[level-membership-for-neonatal-and-perinatal-medicine-category]Chapter 6
Cardiology Secrets
History of Pediatric Cardiology
The first successful ligation of a PDA was by Gross and Hubbard in 1938.
Christiaan Barnard performed the first cardiac transplantation in South Africa in 1967. Infant heart transplantation was attempted unsuccessfully 3 days later by Adrian Kantrowitz in New York City. Neonatal heart transplantation would not be achieved, however, until November 15, 1985, by Leonard Bailey at Loma Linda Medical Center. ∗†
Fetal Echocardiography and Prenatal Conditions that Can Contribute to Neonatal Heart Disease
 Family history of congenital heart disease
Family history of congenital heart disease
 Metabolic disorders (diabetes, phenylketonuria)
Metabolic disorders (diabetes, phenylketonuria)

 Exposure to prostaglandin synthase inhibitors (ibuprofen, salicylic acid)
Exposure to prostaglandin synthase inhibitors (ibuprofen, salicylic acid)

 Autoimmune disease (systemic lupus erythematosus, Sjögren syndrome)
Autoimmune disease (systemic lupus erythematosus, Sjögren syndrome)
 Familial inherited disorders (e.g., Marfan syndrome, Noonan syndrome)
Familial inherited disorders (e.g., Marfan syndrome, Noonan syndrome)

 Abnormal obstetric ultrasound screen
Abnormal obstetric ultrasound screen




 Increased first trimester nuchal translucency
Increased first trimester nuchal translucency


5. What is the incidence of congenital heart disease? What is the recurrence risk if a previous child has congenital heart disease?
Table 6-1 lists common genetic and chromosomal syndromes associated with congenital heart disease.
TABLE 6-1
COMMON GENETIC OR CHROMOSOMAL SYNDROMES ASSOCIATED WITH CONGENITAL HEART DISEASE
| GENETIC OR CHROMOSOMAL SYNDROME | COMMON CARDIAC ANATOMIC LESION |
| Apert syndrome | Ventricular septal defect, coarctation of the aorta, tetralogy of Fallot |
| Beckwith–Wiedemann syndrome | Atrial septal defect, ventricular septal defect, hypertrophic cardiomyopathy |
| CHARGE syndrome | Endocardial cushion defect, ventricular septal defect, double outlet right ventricle, tetralogy of Fallot |
| DiGeorge syndrome | Interrupted aortic arch, truncus arteriosus, tetralogy of Fallot, right aortic arch, ventricular septal defect, aberrant right subclavian artery |
| Ellis–van Creveld syndrome | Atrial septal defect, single/common atrium |
| Holt–Oram syndrome | Atrial septal defect, ventricular septal defect |
| Kartagener syndrome | Mirror image dextrocardia |
| Marfan syndrome | Dilated aortic root, mitral valve prolapse, tricuspid valve prolapse |
| Neurofibromatosis | Atrial septal defect, coarctation of the aorta, interrupted aortic arch, pulmonic stenosis, ventricular septal defect, complete heart block, hypertrophic cardiomyopathy |
| Noonan syndrome | Pulmonary stenosis, hypertrophic cardiomyopathy, tetralogy of Fallot |
| Pentalogy of Cantrell | Atrial septal defect, ventricular septal defect, total anomalous pulmonary venous drainage, tetralogy of Fallot, ectopia cordis |
| Pierre Robin syndrome | Pulmonary stenosis, atrial septal defect |
| Thrombocytopenia absent radius (TAR) syndrome | Atrial septal defect, tetralogy of Fallot |
| Treacher Collins syndrome | Ventricular septal defect, atrial septal defect |
| Tuberous sclerosis | Rhabdomyoma, angioma, coarctation of the aorta, interrupted aortic arch |
| Trisomy 13 | Ventricular septal defect, atrial septal defect, endocardial cushion defect, tetralogy of Fallot |
| Trisomy 18 | Bicuspid aortic valve, pulmonic stenosis, ventricular septal defect, atrial septal defect, endocardial cushion defect, polyvalvular thickening |
| Trisomy 21 | Endocardial cushion defect, ventricular septal defect, atrial septal defect, tetralogy of Fallot, coarctation of the aorta |
| Turner syndrome | Bicuspid aortic valve, coarctation of the aorta, aortic stenosis, ventricular septal defect, atrial septal defect |
| VACTERL syndrome | Ventricular septal defect, atrial septal defect, tetralogy of Fallot |
| Williams syndrome | Supravalvular aortic or pulmonic stenosis |
| Wolf–Hirschhorn syndrome | Atrial septal defect, ventricular septal defect |
Adapted from Drose J. Fetal echocardiography. 1st ed. Philadelphia: Saunders; 1988.
Table 6-2 lists teratogens known to cause congenital heart disease. ∗†‡
TABLE 6-2
TERATOGENS THAT CAUSE CONGENITAL HEART DISEASE
| TERATOGEN | COMMON CARDIAC ANATOMIC LESION |
| Fetal alcohol syndrome | Ventricular septal defect, atrial septal defect, tetralogy of Fallot, coarctation of the aorta |
| Fetal hydantoin (Dilantin) syndrome | Ventricular septal defect, tetralogy of Fallot, pulmonic stenosis, patent ductus arteriosus, atrial septal defect, coarctation of the aorta |
| Fetal trimethadione syndrome | Ventricular septal defect, d-transposition of the great vessels, tetralogy of Fallot, hypoplastic left heart syndrome, double outlet right ventricle, pulmonary atresia, atrial septal defect, aortic stenosis, pulmonic stenosis |
| Fetal carbamazepine syndrome | Ventricular septal defect, tetralogy of Fallot |
| Valproic acid | Ventricular septal defect, coarctation of the aorta, interrupted aortic arch, tetralogy of Fallot, hypoplastic left heart syndrome, aortic stenosis, atrial septal defect, pulmonary atresia |
| Retinoic acid embryopathy | Conotruncal malformations |
| Thalidomide embyropathy | Conotruncal malformations |
| Maternal phenylketonuria (PKU) (fetal effects) | Tetralogy of Fallot, ventricular septal defect, coarctation of the aorta |
| Maternal systemic lupus erythematosus/Sjögren syndrome (fetal effects) | Complete congenital heart block, dilated cardiomyopathy |
| Fetal rubella syndrome | Patent ductus arteriosus, peripheral pulmonary artery stenosis |
| Maternal diabetes | Hypertrophic cardiomyopathy, conotruncal abnormalities |
Table adapted from Drose J. Fetal echocardiography. 1st ed. Philadelphia: Saunders; 1988.
 KEY POINTS: PHYSIOLOGIC VARIABLES IN FETAL AND PERINATAL LIFE
KEY POINTS: PHYSIOLOGIC VARIABLES IN FETAL AND PERINATAL LIFE



Pulmonary vascular resistance = pulmonary artery pressure/pulmonary blood flow
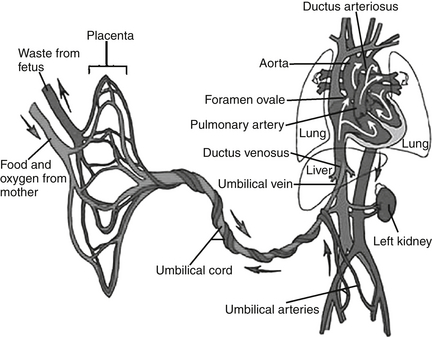
The very high PVR in the fetus results in low pulmonary blood flow, with diversion of the right ventricular blood away from the pulmonary vascular bed and towards the systemic vascular bed through the ductus arteriosus. This dynamic results in increased thickness of the muscular medial layer of the pulmonary arteries. As a newborn’s lungs expand, they inspire oxygen, which is a potent vasodilator that causes the pulmonary vascular resistance to fall. The rise in the PaO2 causes the smooth muscle in the pulmonary circulation to relax, and vasodilation occurs. The PVR falls dramatically during the first week of life. As the smooth muscle of the pulmonary arteries continues to thin, the PVR continues to fall, reaching a nadir at 6 to 8 weeks after delivery. ∗
 KEY POINTS: ECHOCARDIOGRAPHIC ASSESSMENT OF VENTRICULAR FUNCTION AND PULMONARY HYPERTENSION
KEY POINTS: ECHOCARDIOGRAPHIC ASSESSMENT OF VENTRICULAR FUNCTION AND PULMONARY HYPERTENSION14. Name three scenarios in which a right-to-left shunt is seen in the infant with a PDA.



16. How can the modified Bernoulli equation be useful in interpreting pulmonary artery pressures in a neonate?
This formula allows the pulmonary artery pressure to be estimated. Two examples are as follows:
 If the velocity across a tricuspid regurgitant jet is 4 m/sec, the calculated right ventricular pressure will be 64 mmHg [P = 4 × (V max2) which is 4 × (42)= 64 mmHg]. By adding an estimation of right atrial pressure (usually use 5 mmHg), the estimated right ventricular pressure (as well as a pulmonary artery pressure in the absence of pulmonary stenosis) would be 69 mmHg. Therefore this patient has pulmonary hypertension.
If the velocity across a tricuspid regurgitant jet is 4 m/sec, the calculated right ventricular pressure will be 64 mmHg [P = 4 × (V max2) which is 4 × (42)= 64 mmHg]. By adding an estimation of right atrial pressure (usually use 5 mmHg), the estimated right ventricular pressure (as well as a pulmonary artery pressure in the absence of pulmonary stenosis) would be 69 mmHg. Therefore this patient has pulmonary hypertension.

