MITRAL REGURGITATION
ETIOLOGY
MR may result from an abnormality or disease process that affects any one or more of the five functional components of the mitral valve apparatus (leaflets, annulus, chordae tendineae, papillary muscles, and subjacent myocardium) (Table 284-1). Acute MR can occur in the setting of acute myocardial infarction (MI) with papillary muscle rupture (Chap. 295), following blunt chest wall trauma, or during the course of infective endocarditis. With acute MI, the posteromedial papillary muscle is involved much more frequently than the anterolateral papillary muscle because of its singular blood supply. Transient, acute MR can occur during periods of active ischemia and bouts of angina pectoris. Rupture of chordae tendineae can result in “acute-on-chronic MR” in patients with myxomatous degeneration of the valve apparatus.
Chronic MR can result from rheumatic disease, mitral valve prolapse (MVP), extensive mitral annular calcification, congenital valve defects, hypertrophic obstructive cardiomyopathy (HOCM), and dilated cardiomyopathy (Chap. 287). Distinction also should be drawn between primary (degenerative, organic) MR, in which the leaflets and/or chordae tendineae are primarily responsible for abnormal valve function, and functional (secondary) MR, in which the leaflets and chordae tendineae are structurally normal but the regurgitation is caused by annular enlargement, papillary muscle displacement, leaflet tethering, or their combination. The rheumatic process produces rigidity, deformity, and retraction of the valve cusps and commissural fusion, as well as shortening, contraction, and fusion of the chordae tendineae. The MR associated with both MVP and HOCM is usually dynamic in nature. MR in HOCM occurs as a consequence of anterior papillary muscle displacement and systolic anterior motion of the anterior mitral valve leaflet into the narrowed LV outflow tract. Annular calcification is especially prevalent among patients with advanced renal disease and is commonly observed in women >65 years of age with hypertension and diabetes. MR may occur as a congenital anomaly (Chap. 282), most commonly as a defect of the endocardial cushions (atrioventricular cushion defects). A cleft anterior mitral valve leaflet accompanies primum atrial septal defect. Chronic MR is frequently secondary to ischemia and may occur as a consequence of ventricular remodeling, papillary muscle displacement, and leaflet tethering, or with fibrosis of a papillary muscle, in patients with healed MI(s) and ischemic cardiomyopathy. Similar mechanisms of annular dilation and ventricular remodeling contribute to the MR that occurs among patients with nonischemic forms of dilated cardiomyopathy once the LV end-diastolic dimension reaches 6 cm.
Irrespective of cause, chronic severe MR is often progressive, because enlargement of the LA places tension on the posterior mitral leaflet, pulling it away from the mitral orifice and thereby aggravating the valvular dysfunction. Similarly, LV dilation increases the regurgitation, which, in turn, enlarges the LA and LV further, resulting in a vicious circle; hence the aphorism, “mitral regurgitation begets mitral regurgitation.”
PATHOPHYSIOLOGY
The resistance to LV emptying (LV afterload) is reduced in patients with MR. As a consequence, the LV is decompressed into the LA during ejection, and with the reduction in LV size during systole, there is a rapid decline in LV tension. The initial compensation to MR is more complete LV emptying. However, LV volume increases progressively with time as the severity of the regurgitation increases and as LV contractile function deteriorates. This increase in LV volume is often accompanied by a reduced forward CO. LV compliance is often increased, and thus, LV diastolic pressure does not increase until late in the course. The regurgitant volume varies directly with the LV systolic pressure and the size of the regurgitant orifice; the latter, in turn, is influenced by the extent of LV and mitral annular dilation. Because EF rises in severe MR in the presence of normal LV function, even a modest reduction in this parameter (<60%) reflects significant dysfunction.
During early diastole, as the distended LA empties, there is a particularly rapid y descent in the absence of accompanying MS. A brief, early diastolic LA-LV pressure gradient (often generating a rapid filling sound [S3] and mid-diastolic murmur masquerading as MS) may occur in patients with pure, severe MR as a result of the very rapid flow of blood across a normal-sized mitral orifice.
Semiquantitative estimates of LV ejection fraction (LVEF), CO, PA systolic pressure, regurgitant volume, regurgitant fraction (RF), and the effective regurgitant orifice area can be obtained during a careful Doppler echocardiographic examination. These measurements can also be obtained accurately with cardiac magnetic resonance (CMR) imaging, although this technology is not widely available. Left and right heart catheterization with contrast ventriculography is used less frequently. Severe, nonischemic MR is defined by a regurgitant volume ≥60 mL/beat, RF ≥50%, and effective regurgitant orifice area ≥0.40 cm2. Severe ischemic MR, however, is usually associated with an effective regurgitant orifice area of >0.2 cm2. In the latter instance, lesser degrees of MR carry relatively greater prognostic weight.
LA Compliance In acute severe MR, the regurgitant volume is delivered into a normal-sized LA having normal or reduced compliance. As a result, LA pressures rise markedly for any increase in LA volume. The v wave in the LA pressure pulse is usually prominent, LA and pulmonary venous pressures are markedly elevated, and pulmonary edema is common. Because of the rapid rise in LA pressures during ventricular systole, the murmur of acute MR is early in timing and decrescendo in configuration ending well before S2, as a reflection of the progressive diminution in the LV-LA pressure gradient. LV systolic function in acute MR may be normal, hyperdynamic, or reduced, depending on the clinical context.
Patients with chronic severe MR, on the other hand, develop marked LA enlargement and increased LA compliance with little if any increase in LA and pulmonary venous pressures for any increase in LA volume. The LA v wave is relatively less prominent. The murmur of chronic MR is classically holosystolic in timing and plateau in configuration, as a reflection of the near-constant LV-LA pressure gradient. These patients usually complain of severe fatigue and exhaustion secondary to a low forward CO, whereas symptoms resulting from pulmonary congestion are less prominent initially; AF is almost invariably present once the LA dilates significantly.
SYMPTOMS
Patients with chronic mild-to-moderate, isolated MR are usually asymptomatic. This form of LV volume overload is well tolerated. Fatigue, exertional dyspnea, and orthopnea are the most prominent complaints in patients with chronic severe MR. Palpitations are common and may signify the onset of AF. Right-sided heart failure, with painful hepatic congestion, ankle edema, distended neck veins, ascites, and secondary TR, occurs in patients with MR who have associated pulmonary vascular disease and pulmonary hypertension. Acute pulmonary edema is common in patients with acute severe MR.
PHYSICAL FINDINGS
In patients with chronic severe MR, the arterial pressure is usually normal, although the carotid arterial pulse may show a sharp, low-volume upstroke owing to the reduced forward CO. A systolic thrill is often palpable at the cardiac apex, the LV is hyperdynamic with a brisk systolic impulse and a palpable rapid-filling wave (S3), and the apex beat is often displaced laterally.
In patients with acute severe MR, the arterial pressure may be reduced with a narrow pulse pressure, the jugular venous pressure and wave forms may be normal or increased and exaggerated, the apical impulse is not displaced, and signs of pulmonary congestion are prominent.
Auscultation S1 is generally absent, soft, or buried in the holosystolic murmur of chronic, severe MR. In patients with severe MR, the aortic valve may close prematurely, resulting in wide but physiologic splitting of S2. A low-pitched S3 occurring 0.12–0.17 s after the aortic valve closure sound, i.e., at the completion of the rapid-filling phase of the LV, is believed to be caused by the sudden tensing of the papillary muscles, chordae tendineae, and valve leaflets. It may be followed by a short, rumbling, mid-diastolic murmur, even in the absence of structural MS. A fourth heart sound is often audible in patients with acute severe MR who are in sinus rhythm. A presystolic murmur is not ordinarily heard with isolated MR.
A systolic murmur of at least grade III/VI intensity is the most characteristic auscultatory finding in chronic severe MR. It is usually holosystolic (see Fig. 267-5A), but as previously noted, it is decrescendo and ceases in mid to late systole in patients with acute severe MR. The systolic murmur of chronic MR is usually most prominent at the apex and radiates to the axilla. However, in patients with ruptured chordae tendineae or primary involvement of the posterior mitral leaflet with prolapse or flail, the regurgitant jet is eccentric, directed anteriorly, and strikes the LA wall adjacent to the aortic root. In this situation, the systolic murmur is transmitted to the base of the heart and, therefore, may be confused with the murmur of AS. In patients with ruptured chordae tendineae, the systolic murmur may have a cooing or “seagull” quality, whereas a flail leaflet may produce a murmur with a musical quality. The systolic murmur of chronic MR not due to MVP is intensified by isometric exercise (handgrip) but is reduced during the strain phase of the Valsalva maneuver because of the associated decrease in LV preload.
LABORATORY EXAMINATION
ECG In patients with sinus rhythm, there is evidence of LA enlargement, but RA enlargement also may be present when pulmonary hypertension is significant and affects RV function. Chronic severe MR is frequently associated with AF. In many patients, there is no clear-cut ECG evidence of enlargement of either ventricle. In others, the signs of eccentric LV hypertrophy are present.
Echocardiogram TTE is indicated to assess the mechanism of the MR and its hemodynamic severity. LV function can be assessed from LV end-diastolic and end-systolic volumes and EF. Observations can be made regarding leaflet structure and function, chordal integrity, LA and LV size, annular calcification, and regional and global LV systolic function. Doppler imaging should demonstrate the width or area of the color flow MR jet within the LA, the duration and intensity of the continuous wave Doppler signal, the pulmonary venous flow contour, the early peak mitral inflow velocity, and quantitative measures of regurgitant volume, RF, and effective regurgitant orifice area. In addition, the PAPs can be estimated from the TR jet velocity. TTE is also indicated to follow the course of patients with chronic MR and to provide rapid assessment for any clinical change. The echocardiogram in patients with MVP is described in the next section. TEE provides greater anatomic detail than TTE (see Fig. 270e-5). Exercise testing with TTE can be useful to assess exercise capacity as well as any dynamic change in MR severity, PA systolic pressures, and biventricular function, for patients in whom there is a discrepancy between clinical findings and the results of functional testing performed at rest.
Chest X-Ray The LA and LV are the dominant chambers in chronic MR. Late in the course of the disease, the LA may be massively enlarged and forms the right border of the cardiac silhouette. Pulmonary venous congestion, interstitial edema, and Kerley B lines are sometimes noted. Marked calcification of the mitral leaflets occurs commonly in patients with long-standing, combined rheumatic MR and MS. Calcification of the mitral annulus may be visualized, particularly on the lateral view of the chest. Patients with acute severe MR may have asymmetric pulmonary edema if the regurgitant jet is directed predominantly to the orifice of an upper lobe pulmonary vein.
|
TREATMENT |
MITRAL REGURGITATION |
MEDICAL TREATMENT (FIG. 284-4)
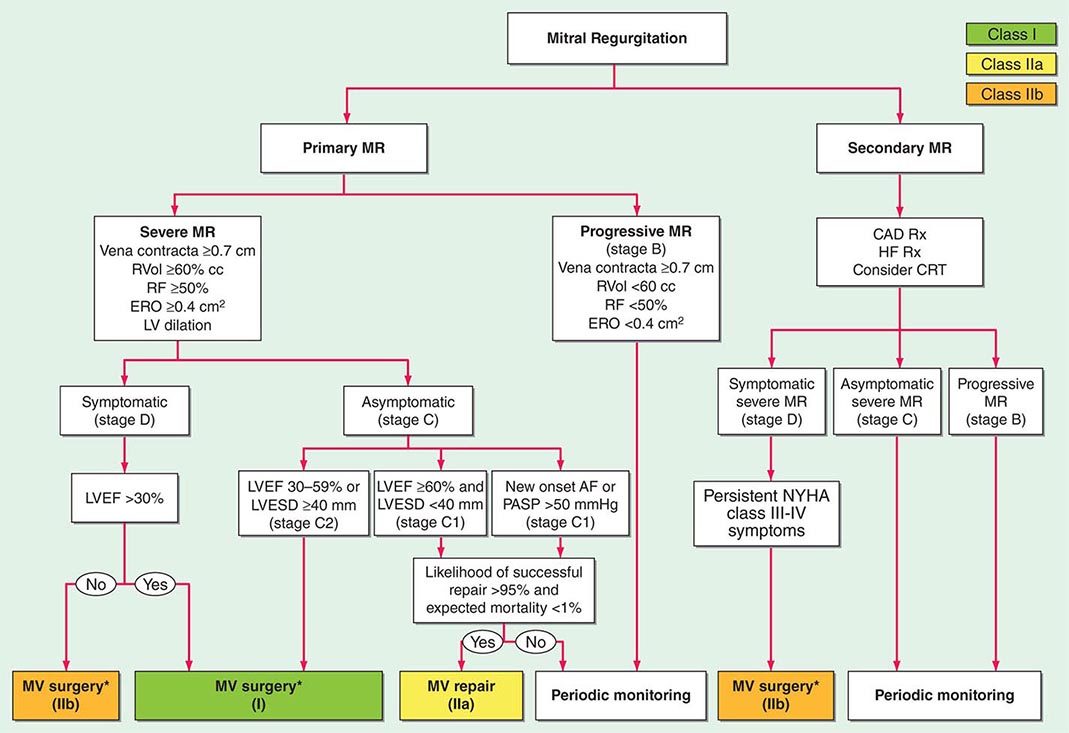
FIGURE 284-4 Management of mitral regurgitation. See legend for Fig. 283-2 for explanation of treatment recommendations (class I, IIa, IIb) and disease stages (B, C1, C2, D). Preoperative coronary angiography should be performed routinely as determined by age, symptoms, and coronary risk factors. Cardiac catheterization and angiography may also be helpful when there is a discrepancy between clinical and noninvasive findings. AF, atrial fibrillation; CAD, coronary artery disease; CRT, cardiac resynchronization therapy; ERO, effective regurgitant orifice; HF, heart failure; LV, left ventricular; LVEF, left ventricular ejection fraction; LVESD, left ventricular end-systolic dimension; MR, mitral regurgitation, MV, mitral valve; MVR, mitral valve replacement; NYHA, New York Heart Association; PASP, pulmonary artery systolic pressure; RF, regurgitant fraction; RVol, regurgitant volume; and Rx, therapy. *Mitral valve repair preferred over MVR when possible. (Adapted from RA Nishimura et al: 2014 AHA/ACC Guideline for the Management of Patients with Valvular Heart Disease. J Am Coll Cardiol doi: 10.1016/j.jacc.2014.02.536, 2014, with permission.)
The management of chronic severe MR depends to some degree on its cause. Warfarin should be provided once AF intervenes with a target INR of 2–3. Novel oral anticoagulants are not approved for this indication. Cardioversion should be considered depending on the clinical context and LA size. In contrast to the acute setting, there are no large, long-term prospective studies to substantiate the use of vasodilators for the treatment of chronic, isolated severe MR with preserved LV systolic function in the absence of systemic hypertension. The severity of MR in the setting of an ischemic or nonischemic dilated cardiomyopathy may diminish with aggressive guideline-directed treatment of heart failure including the use of diuretics, beta blockers, angiotensin-converting enzyme (ACE) inhibitors, digitalis, and biventricular pacing (cardiac resynchronization therapy [CRT]) when otherwise indicated. Asymptomatic patients with severe MR in sinus rhythm with normal LV size and systolic function should avoid isometric forms of exercise.
Patients with acute severe MR require urgent stabilization and preparation for surgery. Diuretics, intravenous vasodilators (particularly sodium nitroprusside), and even intraaortic balloon counterpulsation may be needed for patients with post-MI papillary muscle rupture or other forms of acute severe MR.
SURGICAL TREATMENT
In the selection of patients with chronic, nonischemic, primary or organic, severe MR for surgical treatment, the often slowly progressive nature of the condition must be balanced against the immediate and long-term risks associated with operation. These risks are significantly lower for primary valve repair than for valve replacement (Table 287-2). Repair usually consists of valve reconstruction using a variety of valvuloplasty techniques and insertion of an annuloplasty ring. Repair spares the patient the long-term adverse consequences of valve replacement, including thromboembolic and hemorrhagic complications in the case of mechanical prostheses and late valve failure necessitating repeat valve replacement in the case of bioprostheses. In addition, by preserving the integrity of the papillary muscles, subvalvular apparatus, and chordae tendineae, mitral repair and valvuloplasty maintain LV function to a relatively greater degree.
Surgery for chronic nonischemic severe MR is indicated once symptoms occur, especially if valve repair is feasible (Fig. 284-4). Other indications for early consideration of mitral valve repair include recent-onset AF and pulmonary hypertension defined as a systolic PA pressure ≥50 mmHg at rest or ≥60 mmHg with exercise. Surgical treatment of chronic nonischemic severe MR is indicated for asymptomatic patients when LV dysfunction is progressive with the LVEF falling below 60% and/or end-systolic dimension increasing beyond 40 mm. These aggressive recommendations for surgery are predicated on the outstanding results achieved with mitral valve repair particularly when applied to patients with myxomatous disease such as that associated with prolapse or flail leaflet. Indeed primary valvuloplasty repair of patients younger than 75 years with normal LV systolic function and no CAD can now be performed by experienced surgeons with <1% perioperative mortality risk. The risk of stroke, however, is also approximately 1%. Repair is feasible in up to 95% of patients with myxomatous disease operated on by a high-volume surgeon in a referral center of excellence. Long-term durability is excellent; the incidence of reoperative surgery for failed primary repair is ~1% per year for the first 10 years after surgery. For patients with AF, left or biatrial maze surgery, or radiofrequency, isolation of the pulmonary veins is often performed to reduce the risk of recurrent postoperative AF.
The surgical management of patients with functional, ischemic MR is more complicated and most often involves simultaneous coronary artery revascularization. Current surgical practice includes annuloplasty repair with an undersized, rigid ring or chord-sparing valve replacement for patients with moderate or greater degrees of MR. Valve repair for ischemic MR is associated with lower perioperative mortality rates but higher rates of recurrent MR over time. In patients with ischemic MR and significantly impaired LV systolic function (EF <30%), the risk of surgery is higher, recovery of LV performance is incomplete, and long-term survival is reduced. Referral for surgery must be individualized and made only after aggressive attempts with guideline-directed medical therapy and CRT, when indicated. The routine performance of valve repair in patients with significant MR in the setting of severe, functional, nonischemic dilated cardiomyopathy has not been shown to improve long-term survival compared with optimal medical therapy. Patients with acute severe MR can often be stabilized temporarily with appropriate medical therapy, but surgical correction will be necessary emergently in the case of papillary muscle rupture and within days to weeks in most other settings.
When surgical treatment is contemplated, left and right heart catheterization and left ventriculography may be helpful in confirming the presence of severe MR in patients in whom there is a discrepancy between the clinical and TTE findings that cannot be resolved with TEE or CMR. Coronary angiography identifies patients who require concomitant coronary revascularization.
TRANSCATHETER MITRAL VALVE REPAIR
A transcatheter approach to the treatment of either organic or functional MR may be feasible in selected patients with appropriate anatomy. The proper role of currently available techniques remains under active investigation. One approach involves the deployment of a clip delivered via transseptal puncture that grasps the leading edges of the mitral leaflets in their mid-portion (anterior scallop to posterior scallop or A2-P2; Fig. 284-5). The length and width of the gap between these leading edges dictate patient eligibility. The device is commercially available for the treatment of prohibitive surgical risk patients with severe, degenerative (organic) MR and is undergoing study in the United States for treatment of patients with symptomatic heart failure, reduced LVEF, and severe, functional MR despite guideline-directed medical therapy. A second approach involves the deployment of a device within the coronary sinus that can be adjusted to reduce its circumference, thus secondarily decreasing the circumference of the mitral annulus and the effective orifice area of the valve much like a surgically implanted ring. Variations in the anatomic relationship of the coronary sinus to the mitral annulus and circumflex coronary artery have limited the applicability of this technique. Attempts to reduce the septal-lateral dimension of a dilated annulus using adjustable cords placed across the LV in a subvalvular location have also been investigated.
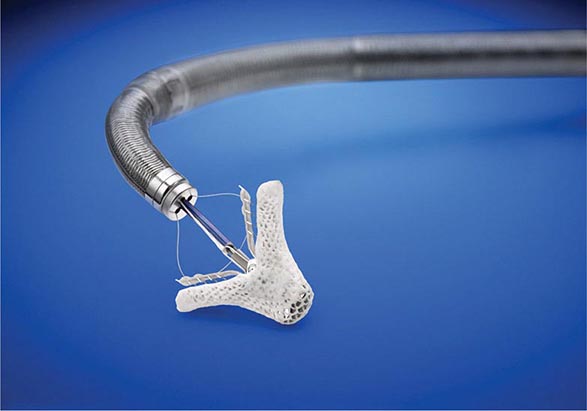
FIGURE 284-5 Clip used to grasp the free edges of the anterior and posterior leaflets in their midsections during transcatheter repair of selected patients with mitral regurgitation. (Courtesy of Abbott Vascular. © 2014 Abbott Laboratories. All rights reserved.)
MITRAL VALVE PROLAPSE
MVP, also variously termed the systolic click-murmur syndrome, Barlow’s syndrome, floppy-valve syndrome, and billowing mitral leaflet syndrome, is a relatively common but highly variable clinical syndrome resulting from diverse pathologic mechanisms of the mitral valve apparatus. Among these are excessive or redundant mitral leaflet tissue, which is commonly associated with myxomatous degeneration and greatly increased concentrations of certain glycosaminoglycans.
In most patients with MVP, the cause is unknown, but in some, it appears to be genetically determined. A reduction in the production of type III collagen has been incriminated, and electron microscopy has revealed fragmentation of collagen fibrils.
MVP is a frequent finding in patients with heritable disorders of connective tissue, including Marfan’s syndrome (Chap. 427), osteogenesis imperfecta, and Ehlers-Danlos syndrome. MVP may be associated with thoracic skeletal deformities similar to but not as severe as those in Marfan’s syndrome, such as a high-arched palate and alterations of the chest and thoracic spine, including the so-called straight back syndrome.
In most patients with MVP, myxomatous degeneration is confined to the mitral valve, although the tricuspid and aortic valves may also be affected. The posterior mitral leaflet is usually more affected than the anterior, and the mitral valve annulus is often dilated. In many patients, elongated, redundant, or ruptured chordae tendineae cause or contribute to the regurgitation.
MVP also may occur rarely as a sequel to acute rheumatic fever, in ischemic heart disease, and in various cardiomyopathies, as well as in 20% of patients with ostium secundum atrial septal defect.
MVP may lead to excessive stress on the papillary muscles, which, in turn, leads to dysfunction and ischemia of the papillary muscles and the subjacent ventricular myocardium. Rupture of chordae tendineae and progressive annular dilation and calcification contribute to valvular regurgitation, which then places more stress on the diseased mitral valve apparatus, thereby creating a vicious circle. ECG changes (see below) and ventricular arrhythmias described in some patients with MVP appear to result from regional ventricular dysfunction related to the increased stress placed on the papillary muscles.
CLINICAL FEATURES
MVP is more common in women and occurs most frequently between the ages of 15 and 30 years; the clinical course is most often benign. MVP may also be observed in older (>50 years) patients, often men, in whom MR is often more severe and requires surgical treatment. There is an increased familial incidence for some patients, suggesting an autosomal dominant form of inheritance with incomplete penetrance. MVP varies in its clinical expression, ranging from only a systolic click and murmur with mild prolapse of the posterior leaflet to severe MR due to chordal rupture and leaflet flail. The degree of myxomatous change of the leaflets can also vary widely. In many patients, the condition progresses over years or decades; in others, it worsens rapidly as a result of chordal rupture or endocarditis.
Most patients are asymptomatic and remain so for their entire lives. However, in North America, MVP is now the most common cause of isolated severe MR requiring surgical treatment. Arrhythmias, most commonly ventricular premature contractions and paroxysmal supraventricular and ventricular tachycardia, as well as AF, have been reported and may cause palpitations, light-headedness, and syncope. Sudden death is a very rare complication and occurs most often in patients with severe MR and depressed LV systolic function. There may be an excess risk of sudden death among patients with a flail leaflet. Many patients have chest pain that is difficult to evaluate; it is often substernal, prolonged, and not related to exertion, but may rarely resemble angina pectoris. Transient cerebral ischemic attacks secondary to emboli from the mitral valve due to endothelial disruption have been reported. Infective endocarditis may occur in patients with MR and/or leaflet thickening.
Auscultation A frequent finding is the mid or late (nonejection) systolic click, which occurs 0.14 s or more after S1 and is thought to be generated by the sudden tensing of slack, elongated chordae tendineae or by the prolapsing mitral leaflet when it reaches its maximal excursion. Systolic clicks may be multiple and may be followed by a high-pitched, mid-late systolic crescendo-decrescendo murmur, which occasionally is “whooping” or “honking” and is heard best at the apex. The click and murmur occur earlier with standing, during the strain phase of the Valsalva maneuver, and with any intervention that decreases LV volume, exaggerating the propensity of mitral leaflet prolapse. Conversely, squatting and isometric exercises, which increase LV volume, diminish MVP; the click-murmur complex is delayed, moves away from S1, and may even disappear. Some patients have a mid-systolic click without a murmur; others have a murmur without a click. Still others have both sounds at different times.
