OVERVIEW AND DEFINITIONS
Sudden cardiac death (SCD) is defined as natural death due to cardiac causes in a person who may or may not have previously recognized heart disease but in whom the time and mode of death are unexpected. The term “sudden,” in the context of SCD, is defined for most clinical and epidemiologic purposes as 1 h or less between a change in clinical status heralding the onset of the terminal clinical event and the cardiac arrest itself. One exception is unwitnessed deaths, in which pathologists may expand the temporal definition to 24 h after the victim was last seen to be alive and stable.
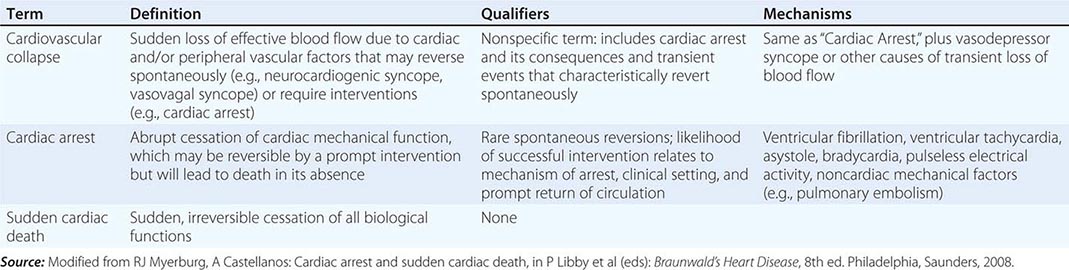
Another exception is the variable interval between cardiac arrest and biological death that results from community-based interventions, following which victims may remain biologically alive for days or even weeks after a cardiac arrest that has resulted in irreversible central nervous system damage. Confusion in terms can be avoided by adhering strictly to definitions of cardiovascular collapse, cardiac arrest, and death (Table 327-1). Although cardiac arrest is often potentially reversible by appropriate and timely interventions, death is biologically, legally, and literally an absolute and irreversible event. Biological death may be delayed by interventions, but the relevant pathophysiologic event remains the sudden and unexpected cardiac arrest. Accordingly, for statistical purposes, deaths that occur during hospitalization or within 30 days after resuscitated cardiac arrest are counted as sudden deaths.
|
DISTINCTION BETWEEN CARDIOVASCULAR COLLAPSE, CARDIAC ARREST, AND DEATH |
The majority of natural deaths are caused by cardiac disorders. However, it is common for underlying heart diseases—often far advanced—to go unrecognized before the fatal event. As a result, up to two-thirds of all SCDs occur as the first clinical expression of previously undiagnosed disease or in patients with known heart disease, the extent of which suggests low individual risk. The magnitude of sudden cardiac death as a public health problem is highlighted by the estimate that ~50% of all cardiac deaths are sudden and unexpected, accounting for a total SCD burden estimated to range from <200,000 to >450,000 deaths each year in the United States. SCD is a direct consequence of cardiac arrest, which may be reversible if addressed promptly. Because resuscitation techniques and emergency rescue systems are available to respond to victims of out-of-hospital cardiac arrest, which was uniformly fatal in the past, understanding the SCD problem has practical clinical importance.
CLINICAL DEFINITION OF FORMS OF CARDIOVASCULAR COLLAPSE
Cardiovascular collapse is a general term connoting loss of sufficient cerebral blood flow to maintain consciousness due to acute dysfunction of the heart and/or peripheral vasculature. It may be caused by vasodepressor syncope (vasovagal syncope, postural hypotension with syncope, neurocardiogenic syncope; Chap. 27), a transient severe bradycardia, or cardiac arrest. The latter is distinguished from the transient forms of cardiovascular collapse in that it usually requires an active intervention to restore spontaneous blood flow. In contrast, vasodepressor syncope and other primary bradyarrhythmic syncopal events are transient and non-life-threatening, with spontaneous return of consciousness.
In the past, the most common electrical mechanism for cardiac arrest was ventricular fibrillation (VF) or pulseless sustained ventricular tachycardia (PVT). These were the initial rhythms recorded in 60–80% of cardiac arrests, with VF being the far more common of the two. Severe persistent bradyarrhythmias, asystole, and pulseless electrical activity (PEA; organized electrical activity, unusually slow, without mechanical response, formerly called electromechanical dissociation [EMD]) caused another 20–30%. Currently, asystole has emerged as the most common mechanism recorded at initial contact (45–50% of cases). PEA accounts for 20–25%, and VF is now present on initial contact in 25–35%. Undoubtedly, a significant proportion of the asystole cases began as VF and deteriorated to asystole because of long response times, but there are data suggesting an absolute reduction in VF as well. Acute low cardiac output states, having a precipitous onset, also may present clinically as a cardiac arrest. These hemodynamic causes include massive acute pulmonary emboli, internal blood loss from a ruptured aortic aneurysm, intense anaphylaxis, and cardiac rupture with tamponade after myocardial infarction (MI).
ETIOLOGY, INITIATING EVENTS, AND CLINICAL EPIDEMIOLOGY
Clinical, epidemiologic, and pathologic studies have provided information on the underlying structural substrates in victims of SCD and identified subgroups at high risk for SCD. In addition, studies of clinical physiology have begun to identify transient functional factors that may convert a long-standing underlying structural abnormality from a stable to an unstable state, leading to the onset of cardiac arrest (Table 327-2).
|
CARDIAC ARREST AND SUDDEN CARDIAC DEATH |
Cardiac disorders constitute the most common causes of sudden natural death. After an initial peak incidence of sudden death between birth and 6 months of age (sudden infant death syndrome [SIDS]), the incidence of sudden death declines sharply and remains low through childhood and adolescence. Among adolescents and young adults, the incidence of SCD is approximately 1 per 100,000 population per year. The incidence begins to increase in adults over age 30 years, reaching a second peak in the age range of 45–75 years, when it approximates 1–2 per 1000 per year among the unselected adult population. Increasing age within this range is associated with increasing risk for sudden cardiac death (Fig. 327-1A). From 1 to 13 years of age, only one of five sudden natural deaths is due to cardiac causes. Between 14 and 21 years of age, the proportion increases to 30%, and it rises to 88% in the middle-aged and elderly.
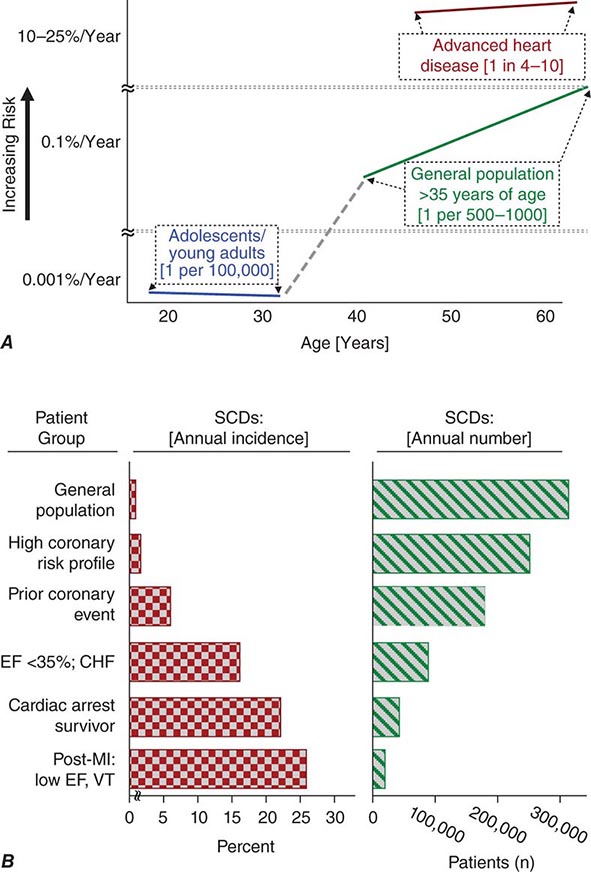
FIGURE 327-1 Panel A demonstrates age-related risk for sudden cardiac death (SCD). For the general population age 35 years and older, SCD risk is 0.1–0.2% per year (1 per 500–1000 population). Among the general population of adolescents and adults younger than age 30 years, the overall risk of SCD is 1 per 100,000 population, or 0.001% per year. The risk of SCD increases dramatically beyond age 35 years. The greatest rate of increase is between 40 and 65 years (vertical axis is discontinuous). Among patients older than 30 years of age, with advanced structural heart disease and markers of high risk for cardiac arrest, the event rate may exceed 25% per year, and age-related risk attenuates. (Modified from RJ Myerburg, A Castellanos: Cardiac arrest and sudden cardiac death, in P Libby et al [eds]: Braunwald’s Heart Disease, 8th ed. Philadelphia, Saunders, 2008.) Panel B demonstrates the incidence of SCD in population subgroups and the relation of total number of events per year to incidence figures. Approximations of subgroup incidence figures and the related population pool from which they are derived are presented. Approximately 50% of all cardiac deaths are sudden and unexpected. The incidence bars on the left (percent/year) indicate the approximate percentage of sudden and nonsudden deaths in each of the population subgroups indicated, ranging from the lowest percentage in unselected adult populations (0.1–2% per year) to the highest percentage in patients with severe left ventricular dysfunction and heart failure (approximately 25% per year). The bars on the right indicate the total number of events per year in each of these groups with the population impact size of each of the subgroups. The highest risk categories identify the smallest number of total annual events, and the lowest incidence category accounts for the largest number of events per year. CHF, congestive heart failure; EF, ejection fraction; MI, myocardial infarction; VT, ventricular tachycardia. (After RJ Myerburg et al: Circulation 85:2, 1992.)
Young and middle-aged men and women have different susceptibilities to SCD, but the sex differences decrease and ultimately disappear with advancing age. The difference in risk for SCD parallels the differences in age-related risks for other manifestations of coronary heart disease (CHD) between men and women. As the gender gap for manifestations of CHD closes in the sixth to eighth decades of life, the excess risk of SCD in males progressively narrows. Despite the lower incidence among younger women, coronary risk factors such as cigarette smoking, diabetes, hyperlipidemia, and hypertension are highly influential, and SCD remains an important clinical and epidemiologic problem. The incidence of SCD among the African-American population appears to be higher than it is among the white population; the reasons remain uncertain.
Genetic factors contribute to the risk of acquiring CHD, and a genetic basis for its expression as SCD is being explored. A genetic hypothesis for at least part of the SCD risk is supported by data suggesting a familial predisposition to SCD as a specific form of expression of CHD. A parental history of SCD as a first cardiac event increases the probability that an acute coronary event in the offspring will express similarly. In a number of less common syndromes, such as hypertrophic cardiomyopathy, congenital long QT interval syndromes, right ventricular dysplasia, and the syndrome of right bundle branch block and nonischemic ST-segment elevations (Brugada syndrome), and other more rare syndromes, there is a specific inherited risk of ventricular arrhythmias and SCD (Chap. 277).
The etiologic structural substrates and functional factors contributing to expression of the SCD syndrome are listed in Table 327-2. Worldwide, and especially in Western cultures, coronary atherosclerotic heart disease is the most common structural abnormality associated with SCD in middle-aged and older adults. Up to 80% of all SCDs in the United States are due to the consequences of coronary atherosclerosis. The nonischemic cardiomyopathies (dilated and hypertrophic, collectively; Chap. 273e) account for another 10–15% of SCDs, and all the remaining diverse etiologies cause only 5–10% of all SCDs. The inherited arrhythmia syndromes (see above and Table 327-2) are proportionally more common causes in adolescents and young adults. For some of these syndromes, such as hypertrophic cardiomyopathy (Chap. 287), the risk of SCD increases significantly after the onset of puberty.
Transient ischemia in a previously scarred or hypertrophied heart, hemodynamic and fluid and electrolyte disturbances, fluctuations in autonomic nervous system activity, and transient electrophysiologic changes caused by drugs or other chemicals (e.g., proarrhythmia) have all been implicated as mechanisms responsible for the transition from electrophysiologic stability to instability. In addition, reperfusion of ischemic myocardium may cause transient electrophysiologic instability and arrhythmias.
PATHOLOGY
Data from postmortem examinations of SCD victims parallel the clinical observations on the prevalence of CHD as the major structural etiologic factor. More than 80% of SCD victims have pathologic findings of CHD. The pathologic description often includes a combination of long-standing, extensive atherosclerosis of the epicardial coronary arteries and unstable coronary artery lesions, which include various permutations of eroded, fissured, or ruptured plaques; platelet aggregates; hemorrhage; and/or thrombosis. As many as 70–75% of males who die suddenly have preexisting healed MIs, whereas only 20–30% have recent acute MIs, despite the prevalence of unstable plaques and thrombi. The latter suggests transient ischemia as the mechanism of onset. Regional or global left ventricular (LV) hypertrophy often coexists with prior MIs.
PREDICTION AND PREVENTION OF CARDIAC ARREST AND SUDDEN CARDIAC DEATH
SCD accounts for approximately one-half the total number of cardiovascular deaths. As shown in Fig. 327-1B, the very-high-risk subgroups consist of more focused populations at higher risk of cardiac arrest or SCD, with better individual prediction, but the representation of such subgroups within the overall population burden of SCD is small. This is indicated by the absolute number of events (“events per year”), in contrast to the percentage per year in the subgroup. To achieve a major population impact, effective prevention of underlying diseases and the development of new epidemiologic and clinical probes that will allow better individual risk prediction by identifying specific high-risk subgroups within the large general populations are needed.
Strategies for predicting and preventing SCD are classified as primary and secondary. Primary prevention refers to the attempt to identify individual patients at specific risk for SCD and institute preventive strategies. Secondary prevention refers to measures taken to prevent recurrent cardiac arrest or death in individuals who have survived a prior cardiac arrest.
The effectiveness of the prevention strategies currently used depends on the magnitude of risk among the various population subgroups. Because the annual incidence of SCD among the unselected adult population is limited to approximately 1 per 1000 population per year (Fig. 327-1) and ~50% of all SCDs due to coronary artery disease occur as the first clinical manifestation of the disease (Fig. 327-2A), the only currently practical strategies are profiling for risk of developing CHD and risk factor control (Fig. 327-2B). The most powerful long-term risk factors include age, cigarette smoking, elevated serum cholesterol, diabetes mellitus, elevated blood pressure, LV hypertrophy, and nonspecific electrocardiographic abnormalities. Markers of inflammation (e.g., levels of C-reactive protein) that may predict plaque destabilization have been added to risk classifications. The presence of multiple risk factors progressively increases incidence, but not sufficiently or specifically enough to warrant therapies targeted to potentially fatal arrhythmias (Fig. 327-1A). However, recent studies suggesting familial clustering of SCD associated with a first acute coronary syndrome offer hope that genetic markers for specific risk may be forthcoming.
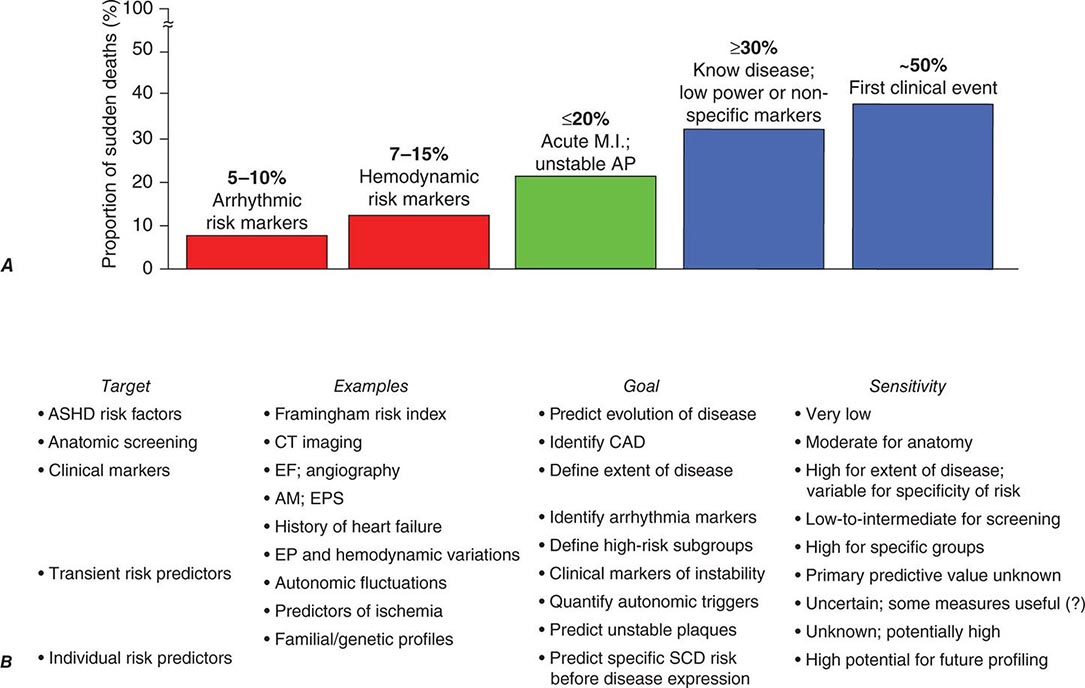
FIGURE 327-2 Population subsets, risk predictors, and distribution of sudden cardiac deaths (SCDs) according to clinical circumstances. A. The population subset with high-risk arrhythmia markers in conjunction with low ejection fraction is a group at high risk of SCD but accounts for <10% of the total SCD burden attributable to coronary artery disease. In contrast, 50% of all SCD victims present with SCD as the first and only manifestation of underlying disease, and up to 30% have known disease but are considered relatively low risk because of the absence of high-risk markers. B. Profiling for individual prediction and prevention of SCD is difficult. The highest absolute numbers of events occur among the general population who may have risk factors for coronary heart disease or expressions of disease that do not predict high risk. This results in a low sensitivity for predicting and preventing SCD. New approaches that include epidemiologic modeling of transient risk factors and genetic predictors of individual patient risk offer hope for greater sensitivity in the future. AM, ambulatory monitoring; AP, angina pectoris; ASHD, arteriosclerotic heart disease; CAD, coronary artery disease; CT, computed tomography; EF, ejection fraction; EP, electrophysiologic; EPS, electrophysiologic study; MI, myocardial infarction. (Modified from RJ Myerburg: J Cardiovasc Electrophysiol 12:369–381, 2001.)
After coronary artery disease has been identified in a patient, additional strategies for risk profiling become available (Fig. 327-2B), but the majority of SCDs occur among the large unselected groups rather than in the specific high-risk subgroups that become evident among populations with established disease (compare events per year with percentage per year in Fig. 327-1B). After a major cardiovascular event, such as acute MI, recent onset of heart failure, or survival after out-of-hospital cardiac arrest, the highest risk of death occurs during the initial 6–18 months after the event and then plateaus toward the baseline risk associated with the extent of underlying disease. However, many of the early deaths are nonsudden, diluting the potential benefit of strategies targeted specifically to SCD. Thus, although post-MI beta blocker therapy has an identifiable benefit for both early SCD and nonsudden mortality risk, a total mortality benefit for implantable cardioverter-defibrillator (ICD) therapy early after MI has not been observed.
Among patients in the acute, convalescent, and chronic phases of MI (Chap. 295), subgroups at high absolute risk of SCD can be identified. During the acute phase, the potential risk of cardiac arrest from onset through the first 48 h used to be as high as 15%, but is now reported in the range of 2.3–4.4% because of early patient awareness of the significance of symptoms and the availability of emergency revascularization strategies. Those who survive acute-phase VF are not at continuing risk for recurrent cardiac arrest indexed to that event. During the convalescent phase after MI (3 days to ~6 weeks), an episode of sustained ventricular tachycardia (VT) or VF, which is usually associated with a large infarct, predicts a natural history mortality risk of >25% at 12 months. At least one-half of the deaths are sudden. Aggressive intervention techniques may reduce this incidence.
During the chronic phase after MI, the longer-term risk for total mortality and SCD mortality is predicted by a number of factors (Fig. 327-2B). The most important for both SCD and nonsudden death is the extent of myocardial damage sustained as a result of the acute MI. This is measured by the magnitude of reduction of the ejection fraction (EF) and/or the occurrence of heart failure. Various studies have demonstrated that ventricular arrhythmias identified by ambulatory monitoring contribute significantly to this risk, especially in patients with an EF <40%. In addition, inducibility of VT or VF during electrophysiologic testing of patients who have ambient ventricular arrhythmias (premature ventricular contractions [PVCs] and nonsustained VT) and an EF <35% is a strong predictor of SCD risk. Patients in this subgroup are now considered candidates for ICDs (see below). Risk falls off sharply with EFs >35% and the absence of ambient arrhythmias after MI, and conversely is high with EFs <30% even without the ambient arrhythmia markers.
The cardiomyopathies (dilated and hypertrophic, Chap. 287) are the second most common category of diseases associated with risk of SCD (Table 327-2). Some risk factors have been identified, largely related to extent of disease, presence of heart failure, documented ventricular arrhythmias, and syncope thought to be due to arrhythmias. The less common causes of SCD include valvular heart disease (primarily aortic) and inflammatory and infiltrative disorders of the myocardium. The latter include viral myocarditis, sarcoidosis, and amyloidosis.
Among adolescents and young adults, rare inherited disorders such as hypertrophic cardiomyopathy, the long QT interval syndromes, right ventricular dysplasia, and the Brugada syndrome have received attention as important causes of SCD, as has acute myocarditis and other less common acquired diseases. Among the subgroup of young competitive athletes, the incidence of SCD may be higher than it is for the general adolescent and young adult population, perhaps up to 1 in 75,000–100,000. Hypertrophic cardiomyopathy (Chap. 287) is the most common cause in the United States.
Secondary prevention strategies should be applied to survivors of cardiac arrest that was not associated with an acute MI or other controllable transient risk factors, such as certain drug exposures and correctable electrolyte imbalances. Multivessel coronary artery disease and dilated cardiomyopathy, especially with markedly reduced left ventricular EF, predict a high risk of recurrence of cardiac arrest or SCD and are indications for specific interventions, such as ICDs (see below). The occurrence of otherwise unexplained syncope or documented life-threatening arrhythmias in patients with long QT syndromes or right ventricular dysplasia are also associated with increased risk of SCD.
CLINICAL CHARACTERISTICS OF CARDIAC ARREST
PRODROME, ONSET, ARREST, DEATH
SCD may be presaged by days to months of increasing angina, dyspnea, palpitations, easy fatigability, and other nonspecific complaints. However, these prodromal symptoms are generally predictive of any major cardiac event; they are not specific for predicting SCD.
The onset of the clinical transition, leading to cardiac arrest, is defined as an acute change in cardiovascular status preceding cardiac arrest by up to 1 h. When the onset is instantaneous or abrupt, the probability that the arrest is cardiac in origin is >95%. Continuous electrocardiogram (ECG) recordings fortuitously obtained at the onset of a cardiac arrest commonly demonstrate a tendency for the heart rate to increase and for advanced grades of PVCs to evolve during the minutes or hours before the event.
The probability of achieving successful resuscitation from cardiac arrest is related to the interval from onset of loss of circulation to return of spontaneous circulation (ROSC), the setting in which the event occurs, the mechanism (VF, VT, PEA, asystole), and the clinical status of the patient before the cardiac arrest. ROSC and survival rates as a result of defibrillation decrease almost linearly from the first minute to 10 min. After 4-5 min, survival rates are no better than 25–30% in out-of-hospital settings without bystander cardiopulmonary resuscitation (CPR). Those settings in which it is possible to institute prompt CPR followed by prompt defibrillation provide a better chance of a successful outcome. The outcome in intensive care units and other in-hospital environments is heavily influenced by the patient’s preceding clinical status. The immediate outcome is good for cardiac arrest occurring in the intensive care unit in the presence of an acute cardiac event or transient metabolic disturbance, but survival among patients with far-advanced chronic cardiac disease or advanced noncardiac diseases (e.g., renal failure, pneumonia, sepsis, diabetes, cancer) is low and not much better in the in-hospital setting. Survival rates after unexpected cardiac arrest in unmonitored areas in a hospital do not differ from witnessed out-of-hospital arrests. Since implementation of community response systems, survival from out-of-hospital cardiac arrest has improved, although it still remains low, under most circumstances. Survival probabilities in public sites exceed those in the home environment, where the majority of cardiac arrests occur.
The success rate for initial resuscitation and survival to hospital discharge after an out-of-hospital cardiac arrest depends heavily on the mechanism of the event. When the mechanism is pulseless VT, the outcome is best; VF is the next most successful; and asystole and PEA, now the most common mechanisms, generate dismal outcome statistics. Advanced age also adversely influences the chances of successful resuscitation.
The probability of progression to biologic death is a function of the mechanism of cardiac arrest and the length of the delay before interventions. VF without CPR within the first 4–6 min has a poor outcome even if defibrillation is successful because of secondary brain damage; the prompt interposition of bystander CPR (basic life support; see below) improves outcome at any point along the time scale, especially when followed by early successful defibrillation. However, there are few survivors among patients who had no life support activities for the first 8 min after onset. Evaluations of deployment of automatic external defibrillators (AEDs) in communities (e.g., police vehicles, large buildings, airports, and stadiums) are beginning to generate encouraging data, but the data for home deployment has been have been less impressive.
Death during the hospitalization after a successfully resuscitated cardiac arrest relates closely to the severity of central nervous system injury. Anoxic encephalopathy and infections subsequent to prolonged respirator dependence account for 60% of the deaths. Another 30% occur as a consequence of low cardiac output states that fail to respond to interventions. Recurrent arrhythmias are the least common cause of death, accounting for only 10% of in-hospital deaths.