17. What is the definition of the echocardiographic term shortening fraction?
The formula to measure shortening fraction is as follows:

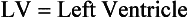
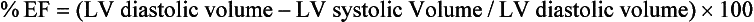
22. During postnatal life, as PVR falls, what changes can be expected in the magnitude of a left-to-right shunt of a large VSD?
With a large-pressure nonrestrictive VSD, the right ventricular and pulmonary artery pressures remain high. The right and left ventricular pressures remain equal with a nonrestrictive VSD. As the pressure across the pulmonary vascular bed will not change and the PVR falls, the pulmonary blood flow (PBF) will increase because there is an inverse relationship between PBF and PVR. PBF = Δ Pressure across the pulmonary vascular bed/PVR. Therefore the magnitude of the left-to-right shunt will increase. ∗†
 KEY POINTS: INNOCENT MURMURS
KEY POINTS: INNOCENT MURMURS23. What are the common innocent murmurs heard in the newborn period?
 Peripheral pulmonary stenosis (PPS): This murmur is a soft, grade 1-2/6 systolic ejection murmur, heard best at the left upper sternal border, with radiation to both axillae and the back. It will usually disappear or be much softer at 6 to 8 weeks after delivery in normal infants and completely disappear by 6 months after delivery.
Peripheral pulmonary stenosis (PPS): This murmur is a soft, grade 1-2/6 systolic ejection murmur, heard best at the left upper sternal border, with radiation to both axillae and the back. It will usually disappear or be much softer at 6 to 8 weeks after delivery in normal infants and completely disappear by 6 months after delivery.

 KEY POINTS: PHYSIOLOGY OF THE CYANOTIC NEONATE
KEY POINTS: PHYSIOLOGY OF THE CYANOTIC NEONATE
1. The hyperoxia test does not rule out some common mixing congenital heart lesions.
2. Reversed differential cyanosis can be seen when the postductal saturations are higher than the preductal saturations. This sign can be seen in d-transposition of the great arteries with a coarctation of the aorta, d-transposition of the great arteries with interrupted aortic arch, and d-transposition of the great arteries with pulmonary hypertension.
28. What is differential cyanosis, and what are the implications (pink upper body and blue lower body)?
The oxygen dissociation curve shows the relationship between oxygen saturation (%) and the partial pressure of oxygen, PO2, in mmHg. This relationship is a sigmoid-shaped curve, with it being fairly flat in the upper range of oxygen saturation (above 85%). Blood pH, temperature, PCO2, 2,3-diphosphoblycerate, and the type of hemoglobin influence the relationship between oxygen saturation and the partial pressure of oxygen ( Fig. 6-2).
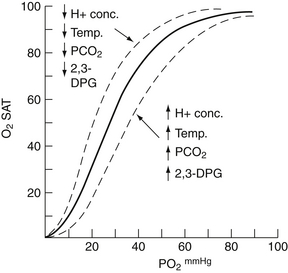
Figure 6-2 Oxygen dissociation curve for normal adult human blood (solid line) and curves showing the effect of either an increase (↑) or a decrease (↓) in hydrogen ion concentration, body temperature, PCO2, and 2,3-DPG level (dotted lines). (From Gessner I, Vitorica B: Pediatric cardiology: a problem-oriented approach. Philadelphia: Saunders; 1993. p. 98.)
32. What is a hyperoxia test, and how is it used in differentiating pulmonary and cardiac causes of cyanosis?
A hyperoxia test attempts to differentiate between pulmonary disease with V/Q mismatch and cyanotic congenital heart disease. Initially, one measures the oxygen saturation in room air. If the oxygen saturation is low, the patient should be placed in 100% FiO2. The patient with pulmonary disease will show an increase in PO2 (to a variable degree). In the patient with a fixed intracardiac mixing lesion, the PO2 does not change significantly. A preductal and postductal arterial blood gas result should be obtained. A preductal arterial blood gas result can be obtained from the right radial artery. A postductal arterial blood gas can be obtained either from an umbilical artery or from a lower extremity artery. In pulmonary disease the preductal arterial PO2 in 100% FiO2 usually exceeds 150 mmHg. If the ductus arteriosus is patent and a right-to-left ductal shunt occurs because of high PVR, the postductal PO2 will be lower than the preductal PO2. In addition, the arterial PCO2 is elevated relative to the patient’s respiratory effort.
33. Which critical cyanotic lesions may not be excluded if the hyperoxia test yields a PO2 after 10 minutes greater than 150 torr?
34. What are the current guidelines for pulse oximetry screening of newborns to detect critical cyanotic congenital heart disease?
As published in Pediatrics in 2011, pulse oximetry assessment of the right hand and a foot is recommended before discharge of all newborns from the hospital. If the oxygen saturation is less than 90% in the right hand or foot, the test is positive and further evaluation is needed. If the oxygen saturation is 95% or greater in the right hand or foot and the difference is 3% or less between the two sites, then the test is negative. If the oxygen saturation is 90% to 95% or greater than 3% difference between the two sites is found, then the test should be repeated in 1 hour up to two times. It is considered a positive screen if these findings are reproduced twice. ∗†‡
 KEY POINTS: CONGESTIVE HEART FAILURE
KEY POINTS: CONGESTIVE HEART FAILUREDuctal-dependent abnormalities:






Non–ductal-dependent abnormalities:
 Total anomalous pulmonary venous return
Total anomalous pulmonary venous return

 AV septal defect (endocardial cushion defect)
AV septal defect (endocardial cushion defect)
 Myocardial dysfunction (cardiomyopathy)
Myocardial dysfunction (cardiomyopathy)





38. What are some heart lesions that can cause heart failure in the infant beyond the newborn period?
Obstruction to systemic blood flow:





 Total anomalous pulmonary venous return, without obstruction
Total anomalous pulmonary venous return, without obstruction
 Single ventricle with excessive PBF
Single ventricle with excessive PBF



Myocardial dysfunction/pericardial disease:
 Anomalous left coronary artery
Anomalous left coronary artery












Compensatory mechanisms include increased heart rate, enhanced stroke volume (Frank–Starling mechanism), sympathetic nerve activation (increased sympathetic tone, renin-angiotensin system activation), increased 2,3-diphosphoglycerate, increased atrial natriuretic peptides, and myocardial hypertrophy. ∗†
 KEY POINTS: TREATMENT OF HYPOTENSION ON THE NEONATE
KEY POINTS: TREATMENT OF HYPOTENSION ON THE NEONATE
1. Dopamine and dobutamine have different physiologic effects in the newborn.
2. Dopamine is a good inotrope to use (depending on dose) if there is a risk of renal ischemia, low cardiac output, or hypotension with decreased systemic vascular resistance.
3. Dobutamine is a good inotrope to use for low cardiac output in patients at risk for myocardial ischemia, pulmonary hypertension, and left ventricular diastolic dysfunction.
42. What is the most frequent primary cause of hypotension in the preterm neonate in the immediate postnatal period?
44. What are the mechanisms of action of the cardiovascular effects of dopamine in the preterm neonate?
45. What are the pros and cons of dopamine and dobutamine in the treatment of hypotension in a preterm neonate?
Dopamine may be the preferred inotrope in the treatment of hypotension secondary to neonatal sepsis because it increases peripheral vascular contractility. However, dobutamine may be preferred in treatment of hypotension secondary to cardiomyopathy, which frequently occurs with perinatal asphyxia. Whereas dopamine has a greater increase in arterial blood pressure than dobutamine, dobutamine has been shown to increase superior vena cava blood flow and may improve end-organ perfusion to a great extent. ∗†‡
 KEY POINTS: CYANOTIC CONGENITAL HEART DISEASE
KEY POINTS: CYANOTIC CONGENITAL HEART DISEASE
1. An unrestrictive atrial communication is crucial in certain cyanotic congenital heart lesions to provide mixing (right-to-left) and/or cardiac output.
2. The most common cyanotic lesion presenting in the newborn period is d-transposition of the great arteries.
3. An electrocardiogram with a left axis shift in a cyanotic newborn baby may be consistent with tricuspid atresia.
4. Cyanotic CHD does not include cyanosis secondary to intrapulmonary right-to-left shunting.
47. What are the five Ts of cyanotic congenital heart disease? What are other principal cyanotic heart lesions that do not begin with a T?








The screening is performed for the following reasons:
 Prenatal screening depicts less than 50% of all CHD.
Prenatal screening depicts less than 50% of all CHD.
 Routine newborn examination misses more than 50% of all infants with CHD.
Routine newborn examination misses more than 50% of all infants with CHD.
 Of CHD-related deaths in the first week of life, 25% of cases do not have a diagnosis of CHD.
Of CHD-related deaths in the first week of life, 25% of cases do not have a diagnosis of CHD.
53. In infants with d-transposition of the great arteries, how does a balloon atrial septostomy improve systemic oxygen saturation?
Progressive hypoxemia from poor intercirculatory mixing is a medical emergency. An atrial septostomy may be necessary to improve hypoxemia even after Prostaglandin E1 (PGE) has maintained ductal patency. The balloon atrial septostomy permits unrestricted bidirectional mixing of fully saturated blood in the left atrium with desaturated blood in the right atrium to achieve a higher net saturation of blood in the systemic circulation. After this procedure, patency of the ductus is generally no longer essential. Variations in oxygen saturation can be expected, although mixing is usually excellent.
57. What are the different types of TAPVR?
 Supracardiac: represents 50% of all total anomalous pulmonary venous return. The pulmonary veins usually drain into the right superior vena cava via a left vertical vein. Although generally nonobstructive, they can be obstructed in some cases (the vertical vein is “pinched” between the left pulmonary artery and the left bronchus, or at superior vena cava insertion).
Supracardiac: represents 50% of all total anomalous pulmonary venous return. The pulmonary veins usually drain into the right superior vena cava via a left vertical vein. Although generally nonobstructive, they can be obstructed in some cases (the vertical vein is “pinched” between the left pulmonary artery and the left bronchus, or at superior vena cava insertion).