LABORATORY EXAMINATION
The ECG most commonly is normal but may show biphasic or inverted T waves in leads II, III, and aVF, and occasionally supraventricular or ventricular premature beats. TTE is particularly effective in identifying the abnormal position and prolapse of the mitral valve leaflets. A useful echocardiographic definition of MVP is systolic displacement (in the parasternal long axis view) of the mitral valve leaflets by at least 2 mm into the LA superior to the plane of the mitral annulus. Color flow and continuous wave Doppler imaging is helpful to evaluate the associated MR and provide semiquantitative estimates of severity. The jet lesion of MR due to MVP is most often eccentric, and assessment of RF and effective regurgitant orifice area can be difficult. TEE is indicated when more accurate information is required and is performed routinely for intraoperative guidance for valve repair. Invasive left ventriculography is rarely necessary but can also show prolapse of the posterior and sometimes of both mitral valve leaflets.
285 |
Tricuspid and Pulmonic Valve Disease |
TRICUSPID STENOSIS
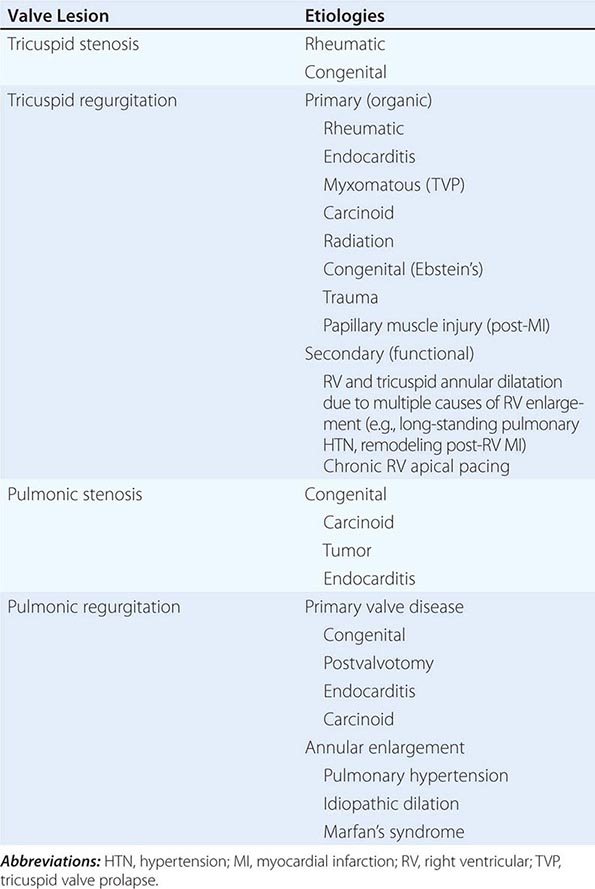
Tricuspid stenosis (TS), which is much less prevalent than mitral stenosis (MS) in North America and Western Europe, is generally rheumatic in origin, and is more common in women than men (Table 285-1). It does not occur as an isolated lesion and is usually associated with MS. Hemodynamically significant TS occurs in 5–10% of patients with severe MS; rheumatic TS is commonly associated with some degree of tricuspid regurgitation (TR). Nonrheumatic causes of TS are rare.
|
CAUSES OF TRICUSPID AND PULMONIC VALVE DISEASES |
PATHOPHYSIOLOGY
A diastolic pressure gradient between the right atrium (RA) and right ventricle (RV) defines TS. It is augmented when the transvalvular blood flow increases during inspiration and declines during expiration. A mean diastolic pressure gradient of 4 mmHg is usually sufficient to elevate the mean RA pressure to levels that result in systemic venous congestion. Unless sodium intake has been restricted and diuretics administered, this venous congestion is associated with hepatomegaly, ascites, and edema, sometimes severe. In patients with sinus rhythm, the RA a wave may be extremely tall and may even approach the level of the RV systolic pressure. The y descent is prolonged. The cardiac output (CO) at rest is usually depressed, and it fails to rise during exercise. The low CO is responsible for the normal or only slightly elevated left atrial (LA), pulmonary artery (PA), and RV systolic pressures despite the presence of MS. Thus, the presence of TS can mask the hemodynamic and clinical features of any associated MS.
SYMPTOMS
Because the development of MS generally precedes that of TS, many patients initially have symptoms of pulmonary congestion and fatigue. Characteristically, patients with severe TS complain of relatively little dyspnea for the degree of hepatomegaly, ascites, and edema that they have. However, fatigue secondary to a low CO and discomfort due to refractory edema, ascites, and marked hepatomegaly are common in patients with advanced TS and/or TR. In some patients, TS may be suspected for the first time when symptoms of right-sided failure persist after an adequate mitral valvotomy.
PHYSICAL FINDINGS
Because TS usually occurs in the presence of other obvious valvular disease, the diagnosis may be missed unless it is considered. Severe TS is associated with marked hepatic congestion, often resulting in cirrhosis, jaundice, serious malnutrition, anasarca, and ascites. Congestive hepatomegaly and, in cases of severe tricuspid valve disease, splenomegaly are present. The jugular veins are distended, and in patients with sinus rhythm, there may be giant a waves. The v waves are less conspicuous, and because tricuspid obstruction impedes RA emptying during diastole, there is a slow y descent. In patients with sinus rhythm, there may be prominent presystolic pulsations of the enlarged liver as well.
On auscultation, an opening snap (OS) of the tricuspid valve may rarely be heard approximately 0.06 s after pulmonic valve closure. The diastolic murmur of TS has many of the qualities of the diastolic murmur of MS, and because TS almost always occurs in the presence of MS, it may be missed. However, the tricuspid murmur is generally heard best along the left lower sternal border and over the xiphoid process, and is most prominent during presystole in patients with sinus rhythm. The murmur of TS is augmented during inspiration, and it is reduced during expiration and particularly during the strain phase of the Valsalva maneuver, when tricuspid transvalvular flow is reduced.
LABORATORY EXAMINATION
The electrocardiogram (ECG) features of RA enlargement (see Fig. 268-8) include tall, peaked P waves in lead II, as well as prominent, upright P waves in lead V1. The absence of ECG evidence of RV hypertrophy (RVH) in a patient with right-sided heart failure who is believed to have MS should suggest associated tricuspid valve disease. The chest x-ray in patients with combined TS and MS shows particular prominence of the RA and superior vena cava without much enlargement of the PA and with less evidence of pulmonary vascular congestion than occurs in patients with isolated MS. On echocardiographic examination, the tricuspid valve is usually thickened and domes in diastole; the transvalvular gradient can be estimated by continuous wave Doppler echocardiography. Severe TS is characterized by a valve area ≤1 cm2 or pressure half-time of ≥190 ms. The RA and inferior vena cava (IVC) are enlarged. Transthoracic echocardiography (TTE) provides additional information regarding the severity of any associated TR, mitral valve structure and function, left ventricle (LV) and RV size and function, and PA pressure. Cardiac catheterization is not routinely necessary for assessment of TS.
TRICUSPID REGURGITATION
In at least 80% of cases, TR is secondary to marked dilation of the tricuspid annulus from RV enlargement due to PA hypertension (Table 285-1). Functional TR may complicate RV enlargement of any cause, however, including an inferior myocardial infarction (MI) that involves the RV. It is commonly seen in the late stages of heart failure due to rheumatic or congenital heart disease with severe PA hypertension (PA systolic pressure >55 mmHg), as well as in ischemic and idiopathic dilated cardiomyopathies. It is reversible in part if PA hypertension can be relieved. Functional TR can also develop from chronic RV apical pacing. Rheumatic fever may produce primary (organic) TR, often associated with TS. Infarction of RV papillary muscles, tricuspid valve prolapse, carcinoid heart disease, endomyocardial fibrosis, radiation, infective endocarditis, and leaflet trauma all may produce TR. Less commonly, TR results from congenitally deformed tricuspid valves, and it occurs with defects of the atrioventricular canal, as well as with Ebstein’s malformation of the tricuspid valve (Chap. 282).
PATHOPHYSIOLOGY
The incompetent tricuspid valve allows blood to flow backward from the RV into the RA, the volume of which is dependent on the driving pressure (i.e., RV systolic pressure) and the size of the regurgitant orifice. The severity and physical signs of TR can vary as a function of PA systolic pressure (in the absence of RV outflow tract stenosis), the dimension of the tricuspid valve annulus, the respiratory cycle–dependent changes in RV preload, and RA compliance. RV filling is increased during inspiration. Forward CO is reduced and does not augment with exercise. Significant degrees of TR will lead to RA enlargement and elevation of the RA and jugular venous pressures with prominent c-v waves in the pulse tracings. Progressively severe TR can lead to “ventricularization” of the RA wave form (see Fig. 267-1B). Severe TR is also characterized by RV dilation (RV volume overload) and eventual systolic dysfunction, the rate of which can be accelerated by a concomitant pressure load from PA hypertension or by myocardial fibrosis from previous injury.
SYMPTOMS
Mild or moderate degrees of TR are usually well tolerated in the absence of other hemodynamic disturbances. Because TR most often coexists with left-sided valve lesions, LV dysfunction, and/or PA hypertension, symptoms related to these lesions may dominate the clinical picture. Fatigue and exertional dyspnea owing to reduced forward CO are early symptoms of isolated, severe TR. As the disease progresses and RV function declines, patients may report cervical pulsations, abdominal fullness/bloating, diminished appetite, and muscle wasting, although with progressive weight gain and painful swelling of the lower extremities.
PHYSICAL FINDINGS
The neck veins in patients with severe TR are distended with prominent c-v waves and rapid y descents (in the absence of TS). TR is more often diagnosed by examination of the neck veins than by auscultation of the heart sounds. Other findings may include marked hepatomegaly with systolic pulsations, ascites, pleural effusions, edema, and a positive hepatojugular reflex. A prominent RV pulsation along the left parasternal region and a blowing holosystolic murmur along the lower left sternal margin, which may be intensified during inspiration (Carvallo’s sign) and reduced during expiration or the strain phase of the Valsalva maneuver, are characteristic findings. The murmur of TR may sometimes be confused with that of MR unless attention is paid to its variation during the respiratory cycle and the extent of RV enlargement is appreciated. Atrial fibrillation (AF) is usually present in the chronic phase of the disease.
LABORATORY EXAMINATION
The ECG may show changes characteristic of the lesion responsible for the TR, e.g., an inferior Q-wave MI suggestive of a prior RV MI, RVH, or a bizarre right bundle branch block type pattern with preexcitation in patients with Ebstein’s anomaly. ECG signs of RA enlargement may be present in patients with sinus rhythm; AF is frequently noted. The chest x-ray may show RA and RV enlargement, depending on the chronicity and severity of TR. TTE is usually definitive with demonstration of RA dilation and RV volume overload and prolapsing, flail, scarred, or displaced/tethered tricuspid leaflets; the diagnosis and assessment of TR can be made by color flow Doppler imaging (see Fig. 270e-8). Severe TR is accompanied by hepatic vein systolic flow reversal. Continuous wave Doppler of the TR velocity profile is useful in estimating PA systolic pressure. Accurate assessment of TR severity, PA pressures, and RV size and systolic function with TTE can be quite challenging in many patients. Real-time three-dimensional echocardiography and cardiac magnetic resonance (CMR) imaging provide alternative imaging modalities, although they are not widely available. In patients with severe TR, the CO is usually markedly reduced, and the RA pressure pulse may exhibit no x descent during early systole but a prominent c-v wave with a rapid y descent. The mean RA and RV end-diastolic pressures are often elevated. Exercise testing can be used to assess functional capacity in patients with asymptomatic severe TR. The prognostic significance of exercise-induced changes in TR severity and RV function has not been well studied.
|
TREATMENT |
TRICUSPID REGURGITATION (FIG. 285-1) |
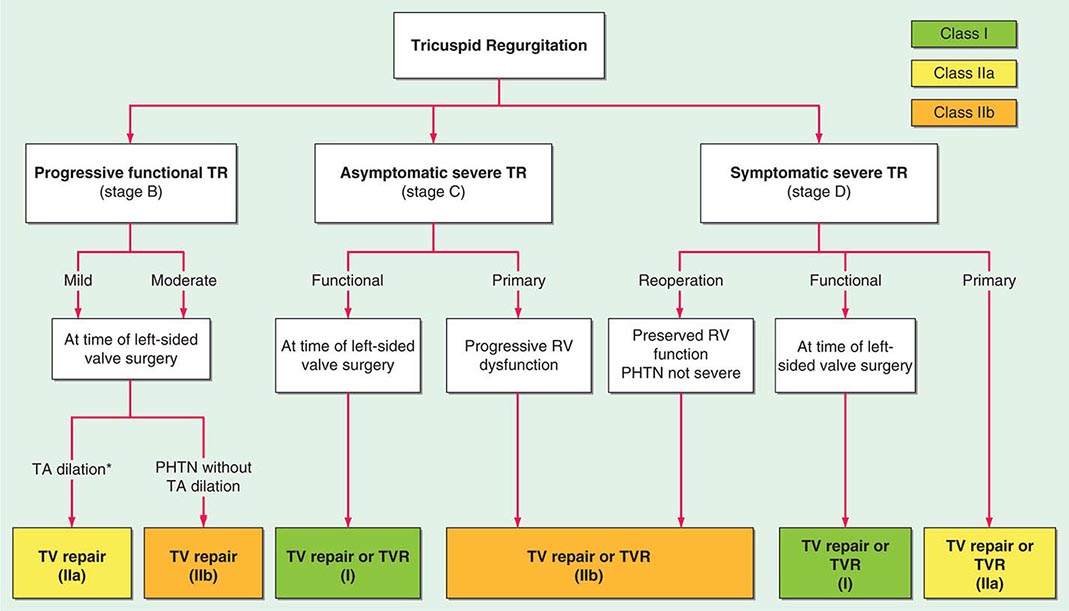
FIGURE 285-1 Management of tricuspid regurgitation. See legend for Fig. 283-2 for explanation of treatment recommendations (Class I, IIa, IIb) and disease stages (B, C, D). Preoperative coronary angiography should be performed routinely as determined by age, symptoms, and coronary risk factors. Cardiac catheterization and angiography may also be helpful when there is a discrepancy between clinical and noninvasive findings. PHTN, pulmonary hypertension; RV, right ventricular; TA, tricuspid annular; TTE, transthoracic echocardiogram; TR, tricuspid regurgitation; TV, tricuspid valve; TVR, tricuspid valve replacement. *TA dilation is defined by >40 mm on TTE (>21 mm/m2) or >70 mm on direct intraoperative measurement. (Adapted from RA Nishimura et al: 2014 AHA/ACC Guideline for the Management of Patients with Valvular Heart Disease. J Am Coll Cardiol doi: 10.1016/j.jacc.2014.02.536, 2014, with permission.)
Diuretics can be useful for patients with severe TR and signs of right heart failure. An aldosterone antagonist may be particularly helpful because many patients have secondary hyperaldosteronism from marked hepatic congestion. Therapies to reduce elevated PA pressures and/or pulmonary vascular resistance, including those targeted at left-sided heart disease, can also be considered for patients with PA hypertension and severe functional TR. Tricuspid valve surgery is recommended for patients with severe TR who are undergoing left-sided valve surgery and is also undertaken frequently for treatment of even moderate TR in patients undergoing left-sided valve surgery who have tricuspid annular dilation (>40 mm), a history of right heart failure, or PA hypertension. Operation most often comprises repair rather than replacement in these settings and has become routine in most major surgical centers. Surgery may also infrequently be required for treatment of severe, primary TR with right heart failure not responsive to standard medical therapy or because of progressively declining RV systolic function. Reported perioperative mortality rates for isolated tricuspid valve surgery (repair and replacement) are high (~8-9%) and likely are influenced by the hazards encountered during reoperation on patients who have undergone previous left-sided valve surgery and have reduced RV function. Indwelling pacemaker or defibrillator leads can also pose technical challenges.
PULMONIC STENOSIS
Pulmonic valve stenosis (PS) is essentially a congenital disorder (Table 285-1). With isolated PS, the valve is typically domed. Dysplastic pulmonic valves are seen as part of the Noonan’s syndrome (Chap. 302), which maps to chromosome 12. Much less common etiologies include carcinoid and obstructing tumors or bulky vegetations. The pulmonic valve is only very rarely affected by the rheumatic process.
PATHOPHYSIOLOGY
PS is defined hemodynamically by a systolic pressure gradient between the RV and main PA. RV hypertrophy develops as a consequence of sustained obstruction to RV outflow, and systolic ejection is prolonged. Compared with the ability of the LV to compensate for the pressure overload imposed by aortic stenosis (AS), RV dysfunction from afterload mismatch occurs earlier in the course of PS and at lower peak systolic pressures, because the RV adapts less well to this type of hemodynamic burden. With normal systolic function and CO, severe PS is defined by a peak systolic gradient across the pulmonic valve of >50 mmHg; moderate PS correlates with a peak gradient of 30–50 mmHg. PS rarely progresses in patients with peak gradients less than 30 mmHg, but may worsen in those with moderate disease due to valve thickening and calcification with age. The RA a wave elevates in relation to the higher pressures needed to fill a noncompliant, hypertrophied RV. A prominent RA v wave signifies functional TR from RV and annular dilation. The CO is maintained until late in the course of the disease.
SYMPTOMS
Patients with mild or even moderate PS are usually asymptomatic and first come to medical attention because of a heart murmur that leads to echocardiography. With severe PS, patients may report exertional dyspnea or early-onset fatigue. Anginal chest pain from RV oxygen supply-demand mismatch and syncope may occur with very severe forms of obstruction, particularly in the presence of a destabilizing trigger such as atrial fibrillation, fever, infection, or anemia.
PHYSICAL FINDINGS
The murmur of mild or moderate PS is mid-systolic in timing, crescendo-decrescendo in configuration, heard best in the left second interspace, and usually introduced by an ejection sound (click) in younger adults whose valves are still pliable. The ejection sound is the only right-sided acoustic event that decreases in intensity with inspiration. This phenomenon reflects premature opening of the pulmonic valve by the elevated RV end-diastolic (postatrial a wave) pressure. The systolic murmur increases in intensity during inspiration. With progressively severe PS, the ejection sound moves closer to the first heart sound and eventually becomes inaudible. A right-sided fourth heart sound may emerge. The systolic murmur peaks later and may persist through the aortic component of the second heart sound (A2). Pulmonic valve closure is delayed, and the pulmonic component of the second heart sound (P2) is reduced or absent. A prominent a wave, indicative of the higher atrial pressure necessary to fill the noncompliant RV, may be seen in the jugular venous pulse. A parasternal or RV lift can be felt with significant pressure overload. Signs of right heart failure, such as hepatomegaly, ascites, and edema, are uncommon but may appear very late in the disease.
LABORATORY EXAMINATION
The ECG will show right axis deviation, RVH, and RA enlargement in adult patients with severe PS. Chest x-ray findings include poststenotic dilation of the main PA in the frontal plane projection and filling of the retrosternal airspace due to RV enlargement on the lateral film. In some patients with RVH, the cardiac apex appears to be lifted off the left hemidiaphragm. The RA may also be enlarged. TTE allows definitive diagnosis and characterization in most cases, with depiction of the valve and assessment of the gradient, RV function, PA pressures (which should be low), and any associated cardiac lesions. TEE may be useful in some patients for improved delineation of the RV outflow tract (RVOT) and assessment of infundibular hypertrophy. Cardiac catheterization is not usually necessary, but if performed, pressures should be obtained from just below and above the pulmonic valve with attention to the possibility that a dynamic component to the gradient may exist. The correlation between Doppler assessment of peak instantaneous gradient and catheterization-measured peak-to-peak gradient is weak. The latter may correlate better with the Doppler mean gradient.
PULMONIC REGURGITATION
Pulmonic regurgitation (PR) may develop as a consequence of primary valve pathology, annular enlargement, or their combination; after surgical treatment of RVOT obstruction in children with such disorders as tetralogy of Fallot; or after pulmonic balloon valvotomy (Table 285-1). Carcinoid usually causes mixed pulmonic valve disease with PR and PS. Long-standing severe PA hypertension from any cause can result in dilation of the pulmonic valve ring and PR.
PATHOPHYSIOLOGY
Severe PR results in RV chamber enlargement and eccentric hypertrophy. As is the case for aortic regurgitation (AR), PR is a state of increased preload and afterload. The reverse pressure gradient from the PA to the RV, which drives the PR, progressively decreases throughout diastole and accounts for the decrescendo nature of the diastolic murmur. As RV diastolic pressure increases, the murmur becomes shorter in duration. The forward CO is preserved during the early stages of the disease, but may not increase normally with exercise and declines over time. A reduction in RV ejection fraction may be an early indicator of hemodynamic compromise. In advanced stages, there is significant enlargement of the RV and RA with marked elevation of the jugular venous pressure.
SYMPTOMS
Mild or moderate degrees of PR do not, by themselves, result in symptoms. Other problems, such as PA hypertension, may dominant the clinical picture. With progressively severe PR and RV dysfunction, fatigue, exertional dyspnea, abdominal fullness/bloating, and lower extremity swelling may be reported.
PHYSICAL FINDINGS
The physical examination hallmark of PR is a high-pitched, decrescendo diastolic murmur (Graham Steell murmur) heard along the left sternal border that can be difficult to distinguish from the more frequently appreciated murmur of aortic regurgitation. The Graham Steell murmur may become louder with inspiration and is usually associated with a loud and sometimes palpable P2 and an RV lift, as would be expected in patients with significant PA hypertension of any cause. Survivors of childhood surgery for tetralogy of Fallot or PS/pulmonary atresia may have an RV-PA conduit that is freely regurgitant because it does not contain a valve. PA pressures in these individuals are not elevated and the diastolic murmur can be misleadingly low pitched and of short duration despite significant degrees of PR and RV volume overload.
LABORATORY EXAMINATION
Depending on both the etiology and severity of PR, the ECG may show findings of RVH and RA enlargement. On chest x-ray, the RV and RA may be enlarged. Pulmonic valve morphology and function can be assessed with transthoracic Doppler echocardiography. PA pressures can be estimated from the tricuspid valve systolic jet velocity. CMR provides greater anatomic detail, particularly in patients with repaired congenital heart disease, and more precise assessment of RV volumes. Cardiac catheterization is not routinely necessary but would be performed as part of a planned transcatheter procedure.
286 |
Multiple and Mixed Valvular Heart Disease |
Many acquired and congenital cardiac lesions may result in stenosis and/or regurgitation of one or more heart valves. For example, rheumatic heart disease can involve the mitral (mitral stenosis [MS], mitral regurgitation [MR], or MS and MR), aortic (aortic stenosis [AS], aortic regurgitation [AR], or AS and AR), and/or tricuspid (tricuspid stenosis [TS], tricuspid regurgitation [TR], or TS and TR) valve, alone or in combination. The common association of functional TR with significant mitral valve disease is discussed in Chap. 285. Severe mitral annular calcification can result in regurgitation (due to decreased annular shortening during systole) and mild stenosis (caused by extension of the calcification onto the leaflets resulting in restricted valve opening). Patients with severe AS may develop functional MR that may not improve after isolated aortic valve replacement (AVR). Chordal rupture has been described infrequently in patients with severe AS. Aortic valve infective endocarditis may secondarily involve the mitral apparatus either by abscess formation and contiguous spread via the intervalvular fibrosa or by “drop metastases” from the aortic leaflets onto the anterior leaflet of the mitral valve. Mediastinal radiation may result in aortic, mitral, and even tricuspid valve disease, most often with mixed stenosis and regurgitation. Carcinoid heart disease may cause mixed lesions of either or both the tricuspid and pulmonic valves. Ergotamines, and the previously used combination of fenfluramine and phentermine, can rarely result in mixed lesions of the aortic and/or mitral valve. Patients with Marfan’s syndrome may have both AR from aortic root dilation and MR due to mitral valve prolapse (MVP). Myxomatous degeneration causing prolapse of multiple valves (mitral, aortic, tricuspid) can also occur in the absence of an identifiable connective tissue disorder. Bicuspid aortic or pulmonic valve disease can result in mixed stenosis and regurgitation.
PATHOPHYSIOLOGY
In patients with multivalvular heart disease, the pathophysiologic derangements associated with the more proximal valve disease can mask the full expression of the attributes of the more distal valve lesion. For example, in patients with rheumatic mitral and aortic valve disease, the reduction in cardiac output (CO) imposed by the mitral valve disease will decrease the magnitude of the hemodynamic derangements related to the severity of the aortic valve lesion (stenotic, regurgitant, or both). Alternatively, the development of atrial fibrillation (AF) during the course of MS can lead to sudden worsening in a patient whose aortic valve disease was not previously felt to be significant. The development of reactive pulmonary vascular disease, sometimes referred to as a “secondary obstructive lesion in series,” can impose an additional challenge in these settings. As CO falls with progressive tricuspid valve disease, the severity of any associated mitral or aortic disease can be underestimated.
One of the most common examples of multivalve disease is that of functional TR in the setting of significant mitral valve disease. Functional TR occurs as a consequence of right ventricular and annular dilation; pulmonary artery (PA) hypertension is often present. The tricuspid leaflets are morphologically normal. Progressive degrees of TR lead to right ventricular volume overload and continued chamber and annular dilation. The TR is usually central in origin; reflux into the right atrium (RA) is expressed as large, systolic c-v waves in the RA pressure pulse. The height of the c-v wave is dependent on RA compliance and the volume of regurgitant flow. The RA wave form may become “ventricularized” in advanced stages of chronic, severe TR with PA hypertension. CO falls and the severity of the associated mitral valve disease may become more difficult to appreciate. Primary rheumatic tricuspid valve disease may occur with rheumatic mitral disease and cause hemodynamic changes reflective of TR, TS, or their combination. With TS, the y descent in the RA pressure pulse is prolonged.
