, patent ductus arteriosus, atrial septal defect |
| Amphetamines | 5-10 | Ventricular septal defect, patent ductus arteriosus, atrial septal defect, transposition of the great vessels |
| Anticonvulsants | 2-3 | Pulmonary stenosis, aortic stenosis, coarctation of aorta, patent ductus arteriosus |
| Trimethadione | 15-30 | Transposition of great arteries, tetralogy of Fallot, hypoplastic left heart syndrome |
| Lithium | 10 | Ebstein’s anomaly, tricuspid atresia, atrial septal defect |
| Sex hormones | 2-4 | Ventricular septal defect, transposition of great arteries, tetralogy of Fallot |
| INFECTIONS | ||
| Rubella | 35 | Peripheral pulmonary artery stenosis, ventricular septal defect, patent ductus arteriosus, atrial septal defect |
| MATERNAL CONDITIONS | ||
| Diabetes | 3-5 | Transposition of great arteries, ventricular septal defect, coarctation of aorta |
| 30-50 | Cardiomegaly, myopathy | |
| Lupus erythematosus | ? | Heart block |
BOX 24-1
Exposure to Environmental Agents During Work and/or Hobby
• Paternal exposure to cold temperature
• Maternal exposure to various solvents, hair dyes, auto body repair work
Drug Exposure
• Diazepam, phenothiazines
• Corticosteroids
• Gastrointestinal drugs
• Paternal exposure to cocaine
Maternal Reproductive History
• Genetic risk factor (family history of congenital heart disease), more than three prior pregnancies and an increased number of miscarriages
• Without genetic risk but with premature births and previous induced abortion
Syndromic Association
• 27.7% of all cases had chromosomal anomalies, heritable syndromes, or an additional major organ system defect (see Table 24-2)
| Population | Incidence of Congenital Heart Disease (%) | Most Common Lesions | ||
|---|---|---|---|---|
| 1 | 2 | 3 | ||
| Trisomy 21 syndrome | 50 | Ventricular septal defect, endocardial cushion defect | Atrial septal defect | Patent ductus arteriosus |
| Trisomy 18 syndrome | 99+ | Ventricular septal defect | Patent ductus arteriosus | Pulmonary stenosis |
| Trisomy 13 syndrome | 90 | Ventricular septal defect | Patent ductus arteriosus | Dextrocardia |
| Turner syndrome | 35 | Coarctation of the aorta | Aortic stenosis | Atrial septal defect |
| DiGeorge syndrome 22q deletion syndrome | 50 | Truncus arteriosus | Tetralogy of Fallot | Interrupted aortic arch |
Since the 1990s, much progress has been made in understanding the genetics of heart disease. Studies in recent years have brought an explosion of information on the identification of potential candidate genes regulating heart development. 5 An example is the relationship between TBX-1 and cardiovascular defects seen in DiGeorge syndrome. 16 Mutations in NOTCH-1 have been found to cause aortic valve disease. 37 Although research defining the signaling cascades that lead to specific forms of congenital heart defects still needs to be done, 27 some information is well established (e.g., 50% of congenital heart defects involve a ventricular septal defect (VSD) alone or in combination with other abnormalities). Studies also demonstrate that preterm infants are two and a half times more likely to have cardiovascular (CV) malformations.32Table 24-3 shows the most common congenital heart defects and their time of presentation.
| *These numbers are intended as a rough guideline because there is considerable overlap. Infants with congenital heart disease are often active initially and appear well for several hours or days after birth. In contrast, infants with respiratory distress often have characteristic symptoms within the first several hours after birth. |
|||||
| From Flyer DC et al: Report of the New England Regional Infant Cardiac Program, Pediatrics 65:377, 1980. | |||||
| 0-6 Days | (%) | 7-13 Days | (%) | 13-20 Days | (%) |
|---|---|---|---|---|---|
| Transposition of great arteries | (17) | Coarctation of the aorta | (19) | Ventricular septal defect | (20) |
| Hypoplastic left ventricle | (12) | Ventricular septal defect | (15) | Transposition of great arteries | (17) |
| Lung disease | (10) | Hypoplastic left ventricle | (11) | Coarctation of the aorta | (16) |
| Tetralogy of Fallot | (9) | Transposition of great arteries | (9) | Tetralogy of Fallot | (8) |
| Coarctation of the aorta | (7) | Tetralogy of Fallot | (6) | Endocardial cushion defect | (6) |
| Ventricular septal defect | (7) | Heterotaxia | (4) | Heterotaxia | (6) |
| Pulmonary atresia (with intact ventricular septum) | (7) | Truncus arteriosus | (4) | Patent ductus arteriosus | (4) |
| Single ventricle | (4) | Total anomalous pulmonary venous return | (3) | ||
| Heterotaxia | (6) | ||||
| Other | (25) | Other | (28) | Other | (20) |
| Total 896 | (100) | Total 210 | (100) | Total 116 | (100) |
PRENATAL DIAGNOSIS
Because of the widespread use of antenatal ultrasound, it is increasingly common for the fetus to be diagnosed with congenital heart disease. Fetal echocardiography has proven to be a valid, reliable, and accurate tool in the prenatal diagnosis of CHD. 9 The recommended timing of a fetal echocardiogram is between 18 to 20 weeks’ gestation, although reasonable images can be obtained as early as 16 weeks. Physical examination alone picks up 30% of cases of CHD. 20 Pulse oximetry is not an accurate screening tool for CHD because of its relatively high false-positive rate, especially when used shortly after birth when the Sp o2 in neonates is low. 20 Recent studies are investigating serum high-sensitivity C-reactive protein (hs-CRP) and brain natriuretic protein (BNP) levels as markers of CHD. Thus far, hs-CRP and BNP levels are useful only as indicators of hypoxia. 33
Pregnant women are encouraged to undergo fetal echocardiography if (1) genetic evaluation determines that the fetus has a chromosomal or genetic syndrome associated with CHD, (2) the mother or a previous child has CHD, or (3) there is a family history of CHD. Box 24-2 lists CHDs that are diagnosed and CHDs that may be missed on fetal echocardiogram. Magnetic resonance angiography is also an important diagnostic tool, especially for lesions that may be missed by fetal echocardiography. Detailed postnatal examinations are critical in determining the full extent of cardiac malformation.
BOX 24-2
Accurately Diagnosed
• Hypoplastic left heart syndrome
• Tricuspid atresia
• Pulmonary atresia
• Truncus arteriosus
• Tetralogy of Fallot
• Atriovenous septal defects
• Large ventricular septal defect (VSD)
• Transposition of the great arteries
May Be Missed
• Coarctation of the aorta
• Small VSD/atrial septal defect (ASD)
• Total anomalous pulmonary venous return (TAPVR)
• Mild aortic or pulmonary stenosis
Data Collection
HISTORY
A family history of congenital heart disease is significant, since a sibling with CHD increases the recurrence risk threefold. Viral exposure during pregnancy (rubella, Coxsackie B, and enteroviruses) and maternal ingestion of alcohol or drugs should be evaluated. Pregnancy, labor, and delivery complications should be carefully examined as risk factors that could affect the cardiovascular system. For example, intrauterine hypoxia and perinatal hypoxia are risk factors for the development of myocardial dysfunction, as well as persistent pulmonary hypertension of the newborn (PPHN). The timing of the onset of symptoms may indicate the type of anomaly (see Table 24-3).
CLINICAL PRESENTATION OF INFANTS WITH SEVERE CARDIAC DISEASE
In many neonates, congenital heart disease is not suspected until after birth when the newborn presents with one or more signs or symptoms (see the Critical Findings box on p. 684). 36 Timing of presentation of signs or symptoms depends on severity of the defect, in utero effects of the defect, and alterations in cardiovascular physiology during transitional circulation (i.e., closure of the ductus arteriosus and the fall in PVR). Despite the presence of many heterogeneous forms of heart disease, a surprisingly limited number of signs and symptoms present in the neonate.
Murmurs
Heart murmurs are a common finding in neonates. Estimates of prevalence of heart murmurs in neonates range from 1% to 70% depending on the study. 8Although cardiac murmurs in the neonatal period do not necessarily indicate heart disease, they must be carefully evaluated. The absence of a murmur does not exclude severe life-threatening cardiac anomalies. Pathologic murmurs tend to appear at characteristic ages (e.g., murmurs associated with semilunar valve stenosis and atrioventricular valve insufficiency tend to be noted very shortly after birth). In contrast, murmurs caused by left-to-right shunt lesions (PDA, VSD) may not be heard until the second to fourth week of life. Therefore the age of the neonate when the murmur is first noted and the character of the murmur give important clues about the nature of the cardiac defect.
Cyanosis
Cyanosis (a bluish discoloration of the skin, nail beds, and mucous membranes) is one of the most common presenting signs of congenital heart disease in the neonate. Depending on the underlying skin complexion, clinically apparent cyanosis is usually not visible until there is more than 3 gm/dL of desaturated hemoglobin in the arterial system. 20 Cyanosis depends on both the severity of hypoxemia (which determines the percent of oxygen saturation) and the hemoglobin concentration. True central cyanosis should be differentiated from acrocyanosis, blueness of the hands and feet only, which is a normal finding in the neonate.
Cyanosis in the newborn must be differentiated between cardiac and respiratory causes. Pulmonary disorders cause cyanosis in the neonate because of intrapulmonary right-to-left shunting. Varying degrees of hypoxemia manifest as cyanosis and can be to the result of primary lung disease (see Chapter 23) and central nervous system abnormalities. Clinical cyanosis can occur without hypoxemia in a neonate with methemoglobinemia and polycythemia.
Respiratory Distress
Most infants with cyanosis from CHD do not have respiratory distress (e.g., tachypnea, intercostal retractions, grunting, nasal flaring, dyspnea, rales, cyanosis). Often the degree of cyanosis is not proportional to the degree of respiratory distress evaluated from the physical and chest x-ray examinations. If cyanosis is present and is caused by a fixed right-to-left shunt (cardiac lesion), increasing inspired oxygen will have little effect on the arterial blood gases. However, if the cyanosis is caused by a diffusion defect in the lungs (pulmonary disorder), the degree of cyanosis often decreases with increasing inspired oxygen.
The hyperoxia test is beneficial in differentiating respiratory disease from cyanotic heart disease. This single test is perhaps the most sensitive and specific tool in the initial evaluation of the neonate with suspected congenital heart disease and is used to investigate the possibility of a fixed (intracardiac) right-to-left shunt. The hyperoxia test is performed by obtaining arterial blood gas measurements (preferably from the right radial artery) when the infant is in room air and then after the infant has been in 100% oxygen for 5 to 10 minutes. If the Pa o2 is greater than 150 mm Hg, the presence of a right-to-left shunt and cyanotic congenital heart disease as the cause of cyanosis is unlikely. Pulse oximetry cannot be used for documentation.
Congestive Heart Failure
Congestive heart failure (CHF) occurs when the heart cannot meet the metabolic demands of the tissues. Signs and symptoms of congestive heart failure reflect decreased cardiac output and decreased tissue perfusion. In the early stages, the neonate may be tachypneic and tachycardic with an increased respiratory effort, rales, hepatomegaly, and delayed capillary refill. Edema caused by CHF is rarely seen in neonates. Diaphoresis, feeding difficulties, and growth failure later become apparent. Finally, congestive heart failure may present acutely with cardiorespiratory collapse, particularly with obstructive defects. Birth asphyxia and anemia must also be considered as causes of congestive heart failure in neonates.
The common symptoms associated with congestive heart failure (see the Critical Findings box on p. 685) can be understood using the physiologic principles previously outlined.
Tachycardia
The heart attempts to compensate for the decrease in cardiac output (CO) by increasing either the heart rate (HR) or the stroke volume (SV) (CO = HR × SV). Because the fetal myocardium has fewer contractile elements and is poorly innervated by the sympathetic nervous system, capacity to increase stroke volume is very limited. Therefore increases in cardiac output are achieved mainly by increasing the heart rate.
Congestive Heart Failure
• Tachycardia
• Cardiac enlargement
• Tachypnea
• Gallop rhythm
• Decreased peripheral pulses and skin mottling in the extremities
• Decreased urine output and edema
• Diaphoresis
• Hepatomegaly
• Decreased activity
• Failure to thrive and feeding problems
• Diminished cardiac output
Cardiac Enlargement
Hypertrophy and dilation of the heart occur in response to the volume or pressure overload. This enlargement is evident on chest x-ray examination.
Gallop Rhythm
The gallop rhythm is an abnormal filling sound caused by dilation of the ventricles. It is heard as a triple rhythm on auscultation.
Decreased Peripheral Pulses/Mottling of the Extremities
Decreased cardiac output results in a compensatory redistribution of blood flow to vital tissues. Peripheral tissue perfusion is decreased, which results in mottling of the skin and decreased pulses.
Decreased Urine Output and Edema
Decreased renal perfusion results in decreased glomerular filtration. The body interprets this as a decrease in intravascular volume and begins to initiate compensatory mechanisms such as vasoconstriction and retention of fluid and sodium. Neonates manifest this as weight gain and periorbital edema.
Diaphoresis
Congestive heart failure leads to an increase in metabolic rate and increased activity of the autonomic nervous system, resulting in diaphoresis. This is representative of the increased workload of the heart in failure.
Hepatomegaly
The right ventricle in congestive heart failure is less compliant and does not adequately empty. This leads to elevated pressures in the right atrium, central venous system, and hepatic system. Hepatomegaly results from hepatic venous congestion.
Decreased Activity and Exercise Intolerance
The decreased perfusion to peripheral tissues and the increased energy needed by the heart in failure leaves little energy for activities such as feeding and crying. The infant in heart failure may sleep the majority of the time.
Failure to Thrive/Feeding Difficulties
Tachypnea compromises the infant’s ability to feed. The basal metabolic rate increases in neonates with congestive heart failure. This necessitates a higher caloric intake (150 kcal or more).
Dysrhythmias
Abnormalities of the cardiac rhythm and murmurs are discussed individually later.
CARDIAC EXAMINATION
See “Specific Conditions” section.
LABORATORY DATA
Arterial Blood Gases
The Pa co2 in cardiac disease is often normal. It is usually increased if a primary pulmonary disease is present. Frequent monitoring of blood gases is unnecessary, but the acid-base balance should be monitored closely.
Four-Extremity Blood Pressure
The measurement of blood pressure should be taken in both arms and both legs. A systolic pressure that is more than 10 mm Hg higher in the upper body compared with the lower body is abnormal and suggests coarctation of the aorta, aortic arch hypoplasia, or interrupted aortic arch. However, this is a highly specific test with low sensitivity; the lack of systolic blood pressure gradient does not conclusively rule out aortic arch abnormalities.
Chest X-ray Examination
Frontal and lateral views (if possible) of the chest should be obtained. In neonates, the size of the heart may be difficult to determine because of the overlying thymus. Chest x-ray examination may be normal even in the presence of life-threatening CHD. However, the degree of pulmonary vascularity helps define the type of CHD present and is characterized as being increased, normal, or decreased. Likewise, the heart size should be evaluated and is described as being increased, normal, or decreased.
Electrocardiogram
Neonatal electrocardiogram (ECG) reflects the hemodynamic relationships that existed in utero. Many forms of CHD have minimal prenatal hemodynamic effects, and therefore the ECG is frequently “normal for age” despite significant structural defects (e.g., transposition of the great arteries, tetralogy of Fallot).
Echocardiogram
The echocardiogram is indispensable in the diagnosis of congenital heart disease. 4 Two-dimensional echocardiography, used to define cardiac anatomy, estimates pressures, measures gradients, and evaluates cardiac function21 and, supplemented with Doppler and color Doppler, has become the primary diagnostic tool in pediatric cardiology. Noninvasive transthoracic echocardiogram is the most commonly used approach. During the procedure, close monitoring is recommended with attention to vital signs, respiratory status, and temperature. Recently, three-dimensional echocardiograms (3D Echos) that offer real-time three-dimensional imaging with 2 Dx-matrix probe and reconstructed 3D imaging using spatiotemporal image correlation (STIC) techniques21 are being used.
Computed Tomography
More practitioners are finding 64-slice multidimensional computed tomography (64-MDCT) to be helpful in imaging some thoracic regions beyond the scope of echocardiograms, particularly after surgical revision. MDCT offers higher spatial resolution with shorter scan times. 31
Magnetic Resonance Imaging
Magnetic resonance imaging (MRI) offers three-dimensional reconstruction and high-resolution images of the heart and great vessels. The MRI is of particular use in evaluating extracardiac vascular abnormalities, such as arch anomalies, vascular rings, and pulmonary arteriovenous anomalies. MRI provides high spatial resolution, excellent soft-tissue definition, a large field of view, and unrestricted demonstration of cardiovascular morphology; however, it does require sedation and a stable patient, and it is expensive. 13
General Treatment Strategy
Optimal management of infants with heart disease requires specialized expertise. Infants are monitored closely for hypoxia, hypoglycemia, acidosis, and congestive heart failure.
The infant must be kept in an incubator or radiant heat warmer in which body temperature is maintained while color changes (pallor and increased cyanosis) may be observed. A cardiorespiratory monitor for continuous cardiac monitoring detects bradycardia, tachycardia, and dysrhythmias. Monitoring of oxygen saturations is helpful in determining adequacy of pulmonary blood flow and/or increased need for oxygen. Respiratory effort is assessed for tachypnea, shallow breathing, apnea, retractions, grunting, and nasal flaring. Observe and document activity level such as feeding behavior, muscle tone, spontaneous movement, and seizure activity.
MANAGEMENT OF CONGESTIVE HEART FAILURE
The medical management of congestive heart failure attempts to reverse the outlined process and helps the heart compensate with increased cardiac output.
Digoxin acts primarily as a positive inotropic (improves contractility) agent but decreases the heart rate and increases urine output (Box 24-3). Digoxin slows conduction at the atrioventricular (AV) node. This drug should be used with caution if acidosis, myocarditis, or obstructive lesions (e.g., tetralogy of Fallot, subvalvular pulmonary stenosis, asymmetric septal hypertrophy) are present. Diuretics such as furosemide (Table 24-4) help decrease total body water (which is increased as a result of congestive heart failure). In general, chronic fluid restriction and low-salt diets are not commonly used in newborns or infants with congestive heart failure.