 Mixed: represents 10% of all total anomalous pulmonary venous return. It represents a combination of the other types of TAPVR ( Fig. 6-3).
Mixed: represents 10% of all total anomalous pulmonary venous return. It represents a combination of the other types of TAPVR ( Fig. 6-3).
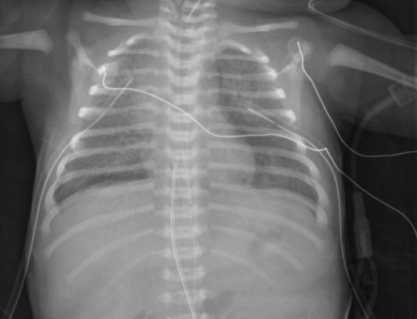
In patients with obstructed TAPVR, cardiac surgery is an emergency and involves anastamosis of the common pulmonary vein to the left atrium. Nonobstructive TAPVR can be managed semi-electively, with the key factor being initial control of CHF.
Infants with HLHS can present within the first few hours to days of life. Clinical symptoms depend on the status of the inter-atrial communication, patency of the ductus, PVR, and potentially the degree of AV valve competency. Common clinical signs are tachypnea, hepatomegaly, pulmonary rales, and a single second heart sound (S2). Cyanosis may be minimal. Closure of the ductus will result in decreased peripheral pulses and a “shock-like” picture ( Fig. 6-4).
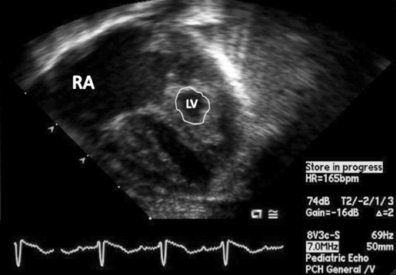
Figure 6-4 Apical four-chamber view of neonate with hypoplastic left heart syndrome. Note the dilated right atrium (RA) and ventricle with severely hypoplastic, non–apex-forming left ventricle (LV; outlined).
67. Under what circumstances can information from an electrocardiogram in a cyanotic neonate help indicate the presence of congenital heart disease?
The key is an abnormal frontal plane axis. A leftward axis for a newborn (i.e., left superior axis −90 to 0 [see Fig. 6-5]) strongly implies a structural anomaly of the heart. Dominant left ventricular forces often accompany this anomaly. The differential diagnosis includes tricuspid atresia, pulmonary valve atresia with intact ventricular septum, critical pulmonary stenosis, or complex single left ventricle.
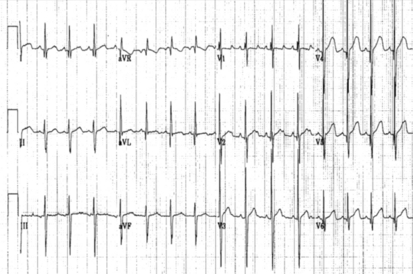
Figure 6-5 12-lead electrocardiogram consistent with left axis deviation. Left axis deviation (frontal QRS plane −45 degrees) may be seen with tricuspid atresia.
68. What are some common chest x-ray findings in infants with cyanotic congenital heart lesions?
 d-transposition of the great arteries with an intact ventricular septum: Cardiomegaly with increased pulmonary vascular markings. Egg-shaped cardiac silhouette, with a narrow, superior mediastinum.
d-transposition of the great arteries with an intact ventricular septum: Cardiomegaly with increased pulmonary vascular markings. Egg-shaped cardiac silhouette, with a narrow, superior mediastinum.
 Tetralogy of Fallot : Normal heart size or smaller than normal. Decreased pulmonary vascular markings, concave main pulmonary artery segment with an upturned apex (boot-shaped heart). This can have a right-sided aortic arch.
Tetralogy of Fallot : Normal heart size or smaller than normal. Decreased pulmonary vascular markings, concave main pulmonary artery segment with an upturned apex (boot-shaped heart). This can have a right-sided aortic arch.



 Ebstein anomaly of the tricuspid valve: In severe cases the heart is enormous, balloon shaped, and occupies almost the entire cardiothoracic area ( Figs. 6-6 and 6-7).
Ebstein anomaly of the tricuspid valve: In severe cases the heart is enormous, balloon shaped, and occupies almost the entire cardiothoracic area ( Figs. 6-6 and 6-7).
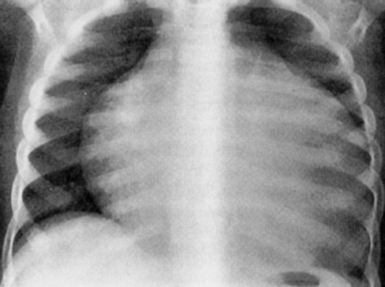
Figure 6-6 AP chest radiograph revealing significant cardiomegaly (“wall-to-wall heart”) consistent with Ebstein’s anomaly of the tricuspid valve.
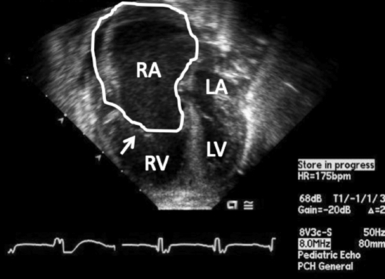
Figure 6-7 Ebstein’s malformation of the tricuspid valve. Four-chamber echocardiogram demonstrating an inferiorly displaced tricuspid valve (arrow). The right atrium (RA) is enlarged (outlined). The tricuspid valve is well below the level of the mitral valve. LA, left atrium; LV, left ventricle; RV, right ventricle.








 KEY POINTS: ACYANOTIC AND OBSTRUCTIVE CONGENITAL HEART LESIONS
KEY POINTS: ACYANOTIC AND OBSTRUCTIVE CONGENITAL HEART LESIONS
1. Neonates with a large left-to-right shunt typically develop CHF at 3 to 6 weeks of age, when the PVR drops and the left-to-right shunting increases with increased flow into the pulmonary arteries. This increase in PBF will result in increased flow across the mitral valve and enlarge the left ventricle.









78. If the apex of the heart is on the opposite side of the patient from the stomach, what is the likelihood that the patient has congenital heart disease?
79. What are the different types of interrupted aortic arch?



VSD and PDA are commonly present.
DiGeorge syndrome (22q11 Deletion Syndrome) is a constellation of clinical symptoms that includes a lack of thymus gland and a lack of parathyroid (hypocalcemia), with certain common conotruncal heart defects. A similar syndrome (velocardiofacial syndrome) may have associated midline facial defects (i.e., cleft palate) with a lower proclivity for thymic deficiency. ∗†‡
 KEY POINTS: VASCULAR RINGS
KEY POINTS: VASCULAR RINGSA complete vascular ring is formed by abnormal vascular structures completely encircling the trachea and esophagus (e.g., double aortic arch and a right aortic arch with a left-sided ligamentum arteriosum). An incomplete vascular ring occurs when there is vascular compression of the trachea and esophagus without completely encircling these structures (e.g., innominate artery compression and pulmonary sling).
Most infants with vascular rings present with symptoms within the first several weeks to months of life, with a double aortic arch and pulmonary sling being symptomatic earlier than the other rings. Infants may hold their heads hyperextended to alleviate the symptoms of airway obstruction. Symptoms of respiratory distress, stridor, “seal bark” cough, apnea, dysphagia, and recurrent respiratory infections may occur. Dysphagia may first be detected when infants transition from liquid formula to solid food ( Fig. 6-8).
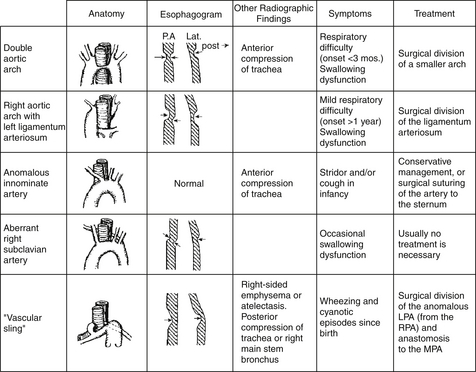
Figure 6-8 Different types of vascular rings. LPA, Left pulmonary artery; RPA, right pulmonary artery; MPA, main pulmonary artery. (From Park MK: Pediatric cardiology for practitioners. 3rd ed. St Louis: Mosby; 1996. p. 246.)
87. How does a surgeon repair some of the more common forms of vascular rings?
 Double aortic arch: A thoracotomy is performed on the side opposite the dominant aortic arch (typically a left thoracotomy). The lesser arch is clamped, divided, and oversewn. The ligamentum arteriosum is always ligated and divided. Dissection around the esophagus and trachea to lyse any residual adhesive bands is performed.
Double aortic arch: A thoracotomy is performed on the side opposite the dominant aortic arch (typically a left thoracotomy). The lesser arch is clamped, divided, and oversewn. The ligamentum arteriosum is always ligated and divided. Dissection around the esophagus and trachea to lyse any residual adhesive bands is performed.

 Pulmonary artery sling: Transection of the left pulmonary artery with translocation of the left pulmonary artery anterior to the trachea to its normal position arising from the main pulmonary artery. A barium swallow will reveal an anterior indentation of the esophagous on the lateral projection. ∗†‡
Pulmonary artery sling: Transection of the left pulmonary artery with translocation of the left pulmonary artery anterior to the trachea to its normal position arising from the main pulmonary artery. A barium swallow will reveal an anterior indentation of the esophagous on the lateral projection. ∗†‡
 KEY POINTS: SEGMENTAL CARDIAC AND CARDIAC MALPOSITION ANALYSIS
KEY POINTS: SEGMENTAL CARDIAC AND CARDIAC MALPOSITION ANALYSIS
1. When using a chest x-ray to evaluate a patient with congenital heart disease, identification of the stomach bubble and liver shadow will help assess abdominal situs.
2. Although electrocardiography may define the location of the sinoatrial node and ventricular mass, echocardiography is the diagnostic test of choice to define the cardiac segments.
 Atrial sidedness: S (solitus), I (inversus), and A (ambiguus)
Atrial sidedness: S (solitus), I (inversus), and A (ambiguus)
 Ventricular looping: D loop, L loop, and X (indeterminate loop)
Ventricular looping: D loop, L loop, and X (indeterminate loop)