Another example of rheumatic, multivalve disease involves the combination of mitral and aortic valve pathology, frequently characterized by MS and AR. In isolated MS, left ventricular (LV) preload and diastolic pressure are reduced as a function of the severity of inflow obstruction. With concomitant AR, however, LV filling is enhanced and diastolic pressure may rise depending on the compliance characteristics of the chamber. Because the CO falls with progressive degrees of MS, transaortic valve flows will decline, masking the potential severity of the aortic valve lesion (AR, AS, or its combination). As noted above, onset of AF in such patients can be especially deleterious.
Functional MR may complicate the course of some patients with severe AS. The mitral valve leaflets and chordae tendineae are usually normal. Incompetence is related to changes in LV geometry (remodeling) and abnormal systolic tethering of the leaflets in the context of markedly elevated LV systolic pressures. Relief of the excess afterload with surgical or transcatheter AVR often, but not always, results in reduction or elimination of the MR. Persistence of significant MR following AVR is associated with impaired functional outcomes and reduced survival. Identification of patients who would benefit from concomitant treatment of their functional MR at time of AVR is quite challenging. Most surgeons advocate for repair of moderate-to-severe or severe functional MR at time of surgical AVR.
In patients with mixed AS and AR, assessment of valve stenosis can be influenced by the magnitude of the regurgitant valve flow. Because transvalvular systolic flow velocities are augmented in patients with AR and preserved LV systolic function, the LV-aortic Doppler-derived pressure gradient and the intensity of the systolic murmur will be elevated to values higher than expected for the true systolic valve orifice size as delineated by planimetry. Uncorrected, the Gorlin formula, which relies on forward CO (systolic transvalvular flow) and the mean pressure gradient for calculation of valve area, is not accurate in the setting of mixed aortic valve disease. Similar considerations apply to patients with mixed mitral valve disease. The peak mitral valve Doppler E wave velocity (v0) is increased in the setting of severe MR because of enhanced early diastolic flow and may not accurately reflect the contribution to left atrial (LA) hypertension from any associated MS. When either AR or MR is the dominant lesion in patients with mixed aortic or mitral valve disease, respectively, the LV is dilated. When AS or MS predominates, LV chamber size will be normal or small. It can sometimes be difficult to ascertain whether stenosis or regurgitation is the dominant lesion in patients with mixed valve disease, although an integrated clinical and noninvasive assessment can usually provide clarification for purposes of patient management and follow-up.
Patients with significant AS, a nondilated LV chamber, and concentric hypertrophy will poorly tolerate the abrupt development of aortic regurgitation, as may occur, for example, with infective endocarditis or after surgical or transcatheter AVR complicated by paravalvular leakage. The noncompliant LV is not prepared to accommodate the sudden volume load, and as a result, LV diastolic pressure rises rapidly and severe heart failure develops. Indeed, paravalvular regurgitation is a significant risk factor for short- to intermediate-term death following transcatheter AVR. Conditions in which the LV may not be able to dilate in response to chronic AR (or MR) include radiation heart disease and, in some patients, the cardiomyopathy associated with obesity and diabetes. Noncompliant ventricles of small chamber size predispose to earlier onset diastolic dysfunction and heart failure in response to any further perturbation in valve function.
SYMPTOMS
Compared with patients with isolated, single-lesion valve disease, patients with multiple or mixed valve disease may develop symptoms at a relatively earlier stage in the natural history of their disease. Symptoms such as exertional dyspnea and fatigue are usually related to elevated filling pressures, reduced CO, or their combination. Palpitations may signify AF and identify mitral valve disease as an important component of the clinical presentation, even when not previously suspected. Chest pain compatible with angina could reflect left or right ventricular oxygen supply/demand mismatch on a substrate of hypertrophy and pressure/volume overload with or without superimposed coronary artery disease. Symptoms related to right heart failure (abdominal fullness/bloating, edema) are late manifestations of advanced disease.
PHYSICAL FINDINGS
Mixed disease of a single valve is most often manifested by systolic and diastolic murmurs, each with the attributes expected for the valve in question. Thus, patients with AS and AR will have characteristic mid-systolic, crescendo-decrescendo and blowing, decrescendo diastolic murmurs at the base of the heart in the second right interspace and along the left sternal edge, respectively. Many patients with significant AR have mid-systolic outflow murmurs even in the absence of valve sclerosis/stenosis, and other findings of AS must be sought. The separate murmurs of AS and AR can occasionally be difficult to distinguish from the continuous murmurs associated with either a patent ductus arteriosus (PDA) or ruptured sinus of Valsalva aneurysm. With mixed aortic valve disease, the systolic murmur should end before, and not envelope or extend through, the second heart sound (S2). The murmur associated with a PDA is heard best to the left of the upper sternum. The continuous murmur heard with a ruptured sinus of Valsalva aneurysm is often first appreciated after an episode of acute chest pain. An early ejection click, which usually defines bicuspid aortic valve disease in young adults, is often not present in patients with congenital, mixed AS and AR. As noted above, both the intensity and duration of these separate murmurs can be influenced by a reduction in CO and transvalvular flow due to coexistent mitral valve disease. In patients with isolated MS and MR, expected findings would include a blowing, holosystolic murmur and a mid-diastolic rumble (with or without an opening snap) best heard at the cardiac apex. An irregularly irregular heart rhythm in such patients would likely signify AF. Findings with TS and TR would mimic those of left-sided MS and MR, save for the expected changes in the murmurs with respiration. The murmurs of pulmonic stenosis and regurgitation behave in a fashion directionally similar to AS and AR; dynamic changes during respiration should be noted. Specific attributes of these cardiac murmurs are reviewed in Chap. 285.
LABORATORY EXAMINATION
The electrocardiogram (ECG) may show evidence of ventricular hypertrophy and/or atrial enlargement. ECG signs indicative of right-sided cardiac abnormalities in patients with left-sided valve lesions should prompt additional assessment for PA hypertension and/or right-sided valve disease. The presence of AF in patients with aortic valve disease may be a clue to the presence of previously unsuspected mitral valve disease in the appropriate context. The chest x-ray can be reviewed for evidence of cardiac chamber enlargement, valve and/or annular calcification, and any abnormalities in the appearance of the pulmonary vasculature. The latter could include enlargement of the main and proximal pulmonary arteries with PA hypertension and pulmonary venous redistribution/engorgement or Kerley B lines with increasing degrees of LA hypertension. An enlarged azygos vein in the frontal projection indicates RA hypertension. Roentgenographic findings not expected based on a single or mixed valve lesion may reflect other valve disease.
Transthoracic echocardiography (TTE) is the most commonly used imaging modality for the diagnosis and characterization of multiple and/or mixed valvular heart disease and may often demonstrate findings not clinically suspected. Transesophageal echocardiography (TEE) may sometimes be required for more accurate assessment of valve anatomy (specifically, the mitral valve) and when infective endocarditis (IE) is considered responsible for the clinical presentation. TTE findings of particular interest include those related to valve morphology and function, calcification, chamber size, ventricular wall thickness, estimated PA systolic pressure, and the dimensions of the great vessels, including the root and ascending aorta, PA, and inferior vena cava. Exercise testing (with or without echocardiography) can be useful when the degree of functional limitation reported by the patient is not adequately explained by the findings on TTE performed at rest. An integrated assessment of the clinical and TTE findings is needed to help determine the dominant valve lesion(s) and establish an appropriate plan for treatment and follow-up. Natural history is usually influenced to a relatively greater degree by the dominant lesion. Exercise testing (with or without echocardiography) can
Cardiac magnetic resonance (CMR) can be used to provide additional anatomic and physiologic information when echocardiography proves suboptimal, but is less well suited to the evaluation of valve morphology. Cardiac computed tomography (CT) has been used to assess intracardiac structures in patients with complicated IE. Coronary CT angiography provides a noninvasive alternative for the assessment of coronary artery anatomy prior to surgery.
Invasive hemodynamic evaluation with right and left heart catheterization may be required to characterize more completely the individual contributions of each lesion in patients with either multiple or mixed valvular heart disease. Measurement of PA pressures and calculation of pulmonary vascular resistance (PVR) can help inform clinical decision-making in certain patient subsets, such as those with advanced mitral and tricuspid valve disease. Attention to the accurate assessment of CO is essential. Coronary angiography (if indicated) can be performed as part of the procedure. Contrast ventriculography and great vessel angiography are performed infrequently.
287 |
Cardiomyopathy and Myocarditis |
DEFINITION AND CLASSIFICATION
Cardiomyopathy is disease of the heart muscle. It is estimated that cardiomyopathy accounts for 5–10% of the heart failure in the 5–6 million patients carrying that diagnosis in the United States. This term is intended to exclude cardiac dysfunction that results from other structural heart disease, such as coronary artery disease, primary valve disease, or severe hypertension; however, in general usage, the phrase ischemic cardiomyopathy is sometimes applied to describe diffuse dysfunction attributed to multivessel coronary artery disease, and nonischemic cardiomyopathy to describe cardiomyopathy from other causes. As of 2006, cardiomyopathies are defined as “a heterogeneous group of diseases of the myocardium associated with mechanical and/or electrical dysfunction that usually (but not invariably) exhibit inappropriate ventricular hypertrophy or dilatation and are due to a variety of causes that frequently are genetic.”1
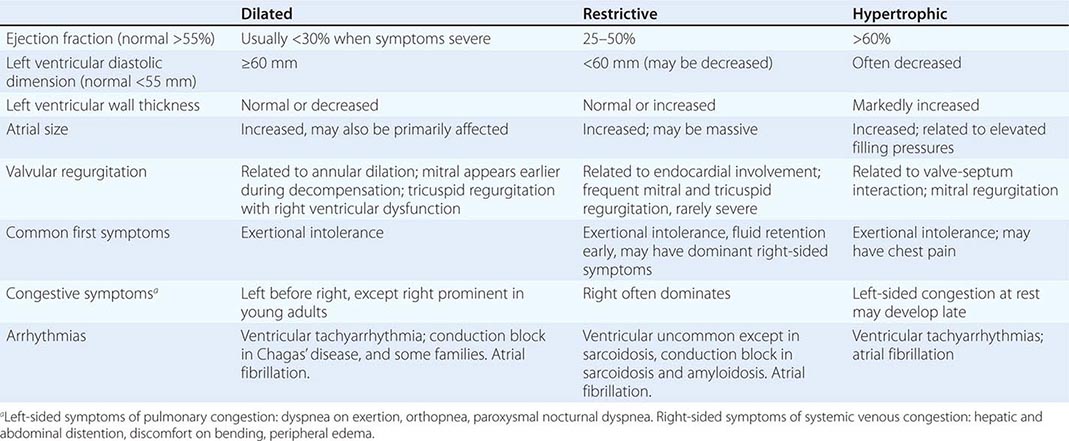
The traditional classification of cardiomyopathies into a triad of dilated, restrictive, and hypertrophic was based initially on autopsy specimens and later on echocardiographic findings. Dilated and hypertrophic cardiomyopathies can be distinguished on the basis of left ventricular wall thickness and cavity dimension; however, restrictive cardiomyopathy can have variably increased wall thickness and chamber dimensions that range from reduced to slightly increased, with prominent atrial enlargement. Restrictive cardiomyopathy is now defined more on the basis of abnormal diastolic function, which is also present but initially less prominent in dilated and hypertrophic cardiomyopathy. Restrictive cardiomyopathy can overlap in presentation, gross morphology, and etiology with both hypertrophic and dilated cardiomyopathies (Table 287-1).
|
PRESENTATION WITH SYMPTOMATIC CARDIOMYOPATHY |
Expanding information renders this classification triad based on phenotype increasingly inadequate to define disease or therapy. Identification of more genetic determinants of cardiomyopathy has suggested a four-way classification scheme of etiology as primary (affecting primarily the heart) and secondary to other systemic disease. The primary causes are then divided into genetic, mixed genetic and acquired, and acquired; however, genetic information is often unavailable at the time of initial presentation, the phenotypic expression of a given mutation varies widely, and genetic predisposition influences the clinical phenotype of acquired cardiomyopathies, as well. Although the proposed genetic classification does not yet guide many current clinical strategies, it will likely become increasingly relevant as classification of disease moves beyond individual organ pathology to more integrated systems approaches.
GENERAL PRESENTATION
For all cardiomyopathies, the early symptoms often relate to exertional intolerance with breathlessness or fatigue, usually from inadequate cardiac reserve during exercise. These symptoms may initially go unnoticed or be attributed to other causes, commonly lung disease or age-dependent exercise limitation. As fluid retention leads to elevation of resting filling pressures, shortness of breath may occur during routine daily activity such as dressing and may manifest as dyspnea or cough when lying down at night. Although often considered the hallmark of congestion, peripheral edema may be absent despite severe fluid retention, particularly in younger patients in whom ascites and abdominal discomfort may dominate. The nonspecific term congestive heart failure describes only the resulting syndrome of fluid retention, which is common to all three types of cardiomyopathy and also to cardiac structural diseases associated with elevated filling pressures. All three types of cardiomyopathy can be associated with atrioventricular valve regurgitation, typical and atypical chest pain, atrial and ventricular tachyarrhythmias, and embolic events (Table 287-1). Initial evaluation begins with a detailed clinical history and examination, looking for clues to cardiac, extracardiac, and familial disease (Table 287-2).
|
INITIAL EVALUATION OF CARDIOMYOPATHY |
aLevel I recommendations from ACC/AHA Practice Guidelines for Chronic Heart Failure in the Adult.
GENETIC ETIOLOGIES OF CARDIOMYOPATHY
![]() Estimates for the prevalence of genetic etiology for cardiomyopathy continue to rise, with increasing attention paid to the family history and the availability of genetic testing. Well-recognized in hypertrophic cardiomyopathy, heritability is also present in at least 30% of dilated cardiomyopathy without other clear etiology. Careful family history should elicit not only known cardiomyopathy and heart failure, but also family members who have had sudden death, often incorrectly attributed to “a massive heart attack,” who have had atrial fibrillation or pacemaker implantation by middle age, or who have muscular dystrophy.
Estimates for the prevalence of genetic etiology for cardiomyopathy continue to rise, with increasing attention paid to the family history and the availability of genetic testing. Well-recognized in hypertrophic cardiomyopathy, heritability is also present in at least 30% of dilated cardiomyopathy without other clear etiology. Careful family history should elicit not only known cardiomyopathy and heart failure, but also family members who have had sudden death, often incorrectly attributed to “a massive heart attack,” who have had atrial fibrillation or pacemaker implantation by middle age, or who have muscular dystrophy.
Most familial cardiomyopathies are inherited in an autosomal dominant pattern, with occasional autosomal recessive and X-linked inheritance (Table 287-3). Missense mutations with amino acid substitutions are the most common in cardiomyopathy. Expressed mutant proteins may interfere with function of the normal allele through a dominant negative mechanism. Mutations introducing a premature stop codon (nonsense) or shift in the reading frame (frameshift) may create a truncated or unstable protein the lack of which causes cardiomyopathy (haploinsufficiency). Deletions or duplications of an entire exon or gene are uncommon causes of cardiomyopathy, except for the dystrophinopathies.
|
SELECTED GENETIC DEFECTS ASSOCIATED WITH CARDIOMYOPATHY |
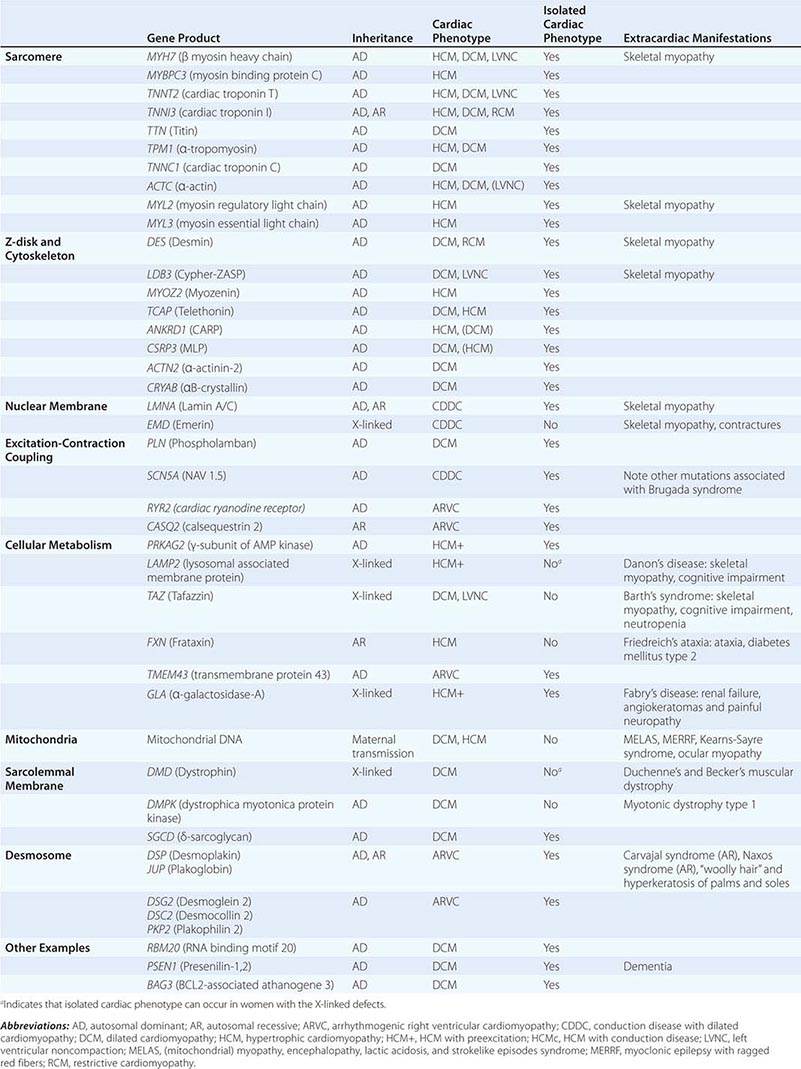
Many different genes have been implicated in human cardiomyopathy (locus heterogeneity), and many mutations within those genes have been associated with disease (allelic heterogeneity). Although most identified mutations are “private” to individual families, several specific mutations are found repeatedly, either due to a founder effect or recurrent mutations at a common residue.
Genetic cardiomyopathy is characterized by age dependence and incomplete penetrance. The defining phenotype of cardiomyopathy is rarely present at birth and, in some individuals, may never manifest. Related individuals who carry the same mutation may differ in the severity of cardiomyopathy and associated consequences of rhythm disorders and need for transplantation, indicating the important role of other genetic, epigenetic, and environmental modifiers in disease expression. Sex appears to play a role, as penetrance and clinical severity may be greater in men for most cardiomyopathies. Clinical disease expression is generally more severe in the 3–5% of individuals who harbor two or more mutations linked to cardiomyopathy. However, the clinical course of a patient usually cannot be predicted based on which mutation is present; thus, current therapy is based on the phenotype rather than the genetic defect. Currently, the greatest utility of genetic testing for cardiomyopathy is to inform family evaluations. However, genetic testing occasionally enables the detection of a disease for which specific therapy is indicated, such as the replacements for defective metabolic enzymes in Fabry’s disease and Gaucher disease.
GENES AND PATHWAYS IN CARDIOMYOPATHY
Mutations in sarcomeric genes, encoding the thick and thin myofilament proteins, are the best characterized. While the majority are associated with hypertrophic cardiomyopathy, an increasing number of sarcomeric mutations have now been implicated in dilated cardiomyopathy, and some in left ventricular noncompaction. Few mutations have been identified in excitation-contraction coupling proteins, perhaps because they are too crucial for survival to allow variation. The most commonly recognized genetic causes of dilated cardiomyopathy are structural mutations of the giant protein titin, encoded TTN, which maintains sarcomere structure and acts as a key signaling molecule.
As cytoskeletal proteins play crucial roles in the structure, connection, and stability of the myocyte, multiple defects in these proteins can lead to cardiomyopathy, usually with a dilated phenotype (Fig. 287-1). For example, desmin forms intermediate filaments that connect the nuclear and plasma membranes, Z-lines, and the intercalated disks between muscle cells. Desmin mutations impair the transmission of force and signaling for both cardiac and skeletal muscle and may cause combined cardiac and skeletal myopathy.
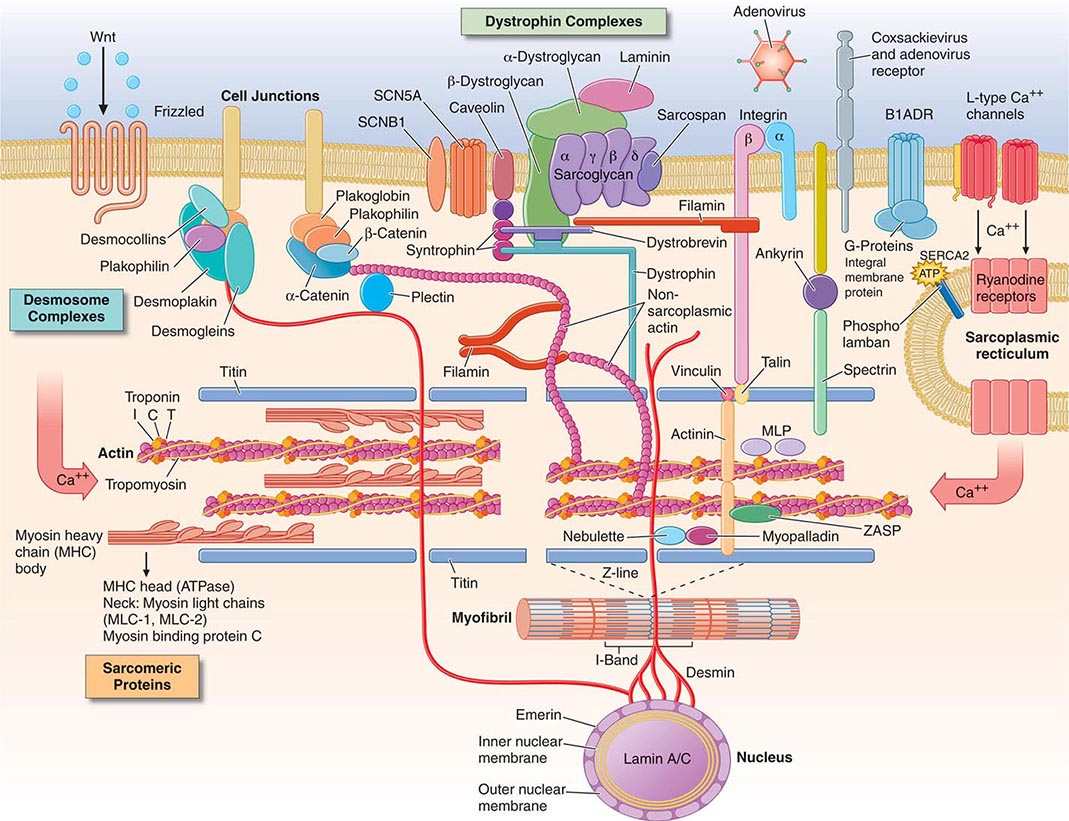
FIGURE 287-1 Drawing of myocyte indicating multiple sites of abnormal gene products associated with cardiomyopathy. Major functional groups include the sarcomeric proteins (actin, myosin, tropomyosin, and the associated regulatory proteins), the dystrophin complex stabilizing and connecting the cell membrane to intracellular structures, the desmosome complexes associated with cell-cell connections and stability, and multiple cytoskeletal proteins that integrate and stabilize the myocyte. ATP, adenosine triphosphate. (Figure adapted from Jeffrey A. Towbin, MD, University of Cincinnati, with permission.)
Sarcolemmal membrane protein defects are associated with dilated cardiomyopathy. The best known is dystrophin, encoded by the × chromosome gene DMD, abnormalities of which cause Duchenne’s and Becker’s muscle dystrophy. (Interestingly, abnormal dystrophin can be acquired when the coxsackie virus cleaves dystrophin during viral myocarditis.) This protein provides a network that supports the sarcolemma and also connects to the sarcomere. The progressive functional defect in both cardiac and skeletal muscle reflects vulnerability to mechanical stress. Dystrophin is associated at the membrane with a complex of other proteins, such as metavinculin, abnormalities of which also cause dilated cardiomyopathy. Defects in the sarcolemmal channel proteins (channelopathies) are generally associated with primary arrhythmias, but mutations in SCN5A, distinct from those that cause the Brugada or long-QT syndromes, have been implicated in dilated cardiomyopathy with conduction disease.
Nuclear membrane protein defects in cardiac and skeletal muscle occur in either autosomal (lamin A/C) or X-linked (emerin) patterns. These defects are associated with a high prevalence of atrial arrhythmias and conduction system disease, which can occur in some family members without or before detectable cardiomyopathy.
Intercalated disks contribute to intracellular connections, allowing mechanical and electrical coupling between cells and also connections to desmin filaments within the cell. Mutations in proteins of the desmosomal complex compromise attachment of the myocytes, which can become disconnected and die, to be replaced by fat and fibrous tissue. These areas are highly arrhythmogenic and may dilate to form aneurysms. Although more often noted in the right ventricle (arrhythmogenic right ventricular dysplasia), this condition can affect both ventricles and has also been termed “arrhythmogenic cardiomyopathy.”