In the setting of acute MI (Chap. 295), it is important to distinguish between primary and secondary cardiac arrests. Primary cardiac arrests are those that occur in the absence of hemodynamic instability, and secondary cardiac arrests are those that occur in patients in whom abnormal hemodynamics dominate the clinical picture before cardiac arrest. The success rate for immediate resuscitation in primary cardiac arrest during acute MI in a monitored setting should exceed 90%. In contrast, as many as 70% of patients with secondary cardiac arrest succumb immediately or during the same hospitalization.
|
TREATMENT |
CARDIAC ARREST |
An individual who collapses suddenly is managed in five stages: (1) initial evaluation and basic life support if cardiac arrest is confirmed, (2) public access defibrillation (when available), (3) advanced life support, (4) postresuscitation care, and (5) long-term management. The initial response, including confirmation of loss of circulation, followed by basic life support and public access defibrillation, can be carried out by physicians, nurses, paramedical personnel, and trained laypersons.
INITIAL EVALUATION AND BASIC LIFE SUPPORT
Confirmation that a sudden collapse with loss of consciousness (LOC) is due to a cardiac arrest includes prompt observations of the state of consciousness, respiratory movements, skin color, and the presence or absence of pulses in the carotid or femoral arteries. For lay responders, the pulse check is no longer recommended because it is unreliable. As soon as a cardiac arrest is suspected, confirmed, or even considered to be impending, calling an emergency rescue system (e.g., 911) is the immediate priority. With the development of AEDs that are easily used by nonconventional emergency responders, an additional layer for response has evolved (see below).
Careful attention to the respiratory status after abrupt LOC is important. Although normal breathing or tachypnea after LOC makes cardiac arrest less likely, gasping respiratory movements may persist during a true cardiac arrest, and their presence should not deter appropriate responses. In fact, continued gasping is considered a good prognostic sign for successful outcome. It is also important to observe for severe stridor with a persistent pulse as a clue to aspiration of a foreign body or food. If this is suspected, a Heimlich maneuver (see below) may dislodge the obstructing body. A precordial blow, or “thump,” delivered firmly with a clenched fist to the junction of the middle and lower thirds of the sternum may occasionally revert VT or VF, but there is concern about converting VT to VF. Therefore, it is recommended to use precordial thumps as a life support technique only when monitoring and defibrillation are available. This conservative application of the technique remains controversial.
The third action during the initial response is to clear the airway. The head is tilted back and the chin lifted so that the oropharynx can be explored to clear the airway. Dentures or foreign bodies are removed, and the Heimlich maneuver is performed if there is reason to suspect that a foreign body is lodged in the oropharynx. If respiratory arrest precipitating cardiac arrest is suspected, a second precordial thump is delivered after the airway is cleared.
Basic life support, more popularly known as CPR, is intended to maintain organ perfusion until definitive interventions can be instituted. The initial and primary element of CPR is maintenance of perfusion until spontaneous circulation can be restored. Closed chest cardiac compression maintains a pump function by sequential filling and emptying of the chambers, with competent valves maintaining forward direction of flow. The palm of one hand is placed over the lower sternum, with the heel of the other resting on the dorsum of the lower hand. The sternum is depressed, with the arms remaining straight, at a rate of 100 per minute. Sufficient force is applied to depress the sternum 4–5 cm, and relaxation is abrupt.
Until recently, providing ventilation of the lungs by mouth-to-mouth respiration was used if no specific rescue equipment was immediately available (e.g., plastic oropharyngeal airways, esophageal obturators, masked Ambu bag). However, ventilatory support during CPR has yielded to evidence that continuous chest compressions (“hands only” CPR) results in better outcomes. Compressions are interrupted only for single shocks from an AED when available, with 2 min of CPR between each single shock.
AUTOMATED EXTERNAL DEFIBRILLATION (AED)
AEDs that are easily used by nonconventional responders, such as nonparamedic firefighters, police officers, ambulance drivers, trained security guards, and minimally trained or untrained laypersons, have been developed. This advance has inserted another level of response into the cardiac arrest paradigm. A number of studies have demonstrated that AED use by nonconventional responders in strategic response systems and public access lay responders can improve cardiac arrest survival rates. The rapidity with which defibrillation/cardioversion is achieved is an important element for successful resuscitation, both for ROSC and for protection of the central nervous system. Chest compressions should be carried out while the defibrillator is being charged. As soon as a diagnosis of VF or VT is established, a biphasic waveform shock of 150–200 J (360 J if a monophasic waveform device is used) should be delivered. If 5 min has elapsed between collapse and first contact with the victim, there is some evidence that 60–90 s of CPR before the first shock may improve probability of survival without neurologic damage. If the initial shock does not successfully revert VT or VF, chest compression at a rate of 100 per minute is resumed for 2 min, and then a second shock is delivered. Multiple shocks given in sequence are no longer recommended, in order to minimize interruptions of chest compressions. This sequence is continued until personnel capable of, and equipped for, advanced life support are available, although not much data support the notion that shocks and chest compressions alone will revert VF after three shocks have failed.
ADVANCED CARDIAC LIFE SUPPORT (ACLS)
ACLS is intended to achieve and maintain organ perfusion and adequate ventilation, control cardiac arrhythmias, and stabilize blood pressure and cardiac output. The activities carried out to achieve these goals include (1) defibrillation/cardioversion and/or pacing, (2) intubation with an endotracheal tube, and (3) insertion of an intravenous line.
As in basic life support, the major emphasis during ACLS is minimizing interruptions of chest compressions until ROSC is achieved. After two or three unsuccessful defibrillation attempts, epinephrine, 1 mg IV, is given and attempts to defibrillate are repeated. The dose of epinephrine may be repeated after intervals of 3–5 min (Fig. 327-3A). Vasopressin (a single 40-unit dose given IV) has been suggested as an alternative to epinephrine.
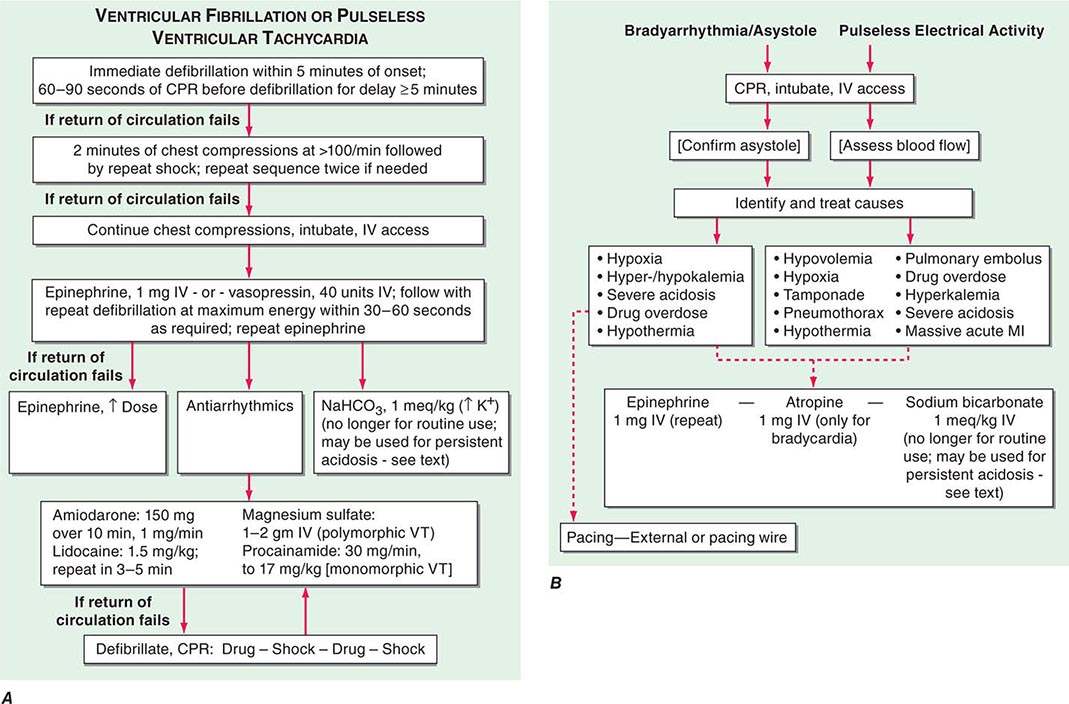
FIGURE 327-3 A. The algorithm of ventricular fibrillation or pulseless ventricular tachycardia begins with and initial defibrillate on attempt. If a single shock fails to restore a pulse, it is followed by 2 min of cardiopulmonary resuscitation (CPR; chest compressions), followed by another single shock. After three such sequences, epinephrine and then antiarrhythmic drugs are added to the protocol. See text for details. B. The algorithms for bradyarrhythmia/asystole (left) or pulseless electrical activity (right) are dominated first by continued life support and a search for reversible causes. Subsequent therapy is nonspecific and is accompanied by a low success rate. See text for details. MI, myocardial infarction; VT, ventricular tachycardia.
If the patient is less than fully conscious upon reversion or if two or three attempts fail, prompt intubation, ventilation, and arterial blood gas analysis should be carried out. Ventilation with O2 (room air if O2 is not immediately available) may promptly reverse hypoxemia and acidosis. Quantitative waveform capnography is now recommended for confirmation and monitoring of endotracheal tube placement. A patient who is persistently acidotic after successful defibrillation and intubation or had acidosis prior to arrest, may be given 1 meq/kg NaHCO3 initially and an additional 50% of the dose repeated every 10–15 min. However, it should not be used routinely.
After initial unsuccessful defibrillation attempts or with persistent/recurrent electrical instability, antiarrhythmic therapy should be instituted. Intravenous amiodarone has emerged as the initial treatment of choice (150 mg over 10 min, followed by 1 mg/min for up to 6 h and 0.5 mg/min thereafter) (Fig. 327-3A). For cardiac arrest due to VF in the early phase of an acute coronary syndrome, a bolus of 1 mg/kg of lidocaine may be given intravenously as an alternative, and the dose may be repeated in 2 min. It also may be tried in patients in whom amiodarone is unsuccessful. Intravenous procainamide (loading infusion of 100 mg/5 min to a total dose of 500–800 mg, followed by continuous infusion at 2–5 mg/min) is now rarely used in this setting but may be tried for persisting, hemodynamically stable arrhythmias. Intravenous calcium gluconate is no longer considered safe or necessary for routine administration. It is used only in patients in whom acute hyperkalemia is known to be the triggering event for resistant VF, in the presence of known hypocalcemia, or in patients who have received toxic doses of calcium channel antagonists.
Cardiac arrest due to bradyarrhythmias or asystole (B/A cardiac arrest) is managed differently (Fig. 327-3B). The patient is promptly intubated, CPR is continued, and an attempt is made to control hypoxemia and acidosis and identify other reversible causes. Epinephrine may be given intravenously or by an intraosseous route. Atropine is no longer considered effective for asystole or PEA, but can be used for bradyarrhythmias. External pacing devices are used to attempt to establish a regular rhythm when atropine fails for a bradyarrhythmia, but chronotropic agents given intravenously are now recognized as an equally effective alternative.
The success rate may be good when B/A arrest is due to acute inferior wall MI or to correctable airway obstruction or drug-induced respiratory depression or with prompt resuscitation efforts. For acute airway obstruction, prompt removal of foreign bodies by the Heimlich maneuver or, in hospitalized patients, by intubation and suctioning of obstructing secretions in the airway is often successful. The prognosis is generally very poor in other causes of this form of cardiac arrest, such as end-stage cardiac or noncardiac diseases. Treatment of PEA is similar to that for bradyarrhythmias, but its outcome is also dismal.
POST-CARDIAC ARREST SYNDROME AND POSTRESUSCITATION CARE
After return of spontaneous or stable assisted circulation, attention shifts to the diagnostic and therapeutic elements of the post-cardiac arrest syndrome. This recently developed clinical classification emerged from the organization of the elements of injury following cardiac arrest into a multidisciplinary continuum. The four components of post-cardiac arrest syndrome include brain injury, myocardial dysfunction, systemic ischemia/reperfusion responses, and control of persistent precipitating factors. The therapeutic goal is to maintain a stable electrical, hemodynamic, and central nervous system status.
Postresuscitation care is determined by the specific clinical circumstances. The most pressing is the presence of anoxic encephalopathy, which is a strong predictor of in-hospital death and postarrest disability. Mild therapeutic hypothermia is indicated for resuscitated cardiac arrest victims who are hemodynamically stable, but remain comatose. Core body temperature is decreased to 32–34°C, by several available techniques (external and/or internal [core]), as soon as practical after resuscitation and maintained for a minimum of 12–24 h. By reducing metabolic demands and cerebral edema, this intervention improves probability of survival with better neurologic outcome.
Primary VF in acute MI (not accompanied by low-output states) (Chap. 295) is generally very responsive to life support techniques and easily controlled after the initial event. In the in-hospital setting, respirator support is usually not necessary or is needed for only a short time, and hemodynamics stabilize promptly after defibrillation or cardioversion. In secondary VF in acute MI (those events in which hemodynamic abnormalities predispose to the potentially fatal arrhythmia), resuscitative efforts are less often successful, and in patients who are successfully resuscitated, the recurrence rate is high. The clinical picture and outcome are dominated by hemodynamic instability and the ability to control hemodynamic dysfunction. Bradyarrhythmias, asystole, and PEA are commonly secondary events in hemodynamically unstable patients.
The outcome after in-hospital cardiac arrest associated with noncardiac diseases is poor, and in the few successfully resuscitated patients, the postresuscitation course is dominated by the nature of the underlying disease. Patients with end-stage cancer, renal failure, acute central nervous system disease, and uncontrolled infections, as a group, have a survival rate of <10% after in-hospital cardiac arrest. Some major exceptions are patients with transient airway obstruction, electrolyte disturbances, proarrhythmic effects of drugs, and severe metabolic abnormalities, most of whom may have a good chance of survival if they can be resuscitated promptly and stabilized while the transient abnormalities are being corrected.
LONG-TERM MANAGEMENT AFTER SURVIVAL OF OUT-OF-HOSPITAL CARDIAC ARREST
Patients who survive cardiac arrest without irreversible damage to the central nervous system and who achieve hemodynamic stability should have diagnostic testing to define appropriate therapeutic interventions for their long-term management. This approach is driven by the fact that survival after out-of-hospital cardiac arrest is followed by a 10–25% mortality rate during the first 2 years after the event, and there are data suggesting that significant survival benefits can be achieved by prescription of an ICD.
Among patients in whom an acute ST elevation MI or transient and reversible myocardial ischemia is identified as the specific mechanism triggering an out-of-hospital cardiac arrest, the management is dictated in part by the transient nature of life-threatening arrhythmia risk during the acute coronary syndrome (ACS) and in part by the extent of permanent myocardial damage that results. Cardiac arrest during the acute ischemic phase is not an ICD indication, but survivors of cardiac arrest not associated with an ACS do benefit. In addition, patients who survive MI with an EF less than 30–35% appear to benefit from ICDs.
For patients with cardiac arrest determined to be due to a treatable transient ischemic mechanism, particularly with higher EFs, catheter interventional, surgical, and/or pharmacologic anti-ischemic therapy is generally accepted for long-term management.
Survivors of cardiac arrest due to other categories of disease, such as the hypertrophic or dilated cardiomyopathies and the various rare inherited disorders (e.g., right ventricular dysplasia, long QT syndrome, Brugada syndrome, catecholaminergic polymorphic VT, and so-called idiopathic VF), are all considered ICD candidates.
PREVENTION OF SCD IN HIGH-RISK INDIVIDUALS WITHOUT PRIOR CARDIAC ARREST
Post-MI patients with EFs <35% and other markers of risk such as ambient ventricular arrhythmias, inducible ventricular tachyarrhythmias in the electrophysiology laboratory, and a history of heart failure are considered candidates for ICDs 40 days or more after the MI. Total mortality benefits in the range of a 20–35% reduction over 2–5 years have been observed in a series of clinical trials. One study suggested that an EF <30% was a sufficient marker of risk to indicate ICD benefit, and another demonstrated benefit for patients with Functional Class 2 or 3 heart failure and EFs ≤35%, regardless of etiology (ischemic or nonischemic) or the presence of ambient or induced arrhythmias (Chaps. 277 and 279). For patients with newly diagnosed heart failure and an EF <35%, the required delay between diagnosis and institution of medical therapy, and subsequent implantation of an ICD, is 90 days. In general, there appears to be a gradient of increasing ICD benefit with EFs ranging lower than the threshold indications. However, patients with very low EFs (e.g., <20%) may receive less benefit.
Decision making for primary prevention in disorders other than coronary artery disease and dilated cardiomyopathy is generally driven by observational data and judgment based on clinical observations. Controlled clinical trials providing evidence-based indicators for ICDs are lacking for these smaller population subgroups. In general, for the rare disorders listed above, indicators of arrhythmic risk such as syncope, documented ventricular tachyarrhythmias, aborted cardiac arrest, or a family history of premature SCD in some conditions, and a number of other clinical or ECG markers, may be used as indicators for ICDs.
SECTION 3 |
NEUROLOGIC CRITICAL CARE |
328 |
Coma |
Coma is among the most common and striking problems in general medicine. It accounts for a substantial portion of admissions to emergency wards and occurs on all hospital services. It demands immediate attention and requires an organized approach.
There is a continuum of states of reduced alertness, the most severe form being coma, defined as a deep sleeplike state from which the patient cannot be aroused. Stupor refers to a higher degree of arousability in which the patient can be transiently awakened by vigorous stimuli, accompanied by motor behavior that leads to avoidance of uncomfortable or aggravating stimuli. Drowsiness, which is familiar to all persons, simulates light sleep and is characterized by easy arousal and the persistence of alertness for brief periods. Drowsiness and stupor are usually accompanied by some degree of confusion (Chap. 34). A precise narrative description of the level of arousal and of the type of responses evoked by various stimuli as observed at the bedside is preferable to ambiguous terms such as lethargy, semicoma, or obtundation.
Several conditions that render patients unresponsive and simulate coma are considered separately because of their special significance. The vegetative state signifies an awake-appearing but nonresponsive state in a patient who has emerged from coma. In the vegetative state, the eyelids may open, giving the appearance of wakefulness. Respiratory and autonomic functions are retained. Yawning, coughing, swallowing, and limb and head movements persist, and the patient may follow visually presented objects, but there are few, if any, meaningful responses to the external and internal environment—in essence, an “awake coma.” The term vegetative is unfortunate because it is subject to misinterpretation. There are always accompanying signs that indicate extensive damage in both cerebral hemispheres, e.g., decerebrate or decorticate limb posturing and absent responses to visual stimuli (see below). In the closely related but less severe minimally conscious state, the patient displays rudimentary vocal or motor behaviors, often spontaneous, but some in response to touch, visual stimuli, or command. Cardiac arrest with cerebral hypoperfusion and head injuries are the most common causes of the vegetative and minimally conscious states (Chaps. 327 and 330). The prognosis for regaining mental faculties once the vegetative state has supervened for several months is very poor, and after a year, almost nil; hence the term persistent vegetative state. Most reports of dramatic recovery, when investigated carefully, are found to yield to the usual rules for prognosis, but there have been rare instances in which recovery has occurred to a severely disabled condition and, in rare childhood cases, to an even better state. The possibility of incorrectly attributing meaningful behavior to patients in the vegetative and minimally conscious states creates inordinate problems and anguish. On the other hand, the question of whether these patients lack any capability for cognition has been reopened by functional imaging studies that have demonstrated, in a small proportion of posttraumatic cases, meaningful cerebral activation in response to verbal and other stimuli.
Apart from the above conditions, several syndromes that affect alertness are prone to be misinterpreted as stupor or coma. Akinetic mutism refers to a partially or fully awake state in which the patient is able to form impressions and think, as demonstrated by later recounting of events, but remains virtually immobile and mute. The condition results from damage in the regions of the medial thalamic nuclei or the frontal lobes (particularly lesions situated deeply or on the orbitofrontal surfaces) or from extreme hydrocephalus. The term abulia describes a milder form of akinetic mutism characterized by mental and physical slowness and diminished ability to initiate activity. It is also usually the result of damage to the frontal lobes and its connections (Chap. 36).
Catatonia is a curious hypomobile and mute syndrome that occurs as part of a major psychosis, usually schizophrenia or major depression. Catatonic patients make few voluntary or responsive movements, although they blink, swallow, and may not appear distressed. There are nonetheless signs that the patient is responsive, although it may take ingenuity on the part of the examiner to demonstrate them. For example, eyelid elevation is actively resisted, blinking occurs in response to a visual threat, and the eyes move concomitantly with head rotation, all of which are inconsistent with the presence of a brain lesion causing unresponsiveness. It is characteristic but not invariable in catatonia for the limbs to retain the postures in which they have been placed by the examiner (“waxy flexibility,” or catalepsy). With recovery, patients often have some memory of events that occurred during their catatonic stupor. Catatonia is superficially similar to akinetic mutism, but clinical evidence of cerebral damage such as Babinski signs and hypertonicity of the limbs is lacking. The special problem of coma in brain death is discussed below.
The locked-in state describes yet another type of pseudocoma in which an awake patient has no means of producing speech or volitional movement but retains voluntary vertical eye movements and lid elevation, thus allowing the patient to signal with a clear mind. The pupils are normally reactive. Such individuals have written entire treatises using Morse code. The usual cause is an infarction or hemorrhage of the ventral pons that transects all descending motor (corticospinal and corticobulbar) pathways. A similar awake but de-efferented state occurs as a result of total paralysis of the musculature in severe cases of Guillain-Barré syndrome (Chap. 460), critical illness neuropathy (Chap. 330), and pharmacologic neuromuscular blockade.
THE ANATOMY AND PHYSIOLOGY OF COMA
Almost all instances of diminished alertness can be traced to widespread abnormalities of the cerebral hemispheres or to reduced activity of a special thalamocortical alerting system termed the reticular activating system (RAS). The proper functioning of this system, its ascending projections to the cortex, and the cortex itself are required to maintain alertness and coherence of thought. It follows that the principal causes of coma are (1) lesions that damage the RAS in the upper midbrain or its projections; (2) destruction of large portions of both cerebral hemispheres; or (3) suppression of reticulocerebral function by drugs, toxins, or metabolic derangements such as hypoglycemia, anoxia, uremia, and hepatic failure.
The proximity of the RAS to midbrain structures that control pupillary function and eye movements permits clinical localization of the cause of coma in many cases. Pupillary enlargement with loss of light reaction and loss of vertical and adduction movements of the eyes suggests that the lesion is in the upper brainstem where the nuclei subserving these functions reside. Conversely, preservation of pupillary light reactivity and of eye movements absolves the upper brainstem and indicates that widespread structural lesions or metabolic suppression of the cerebral hemispheres is responsible for coma.
Coma Due to Cerebral Mass Lesions and Herniations In addition to the fixed restriction of the skull, the cranial cavity is separated into compartments by infoldings of the dura. The two cerebral hemispheres are separated by the falx, and the anterior and posterior fossae by the tentorium. Herniation refers to displacement of brain tissue by an overlying or adjacent mass into a contiguous compartment that it normally does not occupy. Coma and many of its associated signs can be attributed to these tissue shifts, and certain clinical features are characteristic of specific configurations of herniation (Fig. 328-1). They are in essence “false localizing” signs because they derive from compression of brain structures at a distance from the mass.
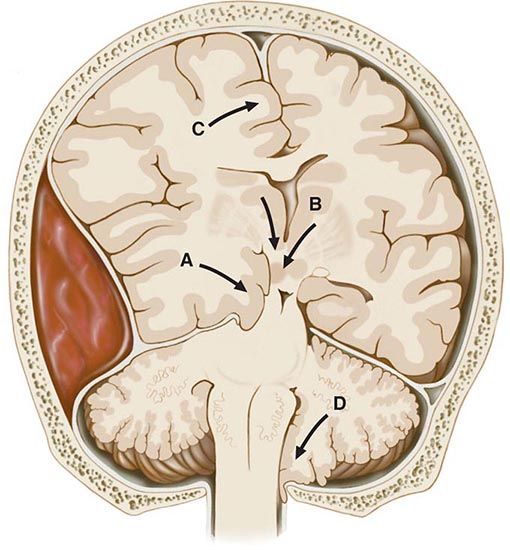
FIGURE 328-1 Types of cerebral herniation: (A) uncal; (B) central; (C) transfalcial; and (D) foraminal.
In the most common form of herniation, brain tissue is displaced from the supratentorial to the infratentorial compartment through the tentorial opening; this is referred to as transtentorial herniation. Uncal transtentorial herniation refers to impaction of the anterior medial temporal gyrus (the uncus) into the tentorial opening just anterior to and adjacent to the midbrain (Fig. 328-1A). The uncus compresses the third nerve as the nerve traverses the subarachnoid space, causing enlargement of the ipsilateral pupil (the fibers subserving parasympathetic pupillary function are located peripherally in the nerve). The coma that follows is due to compression of the midbrain against the opposite tentorial edge by the displaced parahippocampal gyrus (Fig. 328-2). Lateral displacement of the midbrain may compress the opposite cerebral peduncle against the tentorial edge, producing a Babinski sign and hemiparesis contralateral to the hemiparesis that resulted from the mass (the Kernohan-Woltman sign). Herniation may also compress the anterior and posterior cerebral arteries as they pass over the tentorial reflections, with resultant brain infarction. The distortions may also entrap portions of the ventricular system, resulting in hydrocephalus.