Normal cardiac segmental anatomy is {S, D, S}.
Because depolarization occurs in the left ventricle before the right ventricle, the presence/location of the Q waves over the precordium can assist in the anatomic location of the left ventricle and right ventricle. If Q waves are seen in V5 and V6, and lead 1, the left ventricle is D-looped and on the left side. If Q waves are seen in V4R, V1 and V2, but not seen in V5 and V6, it is likely that the ventricles are L-looped (Figs. 6-9 and 6-10).
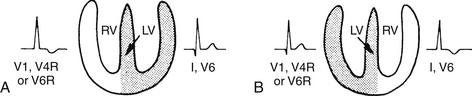
Figure 6-9 Locating the ventricles from the electrocardiogram. The left ventricle (LV) is usually located on the same side as the precordial leads that show Q waves. If V6 shows a Q wave, the LV is on the left side. If V4R and V1 show a Q wave, the LV is to the right of the anatomic right ventricle (RV). Note that SQ waves are also present in V1 in severe right ventricular hypertrophy (RVH). (From Park MK, Guntheroth WG. How to read: pediatric ECGs. 3rd ed. St Louis: Mosby; 1992. p. 253.)
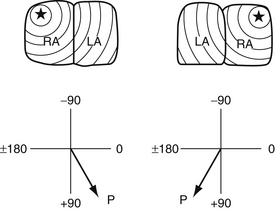
Figure 6-10 Locating the atria by the use of the P axis. When the right atrium (RA) is on the right side, the P axis is in the left lower quadrant (0 to 90 degrees). When the RA is on the left side, the P axis is in the right lower quadrant (90 to −180 degrees). LA, Left atrium. (From Park MK, Guntheroth WG. How to read: pediatric ECGs, 3rd ed. St Louis: Mosby; 1992. p. 252.)
The atrial sidedness follows the visceral organ arrangement.
 If abdominal situs is present (liver on the right and stomach on the left), then the right atrium is on the right.
If abdominal situs is present (liver on the right and stomach on the left), then the right atrium is on the right.


The word heterotaxy means “different arrangement.” There is abnormal relationship and arrangement of the cardiac atria and the thoracoabdominal organs, including the spleen, lungs and intestines. Polysplenia (associated with left atrial isomerism) or asplenia (right atrial isomerism) is present. These patients are at high risk for malrotation of the intestines, which should be investigated with ultrasound or barium study. The cardiac malformations are complex typically, with abnormal venoatrial connections ( Fig. 6-11).
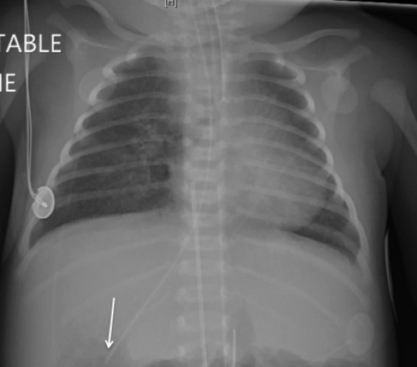
Figure 6-11 AP chest radiograph in a neonate with complex heart disease with heterotaxy. Note the stomach bubble (NG tube; arrow) is on the right side. The umbilical arterial catheter and aortic knob likely confirm a right-sided aortic arch. There is mild interstitial edema.
Mesocardia occurs when the heart occupies the midline of the thorax. ∗†‡
 KEY POINTS: FEEDING NEONATES WITH CHD
KEY POINTS: FEEDING NEONATES WITH CHD98. What is the most common reason that enteral feedings are withheld from neonates with ductal-dependent CHD?
The most commonly reported reason for exercising caution when feeding neonates with ductal-dependent CHD is the theoretical risk of intestinal hypoperfusion, which may lead to necrotizing enterocolitis. The two most common findings seen in neonates with CHD and necrotizing enterocolits are widened pulse pressure and low diastolic blood pressure, which are seen in patients with retrograde diastolic flow in the descending aorta. ∗†‡
Cardiac Surgery
The modified Blalock–Taussig shunt is a Gore-Tex interposition shunt placed between the subclavian artery (right or left) and the right or left pulmonary artery. It is used for congenital heart lesions that require increased PBF in the neonatal and infancy periods. Examples include lesions with a hypoplastic pulmonary annulus, atretic pulmonary valve annulus, or severely hypoplastic main and branch pulmonary arteries. The cardiac malformations in such instances would include severe tetralogy of Fallot, tetralogy of Fallot with pulmonary atresia, tricuspid atresia, pulmonary atresia with VSD, and pulmonary atresia with intact ventricular septum. A modified Blalock–Taussig shunt is used in the first stage of HLHS repair (modified Norwood operation) ( Fig. 6-12).
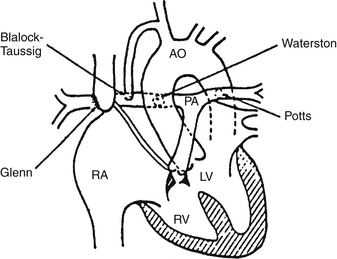
Figure 6-12 Major shunt operations for congenital heart disease. AO, Aorta; PA, pulmonary artery; RA, right atrium; LV, left ventricle; RV, right ventricle. (From Park MK: Pediatric cardiology for practitioners. 3rd ed. St Louis: Mosby; 1996.)











106. What is the Norwood procedure (or modified Norwood, stage I), and what are the two additional procedures for neonates with single-ventricle physiology?
The modified Norwood procedure is performed as follows:
 A. Stage 1: Anastomosis of the proximal main pulmonary artery to the aorta, with aortic arch reconstruction and trans-section and patch closure of the distal main pulmonary artery; a modified right Blalock–Taussig shunt (right subclavian artery to right pulmonary artery) to provide PBF, and lastly, atrial septectomy to allow for adequate left-to-right atrial communication.
A. Stage 1: Anastomosis of the proximal main pulmonary artery to the aorta, with aortic arch reconstruction and trans-section and patch closure of the distal main pulmonary artery; a modified right Blalock–Taussig shunt (right subclavian artery to right pulmonary artery) to provide PBF, and lastly, atrial septectomy to allow for adequate left-to-right atrial communication.

 B. Stage 2: Bidirectional Glenn anastomosis (approximately 4 to 6 months)
B. Stage 2: Bidirectional Glenn anastomosis (approximately 4 to 6 months)
 C. Stage 3: Completion of a Fontan operation (approximately 1 to 2 years)
C. Stage 3: Completion of a Fontan operation (approximately 1 to 2 years)
Junctional ectopic tachycardia (JET) is a common arrhythmia after surgery. The rhythm generally occurs in the first 24 to 48 hours following surgery. Typical electrocardiographic findings include AV dissociation and narrow QRS morphology with a rapid QRS rate (170 to 210 bpm). Treatment for JET involves slowing the ectopic rate to allow restoration of AV synchrony. Core temperature cooling (i.e., 35 to 36° C) has been effective. Amiodarone has also been used to slow the junctional rate so as to allow temporary pacing to restore AV synchrony. Typically, as the infant recovers from surgery with less need for intravenous inotropic support, the JET resolves.
Extracorporeal membrane organization (ECMO) can provide a means to assist the heart and lungs (or both) temporarily. Venoarterial ECMO is used in neonatal patients with either life-threatening pulmonary disease (congenital diaphragmatic hernia, or persistent fetal circulation) or neonates with postoperative ventricular failure. ∗†‡§
 KEY POINTS: PREOPERATIVE STABILIZATION
KEY POINTS: PREOPERATIVE STABILIZATION
1. There are a variety of presentations in an infant with congenital heart disease.
2. The starting dose of prostaglandin E1 (PGE1) is 0.05 to 0.10 μg/kg/min.
3. To maintain patency of the ductus arteriosus, prostaglandin E1 (PGE1) is used in both right- and left-sided heart obstructive lesions; often the dose can be lowered to 0.01 to 0.02 μg/kg/min.
111. What are the principles of preoperative management in the neonate with critical congenital heart disease?
 Initial stabilization: airway management, vascular access, and maintaining patency of a ductus arteriosus with PGE1. Delineation of the anatomic defect should be performed by echocardiography.
Initial stabilization: airway management, vascular access, and maintaining patency of a ductus arteriosus with PGE1. Delineation of the anatomic defect should be performed by echocardiography.

 Evaluation of other congenital heart defects
Evaluation of other congenital heart defects

 Cardiac catheterization if indicated
Cardiac catheterization if indicated
 Surgical management if indicated, when all other systems are optimized
Surgical management if indicated, when all other systems are optimized
113. What are the neonatal presentations of CHD?









Side effects of PGE1 include the following:





Side effects may be more complex in infants weighing less than 2 kg.
116. In what lesions does keeping the ductus open help improve PBF, and in what lesions does keeping the ductus open help improve cardiac output?
Neonates with sepsis will demonstrate similar clinical findings to those of neonates with ductal-dependent systemic CHD. Antibiotic delivery will have little impact in this cohort. Patency of the ductus is critical in the group with CHD to maintain coronary perfusion and prevent myocardial ischemia. ∗†
 KEY POINTS: POSTOPERATIVE MANAGEMENT
KEY POINTS: POSTOPERATIVE MANAGEMENT120. What information is needed to care for the neonate with congenital heart disease after cardiothoracic surgery?
 The underlying anatomic defect and the expectations of surgical repair
The underlying anatomic defect and the expectations of surgical repair
 The clinical/physiologic status of the infant in the preoperative period
The clinical/physiologic status of the infant in the preoperative period
 The anesthetic regimen used during surgery
The anesthetic regimen used during surgery
 The duration of the cardiopulmonary bypass, aortic cross clamp time, and circulatory arrest time
The duration of the cardiopulmonary bypass, aortic cross clamp time, and circulatory arrest time








121. What is the purpose of the postoperative echocardiogram?
The postoperative echocardiogram, most commonly transesophageal, should assess for the following:

 Ventricular function (both right ventricular and left ventricular)
Ventricular function (both right ventricular and left ventricular)
 Adequacy of the semilunar and AV valves
Adequacy of the semilunar and AV valves