BOX 24-3
| Digitalizing Schedule | |
|
Preterm infant
PO route: 20 mcg/kg total dose*
Term infant
PO route: 30 mcg/kg total dose*
|
Total dose is usually divided into three doses giving one half, then one fourth, then one fourth of the total dose q 8 hr. Check electrocardiogram rhythm strip for rate, PR interval, and dysrhythmias before each dose. |
| Maintenance Schedule | |
|
Preterm infant
PO route: 5-10 mcg/kg/day*
|
Total dose should be divided BID. Allow 12-24 hr between last digitalizing and first maintenance doses. It takes about 6 days to “digitalize” a patient with maintenance doses alone. The sign of digitalis effect is usually prolongation of the PR interval. The first sign of digitalis toxicity is usually vomiting, dysrhythmia, or bradycardia. |
|
Term infant
PO route: 5-10 mcg/kg/day
|
Drugs such as quinidine, amiodarone, and diuretics predispose to digoxin toxicity. The clearance of digoxin is directly related to renal function. Dosage must be reduced in patients with impaired renal function. |
| BID, Twice daily. | |
| *Intravenous (IV) dose is 75% of oral (PO) dose. |
|
| Standard Concentrations: Each institution’s concentration may vary; NOT to exceed maximum concentration per pharmacy reference manuals. | ||||
| Neonatal Drug Guidelines Updated May 7, 2002. | ||||
| BID, Twice daily; BP, blood pressure; BUN, blood urea nitrogen; CNS, central nervous system; ETT, endotracheal tube; GI, gastrointestinal; IHSS, idiopathic hypertrophic subaortic stenosis; IV, intravenous; IM, intramuscular; KCl, potassium chloride; kg, kilograms; Maint., maintenance; max, maximum; mcg, micrograms; mg, milligrams; min, minutes; NEC, necrotizing enterocolitis; NS, normal saline; PO, per os; PRN, as needed; q, every; QID, four times a day; TID, three times a day. | ||||
| Drug | Route | Dose | Onset of Action | Comments |
|---|---|---|---|---|
| Atropine | IV | 0.01-0.03 mg/kg/dose PRN (max 0.4 mg) | Seconds | May cause tachycardia, urinary retention, or hyperthermia |
| PO | 0.01-0.03 mg/kg/dose q 4-6 hr (max 0.4 mg) | Minutes | May cause tachycardia | |
| ETT | Give 2-3 times the IV dose followed by NS flush | Minutes | May cause tachycardia | |
| Calcium chloride (10% solution) | IV | 0.2-0.3 mL (20-30 mg)/kg/ dose q 10 min PRN (max 500 mg) | Minutes | Slow infusion; must be IV; potentiates digoxin, bradycardia |
| Calcium gluconate (10% solution) | IV | 1-2 mL/kg/dose (100-200 mg/kg/dose) q 10 min PRN (max 500 mg) | Minutes | Slow infusion (over 10-30 min); must be IV; potentiates digoxin, bradycardia/dysrhythmias |
| Captopril (Capoten) | PO | Younger than 2 months:
Initial dose: 0.1-0.25 mg/kg/dose q 8-24 hr
Titrate: up to 0.5 mg/kg/dose
Older than 2 months:
Initial dose: 0.3 mg/kg/day
Titrate: up to 6 mg/kg/day in 1-4 divided doses
|
15 min + | Hypotension, tachycardia, increased BUN and serum creatinine, hypercalcemia |
| Diazoxide (Hyperstat) | IV | 5 mg/kg/dose q 30 min PRN | 1-2 min | May cause hypotension or hypoglycemia |
| Dobutamine (Dobutrex) | IV | 2-10 mcg/kg/min | Minutes |
Do not use if IHSS or tetralogy of Fallot, may cause ventricular ectopy, tachycardia, or hypertension
Incompatible with alkaline solutions
|
| Dopamine (Intropin) | IV | 5-10 mcg/kg/min | Minutes | Tachydysrhythmia, vasoconstriction, gangrene of extremities, anginal pain, and palpitations can occur; inactivated in alkaline solution |
| Epinephrine (1:10,000) | IV/ETT | 0.1-0.3 mL/kg/dose (max 5 mL/dose) q 3-5 min PRN | Seconds | May cause tachycardia, dysrhythmias, or hypertension; not effective if acidosis is present |
| Esmolol (Brevibloc) | IV |
Loading dose: 500 mcg/kg/min
Continuous infusion: titrate 50-200 mcg/kg/min
|
Minutes | May cause bradycardia, hypotension, bronchoconstriction |
| Furosemide (Lasix) | IV | 1-2 mg/kg/dose | 5-15 min | May cause metabolic alkalosis and hypokalemia; monitor electrolytes—may need KCl supplementation; renal calcification |
| PO | 1-4 mg/kg/dose | 30-60 min | ||
| Hydralazine (Apresoline) | IV PO |
0.1-0.5 mg/kg q 3-6 hr 0.1-0.5 mg/kg q 6 hr; may increase to max of 2 mg/kg/dose q 6 hr |
15-30 min Often days until titrated effect achieved |
May cause lupus-like syndrome, tachycardia, or hypotension |
| Hydrochloro-thiazide (HydroDIURIL) | PO | 1-2 mg/kg q 12 hr | 1-2 hr | May cause electrolyte imbalance, may need KCl supplementation |
| Ibuprofen lysine (NeoProfen) | IV | 10 mg/kg first dose; then 5 mg/kg second and third dose (q 24 hr) |
Most effective in first 3 days of life
Monitor urine output and creatinine levels; discontinue drug if dramatic decrease in urine output
Significantly fewer adverse effects (compared with indomethacin) on renal and mesenteric blood flow; less oliguria and increase in serum creatinine levels
|
|
| Indomethacin (Indocin) | IV | 0.1-0.2 mg/kg/dose; may be repeated q 8 hr for a total of 3 doses |
Less effective if administered after 7 days of age; probably will have no effect after 14 days
Monitor urine output and creatinine levels; discontinue drug if dramatic decrease in urine output
Contraindications: severe renal impairment, active bleeding in the CNS or GI tract, and NEC10
|
|
| Isoproterenol (Isuprel) | IV | 0.1-0.4 mcg/kg/min | 30-60 seconds | May cause tachycardia/ventricular tachydysrhythmia; may also cause subendocardial ischemia |
| Lidocaine (Xylocaine) | IV |
IV bolus: 1-3 mg/kg
IV drip: 30-50 mcg/kg/min
|
May cause dysrhythmia, CNS agitation or depression | |
| Milrinone (Primacor) | IV |
Loading: 50 mcg/kg over 15 min
Maint.: 0.25-0.75 mcg/kg/min
|
May cause ventricular dysrhythmias, ventricular fibrillation, or hypotension | |
| Nitroprusside (Nipride) | IV | 1-10 mcg/kg/min over 10 min to control BP; chronic infusion: 2 mcg/kg/min (protect from light; change solution q 4 hr) | Seconds | May cause hypotension and reflex tachycardia; may cause thiocyanate toxicity, especially if decreased renal function is present |
| Phentolamine (Regitine) | IV | 1-20 mcg/kg/min | 5-10 min | May cause hypotension; commonly used with an inotropic agent |
| PO | 5 mg/kg/day QID | |||
| Phenytoin (Dilantin) | IV |
Load: 10-15 mg/kg over 5 min (slow infusion)
Maint.: 3-5 mg/kg/day BID
|
5-10 min | May cause cardiac depression |
| PO | 3-5 mg/kg/day BID | 2-4 hr | Therapeutic blood levels (5-20 mcg/mL) | |
| Procainamide (Pronestyl) | IV |
Load: 10-15 mg/kg/dose over 5 min (max 100 mg)
Maint.: IV 30-80 mcg/kg/min
|
1-5 min | May cause hypotension or lupus-like syndrome |
| IM | 5-8 mg/kg q 6 hr | 15-30 min | Same as for IV | |
| Propranolol (Inderal) | IV |
Dysrhythmias: 0.01-0.15 mg/kg/dose slow IV q 6-8 hr PRN (max single dose, 10 mg)
Hypercyanotic spell: 0.15-0.25 mg/kg/dose slow IV push q 15 min (max dose 10 mg)
|
2-5 min | May severely decrease cardiac output |
| PO | Dysrhythmias: 0.5-1 mg/kg/dose TID-QID (max daily dose 60 mg) Hypercyanotic spell: 1-2 mg/kg/dose QID | 30-60 min | Same as for IV | |
| Prostaglandin E 1 (Prostin VR) | IV |
Initial dose: 0.05-0.1 mcg/kg/min cont. IV infusion
Maint.: 0.01-0.05 mcg/kg/min cont. IV infusion
|
30 min | May cause apnea, fever, or hypotension |
| Spironolactone (Aldactone) | PO | 1-2 mg/kg/day | 3-5 days | Hyperkalemia, drowsiness, GI upset |
| Tolazoline | IV |
Test: 1-2 mg/kg slow IV push
Maint.: 1-2 mg/kg/hr
|
Minutes | May cause hypotension, GI or pulmonary hemorrhage |
| Adenosine | IV | 30-250 mcg/kg | Slows the spontaneous heart rate and prolongs the PR interval; may cause transient complete heart block and hypotension; half-life is only 9.3 seconds so its effects quickly dissipate | |
Infants with congestive heart failure may be difficult to feed, and the process is often frustrating. They may have trouble sucking, swallowing, and breathing simultaneously. They may have to rest frequently during a feeding, thus prolonging feeding times, and they may fall asleep exhausted before adequate caloric intake is achieved. Because caloric requirements are higher in infants with CHD, the use of higher caloric formulas may be of benefit. Adequate nutrition must be ensured by the following:
• Observing the infant’s ability to nipple feed (a soft, free-flowing [premature] nipple offers the least resistance to sucking and helps the infant conserve energy)
• Providing adequate calories for growth and, if necessary, using alternative feeding methods (i.e., gavage or continuous nasogastric drip) if the infant is sucking poorly
• Anticipating the infant’s hunger and offering feedings before the infant uses energy by crying
• Positioning the infant in a semi-erect position for feeding
• Burping the infant after every half ounce consumed to help minimize vomiting
• Weighing the infant daily and checking for appropriate weight gain
Before discharge from the nursery, the infant should be in stable condition (e.g., feeding well and gaining weight appropriately).
An important fact for families to understand is that many infants gain weight very slowly because of their cardiac defects, regardless of the method of feeding used. The family of an infant in congestive heart failure needs support and teaching. Explanation of the term congestive heart failure should be given early, because it is a frightening term for parents. The term “heart failure” is often interpreted as “heart attack” or “cardiac arrest.” Parents must understand that saying an infant is in heart failure does not imply that the infant’s heart will stop beating. A simple explanation describing heart failure as a condition in which the heart shows signs of being less able to pump sufficient blood to meet all the needs of the body helps decrease anxiety for the family.
SPECIFIC CONDITIONS
Patent Ductus Arteriosus
PHYSIOLOGY
The ductus arteriosus is a normal pathway in the fetal circulatory system and allows blood from the right ventricle and pulmonary arterial system to flow into the descending aorta for ultimate delivery to the placenta (Figure 24-3). Functionally, the patent ductus arteriosus (PDA) closes within a few hours to several days after birth, but this closure is often delayed in premature infants. After birth, as a result of a decrease in the pressure of the pulmonary circulation and an increase in the pressure of the aorta, the blood flow through a PDA is predominantly from the aorta to the pulmonary artery (left-to-right shunt). The hemodynamic changes and the resultant clinical manifestations of a PDA depend on the magnitude of the pulmonary vascular resistance and the size of the ductal lumen.
 |
| FIGURE 24-3
(Modified from Hockenberry MJ, Wilson D: Wong’s essentials of pediatric nursing, ed 8, St Louis, 2009, Mosby.)
|
Approximately 15% of infants with PDAs have additional cardiac defects (e.g., VSD, coarctation of the aorta, aortic stenosis, pulmonary stenosis). PDA can be associated with known syndromes, most commonly rubella.
DATA COLLECTION
History
Asphyxial insult or respiratory distress syndrome (RDS), inability to wean from a ventilator, and an increasing F io2 demand usually accompany PDA.
Physical Findings
Increased flow to the pulmonary circulation and volume overload of the left ventricle are the two major physiologic abnormalities in a PDA.
Cyanosis
Generally, cyanosis is not present in an isolated PDA, because the predominant shunt is from left to right.
Heart Sounds
Infants with a PDA may have an audible murmur as a result of the left-to-right shunting through the ductus during systole. A grade I through III systolic murmur is best heard at the upper left sternal border with radiation to the left axilla and faintly to the back. Although this murmur occasionally may spill into diastole, the classical continuous machinery-like murmur is an unusual occurrence in the newborn period. It is often helpful to briefly disconnect the newborn from the ventilator before auscultating. There are cases of large PDAs in which no murmur is audible.
Pulses
Because of the rapid upstroke and wide pulse pressure, the peripheral pulses are bounding. Pulses are hyperdynamic and easily palpated. Assessment of the pulses should include palpation of palmar, plantar, and popliteal pulses. The presence of an easily palpated pulse in these areas suggests the presence of an aortic run-off lesion, which is most commonly a PDA.
Congestive Heart Failure
Because of the volume overload of the left ventricle, the infant may show signs of congestive heart failure and pulmonary edema (see “Congestive Heart Failure” section).
Laboratory Data
Arterial Blood Gases
Arterial blood gas values are normal.
Chest X-ray Examination
Chest x-ray examination is normal in small shunts. Cardiomegaly is present with increased pulmonary vascularity in large shunts.
Electrocardiogram
The ECG may be normal, demonstrate left ventricular hypertrophy, or demonstrate combined ventricular hypertrophy.
Echocardiogram
Direct imaging is the preferred method both to diagnose patency and to determine the significance of the ductus arteriosus. An increased left atrial/aortic ratio suggests a moderate to large left-to-right shunt (i.e., PDA, VSD). An echocardiogram should be performed before medical or surgical closure of the PDA to rule out a ductal-dependent lesion or other associated anomalies. Color-flow Doppler mapping allows visualization of the PDA and aids in determining the size and direction of the shunt across the PDA (i.e., left to right, right to left, bidirectional).
Cardiac Catheterization
If the echocardiogram has eliminated a ductal-dependent lesion, cardiac catheterization is usually not necessary before treatment.
TREATMENT
Medical Management
Asymptomatic infants with PDAs generally do not require medical management or surgical ligation. These infants should be monitored for evidence of congestive heart failure, failure to thrive, increasing oxygen requirement, or other complications.
Symptomatic infants require ductal closure by either pharmacologic management with prostaglandin inhibitors such as indomethacin or ibuprofen lysine therapy (both are Food and Drug Administration [FDA]–approved cyclo-oxygenase [COX] inhibitors) or surgical ductal ligation. Medical management such as fluid restriction, watchful waiting, and ventilator support is rarely successful, especially in low-birth-weight infants. The current trend is to treat early presymptomatic therapeutic PDA at 2 to 3 days of age after a confirmed echocardiogram.30 Although indomethacin was first reported in the 1970s and became first-line therapy for the treatment of PDA, ibuprofen lysine has been shown to be equally effective in closure rates and may be preferable because of its better toxicity profile. 25Table 24-4 has indomethacin and ibuprofen lysine doses, contraindications, and side effects. Urine output, as well as creatinine levels, should be continuously monitored with both medications. If urine output decreases dramatically, the drug should be discontinued. Ibuprofen appears to have significantly fewer adverse effects on renal and mesenteric blood flow, as well as fewer issues with increased serum creatinine and oliguria. 34
Surgical Treatment
Although surgical ligation of the ductus arteriosus through a lateral thoracotomy incision is a low-risk procedure when performed by an experienced surgical team, this should be reserved for those preterm infants who cannot tolerate or have failed pharmacologic intervention or when it is contraindicated. Questions have been raised about the long-term effects of surgical PDA closure in extremely-low-birth-weight infants and the relationship between ligation and bronchopulmonary dysplasia (BPD), severe retinopathy of prematurity (ROP), and neurosensory impairment. 14
COMPLICATIONS AND RESIDUAL EFFECTS
Complications and residual effects, although rare, include recanalization, recurrent laryngeal or phrenic nerve palsies, and false aneurysms. The surgical mortality rate in the neonatal period is generally less than 1%.
PROGNOSIS AND FOLLOW-UP
Asymptomatic infants have an excellent prognosis, although close follow-up is necessary because if the ductus remains patent beyond infancy, repair is usually recommended. Symptomatic infants with PDA generally experience failure to thrive, continued congestive heart failure, increased oxygen requirements with resultant BPD, or pulmonary infections.
Ventricular Septal Defect
PHYSIOLOGY
VSD is the most common cause of congestive heart failure after the initial neonatal period. VSDs may involve various portions of the ventricular septum and are classified according to the anatomic position that they occupy when viewed from the right ventricle (Figure 24-4). Defects in the membranous septum are the most common type and have been found to close spontaneously 20% of the time. 12 In contrast, 65% of muscular VSDs close spontaneously. 12
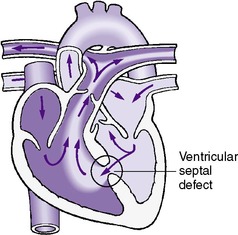 |
| FIGURE 24-4
(Modified from Hockenberry MJ, Wilson D: Wong’s essentials of pediatric nursing, ed 8, St Louis, 2009, Mosby.)
|
A VSD may occur as an isolated anomaly or may be part of a more complex cardiac lesion. Only isolated VSDs are discussed in this section. The effect of the VSD on the circulation depends on both the size of the VSD and the relative PVR. PVR is nearly systemic immediately after birth but rapidly falls to one-fourth to one-third systemic in the first several days of life.
In a small VSD, the left-to-right shunting at the ventricular level is minimal and the infants are asymptomatic.
Larger VSDs may have a moderate to large left-to-right shunt, resulting in congestive heart failure and pulmonary edema. Premature infants tend to have lower pulmonary vascular resistance at birth, allowing greater left-to-right shunting and therefore may be symptomatic. Infants with severe lung disease (e.g., RDS, BPD, pneumonia) may have elevated PVR and therefore minimal left-to-right shunting.
DATA COLLECTION
See Table 24-1 for infants at increased risk.
Physical Findings
Cyanosis
Infants with isolated VSDs are rarely cyanotic in the neonatal period.
Heart Sounds
Most infants with VSDs have a heart murmur. The time when this murmur is first audible depends on the PVR and the size of the defect. The murmur is typically a grade II to III/VI systolic murmur heard best at the lower left sternal border. A diastolic flow rumble at the apex indicates a large left-to-right shunt.
Congestive Heart Failure
Congestive heart failure is unusual in the newborn with an isolated VSD. When it occurs, however, it is a result of the volume overload of the left ventricle (see “Congestive Heart Failure” section).
Laboratory Data
Arterial Blood Gases
Arterial blood gas values are normal.
Chest X-ray Examination
A chest x-ray examination shows a normal to increased heart size with an increased pulmonary blood flow.
Electrocardiogram
The ECG in an infant with a VSD is usually normal but may demonstrate left or bi-ventricular hypertrophy.
Echocardiogram
A two-dimensional echocardiogram can demonstrate the VSD in 90% of the cases. Doppler interrogation of the ventricular septum and color-flow mapping have greatly increased the accuracy of diagnosing even the smallest VSDs noninvasively. The use of color-flow studies is particularly advantageous in identifying the presence of multiple VSDs and the direction of blood flow across a VSD.
Cardiac Catheterization
A cardiac catheterization is diagnostic but not necessary in the neonatal period unless there is some question about the diagnosis or if surgery is being considered.
TREATMENT
Medical Management
Medical management of congestive heart failure associated with VSDs includes digoxin, diuretics, and caloric supplementation. Failure to thrive is an indication for surgical repair of the defect.
Surgical Treatment
Surgical treatment of a VSD consists of either suture closure or patching (using most commonly a synthetic material such as Dacron). The surgical approach is through a median sternotomy incision. The defect is approached through the right atrium and tricuspid valve, thereby avoiding a right ventriculotomy.
If the infant is small (<2 kg) or if multiple muscular VSDs are present, it may be necessary to perform a palliative procedure called pulmonary artery banding to decrease pulmonary blood flow until the infant is older and can undergo debanding and closure of the VSDs. With improvements in surgical technique and technology, VSD closure can be performed safely and effectively in the younger pediatric population. 17
COMPLICATIONS AND RESIDUAL EFFECTS
Complications or residual effects may include (1) a persistent shunt (residual VSD), (2) conduction abnormalities (right bundle-branch block and third-degree heart block), and (3) aortic or tricuspid insufficiency (<1%).
The mortality rate in infants is less than 5%, with higher mortality found in the neonatal period. Contraindications to primary VSD closure include the diagnosis of double-outlet right ventricle and multiple muscular VSDs. The combined risk of pulmonary banding plus later debanding and VSD closure is about 10%.
PROGNOSIS AND FOLLOW-UP
Depending on the anatomic type, approximately 50% of small VSDs may close spontaneously in the first months of life. If a large left-to-right shunt is persistent after 12 to 24 months of age, the infant is susceptible to the development of pulmonary vascular disease.
Coarctation of the Aorta
PHYSIOLOGY
Coarctation of the aorta is a localized constriction of the aorta that usually occurs at the junction of the transverse aortic arch and the descending aorta in the vicinity of the ductus arteriosus (Figure 24-5). However, coarctation can occur anywhere in the aorta from above the aortic valve to the abdominal aorta. The precise location of the coarctation and the presence or absence of associated anomalies affect the clinical presentation. Associated anomalies include PDA, VSD, and bicuspid aortic valve (50%). Coarctation is observed in approximately 10% of infants with Turner syndrome.