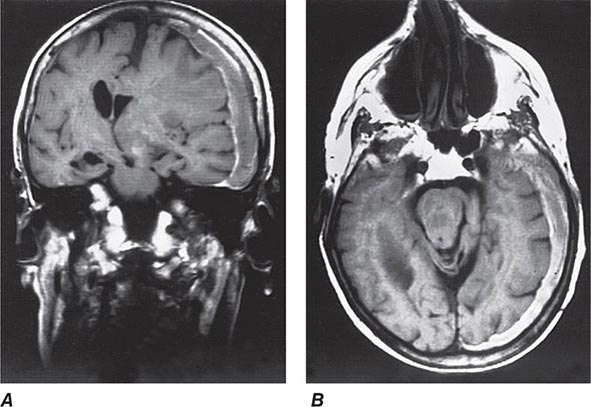
FIGURE 328-2 Coronal (A) and axial (B) magnetic resonance images from a stuporous patient with a left third nerve palsy as a result of a large left-sided subdural hematoma (seen as a gray-white rim). The upper midbrain and lower thalamic regions are compressed and displaced horizontally away from the mass, and there is transtentorial herniation of the medial temporal lobe structures, including the uncus anteriorly. The lateral ventricle opposite to the hematoma has become enlarged as a result of compression of the third ventricle.
Central transtentorial herniation denotes a symmetric downward movement of the thalamic structures through the tentorial opening with compression of the upper midbrain (Fig. 328-1B). Miotic pupils and drowsiness are the heralding signs, in contrast to a unilaterally enlarged pupil of the uncal syndrome. Both uncal and central transtentorial herniations cause progressive compression of the brainstem, with initial damage to the midbrain, then the pons, and finally the medulla. The result is an approximate sequence of neurologic signs that corresponds to each affected level. Other forms of herniation are transfalcial herniation (displacement of the cingulate gyrus under the falx and across the midline, Fig. 328-1C) and foraminal herniation (downward forcing of the cerebellar tonsils into the foramen magnum, Fig. 328-1D), which causes compression of the medulla, respiratory arrest, and death.
A direct relationship between the various configurations of transtentorial herniation and coma is not always found. Drowsiness and stupor can occur with moderate horizontal displacement of the diencephalon (thalamus), before transtentorial herniation is evident. This lateral shift may be quantified on axial images of computed tomography (CT) and magnetic resonance imaging (MRI) scans (Fig. 328-2). In cases of acutely enlarging masses, horizontal displacement of the pineal calcification of 3–5 mm is generally associated with drowsiness, 6–8 mm with stupor, and >9 mm with coma. Intrusion of the medial temporal lobe into the tentorial opening is also apparent on MRI and CT scans as obliteration of the cisterna that surrounds the upper brainstem.
Coma due to Metabolic Disorders Many systemic metabolic abnormalities cause coma by interrupting the delivery of energy substrates (e.g., oxygen, glucose) or by altering neuronal excitability (drugs and alcohol, anesthesia, and epilepsy). The metabolic abnormalities that produce coma may, in milder forms, induce an acute confusional state. Thus, in metabolic encephalopathies, clouded consciousness and coma are in a continuum.
Cerebral neurons are fully dependent on cerebral blood flow (CBF) and the delivery of oxygen and glucose. CBF is ~75 mL per 100 g/min in gray matter and 30 mL per 100 g/min in white matter (mean ~55 mL per 100 g/min); oxygen consumption is 3.5 mL per 100 g/min, and glucose utilization is 5 mg per 100 g/min. Brain stores of glucose are able to provide energy for ~2 min after blood flow is interrupted, and oxygen stores last 8–10 s after the cessation of blood flow. Simultaneous hypoxia and ischemia exhaust glucose more rapidly. The electroencephalogram (EEG) rhythm in these circumstances becomes diffusely slowed, typical of metabolic encephalopathies, and as substrate delivery worsens, eventually brain electrical activity ceases.
Unlike hypoxia-ischemia, which causes neuronal destruction, most metabolic disorders such as hypoglycemia, hyponatremia, hyperosmolarity, hypercapnia, hypercalcemia, and hepatic and renal failure cause only minor neuropathologic changes. The reversible effects of these conditions on the brain are not understood but may result from impaired energy supplies, changes in ion fluxes across neuronal membranes, and neurotransmitter abnormalities. For example, the high ammonia concentration of hepatic coma interferes with cerebral energy metabolism and with the Na+, K+-ATPase pump, increases the number and size of astrocytes, and causes increased concentrations of potentially toxic products of ammonia metabolism; it may also affect neurotransmitters, including the production of putative “false” neurotransmitters that are active at receptor sites. Apart from hyperammonemia, which of these mechanisms is of critical importance is not clear. The mechanism of the encephalopathy of renal failure is also not known. Unlike ammonia, urea does not produce central nervous system (CNS) toxicity, and a multifactorial causation has been proposed for the encephalopathy, including increased permeability of the blood-brain barrier to toxic substances such as organic acids and an increase in brain calcium and cerebrospinal fluid (CSF) phosphate content.
Coma and seizures are common accompaniments of large shifts in sodium and water balance in the brain. These changes in osmolarity arise from systemic medical disorders, including diabetic ketoacidosis, the nonketotic hyperosmolar state, and hyponatremia from any cause (e.g., water intoxication, excessive secretion of antidiuretic hormone, or atrial natriuretic peptides). Sodium levels <125 mmol/L induce confusion, and levels <115 mmol/L are typically associated with coma and convulsions. In hyperosmolar coma, the serum osmolarity is generally >350 mosmol/L. Hypercapnia depresses the level of consciousness in proportion to the rise in carbon dioxide (CO2) tension in the blood. In all of these metabolic encephalopathies, the degree of neurologic change depends to a large extent on the rapidity with which the serum changes occur. The pathophysiology of other metabolic encephalopathies such as those due to hypercalcemia, hypothyroidism, vitamin B12 deficiency, and hypothermia are incompletely understood but must reflect derangements of CNS biochemistry, membrane function, or neurotransmitters.
Epileptic Coma Generalized electrical seizures are associated with coma, even in the absence of motor convulsions (nonconvulsive status epilepticus). The self-limited coma that follows a seizure, the postictal state, may be due to exhaustion of energy reserves or effects of locally toxic molecules that are the by-product of seizures. The postictal state produces continuous, generalized slowing of the background EEG activity similar to that of metabolic encephalopathies.
Toxic (Including Drug-Induced) Coma This common class of encephalopathy is in large measure reversible and leaves no residual damage provided there has not been cardiorespiratory failure. Many drugs and toxins are capable of depressing nervous system function. Some produce coma by affecting both the brainstem nuclei, including the RAS, and the cerebral cortex. The combination of cortical and brainstem signs, which occurs in certain drug overdoses, may lead to an incorrect diagnosis of structural brainstem disease. Overdose of medications that have atropinic actions produces signs such as dilated pupils, tachycardia, and dry skin; opiate overdose produces pinpoint pupils <1 mm in diameter.
Coma due to Widespread Damage to the Cerebral Hemispheres This category, comprising a number of unrelated disorders, results from widespread structural cerebral damage that simulates a metabolic disorder of the cortex. Hypoxia-ischemia is perhaps the best characterized and one in which it is not possible initially to distinguish the acute reversible effects of oxygen deprivation of the brain from the subsequent effects of anoxic neuronal damage. Similar widespread cerebral damage may be produced by disorders that occlude small blood vessels throughout the brain; examples include cerebral malaria, thrombotic thrombocytopenic purpura, and hyperviscosity. Diffuse white matter damage from cranial trauma or inflammatory demyelinating diseases can cause a similar coma syndrome.
APPROACH TO THE PATIENT:
Coma
A video examination of the comatose patient is shown in Chap. 329e.
Acute respiratory and cardiovascular problems should be attended to prior to neurologic assessment. In most instances, a complete medical evaluation, except for vital signs, funduscopy, and examination for nuchal rigidity, may be deferred until the neurologic evaluation has established the severity and nature of coma. The approach to the patient with coma from cranial trauma is discussed in Chap. 457e.
HISTORY
The cause of coma may be immediately evident as in cases of trauma, cardiac arrest, or observed drug ingestion. In the remainder, certain points are useful: (1) the circumstances and rapidity with which neurologic symptoms developed; (2) the antecedent symptoms (confusion, weakness, headache, fever, seizures, dizziness, double vision, or vomiting); (3) the use of medications, drugs, or alcohol; and (4) chronic liver, kidney, lung, heart, or other medical disease. Direct interrogation of family, observers, and ambulance technicians on the scene, in person or by telephone, is an important part of the evaluation when possible.
GENERAL PHYSICAL EXAMINATION
Fever suggests a systemic infection, bacterial meningitis, encephalitis, heat stroke, neuroleptic malignant syndrome, malignant hyperthermia due to anesthetics, or anticholinergic drug intoxication. Only rarely is fever attributable to a lesion that has disturbed hypothalamic temperature-regulating centers (“central fever”). A slight elevation in temperature may follow vigorous convulsions. Hypothermia is observed with exposure that attends alcohol, barbiturate, sedative, or phenothiazine intoxication; hypoglycemia; peripheral circulatory failure; or extreme hypothyroidism. Hypothermia itself causes coma when the temperature is <31°C (87.8°F). Tachypnea may indicate systemic acidosis or pneumonia or, rarely, infiltration of the brain with lymphoma. Aberrant respiratory patterns that reflect brainstem disorders are discussed below. Marked hypertension suggests hypertensive encephalopathy or cerebral hemorrhage or head injury. Hypotension is characteristic of coma from alcohol or barbiturate intoxication, internal hemorrhage, myocardial infarction, sepsis, profound hypothyroidism, or Addisonian crisis. The funduscopic examination can detect subarachnoid hemorrhage (subhyaloid hemorrhages), hypertensive encephalopathy (exudates, hemorrhages, vessel-crossing changes, papilledema), and increased intracranial pressure (ICP) (papilledema). Cutaneous petechiae suggest thrombotic thrombocytopenic purpura, meningococcemia, or a bleeding diathesis associated with an intracerebral hemorrhage. Cyanosis and reddish or anemic skin coloration are other indications of an underlying systemic disease or carbon monoxide as responsible for the coma.
NEUROLOGIC EXAMINATION
The patient should be observed without intervention by the examiner. Tossing about in the bed, reaching up toward the face, crossing legs, yawning, swallowing, coughing, or moaning reflect a drowsy state that is close to normal awakeness. Lack of restless movements on one side or an outturned leg suggests a hemiplegia. Intermittent twitching movements of a foot, finger, or facial muscle may be the only sign of seizures. Multifocal myoclonus almost always indicates a metabolic disorder, particularly uremia, anoxia, drug intoxication (especially with lithium or haloperidol), or a prion disease (Chap. 453e). In a drowsy and confused patient, bilateral asterixis is a certain sign of metabolic encephalopathy or drug intoxication.
Decorticate rigidity and decerebrate rigidity, or “posturing,” describe stereotyped arm and leg movements occurring spontaneously or elicited by sensory stimulation. Flexion of the elbows and wrists and supination of the arm (decorticate posturing) suggests bilateral damage rostral to the midbrain, whereas extension of the elbows and wrists with pronation (decerebrate posturing) indicates damage to motor tracts in the midbrain or caudal diencephalon. The less frequent combination of arm extension with leg flexion or flaccid legs is associated with lesions in the pons. These concepts have been adapted from animal work and cannot be applied with precision to coma in humans. In fact, acute and widespread disorders of any type, regardless of location, frequently cause limb extension, and almost all extensor posturing becomes predominantly flexor as time passes.
LEVEL OF AROUSAL
A sequence of increasingly intense stimuli is used to determine the threshold for arousal and the motor response of each side of the body. The results of testing may vary from minute to minute, and serial examinations are useful. Tickling the nostrils with a cotton wisp is a moderate stimulus to arousal—all but deeply stuporous and comatose patients will move the head away and arouse to some degree. An even greater degree of responsiveness is present if the patient uses his hand to remove an offending stimulus. Pressure on the knuckles or bony prominences and pinprick stimulation are humane forms of noxious stimuli; pinching the skin causes unsightly ecchymoses and is generally not necessary but may be useful in eliciting abduction withdrawal movements of the limbs. Posturing in response to noxious stimuli indicates severe damage to the corticospinal system, whereas abduction-avoidance movement of a limb is usually purposeful and denotes an intact corticospinal system. Posturing may also be unilateral and coexist with purposeful limb movements, reflecting incomplete damage to the motor system.
BRAINSTEM REFLEXES
Assessment of brainstem function is essential to localization of the lesion in coma (Fig. 328-3). The brainstem reflexes that are examined are pupillary size and reaction to light, spontaneous and elicited eye movements, corneal responses, and the respiratory pattern. As a rule, coma due to bilateral hemispheral disease preserves these brainstem activities, particularly the pupillary reactions and eye movements. However, the presence of abnormal brainstem signs does not always indicate that the primary lesion is in the brainstem because hemispheral masses can cause secondary brainstem damage by the earlier described transtentorial herniations.
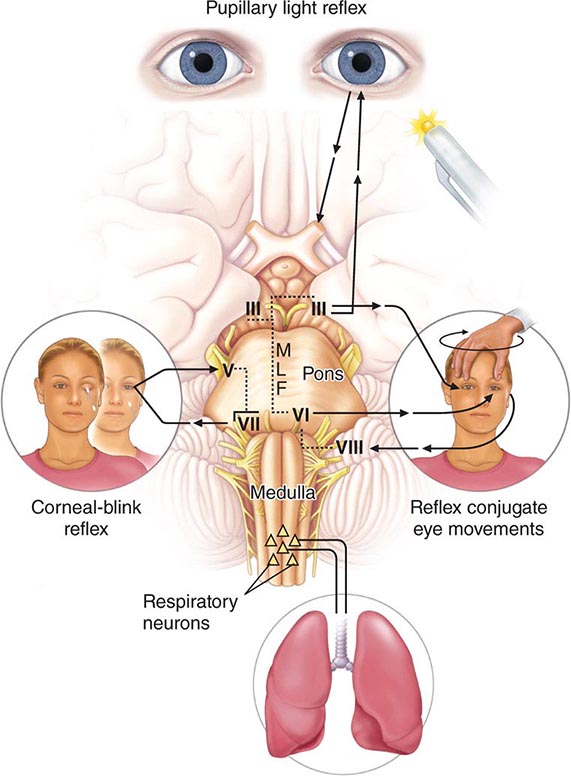
FIGURE 328-3 Examination of brainstem reflexes in coma. Midbrain and third nerve function are tested by pupillary reaction to light, pontine function by spontaneous and reflex eye movements and corneal responses, and medullary function by respiratory and pharyngeal responses. Reflex conjugate, horizontal eye movements are dependent on the medial longitudinal fasciculus (MLF) interconnecting the sixth and contralateral third nerve nuclei. Head rotation (oculocephalic reflex) or caloric stimulation of the labyrinths (oculovestibular reflex) elicits contraversive eye movements (for details see text).
Pupillary Signs Pupillary reactions are examined with a bright, diffuse light (preferably not an ophthalmoscope, which illuminates only a limited part of the retina). Reactive and round pupils of midsize (2.5–5 mm) essentially exclude midbrain damage, either primary or secondary to compression. A response to light may be difficult to appreciate in pupils <2 mm in diameter, and bright room lighting mutes pupillary reactivity. One enlarged and poorly reactive pupil (>6 mm) signifies compression or stretching of the third nerve from the effects of a cerebral mass above. Enlargement of the pupil contralateral to a hemispheral mass may occur but is infrequent. An oval and slightly eccentric pupil is a transitional sign that accompanies early midbrain–third nerve compression. The most extreme pupillary sign, bilaterally dilated and unreactive pupils, indicates severe midbrain damage, usually from compression by a supratentorial mass. Ingestion of drugs with anticholinergic activity, the use of mydriatic eye drops, and direct ocular trauma are among the causes of misleading pupillary enlargement.
Unilateral miosis in coma has been attributed to dysfunction of sympathetic efferents originating in the posterior hypothalamus and descending in the tegmentum of the brainstem to the cervical cord. It is therefore of limited localizing value but is an occasional finding in patients with a large cerebral hemorrhage that affects the thalamus. Reactive and bilaterally small (1–2.5 mm) but not pinpoint pupils are seen in metabolic encephalopathies or in deep bilateral hemispheral lesions such as hydrocephalus or thalamic hemorrhage. Even smaller reactive pupils (<1 mm) characterize narcotic or barbiturate overdoses but also occur with extensive pontine hemorrhage. The response to naloxone and the presence of reflex eye movements (see below) assist in distinguishing between these.
Ocular Movements The eyes are first observed by elevating the lids and observing the resting position and spontaneous movements of the globes. Lid tone, tested by lifting the eyelids and noting their resistance to opening and the speed of closure, is progressively reduced as unresponsiveness progresses. Horizontal divergence of the eyes at rest is normal in drowsiness. As coma deepens, the ocular axes may become parallel again.
Spontaneous eye movements in coma often take the form of conjugate horizontal roving. This finding alone exonerates damage in the midbrain and pons and has the same significance as normal reflex eye movements (see below). Conjugate horizontal ocular deviation to one side indicates damage to the pons on the opposite side or, alternatively, to the frontal lobe on the same side. This phenomenon is summarized by the following maxim: The eyes look toward a hemispheral lesion and away from a brainstem lesion. Seizures also drive the eyes to one side but usually with superimposed clonic movements of the globes. The eyes may occasionally turn paradoxically away from the side of a deep hemispheral lesion (“wrong-way eyes”). The eyes turn down and inward with thalamic and upper midbrain lesions, typically thalamic hemorrhage. “Ocular bobbing” describes brisk downward and slow upward movements of the eyes associated with loss of horizontal eye movements and is diagnostic of bilateral pontine damage, usually from thrombosis of the basilar artery. “Ocular dipping” is a slower, arrhythmic downward movement followed by a faster upward movement in patients with normal reflex horizontal gaze; it usually indicates diffuse cortical anoxic damage.
The oculocephalic reflexes, elicited by moving the head from side to side or vertically and observing eye movements in the direction opposite to the head movement, depend on the integrity of the ocular motor nuclei and their interconnecting tracts that extend from the midbrain to the pons and medulla (Fig. 328-3). The movements, called somewhat inappropriately “doll’s eyes” (which refers more accurately to the reflex elevation of the eyelids with flexion of the neck), are normally suppressed in the awake patient. The ability to elicit them therefore reflects both reduced cortical influence on the brainstem and intact brainstem pathways, indicating that coma is caused by a lesion or dysfunction in the cerebral hemispheres. The opposite, an absence of reflex eye movements, usually signifies damage within the brainstem but can result from overdoses of certain drugs. In this circumstance, normal pupillary size and light reaction distinguishes most drug-induced comas from structural brainstem damage.
Thermal, or “caloric,” stimulation of the vestibular apparatus (oculovestibular response) provides a more intense stimulus for the oculocephalic reflex but provides essentially the same information. The test is performed by irrigating the external auditory canal with cool water in order to induce convection currents in the labyrinths. After a brief latency, the result is tonic deviation of both eyes to the side of cool-water irrigation and nystagmus in the opposite direction. (The acronym “COWS” has been used to remind generations of medical students of the direction of nystagmus—“cold water opposite, warm water same.”) The loss of induced conjugate ocular movements indicates brainstem damage. The presence of corrective nystagmus indicates that the frontal lobes are functioning and connected to the brainstem; thus catatonia or hysterical coma is likely.
By touching the cornea with a wisp of cotton, a response consisting of brief bilateral lid closure is normally observed. The corneal reflex depends on the integrity of pontine pathways between the fifth (afferent) and both seventh (efferent) cranial nerves; in conjunction with reflex eye movements, it is a useful test of pontine function. CNS-depressant drugs diminish or eliminate the corneal responses soon after reflex eye movements are paralyzed but before the pupils become unreactive to light. The corneal (and pharyngeal) response may be lost for a time on the side of an acute hemiplegia.
Respiratory Patterns These are of less localizing value in comparison to other brainstem signs. Shallow, slow, but regular breathing suggests metabolic or drug depression. Cheyne-Stokes respiration in its typical cyclic form, ending with a brief apneic period, signifies bihemispheral damage or metabolic suppression and commonly accompanies light coma. Rapid, deep (Kussmaul) breathing usually implies metabolic acidosis but may also occur with pontomesencephalic lesions. Agonal gasps are the result of lower brainstem (medullary) damage and are recognized as the terminal respiratory pattern of severe brain damage. A number of other cyclic breathing variations have been described but are of lesser significance.
LABORATORY STUDIES AND IMAGING
The studies that are most useful in the diagnosis of coma are chemical-toxicologic analysis of blood and urine, cranial CT or MRI, EEG, and CSF examination. Arterial blood gas analysis is helpful in patients with lung disease and acid-base disorders. The metabolic aberrations commonly encountered in clinical practice are usually exposed by measurement of electrolytes, glucose, calcium, osmolarity, and renal (blood urea nitrogen) and hepatic (NH3) function. Toxicologic analysis may be necessary in any case of acute coma where the diagnosis is not immediately clear. However, the presence of exogenous drugs or toxins, especially alcohol, does not exclude the possibility that other factors, particularly head trauma, are also contributing to the clinical state. An ethanol level of 43 mmol/L (0.2 g/dL) in nonhabituated patients generally causes impaired mental activity; a level of >65 mmol/L (0.3 g/dL) is associated with stupor. The development of tolerance may allow the chronic alcoholic to remain awake at levels >87 mmol/L (0.4 g/dL).
The availability of CT and MRI has focused attention on causes of coma that are detectable by imaging (e.g., hemorrhage, tumor, or hydrocephalus). Resorting primarily to this approach, although at times expedient, is imprudent because most cases of coma (and confusion) are metabolic or toxic in origin. Furthermore, the notion that a normal CT scan excludes an anatomic lesion as the cause of coma is erroneous. Bilateral hemisphere infarction, acute brainstem infarction, encephalitis, meningitis, mechanical shearing of axons as a result of closed head trauma, sagittal sinus thrombosis, and subdural hematoma isodense to adjacent brain are some of the disorders that may not be detected. Nevertheless, if the source of coma remains unknown, a scan should be obtained.
The EEG (Chap. 442e) is useful in metabolic or drug-induced states but is rarely diagnostic. However, it is the essential test to reveal coma that is due to clinically unrecognized, nonconvulsive seizures, and shows fairly characteristic patterns in herpesvirus encephalitis and prion (Creutzfeldt-Jakob) disease. The EEG may be further helpful in disclosing generalized slowing of the background activity, a reflection of the severity of an encephalopathy. Predominant high-voltage slowing (δ or triphasic waves) in the frontal regions is typical of metabolic coma, as from hepatic failure, and widespread fast (β) activity implicates sedative drugs (e.g., benzodiazepines). A special pattern of “alpha coma,” defined by widespread, variable 8- to 12-Hz activity, superficially resembles the normal α rhythm of waking but, unlike normal α activity, is not altered by environmental stimuli. Alpha coma results from pontine or diffuse cortical damage and is associated with a poor prognosis. Normal α activity on the EEG, which is suppressed by stimulating the patient, also alerts the clinician to the locked-in syndrome or to hysteria or catatonia. Still, the most important use of EEG recordings in coma is to reveal clinically inapparent epileptic discharges.
Lumbar puncture is performed less frequently than in the past for coma diagnosis because neuroimaging effectively excludes intracerebral and extensive subarachnoid hemorrhage. However, examination of the CSF remains indispensable in the diagnosis of meningitis and encephalitis. For patients with an altered level of consciousness, it is generally recommended that an imaging study be performed prior to lumbar puncture to exclude a large intracranial mass lesion. Blood culture and antibiotic administration usually precede the imaging study if meningitis is suspected (Chap. 164).
DIFFERENTIAL DIAGNOSIS OF COMA
(Table 328-1) The causes of coma can be divided into three broad categories: those cases without focal neurologic signs (e.g., metabolic and toxic encephalopathies); meningitis syndromes, characterized by fever or stiff neck and an excess of cells in the spinal fluid (e.g., bacterial meningitis, subarachnoid hemorrhage, encephalitis); and diseases associated with prominent focal signs (e.g., stroke, cerebral hemorrhage). Conditions that cause sudden coma include drug ingestion, cerebral hemorrhage, trauma, cardiac arrest, epilepsy, and basilar artery occlusion from an embolism. Coma that appears subacutely is usually related to a preexisting medical or neurologic problem or, less often, to secondary brain swelling surrounding a mass such as tumor or cerebral infarction.
|
DIFFERENTIAL DIAGNOSIS OF COMA |
Abbreviations: CSF, cerebrospinal fluid; CT, computed tomography; MRI, magnetic resonance imaging; RBCs, red blood cells; WBCs, white blood cells.
The diagnosis of coma due to cerebrovascular disease can be difficult (Chap. 446). The most common diseases are (1) basal ganglia and thalamic hemorrhage (acute but not instantaneous onset, vomiting, headache, hemiplegia, and characteristic eye signs); (2) pontine hemorrhage (sudden onset, pinpoint pupils, loss of reflex eye movements and corneal responses, ocular bobbing, posturing, and hyperventilation); (3) cerebellar hemorrhage (occipital headache, vomiting, gaze paresis, and inability to stand and walk); (4) basilar artery thrombosis (neurologic prodrome or warning spells, diplopia, dysarthria, vomiting, eye movement and corneal response abnormalities, and asymmetric limb paresis); and (5) subarachnoid hemorrhage (precipitous coma after sudden severe headache and vomiting). The most common stroke, infarction in the territory of the middle cerebral artery, does not cause coma, but edema surrounding large infarctions may expand over several days and cause coma from mass effect.