 General assessment of the preload (volume) of the ventricular chambers
General assessment of the preload (volume) of the ventricular chambers
122. What are the most common noncardiac causes of respiratory compromise after cardiothoracic surgery?
TABLE 6-3
NONCARDIAC CAUSES OF RESPIRATORY COMPROMISE AFTER CARDIOTHORACIC SURGERY 2
| Central nervous system | Neuromuscular |
| 1. General anesthesia | 1. Residual neuromuscular blockade |
| 2. Administration of analgesics or sedative/hypnotics | 2. Respiratory muscle weakness—from disuse and/or malnutrition |
| 3. Hypoxic-ischemic encephalopathy | Alveolar disease |
| 4. Apnea of prematurity | 1. Acute lung injury from cardiopulmonary bypass |
| Isolated neuropathies 1. Hemidiaphragmatic paresis or paralysis – phrenic nerve injury |
2. Increased lung fluid from left-to-right shunt lesions 3. Atelectasis |
| 2. Vocal cord paralysis – recurrent | 4. Pneumonia |
| Airway abnormalities proximal to the alveoli | 5. Pulmonary hemorrhage |
| 1. Tracheostomy or endotracheal tube obstruction | 6. Pulmonary hypoplasia |
| 2. Post-extubation subglottic edema | Extrinsic lung compression |
| 3. Laryngotracheomalacia | 1. Pleural effusion (transudate vs. exudate) |
| 4. Left mainstem bronchomalacia from long-standing left atrial or left pulmonary artery enlargement | 2. Pneumothorax, hemothorax, chylothorax Chest Wall 1. Midsternal |
| 2. Thoracotomy | |
| 3. Clam-shell incisions |
(Adapted from Newth CJL, Hammer J. Pulmonary issues. In: Chang AC, Hanley FL, Wernovsky G, Wessel DL, editors. Pediatric cardiac intensive care. Baltimore: Williams & Wilkins; 1998. p 352.)
Procedures associated with phrenic nerve injury:









Chylothorax is a rare complication of cardiac surgery with an incidence of approximately 0.85% to 3.8%. Chylothorax may occur as a direct injury to the thoracic duct in surgeries such as coarctation of the aorta repair or PDA ligation. Second, it may occur with thrombosis of the superior vena cava, leading to increased hydrostatic pressure in the superior vena cava and thoracic duct. Third, high central venous pressures after a surgery such as a Glenn palliation for HLHS may cause chylothorax. Most common surgeries that are complicated by chylothoraces are tetralogy of Fallot, Glenn and Fontan palliation, and orthotopic heart transplantation.














129. What consideration should be given to the neonate after surgery for oral or nasogastric feeding?
Although there is no correct answer, the clinician should consider whether the infant was fed before surgery, length of bypass and circulatory arrest times, adequacy of the cardiac output, presence of bowel sounds, minimization of vasoconstrictive agents, and the absence of lactic acidosis. Local systemic monitoring with NIRS may be helpful. ∗†‡§
Cardiac Transplantation
134. Are neonates at an advantage compared with older children with regard to heart transplantation?
Some evidence suggests that infants who receive transplants when younger than 6 months of age have improved survival 10 years after the transplant compared with older children. Newborn infants do not have a mature complement system and do not produce a typical isohemagglutinin response to blood groups. As such, it is possible to perform ABO-incompatible heart transplantation during infancy. ∗†‡§
 KEY POINTS: INTERVENTIONAL CARDIOLOGY
KEY POINTS: INTERVENTIONAL CARDIOLOGY135. Which kinds of congenital heart disease may benefit from a cardiac catheterization interventional procedure?
TABLE 6-4
CONGENITAL HEART DISEASES THAT MAY BENEFIT FROM CARDIAC CATHETERIZATION INTERVENTIONAL PROCEDURES
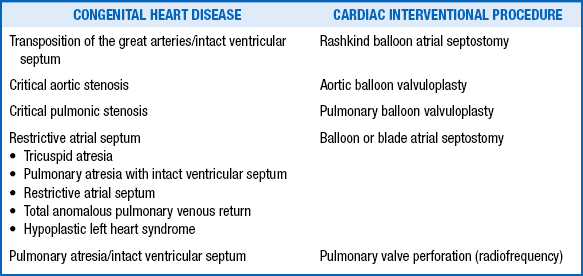
136. Which anomaly of the systemic veins may prevent access to the right side of the heart from the femoral veins?
Although originally developed using fluoroscopy, a balloon atrial septostomy may be performed at the bedside using transthoracic echocardiography guidance. ∗†
 KEY POINTS: INTERPRETING NORMAL AND ABNORMAL ELECTROCARDIOGRAMS IN THE NEWBORN PERIOD
KEY POINTS: INTERPRETING NORMAL AND ABNORMAL ELECTROCARDIOGRAMS IN THE NEWBORN PERIOD Right axis deviation +55-200 degrees (premature infants are generally 65-174 degrees)
Right axis deviation +55-200 degrees (premature infants are generally 65-174 degrees)
 Right ventricular dominance, tall R wave in V1, and deep S wave in V6
Right ventricular dominance, tall R wave in V1, and deep S wave in V6
 Positive T wave in the right precordial leads V3r, V4r, and V1
Positive T wave in the right precordial leads V3r, V4r, and V1
 Longer QTc interval up to 0.46 sec
Longer QTc interval up to 0.46 sec
 QRS duration less than 80 msec
QRS duration less than 80 msec
 Right ventricular forces decrease over the first 6 months of life.
Right ventricular forces decrease over the first 6 months of life.
143. What are the electrocardiographic signs of abnormal right ventricular hypertrophy in a newborn?




145. What are the electrocardiographic abnormalities observed in infants with hyperkalemia?



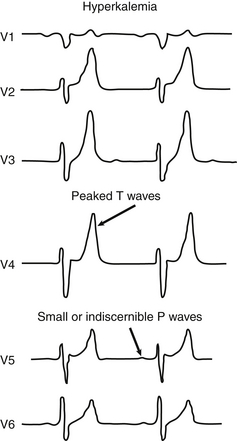



Hypomagnesemia may mimic hypokalemia.
Prolongation of the QT interval may be evident.
149. What electrocardiographic changes are associated with digitalis?


A delta wave is a slurring in the upstroke of the QRS complex; it generally occurs in association with a short PR interval. The delta wave signifies that some or all of the atrial depolarization to the ventricle is by way of a bypass tract rather than solely antegrade through the AV node. This finding is associated with a Wolff–Parkinson–White pattern on the electrocardiogram. Patients with a Wolff–Parkinson–White pattern may develop SVT (also called Wolff–Parkinson–White syndrome) ( Fig. 6-14).
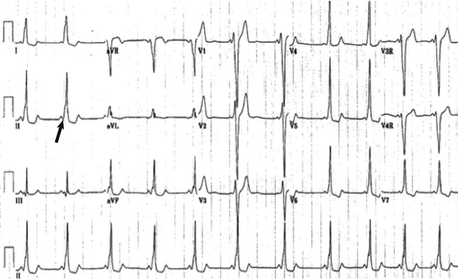
Figure 6-14 12-lead electrocardiogram and lead II rhythm strip. Sinus rhythm with ventricular preexcitation (Wolff–Parkinson–White) pattern (“delta wave” arrow). Notice the very short PR interval.
Anatomic substrate results in a direct electrical communication between the atria and the ventricles rather than through the AV node. Conduction becomes a fusion between AV nodal conduction and antegrade conduction via the bypass tract. Patients with an electrocardiogram having a Wolff–Parkinson–White pattern may have SVT and be considered as having Wolff–Parkinson–White syndrome.
152. Are there certain congenital heart lesions that may be seen with Wolff–Parkinson–White syndrome?










 KEY POINTS: ARRHYTHMIAS
KEY POINTS: ARRHYTHMIAS
1. Maternal connective tissue diseases (systemic lupus erythematosus and Sjögren syndrome) can cause complete congenital heart block or dilated cardiomyopathy (or both) in the fetus and infant.
2. Atrial premature contractions are benign arrhythmias. On rare occasions and if sufficiently frequent, they can trigger SVT.
155. What are congenital anatomic and nonanatomic causes of congenital complete heart block in the fetus?
Congenital complete heart block in the fetus can occur as a result of structural congenital heart disease (L-TGA, left atrial isomerism, or maternal collagen vascular disease). Heart block occurs in women with a variety of connective tissue diseases (e.g., systemic lupus erythematosus, Sjögren syndrome). The incidence is 1/15,000 to 1/20,000 infants. Anti-RO (SSA) and La (SSB) antibodies are found in many of these women, but only a minority of fetuses are affected. Most cases are identified between 18 and 24 weeks’ gestation ( Fig. 6-16).

Figure 6-16 Rhythm strip (lead II) revealing complete heart block. The P waves (arrow) are more frequent than the QRS complex (broken arrow). The atria and ventricle are dissociated with no relationship. The QRS is narrow implying an escape rhythm above the ventricle.
The most common benign arrhythmia in the fetus is a premature atrial contraction. This benign dysrhythmia is commonly detected during fetal monitoring. Blocked premature atrial contractions often cause what appears to be a pause on the monitoring strips, and they occur when the ventricle is refractory and not conducted ( Fig. 6-17).
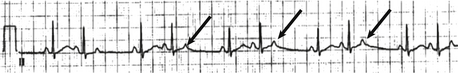
All types of tachyarrhythmias and bradyarrhythmias can be detected. Premature beats account for 80% to 90% of fetal arrhythmias but are generally benign. Re-entrant supraventricular tachyarrhythmias account for 5% of fetal arrhythmias, complete heart block 2.5%, and atrial flutter 1% to 2%. Ventricular arrhythmias are rare ( Fig. 6-18).
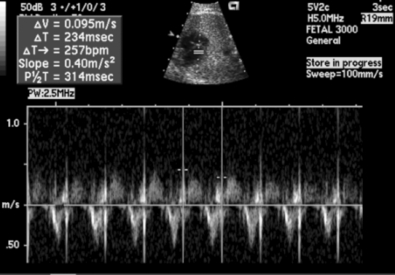
Figure 6-18 Spectral Doppler tracing of left ventricular outflow tract and the mitral valve inflow of a fetus in supraventricular tachycardia (SVT). There is a 1:1 atrioventricular relationship at a heart rate of 257 beats per minute.
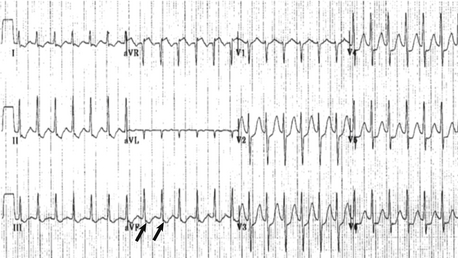
Antiarrhythmic preference is often based on the severity of the symptoms. For straightforward SVT that terminated with intravenous adenosine, beta blockers tend to be relatively safe and effective. In infants with beta-blocker refractory SVT, alternative drugs to consider are flecainide, sotalol, and amiodarone. All three of these drugs have potential pro-arrhythmic side effects and institution of such antiarrhythmics should occur in a hospital (monitor/telemetry) setting. In general flecainide should not be administered to patients with significant structural heart disease. The QTc should be closely followed in patients on either sotalol or amiodarone ( Fig. 6-19).