Owing to the conservation of signaling pathways in multiple systems, we may expect to discover more extracardiac manifestations of genetic abnormalities initially considered to manifest exclusively in the heart. In contrast, the monogenic disorders of metabolism that affect the heart are already clearly recognized to affect multiple organ systems. Currently, it is most important to diagnose defective enzymes for which specific enzyme replacement therapy can now ameliorate the course of disease, such as with alpha-galactosidase A deficiency (Fabry’s disease). Abnormalities of mitochondrial DNA (maternally transmitted) impair energy production with multiple clinical manifestations, including impaired cognitive function and skeletal myopathy. The phenotypic expression is highly variable depending on the distribution of the maternal mitochondria during embryonic development. Heritable systemic diseases, such as familial amyloidosis and hemochromatosis, can affect the heart without mutation of genes expressed in the heart.
For any patient with suspected or proven genetic disease, family members should be considered and evaluated in a longitudinal fashion. Screening includes an echocardiogram and electrocardiogram (ECG). The indications and implications for confirmatory specific genetic testing vary depending on the specific mutation. The profound questions raised by families about diseases shared and passed down merit serious and sensitive discussion, ideally provided by a trained genetic counselor.
DILATED CARDIOMYOPATHY
An enlarged left ventricle with decreased systolic function as measured by left ventricular ejection fraction characterizes dilated cardiomyopathy (Figs. 287-2, 287-3, and 287-4). Systolic failure is more marked than diastolic dysfunction. Although the syndrome of dilated cardiomyopathy has multiple etiologies (Table 287-4), there appear to be common pathways of secondary response and disease progression. When myocardial injury is acquired, some myocytes may die initially, whereas others survive only to have later programmed cell death (apoptosis), and remaining myocytes hypertrophy in response to increased wall stress. Local and circulating factors stimulate deleterious secondary responses that contribute to progression of disease. Dynamic remodeling of the interstitial scaffolding affects diastolic function and the amount of ventricular dilation. Mitral regurgitation commonly develops as the valvular apparatus is distorted and is usually substantial by the time heart failure is severe. Many cases that present “acutely” have progressed silently through these stages over months to years. Dilation and decreased function of the right ventricle may result from the initial injury and occasionally dominate, but more commonly appear later in relation to mechanical interactions with the failing left ventricle and the elevated afterload presented by secondary pulmonary hypertension.
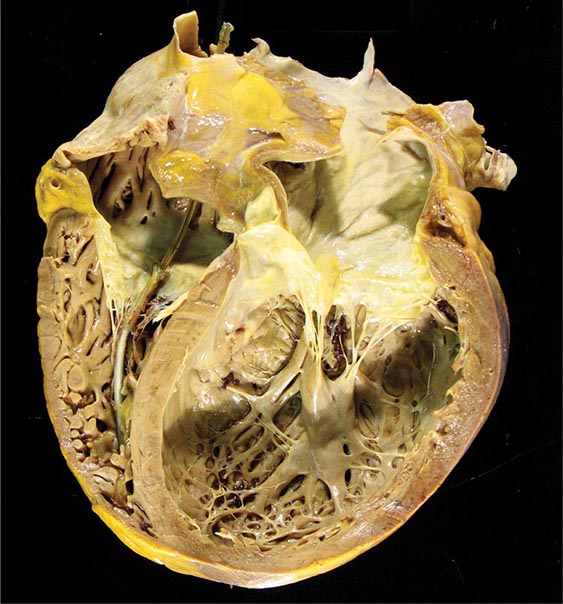
FIGURE 287-2 Dilated cardiomyopathy. This gross specimen of a heart removed at the time of transplantation shows massive left ventricular dilation and moderate right ventricular dilation. Although the left ventricular wall in particular appears thinned, there is significant hypertrophy of this heart, which weighs more than 800 g (upper limit of normal = 360 g). A defibrillator lead is seen traversing the tricuspid valve into the right ventricular apex. (Image courtesy of Robert Padera, MD, PhD, Department of Pathology, Brigham and Women’s Hospital, Boston.)
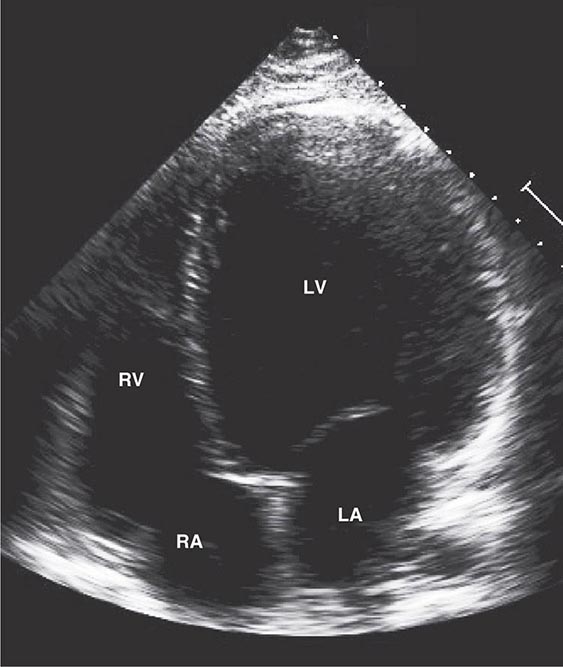
FIGURE 287-3 Dilated cardiomyopathy. This echocardiogram of a young man with dilated cardiomyopathy shows massive global dilation and thinning of the walls of the left ventricle (LV). The left atrium (LA) is also enlarged compared to normal. Note that the echocardiographic and pathologic images are vertically opposite, such that the LV is by convention on the top right in the echocardiographic image and bottom right in the pathologic images. RA, right atrium; RV, right ventricle. (Image courtesy of Justina Wu, MD, Brigham and Women’s Hospital, Boston.)
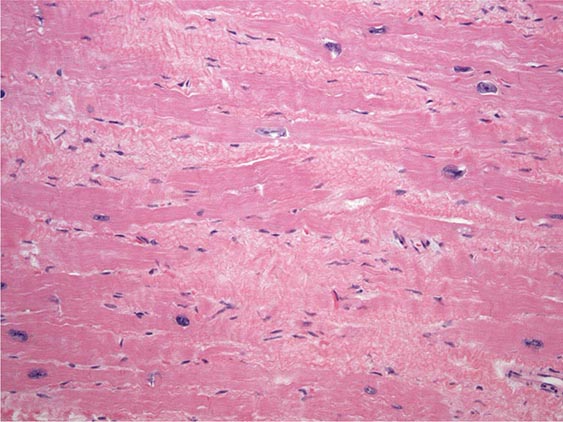
FIGURE 287-4 Dilated cardiomyopathy. Microscopic specimen of a dilated cardiomyopathy showing the nonspecific changes of interstitial fibrosis and myocyte hypertrophy characterized by increased myocyte size and enlarged, irregular nuclei. Hematoxylin and eosin–stained section, 100× original magnification. (Image courtesy of Robert Padera, MD, PhD, Department of Pathology, Brigham and Women’s Hospital, Boston.)
|
MAJOR CAUSES OF DILATED CARDIOMYOPATHY (WITH COMMON EXAMPLES) |
aSome specific cases can be linked now to specific genetic mutation in a familial cardiomyopathy; others with similar phenotypes that appear to be acquired or idiopathic may represent genetic factors not yet identified.
Regardless of the nature and degree of direct cell injury, the resulting functional impairment often includes some contribution from secondary responses that may be modifiable or reversible. Almost half of all patients with new-onset cardiomyopathy demonstrate substantial spontaneous recovery. Even with long-standing disease, some patients have dramatic improvement to near-normal ejection fractions during pharmacologic therapy, particularly notable with the β-adrenergic antagonists coupled with renin-angiotensin system inhibition. For patients in whom left bundle branch block precedes clinical heart failure by many years, cardiac resynchronization pacing may be particularly likely to improve ejection fraction and decrease ventricular size. Interest in the potential for recovery of cardiomyopathy has been further stimulated by occasional “recovery” of left ventricular function after prolonged mechanical circulatory support. The diagnosis and therapy for dilated cardiomyopathy are generally dictated by the stage of heart failure (Chap. 279), with specific aspects discussed for relevant etiologies below.
MYOCARDITIS
Myocarditis (inflammation of the heart) can result from multiple causes but is most commonly attributed to infective agents that can injure the myocardium through direct invasion, production of cardiotoxic substances, or chronic inflammation with or without persistent infection. Myocarditis cannot be assumed from a presentation of decreased systolic function in the setting of an acute infection, as any severe infection causing systemic cytokine release can depress cardiac function transiently. Infectious myocarditis has been reported with almost all types of infective agents but is most commonly associated with viruses and the protozoan Trypanosoma cruzi.
INFECTIVE MYOCARDITIS
The pathogenesis of viral myocarditis has been extensively studied in murine models. After viruses gain entry through the respiratory or gastrointestinal tract, they can infect organs possessing specific receptors, such as the coxsackie-adenovirus receptor on the heart. Viral infection and replication can cause myocardial injury and lysis. For example, the enteroviral protease 2A facilitates viral replication and infection through degradation of the myocyte protein dystrophin, which is crucial for myocyte stability. Activation of viral receptor proteins can also activate host tyrosine kinases, which modify the cytoskeleton to facilitate further viral entry.
The first host response to infection is the nonspecific innate immune response, heavily dependent on Toll-like receptors that recognize common antigenic patterns. Cytokine release is rapid, followed by triggered activation and expansion of specific T- and B-cell populations. This initial response appears to be crucial, as early immunosuppression in animal models can increase viral replication and worsen cardiac injury. However, successful recovery from viral infection depends not only on the efficacy of the immune response to limit viral infection, but also on timely downregulation to prevent overreaction and autoimmune injury to the host.
The secondary acquired immune response is more specifically addressed against the viral proteins and can include both T-cell infiltration and antibodies to viral proteins. If unchecked, the acquired immune response can perpetuate secondary cardiac damage. Ongoing cytokine release activates matrix metalloproteinases that can disrupt the collagen and elastin scaffolding of the heart, potentiating ventricular dilation. Stimulation of profibrotic factors leads to pathologic interstitial fibrosis. Some of the antibodies triggered through co-stimulation or molecular mimicry also recognize targets within the host myocyte, such as the β-adrenergic receptor, troponin, and Na+/K+ ATPase, but it remains unclear whether these antibodies contribute actively to cardiac dysfunction in humans or merely serve as markers of cardiac injury.
It is not known how long the viruses persist in the human heart, whether late persistence of the viral genome continues to be deleterious, or how often a dormant virus can again become pathogenic. Genomes of common viruses have frequently been detected in patients with clinical diagnoses of myocarditis or dilated cardiomyopathy, but there is little information on how often these are present in patients without cardiac disease (see below). Further information is needed to understand the relative timing and contribution of infection, immune responses, and secondary adaptations in the progression of heart failure after viral myocarditis (Fig 287-5).
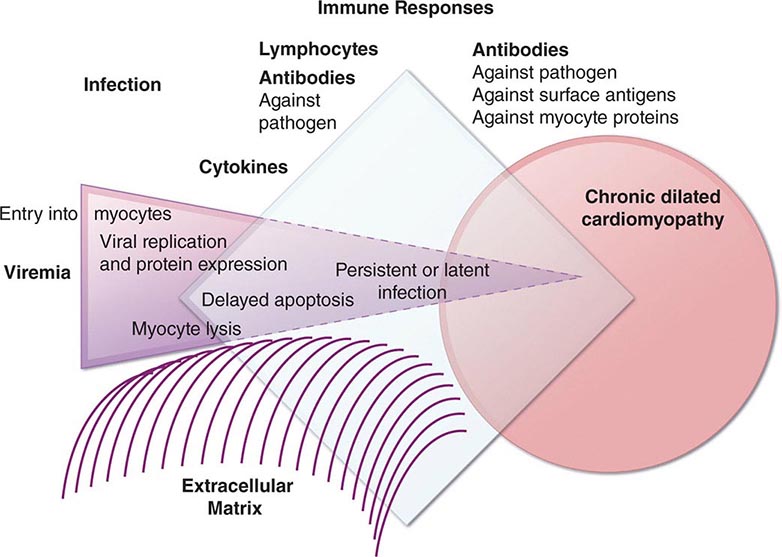
FIGURE 287-5 Schematic diagram demonstrating the possible progression from infection through direct, secondary, and autoimmune responses to dilated cardiomyopathy. Most of the supporting evidence for this sequence is derived from animal models. It is not known to what degree persistent infection and/or ongoing immune responses contribute to ongoing myocardial injury in the chronic phase.
Clinical Presentation of Viral Myocarditis Acute viral myocarditis often presents with symptoms and signs of heart failure. Some patients present with chest pain suggestive of pericarditis or acute myocardial infarction. Occasionally, the presentation is dominated by atrial or ventricular tachyarrhythmias, or by pulmonary or systemic emboli from intracardiac thrombi. Electrocardiographic or echocardiographic abnormalities may also be detected incidentally during evaluation for other diagnoses. The typical patient with presumed viral myocarditis is a young to middle-aged adult who develops progressive dyspnea and weakness within a few days to weeks after a viral syndrome that was accompanied by fever and myalgias.
A small number of patients present with fulminant myocarditis, with rapid progression from a severe febrile respiratory syndrome to cardiogenic shock that may involve multiple organ systems, leading to renal failure, hepatic failure, and coagulopathy. These patients are typically young adults who have recently been dismissed from urgent care settings with antibiotics for bronchitis or oseltamivir for viral syndromes, only to return within a few days in rapidly progressive cardiogenic shock. Prompt triage is vital to provide aggressive support with high-dose intravenous catecholamine therapy and sometimes with temporary mechanical circulatory support. Recognition of patients with this fulminant presentation is potentially life-saving as more than half can survive, with marked improvement demonstrable within the first few weeks. The ejection fraction function of these patients often recovers to near-normal, although residual diastolic dysfunction may limit vigorous exercise for some survivors.
Chronic viral myocarditis is often invoked, but rarely proven, as a diagnosis when no other cause of dilated cardiomyopathy can be identified. However, some cases of otherwise unexplained cardiomyopathy will later be recognized to have a genetic basis, or ultimately found to have resulted from excess alcohol consumption or illicit drugs. There are likely many other causes that cannot yet be identified. The prevalence of previous or persistent viral infection as the cause for chronic dilated cardiomyopathy remains highly controversial.
Laboratory evaluation for myocarditis The initial evaluation for suspected myocarditis includes an ECG, an echocardiogram, and serum levels of troponin and creatine phosphokinase fractions. Magnetic resonance imaging is increasingly used for the diagnosis of myocarditis, which is supported by evidence of increased tissue edema and gadolinium enhancement (Fig. 287-6), particularly in the mid-wall (as distinct from usual coronary artery territories).
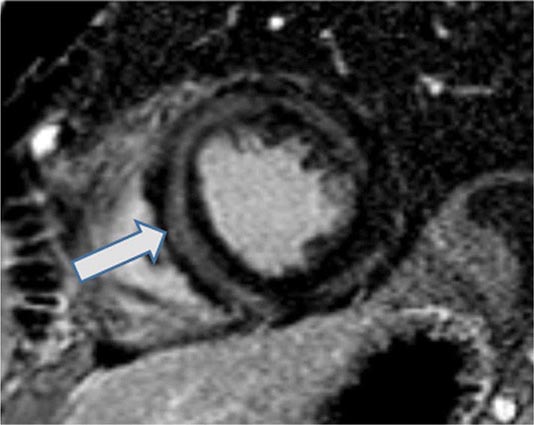
FIGURE 287-6 Magnetic resonance image of myocarditis showing the typical mid-wall location (arrow) for late gadolinium enhancement from cardiac inflammation and scarring. (Image courtesy of Ron Blankstein, MD, and Marcelo Di Carli, MD, Division of Nuclear Medicine, Brigham and Women’s Hospital, Boston.)
Endomyocardial biopsy is not often indicated for the initial evaluation of suspected viral myocarditis unless ventricular tachyarrhythmias suggest possible etiologies of sarcoidosis or giant cell myocarditis. The indications and benefit of endomyocardial biopsy for evaluation of myocarditis or new-onset cardiomyopathy remain controversial.
The Dallas Criteria for myocarditis on endomyocardial biopsy include lymphocytic infiltrate with evidence of myocyte necrosis (Fig. 287-7) and are negative in 80–90% of patients with clinical myocarditis. Negative Dallas Criteria can reflect sampling error or early resolution of lymphocytic infiltrates, but also the insensitivity of the test when inflammation results from cytokines and antibody-mediated injury. Routine histologic examination of endomyocardial biopsy rarely reveals a specific infective etiology, such as toxoplasmosis or Cytomegalovirus. Immunohistochemistry of myocardial biopsy samples is commonly used to identify active lymphocyte subtypes and may also detect upregulation of HLA antigens and the presence of complement components attributed to inflammation, but the specificity and significance of these findings are uncertain.
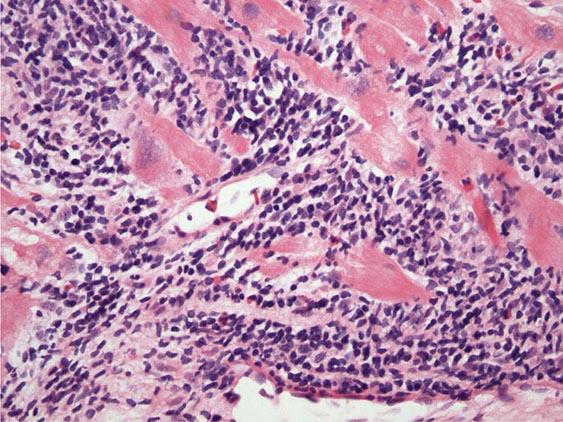
FIGURE 287-7 Acute myocarditis. Microscopic image of an endomyocardial biopsy showing massive infiltration with mononuclear cells and occasional eosinophils associated with clear myocyte damage. The myocyte nuclei are enlarged and reactive. Such extensive involvement of the myocardium would lead to extensive replacement fibrosis even if the inflammatory response could be suppressed. Hematoxylin and eosin–stained section, 200× original magnification. (Image courtesy of Robert Padera, MD, PhD, Department of Pathology, Brigham and Women’s Hospital, Boston.)
An increase in circulating viral titers between acute and convalescent blood samples supports a diagnosis of acute viral myocarditis with potential spontaneous improvement. There is no established role for measuring circulating anti-heart antibodies, which may be the result, rather than a cause, of myocardial injury and have been found also in patients with coronary artery disease and genetic cardiomyopathy.
Patients with recent or ongoing viral syndromes can be classified into three levels of diagnosis:
1. Possible subclinical acute myocarditis is diagnosed when a patient has a typical viral syndrome but no cardiac symptoms, with one or more of the following:
• Elevated biomarkers of cardiac injury (troponin or CK-MB)
• ECG findings suggestive of acute injury
• Reduced left ventricular ejection fraction or regional wall motion
• Abnormality on cardiac imaging, usually echocardiography
2. Probable acute myocarditis is diagnosed when the above criteria are met and accompanied also by cardiac symptoms, such as shortness of breath or chest pain, which can result from pericarditis or myocarditis. When clinical findings of pericarditis (pleuritic chest pain, ECG abnormalities, pericardial rub or effusion) are accompanied by elevated troponin or CK-MB or abnormal cardiac wall motion, the terms perimyocarditis or myopericarditis are sometimes used.
3. Definite myocarditis is diagnosed when there is histologic or immunohistologic evidence of inflammation on endomyocardial biopsy (see below) and does not require any other laboratory or clinical criteria.
SPECIFIC VIRUSES IMPLICATED IN MYOCARDITIS
In humans, viruses are often suspected but rarely proven to be the direct cause of clinical myocarditis. First implicated was the picornavirus family of RNA viruses, principally the enteroviruses, coxsackie virus, echovirus, and poliovirus. Influenza, another RNA virus, is implicated with varying frequency every winter and spring as epitopes change. Of the DNA viruses, adenovirus, vaccinia (smallpox vaccine), and the herpesviruses (varicella zoster, cytomegalovirus, Epstein-Barr virus, and human herpesvirus 6 [HHV6]) are well-recognized to cause myocarditis but also occur commonly in the healthy population. Polymerase chain reaction (PCR) detects viral genomes in the majority of patients with dilated cardiomyopathy, but also in normal “control” hearts. Most often detected are parvovirus B19 and HHV6, which may affect the cardiovascular system, in part, through infection of vascular endothelial cells. However, their contribution to chronic cardiomyopathy is uncertain, as serologic evidence of exposure is present in many children and most adults.
Human immunodeficiency virus (HIV) was associated with an incidence of dilated cardiomyopathy of 1–2%; however, with the advent of highly active antiretroviral therapy (HAART), HIV has been associated with a significantly lower incidence of cardiac disease. Cardiomyopathy in HIV may result from cardiac involvement with other associated viruses, such as cytomegalovirus and hepatitis C, as well as by HIV directly. Antiviral drugs to treat chronic HIV can cause cardiomyopathy, both directly and through drug hypersensitivity. The clinical picture may be complicated by pericardial effusions and pulmonary hypertension. There is a high frequency of lymphocytic myocarditis found at autopsy, and viral particles have been demonstrated in the myocardium in some cases, consistent with direct causation.
Hepatitis C has been repeatedly implicated in cardiomyopathy, particularly in Germany and Asia. Cardiac dysfunction may improve after interferon therapy. As this cytokine itself often depresses cardiac function transiently, careful coordination of administration and ongoing clinical evaluation are critical. Involvement of the heart with hepatitis B is uncommon, but can be seen when associated with systemic vasculitis (polyarteritis nodosa).
Additional viruses implicated specifically in myocarditis include mumps, respiratory syncytial virus, the arboviruses (dengue fever and yellow fever), and arenaviruses (Lassa fever). However, for any serious infection, the systemic inflammatory response can cause nonspecific depression of cardiac function, which is generally reversible if the patient survives.
THERAPY
There is currently no specific therapy recommended during any stage of viral myocarditis. During acute infection, therapy with anti-inflammatory or immunosuppressive medications is avoided, as their use has been shown to increase viral replication and myocardial injury in animal models. Therapy with specific antiviral agents (such as oseltamivir) has not been studied in relation to cardiac involvement. There is ongoing investigation into the impact of antiviral therapy to treat chronic viral persistence identified from endomyocardial biopsy. Large trials of immunosuppressive therapy for Dallas Criteria–positive myocarditis have been negative. There are some initial encouraging results and ongoing investigations with immunosuppressive therapy for immune-mediated myocarditis defined by immunohistologic criteria on biopsy or circulating anti-heart antibodies in the absence of myocardial viral genomes. However, neither antiviral nor anti-inflammatory therapies are currently recommended. Until we have a better understanding of the different phases of viral myocarditis and its sequelae and the effects of timed or targeted therapies, treatment will continue to be directed to the clinical cardiovascular stage of the disease, for dilated cardiomyopathy in general.
Parasitic Myocarditis Chagas’ disease is the third most common parasitic infection in the world and the most common infective cause of cardiomyopathy. The protozoan T. cruzi is transmitted by the bite of the reduviid bug, endemic in the rural areas of South and Central America. Transmission can also occur through blood transfusion, organ donation, from mother to fetus, and occasionally orally. While programs to eradicate the insect vector have decreased the prevalence from about 16 million to less than 10 million in South America, cases are increasingly recognized in Western developed countries. Approximately 100,000 affected individuals are currently living in the United States, most of whom contracted the disease in endemic areas.
Multiple pathogenic mechanisms are implicated. The parasite itself can cause myocyte lysis and primary neuronal damage, and specific immune responses may recognize the parasites or related antigens and lead to chronic immune activation in the absence of detectable parasites. Molecular techniques have revealed persistent parasite DNA fragments in infected individuals. Further evidence for persistent infection is the eruption of parasitic skin lesions during immunosuppression after cardiac transplantation. As with viral myocarditis, the relative roles of persistent infection and of secondary autoimmune injury have not been resolved (Fig. 287-5). An additional factor in the progression of Chagas’ disease is the autonomic dysfunction and microvascular damage that may contribute to cardiac and gastrointestinal disease.
The acute phase of Chagas’ disease with parasitemia is usually unrecognized, but in fewer than 5% of cases, it presents clinically within a few weeks of infection, with nonspecific symptoms or occasionally with acute myocarditis and meningoencephalitis. In the absence of antiparasitic therapy, the silent stage progresses slowly over 10–30 years in almost half of patients to manifest in the cardiac and gastrointestinal systems in the chronic stages. Features typical of Chagas’ disease are conduction system abnormalities, particularly sinus node and atrioventricular (AV) node dysfunction and right bundle branch block. Atrial fibrillation and ventricular tachyarrhythmias also occur. Small ventricular aneurysms are common, particularly at the ventricular apex. These dilated ventricles are particularly thrombogenic, giving rise to pulmonary and systemic emboli. Xenodiagnosis, detection of the parasite itself, is rarely performed. The serologic tests for specific IgG antibodies against the trypanosome lack sufficient specificity and sensitivity, thereby requiring two separate positive tests required to make a diagnosis.
Treatment of the advanced stages focuses on clinical manifestations of the disease and includes heart failure medications, pacemaker-defibrillators, and anticoagulation. Increasing attention is directed to antiparasitic therapy even in chronic disease without obvious active infection. The most common effective antiparasitic therapies are benznidazole and nifurtimox, both associated with multiple severe reactions, including dermatitis, gastrointestinal distress, and neuropathy. Survival is less than 30% at 5 years after the onset of overt clinical heart failure. Patients without major extracardiac disease have occasionally undergone transplantation, after which they may require lifelong therapy to suppress reactivation of infection.