The syndrome of acute hydrocephalus accompanies many intracranial diseases, particularly subarachnoid hemorrhage. It is characterized by headache and sometimes vomiting that may progress quickly to coma with extensor posturing of the limbs, bilateral Babinski signs, small unreactive pupils, and impaired oculocephalic movements in the vertical direction.
The majority of medical causes of coma can be established without a neuroimaging study but if the history and examination do not indicate the cause of coma, CT or MRI is needed. Sometimes imaging results can be misleading such as when small subdural hematomas or old strokes are found, but the patient’s coma is due to intoxication.
BRAIN DEATH
This is a state of irreversible cessation of all cerebral function with preservation of cardiac activity and maintenance of respiratory and somatic function by artificial means. It is the only type of brain damage recognized as equivalent to death. Criteria have been advanced for the diagnosis of brain death, and it is essential to adhere to standards endorsed by the local medical community. Ideal criteria are simple, can be assessed at the bedside, and allow no chance of diagnostic error. They contain three essential elements: (1) widespread cortical destruction that is reflected by deep coma and unresponsiveness to all forms of stimulation; (2) global brainstem damage demonstrated by absent pupillary light reaction and by the loss of oculovestibular and corneal reflexes; and (3) destruction of the medulla, manifested by complete and irreversible apnea. The heart rate is invariant and does not accelerate to atropine. Diabetes insipidus is usually present but may only develop hours or days after the other clinical signs of brain death. The pupils are usually midsized but may be enlarged; they should not, however, be small. Loss of deep tendon reflexes is not required because the spinal cord remains functional. Babinski signs are generally absent and the toe response is instead, often flexor.
Demonstration that apnea is due to structural medullary damage requires that the PCO2 be high enough to stimulate respiration during a test of spontaneous breathing. Apnea testing can be done safely by the use of diffusion oxygenation prior to removing the ventilator. This is accomplished by preoxygenation with 100% oxygen, which is then sustained during the test by oxygen administered through a tracheal cannula. CO2 tension increases ~0.3–0.4 kPa/min (2–3 mmHg/min) during apnea. At the end of a period of observation, typically several minutes, arterial PCO2 should be at least >6.6–8.0 kPa (50–60 mmHg) for the test to be valid. Apnea is confirmed if no respiratory effort has been observed in the presence of a sufficiently elevated PCO2. Other techniques, including the administration of CO2 to accelerate the test, are used in special circumstances. The apnea test is usually stopped if there is serious cardiovascular instability.
An isoelectric EEG may be used as a confirmatory test for total cerebral damage. Radionuclide brain scanning, cerebral angiography, or transcranial Doppler measurements may also be included to demonstrate the absence of CBF, but they have not been as extensively correlated with pathologic changes.
The possibility of profound drug-induced or hypothermic depression of the nervous system must be excluded, and some period of observation, usually 6–24 h, is desirable, during which the clinical signs of brain death are sustained. It is advisable to delay clinical testing for at least 24 h if a cardiac arrest has caused brain death or if the inciting disease is not known.
Although it is largely accepted in Western society that the respirator can be disconnected from a brain-dead patient and that organ donation is subsequently possible, problems frequently arise because of poor communication and inadequate preparation of the family by the physician. Reasonable medical practice, ideally with the agreement of the family, also allows the removal of support or transfer out of an intensive care unit of patients who are not brain dead but whose neurologic conditions are nonetheless hopeless.
PROGNOSIS
One hopes to avoid the difficult outcome of a patient who is left severely disabled or vegetative. Children and young adults may have ominous early clinical findings such as abnormal brainstem reflexes and yet recover; temporization in offering a prognosis in this group of patients is wise. Metabolic comas have a far better prognosis than traumatic ones. All systems for estimating prognosis in adults should be taken as approximations, and medical judgments must be tempered by factors such as age, underlying systemic disease, and general medical condition. In an attempt to collect prognostic information from large numbers of patients with head injury, the Glasgow Coma Scale was devised; empirically, it has predictive value in cases of brain trauma (see Table 457e-2). For anoxic and metabolic coma, clinical signs such as the pupillary and motor responses after 1 day, 3 days, and 1 week have been shown to have predictive value. Other studies suggest that the absence of corneal responses may have the most discriminative value. The absence of the cortical waves of the somatosensory evoked potentials has also proved a strong indicator of poor outcome in coma from any cause.
The uniformly poor outcome of the prolonged vegetative state has already been mentioned, but recent reports that a small number of such patients display consistent cortical activation on functional MRI in response to salient stimuli have begun to alter the perception of the possible internal mental milieu of such individuals. These findings do not change the poor prognosis. For example, in one series, about 10% of vegetative patients after traumatic brain injury could activate their frontal or temporal lobes in response to requests by an examiner to imagine certain visuospatial tasks. In one case, a rudimentary form of communication could be established. There are also reports in exceptional patients of improvement in cognitive function with the implantation of thalamic-stimulating electrodes. It is prudent to avoid generalizations from these findings.
329e |
Examination of the Comatose Patient |
This chapter features a video illustrating the examination of a comatose patient. Proper techniques are demonstrated and supplemented with a discussion of interpretation of findings and implications for management. Also included is an overview of coma and its anatomic basis.
330 |
Neurologic Critical Care, Including Hypoxic-Ischemic Encephalopathy, and Subarachnoid Hemorrhage |
Life-threatening neurologic illness may be caused by a primary disorder affecting any region of the neuraxis or may occur as a consequence of a systemic disorder such as hepatic failure, multisystem organ failure, or cardiac arrest (Table 330-1). Neurologic critical care focuses on preservation of neurologic tissue and prevention of secondary brain injury caused by ischemia, hemorrhage, edema, herniation, and elevated intracranial pressure (ICP). Management of other organ systems proceeds concurrently and may need to be modified in order to maintain the overall focus on neurologic issues.
|
NEUROLOGIC DISORDERS IN CRITICAL ILLNESS |
PATHOPHYSIOLOGY
Brain Edema Swelling, or edema, of brain tissue occurs with many types of brain injury. The two principal types of edema are vasogenic and cytotoxic. Vasogenic edema refers to the influx of fluid and solutes into the brain through an incompetent blood-brain barrier (BBB). In the normal cerebral vasculature, endothelial tight junctions associated with astrocytes create an impermeable barrier (the BBB), through which access into the brain interstitium is dependent upon specific transport mechanisms. The BBB may be compromised in ischemia, trauma, infection, and metabolic derangements. Vasogenic edema results from abnormal permeability of the BBB, and typically develops rapidly following injury. Cytotoxic edema results from cellular swelling, membrane breakdown, and ultimately cell death. Clinically significant brain edema usually represents a combination of vasogenic and cytotoxic components. Edema can lead to increased ICP as well as tissue shifts and brain displacement or herniation from focal processes (Chap. 328). These tissue shifts can cause injury by mechanical distention and compression in addition to the ischemia of impaired perfusion consequent to the elevated ICP.
Ischemic Cascade and Cellular Injury When delivery of substrates, principally oxygen and glucose, is inadequate to sustain cellular function, a series of interrelated biochemical reactions known as the ischemic cascade is initiated (see Fig. 446-2). The release of excitatory amino acids, especially glutamate, leads to influx of calcium and sodium ions, which disrupt cellular homeostasis. An increased intracellular calcium concentration may activate proteases and lipases, which then lead to lipid peroxidation and free radical–mediated cell membrane injury. Cytotoxic edema ensues, and ultimately necrotic cell death and tissue infarction occur. This pathway to irreversible cell death is common to ischemic stroke, global cerebral ischemia, and traumatic brain injury.
Penumbra refers to areas of ischemic brain tissue that have not yet undergone irreversible infarction, implying that these regions are potentially salvageable if ischemia can be reversed. Factors that may exacerbate ischemic brain injury include systemic hypotension and hypoxia, which further reduce substrate delivery to vulnerable brain tissue, and fever, seizures, and hyperglycemia, which can increase cellular metabolism, outstripping compensatory processes. Clinically, these events are known as secondary brain insults because they lead to exacerbation of the primary brain injury. Prevention, identification, and treatment of secondary brain insults are fundamental goals of management.
An alternative pathway of cellular injury is apoptosis. This process implies programmed cell death, which may occur in the setting of ischemic stroke, global cerebral ischemia, traumatic brain injury, and possibly intracerebral hemorrhage. Apoptotic cell death can be distinguished histologically from the necrotic cell death of ischemia and is mediated through a different set of biochemical pathways; apoptotic cell death occurs without cerebral edema and therefore is often not seen on brain imaging. At present, interventions for prevention and treatment of apoptotic cell death remain less well defined than those for ischemia. Excitotoxicity and mechanisms of cell death are discussed in more detail in Chap. 444e.
Cerebral Perfusion and Autoregulation Brain tissue requires constant perfusion in order to ensure adequate delivery of substrate. The hemodynamic response of the brain has the capacity to preserve perfusion across a wide range of systemic blood pressures. Cerebral perfusion pressure (CPP), defined as the mean systemic arterial pressure (MAP) minus the ICP, provides the driving force for circulation across the capillary beds of the brain. Autoregulation refers to the physiologic response whereby cerebral blood flow (CBF) is regulated via alterations in cerebrovascular resistance in order to maintain perfusion over wide physiologic changes such as neuronal activation or changes in hemodynamic function. If systemic blood pressure drops, cerebral perfusion is preserved through vasodilation of arterioles in the brain; likewise, arteriolar vasoconstriction occurs at high systemic pressures to prevent hyperperfusion, resulting in fairly constant perfusion across a wide range of systemic blood pressures (Fig. 330-1). At the extreme limits of MAP or CPP (high or low), flow becomes directly related to perfusion pressure. These autoregulatory changes occur in the microcirculation and are mediated by vessels below the resolution of those seen on angiography. CBF is also strongly influenced by pH and PaCO2. CBF increases with hypercapnia and acidosis and decreases with hypocapnia and alkalosis because of pH related changes in cerebral vascular resistance. This forms the basis for the use of hyperventilation to lower ICP, and this effect on ICP is mediated through a decrease in both CBF and intracranial blood volume. Cerebral autoregulation is a complex process critical to the normal homeostatic functioning of the brain, and this process may be disordered focally and unpredictably in disease states such as traumatic brain injury and severe focal cerebral ischemia.
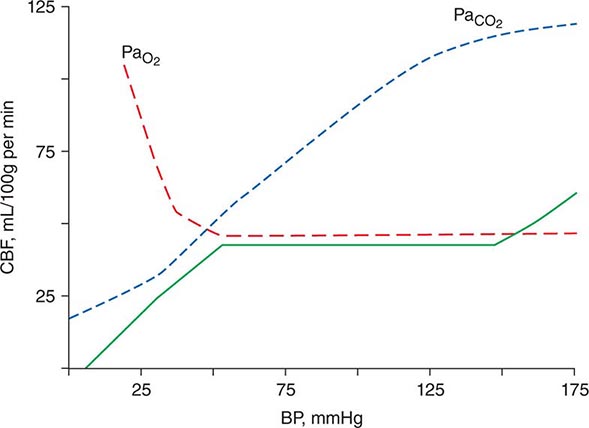
FIGURE 330-1 Autoregulation of cerebral blood flow (solid line). Cerebral perfusion is constant over a wide range of systemic blood pressure. Perfusion is increased in the setting of hypoxia or hypercarbia. BP, blood pressure; CBF, cerebral blood flow. (Reprinted with permission from HM Shapiro: Anesthesiology 43:447, 1975. Copyright 1975, Lippincott Company.).
Cerebrospinal Fluid and Intracranial Pressure The cranial contents consist essentially of brain, cerebrospinal fluid (CSF), and blood. CSF is produced principally in the choroid plexus of each lateral ventricle, exits the brain via the foramens of Luschka and Magendie, and flows over the cortex to be absorbed into the venous system along the superior sagittal sinus. In adults, approximately 150 mL of CSF are contained within the ventricles and surrounding the brain and spinal cord; the cerebral blood volume is also ~150 mL. The bony skull offers excellent protection for the brain but allows little tolerance for additional volume. Significant increases in volume eventually result in increased ICP. Obstruction of CSF outflow, edema of cerebral tissue, or increases in volume from tumor or hematoma may increase ICP. Elevated ICP diminishes cerebral perfusion and can lead to tissue ischemia. Ischemia in turn may lead to vasodilation via autoregulatory mechanisms designed to restore cerebral perfusion. However, vasodilation also increases cerebral blood volume, which in turn then increases ICP, lowers CPP, and provokes further ischemia (Fig. 330-2). This vicious cycle is commonly seen in traumatic brain injury, massive intracerebral hemorrhage, and large hemispheric infarcts with significant tissue shifts.
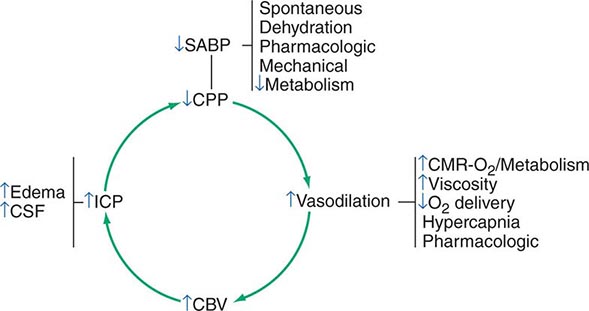
FIGURE 330-2 Ischemia and vasodilatation. Reduced cerebral perfusion pressure (CPP) leads to increased ischemia, vasodilation, increased intracranial pressure (ICP), and further reductions in CPP, a cycle leading to further neurologic injury. CBV, cerebral blood volume; CMR, cerebral metabolic rate; CSF, cerebrospinal fluid; SABP, systolic arterial blood pressure. (Adapted from MJ Rosner et al: J Neurosurg 83:949, 1995; with permission.)
APPROACH TO THE PATIENT:
Severe Central Nervous System Dysfunction
Critically ill patients with severe central nervous system (CNS) dysfunction require rapid evaluation and intervention in order to limit primary and secondary brain injury. Initial neurologic evaluation should be performed concurrent with stabilization of basic respiratory, cardiac, and hemodynamic parameters. Significant barriers may exist to neurologic assessment in the critical care unit, including endotracheal intubation and the use of sedative or paralytic agents to facilitate procedures.
An impaired level of consciousness is common in critically ill patients. The essential first task in assessment is to determine whether the cause of dysfunction is related to a diffuse, usually metabolic, process or whether a focal, usually structural, process is implicated. Examples of diffuse processes include metabolic encephalopathies related to organ failure, drug overdose, or hypoxia-ischemia. Focal processes include ischemic and hemorrhagic stroke and traumatic brain injury, especially with intracranial hematomas. Because these two categories of disorders have fundamentally different causes, treatments, and prognoses, the initial focus is on making this distinction rapidly and accurately. The approach to the comatose patient is discussed in Chap. 328; etiologies are listed in Table 328-1.
Minor focal deficits may be present on the neurologic examination in patients with metabolic encephalopathies. However, the finding of prominent focal signs such as pupillary asymmetry, hemiparesis, gaze palsy, or paraplegia should suggest the possibility of a structural lesion. All patients with a decreased level of consciousness associated with focal findings should undergo an urgent neuroimaging procedure, as should all patients with coma of unknown etiology. Computed tomography (CT) scanning is usually the most appropriate initial study because it can be performed quickly in critically ill patients and demonstrates hemorrhage, hydrocephalus, and intracranial tissue shifts well. Magnetic resonance imaging (MRI) may provide more specific information in some situations, such as acute ischemic stroke (diffusion-weighted imaging [DWI]) and cerebral venous sinus thrombosis (magnetic resonance venography [MRV]). Any suggestion of trauma from the history or examination should alert the examiner to the possibility of cervical spine injury and prompt an imaging evaluation using plain x-rays, CT, or MRI.
Acute brainstem ischemia due to basilar artery thrombosis may cause brief episodes of spontaneous extensor posturing superficially resembling generalized seizures. Coma of sudden onset, accompanied by these movements and cranial nerve abnormalities, necessitates emergency imaging. A noncontrast CT scan of the brain may reveal a hyperdense basilar artery indicating thrombus in the vessel, and subsequent CT or MR angiography can assess basilar artery patency.
Other diagnostic studies are best used in specific circumstances, usually when neuroimaging studies fail to reveal a structural lesion and the etiology of the altered mental state remains uncertain. Electroencephalography (EEG) can be important in the evaluation of critically ill patients with severe brain dysfunction. The EEG of metabolic encephalopathy typically reveals generalized slowing. One of the most important uses of EEG is to help exclude inapparent seizures, especially nonconvulsive status epilepticus. Untreated continuous or frequently recurrent seizures may cause neuronal injury, making the diagnosis and treatment of seizures crucial in this patient group. Lumbar puncture (LP) may be necessary to exclude infectious or inflammatory processes, and an elevated opening pressure may be an important clue to cerebral venous sinus thrombosis. In patients with coma or profound encephalopathy, it is preferable to perform a neuroimaging study prior to LP. If bacterial meningitis is suspected, an LP may be performed first or antibiotics may be empirically administered before the diagnostic studies are completed. Standard laboratory evaluation of critically ill patients should include assessment of serum electrolytes (especially sodium and calcium), glucose, renal and hepatic function, complete blood count, and coagulation. Serum or urine toxicology screens should be performed in patients with encephalopathy of unknown cause. EEG, LP, and other specific laboratory tests are most useful when the mechanism of the altered level of consciousness is uncertain; they are not routinely performed in clear-cut cases of stroke or traumatic brain injury.
Monitoring of ICP can be an important tool in selected patients. In general, patients who should be considered for ICP monitoring are those with primary neurologic disorders, such as stroke or traumatic brain injury, who are at significant risk for secondary brain injury due to elevated ICP and decreased CPP. Included are patients with the following: severe traumatic brain injury (Glasgow Coma Scale [GCS] score ≤8 [see Table 457e-2]); large tissue shifts from supratentorial ischemic or hemorrhagic stroke; or hydrocephalus from subarachnoid hemorrhage (SAH), intraventricular hemorrhage, or posterior fossa stroke. An additional disorder in which ICP monitoring can add important information is fulminant hepatic failure, in which elevated ICP may be treated with barbiturates or, eventually, liver transplantation. In general, ventriculostomy is preferable to ICP monitoring devices that are placed in the brain parenchyma, because ventriculostomy allows CSF drainage as a method of treating elevated ICP. However, parenchymal ICP monitoring is most appropriate for patients with diffuse edema and small ventricles (which may make ventriculostomy placement more difficult) or any degree of coagulopathy (in which ventriculostomy carries a higher risk of hemorrhagic complications) (Fig 330-3).
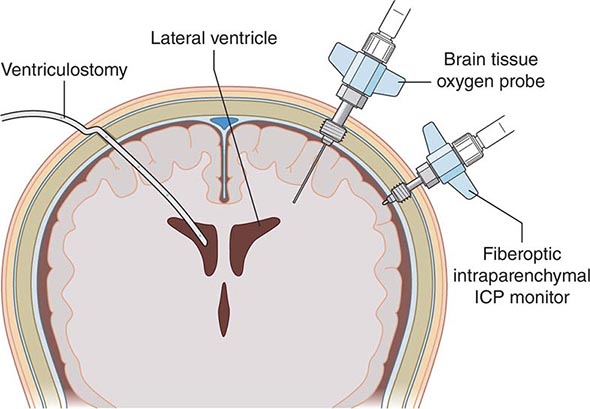
FIGURE 330-3 Intracranial pressure and brain tissue oxygen monitoring. A ventriculostomy allows for drainage of cerebrospinal fluid to treat elevated intracranial pressure (ICP). Fiberoptic ICP and brain tissue oxygen monitors are usually secured using a screwlike skull bolt. Cerebral blood flow and microdialysis probes (not shown) may be placed in a manner similar to the brain tissue oxygen probe.
Treatment of Elevated ICP Elevated ICP may occur in a wide range of disorders, including head trauma, intracerebral hemorrhage, SAH with hydrocephalus, and fulminant hepatic failure. Because CSF and blood volume can be redistributed initially, by the time elevated ICP occurs, intracranial compliance is severely impaired. At this point, any small increase in the volume of CSF, intravascular blood, edema, or a mass lesion may result in a significant increase in ICP and a decrease in cerebral perfusion. This is a fundamental mechanism of secondary ischemic brain injury and constitutes an emergency that requires immediate attention. In general, ICP should be maintained at <20 mmHg and CPP should be maintained at ≥60 mmHg.
Interventions to lower ICP are ideally based on the underlying mechanism responsible for the elevated ICP (Table 330-2). For example, in hydrocephalus from SAH, the principal cause of elevated ICP is impairment of CSF drainage. In this setting, ventricular drainage of CSF is likely to be sufficient and most appropriate. In head trauma and stroke, cytotoxic edema may be most responsible, and the use of osmotic agents such as mannitol or hypertonic saline becomes an appropriate early step. As described above, elevated ICP may cause tissue ischemia, and, if cerebral autoregulation is intact, the resulting vasodilation can lead to a cycle of worsening ischemia. Paradoxically, administration of vasopressor agents to increase mean arterial pressure may actually lower ICP by improving perfusion, thereby allowing autoregulatory vasoconstriction as ischemia is relieved and ultimately decreasing intracranial blood volume.
|
STEPWISE APPROACH TO TREATMENT OF ELEVATED INTRACRANIAL PRESSURE (ICP)a |
Insert ICP monitor—ventriculostomy versus parenchymal device
General goals: maintain ICP <20 mmHg and CPP ≥60 mmHg. For ICP >20–25 mmHg for >5 min:
1. Elevate head of the bed; midline head position
2. Drain CSF via ventriculostomy (if in place)
3. Osmotherapy—mannitol 25–100 g q4h as needed (maintain serum osmolality <320 mosmol) or hypertonic saline (30 mL, 23.4% NaCl bolus)
4. Glucocorticoids—dexamethasone 4 mg q6h for vasogenic edema from tumor, abscess (avoid glucocorticoids in head trauma, ischemic and hemorrhagic stroke)
5. Sedation (e.g., morphine, propofol, or midazolam); add neuromuscular paralysis if necessary (patient will require endotracheal intubation and mechanical ventilation at this point, if not before)
6. Hyperventilation—to PaCO2 30–35 mmHg (short-term use or skip this step)
7. Pressor therapy—phenylephrine, dopamine, or norepinephrine to maintain adequate MAP to ensure CPP ≥60 mmHg (maintain euvolemia to minimize deleterious systemic effects of pressors). May adjust target CPP in individual patients based on autoregulation status.
8. Consider second-tier therapies for refractory elevated ICP
a. Decompressive craniectomy
b. High-dose barbiturate therapy (“pentobarb coma”)
c. Hypothermia to 33°C
aThroughout ICP treatment algorithm, consider repeat head computed tomography to identify mass lesions amenable to surgical evacuation. May alter order of steps based on directed treatment to specific cause of elevated ICP.
Abbreviations: CPP, cerebral perfusion pressure; CSF, cerebrospinal fluid; MAP, mean arterial pressure; PaCO2, arterial partial pressure of carbon dioxide.
Early signs of elevated ICP include drowsiness and a diminished level of consciousness. Neuroimaging studies may reveal evidence of edema and mass effect. Hypotonic IV fluids should be avoided, and elevation of the head of the bed is recommended. Patients must be carefully observed for risk of aspiration and compromise of the airway as the level of alertness declines. Coma and unilateral pupillary changes are late signs and require immediate intervention. Emergent treatment of elevated ICP is most quickly achieved by intubation and hyperventilation, which causes vasoconstriction and reduces cerebral blood volume. To avoid provoking or worsening cerebral ischemia, hyperventilation, if used at all, is best administered only for short periods of time until a more definitive treatment can be instituted. Furthermore, the effects of hyperventilation on ICP are short-lived, often lasting only for several hours because of the buffering capacity of the cerebral interstitium, and rebound elevations of ICP may accompany abrupt discontinuation of hyperventilation. As the level of consciousness declines to coma, the ability to follow the neurologic status of the patient by examination lessens and measurement of ICP assumes greater importance. If a ventriculostomy device is in place, direct drainage of CSF to reduce ICP is possible. Finally, high-dose barbiturates, decompressive hemicraniectomy, and hypothermia are sometimes used for refractory elevations of ICP, although these have significant side effects and have not been proven to improve outcome.
Secondary Brain Insults Patients with primary brain injuries, whether due to trauma or stroke, are at risk for ongoing secondary ischemic brain injury. Because secondary brain injury can be a major determinant of a poor outcome, strategies for minimizing secondary brain insults are an integral part of the critical care of all patients. Although elevated ICP may lead to secondary ischemia, most secondary brain injury is mediated through other clinical events that exacerbate the ischemic cascade already initiated by the primary brain injury. Episodes of secondary brain insults are usually not associated with apparent neurologic worsening. Rather, they lead to cumulative injury limiting eventual recovery, which manifests as a higher mortality rate or worsened long-term functional outcome. Thus, close monitoring of vital signs is important, as is early intervention to prevent secondary ischemia. Avoiding hypotension and hypoxia is critical, as significant hypotensive events (systolic blood pressure <90 mmHg) as short as 10 min in duration have been shown to adversely influence outcome after traumatic brain injury. Even in patients with stroke or head trauma who do not require ICP monitoring, close attention to adequate cerebral perfusion is warranted. Hypoxia (pulse oximetry saturation <90%), particularly in combination with hypotension, also leads to secondary brain injury. Likewise, fever and hyperglycemia both worsen experimental ischemia and have been associated with worsened clinical outcome after stroke and head trauma. Aggressive control of fever with a goal of normothermia is warranted but may be difficult to achieve with antipyretic medications and cooling blankets. The value of newer surface or intravascular temperature control devices for the management of refractory fever is under investigation. The use of IV insulin infusion is encouraged for control of hyperglycemia because this allows better regulation of serum glucose levels than SC insulin. A reasonable goal is to maintain the serum glucose level at <10.0 mmol/L (<180 mg/dL), although episodes of hypoglycemia appear equally detrimental and the optimal targets remain uncertain. New cerebral monitoring tools that allow continuous evaluation of brain tissue oxygen tension, CBF, and metabolism (via microdialysis) may further improve the management of secondary brain injury.