Cor Pulmonale
Cor pulmonale may result from any chronic pathology that causes PVR to remain elevated after birth.













The chronic management of cor pulmonale depends on the underlying etiology. Factors that worsen PVR, such as hypoxia and acidosis, should be avoided. Intralipid administration as part of hyperalimentation may also increase pulmonary artery pressure and should be used only with great caution. Diuretics are commonly used in infants with BPD, although the evidence for their long-term benefit is not well established. A variety of pulmonary vasodilators are available with different actions and side effect profiles. ∗
 KEY POINTS: ENDOCARDITIS
KEY POINTS: ENDOCARDITISEarly signs of endocarditis in neonates may be very subtle; heart murmurs, skin abscesses, and hepatomegaly are the most common signs found in neonatal patients. The findings that one sees in children with subacute bacterial endocarditis—splenomegaly, petechiae, and splinter hemorrhages—are not usually seen in the newborn infant. ∗
 KEY POINTS: CARDIOMYOPATHY IN THE NEONATE
KEY POINTS: CARDIOMYOPATHY IN THE NEONATE171. What are the three types of cardiomyopathy in the neonate and the echocardiographic finding with each type?
Table 6-5 lists these three types of cardiomyopathy.
TABLE 6-5
ECHOCARDIOGRAPHIC FINDINGS IN THE THREE TYPES OF NEONATAL CARDIOMYOPATHY
| TYPE OF CARDIOMYOPATHY | FUNCTION SEEN ON ECHO | OTHER ECHO FINDINGS |
| Dilated cardiomyopathy | Globular left ventricle Globular and poorly contracting right ventricular size, and contractility possibly normal or similarly depressed |
Endocardial fibroelastosis |
| Hypertrophic cardiomyopathy | Marked left or biventricular hypertrophy with normal/hyperdynamic systolic function | +/− Presence of asymmetric septal hypertrophy (left ventricle) The left ventricular cavity smaller than normal Ventricular filling impaired by diastolic relaxation abnormalities |
| Restrictive cardiomyopathy | Normal ventricular size and contractility | Abnormal diastolic filling Markedly decreased ventricular compliance |
172. Which metabolic disease is associated with hypertrophic cardiomyopathy, short PR interval, and huge QRS voltage?
Pompe disease (glycogen storage disease type II or acid maltase deficiency).
CHF, feeding intolerance, tachypnea, tachycardia, arrhythmias, and sudden cardiac death.
The natural history of DCM in infants and children relates to the diverse nature of the disorder and the age at presentation. For all children the median age at diagnosis is 1.5 years with a 1-year and 5-year survival rate at 87% and 77%, respectively. Transplantation is generally not offered for patients with neuromuscular disorders or inborn errors of metabolism.∗†‡§
Cardiac Tumors




Bradycardia, SVT, Wolff–Parkinson–White syndrome, and ventricular tachycardia.