African trypanosomiasis infection results from the tsetse fly bite and can occur in travelers exposed during trips to Africa. The West African form is caused by Trypanosoma brucei gambiense and progresses silently over years. The East African form caused by T. brucei rhodesiense can progress rapidly through perivascular infiltration to myocarditis and heart failure, with frequent arrhythmias. The diagnosis is made by identification of trypanosomes in blood, lymph nodes, or other affected sites. Antiparasitic therapy has limited efficacy and is determined by the specific type and the stage of infection (hemolymphatic or neurologic).
Toxoplasmosis is contracted through undercooked infected beef or pork, transmission from feline feces, organ transplantation, transfusion, or maternal-fetal transmission. Immunocompromised hosts are most likely to experience reactivation of latent infection from cysts. The cysts have been found in up to 40% of autopsies of patients dying from HIV infection. Toxoplasmosis may present with encephalitis or chorioretinitis and, in the heart, can cause myocarditis, pericardial effusion, constrictive pericarditis, and heart failure. The diagnosis in an immunocompetent patient is made when the IgM is positive and the IgG becomes positive later. Active toxoplasmosis may be suspected in an immunocompromised patient with myocarditis and a positive IgG titer for toxoplasmosis, particularly when avidity testing identifies high specificity of the antibody. Fortuitous sampling occasionally reveals the cysts in the myocardium. Combination therapy can include pyrimethamine and sulfadiazine or clindamycin.
Trichinellosis is caused by Trichinella spiralis larva ingested with undercooked meat. Larvae migrating into skeletal muscles cause myalgias, weakness, and fever. Periorbital and facial edema and conjunctival and retinal hemorrhage may also be seen. Although the larva may occasionally invade the myocardium, clinical heart failure is rare and, when observed, attributed to the eosinophilic inflammatory response. The diagnosis is made from the specific serum antibody and is further supported by the presence of eosinophilia. Treatment includes antihelminthic drugs (albendazole, mebendazole) and glucocorticoids if inflammation is severe.
Cardiac involvement with Echinococcus is rare, but cysts can form and rupture in the myocardium and pericardium.
Bacterial Infections Most bacterial infections can involve the heart occasionally through direct invasion and abscess formation, but do so rarely. More commonly, systemic inflammatory responses depress contractility in severe infection and sepsis. Diphtheria specifically affects the heart in almost one-half of cases, and cardiac involvement is the most common cause of death in patients with this infection. The prevalence of vaccines has shifted the incidence of diphtheria from children worldwide to countries without routine immunization and to older populations who have lost their immunity. The bacillus releases a toxin that impairs protein synthesis and may particularly affect the conduction system. The specific antitoxin should be administered as soon as possible, with higher priority than antibiotic therapy. Other systemic bacterial infections that can involve the heart include brucellosis, chlamydophila, legionella, meningococcus, mycoplasma, psittacosis, and salmonellosis, for which specific treatment is directed at the systemic infection.
Clostridial infections cause myocardial damage from the released toxin. Gas bubbles can be detected in the myocardium, and occasionally abscesses can form in the myocardium and pericardium. Streptococcal infection with β-hemolytic streptococci is most commonly associated with acute rheumatic fever and is characterized by inflammation and fibrosis of cardiac valves and systemic connective tissue, but it can also lead to a myocarditis with focal or diffuse infiltrates of mononuclear cells.
Tuberculosis can involve the myocardium directly as well as through tuberculous pericarditis, but rarely does so when the disease is treated with antibiotics. Whipple’s disease is caused by Tropheryma whipplei. The usual manifestations are in the gastrointestinal tract, but pericarditis, coronary arteritis, valvular lesions, and occasionally clinical heart failure may also occur. Multidrug antituberculous regimens are effective, but the disease tends to relapse even with appropriate treatment.
Other Infections Spirochetal myocarditis has been diagnosed from myocardial biopsies containing Borrelia burgdorferi that causes Lyme disease. Lyme carditis most often presents with arthritis and conduction system disease that resolves within 1–2 weeks of antibiotic treatment, only rarely implicated in chronic heart failure.
Fungal myocarditis can occur due to hematogenous or direct spread of infection from other sites, as has been described for aspergillosis, actinomycosis, blastomycosis, candidiasis, coccidioidomycosis, cryptococcosis, histoplasmosis, and mucormycosis. However, cardiac involvement is rarely the dominant clinical feature of these infections.
The rickettsial infections, Q fever, Rocky Mountain spotted fever, and scrub typhus are frequently accompanied by ECG changes, but most clinical manifestations relate to systemic vascular involvement.
NONINFECTIVE MYOCARDITIS
Myocardial inflammation can occur without apparent preceding infection. The paradigm of noninfective inflammatory myocarditis is cardiac transplant rejection, from which we have learned that myocardial depression can develop and reverse quickly, that noncellular mediators such as antibodies and cytokines play a major role in addition to lymphocytes, and that myocardial antigens are exposed by prior physical injury and viral infection.
The most commonly diagnosed noninfective inflammation is granulomatous myocarditis, including both sarcoidosis and giant cell myocarditis. Sarcoidosis, as discussed in Chap. 390, is a multisystem disease most commonly affecting the lungs. Although classically presenting with higher prevalence in young African-American men, the epidemiology appears to be changing, with increasing recognition of sarcoidosis in Caucasian patients in nonurban areas. Patients with pulmonary sarcoid are at high risk for cardiac involvement, but cardiac sarcoidosis also occurs without clinical lung disease. Regional clustering of the disease supports the suspicion that the granulomatous reaction is triggered by an infectious or environmental allergen not yet identified.
The sites and density of cardiac granulomata, the time course, and the degree of extracardiac involvement are remarkably variable. Patients may present with rapid-onset heart failure and ventricular tachyarrhythmias, conduction block, chest pain syndromes, or minor cardiac findings in the setting of ocular involvement, an infiltrative skin rash, or a nonspecific febrile illness. They may also present less acutely after months to years of fluctuating cardiac symptoms. When ventricular tachycardia or conduction block dominates the initial presentation of heart failure without coronary artery disease, suspicion should be high for these granulomatous myocarditides.
Depending on the time course, the ventricles may appear restrictive or dilated. There is often right ventricular predominance of both dilation and ventricular arrhythmias, sometimes initially attributed to arrhythmogenic right ventricular dysplasia. Small ventricular aneurysms are common. Computed tomography of the chest often reveals pulmonary lymphadenopathy even in the absence of clinical lung disease. Metabolic imaging (positron emission tomography (PET]) of the whole chest can highlight active sarcoid lesions that are avid for glucose. Magnetic resonance imaging (MRI) of the heart can identify areas likely to be inflammatory. To rule out chronic infections, such as tuberculosis or histoplasmosis as the cause of adenopathy, the diagnosis usually requires pathologic confirmation. Biopsy of enlarged mediastinal nodes may provide the highest yield. The scattered granulomata of sarcoidosis can easily be missed on cardiac biopsy (Fig. 287-8).
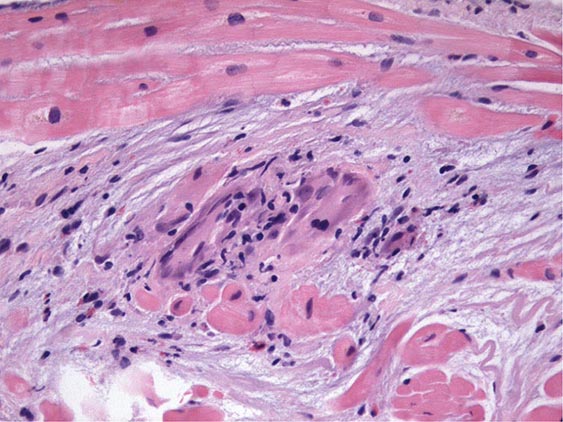
FIGURE 287-8 Sarcoidosis. Microscopic image of an endomyocardial biopsy showing a noncaseating granuloma and associated interstitial fibrosis typical of sarcoidosis. No microorganisms were present on special stains, and no foreign material was identified. Hematoxylin and eosin–stained section, 200× original magnification. (Image courtesy of Robert Padera, MD, PhD, Department of Pathology, Brigham and Women’s Hospital, Boston.)
Immunosuppressive treatment for sarcoidosis is initiated with high-dose glucocorticoids, which are often more effective for arrhythmias than for the heart failure. Patients with sarcoid lesions that persist or recur during tapering of corticosteroids are considered candidates for other immunosuppressive therapies, frequently with agents also used for cardiac transplantation. Pacemakers and implantable defibrillators are generally indicated to prevent life-threatening heart block or ventricular tachycardia, respectively. Because the inflammation often resolves into extensive fibrosis that impairs cardiac function and provides pathways for reentrant arrhythmias, the prognosis for improvement is best when the granulomata are not extensive and the ejection fraction is not severely reduced.
Giant cell myocarditis is less common than sarcoidosis, but accounts for 10–20% of biopsy-positive cases of myocarditis. Giant cell myocarditis typically presents with rapidly progressive heart failure and tachyarrhythmias. Diffuse granulomatous lesions are surrounded by extensive inflammatory infiltrate unlikely to be missed on endomyocardial biopsy, often with extensive eosinophilic infiltration. Associated conditions are thymomas, thyroiditis, pernicious anemia, other autoimmune diseases, and occasionally recent infections. Glucocorticoid therapy is less effective than for sarcoidosis and is sometimes combined with other immunosuppressive agents. The course is generally of rapid deterioration requiring urgent transplantation. Although the severity of presentation and myocardial histology are more fulminant than with sarcoidosis, the occasional finding of giant cell myocarditis after sarcoidosis suggests that they may in some cases represent different stages of the same disease spectrum.
Eosinophilic myocarditis can be an important manifestation of the hypereosinophilic syndrome, which in Western countries is often idiopathic, although in Mediterranean and African countries, it is likely a consequence of antecedent infection. It may also be seen with systemic eosinophilic syndromes such as Churg-Strauss syndrome or malignancies. Hypersensitivity myocarditis is often an unexpected diagnosis, made when the biopsy reveals infiltration with lymphocytes and mononuclear cells with a high proportion of eosinophils. Most commonly, the reaction is attributed to antibiotics, particularly those taken chronically, but thiazides, anticonvulsants, indomethacin, and methyldopa have also been implicated. Occasional associations with the smallpox vaccine have been reported. Although the circulating eosinophil count may be slightly elevated in hypersensitivity myocarditis, it does not reach the high levels of the hypereosinophilic syndrome. High-dose glucocorticoids and discontinuation of the trigger agent can be curative for hypersensitivity myocarditis.
Myocarditis is often associated with systemic inflammatory diseases, such as polymyositis and dermatomyositis, which affect skeletal and cardiac muscle. Although noninfective inflammatory myocarditis is sometimes included in the differential diagnosis of cardiac findings in patients with connective tissue disease such as systemic lupus erythematosus, pericarditis, vasculitis, pulmonary hypertension, and accelerated coronary artery disease are more common cardiac manifestations of connective tissue disease.
Peripartum cardiomyopathy (PPCM) develops during the last trimester or within the first 6 months after pregnancy, with a frequency between 1:2000 and 1:15,000 deliveries. Risk factors are increased maternal age, increased parity, twin pregnancy, malnutrition, use of tocolytic therapy for premature labor, and preeclampsia or toxemia of pregnancy. Heart failure early after delivery was previously common in Nigeria, when the custom for new mothers included salt ingestion while reclining on a warm bed, which likely impaired mobilization of the excess circulating volume after delivery. In the Western world, lymphocytic myocarditis has sometimes been found on myocardial biopsy. This inflammation has been hypothesized to reflect increased susceptibility to viral myocarditis or an autoimmune myocarditis due to cross-reactivity of anti-uterine antibodies against cardiac muscle. Another proposed mechanism invokes an abnormal prolactin cleavage fragment, which is induced by oxidative stress and may trigger myocardial apoptosis; this observation has led to preliminary investigation of bromocriptine as possible therapy.
Very recently, PPCM has been found to be associated with increased antiangiogenic signaling, a process that is exacerbated by preeclampsia. In animal models of this disease, proangiogenic therapies have proven curative.
As the increased circulatory demand of pregnancy can aggravate other cardiac disease that was clinically unrecognized, it is crucial to the diagnosis of PPCM that there be no evidence for a preexisting cardiac disorder. By contrast, heart failure presenting earlier in pregnancy has been termed pregnancy-associated cardiomyopathy (PACM). Both PPCM and PACM have been found in some families with other presentations of dilated cardiomyopathy, in some cases with known sarcomeric protein mutations. Pregnancy may, thus, represent an environmental trigger for accelerated phenotypic expression of genetic cardiomyopathy.
TOXIC CARDIOMYOPATHY
Cardiotoxicity has been reported with multiple environmental and pharmacologic agents. Often these associations are seen only with very high levels of exposure or acute overdoses, in which acute electrocardiographic and hemodynamic abnormalities may reflect both direct drug effect and systemic toxicity.
Alcohol is the most common toxin implicated in chronic dilated cardiomyopathy. Excess consumption may contribute to more than 10% of cases of heart failure, including exacerbation of cases with other primary etiologies such as valvular disease or previous infarction. Toxicity is attributed both to alcohol and to its primary metabolite, acetaldehyde. Polymorphisms of the genes encoding alcohol dehydrogenase and the angiotensin-converting enzyme increase the likelihood of alcoholic cardiomyopathy in an individual with excess consumption. Superimposed vitamin deficiencies and toxic alcohol additives are rarely implicated. The alcohol consumption necessary to produce cardiomyopathy in an otherwise normal heart has been estimated to be five to six drinks (about 4 ounces of pure ethanol) daily for 5–10 years, but frequent binge drinking may also be sufficient. Many patients with alcoholic cardiomyopathy are fully functional in their daily lives without apparent stigmata of alcoholism. The cardiac impairment in severe alcoholic cardiomyopathy is the sum of both permanent damage and a substantial component that is reversible after cessation of alcohol consumption. Atrial fibrillation occurs commonly both early in the disease (“holiday heart”) and in advanced stages. Medical therapy includes neurohormonal antagonists and diuretics as needed for fluid management. Withdrawal should be supervised to avoid exacerbations of heart failure or arrhythmias, and ongoing support arranged. Even with severe disease, marked improvement can occur within 3–6 months of abstinence. Implantable defibrillators are generally deferred until an adequate period of abstinence, after which they may not be necessary if the ejection fraction has improved. With continued consumption, the prognosis is grim.
Cocaine, amphetamines, and related catecholaminergic stimulants can produce chronic cardiomyopathy as well as acute ischemia and tachyarrhythmias. Pathology reveals microinfarcts consistent with small vessel ischemia, similar to those seen with pheochromocytoma.
Chemotherapy agents are the most common drugs implicated in toxic cardiomyopathy. Judicious use of these drugs requires balancing the risks of the malignancy and the risks of cardiotoxicity, as many cancers have a chronic course with better prognosis than heart failure.
Anthracyclines cause characteristic histologic changes of vacuolar degeneration and myofibrillar loss. Generation of reactive oxygen species involving heme compounds is currently the favored explanation for myocyte injury and fibrosis. Disruption of the large titin protein may contribute to loss of sarcomere organization. Risk for cardiotoxicity increases with higher doses, preexisting cardiac disease, and concomitant chest irradiation. There are three different presentations of anthracycline-induced cardiomyopathy. (1) Heart failure can develop acutely during administration of a single dose, but may clinically resolve in a few weeks. (2) Early-onset doxorubicin cardiotoxicity develops in about 3% of patients during or shortly after a chronic course, relating closely to total dose. It may be rapidly progressive, but may also resolve to good, but not normal, ventricular function. (3) The chronic presentation differs according to whether therapy was given before or after puberty. Patients who received doxorubicin while still growing may have impaired development of the heart, which leads to clinical heart failure by the time the patient reaches the early twenties. Late after adult exposure, patients may develop the gradual onset of symptoms or an acute onset precipitated by a reversible second insult, such as influenza or atrial fibrillation. Doxorubicin cardiotoxicity leads to a relatively nondilated ventricle, perhaps due to the accompanying fibrosis. Thus, the stroke volume may be severely reduced with an ejection fraction of 30–40%, which would be well tolerated in a patient with a larger ventricle typical of other cardiomyopathies with systolic dysfunction. Therapy includes angiotensin-converting enzyme inhibitors and β-adrenergic blocking agents used for other causes of heart failure, with careful suppression of “inappropriate” sinus tachycardia, and attention to postural hypotension that can occur in these patients. Once thought to have an inexorable downward course, some patients with doxorubicin cardiotoxicity improve under careful management to near-normal clinical function for many years.
Trastuzumab (Herceptin) is a monoclonal antibody that interferes with cell surface receptors crucial for some tumor growth and for cardiac adaptation. The incidence of cardiotoxicity is lower than for anthracyclines but enhanced by coadministration with them. Although considered to be more often reversible, trastuzumab cardiotoxicity does not always resolve, and some patients progress to clinical heart failure and death. As with anthracycline cardiotoxicity, therapy is as usual for heart failure, but it is not clear whether the spontaneous rate of improvement is enhanced by neurohormonal antagonists.
Cardiotoxicity with cyclophosphamide and ifosfamide generally occurs acutely and with very high doses. 5-Fluorouracil, cisplatin, and some other alkylating agents can cause recurrent coronary spasm that occasionally leads to depressed contractility. Acute administration of interferon-α can cause hypotension and arrhythmias. Clinical heart failure occurring during repeated chronic administration usually resolves after discontinuation.
Many small-molecule tyrosine kinase inhibitors are under development for different malignancies. Although these agents are “targeted” at specific tumor receptors or pathways, the biologic conservation of signaling pathways can cause these inhibitors to have “off-target” effects that include the heart and vasculature. Recognition of cardiotoxicity during therapy with these agents is complicated because they occasionally cause peripheral fluid accumulation (ankle edema, periorbital swelling, pleural effusions) due to local factors rather than elevated central venous pressures. Therapeutic approaches include withdrawal of the tyrosine kinase inhibitor (when possible) and substitution with a congener (when available), as well as conventional treatment for heart failure. Prophylactic treatment with beta blockers and angiotensin-converting enzyme inhibitors prior to and during chemotherapy is a topic of ongoing investigation.
Other therapeutic drugs that can cause cardiotoxicity during chronic use include hydroxychloroquine, chloroquine, emetine, and antiretroviral therapies.
Toxic exposures can cause arrhythmias or respiratory injury acutely during accidents. Chronic exposures implicated in cardiotoxicity include hydrocarbons, fluorocarbons, arsenicals, lead, and mercury.
METABOLIC CAUSES OF CARDIOMYOPATHY
Endocrine disorders affect multiple organ systems, including the heart. Hyperthyroidism and hypothyroidism do not often cause clinical heart failure in an otherwise normal heart, but commonly exacerbate heart failure. Clinical signs of thyroid disease may be masked, so tests of thyroid function are part of the routine evaluation of cardiomyopathy. Hyperthyroidism should always be considered with new-onset atrial fibrillation or ventricular tachycardia or atrial fibrillation in which the rapid ventricular response is difficult to control. The most common current reason for thyroid abnormalities in the cardiac population is the treatment of tachyarrhythmias with amiodarone, a drug with substantial iodine content. Hypothyroidism should be treated with very slow escalation of thyroid supplements to avoid exacerbating tachyarrhythmias and heart failure. Hyperthyroidism and heart failure are a dangerous combination that merits very close supervision, often hospitalization, during titration of antithyroid medications, during which decompensation of heart failure may occur precipitously and fatally.
Pheochromocytoma is rare, but should be considered when a patient has heart failure and very labile blood pressure and heart rate, sometimes with episodic palpitations (Chap. 407). Patients with pheochromocytoma often have postural hypotension. In addition to α-adrenergic receptor antagonists, definitive therapy requires surgical extirpation. Very high renin states, such as those caused by renal artery stenosis, can lead to modest depression in ejection fraction with little or no ventricular dilation and markedly labile symptoms with flash pulmonary edema, related to sudden shifts in vascular tone and intravascular volume.
Controversies remain regarding whether diabetes and obesity are sufficient to cause cardiomyopathy. Most heart failure in diabetes results from epicardial coronary disease, with further increase in coronary artery risk due to accompanying hypertension and renal dysfunction. Cardiomyopathy may result in part from insulin resistance and increased advanced-glycosylation end products, which impair both systolic and diastolic function. However, much of the dysfunction can be attributed to scattered focal ischemia resulting from distal coronary artery tapering and limited microvascular perfusion even without proximal focal stenoses. Diabetes is a typical factor in heart failure with “preserved” ejection fraction, along with hypertension, advanced age, and female gender.
The existence of a cardiomyopathy due to obesity is generally accepted. In addition to cardiac involvement from associated diabetes, hypertension, and vascular inflammation of the metabolic syndrome, obesity alone is associated with impaired excretion of excess volume load, which, over time, can lead to increased wall stress and secondary adaptive neurohumoral responses. Fluid retention may be aggravated by large fluid intake and the rapid clearance of natriuretic peptides by adipose tissue. In the absence of another obvious cause of cardiomyopathy in an obese patient with systolic dysfunction without marked ventricular dilation, effective weight reduction is often associated with major improvement in ejection fraction and clinical function. Improvement in cardiac function has been described after successful bariatric surgery, although all major surgical therapy poses increased risk for patients with heart failure. Postoperative malabsorption and nutritional deficiencies, such as calcium and phosphate deficiencies, may be particularly deleterious for patients with cardiomyopathy.
Nutritional deficiencies can occasionally cause dilated cardiomyopathy but are not commonly implicated in developed Western countries. Beri-beri heart disease due to thiamine deficiency can result from poor nutrition in undernourished populations and in patients deriving most of their calories from alcohol, and has been reported in teenagers subsisting only on highly processed foods. This disease is initially a vasodilated state with very high output heart failure that can later progress to a low output state; thiamine repletion can lead to prompt recovery of cardiovascular function. Abnormalities in carnitine metabolism can cause dilated or restrictive cardiomyopathies, usually in children. Deficiency of trace elements such as selenium can cause cardiomyopathy (Keshan’s disease).
Calcium is essential for excitation-contraction coupling. Chronic deficiencies of calcium, such as can occur with hypoparathyroidism (particularly postsurgical) or intestinal dysfunction (from diarrheal syndromes and following extensive resection), can cause severe chronic heart failure that responds over days or weeks to vigorous calcium repletion. Phosphate is a component of high-energy compounds needed for efficient energy transfer and multiple signaling pathways. Hypophosphatemia can develop during starvation and early refeeding following a prolonged fast, and occasionally during hyperalimentation. Magnesium is a cofactor for thiamine-dependent reactions and for the sodium-potassium adenosine triphosphatase (ATPase), but hypomagnesemia rarely becomes sufficiently profound to cause clinical cardiomyopathy.
Hemochromatosis is variably classified as a metabolic or storage disease (Chap. 428). It is included among the causes of restrictive cardiomyopathy, but the clinical presentation is often that of a dilated cardiomyopathy. The autosomal recessive form is related to the HFE gene. With up to 10% of the population heterozygous for one mutation, the clinical prevalence might be as high as 1 in 500. The lower observed rates highlight the limited penetrance of the disease, suggesting the role of additional genetic and environmental factors for clinical expression. Hemochromatosis can also be acquired from iron overload due to hemolytic anemia and transfusions. Excess iron is deposited in the perinuclear compartment of cardiomyocytes, with resulting disruption of intracellular architecture and mitochondrial function. Diagnosis is easily made from measurement of serum iron and transferrin saturation, with a threshold of >60% for men and >45–50% for women. MRI can help to quantitate iron stores in the liver and heart, and endomyocardial biopsy tissue can be stained for iron (Fig. 287-9), which is particularly important if the patient has another cause for cardiomyopathy. If diagnosed early, hemochromatosis can often be managed by repeated phlebotomy to remove iron. For more severe iron overload, iron chelation therapy with desferrioxamine (deferoxamine) or deferasirox can help to improve cardiac function if myocyte loss and replacement fibrosis are not too severe.
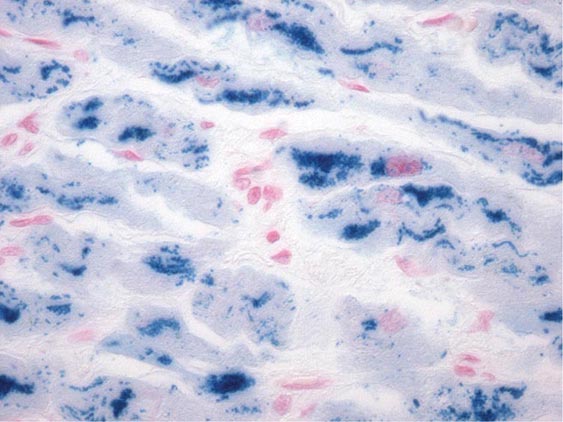
FIGURE 287-9 Hemochromatosis. Microscopic image of an endomyocardial biopsy showing extensive iron deposition within the cardiac myocytes with the Prussian blue stain (400× original magnification). (Image courtesy of Robert Padera, MD, PhD, Department of Pathology, Brigham and Women’s Hospital, Boston.)
Inborn disorders of metabolism occasionally present with dilated cardiomyopathy, although they are most often associated with restrictive cardiomyopathy (Table 287-4).