CRITICAL CARE DISORDERS OF THE CENTRAL NERVOUS SYSTEM
HYPOXIC-ISCHEMIC ENCEPHALOPATHY
This occurs from lack of delivery of oxygen to the brain because of extreme hypotension (hypoxia-ischemia) or hypoxia due to respiratory failure. Causes include myocardial infarction, cardiac arrest, shock, asphyxiation, paralysis of respiration, and carbon monoxide or cyanide poisoning. In some circumstances, hypoxia may predominate. Carbon monoxide and cyanide poisoning are sometimes termed histotoxic hypoxia because they cause a direct impairment of the respiratory chain.
Clinical Manifestations Mild degrees of pure hypoxia, such as occur at high altitudes, cause impaired judgment, inattentiveness, motor incoordination, and, at times, euphoria. However, with hypoxia-ischemia, such as occurs with circulatory arrest, consciousness is lost within seconds. If circulation is restored within 3–5 min, full recovery may occur, but if hypoxia-ischemia lasts beyond 3–5 min, some degree of permanent cerebral damage usually results. Except in extreme cases, it may be difficult to judge the precise degree of hypoxia-ischemia, and some patients make a relatively full recovery after even 8–10 min of global cerebral ischemia. The brain is more tolerant to pure hypoxia than it is to hypoxia-ischemia. For example, a PaO2 as low as 20 mmHg (2.7 kPa) can be well tolerated if it develops gradually and normal blood pressure is maintained, whereas short durations of very low or absent cerebral circulation usually result in permanent impairment.
Clinical examination at different time points after a hypoxic-ischemic insult (especially cardiac arrest) is useful in assessing prognosis for long-term neurologic outcome. The prognosis is better for patients with intact brainstem function, as indicated by normal pupillary light responses and intact oculocephalic (doll’s eyes), oculovestibular (caloric), and corneal reflexes. Absence of these reflexes and the presence of persistently dilated pupils that do not react to light are grave prognostic signs. A low likelihood of a favorable outcome from hypoxic-ischemic coma is strongly suggested by an absent pupillary light reflex or extensor or absent motor response to pain on day 3 following the injury, excluding patients with metabolic disturbances and those treated with high-dose barbiturates or hypothermia, which confound interpretation of these signs. Electrophysiologically, the bilateral absence of the N20 component of the somatosensory evoked potential (SSEP) in the first several days also conveys a poor prognosis. A very elevated serum level (>33 μg/L) of the biochemical marker neuron-specific enolase (NSE) is indicative of brain damage after resuscitation from cardiac arrest and predicts a poor outcome. However, at present, SSEPs and NSE levels may be difficult to obtain in a timely fashion, with SSEP testing requiring substantial expertise in interpretation and NSE measurements not yet standardized. Recent studies suggest that the administration of mild hypothermia after cardiac arrest (see “Treatment”) may affect the time points when these clinical and electrophysiologic predictors become reliable in identifying patients with a very low likelihood of clinically meaningful recovery. For example, the false-positive rate for incorrect prediction of poor neurologic outcome may be as high as 21% (95% confidence interval [CI] 8–43%) for patients treated with mild hypothermia who exhibit 3-day motor function no better than extensor posturing. Long-term consequences of hypoxic-ischemic encephalopathy include persistent coma or a vegetative state (Chap. 328), dementia, visual agnosia (Chap. 36), parkinsonism, choreoathetosis, cerebellar ataxia, myoclonus, seizures, and an amnestic state, which may be a consequence of selective damage to the hippocampus.
Pathology Principal histologic findings are extensive multifocal or diffuse laminar cortical necrosis (Fig. 330-4), with frequent involvement of the hippocampus. The hippocampal CA1 neurons are vulnerable to even brief episodes of hypoxia-ischemia, perhaps explaining why selective persistent memory deficits may occur after brief cardiac arrest. Scattered small areas of infarction or neuronal loss may be present in the basal ganglia, hypothalamus, or brainstem. In some cases, extensive bilateral thalamic scarring may affect pathways that mediate arousal, and this pathology may be responsible for the persistent vegetative state. A specific form of hypoxic-ischemic encephalopathy, so-called watershed infarcts, occurs at the distal territories between the major cerebral arteries and can cause cognitive deficits, including visual agnosia, and weakness that is greater in proximal than in distal muscle groups.
FIGURE 330-4 Cortical laminar necrosis in hypoxic-ischemic encephalopathy. T1-weighted postcontrast magnetic resonance imaging shows cortical enhancement in a watershed distribution consistent with laminar necrosis.
Diagnosis Diagnosis is based on the history of a hypoxic-ischemic event such as cardiac arrest. Blood pressure <70 mmHg systolic or PaO2 <40 mmHg is usually necessary, although both absolute levels and duration of exposure are important determinants of cellular injury. Carbon monoxide intoxication can be confirmed by measurement of carboxyhemoglobin and is suggested by a cherry red color of the venous blood and skin, although the latter is an inconsistent clinical finding.
METABOLIC ENCEPHALOPATHIES
Altered mental states, variously described as confusion, delirium, disorientation, and encephalopathy, are present in many patients with severe illness in an intensive care unit (ICU). Older patients are particularly vulnerable to delirium, a confusional state characterized by disordered perception, frequent hallucinations, delusions, and sleep disturbance. This is often attributed to medication effects, sleep deprivation, pain, and anxiety. The presence of delirium is associated with worsened outcome in critically ill patients, even in those without an identifiable CNS pathology such as stroke or brain trauma. In these patients, the cause of delirium is often multifactorial, resulting from organ dysfunction, sepsis, and especially the use of medications given to treat pain, agitation, or anxiety. Critically ill patients are often treated with a variety of sedative and analgesic medications, including opiates, benzodiazepines, neuroleptics, and sedative-anesthetic medications, such as propofol. In critically ill patients requiring sedation, use of the centrally acting α2 agonist dexmedetomidine may reduce delirium and shorten the duration of mechanical ventilation compared to the use of benzodiazepines such as lorazepam or midazolam. The presence of family members in the ICU may also help to calm and orient agitated patients, and in severe cases, low doses of neuroleptics (e.g., haloperidol 0.5–1 mg) can be useful. Current strategies focus on limiting the use of sedative medications when this can be done safely.
In the ICU setting, several metabolic causes of an altered level of consciousness predominate. Hypercarbic encephalopathy can present with headache, confusion, stupor, or coma. Hypoventilation syndrome occurs most frequently in patients with a history of chronic CO2 retention who are receiving oxygen therapy for emphysema or chronic pulmonary disease (Chap. 318). The elevated PaCO2 leading to CO2 narcosis may have a direct anesthetic effect, and cerebral vasodilation from increased PaCO2 can lead to increased ICP. Hepatic encephalopathy is suggested by asterixis and can occur in chronic liver failure or acute fulminant hepatic failure. Both hyperglycemia and hypoglycemia can cause encephalopathy, as can hypernatremia and hyponatremia. Confusion, impairment of eye movements, and gait ataxia are the hallmarks of acute Wernicke’s disease (see below).
SEPSIS-ASSOCIATED ENCEPHALOPATHY
Pathogenesis In patients with sepsis, the systemic response to infectious agents leads to the release of circulating inflammatory mediators that appear to contribute to encephalopathy. Critical illness, in association with the systemic inflammatory response syndrome (SIRS), can lead to multisystem organ failure. This syndrome can occur in the setting of apparent sepsis, severe burns, or trauma, even without clear identification of an infectious agent. Many patients with critical illness, sepsis, or SIRS develop encephalopathy without obvious explanation. This condition is broadly termed sepsis-associated encephalopathy. Although the specific mediators leading to neurologic dysfunction remain uncertain, it is clear that the encephalopathy is not simply the result of metabolic derangements of multiorgan failure. The cytokines tumor necrosis factor, interleukin (IL)-1, IL-2, and IL-6 are thought to play a role in this syndrome.
Diagnosis Sepsis-associated encephalopathy presents clinically as a diffuse dysfunction of the brain without prominent focal findings. Confusion, disorientation, agitation, and fluctuations in level of alertness are typical. In more profound cases, especially with hemodynamic compromise, the decrease in level of alertness can be more prominent, at times resulting in coma. Hyperreflexia and frontal release signs such as a grasp or snout reflex (Chap. 36) can be seen. Abnormal movements such as myoclonus, tremor, or asterixis can occur. Sepsis-associated encephalopathy is quite common, occurring in the majority of patients with sepsis and multisystem organ failure. Diagnosis is often difficult because of the multiple potential causes of neurologic dysfunction in critically ill patients and requires exclusion of structural, metabolic, toxic, and infectious (e.g., meningitis or encephalitis) causes. The mortality rate of patients with sepsis-associated encephalopathy severe enough to produce coma approaches 50%, although this principally reflects the severity of the underlying critical illness and is not a direct result of the encephalopathy. Patients dying from severe sepsis or septic shock may have elevated levels of the serum brain injury biomarker S-100β and neuropathologic findings of neuronal apoptosis and cerebral ischemic injury. Successful treatment of the underlying critical illness almost always results in substantial improvement of the encephalopathy. However, although severe disability to the level of chronic vegetative or minimally conscious states is uncommon, long-term cognitive dysfunction clinically similar to dementia is being increasingly recognized in some survivors.
CENTRAL PONTINE MYELINOLYSIS
This disorder typically presents in a devastating fashion as quadriplegia and pseudobulbar palsy. Predisposing factors include severe underlying medical illness or nutritional deficiency; most cases are associated with rapid correction of hyponatremia or with hyperosmolar states. The pathology consists of demyelination without inflammation in the base of the pons, with relative sparing of axons and nerve cells. MRI is useful in establishing the diagnosis (Fig. 330-5) and may also identify partial forms that present as confusion, dysarthria, and/or disturbances of conjugate gaze without quadriplegia. Occasional cases present with lesions outside of the brainstem. Therapeutic guidelines for the restoration of severe hyponatremia should aim for gradual correction, i.e., by ≤10 mmol/L (10 meq/L) within 24 h and 20 mmol/L (20 meq/L) within 48 h.
FIGURE 330-5 Central pontine myelinolysis. Axial T2-weighted magnetic resonance scan through the pons reveals a symmetric area of abnormal high signal intensity within the basis pontis (arrows).
WERNICKE’S DISEASE
Wernicke’s disease is a common and preventable disorder due to a deficiency of thiamine (Chap. 96e). In the United States, alcoholics account for most cases, but patients with malnutrition due to hyperemesis, starvation, renal dialysis, cancer, AIDS, or rarely gastric surgery are also at risk. The characteristic clinical triad is that of ophthalmoplegia, ataxia, and global confusion. However, only one-third of patients with acute Wernicke’s disease present with the classic clinical triad. Most patients are profoundly disoriented, indifferent, and inattentive, although rarely they have an agitated delirium related to ethanol withdrawal. If the disease is not treated, stupor, coma, and death may ensue. Ocular motor abnormalities include horizontal nystagmus on lateral gaze, lateral rectus palsy (usually bilateral), conjugate gaze palsies, and rarely ptosis. Gait ataxia probably results from a combination of polyneuropathy, cerebellar involvement, and vestibular paresis. The pupils are usually spared, but they may become miotic with advanced disease.
Wernicke’s disease is usually associated with other manifestations of nutritional disease, such as polyneuropathy. Rarely, amblyopia or myelopathy occurs. Tachycardia and postural hypotension may be related to impaired function of the autonomic nervous system or to the coexistence of cardiovascular beriberi. Patients who recover show improvement in ocular palsies within hours after the administration of thiamine, but horizontal nystagmus may persist. Ataxia improves more slowly than the ocular motor abnormalities. Approximately half recover incompletely and are left with a slow, shuffling, wide-based gait and an inability to tandem walk. Apathy, drowsiness, and confusion improve more gradually. As these symptoms recede, an amnestic state with impairment in recent memory and learning may become more apparent (Korsakoff’s psychosis). Korsakoff’s psychosis is frequently persistent; the residual mental state is characterized by gaps in memory, confabulation, and disordered temporal sequencing.
Pathology Periventricular lesions surround the third ventricle, aqueduct, and fourth ventricle, with petechial hemorrhages in occasional acute cases and atrophy of the mammillary bodies in most chronic cases. There is frequently endothelial proliferation, demyelination, and some neuronal loss. These changes may be detected by MRI scanning (Fig. 330-6). The amnestic defect is related to lesions in the dorsal medial nuclei of the thalamus.
FIGURE 330-6 Wernicke’s disease. Coronal T1-weighted postcontrast magnetic resonance imaging reveals abnormal enhancement of the mammillary bodies (arrows), typical of acute Wernicke’s encephalopathy.
Pathogenesis Thiamine is a cofactor of several enzymes, including transketolase, pyruvate dehydrogenase, and α-ketoglutarate dehydrogenase. Thiamine deficiency produces a diffuse decrease in cerebral glucose utilization and results in mitochondrial damage. Glutamate accumulates due to impairment of α-ketoglutarate dehydrogenase activity and, in combination with the energy deficiency, may result in excitotoxic cell damage.
CRITICAL CARE DISORDERS OF THE PERIPHERAL NERVOUS SYSTEM
Critical illness with disorders of the peripheral nervous system (PNS) arises in two contexts: (1) primary neurologic diseases that require critical care interventions such as intubation and mechanical ventilation, and (2) secondary PNS manifestations of systemic critical illness, often involving multisystem organ failure. The former include acute polyneuropathies such as Guillain-Barré syndrome (Chap. 460), neuromuscular junction disorders including myasthenia gravis (Chap. 461) and botulism (Chap. 178), and primary muscle disorders such as polymyositis (Chap. 462e). The latter result either from the systemic disease itself or as a consequence of interventions.
General principles of respiratory evaluation in patients with PNS involvement, regardless of cause, include assessment of pulmonary mechanics, such as maximal inspiratory force (MIF) and vital capacity (VC), and evaluation of strength of bulbar muscles. Regardless of the cause of weakness, endotracheal intubation should be considered when the MIF falls to <–25 cmH2O or the VC is <1 L. Also, patients with severe palatal weakness may require endotracheal intubation in order to prevent acute upper airway obstruction or recurrent aspiration. Arterial blood gases and oxygen saturation from pulse oximetry are used to follow patients with potential respiratory compromise from PNS dysfunction. However, intubation and mechanical ventilation should be undertaken based on clinical assessment rather than waiting until oxygen saturation drops or CO2 retention develops from hypoventilation. Noninvasive mechanical ventilation may be considered initially in lieu of endotracheal intubation but is generally insufficient in patients with severe bulbar weakness or ventilatory failure with hypercarbia. Principles of mechanical ventilation are discussed in Chap. 323.
NEUROPATHY
Although encephalopathy may be the most obvious neurologic dysfunction in critically ill patients, dysfunction of the PNS is also quite common. It is typically present in patients with prolonged critical illnesses lasting several weeks and involving sepsis; clinical suspicion is aroused when there is failure to wean from mechanical ventilation despite improvement of the underlying sepsis and critical illness. Critical illness polyneuropathy refers to the most common PNS complication related to critical illness; it is seen in the setting of prolonged critical illness, sepsis, and multisystem organ failure. Neurologic findings include diffuse weakness, decreased reflexes, and distal sensory loss. Electrophysiologic studies demonstrate a diffuse, symmetric, distal axonal sensorimotor neuropathy, and pathologic studies have confirmed axonal degeneration. The precise mechanism of critical illness polyneuropathy remains unclear, but circulating factors such as cytokines, which are associated with sepsis and SIRS, are thought to play a role. It has been reported that up to 70% of patients with the sepsis syndrome have some degree of neuropathy, although far fewer have a clinical syndrome profound enough to cause severe respiratory muscle weakness requiring prolonged mechanical ventilation or resulting in failure to wean. Aggressive glycemic control with insulin infusions appears to decrease the risk of critical illness polyneuropathy. Treatment is otherwise supportive, with specific intervention directed at treating the underlying illness. Although spontaneous recovery is usually seen, the time course may extend over weeks to months and necessitate long-term ventilatory support and care even after the underlying critical illness has resolved.
DISORDERS OF NEUROMUSCULAR TRANSMISSION
A defect in neuromuscular transmission may be a source of weakness in critically ill patients. Botulism (Chap. 178) may be acquired by ingesting botulinum toxin from improperly stored food or may arise from an anaerobic abscess from Clostridium botulinum (wound botulism). Infants can present with generalized weakness from gut-derived Clostridium infection, especially if they are fed honey. Diplopia and dysphagia are early signs of foodborne botulism. Treatment is mostly supportive, although use of antitoxin early in the course may limit the duration of the neuromuscular blockade. General ICU care is similar to patients with Guillain-Barré syndrome or myasthenia gravis with focused care to avoid ulcer formation at pressure points, deep venous thromboprophylaxis, and infection prevention. Public health officers should be rapidly informed when the diagnosis is made to prevent further exposure to others from the tainted food or source of wound botulism (such as injection drug use).
Undiagnosed myasthenia gravis (Chap. 461) may be a consideration in weak ICU patients; however, persistent weakness secondary to impaired neuromuscular junction transmission is almost always due to administration of drugs. A number of medications impair neuromuscular transmission; these include antibiotics, especially aminoglycosides, and beta-blocking agents. In the ICU, the nondepolarizing neuromuscular blocking agents (nd-NMBAs), also known as muscle relaxants, are most commonly responsible. Included in this group of drugs are such agents as pancuronium, vecuronium, rocuronium, and cisatracurium. They are often used to facilitate mechanical ventilation or other critical care procedures, but with prolonged use persistent neuromuscular blockade may result in weakness even after discontinuation of these agents hours or days earlier. Risk factors for this prolonged action of neuromuscular blocking agents include female sex, metabolic acidosis, and renal failure.
Prolonged neuromuscular blockade does not appear to produce permanent damage to the PNS. Once the offending medications are discontinued, full strength is restored, although this may take days. In general, the lowest dose of neuromuscular blocking agent should be used to achieve the desired result and, when these agents are used in the ICU, a peripheral nerve stimulator should be used to monitor neuromuscular junction function.
MYOPATHY
Critically ill patients, especially those with sepsis, frequently develop muscle weakness and wasting, often in the face of seemingly adequate nutritional support. Critical illness myopathy is an overall term that describes several different discrete muscle disorders that may occur in critically ill patients. The assumption has been that a catabolic myopathy may develop as a result of multiple factors, including elevated cortisol and catecholamine release and other circulating factors induced by the SIRS. In this syndrome, known as cachectic myopathy, serum creatine kinase levels and electromyography (EMG) are normal. Muscle biopsy shows type II fiber atrophy. Panfascicular muscle fiber necrosis may also occur in the setting of profound sepsis. This less common acute necrotizing intensive care myopathy is characterized clinically by weakness progressing to a profound level over just a few days. There may be associated elevations in serum creatine kinase and urine myoglobin. Both EMG and muscle biopsy may be normal initially but eventually show abnormal spontaneous activity and panfascicular necrosis with an accompanying inflammatory reaction. Acute rhabdomyolysis can occur from alcohol ingestion or from compartment syndromes.
A thick-filament myopathy may occur in the setting of glucocorticoid and nd-NMBA use. The most frequent scenario in which this is encountered is the asthmatic patient who requires high-dose glucocorticoids and nd-NMBA to facilitate mechanical ventilation. This muscle disorder is not due to prolonged action of nd-NMBAs at the neuromuscular junction but, rather, is an actual myopathy with muscle damage; it has occasionally been described with high-dose glucocorticoid use or sepsis alone. Clinically this syndrome is most often recognized when a patient fails to wean from mechanical ventilation despite resolution of the primary pulmonary process. Pathologically, there may be loss of thick (myosin) filaments. Thick-filament critical illness myopathy has a good prognosis. If patients survive their underlying critical illness, the myopathy invariably improves and most patients return to normal. However, because this syndrome is a result of true muscle damage, not just prolonged blockade at the neuromuscular junction, this process may take weeks or months, and tracheotomy with prolonged ventilatory support may be necessary. Some patients do have residual long-term weakness, with atrophy and fatigue limiting ambulation. At present, it is unclear how to prevent this myopathic complication, except by avoiding use of nd-NMBAs, a strategy not always possible. Monitoring with a peripheral nerve stimulator can help to avoid the overuse of these agents. However, this is more likely to prevent the complication of prolonged neuromuscular junction blockade than it is to prevent this myopathy.
SUBARACHNOID HEMORRHAGE
Subarachnoid hemorrhage (SAH) renders the brain critically ill from both primary and secondary brain insults. Excluding head trauma, the most common cause of SAH is rupture of a saccular aneurysm. Other causes include bleeding from a vascular malformation (arteriovenous malformation or dural arteriovenous fistula) and extension into the subarachnoid space from a primary intracerebral hemorrhage. Some idiopathic SAHs are localized to the perimesencephalic cisterns and are benign; they probably have a venous or capillary source, and angiography is unrevealing.
SACCULAR (“BERRY”) ANEURYSM
Autopsy and angiography studies have found that about 2% of adults harbor intracranial aneurysms, for a prevalence of 4 million persons in the United States; the aneurysm will rupture, producing SAH, in 25,000–30,000 cases per year. For patients who arrive alive at hospital, the mortality rate over the next month is about 45%. Of those who survive, more than half are left with major neurologic deficits as a result of the initial hemorrhage, cerebral vasospasm with infarction, or hydrocephalus. If the patient survives but the aneurysm is not obliterated, the rate of rebleeding is about 20% in the first 2 weeks, 30% in the first month, and about 3% per year afterward. Given these alarming figures, the major therapeutic emphasis is on preventing the predictable early complications of the SAH.
Unruptured, asymptomatic aneurysms are much less dangerous than a recently ruptured aneurysm. The annual risk of rupture for aneurysms <10 mm in size is ~0.1%, and for aneurysms ≥10 mm in size is ~0.5–1%; the surgical morbidity rate far exceeds these percentages. Because of the longer length of exposure to risk of rupture, younger patients with aneurysms >10 mm in size may benefit from prophylactic treatment. As with the treatment of asymptomatic carotid stenosis, this risk-benefit ratio strongly depends on the complication rate of treatment.
Giant aneurysms, those >2.5 cm in diameter, occur at the same sites (see below) as small aneurysms and account for 5% of cases. The three most common locations are the terminal internal carotid artery, middle cerebral artery (MCA) bifurcation, and top of the basilar artery. Their risk of rupture is ~6% in the first year after identification and may remain high indefinitely. They often cause symptoms by compressing the adjacent brain or cranial nerves.
Mycotic aneurysms are usually located distal to the first bifurcation of major arteries of the circle of Willis. Most result from infected emboli due to bacterial endocarditis causing septic degeneration of arteries and subsequent dilation and rupture. Whether these lesions should be sought and repaired prior to rupture or left to heal spontaneously with antibiotic treatment is controversial.
Pathophysiology Saccular aneurysms occur at the bifurcations of the large- to medium-sized intracranial arteries; rupture is into the subarachnoid space in the basal cisterns and often into the parenchyma of the adjacent brain. Approximately 85% of aneurysms occur in the anterior circulation, mostly on the circle of Willis. About 20% of patients have multiple aneurysms, many at mirror sites bilaterally. As an aneurysm develops, it typically forms a neck with a dome. The length of the neck and the size of the dome vary greatly and are important factors in planning neurosurgical obliteration or endovascular embolization. The arterial internal elastic lamina disappears at the base of the neck. The media thins, and connective tissue replaces smooth-muscle cells. At the site of rupture (most often the dome), the wall thins, and the tear that allows bleeding is often ≤0.5 mm long. Aneurysm size and site are important in predicting risk of rupture. Those >7 mm in diameter and those at the top of the basilar artery and at the origin of the posterior communicating artery are at greater risk of rupture.
Clinical Manifestations Most unruptured intracranial aneurysms are completely asymptomatic. Symptoms are usually due to rupture and resultant SAH, although some unruptured aneurysms present with mass effect on cranial nerves or brain parenchyma. At the moment of aneurysmal rupture with major SAH, the ICP suddenly rises. This may account for the sudden transient loss of consciousness that occurs in nearly half of patients. Sudden loss of consciousness may be preceded by a brief moment of excruciating headache, but most patients first complain of headache upon regaining consciousness. In 10% of cases, aneurysmal bleeding is severe enough to cause loss of consciousness for several days. In ~45% of cases, severe headache associated with exertion is the presenting complaint. The patient often calls the headache “the worst headache of my life”; however, the most important characteristic is sudden onset. Occasionally, these ruptures may present as headache of only moderate intensity or as a change in the patient’s usual headache pattern. The headache is usually generalized, often with neck stiffness, and vomiting is common.
Although sudden headache in the absence of focal neurologic symptoms is the hallmark of aneurysmal rupture, focal neurologic deficits may occur. Anterior communicating artery or MCA bifurcation aneurysms may rupture into the adjacent brain or subdural space and form a hematoma large enough to produce mass effect. The deficits that result can include hemiparesis, aphasia, and abulia.