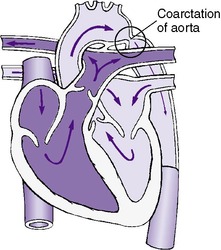 |
| FIGURE 24-5
(Modified from Hockenberry MJ, Wilson D: Wong’s essentials of pediatric nursing, ed 8, St Louis, 2009, Mosby.)
|
DATA COLLECTION
Physical Findings
In utero, the majority of systemic blood flow to the lower body is via the ductus arteriosus. After ductal closure, the neonate with coarctation becomes critically ill because the left ventricle must suddenly generate adequate pressure to pump the entire cardiac output past a significant point of obstruction. Newborns with critical coarctation of the aorta will then have signs and symptoms of congestive heart failure and low cardiac output. Severe coarctation of the aorta is a medical and surgical emergency.
Cyanosis
Generally, cyanosis is not present in the newborn with isolated coarctation of the aorta.
Heart Sounds
A cardiac murmur generally is not found in an isolated, severe coarctation of the aorta. If other cardiac defects are present, however, a murmur may be heard. A soft grade I to II/VI systolic murmur may be present at the left sternal border, radiating to the left axilla and to the back. A murmur heard only in the back is strongly suggestive of coarctation. A gallop rhythm sometimes is present and is associated with congestive heart failure. The murmurs of associated anomalies, however, usually are dominant.
Pulses and Blood Pressure
The blood pressure proximal to the area of obstruction is higher than the blood pressure distal to the area of obstruction.
The most consistent physical finding in infants with critical coarctation of the aorta is a higher systolic blood pressure (>15 mm Hg) in the upper extremities than in the lower extremities. This blood pressure must be measured with the appropriate-size cuff. In addition, pulses are easily palpable in one or both upper extremities but are difficult to palpate or are absent in the lower extremities. As mentioned earlier, pulses should be carefully evaluated in all extremities and blood pressures obtained in both arms and both legs.
Congestive Heart Failure
Congestive heart failure is a common finding in infants with severe coarctation and is the result of pressure overload on the left ventricle (see “Congestive Heart Failure” section).
Laboratory Data
Arterial Blood Gases
Arterial blood gas values are normal.
Chest X-ray Examination
Cardiomegaly may be seen on the x-ray film. Pulmonary vascularity is normal unless associated anomalies are present.
Electrocardiogram
Right ventricular hypertrophy is frequently present. Left ventricular hypertrophy or combined ventricular hypertrophy is rarely seen in the newborn period. The ECG may be normal.
Echocardiogram
The area of coarctation can often be visualized using two-dimensional techniques and color-flow mapping. However, cautious interpretation of the findings is suggested if a PDA is present.
Cardiac Catheterization
Cardiac catheterization can be diagnostic when performed before a surgical procedure to evaluate for other associated anomalies; however, it is rarely necessary.
TREATMENT
Medical Management
Congestive heart failure should be treated immediately and aggressively for stabilization before surgery. Intractable congestive heart failure, acidosis, oliguria, and hypertension are indications for corrective surgery as soon as possible. (See “General Treatment Strategy” discussion in “Congenital Heart Disease” section.) Medical management consists of continuous intravenous infusion of prostaglandin E 1 (PGE 1) to keep the ductus arteriosus open, dopamine and/or dobutamine for inotropic support, and correction of metabolic acidosis, hypoglycemia, and anemia. Since the introduction of PGE 1, emergency surgical repair is rarely necessary. Balloon angioplasty is rarely performed as a palliative emergency procedure.
Surgical Treatment
The two most common surgical procedures are resection of the coarctation with end-to-end anastomosis and the subclavian flap aortoplasty. With the former, the coarcted segment is resected and the ends of the aorta reanastomosed. With the latter, a longitudinal incision is made in the aorta across the coarctated site and continued to the end of the distally divided left subclavian artery. The left subclavian artery is used as a patch or flap to increase the diameter of the aorta. Both procedures are performed through a lateral thoracotomy incision and have been highly successful in relieving coarctation and providing for future growth of the aorta. Absorbable suture material often is used with the intention of decreasing the incidence of recoarctation from rigid suture lines.
COMPLICATIONS AND RESIDUAL EFFECTS
Complications and residual effects include (1) diminished or absent pulses in the left arm, (2) persistent hypertension, (3) Horner syndrome, (4) paraplegia (<0.5%), (5) mesenteric vasculitis, and (6) residual coarctation.
The overall operative mortality rate is as high as 20% in infancy. The high mortality rate is usually related to preoperative status and the presence of associated lesions.
PROGNOSIS AND FOLLOW-UP
Infants with mild coarctation require minimal care initially. If these patients are medically managed, close follow-up is mandatory, with repair likely at a later date.
Infants with severe coarctation require prompt medical and surgical treatment. If this therapy is instituted early, the prognosis generally is favorable. Untreated infants with severe coarctation often have a rapidly deteriorating clinical course with left intractable congestive heart failure, and the prognosis is guarded. After surgical repair, frequent follow-up is necessary to ensure adequate coarctation repair. Cardiac catheterization may be necessary several months to years after the surgical procedure is completed if recoarctation is suspected (20%). Balloon angioplasty can be performed if significant residual obstruction is found.
Critical Aortic Stenosis
PHYSIOLOGY
Obstruction of the left ventricular outlet may occur below the aortic valve, at the aortic valve, or above the aortic valve (subvalvular, valvular, or supravalvular aortic stenosis) (Figure 24-6). Valvular aortic stenosis is the most common type and is discussed here. A pressure gradient (the pressure difference from the left ventricle to the ascending aorta) of at least 60 mm Hg or more is indicative of significant aortic stenosis in the newborn.
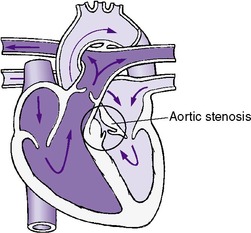 |
| FIGURE 24-6
(Modified from Hockenberry MJ, Wilson D: Wong’s essentials of pediatric nursing, ed 8, St Louis, 2009, Mosby.)
|
DATA COLLECTION
Physical Findings
Although most infants with aortic stenosis are asymptomatic in the neonatal period, a neonate with critical or severe aortic stenosis needs emergent treatment. The infant with critical aortic stenosis has pale gray, cool skin with decreased perfusion and peripheral pulses.
Cyanosis
Cyanosis is generally not present in isolated valvular aortic stenosis.
Heart Sounds
A grade II to IV/VI harsh systolic murmur is typically heard in the upper right sternal border, radiating to the upper left sternal border and faintly to the neck. The intensity of the murmur is unrelated to the severity of the obstruction. An ejection click may be heard at the apex. A suprasternal notch thrill is sometimes palpable.
Congestive Heart Failure
Infants with critical aortic stenosis have congestive heart failure caused by a pressure overload of the left ventricle (see “Congestive Heart Failure” section).
Laboratory Data
Arterial Blood Gases
Arterial blood gas values are generally normal.
Chest X-ray Examination
A chest x-ray examination shows cardiomegaly with normal pulmonary vascularity.
Electrocardiogram
The ECG may be normal or demonstrate left ventricular hypertrophy. There is poor correlation between an electrocardiographic abnormality and the degree of aortic stenosis present.
Echocardiogram
The aortic valve is usually thickened and appears to close abnormally on an echocardiogram. Doppler interrogation can accurately estimate the systolic pressure gradient from the left ventricle to the ascending aorta and identify the level or levels of obstruction.
Cardiac Catheterization
Cardiac catheterization is diagnostic and may or may not be performed in cases of critical aortic stenosis. Some centers are performing balloon dilation of the aortic valve during the cardiac catheterization.
TREATMENT
Medical Management
Initial medical management includes treatment of low output state. Positive end-expiratory pressure (PEEP) is helpful to overcome pulmonary venous desaturation from pulmonary edema. Inspired oxygen should be limited to F io2 of 0.5 to 0.6 unless severe hypoxemia is present. Surgical intervention is necessary for critical aortic stenosis in the newborn (see “General Treatment Strategy” section). However, balloon dilatation of aortic valve stenosis in the cardiac catheterization laboratory has been a successful alternative to surgical intervention in selected newborns.
Surgical Treatment
Aortic valvotomy through a median sternotomy incision is the surgical procedure for correcting critical aortic stenosis in infants. This procedure can usually be accomplished in the newborn with inflow occlusion and circulatory arrest for 1 to 2 minutes. In older infants, cardiopulmonary bypass should be performed. The fused commissures of the valve are incised, permitting the leaflets to open freely during systole.
COMPLICATIONS AND RESIDUAL EFFECTS
Complications and residual effects include aortic insufficiency and residual aortic stenosis. The mortality rate in infancy ranges from 5% to 50%, with the highest risk involving the newborn with critical obstruction.
PROGNOSIS AND FOLLOW-UP
All patients with critical aortic stenosis will require lifelong follow-up. Further surgical or catheter intervention is frequently necessary.
Critical Pulmonary Stenosis with Intact Ventricular Septum
PHYSIOLOGY
In critical pulmonary stenosis with intact ventricular septum, the flow to the pulmonary artery from the right ventricle is obstructed. The obstruction may occur below the valve in the infundibular area, above the valve, or at the valve (subvalvular, valvular, or supravalvular). In valvular stenosis, the orifice of the pulmonary valve is markedly narrowed and the valvular tissue may assume the shape of a cone (Figure 24-7). The pulmonary artery distal to this area of stenosis may be dilated. Because the ventricular septum is intact, the right ventricle is subjected to a marked increase in pressure and becomes hypertrophied. A pressure gradient from the right ventricle to the pulmonary artery of 50 mm Hg or more is indicative of significant pulmonary stenosis in the newborn.
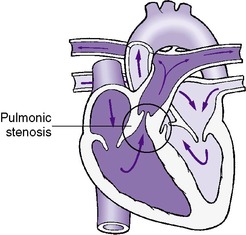 |
| FIGURE 24-7
(Modified from Hockenberry MJ, Wilson D: Wong’s essentials of pediatric nursing, ed 8, St Louis, 2009, Mosby.)
|
DATA COLLECTION
Physical Findings
Cyanosis
Cyanosis is generally not present in an isolated lesion but may occur in the presence of a right-to-left atrial shunt.
Heart Sounds
A harsh grade II to III/VI systolic murmur is heard in the upper left sternal border, radiating to both axillae and faintly to the back. Diastole is quiet. A murmur of tricuspid insufficiency (grade I/VI, soft, systolic murmur at the lower left sternal border) may be heard. An ejection click also may be heard at the left sternal border.
Congestive Heart Failure
The infant with critical pulmonary stenosis typically has signs and symptoms of right-sided congestive heart failure resulting from excessive pressure overload (see “Congestive Heart Failure” section).
Laboratory Data
Arterial Blood Gases
Arterial blood gas values generally are normal unless there is an atrial right-to-left shunt.
Chest X-ray Examination
The chest x-ray examination may be normal but usually demonstrates cardiomegaly with normal or decreased pulmonary vascularity.
Electrocardiogram
The ECG may be normal or demonstrate right ventricular hypertrophy.
Echocardiogram
An abnormal pulmonary valve pattern on a two-dimensional echocardiogram is diagnostic. Doppler interrogation and color-flow mapping can accurately estimate the systolic pressure gradient from the right ventricle to the pulmonary artery and identify the level or levels of obstruction.
Cardiac Catheterization
Infants suspected of having critical pulmonary stenosis with an intact ventricular septum usually undergo cardiac catheterization as soon as possible. Catheter balloon valvotomy has become the treatment of choice for this defect. Successful balloon valvotomy is associated with excellent clinical results, and the need for repeat surgical procedures is quite low.
TREATMENT
Medical Management
PGE 1 has been used successfully to maintain the patency of the ductus arteriosus, thereby allowing adequate pulmonary blood flow until surgery or balloon dilation is performed. If balloon dilation has been successful, surgical intervention may be postponed or may not be necessary at all.
Surgical Treatment
The degree of pulmonary stenosis and the size of the pulmonary arteries determine surgical approach. If the right ventricle and pulmonary arteries are of adequate size, then pulmonary valvulotomy through a median sternotomy incision is the preferable procedure. This involves incising the pulmonary valve commissures, allowing the leaflets to open freely during systole. Like aortic valvulotomy, this procedure can often be performed under inflow occlusion.
COMPLICATIONS AND RESIDUAL EFFECTS
Complications and residual effects include pulmonary insufficiency and residual pulmonary stenosis. The mortality rate for pulmonary valvulotomy is 17% in newborns.
If the right ventricle and pulmonary arteries are too small to allow antegrade flow, then a palliative procedure such as the Blalock-Taussig operation is performed. This procedure consists of bringing down the subclavian artery opposite the aortic arch and anastomosing it to the ipsilateral pulmonary artery or placing a Gore-Tex or Dacron tube graft (conduit) between the subclavian artery and the pulmonary artery.
Complications and residual side effects of Blalock-Taussig shunts include (1) diminished or absent pulses in the affected arm, (2) congestive heart failure from an overlarge shunt, and (3) inadequacy of the shunt. The mortality in this group is higher than in infants with adequate-size right ventricles and pulmonary arteries.
PROGNOSIS AND FOLLOW-UP
If a palliative shunt has been used, follow-up catheterization and surgical procedures should be anticipated either when the shunt becomes nonfunctional or when total repair is expected. If the lesion has been primarily corrected surgically in the neonatal period, repeated catheterization may be performed several months later to evaluate the residual obstruction if it is suspected. However, evaluation by echocardiogram may be sufficient without catheterization.
Atrioventricular Septal Defect, Endocardial Cushion Defect (Atrioventricular Canal)
PHYSIOLOGY
Nearly 70% of infants with complete atrioventricular canal have trisomy 21 (Down syndrome). The complete type of AV septal cushion defect is characterized by a large central hole in the endocardial cushion of the heart with free communication among all four chambers. The anterior leaflet of the mitral valve and the septal leaflet of the tricuspid valve both have clefts and are continuous with each other through the defect. Thus the AV valves are represented by a valve common to both sides of the heart (Figure 24-8).
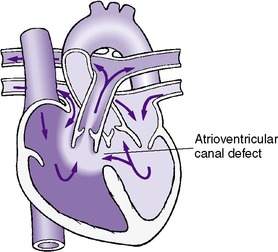 |
| FIGURE 24-8
(Modified from Hockenberry MJ, Wilson D: Wong’s essentials of pediatric nursing, ed 8, St Louis, 2009, Mosby.)
|
These infants usually have a left-to-right shunt at both the atrial and ventricular levels. AV valve insufficiency also may be present. The symptomatology depends on the degree of shunting at the atrial and ventricular levels and the amount of AV valve insufficiency present.
DATA COLLECTION
Physical Findings
Cyanosis
There may be some degree of cyanosis, particularly in the immediate neonatal period before the pulmonary vascular resistance has fallen.
Heart Sounds
Often there is no murmur audible. If AV valve insufficiency is present, a blowing, systolic, apical murmur with radiation to the left axilla or a VSD murmur is heard.
Congestive Heart Failure
Because of the large left-to-right shunt, which increases as pulmonary vascular resistance falls, these neonates typically present early in life with congestive heart failure. Congestive heart failure may be present because of volume overload of the ventricles as a result of AV valve insufficiency (see “Congestive Heart Failure” section).
Laboratory Data
Arterial Blood Gases
The Pa co2 may be elevated if there is severe AV valve insufficiency and pulmonary edema. The pH is usually normal. The Pa o2 usually is normal also.
Chest X-ray Examination
The heart size may be normal or increased. The pulmonary vascularity is generally increased.
Electrocardiogram
An ECG with a left axis deviation, counterclockwise loop in the frontal plane, and superior axis suggests AV septal defect.
Echocardiogram
A two-dimensional echocardiogram with Doppler and color-flow mapping is diagnostic and demonstrates the atrial septal defect (ASD), VSD, and common AV valve and the degree of AV valve insufficiency.
Cardiac Catheterization
Cardiac catheterization is diagnostic but generally not performed in the neonatal period in a typical AV septal defect. An echocardiographic diagnosis may be sufficient without cardiac catheterization.
TREATMENT
Medical Management
Medical treatment for congestive heart failure includes digoxin and diuretics. Other measures including providing a high level of supplemental oxygen, nitric oxide, and maintaining a mild respiratory alkalosis.
Surgical Treatment
If the infant does not respond to medical treatment and exhibits congestive heart failure, severe AV valve regurgitation, or pulmonary hypertension, surgical repair is necessary. The surgical procedure through a median sternotomy incision involves closing the ASD and VSD, separating the common leaflets of the mitral and tricuspid valves, and reconstructing the mitral valve.
COMPLICATIONS AND RESIDUAL EFFECTS
Complications and residual effects include (1) persistent shunt (residual ASD or VSD), (2) conduction abnormalities, (3) dysrhythmias and third-degree heart block, (4) mitral regurgitation, and (5) tricuspid regurgitation.
PROGNOSIS AND FOLLOW-UP
In the complete AV septal defect, congestive heart failure is a frequent problem and early surgical intervention generally is necessary. The prognosis after surgical repair in the neonatal period is guarded, with a generally favorable outcome if the surgery can be postponed until the infant is older than 6 months. The prognosis is also guarded if pulmonary hypertension persists after surgical intervention.
Ebstein’s Anomaly
PHYSIOLOGY
Ebstein’s anomaly is an uncommon but important anatomic heart defect when presenting in the neonatal period. 26 Anatomically, there is a downward displacement of the tricuspid valve into the body of the right ventricle. The resultant right ventricular cavity is smaller than normal and, because the elevated pulmonary vascular resistance is normally present in the newborn period, the cardiac output from the right ventricle to the pulmonary artery is decreased. This cardiac output generally increases as the pulmonary vascular resistance decreases after birth. Tricuspid insufficiency is present in varying degrees in the infant. There is a right-to-left shunt at the atrial level via the foramen ovale.
DATA COLLECTION
Physical Findings
Cyanosis
Varying degrees of cyanosis are present, depending on the amount of right-to-left shunting at the foramen ovale and the amount of blood that enters the pulmonary circulation by the right ventricle. In severe cases, the amount of pulmonary blood flow is markedly decreased, and these infants may be deeply cyanotic.
Heart Sounds
The second heart sound, S 2, is normal in the mildly affected infant, but the pulmonary component of S 2 may be diminished or inaudible in severely affected patients. A nonspecific systolic murmur is usually present and varies from a grade I/VI to a grade V/VI, representing tricuspid insufficiency. Diastolic murmurs, ejection clicks, and triple or quadruple rhythms are frequently heard.
Congestive Heart Failure
Newborns who are symptomatic usually have congestive heart failure resulting from volume overload of the left ventricle (see “Congestive Heart Failure” section).
Laboratory Data
Arterial Blood Gases
The Pa o2 may be normal to very low, depending on the amount of shunting at the atrial level. Pa o2 values in the low 20s are not uncommon.
Chest X-ray Examination
The chest x-ray examination shows cardiomegaly with decreased pulmonary vascularity. Massive cardiomegaly generally indicates severe tricuspid insufficiency.
Electrocardiogram
An ECG shows abnormal P waves and various degrees of heart block. The QRS complex generally demonstrates a right bundle-branch block pattern. Wolff-Parkinson-White (WPW) (preexcitation) syndrome frequently is present, and dysrhythmias are common.
Echocardiogram
A two-dimensional echocardiogram is diagnostic. Doppler interrogation and color-flow mapping are very useful in evaluating the amount of antegrade blood flow through the pulmonary valve, shunting at the arterial level, and the degree of tricuspid insufficiency present.
Cardiac Catheterization
There is an increased risk for dysrhythmias during catheterization. This procedure is not generally performed in the neonatal period unless a question about the differential diagnosis exists (to rule out pulmonary atresia).
TREATMENT
Medical Management
Medical management is aimed at supporting the neonate through the initial period of transitional circulation. Because of elevated pulmonary vascular resistance, pulmonary blood flow may be quite severely limited with profound hypoxemia and acidosis. PGE 1 is used to maintain a patent ductus arteriosus. Other measures include providing a high level of supplemental oxygen and maintaining a mild respiratory alkalosis. Both of these measures help decrease pulmonary vascular resistance and promote antegrade pulmonary blood flow. Recently, oral sildenafil (Viagra) is being used in symptomatic neonates. This pulmonary vasodilator reduces right ventricular afterload and helps improve forward flow of blood across the pulmonary valve. 1Ebstein’s anomaly is often associated with WPW syndrome and supraventricular tachycardia.