FAMILIAL DILATED CARDIOMYOPATHY
The genetic basis for cardiomyopathy is discussed in the section, “Genetic Etiologies of Cardiomyopathy.” The recognized frequency of familial involvement in dilated cardiomyopathy has increased to over 30%. Mutations in TTN, encoding the giant sarcomeric protein titin, are the most common cause of dilated cardiomyopathy, accounting for up to 25% of familial disease. On average, men with TTN mutations develop cardiomyopathy a decade before women, without distinctive clinical features. Mutations in thick and thin filament genes account for ~8% of dilated cardiomyopathy and may manifest in early childhood.
The most recognizable familial cardiomyopathy syndromes with extracardiac manifestations are the muscular dystrophies. Both Duchenne’s and the milder Becker’s dystrophy result from abnormalities in the X-linked dystrophin gene of the sarcolemmal membrane. Skeletal myopathy is present in multiple other genetic cardiomyopathies (Table 287-3), some of which are associated with creatine kinase elevations.
Families with a history of atrial arrhythmias, conduction system disease, and cardiomyopathy may have abnormalities of the nuclear membrane lamin proteins. While all dilated cardiomyopathies carry a risk of sudden death, a family history of cardiomyopathy with sudden death raises suspicion for a particularly arrhythmogenic mutation; affected family members may be considered for implantable defibrillators even before meeting the reduced ejection fraction threshold for primary prevention of sudden death.
A prominent family history of sudden death or ventricular tachycardia before clinical cardiomyopathy suggests genetic defects in the desmosomal proteins (Fig. 287-10). Originally described as affecting the right ventricle (arrhythmogenic right ventricular dysplasia [ARVD]), this disorder (arrhythmogenic ventricular dysplasia) can affect either or both ventricles. Patients often present first with ventricular tachycardia. Genetic defects in proteins of the desmosomal complex disrupt myocyte junctions and adhesions, leading to replacement of myocardium by deposits of fat. Thin ventricular walls may be recognized on echocardiography but are better visualized on MRI. Because desmosomes are also important for elasticity of hair and skin, some of the defective desmosomal proteins are associated with striking “woolly hair” and thickened skin on the palms and soles. Implantable defibrillators are usually indicated to prevent sudden death. There is variable progression to right, left, or biventricular failure.
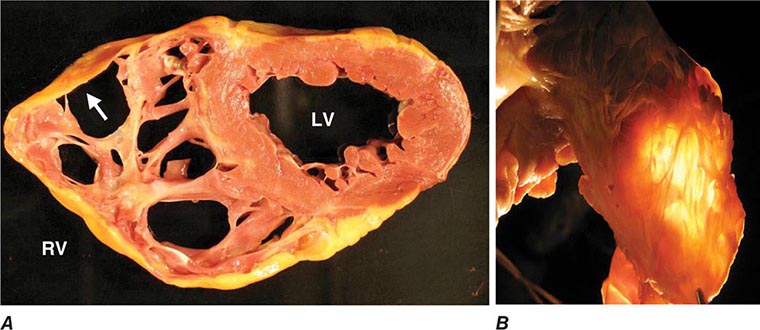
FIGURE 287-10 Arrhythmogenic right ventricular dysplasia. A. Cross-sectional slice of a pathology specimen removed at transplantation, showing severe dysplasia of the right ventricle (RV) with extensive fatty replacement of right ventricular myocardium. B. The remarkably thin right ventricular free wall is revealed by transillumination. LV, left ventricle. (Images courtesy of Gayle Winters, MD, and Richard Mitchell, MD, PhD, Division of Pathology, Brigham and Women’s Hospital, Boston.)
Left ventricular noncompaction is a condition of unknown prevalence that is increasingly revealed with the refinement of imaging techniques. The diagnostic criteria include the presence of multiple trabeculations in the left ventricle distal to the papillary muscles, creating a “spongy” appearance of the apex. Noncompaction has been associated with multiple genetic variants in the sarcomeric and other genes, such as TAZ (encoding tafazzin). The diagnosis may be made incidentally or in patients previously diagnosed with cardiomyopathy, in whom the criteria for noncompaction may appear and resolve with changing left ventricular size and function. The three cardinal clinical features are ventricular arrhythmias, embolic events, and heart failure. Treatment generally includes anticoagulation and early consideration for an implantable defibrillator, in addition to neurohormonal antagonists as indicated by stage of disease.
Some families inherit a susceptibility to viral-induced myocarditis. This propensity may relate to abnormalities in cell surface receptors, such as the coxsackie-adenovirus receptor, that bind viral proteins. Some may have partial homology with viral proteins such that an autoimmune response is triggered against the myocardium.
Prognosis and therapy of familial dilated cardiomyopathy are dictated primarily by the stage of clinical disease and the risk for sudden death. In some cases, the familial etiology facilitates prognostic decisions, particularly regarding the likelihood of recovery after a new diagnosis, which is unlikely for familial disease. The rate of progression of disease, once manifest, is, to some extent, heritable, although marked variation can be seen. However, there have been cases of remarkable clinical remission after acute presentation, likely after a reversible additional insult, such as prolonged tachycardia or infective myocarditis.
TAKOTSUBO CARDIOMYOPATHY
The apical ballooning syndrome, or stress-induced cardiomyopathy, occurs typically in older women after sudden intense emotional or physical stress. The ventricle shows global ventricular dilation with basal contraction, forming the shape of the narrow-necked jar (takotsubo) used in Japan to trap octopi. Originally described in Japan, it is increasingly recognized elsewhere during emergency cardiac catheterization and intensive care unit admissions for noncardiac conditions. Presentations include pulmonary edema, hypotension, and chest pain with ECG changes mimicking an acute infarction. The left ventricular dysfunction extends beyond a specific coronary artery distribution and generally resolves within days to weeks. Animal models and ventricular biopsies suggest that this acute cardiomyopathy may result from intense sympathetic activation with heterogeneity of myocardial autonomic innervation, diffuse microvascular spasm, and/or direct catecholamine toxicity. Coronary angiography may be required to rule out acute coronary occlusion. No therapies have been proven beneficial, but reasonable strategies include nitrates for pulmonary edema, intraaortic balloon pump if needed for low output, combined alpha and beta blockers rather than selective beta blockade if hemodynamically stable, and magnesium for arrhythmias related to QT prolongation. Anticoagulation is generally withheld due to the occasional occurrence of ventricular rupture. While the prognosis is generally good, recurrences have been described in up to 10% of patients.
IDIOPATHIC DILATED CARDIOMYOPATHY
Idiopathic dilated cardiomyopathy is a diagnosis of exclusion, when all other known factors have been excluded. Approximately two-thirds of dilated cardiomyopathies are still labeled as idiopathic; however, a substantial proportion of these may reflect unrecognized genetic disease. Continued reconsideration of etiology during chronic heart failure management often reveals specific causes later in a patient’s course.
OVERLAPPING TYPES OF CARDIOMYOPATHY
The limitations of our phenotypic classification are revealed through the multiple overlaps between the etiologies and presentations of the three types. Cardiomyopathy with reduced systolic function but without severe dilation can represent early dilated cardiomyopathy, “minimally dilated cardiomyopathy,” or restrictive diseases without marked increases in ventricular wall thickness. For example, sarcoidosis and hemochromatosis can present as dilated or restrictive disease. Early stages of amyloidosis are often mistaken for hypertrophic cardiomyopathy. Progression of hypertrophic cardiomyopathy into a “burned-out” phase occurs occasionally, with decreased contractility and modest ventricular dilation. Overlaps are particularly common with the inherited metabolic disorders, which can present as any of the three major phenotypes (Fig. 287-4).
DISORDERS OF METABOLIC PATHWAYS
Multiple genetic disorders of metabolic pathways can cause myocardial disease, due to infiltration of abnormal products or cells containing them between the myocytes, and storage disease, due to their accumulation within cells (see HPIM 18e, Table 238-4, and 287-5). The restrictive phenotype is most common, but mildly dilated cardiomyopathy may occur. Hypertrophic cardiomyopathy may be mimicked by the myocardium thickened with these abnormal products causing “pseudohypertrophy.” Most of these diseases are diagnosed during childhood.
|
CAUSES OF RESTRICTIVE CARDIOMYOPATHIES |
aCan be familial.
Fabry’s disease results from a deficiency of the lysosomal enzyme alpha-galactosidase A caused by one of more than 160 mutations. This disorder of glycosphingolipid metabolism is an X-linked recessive disorder that may also cause clinical disease in female carriers. Glycolipid accumulation may be limited to the cardiac tissues or may also involve the skin and kidney. Electron microscopy of endomyocardial biopsy tissue shows diagnostic vesicles containing concentric lamellar figures (Fig. 287-11). Diagnosis is crucial because enzyme replacement can reduce abnormal deposits and improve cardiac and clinical function. The magnitude of clinical impact has not been well-established for this therapy, which requires frequent infusions of the enzyme at a cost of over $100,000 a year. Enzyme replacement can also improve the course of Gaucher’s disease, in which cerebroside-rich cells accumulate in multiple organs due to a deficiency of beta-glucosidase. Cerebroside-rich cells infiltrate the heart, which can also lead to a hemorrhagic pericardial effusion and valvular disease.
FIGURE 287-11 Fabry’s disease. Transmission electron micrograph of a right ventricular endomyocardial biopsy specimen at high magnification showing the characteristic concentric lamellar inclusions of glycosphingolipids accumulating as a result of deficiency of the lysosomal enzyme alpha-galactosidase A. Image taken at 15,000× original magnification. (Image courtesy of Robert Padera, MD, PhD, Department of Pathology, Brigham and Women’s Hospital, Boston.)
Glycogen storage diseases lead to accumulation of lysosomal storage products and intracellular glycogen accumulation, particularly with glycogen storage disease type III, due to a defective debranching enzyme. There are more than 10 types of mucopolysaccharidoses, in which autosomal dominant or X-linked deficiencies of lysosomal enzymes lead to the accumulation of glycosaminoglycans in the skeleton, nervous system, and occasionally the heart. With characteristic facies, short stature, and frequent cognitive impairment, most individuals are diagnosed early in childhood and die before adulthood.
Carnitine is an essential cofactor in long-chain fatty acid metabolism. Multiple defects have been described that lead to carnitine deficiency, causing intracellular lipid inclusions and restrictive or dilated cardiomyopathy, often presenting in children. Fatty acid oxidation requires many metabolic steps with specific enzymes that can be deficient, with complex interactions with carnitine. Depending on the defect, cardiac and skeletal myopathy can be ameliorated with replacement of fatty acid intermediates and carnitine.
Two monogenic metabolic cardiomyopathies have recently been described as causes of increased ventricular wall thickness without an increase of muscle subunits or an increase in contractility. Mutations in the gamma-2 regulatory subunit of the adenosine monophosphate (AMP)-activated protein kinase important for glucose metabolism (PRKAG2) have been associated with a high prevalence of conduction abnormalities, such as AV block and ventricular preexcitation (Wolff-Parkinson-White syndrome). Several defects have been reported in an X-linked lysosome-associated membrane protein (LAMP2). This defect can be maternally transmitted or sporadic and has occasionally been isolated to the heart, although it often leads to a syndrome of skeletal myopathy, mental retardation, and hepatic dysfunction referred to as Danon’s disease. Extreme left ventricular hypertrophy appears early, often in childhood, and can progress rapidly to end-stage heart failure with low ejection fraction. Electron microscopy of these metabolic disorders shows that the myocytes are enlarged by multiple intracellular vacuoles of metabolic by-products.
RESTRICTIVE CARDIOMYOPATHY
The least common of the physiologic triad of cardiomyopathies is restrictive cardiomyopathy, which is dominated by abnormal diastolic function, often with mildly decreased contractility and ejection fraction (usually >30–50%). Both atria are enlarged, sometimes massively. Modest left ventricular dilation can be present, usually with an end-diastolic dimension <6 cm. End-diastolic pressures are elevated in both ventricles, with preservation of cardiac output until late in the disease. Subtle exercise intolerance is usually the first symptom but is often not recognized until after clinical presentation with congestive symptoms. The restrictive diseases often present with relatively more right-sided symptoms, such as edema, abdominal discomfort, and ascites, although filling pressures are elevated in both ventricles. The cardiac impulse is less displaced than in dilated cardiomyopathy and less dynamic than in hypertrophic cardiomyopathy. A fourth heart sound is more common than a third heart sound in sinus rhythm, but atrial fibrillation is common. Jugular venous pressures often show rapid Y descents and may increase during inspiration (positive Kussmaul’s sign). Most restrictive cardiomyopathies are due to infiltration of abnormal substances between myocytes, storage of abnormal metabolic products within myocytes, or fibrotic injury (Table 287-5). The differential diagnosis should include constrictive pericardial disease, which may also be dominated by right-sided heart failure.
INFILTRATIVE DISEASE
Amyloidosis is the major cause of restrictive cardiomyopathy (Figs. 287-12, 287-13, and 287-14). Several proteins can self-assemble to form the beta-sheets of amyloid proteins, which deposit with different consequences depending on the type of protein. The systemic amyloidoses are discussed in Chap. 137. In addition to cardiac infiltration, neurologic involvement occurs commonly with primary amyloidosis (immunoglobulin light chains) and with familial amyloidosis (genetic abnormalities of transthyretin). There are over 100 identified mutations in transthyretin on chromosome 13, among which the V122I transthyretin mutation has been identified in about 4% of African Americans and in 10% of African Americans with heart failure and may contribute importantly to heart failure in general in the elderly African-American population. Organ dysfunction was previously attributed solely to physical disruption from the infiltrating amyloid fibrils, but newer information suggests additional direct toxicity from the immunoglobulin light chain and abnormal transthyretin protein aggregates themselves. In senile amyloidosis, there is abnormal accumulation of normal transthyretin or natriuretic peptide folding, detected in 10% of people over 80 years and half of those over 90 years but often without apparent clinical disease. Men show a greater burden of amyloid deposition and 20-fold greater likelihood of clinical disease with senile amyloidosis. The aging of the population will soon render senile amyloidosis the most common of the amyloidoses.
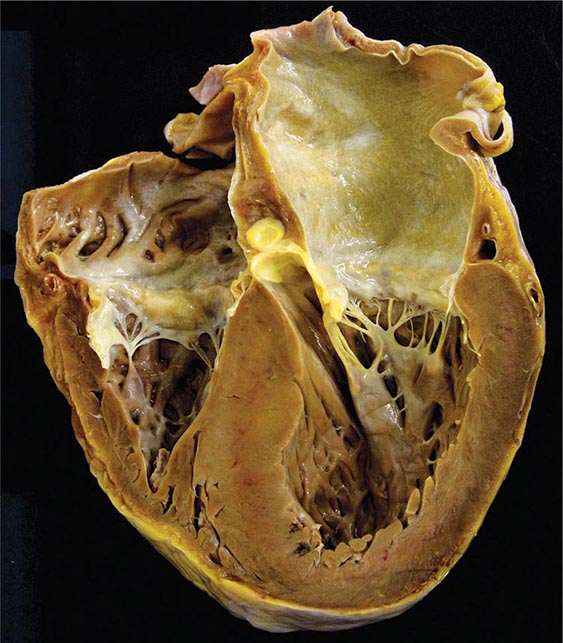
FIGURE 287-12 Restrictive cardiomyopathy—amyloidosis. Gross specimen of a heart with amyloidosis. The heart is firm and rubbery with a waxy cut surface. The atria are markedly dilated, and the left atrial endocardium, normally smooth, has yellow-brown amyloid deposits that give texture to the surface. (Image courtesy of Robert Padera, MD, PhD, Department of Pathology, Brigham and Women’s Hospital, Boston.)
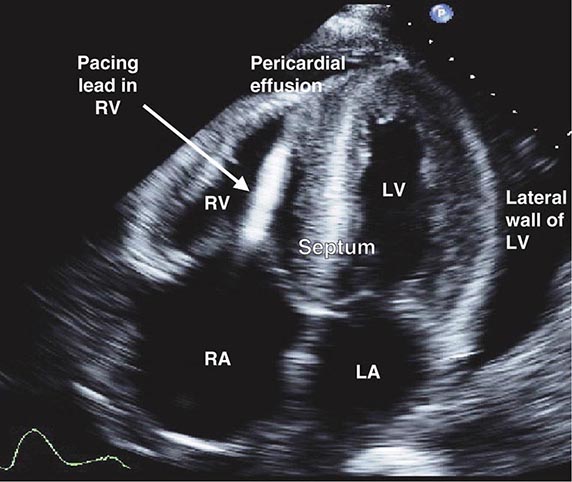
FIGURE 287-13 Restrictive cardiomyopathy—amyloidosis. Echocardiogram showing thickened walls of both ventricles without major chamber dilation. The atria are markedly dilated, consistent with chronically elevated ventricular filling pressures. In this example, there is a characteristic hyperrefractile “glittering” of the myocardium typical of amyloid infiltration, which is often absent (especially with more recent echocardiographic systems of better resolution). The mitral and tricuspid valves are thickened. A pacing lead is visible in the right ventricle (RV), and a pericardial effusion is evident. Note that the echocardiographic and pathologic images are vertically opposite, such that the left ventricle (LV) is by convention on the top right in the echocardiographic image and bottom right in the pathologic images. LA, left atrium; RA, right atrium. (Image courtesy of Justina Wu, MD, Brigham and Women’s Hospital, Boston.)
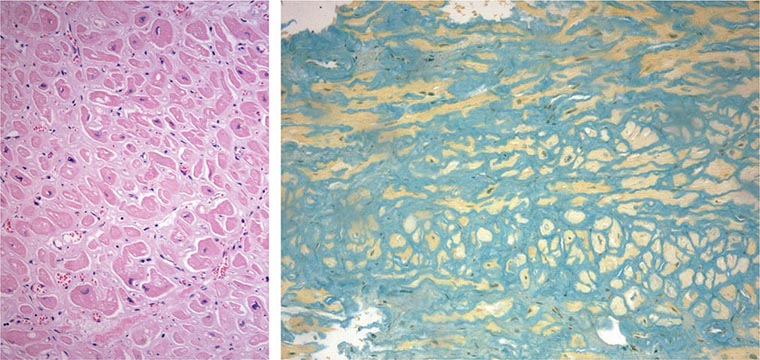
FIGURE 287-14 Amyloidosis—microscopic images of amyloid involving the myocardium. The left panel (hematoxylin and eosin stain) shows glassy, grey-pink amorphous material infiltrating between cardiomyocytes, which stain a darker pink. The right panel shows a sulfated blue stain that highlights the amyloid green and stains the cardiac myocytes yellow. (The Congo red stain can also be used to highlight amyloid; under polarized light, amyloid will have an apple-green birefringence when stained with Congo red.) Images at 100× original magnification. (Image courtesy of Robert Padera, MD, PhD, Department of Pathology, Brigham and Women’s Hospital, Boston.)
Cardiac amyloid is classically suspected from thickened ventricular walls with an ECG that shows low voltage. However, low voltage is not always present and is less common in familial or senile amyloidosis than in primary AL amyloidosis. A characteristic refractile brightness in the septum on echocardiography is suggestive of the diagnosis, but neither sensitive nor specific. Both atria are dilated, often dramatically, and diastolic dysfunction may be more obvious than in left ventricular hypertrophy from other causes. Amyloid infiltration can also be detected with gadolinium enhancement in MRI. The diagnosis of primary or familial amyloidosis can sometimes be made from biopsies of an abdominal fat pad or the rectum, but cardiac amyloidosis is most reliably identified from a biopsy of the heart, in which amyloid fibrils infiltrate the myocardium diffusely, particularly around the conduction system and coronary vessels (Fig. 287-14). Diagnosis of the type of amyloid protein requires immunohistochemistry of biopsied tissue rather than serum or urine electrophoresis, which can lead to incorrect classification.
Therapy for all types of amyloid is predominantly for symptoms of fluid retention, which often requires high doses of loop diuretics. Digoxin bound to the amyloid fibrils can reach toxic levels, and should therefore be used only in very low doses, if at all. There is no evidence regarding use of neurohormonal antagonists in amyloid heart disease, where the possible theoretical benefit has to be balanced against the possibility of aggravating postural hypotension and diminishing the crucial heart rate reserve. The risk of intracardiac thrombi may warrant chronic anticoagulation.
The prognosis is worst for primary amyloid, with a median survival of 6–12 months after symptoms of heart failure. If present, multiple myeloma is treated with chemotherapy, the extent of which is often limited by the potential of worsening cardiac dysfunction. Immunoglobulin-associated amyloid has occasionally been treated with sequential heart transplantation and delayed bone marrow transplant, with frequent recurrence of amyloid in the transplanted heart. Abnormal transthyretin-associated cardiac amyloid has a somewhat better prognosis and can be treated in selected patients with heart and liver transplantation. Senile cardiac amyloid has the slowest progression and best overall prognosis.
FIBROTIC RESTRICTIVE CARDIOMYOPATHY
Progressive fibrosis can cause restrictive myocardial disease without ventricular dilation. Thoracic radiation, common for breast and lung cancer or mediastinal lymphoma, can produce early or late restrictive cardiomyopathy. Patients with radiation cardiomyopathy may present with a possible diagnosis of constrictive pericarditis, as the two conditions often coexist. Careful hemodynamic evaluation and, often, endomyocardial biopsy should be performed if considering pericardial stripping surgery, which is unlikely to be successful in the presence of underlying restrictive cardiomyopathy.
Scleroderma causes small vessel spasm and ischemia that can lead to a small, stiff heart with reduced ejection fraction without dilation. The pulmonary hypertension associated with scleroderma may lead to more clinical right heart failure because of concomitant fibrotic disease of the right ventricle. Doxorubicin causes direct myocyte injury usually leading to dilated cardiomyopathy, but the limited degree of dilation may result from increased fibrosis, which restricts remodeling.
ENDOMYOCARDIAL DISEASE
The physiologic picture of elevated filling pressures with atrial enlargement and preserved ventricular contractility with normal or reduced ventricular volumes can result from extensive fibrosis of the endocardium, without transmural myocardial disease. For patients who have not lived in the equatorial regions, this picture is rare, and when seen is often associated with a history of chronic hypereosinophilic syndrome (Löffler’s endocarditis), which is more common in men than women. In this disease, persistent hypereosinophilia of >1500 eos/mm3 for at least 6 months can cause an acute phase of eosinophilic injury in the endocardium (see earlier discussion of eosinophilic myocarditis), with systemic illness and injury to other organs. There is usually no obvious cause, but the hypereosinophilia can occasionally be explained by allergic, parasitic, or malignant disease. It is postulated to be followed by a period in which cardiac inflammation is replaced by evidence of fibrosis with superimposed thrombosis. In severe disease, the dense fibrotic layer can obliterate the ventricular apices and extend to thicken and tether the AV valve leaflets. The clinical disease may present with heart failure, embolic events, and atrial arrhythmias. While plausible, the sequence of transition from eosinophilic myocarditis or Löffler’s endocarditis to endomyocardial fibrosis has not been clearly demonstrated.
In tropical countries, up to one-quarter of heart failure may be due to endomyocardial fibrosis, affecting either or both ventricles. This condition shares with the previous condition the partial obliteration of the ventricular apex with fibrosis extending into the valvular inflow tract and leaflets; however, it is not clear that the etiologies are the same for all cases. Pericardial effusions frequently accompany endomyocardial fibrosis but are not common in Löffler’s endocarditis. For endomyocardial fibrosis, there is no gender difference, but a higher prevalence in African-American populations. While tropical endomyocardial fibrosis could represent the end-stage of previous hypereosinophilic disease triggered by endemic parasites, neither prior parasitic infection nor hypereosinophilia is usually documented. Geographic nutritional deficiencies have also been proposed as an etiology.
Medical treatment focuses on glucocorticoids and chemotherapy to suppress hypereosinophilia when present. Fluid retention may become increasingly resistant to diuretic therapy. Anticoagulation is recommended. Atrial fibrillation is associated with worse symptoms and prognosis, but may be difficult to suppress. Surgical resection of the apices and replacement of the fibrotic valves can improve symptoms, but surgical morbidity and mortality and later recurrence rates are high.
The serotonin secreted by carcinoid tumors can produce fibrous plaques in the endocardium and right-sided cardiac valves, occasionally affecting left-sided valves, as well. Valvular lesions may be stenotic or regurgitant. Systemic symptoms include flushing and diarrhea. Liver disease from hepatic metastases may play a role by limiting hepatic function and thereby allowing more serotonin to reach the venous circulation.
HYPERTROPHIC CARDIOMYOPATHY
Hypertrophic cardiomyopathy is defined as left ventricular hypertrophy that develops in the absence of causative hemodynamic factors, such as hypertension, aortic valve disease, or systemic infiltrative or storage diseases (Figs. 287-15 and 287-16). It has previously been termed hypertrophic obstructive cardiomyopathy (HOCM), asymmetric septal hypertrophy (ASH), and idiopathic hypertrophic subaortic stenosis (IHSS). However, the accepted terminology is now hypertrophic cardiomyopathy with or without obstruction. Prevalence in North America, Japan, and China is about 1:500. It is the leading cause of sudden death in the young and is an important cause of heart failure. Although pediatric presentation is associated with increased early morbidity and mortality, the prognosis for patients diagnosed as adults is generally favorable.