Occasionally, prodromal symptoms suggest the location of a progressively enlarging unruptured aneurysm. A third cranial nerve palsy, particularly when associated with pupillary dilation, loss of ipsilateral (but retained contralateral) light reflex, and focal pain above or behind the eye, may occur with an expanding aneurysm at the junction of the posterior communicating artery and the internal carotid artery. A sixth nerve palsy may indicate an aneurysm in the cavernous sinus, and visual field defects can occur with an expanding supraclinoid carotid or anterior cerebral artery aneurysm. Occipital and posterior cervical pain may signal a posterior inferior cerebellar artery or anterior inferior cerebellar artery aneurysm (Chap. 446). Pain in or behind the eye and in the low temple can occur with an expanding MCA aneurysm. Thunderclap headache is a variant of migraine that simulates an SAH. Before concluding that a patient with sudden, severe headache has thunderclap migraine, a definitive workup for aneurysm or other intracranial pathology is required.
Aneurysms can undergo small ruptures and leaks of blood into the subarachnoid space, so-called sentinel bleeds. Sudden unexplained headache at any location should raise suspicion of SAH and be investigated, because a major hemorrhage may be imminent.
The initial clinical manifestations of SAH can be graded using the Hunt-Hess or World Federation of Neurosurgical Societies classification schemes (Table 330-3). For ruptured aneurysms, prognosis for good outcomes falls as the grade increases. For example, it is unusual for a Hunt-Hess grade 1 patient to die if the aneurysm is treated, but the mortality rate for grade 4 and 5 patients may be as high as 80%.
|
GRADING SCALES FOR SUBARACHNOID HEMORRHAGE |
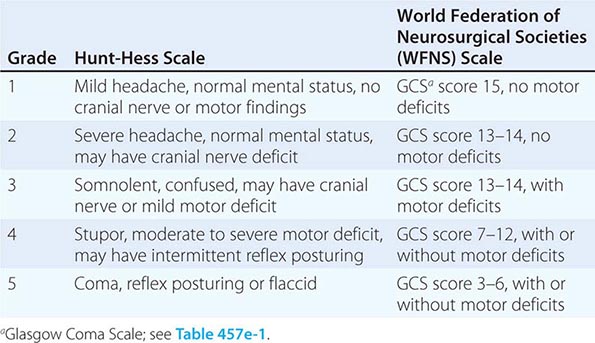
Delayed Neurologic Deficits There are four major causes of delayed neurologic deficits: rerupture, hydrocephalus, vasospasm, and hyponatremia.
1. Rerupture. The incidence of rerupture of an untreated aneurysm in the first month following SAH is ~30%, with the peak in the first 7 days. Rerupture is associated with a 60% mortality rate and poor outcome. Early treatment eliminates this risk.
2. Hydrocephalus. Acute hydrocephalus can cause stupor and coma and can be mitigated by placement of an external ventricular drain. More often, subacute hydrocephalus may develop over a few days or weeks and causes progressive drowsiness or slowed mentation (abulia) with incontinence. Hydrocephalus is differentiated from cerebral vasospasm with a CT scan, CT angiogram, transcranial Doppler (TCD) ultrasound, or conventional x-ray angiography. Hydrocephalus may clear spontaneously or require temporary ventricular drainage. Chronic hydrocephalus may develop weeks to months after SAH and manifest as gait difficulty, incontinence, or impaired mentation. Subtle signs may be a lack of initiative in conversation or a failure to recover independence.
3. Vasospasm. Narrowing of the arteries at the base of the brain following SAH causes symptomatic ischemia and infarction in ~30% of patients and is the major cause of delayed morbidity and death. Signs of ischemia appear 4–14 days after the hemorrhage, most often at 7 days. The severity and distribution of vasospasm determine whether infarction will occur.
4. Delayed vasospasm is believed to result from direct effects of clotted blood and its breakdown products on the arteries within the subarachnoid space. In general, the more blood that surrounds the arteries, the greater the chance of symptomatic vasospasm. Spasm of major arteries produces symptoms referable to the appropriate vascular territory (Chap. 446). All of these focal symptoms may present abruptly, fluctuate, or develop over a few days. In most cases, focal spasm is preceded by a decline in mental status.
5. Vasospasm can be detected reliably with conventional x-ray angiography, but this invasive procedure is expensive and carries the risk of stroke and other complications. TCD ultrasound is based on the principle that the velocity of blood flow within an artery will rise as the lumen diameter is narrowed. By directing the probe along the MCA and proximal anterior cerebral artery (ACA), carotid terminus, and vertebral and basilar arteries on a daily or every-other-day basis, vasospasm can be reliably detected and treatments initiated to prevent cerebral ischemia (see below). CT angiography is another method that can detect vasospasm.
6. Severe cerebral edema in patients with infarction from vasospasm may increase the ICP enough to reduce cerebral perfusion pressure. Treatment may include mannitol, hyperventilation, and hemicraniectomy; moderate hypothermia may have a role as well.
7. Hyponatremia. Hyponatremia may be profound and can develop quickly in the first 2 weeks following SAH. There is both natriuresis and volume depletion with SAH, so that patients become both hyponatremic and hypovolemic. Both atrial natriuretic peptide and brain natriuretic peptide have a role in producing this “cerebral salt-wasting syndrome.” Typically, it clears over the course of 1–2 weeks and, in the setting of SAH, should not be treated with free-water restriction as this may increase the risk of stroke (see below).
Laboratory Evaluation and Imaging (Fig. 330-7) The hallmark of aneurysmal rupture is blood in the CSF. More than 95% of cases have enough blood to be visualized on a high-quality noncontrast CT scan obtained within 72 h. If the scan fails to establish the diagnosis of SAH and no mass lesion or obstructive hydrocephalus is found, a lumbar puncture should be performed to establish the presence of subarachnoid blood. Lysis of the red blood cells and subsequent conversion of hemoglobin to bilirubin stains the spinal fluid yellow within 6–12 h. This xanthochromic spinal fluid peaks in intensity at 48 h and lasts for 1–4 weeks, depending on the amount of subarachnoid blood.
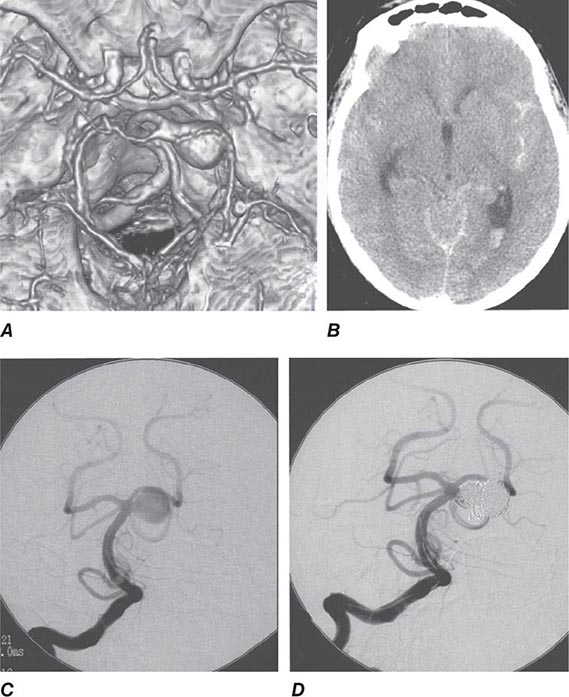
FIGURE 330-7 Subarachnoid hemorrhage. A. Computed tomography (CT) angiography revealing an aneurysm of the left superior cerebellar artery. B. Noncontrast CT scan at the level of the third ventricle revealing subarachnoid blood (bright) in the left sylvian fissure and within the left lateral ventricle. C. Conventional anteroposterior x-ray angiogram of the right vertebral and basilar artery showing the large aneurysm. D. Conventional angiogram following coil embolization of the aneurysm, whereby the aneurysm body is filled with platinum coils delivered through a microcatheter navigated from the femoral artery into the aneurysm neck.
The extent and location of subarachnoid blood on a noncontrast CT scan help locate the underlying aneurysm, identify the cause of any neurologic deficit, and predict delayed vasospasm. A high incidence of symptomatic vasospasm in the MCA and ACA has been found when early CT scans show subarachnoid clots >5 × 3 mm in the basal cisterns, or layers of blood >1 mm thick in the cerebral fissures. CT scans less reliably predict vasospasm in the vertebral, basilar, or posterior cerebral arteries.
Lumbar puncture prior to an imaging procedure is indicated only if a CT scan is not available at the time of the suspected SAH. Once the diagnosis of hemorrhage from a ruptured saccular aneurysm is suspected, four-vessel conventional x-ray angiography (both carotids and both vertebrals) is generally performed to localize and define the anatomic details of the aneurysm and to determine if other unruptured aneurysms exist (Fig. 330-7C). At some centers, the ruptured aneurysm can be treated using endovascular techniques at the time of the initial angiogram as a way to expedite treatment and minimize the number of invasive procedures. CT angiography is an alternative method for locating the aneurysm and may be sufficient to plan definitive therapy.
Close monitoring (daily or twice daily) of electrolytes is important because hyponatremia can occur precipitously during the first 2 weeks following SAH (see above).
The electrocardiogram (ECG) frequently shows ST-segment and T-wave changes similar to those associated with cardiac ischemia. A prolonged QRS complex, increased QT interval, and prominent “peaked” or deeply inverted symmetric T waves are usually secondary to the intracranial hemorrhage. There is evidence that structural myocardial lesions produced by circulating catecholamines and excessive discharge of sympathetic neurons may occur after SAH, causing these ECG changes and a reversible cardiomyopathy sufficient to cause shock or congestive heart failure. Echocardiography reveals a pattern of regional wall motion abnormalities that follow the distribution of sympathetic nerves rather than the major coronary arteries, with relative sparing of the ventricular wall apex. The sympathetic nerves themselves appear to be injured by direct toxicity from the excessive catecholamine release. An asymptomatic troponin elevation is common. Serious ventricular dysrhythmias occurring in-hospital are unusual.
SECTION 4 |
ONCOLOGIC EMERGENCIES |
331 |
Oncologic Emergencies |
Emergencies in patients with cancer may be classified into three groups: pressure or obstruction caused by a space-occupying lesion, metabolic or hormonal problems (paraneoplastic syndromes, Chap. 121), and treatment-related complications.
STRUCTURAL-OBSTRUCTIVE ONCOLOGIC EMERGENCIES
SUPERIOR VENA CAVA SYNDROME
Superior vena cava syndrome (SVCS) is the clinical manifestation of superior vena cava (SVC) obstruction, with severe reduction in venous return from the head, neck, and upper extremities. Malignant tumors, such as lung cancer, lymphoma, and metastatic tumors, are responsible for the majority of SVCS cases. With the expanding use of intravascular devices (e.g., permanent central venous access catheters, pacemaker/defibrillator leads), the prevalence of benign causes of SVCS is increasing now, accounting for at least 40% of cases. Lung cancer, particularly of small-cell and squamous cell histologies, accounts for approximately 85% of all cases of malignant origin. In young adults, malignant lymphoma is a leading cause of SVCS. Hodgkin’s lymphoma involves the mediastinum more commonly than other lymphomas but rarely causes SVCS. When SVCS is noted in a young man with a mediastinal mass, the differential diagnosis is lymphoma versus primary mediastinal germ cell tumor. Metastatic cancers to the mediastinal lymph nodes, such as testicular and breast carcinomas, account for a small proportion of cases. Other causes include benign tumors, aortic aneurysm, thyromegaly, thrombosis, and fibrosing mediastinitis from prior irradiation, histoplasmosis, or Behçet’s syndrome. SVCS as the initial manifestation of Behçet’s syndrome may be due to inflammation of the SVC associated with thrombosis.
Patients with SVCS usually present with neck and facial swelling (especially around the eyes), dyspnea, and cough. Other symptoms include hoarseness, tongue swelling, headaches, nasal congestion, epistaxis, hemoptysis, dysphagia, pain, dizziness, syncope, and lethargy. Bending forward or lying down may aggravate the symptoms. The characteristic physical findings are dilated neck veins; an increased number of collateral veins covering the anterior chest wall; cyanosis; and edema of the face, arms, and chest. Facial swelling and plethora are typically exacerbated when the patient is supine. More severe cases include proptosis, glossal and laryngeal edema, and obtundation. The clinical picture is milder if the obstruction is located above the azygos vein. Symptoms are usually progressive, but in some cases, they may improve as collateral circulation develops.
Signs and symptoms of cerebral and/or laryngeal edema, though rare, are associated with a poorer prognosis and require urgent evaluation. Seizures are more likely related to brain metastases than to cerebral edema from venous occlusion. Patients with small-cell lung cancer and SVCS have a higher incidence of brain metastases than those without SVCS.
Cardiorespiratory symptoms at rest, particularly with positional changes, suggest significant airway and vascular obstruction and limited physiologic reserve. Cardiac arrest or respiratory failure can occur, particularly in patients receiving sedatives or undergoing general anesthesia.
Rarely, esophageal varices may develop. These are “downhill” varices based on the direction of blood flow from cephalad to caudad (in contrast to “uphill” varices associated with caudad to cephalad flow from portal hypertension). If the obstruction to the SVC is proximal to the azygous vein, varices develop in the upper one-third of the esophagus. If the obstruction involves or is distal to the azygous vein, varices occur in the entire length of the esophagus. Variceal bleeding may be a late complication of chronic SVCS.
Superior vena cava obstruction may lead to bilateral breast edema with bilateral enlarged breast. Unilateral breast dilatation may be seen as a consequence of axillary or subclavian vein blockage.
The diagnosis of SVCS is a clinical one. The most significant chest radiographic finding is widening of the superior mediastinum, most commonly on the right side. Pleural effusion occurs in only 25% of patients, often on the right side. The majority of these effusions are exudative and occasionally chylous. However, a normal chest radiograph is still compatible with the diagnosis if other characteristic findings are present. Computed tomography (CT) provides the most reliable view of the mediastinal anatomy. The diagnosis of SVCS requires diminished or absent opacification of central venous structures with prominent collateral venous circulation. Magnetic resonance imaging (MRI) has no advantages over CT. Invasive procedures, including bronchoscopy, percutaneous needle biopsy, mediastinoscopy, and even thoracotomy, can be performed by a skilled clinician without any major risk of bleeding. Endobronchial or esophageal ultrasound-guided needle aspiration may establish the diagnosis safely. For patients with a known cancer, a detailed workup usually is not necessary, and appropriate treatment may be started after obtaining a CT scan of the thorax. For those with no history of malignancy, a detailed evaluation is essential to rule out benign causes and determine a specific diagnosis to direct the appropriate therapy.
|
TREATMENT |
SUPERIOR VENA CAVA SYNDROME |
The one potentially life-threatening complication of a superior mediastinal mass is tracheal obstruction. Upper airway obstruction demands emergent therapy. Diuretics with a low-salt diet, head elevation, and oxygen may produce temporary symptomatic relief. Glucocorticoids may be useful at shrinking lymphoma masses; they are of no benefit in patients with lung cancer.
Radiation therapy is the primary treatment for SVCS caused by non-small-cell lung cancer and other metastatic solid tumors. Chemotherapy is effective when the underlying cancer is small-cell carcinoma of the lung, lymphoma, or germ cell tumor. SVCS recurs in 10–30% of patients; it may be palliated with the use of intravascular self-expanding stents (Fig. 331-1). Early stenting may be necessary in patients with severe symptoms; however, the prompt increase in venous return after stenting may precipitate heart failure and pulmonary edema. Other complications of stent placement include hematoma at the insertion site, SVC perforation, stent migration in the right ventricle, stent fracture, and pulmonary embolism. Surgery may provide immediate relief for patients in whom a benign process is the cause.
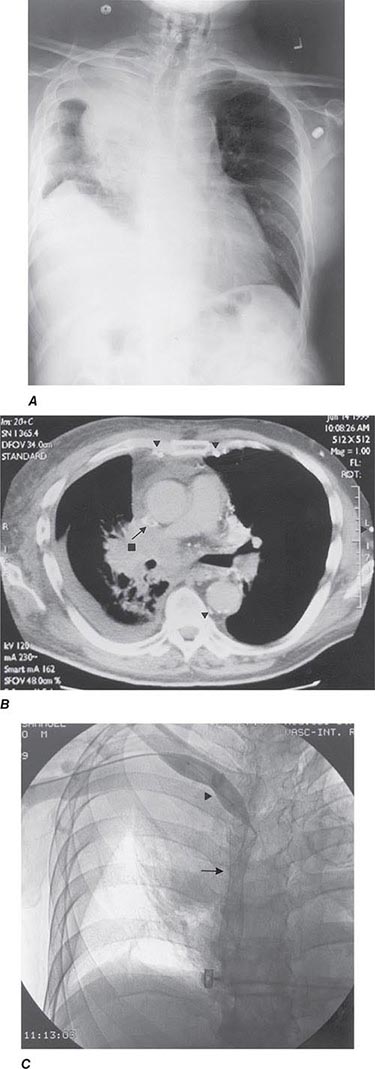
FIGURE 331-1 Superior vena cava syndrome (SVCS). A. Chest radiographs of a 59-year-old man with recurrent SVCS caused by non-small-cell lung cancer showing right paratracheal mass with right pleural effusion. B. Computed tomography of same patient demonstrating obstruction of the superior vena cava with thrombosis (arrow) by the lung cancer (square) and collaterals (arrowheads). C. Balloon angioplasty (arrowhead) with Wallstent (arrow) in same patient.
Clinical improvement occurs in most patients, although this improvement may be due to the development of adequate collateral circulation. The mortality associated with SVCS does not relate to caval obstruction but rather to the underlying cause.
SVCS AND CENTRAL VENOUS CATHETERS IN ADULTS
The use of long-term central venous catheters has become common practice in patients with cancer. Major vessel thrombosis may occur. In these cases, catheter removal should be combined with anticoagulation to prevent embolization. SVCS in this setting, if detected early, can be treated by fibrinolytic therapy without sacrificing the catheter. The routine use of low-dose warfarin or low-molecular-weight heparin to prevent thrombosis related to permanent central venous access catheters in cancer patients is not recommended.
PERICARDIAL EFFUSION/TAMPONADE
Malignant pericardial disease is found at autopsy in 5–10% of patients with cancer, most frequently with lung cancer, breast cancer, leukemias, and lymphomas. Cardiac tamponade as the initial presentation of extrathoracic malignancy is rare. The origin is not malignancy in about 50% of cancer patients with symptomatic pericardial disease, but it can be related to irradiation, drug-induced pericarditis, hypothyroidism, idiopathic pericarditis, infection, or autoimmune diseases. Two types of radiation pericarditis occur: an acute inflammatory, effusive pericarditis occurring within months of irradiation, which usually resolves spontaneously, and a chronic effusive pericarditis that may appear up to 20 years after radiation therapy and is accompanied by a thickened pericardium.
Most patients with pericardial metastasis are asymptomatic. However, the common symptoms are dyspnea, cough, chest pain, orthopnea, and weakness. Pleural effusion, sinus tachycardia, jugular venous distention, hepatomegaly, peripheral edema, and cyanosis are the most frequent physical findings. Relatively specific diagnostic findings, such as paradoxical pulse, diminished heart sounds, pulsus alternans (pulse waves alternating between those of greater and lesser amplitude with successive beats), and friction rub are less common than with nonmalignant pericardial disease. Chest radiographs and electrocardiogram (ECG) reveal abnormalities in 90% of patients, but half of these abnormalities are nonspecific. Echocardiography is the most helpful diagnostic test. Pericardial fluid may be serous, serosanguineous, or hemorrhagic, and cytologic examination of pericardial fluid is diagnostic in most patients. Measurements of tumor markers in the pericardial fluid are not helpful in the diagnosis of malignant pericardial fluid. Pericardioscopy (not widely available) with targeted pericardial and epicardial biopsy may differentiate neoplastic and benign pericardial disease. A combination of cytology, pericardial and epicardial biopsy, and guided pericardioscopy gives the best diagnostic yield. CT scan findings of irregular pericardial thickening and mediastinal lymphadenopathy suggest this is a malignant pericardial effusion. Cancer patients with pericardial effusion containing malignant cells on cytology have a very poor survival, about 7 weeks.
INTESTINAL OBSTRUCTION
Intestinal obstruction and reobstruction are common problems in patients with advanced cancer, particularly colorectal or ovarian carcinoma. However, other cancers, such as lung or breast cancer and melanoma, can metastasize within the abdomen, leading to intestinal obstruction. Metastatic disease from colorectal, ovarian, pancreatic, gastric, and occasionally breast cancer can lead to peritoneal carcinomatosis, with infiltration of the omentum and peritoneal surface, thus limiting bowel motility. Typically, obstruction occurs at multiple sites in peritoneal carcinomatosis. Melanoma has a predilection to involve the small bowel; this involvement may be isolated, and resection may result in prolonged survival. Intestinal pseudoobstruction is caused by infiltration of the mesentery or bowel muscle by tumor, involvement of the celiac plexus, or paraneoplastic neuropathy in patients with small-cell lung cancer. Paraneoplastic neuropathy is associated with IgG antibodies reactive to neurons of the myenteric and submucosal plexuses of the jejunum and stomach. Ovarian cancer can lead to authentic luminal obstruction or to pseudoobstruction that results when circumferential invasion of a bowel segment arrests the forward progression of peristaltic contractions.
The onset of obstruction is usually insidious. Pain is the most common symptom and is usually colicky in nature. Pain can also be due to abdominal distention, tumor masses, or hepatomegaly. Vomiting can be intermittent or continuous. Patients with complete obstruction usually have constipation. Physical examination may reveal abdominal distention with tympany, ascites, visible peristalsis, high-pitched bowel sounds, and tumor masses. Erect plain abdominal films may reveal multiple air-fluid levels and dilation of the small or large bowel. Acute cecal dilation to >12–14 cm is considered a surgical emergency because of the high likelihood of rupture. CT scan is useful in defining the extent of disease and the exact nature of the obstruction and differentiating benign from malignant causes of obstruction in patients who have undergone surgery for malignancy. Malignant obstruction is suggested by a mass at the site of obstruction or prior surgery, adenopathy, or an abrupt transition zone and irregular bowel thickening at the obstruction site. Benign obstruction is more likely when CT shows mesenteric vascular changes, a large volume of ascites, or a smooth transition zone and smooth bowel thickening at the obstruction site. In challenging patients with obstructive symptoms, particularly low-grade small-bowel obstruction (SBO), CT enteroclysis often can help establish the diagnosis by providing distention of small-bowel loops. In this technique, water-soluble contrast is infused through a nasoenteric tube into the duodenum or proximal small bowel followed by CT images. The prognosis for the patient with cancer who develops intestinal obstruction is poor; median survival is 3–4 months. About 25–30% of patients are found to have intestinal obstruction due to causes other than cancer. Adhesions from previous operations are a common benign cause. Ileus induced by vinca alkaloids, narcotics, or other drugs is another reversible cause.
URINARY OBSTRUCTION
Urinary obstruction may occur in patients with prostatic or gynecologic malignancies, particularly cervical carcinoma; metastatic disease from other primary sites such as carcinomas of the breast, stomach, lung, colon, and pancreas; or lymphomas. Radiation therapy to pelvic tumors may cause fibrosis and subsequent ureteral obstruction. Bladder outlet obstruction is usually due to prostate and cervical cancers and may lead to bilateral hydronephrosis and renal failure.
Flank pain is the most common symptom. Persistent urinary tract infection, persistent proteinuria, or hematuria in patients with cancer should raise suspicion of ureteral obstruction. Total anuria and/or anuria alternating with polyuria may occur. A slow, continuous rise in the serum creatinine level necessitates immediate evaluation. Renal ultrasound is the safest and cheapest way to identify hydronephrosis. The function of an obstructed kidney can be evaluated by a nuclear scan. CT scan can reveal the point of obstruction and identify a retroperitoneal mass or adenopathy.
MALIGNANT BILIARY OBSTRUCTION
This common clinical problem can be caused by a primary carcinoma arising in the pancreas, ampulla of Vater, bile duct, or liver or by metastatic disease to the periductal lymph nodes or liver parenchyma. The most common metastatic tumors causing biliary obstruction are gastric, colon, breast, and lung cancers. Jaundice, light-colored stools, dark urine, pruritus, and weight loss due to malabsorption are usual symptoms. Pain and secondary infection are uncommon in malignant biliary obstruction. Ultrasound, CT scan, or percutaneous transhepatic or endoscopic retrograde cholangiography will identify the site and nature of the biliary obstruction.
SPINAL CORD COMPRESSION
Malignant spinal cord compression (MSCC) is defined as compression of the spinal cord and/or cauda equina by an extradural tumor mass. The minimum radiologic evidence for cord compression is indentation of the theca at the level of clinical features. Spinal cord compression occurs in 5–10% of patients with cancer. Epidural tumor is the first manifestation of malignancy in about 10% of patients. The underlying cancer is usually identified during the initial evaluation; lung cancer is the most common cause of MSCC.
Metastatic tumor involves the vertebral column more often than any other part of the bony skeleton. Lung, breast, and prostate cancer are the most frequent offenders. Multiple myeloma also has a high incidence of spine involvement. Lymphomas, melanoma, renal cell cancer, and genitourinary cancers also cause cord compression. The thoracic spine is the most common site (70%), followed by the lumbosacral spine (20%) and the cervical spine (10%). Involvement of multiple sites is most frequent in patients with breast and prostate carcinoma. Cord injury develops when metastases to the vertebral body or pedicle enlarge and compress the underlying dura. Another cause of cord compression is direct extension of a paravertebral lesion through the intervertebral foramen. These cases usually involve a lymphoma, myeloma, or pediatric neoplasm. Parenchymal spinal cord metastasis due to hematogenous spread is rare. Intramedullary metastases can be seen in lung cancer, breast cancer, renal cancer, melanoma, and lymphoma and are frequently associated with brain metastases and leptomeningeal disease.