Surgical Treatment
Surgical treatment for Ebstein’s anomaly is controversial and generally reserved for the severely symptomatic patient. The procedure, performed through a median sternotomy incision, involves repositioning the tricuspid valve and an anuloplasty to improve the competency of the valve. In addition, plication of the atrialized ventricle is performed. Replacing the tricuspid valve may be necessary.
COMPLICATIONS AND RESIDUAL EFFECTS
High mortality rate in neonates with Ebstein’s anomaly is associated with pulmonary hypoplasia because of the massively enlarged right heart in utero that prevented pulmonary development.
Complications and residual effects include tricuspid insufficiency and dysrhythmias. The mortality in infancy is unknown because of insufficient data.
PROGNOSIS AND FOLLOW-UP
The prognosis for mild Ebstein’s anomaly is generally favorable. Infants with severe Ebstein’s anomaly generally improve as the pulmonary vascular resistance decreases and right ventricular output increases. Although surgery has been used successfully in the more severe forms of Ebstein’s anomaly, the prognosis is less favorable in patients requiring surgical intervention.
The prognosis for neonates presenting with profound cyanosis caused by Ebstein’s anomaly is quite grave.
Persistent Pulmonary Hypertension in the Newborn
PHYSIOLOGY
Infants with abnormally elevated PVR have persistent pulmonary hypertension of the newborn (PPHN) or persistent fetal circulation. These infants are generally hypoxic and acidotic but usually do not have severe pulmonary parenchymal disease or underlying cardiac disease. These infants have a right-to-left shunt at the ductal and atrial levels.
DATA COLLECTION
History
PPHN is usually associated with severe antepartum or peripartum conditions that involve hypoxia reflected by low Apgar scores. These infants are generally term or late preterm and are symptomatic within the first hours after birth. Associated findings may include polycythemia, hypoglycemia, or an anatomic abnormality such as congenital diaphragmatic hernia.
Physical Findings
Cyanosis
The milder cases of PPHN have minimal transient tachypnea and cyanosis associated with stress (crying or feeding). Severe cases demonstrate marked cyanosis, tachypnea, acidosis, and decreased peripheral perfusion.
Heart Sounds
A loud pulmonary component of S 2 and occasionally the systolic ejection murmur of tricuspid regurgitation (TR) are heard.
Congestive Heart Failure
Infants with PPHN may have congestive heart failure because of pressure overload of the right ventricle (see “Congestive Heart Failure” section).
Laboratory Data
Arterial Blood Gases
Arterial blood gas values demonstrate acidosis, hypoxia, and increased Pa co2. If a blood gas measurement is obtained simultaneously from the right radial artery (preductal) and from the descending aorta with an umbilical artery catheter (UAC) (postductal), the right-to-left shunt at the ductal level can be documented. If blood gas measurements are repeated after intubation and pharmacologic intervention (see “Treatment” section), the degree of hypoxia is often reduced.
Pulse Oximetry
Simultaneous preductal and postductal transcutaneous oxygen measurements may also be used.
Chest X-ray Examination
The chest x-ray examination demonstrates mild to moderate cardiomegaly with normal pulmonary vascular markings. The lung fields may be clear.
Electrocardiogram
The ECG frequently is normal but may demonstrate right ventricular hypertrophy and signs of myocardial ischemia.
Echocardiogram
An echocardiogram helps evaluate cardiac structures and rule out cyanotic lesions. Evaluating the right ventricular and pulmonary artery pressures by Doppler interrogation and the degree of right-to-left shunting at the atrial and ductal levels is helpful.
Cardiac Catheterization
Cardiac catheterization usually is not performed.
TREATMENT
Medical Management
See Chapter 23.
d-Transposition of the Great Arteries
PHYSIOLOGY
d-Transposition of the great arteries (Figure 24-9) is one of the most common forms of serious heart disease. The aorta arises from the right ventricle, receives unoxygenated systemic venous blood, and returns this blood to the systemic arterial circulation. The pulmonary artery arises from the left ventricle, receives oxygenated pulmonary venous blood, and returns this blood to the pulmonary circulation. This creates a situation of “parallel circulations.” The d-transposition anomaly can occur by itself or can be associated with other defects (e.g., PDA, ASD, VSD, pulmonary stenosis). Some patients with transposition have an associated ventricular septal defect, which allows for mixing between the parallel systemic and pulmonary circulations.
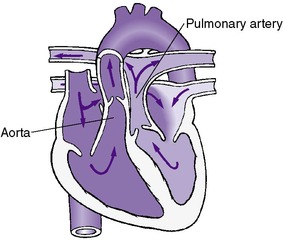 |
| FIGURE 24-9
(Modified from Hockenberry MJ, Wilson D: Wong’s essentials of pediatric nursing, ed 8, St Louis, 2009, Mosby.)
|
DATA COLLECTION
History
Transposition of the great arteries is more prevalent in males and is typically found in infants who are full term.
Physical Findings
The major physiologic abnormalities in d-transposition of the great arteries are an oxygen deficiency in the tissues and excessive workload of the right and left ventricles. The only mixing of oxygenated and unoxygenated blood occurs in the presence of associated lesions (e.g., patent foramen ovale, ASD, VSD, PDA, collateral circulation). The extent of the mixing depends on the number, size, and position of the anatomic communications, the pressure differential between the two systems, and changes in the systemic and pulmonary vascular resistances.
Cyanosis
These infants are usually cyanotic within the first hours of life, leading to their early diagnosis. Cyanosis is present in varying degrees, depending on the amount of intracardiac mixing present. Cyanosis may be mild if the mixing occurs through a significant VSD or PDA. Cyanosis is profound with intact ventricular septum or a closing PDA. Oxygen therapy will be of limited benefit. Only a certain amount of oxygenated blood can reach the systemic circulation, and administration of additional oxygen does not improve this situation. Enlargement of the interatrial communication by balloon septostomy (Rashkind procedure) during cardiac catheterization is commonly performed to establish adequate intercirculatory mixing for these infants.
After palliative procedures, the infant will continue to be cyanotic, especially in times of stress (crying, feeding, or exposure to cold temperatures). If the Pa o2, measured at rest in room air, is not greater than 35 mm Hg or if persistent metabolic acidosis is present, inadequate intracardiac mixing should be suspected.
Heart Sounds
The aorta arises from the anterior (right) ventricle, and the closure of the aortic valve is easily heard. The S 2 is single with an increased intensity. Murmurs, if present, are usually those of associated lesions.
Congestive Heart Failure
The infant may show signs of congestive heart failure, but this is rare unless there is a large VSD or PDA present (see “Congestive Heart Failure” section).
Laboratory Data
Arterial Blood Gases
In neonates with transposition of the great arteries and an intact ventricular septum, a very low Pa o2 (15-20 torr) with normal Pa co2 and mild metabolic acidosis are often seen.
Chest X-ray Examination
The chest x-ray examination may be normal or demonstrate either decreased or increased pulmonary vascularity. The cardiac silhouette may assume the shape of an “egg on a string.” However, this finding is not diagnostic.
Electrocardiogram
The ECG may be normal or demonstrate right ventricular hypertrophy.
Echocardiogram
The echocardiogram is extremely useful in establishing the diagnosis and evaluating associated lesions in infants with transposition of the great arteries.
Cardiac Catheterization
Cardiac catheterization is diagnostic. A balloon septostomy is commonly performed to improve interatrial mixing.
TREATMENT
Medical Management
Serial venous and arterial pH measurements should be obtained to rule out the presence of a persistent metabolic acidosis that would suggest inadequate intracardiac mixing. Hyperventilation and treatment with sodium bicarbonate are important to promote alkalosis. PGE 1 infusion is used to maintain ductal patency.
Surgical Treatment
The arterial switch procedure in most centers is the treatment of choice for d-transposition of the great arteries. This procedure, through a median sternotomy incision, involves transection of the main pulmonary artery and the aorta above the respective valves. The pulmonary artery is anastomosed to the right ventricle, and the aorta is anastomosed to the left ventricle (the aortic valve becomes a functional pulmonary valve, and the pulmonary valve becomes a functional aortic valve). The coronary arteries are resected with a button of surrounding tissue and reanastomosed to the supravalvular area of the ascending aorta.
It is essential in performing this procedure that the left ventricular (LV) pressure is systemic. In infants with a VSD, the LV pressure tends to remain elevated; therefore this procedure may be postponed for several days or even months. However, once the LV pressure decreases below that of the right ventricle, the morbidity and mortality of surgery increase dramatically. Therefore infants with an intact ventricular septum require surgery within the first few days of life.
COMPLICATIONS AND RESIDUAL EFFECTS
Complications and residual effects of the arterial switch procedure include (1) dysrhythmias, (2) myocardial ischemia and infarction, and (3) aortic or pulmonary supravalvular stenosis.
PROGNOSIS AND FOLLOW-UP
Without treatment, 30% of these infants die within the first week of life, 50% die within the first month, 70% die within the first 6 months, and 90% die within the first year. With treatment, the mortality rate is reduced to approximately 5% or less.
Tetralogy of Fallot
PHYSIOLOGY
Tetralogy of Fallot is the most common cyanotic congenital heart defect. The four components of tetralogy of Fallot are VSD, overriding of the ascending aorta, obstruction of the right ventricular outflow tract, and right ventricular hypertrophy (Figure 24-10).
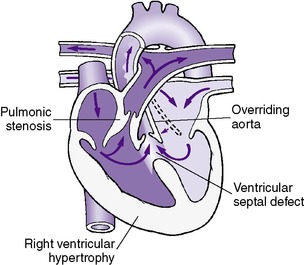 |
| FIGURE 24-10
(Modified from Hockenberry MJ, Wilson D: Wong’s essentials of pediatric nursing, ed 8, St Louis, 2009, Mosby.)
|
DATA COLLECTION
Physical Findings
Symptomatology in these infants relates to the degree of right ventricular outflow tract obstruction. Newborns who are symptomatic usually have severe right ventricular outflow tract obstruction.
Cyanosis
The predominant intracardiac shunt is right to left; therefore most infants with tetralogy of Fallot are cyanotic. However, if the right ventricular outflow obstruction is only mild or moderate, the intracardiac shunt is mainly left to right and the infant initially will not be cyanotic.
Infants with tetralogy of Fallot occasionally have hypercyanotic (or “tet”) spells. These spells consist of cyanosis, irritability, pallor, tachypnea, flaccidity, and possible loss of consciousness. They may be the result of a transient increase in the obstruction of the right ventricular outflow tract (usually the muscular infundibular area) and usually resolve with knee-chest positioning, oxygen, propranolol, or morphine.
Heart Sounds
A grade II to IV/VI harsh systolic murmur at the mid to upper left sternal border is usually present but is diminished or absent during a hypercyanotic spell. The S 2 is usually loud and single (representing aortic closure).
Congestive Heart Failure
Congestive heart failure is uncommon in tetralogy of Fallot.
Laboratory Data
Arterial Blood Gases
The Pa co2 and pH are normal. The Pa o2 is normal if the pulmonary stenosis is mild and there is little right-to-left shunting at the ventricular level. If the pulmonary stenosis, however, is more severe, the amount of right-to-left shunting increases and the Pa o2 falls.
Chest X-ray Examination
The classic chest x-ray examination in tetralogy of Fallot shows the shape of a boot with a normal-size heart. However, the classic chest x-ray pattern described is not common in the newborn. Pulmonary vascularity is either normal or decreased.
Electrocardiogram
The ECG demonstrates right ventricular hypertrophy.
Echocardiogram
The echocardiogram is suggestive when the overriding aorta can be demonstrated. Echocardiograms help identify the pulmonary valve to rule out pulmonary atresia. Doppler interrogation helps define the degree and level of pulmonary stenosis. Color-flow mapping identifies the VSD, as well as the direction of blood flow across the VSD.
Cardiac Catheterization
Cardiac catheterization is diagnostic and performed in the newborn when there is a question about the differential diagnosis (pulmonary atresia).
TREATMENT
Medical Management
Immediate medical management involves establishing adequate pulmonary blood flow with PGE 1 infusion. Digoxin is not routinely used because it may increase the amount of infundibular obstruction present. Propranolol is the preferred drug for treating hypercyanotic spells, although morphine has been used successfully (see “General Treatment Strategy” section).
Surgical Treatment
Total correction of tetralogy of Fallot involves intracardiac repair with patch closure of the large VSD and relief of the right ventricular outflow obstruction performed through a median sternotomy incision. Often a pericardial patch across the pulmonary valve annulus is necessary. Contraindications include small size of the infant, anomalous left anterior descending coronary artery, and hypoplastic pulmonary arteries.
Total surgical repair of tetralogy of Fallot is not usually carried out in the neonatal period. Surgery is usually performed electively within the first year of life. 15 More recently, repair at 3 to 6 months is recommended with successful outcomes. 11 If surgical intervention in infancy is warranted (i.e., the infant is severely hypoxic because of inadequate pulmonary blood flow), a systemic-to-pulmonary shunt is performed. The Blalock-Taussig operation is usually preferred (see description of the Blalock-Taussig operation on p. 697).
COMPLICATIONS AND RESIDUAL EFFECTS
Complications and residual effects include (1) diminished or absent pulses in the affected arm, (2) congestive heart failure from an overly large shunt, and (3) inadequate shunt. Mortality rate in infancy is 10%.
PROGNOSIS AND FOLLOW-UP
Surgery is recommended if the infant is symptomatic or refractory to medical care. Tetralogy of Fallot without surgery carries a grave prognosis. Long-term follow-up after corrective surgery has indicated impaired neurodevelopment, both in intellectual and behavioral functioning. 22
Pulmonary Atresia with Intact Ventricular Septum
PHYSIOLOGY
Pulmonary atresia is characterized by complete agenesis of the pulmonary valve. This lesion produces severe signs or symptoms soon after birth and is not compatible with life unless there is an associated interatrial communication and an additional pathway of entry for blood into the pulmonary circulation (through a PDA or collateral blood flow). Because flow to the lungs may depend on a PDA, death may occur when this structure closes. The right ventricle is usually hypoplastic but may be normal or dilated, depending on the degree of tricuspid insufficiency present. The presence of sinusoidal connections between the right ventricle and the coronary arteries is associated with poorer long-term survival. 8
DATA COLLECTION
Physical Findings
Cyanosis
Cyanosis is always present in varying degrees, depending on the amount of pulmonary blood flow from the PDA and degree of atrial right-to-left shunting.
Heart Sounds
The S 2 is single, and a soft systolic murmur is heard as a result of either the PDA or tricuspid insufficiency in about one half of the infants with pulmonary atresia.
Congestive Heart Failure
Congestive heart failure is usually present with moderate to severe tricuspid insufficiency (see “Congestive Heart Failure” section).
Laboratory Data
Arterial Blood Gases
The pH and Pa co2 are usually within normal range. The Pa o2, however, usually is very low (20 to 30 torr), unless there is a large shunt at the ductal or bronchial collateral level. In some cases, the amount of pulmonary blood flow is insufficient, and the pH may be low, reflecting metabolic acidosis.
Chest X-ray Examination
The heart appears enlarged on x-ray examination if tricuspid insufficiency is present. Pulmonary vascularity is either decreased or normal, depending on the amount of shunting through the PDA or collateral blood flow.
Electrocardiogram
The ECG is usually normal but may demonstrate left ventricular hypertrophy.
Echocardiogram
The two-dimensional echocardiogram with Doppler and color-flow mapping can identify absence of blood flow across the pulmonary valve and is diagnostic.
Cardiac Catheterization
Cardiac catheterization is diagnostic and may be performed if the diagnosis is suspected. A balloon atrial septostomy may be performed at the time of catheterization. Catheterization is also used to define the coronary artery anatomy before surgery.
TREATMENT
Medical Management
PGE 1 is used to maintain patency of the ductus arteriosus until surgical intervention (see “General Treatment Strategy” section).
Surgical Treatment
In most medical centers, a systemic-to-pulmonary shunt such as the Blalock-Taussig operation is performed through a lateral thoracotomy incision. However, some institutions are performing a pulmonary valvulotomy or a pulmonary outflow patch procedure in addition to a shunt. This establishes an open pathway through the atretic valve area between the pulmonary artery and the right ventricle. Antegrade blood flow through the right ventricle and pulmonary artery then promotes growth of these areas. The pulmonary valvotomy and pulmonary outflow patch procedures are performed through a median sternotomy incision.
COMPLICATIONS AND RESIDUAL EFFECTS
Complications and residual effects of the Blalock-Taussig operation include (1) diminished or absent pulses in the affected arm, (2) congestive heart failure from an overlarge shunt, and (3) inadequate shunt. The mortality rate in infants is 25% or higher.
PROGNOSIS AND FOLLOW-UP
Pulmonary atresia is fatal without surgical intervention. If a palliative shunt is performed, catheterization and further surgical procedures should be anticipated when the shunt becomes inadequate. If primary surgical correction is undertaken in the newborn period, catheterization should be anticipated to evaluate residual obstruction. Despite the development of newer surgical techniques, the prognosis in these infants is guarded.
Total Anomalous Pulmonary Venous Return
PHYSIOLOGY
Total anomalous pulmonary venous return (TAPVR) occurs when all pulmonary veins drain into the systemic venous system with complete mixing of pulmonary and systemic venous return. The presence of an ASD is necessary to sustain life (Figure 24-11). The four main varieties of TAPVR are as follows:
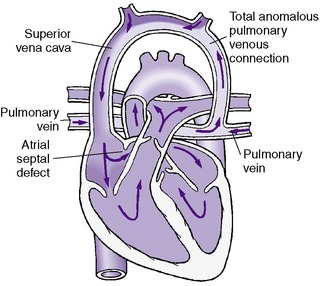 |
| FIGURE 24-11
(Modified from Hockenberry MJ, Wilson D: Wong’s essentials of pediatric nursing, ed 8, St Louis, 2009, Mosby.)
|
• Supracardiac (most common), in which the drainage is to the superior vena cava through the innominate vein
• Cardiac, in which the pulmonary veins drain into the coronary sinus or directly into the right atrium
• Infracardiac, in which the four veins join behind the heart, pass through the diaphragm, and connect to the portal venous system or a systemic vein
• Mixed
Each of the various types of anomalous drainage can occur with or without obstruction along the pulmonary venous pathway. The presence or absence of obstruction profoundly affects the clinical course.
DATA COLLECTION
Physical Findings
Cyanosis
Infants with obstructed or unobstructed TAPVR are frequently cyanotic. Because all pulmonary venous return (oxygenated blood) ultimately enters the right atrium (as opposed to the left atrium), a right-to-left shunt at the atrial level is necessary to sustain life.
Heart Sounds
Murmurs are rarely heard in infants with TAPVR and, when present, are nonspecific.
Congestive Heart Failure
Infants with unobstructed TAPVR usually show signs of congestive heart failure resulting from volume overload of the right ventricle. Infants with obstructed TAPVR generally do not demonstrate evidence of congestive heart failure but typically demonstrate pulmonary venous congestion (see “Congestive Heart Failure” section).
Laboratory Data
Arterial Blood Gases
The pH and Pa co2 are usually normal. The Pa o2 may be within the normal range if there is a large amount of pulmonary blood flow (always associated with severe congestive heart failure). If the pulmonary blood flow is limited, secondary to obstruction of blood flow, the Pa o2 may be low, but this is rare.
Chest X-ray Examination
If the TAPVR is obstructed, the chest x-ray examination will demonstrate pulmonary venous congestion without cardiomegaly. If the TAPVR is unobstructed, the chest x-ray examination will demonstrate a marked increase in pulmonary vascularity and cardiomegaly.
Electrocardiogram
An ECG may demonstrate right axis deviation, right ventricular hypertrophy, and right atrial enlargement.
Echocardiogram
An echocardiogram is diagnostic, but it is sometimes difficult to visualize the pulmonary veins by this method. The diagnosis of TAPVR is strongly suggested when an extra vascular structure is seen behind the small left atrium. With color-flow mapping, the right-to-left shunting across the atrial septum, as well as the anomalous venous return as it enters through the atrium, superior vena cava, or coronary sinus, can be visualized.
Cardiac Catheterization
Infants suspected of having TAPVR may undergo cardiac catheterization to define the type of TAPVR and presence or absence of obstruction. A Rashkind balloon septostomy may be performed at that time to improve intraatrial mixing.