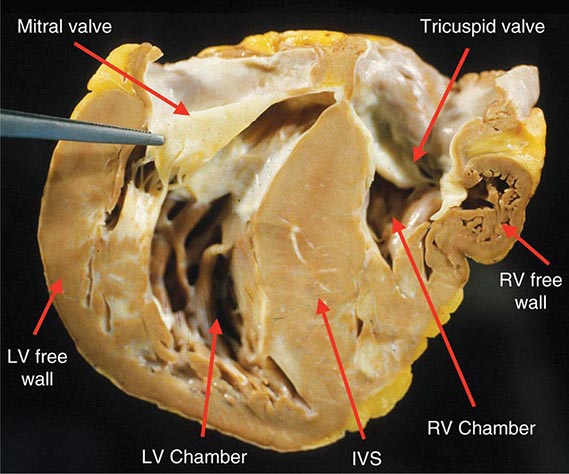
FIGURE 287-15 Hypertrophic cardiomyopathy. Gross specimen of a heart with hypertrophic cardiomyopathy removed at the time of transplantation, showing asymmetric septal hypertrophy (septum much thicker than left ventricular free wall) with the septum bulging into the left ventricular outflow tract causing obstruction. The forceps are retracting the anterior leaflet of the mitral valve, demonstrating the characteristic plaque of systolic anterior motion, manifest as endocardial fibrosis on the interventricular septum in a mirror-image pattern to the valve leaflet. There is patchy replacement fibrosis, and small thick-walled arterioles can be appreciated grossly, especially in the interventricular septum. IVS, interventricular septum; LV, left ventricle; RV, right ventricle. (Image courtesy of Robert Padera, MD, PhD, Department of Pathology, Brigham and Women’s Hospital, Boston.)
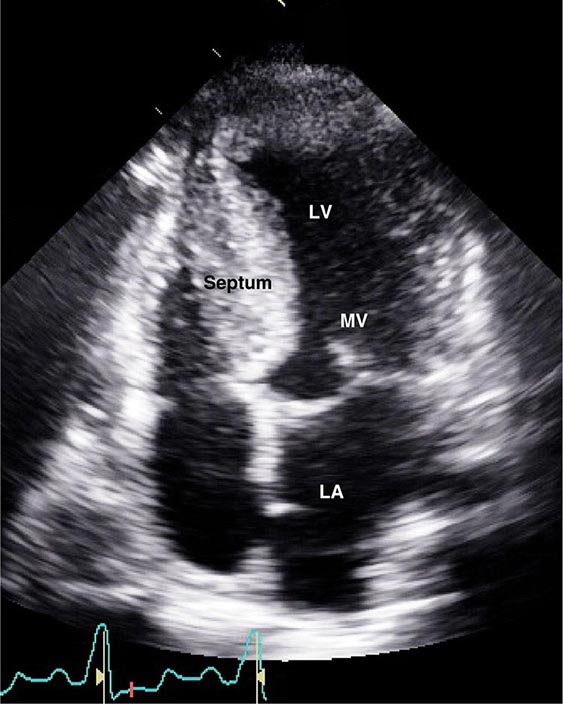
FIGURE 287-16 Hypertrophic cardiomyopathy. This echocardiogram of hypertrophic cardiomyopathy shows asymmetric hypertrophy of the septum compared to the lateral wall of the left ventricle (LV). The mitral valve (MV) is moving anteriorly toward the hypertrophied septum in systole. The left atrium (LA) is enlarged. Note that the echocardiographic and pathologic images are vertically opposite, such that the LV is by convention on the top right in the echocardiographic image and bottom right in the pathologic images. (Image courtesy of Justina Wu, MD, Brigham and Women’s Hospital, Boston.)
The clustering of hypertrophic cardiomyopathy within families has been appreciated since recognition of the disease approximately 55 years ago. Echocardiographic screening of families revealed an autosomal dominant pattern of inheritance. Initial genetic studies using linkage analysis in large families identified disease-causing mutations in sarcomeric genes. A sarcomere mutation is present in ~60% of patients with hypertrophic cardiomyopathy and is more common in those with familial disease and characteristic asymmetric septal hypertrophy. More than nine different sarcomere genes with over 1400 mutations have been implicated, although ∼80% of patients have a mutation in either MYH7 or MYBPC3 (Table 287-3), most of which are unique to individual families (“private” mutations).
Hypertrophic cardiomyopathy is characterized by age-dependent and incomplete penetrance. The defining phenotype of left ventricular hypertrophy is rarely present at birth and usually develops later in life. Accordingly, screening of family members should begin in adolescence and extend through adulthood. In MYBPC3 mutation carriers, the average age of disease development is 40 years, while 30% remain free from hypertrophy after 70 years. Related individuals who carry the same mutation may have a different extent and pattern of hypertrophy (e.g., asymmetric versus concentric), occurrence of outflow tract obstruction, and associated clinical outcomes (e.g., sudden death, atrial fibrillation).
At the level of the sarcomere, hypertrophic cardiomyopathy mutations lead to enhanced calcium sensitivity, maximal force generation, and ATPase activity. Calcium handling is affected through modification of regulatory proteins. Sarcomere mutations lead to abnormal energetics and impaired relaxation, both directly and as a result of hypertrophy. Hypertrophic cardiomyopathy is characterized by misalignment and disarray of the enlarged myofibrils and myocytes (Fig. 287-17), which can also occur to a lesser extent in other cardiac diseases. Although hypertrophy is the defining feature of hypertrophic cardiomyopathy, fibrosis and microvascular disease are also present. Interstitial fibrosis is detectable before overt hypertrophy develops and likely results from early activation of profibrotic pathways. In the majority of patients with overt cardiomyopathy, focal areas of replacement fibrosis can be readily detected with MRI. These areas of “scar” may represent substrate for the development of ventricular arrhythmias. Increased thickness and decreased luminal area of the intramural vessels in hypertrophied myocardium contribute to microvascular ischemia and angina. Microinfarction of hypertrophied myocardium is a hypothesized mechanism for replacement scar formation.
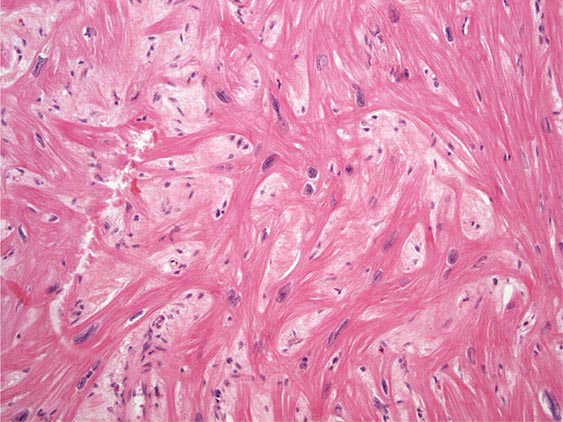
FIGURE 287-17 Hypertrophic cardiomyopathy. Microscopic image of hypertrophic cardiomyopathy showing the characteristic disordered myocyte architecture with swirling and branching rather than the usual parallel arrangement of myocyte fibers. Myocyte nuclei vary markedly in size and interstitial fibrosis is present. (Image courtesy of Robert Padera, MD, PhD, Department of Pathology, Brigham and Women’s Hospital, Boston.)
Macroscopically, hypertrophy is typically manifest as nonuniform ventricular thickening (Fig. 287-15). The interventricular septum is the typical location of maximal hypertrophy, although other patterns of hypertrophic remodeling include concentric and midventricular. Hypertrophy confined to the ventricular apex (apical hypertrophic cardiomyopathy) is less often familial and has a different genetic substrate, with sarcomere mutations present in only ~15%. Left ventricular outflow tract obstruction represents the most common focus of diagnosis and intervention, although diastolic dysfunction, myocardial fibrosis, and microvascular ischemia also contribute to contractile dysfunction and elevated intracardiac pressures. Obstruction is present in ~30% of patients at rest and can be provoked by exercise in another ~30%. Systolic obstruction is initiated by drag forces, which push an anteriorly displaced and enlarged anterior mitral leaflet into contact with the hypertrophied ventricular septum. Mitral leaflet coaptation may ensue, leading to posteriorly directed mitral regurgitation. In order to maintain stroke volume across outflow tract obstruction, the ventricle generates higher pressures, leading to higher wall stress and myocardial oxygen demand. Smaller chamber size and increased contractility exacerbate the severity of obstruction. Conditions of low preload, such as dehydration, and low afterload, such as arterial vasodilation, may lead to transient hypotension and near-syncope. The systolic ejection murmur of left ventricular outflow tract obstruction is harsh and late peaking and can be enhanced by bedside maneuvers that diminish ventricular volume and transiently worsen obstruction, such as standing from a squatting position or the Valsalva maneuver.
DIAGNOSIS
The substantial variability of hypertrophic cardiomyopathy pathology is reflected in the diversity of clinical presentations. Patients may be diagnosed after undergoing evaluations triggered by the abnormal physical findings (murmur) or symptoms of exertional dyspnea, angina, or syncope. Alternatively, diagnosis may follow evaluations prompted by the detection of disease in family members. Cardiac imaging (Fig. 287-16) is central to diagnosis due to the insensitivity of examination and ECG and the need to exclude other causes for hypertrophy. The identification of a disease-causing mutation in a proband can focus family evaluations on mutation carriers, but this strategy requires a high degree of certainty that the mutation is truly pathogenic and not a benign DNA variant. Biopsy is not needed to diagnose hypertrophic cardiomyopathy but can be used to exclude infiltrative and metabolic diseases. Rigorous athletic training (athlete’s heart) may cause intermediate degrees of physiologic hypertrophy difficult to differentiate from mild hypertrophic cardiomyopathy. Unlike hypertrophic cardiomyopathy, hypertrophy in the athlete’s heart regresses with cessation of training, and is accompanied by supernormal exercise capacity (VO2max >50 mL/kg/min), mild ventricular dilation, and normal diastolic function.
|
TREATMENT |
HYPERTROPHIC CARDIOMYOPATHY |
Management focuses on treatment of symptoms and prevention of sudden death and stroke (Fig. 287-18). Left ventricular outflow tract obstruction can be controlled medically in the majority of patients. β-Adrenergic blocking agents and L-type calcium channel blockers (e.g., verapamil) are first-line agents that reduce the severity of obstruction by slowing heart rate, enhancing diastolic filling, and decreasing contractility. Persistent symptoms of exertional dyspnea or chest pain can sometimes be controlled with the addition of disopyramide, an antiarrhythmic agent with potent negative inotropic properties. Patients with or without obstruction may develop heart failure symptoms due to fluid retention and require diuretic therapies for venous congestion. Severe medically refractory symptoms develop in ~5% of patients, for whom surgical myectomy or alcohol septal ablation may be effective. Developed over 50 years ago, surgical myectomy effectively relieves outflow tract obstruction by excising part of the septal myocardium involved in the dynamic obstruction. In selected patients, perioperative mortality is extremely low with excellent long-term survival free from recurrent obstruction and symptoms. Mitral valve repair or replacement is usually unnecessary as associated eccentric mitral regurgitation resolves with myectomy alone. Alcohol septal ablation in patients with suitable coronary anatomy can relieve outflow tract obstruction via a controlled infarction of the proximal septum, which produces similar periprocedural outcomes and gradient reduction as surgical myomectomy. Until long-term outcomes are demonstrated for this procedure, it is relegated primarily to patients who wish to avoid surgery or who have limiting comorbidities. Neither procedure has been shown to improve outcomes other than symptoms. With both procedures, the most common complication is the development of complete heart block necessitating permanent pacing. However, ventricular pacing as a primary therapy for outflow tract obstruction is ineffective and not generally advised.
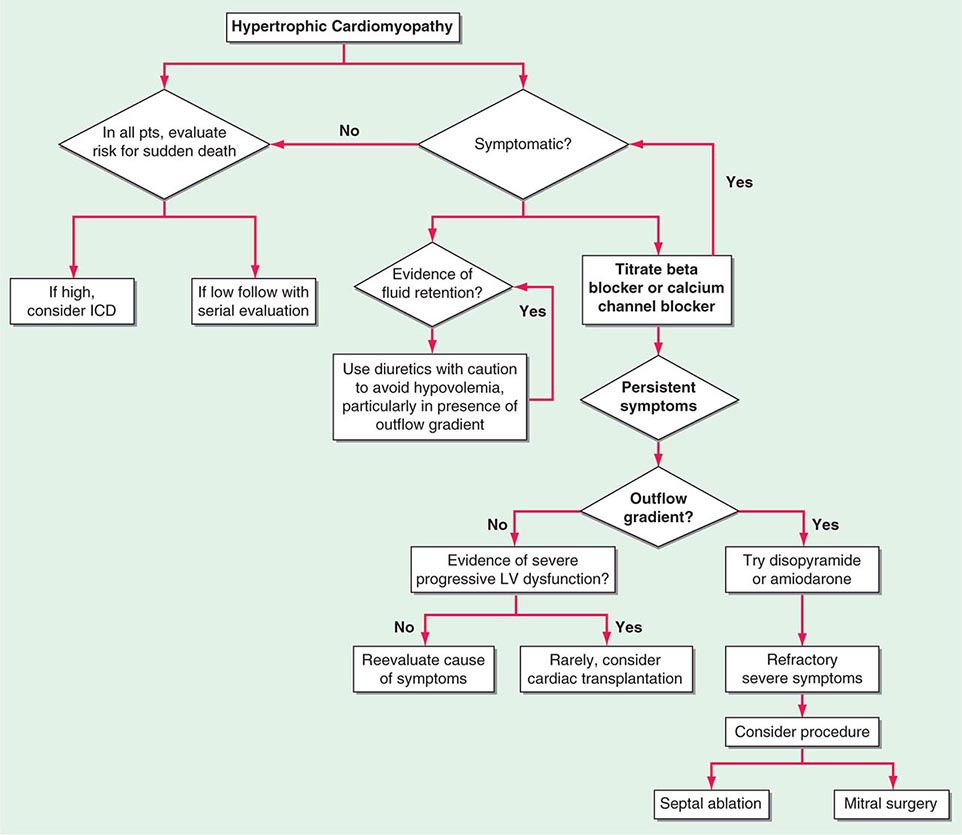
FIGURE 287-18 Treatment algorithm for hypertrophic cardiomyopathy depending on the presence and severity of symptoms and the presence of an intraventricular gradient with obstruction to outflow. Note that all patients with hypertrophic cardiomyopathy should be evaluated for atrial fibrillation and risk of sudden death, whether or not they require treatment for symptoms. ICD, implantable cardioverter-defibrillator; LV, left ventricular.
Patients with hypertrophic cardiomyopathy have an increased risk of sudden cardiac death from ventricular tachyarrhythmias. Vigorous physical activity and competitive sport are prohibited. Factors that increase the risk of sudden death from a baseline of 0.5% per year are presented in Table 287-6. As sudden death has not been reduced by medical or procedural interventions, an implantable cardioverter-defibrillator is advised for patients with two or more risk factors and is advised on a selected basis for patient with one risk factor. Nevertheless, the positive predictive value of most risk factors is low, and many patients receiving a defibrillator never receive an appropriate therapy. Long-term use of a defibrillator may be associated with serious device-related complications, particularly in young active patients. Refinement of sudden death risk through the application of contemporary technologies such as cardiac MRI is ongoing.
|
RISK FACTORS FOR SUDDEN DEATH IN HYPERTROPHIC CARDIOMYOPATHY |
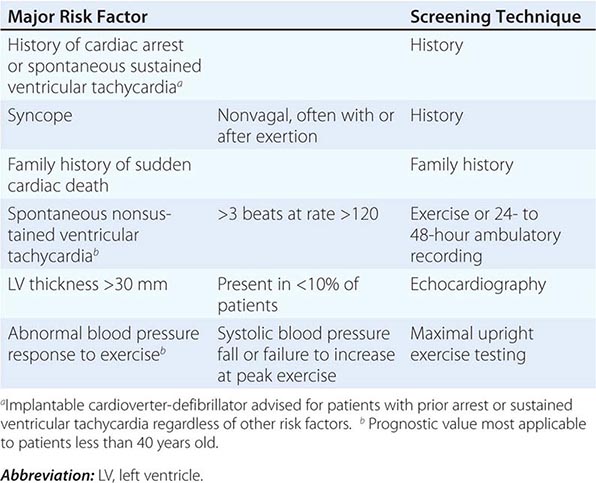
Atrial fibrillation is common in patients with hypertrophic cardiomyopathy and may lead to hemodynamic deterioration and embolic stroke. Rapid ventricular response is poorly tolerated and may worsen outflow tract obstruction. β-Adrenergic blocking agents and L-type calcium channel blockers slow AV nodal conduction and improve symptoms; cardiac glycosides should be avoided, as they may increase contractility and worsen obstruction. Symptoms exacerbated by atrial fibrillation may persist despite adequate rate control due to loss of AV synchrony and may require restoration of sinus rhythm. Disopyramide and amiodarone are the preferred antiarrhythmic agents, with radiofrequency ablation considered for medically refractory cases. Anticoagulation to prevent embolic stroke in atrial fibrillation is recommended.
PROGNOSIS The general prognosis for hypertrophic cardiomyopathy is good, better than in early studies of referral populations. For patients diagnosed as adults, survival is comparable to an age-matched population without cardiomyopathy. The sudden death risk is less than 1% per year; however, up to 1 in 20 patients will progress to overt systolic dysfunction with a reduced ejection fraction with or without dilated remodeling (“burned out” or end-stage hypertrophic cardiomyopathy). These patients suffer from low cardiac output and have a high risk of death from progressive heart failure and sudden death unless they undergo cardiac transplantation.
288 |
Pericardial Disease |
NORMAL FUNCTIONS OF THE PERICARDIUM
The normal pericardium is a double-layered sac; the visceral pericardium is a serous membrane that is separated by a small quantity (15–50 mL) of fluid, an ultrafiltrate of plasma, from the fibrous parietal pericardium. The normal pericardium, by exerting a restraining force, prevents sudden dilation of the cardiac chambers, especially the right atrium and ventricle, during exercise and with hypervolemia. It also restricts the anatomic position of the heart, and probably retards the spread of infections from the lungs and pleural cavities to the heart. Nevertheless, total absence of the pericardium, either congenital or after surgery, does not produce obvious clinical disease. In partial left pericardial defects, the main pulmonary artery and left atrium may bulge through the defect; very rarely, herniation and subsequent strangulation of the left atrium may cause sudden death.
ACUTE PERICARDITIS
Acute pericarditis, by far the most common pathologic process involving the pericardium (Table 288-1), has four principal diagnostic features:
1. Chest pain is usually present in acute infectious pericarditis and in many of the forms presumed to be related to hypersensitivity or autoimmunity. The pain of acute pericarditis is often severe, retrosternal, and left precordial, and referred to the neck, arms, or left shoulder. Frequently the pain is pleuritic, consequent to accompanying pleural inflammation (i.e., sharp and aggravated by inspiration and coughing), but sometimes it is steady, constricting, radiates into either arm or both arms, and resembles that of myocardial ischemia; therefore, confusion with acute myocardial infarction (AMI) is common. Characteristically, however, pericardial pain may be relieved by sitting up and leaning forward and is intensified by lying supine (Chap. 19). Pain is often absent in slowly developing tuberculous, postirradiation, and neoplastic, uremic, and constrictive pericarditis.
The differentiation of AMI from acute pericarditis may become perplexing when, with acute pericarditis, serum biomarkers of myocardial damage such as troponin and creatine kinase-MB rise, presumably because of concomitant involvement of the epicardium in the inflammatory process (an epi-myocarditis) with resulting myocyte necrosis. However, these elevations, if they occur, are quite modest given the extensive electrocardiographic ST-segment elevation in pericarditis. This dissociation is useful in differentiating between these conditions.
2. A pericardial friction rub is audible at some point in about 85% of patients with acute pericarditis, may have up to three components per cardiac cycle, is high-pitched, and is described as rasping, scratching, or grating (Chap. 267). It is heard most frequently at end expiration with the patient upright and leaning forward.
3. The electrocardiogram (ECG) in acute pericarditis without massive effusion usually displays changes secondary to acute subepicardial inflammation (Fig. 288-1). It typically evolves through four stages. In stage 1, there is widespread elevation of the ST segments, often with upward concavity, involving two or three standard limb leads and V2 to V6, with reciprocal depressions only in aVR and sometimes V1. Also, there is depression of the PR segment below the TP segment, reflecting atrial involvement. Usually there are no significant changes in QRS complexes. After several days, the ST segments return to normal (stage 2), and only then, or even later, do the T waves become inverted (stage 3). Weeks or months after the onset of acute pericarditis, the ECG returns to normal (stage 4). In contrast, in AMI, ST elevations are convex, and reciprocal depression is usually more prominent; these changes may return to normal within a day or two. Q waves may develop, with loss of R-wave amplitude, and T-wave inversions are usually seen within hours before the ST segments have become isoelectric (Chaps. 294 and 295).
4. Pericardial effusion is usually associated with pain and/or the ECG changes mentioned above, as well as electrical alternans. Pericardial effusion is especially important clinically when it develops within a relatively short time because it may lead to cardiac tamponade (see below). Differentiation from cardiac enlargement may be difficult on physical examination, but heart sounds may be fainter with pericardial effusion. The friction rub and the apex impulse may disappear. The base of the left lung may be compressed by pericardial fluid, producing Ewart’s sign, a patch of dullness and increased fremitus (and egophony) beneath the angle of the left scapula. The chest roentgenogram may show enlargement of the cardiac silhouette, with a “water bottle” configuration, but may be normal.
|
CLASSIFICATION OF PERICARDITIS |
aAn autosomal recessive syndrome characterized by growth failure, muscle hypotonia, hepatomegaly, ocular changes, enlarged cerebral ventricles, mental retardation, ventricular hypertrophy, and chronic constrictive pericarditis
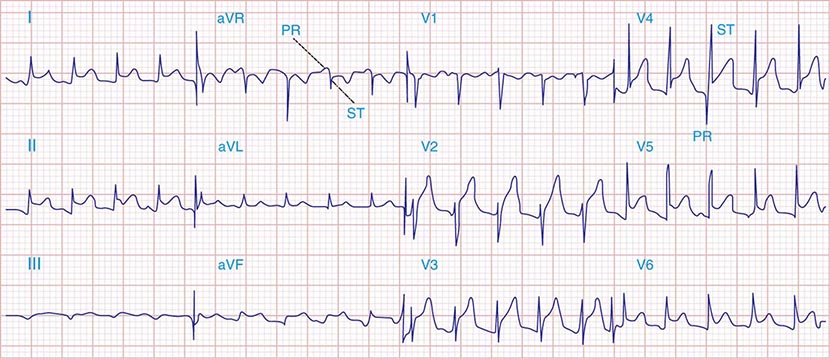
FIGURE 288-1 Acute pericarditis. There are diffuse ST-segment elevations (in this case in leads I, II, aVF, and V2 to V6) due to a ventricular current of injury. There is PR-segment deviation (opposite in polarity to the ST segment) due to a concomitant atrial injury current.
Diagnosis Echocardiography (Chap. 270e) is the most widely used imaging technique. It is sensitive, specific, simple, noninvasive, may be performed at the bedside, and can identify accompanying cardiac tamponade (see below) (Fig. 288-2). The presence of pericardial fluid is recorded by two-dimensional transthoracic echocardiography as a relatively echo-free space between the posterior pericardium and left ventricular epicardium in patients with small effusions and as a space between the anterior right ventricle and the parietal pericardium just beneath the anterior chest wall. In patients with large effusions, the heart may swing freely within the pericardial sac. When severe, the extent of this motion alternates and may be associated with electrical alternans (Fig. 288-3). Echocardiography allows localization and identification of the quantity of pericardial fluid.
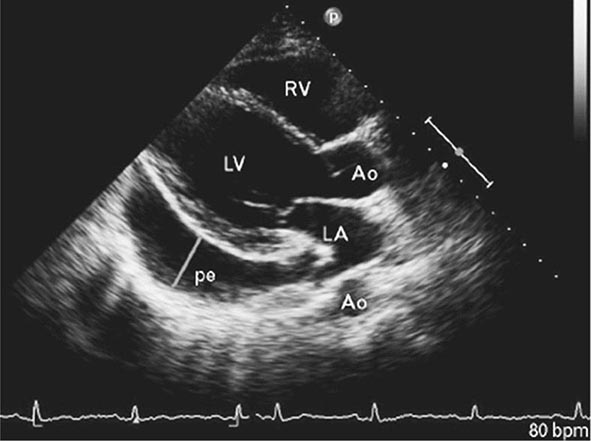
FIGURE 288-2 Two-dimensional echocardiogram in lateral view in a patient with a large pericardial effusion. Ao, aorta; LV, left ventricle; pe, pericardial effusion; RV, right ventricle. (From M Imazio: Curr Opin Cardiol 27:308, 2012.)
FIGURE 288-3 Electrical alternans. This tracing was obtained from a patient with a large pericardial effusion with tamponade. (Reproduced from DM Mirvis, AL Goldberger: Electrocardiography, in RO Bonow et al [eds]: Braunwald’s Heart Disease, 9th ed. Philadelphia: Elsevier, 2012.)
The diagnosis of pericardial fluid or thickening may be confirmed by computed tomography (CT) or magnetic resonance imaging (MRI). These techniques may be superior to echocardiography in detecting loculated pericardial effusions, pericardial thickening, and the identification of pericardial masses.