Expanding extradural tumors induce injury through several mechanisms. Obstruction of the epidural venous plexus leads to edema. Local production of inflammatory cytokines enhances blood flow and edema formation. Compression compromises blood flow, leading to ischemia. Production of vascular endothelial growth factor is associated with spinal cord hypoxia and has been implicated as a potential cause of damage after spinal cord injury.
The most common initial symptom in patients with spinal cord compression is localized back pain and tenderness due to involvement of vertebrae by tumor. Pain is usually present for days or months before other neurologic findings appear. It is exacerbated by movement and by coughing or sneezing. It can be differentiated from the pain of disk disease by the fact that it worsens when the patient is supine. Radicular pain is less common than localized back pain and usually develops later. Radicular pain in the cervical or lumbosacral areas may be unilateral or bilateral. Radicular pain from the thoracic roots is often bilateral and is described by patients as a feeling of tight, band-like constriction around the thorax and abdomen. Typical cervical radicular pain radiates down the arm; in the lumbar region, the radiation is down the legs. Lhermitte’s sign, a tingling or electric sensation down the back and upper and lower limbs upon flexing or extending the neck, may be an early sign of cord compression. Loss of bowel or bladder control may be the presenting symptom but usually occurs late in the course. Occasionally patients present with ataxia of gait without motor and sensory involvement due to involvement of the spinocerebellar tract.
On physical examination, pain induced by straight leg raising, neck flexion, or vertebral percussion may help to determine the level of cord compression. Patients develop numbness and paresthesias in the extremities or trunk. Loss of sensibility to pinprick is as common as loss of sensibility to vibration or position. The upper limit of the zone of sensory loss is often one or two vertebrae below the site of compression. Motor findings include weakness, spasticity, and abnormal muscle stretching. An extensor plantar reflex reflects significant compression. Deep tendon reflexes may be brisk. Motor and sensory loss usually precedes sphincter disturbance. Patients with autonomic dysfunction may present with decreased anal tonus, decreased perineal sensibility, and a distended bladder. The absence of the anal wink reflex or the bulbocavernosus reflex confirms cord involvement. In doubtful cases, evaluation of postvoiding urinary residual volume can be helpful. A residual volume of >150 mL suggests bladder dysfunction. Autonomic dysfunction is an unfavorable prognostic factor. Patients with progressive neurologic symptoms should have frequent neurologic examinations and rapid therapeutic intervention. Other illnesses that may mimic cord compression include osteoporotic vertebral collapse, disk disease, pyogenic abscess or vertebral tuberculosis, radiation myelopathy, neoplastic leptomeningitis, benign tumors, epidural hematoma, and spinal lipomatosis.
Cauda equina syndrome is characterized by low back pain; diminished sensation over the buttocks, posterior-superior thighs, and perineal area in a saddle distribution; rectal and bladder dysfunction; sexual impotence; absent bulbocavernous, patellar, and Achilles’ reflexes; and variable amount of lower-extremity weakness. This reflects compression of nerve roots as they form the cauda equina after leaving the spinal cord. The majority of cauda equine tumors are primary tumors of glial or nerve sheath origin; metastases are very rare.
Patients with cancer who develop back pain should be evaluated for spinal cord compression as quickly as possible (Fig. 331-2). Treatment is more often successful in patients who are ambulatory and still have sphincter control at the time treatment is initiated. Patients should have a neurologic examination and plain films of the spine. Those whose physical examination suggests cord compression should receive dexamethasone (6 mg intravenously every 6 h), starting immediately.
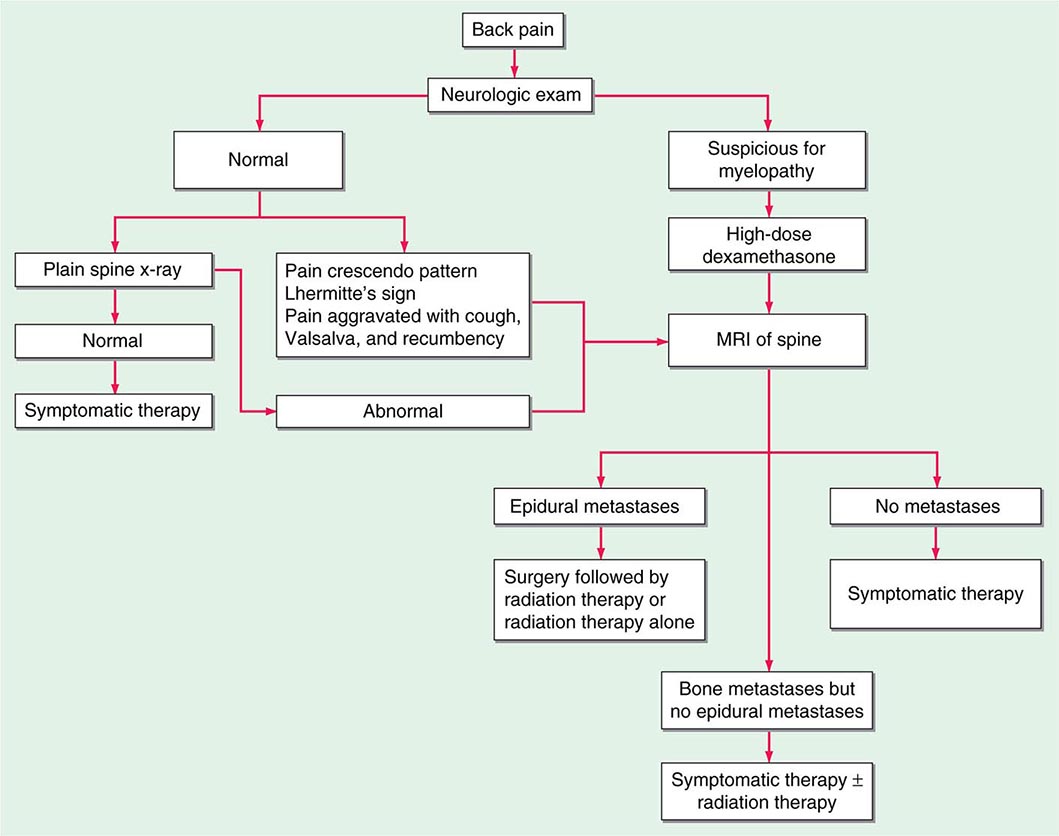
FIGURE 331-2 Management of cancer patients with back pain.
Erosion of the pedicles (the “winking owl” sign) is the earliest radiologic finding of vertebral tumor. Other radiographic changes include increased intrapedicular distance, vertebral destruction, lytic or sclerotic lesions, scalloped vertebral bodies, and vertebral body collapse. Vertebral collapse is not a reliable indicator of the presence of tumor; about 20% of cases of vertebral collapse, particularly those in older patients and postmenopausal women, are due not to cancer but to osteoporosis. Also, a normal appearance on plain films of the spine does not exclude the diagnosis of cancer. The role of bone scans in the detection of cord compression is not clear; this method is sensitive but less specific than spinal radiography.
The full-length image of the cord provided by MRI is the imaging procedure of choice. Multiple epidural metastases are noted in 25% of patients with cord compression, and their presence influences treatment plans. On T1-weighted images, good contrast is noted between the cord, cerebrospinal fluid, and extradural lesions. Owing to its sensitivity in demonstrating the replacement of bone marrow by tumor, MRI can show which parts of a vertebra are involved by tumor. MRI also visualizes intraspinal extradural masses compressing the cord. T2-weighted images are most useful for the demonstration of intramedullary pathology. Gadolinium-enhanced MRI can help to delineate intramedullary disease. MRI is as good as or better than myelography plus postmyelogram CT scan in detecting metastatic epidural disease with cord compression. Myelography should be reserved for patients who have poor MRIs or who cannot undergo MRI promptly. CT scan in conjunction with myelography enhances the detection of small areas of spinal destruction.
In patients with cord compression and an unknown primary tumor, a simple workup including chest radiography, mammography, measurement of prostate-specific antigen, and abdominal CT usually reveals the underlying malignancy.
INCREASED INTRACRANIAL PRESSURE
About 25% of patients with cancer die with intracranial metastases. The cancers that most often metastasize to the brain are lung and breast cancers and melanoma. Brain metastases often occur in the presence of systemic disease, and they frequently cause major symptoms, disability, and early death. The initial presentation of brain metastases from a previously unknown primary cancer is common. Lung cancer is most commonly the primary malignancy. Chest/abdomen CT scans and brain MRI as the initial diagnostic studies can identify a biopsy site in most patients.
The signs and symptoms of a metastatic brain tumor are similar to those of other intracranial expanding lesions: headache, nausea, vomiting, behavioral changes, seizures, and focal, progressive neurologic changes. Occasionally the onset is abrupt, resembling a stroke, with the sudden appearance of headache, nausea, vomiting, and neurologic deficits. This picture is usually due to hemorrhage into the metastasis. Melanoma, germ cell tumors, and renal cell cancers have a particularly high incidence of intracranial bleeding. The tumor mass and surrounding edema may cause obstruction of the circulation of cerebrospinal fluid, with resulting hydrocephalus. Patients with increased intracranial pressure may have papilledema with visual disturbances and neck stiffness. As the mass enlarges, brain tissue may be displaced through the fixed cranial openings, producing various herniation syndromes.
CT scan and MRI are equally effective in the diagnosis of brain metastases. CT scan with contrast should be used as a screening procedure. The CT scan shows brain metastases as multiple enhancing lesions of various sizes with surrounding areas of low-density edema. If a single lesion or no metastases are visualized by contrast-enhanced CT, MRI of the brain should be performed. Gadolinium-enhanced MRI is more sensitive than CT at revealing meningeal involvement and small lesions, particularly in the brainstem or cerebellum.
Intracranial hypertension (“pseudotumor cerebri”) secondary to tretinoin therapy has been reported.
NEOPLASTIC MENINGITIS
Tumor involving the leptomeninges is a complication of both primary central nervous system (CNS) tumors and tumors that metastasize to the CNS. The incidence is estimated at 3–8% of patients with cancer. Melanoma, breast and lung cancer, lymphoma (including AIDS-associated), and acute leukemia are the most common causes. Synchronous intraparenchymal brain metastases are evident in 11–31% of patients with neoplastic meningitis. Leptomeningeal seeding is frequent in patients undergoing resection of brain metastases or receiving stereotactic radiotherapy for brain metastases.
Patients typically present with multifocal neurologic signs and symptoms, including headache, gait abnormality, mental changes, nausea, vomiting, seizures, back or radicular pain, and limb weakness. Signs include cranial nerve palsies, extremity weakness, paresthesia, and decreased deep tendon reflexes.
Diagnosis is made by demonstrating malignant cells in the CSF; however, up to 40% of patients may have false-negative CSF cytology. An elevated CSF protein level is nearly always present (except in HTLV-1–associated adult T cell leukemia). Patients with neurologic signs and symptoms consistent with neoplastic meningitis who have a negative CSF cytology but an elevated CSF protein level should have the spinal tap repeated at least three times for cytologic examination before the diagnosis is rejected. MRI findings suggestive of neoplastic meningitis include leptomeningeal, subependymal, dural, or cranial nerve enhancement; superficial cerebral lesions; intradural nodules; and communicating hydrocephalus. Spinal cord imaging by MRI is a necessary component of the evaluation of nonleukemia neoplastic meningitis because ~20% of patients have cord abnormalities, including intradural enhancing nodules that are diagnostic for leptomeningeal involvement. Cauda equina lesions are common, but lesions may be seen anywhere in the spinal canal. The value of MRI for the diagnosis of leptomeningeal disease is limited in patients with hematopoietic malignancy. Radiolabeled CSF flow studies are abnormal in up to 70% of patients with neoplastic meningitis; ventricular outlet obstruction, abnormal flow in the spinal canal, or impaired flow over the cerebral convexities may affect distribution of intrathecal chemotherapy, resulting in decreased efficacy or increased toxicity. Radiation therapy may correct CSF flow abnormalities before use of intrathecal chemotherapy. Neoplastic meningitis can also lead to intracranial hypertension and hydrocephalus. Placement of a ventriculoperitoneal shunt may effectively palliate symptoms in these patients.
The development of neoplastic meningitis usually occurs in the setting of uncontrolled cancer outside the CNS; thus, prognosis is poor (median survival 10–12 weeks). However, treatment of the neoplastic meningitis may successfully alleviate symptoms and control the CNS spread.
SEIZURES
Seizures occurring in a patient with cancer can be caused by the tumor itself, by metabolic disturbances, by radiation injury, by cerebral infarctions, by chemotherapy-related encephalopathies, or by CNS infections. Metastatic disease to the CNS is the most common cause of seizures in patients with cancer. However, seizures occur more frequently in primary brain tumors than in metastatic brain lesions. Seizures are a presenting symptom of CNS metastasis in 6–29% of cases. Approximately 10% of patients with CNS metastasis eventually develop seizures. Tumors that affect the frontal, temporal, and parietal lobes are more commonly associated with seizures than are occipital lesions. The presence of frontal lesions correlates with early seizures, and the presence of hemispheric symptoms increases the risk for late seizures. Both early and late seizures are uncommon in patients with posterior fossa and sellar lesions. Seizures are common in patients with CNS metastases from melanoma and low-grade primary brain tumors. Very rarely, cytotoxic drugs such as etoposide, busulfan, ifosfamide, and chlorambucil cause seizures. Another cause of seizures related to drug therapy is reversible posterior leukoencephalopathy syndrome (RPLS). RPLS is associated rarely with administration of cisplatin, 5-fluorouracil, bleomycin, vinblastine, vincristine, etoposide, paclitaxel, ifosfamide, cyclophosphamide, doxorubicin, cytarabine, methotrexate, oxaliplatin, cyclosporine, tacrolimus, and vascular endothelial growth factor inhibitors including bevacizumab, aflibercept, sunitinib, sorafenib, pazopanib, and axitinib. RPLS occurs in patients undergoing allogeneic bone marrow or solid-organ transplantation. RPLS is characterized by headache, altered consciousness, generalized seizures, visual disturbances, hypertension, and posterior cerebral white matter vasogenic edema on CT/MRI. Seizures may begin focally but are typically generalized.
PULMONARY AND INTRACEREBRAL LEUKOSTASIS
Hyperleukocytosis and the leukostasis syndrome associated with it is a potentially fatal complication of acute leukemia (particularly myeloid leukemia) that can occur when the peripheral blast cell count is >100,000/mL. The frequency of hyperleukocytosis is 5–13% in acute myeloid leukemia (AML) and 10–30% in acute lymphoid leukemia; however, leukostasis is rare in lymphoid leukemia. At such high blast cell counts, blood viscosity is increased, blood flow is slowed by aggregates of tumor cells, and the primitive myeloid leukemic cells are capable of invading through the endothelium and causing hemorrhage. Brain and lung are most commonly affected. Patients with brain leukostasis may experience stupor, headache, dizziness, tinnitus, visual disturbances, ataxia, confusion, coma, or sudden death. On examination, papilledema, retinal vein distension, retinal hemorrhages, and focal deficit may be present. Administration of 600 cGy of whole-brain irradiation can protect against this complication and can be followed by rapid institution of antileukemic therapy. Hydroxyurea, 3–5 g, can rapidly reduce a high blast cell count while the accurate diagnostic workup is in progress. Pulmonary leukostasis may present as respiratory distress and hypoxemia, and progress to respiratory failure. Chest radiographs may be normal but usually show interstitial or alveolar infiltrates. Hyperleukocytosis rarely may cause acute leg ischemia, renal vein thrombosis, myocardial ischemia, bowel infraction, and priapism. Arterial blood gas results should be interpreted cautiously. Rapid consumption of plasma oxygen by the markedly increased number of white blood cells can cause spuriously low arterial oxygen tension. Pulse oximetry is the most accurate way of assessing oxygenation in patients with hyperleukocytosis. Leukapheresis may be helpful in decreasing circulating blast counts. Treatment of the leukemia can result in pulmonary hemorrhage from lysis of blasts in the lung, called leukemic cell lysis pneumopathy. Intravascular volume depletion and unnecessary blood transfusions may increase blood viscosity and worsen the leukostasis syndrome. Leukostasis is very rarely a feature of the high white cell counts associated with chronic lymphoid or chronic myeloid leukemia.
When acute promyelocytic leukemia is treated with differentiating agents like tretinoin and arsenic trioxide, cerebral or pulmonary leukostasis may occur as tumor cells differentiate into mature neutrophils. This complication can be largely avoided by using cytotoxic chemotherapy or arsenic together with the differentiating agents.
HEMOPTYSIS
Hemoptysis may be caused by nonmalignant conditions, but lung cancer accounts for a large proportion of cases. Up to 20% of patients with lung cancer have hemoptysis some time in their course. Endobronchial metastases from carcinoid tumors, breast cancer, colon cancer, kidney cancer, and melanoma may also cause hemoptysis. The volume of bleeding is often difficult to gauge. Massive hemoptysis is defined as >200–600 mL of blood produced in 24 h. However, any hemoptysis should be considered massive if it threatens life. When respiratory difficulty occurs, hemoptysis should be treated emergently. The first priorities are to maintain the airway, optimize oxygenation, and stabilize the hemodynamic status. If the bleeding side is known, the patient should be placed in a lateral decubitus position, with the bleeding side down to prevent aspiration into the unaffected lung, and given supplemental oxygen. If large-volume bleeding continues or the airway is compromised, the patient should be intubated and undergo emergency bronchoscopy. If the site of bleeding is detected, either the patient undergoes a definitive surgical procedure or the lesion is treated with a neodymium:yttrium-aluminum-garnet (Nd:YAG) laser, argon plasma coagulation, or electrocautery. In stable patients, multidetector CT angiography delineates bronchial and nonbronchial systemic arteries and identifies the source of bleeding and underlying pathology with high sensitivity. Massive hemoptysis usually originates from the high-pressure bronchial circulation. Bronchial artery embolization is considered a first-line definite procedure for managing hemoptysis. Bronchial artery embolization may control brisk bleeding in 75–90% of patients, permitting the definitive surgical procedure to be done more safely.
Embolization without definitive surgery is associated with rebleeding in 20–50% of patients. Recurrent hemoptysis usually responds to a second embolization procedure. A postembolization syndrome characterized by pleuritic pain, fever, dysphagia, and leukocytosis may occur; it lasts 5–7 days and resolves with symptomatic treatment. Bronchial or esophageal wall necrosis, myocardial infarction, and spinal cord infarction are rare complications. Surgery, as a salvage strategy, is indicated after failure of embolization and is associated with better survival when performed in a nonurgent setting.
Pulmonary hemorrhage with or without hemoptysis in hematologic malignancies is often associated with fungal infections, particularly Aspergillus sp. After granulocytopenia resolves, the lung infiltrates in aspergillosis may cavitate and cause massive hemoptysis. Thrombocytopenia and coagulation defects should be corrected, if possible. Surgical evaluation is recommended in patients with aspergillosis-related cavitary lesions.
Bevacizumab, an antibody to vascular endothelial growth factor (VEGF) that inhibits angiogenesis, has been associated with life-threatening hemoptysis in patients with non-small-cell lung cancer, particularly of squamous cell histology. Non-small-cell lung cancer patients with cavitary lesions or previous hemoptysis (≥2.5 mL) within the past 3 months have higher risk for pulmonary hemorrhage.
AIRWAY OBSTRUCTION
Airway obstruction refers to a blockage at the level of the mainstem bronchi or above. It may result either from intraluminal tumor growth or from extrinsic compression of the airway. The most common cause of malignant upper airway obstruction is invasion from an adjacent primary tumor, most commonly lung cancer, followed by esophageal, thyroid, and mediastinal malignancies including lymphomas. Extrathoracic primary tumors such as renal, colon, or breast cancer can cause airway obstruction through endobronchial and/or mediastinal lymph node metastases. Patients may present with dyspnea, hemoptysis, stridor, wheezing, intractable cough, postobstructive pneumonia, or hoarseness. Chest radiographs usually demonstrate obstructing lesions. CT scans reveal the extent of tumor. Cool, humidified oxygen, glucocorticoids, and ventilation with a mixture of helium and oxygen (Heliox) may provide temporary relief. If the obstruction is proximal to the larynx, a tracheostomy may be lifesaving. For more distal obstructions, particularly intrinsic lesions incompletely obstructing the airway, bronchoscopy with mechanical debulking and dilatation or ablational treatments including laser treatment, photodynamic therapy, argon plasma coagulation, electrocautery, or stenting can produce immediate relief in most patients (Fig. 331-3). However, radiation therapy (either external-beam irradiation or brachytherapy) given together with glucocorticoids may also open the airway. Symptomatic extrinsic compression may be palliated by stenting. Patients with primary airway tumors such as squamous cell carcinoma, carcinoid tumor, adenocystic carcinoma, or non-small-cell lung cancer, if resectable, should have surgery.
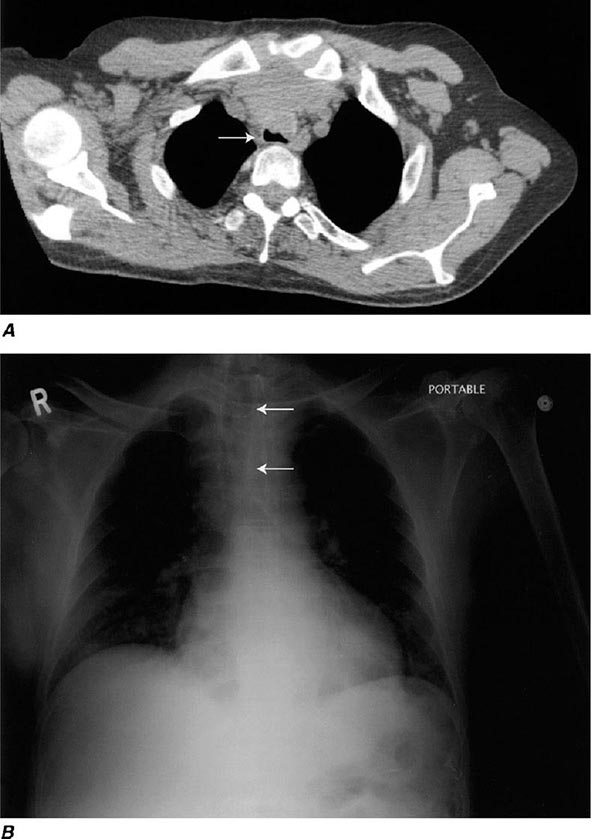
FIGURE 331-3 Airway obstruction. A. Computed tomography scan of a 62-year-old man with tracheal obstruction caused by renal carcinoma showing paratracheal mass with tracheal invasion/obstruction (arrow). B. Chest x-ray of same patient after stent (arrows) placement.
METABOLIC EMERGENCIES
HYPERCALCEMIA
Hypercalcemia is the most common paraneoplastic syndrome. Its pathogenesis and management are discussed fully in Chaps. 121 and 424.
SYNDROME OF INAPPROPRIATE SECRETION OF ANTIDIURETIC HORMONE (SIADH)
Hyponatremia is a common electrolyte abnormality in cancer patients, and SIADH is the most common cause among patients with cancer. SIADH is discussed fully in Chaps. 121 and 401e.
LACTIC ACIDOSIS
Lactic acidosis is a rare and potentially fatal metabolic complication of cancer. Lactic acidosis associated with sepsis and circulatory failure is a common preterminal event in many malignancies. Lactic acidosis in the absence of hypoxemia may occur in patients with leukemia, lymphoma, or solid tumors. In some cases, hypoglycemia also is present. Extensive involvement of the liver by tumor is often present. In most cases, decreased metabolism and increased production by the tumor both contribute to lactate accumulation. Tumor cell overexpression of certain glycolytic enzymes and mitochondrial dysfunction can contribute to its increased lactate production. HIV-infected patients have an increased risk of aggressive lymphoma; lactic acidosis that occurs in such patients may be related either to the rapid growth of the tumor or from toxicity of nucleoside reverse transcriptase inhibitors. Symptoms of lactic acidosis include tachypnea, tachycardia, change of mental status, and hepatomegaly. The serum level of lactic acid may reach 10–20 mmol/L (90–180 mg/dL). Treatment is aimed at the underlying disease. The danger from lactic acidosis is from the acidosis, not the lactate. Sodium bicarbonate should be added if acidosis is very severe or if hydrogen ion production is very rapid and uncontrolled. Other treatment options include renal replacement therapy, such as hemodialysis, and thiamine replacement. The prognosis is poor regardless of the treatment offered.
HYPOGLYCEMIA
Persistent hypoglycemia is occasionally associated with tumors other than pancreatic islet cell tumors. Usually these tumors are large; tumors of mesenchymal origin, hepatomas, or adrenocortical tumors may cause hypoglycemia. Mesenchymal tumors are usually located in the retroperitoneum or thorax. Obtundation, confusion, and behavioral aberrations occur in the postabsorptive period and may precede the diagnosis of the tumor. These tumors often secrete incompletely processed insulin-like growth factor II (IGF-II), a hormone capable of activating insulin receptors and causing hypoglycemia. Tumors secreting incompletely processed big IGF-II are characterized by an increased IGF-II to IGF-I ratio, suppressed insulin and C-peptide level, and inappropriately low growth hormone and β-hydroxybutyrate concentrations. Rarely, hypoglycemia is due to insulin secretion by a non-islet cell carcinoma. The development of hepatic dysfunction from liver metastases and increased glucose consumption by the tumor can contribute to hypoglycemia. If the tumor cannot be resected, hypoglycemia symptoms may be relieved by the administration of glucose, glucocorticoids, or glucagon.