TREATMENT
Medical Management
Obstructed TAPVR is a surgical emergency. Nonobstructed TAPVR may be medically treated temporarily (prostaglandin and ventilatory support), although surgery at the time of diagnosis is generally recommended (see “General Treatment Strategy” section).
Surgical Treatment
Surgical correction of TAPVR depends on the variety. Supracardiac and infracardiac varieties require surgical reimplantation of the common vein into the left atrium. Intracardiac TAPVR can usually be surgically repaired by realigning the atrial septum during closure of the ASD and directing the anomalous veins to the left atrial side. All repairs are performed through a median sternotomy incision.
COMPLICATIONS AND RESIDUAL EFFECTS
Complications and residual effects include pulmonary venous obstruction and dysrhythmias. The mortality rate varies from 5% to 25% in infancy, depending on the anatomic type.
PROGNOSIS AND FOLLOW-UP
Infants with nonobstructed TAPVR generally do well if the lesion is recognized early and early corrective surgery is performed. The prognosis for obstructed TAPVR is less favorable despite early surgical intervention.
Tricuspid Atresia
PHYSIOLOGY
In tricuspid atresia, there is complete agenesis of the tricuspid valve with no direct communication between the right atrium and right ventricle. Systemic venous blood entering the right atrium is shunted through a patent foramen ovale or ASD into the left atrium. If a large VSD is present, the right ventricle and pulmonary arteries may be normal in size. If the ventricular septum is intact but a large PDA is present, the right ventricular cavity may be hypoplastic and the pulmonary arteries are usually slightly decreased or normal in size (Figure 24-12). About 30% of these infants will have transposition of the great arteries.
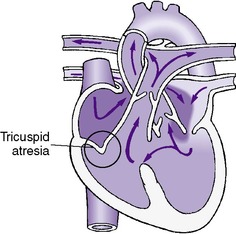 |
| FIGURE 24-12
(Modified from Hockenberry MJ, Wilson D: Wong’s essentials of pediatric nursing, ed 8, St Louis, 2009, Mosby.)
|
DATA COLLECTION
Physical Findings
Cyanosis
The degree of cyanosis varies. Newborns will have marked cyanosis if the pulmonary blood flow is compromised.
Heart Sounds
A single S 2 is present in infants with tricuspid atresia. Murmurs of associated shunts (VSD and PDA) may be present.
Congestive Heart Failure
Congestive heart failure may be present with a large shunt (PDA or VSD) (see “Congestive Heart Failure” section).
Laboratory Data
Arterial Blood Gases
The pH and Pa co2 usually are normal. The Pa o2 may vary from near normal if there is a large VSD or PDA to extremely low if there is limited shunting into the pulmonary system.
Chest X-ray Examination
A chest x-ray examination is nondiagnostic and may show a normal heart size or cardiomegaly. Pulmonary vascularity may be normal, decreased, or increased, depending on the degree of pulmonary blood flow.
Electrocardiogram
An ECG usually demonstrates left axis deviation with a counterclockwise loop, a superior axis in the frontal plane, and left ventricular electrical dominance.
Echocardiogram
Absence of the tricuspid valve and presence of a hypoplastic right ventricle are diagnostic of tricuspid atresia. Color-flow mapping can identify the right-to-left shunt at the atrial level and the presence of a VSD or PDA.
Cardiac Catheterization
An infant suspected of having tricuspid atresia usually undergoes cardiac catheterization and a balloon septostomy. A balloon septostomy is performed to improve intraatrial mixing.
TREATMENT
Medical Management
Immediate medical management is aimed primarily at maintaining adequate pulmonary blood flow. In the usual case of severely limited pulmonary blood flow, PGE 1 infusion maintains pulmonary perfusion via the ductus arteriosus.
Surgical Treatment
The preferred procedure in the neonatal period is a systemic-to-pulmonary shunt such as the Blalock-Taussig operation performed through a lateral thoracotomy incision.
Definitive “repair” of tricuspid atresia is accomplished occasionally with a single operation (Fontan procedure) or, more frequently, a staged procedure (Glenn followed by a Fontan). These operations involve anastomosis of the systemic venous return directly to the pulmonary artery. Any associated defects present are also repaired. Definitive repair is performed through a median sternotomy incision.
COMPLICATIONS AND RESIDUAL EFFECTS
Complications and residual effects include heart failure, pleural effusions, renal or liver failure, persistent shunts, conduit obstruction, dysrhythmia, and protein-losing enteropathy. The mortality rate in infancy is unknown, but in older children it is approximately 10% to 25%.
PROGNOSIS AND FOLLOW-UP
The prognosis for tricuspid atresia is guarded. The Fontan procedure may improve this prognosis.
Truncus Arteriosus
PHYSIOLOGY
Truncus arteriosus is characterized by one great artery arising from the left and right ventricles, overriding a VSD. This common artery has one valve and gives rise to (in order) the coronary arteries, the pulmonary arteries, and the brachiocephalic arteries (Figure 24-13). A coexisting VSD is present in more than 98% of cases. Truncus arteriosus is classified into three types, depending on the origins of the pulmonary arteries:
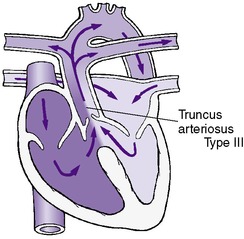 |
| FIGURE 24-13
(Modified from Hockenberry MJ, Wilson D: Wong’s essentials of pediatric nursing, ed 8, St Louis, 2009, Mosby.)
|
1. Type I—A short, main pulmonary artery arises from the common trunk that bifurcates into the right and left pulmonary arteries.
2. Type II—The right and left pulmonary arteries arise directly from the posterior surface of the common trunk.
3. Type III—The right and left pulmonary arteries arise directly from the lateral walls of the common trunk.
The ductus arteriosus is absent in approximately 50% of infants with truncus arteriosus. Between 30% and 35% have a right aortic arch. Extracardiac anomalies are present in 20% to 40% of cases.
DATA COLLECTION
Physical Findings
In truncus arteriosus, the common trunk receives a mixture of unoxygenated blood from the right ventricle and oxygenated blood from the left ventricle. Blood flow to the lungs varies with the type of truncus but is usually increased and at systemic level pressure.
Cyanosis
Cyanosis may be present at birth but varies in intensity according to the amount of pulmonary blood flow. Minimal cyanosis indicates adequate pulmonary blood flow.
Heart Sounds
The first heart sound, S 1, is normal, but the S 2 is single and loud because of the single valve of the common trunk. A loud systolic ejection click is frequently heard.
A loud pansystolic murmur maximal at the lower left sternal border that radiates to the entire precordium is commonly heard. A mid-diastolic rumble may be present. If the truncal valve is insufficient, a blowing diastolic murmur may be heard. A wide pulse pressure also may be present.
Congestive Heart Failure
Congestive heart failure may be present shortly after birth or appear between 2 and 3 weeks of age. The presence of congestive heart failure depends on the amount of pulmonary blood flow. Persistently high pulmonary arteriolar resistance in the first few weeks of life decreases pulmonary blood flow, and congestive heart failure may not be present. However, if the truncal valve is severely abnormal, congestive heart failure may be present shortly after birth (see “Congestive Heart Failure” section).
Laboratory Data
Arterial Blood Gases
The pH and Pa co2 are usually normal. If there is no obstruction to pulmonary blood flow, the Pa o2 may be near normal (usually associated with severe congestive heart failure). If the pulmonary blood flow is restricted, the Pa o2 may be extremely low.
Chest X-ray Examination
Cardiomegaly, displayed pulmonary arteries, and increased vascular markings are typical findings on the chest x-ray examination.
Electrocardiogram
Combined ventricular hypertrophy is most often seen in an ECG. Left atrial enlargement is also commonly found.
Echocardiogram
A two-dimensional echocardiogram is helpful in establishing the diagnosis and in differentiating tetralogy of Fallot from truncus arteriosus. In addition, the echocardiogram is used to identify the number of truncal valve leaflets, the presence of truncal valve insufficiency, or stenosis.
Cardiac Catheterization
A cardiac catheterization is diagnostic and is usually performed on an infant suspected of having truncus arteriosus.
TREATMENT
Medical Management
Medical management of these infants consists of stabilizing and treating congestive heart failure when present. Calcium should be closely monitored because of the possibility of DiGeorge syndrome or 22q deletion syndrome.
Surgical Treatment
Repairing of truncus arteriosus is rare in the newborn period and is usually carried out at 6 weeks to 6 months of age. It consists of separating the pulmonary artery from the common trunk, closing the VSD with a patch, and inserting a right ventricular-to-pulmonary artery valved conduit. The use of homograft conduits for repair of truncus arteriosus has become more common. Total repair of truncus arteriosus is performed through a median sternotomy incision.
COMPLICATIONS AND RESIDUAL EFFECTS
Complications and side effects include pulmonary vascular disease, residual shunts, truncal valve insufficiency, and conduit obstruction. The mortality rate is 40% to 50% in infancy.
PROGNOSIS AND FOLLOW-UP
The natural history depends on the amount of pulmonary blood flow and the competency of the truncal valve. Without treatment, more than half of these infants die before 3 months of age. Survival past 1 year of age ranges from 15% to 30%. Truncus arteriosus is often associated with DiGeorge syndrome, which has a wide spectrum of clinical manifestations.
Hypoplastic Left Heart Syndrome
PHYSIOLOGY
Hypoplastic left heart syndrome represents a clinical spectrum that includes severe coarctation of the aorta, severe aortic valve stenosis or atresia, and severe mitral valve stenosis or atresia (Figure 24-14). The left ventricle and ascending aorta are hypoplastic. Coronary blood flow occurs in a retrograde fashion into the small ascending aorta through the PDA. The resultant poor myocardial perfusion leads to rapid decompensation.
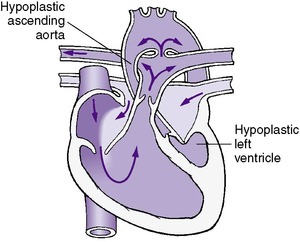 |
| FIGURE 24-14
(Modified from Hockenberry MJ, Wilson D: Wong’s essentials of pediatric nursing, ed 8, St Louis, 2009, Mosby.)
|
DATA COLLECTION
Physical Findings
Cyanosis
These infants are usually not truly cyanotic but, rather, have severe pallor and a grayish skin color as a result of marked poor perfusion, vasoconstriction, and congestive heart failure.
Heart Sounds
A nonspecific systolic murmur is heard in approximately two thirds of infants with hypoplastic left heart syndrome.
Congestive Heart Failure
Congestive heart failure is present in all cases as a result of right ventricular volume and pressure overload (see “Congestive Heart Failure” section).
Laboratory Data
Arterial Blood Gases
The arterial blood gas may represent the single best indicator of hemodynamic stability. Low arterial saturation (75% to 80%) with normal pH indicates an acceptable balance of systemic and pulmonary blood flow with adequate peripheral perfusion. Elevated oxygen saturation (>90%) with acidosis represents significantly increased pulmonary and decreased systemic flow with probable myocardial dysfunction.
Chest X-ray Examination
Cardiomegaly with increased pulmonary vascularity and pulmonary edema is seen on the x-ray examination.
Electrocardiogram
An ECG frequently demonstrates right axis deviation and right ventricular hypertrophy. However, the ECG may be normal.
Echocardiogram
An echocardiogram is usually diagnostic with a small left ventricular cavity and ascending aorta, mitral and aortic valve atresia or hypoplasia, and a dilated right ventricle (RV).
Cardiac Catheterization
Cardiac catheterization carries a high risk in infants with hypoplastic left heart syndrome and is usually not necessary if the echocardiogram is diagnostic. If there is a question about the differential diagnosis, a heart catheterization is indicated.
TREATMENT
Medical Management
Pharmacologic maintenance of ductal patency with PGE 1 and ventilatory support to increase pulmonary resistance is used. A mild respiratory alkalosis (pH 7.35) is helpful. Hyperventilation and supplemental oxygen have not been found to be beneficial. Small to moderate doses of inotropes may be useful in the treatment of hypotension. Surgical intervention offers the only chance of survival.
Subatmospheric Oxygen
When pulmonary blood flow is excessive and is compromising systemic perfusion, specific management techniques can be used to increase PVR and thus create a balance between systemic and pulmonary perfusion. This increase in PVR can be accomplished clinically by administration of inspired CO 2, permissive hypercapnia, hypoventilation, or administration of subatmospheric concentrations of inspired oxygen. The administration of CO 2 or subatmospheric concentrations of oxygen is preferred because of the risk for atelectasis with hypoventilation or permissive hypercapnia. The choice between the two is based on institutional or caregiver preference.
Surgical Treatment
Recent advances in surgical treatment have made it possible to treat this lesion with a multistaged approach. The Norwood procedure is performed initially, consisting of enlargement of the atrial septal defect, ligation of the PDA, anastomosis of the pulmonary artery to the ascending aorta and the aortic arch, and creation of an aortopulmonary shunt (Blalock-Taussig shunt) to maintain pulmonary blood flow. In the second stage, the aortopulmonary shunt is removed and an anastomosis is made between the superior vena cava and the pulmonary artery; it is called a bidirectional Glenn shunt and is performed at 6 to 12 months of age. The final stage is the Fontan procedure, which connects the inferior vena cava to the pulmonary artery; this is generally done at 18 to 36 months of age. Cardiac transplantation is an alternative surgical option. In some centers, the Norwood procedure is performed as a bridge to transplantation, allowing the infant to survive until a donor heart is available.
Heart Transplantation in Infants
Approximately 10% of infants born with congenital heart disease have severe, complex lesions that preclude corrective surgery. For some of these infants, heart transplantation may offer the only chance of long-term survival. Hypoplastic left heart syndrome is the most common indication for heart transplantation in early infancy. Cardiomyopathies are another indication.
Heart transplantation in infancy is severely limited by donor availability. There is a scarcity of donor hearts in this age and size group, and 31% of infants younger than 6 months on transplant lists die waiting for donors. 2
Dysrhythmias
When evaluating an infant with a dysrhythmia, it is essential to assess simultaneously the electrophysiology and hemodynamic status. A neonate with poor perfusion and hypotension should first be treated for shock. A 12-lead ECG can then be done for definitive diagnosis of the type of dysrhythmia. When analyzing the ECG for the mechanism of dysrhythmia, a notation should be made in three main areas: (1) rate; (2) rhythm; and (3) QRS morphology.
PHYSIOLOGY
The development of the cardiac conduction system continues after birth with a steady increase in the sympathetic innervation of the heart. This accounts for the observed heart rate variability and the high frequency of benign dysrhythmias in the newborn. Premature ventricular beats (Figure 24-15), brief episodes of ectopic atrial rhythms, wandering atrial pacemakers (Figure 24-16), and even brief episodes of sinus arrest are all frequently seen in the newborn period. The majority of these dysrhythmias do not require immediate treatment; however, if they persist, the presence of congenital heart disease, sepsis, drug toxicity, persistent hypoxia, adrenal insufficiency, disorders of electrolyte and acid-base balance, hypoglycemia, and hypocalcemia should be considered.
 |
| FIGURE 24-15 |
All cardiac tissue is capable of generating a spontaneous depolarization. However, the sinoatrial (SA) node, atrioventricular (AV) node, and His-Purkinje system consist of specialized conductive tissue with rapid spontaneous depolarization. The SA node is the normal pacemaker of the heart because it has the fastest rate of spontaneous depolarization. If, however, the spontaneous depolarization of the SA node is delayed or slower than normal, an escape rhythm (Figure 24-16) is generated by either the AV node or His-Purkinje system (these rhythms are called nodal escape or ventricular escape, respectively). Dysrhythmias also can originate from an automatic “ectopic” pacemaker located anywhere in the heart. These ectopic pacemakers become more active in the presence of hypoxia, acidosis, digoxin toxicity, abnormal sympathetic nervous system stimulation, increased wall tension (congestive heart failure), or altered electrolyte balance. Drug therapy for dysrhythmias is based on the ability of certain medications to alter the electrophysiologic properties of cardiac tissue. One class of antidysrhythmic drugs directly increases the automaticity of certain cardiac fibers. Examples of such drugs are quinidine, procainamide, lidocaine, and phenytoin (see Table 24-4). Other drugs directly or indirectly affect the autonomic nervous system activity. Propranolol is a beta-adrenergic blocker and works in this fashion. Digoxin exerts its chronotropic activity by altering the sympathetic and parasympathetic nervous system response within the heart.
BENIGN DYSRHYTHMIAS: SINUS BRADYCARDIA, SINUS TACHYCARDIA, AND SINUS DYSRHYTHMIA
Of normal premature infants, 35% to 40% have brief episodes of sinus bradycardia (Figure 24-17), sinus tachycardia, or sinus dysrhythmia (Figure 24-18) that are benign and require no treatment. Healthy premature and term infants may have heart rates that range from 90 to 200 beats/min. Sustained heart rates (>15 seconds) above or below this range should be evaluated with a 12-lead ECG and rhythm strip. These are important, because artifact created by the bedside monitors often makes accurate interpretations of dysrhythmias impossible.
 |
| FIGURE 24-17 |
 |
| FIGURE 24-18 |
SUPRAVENTRICULAR TACHYCARDIA
Supraventricular tachycardia (SVT) (Figure 24-19) is the most common tachydysrhythmia in the newborn period. SVT is the result of dual AV nodal pathways, rapid conduction through an accessory bundle (WPW syndrome), or the existence of an ectopic atrial pacemaker. SVT is occasionally associated with Ebstein’s anomaly of the tricuspid valve, d-transposition of the great vessels, cardiomyopathy, or myocarditis. These lesions are present in 10% to 25% of infants with SVT and should be excluded with the appropriate evaluation. Newborns with prolonged SVT have a history of gradually developing congestive heart failure with findings of anxiety, restlessness, tachypnea, and poor feeding. These symptoms develop after 12 to 24 hours of SVT. SVT often starts and ceases abruptly.
 |
| FIGURE 24-19 |
Criteria for SVT include (1) persistent ventricular rate over 200 to 220 beats/min, (2) a fixed and regular R-R interval, and (3) little variability in heart rate with various activities (e.g., crying, feeding, apnea).
Treatment
Various maneuvers may be used to attempt to convert the infant to normal sinus rhythm (NSR). Vagal maneuvers (unilateral carotid pressure, gagging, rectal stimulation) may be attempted but rarely work. Ocular compression should never be used. Stimulation of the diving reflex using an ice bag applied to the infant’s face may be attempted. (Caution must be used in this procedure to ensure adequate ventilation for the infant.) For infants without WPW syndrome, digoxin is the initial therapy. Parenteral digitalization usually abolishes this dysrhythmia within 12 hours. Propranolol is used as drug therapy for infants with SVT caused by WPW syndrome in the absence of congestive heart failure. In premature infants, propranolol may cause apnea and hypoglycemia. Adenosine, a purinergic agonist, is an especially effective antidysrhythmic drug for treatment of SVT. Adenosine slows the sinus rate and produces transient AV block, interrupting the SVT. Overdrive atrial pacing has been successful in converting SVT to NSR. However, direct-current (DC) cardioversion (1 to 2 watt-seconds/kg) is the most effective mode of treatment. The defibrillator must always be in the synchronous mode. If cardioversion is successful, maintenance drug therapy should be initiated. Other antidysrhythmic drugs such as digoxin have been used to treat this disorder. Recently, however, it has been suggested that digoxin not be used in WPW syndrome and that this disorder be ruled out before digoxin is used. If not contraindicated, digoxin should be administered using standard doses (see Box 24-3).
Propranolol administered intravenously (IV) may be used if the patient is not in congestive heart failure, although it is very risky and not usually recommended. Beta-blocking agents may inhibit circulating catecholamines, which are needed for the maintenance of adequate cardiac output in the face of congestive heart failure. Esmolol is another beta-blocking agent that can be administered IV for SVT (seeTable 24-4).
If the SVT fails to convert using the methods outlined previously, other drugs such as amiodarone, flecainide, or procainamide may be necessary. After conversion to NSR, maintenance drug therapy should be continued for 6 to 12 months or longer. Relapses during the first 48 hours are common (70%) and should be anticipated.