CARDIAC TAMPONADE
The accumulation of fluid in the pericardial space in a quantity sufficient to cause serious obstruction of the inflow of blood into the ventricles results in cardiac tamponade. This complication may be fatal if it is not recognized and treated promptly. The most common causes of tamponade are idiopathic pericarditis and pericarditis secondary to neoplastic disease. Tamponade may also result from bleeding into the pericardial space after leakage from an aortic dissection, cardiac operations, trauma, and treatment of patients with acute pericarditis with anticoagulants.
The three principal features of tamponade (Beck’s triad) are hypotension, soft or absent heart sounds, and jugular venous distention with a prominent x descent but an absent y descent. The limitations of ventricular filling are responsible for a reduction of cardiac output. The quantity of fluid necessary to produce cardiac tamponade may be as small as 200 mL when the fluid develops rapidly to as much as >2000 mL in slowly developing effusions when the pericardium has had the opportunity to stretch and adapt to an increasing volume. Tamponade may also develop more slowly, and in these circumstances, the clinical manifestations can resemble those of heart failure, including dyspnea, orthopnea, and hepatic engorgement.
A high index of suspicion for cardiac tamponade is required because in many instances no obvious cause for pericardial disease is apparent, and this diagnosis should be considered in any patient with otherwise unexplained enlargement of the cardiac silhouette, hypotension, and elevation of jugular venous pressure. There may be reduction in amplitude of the QRS complexes, and electrical alternans of the P, QRS, or T waves should raise the suspicion of cardiac tamponade (Fig. 288-3).
Table 288-2 lists the features that distinguish acute cardiac tamponade from constrictive pericarditis.
|
FEATURES THAT DISTINGUISH CARDIAC TAMPONADE FROM CONSTRICTIVE PERICARDITIS AND SIMILAR CLINICAL DISORDERS |
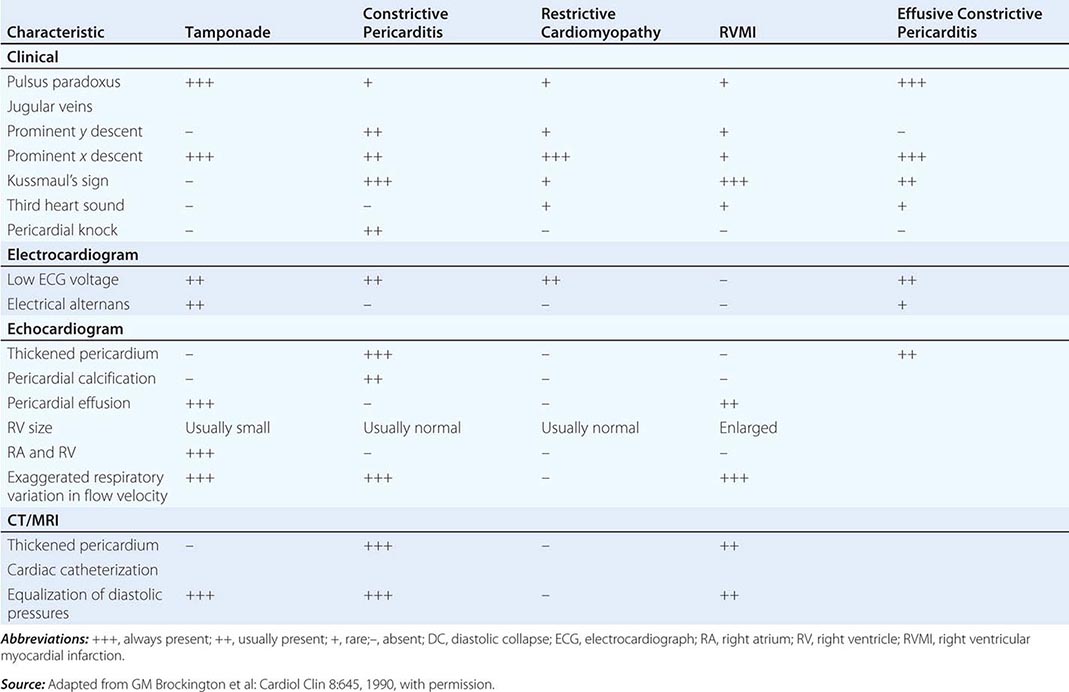
Paradoxical Pulse This important clue to the presence of cardiac tamponade consists of a greater than normal (10 mmHg) inspiratory decline in systolic arterial pressure. When severe, it may be detected by palpating weakness or disappearance of the arterial pulse during inspiration, but usually sphygmomanometric measurement of systolic pressure during slow respiration is required.
Because both ventricles share a tight incompressible covering, i.e., the pericardial sac, the inspiratory enlargement of the right ventricle in cardiac tamponade compresses and reduces left ventricular volume; leftward bulging of the interventricular septum reduces further the left ventricular cavity as the right ventricle enlarges during inspiration. Thus, in cardiac tamponade, the normal inspiratory augmentation of right ventricular volume causes an exaggerated reduction of left ventricular volume, stroke volume, and systolic pressure. Paradoxical pulse also occurs in approximately one-third of patients with constrictive pericarditis (see below), and in some cases of hypovolemic shock, acute and chronic obstructive airway disease, and pulmonary embolus. Right ventricular infarction (Chap. 295) may resemble cardiac tamponade with hypotension, elevated jugular venous pressure, an absent y descent in the jugular venous pulse, and, occasionally, a paradoxical pulse (Table 288-2).
Low-pressure tamponade refers to mild tamponade in which the intrapericardial pressure is increased from its slightly subatmospheric levels to +5 to +10 mmHg; in some instances, hypovolemia coexists. As a consequence, the central venous pressure is normal or only slightly elevated, whereas arterial pressure is unaffected and there is no paradoxical pulse. These patients are asymptomatic or complain of mild weakness and dyspnea. The diagnosis is aided by echocardiography, and both hemodynamic and clinical manifestations improve after pericardiocentesis.
Diagnosis Because immediate treatment of cardiac tamponade may be lifesaving, prompt measures to establish the diagnosis by echocardiography should be undertaken. When pericardial effusion causes tamponade, Doppler ultrasound shows that tricuspid and pulmonic valve flow velocities increase markedly during inspiration, whereas pulmonic vein, mitral, and aortic flow velocities diminish (as in constrictive pericarditis, see below) (Fig. 288-4). In tamponade, there is late diastolic inward motion (collapse) of the right ventricular free wall and the right atrium. Transesophageal echocardiography, CT, or cardiac MRI may be necessary to diagnose a loculated effusion responsible for cardiac tamponade.
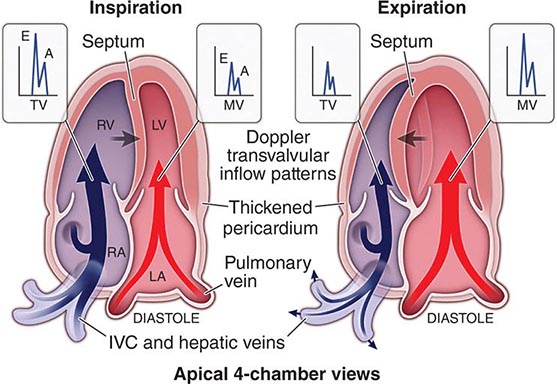
FIGURE 288-4 Constrictive pericarditis. Doppler schema of respirophasic changes in mitral and tricuspid inflow. Reciprocal patterns of ventricular filling are assessed on pulsed Doppler examination of mitral valve (MV) and tricuspid valve (TV) inflow. IVC, inferior vena cava; LA, left atrium; LV, left ventricle; RA, right atrium; RV, right ventricle. (Courtesy of Bernard E. Bulwer, MD; with permission.)
VIRAL OR IDIOPATHIC ACUTE PERICARDITIS
In many instances, acute pericarditis occurs in association with illnesses of known or presumed viral origin and probably is caused by the same agent. Commonly, there is an antecedent infection of the respiratory tract, and viral isolation and serologic studies are negative. In some cases, coxsackievirus A or B or the virus of influenza, echovirus, mumps, herpes simplex, chickenpox, adenovirus, or cytomegalovirus has been isolated from pericardial fluid and/or appropriate elevations in viral antibody titers have been noted. Pericardial effusion is a common cardiac manifestation of HIV; it is usually secondary to infection (often mycobacterial) or neoplasm, most often lymphoma. Frequently, a viral cause cannot be established, and the term idiopathic acute pericarditis is then appropriate.
Viral or idiopathic acute pericarditis occurs at all ages but is more common in young adults and is often associated with pleural effusions and pneumonitis. The almost simultaneous development of fever and precordial pain, often 10–12 days after a presumed viral illness, constitutes an important feature in the differentiation of acute pericarditis from AMI, in which chest pain precedes fever. The constitutional symptoms are usually mild to moderate, and a pericardial friction rub is often audible. The disease ordinarily runs its course in a few days to 4 weeks. The ST-segment alterations in the ECG usually disappear after 1 or more weeks, but the abnormal T waves may persist for several years and be a source of confusion in persons without a clear history of pericarditis. Pleuritis and pneumonitis frequently accompany viral or idiopathic acute pericarditis. Accumulation of some pericardial fluid is common, and both tamponade and constrictive pericarditis are possible, but infrequent, complications.
The most frequent complication is recurrent (relapsing) pericarditis, which occurs in about one-fourth of patients with acute idiopathic pericarditis. In a smaller number, there are multiple recurrences. For treatment, see earlier section on treatment of acute pericarditis.
Postcardiac Injury Syndrome Acute pericarditis may appear in a variety of circumstances that have one common feature—previous injury to the myocardium with blood in the pericardial cavity. The syndrome may develop after a cardiac operation (postpericardiotomy syndrome), after blunt or penetrating cardiac trauma (Chap. 289e), or after perforation of the heart with a catheter. Rarely, it follows AMI.
The clinical picture mimics acute viral or idiopathic pericarditis. The principal symptom is the pain of acute pericarditis, which usually develops 1–4 weeks after the cardiac injury but earlier (1–3 days) after AMI. Recurrences are common and may occur up to 2 years or more following the injury. Fever, pleuritis, and pneumonitis are the outstanding features, and the bout of illness usually subsides in 1 or 2 weeks. The pericarditis may be of the fibrinous variety, or it may be a pericardial effusion, which is often serosanguineous but rarely causes tamponade. ECG changes typical of acute pericarditis may also occur. This syndrome is probably the result of a hypersensitivity reaction to antigen(s) that originate from injured myocardial tissue and/or pericardium.
Often no treatment is necessary aside from aspirin and analgesics. When the illness is severe or followed by a series of disabling recurrences, therapy with an NSAID, colchicine, or a glucocorticoid, such as described for treatment of acute pericarditis, is usually effective.
DIFFERENTIAL DIAGNOSIS
Because there is no specific test for acute idiopathic pericarditis, the diagnosis is one of exclusion. Consequently, all other disorders that may be associated with acute fibrinous pericarditis must be considered. A common diagnostic error is mistaking acute viral or idiopathic pericarditis for AMI and vice versa. When acute fibrinous pericarditis is associated with AMI (Chap. 295), it is characterized by fever, pain, and a friction rub in the first 4 days after the development of the infarct. ECG abnormalities (such as the appearance of Q waves, brief ST-segment elevations with reciprocal changes, and earlier T-wave changes in AMI) and the extent of the elevations of markers of myocardial necrosis (higher in AMI) are helpful in differentiating pericarditis from AMI.
Pericarditis secondary to postcardiac injury is differentiated from acute idiopathic pericarditis chiefly by timing. If it occurs within a few days or weeks of an AMI, a chest blow, a cardiac perforation, or a cardiac operation, it may be justified to conclude that the two are probably related.
It is important to distinguish pericarditis due to collagen vascular disease from acute idiopathic pericarditis. Most important in the differential diagnosis is the pericarditis due to systemic lupus erythematosus (SLE; Chap. 378) or drug-induced (procainamide or hydralazine) lupus. When pericarditis occurs in the absence of any obvious underlying disorder, the diagnosis of SLE may be suggested by a rise in the titer of antinuclear antibodies. Acute pericarditis is an occasional complication of rheumatoid arthritis, scleroderma, and polyarteritis nodosa, and other evidence of these diseases is usually obvious.
Pyogenic (purulent) pericarditis is usually secondary to cardiothoracic operations, by extension of infection from the lungs or pleural cavities, from rupture of the esophagus into the pericardial sac, or from rupture of a ring abscess in a patient with infective endocarditis. It may also complicate the viral, pyogenic, mycobacterial, and fungal infections that occur with HIV infection. It is generally accompanied by fever, chills, septicemia, and evidence of infection elsewhere and generally has a poor prognosis. The diagnosis is made by examination of the pericardial fluid. It requires drainage as well as vigorous antibiotic treatment.
Pericarditis of renal failure occurs in up to one-third of patients with chronic uremia (uremic pericarditis), and is also seen in patients undergoing chronic dialysis who have normal levels of blood urea and creatinine (dialysis-associated pericarditis). These two forms of pericarditis may be fibrinous and are generally associated with serosanguineous effusions. A pericardial friction rub is common, but pain is usually absent or mild. Treatment with an NSAID and intensification of dialysis are usually adequate. Occasionally, tamponade occurs and pericardiocentesis is required. When the pericarditis of renal failure is recurrent or persistent, a pericardial window should be created or pericardiectomy may be necessary.
Pericarditis due to neoplastic diseases results from extension or invasion of metastatic tumors (most commonly carcinoma of the lung and breast, malignant melanoma, lymphoma, and leukemia) to the pericardium; pain, atrial arrhythmias, and tamponade are complications that occur occasionally. Diagnosis is made by pericardial fluid cytology or pericardial biopsy. Mediastinal irradiation for neoplasm may cause acute pericarditis and/or chronic constrictive pericarditis. Unusual causes of acute pericarditis include syphilis, fungal infection (histoplasmosis, blastomycosis, aspergillosis, and candidiasis), and parasitic infestation (amebiasis, toxoplasmosis, echinococcosis, and trichinosis).
CHRONIC PERICARDIAL EFFUSIONS
Chronic pericardial effusions are sometimes encountered in patients without an antecedent history of acute pericarditis. They may cause few symptoms per se, and their presence may be detected by finding an enlarged cardiac silhouette on a chest roentgenogram. Tuberculosis is a common cause. Myxedema may be responsible for chronic pericardial effusion that is sometimes massive but rarely, if ever, causes cardiac tamponade. The cardiac silhouette may be markedly enlarged, and an echocardiogram distinguishes cardiomegaly from pericardial effusion. The diagnosis of myxedema can be confirmed by tests of thyroid function (Chap. 405). Myxedematous pericardial effusion responds to thyroid hormone replacement. Neoplasms, SLE, rheumatoid arthritis, mycotic infections, radiation therapy to the chest, pyogenic infections, and chylopericardium may also cause chronic pericardial effusion and should be considered and specifically sought in such patients.
Aspiration and analysis of the pericardial fluid are often helpful in diagnosis. Pericardial fluid should be analyzed as described in pericardiocentesis. Grossly sanguineous pericardial fluid results most commonly from a neoplasm, tuberculosis, renal failure, or slow leakage from an aortic dissection. Pericardiocentesis may resolve large effusions, but pericardiectomy may be required in patients with recurrence. Intrapericardial instillation of sclerosing agents may be used to prevent reaccumulation of fluid.
CHRONIC CONSTRICTIVE PERICARDITIS
This disorder results when the healing of an acute fibrinous or serofibrinous pericarditis or the resorption of a chronic pericardial effusion is followed by obliteration of the pericardial cavity with the formation of granulation tissue. The latter gradually contracts and forms a firm scar encasing the heart, which may be calcified. In developing nations where the condition is prevalent, a high percentage of cases are of tuberculous origin, but this is now an uncommon cause in North America. Chronic constrictive pericarditis may follow acute or relapsing viral or idiopathic pericarditis, trauma with organized blood clot, or cardiac surgery of any type or result from mediastinal irradiation, purulent infection, histoplasmosis, neoplastic disease (especially breast cancer, lung cancer, and lymphoma), rheumatoid arthritis, SLE, or chronic renal failure treated by chronic dialysis. In many patients, the cause of the pericardial disease is undetermined, and in these patients, an asymptomatic or forgotten bout of viral pericarditis, acute or idiopathic, may have been the inciting event.
The basic physiologic abnormality in patients with chronic constrictive pericarditis is the inability of the ventricles to fill because of the limitations imposed by the rigid, thickened pericardium. Ventricular filling is unimpeded during early diastole but is reduced abruptly when the elastic limit of the pericardium is reached, whereas in cardiac tamponade, ventricular filling is impeded throughout diastole. In both conditions, ventricular end-diastolic and stroke volumes are reduced and the end-diastolic pressures in both ventricles and the mean pressures in the atria, pulmonary veins, and systemic veins are all elevated to similar levels (i.e., within 5 mmHg of one another). Despite these hemodynamic changes, systolic function may be normal or only slightly impaired. However, in advanced cases, the fibrotic process may extend into the myocardium and cause myocardial scarring and atrophy, and venous congestion may then be due to the combined effects of the pericardial and myocardial lesions.
In constrictive pericarditis, the right and left atrial pressure pulses display an M-shaped contour, with prominent x and y descents. The y descent, which is absent or diminished in cardiac tamponade, is the most prominent deflection in constrictive pericarditis; it reflects rapid early filling of the ventricles. The y descent is interrupted by a rapid rise in atrial pressure during early diastole, when ventricular filling is impeded by the constricting pericardium. These characteristic changes are transmitted to the jugular veins, where they may be recognized by inspection. In constrictive pericarditis, the ventricular pressure pulses in both ventricles exhibit characteristic “square root” signs during diastole. These hemodynamic changes, although characteristic, are not pathognomonic of constrictive pericarditis and may also be observed in restrictive cardiomyopathies (Chap. 287, Table 287-2).
CLINICAL AND LABORATORY FINDINGS
Weakness, fatigue, weight gain, increased abdominal girth, abdominal discomfort, and edema are common. The patient often appears chronically ill, and in advanced cases, anasarca, skeletal muscle wasting, and cachexia may be present. Exertional dyspnea is common, and orthopnea may occur, although it is usually not severe. Acute left ventricular failure (acute pulmonary edema) is very uncommon. The cervical veins are distended and may remain so even after intensive diuretic treatment, and venous pressure may fail to decline during inspiration (Kussmaul’s sign). The latter is common in chronic pericarditis but may also occur in tricuspid stenosis, right ventricular infarction, and restrictive cardiomyopathy.
The pulse pressure is normal or reduced. A paradoxical pulse can be detected in about one-third of cases. Congestive hepatomegaly is pronounced and may impair hepatic function and cause jaundice; ascites is common and is usually more prominent than dependent edema. The apical pulse is reduced and may retract in systole (Broadbent’s sign). The heart sounds may be distant; an early third heart sound (i.e., a pericardial knock, occurring at the cardiac apex 0.09–0.12 s after aortic valve closure) with the abrupt cessation of ventricular filling is often conspicuous.
The ECG frequently displays low voltage of the QRS complexes and diffuse flattening or inversion of the T waves. Atrial fibrillation is present in about one-third of patients. The chest roentgenogram shows a normal or slightly enlarged heart. Pericardial calcification is most common in tuberculous pericarditis. Pericardial calcification may, however, occur in the absence of constriction, and constriction may occur without calcification.
Inasmuch as the usual physical signs of cardiac disease (murmurs, cardiac enlargement) may be inconspicuous or absent in chronic constrictive pericarditis, hepatic enlargement and dysfunction associated with jaundice and intractable ascites may lead to a mistaken diagnosis of hepatic cirrhosis. This error can be avoided if the neck veins are inspected and found to be distended.
The transthoracic echocardiogram typically shows pericardial thickening, dilation of the inferior vena cava and hepatic veins, and a sharp halt in ventricular filling in early diastole, with normal ventricular systolic function and flattening of the left ventricular posterior wall. There is a distinctive pattern of transvalvular flow velocity on Doppler echocardiography. During inspiration, there is an exaggerated reduction in blood flow velocity in the pulmonary veins and across the mitral valve and a leftward shift of the ventricular septum; the opposite occurs during expiration. Diastolic flow velocity in the inferior vena cava into the right atrium and across the tricuspid valve increases in an exaggerated manner during inspiration and declines during expiration (Fig. 288-4). However, echocardiography cannot definitively exclude the diagnosis of constrictive pericarditis. CT and MRI scanning (Fig. 288-5) are more accurate than echocardiography in establishing or excluding the presence of a thickened pericardium.
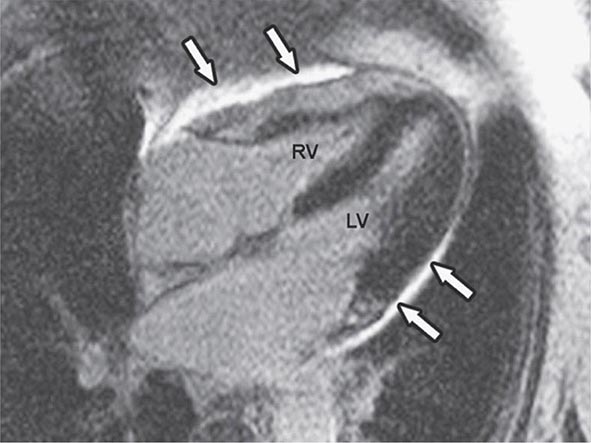
FIGURE 288-5 Magnetic resonance imaging in chronic constrictive pericarditis. The arrows point to a thickened pericardium, which shows late enhancement after gadolinium, characteristic of intense inflammation. LV, left ventricle; RV, right ventricle. (From RY Kwong: Cardiovascular magnetic resonance imaging, in RO Bonow et al [eds]: Braunwald’s Heart Disease, 9th ed. Philadelphia: Elsevier, 2012.)
DIFFERENTIAL DIAGNOSIS
Like chronic constrictive pericarditis, cor pulmonale (Chap. 279) may be associated with severe systemic venous hypertension but little pulmonary congestion; the heart is usually not enlarged, and a paradoxical pulse may be present. However, in cor pulmonale, advanced parenchymal pulmonary disease is usually apparent and venous pressure falls during inspiration (i.e., Kussmaul’s sign is negative). Tricuspid stenosis (Chap. 283) may also simulate chronic constrictive pericarditis; congestive hepatomegaly, splenomegaly, ascites, and venous distention may be equally prominent. However, in tricuspid stenosis, a characteristic murmur and the murmur of accompanying mitral stenosis are usually present.
Because constrictive pericarditis can be corrected surgically, it is important to distinguish chronic constrictive pericarditis from restrictive cardiomyopathy (Chap. 287), which has a similar physiologic abnormality (i.e., restriction of ventricular filling). The differentiating features are summarized in Table 288-2. When a patient has progressive, disabling, and unresponsive congestive heart failure and displays any of the features of constrictive heart disease, Doppler echocardiography to record respiratory effects on transvalvular flow and an MRI or CT scan should be obtained to detect or exclude constrictive pericarditis, because the latter is usually correctable.
Subacute Effusive-Constrictive Pericarditis This form of pericardial disease is characterized by the combination of a tense effusion in the pericardial space and constriction of the heart by thickened pericardium. It shares a number of features with both chronic pericardial effusion producing cardiac compression and with pericardial constriction. It may be caused by tuberculosis (see below), multiple attacks of acute idiopathic pericarditis, radiation, traumatic pericarditis, renal failure, scleroderma, and neoplasms. The heart is generally enlarged, and a paradoxical pulse and a prominent x descent (without a prominent y descent) are present in the atrial and jugular venous pressure pulses. After pericardiocentesis, the physiologic findings may change from those of cardiac tamponade to those of pericardial constriction. Furthermore, the intrapericardial pressure and the central venous pressure may decline, but not to normal. The diagnosis can be established by pericardiocentesis followed by pericardial biopsy. Wide excision of both the visceral and parietal pericardium is usually effective therapy.
Tuberculous Pericardial Disease This chronic infection is a common cause of chronic pericardial effusion, although less so in North America than in the developing world where active tuberculosis is endemic. The clinical picture is that of a chronic, systemic illness in a patient with pericardial effusion. It is important to consider this diagnosis in a patient with known tuberculosis, with HIV, and with fever, chest pain, weight loss, and enlargement of the cardiac silhouette of undetermined origin. If the etiology of chronic pericardial effusion remains obscure despite detailed analysis of the pericardial fluid (see above), a pericardial biopsy, preferably by a limited thoracotomy, should be performed. If definitive evidence is still lacking but the specimen shows granulomas with caseation, antituberculous chemotherapy (Chap. 202) is indicated.
If the biopsy specimen shows a thickened pericardium after 2–4 weeks of antituberculin therapy, pericardiectomy should be carried out to prevent the development of constriction. Tubercular cardiac constriction should be treated surgically while the patient is receiving antituberculous chemotherapy.
OTHER DISORDERS OF THE PERICARDIUM
Pericardial cysts appear as rounded or lobulated deformities of the cardiac silhouette, most commonly at the right cardiophrenic angle. They usually do not cause symptoms, and their major clinical significance lies in the possibility of confusion with a tumor, ventricular aneurysm, or massive cardiomegaly. Tumors involving the pericardium are most commonly secondary to malignant neoplasms originating in or invading the mediastinum, including carcinoma of the bronchus and breast, lymphoma, and melanoma. Mesothelioma is the most common primary malignant tumor. The usual clinical picture of malignant pericardial tumor is an insidiously developing, often bloody pericardial effusion. Surgical exploration is required to establish a definitive diagnosis and to carry out definitive or, more commonly, palliative treatment.