Hypoglycemia can be artifactual; hyperleukocytosis from leukemia, myeloproliferative diseases, leukemoid reactions, or colony-stimulating factor treatment can increase glucose consumption in the test tube after blood is drawn, leading to pseudohypoglycemia.
ADRENAL INSUFFICIENCY
In patients with cancer, adrenal insufficiency may go unrecognized because the symptoms, such as nausea, vomiting, anorexia, and orthostatic hypotension, are nonspecific and may be mistakenly attributed to progressive cancer or to therapy. Primary adrenal insufficiency may develop owing to replacement of both glands by metastases (lung, breast, colon, or kidney cancer; lymphoma), to removal of both glands, or to hemorrhagic necrosis in association with sepsis or anticoagulation. Impaired adrenal steroid synthesis occurs in patients being treated for cancer with mitotane, ketoconazole, or aminoglutethimide or undergoing rapid reduction in glucocorticoid therapy. Rarely, metastatic replacement causes primary adrenal insufficiency as the first manifestation of an occult malignancy. Metastasis to the pituitary or hypothalamus is found at autopsy in up to 5% of patients with cancer, but associated secondary adrenal insufficiency is rare. On the other hand, ipilimumab, an anti-CTLA-4 antibody used for treatment of malignant melanoma, may cause autoimmunity including autoimmune-like enterocolitis, hypophysitis, and hepatitis. Autoimmune hypophysitis may present with headache, visual field defects, and pituitary hormone deficiencies manifesting as hypopituitarism, adrenal insufficiency (including adrenal crisis), or hypothyroidism. Anti-CTLA-4-associated hypophysitis symptoms occur at an average of 6–12 weeks after initiation of therapy. The treatment of severe autoimmune toxicity is glucocorticoids. Almost all patients with hypophysitis respond to withdrawal of ipilimumab and glucocorticoid therapy in several days. However, pituitary dysfunction may resolve or may be permanent, requiring long-term therapy and thyroid and testosterone replacement. Peripheral Addison’s disease can also be observed with anti-CTLA-4 antibodies. Megestrol acetate, used to manage cancer and HIV-related cachexia, may suppress plasma levels of cortisol and adrenocorticotropic hormone (ACTH). Patients taking megestrol may develop adrenal insufficiency, and even those whose adrenal dysfunction is not symptomatic may have inadequate adrenal reserve if they become seriously ill. Paradoxically, some patients may develop Cushing’s syndrome and/or hyperglycemia because of the glucocorticoid-like activity of megestrol acetate. Cranial irradiation for childhood brain tumors may affect the hypothalamus-pituitary-adrenal axis, resulting in secondary adrenal insufficiency.
Acute adrenal insufficiency is potentially lethal. Treatment of suspected adrenal crisis is initiated after the sampling of serum cortisol and ACTH levels (Chap. 406).
TREATMENT-RELATED EMERGENCIES
TUMOR LYSIS SYNDROME
Tumor lysis syndrome (TLS) is characterized by hyperuricemia, hyperkalemia, hyperphosphatemia, and hypocalcemia and is caused by the destruction of a large number of rapidly proliferating neoplastic cells. Acidosis may also develop. Acute renal failure occurs frequently.
TLS is most often associated with the treatment of Burkitt’s lymphoma, acute lymphoblastic leukemia, and other rapidly proliferating lymphomas, but it also may be seen with chronic leukemias and, rarely, with solid tumors. This syndrome has been seen in patients with chronic lymphocytic leukemia after treatment with nucleosides like fludarabine. TLS has been observed with administration of glucocorticoids, hormonal agents such as letrozole and tamoxifen, and monoclonal antibodies such as rituximab and gemtuzumab. TLS usually occurs during or shortly (1–5 days) after chemotherapy. Rarely, spontaneous necrosis of malignancies causes TLS.
Hyperuricemia may be present at the time of chemotherapy. Effective treatment kills malignant cells and leads to increased serum uric acid levels from the turnover of nucleic acids. Owing to the acidic local environment, uric acid can precipitate in the tubules, medulla, and collecting ducts of the kidney, leading to renal failure. Lactic acidosis and dehydration may contribute to the precipitation of uric acid in the renal tubules. The finding of uric acid crystals in the urine is strong evidence for uric acid nephropathy. The ratio of urinary uric acid to urinary creatinine is >1 in patients with acute hyperuricemic nephropathy and <1 in patients with renal failure due to other causes.
Hyperphosphatemia, which can be caused by the release of intracellular phosphate pools by tumor lysis, produces a reciprocal depression in serum calcium, which causes severe neuromuscular irritability and tetany. Deposition of calcium phosphate in the kidney and hyperphosphatemia may cause renal failure. Potassium is the principal intracellular cation, and massive destruction of malignant cells may lead to hyperkalemia. Hyperkalemia in patients with renal failure may rapidly become life-threatening by causing ventricular arrhythmias and sudden death.
The likelihood that TLS will occur in patients with Burkitt’s lymphoma is related to the tumor burden and renal function. Hyperuricemia and high serum levels of lactate dehydrogenase (LDH >1500 U/L), both of which correlate with total tumor burden, also correlate with the risk of TLS. In patients at risk for TLS, pretreatment evaluations should include a complete blood count, serum chemistry evaluation, and urine analysis. High leukocyte and platelet counts may artificially elevate potassium levels (“pseudohyperkalemia”) due to lysis of these cells after the blood is drawn. In these cases, plasma potassium instead of serum potassium should be followed. In pseudohyperkalemia, no electrocardiographic abnormalities are present. In patients with abnormal baseline renal function, the kidneys and retroperitoneal area should be evaluated by sonography and/or CT to rule out obstructive uropathy. Urine output should be watched closely.
|
TREATMENT |
TUMOR LYSIS SYNDROME |
Recognition of risk and prevention are the most important steps in the management of this syndrome (Fig. 331-4). The standard preventive approach consists of allopurinol, urinary alkalinization, and aggressive hydration. Urinary alkalization with sodium bicarbonate is controversial. It increases uric acid solubility, but decreases calcium phosphate solubility. If it is used, it should be discontinued when hyperphosphatemia develops. Intravenous allopurinol may be given in patients who cannot tolerate oral therapy. In some cases, uric acid levels cannot be lowered sufficiently with the standard preventive approach. Rasburicase (recombinant urate oxidase) can be effective in these instances, particularly when renal failure is present. Urate oxidase is missing from primates and catalyzes the conversion of poorly soluble uric acid to readily soluble allantoin. Rasburicase acts rapidly, decreasing uric acid levels within hours; however, it may cause hypersensitivity reactions such as bronchospasm, hypoxemia, and hypotension. Rasburicase should also be administered to high-risk patients for TLS prophylaxis. Rasburicase is contraindicated in patients with glucose-6-phosphate dehydrogenase deficiency who are unable to break down hydrogen peroxide, an end product of the urate oxidase reaction. Rasburicase is known to cause ex vivo enzymatic degradation of uric acid in test tube at room temperature. This leads to spuriously low uric acid levels during laboratory monitoring of the patient with TLS. Samples must be cooled immediately to deactivate the urate oxidase. Despite aggressive prophylaxis, TLS and/or oliguric or anuric renal failure may occur. Care should be taken to prevent worsening of symptomatic hypocalcemia by induction of alkalosis during bicarbonate infusion. Administration of sodium bicarbonate may also lead to urinary precipitation of calcium phosphate, which is less soluble at alkaline pH. Dialysis is often necessary and should be considered early in the course. Hemodialysis is preferred. Hemofiltration offers a gradual, continuous method of removing cellular by-products and fluid. The prognosis is excellent, and renal function recovers after the uric acid level is lowered to ≤10 mg/dL.
FIGURE 331-4 Management of patients at high risk for the tumor lysis syndrome. *See text.
HUMAN ANTIBODY INFUSION REACTIONS
The initial infusion of human or humanized antibodies (e.g., rituximab, gemtuzumab, trastuzumab, alemtuzumab, panitumumab, brentuximab vedotin) is associated with fever, chills, nausea, asthenia, and headache in up to half of treated patients. Bronchospasm and hypotension occur in 1% of patients. Severe manifestations including pulmonary infiltrates, acute respiratory distress syndrome, and cardiogenic shock occur rarely. Laboratory manifestations include elevated hepatic aminotransferase levels, thrombocytopenia, and prolongation of prothrombin time. The pathogenesis is thought to be activation of immune effector processes (cells and complement) and release of inflammatory cytokines, such as tumor necrosis factor α, interferon gamma, interleukin 6, and interleukin 10 (cytokine release syndrome [CRS]). Although its origins are not completely understood, CRS is believed to be due to activation of a variety of cell types including monocytes/macrophages and T and B lymphocytes. Severe reactions from rituximab have occurred with high numbers (>50 × 109 lymphocytes) of circulating cells bearing the target antigen (CD20) and have been associated with a rapid fall in circulating tumor cells, mild electrolyte evidence of TLS, and very rarely, death. In addition, increased liver enzymes, D-dimer, and LDH and prolongation of the prothrombin time may occur. Diphenhydramine, hydrocortisone, and acetaminophen can often prevent or suppress the infusion-related symptoms. If they occur, the infusion is stopped and restarted at half the initial infusion rate after the symptoms have abated. Severe CRS may require intensive support for acute respiratory distress syndrome (ARDS) and resistant hypotension.
HEMOLYTIC-UREMIC SYNDROME
Hemolytic-uremic syndrome (HUS) and, less commonly, thrombotic thrombocytopenic purpura (TTP) (Chap. 341) may rarely occur after treatment with antineoplastic drugs, including mitomycin, gemcitabine, cisplatin, and bleomycin, and with VEGF inhibitors. It occurs most often in patients with gastric, lung, colorectal, pancreatic, and breast carcinoma. In one series, 35% of patients were without evident cancer at the time this syndrome appeared. Secondary HUS/TTP has also been reported as a rare but sometimes fatal complication of bone marrow transplantation.
HUS usually has its onset 4–8 weeks after the last dose of chemotherapy, but it is not rare to detect it several months later. HUS is characterized by microangiopathic hemolytic anemia, thrombocytopenia, and renal failure. Dyspnea, weakness, fatigue, oliguria, and purpura are also common initial symptoms and findings. Systemic hypertension and pulmonary edema frequently occur. Severe hypertension, pulmonary edema, and rapid worsening of hemolysis and renal function may occur after a blood or blood product transfusion. Cardiac findings include atrial arrhythmias, pericardial friction rub, and pericardial effusion. Raynaud’s phenomenon is part of the syndrome in patients treated with bleomycin.
Laboratory findings include severe to moderate anemia associated with red blood cell fragmentation and numerous schistocytes on peripheral smear. Reticulocytosis, decreased plasma haptoglobin, and an LDH level document hemolysis. The serum bilirubin level is usually normal or slightly elevated. The Coombs’ test is negative. The white cell count is usually normal, and thrombocytopenia (<100,000/μL) is almost always present. Most patients have a normal coagulation profile, although some have mild elevations in thrombin time and in levels of fibrin degradation products. The serum creatinine level is elevated at presentation and shows a pattern of subacute worsening within weeks of the initial azotemia. The urinalysis reveals hematuria, proteinuria, and granular or hyaline casts; and circulating immune complexes may be present.
The basic pathologic lesion appears to be deposition of fibrin in the walls of capillaries and arterioles, and these deposits are similar to those seen in HUS due to other causes. These microvascular abnormalities involve mainly the kidneys and rarely occur in other organs. The pathogenesis of cancer treatment–related HUS is not completely understood, but probably the most important factor is endothelial damage. Primary forms of HUS/TTP are related to a decrease in processing of von Willebrand factor by a protease called ADAMTS13.
The case fatality rate is high; most patients die within a few months. There is no consensus on the optimal treatment for chemotherapy-induced HUS. Treatment modalities for HUS/TTP including immunocomplex removal (plasmapheresis, immunoadsorption, or exchange transfusion), antiplatelet/anticoagulant therapies, immunosuppressive therapies, and plasma exchange have varying degrees of success. The outcome with plasma exchange is generally poor, as in many other cases of secondary TTP. Rituximab is successfully used in patients with chemotherapy-induced HUS as well as in ADAMTS13-deficient TTP.
NEUTROPENIA AND INFECTION
These remain the most common serious complications of cancer therapy. They are covered in detail in Chap. 104.
PULMONARY INFILTRATES
Patients with cancer may present with dyspnea associated with diffuse interstitial infiltrates on chest radiographs. Such infiltrates may be due to progression of the underlying malignancy, treatment-related toxicities, infection, and/or unrelated diseases. The cause may be multifactorial; however, most commonly they occur as a consequence of treatment. Infiltration of the lung by malignancy has been described in patients with leukemia, lymphoma, and breast and other solid cancers. Pulmonary lymphatics may be involved diffusely by neoplasm (pulmonary lymphangitic carcinomatosis), resulting in a diffuse increase in interstitial markings on chest radiographs. The patient is often mildly dyspneic at the onset, but pulmonary failure develops over a period of weeks. In some patients, dyspnea precedes changes on the chest radiographs and is accompanied by a nonproductive cough. This syndrome is characteristic of solid tumors. In patients with leukemia, diffuse microscopic neoplastic peribronchial and peribronchiolar infiltration is frequent but may be asymptomatic. However, some patients present with diffuse interstitial infiltrates, an alveolar capillary block syndrome, and respiratory distress. In these situations, glucocorticoids can provide symptomatic relief, but specific chemotherapy should always be started promptly.
Several cytotoxic agents, such as bleomycin, methotrexate, busulfan, nitrosoureas, gemcitabine, mitomycin, vinorelbine, docetaxel, paclitaxel, fludarabine, pentostatin, and ifosfamide may cause pulmonary damage. The most frequent presentations are interstitial pneumonitis, alveolitis, and pulmonary fibrosis. Some cytotoxic agents, including methotrexate and procarbazine, may cause an acute hypersensitivity reaction. Cytosine arabinoside has been associated with noncardiogenic pulmonary edema. Administration of multiple cytotoxic drugs, as well as radiotherapy and preexisting lung disease, may potentiate the pulmonary toxicity. Supplemental oxygen may potentiate the effects of drugs and radiation injury. Patients should always be managed with the lowest FIO2 that is sufficient to maintain hemoglobin saturation.
The onset of symptoms may be insidious, with symptoms including dyspnea, nonproductive cough, and tachycardia. Patients may have bibasilar crepitant rales, end-inspiratory crackles, fever, and cyanosis. The chest radiograph generally shows an interstitial and sometimes an intraalveolar pattern that is strongest at the lung bases and may be symmetric. A small effusion may occur. Hypoxemia with decreased carbon monoxide diffusing capacity is always present. Glucocorticoids may be helpful in patients in whom pulmonary toxicity is related to radiation therapy or to chemotherapy. Treatment is otherwise supportive.
Molecular targeted agents, imatinib, erlotinib, and gefitinib are potent inhibitors of tyrosine kinases. These drugs may cause interstitial lung disease (ILD). In the case of gefitinib, preexisting fibrosis, poor performance status, and prior thoracic irradiation are independent risk factors; this complication has a high fatality rate. In Japan, incidence of interstitial lung disease associated with gefitinib was about 4.5% compared to 0.5% in the United States. Temsirolimus and everolimus, both esters a derivative of rapamycin, are agents that block the effects of mammalian target of rapamycin (mTOR), an enzyme that has an important role in regulating the synthesis of proteins that control cell division. It may cause ground-glass opacities in the lung with or without diffuse interstitial disease and lung parenchymal consolidation. Patients may be asymptomatic with only radiologic findings or may be symptomatic. Symptoms include cough, dyspnea, and/or hypoxemia, and sometimes patients present with systemic symptoms such as fever and fatigue. The incidence of everolimus-induced interstitial lung disease also appears to be higher in Japanese patients. Treatment includes dose reduction or withdrawal and, in some cases, the addition of glucocorticoids.
Radiation pneumonitis and/or fibrosis is a relatively frequent side effect of thoracic radiation therapy. It may be acute or chronic. Radiation-induced lung toxicity is a function of the irradiated lung volume, dose per fraction, and radiation dose. The larger the irradiated lung field, the higher is the risk for radiation pneumonitis. The use of concurrent chemoradiation, particularly regimens including paclitaxel, increases pulmonary toxicity. Radiation pneumonitis usually develops 2–6 months after completion of radiotherapy. The clinical syndrome, which varies in severity, consists of dyspnea, cough with scanty sputum, low-grade fever, and an initial hazy infiltrate on chest radiographs. The infiltrate and tissue damage usually are confined to the radiation field. The patients subsequently may develop a patchy alveolar infiltrate and air bronchograms, which may progress to acute respiratory failure that is sometimes fatal. A lung biopsy may be necessary to make the diagnosis. Asymptomatic infiltrates found incidentally after radiation therapy need not be treated. However, prednisone should be administered to patients with fever or other symptoms. The dosage should be tapered slowly after the resolution of radiation pneumonitis, because abrupt withdrawal of glucocorticoids may cause an exacerbation of pneumonia. Delayed radiation fibrosis may occur years after radiation therapy and is signaled by dyspnea on exertion. Often it is mild, but it can progress to chronic respiratory failure. Therapy is supportive.
Classical radiation pneumonitis that leads to pulmonary fibrosis is due to radiation-induced production of local cytokines such as platelet-derived growth factor β, tumor necrosis factor, interleukins, and transforming growth factor β in the radiation field. An immunologically mediated sporadic radiation pneumonitis occurs in about 10% of patients; bilateral alveolitis mediated by T cells results in infiltrates outside the radiation field. This form of radiation pneumonitis usually resolves without sequelae.
Pneumonia is a common problem in patients undergoing treatment for cancer. Bacterial pneumonia typically causes a localized infiltrate on chest radiographs. Therapy is tailored to the causative organism. When diffuse interstitial infiltrates appear in a febrile patient, the differential diagnosis is extensive and includes pneumonia due to infection with Pneumocystis carinii; viral infections including cytomegalovirus, adenovirus, herpes simplex virus, herpes zoster, respiratory syncytial virus, or intracellular pathogens such as Mycoplasma and Legionella; effects of drugs or radiation; tumor progression; nonspecific pneumonitis; and fungal disease. Detection of opportunistic pathogens in pulmonary infections is still a challenge. Diagnostic tools include chest radiographs, CT scans, bronchoscopy with bronchoalveolar lavage, brush cytology, transbronchial biopsy, fine-needle aspiration, and open lung biopsy. In addition to the culture, evaluation of bronchoalveolar lavage fluid for P. carinii by polymerase chain reaction (PCR) and serum galactomannan test improve the diagnostic yield. Patients with cancer who are neutropenic and have fever and local infiltrates on chest radiograph should be treated initially with broad-spectrum antibiotics. A new or persistent focal infiltrate not responding to broad-spectrum antibiotics argues for initiation of empiric antifungal therapy. When diffuse bilateral infiltrates develop in patients with febrile neutropenia, broad-spectrum antibiotics plus trimethoprim-sulfamethoxazole, with or without erythromycin, should be initiated. Addition of an antiviral agent is necessary in some settings, such as patients undergoing allogeneic hematopoietic stem cell transplantation. If the patient does not improve in 4 days, open lung biopsy is the procedure of choice. Bronchoscopy with bronchoalveolar lavage may be used in patients who are poor candidates for surgery.
In patients with pulmonary infiltrates who are afebrile, heart failure and multiple pulmonary emboli are in the differential diagnosis.
NEUTROPENIC ENTEROCOLITIS
Neutropenic enterocolitis (typhlitis) is the inflammation and necrosis of the cecum and surrounding tissues that may complicate the treatment of acute leukemia. Nevertheless, it may involve any segment of the gastrointestinal tract including small intestine, appendix, and colon. This complication has also been seen in patients with other forms of cancer treated with taxanes, 5-fluorouracil, irinotecan, vinorelbine, cisplatin, carboplatin, and high-dose chemotherapy (Fig. 331-5). It also has been reported in patients with AIDS, aplastic anemia, cyclic neutropenia, idiosyncratic drug reactions involving antibiotics, and immunosuppressive therapies. The patient develops right lower quadrant abdominal pain, often with rebound tenderness and a tense, distended abdomen, in a setting of fever and neutropenia. Watery diarrhea (often containing sloughed mucosa) and bacteremia are common, and bleeding may occur. Plain abdominal films are generally of little value in the diagnosis; CT scan may show marked bowel wall thickening, particularly in the cecum, with bowel wall edema, mesenteric stranding, and ascites, and may help to differentiate neutropenic colitis from other abdominal disorders such as appendicitis, diverticulitis, and Clostridium difficile–associated colitis in this high-risk population. Patients with bowel wall thickness >10 mm on ultrasonogram have higher mortality rates. However, bowel wall thickening is significantly more prominent in patients with C. difficile colitis. Pneumatosis intestinalis is a more specific finding, seen only in those with neutropenic enterocolitis and ischemia. The combined involvement of the small and large bowel suggests a diagnosis of neutropenic enterocolitis. Rapid institution of broad-spectrum antibiotics, bowel rest, and nasogastric suction may reverse the process. Use of myeloid growth factors improved outcome significantly. Surgical intervention is reserved for severe cases of neutropenic enterocolitis with evidence of perforation, peritonitis, gangrenous bowel, or gastrointestinal hemorrhage despite correction of any coagulopathy.
FIGURE 331-5 Abdominal computed tomography (CT) scans of a 72-year-old woman with neutropenic enterocolitis secondary to chemotherapy. A. Air in inferior mesenteric vein (arrow) and bowel wall with pneumatosis intestinalis. B. CT scan of upper abdomen demonstrating air in portal vein (arrows).
C. difficile colitis is increasing in incidence. Newer strains of C. difficile produce about 20 times more of toxins A and B compared to previously studied strains. C. difficile risk is also increased with chemotherapy. Antibiotic coverage for C. difficile should be added if pseudomembranous colitis cannot be excluded.
HEMORRHAGIC CYSTITIS
Hemorrhagic cystitis can develop in patients receiving cyclophosphamide or ifosfamide. Both drugs are metabolized to acrolein, which is a strong chemical irritant that is excreted in the urine. Prolonged contact or high concentrations may lead to bladder irritation and hemorrhage. Symptoms include gross hematuria, frequency, dysuria, burning, urgency, incontinence, and nocturia. The best management is prevention. Maintaining a high rate of urine flow minimizes exposure. In addition, 2-mercaptoethanesulfonate (mesna) detoxifies the metabolites and can be coadministered with the instigating drugs. Mesna usually is given three times on the day of ifosfamide administration in doses that are each 20% of the total ifosfamide dose. If hemorrhagic cystitis develops, the maintenance of a high urine flow may be sufficient supportive care. If conservative management is not effective, irrigation of the bladder with a 0.37–0.74% formalin solution for 10 min stops the bleeding in most cases. N-Acetylcysteine may also be an effective irrigant. Prostaglandin (carboprost) can inhibit the process. In extreme cases, ligation of the hypogastric arteries, urinary diversion, or cystectomy may be necessary.
Hemorrhagic cystitis also occurs in patients who undergo bone marrow transplantation (BMT). In the BMT setting, early-onset hemorrhagic cystitis is related to drugs in the treatment regimen (e.g., cyclophosphamide), and late-onset hemorrhagic cystitis is usually due to the polyoma virus BKV or adenovirus type 11. BKV load in urine alone or in combination with acute graft-versus-host disease correlates with development of hemorrhagic cystitis. Viral causes are usually detected by PCR-based diagnostic tests. Treatment of viral hemorrhagic cystitis is largely supportive, with reduction in doses of immunosuppressive agents, if possible. No antiviral therapy is approved, although cidofovir is reported to be effective in a small series. Hyperbaric oxygen therapy has been used successfully in patients with BKV-associated and cyclophosphamide-induced hemorrhagic cystitis during hematopoietic stem cell transplantation, as well as in hemorrhagic radiation cystitis.
HYPERSENSITIVITY REACTIONS TO ANTINEOPLASTIC DRUGS
Many antineoplastic drugs may cause hypersensitivity reaction. These reactions are unpredictable and potentially life-threatening. Most reactions occur during or within hours of parenteral drug administration. Taxanes, platinum compounds, asparaginase, etoposide, procarbazine, and biologic agents, including rituximab, bevacizumab, trastuzumab, gemtuzumab, cetuximab, and alemtuzumab, are more commonly associated with acute hypersensitivity reactions than are other agents. Acute hypersensitivity reactions to some drugs, such as taxanes, occur during the first or second dose administered. Hypersensitivity to platinum compounds occurs after prolonged exposure. Skin testing may identify patients with high risk for hypersensitivity after carboplatin exposure. Premedication with histamine H1 and H2 receptor antagonists and glucocorticoids reduces the incidence of hypersensitivity reaction to taxanes, particularly paclitaxel. Despite premedication, hypersensitivity reactions may still occur. In these cases, rapid desensitization in the intensive care unit setting or re-treatment may be attempted with care, but the use of alternative agents may be required. Candidate patients for desensitization include those who have mild to severe hypersensitivity type I, with mast cell–mediated and IgE-dependent reactions occurring during a chemotherapy infusion or shortly thereafter.