Fetal SVT is uncommon but, when present, can be associated with severe congestive heart failure and hydrops fetalis. Fetal SVT requires aggressive management, including conversion with maternally administered propranolol and digoxin. A favorable outcome usually can be expected for fetal SVT. Failure to control the fetal SVT in the presence of fetal hydrops is an indication for delivery.
ATRIAL FLUTTER AND FIBRILLATION
The presence of atrial flutter (Figure 24-20) is often suggestive of a serious organic heart disease (endocardial fibroelastosis, Ebstein’s anomaly of the tricuspid valve, or complex heart defects). Atrial flutter is diagnosed when (1) the atrial rate is greater than 220 beats/min; (2) the P waves are very regular; and (3) there is a characteristic sawtooth pattern, indicating a flutter wave. The ventricular rate will vary depending on the degree of AV block present. Atrial fibrillation is extremely rare and almost always indicates a serious organic heart disease. The prognosis for atrial flutter and fibrillation tends to be less favorable than that of SVT. These tachydysrhythmias have been reported to be more difficult to treat in utero, more likely to be associated with structural heart defects, and more likely to develop hydrops fetalis. Some cases of atrial flutter have been found in infants with an intracardiac catheter (in right atrium by echo) and/or those receiving broad-spectrum antimicrobials. 23
 |
| FIGURE 24-20 |
Treatment
The treatment of atrial flutter or fibrillation is DC cardioversion or overdrive atrial pacing followed by maintenance therapy with digoxin.
VENTRICULAR TACHYCARDIA
Ventricular tachycardia is relatively rare and is usually seen with severe medical illnesses such as hypoxemia, shock, electrolyte disturbances, and digoxin toxicity.
Treatment
Ventricular tachycardia is best treated with immediate DC cardioversion. Lidocaine may be used as a bolus (1 to 2 mg/kg IV) or as a continuous IV infusion of 20 to 30 mcg/kg/min. After conversion, maintenance therapy should be initiated using phenytoin, propranolol, lidocaine, procainamide, or amiodarone. Phenytoin, 2 to 4 mg/kg, may be effective if the dysrhythmia is the result of digoxin toxicity.
COMPLETE ATRIOVENTRICULAR BLOCK
In complete heart block, the ventricular rate is slower than the atrial rate and there is no association between the ventricular and atrial rates. Complete heart block can be seen in infants with myocarditis or endocardial fibroelastosis. There is a strong association between congenital heart block and maternal collagen diseases such as systemic lupus erythematosus (SLE). Often these mothers have no signs or symptoms of lupus, but laboratory confirmation is often possible.
Treatment
No treatment is necessary unless the ventricular rate falls below 55 beats/min or the infant becomes symptomatic, in which case a pacemaker is necessary. Isoproterenol may increase the ventricular rate until a pacemaker is placed.
SICK SINUS SYNDROME
Sick sinus syndrome (SSS) is a broad term used to describe dysrhythmias resulting from abnormal sinus node function and includes a wide array of bradydysrhythmias, including sinus bradycardia, sinus pause/arrest, sinoatrial exit block, and slow escape rhythms, including junctional bradycardia. Although most commonly acquired during surgical repair for CHD (because of the proximity to the sinus node), SSS can be congenital. Mutations in the cardiac sodium channel gene SCN5A can result in congenital SSS. 37
PARENT TEACHING
If the diagnosis of CHD is made prenatally by fetal echocardiography, parent education begins before the birth of the infant (see the Parent Teaching box above). Expectant parents should be referred to a pediatric facility experienced in providing complex medical and surgical care for infants with CHD. Arrangements should be made for parents to meet with key members of the medical and surgical team and tour the intensive care units. The period of diagnosis, hospitalization, and early caregiving at home is extremely stressful.
Key Points for Parents of Newborns with Heart Disease
• Refer to a pediatric facility experienced with infants with heart disease.
• Reassure and support family for understanding that “this is not their fault.”
• Explain all tubes, monitors, and equipment to decrease anxiety.
• Help parents accept and understand their infant’s diagnosis.
• Encourage parental bonding with their infant and participation in care.
• Detail home care, to include medications, signs and symptoms, when to call physician.
• Encourage normal activity.
• Explain the necessity of subacute bacterial endocarditis protection.
• Supply parents with resource information—booklets, brochures, Internet resources.
The diagnosis of CHD in their infant is a frightening experience and causes much distress to parents. When an infant is born with a heart defect, the parents may grieve over the loss of the healthy newborn they had anticipated and experience shock, denial, guilt, anger, despair, or confusion. Therefore comprehensive teaching, reassurance, and support are essential for the well-being of both the infant and the family. Understanding the heart defect aids in decreasing anxiety as well as allowing parents to provide good care after discharge.
Explain the infant’s heart defect to the parents. Draw or show a picture of the heart defect, explaining briefly and simply the normal circulation of the heart and how the circulation of their infant’s heart differs from normal. This explanation should be repeated often for parental understanding and retention. Careful explanation of all tubes, monitors, equipment, and procedures in the nursery also helps decrease parental anxiety.
Heart defects are not visible lesions. Most of these infants will appear quite normal and healthy. Thus it may be difficult for some parents to accept that anything is wrong with their infant. In addition, parents are under great emotional and sometimes physical stress (from labor and delivery), which decreases their ability to hear and retain explanations about the defect. Patience and repetition of explanations is important.
Some parents may be unable at first to respond to their newborn with a heart defect. Health care providers should facilitate bonding and decrease the parent’s fear of holding or caring for their infant by encouraging interaction with the infant and enabling parents to participate in their infant’s care. The parents’ confidence in caring for their infant at home should be established in the nursery. Parents must feel comfortable caring for their infant and have the opportunity to demonstrate their ability to do so before discharge from the hospital.
Teaching home care of the infant before discharge should be detailed and include medications, signs to observe, and guidelines for care. It is critical that these be written instructions that can be referred to often. Parents should telephone the physician if the infant demonstrates (1) poor feeding for 1 to 2 days or sweating with feeds, (2) vomiting most of feedings for a 12- to 24-hour period, (3) fast or labored breathing for several hours, (4) decreased activity level, (5) weight loss or failure to gain weight, and (6) frequent respiratory illnesses. 28
All medications should be explained in detail, including their purpose, action, and administration. Parents should be made aware of the potential adverse effects (side effects) of all of their infant’s medications. Parents should be observed giving medications in the nursery before the infant is discharged.
Cyanotic heart disease is particularly disturbing to parents because their infant’s skin color is “blue.” Parents should be cautioned that their infant will appear blue, especially around the mouth, mucous membranes, hands, and feet, and the blueness will increase with activity such as crying, feeding, and bowel movements. Parents should notify the physician about any of the previously listed symptoms in addition to (1) greatly increased cyanosis, especially if associated with fast or labored breathing, (2) decreased movement in any or all of the extremities, (3) decreased responsiveness or eyes deviating to one side, and (4) seizure activity such as jerking motions or stiffness followed by the infant becoming floppy or limp. 28
Many parents will develop a narrow, disease-oriented focus. Emphasize to parents that their infant should be treated as normally as possible. There is no activity restriction for infants with heart disease, because infants “self-limit” according to their capacity. It is difficult for parents with a firstborn infant with heart disease to differentiate “normal baby problems” from cardiac-related problems. For these parents, as well as other parents of children with cardiac defects, it is particularly important to have open communication among the family, primary care provider, and cardiologist. Parents should be encouraged to call these medical personnel as needed for support, answers to questions, and reassurance. Support groups of parents whose children have heart defects provide information, empathy, and practical tips to parents dealing with medical or surgical interventions for their child’s heart defect.
Active participation in care helps alleviate some of the parents’ stress. Parental participation enables opportunities for parents to practice under the guidance of professionals and enables professional assessment of parental abilities, individualizing care, and building on existing competencies. Parents should be encouraged to provide comfort measures such as touch and to assist with diaper changes, positioning, and oral care, even during the critical phase of illness. During the postoperative and convalescent phase of hospitalization, parents should assume more of the infant’s care, such as feeding, care of the incision, and medication administration. Before discharge, parents should be encouraged to room with and completely care for their infant, with nursing assistance available as needed.
Normal Newborn Care and Maintenance
Emphasis on infection prevention strategies, such as handwashing before handling the infant, avoiding ill contacts, and avoiding large crowds, is especially important to teach parents. The trip home should occur in a proper car seat with perhaps a small blanket placed over the chest to ensure that the safety straps do not rub the wound. Standard pediatric immunizations are delayed until just after surgery. Provide information about the infant’s prognosis and follow-up care and anticipatory guidance about special growth and development considerations. Provide explicit instructions on when to call the health care provider.
Infectious Endocarditis Protection
Infants with congenital heart disease are at increased risk for developing adverse outcomes associated with infectious endocarditis (IE), also known as bacterial endocarditis (BE). Recently, the guidelines for the prevention of IE have changed dramatically. The American Heart Association’s Endocarditis Committee extensively reviewed published studies and found no conclusive evidence to link dental or gastrointestinal (GI) or genitourinary (GU) tract procedures with the development of IE in most patients with congenital heart defects. 6Antibiotic prophylaxis with dental procedures is recommended only for patients with cardiac conditions (e.g., prosthetic valves, previous endocarditis, cardiac transplant) associated with the highest risk for adverse outcomes from endocarditis. Antibiotic prophylaxis is also recommended for the following categories of CHD: (1) unrepaired cyanotic CHD; (2) completely repaired CHD with prosthetic material, such as conduits; and (3) repaired CHD with residual defects adjacent to prosthetic material.6 Parents are encouraged to speak with their pediatric cardiologist regarding any questions related to IE antibiotic prophylaxis.
Activity
Normal newborn activity is encouraged after discharge. Special precautions are indicated for handling. Teach parents to avoid picking the infant up under the arms until the wound and underlying structures are healed.
Supporting Ongoing Development
Infants are at risk for neurodevelopmental abnormalities for a number of reasons. The incidence of brain abnormalities is higher than in the general population. Review normal developmental milestones, provide pragmatic strategies to maximize infant development, and fully inform parents about their infant’s increased risk for developmental delay. Referral to early intervention programs may be helpful.
Parenting an infant with CHD requires adaptation, coping, and evolution—from diagnosis to discharge from the hospital and through caretaking and parenting at home. Parents need an outlet so that they feel free to share their thoughts and concerns in a nonjudgmental environment. Consultation with a mental health professional may enable the family to recognize and build on strengths that will help them cope with this enormous challenge.
FUTURE RESEARCH
Improvements in the diagnosis and treatment of CHD have drastically reduced the morbidity and mortality associated with these defects. Future research lies in increasing knowledge about the genetic contributions to CHD. Genetic tests are available for many syndromes strongly associated with structural CHD (e.g., Noonan syndrome, pulmonary valve stenosis) and most chromosomal abnormalities associated with CHD (e.g., Down syndrome, complete atrioventricular septal defect [AVSD]). 37 There is a genetic test (fluorescent in situ hybridization [FISH]) for DiGeorge syndrome, which is often associated with interrupted aortic arch (IAA), tetralogy of Fallot (TOF), or truncus arteriosus. Further research also is being done on the development of surgical and technologic approaches for cardiac surgery of the fetus, as well as reduction of mortality associated with the pediatric cardiac surgery patient. Tissue engineering is also an emerging area of inquiry with the focus on bioengineering of pediatric heart valves and vascular tissue.
REFERENCES
1. Aggarwal, S.; Chintala, K.; Humes, R.A., Sildenafil use in a symptomatic neonate with severe Ebstein’s anomaly of the tricuspid valve, Am J Perinatol 25 (2) ( 2008) 125.
2. In: (Editors: Allen, H.D.; Driscoll, D.J.; Shaddy, R.E.; et al.) Moss and Adams heart disease in infants, children, and adolescents: including the fetus and young adultsed 7 ( 2008)Lippincott Williams & Wilkins, Philadelphia.
3. American Heart Association, Children with congenital or acquired heart disease. ( 2008)The Association, Dallas; Accessed October 4, 2009, fromwww.americanheart.org.
4. Bakiler, A.R.; Ozer, E.A.; Kanik, A.; et al., Accuracy of prenatal diagnosis of congenital heart disease with fetal echocardiography, Fetal Diagn Ther 22 (2007) 241.
5. Bentham, J.; Bhattacharya, S., Genetic mechanisms controlling cardiovascular development, Ann N Y Acad Sci 1123 (1) ( 2008) 10.
6. Bobhate, P.; Pinto, R., Summary of the new guidelines for prevention of infective endocarditis: implication for developing countries, Ann Ped Cardiology 56 (2008) 58.
7. Castanada, A., Congenital heart disease: a surgical-historical perspective, Ann Thorac Surg 79 (5) ( 2005) S2217.
8. Cloherty, J.P.; Eichenwald, E.C.; Stark, A.R., In: Manual of neonatal careed 5 ( 2004)Lippincott Williams & Wilkins, Philadelphia.
9. Dice, J.E.; Bhatia, J., Patent ductus arteriosus: an overview, J Pediatr Pharmacol Ther 12 (2007) 138.
10. Donze, A.; Smith, J.R.; Bryosky, K., Safety and efficacy of ibuprofen versus indomethacin for the treatment of patent ductus arteriosus in the preterm infant: reviewing the evidence, Neonatal Netw 26 (3) ( 2007) 187.
11. Gaca, A.M.; Jaggers, J.J.; Dudley, L.T.; et al., Repair of congenital heart disease: a primer—part 2, Radiology 248 (2008) 44.
12. Garne, E., Atrial and ventricular septal defects: epidemiology and spontaneous closure, J Matern Fetal Neonatal Med 19 (5) ( 2006) 271.
13. Gutierrez, F.R.; Ho, M.; Siegel, M.J., Practical application of magnetic resonance in congenital heart disease, Magn Reson Imaging Clin N Am 16 (3) ( 2008) 403.
14. Kabra, N.S.; Schmidt, B.; Roberts, R.S.; et al., Neurosensory impairment after surgical closure of patent ductus arteriosus in extremely low birth weight infants: results of the trial of indomethacin prophylaxis in preterms, J Pediatr 150 (2007) 229.
15. Karamlou, T.; McCrindle, B.W.; Williams, W.G., Surgery insight: late complications following repair of tetralogy of Fallot and related surgical strategies for management, Nat Clin Pract Cardiovasc Med 3 (2006) 611.
16. Kirk, E.P.; Sunde, M.; Costa, M.W.; et al., Mutations in cardiac T-box factor gene TBX0 are associated with diverse cardiac pathologies, including defects of septation and valvulogenesis and cardiomyopathy, Am J Hum Genet 81 (2) ( 2007) 280.
17. Kohon, B.; Butler, H.; Kirshbom, P.; et al., Closure of symptomatic ventricular septal defects: how early is too early?Pediatr Cardiol 29 (1) ( 2008) 36.
18. Lee, W.; Comstock, C.H., Prenatal diagnosis of congenital heart disease: where are we now?Ultrasound Clinics 1 (2006) 2.
19. Lopez, V.; Keen, C.L.; Lanoue, L., Prenatal zinc deficiency: influence on heart morphology and distribution of key heart proteins in a rat model, Biol Trace Elem Res 122 (3) ( 2008) 238.
20. Mahle, W.T., Physical examination and pulse oximetry in newborn infants: out with the old, in with the new?J Pediatr 152 (2008) 747.
21. Maulik, D., Echocardiography in detection of fetal heart abnormalities, J Matern Fetal Neonatal Med 19 (2006) 9.
22. Miller, S.P.; McQuillen, P.S.; Hamrick, S.; et al., Abnormal brain development in newborns with congenital heart disease, N Engl J Med 357 (2007) 1928.
23. Obidi, E.; Touba, P.; Sharma, J., Atrial flutter in a premature infant with a structurally normal heart, J Matern Fetal Neonatal Med 19 (2006) 113.
24. O’Connor, M.; McDaniel, N.; Brady, W.J., The pediatric electrocardiogram. III. Congenital heart disease and other cardiac syndromes, Am J Emerg Med 26 (4) ( 2008) 497.
25. Ohlsson, A.; Walia, R.; Shah, S., Ibuprofen for the treatment of patent ductus arteriosus in preterm and/or low birth weight infants, Cochrane Database Syst Rev 1 (2008); CD003481.
26. Pashia, S.E., Ebstein’s anomaly, Neonatal Netw 26 (3) ( 2007) 197.
27. Pierpont, M.E.; Basson, C.T.; Benson Jr, D.W.; et al., Genetic basis for congenital heart defects: current knowledge, Circulation 115 (2007) 3015.
28. Pye, S.; Green, A., Parent education after newborn congenital heart surgery, Adv Neonatal Care 3 (2003) 147.
29. Rao, P.S., Perinatal circulatory physiology: its influence on clinical manifestations of neonatal heart disease. I, Neonatol Today 3 (2) ( 2008) 6.
30. Sekar, K.C.; Corff, K.E., Treatment of patent ductus arteriosus: indomethacin or ibuprofen?J Perinatol 28 (suppl 1) ( 2008) S60.
31. Spevak, P.J.; Johnson, P.T.; Fishman, E.K., Review of surgically corrected congenital heart defects: utility of 64-MDCT, AJR Am J Roentgenol 191 (3) ( 2008) 854.
32. Tanner, K.; Sabrine, N.; Wren, C., Cardiovascular malformations among preterm infants, Pediatrics 116 (6) ( 2005) 1536.
33. Tomita, H.; Takamuro, M.; Soda, W.; et al., Increased serum high-sensitivity C-reactive protein is related to hypoxia and brain natriuretic peptide in congenital heart disease, Pediatr Int 50 (2008) 436.
34. Turck, C.J.; Marsh, W.; Stevenson, J.G.; et al., Pharmacoeconomics of surgical intervention versus cyclooxygenase inhibitors for the treatment of patent ductus arteriosus, J Pediatr Pharmocol Ther 12 (3) ( 2007) 183.
35. Wooley, C.F.; Miller, P.J., William Osler, Maude Abbott, Paul Dudley White, and Helen Taussig: The origins of congenital heart disease in North America, Am Heart Hosp J 6 (1) ( 2008) 51.
36. Wren, C.; Reinhardt, Z.; Khawaja, K., Twenty-year trends in diagnosis of life-threatening neonatal cardiovascular malformations, Arch Dis Child Fetal Neonatal Ed 93 (1) ( 2008) F33.
37. Zeigler, V.L., Congenital heart disease and genetics, Crit Care Nurs Clin North Am 20 (2) ( 2008) 159.
RESOURCES FOR PARENTS
American Heart Association: Children with congenital or acquired heart disease, www.americanheart.org.
Congenital Heart Defects.com, sponsored by Baby Hearts Press, www.congenitalheartdefects.com.
Congenital Heart Information Network, www.tchin.org.
Kids With Heart National Association for Children’s Heart Disorders, www.kidswithheart.org.
Little Hearts, Inc, national nonprofit organization, www.littlehearts.org.
Pediheart Organization, www.pediheart.org.
[/level-membership-for-neonatal-and-perinatal-medicine-category][not-level-membership-for-neonatal-and-perinatal-medicine-category]
Patricia M. Kenney, Deandra Hoover, Luther C. Williams and Victor Iskersky
Congenital heart disease (CHD) is the most common life-threatening birth defect encountered in the neonatal intensive care unit (NICU). Although the incidence of these conditions has remained constant at approximately 1% of all infants born in the United States, the methods of diagnosis and treatment have undergone tremendous change over the past several decades. 37 It is the responsibility of the practitioner to recognize the presence of CHD and to provide an accurate diagnose and treatment. This chapter reviews the physiology of neonatal circulation, the pathophysiology of congenital heart disease, and the most current evidence-based treatments.