∗Neill CA, Clark EB. The developing heart: a history of pediatric cardiology. London: Kluwer Academic Associates; 1995.
†Bailey LL. Origins of neonatal heart transplantation: an historical perspective. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu 2011;14(1):98–100.
∗Rychik J, Ayres N, Cuneo B, et al. American Society of Echocardiography guidelines and standards for performance of the fetal echocardiogram. J Am Soc Echocardiogr 2004;17:803–10.
†Drose J. Fetal echocardiography. 1st ed. Philadelphia: Saunders; 1988.
‡Gelb B. Genetic basis of congenital heart disease. Curr Opin Cardiol 2004;19:110–115.
∗Park MK. Pediatric cardiology for the practitioners. 3rd ed. St Louis: Mosby; 1996.
∗Snider AR, Serwer GA, Ritter SB. Echocardiography in pediatric heart disease. 2nd ed. St Louis: Mosby; 1997.
†Lai WW, Mertens LL, Cohen MS, et al. Echocardiography in pediatric and congenital heart disease from fetus to adult. West Sussex: Wiley-Blackwell; 2009.
∗Rudolph A. Congenital diseases of the heart: clinical-physiological considerations. Armonk, New York: Futura; 2001. p. 81–5.
†Yap SH, Anania N, Alboliras ET, et al. Reversed differential cyanosis in the newborn: a clinical finding in the supracardiac total anomalous pulmonary venous connection. Pediatr Cardiol 2009;30:359–362.
†Kemper AR, Mahle WT, Martin GR, et al. Strategies for implementing screening for critical congenital heart disease. Pediatrics 2011;128:1259–67.
∗Silberbach M, Hannon D. Presentation of congenital heart disease in the neonate and young infant. Pediatr Rev 2007;28(4):123–31.
†Anderson RH, Baker EJ, Penny DJ, et al. Paediatric cardiology. 3rd ed. Philadelphia: Churchill Livingstone Elsevier; 2010.
∗Nichols DG, Cameron DE, Greeley WJ, et al. Critical heart disease in infants and children. 1st ed. St Louis: Mosby; 1995.
†Short BL, Meurs KV, Evans JR, et al. Summary Proceedings from the Cardiology Group on Cardiovascular Instability in Preterm Infants. Pediatrics 2006;117:S34–39.
†Engle WD, LeFlore JL. Hypotension in the neonate. Neoreviews 2002;3:e137.
∗Silberbach M, Hannon D. Presentation of congenital heart disease in the neonate and young infant. Pediatr Rev 2007;28(4):123–31.
†Rao PS. Diagnosis and management of cyanotic congenital heart disease: Part I. Indian J Pediatr 2009;76(1):57–70.
†Rao PS. Diagnosis and management of cyanotic congenital heart disease: Part II. Indian J Pediatr 2009;76(3):297–308
∗Anderson RH, Baker EJ, Penny DJ, et al. Paediatric cardiology. 3rd ed. Philadelphia: Churchill Livingstone Elsevier; 2010.
†Rosenthal E. Coarctation of the aorta from fetus to adult: curable condition or life-long process? Heart 2005;91:1495–1502.
†Greenberg F. DiGeorge syndrome: an historical review of clinical and cytogenic features. J Med Genet 1993;30(3):803–806.
∗Moes CAF, Freedom RM. Rings, slings and other things: vascular structures contributing to a neonatal noose. In: Freedom RM, Benson LN, Smallhorn JF, editors. Neonatal heart disease. New York: Springer-Verlag; 1992. p.731–49.
†Woods RK, Sharp RJ, Holcomb GW 3rd, et al. Vascular anomalies and tracheoesophageal compression: a single institution’s 25 year experience. Ann Thorac Surg 2001;72(2):434–8 [discussion 438–9].
†Russell HM, Backer CL. Pediatric thoracic problems: patent ductus arteriosus, vascular rings, congenital tracheal stenosis, and pectus deformities. Surg Clin N Am 2010;90:1091–1113.
∗Van Praagh R, Weinberg PM, Foran RB, et al. Malposition of the heart. In: Adams FH, Emmanoulides GC, RT Reimenschneider TH, editors. Moss’ heart disease in infants, children and adolescents. 5th ed. Baltimore: Williams and Wilkins; 1994.
†Jacobs JP, Anderson RH, Weinberg PM, et al. The nomenclature, definition and classification of cardiac structure in the setting of heterotaxy. Cardiol Young 2007;17(Suppl. 2):1–28.
†Cohen MS, Anderson RH, Cohen MI, et al. Controversies, genetics, diagnostic assessment, and outcomes relating to the heterotaxy syndrome. Cardiol Young 2007;17(2):29–43.
∗Howley LW, Kaufman J, Wymore E, et al. Enteral feeding in neonates with prostaglandin-dependent congenital cardiac disease: international survey on current trends and variations in practice. Cardiol Young 2012;22:121–127.
†Medoff-Cooper B, Naim M, Torowicz D, et al. Feeding, growth, and nutrition in children with congenitally malformed hearts. Cardiol Young 2010;20(Suppl. 3):149–153.
†Kogon BE, Ramaswamy V, Todd K, et al. Feeding difficulty in newborns following congenital heart surgery. Congenit Heart Dis 2007;2:332–37.
∗Chang AC, Hanley FL, Wernovsky G, et al. Pediatric cardiac intensive care. Baltimore: Williams & Wilkins; 1998.
†Anderson RH, Baker EJ, Penny DJ, et al. Paediatric cardiology. 3rd ed. Philadelphia: Churchill Livingstone Elsevier; 2010.
†Gazit AZ, Huddleston CB, Checcia PA, et al. Care of the pediatric cardiac surgery patient part I. Curr Probl Surg 2010;47:185–250.
§Gazit AZ, Huddleston CB, Checcia PA, et al. Care of the pediatric cardiac surgery patient part I. Curr Probl Surg 2010;47:261–376.
∗Chang AC, Hanley FL, Wernovsky G, et al. Pediatric cardiac intensive care. Baltimore: Williams & Wilkins; 1998.
†Brooks PA, Penny DJ. Management of the sick neonate with suspected heart disease. Early Hum Dev 2008;84:155–159.
∗Adapted from Marino BS, Wernovsky G. Preoperative and postoperative care of the infant with critical congenital heart disease. In: Avery GB, Fletcher MA, MacDonald MG, editors. Neonatology: athophysiology and management of the newborn. Philadelphia: Lippincott Williams and Wilkins; 1999. p. 664.
∗Marino BS, Wernovsky G. Preoperative and postoperative care of the infant with critical congenital heart disease. In: Avery GB, Fletcher MA, MacDonald MG, editors. Neonatology: pathophysiology and management of the newborn. Philadelphia: Lippincott: Williams and Wilkins; 1999. p. 664.
†Newth CJL, Hammer J. Pulmonary issues. In: Chang AC, Hanley FL, Wernovsky G, Wessel DL, editors. Pediatric cardiac intensive care. Baltimore: Williams and Wilkins; 1998. p. 352.
†Ricci Z, Garisto C, Favia I, et al. Cerebral NIRS as a marker of superior vena cava oxygen saturation in neonate with congenital heart disease. Paediatr Anaesth 2010;20:1040–45.
§Hirsch JC, Charpie JR, Ohye R, et al. Near-infrared spectroscopy: what we know and we need to know—a systematic review of congenital heart disease literature. J Thorac Cardiovasc Surg 2009;137:154–59.
∗Canter CE, Shaddy RE, Bernstein D, et al. Indications for heart transplantation in pediatric heart disease. Circulation 2007;115:658–676.
†Anderson RH, Baker EJ, Penny DJ, et al. Paediatric cardiology. 3rd ed. Philadelphia: Churchill Livingstone Elsevier; 2010.
†Bernstein D. Heart transplantation in neonates: achievements, challenges, and controversies for the future. Neo Rev 2000;8:e152–8.
§Saczkowski R, Dacey C, Bernier P. Does ABO-incompatible and ABO-compatible neonatal heart transplant have equivalent survival? Interact Cardiovasc Thorac Surg 2010;10:1026–33.
∗Kutty S, Zahn E. Interventional therapy for neonates with critical congenital heart disease. Catheterization and cardiovascular interventions. 2008;72:663–74.
†Freedom RM, Mawson JB, Yoo SJ, et al. Congenital heart disease: textbook of angiography. Armonk, NY: Futura; 1997.
∗Schwartz PJ, Garson A Jr., Paul T, et al. Guidelines for the interpretation of the neonatal electrocardiogram. Eur Heart J. 2002;23:1329–44.
†Taeusch HW, Ballard RA, Gleason CA. Avery’s disease of the newborn. 8th ed. Philadelphia: Elsevier Saunders; 2005.
∗Fischbach PS, Frias PA, Strieper MJ, et al. Natural history and current therapy for complete heart block in children and patients with congenital heart disease. Congenit Heart Dis 2007;4:224–234.
†Kothari DS, Skinner JR. Neonatal tachycardias: an update. Arch Dis Child Fetal Neonatol 2006;91:F136–F144.
†Perry JC, Garson A. Supraventricular tachycardia due to Wolff-Parkinson-White syndrome I children: early disappearance and late recurrence. J Am Coll Cardiol 1991;16:1215–20.
§Texter KM, Kertesz NJ, Friedman RA, et al. Atrial flutter in infants. J Am Coll Cardiol 2006;48:1040–6.
∗Nichols DG, Cameron DE, Greeley WJ, et al. Critical heart disease in infants and children. St Louis: Mosby; 1995.
∗Pearlman SA, Higgins S, Eppes S, et al. Infective endocarditis in the premature infant. Clin Pediatr 1998;37(12):741–6.
∗Keren A, Popp RL. Assignment of patients into the classification of cardiomyopathies. Circulation 1992;86:1622–1633.
†Gilbert-Barness E. Review: metabolic cardiomyopathy and conduction system defects in children. Ann Clin Lab Sci 2004;34:15–34.
†Towbin J, Lowe AM, Colan SD, et al. Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA 2006;296:1867–76.
§Daubney PEF, Nugent AW, Chondros P, et al. Clinical features and outcomes of childhood dilated cardiomyopathy. Results from a national population based study. Circulation 2006;114:2671–78.
∗Becker AE. Primary heart tumors in the pediatric age group: a review of salient pathologic features relevant for clinicians. Pediatr Cardiol 2000;21:317–23.
†Isaacs H Jr. Fetal and neonatal cardiac tumors. Pediatr Cardiol 2004;25:252–73.
[/level-membership-for-neonatal-and-perinatal-medicine-category][not-level-membership-for-neonatal-and-perinatal-medicine-category]Chapter 6
Cardiology Secrets
History of Pediatric Cardiology
The first successful ligation of a PDA was by Gross and Hubbard in 1938.
Christiaan Barnard performed the first cardiac transplantation in South Africa in 1967. Infant heart transplantation was attempted unsuccessfully 3 days later by Adrian Kantrowitz in New York City. Neonatal heart transplantation would not be achieved, however, until November 15, 1985, by Leonard Bailey at Loma Linda Medical Center. ∗†
Fetal Echocardiography and Prenatal Conditions that Can Contribute to Neonatal Heart Disease
Family history of congenital heart disease
Metabolic disorders (diabetes, phenylketonuria)
Exposure to prostaglandin synthase inhibitors (ibuprofen, salicylic acid)
Autoimmune disease (systemic lupus erythematosus, Sjögren syndrome)
Familial inherited disorders (e.g., Marfan syndrome, Noonan syndrome)
Abnormal obstetric ultrasound screen
Increased first trimester nuchal translucency
5. What is the incidence of congenital heart disease? What is the recurrence risk if a previous child has congenital heart disease?
Table 6-1 lists common genetic and chromosomal syndromes associated with congenital heart disease.
TABLE 6-1
COMMON GENETIC OR CHROMOSOMAL SYNDROMES ASSOCIATED WITH CONGENITAL HEART DISEASE
| GENETIC OR CHROMOSOMAL SYNDROME | COMMON CARDIAC ANATOMIC LESION |
| Apert syndrome | Ventricular septal defect, coarctation of the aorta, tetralogy of Fallot |
| Beckwith–Wiedemann syndrome | Atrial septal defect, ventricular septal defect, hypertrophic cardiomyopathy |
| CHARGE syndrome | Endocardial cushion defect, ventricular septal defect, double outlet right ventricle, tetralogy of Fallot |
| DiGeorge syndrome | Interrupted aortic arch, truncus arteriosus, tetralogy of Fallot, right aortic arch, ventricular septal defect, aberrant right subclavian artery |
| Ellis–van Creveld syndrome | Atrial septal defect, single/common atrium |
| Holt–Oram syndrome | Atrial septal defect, ventricular septal defect |
| Kartagener syndrome | Mirror image dextrocardia |
| Marfan syndrome | Dilated aortic root, mitral valve prolapse, tricuspid valve prolapse |
| Neurofibromatosis | Atrial septal defect, coarctation of the aorta, interrupted aortic arch, pulmonic stenosis, ventricular septal defect, complete heart block, hypertrophic cardiomyopathy |
| Noonan syndrome | Pulmonary stenosis, hypertrophic cardiomyopathy, tetralogy of Fallot |
| Pentalogy of Cantrell | Atrial septal defect, ventricular septal defect, total anomalous pulmonary venous drainage, tetralogy of Fallot, ectopia cordis |
| Pierre Robin syndrome | Pulmonary stenosis, atrial septal defect |
| Thrombocytopenia absent radius (TAR) syndrome | Atrial septal defect, tetralogy of Fallot |
| Treacher Collins syndrome | Ventricular septal defect, atrial septal defect |
| Tuberous sclerosis | Rhabdomyoma, angioma, coarctation of the aorta, interrupted aortic arch |
| Trisomy 13 | Ventricular septal defect, atrial septal defect, endocardial cushion defect, tetralogy of Fallot |
| Trisomy 18 | Bicuspid aortic valve, pulmonic stenosis, ventricular septal defect, atrial septal defect, endocardial cushion defect, polyvalvular thickening |
| Trisomy 21 | Endocardial cushion defect, ventricular septal defect, atrial septal defect, tetralogy of Fallot, coarctation of the aorta |
| Turner syndrome | Bicuspid aortic valve, coarctation of the aorta, aortic stenosis, ventricular septal defect, atrial septal defect |