CONGENITAL HEART DISEASE: OVERVIEW
History
In 1892, Dr. William Osler wrote that congenital heart disease was of “limited clinical interest as in a large proportion of cases the anomaly is not compatible with life, and in others, nothing can be done to remedy the defect or even relieve the symptoms.”8 Osler encouraged Dr. Maude Abbott in her CHD finding, and she, along with Dr. Helen Taussig and Dr. Alfred Blalock, suggested surgery to help these “blue babies.”35 This opened up the field of surgical treatment of cyanotic malformations of the heart. In 1938, Dr. Robert Gross was the first to successfully ligate a patent ductus arteriosus (PDA) in a 7-year-old girl at Boston’s Children’s Hospital. The first successful United States heart transplant was done at Stanford University by Dr. Norman Shumway in 1968. 7 What remarkable progress has been made in the area of congenital heart disease. This is the direct result of advances in pediatric and fetal cardiology, cardiac surgery, neonatology, and neonatal intensive care nursing.
Incidence and Survival
Each year, approximately 40,000 babies, or just under 1% of all babies born in the United States, are diagnosed with congenital heart disease. 3 Highly sensitive echocardiography has led to the detection of more trivial forms of congenital heart disease such as tiny ventricular septal defects. This inclusion has led to the higher incidence figures in recent years. The incidence of moderate to severe structural congenital heart defects (in liveborn infants) is 6 to 8 per 1000 live births. 8 Of these infants, approximately 3 per 1000 live births will have CHD that results in death or requires cardiac surgery during the first year of life. 25 Advances in diagnostic imaging, cardiac surgery, and neonatal intensive care have led to significant decreases in mortality rates. The death rate from all congenital heart defects declined nearly 32% between 1994 and 2004. 37
Embryology
The heart is one of the earliest differentiating and functioning organs. In human embryos, the heart begins to beat at about 22 to 23 days of life. Blood begins flowing through the heart in the fourth week of life. The heart develops from the cardiogenic mesoderm that originally lies above the cranial end of the neural tube. The heart forms initially as a simple paired tube inside the pericardial cavity. When the embryonic disk folds, the heart is carried into the correct anatomic position in the chest cavity. A key aspect of heart development is the septation of the heart into separate chambers. This complex process converts this simple tube into a four-chambered heart. Cardiogenesis is such as intricate, complex process that it is of little wonder that congenital heart defects occur.
Physiology
The physiologic changes that occur during the transition from intrauterine to extrauterine life have been well documented. To develop a clear understanding of the various congenital heart defects, knowledge of the basic principles of fetal circulation must be established.
FETAL CIRCULATION
Fetal circulation is intended to use the placenta for gas exchange, whereas postnatal circulation uses the lungs for gas exchange (Figure 24-1). Highly oxygenated blood from the mother enters the fetal circulation through the vein in the umbilical cord. This blood enters the inferior vena cava via the ductus venosus. Nearly one half of the umbilical venous blood goes through the liver and reaches the inferior vena cava via the hepatic veins. Inside the fetal heart, blood enters the right atrium, the chamber on the upper right side of the heart. Most of the blood flows to the left side through the foramen ovale, a special fetal opening between the left and right atria. Blood then passes into the left ventricle and then to the aorta, the large artery coming from the heart. From the aorta, blood is sent to the head and upper extremities. Therefore the brain (via the brachiocephalic vessels) and the heart (via the coronary arteries) are perfused with relatively highly oxygenated blood. After circulation, the blood returns to the right atrium through the superior vena cava. Nearly a third of the blood entering the right atrium stays in the right side of the heart, eventually flowing into the pulmonary artery.
|
|
| FIGURE 24-1
(Modified from Patton KT, Thibodeau GA: Anatomy and physiology, ed 7, St Louis, 2010, Mosby.)
|
Fetal lungs are not used for breathing. The work of exchanging oxygen and carbon dioxide is done by the placenta. Because of high pulmonary vascular resistance (PVR), fetal circulation shunts most of the blood away from the lungs. Blood is shunted from the pulmonary artery to the aorta through a connecting blood vessel called the ductus arteriosus.29 Blood then enters the placental circulation and is resaturated.
CHANGES THAT OCCUR IN THE FETAL CIRCULATION WITH BIRTH
In utero, systemic vascular resistance (SVR) is low, primarily because of low resistance in the placenta. Conversely, the PVR is high, with the constricted and hypertrophied pulmonary arterioles being relatively resistant to blood flow. At birth, the placenta is removed from the circulation, thereby greatly increasing the SVR.
Both the labor process and the first few breaths of life begin the termination of fetal circulation and the transition to newborn circulation. Initiation of respirations produces increased oxygen tension, which decreases PVR and increases pulmonary blood flow. The ductus arteriosus is extremely sensitive to the oxygen content of the blood. As the neonatal Pa o2 rises, the connection between the aorta and the pulmonary artery (ductus arteriosus) is no longer needed and begins to close. Functional closure occurs at approximately 72 hours of life, and anatomic closure usually occurs between 1 and 2 weeks after birth. The circulation in the lungs then increases, and more blood flows into the left atrium of the heart. This increased pressure in the left atrium causes the foramen ovale to close. Anatomic closure of the foramen ovale can take several months. Finally, with the clamping of the umbilical cord, umbilical venous flow ceases and the ductus venosus begins to close, with anatomic closure taking approximately 1 to 2 weeks. 29
Once these changes occur, the newborn’s circulation resembles that of an adult (Figure 24-2). Desaturated blood returns to the heart by the inferior and superior venae cavae and enters the right atrium, right ventricle, pulmonary artery, and pulmonary circulation in which oxygen and carbon dioxide are exchanged. The saturated blood then returns to the heart through the pulmonary venous system and enters the left atrium, left ventricle, and ultimately the aorta and systemic arterial system. However, PVR and pressures in the right ventricle and pulmonary system remain elevated in the neonate because of the hypertrophy of the pulmonary vessels. This hypertrophy slowly resolves so that pulmonary vascular resistance and right heart pressures decrease to lower levels between 1 and 2 months of age.
|
|
| FIGURE 24-2
(Modified from Hockenberry MJ, Wilson D: Wong’s essentials of pediatric nursing, ed 8, St Louis, 2009, Mosby.)
|
Etiology
The development of the cardiovascular system represents a complicated interaction between form and function. In most cases, the cause(s) of abnormal cardiac development is unknown. Traditionally, the etiology of congenital heart defects has been viewed as multifactorial, involving a complex interaction between genetic and environmental factors. Studies are being done on maternal zinc deficiency and its association with an increased risk for fetal heart malformations. These studies postulate that the mechanism of action is the suppression of HNK-1 expression. 19Table 24-1 lists the most common environmental risk factors and their associated cardiac malformations. Box 24-1 lists less common risk factors associated with CHD. Certain chronic illnesses in the mother contribute to the risk for CHD. For example, women with diabetes are at increased risk for having an infant with a heart defect. However, this risk can be greatly reduced by strict control of maternal blood sugar levels. More recent studies have found that the single greatest risk factor for congenital heart defects is genetic. Table 24-2 shows chromosomal syndromes and their associated congenital heart defects.
| Potential Teratogen | Frequency of Cardiovascular Disease (%) | Most Common Malformations |
|---|---|---|
| DRUGS | ||
| Alcohol | 25-30 | Ventricular septal defect, patent ductus arteriosus, atrial septal defect |
| Amphetamines | 5-10 | Ventricular septal defect, patent ductus arteriosus, atrial septal defect, transposition of the great vessels |
| Anticonvulsants | 2-3 | Pulmonary stenosis, aortic stenosis, coarctation of aorta, patent ductus arteriosus |
| Trimethadione | 15-30 | Transposition of great arteries, tetralogy of Fallot, hypoplastic left heart syndrome |
| Lithium | 10 | Ebstein’s anomaly, tricuspid atresia, atrial septal defect |
| Sex hormones | 2-4 | Ventricular septal defect, transposition of great arteries, tetralogy of Fallot |
| INFECTIONS | ||
| Rubella | 35 | Peripheral pulmonary artery stenosis, ventricular septal defect, patent ductus arteriosus, atrial septal defect |
| MATERNAL CONDITIONS | ||
| Diabetes | 3-5 | Transposition of great arteries, ventricular septal defect, coarctation of aorta |
| 30-50 | Cardiomegaly, myopathy | |
| Lupus erythematosus | ? | Heart block |
BOX 24-1
Exposure to Environmental Agents During Work and/or Hobby
• Paternal exposure to cold temperature
• Maternal exposure to various solvents, hair dyes, auto body repair work
Drug Exposure
• Diazepam, phenothiazines
• Corticosteroids
• Gastrointestinal drugs
• Paternal exposure to cocaine
Maternal Reproductive History
• Genetic risk factor (family history of congenital heart disease), more than three prior pregnancies and an increased number of miscarriages
• Without genetic risk but with premature births and previous induced abortion
Syndromic Association
• 27.7% of all cases had chromosomal anomalies, heritable syndromes, or an additional major organ system defect (see Table 24-2)
| Population | Incidence of Congenital Heart Disease (%) | Most Common Lesions | ||
|---|---|---|---|---|
| 1 | 2 | 3 | ||
| Trisomy 21 syndrome | 50 | Ventricular septal defect, endocardial cushion defect | Atrial septal defect | Patent ductus arteriosus |
| Trisomy 18 syndrome | 99+ | Ventricular septal defect | Patent ductus arteriosus | Pulmonary stenosis |
| Trisomy 13 syndrome | 90 | Ventricular septal defect | Patent ductus arteriosus | Dextrocardia |
| Turner syndrome | 35 | Coarctation of the aorta | Aortic stenosis | Atrial septal defect |
| DiGeorge syndrome 22q deletion syndrome | 50 | Truncus arteriosus | Tetralogy of Fallot | Interrupted aortic arch |
Since the 1990s, much progress has been made in understanding the genetics of heart disease. Studies in recent years have brought an explosion of information on the identification of potential candidate genes regulating heart development. 5 An example is the relationship between TBX-1 and cardiovascular defects seen in DiGeorge syndrome. 16 Mutations in NOTCH-1 have been found to cause aortic valve disease. 37 Although research defining the signaling cascades that lead to specific forms of congenital heart defects still needs to be done, 27 some information is well established (e.g., 50% of congenital heart defects involve a ventricular septal defect (VSD) alone or in combination with other abnormalities). Studies also demonstrate that preterm infants are two and a half times more likely to have cardiovascular (CV) malformations.32Table 24-3 shows the most common congenital heart defects and their time of presentation.
| *These numbers are intended as a rough guideline because there is considerable overlap. Infants with congenital heart disease are often active initially and appear well for several hours or days after birth. In contrast, infants with respiratory distress often have characteristic symptoms within the first several hours after birth. |
|||||
| From Flyer DC et al: Report of the New England Regional Infant Cardiac Program, Pediatrics 65:377, 1980. | |||||
| 0-6 Days | (%) | 7-13 Days | (%) | 13-20 Days | (%) |
|---|---|---|---|---|---|
| Transposition of great arteries | (17) | Coarctation of the aorta | (19) | Ventricular septal defect | (20) |
| Hypoplastic left ventricle | (12) | Ventricular septal defect | (15) | Transposition of great arteries | (17) |
| Lung disease | (10) | Hypoplastic left ventricle | (11) | Coarctation of the aorta | (16) |
| Tetralogy of Fallot | (9) | Transposition of great arteries | (9) | Tetralogy of Fallot | (8) |
| Coarctation of the aorta | (7) | Tetralogy of Fallot | (6) | Endocardial cushion defect | (6) |
| Ventricular septal defect | (7) | Heterotaxia | (4) | Heterotaxia | (6) |
| Pulmonary atresia (with intact ventricular septum) | (7) | Truncus arteriosus | (4) | Patent ductus arteriosus | (4) |
| Single ventricle | (4) | Total anomalous pulmonary venous return | (3) | ||
| Heterotaxia | (6) | ||||
| Other | (25) | Other | (28) | Other | (20) |
| Total 896 | (100) | Total 210 | (100) | Total 116 | (100) |
PRENATAL DIAGNOSIS
Because of the widespread use of antenatal ultrasound, it is increasingly common for the fetus to be diagnosed with congenital heart disease. Fetal echocardiography has proven to be a valid, reliable, and accurate tool in the prenatal diagnosis of CHD. 9 The recommended timing of a fetal echocardiogram is between 18 to 20 weeks’ gestation, although reasonable images can be obtained as early as 16 weeks. Physical examination alone picks up 30% of cases of CHD. 20 Pulse oximetry is not an accurate screening tool for CHD because of its relatively high false-positive rate, especially when used shortly after birth when the Sp o2 in neonates is low. 20 Recent studies are investigating serum high-sensitivity C-reactive protein (hs-CRP) and brain natriuretic protein (BNP) levels as markers of CHD. Thus far, hs-CRP and BNP levels are useful only as indicators of hypoxia. 33
Pregnant women are encouraged to undergo fetal echocardiography if (1) genetic evaluation determines that the fetus has a chromosomal or genetic syndrome associated with CHD, (2) the mother or a previous child has CHD, or (3) there is a family history of CHD. Box 24-2 lists CHDs that are diagnosed and CHDs that may be missed on fetal echocardiogram. Magnetic resonance angiography is also an important diagnostic tool, especially for lesions that may be missed by fetal echocardiography. Detailed postnatal examinations are critical in determining the full extent of cardiac malformation.
BOX 24-2
Accurately Diagnosed
• Hypoplastic left heart syndrome
• Tricuspid atresia
• Pulmonary atresia
• Truncus arteriosus
• Tetralogy of Fallot
• Atriovenous septal defects
• Large ventricular septal defect (VSD)
• Transposition of the great arteries
May Be Missed
• Coarctation of the aorta
• Small VSD/atrial septal defect (ASD)
• Total anomalous pulmonary venous return (TAPVR)
• Mild aortic or pulmonary stenosis
Data Collection
HISTORY
A family history of congenital heart disease is significant, since a sibling with CHD increases the recurrence risk threefold. Viral exposure during pregnancy (rubella, Coxsackie B, and enteroviruses) and maternal ingestion of alcohol or drugs should be evaluated. Pregnancy, labor, and delivery complications should be carefully examined as risk factors that could affect the cardiovascular system. For example, intrauterine hypoxia and perinatal hypoxia are risk factors for the development of myocardial dysfunction, as well as persistent pulmonary hypertension of the newborn (PPHN). The timing of the onset of symptoms may indicate the type of anomaly (see Table 24-3).
CLINICAL PRESENTATION OF INFANTS WITH SEVERE CARDIAC DISEASE
In many neonates, congenital heart disease is not suspected until after birth when the newborn presents with one or more signs or symptoms (see the Critical Findings box on p. 684). 36 Timing of presentation of signs or symptoms depends on severity of the defect, in utero effects of the defect, and alterations in cardiovascular physiology during transitional circulation (i.e., closure of the ductus arteriosus and the fall in PVR). Despite the presence of many heterogeneous forms of heart disease, a surprisingly limited number of signs and symptoms present in the neonate.
Murmurs
Heart murmurs are a common finding in neonates. Estimates of prevalence of heart murmurs in neonates range from 1% to 70% depending on the study. 8Although cardiac murmurs in the neonatal period do not necessarily indicate heart disease, they must be carefully evaluated. The absence of a murmur does not exclude severe life-threatening cardiac anomalies. Pathologic murmurs tend to appear at characteristic ages (e.g., murmurs associated with semilunar valve stenosis and atrioventricular valve insufficiency tend to be noted very shortly after birth). In contrast, murmurs caused by left-to-right shunt lesions (PDA, VSD) may not be heard until the second to fourth week of life. Therefore the age of the neonate when the murmur is first noted and the character of the murmur give important clues about the nature of the cardiac defect.
Cyanosis
Cyanosis (a bluish discoloration of the skin, nail beds, and mucous membranes) is one of the most common presenting signs of congenital heart disease in the neonate. Depending on the underlying skin complexion, clinically apparent cyanosis is usually not visible until there is more than 3 gm/dL of desaturated hemoglobin in the arterial system. 20 Cyanosis depends on both the severity of hypoxemia (which determines the percent of oxygen saturation) and the hemoglobin concentration. True central cyanosis should be differentiated from acrocyanosis, blueness of the hands and feet only, which is a normal finding in the neonate.
Cyanosis in the newborn must be differentiated between cardiac and respiratory causes. Pulmonary disorders cause cyanosis in the neonate because of intrapulmonary right-to-left shunting. Varying degrees of hypoxemia manifest as cyanosis and can be to the result of primary lung disease (see Chapter 23) and central nervous system abnormalities. Clinical cyanosis can occur without hypoxemia in a neonate with methemoglobinemia and polycythemia.
Respiratory Distress
Most infants with cyanosis from CHD do not have respiratory distress (e.g., tachypnea, intercostal retractions, grunting, nasal flaring, dyspnea, rales, cyanosis). Often the degree of cyanosis is not proportional to the degree of respiratory distress evaluated from the physical and chest x-ray examinations. If cyanosis is present and is caused by a fixed right-to-left shunt (cardiac lesion), increasing inspired oxygen will have little effect on the arterial blood gases. However, if the cyanosis is caused by a diffusion defect in the lungs (pulmonary disorder), the degree of cyanosis often decreases with increasing inspired oxygen.
The hyperoxia test is beneficial in differentiating respiratory disease from cyanotic heart disease. This single test is perhaps the most sensitive and specific tool in the initial evaluation of the neonate with suspected congenital heart disease and is used to investigate the possibility of a fixed (intracardiac) right-to-left shunt. The hyperoxia test is performed by obtaining arterial blood gas measurements (preferably from the right radial artery) when the infant is in room air and then after the infant has been in 100% oxygen for 5 to 10 minutes. If the Pa o2 is greater than 150 mm Hg, the presence of a right-to-left shunt and cyanotic congenital heart disease as the cause of cyanosis is unlikely. Pulse oximetry cannot be used for documentation.
Congestive Heart Failure
Congestive heart failure (CHF) occurs when the heart cannot meet the metabolic demands of the tissues. Signs and symptoms of congestive heart failure reflect decreased cardiac output and decreased tissue perfusion. In the early stages, the neonate may be tachypneic and tachycardic with an increased respiratory effort, rales, hepatomegaly, and delayed capillary refill. Edema caused by CHF is rarely seen in neonates. Diaphoresis, feeding difficulties, and growth failure later become apparent. Finally, congestive heart failure may present acutely with cardiorespiratory collapse, particularly with obstructive defects. Birth asphyxia and anemia must also be considered as causes of congestive heart failure in neonates.
The common symptoms associated with congestive heart failure (see the Critical Findings box on p. 685) can be understood using the physiologic principles previously outlined.
Tachycardia
The heart attempts to compensate for the decrease in cardiac output (CO) by increasing either the heart rate (HR) or the stroke volume (SV) (CO = HR × SV). Because the fetal myocardium has fewer contractile elements and is poorly innervated by the sympathetic nervous system, capacity to increase stroke volume is very limited. Therefore increases in cardiac output are achieved mainly by increasing the heart rate.
Congestive Heart Failure
• Tachycardia
• Cardiac enlargement
• Tachypnea
• Gallop rhythm
• Decreased peripheral pulses and skin mottling in the extremities
• Decreased urine output and edema
• Diaphoresis
• Hepatomegaly
• Decreased activity
• Failure to thrive and feeding problems
• Diminished cardiac output
Cardiac Enlargement
Hypertrophy and dilation of the heart occur in response to the volume or pressure overload. This enlargement is evident on chest x-ray examination.
Gallop Rhythm
The gallop rhythm is an abnormal filling sound caused by dilation of the ventricles. It is heard as a triple rhythm on auscultation.
Decreased Peripheral Pulses/Mottling of the Extremities
Decreased cardiac output results in a compensatory redistribution of blood flow to vital tissues. Peripheral tissue perfusion is decreased, which results in mottling of the skin and decreased pulses.
Decreased Urine Output and Edema
Decreased renal perfusion results in decreased glomerular filtration. The body interprets this as a decrease in intravascular volume and begins to initiate compensatory mechanisms such as vasoconstriction and retention of fluid and sodium. Neonates manifest this as weight gain and periorbital edema.
Diaphoresis
Congestive heart failure leads to an increase in metabolic rate and increased activity of the autonomic nervous system, resulting in diaphoresis. This is representative of the increased workload of the heart in failure.
Hepatomegaly
The right ventricle in congestive heart failure is less compliant and does not adequately empty. This leads to elevated pressures in the right atrium, central venous system, and hepatic system. Hepatomegaly results from hepatic venous congestion.
Decreased Activity and Exercise Intolerance
The decreased perfusion to peripheral tissues and the increased energy needed by the heart in failure leaves little energy for activities such as feeding and crying. The infant in heart failure may sleep the majority of the time.
Failure to Thrive/Feeding Difficulties
Tachypnea compromises the infant’s ability to feed. The basal metabolic rate increases in neonates with congestive heart failure. This necessitates a higher caloric intake (150 kcal or more).
Dysrhythmias
Abnormalities of the cardiac rhythm and murmurs are discussed individually later.
CARDIAC EXAMINATION
See “Specific Conditions” section.
LABORATORY DATA
Arterial Blood Gases
The Pa co2 in cardiac disease is often normal. It is usually increased if a primary pulmonary disease is present. Frequent monitoring of blood gases is unnecessary, but the acid-base balance should be monitored closely.
Four-Extremity Blood Pressure
The measurement of blood pressure should be taken in both arms and both legs. A systolic pressure that is more than 10 mm Hg higher in the upper body compared with the lower body is abnormal and suggests coarctation of the aorta, aortic arch hypoplasia, or interrupted aortic arch. However, this is a highly specific test with low sensitivity; the lack of systolic blood pressure gradient does not conclusively rule out aortic arch abnormalities.
Chest X-ray Examination
Frontal and lateral views (if possible) of the chest should be obtained. In neonates, the size of the heart may be difficult to determine because of the overlying thymus. Chest x-ray examination may be normal even in the presence of life-threatening CHD. However, the degree of pulmonary vascularity helps define the type of CHD present and is characterized as being increased, normal, or decreased. Likewise, the heart size should be evaluated and is described as being increased, normal, or decreased.
Electrocardiogram
Neonatal electrocardiogram (ECG) reflects the hemodynamic relationships that existed in utero. Many forms of CHD have minimal prenatal hemodynamic effects, and therefore the ECG is frequently “normal for age” despite significant structural defects (e.g., transposition of the great arteries, tetralogy of Fallot).
Echocardiogram
The echocardiogram is indispensable in the diagnosis of congenital heart disease. 4 Two-dimensional echocardiography, used to define cardiac anatomy, estimates pressures, measures gradients, and evaluates cardiac function21 and, supplemented with Doppler and color Doppler, has become the primary diagnostic tool in pediatric cardiology. Noninvasive transthoracic echocardiogram is the most commonly used approach. During the procedure, close monitoring is recommended with attention to vital signs, respiratory status, and temperature. Recently, three-dimensional echocardiograms (3D Echos) that offer real-time three-dimensional imaging with 2 Dx-matrix probe and reconstructed 3D imaging using spatiotemporal image correlation (STIC) techniques21 are being used.
Computed Tomography
More practitioners are finding 64-slice multidimensional computed tomography (64-MDCT) to be helpful in imaging some thoracic regions beyond the scope of echocardiograms, particularly after surgical revision. MDCT offers higher spatial resolution with shorter scan times. 31
Magnetic Resonance Imaging
Magnetic resonance imaging (MRI) offers three-dimensional reconstruction and high-resolution images of the heart and great vessels. The MRI is of particular use in evaluating extracardiac vascular abnormalities, such as arch anomalies, vascular rings, and pulmonary arteriovenous anomalies. MRI provides high spatial resolution, excellent soft-tissue definition, a large field of view, and unrestricted demonstration of cardiovascular morphology; however, it does require sedation and a stable patient, and it is expensive. 13
General Treatment Strategy
Optimal management of infants with heart disease requires specialized expertise. Infants are monitored closely for hypoxia, hypoglycemia, acidosis, and congestive heart failure.
The infant must be kept in an incubator or radiant heat warmer in which body temperature is maintained while color changes (pallor and increased cyanosis) may be observed. A cardiorespiratory monitor for continuous cardiac monitoring detects bradycardia, tachycardia, and dysrhythmias. Monitoring of oxygen saturations is helpful in determining adequacy of pulmonary blood flow and/or increased need for oxygen. Respiratory effort is assessed for tachypnea, shallow breathing, apnea, retractions, grunting, and nasal flaring. Observe and document activity level such as feeding behavior, muscle tone, spontaneous movement, and seizure activity.
MANAGEMENT OF CONGESTIVE HEART FAILURE
The medical management of congestive heart failure attempts to reverse the outlined process and helps the heart compensate with increased cardiac output.
Digoxin acts primarily as a positive inotropic (improves contractility) agent but decreases the heart rate and increases urine output (Box 24-3). Digoxin slows conduction at the atrioventricular (AV) node. This drug should be used with caution if acidosis, myocarditis, or obstructive lesions (e.g., tetralogy of Fallot, subvalvular pulmonary stenosis, asymmetric septal hypertrophy) are present. Diuretics such as furosemide (Table 24-4) help decrease total body water (which is increased as a result of congestive heart failure). In general, chronic fluid restriction and low-salt diets are not commonly used in newborns or infants with congestive heart failure.
BOX 24-3
| Digitalizing Schedule | |
|
Preterm infant
PO route: 20 mcg/kg total dose*
Term infant
PO route: 30 mcg/kg total dose*
|
Total dose is usually divided into three doses giving one half, then one fourth, then one fourth of the total dose q 8 hr. Check electrocardiogram rhythm strip for rate, PR interval, and dysrhythmias before each dose. |
| Maintenance Schedule | |
|
Preterm infant
PO route: 5-10 mcg/kg/day*
|
Total dose should be divided BID. Allow 12-24 hr between last digitalizing and first maintenance doses. It takes about 6 days to “digitalize” a patient with maintenance doses alone. The sign of digitalis effect is usually prolongation of the PR interval. The first sign of digitalis toxicity is usually vomiting, dysrhythmia, or bradycardia. |
|
Term infant
PO route: 5-10 mcg/kg/day
|
Drugs such as quinidine, amiodarone, and diuretics predispose to digoxin toxicity. The clearance of digoxin is directly related to renal function. Dosage must be reduced in patients with impaired renal function. |
| BID, Twice daily. | |
| *Intravenous (IV) dose is 75% of oral (PO) dose. |
|
| Standard Concentrations: Each institution’s concentration may vary; NOT to exceed maximum concentration per pharmacy reference manuals. | ||||
| Neonatal Drug Guidelines Updated May 7, 2002. | ||||
| BID, Twice daily; BP, blood pressure; BUN, blood urea nitrogen; CNS, central nervous system; ETT, endotracheal tube; GI, gastrointestinal; IHSS, idiopathic hypertrophic subaortic stenosis; IV, intravenous; IM, intramuscular; KCl, potassium chloride; kg, kilograms; Maint., maintenance; max, maximum; mcg, micrograms; mg, milligrams; min, minutes; NEC, necrotizing enterocolitis; NS, normal saline; PO, per os; PRN, as needed; q, every; QID, four times a day; TID, three times a day. | ||||
| Drug | Route | Dose | Onset of Action | Comments |
|---|---|---|---|---|
| Atropine | IV | 0.01-0.03 mg/kg/dose PRN (max 0.4 mg) | Seconds | May cause tachycardia, urinary retention, or hyperthermia |
| PO | 0.01-0.03 mg/kg/dose q 4-6 hr (max 0.4 mg) | Minutes | May cause tachycardia | |
| ETT | Give 2-3 times the IV dose followed by NS flush | Minutes | May cause tachycardia | |
| Calcium chloride (10% solution) | IV | 0.2-0.3 mL (20-30 mg)/kg/ dose q 10 min PRN (max 500 mg) | Minutes | Slow infusion; must be IV; potentiates digoxin, bradycardia |
| Calcium gluconate (10% solution) | IV | 1-2 mL/kg/dose (100-200 mg/kg/dose) q 10 min PRN (max 500 mg) | Minutes | Slow infusion (over 10-30 min); must be IV; potentiates digoxin, bradycardia/dysrhythmias |
| Captopril (Capoten) | PO | Younger than 2 months:
Initial dose: 0.1-0.25 mg/kg/dose q 8-24 hr
Titrate: up to 0.5 mg/kg/dose
Older than 2 months:
Initial dose: 0.3 mg/kg/day
Titrate: up to 6 mg/kg/day in 1-4 divided doses
|
15 min + | Hypotension, tachycardia, increased BUN and serum creatinine, hypercalcemia |
| Diazoxide (Hyperstat) | IV | 5 mg/kg/dose q 30 min PRN | 1-2 min | May cause hypotension or hypoglycemia |
| Dobutamine (Dobutrex) | IV | 2-10 mcg/kg/min | Minutes |
Do not use if IHSS or tetralogy of Fallot, may cause ventricular ectopy, tachycardia, or hypertension
Incompatible with alkaline solutions
|
| Dopamine (Intropin) | IV | 5-10 mcg/kg/min | Minutes | Tachydysrhythmia, vasoconstriction, gangrene of extremities, anginal pain, and palpitations can occur; inactivated in alkaline solution |
| Epinephrine (1:10,000) | IV/ETT | 0.1-0.3 mL/kg/dose (max 5 mL/dose) q 3-5 min PRN | Seconds | May cause tachycardia, dysrhythmias, or hypertension; not effective if acidosis is present |
| Esmolol (Brevibloc) | IV |
Loading dose: 500 mcg/kg/min
Continuous infusion: titrate 50-200 mcg/kg/min
|
Minutes | May cause bradycardia, hypotension, bronchoconstriction |
| Furosemide (Lasix) | IV | 1-2 mg/kg/dose | 5-15 min | May cause metabolic alkalosis and hypokalemia; monitor electrolytes—may need KCl supplementation; renal calcification |
| PO | 1-4 mg/kg/dose | 30-60 min | ||
| Hydralazine (Apresoline) | IV PO |
0.1-0.5 mg/kg q 3-6 hr 0.1-0.5 mg/kg q 6 hr; may increase to max of 2 mg/kg/dose q 6 hr |
15-30 min Often days until titrated effect achieved |
May cause lupus-like syndrome, tachycardia, or hypotension |
| Hydrochloro-thiazide (HydroDIURIL) | PO | 1-2 mg/kg q 12 hr | 1-2 hr | May cause electrolyte imbalance, may need KCl supplementation |
| Ibuprofen lysine (NeoProfen) | IV | 10 mg/kg first dose; then 5 mg/kg second and third dose (q 24 hr) |
Most effective in first 3 days of life
Monitor urine output and creatinine levels; discontinue drug if dramatic decrease in urine output
Significantly fewer adverse effects (compared with indomethacin) on renal and mesenteric blood flow; less oliguria and increase in serum creatinine levels
|
|
| Indomethacin (Indocin) | IV | 0.1-0.2 mg/kg/dose; may be repeated q 8 hr for a total of 3 doses |
Less effective if administered after 7 days of age; probably will have no effect after 14 days
Monitor urine output and creatinine levels; discontinue drug if dramatic decrease in urine output
Contraindications: severe renal impairment, active bleeding in the CNS or GI tract, and NEC10
|
|
| Isoproterenol (Isuprel) | IV | 0.1-0.4 mcg/kg/min | 30-60 seconds | May cause tachycardia/ventricular tachydysrhythmia; may also cause subendocardial ischemia |
| Lidocaine (Xylocaine) | IV |
IV bolus: 1-3 mg/kg
IV drip: 30-50 mcg/kg/min
|
May cause dysrhythmia, CNS agitation or depression | |
| Milrinone (Primacor) | IV |
Loading: 50 mcg/kg over 15 min
Maint.: 0.25-0.75 mcg/kg/min
|
May cause ventricular dysrhythmias, ventricular fibrillation, or hypotension | |
| Nitroprusside (Nipride) | IV | 1-10 mcg/kg/min over 10 min to control BP; chronic infusion: 2 mcg/kg/min (protect from light; change solution q 4 hr) | Seconds | May cause hypotension and reflex tachycardia; may cause thiocyanate toxicity, especially if decreased renal function is present |
| Phentolamine (Regitine) | IV | 1-20 mcg/kg/min | 5-10 min | May cause hypotension; commonly used with an inotropic agent |
| PO | 5 mg/kg/day QID | |||
| Phenytoin (Dilantin) | IV |
Load: 10-15 mg/kg over 5 min (slow infusion)
Maint.: 3-5 mg/kg/day BID
|
5-10 min | May cause cardiac depression |
| PO | 3-5 mg/kg/day BID | 2-4 hr | Therapeutic blood levels (5-20 mcg/mL) | |
| Procainamide (Pronestyl) | IV |
Load: 10-15 mg/kg/dose over 5 min (max 100 mg)
Maint.: IV 30-80 mcg/kg/min
|
1-5 min | May cause hypotension or lupus-like syndrome |
| IM | 5-8 mg/kg q 6 hr | 15-30 min | Same as for IV | |
| Propranolol (Inderal) | IV |
Dysrhythmias: 0.01-0.15 mg/kg/dose slow IV q 6-8 hr PRN (max single dose, 10 mg)
Hypercyanotic spell: 0.15-0.25 mg/kg/dose slow IV push q 15 min (max dose 10 mg)
|
2-5 min | May severely decrease cardiac output |
| PO | Dysrhythmias: 0.5-1 mg/kg/dose TID-QID (max daily dose 60 mg) Hypercyanotic spell: 1-2 mg/kg/dose QID | 30-60 min | Same as for IV | |
| Prostaglandin E 1 (Prostin VR) | IV |
Initial dose: 0.05-0.1 mcg/kg/min cont. IV infusion
Maint.: 0.01-0.05 mcg/kg/min cont. IV infusion
|
30 min | May cause apnea, fever, or hypotension |
| Spironolactone (Aldactone) | PO | 1-2 mg/kg/day | 3-5 days | Hyperkalemia, drowsiness, GI upset |
| Tolazoline | IV |
Test: 1-2 mg/kg slow IV push
Maint.: 1-2 mg/kg/hr
|
Minutes | May cause hypotension, GI or pulmonary hemorrhage |
| Adenosine | IV | 30-250 mcg/kg | Slows the spontaneous heart rate and prolongs the PR interval; may cause transient complete heart block and hypotension; half-life is only 9.3 seconds so its effects quickly dissipate | |
Infants with congestive heart failure may be difficult to feed, and the process is often frustrating. They may have trouble sucking, swallowing, and breathing simultaneously. They may have to rest frequently during a feeding, thus prolonging feeding times, and they may fall asleep exhausted before adequate caloric intake is achieved. Because caloric requirements are higher in infants with CHD, the use of higher caloric formulas may be of benefit. Adequate nutrition must be ensured by the following:
• Observing the infant’s ability to nipple feed (a soft, free-flowing [premature] nipple offers the least resistance to sucking and helps the infant conserve energy)
• Providing adequate calories for growth and, if necessary, using alternative feeding methods (i.e., gavage or continuous nasogastric drip) if the infant is sucking poorly
• Anticipating the infant’s hunger and offering feedings before the infant uses energy by crying
• Positioning the infant in a semi-erect position for feeding
• Burping the infant after every half ounce consumed to help minimize vomiting
• Weighing the infant daily and checking for appropriate weight gain
Before discharge from the nursery, the infant should be in stable condition (e.g., feeding well and gaining weight appropriately).
An important fact for families to understand is that many infants gain weight very slowly because of their cardiac defects, regardless of the method of feeding used. The family of an infant in congestive heart failure needs support and teaching. Explanation of the term congestive heart failure should be given early, because it is a frightening term for parents. The term “heart failure” is often interpreted as “heart attack” or “cardiac arrest.” Parents must understand that saying an infant is in heart failure does not imply that the infant’s heart will stop beating. A simple explanation describing heart failure as a condition in which the heart shows signs of being less able to pump sufficient blood to meet all the needs of the body helps decrease anxiety for the family.
SPECIFIC CONDITIONS
Patent Ductus Arteriosus
PHYSIOLOGY
The ductus arteriosus is a normal pathway in the fetal circulatory system and allows blood from the right ventricle and pulmonary arterial system to flow into the descending aorta for ultimate delivery to the placenta (Figure 24-3). Functionally, the patent ductus arteriosus (PDA) closes within a few hours to several days after birth, but this closure is often delayed in premature infants. After birth, as a result of a decrease in the pressure of the pulmonary circulation and an increase in the pressure of the aorta, the blood flow through a PDA is predominantly from the aorta to the pulmonary artery (left-to-right shunt). The hemodynamic changes and the resultant clinical manifestations of a PDA depend on the magnitude of the pulmonary vascular resistance and the size of the ductal lumen.
|
|
| FIGURE 24-3
(Modified from Hockenberry MJ, Wilson D: Wong’s essentials of pediatric nursing, ed 8, St Louis, 2009, Mosby.)
|
Approximately 15% of infants with PDAs have additional cardiac defects (e.g., VSD, coarctation of the aorta, aortic stenosis, pulmonary stenosis). PDA can be associated with known syndromes, most commonly rubella.
DATA COLLECTION
History
Asphyxial insult or respiratory distress syndrome (RDS), inability to wean from a ventilator, and an increasing F io2 demand usually accompany PDA.
Physical Findings
Increased flow to the pulmonary circulation and volume overload of the left ventricle are the two major physiologic abnormalities in a PDA.
Cyanosis
Generally, cyanosis is not present in an isolated PDA, because the predominant shunt is from left to right.
Heart Sounds
Infants with a PDA may have an audible murmur as a result of the left-to-right shunting through the ductus during systole. A grade I through III systolic murmur is best heard at the upper left sternal border with radiation to the left axilla and faintly to the back. Although this murmur occasionally may spill into diastole, the classical continuous machinery-like murmur is an unusual occurrence in the newborn period. It is often helpful to briefly disconnect the newborn from the ventilator before auscultating. There are cases of large PDAs in which no murmur is audible.
Pulses
Because of the rapid upstroke and wide pulse pressure, the peripheral pulses are bounding. Pulses are hyperdynamic and easily palpated. Assessment of the pulses should include palpation of palmar, plantar, and popliteal pulses. The presence of an easily palpated pulse in these areas suggests the presence of an aortic run-off lesion, which is most commonly a PDA.
Congestive Heart Failure
Because of the volume overload of the left ventricle, the infant may show signs of congestive heart failure and pulmonary edema (see “Congestive Heart Failure” section).
Laboratory Data
Arterial Blood Gases
Arterial blood gas values are normal.
Chest X-ray Examination
Chest x-ray examination is normal in small shunts. Cardiomegaly is present with increased pulmonary vascularity in large shunts.
Electrocardiogram
The ECG may be normal, demonstrate left ventricular hypertrophy, or demonstrate combined ventricular hypertrophy.
Echocardiogram
Direct imaging is the preferred method both to diagnose patency and to determine the significance of the ductus arteriosus. An increased left atrial/aortic ratio suggests a moderate to large left-to-right shunt (i.e., PDA, VSD). An echocardiogram should be performed before medical or surgical closure of the PDA to rule out a ductal-dependent lesion or other associated anomalies. Color-flow Doppler mapping allows visualization of the PDA and aids in determining the size and direction of the shunt across the PDA (i.e., left to right, right to left, bidirectional).
Cardiac Catheterization
If the echocardiogram has eliminated a ductal-dependent lesion, cardiac catheterization is usually not necessary before treatment.
TREATMENT
Medical Management
Asymptomatic infants with PDAs generally do not require medical management or surgical ligation. These infants should be monitored for evidence of congestive heart failure, failure to thrive, increasing oxygen requirement, or other complications.
Symptomatic infants require ductal closure by either pharmacologic management with prostaglandin inhibitors such as indomethacin or ibuprofen lysine therapy (both are Food and Drug Administration [FDA]–approved cyclo-oxygenase [COX] inhibitors) or surgical ductal ligation. Medical management such as fluid restriction, watchful waiting, and ventilator support is rarely successful, especially in low-birth-weight infants. The current trend is to treat early presymptomatic therapeutic PDA at 2 to 3 days of age after a confirmed echocardiogram.30 Although indomethacin was first reported in the 1970s and became first-line therapy for the treatment of PDA, ibuprofen lysine has been shown to be equally effective in closure rates and may be preferable because of its better toxicity profile. 25Table 24-4
Buy Membership for Neonatal and Perinatal Medicine Category to continue reading. Learn more here
[/not-level-membership-for-neonatal-and-perinatal-medicine-category]