| VACTERL syndrome | Ventricular septal defect, atrial septal defect, tetralogy of Fallot |
| Williams syndrome | Supravalvular aortic or pulmonic stenosis |
| Wolf–Hirschhorn syndrome | Atrial septal defect, ventricular septal defect |
Adapted from Drose J. Fetal echocardiography. 1st ed. Philadelphia: Saunders; 1988.
Table 6-2 lists teratogens known to cause congenital heart disease. ∗†‡
TABLE 6-2
TERATOGENS THAT CAUSE CONGENITAL HEART DISEASE
| TERATOGEN | COMMON CARDIAC ANATOMIC LESION |
| Fetal alcohol syndrome | Ventricular septal defect, atrial septal defect, tetralogy of Fallot, coarctation of the aorta |
| Fetal hydantoin (Dilantin) syndrome | Ventricular septal defect, tetralogy of Fallot, pulmonic stenosis, patent ductus arteriosus, atrial septal defect, coarctation of the aorta |
| Fetal trimethadione syndrome | Ventricular septal defect, d-transposition of the great vessels, tetralogy of Fallot, hypoplastic left heart syndrome, double outlet right ventricle, pulmonary atresia, atrial septal defect, aortic stenosis, pulmonic stenosis |
| Fetal carbamazepine syndrome | Ventricular septal defect, tetralogy of Fallot |
| Valproic acid | Ventricular septal defect, coarctation of the aorta, interrupted aortic arch, tetralogy of Fallot, hypoplastic left heart syndrome, aortic stenosis, atrial septal defect, pulmonary atresia |
| Retinoic acid embryopathy | Conotruncal malformations |
| Thalidomide embyropathy | Conotruncal malformations |
| Maternal phenylketonuria (PKU) (fetal effects) | Tetralogy of Fallot, ventricular septal defect, coarctation of the aorta |
| Maternal systemic lupus erythematosus/Sjögren syndrome (fetal effects) | Complete congenital heart block, dilated cardiomyopathy |
| Fetal rubella syndrome | Patent ductus arteriosus, peripheral pulmonary artery stenosis |
| Maternal diabetes | Hypertrophic cardiomyopathy, conotruncal abnormalities |
Table adapted from Drose J. Fetal echocardiography. 1st ed. Philadelphia: Saunders; 1988.
Pulmonary vascular resistance = pulmonary artery pressure/pulmonary blood flow
The very high PVR in the fetus results in low pulmonary blood flow, with diversion of the right ventricular blood away from the pulmonary vascular bed and towards the systemic vascular bed through the ductus arteriosus. This dynamic results in increased thickness of the muscular medial layer of the pulmonary arteries. As a newborn’s lungs expand, they inspire oxygen, which is a potent vasodilator that causes the pulmonary vascular resistance to fall. The rise in the PaO2 causes the smooth muscle in the pulmonary circulation to relax, and vasodilation occurs. The PVR falls dramatically during the first week of life. As the smooth muscle of the pulmonary arteries continues to thin, the PVR continues to fall, reaching a nadir at 6 to 8 weeks after delivery. ∗
14. Name three scenarios in which a right-to-left shunt is seen in the infant with a PDA.
16. How can the modified Bernoulli equation be useful in interpreting pulmonary artery pressures in a neonate?
This formula allows the pulmonary artery pressure to be estimated. Two examples are as follows:
If the velocity across a tricuspid regurgitant jet is 4 m/sec, the calculated right ventricular pressure will be 64 mmHg [P = 4 × (V max2) which is 4 × (42)= 64 mmHg]. By adding an estimation of right atrial pressure (usually use 5 mmHg), the estimated right ventricular pressure (as well as a pulmonary artery pressure in the absence of pulmonary stenosis) would be 69 mmHg. Therefore this patient has pulmonary hypertension.
17. What is the definition of the echocardiographic term shortening fraction?
The formula to measure shortening fraction is as follows:
22. During postnatal life, as PVR falls, what changes can be expected in the magnitude of a left-to-right shunt of a large VSD?
With a large-pressure nonrestrictive VSD, the right ventricular and pulmonary artery pressures remain high. The right and left ventricular pressures remain equal with a nonrestrictive VSD. As the pressure across the pulmonary vascular bed will not change and the PVR falls, the pulmonary blood flow (PBF) will increase because there is an inverse relationship between PBF and PVR. PBF = Δ Pressure across the pulmonary vascular bed/PVR. Therefore the magnitude of the left-to-right shunt will increase. ∗†
23. What are the common innocent murmurs heard in the newborn period?
Peripheral pulmonary stenosis (PPS): This murmur is a soft, grade 1-2/6 systolic ejection murmur, heard best at the left upper sternal border, with radiation to both axillae and the back. It will usually disappear or be much softer at 6 to 8 weeks after delivery in normal infants and completely disappear by 6 months after delivery.
KEY POINTS: PHYSIOLOGY OF THE CYANOTIC NEONATE
1. The hyperoxia test does not rule out some common mixing congenital heart lesions.
2. Reversed differential cyanosis can be seen when the postductal saturations are higher than the preductal saturations. This sign can be seen in d-transposition of the great arteries with a coarctation of the aorta, d-transposition of the great arteries with interrupted aortic arch, and d-transposition of the great arteries with pulmonary hypertension.
28. What is differential cyanosis, and what are the implications (pink upper body and blue lower body)?
The oxygen dissociation curve shows the relationship between oxygen saturation (%) and the partial pressure of oxygen, PO2, in mmHg. This relationship is a sigmoid-shaped curve, with it being fairly flat in the upper range of oxygen saturation (above 85%). Blood pH, temperature, PCO2, 2,3-diphosphoblycerate, and the type of hemoglobin influence the relationship between oxygen saturation and the partial pressure of oxygen ( Fig. 6-2).
Figure 6-2 Oxygen dissociation curve for normal adult human blood (solid line) and curves showing the effect of either an increase (↑) or a decrease (↓) in hydrogen ion concentration, body temperature, PCO2, and 2,3-DPG level (dotted lines). (From Gessner I, Vitorica B: Pediatric cardiology: a problem-oriented approach. Philadelphia: Saunders; 1993. p. 98.)
32. What is a hyperoxia test, and how is it used in differentiating pulmonary and cardiac causes of cyanosis?
A hyperoxia test attempts to differentiate between pulmonary disease with V/Q mismatch and cyanotic congenital heart disease. Initially, one measures the oxygen saturation in room air. If the oxygen saturation is low, the patient should be placed in 100% FiO2. The patient with pulmonary disease will show an increase in PO2 (to a variable degree). In the patient with a fixed intracardiac mixing lesion, the PO2 does not change significantly. A preductal and postductal arterial blood gas result should be obtained. A preductal arterial blood gas result can be obtained from the right radial artery. A postductal arterial blood gas can be obtained either from an umbilical artery or from a lower extremity artery. In pulmonary disease the preductal arterial PO2 in 100% FiO2 usually exceeds 150 mmHg. If the ductus arteriosus is patent and a right-to-left ductal shunt occurs because of high PVR, the postductal PO2 will be lower than the preductal PO2. In addition, the arterial PCO2 is elevated relative to the patient’s respiratory effort.
33. Which critical cyanotic lesions may not be excluded if the hyperoxia test yields a PO2 after 10 minutes greater than 150 torr?
34. What are the current guidelines for pulse oximetry screening of newborns to detect critical cyanotic congenital heart disease?
As published in Pediatrics in 2011, pulse oximetry assessment of the right hand and a foot is recommended before discharge of all newborns from the hospital. If the oxygen saturation is less than 90% in the right hand or foot, the test is positive and further evaluation is needed. If the oxygen saturation is 95% or greater in the right hand or foot and the difference is 3% or less between the two sites, then the test is negative. If the oxygen saturation is 90% to 95% or greater than 3% difference between the two sites is found, then the test should be repeated in 1 hour up to two times. It is considered a positive screen if these findings are reproduced twice. ∗†‡
Ductal-dependent abnormalities:
Non–ductal-dependent abnormalities:
Total anomalous pulmonary venous return
AV septal defect (endocardial cushion defect)
Myocardial dysfunction (cardiomyopathy)
38. What are some heart lesions that can cause heart failure in the infant beyond the newborn period?
Obstruction to systemic blood flow:
Total anomalous pulmonary venous return, without obstruction
Single ventricle with excessive PBF
Myocardial dysfunction/pericardial disease:
Anomalous left coronary artery
Compensatory mechanisms include increased heart rate, enhanced stroke volume (Frank–Starling mechanism), sympathetic nerve activation (increased sympathetic tone, renin-angiotensin system activation), increased 2,3-diphosphoglycerate, increased atrial natriuretic peptides, and myocardial hypertrophy. ∗†
KEY POINTS: TREATMENT OF HYPOTENSION ON THE NEONATE
1. Dopamine and dobutamine have different physiologic effects in the newborn.
2. Dopamine is a good inotrope to use (depending on dose) if there is a risk of renal ischemia, low cardiac output, or hypotension with decreased systemic vascular resistance.
3. Dobutamine is a good inotrope to use for low cardiac output in patients at risk for myocardial ischemia, pulmonary hypertension, and left ventricular diastolic dysfunction.
42. What is the most frequent primary cause of hypotension in the preterm neonate in the immediate postnatal period?
44. What are the mechanisms of action of the cardiovascular effects of dopamine in the preterm neonate?
45. What are the pros and cons of dopamine and dobutamine in the treatment of hypotension in a preterm neonate?
Dopamine may be the preferred inotrope in the treatment of hypotension secondary to neonatal sepsis because it increases peripheral vascular contractility. However, dobutamine may be preferred in treatment of hypotension secondary to cardiomyopathy, which frequently occurs with perinatal asphyxia. Whereas dopamine has a greater increase in arterial blood pressure than dobutamine, dobutamine has been shown to increase superior vena cava blood flow and may improve end-organ perfusion to a great extent. ∗†‡
KEY POINTS: CYANOTIC CONGENITAL HEART DISEASE
1. An unrestrictive atrial communication is crucial in certain cyanotic congenital heart lesions to provide mixing (right-to-left) and/or cardiac output.
2. The most common cyanotic lesion presenting in the newborn period is d-transposition of the great arteries.
3. An electrocardiogram with a left axis shift in a cyanotic newborn baby may be consistent with tricuspid atresia.
4. Cyanotic CHD does not include cyanosis secondary to intrapulmonary right-to-left shunting.
47. What are the five Ts of cyanotic congenital heart disease? What are other principal cyanotic heart lesions that do not begin with a T?
The screening is performed for the following reasons:
Prenatal screening depicts less than 50% of all CHD.
Routine newborn examination misses more than 50% of all infants with CHD.
Of CHD-related deaths in the first week of life, 25% of cases do not have a diagnosis of CHD.
53. In infants with d-transposition of the great arteries, how does a balloon atrial septostomy improve systemic oxygen saturation?
Progressive hypoxemia from poor intercirculatory mixing is a medical emergency. An atrial septostomy may be necessary to improve hypoxemia even after Prostaglandin E1 (PGE) has maintained ductal patency. The balloon atrial septostomy permits unrestricted bidirectional mixing of fully saturated blood in the left atrium with desaturated blood in the right atrium to achieve a higher net saturation of blood in the systemic circulation. After this procedure, patency of the ductus is generally no longer essential. Variations in oxygen saturation can be expected, although mixing is usually excellent.
57. What are the different types of TAPVR?
Supracardiac: represents 50% of all total anomalous pulmonary venous return. The pulmonary veins usually drain into the right superior vena cava via a left vertical vein. Although generally nonobstructive, they can be obstructed in some cases (the vertical vein is “pinched” between the left pulmonary artery and the left bronchus, or at superior vena cava insertion).
Mixed: represents 10% of all total anomalous pulmonary venous return. It represents a combination of the other types of TAPVR ( Fig. 6-3).
[/not-level-membership-for-neonatal-and-perinatal-medicine-category]
